The Child with Endocrine Dysfunction
On completion of this chapter the reader will be able to:
 Differentiate between the disorders caused by hypopituitary and hyperpituitary dysfunction.
Differentiate between the disorders caused by hypopituitary and hyperpituitary dysfunction.
Describe the manifestations of thyroid hypofunction and hyperfunction and the management of children with the disorders.
Distinguish between the manifestations of adrenal hypofunction and hyperfunction.
Differentiate among the various categories of diabetes mellitus.
Discuss the management and nursing care of the child with diabetes mellitus in the acute care setting.
Distinguish between a hypoglycemic and a hyperglycemic reaction.
Design a teaching plan for a child with diabetes mellitus.
Formulate a teaching plan for instructing the parents of a child with diabetes mellitus.
RELATED TOPICS and ADDITIONAL RESOURCES
 IN TEXT
IN TEXTAdministration of Medication, Ch. 22
Altered Growth and Maturation, Ch. 17
Biologic Development (Adolescence), Ch. 16
Chronic Renal Failure, Ch. 27
Compliance, Ch. 22
Congenital Hypothyroidism, Ch. 9
Genetic Evaluation and Counseling, Ch. 9
Impact of Chronic Illness, Disability, or Death on the Child and Family, Ch. 18
Metabolic Complications, Ch. 9
Surgical Procedures, Ch. 22
The endocrine system consists of three components: (1) the cell, which sends a chemical message by means of a hormone; (2) the target cells, or end organs, which receive the chemical message; and (3) the environment through which the chemical is transported (blood, lymph, extracellular fluids) from the site of synthesis to the sites of cellular action. The endocrine system controls or regulates metabolic processes governing energy production, growth, fluid and electrolyte balance, response to stress, and sexual reproduction (Baxter and Ribeiro, 2001). The endocrine glands, which are distributed throughout the body, are listed in Table 29-1; also listed are several additional structures sometimes considered endocrine glands, although they are not usually included.
TABLE 29-1
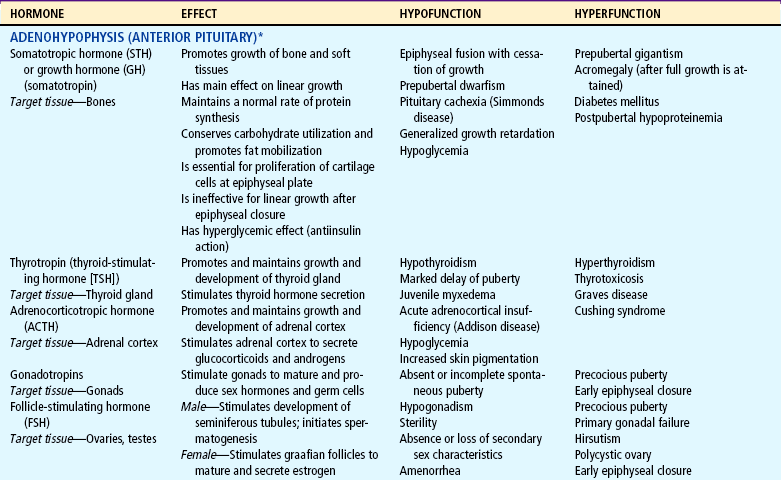

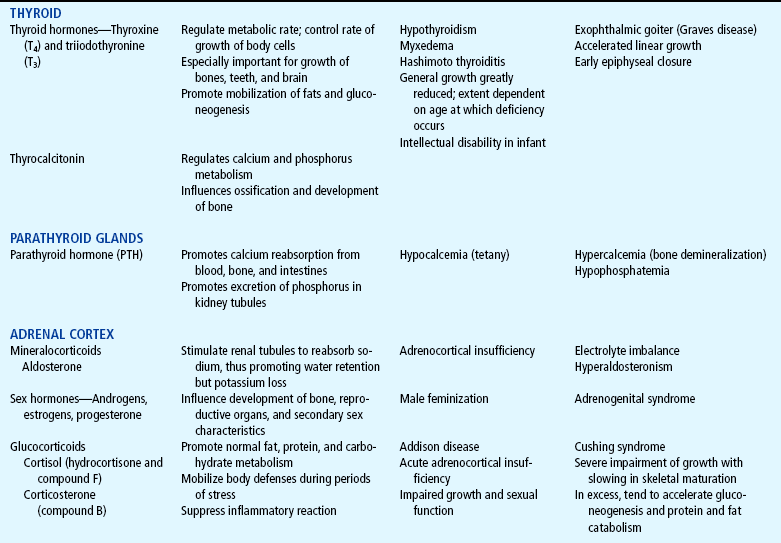

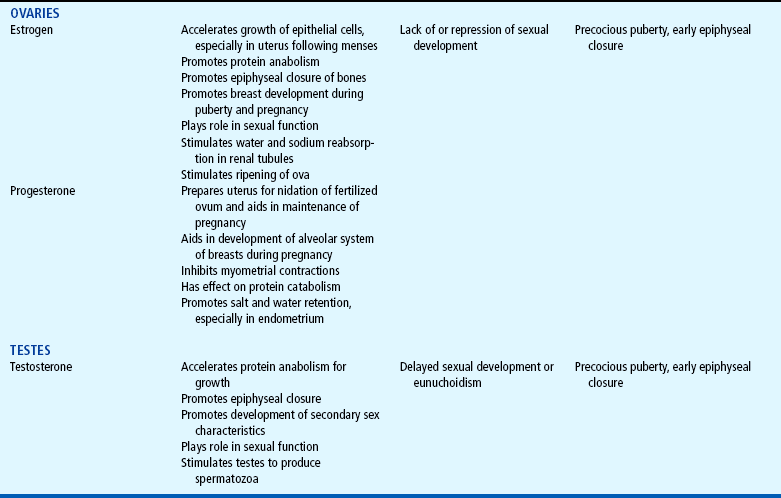
*For each anterior pituitary hormone there is a corresponding hypothalamic-releasing factor. A deficiency in these factors caused by inhibiting anterior pituitary hormone synthesis produces the same effects. (See text for more detailed information.)
†In the male, LH is sometimes known as interstitial cell—stimulating hormone (ICSH).
DISORDERS OF PITUITARY FUNCTION
Deficiencies of the anterior pituitary hormones may be due to organic defects or have an idiopathic etiology and may occur as a single hormonal problem or in combination with other hormonal deficiencies. The clinical manifestations depend on the hormones involved and the age of onset. If the tropic hormones are involved, the resulting disorder reflects the altered stimulus to the target gland. For example, if thyroid-stimulating hormone (TSH) is deficient, thyroid hormone (TH) is also deficient and the child displays the manifestations of hypothyroidism.
An overproduction of the anterior pituitary hormones can result in gigantism (caused by excess growth hormone [GH] production during childhood), hyperthyroidism, hypercortisolism (Cushing syndrome), and precocious puberty from excessive gonadotropins. Overproduction may be caused by hyperplasia of the pituitary cells—which may eventually progress to a tumor (adenoma)–or a primary hypothalamic defect that results in an excess of the hormone’s releasing factor. Although the initial clinical manifestations are a result of pituitary oversecretion, eventually pituitary insufficiency occurs, and the signs of panhypopituitarism become evident.
HYPOPITUITARISM
Hypopituitarism is diminished or deficient secretion of pituitary hormones. The consequences of the condition depend on the degree of dysfunction and lead to gonadotropin deficiency with absence or regression of secondary sex characteristics; GH deficiency, in which children display retarded somatic growth; TSH deficiency, which produces hypothyroidism; and corticotropin deficiency, which results in manifestations of adrenal hypofunction. Hypopituitarism can result from any of the conditions listed in Box 29-1. The most common organic cause of pituitary undersecretion is tumors in the pituitary or hypothalamic region, especially the craniopharyngiomas.
BOX 29-1 Clinical Manifestations of Hypopituitarism
Presenting complaint—short stature
Premature aging common in later life
Height may be retarded more than weight
Tend to be relatively inactive
Less likely to participate in aggressive, sporting-type activities
Bone age nearly always retarded but closely related to height age
Usually primary teeth appear at expected age; eruption of permanent teeth delayed
Teeth are overcrowded and malpositioned (because of underdeveloped jaw)
Constitutional growth delay refers to individuals (usually boys) with delayed linear growth, generally beginning as a toddler, and skeletal and sexual maturation that is behind that of age-mates (Miller and Zimmerman, 2004; Halac and Zimmerman, 2004). Typically these children will reach normal adult height. Often there is a history of a similar pattern of growth in one of the child’s parents or other family members. The untreated child will proceed through normal changes as expected on the basis of bone age. These changes, although occurring later than in the average child, will appear in normal sequence and manner, and treatment with GH is not usually indicated. However, its use has become controversial, especially in relation to parental and child requests for treatment to accelerate growth.
Diagnostic Evaluation
Only a small number of children with delayed growth or short stature have hypopituitary dwarfism. In the majority of instances the cause is constitutional delay. Diagnostic evaluation is aimed at isolating organic causes, which, in addition to GH deficiency, may include hypothyroidism, oversecretion of cortisol, gonadal aplasia, chronic illness, nutritional inadequacy, Russell-Silver dwarfism, or hypochondroplasia.
A complete diagnostic evaluation should include a family history, a history of the child’s growth patterns and previous health status, physical examination, psychosocial evaluation, radiographic surveys, and endocrine studies. Accurate measurement of height (using a calibrated stadiometer) and weight and comparison with standard growth charts are essential. Multiple height measures reflect a more accurate assessment of abnormal growth patterns (Box 29-2) (Hall, 2000).
A skeletal survey in children less than 3 years of age and radiographic examination of the hand-wrist for centers of ossification (bone age) (Box 29-3) in older children are important in evaluating growth.
Definitive diagnosis is based on absent or subnormal reserves of pituitary GH. Because GH levels are normally so low in children that differentiation from abnormal concentrations is unreliable, GH secretion should be stimulated, followed by measurement of blood levels. Exercise is a natural and benign stimulus for GH release, and elevated levels can be detected after 20 minutes of strenuous exercise in normal children. Also, GH levels are elevated 45 to 90 minutes after the onset of sleep.
Initial assessment of the serum insulin-like growth factor-I (IGF-I) and IGF binding protein 3 (IGFBP3) indicates a need for further evaluation of GH dysfunction if levels are less than −1 SD below the mean for age. It is recommended that GH stimulation tests be reserved for children with low serum IGF-I and IGFBP3 levels and poor growth (Hochberg, 1999).
Recent studies have shown that traditional GH stimulation test results can be substantially lower than previously accepted (Hilczer, Smyczynska, and Lewinski, 2006; Guyda, 2000; Mauras, Walton, Nicar, and others, 2000). GH-dependent growth factors may be more sensitive indicators of GH deficiency than GH stimulation tests. Increasingly sensitive radioimmunoassays for GH levels have been developed.
Therapeutic Management
Treatment of GH deficiency caused by organic lesions is directed toward correction of the underlying disease process (e.g., surgical removal or irradiation of a tumor). The definitive treatment of GH deficiency is replacement of GH, which is successful in 80% of affected children. A Cochrane Review of nine randomized controlled trials confirmed that GH therapy can increase short-term grown and improve final height (Bryant, Cave, and Milne, 2003). Reiter, Price, Wilton, and others (2006) found final height within the midparental height range in 1258 patients with idiopathic GH deficiency treated at an early age.
The decision to stop GH therapy is made jointly by the child, family, and health care team. Radiologic evidence of epiphyseal closure is a criterion for ending therapy. Dosage is increased as the time of epiphyseal closure nears to optimize use of the GH. Children with other hormone deficiencies require replacement therapy to correct the specific disorders. This may involve administration of thyroid extract, cortisone, testosterone, or estrogens and progesterone. Treatment with the sex hormones is usually begun during adolescence to promote normal sexual maturation.
Nursing Care Management
The principal nursing consideration is identifying children with growth problems. Despite the fact that the majority of growth problems are not a result of organic causes, any delay in normal growth and sexual development poses special emotional adjustments for these children.
The nurse may be a key person in helping establish a diagnosis. For example, if serial height and weight records are not available, the nurse can question parents about the child’s growth compared with that of siblings, peers, or relatives. Investigating clothing sizes is often helpful in determining growth at different ages. Parents may comment that the child wears out clothes before growing out of them or that, if the clothing fits the body, it often is too long in the sleeves or legs.
Because the behavioral or physical changes that suggest a tumor are insidious, they are frequently overlooked. It is important to correlate the onset of any positive findings with the initial evidence of growth retardation. For example, visual problems and headache are not uncommon in school-age children and can coincidentally occur after a growth problem is recognized. In fact, headache may represent the emotional trauma caused by short stature rather than be a symptom of a tumor. This line of questioning should be pursued cautiously to avoid alarming parents unduly about the possibility of a brain tumor.
Part of a nurse’s role in helping establish a diagnosis is assisting with diagnostic tests. Preparation of the child and family is especially important if a number of tests are being performed, and the child requires particular attention during provocative testing. Blood samples are usually taken every 30 minutes for a 3-hour period. Children also have difficulty overcoming hypoglycemia generated by tests with insulin, so they must be observed carefully for signs of hypoglycemia, whereas those receiving glucagon are at risk of nausea and vomiting.
Child and Family Support.: Children undergoing hormone replacement require additional support. The nurse should provide education for patient self-management during the school-age years.* Nursing functions include family education concerning medication preparation and storage, injection sites, injection technique, and syringe disposal (see Chapter 22). Administration of GH is facilitated by family routines that include a specific time of day for the injection. Younger children may enjoy using a calendar and colorful stickers to designate received injections.
Even when hormone replacement is successful, these children attain their eventual adult height at a slower rate than their peers; therefore they need assistance in setting realistic expectations regarding improvement. Both sexes need guidance toward appropriate vocational goals. Because these children appear younger than their chronologic age, others frequently relate to them in infantile or childish ways. Children having school problems need special counseling. Parents and teachers benefit from guidance directed toward setting realistic expectations for the child based on age and abilities. For example, in the home such children should have the same age-appropriate responsibilities as their siblings. As they approach adolescence, they should be encouraged to participate in group activities with peers. They should wear styles that accentuate their actual age, not their size. If abilities and strengths are emphasized rather than physical size, such children are more likely to develop a positive self-image.
Professionals and families can find resources for research, education, support, and advocacy from the Human Growth Foundation.* The treatment is expensive, but the cost is often partially covered by insurance if the child has a documented deficiency. Children with panhypopituitarism should be advised to wear medical identification at all times.
PITUITARY HYPERFUNCTION
Excess GH before closure of the epiphyseal shafts results in proportional overgrowth of the long bones until the individual reaches a height of 2.4 m (8 feet) or more. Vertical growth is accompanied by rapid and increased development of muscles and viscera. Weight is increased but is usually in proportion to height. Proportional enlargement of head circumference also occurs and may result in delayed closure of the fontanels in young children. Children with a pituitary-secreting tumor may also demonstrate signs of increasing intracranial pressure, especially headache.
If oversecretion of GH occurs after epiphyseal closure, growth is in the transverse direction, producing a condition known as acromegaly. Typical facial features include overgrowth of the head, lips, nose, tongue, jaw, and paranasal and mastoid sinuses; separation and malocclusion of the teeth in the enlarged jaw; disproportion of the face to the cerebral division of the skull; increased facial hair; thickened, deeply creased skin; and increased tendency toward hyperglycemia and diabetes mellitus (DM).
Diagnostic Evaluation
Diagnosis is based on a history of excessive growth during childhood and evidence of increased levels of GH. Radiographic studies may reveal a tumor in an enlarged sella turcica, normal bone age, enlargement of bones (such as the paranasal sinuses), and evidence of joint changes. Endocrine studies to confirm excess of other hormones, specifically thyroid, cortisol, and sex hormones, should also be included in the differential diagnosis.
Therapeutic Management
If a lesion is present, surgical treatment by cryosurgery or hypophysectomy is performed to remove the tumor when feasible. Other therapies aimed at destroying pituitary tissue include external irradiation and radioactive implants. Depending on the extent of surgical extirpation and degree of pituitary insufficiency, hormone replacement with thyroid extract, cortisone, and sex hormones may be necessary.
Nursing Care Management
The primary nursing consideration is early identification of children with excessive growth rates. Although medical management is unable to reduce growth already attained, further growth can be retarded. The earlier the treatment, the more control there is in predetermining a normal adult height. Nurses in ambulatory settings who are frequently involved in growth screening should refer children who demonstrate excessive linear growth for a medical evaluation. They should also observe for signs of a tumor, especially headache, and evidence of concurrent hormonal excesses, particularly the gonadotropins, which cause sexual precocity.
Children with excessive growth rates require as much emotional support as those with short stature. Children and their parents need an opportunity to express their thoughts. A compassionate nurse can be supportive to these children, especially before adolescence when they are larger than their peers. The nurse can emphasize to a tall girl that as boys grow older, they become taller and she will not always be looking down at them.
PRECOCIOUS PUBERTY
Manifestations of sexual development before age 9 years in boys or age 8 years in girls have traditionally been considered precocious development, and these children were recommended for further evaluation (Midyett, Moore, and Jacobson, 2003; Kempers and Otten, 2002). Recent examination of the age limit for defining when puberty is precocious reveals that the onset of puberty in girls is occurring earlier than previous studies have documented (Slyper, 2006; Biro, Huang, Crawford, and others, 2006). Mean onset of puberty was 10.2 and 9.6 years in Caucasian and African-American girls, respectively. Based on these findings, precocious puberty evaluation for a pathologic cause should be performed for Caucasian girls younger than 7 years of age or for African-American girls younger than 6 years of age. No change in the guidelines for evaluation of precocious puberty in boys is recommended. However, recent data suggest that boys may be beginning maturation earlier as well (Slyper, 2006; Herman-Giddens, 2006).
Normally the hypothalamic-releasing factors stimulate secretion of the gonadotropic hormones from the anterior pituitary at the time of puberty. In the male, interstitial cell—stimulating hormone stimulates Leydig cells of the testes to secrete testosterone; in the female, follicle-stimulating hormone (FSH) and luteinizing hormone stimulate the ovarian follicles to secrete estrogens (Nebesio and Eugster, 2007). This sequence of events is known as the hypothalamic-pituitary-gonadal axis. If for some reason the cycle undergoes premature activation, the child will display evidence of advanced or precocious puberty. Causes of precocious puberty are found in Box 29-4.
BOX 29-4 Causes of Precocious Puberty
Idiopathic, with or without hypothalamic hamartoma
After effective treatment of longstanding pseudoisosexual precocity
Modified from Root AW: Precocious puberty, Pediatr Rev 21(1):10-19, 2000.
Isosexual precocious puberty is more common among girls than boys. Approximately 80% of children with precocious puberty have central precocious puberty (CPP), in which pubertal development is activated by the hypothalamic gonadotropin-releasing hormone (GnRH) (Greiner and Kerrigan, 2006). This produces early maturation and development of the gonads with secretion of sex hormones, development of secondary sex characteristics, and sometimes production of mature sperm and ova (Lee, 1999; Root, 2000). CPP may be the result of congenital anomalies; infectious, neoplastic, or traumatic insults to the central nervous system (CNS); or treatment of longstanding sex hormone exposure (Trivin, Couto-Silva, Sainte-Rose, and others, 2006). CPP occurs more frequently in girls and is usually idiopathic, with 95% demonstrating no causative factor (Nebesio and Eugster, 2007; Greiner and Kerrigan, 2006; Root, 2000). A CNS insult or structural abnormality is found in more than 90% of boys with CPP (Root, 2000).
Peripheral precocious puberty (PPP) includes early puberty resulting from hormone stimulation other than the hypothalamic GnRH—stimulated pituitary gonadotropin release. Isolated manifestations that are usually associated with puberty may be seen as variations in normal sexual development (Greiner and Kerrigan, 2006). They appear without other signs of pubescence and are probably caused by unusual end-organ sensitivity to prepubertal levels of estrogen or androgen. Included are premature thelarche (development of breasts in prepubertal girls), premature pubarche (premature adrenarche, early development of sexual hair), and premature menarche (isolated menses without other evidence of sexual development).
Therapeutic Management
Treatment of precocious puberty is directed toward the specific cause when known. Precocious puberty of central (hypothalamic-pituitary) origin is managed with monthly injections of a synthetic analog of luteinizing hormone—releasing hormone, which regulates pituitary secretions (Greiner and Kerrigan, 2006; Muir, 2006). The available preparation, leuprolide acetate (Lupron Depot), is given in a dosage of 0.2 to 0.3 mg/kg intramuscularly once every 4 weeks. Breast development regresses or does not advance, and growth returns to normal rates, enhancing predicted height. Studies suggest that not all patients attain adult targeted heights and the addition of GH therapy may be warranted (Walvoord and Pescovitz, 1999). Treatment is discontinued at a chronologically appropriate time, allowing pubertal changes to resume. Psychologic management of the patient and family is an important aspect of care. Both parents and the affected child should be taught the injection procedure.
Nursing Care Management
Psychologic support and guidance of the child and family are the most important aspects of management. Parents need anticipatory guidance, support and information resources, and reassurance of the benign nature of the condition (Greiner and Kerrigan, 2006; O’Sullivan and O’Sullivan, 2002). Dress and activities for the physically precocious child should be appropriate to the chronologic age. Sexual interest is not usually advanced beyond the child’s chronologic age, and parents need to understand that the child’s mental age is congruent with the chronologic age and that the child’s normal, overt manifestations of affection are age-appropriate and do not represent sexual advances.
Although the child’s heterosexual behavior is appropriate for the chronologic age, the nurse should emphasize to parents that the child is fertile. Usually no form of contraception is necessary unless the child is sexually active. In this situation proper counseling is important because hormonal forms of birth control, such as estrogen pills, will prematurely initiate epiphyseal closure, resulting in stunted linear growth.
DIABETES INSIPIDUS
The principal disorder of posterior pituitary hypofunction is diabetes insipidus (DI), also known as neurogenic DI, resulting from undersecretion of antidiuretic hormone (ADH), or vasopressin (Pitressin), and producing a state of uncontrolled diuresis (Wong and Verbalis, 2002). This disorder is not to be confused with nephrogenic DI, a rare hereditary disorder affecting primarily males and caused by unresponsiveness of the renal tubules to the hormone.
Neurogenic DI may result from a number of different causes. Primary causes are familial or idiopathic; of the total cases, approximately 45% to 50% are idiopathic. Secondary causes include trauma (accidental or surgical), tumors, granulomatous disease, infections (meningitis or encephalitis), and vascular anomalies (aneurysm). Certain drugs, such as alcohol or phenytoin (diphenylhydantoin), can cause a transient polyuria.
The cardinal signs of DI are polyuria and polydipsia. In the older child, signs such as excessive urination accompanied by a compensatory insatiable thirst may be so intense that the child does little more than go to the toilet and drink fluids (Cheetham and Baylis, 2002). Frequently the first sign is enuresis. In the infant the initial symptom is irritability that is relieved with feedings of water but not milk. The infant is also prone to dehydration, electrolyte imbalance, hyperthermia, azotemia, and potential circulatory collapse.
Dehydration is usually not a serious problem in older children, who are able to drink larger quantities of water. However, any period of unconsciousness, such as after trauma or anesthesia, may be life threatening because the voluntary demand for fluid is absent. During such instances careful monitoring of urine volumes, blood concentration, and intravenous (IV) fluid replacement is essential to prevent dehydration.
Diagnostic Evaluation
The simplest test used to diagnose this condition is restriction of oral fluids and observation of consequent changes in urine volume and concentration. Normally, reducing fluids results in concentrated urine and diminished volume. In DI, fluid restriction has little or no effect on urine formation but causes weight loss from dehydration. Accurate results from this procedure require strict monitoring of fluid intake and urinary output, measurement of urine concentration (specific gravity or osmolality), and frequent weight checks. A weight loss between 3% and 5% indicates significant dehydration and requires termination of the fluid restriction.
If this test is positive, the child should be given a test dose of injected aqueous vasopressin, which should alleviate the polyuria and polydipsia. Unresponsiveness to exogenous vasopressin usually indicates nephrogenic DI. An important diagnostic consideration is to differentiate DI from other causes of polyuria and polydipsia, especially DM. DI may be the early sign of an evolving cerebral process (De Buyst, Massa, Christophe, and others, 2007).
Therapeutic Management
The usual treatment is hormone replacement, either with an intramuscular or subcutaneous injection of vasopressin tannate in peanut oil or with a nasal spray of aqueous lysine vasopressin (Verbalis, 2003). The injectable form has the advantage of lasting 48 to 72 hours, which affords the child a full night’s sleep. However, it has the disadvantage of requiring frequent injections and proper preparation of the drug.
Nursing Care Management
The initial objective is identification of the disorder. Because an early sign may be sudden enuresis in a child who is toilet trained, excessive thirst with bed-wetting is an indication for further investigation. Another clue is persistent irritability and crying in an infant that is relieved only by bottle-feedings of water. After head trauma or certain neurosurgical procedures, the development of DI can be anticipated; therefore these patients must be closely monitored.
Assessment includes measurement of body weight, serum electrolytes, blood urea nitrogen, hematocrit, and urine specific gravity taken before surgery and every other day after the procedure. Fluid intake and output should be carefully measured and recorded. Alert patients are able to adjust intake to urine losses, but unconscious or very young patients require closer fluid observation. In children who are not toilet trained, collection of urine specimens may require application of a urine-collecting device.
After confirmation of the diagnosis, parents need a thorough explanation regarding the condition with specific clarification that DI is a different condition from DM. They must realize that treatment is lifelong. If children are to receive the injectable vasopressin, ideally two caregivers should be taught the correct procedure for preparation and administration of the drug. Once children are old enough, they should be encouraged to assume full responsibility for their care.
For emergency purposes, these children should wear medical alert identification. Older children should carry the nasal spray with them for temporary relief of symptoms. School personnel need to be aware of the problem so they can grant children unrestricted use of the lavatory. Failure to permit this may result in embarrassing accidents that often lead to a child’s unwillingness to attend school.
SYNDROME OF INAPPROPRIATE ANTIDIURETIC HORMONE
The disorder that results from oversecretion of the posterior pituitary hormone, or ADH, is known as syndrome of inappropriate antidiuretic hormone (SIADH). It is observed with increased frequency in a variety of conditions, especially those involving infections, tumors, or other CNS disease or trauma (Lin, Liu, and Lim, 2005).
The manifestations are directly related to fluid retention and hypotonicity. Excess ADH causes most of the filtered water to be reabsorbed from the kidneys back into central circulation. Serum osmolality is low, and urine osmolality is inappropriately elevated. When serum sodium levels are diminished to 120 mEq/L, affected children display anorexia, nausea (and sometimes vomiting), stomach cramps, irritability, and personality changes. With progressive reduction in sodium, other neurologic signs, stupor, and convulsions may be evident. The symptoms usually disappear when the underlying disorder is corrected.
The immediate management consists of restricting fluids. Subsequent management depends on the cause and severity. Fluids continue to be restricted to one-fourth to one-half maintenance. When there are no fluid abnormalities but SIADH can be anticipated, fluids are often restricted expectantly at two-thirds to three-fourths maintenance.
Nursing Care Management
The first goal of nursing management is recognizing the presence of SIADH from symptoms described in patients at risk, especially those in the pediatric intensive care unit.
Accurately measuring intake and output, noting daily weight, and observing for signs of fluid overload are primary nursing functions, especially in the child receiving IV fluids. Seizure precautions are implemented, and the child and family need education regarding the rationale for fluid restrictions. The rare child with chronic SIADH will be placed on long-term ADH-antagonizing medication, and the child and family will require instructions for its administration.
DISORDERS OF THYROID FUNCTION
The thyroid gland secretes two types of hormones: TH, which consists of the hormones thyroxine(T4) and triiodothyronine (T3), and calcitonin. The secretion of thyroid hormones is controlled by TSH from the anterior pituitary, which in turn is regulated by thyrotropin-releasing factor (TRF) from the hypothalamus as a negative feedback response. Consequently, hypothyroidism or hyperthyroidism may result from a defect in the target gland or from a disturbance in the secretion of TSH or TRF. Because the functions of T3 and T4 are qualitatively the same, the term thyroid hormone (TH) is used throughout the discussion.
The synthesis of TH depends on available sources of dietary iodine and tyrosine. The thyroid is the only endocrine gland capable of storing excess amounts of hormones for release as needed. During circulation in the bloodstream, T4 and T3 are bound to carrier proteins (thyroxine-binding globulin). They must be unbound before they are able to exert their metabolic effect.
The main physiologic action of TH is to regulate the basal metabolic rate and thereby control the processes of growth and tissue differentiation. Unlike GH, TH is involved in many more diverse activities that influence the growth and development of body tissues. Therefore a deficiency of TH exerts a more profound effect on growth than that seen in hypopituitarism.
Calcitonin helps maintain blood calcium levels by decreasing the calcium concentration. Its effect is the opposite of parathyroid hormone (PTH) in that it inhibits skeletal demineralization and promotes calcium deposition in the bone.
JUVENILE HYPOTHYROIDISM
Hypothyroidism is one of the most common endocrine problems of childhood. It may be either congenital (see Chapter 9) or acquired and represents a deficiency in secretion of TH (Foley, 2001). Hypothyroidism from dietary insufficiency of iodine is now rare in the United States, since iodized salt is a readily available source of the nutrient.
Beyond infancy, primary hypothyroidism may be caused by a number of defects. For example, a congenital hypoplastic thyroid gland may provide sufficient amounts of TH during the first year or two but be inadequate when rapid body growth increases demands on the gland. A partial or complete thyroidectomy for cancer or thyrotoxicosis can leave insufficient thyroid tissue to furnish hormones for body requirements. Radiotherapy for Hodgkin disease or other malignancies may lead to hypothyroidism (Pizzo and Poplack, 2006). Infectious processes may cause hypothyroidism. It can also occur when dietary iodine is deficient.
Clinical manifestations depend on the extent of dysfunction and the child’s age at onset. Primary congenital hypothyroidism is characterized by low levels of circulating thyroid hormones and raised levels of TSH at birth (Macchia, 2000). The GnRH test and baseline measurement of gonadotropin and sex hormone serum concentrations at 3 months of age are promising options for assessment of hypothalamic-pituitary-gonadal function in infants with congenital hypothyroidism (van Tijn, Schroor, Delemarre-van de Waal, and others, 2007). The presenting symptoms are decelerated growth from chronic deprivation of TH or thyromegaly. Impaired growth and development are less severe when hypothyroidism is acquired at a later age, and, because brain growth is nearly complete by 2 to 3 years of age, intellectual disability and neurologic sequelae are not associated with juvenile hypothyroidism. Other manifestations are myxedematous skin changes (dry skin, puffiness around the eyes, sparse hair), constipation, sleepiness, and mental decline (Box 29-5).
Therapy is TH replacement, the same as for hypothyroidism in the infant, although the prompt treatment needed in the infant is not required in the child. In children with severe symptoms, the restoration of euthyroidism is achieved more gradually with administration of increasing amounts of l-thyroxine over a period of 4 to 8 weeks to avoid symptoms of hyperthyroidism, which can occur with treatment of chronic hypothyroidism. Researchers have found that children treated early continue to have mild delays in reading, comprehension, and arithmetic but catch up by grade six (Rovet and Ehrlich, 2000). However, adolescents may demonstrate problems with memory, attention, and visuospatial processing.
Nursing Care Management
The importance of early recognition in the infant is discussed in Chapter 9. Growth cessation or retardation in a child whose growth has previously been normal should alert the observer to the possibility of hypothyroidism. After diagnosis and implementation of thyroxine therapy, the importance of compliance and periodic monitoring of response to therapy should be stressed to parents. Children should learn to take responsibility for their own health as soon as they are old enough, at about 9 or 10 years of age.
GOITER
A goiter is an enlargement or hypertrophy of the thyroid gland. It may occur with deficient (hypothyroid), excessive (hyperthyroid), or normal (euthyroid) TH secretion. It can be congenital or acquired. Congenital disease usually occurs as a result of maternal administration of antithyroid drugs or iodides during pregnancy. Acquired disease can result from increased secretion of pituitary TSH in response to decreased circulating levels of TH or from infiltrative neoplastic or inflammatory processes. In areas where dietary iodine (essential for TH production) is deficient, goiter can be endemic.
Enlargement of the thyroid gland may be mild and noticeable only when there is an increased demand for TH (e.g., during periods of rapid growth). Where iodine deficiency is severe, a large percentage of the population displays goiters. Enlargement of the thyroid at birth can be sufficient to cause severe respiratory distress. Sporadic goiter is usually caused by lymphocytic thyroiditis, and intrinsic biochemical defects in synthesis of the hormones are associated with goiters. TH replacement is necessary to treat the hypothyroidism and reverse the TSH effect on the gland.
Nursing Care Management
Large goiters are identified by their obvious appearance. Smaller nodules may be evident only on palpation. Nurses in ambulatory settings need to be aware of the possibility of goiters and report such findings. Benign enlargement of the thyroid gland may occur during adolescence and should not be confused with pathologic states. Nodules rarely are caused by a cancerous tumor but always require evaluation. Questions regarding exposure to radiation should be included in the assessment.
Immediate surgery to remove part of the gland may be lifesaving in infants born with a goiter. When thyroid replacement is necessary, parents have the same needs regarding its administration as discussed for the parents of children who have hypothyroidism (see Chapter 9).
LYMPHOCYTIC THYROIDITIS
Lymphocytic thyroiditis (Hashimoto disease, juvenile autoimmune thyroiditis) is the most common cause of thyroid disease in children and adolescents and accounts for the largest percentage of juvenile hypothyroidism (Szymborska and Staroszczyk, 2000). It accounts for many of the enlarged thyroid glands formerly designated thyroid hyperplasia of adolescence or adolescent goiter. Although it can occur during the first 3 years of life, it occurs more frequently after age 6. It reaches a peak incidence during adolescence, and there is evidence that the disease is self-limiting. The presence of a goiter and elevated thyroglobulin antibody with progressive increase in both thyroid peroxidase antibody and TSH may be predictive factors for future development of hypothyroidism (Radetti, Gottardi, Bona, and others, 2006).
The presence of the enlarged thyroid gland is usually detected by the practitioner during a routine examination, although it may be noted by parents when the youngster swallows. In most children the entire gland is enlarged symmetrically (though it may be asymmetric) and is firm, freely movable, and nontender. There may be manifestations of moderate tracheal compression (sense of fullness, hoarseness, and dysphagia), but it is extremely rare for a nontoxic diffuse goiter to enlarge to the extent that it causes mechanical obstruction. Most children are euthyroid, but some display symptoms of hypothyroidism. Other signs suggestive of hyperthyroidism are found in Box 29-6.
Diagnostic Evaluation
Thyroid function tests are usually normal, although TSH levels may be slightly or moderately elevated. With progressive disease the T4 decreases, followed by a decrease in T3 levels and an increase in TSH. A variety of abnormalities in radioactive iodine uptake may be noted. The majority of children have serum antibody titers to thyroid antigens, but fewer children have a positive red blood cell hemagglutination test result. When both tests are used, almost all children with thyroid autoimmunity are detected. However, levels in children are lower than in adults; therefore repeated measurements may be needed in doubtful cases, since titers may increase later in the disease.
Therapeutic Management
In many cases the goiter is transient and asymptomatic and regresses spontaneously within a year or two. Therapy of a nontoxic diffuse goiter is usually simple, uncomplicated, and effective. Oral administration of TH decreases the size of the gland significantly and provides the feedback needed to suppress TSH stimulation, and the hyperplastic thyroid gland gradually regresses in size. Surgery is contraindicated in this disorder. Untreated patients should be evaluated periodically.
HYPERTHYROIDISM
The largest percentage of hyperthyroidism in childhood is caused by Graves disease, which is usually associated with an enlarged thyroid gland and exophthalmos (Ma, Xie, Kuang, and others, 2006; Streetman and Khanderia, 2004; Thompson, 2002). Most cases of Graves disease in children occur between ages 6 and 15, with a peak incidence at 12 to 14 years of age, but the disease may be present at birth in children of thyrotoxic mothers. The incidence is five times higher in girls than in boys.
The hyperthyroidism of Graves disease is apparently caused by an autoimmune response to TSH receptors, but no specific etiology has been identified. There is definitive evidence for familial association, with a high concordance incidence in twins. Patients with Graves disease possess the histocompatibility antigens A1, B8, and DR3 (Dallas and Foley, 2003; Simmonds, Howson, Heward, and others, 2005).
The development of manifestations is highly variable. Signs and symptoms develop gradually, with an interval between onset and diagnosis of approximately 6 to 12 months. The principal clinical features are excessive motion—irritability, hyperactivity, short attention span, tremors, insomnia, and emotional lability. Clinical manifestations are presented in Box 29-7.
BOX 29-7 Clinical Manifestations of Hyperthyroidism (Graves Disease)
Emotional lability
Physical restlessness, characteristically at rest
Decelerated school performance
Exophthalmos (protruding eyeballs), observed in many children, is accompanied by a wide-eyed staring expression, increased blinking, lid lag, lack of convergence, and absence of wrinkling of the forehead when looking upward. As protrusion of the eyeball increases, the child may not be able to completely cover the cornea with the lid. Visual disturbances may include blurred vision and loss of visual acuity. Ophthalmopathy can develop long before or after the onset of hyperthyroidism. A consistent pathogenic link between them has not been identified. It is now thought that Graves ophthalmopathy is a disorder of autoimmune origin caused by a complex interplay of endogenous and environmental factors (Bartalena, Tanda, Piantanida, and others, 2003).
Diagnostic Evaluation
The presence of a thyroid mass in a child requires a thorough history, including inquiry into prior irradiation to the head and neck and exposure to a goitrogen. The diagnosis is established on the basis of increased levels of T4 and T3. TSH is suppressed to unmeasurable levels (Ma, Xie, Kuang, and others, 2006). Other tests are rarely indicated.
Therapeutic Management
Therapy for hyperthyroidism is controversial, but all methods are directed toward retarding the rate of hormone secretion. The three acceptable modes available are the antithyroid drugs, which interfere with the biosynthesis of TH, including propylthiouracil (PTU) and methimazole (MTZ, Tapazole); subtotal thyroidectomy; and ablation with radioiodine (131I iodide) (Streetman and Khanderia, 2004; Rivkees and Cornelius, 2003). Each is effective, but each has advantages and disadvantages.
When affected children exhibit signs and symptoms of hyperthyroidism (e.g., increased weight loss, pulse, pulse pressure, and blood pressure), their activity should be limited to classwork only. Vigorous exercise is restricted until thyroid levels are decreased to normal or near-normal values.
Thyrotoxicosis (thyroid “crisis” or thyroid “storm”) may occur from sudden release of the hormone. Although thyrotoxicosis is unusual in children, a crisis can be life threatening. These “storms” are evidenced by the acute onset of severe irritability and restlessness, vomiting, diarrhea, hyperthermia, hypertension, severe tachycardia, and prostration. There may be rapid progression to delirium, coma, and even death. A crisis may be precipitated by acute infection, surgical emergencies, or discontinuation of antithyroid therapy. Treatment, in addition to antithyroid drugs, is administration of β-adrenergic blocking agents (propranolol), which provide relief from the adrenergic hyperresponsiveness that produces the disturbing side effects of the reaction. Therapy is usually required for 2 to 3 weeks.
The American Thyroid Association* has an extensive website with information related to prevention, treatment, and cure of thyroid disease.
Nursing Care Management
The initial nursing objective is identification of children with hyperthyroidism. Because the clinical manifestations often appear gradually, the goiter and ophthalmic changes may not be noticed, and the excessive activity may be attributed to behavioral problems. Nurses in ambulatory settings, particularly schools, need to be alert to signs that suggest this disorder, especially weight loss despite an excellent appetite, academic difficulties resulting from a short attention span and inability to sit still, unexplained fatigue and sleeplessness, and difficulty with fine motor skills such as writing. Exophthalmos may develop long before the onset of signs and symptoms of hyperthyroidism and may be the only presenting sign (Thompson, 2002). Exophthalmos is less common in adults than children (Jospe, 2001).
Much of these children’s care is related to treating physical symptoms before a response to drug therapy is achieved. These children need a quiet, unstimulating environment that is conducive to rest. Sometimes hospitalization is necessary during the immediate treatment phase to remove a child from a troubled home. A regular routine is beneficial in providing frequent rest periods, minimizing the stress of coping with unexpected demands, and meeting the children’s needs promptly. Physical activity is restricted. For example, school physical education classes are discontinued.
Emotional lability is often manifested by sudden episodes of crying or elation. Such behavior, coupled with irritability, disrupts interpersonal relationships, creating difficulties within and outside the home. Parents need help in understanding the uncontrollable nature of these outbursts and ways of minimizing them through decreased environmental stimulation, stress, and frustration. The child should be encouraged to express feelings about behavior and its effect on others. The nurse can encourage the child to concentrate on friendship with one special peer rather than a group until the condition is stabilized.
Heat intolerance may produce considerable family conflict. Preferring a cooler environment than others, the child is likely to open windows, complain about the heat, wear minimum clothing, and remove blankets while sleeping. Although the child should dress in accordance with climatic conditions, the use of light cotton clothing in the home, good ventilation, air conditioning or fans, frequent baths, and adequate hydration is helpful in providing comfort. Hygiene should be stressed because of excessive sweating.
Dietary requirements should be adjusted to meet the child’s increased metabolic rate. Although the need for calories is increased, these should be provided in wholesome foods rather than “junk” foods. The child may require vitamin supplements to meet daily requirement. Rather than three large meals, the child’s appetite may be better satisfied by five or six moderate meals throughout the day. Family members should refrain from making remarks about the child’s appetite because the child may voluntarily restrict his or her eating to avoid such attention.
Once therapy is instituted, the nurse explains the drug regimen, emphasizing the importance of observing for side effects of antithyroid drugs. Untoward effects of propylthiouracil and related compounds include urticarial rash, fever, arthritis, or arthralgia. There may be enlargement of the salivary and cervical lymph glands, a diminished sense of taste, hepatitis, and edema of the lower extremities. Parents should also be aware of the signs of hypothyroidism, which can occur from overdose of the drugs. The most common indications are lethargy and somnolence.
DISORDERS OF PARATHYROID FUNCTION
The parathyroid glands secrete PTH, the main function of which, along with vitamin D and calcitonin, is homeostasis of serum calcium concentration (Perheentupa, 2003). The effect of PTH on calcium is opposite that of calcitonin. The net result of the integrated action of PTH and vitamin D is maintenance of serum calcium levels within a narrow normal range and the mineralization of bone. Secretion of PTH is controlled by a negative feedback system involving the serum calcium ion concentration. Low ionized calcium levels stimulate PTH secretion, causing absorption of calcium by the target tissues; high ionized calcium concentrations suppress PTH.
HYPOPARATHYROIDISM
Hypoparathyroidism is a spectrum of disorders that result in deficient PTH. Congenital hypoparathyroidism may be caused by a specific defect in the synthesis or cellular processing of PTH or by aplasia or hypoplasia of the gland (Perheentupa, 2003).
Hypoparathyroidism can also occur secondary to other causes. Postoperative hypoparathyroidism may follow thyroidectomy with acute or gradual onset and be transient or permanent. Two forms of transient hypoparathyroidism may be present in the newborn, both of which are the result of a relative PTH deficiency. One type is caused by maternal hyperparathyroidism or maternal DM. A more common, later form appears almost exclusively in infants fed a milk formula with a high phosphate-to-calcium ratio.
Clinical signs of hypoparathyroidism are found in Box 29-8. Muscle cramps are an early symptom, progressing to numbness, stiffness, and tingling in the hands and feet. A positive Chvostek or Trousseau sign or laryngeal spasms may be present. Convulsions with loss of consciousness may occur. These episodes may be preceded by abdominal discomfort, tonic rigidity, head retraction, and cyanosis. Headaches and vomiting with increased intracranial pressure and papilledema may occur and may suggest a brain tumor (Behrman, Kliegman, and Jenson, 2004).
BOX 29-8 Clinical Manifestations of Hypoparathyroidism
Short stature
Short, stubby fingers and toes
Dimpling of skin over knuckles
Dry, scaly, coarse skin with eruptions
Nails thin and brittle with characteristic transverse grooves
Laryngospasm (laryngeal stridor)
Positive Chvostek sign or Trousseau sign (see Nursing Alert on p. 1035)
Diagnostic Evaluation
The diagnosis of hypoparathyroidism is made on the basis of clinical manifestations associated with decreased serum calcium and increased serum phosphorus. Levels of plasma PTH are low in idiopathic hypoparathyroidism but high in pseudohypoparathyroidism. End-organ responsiveness is tested by the administration of PTH with measurement of urinary cyclic adenosine monophosphate (cAMP). Kidney function tests are included in the differential diagnosis to rule out renal insufficiency. Although bone radiographs are usually normal, they may demonstrate increased bone density and suppressed growth.
Therapeutic Management
The objective of treatment is to maintain normal serum calcium and phosphate levels with minimum complications. Acute or severe tetany is corrected immediately by IV and oral administration of calcium gluconate and follow-up daily doses to achieve normal levels. Twice-daily serum calcium measurements are taken to monitor the efficacy of therapy and prevent hypercalcemia. When diagnosis is confirmed, vitamin D therapy is begun. Vitamin D therapy is somewhat difficult to regulate because the drug has a prolonged onset and a long half-life. Some authorities advocate beginning with a lower dose with stepwise increases and careful monitoring of serum calcium until stable levels are achieved. Others prefer rapid induction with higher doses and rapid reduction to lower maintenance levels (Behrman, Kliegman, and Jenson, 2004).
Long-term management consists of administration of massive doses of vitamin D, and oral calcium supplementation may be useful in maintaining adequate serum calcium levels, although it is not essential. Blood calcium and phosphorus are monitored frequently until the levels have stabilized; they are then monitored monthly and less often until the child is seen at 6-month intervals. Renal function, blood pressure, and serum vitamin D levels are measured every 6 months. Serum magnesium levels are measured every 3 to 6 months to permit detection of hypomagnesemia, which may raise the requirement for vitamin D.
Nursing Care Management
The initial objective is recognition of hypocalcemia. Unexplained convulsions, irritability (especially to external stimuli), gastrointestinal symptoms (diarrhea, vomiting, cramping), and positive signs of tetany should lead the nurse to suspect this disorder. Much of the initial nursing care is related to the physical manifestations and includes institution of seizure and safety precautions; reduction of environmental stimuli (e.g., avoiding sudden or loud noise, bright lights, stimulating activities); and observation for signs of laryngospasm such as stridor, hoarseness, and a feeling of tightness in the throat. A tracheostomy set and injectable calcium gluconate should be located near the bedside for emergency use. The administration of calcium gluconate requires precautions against extravasation of the drug and tissue destruction.
After initiating treatment, the nurse discusses with the parents the need for continuous daily administration of calcium salts and vitamin D. Because vitamin D toxicity can be a serious consequence of therapy, parents are advised to watch for signs that include weakness, fatigue, lassitude, headache, nausea, vomiting, and diarrhea. Early renal impairment is manifested by polyuria, polydipsia, and nocturia.
HYPERPARATHYROIDISM
Hyperparathyroidism is rare in childhood but can be primary or secondary. The most common cause of primary hyperparathyroidism is adenoma of the gland (Behrman, Kliegman, and Jenson, 2004). The most common causes of secondary hyperparathyroidism are chronic renal disease, renal osteodystrophy, and congenital anomalies of the urinary tract. The common factor is hypercalcemia. The clinical signs of hyperparathyroidism are listed in Box 29-9.
Diagnostic Evaluation
Blood studies to identify elevated calcium and decreased phosphorus levels are routinely performed. Measurement of PTH, as well as several tests to isolate the cause of the hypercalcemia, such as renal function studies, should be included. Other procedures used to substantiate the physiologic consequences of the disorder include electrocardiography and radiographic bone surveys.
Therapeutic Management
Treatment depends on the cause of hyperparathyroidism. The treatment of primary hyperparathyroidism is surgical removal of the tumor or hyperplastic tissue. Treatment of secondary hyperparathyroidism is directed at the underlying contributing cause, which subsequently restores the serum calcium balance. However, in some instances such as in chronic renal failure the underlying disorder is irreversible. In this case treatment is aimed at raising serum calcium levels to inhibit the stimulatory effect of low levels on the parathyroids. This includes oral administration of calcium salts, high doses of vitamin D to enhance calcium absorption, a low-phosphorus diet, and administration of a phosphorus-mobilizing aluminum hydroxide to reduce phosphate absorption.
Nursing Care Management
The initial nursing objective is recognition of the disorder. Because secondary hyperparathyroidism is a consequence of chronic renal failure, the nurse is always alert to signs that suggest this complication, especially bone pain and fractures. Because urinary symptoms are the earliest indication, assessment of other body systems for evidence of high calcium levels is indicated when polyuria and polydipsia coexist. Clues to the possibility of hyperparathyroidism include change in behavior, especially inactivity; unexplained gastrointestinal symptoms; and cardiac irregularities.
DISORDERS OF ADRENAL FUNCTION
The adrenal cortex secretes three main groups of hormones collectively called steroids and classified according to their biologic activity: (1) glucocorticoids (cortisol, corticosterone), (2) mineralocorticoids (aldosterone), and (3) sex steroids (androgens, estrogens, and progestins). Alterations in the levels of these hormones produce significant dysfunction in a variety of body tissues and organs. Because the adrenocortical cells are capable of producing any of the steroids, pathologic conditions may result in a deficiency or an excess of more than one type of hormone. However, most are rare in children.
The adrenal medulla secretes the catecholamines epinephrine and norepinephrine. Both hormones have essentially the same effects on various organs as those caused by direct sympathetic stimulation, except that the hormonal effects last several times longer. Catecholamine-secreting tumors are the primary cause of adrenal medullary hyperfunction.
ACUTE ADRENOCORTICAL INSUFFICIENCY
The acute form of adrenocortical insufficiency (adrenal crisis) may have a number of causes during childhood. Although a rare disorder, some of the more common etiologic factors include hemorrhage into the gland from trauma, which may be caused by a prolonged, difficult labor; fulminating infections, such as meningococcemia, which result in hemorrhage and necrosis (Waterhouse-Friderichsen syndrome); abrupt withdrawal of exogenous sources of cortisone or failure to increase exogenous supplies during stress; or congenital adrenogenital hyperplasia of the salt-losing type.
Early symptoms of adrenocortical insufficiency include increased irritability, headache, diffuse abdominal pain, weakness, nausea and vomiting, and diarrhea. Other clinical signs are found in Box 29-10. In the newborn, adrenal crisis is accompanied by extreme hyperpyrexia (high temperature), tachypnea, cyanosis, and seizures. Usually there is no evidence of infection or purpura. However, hemorrhage into the adrenal gland may be evident as a palpable retroperitoneal mass.
BOX 29-10 Clinical Manifestations of Acute Adrenocortical Insufficiency
GENERALIZED HEMORRHAGIC MANIFESTATIONS (WATERHOUSE-FRIDERICHSEN SYNDROME)
Fever (increases as condition worsens)
Diagnostic Evaluation
There is no rapid, definitive test for confirmation of acute adrenocortical insufficiency. Routine procedures such as measurement of plasma cortisol levels are too time-consuming to be practical. Therefore diagnosis is usually made based on clinical presentation, especially when a fulminating sepsis is accompanied by hemorrhagic manifestations and signs of circulatory collapse despite adequate antibiotic therapy. Because there is no real danger in administering a cortisol preparation for a short period, treatment should be instituted immediately. Improvement with cortisol therapy confirms the diagnosis.
Therapeutic Management
Treatment involves replacement of cortisol, replacement of body fluids to combat dehydration and hypovolemia, administration of glucose solutions to correct hypoglycemia, and specific antibiotic therapy in the presence of infection. Initially IV hydrocortisone (Solu-Cortef) is administered. Normal saline containing 5% glucose is given parenterally to replace lost fluid, electrolytes, and glucose. If hemorrhage has been severe, whole blood may be replaced. In the event that these measures do not reverse the circulatory collapse, vasopressors are used for immediate vasoconstriction and elevation of blood pressure.
Once the child’s condition is stabilized, oral doses of cortisone, fluids, and salt are given, similar to the regimen used for chronic adrenal insufficiency. To maintain sodium retention, aldosterone is replaced by synthetic salt-retaining steroids.
Nursing Care Management
Because of the abrupt onset and potentially fatal outcome of this condition, prompt recognition is essential. Vital signs and blood pressure are taken every 15 minutes to monitor the hyperpyrexia and shocklike state. Seizure precautions are instituted, since convulsions from the elevated temperature are not uncommon. As soon as therapy is instituted, the nurse should monitor the child’s response to fluid and cortisol replacement. Too rapid administration of fluids can precipitate cardiac failure, whereas overdosage with cortisol produces hypotension and a sudden fall in temperature.
Once the acute phase is over and the hypovolemia is corrected, the child is given oral fluids, such as small quantities of ginger ale, fruit juice, or salted broth. Too rapid ingestion of oral fluids may induce vomiting, which increases dehydration. Therefore the nurse should plan a gradual schedule for reintroducing liquids. For children who refuse to drink, the prospect of having the IV infusion removed once oral fluids are increased is often a motivating factor.
The sudden, severe nature of this disorder necessitates a great deal of emotional support for the child and family. The child may be placed in an intensive care unit where the surroundings are strange and frightening. Despite the need for emergency intervention, the nurse must be sensitive to the family’s psychologic needs and prepare them for each procedure, even if this is a brief statement such as “The intravenous infusion is necessary to replace fluid that the child is losing.” Because recovery within 24 hours is often dramatic, the nurse should keep the parents apprised of the child’s condition, emphasizing signs of improvement, such as a lowered temperature and elevated blood pressure. If paralysis occurs, the nurse should assure them that this condition is temporary and quickly reversed.
CHRONIC ADRENOCORTICAL INSUFFICIENCY (ADDISON DISEASE)
Chronic adrenocortical insufficiency is rare in children. When it does occur, it is usually caused by a destructive lesion of the adrenal gland or neoplasms, or the cause is idiopathic. At one time, generalized tuberculosis was the leading cause of adrenal gland destruction.
Evidence of this disorder is usually gradual in onset, since 90% of adrenal tissue must be nonfunctional before signs of insufficiency are manifested. However, during periods of stress, when demands for additional cortisol are increased, symptoms of acute insufficiency may appear in a previously well child (Box 29-11).
Definitive diagnosis is based on measurements of functional cortisol reserve. The cortisol and urinary 17-hydroxycorticosteroid levels are low and fail to rise while plasma adrenocorticotropic hormone (ACTH) levels are elevated with corticotropin (ACTH) stimulation, the definitive test for the disease.
Therapeutic Management
Treatment involves replacement of glucocorticoids (cortisol) and mineralocorticoids (aldosterone). Some children are able to be maintained solely on oral supplements of cortisol (cortisone or hydrocortisone preparations) with a liberal intake of salt. During stressful situations, such as fever, infection, emotional upset, or surgery, the dosage must be tripled to accommodate the body’s increased need for glucocorticoids. Failure to meet this requirement will precipitate an acute crisis. Overdosage produces appearance of cushingoid signs.
Children with more severe states of chronic adrenal insufficiency require mineralocorticoid replacement to maintain fluid and electrolyte balance. Other forms of therapy include monthly injections of desoxycorticosterone acetate or implantation of desoxycorticosterone acetate pellets subcutaneously every 9 to 12 months.
Nursing Care Management
Once the disorder is diagnosed, parents need guidance concerning drug therapy. They must be aware of the continuous need for cortisol replacement. Sudden termination of the drug because of inadequate supplies or inability to ingest the oral form because of vomiting places the child in danger of an acute adrenal crisis. Therefore parents should always have a spare supply of the medication in the home. Ideally they will have a prefilled syringe of hydrocortisone and be instructed in proper technique for intramuscular administration of the drug in case of crisis. Unnecessary administration of cortisone will not harm the child, but if it is needed, it may be lifesaving. Any evidence of acute insufficiency should be reported to the practitioner immediately.
Parents also need to be aware of side effects of the drugs. Undesirable side effects of cortisone include gastric irritation, which is minimized by ingestion with food or the use of an antacid; increased excitability and sleeplessness; weight gain, which may require dietary management to prevent obesity; and, rarely, behavioral changes, including depression or euphoria. Parents should be aware of signs of overdose and report these to the practitioner. In addition, the drug has a bitter taste, which creates a challenge for nurses and parents in its administration.
Because the body cannot supply endogenous sources of cortical hormones during times of stress, the home environment should be stable and relatively unstressful. Parents need to be aware that during periods of emotional or physical crisis the child requires additional hormone replacement. The child should wear medical identification, such as a bracelet, to permit medical personnel to adjust requirements during emergency care.
CUSHING SYNDROME
Cushing syndrome is a characteristic group of manifestations caused by excessive circulating free cortisol. It can result from a variety of causes, which generally fall into one of five categories (Box 29-12). Cushing syndrome in young children may be due to an adrenal tumor (Moshang, 2003).
Cushing syndrome is uncommon in children. When seen, it is often caused by excessive or prolonged steroid therapy that produces a cushingoid appearance (Fig. 29-1). This condition is reversible once the steroids are gradually discontinued. Abrupt withdrawal will precipitate acute adrenal insufficiency. Gradual withdrawal of exogenous supplies is necessary to allow the anterior pituitary an opportunity to secrete increasing amounts of ACTH to stimulate the adrenals to produce cortisol.
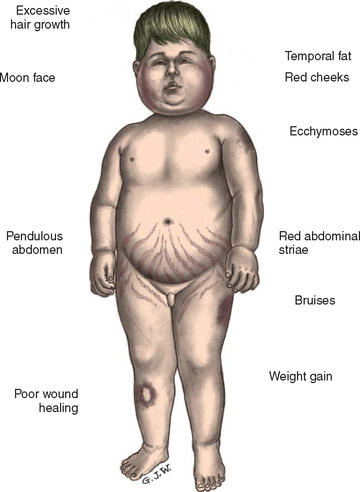
Clinical Manifestations
Because the actions of cortisol are widespread, clinical manifestations are equally profound and diverse. Those symptoms that produce changes in physical appearance occur early in the disorder and are of considerable concern to school-age and older children. The physiologic disturbances, such as hyperglycemia, susceptibility to infection, hypertension, and hypokalemia, may have life-threatening consequences unless recognized early and treated successfully. Children with short stature may be responding to increased cortisol levels, resulting in Cushing syndrome. Cortisol inhibits the action of GH.
Diagnostic Evaluation
Several tests are helpful in confirming excess cortisol levels. They include fasting blood glucose levels for hyperglycemia, serum electrolyte levels for hypokalemia and alkalosis, 24-hour urinary levels of elevated 17-hydroxycorticoids and 17-ketosteroids, and radiographic studies of bone for evidence of osteoporosis and of the skull for enlargement of the sella turcica. Another procedure used to establish a more definitive diagnosis is the dexamethasone (cortisone) suppression test (Nieman and Ilias, 2005). Administration of an exogenous supply of cortisone normally suppresses ACTH production. However, in individuals with Cushing syndrome, cortisol levels remain elevated. This test is helpful in differentiating between children who are obese and those who appear to have cushingoid features.
Therapeutic Management
Treatment depends on the cause. In most cases surgical intervention involves bilateral adrenalectomy and postoperative replacement of the cortical hormones (the therapy for this is the same as that outlined for chronic adrenal insufficiency). If a pituitary tumor is found, surgical extirpation or irradiation may be chosen. In either of these instances, treatment of panhypopituitarism with replacement of GH, thyroid extract, ADH, gonadotropins, and steroids may be necessary for an indefinite period (Nieman and Ilias, 2005).
Nursing Care Management
Nursing care also depends on the cause. When cushingoid features are caused by steroid therapy, the effects may be lessened with administration of the drug early in the morning and on an alternate-day basis. Giving the drug early in the day maintains the normal diurnal pattern of cortisol secretion. If given during the evening, it is more likely to produce symptoms because endogenous cortisol levels are already low and the additional supply exerts more pronounced effects. An alternate-day schedule allows the anterior pituitary an opportunity to maintain more normal hypothalamic-pituitary-adrenal control mechanisms.
If an organic cause is found, nursing care is related to the treatment regimen. Although a bilateral adrenalectomy permanently solves one condition, it reciprocally produces another syndrome. Before surgery, parents need to be adequately informed of the operative benefits and disadvantages. Postoperative teaching regarding drug replacement is the same as discussed in the previous section.
Anorexia and nausea and vomiting are common and may be improved with the use of nasogastric decompression. Muscle and joint pain may be severe, requiring use of analgesics. The psychologic depression can be profound and may not improve for months. Parents should be aware of the physiologic reasons behind these symptoms in order to be supportive of the child.
CONGENITAL ADRENAL HYPERPLASIA
Congenital adrenal hyperplasia (CAH) is a family of disorders caused by decreased enzyme activity required for cortisol production in the adrenal cortex. The most common defect is 21-hydroxylase deficiency, which constitutes more than 90% of all cases of CAH (American Academy of Pediatrics, 2000; Levine, 2000). This deficiency occurs in approximately 1 per 12,000 to 15,000 births and causes overproduction of the adrenal androgens, resulting in virilization of the female fetus.
Excessive androgens cause masculinization of the urogenital system at approximately the tenth week of fetal development. The most pronounced abnormalities occur in the girl, who is born with varying degrees of ambiguous genitalia. Masculinization of external genitalia causes the clitoris to enlarge so that it appears as a small phallus. Fusion of the labia produces a saclike structure resembling the scrotum without testes. However, no abnormal changes occur in the internal sexual organs, although the vaginal orifice is usually closed by the fused labia. The label ambiguous genitalia should be applied to any infant with hypospadias or micropenis and no palpable gonads, and a diagnostic evaluation for CAH should be contemplated. Males do not display genital abnormalities at birth (New and Ghizzoni, 2003).
Increased pigmentation of skin creases and genitalia caused by increased ACTH may be a subtle sign of adrenal insufficiency. A salt-wasting crisis frequently occurs, usually within the first few weeks of life (Behrman, Kliegman, and Jenson, 2004). Infants fail to gain weight, and hyponatremia and hyperkalemia may be significant. Cardiac arrest can occur.
Untreated CAH results in early sexual maturation, with enlargement of the external sexual organs; development of axillary, pubic, and facial hair; deepening of the voice; acne; and marked increase in musculature with changes toward an adult male physique. However, in contrast to precocious puberty, breasts do not develop in the female, and she remains amenorrheic and infertile. In the male the testes remain small, and spermatogenesis does not occur. In both sexes linear growth is accelerated, and epiphyseal closure is premature, resulting in short stature by the end of puberty.
Diagnostic Evaluation
Clinical diagnosis is initially based on congenital abnormalities that lead to difficulty in assigning sex to the newborn and on signs and symptoms of adrenal insufficiency or hypertension. Definitive diagnosis is confirmed by evidence of increased 17-ketosteroid levels in most types of CAH (Levine, 2000). Usually the level of 17-hydroxycorticoids is low or near normal. In complete 21-hydroxylase deficiency, blood electrolytes demonstrate loss of sodium and chloride and elevation of potassium. In older children bone age is advanced, and linear growth is increased. Chromosome typing for positive sex determination and to rule out any other genetic abnormality (e.g., Turner syndrome) is always done in any case of ambiguous genitalia.
Another test that can be used to visualize the presence of pelvic structures is ultrasonography, a noninvasive, painless imaging technique that does not require anesthesia or sedation. It is especially useful in CAH because it readily identifies the absence or presence of female reproductive organs in a newborn or child with ambiguous genitalia. Because ultrasonography yields immediate results, it has the advantage of determining the child’s gender long before the more complex laboratory results for chromosome analysis or steroid levels are available.
Therapeutic Management
The initial medical objective is to confirm the diagnosis and assign a sex to the child, usually according to the genotype. In both sexes cortisone is administered to suppress the abnormally high secretions of ACTH. If cortisone is begun early enough, it is very effective. Cortisone depresses the secretion of ACTH by the adenohypophysis, which in turn inhibits the secretion of adrenocorticosteroids, which stems the progressive virilization. The signs and symptoms of masculinization in the female gradually disappear, and excessive early linear growth is slowed. Puberty occurs normally at the appropriate age.
The recommended oral dosage is divided to simulate the normal diurnal pattern of ACTH secretion. Because these children are unable to produce cortisol in response to stress, it is necessary to increase the dosage during episodes of infection, fever, or other stresses. Acute emergencies require immediate IV or intramuscular administration. Emergency situations include bacterial and viral infections, vomiting, surgery, fractures, major injuries, and sometimes insect stings.
Children with the salt-losing type of CAH require aldosterone replacement, as outlined under chronic adrenal insufficiency, and supplementary dietary salt. Frequent laboratory tests are conducted to assess the effects on electrolytes, hormonal profiles, and renin levels. The frequency of testing is individualized to the child.
Depending on the degree of masculinization in the female, reconstructive surgery may be required to reduce the size of the clitoris, separate the labia, and create a vaginal orifice (Miranda, Oliveira-Filho, Lemos-Marini, and others, 2004). Surgery is performed when the infant is physically able to withstand the procedure but before she is old enough to be aware of the abnormal genitalia. Plastic surgery is generally done in stages and yields excellent cosmetic results. Reports concerning sexual satisfaction after partial clitoridectomy indicate that the capacity for orgasm and sexual gratification is not necessarily impaired.
Unfortunately, not all children with CAH are diagnosed at birth and raised in accordance with their genetic sex. Particularly in the case of affected females, masculinization of the external genitalia may have led to sex assignment as a male. In males, diagnosis is usually delayed until early childhood, when signs of virilism appear. In these situations it is advisable to continue rearing the child as a male in accordance with assigned sex and phenotype. Hormone replacement may be required to permit linear growth and to initiate male pubertal changes. Surgery is usually indicated to remove the female organs and reconstruct the phallus for satisfactory sexual relations. These individuals are not fertile.
Nursing Care Management
Of major importance is recognition of ambiguous genitalia in newborns. If there is any question regarding assignment of sex, the parents need to be told immediately to prevent the embarrassing situation of informing family members of the child’s sex and then having to change the announcement.
As soon as the sex is determined, parents should be informed of the findings and encouraged to choose an appropriate name, and the child should be identified as a male or female, with no reference to ambiguous sex. If the appearance of the enlarged genitalia in a girl concerns the parents, they should be encouraged to discuss their feelings. Suggesting ways to avoid questioning remarks from visitors, such as diapering the child in a separate room, is also helpful. If surgery is anticipated, showing parents before-and-after photographs of reconstruction helps to reinforce the expected cosmetic benefits.
In general, rearing the genetically female child as a girl is preferred because of the success of surgical intervention and the satisfactory results with hormones in reversing virilism and providing a prospect of normal puberty and the ability to conceive. This is in contrast to the choice of rearing the child as a boy, in which case the child is sterile and may never be able to function satisfactorily in heterosexual relationships. If the parents persist in their decision to assign a male sex to a genetically female child, a psychologic consultation should be requested to explore their motivations and ensure their understanding of the future consequences for the child.
Nursing care management regarding cortisol and aldosterone replacement are the same as those discussed for chronic adrenocortical insufficiency. Because infants are especially prone to dehydration and salt-losing crises, parents need to be aware of signs of dehydration and the urgency of immediate medical intervention to stabilize the child’s condition. Parents should have injectable hydrocortisone available and know how to prepare and administer the intramuscular injection (see Chapter 22).
In the unfortunate situation in which the sex is erroneously assigned and the correct sex determined later, parents need a great deal of help in understanding the reason for the incorrect sex identification and the options for sex reassignment or medical-surgical intervention. Because children become aware of their sexual identity by 18 months to 2 years of age, it is believed that any reassignment after this period can cause tremendous psychologic conflicts in the child. Therefore sex rearing should be continued as previously established with medical-surgical intervention as required.
PHEOCHROMOCYTOMA
Pheochromocytoma is a rare tumor characterized by secretion of catecholamines. The tumor most commonly arises from the chromaffin cells of the adrenal medulla but may occur wherever these cells are found, such as along the paraganglia of the aorta or thoracolumbar sympathetic chain (Pacak, Eisenhofer, Ahlman, and others, 2007). Approximately 10% of these tumors are located in extraadrenal sites. In children they are frequently bilateral or multiple and are generally benign. Often there is a familial transmission of the condition as an autosomal dominant trait (Behrman, Kliegman, and Jenson, 2004).
The clinical manifestations of pheochromocytoma are caused by an increased production of catecholamines, producing hypertension, tachycardia, headache, decreased gastrointestinal activity with resultant constipation, increased metabolism with anorexia, weight loss, hyperglycemia, polyuria, polydipsia, hyperventilation, nervousness, heat intolerance, and diaphoresis. In severe cases, signs of congestive heart failure are evident.
Diagnostic Evaluation
The clinical manifestations mimic those of other disorders, such as hyperthyroidism or DM.
Therapeutic Management
Definitive treatment consists of surgical removal of the tumor. In children the tumors may be bilateral, requiring a bilateral adrenalectomy and lifelong glucocorticoid and mineralocorticoid therapy. The major complications that can occur during surgery are severe hypertension, tachyarrhythmias, and hypotension. The first two are caused by excessive release of catecholamines during manipulation of the tumor, and the latter results from catecholamine withdrawal and hypovolemic shock.
Preoperative medication to inhibit the effects of catecholamines is begun 1 to 3 weeks before surgery to prevent these complications. The major group of drugs used is the α-adrenergic blocking agents with or without β-adrenergic blocking agents. The most commonly used α-adrenergic blocker is phenoxybenzamine (Dibenzyline), a long-acting medication given orally every 12 hours. The shorter-acting phentolamine (Regitine) is equally effective but less satisfactory for long-term use, although it is useful for acute hypertension. To control catecholamine release when α-adrenergic blocking agents are inadequate, the child is given β-adrenergic blocking agents.
Success of therapy is judged by lowering of blood pressure to normal, absence of hypertensive attacks (flushing or blanching, fainting, headache, palpitations, tachycardia, nausea and vomiting, profuse sweating), heat tolerance, decrease in perspiration, and disappearance of hyperglycemia. A disadvantage of these drugs is their inability to block the effects of catecholamines on beta receptors.
Nursing Care Management
An initial nursing objective is identification of children with this disorder. Outstanding clues are hypertension and hypertensive attacks. Because of behavioral changes (nervousness, excitability, overactivity, even psychosis), increased cardiac and respiratory activity may appear to be related to an acute anxiety attack. Therefore a careful history of the onset of symptoms and association with stressful events is helpful in distinguishing between an organic and a psychologic cause for the symptoms.
Preoperative nursing care involves frequent monitoring of vital signs and observation for evidence of hypertensive attacks and congestive heart failure. Therapeutic effects are evidenced by normal vital signs and absence of glycosuria. Daily blood glucose levels, urine acetone, and any signs of hyperglycemia are noted and reported immediately.
The environment is made conducive to rest and free of emotional stress. This requires adequate preparation during hospital admission and before surgery. Parents are encouraged to room-in with their child and to participate in care. Play activities need to be tailored to the child’s energy level without being overly strenuous or challenging because these can increase metabolic rate and promote frustration and anxiety.
After surgery the child is observed for signs of shock from removal of excess catecholamines. If a bilateral adrenalectomy was performed, the nursing interventions are those discussed for chronic adrenocortical insufficiency.
DISORDERS OF PANCREATIC HORMONE SECRETION: DIABETES MELLITUS
DM is a chronic disorder of metabolism characterized by a partial or complete deficiency of the hormone insulin. It is the most common metabolic disease, resulting in metabolic adjustment or physiologic change in almost all areas of the body. Approximately one in three children born in the United States will develop diabetes. The odds are higher for African-American and Hispanic children: nearly 50% of them will develop diabetes (Urrutia-Rojas and Menchaca, 2006). DM in children can occur at any age but has a peak incidence between ages 10 and 15 years, with 75% diagnosed before 18 years of age. The incidence in boys is slightly higher than in girls (1:1 to 1.2:1).
Traditionally DM had been classified according to the type of treatment needed. The old categories were insulin-dependent diabetes mellitus (IDDM), or type I; and non—insulin-dependent diabetes mellitus (NIDDM), or type II. In 1997 these terms were eliminated because treatment can vary (some people with NIDDM require insulin) and because the terms do not indicate the underlying problem. The new terms are type 1 and type 2, using Arabic symbols to avoid confusion (e.g., type II could be read as type eleven) (American Diabetes Association, 2001). The characteristics of type 1 DM and type 2 DM are outlined in Table 29-2.
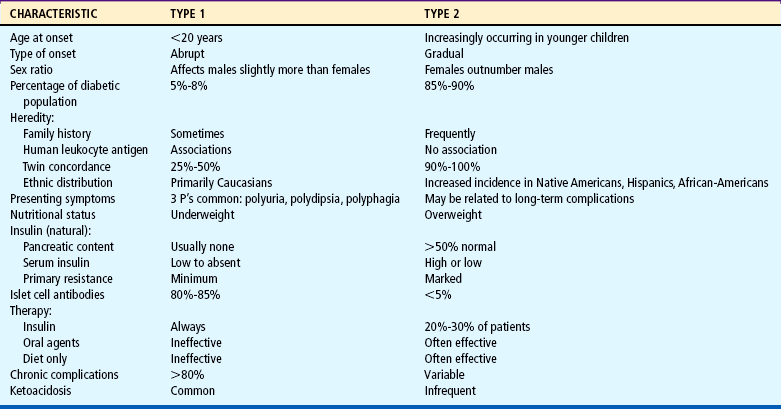
Type 1 DM is more prominent in Caucasians, with an incidence of 20 per 100,000; the incidence in African-Americans is 11 per 100,000; the incidence in Hispanics is 15.2 per 100,000; and the incidence in Cubans is 2.6 per 100,000. Native Americans tend to develop type 2 DM rather than type 1 DM even when diagnosed in childhood. The Pima Tribe reports a greater than 55% incidence of type 2 DM. Type 1 diabetes is characterized by destruction of the pancreatic β cells, which produce insulin; this usually leads to absolute insulin deficiency. Type 1 diabetes has two forms. Immune-mediated DM results from an autoimmune destruction of the β cells; it typically starts in children or young adults who are slim, but it can arise in adults of any age. Idiopathic type 1 refers to rare forms of the disease that have no known cause.
Type 2 diabetes usually arises because of insulin resistance, in which the body fails to use insulin properly, combined with relative (rather than absolute) insulin deficiency. People with type 2 can range from predominantly insulin resistant with relative insulin deficiency to predominantly deficient in insulin secretion with some insulin resistance. It typically occurs in those who are over 45, are overweight and sedentary, and have a family history of diabetes.
The symptomatology of diabetes is more readily recognizable in children than in adults, so it is surprising that the diagnosis may sometimes be missed or delayed. Diabetes is a great imitator; influenza, gastroenteritis, and appendicitis are the conditions most often diagnosed when it turns out that the disease is really diabetes (Box 29-13).
BOX 29-13 Clinical Manifestations of Type 1 Diabetes Mellitus
Pathophysiology
Insulin is needed to support the metabolism of carbohydrates, fats, and proteins, primarily by facilitating the entry of these substances into the cell. Insulin is needed for the entry of glucose into the muscle and fat cells, prevention of mobilization of fats from fat cells, and storage of glucose as glycogen in the cells of liver and muscle. Insulin is not needed for the entry of glucose into nerve cells or vascular tissue. The chemical composition and molecular structure of insulin are such that it fits into receptor sites on the cell membrane. Here it initiates a sequence of poorly defined chemical reactions that alter the cell membrane to facilitate the entry of glucose into the cell and stimulate enzymatic systems outside the cell that metabolize the glucose for energy production.
With a deficiency of insulin, glucose is unable to enter the cell, and its concentration in the bloodstream increases. The increased concentration of glucose (hyperglycemia) produces an osmotic gradient that causes the movement of body fluid from the intracellular space to the interstitial space, then to the extracellular space and into the glomerular filtrate in order to “dilute” the hyperosmolar filtrate. Normally the renal tubular capacity to transport glucose is adequate to reabsorb all the glucose in the glomerular filtrate. When the glucose concentration in the glomerular filtrate exceeds the renal threshold (6180 mg/dl), glucose spills into the urine (glycosuria) along with an osmotic diversion of water (polyuria), a cardinal sign of diabetes. The urinary fluid losses cause the excessive thirst (polydipsia) observed in diabetes. This water “washout” results in a depletion of other essential chemicals, especially potassium.
Protein is also wasted during insulin deficiency. Because glucose is unable to enter the cells, protein is broken down and converted to glucose by the liver (glucogenesis); this glucose then contributes to the hyperglycemia. These mechanisms are similar to those seen in starvation when substrate (glucose) is absent. The body is actually in a state of starvation during insulin deficiency. Without the use of carbohydrates for energy, fat and protein stores are depleted as the body attempts to meet its energy needs. The hunger mechanism is triggered, but increased food intake (polyphagia) enhances the problem by further elevating blood glucose.
Ketoacidosis.: When insulin is absent or insulin sensitivity is altered, glucose is unavailable for cellular metabolism, and the body chooses alternate sources of energy, principally fat. Consequently fats break down into fatty acids, and glycerol in the fat cells is converted by the liver to ketone bodies (β-hydroxybutyric acid, acetoacetic acid, acetone). Any excess is eliminated in the urine (ketonuria) or the lungs (acetone breath). The ketone bodies in the blood (ketonemia) are strong acids that lower serum pH, producing ketoacidosis.
Ketones are organic acids that readily produce excessive quantities of free hydrogen ions, causing a fall in plasma pH. Then chemical buffers in the plasma, principally bicarbonate, combine with the hydrogen ions to form carbonic acid, which readily dissociates into water and carbon dioxide. The respiratory system attempts to eliminate the excess carbon dioxide by increased depth and rate’Kussmaul respirations, or the hyperventilation characteristic of metabolic acidosis. The ketones are buffered by sodium and potassium in the plasma. The kidney attempts to compensate for the increased pH by increasing tubular secretion of hydrogen and ammonium ions in exchange for fixed base, thus depleting the base buffer concentration.
With cellular death, potassium is released from the cell (intracellular fluid) into the bloodstream (extracellular fluid) and excreted by the kidney, where the loss is accelerated by osmotic diuresis. The total body potassium is then decreased, even though the serum potassium level may be elevated as a result of the decreased fluid volume in which it circulates. Alteration in serum and tissue potassium can lead to cardiac arrest.
If these conditions are not reversed by insulin therapy in combination with correction of the fluid deficiency and electrolyte imbalance, progressive deterioration occurs, with dehydration, electrolyte imbalance, acidosis, coma, and death. Diabetic ketoacidosis (DKA) should be diagnosed promptly in a seriously ill patient and therapy instituted in an intensive care unit.
Long-Term Complications: Long-term complications of diabetes involve both the microvasculature and the macrovasculature. The principal microvascular complications are nephropathy, retinopathy, and neuropathy. Microvascular disease develops during the first 30 years of diabetes, beginning in the first 10 to 15 years after puberty, with renal involvement evidenced by proteinuria and clinically apparent retinopathy.
With poor diabetic control, vascular changes can appear as early as 2½ to 3 years after diagnosis; however, with good to excellent control, changes can be postponed for 20 or more years. Intensive insulin therapy appears to delay the onset and slow the progression of clinically important retinopathy, including vision-threatening lesions, nephropathy, and neuropathy. Hypertension and atherosclerotic cardiovascular disease are also major causes of morbidity and mortality in patients with DM (Karnik, Fields, and Shannon, 2007). The postpubertal duration, not the total duration, of type 1 DM is implicated as a risk factor for the development of microvascular disease (Schultz, Konopelska-Bahu, Dalton, and others, 1999). The process appears to be one of glycosylation, wherein proteins from the blood become deposited in the walls of small vessels (e.g., glomeruli), where they become trapped by “sticky” glucose compounds (glycosyl radicals). The buildup of these substances over time causes narrowing of the vessels, with subsequent interference with microcirculation to the affected areas (Beisswenger, Szwergold, and Yeo, 2001). Macrovascular disease develops after 25 years of diabetes and creates the predominant problems in patients with type 2 DM.
Other complications have been observed in children with type 1 DM. Hyperglycemia appears to influence thyroid function, and altered function is frequently observed at the time of diagnosis and in poorly controlled diabetes. Limited mobility of small joints of the hand occurs in 30% of 7- to 18-year-old children with type 1 DM and appears to be related to changes in the skin and soft tissues surrounding the joint as a result of glycosylation.
Diagnostic Evaluation
Three groups of children who should be considered as candidates for diabetes are (1) children who have glycosuria, polyuria, and a history of weight loss or failure to gain despite a voracious appetite; (2) those with transient or persistent glycosuria; and (3) those who display manifestations of metabolic acidosis, with or without stupor or coma. In every case diabetes must be considered if there is glycosuria, with or without ketonuria, and unexplained hyperglycemia.
Glycosuria by itself is not diagnostic of diabetes. Other sugars, such as galactose, can produce a positive result with certain test strips, and a mild degree of glycosuria can be caused by other conditions, such as infection, trauma, emotional or physical stress, hyperalimentation, and some renal or endocrine diseases.
An 8-hour fasting blood glucose level of 126 mg/dl or more, a random blood glucose value of 200 mg/dl or more accompanied by classic signs of diabetes, or an oral glucose tolerance test (OGTT) finding of 200 mg/dl or more in the 2-hour sample is almost certain to indicate diabetes (American Diabetes Association, 2005; Hoffman, 2003). Postprandial blood glucose determinations and the traditional OGTTs have yielded low detection rates in children and are not usually necessary for establishing a diagnosis. Serum insulin levels may be normal or moderately elevated at the onset of diabetes; delayed insulin response to glucose indicates impaired glucose tolerance.
Ketoacidosis must be differentiated from other causes of acidosis or coma, including hypoglycemia, uremia, gastroenteritis with metabolic acidosis, salicylate intoxication encephalitis, and other intracranial lesions. DKA is a state of relative insulin insufficiency and may include the presence of hyperglycemia (blood glucose level ≥330 mg/dl), ketonemia (strongly positive), acidosis (pH, <7.30 and bicarbonate, <15 mmol/L), glycosuria, and ketonuria (Magee and Bhatt, 2001). Tests used to determine glycosuria and ketonuria are the glucose oxidase tapes (Keto-Diastix).
Therapeutic Management
The management of the child with type 1 DM consists of a multidisciplinary approach involving the family; the child (when appropriate); and professionals, including a pediatric endocrinologist, diabetes nurse educator, nutritionist, and exercise physiologist. Often psychologic support from a mental health professional is also needed. Communication among the team members is essential and extends to other individuals in the child’s life, such as teachers, school nurse, school guidance counselor, and coach.
The definitive treatment is replacement of insulin that the child is unable to produce. However, insulin needs are also affected by emotions, nutritional intake, activity, and other life events such as illnesses and puberty. The complexity of the disease and its management requires that the child and family incorporate diabetes needs into their lifestyle. Medical and nutritional guidance are primary, but management also includes continuing diabetes education, family guidance, and emotional support.
Insulin Therapy.: Insulin replacement is the cornerstone of management of type 1 DM. Insulin dosage is tailored to each child based on home blood glucose monitoring. The goal of insulin therapy is maintaining near-normal blood glucose values while avoiding too frequent episodes of hypoglycemia. The goals of treatment are to maintain near-normal glucose levels of less than 126 mg/dl, and glycosylated hemoglobin (hemoglobin A1c) of 7% or less (Hannon, Gungor, and Arslanian, 2006). Glycemic control decreases the likelihood of long-term complications in patients with DM (Petitti, Imperatore, Palla, and others, 2007). Insulin is administered as two or more injections per day or as continuous subcutaneous infusion using a portable insulin pump.
Healthy pancreatic cells secrete insulin at a low but steady basal rate with superimposed bursts of increased secretion that coincide with intake of nutrients. Consequently insulin levels in the blood increase and decrease coincidentally with rises and falls in blood glucose levels. In addition, insulin is secreted directly into the portal circulation; therefore the liver, which is the major site of glucose disposal, receives the largest concentration of insulin. No matter which method of insulin replacement is used, this normal pattern cannot be duplicated. Subcutaneous injection results in absorption of the drug into the general circulation, thus reducing the concentrations of insulin to which the liver is exposed.
Insulin Preparations.: Insulin is available in highly purified pork preparations and in human insulin biosynthesized by and extracted from bacterial or yeast cultures. Most clinicians suggest human insulin as the treatment of choice. Insulin is available in rapid-, intermediate-, and long-acting preparations, and all are packaged in the strength of 100 units/ml. Some insulins are available as premixed insulins, such as 70/30 and 50/50 ratios, the first number indicating the percentage of intermediate-acting and the second number the percentage of rapid-acting insulin. The different types of insulin are found in Box 29-14.
There are four types of insulin, based on the following criteria:
However, each person responds to insulin in his or her own way. That is why onset, peak time, and duration are given as ranges.
Rapid-acting insulin (e.g., NovoLog) reaches the blood within 15 minutes after injection. The insulin peaks 30 to 90 minutes later and may last as long as 5 hours.
Short-acting (regular) insulin (e.g., Novolin R) usually reaches the blood within 30 minutes after injection. The insulin peaks 2 to 4 hours later and stays in the blood for about 4 to 8 hours.
Intermediate-acting insulins (e.g., Novolin N) reach the blood 2 to 6 hours after injection. The insulins peak 4 to 14 hours later and stay in the blood for about 14 to 20 hours.
Long-acting insulin (e.g., Lantus) takes 6 to 14 hours to start working. It has no peak or a very small peak 10 to 16 hours after injection. The insulin stays in the blood between 20 and 24 hours.
Some insulins come mixed together (e.g., Novolin 70/30). For example, you can buy regular insulin and NPH insulins already mixed in one bottle, which makes it easier to inject two kinds of insulin at the same time. However, you cannot adjust the amount of one insulin without also changing how much you get of the other insulin.
Adapted from American Diabetes Association: Resource guide 2005, retrieved April 10, 2008, from http://www.diabetes.org/rg2005/insulin.jsp.
Dosage.: Conventional management has consisted of a twice-daily insulin regimen of a combination of rapid-acting and intermediate-acting insulin drawn up into the same syringe and injected before breakfast and before the evening meal. The amount of morning regular insulin is determined by patterns in the late morning and lunchtime blood glucose values. The morning intermediate-acting dosage is determined by patterns in the late afternoon and supper blood glucose values. Fasting blood glucose patterns at breakfast help determine the evening dose of intermediate insulin, and the blood glucose patterns at bedtime help determine the evening dose of rapid-acting (regular) insulin. For some children, better morning glucose control is achieved by a later (bedtime) injection of intermediate-acting insulin.
Regular insulin is best administered at least 30 minutes before meals. This allows sufficient time for absorption and results in a significantly greater reduction in the postprandial rise in blood glucose than if the meal were eaten immediately after the insulin injection. Intensive therapy consists of multiple injections throughout the day with a once- or twice-daily dose of long-acting (Ultralente) insulin to simulate the basal insulin secretion and injections of rapid-acting insulin before each meal. A multiple daily injection program reduces microvascular complications of diabetes in young, healthy patients who have type 1 DM.
The precise dose of insulin needed cannot be predicted. Therefore the total dosage and percentage of regular- to intermediate-acting insulin should be determined empirically for each child. Usually 60% to 75% of the total daily dose is given before breakfast, and the remainder before the evening meal. Furthermore insulin requirements do not remain constant but change continuously during growth and development; the need varies according to the child’s activity level and pubertal status. For example, less insulin is required during spring and summer months, when the child is more active. Illness also alters insulin requirements. Some children require more frequent insulin administration. This includes children with difficult-to-control diabetes and children during the adolescent growth spurt.
Methods of Administration.: Daily insulin is administered subcutaneously by twice-daily injections, by multiple-dose injections, or by means of an insulin infusion pump. The insulin pump is an electromechanical device designed to deliver fixed amounts of regular or lispro insulin continuously (basal rate), thereby more closely imitating the release of the hormone by the islet cells (Olohan and Zappitelli, 2003). Although the pump delivers a programmed amount of basal insulin, the child or parent must program a dose for the pump to deliver before each meal.
The system consists of a syringe to hold the insulin, a plunger, and a computerized mechanism to drive the plunger. The insulin flows from the syringe through a catheter to a needle inserted into subcutaneous tissue (the abdomen or thigh), and the lightweight device is worn on a belt or a shoulder holster. The needle and catheter are changed every 48 to 72 hours by the child or parent, using aseptic technique, and then taped in place.
Although the pump provides more consistent insulin delivery, it has certain disadvantages. Pump therapy is expensive and requires commitment from the parent and child. A certain level of math skills is required to calculate infusion rates. It should also not be removed for more than 1 hour at a time, which may limit some activities. Skin infections are common; and, as with any other mechanical device, it is subject to malfunction. However, the pumps are equipped with alarms that signal problems, such as a depleted battery, an occluded needle or tubing, or a microprocessor malfunction.
Monitoring.: Daily monitoring of blood glucose levels is an essential aspect of appropriate DM management. Plasma blood glucose and hemoglobin A1c goal ranges are found in Table 29-3.
TABLE 29-3
Plasma Blood Glucose and Hemoglobin A1c Goals for Type 1 Diabetes Mellitus by Age-Group
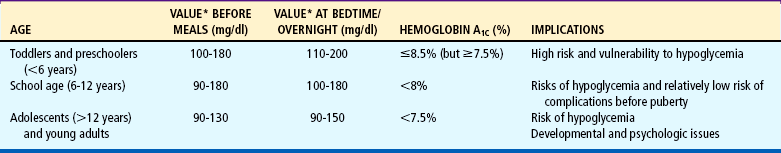
*Plasma blood glucose goal range.
Modified from American Diabetes Association: Standards of medical care in diabetes, Diabetes Care 28(Suppl):S4–36, 2005.
Blood Glucose.: Self-monitoring of blood glucose (SMBG) has improved diabetes management and is used successfully by children from the onset of their diabetes. By testing their own blood, children are able to change their insulin regimen to maintain their glucose level in the euglycemic (normal) range of 80 to 120 mg/dl. Diabetes management depends to a great extent on SMBG. In general, children tolerate the testing well.
Glycosylated Hemoglobin.: The measurement of glycosylated hemoglobin (hemoglobin A1c) levels is a satisfactory method for assessing control of the diabetes. As red blood cells circulate in the bloodstream, glucose molecules gradually attach to the hemoglobin A molecules and remain there for the lifetime of the red blood cell, approximately 120 days. The attachment is not reversible; therefore this glycosylated hemoglobin reflects the average blood glucose levels over the previous 2 to 3 months. The test is a satisfactory method for assessing control, detecting incorrect testing, monitoring effectiveness of changes in treatment, defining patients’ goals, and detecting nonadherence. Nondiabetic hemoglobin A1c values are generally between 4% and 6% but can vary by laboratory. Diabetes control for children depends on age, with hemoglobin A1c levels of 6.5% to 8% indicating a slightly elevated but acceptable range (American Diabetes Association, 2005). Hemoglobin A1c levels of less than 7% are a well-established goal at most care centers.
Nutrition.: Essentially the nutritional needs of children with diabetes are no different from those of healthy children. Children with diabetes need no special foods or supplements. They need sufficient calories to balance daily expenditure for energy and to satisfy the requirement for growth and development. Unlike the child without diabetes, whose insulin is secreted in response to food intake, insulin injected subcutaneously has a relatively predictable time of onset, peak effect, duration of action, and absorption rate depending on the type of insulin used. Consequently the timing of food consumption must be regulated to correspond to the timing and action of the insulin prescribed.
Meals and snacks must be eaten according to peak insulin action, and the total number of calories and proportions of basic nutrients must be consistent from day to day. The constant release of insulin into the circulation makes the child prone to hypoglycemia between the three daily meals unless a snack is provided between meals and at bedtime. The distribution of calories should be calculated to fit the activity pattern of each child. For example, a child who is more active in the afternoon will need a larger snack at that time. This larger snack might also be split to allow some food at school and some food after school. Food intake should be altered to balance food, insulin, and exercise. Extra food is needed for increased activity.
Concentrated sweets are discouraged, and because of the increased risk for atherosclerosis in persons with DM, fat is reduced to 30% or less of the total caloric requirement. Dietary fiber has become increasingly important in dietary planning because of its influence on digestion, absorption, and metabolism of many nutrients. It has been found to diminish the rise in blood glucose after meals.
Correctly used, the diet allows for flexibility and the incorporation of preferred foods in most instances. For the growing child, food restriction should never be used for diabetes control, although caloric restrictions may be imposed for weight control if the child is overweight. In general, the child’s appetite should be the guide for the amount of calories needed, with the total caloric intake adjusted to appetite and activity.
Exercise.: Exercise is encouraged and never restricted unless indicated by other health conditions. Exercise lowers blood glucose levels, depending on the intensity and duration of the activity. Consequently exercise should be included as part of diabetes management, and the type and amount of exercise should be planned around the child’s interests and capabilities. However, in most instances children’s activities are unplanned, and the resulting decrease in blood glucose can be compensated for by providing extra snacks before (and, if the exercise is prolonged, during) the activity. In addition to a feeling of well-being, regular exercise aids in utilization of food and often results in a reduction of insulin requirements.
Hypoglycemia.: Occasional episodes of hypoglycemia are an integral part of insulin therapy, and an objective of diabetes management is to achieve the best possible glycemic control while minimizing the frequency and severity of hypoglycemia. Even with good control, a child may frequently experience mild symptoms of hypoglycemia. If the signs and symptoms are recognized early and promptly relieved by appropriate therapy, the child’s activity should be interrupted for no more than a few minutes
The signs and symptoms of hypoglycemia are caused by both increased adrenergic activity and impaired brain function. The increased adrenergic nervous system activity plus increased secretion of catecholamines produces nervousness, pallor, tremulousness, palpitations, sweating, and hunger. Weakness, dizziness, headache, drowsiness, irritability, loss of coordination, seizures, and coma are more severe responses and reflect CNS glucose deprivation and the body’s attempts to elevate the serum glucose levels.
It is often difficult to distinguish between hyperglycemia and a hypoglycemic reaction (Table 29-4). Because the symptoms are similar and usually begin with changes in behavior, the simplest way to differentiate between the two is to test the blood glucose level. The blood glucose level is low in hypoglycemia, whereas in hyperglycemia the glucose level is significantly elevated. Urinary ketones may be present after hypoglycemia as a result of starvation ketone production. In doubtful situations it is safer to give the child some simple carbohydrate. This will help alleviate the symptoms in the case of hypoglycemia but will do little harm if the child is hyperglycemic.
TABLE 29-4
Comparison of Manifestations of Hypoglycemia and Hyperglycemia
| VARIABLE | HYPOGLYCEMIA | HYPERGLYCEMIA |
| Onset | Rapid (minutes) | Gradual (days) |
| Mood | Labile, irritable, nervous, weepy | Lethargic |
| Mental status | Difficulty concentrating, speaking, focusing, coordinating | Dulled sensorium |
| Nightmares | Confusion | |
| Inward feeling | Shaky feeling | Thirst |
| Hunger | Weakness | |
| Headache | Nausea and vomiting | |
| Dizziness | Abdominal pain | |
| Skin | Pallor | Flushed |
| Sweating | Signs of dehydration | |
| Mucous membranes | Normal | Dry, crusty |
| Respirations | Shallow, normal | Deep, rapid (Kussmaul) |
| Pulse | Tachycardia, palpitations | Less rapid, weak |
| Breath odor | Normal | Fruity, acetone |
| Neurologic | Tremors | Diminished reflexes Paresthesia |
| Ominous signs | Late—Hyperreflexia, dilated pupils, seizure | Acidosis, coma |
| Shock, coma | ||
| Blood: | ||
| Glucose | Low: <60 mg/dl | High: ≥250 mg/dl |
| Ketones | Negative | High, large |
| Osmolarity | Normal | High |
| pH | Normal | Low (≤7.25) |
| Hematocrit | Normal | High |
| Bicarbonate | Normal | <20 mEq/L |
| Urine: | ||
| Output | Normal | Polyuria (early) to oliguria (late) |
| Glucose | Negative | Enuresis, nocturia |
| Ketones | Negative or trace | High |
| Visual | Diplopia | Blurred vision |
Children are usually able to detect the onset of hypoglycemia, but some are too young to implement treatment. Parents should become adept at recognizing the onset of symptoms—for example, a change in a child’s behavior, such as tearfulness or euphoria. In the majority of cases, 10 to 15 g of simple carbohydrate, such as 1 tablespoon of table sugar, will elevate the blood glucose level and alleviate the symptoms. The simpler the carbohydrate, the more rapidly it will be absorbed (8 ounces of milk equals 15 g of carbohydrate). The rapid-releasing sugar is followed by a complex carbohydrate such as a slice of bread or a cracker and by a protein such as peanut butter or milk.
For a mild reaction, milk or fruit juice is a good food to use in children. Milk supplies them with lactose or milk sugar, as well as a more prolonged action from the protein and fat (aids in decreased absorption). Other glucose sources include Insta-Glucose (cherry-flavored glucose), carbonated drinks (not sugarless), sherbet, gelatin, or cake icing. All children with diabetes should carry with them glucose tabs, Insta-Glucose, sugar cubes, or sugar-containing candy such as LifeSavers or Charms. A difficulty with candies or icing is that the child may learn to fake a reaction to get the sweets; therefore commercial treatment products such as Insta-Glucose or glucose tabs may be preferred.
Glucagon is sometimes prescribed for home treatment of hypoglycemia. It is available as an emergency kit that must be mixed at the time of use and is administered intramuscularly or subcutaneously. Glucagon functions by releasing stored glycogen from the liver and requires about 15 to 20 minutes to elevate the blood glucose level.
Once the child is responsive, the lost glycogen stores are replaced by small amounts of sugar-containing fluid administered frequently until the child feels comfortable trying solid foods.
Morning Hyperglycemia.: The management of elevated morning blood glucose levels depends on whether the increase is a true dawn phenomenon, insulin waning, or a rebound hyperglycemia (the Somogyi effect). Insulin waning is a progressive rise in blood glucose levels from bedtime to morning. It is treated by increasing the nocturnal insulin dose. The true dawn phenomenon shows a relatively normal blood glucose level until about 3 am, when the level begins to rise. The Somogyi effect may occur at any time but often entails an elevated blood glucose level at bedtime and a drop at 2 amwith a rebound rise following. The treatment for this phenomenon is decreasing the nocturnal insulin dose to prevent the 2 am hypoglycemia. The rebound rise in the blood glucose level is a result of counterregulatory hormones (epinephrine, GH, and corticosteroids), which are stimulated by hypoglycemia. More frequent blood monitoring (especially at times of anticipated peak insulin action) will usually identify these conditions. Trace amounts of urinary ketones aid in identifying undetected hypoglycemia.
Illness Management.: Illness alters diabetes management, and maintaining control is usually related to the seriousness of the illness. In the well-controlled child an illness will run its course as it does in the unaffected child. The goals during an illness are to restore euglycemia, treat urinary ketones, and maintain hydration. Blood glucose levels and urinary ketones should be monitored every 3 hours. Some hyperglycemia and ketonuria are expected in most illnesses, even with diminished food intake, and are an indication for increased insulin. Insulin should never be omitted during an illness, although dosage requirements may increase, decrease, or remain unchanged, depending on the severity of the illness and the child’s appetite. Often the child will need supplemental insulin between usual dose times. If the child vomits more than once, if blood glucose levels remain above 240 mg/dl, or if urinary ketones remain high, the health care practitioner should be notified. Simple carbohydrates may be substituted for carbohydrate-containing exchanges in the meal plan. Although insulin and diet are important tools in sick-day care, fluids are the most important intervention. Fluids must be encouraged to prevent dehydration and to flush out ketones.
Therapeutic Management of Diabetic Ketoacidosis
DKA, the most complete state of insulin deficiency, is a life-threatening situation. Management consists of rapid assessment, adequate insulin to reduce the elevated blood glucose level, fluids to overcome dehydration, and electrolyte replacement (especially potassium).
Because DKA constitutes an emergency situation, the child should be admitted to an intensive care facility for management. The priority is to obtain a venous access for administration of fluids, electrolytes, and insulin. The child should be weighed, measured, and placed on a cardiac monitor. Blood glucose and ketone levels are determined at the bedside, and samples are obtained for laboratory measurement of glucose, electrolytes, blood urea nitrogen, arterial pH, Po2, Pco2, hemoglobin, hematocrit, white blood cell count and differential, calcium, and phosphorus.
Oxygen may be administered to patients who are cyanotic and in whom arterial oxygen is less than 80%. Gastric suction is applied to unconscious children to avoid the possibility of pulmonary aspiration. Antibiotics may be administered to febrile children after appropriate specimens are obtained for culture. A Foley catheter may or may not be inserted for urine samples and measurement. Unless the child is unconscious, a collection bag is usually sufficient for accurate assessments.
Fluid and Electrolyte Therapy.: All patients with DKA suffer from dehydration (10% of total body weight in severe ketoacidosis) because of the osmotic diuresis, accompanied by depletion of electrolytes, sodium, potassium, chloride, phosphate, and magnesium. Serum pH and bicarbonate reflect the degree of acidosis. Prompt and adequate fluid therapy restores tissue perfusion and suppresses the elevated levels of stress hormones.
The initial hydrating solution is 0.9% saline solution. Traditionally deficits have been replaced at a rate of 50% over the first 8 to 12 hours and the remaining 50% over the next 16 to 24 hours. Current trends suggest more cautious fluid management to reduce the risk of cerebral edema. The fluid deficit is replaced evenly over a period of 24 to 48 hours.
Serum potassium levels may be normal on admission, but after fluid and insulin administration the rapid return of potassium to the cells can seriously deplete serum levels, with the attendant risk of cardiac arrhythmias. As soon as the child has established renal function (is voiding at least 25 ml/hr) and insulin has been given, vigorous potassium replacement is implemented. The cardiac monitor is used as a guide to therapy, and configuration of T waves should be observed every 30 to 60 minutes to determine changes that might indicate alterations in potassium concentration (widening of the QT interval and the appearance of a U wave following a flattened T wave indicate hypokalemia; an elevated and spreading T wave and shortening of the QT interval indicate hyperkalemia).
Insulin should not be given until urinary ketones and a blood glucose level have been obtained. Continuous IV regular insulin is given at a dosage of 0.1 units/kg/hr. Insulin therapy should be started after the initial rehydration bolus, since serum glucose levels fall rapidly after volume expansion. Blood glucose levels should decrease by 50 to 100 mg/dl/hr. When blood glucose levels fall to 250 to 300 mg/dl, dextrose is added to the IV solution. The goal is to maintain blood glucose levels between 120 and 240 mg/dl by adding 5% to 10% dextrose. Sodium bicarbonate is used conservatively; it is used for pH less than 7.0, severe hyperkalemia, or cardiac instability. Because sodium bicarbonate has been associated with increased risk for cerebral edema, children receiving this substance must be carefully monitored for changes in level of consciousness (Glaser, Barnett, McCaslin, and others, 2001).
When the critical period is over, the task of regulating insulin dosage in relation to diet and activity is started. Children should be actively involved in their own care and are given responsibility according to their ability and the guidance of the nurse.
Nursing Care Management
Children with DM may be admitted to the hospital at the time of their initial diagnosis; during illness or surgery; or for episodes of ketoacidosis, which may be precipitated by any of a variety of factors (see the Evidence-Based Practice box evaluating hospitalization compared to outpatient care for newly diagnosed children with type 1 DM). Many children are able to keep the disease under control with periodic assessment and adjustment of insulin, diet, and activity as needed under the supervision of a practitioner. Under most circumstances these children can be managed well at home and require hospitalization only for a serious illness or upset.
However, a small number of children with diabetes exhibit a degree of metabolic lability and have repeated episodes of DKA that require hospitalization, which interferes with education and social development. These children appear to display a characteristic personality structure. They tend to be unusually passive and nonassertive and to come from families that are inclined to smooth over conflicts without resolution. Children in this type of setting experience emotional arousal with little, if any, opportunity or ability to resolve it. Other children from psychosocially dysfunctional families display behavioral and personality problems. This emotional stress causes an increased production of endogenous catecholamines, which stimulate fat breakdown, leading to ketonemia and ketonuria.
Hospital Management.: The child with DKA requires intensive nursing care. Vital signs should be observed and recorded frequently. Hypotension caused by the contracted blood volume of the dehydrated state may cause decreased peripheral blood flow, which can be particularly hazardous to the heart, lungs, and kidneys. An elevated temperature may indicate infection and should be reported so that treatment can be implemented immediately.
Careful and accurate records should be maintained, including vital signs (pulse, respiration, temperature, blood pressure), weight, IV fluids, electrolytes, insulin, blood glucose level, and intake and output. A urine collection device or retention catheter is used to obtain the urine measurements, which include volume, specific gravity, and glucose and ketone values. The volume relative to the glucose content is important because 5% glucose in a 300-ml sample is a significantly greater amount than a similar reading from a 75-ml sample. A diabetic flow sheet maintained at the bedside provides an ongoing record of the vital signs, urine and blood tests, amount of insulin given, and intake and output. The level of consciousness is assessed and recorded at frequent intervals. The comatose child generally regains consciousness fairly soon after initiation of therapy but is managed like any unconscious child until then.
When the critical period is over, the task of regulating insulin dosage to diet and activity is begun. The same meticulous records of intake and output, urine glucose and acetone levels, and insulin administration are maintained. Capable children should be actively involved in their own care and are given responsibility for keeping the intake and output record, testing the blood and urine, and, when appropriate, administering their own insulin—all under the supervision and guidance of the nurse (see Nursing Process box and Nursing Care Plan).
Child and Family Education.: Several organizations are prepared to assist with education and dissemination of knowledge about diabetes. The American Diabetes Association,* Canadian Diabetes Association,† Juvenile Diabetes Research Foundation International,‡ and American Association of Diabetes Educators§ are valuable resources for a wide variety of educational materials. The National Institute of Diabetes and Digestive and Kidney Diseases* publishes a number of comprehensive annotated bibliographies, including “Educational Materials for and About Young People with Diabetes,” a compilation of resource materials for children, siblings, parents, teachers, and health professionals, and “Sports and Exercise for People with Diabetes.”
The Child with Diabetes Mellitus
Assess for signs and symptoms of type 1 diabetes mellitus (DM) in children (see Box 29-13). Common symptoms include polyphagia, polyuria, polydipsia, weight loss, and fatigue.
DIAGNOSIS (PROBLEM IDENTIFICATION)
After a thorough assessment, several nursing diagnoses are evident (see Nursing Care Plan, p. 1052). Other nursing diagnoses include:
The effectiveness of nursing interventions for the family and child with type 1 DM is determined by continual assessment and evaluation of care based on the following guidelines:
Interview family to determine their understanding of the disease; have child and family demonstrate and discuss the needed assessment and therapeutic techniques.
Interview family regarding their understanding of control; analyze and evaluate management records.
Discuss the child’s disease with him or her.
Interview family and child regarding their feelings and concerns about the disease.
Medical Identification.: One of the first things the nurse should call to the parents’ attention is the need for the child to wear some means of medical identification. Usually recommended is the MedicAlert identification, a stainless steel or silver- or gold-plated identification bracelet that is visible and immediately recognizable. It contains a collect telephone number that medical personnel can call around the clock for medical records and personal information.
Nature of Diabetes.: The better the parents understand the pathophysiology of diabetes and the function and action of insulin and glucagon in relation to caloric intake and exercise, the better they will understand the disease and its effects on the child. Parents need answers to a number of questions (voiced or unvoiced) to increase their confidence in coping with the disease. For example, they may want to know about the various procedures performed on their child and treatment rationale, such as what is being put in the IV bottle and the expected effect.
Meal Planning.: Normal nutrition is a major aspect of the family education program. Diet instruction is usually conducted by the nutritionist, with reinforcement and guidance from the nurse. The emphasis is on adequate intake for age, consistent menus, complex carbohydrates, and consistent eating times. The family is taught how the meal plan relates to the requirements of growth and development, the disease process, and the insulin regimen. Meals and snacks are modified based on the child’s preferences and current menu, preserving cultural patterns and preferences as much as possible. Extensive exchange lists are available that include foods compatible with most lifestyles.
Learning about foods within specific food groups helps in making choices. Weights and measures of foods are used as eye-training devices for defining serving sizes and should be practiced for about 3 months, with gradual progression to estimation of food portions. Even when the child and family become competent in estimating portion sizes, reassessment should take place weekly or monthly and when there is any change of brands.
Family members should also be guided in reading labels for the nutritional value of foods and food content. They need to become familiar with the carbohydrate content of food groups. Substitution with foods of equal carbohydrate content is the skill needed for successful carbohydrate counting. Substitution might be necessary if a food is not available in sufficient quantity or for the teenager who wishes to eat fast food with peers. The use of a multiple daily injection program lends flexibility to the timing of meals.
Lists of popular fast-food items and items served at the major fast-food chains can be obtained from the restaurants to help guide food selections. It is important that the child know the nutritional value of these items (the major chains are remarkably uniform), but the child should be cautioned to avoid high-fat and high-sugar/high-carbohydrate items; for example, the child could choose a plain hamburger instead of a double cheeseburger.
Children should use sugar substitutes in moderation in items such as soft drinks. Artificial sweeteners have been shown to be safe, but if there is any question about amounts, the physician, dietitian, or nurse specialist can provide guidelines based on body weight. Sugar-free chewing gum and candies made with sorbitol may be used in moderation by children with DM. Although sorbitol is less cariogenic than other varieties of sugar substitutes, it is an alcohol sugar that is metabolized to fructose and then to glucose. Furthermore, large amounts can cause osmotic diarrhea. Most dietetic foods contain sorbitol. They are more expensive than regular foods. Also, while a product may be sugar free, it is not necessarily carbohydrate free.




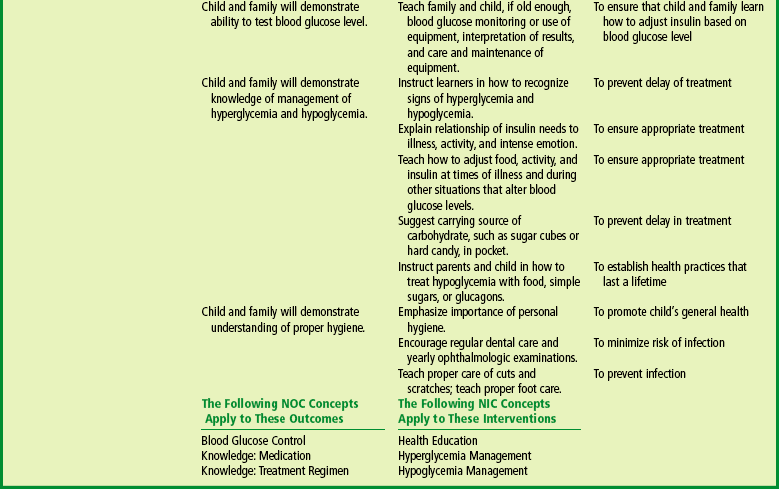
Traveling.: Traveling requires planning, especially when a trip involves crossing time zones. A number of tips are included in pamphlets available free of charge. Suggestions for traveling encompass what will be needed from the practitioner before leaving, what and how much to take along, needs in transit, what to consider at the destination, and planning for when the child returns home. Planning is needed no matter what type of travel is considered—automobile, plane, bus, or train.
Insulin.: Families need to understand the treatment method and the insulin prescribed, including the effective duration, onset, and peak action. They also need to know the characteristics of the various types of insulins, the proper mixing and dilution of insulins, and how to substitute another type when their usual brand is not available (insulin is a nonprescription drug). Insulin need not be refrigerated but should be maintained at a temperature between 15° and 29.4° C (59° and 85° F). Freezing renders insulin inactive.
Insulin bottles that have been “opened” (i.e., the stopper has been punctured) should be stored at room temperature or refrigerated for up to 28 to 30 days. After 1 month these vials should be discarded. Unopened vials should be refrigerated and are good until the expiration date on the label. Diabetic supplies should not be left in a hot environment.
Injection Procedure.: Learning to give insulin injections is a source of anxiety for both parents and children. It is helpful for the learner to know that this important aspect of care will become as routine as brushing the teeth. First, the basic injection technique is taught, using an orange or similar item and sterile normal saline for practice. To gain children’s confidence, the nurse can demonstrate the technique by giving a skillful injection to the parent and then having the parent return the demonstration by giving the nurse an injection. With practice and confidence, the parents will soon be able to give the insulin injection to their children, and their children will trust them. Another effective strategy is to instruct the children and then have them teach the technique to the parents while the nurse observes. Both parents should participate, and as little time as possible should elapse between instruction and the actual injection, especially with parents and teenage learners.
Insulin can be injected into any area in which there is adipose (fat) tissue over muscle; the drug is injected at a 90-degree angle. Newly diagnosed children may have lost adipose tissue, and care should be exerted not to inject intramuscularly. The pinch technique is the most effective method for tenting the skin to allow easy entrance of the needle to subcutaneous tissues in children. The site selected will sometimes depend on whether children or parents administer the insulin. The arms, thighs, hips, and abdomen are usual injection sites for insulin. The children can reach the thighs, abdomen, and part of the hip and arm easily but may require help to inject other sites. For example, a parent can pinch a loose fold of skin of the arm while the child injects the insulin.
The parents and child are helped to work out a rotation pattern to various areas of the body to enhance absorption, since insulin absorption is slowed by fat pads that develop in overused injection areas. The most efficient rotation plan involves giving about four to six injections in one area (each injection about 2.5 cm [1 inch] apart, or the diameter of the insulin vial from the previous injection) and then moving to another area.
It is important to remember that the absorption rate varies in different parts of the body (Table 29-5). The methodical use of one anatomic area and then movement to another (as described in the previous paragraph) minimizes variations in absorption rates. However, absorption is also altered by vigorous exercise, which enhances absorption from exercised muscles; therefore it is recommended that a site be chosen other than the exercising extremity (e.g., avoiding legs and arms when playing in a tennis tournament).
TABLE 29-5
Onset and Duration of Action Related to Injection Site
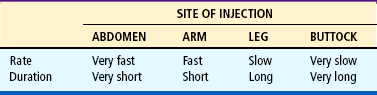
From Albisser AM, Sperlich M: Adjusting insulins, Diabetes Educ 18(3):211–218, 1992.
Injection sites for an entire month can be determined in advance on a simple chart. For example, the “paper doll” (body outline) can be constructed and insulin sites marked by the child. After injection, the child places the date on the appropriate site. To keep in practice, it is a good idea for the parent to give two or three injections a week in areas that are difficult for the child to reach. The same basic methodology is used when teaching children to give their own insulin injections (Fig. 29-2). They should practice first on an orange or a doll, building courage gradually. Other devices are available for insulin injection and may offer advantages to some children. Children who do not wish to give themselves injections can be taught to use a syringe-loaded injector (Inject-Ease). With the device, puncture is always automatic. Adolescents respond well to a self-contained and compact device resembling a fountain pen (NovoPen), which eliminates conventional vials and syringes. Preloaded pens may also cause less pain, since the needle is not blunted by piercing the rubber top of the insulin vial (Lteif and Schwenk, 1999).
Continuous Subcutaneous Insulin Infusion.: Some children are considered candidates for use of a portable insulin pump, and even some young children with unsatisfactory metabolic control can benefit from its use. The child and the parents are taught to operate the device, including the mechanics of the pump, battery changes, and alarm systems. A number of devices are on the market that vary in the basal rates they are able to deliver and in the cost of the equipment. Families can investigate the various devices and select the model that best suits their needs. Product information is available from pump manufacturers and distributors.*
Parents and children learn (1) the technical aspects of the pump and self-monitoring of blood glucose; (2) prevention and treatment for hyperglycemia, sick-day management, and meal planning; (3) the effects of exercise, stress, and diet on blood glucose levels; and (4) decision-making strategies to evaluate blood glucose patterns and make adjustments in all aspects of the regimen.
Numerous blood glucose measurements (at least four times per day) are an essential part of infusion pump use. Intensive education and supervision are critical to obtaining maximum efficiency and control. This is particularly important if the family has been accustomed to a conventional insulin regimen. They must realize that simply wearing the pump will not normalize blood glucose. The pump is merely an insulin delivery device, and frequent, routine blood glucose determinations are necessary to adjust the insulin delivery rate.
The major problem with use of the insulin pump is inflammation from irritation or infection at the insertion site. The site should be cleaned thoroughly before the needle is inserted and then covered with a transparent dressing. The site is changed and rotated every 48 to 72 hours (this may vary) or at the first sign of inflammation. Nurses working where pumps are part of the therapeutic regimen should become familiar with the operation of the specific device being used and the protocol of disease management. Others should be aware of this management technique and be prepared to assist patients using the pump.
Monitoring.: Nurses should also be prepared to teach and supervise blood glucose monitoring. SMBG is associated with few complications, and although it does not necessarily lead to improved metabolic control, it provides a more accurate assessment of blood glucose levels than can be obtained with the historical urine testing. Blood glucose monitoring has the added advantage that it can be performed anywhere (see Atraumatic Care box).
Blood for testing can be obtained by two different methods: manually or with a mechanical bloodletting device. A mechanical device is recommended for children, although the child and family should learn to use both methods in the event of mechanical failure. Several lancet devices are available, and each provides a means for obtaining a large drop of blood for testing (Fig. 29-3).
 ATRAUMATIC CARE
ATRAUMATIC CAREMinimizing Pain of Blood Glucose Monitoring
To enhance blood flow to the finger, hold it under warm water for a few seconds before the puncture.
When obtaining blood samples, use the ring finger or thumb (blood flows more easily to these areas), and puncture the finger just to the side of the finger pad (more blood vessels and fewer nerve endings).
To prevent a deep puncture, press the platform of the lancet device lightly against the skin and avoid steadying the finger against a hard surface.
Use lancet devices with adjustable-depth tips. Begin with the shallowest setting.
Use glucose monitors that require small blood samples (e.g., Ascensia Elite) to avoid repeated punctures.
The blood sample may be obtained from fingertips or alternate sites such as the forearm. Alternate site testing requires a meter that can test a small volume of blood. Not all meters are capable of this.
Signs of redness and soreness at the site of finger puncture should be examined by the practitioner. It may be evidence of poor technique, poor hygiene, or poor skin healing relative to poor control. Many types of blood-testing meters are available for home use. Newer technology has brought about improvements in meter size and ease of use. The family should be shown features of several meters, including advantages and disadvantages, and allowed to choose equipment that best meets their needs.
The least expensive testing method uses a reagent strip to which blood is applied (Fig. 29-4). After blotting, the color change is compared against a color scale for an estimation of the blood glucose level. The strips can be cut in half (although not all professionals recommend this) to obtain two readings per strip. This method is not accepted practice but may be necessary for some families or situations.
Urine Testing.: Testing for urinary ketones is recommended during times of illness or when blood glucose values are elevated. Information on a specific ketone-testing product should include correct procedure, storage, and product expiration. Families need a clear understanding of home management of ketones: fluids and additional insulin as directed by the health care team.
Signs of Hyperglycemia.: Severe hyperglycemia is most often caused by illness, growth, emotional upset, or missed insulin doses. Emotional stress from school finals or examinations or physical response to immunizations are examples of causes of hyperglycemia. With careful glucose monitoring, any elevation can be managed by adjustment of insulin or food intake. Parents should understand how to adjust food, activity, and insulin at the time of illness or when the child is treated for an illness with a medication known to raise the blood glucose level (e.g., steroids). The hyperglycemia is managed by increasing insulin soon after the increased glucose level is noted. Health care professionals should be aware that adolescent girls often become hyperglycemic around the time of their menses and should be advised to increase insulin dosages if necessary.
Signs of Hypoglycemia.: Hypoglycemia is caused by imbalances of food intake, insulin, and activity. Ideally hypoglycemia should be prevented, and parents need to be prepared to prevent, recognize, and treat the problem. They should be familiar with the signs of hypoglycemia and instructed in treatment, including care of the child with seizures. Early signs are adrenergic, including sweating and trembling, which help raise the blood glucose level, much like the reaction when an individual is startled or anxious. The second set of symptoms that follow an untreated adrenergic reaction is neuroglycopenic (also called brain hypoglycemia). These symptoms typically include difficulty with balance, memory, attention, or concentration; dizziness or lightheadedness; and slurred speech. Severe and prolonged hypoglycemia leads to seizures, coma, and possible death (Cryer, 2000). Hypoglycemia can be managed effectively as outlined in the Emergency Treatment box.
It is advisable for parents to plan for anticipated excitement or exercise. In addition, gastroenteritis may decrease insulin needs slightly as a result of poor appetite, vomiting, or diarrhea. If the blood glucose level is low but urinary ketones are present, the family should be aware of the increased need for simple carbohydrates and liquids.
Hygiene.: All aspects of personal hygiene should be emphasized for the child with diabetes. The child should be cautioned against wearing shoes without socks, wearing sandals, or walking barefoot. Correct nail and extremity care tailored to the individual child (with the guidance of a podiatrist) can begin health practices that last a lifetime. Eyes should be checked once a year unless the child wears glasses, and then as directed by the ophthalmologist. Regular dental care is emphasized, and cuts and scratches should be treated with plain soap and water unless otherwise indicated. Diaper rash in infants and candidal infections in teens may indicate poor diabetes control.
Exercise.: Exercise is an important component of the treatment plan. If the child is more active at one time of the day than at another time, food or insulin can be altered to meet that activity pattern. Food should be increased in the summer, when children tend to be more active. Decreased activity on return to school may require a decrease in food intake or increase in insulin dosage. The child who is active in team sports will need a snack about a half hour before the anticipated activity. Races or other competition may call for a slightly higher food intake than at practice times.
Food intake will usually need to be repeated for prolonged activity periods, often as frequently as every 45 minutes to 1 hour. Families should be informed that if increased food is not tolerated, decreased insulin is the next course of action. If the timing of the exercise is changed so that the supper meal is delayed, the insulin in the second or third dose of the day may be moved back to precede the mealtime. Sugar may sometimes be needed during exercise periods for quick response. Elevated blood glucose levels after extreme activity may represent the body’s adrenergic response to exercise. If the blood glucose level is elevated (≥240 mg/dl) before planned exercise, urinary ketones should be checked and the activity may need be postponed until the blood glucose is controlled.
Record Keeping.: Home records are an invaluable aid to diabetes self-management. The nurse and family devise a method to chart insulin administered, blood glucose values, urine ketone results, and other factors and events that affect diabetes control. The child and family are encouraged to observe for patterns of blood glucose responses to events such as exercise. If lapses in management occur (such as eating a candy bar), the child should be encouraged to note this and not be criticized for the transgression.
Self-Management: Self-management is the key to close control. Being able to make changes when they are needed rather than waiting until the next contact with health care professionals is important for self-management and gives the individual and family the feeling they have control over the disease. Psychologically this helps family members feel they are useful and participating members of the team. Allowing the child to learn to look at records objectively promotes independence in self-management support. As children grow and assume more responsibility for self-management, they develop confidence in their ability to manage their disease and confidence in themselves as persons. They learn to respond to the disease and to make more accurate interpretations and changes in treatment when they become adults.
Puberty is associated with decreased sensitivity to insulin that normally would be compensated for by an increased insulin secretion. Health care professionals should anticipate that pubertal patients will have more difficulty maintaining glycemic control. Insulin doses commonly need to be increased, often dramatically (McConnell, Harper, Campbell, and others, 2001). Patients should be taught to give themselves additional doses of rapid-acting insulin (5% to 10% of their daily dose) when their blood glucose levels are increased. The use of supplemental rapid-acting insulin is preferred to withholding food in the adolescent.
Child or Adolescent and Family Support.: Just as the physiologic responses affect the child, the parents and other family members of the child with newly diagnosed DM experience various emotional responses to the crisis. Care in the acute setting is short but may create fears and frustrations. The prospect of a chronic illness in their child engenders all the feelings and concerns that are faced by parents of children with other chronic illnesses (see Chapter 18). The threat of complications and death is always present, as well as the continuing drain on emotional and financial resources.
Certain fears may develop as a result of past experiences with the disease. A severe insulin reaction with seizures can contribute to fear of repetition. Once parents observe a seizure or the adolescent has one in a public place, the desire to maintain better control is reinforced. They must understand how to prevent problems and how to handle problems calmly and coolly if they occur, and they must understand the complexities of the body, the disease, and its complications. Young children usually adjust well to problems related to the disease. With toddlers and preschoolers, insulin injections and glucose testing may be difficult at first. However, they usually accept the procedures when the parents use a matter-of-fact approach, without calling attention to a “hurt,” and treat the procedure like any other routine part of the child’s life. After the injection, time with some special and positive attention, such as reading or talking, or another pleasant activity, is one way to convert children who initially refuse injections to those who accept them.
In the years before adolescence children probably accept their condition most easily. They are able to understand the basic concepts related to their disease and its treatment. They are able to test blood glucose and urine, recognize food groups, give injections, keep records, and distinguish fear or excitement from hypoglycemia. They understand how to recognize, prevent, and treat hypoglycemia. However, they still need considerable parental involvement.
Adolescents appear to have the most difficulty adjusting. Adolescence is a time of stress in trying to be perfect and like one’s peers, and no matter what others say, having diabetes is being different. Some adolescents are more upset about not being able to have a candy bar than about injections, diet, and other aspects of management. If children can accept the difference as a part of life—in other words, that each person is different in some way—then, with adequate parental support, they should be able to adjust well (see Critical Thinking Exercise).
Camping and other special group activities are useful. At diabetes camp, children learn that they are not alone. As a result, they become more independent and resourceful in other settings. Useful information about such camps and organizations can be obtained from the American Diabetes Association. A list of accredited camps specifically for children and teenagers with diabetes is also available from the American Camping Association.*
Rebecca, a 15-year-old with a 3-year history of type 1 diabetes mellitus (DM), has been admitted to the pediatric intensive care unit for treatment of diabetic ketoacidosis (DKA). This is her fifth hospital admission for DKA in the past year. Rebecca’s parents are divorced, and she has four younger siblings, none of whom has diabetes. Rebecca’s mother has maintained two jobs for the past 5 years and frequently leaves Rebecca in charge of the household. In anticipation of her discharge, you are planning a patient education program for Rebecca and her mother. What important issues regarding Rebecca’s unstable diabetes management must you consider to plan the education program?
1. Evidence—Is there sufficient evidence to draw conclusions about Rebecca’s recurrent episodes of DKA?
2. Assumptions—Describe an underlying assumption about each of the following:
3. What priorities for nursing care should be established for Rebecca?
The endocrine system has three components: the cell, which sends a chemical message via a hormone; target cells, which receive the message; and the environment through which the chemical is transported from the site of synthesis to the sites of cellular action.
Pituitary dysfunction is manifested primarily by growth disturbance.
The main physiologic action of TH is to regulate the basal metabolic rate and control the processes of growth and tissue differentiation.
Disorders of thyroid function include hypothyroidism, autoimmune thyroiditis, goiter, and hyperthyroidism.
Therapy for hyperthyroidism is directed at retarding the rate of hormone secretion and may include drug therapy, thyroidectomy, or radioiodine therapy.
Classic forms of hypoparathyroidism in childhood are idiopathic (deficient production of PTH) and pseudohypoparathyroidism (increased PTH production with end-organ unresponsiveness to PTH).
The adrenal cortex secretes three important groups of hormones: glucocorticoids, mineralocorticoids, and sex steroids.
Disorders of adrenal function include acute adrenocortical insufficiency, chronic adrenocortical insufficiency, Cushing syndrome, and CAH.
Five categories of Cushing syndrome are pituitary, adrenal, ectopic, iatrogenic, and food dependent.
Management of CAH includes assignment of a sex according to genotype, administration of cortisone, and, possibly, reconstructive surgery.
DM is categorized as type 1 diabetes and type 2 diabetes.
The focus of type 1 DM is insulin replacement, diet, and exercise.
Education of families includes explanation of diabetes, meal planning, administering insulin injections, monitoring general hygienic practices, promoting exercise, record keeping, and observing for complications.
 answers to CRITICAL THINKING EXERCISE
answers to CRITICAL THINKING EXERCISE
1. Yes. Rebecca has had five hospital admissions for DKA in the past year. Numerous factors must be involved with her unstable disease.
a. The normal tasks of adolescence can play a significant role in blood glucose instability.
b. Adolescent girls with diabetes have frequent fluctuations of blood glucose levels immediately before, during, or after their menses.
c. Rebecca’s personal loss from the divorce, her mother’s absence because of a heavy work schedule, and the added responsibilities of the household may cause significant stress, resulting in elevated blood glucose levels.
d. Careful, frequent, consistent monitoring of blood glucose levels is essential for effective insulin management during adolescence.
3. The first priority would be to focus directly on the issues of hyperglycemia. Determination of Rebecca’s practice of monitoring and managing her diabetes at home is essential. Areas of diabetes management that should be emphasized include careful dietary management, an appropriate exercise program, conscientious self-testing of blood glucose, appropriate administration of daily insulin, and adherence to sliding-scaling insulin therapy. Discussion of the emotional stressors she identifies at this time is appropriate.
4. Yes, Rebecca’s history of DKA over the past year supports her inability to monitor and manage her diabetes.
5. It would be appropriate to make a referral for special support services for counseling and possible home care follow-up to assess her diabetes management skills at home.
REFERENCES
American Academy of Pediatrics, Section on Endocrinology and Committee on Genetics. Technical report: congenital adrenal hyperplasia. Pediatrics. 2000;106(6):1511–1518.
American Diabetes Association. Care of children and adolescents with type 1 diabetes. Diabetes Care. 2005;28:186–212.
American Diabetes Association. Report of the Expert Committee on the Diagnosis and Classification of Diabetes Mellitus. Diabetes Care. 2001;24(Suppl 1):S5–S20.
Bartalena, L, Tanda, ML, Piantanida, E, et al. Oxidative stress and Graves’ ophthalmopathy: in vitro studies and therapeutic implications. Biofactors. 2003;19(3-4):155–163.
Baxter, JD, Ribeiro, RCJ. Introduction to endocrinology. In Greenspan FS, Gardner DG, eds.: Basic and clinical endocrinology, ed 6, New York: Lange Medical Books/McGraw-Hill, 2001.
Behrman, RE, Kliegman, RM, Jenson, HB. Nelson textbook of pediatrics, ed 17. Philadelphia: Saunders, 2004.
Beisswenger, P, Szwergold, B, Yeo, K. Glycated proteins in diabetes. Clin Lab Med. 2001;21(10):53–78.
Biro, FM, Huang, B, Crawford, PB, et al. Pubertal correlates in black and white girls. J Pediatr. 2006;148(2):234–240.
Bryant, J, Cave, C, Milne, R, Recombinant growth hormone for idiopathic short stature in children and adolescents. Cochrane Database Syst Rev 2003;2:CD004440, 10.1002/14651858.CD004440
Cheetham, T, Baylis, PH. Diabetes insipidus in children: pathophysiology, diagnoses and management. Paediatr Drugs. 2002;4(12):785–796.
Cryer, P. Glucose counterregulatory hormones: physiology, pathophysiology, and relevance to clinical hypoglycemia. In Le Roith D, Taylor S, Olefsky J, eds.: Diabetes mellitus: a fundamental and clinical text, ed 5, Philadelphia: Lippincott Williams & Wilkins, 2000.
Dallas, JS, Foley, TP. Hyperthyroidism. In Lifshitz F, ed.: Pediatric endocrinology, ed 4, New York: Marcel Dekker, 2003.
De Buyst, J, Massa, G, Christophe, C, et al. Clinical, hormonal and imaging findings in 27 children with central diabetes insipidus. Eur J Pediatr. 2007;166(1):43–49.
Foley, TP. Hypothyroidism. In Hoekelman RA, Adam HM, Nelson NM, et al, eds.: Primary pediatric care, ed 4, St Louis: Mosby, 2001.
Glaser, N, Barnett, P, McCaslin, I, et al. Risk factors for cerebral edema in children with diabetic ketoacidosis: the Pediatric Emergency Medicine Collaborative Research Committee of the American Academy of Pediatrics. N Engl J Med. 2001;344(4):264–269.
Greiner, MV, Kerrigan, JR. Puberty: timing is everything. Pediatr Ann. 2006;35(12):916–922.
Guyda, HJ. Growth hormone testing and the short child. Pediatr Res. 2000;48(5):579–580.
Halac, I, Zimmerman, D. Evaluating short stature in children. Pediatr Ann. 2004;33(3):171–176.
Hall, DMB. Growth monitoring. Arch Dis Child. 2000;82(1):10–15.
Hannon, TS, Gungor, N, Arslanian, SA. Type 2 diabetes in children and adolescents: a review for the primary care provider. Pediatr Ann. 2006;35(12):880–887.
Herman-Giddens, ME. Recent data on pubertal milestones in United States children: the secular trend toward earlier development. Int J Androl. 2006;29(1):241–246.
Hilczer, M, Smyczynska, J, Lewinski, A. Limitations of clinical utility of growth hormone stimulating tests in diagnosing children with short stature. Endocrinol Regul. 2006;40(3):69–75.
Hochberg, Z. Practical algorithms in pediatric endocrinology. Basel, Switzerland: Karger, 1999.
Hoffman, RP. Juvenile diabetes: avoiding problems during therapy. Clin Advisor. 2003:70–75.
Hoffman, RP. Eating disorders in adolescents with type 1 diabetes: a closer look at a complicated condition. Postgrad Med. 2001;109(4):67–69. [73-74,].
Jospe, N. Hyperthyroidism. In Hoekelman RA, Adam HM, Nelson NM, et al, eds.: Primary pediatric care, ed 4, St Louis: Mosby, 2001.
Karnik, AA, Fields, AV, Shannon, RP. Diabetic cardiomyopathy. Curr Hypertens Rep. 2007;9(6):467–473.
Kempers, MJ, Otten, BJ. Idiopathic precocious puberty versus puberty in adopted children: auxological response to gonadotrophin-releasing hormone agonist treatment and final height. Eur J Endocrinol. 2002;147(5):609–616.
Lee, PA. Central precocious puberty: an overview of diagnosis, treatment, and outcome. Endocrinol Metab Clin North Am. 1999;28(4):901–918.
Levine, LS. Congenital adrenal hyperplasia. Pediatr Rev. 2000;21(5):159–170.
Lin, M, Liu, SJ, Lim, IT. Disorders of water imbalance. Emerg Med Clin North Am. 2005;23(3):749–770.
Lteif, AN, Schwenk, WF. Accuracy of pen injectors versus insulin syringes in children with type 1 diabetes. Diabetes Care. 1999;22(10):137–140.
Ma, C, Xie, JW, Kuang, AR, et al, Radioiodine treatment for pediatric Grave’s disease (protocol). Cochrane Database Syst Rev 2006;4:CD006294, 10.1002/14651858
Macchia, PE. Recent advances in understanding the molecular basis of primary congenital hypothyroidism. Mol Med Today. 2000;6(1):36–42.
Magee, MF, Bhatt, BA. Management of decompensated diabetes: diabetic ketoacidosis and hyperglycemic hyperosmolar syndrome. Crit Care Clin. 2001;17(1):75–106.
Majzoub, JA, Muglia, LJ. Disorders of water homeostasis. In Lifshitz F, ed.: Pediatric endocrinology, ed 4, New York: Marcel Dekker, 2003.
Mauras, N, Walton, P, Nicar, M, et al. Growth hormone stimulation testing in both short and normal statured children: use of an immunofunctional assay. Pediatr Res. 2000;48(5):614–618.
McConnell, EM, Harper, R, Campbell, M, et al. Achieving optimal diabetic control in adolescence: the continuing enigma. Diabetes Metab Res Rev. 2001;17(10):67–74.
Midyett, LK, Moore, WV, Jacobson, JD. Are pubertal changes in girls before age 8 benign? Pediatrics. 2003;111(1):47–51.
Miller, BS, Zimmerman, D. Idiopathic short stature in children. Pediatr Ann. 2004;33(3):177–181.
Miranda, ML, Oliveira-Filho, AG, Lemos-Marini, SH, et al. Labioscrotal island flap in feminizing genitoplasty. J Pediatr Surg. 2004;39(7):1030–1033.
Moshang, T. Cushing’s disease, 70 years later … and the beat goes on (editorial). J Clin Endocrinol Metab. 2003;88(1):31–33.
Muir, A. Precocious puberty. Pediatr Rev. 2006;27(10):373–381.
Nebesio, TD, Eugster, EA. Current concepts in normal and abnormal puberty. Curr Prob Pediatr Adolesc Health Care. 2007;37(2):50–72.
Nieman, LK, Ilias, I. Evaluation and treatment of Cushing’s syndrome. Am J Med. 2005;118(12):1340–1346.
New, MI, Ghizzoni, L. Update on congenital adrenal hyperplasia. In Lifshitz F, ed.: Pediatric endocrinology, ed 4, New York: Marcel Dekker, 2003.
Olohan, K, Zappitelli, D. The insulin pump. Am J Nurs. 2003;103(4):48–56.
O’Sullivan, E, O’Sullivan, M. Precocious puberty: a parent’s perspective. Arch Dis Childhood. 2002;86:320–321.
Pacak, K, Eisenhofer, G, Ahlman, H, et al. Pheochromocytoma: recommendations for clinical practice from the First International Symposium. Nat Clin Pract Endocrinol Metab. 2007;3(2):92–102.
Perheentupa, J. Hypoparathyroidism and mineral homeostasis. In Lifshitz F, ed.: Pediatric endocrinology, ed 4, New York: Marcel Dekker, 2003.
Petitti, BD, Imperatore, G, Palla, SL, et al. Serum lipids and glucose control: the SEARCH for Diabetes in Youth study. Arch Pediatr Adolesc Med. 2007;161(2):159–165.
Pizzo, PA, Poplack, DG. Principles and theories of pediatric oncology. Philadelphia: Lippincott Williams & Wilkins, 2006.
Radetti, G, Gottardi, E, Bona, G, et al. The natural history of euthyroid Hashimoto’s thyroiditis in children. J Pediatr. 2006;149(6):827–832.
Reiter, EO, Price, DA, Wilton, P, et al. Effect of growth hormone (GH) treatment on the near-final height of 1258 patients with idiopathic GH deficiency: analysis of a large international database. J Clin Endocrinol Metab. 2006;91(6):2047–2054.
Rivkees, SA, Cornelius, EA. Influence of iodine-131 dose on the outcome of hyperthyroidism in children. Pediatrics. 2003;111(4):745–748.
Root, AW. Precocious puberty. Pediatr Rev. 2000;21(1):10–19.
Rovet, JF, Ehrlich, R. Psychoeducational outcome in children with early-treated congenital hypothyroidism. Pediatrics. 2000;105(3):515–522.
Schultz, CJ, Konopelska-Bahu, T, Dalton, RN, et al. Microalbuminuria prevalence varies with age, sex, and puberty in children with type 1 diabetes followed from diagnosis in a longitudinal study. Diabetes Care. 1999;22(3):495–502.
Simmonds, MJ, Howson, JM, Heward, JM, et al. Regression mapping of association between the human leukocyte antigen region and Graves disease. Am J Human Genet. 2005;76(1):157–163.
Slyper, AH. The pubertal timing controversy in the USA, and a review of possible causative factors for the advance in timing of onset of puberty. Clin Endocrinol (Oxf). 2006;65(1):1–8.
Streetman, DD, Khanderia, U. Diagnosis and treatment of Graves disease. Am J Nurse Pract. 2004;8(1):27–36.
Szymborska, M, Staroszczyk, B. Thyroiditis in children. Med Wieku Rozwoj. 2000;IV(4):383–391.
Thompson, GB. Surgical management in Graves’ disease. Panminerva Med. 2002;44(4):287–293.
Trivin, C, Couto-Silva, AC, Sainte-Rose, Z, et al. Presentation and evolution of organic central precocious puberty according to the type of CNS lesion. Clin Endocrinol (Oxford). 2006;65(2):239–245.
Urrutia-Rojas, X, Menchaca, J. Prevalence of risk for type 2 diabetes in school children. J Sch Health. 2006;76(5):189–194.
Van Tijn, DA, Schroor, EJ, Delemarre-van de Waal, HA, et al. Early assessment of hypothalamic-pituitary-gonadal function in patients with congenital hypothyroidism of central origin. J Clin Endocrinol Metab. 2007;92(1):104–109.
Verbalis, JG. Diabetes insipidus. Rev Endocr Metab Disord. 2003;4(2):177–185.
Walvoord, EC, Pescovitz, OH. Combined use of growth hormone and gonadotropin-releasing hormone analogues in precocious puberty: theoretic and practical considerations. Pediatrics. 1999;104(4 Pt 2):1010–1014.
Wong, LL, Verbalis, JG. Systemic diseases associated with disorders of water homeostasis. Endocrinol Metab Clin North Am. 2002;31(1):121–140.
*Home care instructions on giving subcutaneous injections are available in Wilson D, Hockenberry M: Wong’s clinical manual of pediatric nursing, ed 7, St Louis, 2008, Mosby.
*997 Glen Cove Ave., Suite 5, Glen Head, NY 11545; (800) 451-6434; e-mail: hgf1@hgfound.org; http://www.hgfound.org.
*6066 Leesburg Pike, Suite 550, Falls Church, VA 22041; (800) THYROID; e-mail: thyroid@thyroid.org; http://www.thyroid.org
*1701 N. Beauregard St., Alexandria, VA 22311; (800) 342-2383; http://www.diabetes.org.
†1400-522 University Ave., Toronto, ON M5G 2R5; (800) 226-8464; http://www.diabetes.ca.
‡120 Wall St., New York, NY 10005; (800) 533-CURE; http://www.jdrf.org.
§200 W. Madison St., Suite 800, Chicago, IL 60606; (800) 338-3633; e-mail: education@aadenet.org; http://www.diabeteseducator.org.
*Office of Communications and Public Liaison, NIDDK, NIH, Building 31, Room 9A06, 31 Center Drive, MSC 2560, Bethesda, MD 20892-2560; (301) 496-3583; http://www.niddk.nih.gov.
*Medtronic MiniMed, http://www.minimed.com; Disetronic, http://www.disetronic-usa.com; Animas, http://www.animascorp.com.
*5000 State Road 67 N., Martinsville, IN 46151; (765) 342-8456; http://www.acacamps.org.