ALTERATIONS OF ADRENAL FUNCTION
Disorders of the Adrenal Cortex
Disorders of the adrenal cortex are related to either hyperfunction or hypofunction. Hyperfunction that causes increased levels of circulating cortisol leads to Cushing disease, or Cushing syndrome; hyperfunction that causes increased secretion of adrenal androgens and estrogens leads to virilization or feminization; and hyperfunction that causes increased levels of aldosterone leads to hyperaldosteronism, which may be primary or secondary. Hypofunction of the adrenal cortex leads to Addison disease.
Adrenocortical Hyperfunction: Cushing Disease, Cushing Syndrome
Cushing disease is caused by excessive anterior pituitary secretion of ACTH. Cushing syndrome is an uncommon disorder and occurs whenever there is an excessive level of cortisol regardless of the cause. Cushing syndrome is the most common complication of Cushing disease. Cushing-like syndrome may develop as a result of the exogenous administration of glucocorticoids.244 Endogenous Cushing syndrome is divided into two main forms: corticotropin dependent and corticotropin independent. Corticotropin-dependent Cushing syndrome is the most common (80% to 85% of cases), primarily because of ACTH-secreting pituitary tumors and is more common in women. The remaining cases are usually derived from ACTH-secreting carcinoid tumors or small-cell carcinoma of the lung. Corticotropin-independent Cushing syndrome is less common (15% to 20% of cases) and is usually caused by an adrenal tumor or, more rarely, corticotropin-independent macronodular adrenal hyperplasia, primary pigmented nodular adrenal disease.245 Adrenal tumors, rather than pituitary tumors, are more common in children, especially girls.246 In Cushing disease there is a loss of normal feedback inhibition by cortisol because there appears to be a higher set point for cortisol feedback on corticotropin-releasing hormone (CRH) and ACTH secretion.
PATHOPHYSIOLOGY Although the origin of Cushing disease remains incompletely understood, the vast majority of individuals with Cushing disease have a pituitary microadenoma, which secretes ACTH.247 Ectopic ACTH-secreting tumors are nonpituitary tumors that synthesize and hypersecrete ACTH, leading to hypercortisolism. Some tumors may hypersecrete CRH, which results in oversecretion of ACTH from the pituitary. Tumors associated with episodic secretion of ACTH and hypercortisolism include small cell carcinomas of the lung, thymoma, pancreatic cell tumors, carcinoid tumors, medullary carcinoma of the thyroid, and pheochromocytoma tumors. Even though the secretion of ectopic ACTH from the neoplasm is not under hypothalamic-pituitary control, the normal pituitary release of ACTH is inhibited by the elevated levels of cortisol. However, cortisol fails to inhibit the release of ACTH from the ectopic source. Autonomous secretion of cortisol can be the result of either an adrenal adenoma or, less commonly, adrenal cortical carcinoma.248
Elevated cortisol levels suppress CRH and ACTH release secretion from the hypothalamus and anterior pituitary, respectively, which leads to low levels of ACTH. Low levels of ACTH cause atrophy of the remaining normal portions of the adrenal cortex, which over time will alter the cortisol-secreting activity of normal cells. The normal diurnal variation in cortisol secretion is lost in individuals with hypercortisolism regardless of the underlying cause.
CLINICAL MANIFESTATIONS Most of the clinical signs and symptoms of Cushing syndrome are caused by hypercortisolism.249 Weight gain is the most common feature and results from the accumulation of adipose tissue in the trunk, facial, and cervical areas. These characteristic patterns of fat deposition have been described as “truncal [central] obesity,” “moon face,” and “buffalo hump” (Figures 21-21 and 21-22). Transient weight gain from sodium and water retention may be present because of the mineralocorticoid effects of cortisol, exhibited when cortisol is present in high levels.
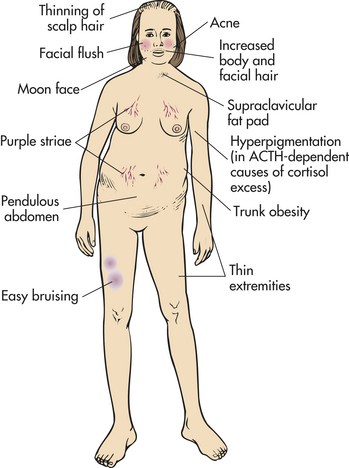
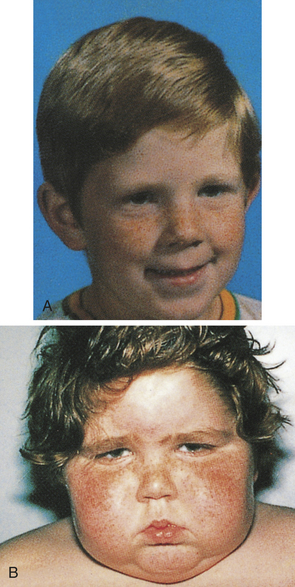
Figure 21-22 Cushing syndrome. A, Patient before onset of Cushing syndrome. B, Patient 4 months later. Moon facies is clearly demonstrated. (From Zitelli BJ, Davis HW: Atlas of pediatric physical diagnosis, ed 4, London, 2002, Gower.)
Glucose intolerance occurs because of cortisol-induced insulin resistance and increased gluconeogenesis and glycogen storage by the liver. Overt diabetes mellitus develops in approximately 20% of individuals with hypercortisolism. Polyuria, which is sometimes seen in hypercortisolism, is a manifestation of hyperglycemia and resultant glycosuria.
Protein wasting is commonly observed in hypercortisolism and is caused by the catabolic effects of cortisol on peripheral tissues. Muscle wasting leads to muscle weakness and is especially obvious in the muscles of the extremities with thinning of the limbs. Hypercortisolism increases bone resorption, inhibits bone formation, decreases intestinal calcium absorption, and increases renal calcium excretion. This leads to osteoporosis, with pathologic fractures, vertebral compression fractures, bone and back pain, kyphosis, and reduced height. Hypercalciuria may result in renal stones, which are experienced by approximately 20% of individuals with this disease. Loss of collagen also leads to thin, weakened integumentary tissues through which capillaries are more visible; the tissues are easily stretched by adipose deposits. Together these changes account for the characteristic purple striae most often observed in the truncal area. Loss of collagenous support around small vessels makes them susceptible to rupture, leading to easy bruising, even with minor trauma. Thin, atrophied skin is also easily damaged, leading to skin breaks and ulcerations.
Hyperpigmentation in Cushing syndrome is associated with very high serum levels of ACTH, believed to be caused by increased melanocyte-stimulating hormones resulting from excess conversion of pro-opiomelanocortin when ACTH is elevated.250 The pigmentation involves the mucous membranes, hair, and skin, all of which acquire a characteristic brownish or bronze color.
Cortisol has a permissive effect on the actions of the catecholamines. With elevated cortisol levels, vascular sensitivity to catecholamines is increased significantly, leading to vasoconstriction and hypertension. Metabolic syndrome with abdominal obesity, hypertension, glucose intolerance, and dyslipidemias is a common complication (see p. 751). Chronically elevated cortisol levels also cause suppression of the immune system and increased susceptibility to infections. Consequently, individuals with hypercortisolism experience poor wound healing and are particularly susceptible to superficial fungal infections.
Approximately 50% of individuals with Cushing syndrome experience alterations in their mental status and include effects of cortisol on hippocampal neurons and the implications for learning and memory and other neurologic functions when cortisol is elevated. These may range from irritability and depression to severe psychiatric disturbances such as schizophrenia.251,252 The effects of glucocorticoids on mood are complex.
Females may experience symptoms of increased adrenal androgen levels, increased hair growth (especially facial hair), acne, and oligomenorrhea. Androgen levels rarely become high enough to cause changes of the voice, recession of the hairline, and clitoral hypertrophy unless an adrenal carcinoma is involved. Infertility is common in men and women.253 Routine laboratory examinations may reveal hyperglycemia, glycosuria, hypokalemia, and metabolic alkalosis.
EVALUATION AND TREATMENT A variety of laboratory tests must be used to diagnose hypercortisolism and to determine the underlying disorder.254 These include urinary free cortisol higher than 50 mcg per 24 hours, abnormal dexamethasone suppressibility of either urinary or serum cortisol, and simultaneous measurement of ACTH and cortisol. Late evening salivary cortisol levels are used as a screening test and to document alterations in the diurnal variation of cortisol. Visualizing procedures, including pituitary MRI or abdominal scanning, are essential in the evaluation. Selective catheterization of the veins draining the pituitary (inferior petrosal sinus sampling) is very helpful in determining the cause of hypercortisolism and in localizing pituitary tumors.249 The diagnosis and evaluation of hypercortisolism is one of the most challenging problems in endocrinology.255
Without treatment, approximately 50% of individuals with Cushing syndrome die within 5 years of onset. Major causes of death are overwhelming infection, suicide, complications from generalized arteriosclerosis, and hypertensive disease. Treatment is specific for the cause of hypercorticoadrenalism and includes medication, radiation therapy, and surgery.256,257 Therefore, it is essential to differentiate between pituitary, adrenal, and ectopic causes of the hypercortisolism.
Hyperaldosteronism
Hyperaldosteronism is characterized by excessive aldosterone secretion by the adrenal cortex. The excessive secretion can result from a primary adrenal disorder, such as an aldosterone-secreting adenoma, or from excessive stimulation of the normal adrenal cortex by substances such as angiotensin II, ACTH, or elevated potassium. Both primary and secondary forms of hyperaldosteronism can occur in individuals. Primary aldosteronism (primary hyperaldosteronism) refers to an excessive secretion of aldosterone caused by an abnormality of the adrenal cortex. Several different subtypes have been identified.258 Secondary aldosteronism (secondary hyperaldosteronism) involves excessive aldosterone secretion from an extra-adrenal stimulus, most often angiotensin II through a renin-dependent mechanism.
Primary aldosteronism (Conn disease, primary hyperaldosteronism) presents a clinical picture of hypertension, hypokalemia, renal potassium wasting, and neuromuscular manifestations. The most common cause of primary aldosteronism are a benign, single aldosterone-producing adrenal adenoma (30% to 40% of cases) and idiopathic bilateral adrenal hyperplasia (60%). Other rare tumors account for the remainder of cases.259 The incidence of primary hyperaldosteronism is not known, but 5% to 13% of individuals with hypertension have primary aldosteronism.260
Secondary aldosteronism can be expected to result from sustained elevated renin release and activation of angiotensin II because aldosterone secretion is normally stimulated by the renin-angiotensin system. (Factors that affect renin and aldosterone secretion are summarized in Table 21-15.) Increased renin-angiotensin secretion occurs in a variety of situations. In general, these include decreased circulating blood volume (e.g., in dehydration, shock, or hypoalbuminemia) and decreased delivery of blood to the kidneys (e.g., renal artery stenosis, heart failure, or hepatic cirrhosis). In many of these instances the activation of the renin-angiotensin system and subsequent aldosterone secretion may be seen as compensatory, although in some instances (e.g., congestive heart failure) the increased circulating volume may further worsen the condition.
Table 21-15
Physiologic Factors Affecting Renin and Aldosterone Secretion
| Factors | Renin Secretion |
| Age | Highest in infants; lowest in older adults |
| Menstrual cycle | Highest in luteal phase (see Chapter 22) |
| Sodium intake | Increased by salt restriction |
| Decreased by salt loading | |
| Potassium status | Increased by K+ excess |
| Posture | Increased with erect posture |
| Sympathetic nervous system | Renin increased by catecholamine stimulation |
| Time of sampling | Highest before noon; lowest in evening |
Increased estrogen levels associated with pregnancy and use of oral contraceptives also increase renin-angiotensin levels, apparently by stimulating renin substrate production by the liver. These pregnancy-induced changes, however, may represent adaptation to pregnancy and are therefore not representative pathophysiologic alterations.
Other causes of secondary hyperaldosteronism include Bartter syndrome, which is a heterogeneous autosomal recessive disorder associated with reduced or absent salt transport by the thick ascending limb of the loop of Henle. Symptoms include salt wasting and low blood pressure, hypokalemia, metabolic acidosis, and hypercalciuria.261 Renin-secreting tumors of the kidney also cause secondary hyperaldosteron-ism. Diuretic use is perhaps the most common cause of secondary hyperaldosteronism. (Renal disorders are discussed in Chapter 36.) Apparent mineralocorticoid excess can be seen in individuals who consume excess licorice (glycyrrhizic acid) or chewing tobacco.
PATHOPHYSIOLOGY In primary hyperaldosteronism, pathophysiologic alterations are caused by excessive aldosterone secretion and the fluid and electrolyte imbalances that ensue. Hyperaldosteronism promotes increased sodium reabsorption with corresponding hypervolemia (see Chapter 3). The extracellular fluid volume overload and suppression of normal feedback mechanisms of renin secretion are characteristic of primary disorders.
Edema usually does not occur with primary aldosteronism, possibly because of the renal tubular “escape” phenomenon that is activated in chronic hyperaldosteronism. The escape phenomenon changes or resets the rate of sodium excretion and prevents more severe sodium retention. The escape phenomenon operates in the proximal tubules and causes additional sodium to pass to the distal tubules, where the sodium is, to some extent, reabsorbed in exchange for potassium. This mechanism, while protecting from excessive sodium reabsorption and edema, increases urinary losses of potassium and causes hypokalemia (Figure 21-23). Metabolic syndrome (hypertension, obesity, dyslipidemia, and hyperglycemia) also is associated with primary aldosteronism and may be related to insulin resistance that is related to aldosterone action on insulin receptor function.262
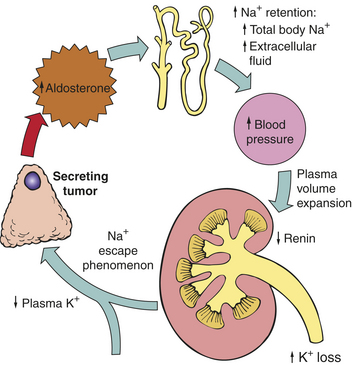
Figure 21-23 Primary hyperaldosteronism. Pathophysiology of mineralocorticoid excess syndromes in primary hyperaldosteronism.
In secondary hyperaldosteronism the effect of increased extracellular volume on renin secretion may vary. If renin secretion is being stimulated by variables other than pressure-initiated cellular changes at the juxtaglomerular apparatus (see Chapter 35), increased circulating blood volume may not decrease renin secretion through feedback mechanisms. This physiologic process is normal in pregnancy and related to increased plasma estrogen.
In Bartter syndrome a state of hypokalemia develops because of defective renal tubular reabsorptive mechanisms. The hypokalemic state may induce the formation of prostaglandins (especially PGE2) by the renal cells, which stimulates renin and hence aldosterone secretion. The stimulatory effect on aldosterone secretion is offset somewhat by the aldosterone-suppressing effects of hypokalemia.
Potassium secretion also is promoted by aldosterone, so that with excessive circulating levels of aldosterone, hypokalemia occurs (see Chapter 3). In hyperaldosteronism, hypokalemic alkalosis, changes in myocardial conduction, and skeletal muscle alterations may be seen, particularly with severe potassium depletion (i.e., the renal tubules may become insensitive to ADH, thus promoting excessive loss of free water). Rarely, this may result in mild hypernatremia because water is not able to follow the sodium that is reabsorbed.
CLINICAL MANIFESTATIONS Hypertension, and less commonly hypokalemia, and metabolic alkalosis are
the hallmarks of hyperaldosteronism.263 Hypertension may result from increased intravascular volume or from a state of aldosterone-mediated vasoconstriction, although the latter mechanism requires very high levels of aldosterone. If hypertension is sustained, the long-term effects of elevated arterial pressure become evident, which include the development of left ventricular dilation and hypertrophy. Because of the increased arterial pressure, renin secretion is typically suppressed, although it is elevated in secondary hyperaldosteronism, which provides a means to clearly differentiate between these conditions.
Aldosterone-stimulated potassium loss can be variable. Serum potassium levels below 3.0 mEq/L result in the typical manifestations of hypokalemia. Hypokalemic alkalosis is caused by the movement of potassium from the intercellular to extracellular space in exchange for hydrogen ions as well as renal loss of hydrogen ions to facilitate sodium reabsorption (see Chapter 3).
EVALUATION AND TREATMENT A variety of clinical and laboratory measurements are useful in the assessment of hyper aldosteronism.264 These include blood pressure, serum and urinary electrolyte levels, serum and urinary levels of aldosterone and renin, plasma aldosterone concentration to plasma renin activity ratio, and aldosterone suppression testing. Blood pressure is elevated, serum sodium may be normal or elevated, serum potassium may be normal or depressed, and urinary potassium is elevated (i.e., more than 30 mmol/day). Serum aldosterone, as measured by radioimmunoassay, usually is greater than 15 ng/dl. Plasma renin activity is generally less than 1 ng/ml/hr for individuals with primary aldosteronism. A plasma aldosterone (in nanograms/dl)-to-renin (in nanograms/ml/hr) activity ratio of ≥ 20ng/dl is very suspicious for primary hyperaldosteronism. Serum aldosterone and plasma renin activity both must be measured under controlled situations and after careful dietary regulation of sodium and potassium intake (see Table 21-15). Aldosterone suppression testing commonly is accomplished with fludrocortisone acetate (Florinef) or salt loading. Imaging techniques, such as CT and nuclear magnetic resonance (NMR), may be used to localize an aldosterone-secreting adenoma. Selective venous catheterization of both adrenal veins is also useful.
Treatment includes management of hypertension and hypokalemia, as well as correction of any underlying causal abnormalities. If an aldosterone-secreting adenoma is present, it is generally approached surgically; however, medical management with spironolactone or eplerenone, a new drug without the side effects of spironolactone, is a viable option in complicated cases.265
Adrenocortical Hypofunction
Hypocortisolism (low levels of cortisol secretion) develops because of either inadequate stimulation of the adrenal glands by ACTH or a primary inability of the adrenals to produce and secrete the adrenocortical hormones. In some syndromes, however, there is partial dysfunction of the adrenal cortex so that only synthesis of cortisol and aldosterone or the adrenal androgens is affected. Hypofunction of the adrenal cortex may affect glucocorticoid or mineralocorticoid secretion or a combination of both.
Primary adrenal insufficiency is termed Addison disease. Addison disease is relatively rare, occurring most often in adults 30 to 60 years of age, although it may appear at any time throughout the life span. Addison disease, caused by autoimmune mechanisms, is more common in women.
The most common cause of Addison disease in the United States is autoimmune destruction of the adrenal cortex. Other causes include infections (tuberculosis, fungal, human immunodeficiency virus [HIV]), infiltrative diseases (amyloidosis, metastatic carcinoma), or bilateral adrenal hemorrhage. Adrenoleukodystrophy and adrenomyeloneuropathy are two rare types of X-linked adrenal deficiency that lead to symptoms of hypocortisolism and progressive neurologic symptoms.
PATHOPHYSIOLOGY Addison disease is characterized by elevated serum ACTH levels with inadequate corticosteroid synthesis and output. Before clinical manifestations of hypocortisolism are evident, more than 90% of total adrenocortical tissue must be destroyed.
Idiopathic Addison Disease: Idiopathic Addison disease (organ-specific autoimmune adrenalitis), which causes adrenal atrophy and hypofunction, generally is recognized as an organ-specific autoimmune disease. (Autoimmunity is discussed in Chapter 8.) It may occur in childhood (type 1) or adulthood (type 2). Autoantibodies are present in 50% to 70% of individuals with idiopathic Addison disease, and this percentage increases in younger persons and in those with other autoimmune diseases. The autoantibodies are specific for the cells of the adrenal cortex and 21-hydroxylase (the enzyme that produces cortisol and aldosterone).266 A combination of cell membrane and cytoplasmic antibodies and cell-mediated immune mechanisms contributes to the pathologic findings of the disease. Apparently a genetic defect in immune surveillance mechanisms causes a deficiency of immune-suppressor cells. This deficiency allows the proliferation of immunocytes directed against specific antigens within the adrenocortical cells.
Idiopathic Addison disease often is associated with other autoimmune diseases and in such cases is known as autoimmune polyendocrine syndrome (APS). APSI (APS type I) is inherited as autosomal recessive and includes Addison disease, hypoparathyroidism, mucocutaneous candidias, and other less common symptoms. APSII (APS type II) is more common and involves Addision disease, immune thyroid disease, diabetes mellitus, celiac disease, and hypogonadism.267 (Mechanisms of inheritance are described in Chapter 4.)
The adrenal glands in idiopathic Addison disease are smaller than normal and may be misshapen. Microscopically, gland atrophy is evident throughout the cortex, although the medulla appears intact. Extensive diffuse cortical lymphocytic infiltrate supports the immune component of the disease process.
Secondary Hypocortisolism: Secondary hypocortisolism is characterized by low to absent ACTH levels, which cause inadequate adrenal stimulation, adrenal atrophy, and ultimately decreased corticosteroidogenesis. The exogenous administration of glucocorticoids for nonendocrine disease results in this form of hypocortisolism. Successful surgical removal of cortisol-secreting tumors also results in postoperative hypocortisolism. In these cases, increased glucocorticoid levels suppress ACTH production through normal feedback mechanisms. With decreased ACTH levels, corticosteroid synthesis by remaining adrenal tissue is suppressed. Pituitary hypofunction (as occurs in postpartum pituitary infarction [Sheehan syndrome] and panhypopituitarism, hypophysectomy, or isolated ACTH deficiency) causes inadequate ACTH production and secretion and absence of pituitary responsiveness to normal stimulatory mechanisms. In all instances of low ACTH levels, adrenal atrophy occurs, and endogenous adrenal steroidogenesis is depressed.
Clinical manifestations of secondary hypocortisolism are similar to those of Addison disease. One difference is that with the typically low levels of ACTH seen in secondary hypocortisolism, hyperpigmentation does not occur. Second, the renin-angiotensin system is usually normal in these individuals; therefore, aldosterone and potassium levels also tend to be normal.
CLINICAL MANIFESTATIONS The symptoms of Addison disease are primarily a result of hypocortisolism and hypoaldosteronism and include weakness and fatigue, anorexia, weight loss, nausea, diarrhea, and orthostatic hypotension. Hyperpigmentation is associated with elevated ACTH levels. Decreased adrenal androgen secretion is usually not clinically obvious in men because the adrenals are not a major source of male androgens. Women may experience a loss of some secondary sex characteristics, such as pubic and axillary hair, normally maintained by the adrenal androgens.268 Disturbances in mood and motivation are common.269 The symptoms of Addison disease are summarized in Table 21-16.
Table 21-16
Clinical Manifestations and Pathophysiologic Mechanisms of Addison Disease
| Clinical Manifestations | Pathophysiologic Mechanism |
| Weakness and easy fatigability that worsens as the day progresses, seen especially after exposure to stressors | Not known, may be related to hypoglycemia, hypotension, decreased metabolism of proteins |
| Gastrointestinal disturbances: anorexia, nausea, vomiting, diarrhea, abdominal pain, weight loss | May be associated with celiac disease or electrolyte abnormalities |
| Hypoglycemia, manifested by fatigue, mental confusion, apathy, psychosis | Absence of cortisol leads to decreased gluconeogenesis, decreased glycogen storage by liver, decreased metabolism of proteins, increased insulin sensitivity |
| Hyperpigmentation | Elevations of ACTH that lead to stimulation of melanocytes |
| Vitiligo (white patchy areas of depigmented skin) | Autoimmune destruction of melanocytes |
| Addisonian crisis: severe hypotension and vascular collapse | Combined effects of hypocortisolism, hypoaldosteronism, extracellular volume depletion, and some precipitating stressor (e.g., infection, vomiting, diarrhea); decreased vasomotor tone caused by cortisol deficiency |
EVALUATION AND TREATMENT Serum and urine levels of cortisol are depressed with hypocortisolism. In primary adrenal insufficiency (Addison disease), ACTH levels are clearly elevated, and ACTH levels are low in secondary adrenal insufficiency. ACTH levels can be interpreted only in the face of a simultaneous measurement of cortisol. Individuals may develop azotemia caused by dehydration, and hyponatremia is common. Hyperkalemia is seen only in Addison disease, but hypoglycemia may be seen in hypocortisolism from any cause. Anemia, eosinophilia, and lymphocytosis are also common with symptoms of fatigue. The ACTH stimulation test may be used to evaluate adrenocortical function. This is achieved by administering ACTH and monitoring the serum cortisol levels.
The treatment of Addison disease involves glucocorticoid and possibly mineralocorticoid replacement therapy, together with dietary modifications. All individuals with hypocortisolism require lifetime daily glucocorticoid replacement therapy. In the event of acute stressors, additional cortisol must be administered to approximate the amount of cortisol that might be expected to be secreted if normal adrenal function were present (approximately 100 to 300 mg/day). Fludrocortisone is used for mineralocorticoid replacement therapy.270
The individual’s diet should include at least 150 mEq of sodium per day, with sodium intake increased in the event of excessive sweating or diarrhea. Treatment also must include correction of any underlying disorders.
Hypersecretion of Adrenal Androgens and Estrogens
Hypersecretion of adrenal androgens and estrogens may be caused by adrenal tumors, either adenomas or carcinomas, Cushing syndrome, or defects in steroid synthesis. The clinical syndrome that results depends on the hormone secreted, the gender of the individual, and the ages at which the hypersecretion was initiated. Hypersecretion of estrogens causes feminization, the development of female sex characteristics. Hypersecretion of androgens causes virilization, the development of male sex characteristics (Figure 21-24).
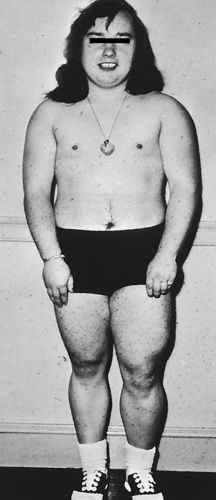
Figure 21-24 Virilization. Virilization of a young girl by an androgen-secreting tumor of the adrenal cortex. Masculine features include lack of breast development, increased muscle bulk, and hirsutism. (From Thibodeau GA: Anatomy & physiology, St Louis, 1987, Mosby.)
The effects of an estrogen-secreting tumor are most evident in men and result in gynecomastia (breast enlargement) (98% of cases), testicular atrophy, and decreased libido. In female children such tumors may lead to early development of secondary sex characteristics. An androgen-secreting tumor indicates changes more easily observed in women, including hirsutism, clitoral enlargement, deepening of the voice, amenorrhea, acne, and breast atrophy. In children, virilizing tumors promote precocious sexual development and bone aging. Treatment of androgen-secreting tumors usually involves surgical excision.
Disorders of the Adrenal Medulla
No known physiologic alterations are associated with hypofunction of the adrenal medulla. Bilateral adrenalectomy, for example, is followed by a rapid decrease in urinary excretion of epinephrine, but excretion of norepinephrine remains relatively stable. Pathophysiologic alterations are instead associated with hyperfunctioning of the adrenal medulla.
Adrenal Medulla Hyperfunction
Adrenomedullary hyperfunction is caused by tumors derived from the chromaffin cells of the adrenal medulla. These tumors, known as pheochromocytomas, secrete catecholamines on a continual or episodic basis (Figure 21-25). Less than 10% of these tumors are malignant; those that are malignant may metastasize to the lungs, liver, bones, or paraaortic lymph nodes. Most pheochromocytomas produce norepinephrine, although large tumors secrete epinephrine and norepinephrine.
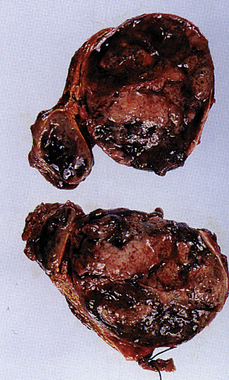
Figure 21-25 Pheochromocytoma. Gross appearance of adrenal pheochromocytoma. (From Rosai J: Akerman’s surgical pathology, ed 8, St Louis, 1996, Mosby.)
The true incidence of pheochromocytoma in the general population is not known. About one tenth of one percent of the adult hypertensive population has a pheochromocytoma.271 The tumors are most common in people 40 to 60 years of age, with men and women equally affected. Familial forms of the disease are associated with mutations in the RET proto-oncogene, the Von Hippel-Lindau (VHL) gene, a tumor-suppressor gene that encodes succinate dehydrogenase.272
PATHOPHYSIOLOGY Pheochromocytomas cause excessive production of catecholamines attributable to autonomous functioning of the tumor. Approximately 5% of people with pheochromocytomas have no symptoms because the tumor appears to be nonfunctioning; however, these tumors can release catecholamines in response to stressors, such as abdominal surgery.
CLINICAL MANIFESTATIONS The clinical manifestations of a pheochromocytoma are related to the chronic effects of catecholamine secretion and include persistent hypertension associated with diaphoresis, tachycardia, palpitations, and severe headache.273 Hypertension is a result of increased peripheral vascular resistance and may be sustained or paroxysmal. Headaches appear because of sudden changes in catecholamine levels in the blood, affecting cerebral blood flow. Hypermetabolism is related to chronic activation of sympathetic receptors in adipocytes, hepatocytes, and other tissues. Glucose intolerance may occur because of catecholamine-induced inhibition of insulin release by the pancreas. Complaints of warmth, heat intolerance, weight loss, and constipation are common despite a normal or an increased appetite.
An acute episode of hypertension related to hypersecretion of catecholamines may follow specific events. Exercise, excessive ingestion of tyrosine-containing foods (aged cheese, red wine, beer, yogurt), ingestion of caffeine-containing foods, external pressure on the tumor, and induction of anesthesia all can increase secretion of catecholamines by the tumor.
These tumors tend to be extremely vascular and can rupture. Such an event can cause massive and potentially fatal hemorrhage. Rupture of a pheochromocytoma is characterized by a sudden, unexplained decrease in blood pressure; sudden, severe abdominal pain; and a rigid abdomen.
EVALUATION AND TREATMENT A diagnosis of pheochromocytoma is made when increased catecholamine production is demonstrated in the blood or urine. Individuals with this disorder can have total urine catecholamine levels greater than 250 mg/day. After elevation of urinary or plasma catecholamines is documented, the site of the tumor is determined using MRI; because of the possibility of metastasis, whole-body scanning may be done.
The usual treatment of pheochromocytoma is laparoscopic surgical excision of the tumor with adjunctive radiopharmaceuticals or chemotherapy. Medical therapy is used to stabilize blood pressure before surgery. Drugs used include α-adrenergic blocking agents and, possibly later, β-adrenergic blocking agents. Open resection is completed for large tumors or when metastasis is suspected.274
Acromegaly 733
ACTH deficiency 731
Addison disease (primary adrenal insufficiency) 770
Advanced glycosylation end product (AGE) 759
Aldose reductase 758
Amylin 752
Autoimmune thyroiditis (Hashimoto disease, chronic lymphocyte thyroiditis) 741
Bartter syndrome 769
Beta cell dysfunction 751
Congenital hypothyroidism 741
Cushing disease 765
Cushing syndrome 765
Dawn phenomenon 758
Diabetes insipidus (DI) 730
Diabetes mellitus 745
Diabetic ketoacidosis (DKA) 755
Diabetic neuropathy 762
Diabetic retinopathy 759
Dipeptidyl peptidase VI (DPP-IV) 752
Distal symmetric polyneuropathy 762
Dwarfism 731
Exopthalmos 737
Familial hypocalciuric hypercalcemia (FHH) 743
Feminization 771
FSH deficiency 732
Gestational diabetes mellitus (GDM) 754
GH deficiency 732
Ghrelin 752
Giantism 733
Glucagon 752
Glucagon-like peptide (GLP-1) 752
Glucose-dependent insulinotropic polypeptide (GIP) 752
Glycosylated hemoglobin 749
Graves disease 736
Hemoglobin A1c 749
Hyperaldosteronism 768
Hyperosmolar hyperglycemic nonketotic syndrome (HHNKS) 757
Hyperparathyroidism 742
Hyperprolactinemia 735
Hypocortisolism 770
Hypoglycemia 754
Hypoparathyroidism 744
Hypopituitarism 731
Hypothyroidism 739
Iatrogenic hypothyroidism 741
Idiopathic Addison disease (organ-specific autoimmune adrenalitis) 770
Incretin 752
Insulin resistance 750
Iodine deficiency (endemic goiter) 741
LH deficiency 732
Maculopathy 759
Maturity-onset diabetes of youth (MODY) 753
Metabolic syndrome 751
Myxedema 739
Myxedema coma 741
Nephrogenic diabetes insipidus 730
Neurogenic diabetes insipidus 730
Nonenzymatic glycosylation 759
Painless thyroiditis 741
Panhypopituitarism 731
Pheochromocytoma 772
Pituitary adenoma 733
Polyol pathway 758
Postpartum thyroiditis 741
Prediabetes 745
Pretibial myxedema (Graves dermopathy) 737
Primary adrenal insufficiency (Addison disease) 770
Primary aldosteronism (primary hyperaldosteronism) 768
Primary hyperparathyroidism 742
Primary hyperthyroidism 736
Primary hypothyroidism 741
Prolactinoma 735
Protein kinase C (PKC) 758
Pseudohyperparathyroidism 743
Secondary aldosteronism (secondary hyperaldosteronism) 768
Secondary hyperparathyroidism 743
Secondary hyperthyroidism 736
Secondary hypocortisolism 770
Secondary (central) hypothryoidism 739
Sheehan syndrome 731
Somogyi effect 758
Subacute thyroiditis 741
Subclinical hypothyroidism 741
Syndrome of inappropriate antidiuretic hormone (SIADH) secretion 729
Thyroid carcinoma 742
Thyrotoxic crisis (thyroid storm) 738
Thyrotoxicosis 736
Toxic adenoma 738
Toxic multinodular goiter 738
TSH deficiency 732
Type 1 diabetes mellitus 745
Type 2 diabetes mellitus 750
Virilization 771
REFERENCES
1. Kim, T.J., Travers, S. Case report: thyroid hormone resistance and its therapeutic challenges. Curr Opin Pediatr. 2008;20(4):490–493.
2. Tonacchera, M., et al. Identification of TSH receptor mutations in three families with resistance to TSH. Clin Endocrinol. 2007;67(5):712–718.
3. Chen, H., Hewison, M., Adams, J.S. Control of estradiol-directed gene transactivation by an intracellular estrogen-binding protein and an estrogen response element-binding protein. Mol Endocrinol. 2008;22(3):559–569.
4. Visser, W.E., et al. Thyroid hormone transport in and out of cells. Trends Endocrinol Metab. 2008;19(2):50–56.
5. Frank, E., Landgraf, R. The vasopressin system—from antidiuresis to psychopathology. Eur J Pharmacol. 2008;583(2-3):226–242.
6. Ellison, D.H., Berl, T. Clinical practice. The syndrome of inappropriate antidiuresis. N Engl J Med. 2007;356(20):2064–2072.
7. Multz, A.S. Vasopressin dysregulation and hyponatremia in hospitalized patients. J Intensive Care Med. 2007;22(4):216–223.
8. Decaux, G., Musch, W. Clinical laboratory evaluation of the syndrome of inappropriate secretion of antidiuretic hormone. Clin J Am Soc Nephrol. 2008;3(4):1175–1184.
9. Ali, F., et al. Therapeutic potential of vasopressin receptor antagonists. Drugs. 2007;67(6):847–858.
10. Soupart, A., et al. Successful long-term treatment of hyponatremia in syndrome of inappropriate antidiuretic hormone secretion with satavaptan (SR121463B), an orally active nonpeptide vasopressin V2-receptor antagonist. Clin J Am Soc Nephrol. 2006;1(6):1154–1160.
11. Ball, S.G. Vasopressin and disorders of water balance: the physiology and pathophysiology of vasopressin. Ann Clin Biochem. 2007;44(Pt 5):417–431.
12. Makaryus, A.N., McFarlane, S.I. Diabetes insipidus: diagnosis and treatment of a complex disease. Cleve Clin J Med. 2006;73(1):65–71.
13. Loh, J.A., Verbalis, J.G. Disorders of water and salt metabolism associated with pituitary disease. Endocrin Metab Clin North Am. 2008;37(1):213–234. [x].
14. Sands, J.M., et al. Nephrogenic diabetes insipidus. Ann Intern Med. 2006;144(3):186–194.
15. Einaudi, S., Bondone, C. The effects of head trauma on hypothalamic-pituitary function in children and adolescents. Curr Opin Pediatr. 2007;19(4):465–470.
16. Khanna, A. Acquired nephrogenic diabetes insipidus. Semin Nephrol. 2006;26(3):244–248.
17. Robben, J.H., Knoers, N.V., Deen, P.M. Cell biological aspects of the vasopressin type-2 receptor and aquaporin 2 water channel in nephrogenic diabetes insipidus. Am J Physiol Renal Physiol. 2006;291(2):F257–F270.
18. Spanakis, E., Milord, E., Gragnoli, C. AVPR2 variants and mutations in nephrogenic diabetes insipidus: review and missense mutation significance. J Cell Physiol. 2008;217(3):605–617.
19. Schrier, R.W. Aquaporin-related disorders of water homeostasis. Drug News Perspect. 2007;20(7):447–453.
20. Agha, A., Phillips, J., Thompson, C.J. Hypopituitarism following traumatic brain injury (TBI). Br J Neurosurg. 2007;21(2):210–216.
21. Schneider, H.J., et al. Hypopituitarism [see comment]. Lancet. 2007;369(9571):1461–1470.
22. Toogood, A.A., Stewart, P.M. Hypopituitarism: clinical features, diagnosis, and management. Endocrinol Metab Clin North Am. 2008;37(1):235–261. [x].
23. Rupp, D., Molitch, M. Pituitary stalk lesions. Curr Opin Endocrinol Diabetes Obes. 2008;15(4):339–345.
24. Mullis, P.E. Genetics of growth hormone deficiency. Endocrinol Metab Clin North Am. 2007;36(1):17–36.
25. Aikawa, S., et al. High level expression of Prop-1 gene in gonadotropic cell lines. J Repro Develop. 2006;52(2):195–201.
26. Behan, L.A., Agha, A. Endocrine consequences of adult traumatic brain injury. Hormone Res. 2007;68(Suppl 5):18–21.
27. Kaplun, J., et al. Sequential pituitary MR imaging in Sheehan syndrome: report of 2 cases. AJNR Am J Neuroradiol. 2008;29(5):941–943.
28. Richmond, E.J., Rogol, A.D. Growth hormone deficiency in children. Pituitary. 2008;11(2):115–120.
29. Buzi, F., et al. Growth hormone receptor polymorphisms. Endocr Dev. 2007;11:28–35.
30. Mullis, P.E. Genetics of growth hormone deficiency. Endocrinol Metab Clin North Am. 2007;36(1):17–36.
31. Clemmons, D.R. Value of insulin-like growth factor system markers in the assessment of growth hormone status. Endocrinol Metab Clin North Am. 2007;36(1):109–129.
32. Thorner, M.O., Nass, R. Human studies of growth hormone and aging. Pediatr Endocrinol Rev. 2007;4(3):233–234.
33. Johannsson, G. Management of adult growth hormone deficiency. Endocrinol Metab Clin North Am. 2007;36(1):203–220.
34. Auernhammer, C.J., Vlotides, G. Anterior pituitary hormone replacement therapy—a clinical review. Pituitary. 2007;10(1):1–15.
35. Ezzat, S., et al. The prevalence of pituitary adenomas: a systemic review. Cancer. 2004;101(3):613–619.
36. Zhang, H.W., et al. Diagnosis and treatment of pituitary microadenoma: report of 80 cases. Neurol Res. 2008;30(6):587–593.
37. Li-Ng, M., Sharma, M. Invasive pituitary adenoma. J Clin Endocrinol Metab. 2008;93(9):3284–3285.
38. Kanou, Y., et al. Clinical implications of dynamic MRI for pituitary adenomas: clinical and histologic analysis. J Clin Neurosci. 2002;9(6):659–663.
39. Zhang, Y., et al. Endoscopic transsphenoidal treatment of pituitary adenomas. Neurol Res. 2008;30(6):581–586.
40. Ben-Shlomo, A., Melmed, S. Acromegaly. Endocrinol Metab Clin North Am. 2008;37(1):101–122. [viii].
41. Cordero, R.A., Barkan, A.L. Current diagnosis of acromegaly. Rev Endocr Metab Disord. 2008;9(1):13–19.
42. Ayuk, J., Sheppard, M.C. Does acromegaly enhance mortality? Rev Endocr Metab Disord. 2008;9(1):33–39.
43. Katznelson, L. An update on treatment strategies for acromegaly. Exp Opin Pharmacother. 2008;9(13):2273–2280.
44. Vallette, S., Serri, O. Octreotide LAR for the treatment of acromegaly. Exp Opin Drug Metab Toxicol. 2008;4(6):783–793.
45. Roelfsema, F., et al. The role of pegvisomant in the treatment of acromegaly. Exp Opin Biol Ther. 2008;8(5):691–704.
46. Prabhakar, V.K., Davis, J.R. Hyperprolactinaemia. Best Pract Res Clin Obstet Gynaecol. 2008;22(2):341–353.
47. Mancini, T., Casanueva, F.F., Giustina, A. Hyperprolactinemia and prolactinomas. Endocrinol Metab Clin North Am. 2008;37(1):67–99. [viii].
48. Chahal, J., Schlechte, J. Hyperprolactinemia. Pituitary. 2008;11(2):141–146.
49. Kars, M., et al. Cabergoline and cardiac valve disease in prolactinoma patients: additional studies during long-term treatment are required. Eur J Endocrinol. 2008;159(4):363–367.
50. Nayak, B., Hodak, S.P. Hyperthyroidism. Endocrinol Metab Clin North Am. 2007;36(3):617–656. [v].
51. Brent, G.A. Clinical practice. Graves’ disease. N Engl J Med. 2008;358(24):2594–2605.
52. Caturegli, P., et al. Autoimmune thyroid diseases. Curr Opin Rheumatol. 2007;19(1):44–48.
53. Fatourechi, V. Pretibial myxedema: pathophysiology and treatment options. Am J Clin Dermatol. 2005;6(5):295–309.
54. Mota, A., Borrione, P., Santiago, M. Thyroid acropachy. J Clin Rheumatol. 2007;13(6):360.
55. Khoo, T.K., Bahn, R.S. Pathogenesis of Graves’ ophthalmopathy: the role of autoantibodies. Thyroid. 2007;17(10):1013–1018.
56. Wiersinga, W.M. Management of Graves’ ophthalmopathy. Nat Clin Pract Endocrinol Metab. 2007;3(5):396–404.
57. Cerci, C., et al. Thyroid cancer in toxic and non-toxic multinodular goiter. J Postgrad Med. 2007;53(3):157–160.
58. Tunca, F., et al. The preoperative exclusion of thyroid carcinoma in multinodular goiter: dynamic contrast-enhanced magnetic resonance imaging versus ultrasonography-guided fine-needle aspiration biopsy. Surgery. 2007;142(6):992–1002.
59. Porterfield, J.R., Jr., et al. Evidence-based management of toxic multinodular goiter (Plummer’s disease). World J Surg. 2008;32(7):1278–1284.
60. Kearney, T., Dang, C. Diabetic and endocrine emergencies. Postgrad Med J. 2007;83(976):79–86.
61. Ngo, S.Y., Chew, H.C. When the storm passes unnoticed—a case series of thyroid storm. Resuscitation. 2007;73(3):485–490.
62. Dahl, P., Danzi, S., Klein, I. Thyrotoxic cardiac disease. Curr Heart Fail Rep. 2008;5(3):170–176.
63. Kung, A.W. Neuromuscular complications of thyrotoxicosis. Clin Endocrinol. 2007;67(5):645–650.
64. Devdhar, M., Ousman, Y.H., Burman, K.D. Hypothyroidism. Endocrinol Metab Clin North Am. 2007;36(3):595–615. [v].
65. Yamada, M., Masatomo, M. Mechanisms related to the pathophysiology and management of central hypothyroidism. Nat Clin Pract Endocrinol Metab. 2008;4(12):683–694.
66. Kwaku, M.P., Burman, K.D. Myxedema coma. J Intensive Care Med. 2007;22(4):224–231.
67. Vaidya, B., Pearce, S.H. Management of hypothyroidism in adults. BMJ. 2008;337:a801.
68. Beynon, J., Akhtar, S., Kearney, T. Predictors of outcome in myxoedema coma. Crit Care (Lond). 2008;12(1):111.
69. Herrick, B. Subclinical hypothyroidism. Am Family Phys. 2008;77(7):953–955.
70. Villar H C et al: Thyroid hormone replacement for subclinical hypothyroidism, Cochrane Database Syst Rev (3):CD003419.
71. Takami, H.E., Miyabe, R., Kameyama, K. Hashimoto’s thyroiditis. World J Surg. 2008;32(5):688–692.
72. Zeitlin, A.A., et al. Analysis of HLA class II genes in Hashimoto’s thyroiditis reveals differences compared to Graves’ disease. Genes Immun. 2008;9(4):358–363.
73. Duntas, L.H. Environmental factors and autoimmune thyroiditis. Nat Clin Pract Endocrinol Metab. 2008;4(8):454–460.
74. Kimura, H., Caturegli, P. Chemokine orchestration of autoimmune thyroiditis. Thyroid. 2007;17(10):1005–1011.
75. Wang, S.H., Baker, J.R. The role of apoptosis in thyroid autoimmunity. Thyroid. 2007;17(10):975–979.
76. Nishihara, E., et al. Clinical characteristics of 852 patients with subacute thyroiditis before treatment. Intern Med. 2008;47(8):725–729.
77. Lucas, A., et al. Postpartum thyroiditis: long-term follow-up. Thyroid. 2005;15(10):1177–1181.
78. Harris, K.B., Pass, K.A. Increase in congenital hypothyroidism in New York state and in the United States. Mol Genet Metab. 2007;91(3):268–277.
79. Gruters, A., Krude, H. Update on the management of congenital hypothyroidism. Hormone Res. 2007;68(Suppl 5):107–111.
80. Arenz, S., et al. Intellectual outcome, motor skills and BMI of children with congenital hypothyroidism: a population-based study. Acta Paediatr. 2008;97(4):447–450.
81. van der Sluijs Veer, L., et al. Quality of life, developmental milestones, and self-esteem of young adults with congenital hypothyroidism diagnosed by neonatal screening. J Clin Endocrinol Metab. 2008;93(7):2654–2661.
82. American Cancer Society, Inc., Surveillance and Health Policy Research: Estimated New Cancer Cases and Deaths by Sex, U.S. 2009 available at: http://www.cancer.org/downloads/stt/CFF2009_EstCD_3.pdf.
83. Fitzgibbons, S.C., Brams, D.M., Wei, J.P. The treatment of thyroid cancer. Am Surgeon. 2008;74(5):389–399.
84. Brown, R.L. Standard and emerging therapeutic approaches for thyroid malignancies. Semin Oncol. 2008;35(3):298–308.
85. DeLellis, R.A., Mazzaglia, P., Mangray, S. Primary hyperparathyroidism: a current perspective. Arc Pathol Lab Med. 2008;132(8):1251–1262.
86. Rodgers, S.E., Lew, J.I., Solorzano, C.C. Primary hyperparathyroidism. Curr Opin Oncol. 2008;20(1):52–58.
87. Sitges-Serra, A., Bergenfelz, A. Clinical update: sporadic primary hyperparathyroidism. Lancet. 2007;370(9586):468–470.
88. Goodman, W.G., Quarles, L.D. Development and progression of secondary hyperparathyroidism in chronic kidney disease: lessons from molecular genetics. Kidney Int. 2008;74(3):276–288.
89. Hruska, K.A., et al. Hyperphosphatemia of chronic kidney disease. Kidney Int. 2008;74(2):148–157.
90. de Francisco, A.L. New strategies for the treatment of hyperparathyroidism incorporating calcimimetics. Exp Opin Pharmacother. 2008;9(5):795–811.
91. Komaba, H., Tanaka, M., Fukagawa, M. Treatment of chronic kidney disease-mineral and bone disorder (CKD-MBD). Intern Med. 2008;47(11):989–994.
92. Evenepoel, P. Calcimimetics in chronic kidney disease: evidence, opportunities and challenges. Kidney Int. 2008;74(3):265–275.
93. Schlieper, G., Floege, J. Calcimimetics in CKD-results from recent clinical studies. Pediatr Nephrol. 2008;23(10):1721–1728.
94. Shoback, D. Clinical practice, Hypoparathyroidism. N Engl J Med. 2008;359(4):391–403.
95. Asari, R., et al. Hypoparathyroidism after total thyroidectomy: a prospective study. Arch Surg. 2008;143:132–137.
96. Kobrynski, L.J., Sullivan, K.E. Velocardiofacial syndrome, DiGeorge syndrome: the chromosome 22q11.2 deletion syndromes. Lancet. 2007;370(9596):1443–1452.
97. Angelopoulos, N.G., Goula, A., Tolis, G. Sporadic hypoparathyroidism treated with teriparatide: a case report and literature review. Exp Clin Endocrinol Diabetes. 2007;115(1):50–54.
98. American Diabetes Association. Diagnosis and classification of diabetes mellitus. Diabetes Care. 2007;30(Suppl 1):S42–S47.
99. Center for Disease Control and Prevention: Diabetes data and trends. Available at http://apps.nccd.cdc.gov/DDTSTRS/default.aspx.
100. Daneman, D. Type 1 diabetes. Lancet. 2006;367:847–858.
101. Eisenbarth, G.S. Update in type 1 diabetes. J Clin Endocrinol Metab. 2007;92(7):2403–2407.
102. Jahromi, M.M., Eisenbarth, G.S. Cellular and molecular pathogenesis of type 1A diabetes. Cell Mol Life Sci. 2007;64(7-8):865–872.
103. Richer, M.J., Horwitz, M.S. Viral infections in the pathogenesis of autoimmune diseases: focus on type 1 diabetes. Front Biosci. 2008;13:4241–4257.
104. Kaminitz, A., et al. The vicious cycle of apoptotic beta-cell death in type 1 diabetes. Immunol Cell Biol. 2007;85(8):582–589.
105. Chistiakov, D.A., Voronova, N.V., Chistiakov, P.A. The crucial role of IL-2/IL-2RA-mediated immune regulation in the pathogenesis of type 1 diabetes, an evidence coming from genetic and animal model studies. Immunol Lett. 2008;118(1):1–5.
106. Meier, J.J. Beta cell mass in diabetes: a realistic therapeutic target? Diabetologia. 2008;51(5):703–713.
107. Sherr, J., et al. Prevention of type 1 diabetes: the time has come. Nat Clin Pract Endocrinol Metab. 2008;4(6):334–343.
108. Aguilar-Bryan, L., Bryan, J. Neonatal diabetes mellitus. Endocr Rev. 2008;29(3):265–291.
109. Hills, C.E., Brunskill, N.J. Intracellular signalling by C-peptide. Exp Diabetes Res. 2008;2008:635158.
110. Ekberg, K., Johansson, B.L. Effect of C-peptide on diabetic neuropathy in patients with type 1 diabetes. Exp Diabetes Res. 2008;2008:457912.
111. Forst, T., et al. Role of C-Peptide in the regulation of microvascular blood flow. Exp Diabetes Res. 2008;2008:176245.
112. Rebsomen, L., et al. C-Peptide effects on renal physiology and diabetes. Exp Diabetes Res. 2008;2008:281536.
113. Sima, A.A., Kamiya, H. Is C-peptide replacement the missing link for successful treatment of neurological complications in type 1 diabetes? Curr Drug Targets. 2008;9(1):37–46.
114. Wilkin, T.J. Diabetes: 1 and 2, or one and the same? Progress with the accelerator hypothesis. Pediatr Diabetes. 2008;9(3 Pt 2):23–32.
115. Pozilli, P. Immuno-intervention and preservation of beta-cell function in type 1 diabetes. Diabetes Metab Res Rev. 2007;23:255–256.
116. Diabetes Control and Complications Trial Research Group. The effect of intensive treatment of diabetes on the development and progression of long-term complications in insulin-dependent diabetes mellitus. N Engl J Med. 1993;329(14):977–986.
117. Danne, T., Lange, K., Kordonouri, O. New developments in the treatment of type 1 diabetes in children. Arch Dis Child. 2007;92(11):1015–1019.
118. Unger, J. Management of type 1 diabetes. Prim Care. 2007;34(4):791–808. [viii].
119. American Diabetes AssociationExecutive Summary. Standards of medical care in diabetes—2008. Diabetes Care. 2008;31:S5–S11.
120. Shalitin, S., Phillip, M. The role of new technologies in treating children and adolescents with type 1 diabetes mellitus. Pediatr Diabetes. 2007;8(Suppl 6):72–79.
121. Cali, A.M., Caprio, S. Prediabetes and type 2 diabetes in youth: an emerging epidemic disease? Curr Opin Endocrinol Diabetes Obes. 2008;15(2):123–127.
122. Peterson, K., et al. Management of type 2 diabetes in youth: an update. Am Fam Physician. 2007;76(5):634–637.
123. Shaw, J. Epidemiology of childhood type 2 diabetes and obesity. Pediatr Diabetes. 2007;8(Suppl 9):8–15.
124. Sattar, N., Wannamethee, S.G., Forouhi, N.G. Novel biochemical risk factors for type 2 diabetes: pathogenic insights or prediction possibilities? Diabetologia. 2008;51(6):926–940.
125. Jafar-Mohammadi, B., McCarthy, M.I. Genetics of type 2 diabetes mellitus and obesity—a review. Ann Med. 2008;40(1):2–10.
126. Lindgren, C.M., McCarthy, M.I. Mechanisms of disease: genetic insights into the etiology of type 2 diabetes and obesity. Nat Clin Pract Endocrinol Metab. 2008;4(3):156–163.
127. Moore, A.F., Florez, J.C. Genetic susceptibility to type 2 diabetes and implications for antidiabetic therapy. Annu Rev Med. 2008;59:95–111.
128. Romao, I., Roth, J. Genetic and environmental interactions in obesity and type 2 diabetes. J Am Diet Assoc. 2008;108(4 Suppl 1):S24–S28.
129. Florez, J.C. Newly identified loci highlight beta cell dysfunction as a key cause of type 2 diabetes: where are the insulin resistance genes? Diabetologia. 2008;51(7):1100–1110.
130. Maedler, K. Beta cells in type 2 diabetes—a crucial contribution to pathogenesis. Diabetes Obes Metab. 2008;10(5):408–420.
131. Muoio, D.M., Newgard, C.B. Mechanisms of disease: molecular and metabolic mechanisms of insulin resistance and beta-cell failure in type 2 diabetes. Nat Rev Mol Cell Biol. 2008;9(3):193–205.
132. Meier, J.J. Beta cell mass in diabetes: a realistic therapeutic target? Diabetologia. 2008;51(5):703–713.
133. Guilherme, A., et al. Adipocyte dysfunctions linking obesity to insulin resistance and type 2 diabetes. Nat Rev Mol Cell Biol. 2008;9(5):367–377.
134. Hojlund, K., et al. Mitochondrial dysfunction in type 2 diabetes and obesity. Endocrinol Metab Clin North Am. 2008;37(3):713–731.
135. Schenk, S., Saberi, M., Olefsky, J.M. Insulin sensitivity: modulation by nutrients and inflammation. J Clin Invest. 2008;118(9):2992–3002.
136. Glund, S., Krook, A. Role of interleukin-6 signalling in glucose and lipid metabolism. Acta Physiologica. 2008;192(1):37–48.
137. Guest, C.B., et al. The implication of proinflammatory cytokines in type 2 diabetes. Front Biosci. 2008;13:5187–5194.
138. Heilbronn, L.K., Campbell, L.V. Adipose tissue macrophages, low grade inflammation and insulin resistance in human obesity. Curr Pharm Des. 2008;14(12):1225–1230.
139. Poitout, V., Robertson, R.P. Glucolipotoxicity: fuel excess and beta-cell dysfunction. Endocr Rev. 2008;29(3):351–366.
140. Donath, M.Y., et al. Cytokines and beta-cell biology: from concept to clinical translation. Endocr Rev. 2008;29(3):334–350.
141. Scheuner, D., Kaufman, R.J. The unfolded protein response: a pathway that links insulin demand with β-cell failure and diabetes. Endocr Rev. 2008;29(3):317–333.
142. Burcelin, R., Knauf, C., Cani, P.D. Pancreatic alpha-cell dysfunction in diabetes. Diabetes Metab. 2008;34(Suppl 2):S49–S55.
143. Göke, B., Islet cell function: alpha and beta cells—partners towards normoglycaemia. Int J Clin Pract Suppl. 2008(159):2–7.
144. Lebovitz, H.E. New treatments of diabetes: the beta-amylin agonists. Annales d Endocrinologie. 2008;69(2):147–150.
145. Haataja, L., et al. Islet amyloid in type 2 diabetes, and the toxic oligomer hypothesis. Endocr Rev. 2008;29(3):303–316.
146. Khemtemourian, L., et al. Recent insights in islet amyloid polypeptide-induced membrane disruption and its role in beta-cell death in type 2 diabetes mellitus. Exp Diabetes Res. 2008;2008:421287.
147. Salehi, M., Aulinger, B.A., D’Allesio, D.A. Targeting β-cell mass in type 2 diabetes: promise and limitations of new drugs based on incretins. Endocr Rev. 2008;29(3):367–379.
148. Madsbad, S., et al. Glucagon-like peptide receptor agonists and dipeptidyl peptidase-4 inhibitors in the treatment of diabetes: a review of clinical trials. Curr Opin Clin Nutr Metab Care. 2008;11(4):491–499.
149. McIntosh, C.H. Dipeptidyl peptidase IV inhibitors and diabetes therapy. Front Biosci. 2008;13:1753–1773.
150. Richter, B., et al. Dipeptidyl peptidase-4 (DPP-4) inhibitors for type 2 diabetes mellitus. Cochrane Database Syst Rev. (2):2008. [CD006739].
151. Dezaki, K., Sone, H., Yada, T. Ghrelin is a physiological regulator of insulin release in pancreatic islets and glucose homeostasis. Pharmacol Ther. 2008;118(2):239–249.
152. Crandall, J.P., et al. The prevention of type 2 diabetes. Nat Clin Pract Endocrinol Metab. 2008;4(7):382–393.
153. Cummings, S., Apovian, C.M., Khaodhiar, L. Obesity surgery: evidence for diabetes prevention/management. J Am Diet Assoc. 2008;108(4 Suppl 1):S40–S44.
154. Moo, T.A., Rubino, F. Gastrointestinal surgery as treatment for type 2 diabetes. Curr Opin Endocrinol Diabetes Obes. 2008;15(2):153–158.
155. Schernthaner, G., Morton, J.M. Bariatric surgery in patients with morbid obesity and type 2 diabetes. Diabetes Care. 2008;31(Suppl 2):S297–S302.
156. Tibaldi, J. Initiating and intensifying insulin therapy in type 2 diabetes mellitus. Am J Med. 2008;121(6 Suppl):S20–S29.
157. Leibiger, I.B., Berggren, P.O. Insulin signaling in the pancreatic beta-cell. Annu Rev Nutr. 2008;28:233–251.
158. Fajans, S.S., Bell, G.I., Polonsky, K.S. Molecular mechanisms and clinical pathophysiology of maturity-onset diabetes of the young. N Engl J Med. 2001;345:971–980.
159. Holmkvist, J., et al. Common variants in maturity-onset diabetes of the young genes and future risk of type 2 diabetes. Diabetes. 2008;57(6):1738–1744.
160. Vaxillaire, M., Froguel, P. Monogenic diabetes in the young, pharmacogenetics and relevance to multifactorial forms of type 2 diabetes. Endocr Rev. 2008;29(3):254–264.
161. Winckler, W., et al. Evaluation of common variants in the six known maturity-onset diabetes of the young (MODY) genes for association with type 2 diabetes. Diabetes. 2007;56(3):685–693.
162. Bentley-Lewis, R., et al. Gestational diabetes mellitus: postpartum opportunities for the diagnosis and prevention of type 2 diabetes mellitus. Nat Clin Pract Endocrinol Metab. 2008;4(10):552–558.
163. Galtier, F., et al. Optimizing the outcome of pregnancy in obese women: from pregestational to long-term management. Diabetes Metab. 2008;34(1):19–25.
164. Khandelwal, M. GDM: postpartum management to reduce long-term risks. Curr Diabetes Rep. 2008;8(4):287–293.
165. Pickup, J.C., Sutton, A.J. Severe hypoglycaemia and glycaemic control in type 1 diabetes: meta-analysis of multiple daily insulin injections compared with continuous subcutaneous insulin infusion. Diabetic Med. 2008;25(7):765–774.
166. Boyle, P.J., Zrebiec, J. Impact of therapeutic advances on hypoglycaemia in type 2 diabetes. Diabetes Metab Res Rev. 2008;24(4):257–285.
167. Amiel, S.A., et al. Hypoglycaemia in type 2 diabetes. Diabetic Med. 2008;25(3):245–254.
168. Qayyum, R., et al. Systematic review: comparative effectiveness and safety of premixed insulin analogues in type 2 diabetes. Ann Intern Med. 2008;149(8):549–559.
169. Hoffman, R.P. Sympathetic mechanisms of hypoglycemic counterregulation. Curr Diabetes Rev. 2007;3(3):185–193.
170. McCrimmon, R. The mechanisms that underlie glucose sensing during hypoglycaemia in diabetes. Res Diabetic Med. 2008;25(5):513–522.
171. Rossetti, P., et al. Prevention of hypoglycemia while achieving good glycemic control in type 1 diabetes: the role of insulin analogs. Diabetes Care. 2008;31(Suppl 2):S113–S120.
172. Kitabchi, A.E., et al. Thirty years of personal experience in hyperglycemic crises: diabetic ketoacidosis and hyperglycemic hyperosmolar state. J Clin Endocrinol Metab. 2008;93(5):1541–1552.
173. Davis, S.N., Umpierrez, G.E. Diabetic ketoacidosis in type 2 diabetes mellitus—pathophysiology and clinical presentation. Nat Clin Pract Endocrinol Metab. 2007;3(11):730–731.
174. Pinhas-Hamiel, O., Zeitler, P. Acute and chronic complications of type 2 diabetes mellitus in children and adolescents. Lancet. 2007;369(9575):1823–1831.
175. Balasubramanyam, A., et al. Syndromes of ketosis-prone diabetes mellitus. Endocrin Rev. 2008;29:292–302.
176. Kitabchi, A.E., Nyenwe, E.A. Hyperglycemic crises in diabetes mellitus: diabetic ketoacidosis and hyperglycemic hyperosmolar state. Endocrinol Metab Clin North Am. 2006;35(4):725–751. [viii].
177. Levin, D.L. Cerebral edema in diabetic ketoacidosis. Pediatr Crit Care Med. 2008;9(3):320–329.
178. Keenan, C.R., Murin, S., White, R.H. High risk for venous thromboembolism in diabetics with hyperosmolar state: comparison with other acute medical illnesses. J Thromb Haemostasis. 2007;5(6):1185–1190.
179. Guillod, L., et al. Nocturnal hypoglycaemias in type 1 diabetic patients: what can we learn with continuous glucose monitoring? Diabetes Metab. 2007;33(5):360–365.
180. Hoi-Hansen, T., Pedersen-Bjergaard, U., Thorsteinsson, B. The Somogyi phenomenon revisited using continuous glucose monitoring in daily life. Diabetologia. 2005;48(11):2437–2438.
181. Carroll, M.F., Schade, D.S. The dawn phenomenon revisited: implications for diabetes therapy. Endocr Pract. 2005;11(1):55–64.
182. Aronson, D. Hyperglycemia and the pathobiology of diabetic complications. Adv Cardiol. 2008;45:1–16.
183. Graves, D.T., Kayal, R.A. Diabetic complications and dysregulated innate immunity. Front Biosci. 2008;13:1227–1239.
184. Kakehi, T., Yabe-Nishimura, C. NOX enzymes and diabetic complications. Semin Immunopathol. 2008;30(3):301–314.
185. Lorenzi, M. The polyol pathway as a mechanism for diabetic retinopathy: attractive, elusive, and resilient. Exp Diabetes Res. 2007. [61038, 2007].
186. Oates, P.J. Aldose reductase, still a compelling target for diabetic neuropathy. Curr Drug Targets. 2008;9(1):14–36.
187. Noh, H., King, G.L. The role of protein kinase C activation in diabetic nephropathy. Kidney Int. 2007;106(Suppl):S49–S53.
188. Das Evcimen, N., King, G.L. The role of protein kinase C activation and the vascular complications of diabetes. Pharmacol Res. 2007;55(6):498–510.
189. Jandeleit-Dahm, K., Cooper, M.E. The role of AGEs in cardiovascular disease. Curr Pharm Des. 2008;14(10):979–986.
190. Meerwaldt, R., et al. The clinical relevance of assessing advanced glycation endproducts accumulation in diabetes. Cardiovasc Diabetol. 2008;7:29.
191. Yamagishi, S., et al. Advanced glycation end products (AGEs) and cardiovascular disease (CVD) in diabetes. Cardiovasc Hematol Agents Med Chem. 2007;5(3):236–240.
192. Yan, S.F., Ramasamy, R., Schmidt, A.M. Mechanisms of disease: advanced glycation end-products and their receptor in inflammation and diabetes complications. Nat Clin Pract Endocrinol Metab. 2008;4(5):285–293.
193. Forbes, J.M., Coughlan, M.T., Cooper, M.E. Oxidative stress as a major culprit in kidney disease in diabetes. Diabetes. 2008;57(6):1446–1454.
194. Son, S.M. Role of vascular reactive oxygen species in development of vascular abnormalities in diabetes. Diabetes Res Clin Pract. 2007;77(Suppl 1):S65–S70.
195. Copeland, R.J., Bullen, J.W., Hart, G.W. Cross-talk GlcNAcylation and phosphorylation: roles in insulin resistance and glucose toxicity. Am J Physiol Endocrinol Metab. 2008;295(1):E17–E28.
196. Fulop, N., Marchase, R.B., Chatham, J.C. Role of protein O-linked N-acetyl-glucosamine in mediating cell function and survival in the cardiovascular system. Cardiovasc Res. 2008;73(2):288–297.
197. Krentz, A.J., Clough, G., Byrne, C.D. Interactions between microvascular and macrovascular disease in diabetes: pathophysiology and therapeutic implications. Diabetes Obes Metab. 2007;9(6):781–791.
198. Diabetes Control and Complications Trial Research Group. The effect of intensive treatment of diabetes on the development and progression of long-term complications in insulin-dependent diabetes mellitus. N Engl J Med. 1993;329(14):977–986.
199. Lang, G.E. Pharmacological treatment of diabetic retinopathy. Ophthalmologica. 2008;221(2):112–117.
200. Cheung, N., Wong, T.Y. Diabetic retinopathy and systemic vascular complications. Prog Retin Eye Res. 2008;27(2):161–176.
201. Gunduz, K., Bakri, S.J. Management of proliferative diabetic retinopathy. Compr Ophthalmol Update. 2007;8(5):245–256.
202. Dronavalli, S., Duka, I., Bakris, G.L. The pathogenesis of diabetic nephropathy. Nat Clin Pract Endocrinol Metab. 2008;4(8):444–452.
203. Fioretto, P., Mauer, M. Histopathology of diabetic nephropathy. Semin Nephrol. 2007;27(2):195–207.
204. Forbes, J.M., Fukami, K., Cooper, M.E. Diabetic nephropathy: where hemodynamics meet metabolism. Exp Clin Endocrinol Diabetes. 2007;115(2):69–84.
205. Navarro-Gonzalez, J.F., Mora-Fernandez, C. The role of inflammatory cytokines in diabetic nephropathy. J Am Soc Nephrol. 2008;19(3):433–442.
206. Yamagishi, S., et al. Molecular mechanisms of diabetic nephropathy and its therapeutic intervention. Curr Drug Targets. 2007;8(8):952–959.
207. Rogus, J.J., et al. High-density single nucleotide polymorphism genome-wide linkage scan for susceptibility genes for diabetic nephropathy in type 1 diabetes: discordant sibpair approach. Diabetes. 2008;57(9):2519–2526.
208. Harris, R.D., et al. Global glomerular sclerosis and glomerular arteriolar hyalinosis in insulin dependent diabetes. Kidney Int. 1991;40(1):107–114.
209. Battisti, W.P., Palmisano, J., Keane, W.E. Dyslipidemia in patients with type 2 diabetes. Relationships between lipids, kidney disease and cardiovascular disease. Clin Chem Lab Med. 2003;41(9):1174–1181.
210. Khosla, N., Sarafidis, P.A., Bakris, G.L. Microalbuminuria. Clin Labor Med. 2006;26(3):635–653.
211. Stachell, S.C., Tooke, J.E. What is the mechanism of microalbuminuria in diabetes: a role for the glomerular endothelium? Diabetologia. 2008;51(5):714–725.
212. Steffes, M.W., Mauer, S.M. Diabetic nephropathy: a disease causing and complicated by hypertension. Clin Chem. 1991;37(10 Pt 2):1838–1842.
213. Basi, S., Lewis, J.B. Microalbuminuria as a target to improve cardiovascular and renal outcomes in diabetic patients. Curr Diab Rep. 2007;7(6):439–442.
214. Aso, Y. Cardiovascular disease in patients with diabetic nephropathy. Curr Mol Med. 2008;8(6):533–543.
215. Sonkodi, S., Mogyorosi, A. Treatment of diabetic nephropathy with angiotensin II blockers. Nephrol Dial Transplant. 2003;18(Suppl 5):v21–23.
216. Vinik, A.I., Mehrabyan, A. Diabetic neuropathies. Med Clin North Am. 2004;88(4):947–999. [xi].
217. Sima, A.A. The heterogeneity of diabetic neuropathy. Front Biosci. 2008;13:4809–4816.
218. Zochodne, D.W. Diabetes mellitus and the peripheral nervous system: manifestations and mechanisms. Muscle Nerve. 2007;36(2):144–166.
219. Zochodne, D.W., Ramji, N., Toth, C. Neuronal targeting in diabetes mellitus: a story of sensory neurons and motor neurons. Neuroscientist. 2008;14(4):311–318.
220. Casellini, C.M., Vinik, A.I. Clinical manifestations and current treatment options for diabaetic neuropathies. Endocr Pract. 2007;13(5):550–566.
221. Chandrasekharan, B., Srinivasan, S. Diabetes and the enteric nervous system. Neurogastroenterol Motil. 2007;19(12):951–960.
222. Vinik, A.I., et al. Diabetic autonomic neuropathy. Diabetes Care. 2003;26(5):1553–1579.
223. Biessels, G.J., et al. Cognitive dysfunction and diabaetes: implications for primary care. Prim Care Diabetes. 2007;1(4):187–193.
224. Diabetes Control and Complications Trial/Epidemiology of Diabetes Interventions and Complications Study Research Group, et al. Long-term effect of diabetes and its treatment on cognitive function. N Engl J Med. 2007;356(18):1842–1852.
225. La Fonaine, J., et al. Levels of endothelial nitric oxide synthase and calcitonin gene-related peptide in the Charcot foot: a pilot study. J Foot Ankle Surg. 2008;47(5):424–429.
226. Ohkabo, Y., et al. Intensive insulin therapy prevents the progression of diabetes microvascular complications in Japanese patients with non-insulin–dependent diabetes mellitus: a randomized, prospective 6-year study. Diabetes Res Clin Pract. 1995;28(2):103–117.
227. UK Prospective Diabetes Study (UKPDS) Group. Intensive blood-glucose control with sulphonylureas or insulin compared with conventional treatment and risk of complications in patients with type 2 diabetes (UKPDS 33). Lancet. 1998;352(9131):837–853.
228. Chiarelli, F., Mohn, A. Angiopathy in children with diabetes. Minerva Pediatr. 2002;54(3):187–201.
229. Coccheri, S. Approaches to prevention of cardiovascular complications and events in diabetes mellitus. Drugs. 2007;67(7):997–1026.
230. Sowers, J.R. Insulin resistance and hypertension. Am J Physiol Heart Circ Physiol. 2004;286(5):H1597–H1602.
231. Orasanu, G., Plutzky, J. The pathologic continuum of diabetic vascular disease. J Am Coll Cardiol. 2009;53(5 Suppl):S35–42.
232. Retnakaran, R., Zinman, B. Type 1 diabetes, hyperglycaemia, and the heart. Lancet. 2008;371(9626):1790–1799.
233. Natarajan, A., Zaman, A.G., Marshall, S.M. Platelet hyperactivity in type 2 diabetes: role of antiplatelet agents. Diab Vasc Dis Res. 2008;5(2):138–144.
234. Guha, A., Harmancey, R., Taegtmeyer, H. Nonischemic heart failure in diabetes mellitus. Curr Opin Cardiol. 2008;23(3):241–248.
235. Bell, D.S. Stroke in the diabetic patient. Diabetes Care. 1994;17(3):213–219.
236. Furie, K., Inzucchi, S.E. Diabetes mellitus, insulin resistance, hyperglycemia, and stroke. Curr Neurol Neurosci Rep. 2008;8(1):12–19.
237. Gazis, A., et al. Mortality in patients with diabetic neuropathic osteoarthropathy (Charcot foot). Diabetes Med. 2004;21(11):1243–1246.
238. Al-Delaimy, W.K., et al. Effect of type 2 diabetes and its duration on the risk of peripheral arterial disease among men. Am J Med. 2004;116(4):236–240.
239. Mohler, E.R. 3rd: Therapy insight: peripheral arterial disease and diabetes-from pathogenesis to treatment guidelines. Nat Clin Pract Cardiovasc Med. 2007;4(3):151–162.
240. Bowering, C.K. Diabetic foot ulcers: pathophysiology, assessment, and therapy. Can Fam Physician. 2001;47:1107–1116.
241. Jude, E.B., Unsworth, P.F. Optimal treatment of infected diabetic foot ulcers, Drugs Aging. 2004;21(13):833–850.
242. Gardner, S.E., Grantz, R.A. Wound bioburden and infection-related complications in diabetic foot ulcers. Biol Res Nurs. 2008;10(1):44–53.
243. Peleg, A.Y., et al. Common infections in diabetes: pathogenesis, management and relationship to glycaemic control. Diabetes Metab Res Rev. 2007;23(1):3–13.
244. Bolland, M.J., et al. Cushing’s syndrome due to interaction between inhaled corticosteroids and itraconazole. Ann Pharmacother. 2004;38(1):46–49.
245. Newell-Price, J., et al. Cushing’s syndrome. Lancet. 2006;367:1605–1617.
246. Stratakis, C.A. Cushing syndrome caused by adrenocortical tumors and hyperplasias (corticotropin-independent Cushing syndrome). Endocr Dev. 2008;13:117–132.
247. Aghi, M.K. Management of recurrent and refractory Cushing disease. Nat Clin Pract Endocrinol Metab. 2008;4(10):560–568.
248. Pivonello, R., et al. Cushing’s syndrome. Endocrinol Metab Clin North Am. 2008;37(1):135–149. [ix].
249. Schuff, K.G. Issues in the diagnosis of Cushing’s syndrome for the primary care physician. Prim Care. 2003;30(4):791–799.
250. Newell-Price, J. Proopiomelanocortin gene expression and DNA methylation: implications for Cushing’s syndrome and beyond. Endocrinology. 2003;177(3):365–372.
251. Findling, J.W., Raff, H. Diagnosis and differential diagnosis of Cushing’s syndrome. Endocrinol Metab Clin North Am. 2001;30(3):729–747.
252. Sonimo, N., Fava, G.A. Psychiatric disorders associated with Cushing’s syndrome. Epidemiology, pathophysiology, and treatment. CNS Drugs. 2001;15(5):361–373.
253. Pecori Giraldi, F., Moro, M, ., Cavagnini, F. Gender-related differences in the presentation and course of Cushing’s disease. J Clin Endocrinol Metab. 2003;88:1554–1558.
254. Reimondo, G., et al. Laboratory differentiation of Cushing’s syndrome. Clin Chim Acta. 2008;388(1-2):5–14.
255. Davies, J.S., et al. Diagnostic dilemmas in Cushing’s syndrome. Ann Clin Biochem. 2000;37(Pt1):85–89.
256. Biller, B.M., et al. Treatment of adrencorticotropin-dependent Cushing’s syndrome: a concensus statement. J Clin Endocrinol Metab. 2008;93(7):2454–2462.
257. Shalet, S., Mukherjee, A. Pharmacological treatment of hypercortisolism. Curr Opin Endocrinol Diabetes Obes. 2008;15(3):234–238.
258. Young, W.F. Primary aldosteronism: renaissance of a syndrome. Clin Endocrinol Oxf. 2007;66(5):607–618.
259. Karagiannis, A., et al. Medical treatment as an alternative to adrenalectomy in patients with aldosterone-producing adenoma. Endocr Relat Cancer. 2008;15(3):693–700.
260. Young, W.F., Jr. Minireview: primary aldosteronism—changing concepts in diagnosis and treatment. Endocrinology. 2003;144(6):2208–2213.
261. Herbert, S.C. Bartter syndrome. Curr Opin Nephrol Hypertens. 2003;12(4):527–532.
262. Fallo, F., et al. The metabolic syndrome in primary aldosteronism. Curr Diab Rep. 2008;8(1):43–47.
263. Mattsson, C., Young, W.F., Jr. Primary aldosteronism: diagnostic and treatment strategies. Nat Clin Pract Nephrol. 2006;2(4):198–208.
264. Boscaro, M., et al. Diagnosis and management of primary aldosteronism. Curr Opin Endocrinol Diabetes Obes. 2008;15(4):332–338.
265. Rossi, G.P., Pessina, A.C., Heagerty, A.M. Primary aldosteronism: an update on screening, diagnosis and treatment. J Hypertens. 2008;26(4):613–621.
266. Falorni, A., et al. Italian Addison network study: update of diagnostic criteria for the etiological classification of primary adrenal insufficiency. J Clin Endocrinol Metab. 2004;89(4):1598–1604.
267. Larsen, R.P., et al. Williams textbook of endocrinology, ed 10. Philadelphia: Saunders; 2003.
268. Nieman, L.K. Chanco Turner ML: Addison’s disease. Clin Dermatol. 2006;24(4):276–280.
269. Anglin, R.E., Rosebush, P.I., Mazurek, M.F. The neuropsychiatric profile of Addison’s disease, revisiting a forgotten phenomenon. J Neuropsychiatry Clin Neurosci. 2006;18(4):450–459.
270. Lovas, K., Husebye, E.S. Replacement therapy for Addison’s disease: recent developments. Expert Opin Invest Drugs. 2008;17(4):497–509.
271. Opocher, G., et al. Clinical and genetic aspects of phaeochromocytoma. Horm Res. 2003;59(Suppl 1):59–61.
272. Mittendorf, E.A., et al. Pheochromocytoma: advances in genetics, diagnosis, localization, and treatment. Hematol Oncol Clin North Am. 2007;21(3):509–525.
273. Bravo, E.K.L., Pheochromocytoma. Cardiol Rev. 2002;10(1):44–50.
274. Chrisoulidou, A., et al. The diagnosis and management of malignant phaeochromocytoma and paraganglioma. Endocr Relat Cancer. 2007;14(3):569–585.