The Extracellular Matrix
Learning objectives
After reading this chapter you should be able to:
 Describe the composition, structure and function of the extracellular matrix (ECM) and its components, including collagens, noncollagenous proteins and proteoglycans.
Describe the composition, structure and function of the extracellular matrix (ECM) and its components, including collagens, noncollagenous proteins and proteoglycans.
Outline the sequence of steps in the biosynthesis and post-translational modification of collagens and elastin, including the structure and synthesis of crosslinks.
Discuss the functional roles of the ECM in tissues.
Describe the pathways of biosynthesis and turnover of proteoglycans.
Discuss the structure and function of integrins as receptors for ECM components.
Introduction
The extracellular matrix (ECM) is a complex network of secreted macromolecules located in the extracellular space. Historically, the ECM has been described as simply providing a three-dimensional framework for the organization of tissues and organs; however, it has become increasingly clear that it plays a central role in regulating basic cellular processes, including proliferation, differentiation, migration and even survival. The macromolecular network of the ECM is made up of collagens, elastin, glycoproteins and proteoglycans that are secreted by a variety of cell types including fibroblasts, chondrocytes, osteoblasts and others. The components of the ECM are in intimate contact with their cells of origin and form a three-dimensional gelatinous bed in which the cells thrive. Proteins in the ECM are also bound to the cell surface, so that they transmit mechanical signals resulting from stretching and compression of tissues. The relative abundance, distribution, and molecular organization of ECM components vary enormously, depending upon tissue type, developmental stage and pathologic status. Variations in the composition, accumulation and organization of the ECM dramatically impact the structural and functional properties of the tissue. Changes in these ECM characteristics are associated with chronic diseases, such as arthritis, atherosclerosis, cancer, and fibrosis.
Collagens
Collagens are the major proteins in the ECM
The collagens are a family of proteins that comprise about 30% of total protein mass in the body. As the primary structural components of the ECM in connective tissues, collagens have an important role in tissue architecture and integrity, and in mediating a variety of cell–cell and cell–matrix interactions. To date, more than 25 different types of collagens have been identified. They are composed of related, but distinct, peptide chains and vary greatly in their distribution, organization and function in tissues.
Triple-helical structure of collagens
The left-handed triple helical structure of collagen is unique among proteins
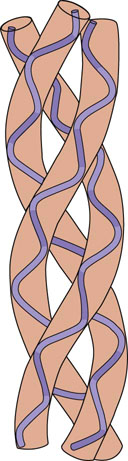
The collagens are heterotrimeric proteins composed of three individual peptide chains. The structural hallmark of collagens is their triple-helical structure, formed by folding of the three component peptide chains. X-ray diffraction analysis indicates that three left-handed helical chains are wrapped around one another in a rope-like fashion, to form a superhelix structure (Fig. 29.1). The left-handed helix is more extended than the α-helix of globular proteins, having nearly twice the rise per turn and only three, rather than 3.6, amino acids per turn of the helix. Every third amino acid is glycine, because only this amino acid, with the smallest side chain, fits into the crowded central core. The characteristic, repeating sequence of collagen is Gly-X-Y, where X and Y can be any amino acid but most often X is proline and Y is hydroxyproline. Because of their restricted rotation and bulk, proline and hydroxyproline confer rigidity to the helix. The intra- and interchain helices are stabilized by hydrogen bonds, largely between peptide NH and C O groups. The side chains of the X and Y amino acids point outward from the helix, and thus are on the surface of the protein, where they form lateral interactions with other triple helices or proteins.
O groups. The side chains of the X and Y amino acids point outward from the helix, and thus are on the surface of the protein, where they form lateral interactions with other triple helices or proteins.
Types of collagen
Some representative collagens are listed in Table 29.1. The collagen family of proteins can be divided into two main types: the fibril-forming (fibrillar) and the nonfibrillar collagens.
Table 29.1
Members of the collagen family. Classification and distribution of different collagen types
| Type | Class | Distribution |
| I | Fibrillar | Widely distributed including skin, tendon, bone, heart |
| II | Fibrillar | Cartilage, developing cornea and vitreous humor |
| III | Fibrillar | Extensible connective tissue, e.g. skin, lung and vascular system |
| IV | Network | Basement membranes, kidney, vascular wall |
| V | Fibrillar | Liver, cornea and mucosa |
| VI | Beaded filament | Most connective tissue |
| IX | Facit | Cartilage, vitreous humor |
| XI | Fibril forming | Cartilage, bone, placenta |
| XII | Facit | Embryonic tendon and skin |
| XIII | Transmembrane domain | Widely distributed |
| XIV | Facit | Fetal skin and tendons |
FACIT, fibril-associated collagen with interrupted triple helices.
Fibril-forming collagens
Fibrillar collagens provide tensile strength to tendons, ligaments and skin
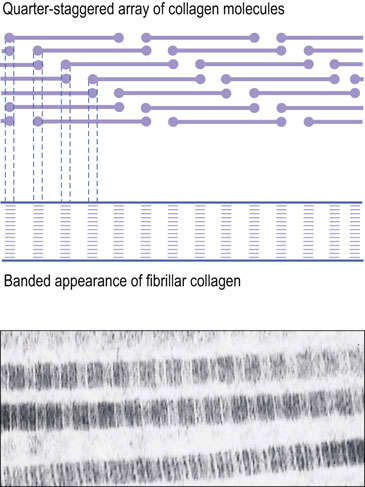
Fibril-forming collagens include types I, II, III, V, and XI (see Table 29.1). Collagen fibrils can be formed from a mixture of different fibrillar collagens. For instance, dermal collagen fibrils are hybrids of type I and type III collagen, and fibrils in corneal stroma are hybrids of type I and type IV collagen. Type I is the most abundant fibrillar collagen and occurs in a wide variety of tissues; others have a more limited tissue distribution (see Table 29.1). Type I and related fibrillar collagens form well-organized, banded fibrils and provide high-tensile strength to skin, tendons, and ligaments. As indicated above, collagens are heterotrimers composed of three α-helical peptide chains (see Fig. 29.1). The type I collagen heterotrimer is composed of two α1(I) chains and one α2(I) chain. Each of these peptide chains contains about 1000 amino acids and has a triple-helical domain structure along almost the entire length of the molecule. The collagen fibrils are formed by lateral association of triple helices in a ‘quarter-staggered’ alignment in which each molecule is displaced by about one-quarter of its length relative to its nearest neighbor (Fig. 29.2). The quarter-staggered array is responsible for the banded appearance of collagen fibrils in connective tissues. The fibrils are stabilized by both noncovalent forces and interchain crosslinks derived from lysine residues (see below).
Nonfibrillar collagens
Non-fibrillar, lattice-forming collagens are major structural components of basement membranes
 Clinical box Osteogenesis imperfecta (incidence 1 in 30,000–50,000)
Clinical box Osteogenesis imperfecta (incidence 1 in 30,000–50,000)
A 6-year-old boy was seen in the casualty department with broken tibia and fibula occurring during a soccer game. His 6-foot-tall father explained that he had broken his legs four times while at school. The father's teeth were slightly transparent and discolored.
Osteogenesis imperfecta (OI), also called brittle bone disease, is a congenital disease caused by multiple genetic defects in the synthesis of type I collagen. It is characterized by fragile bones, thin skin, abnormal teeth and weak tendons. The majority of individuals with this disease have mutations in genes encoding α1(I) or α2(I) collagen chains. Many of these mutations are single-base substitutions that convert glycine in the Gly-X-Y repeat to bulky amino acids, preventing the correct folding of the collagen chains into a triple helix and their assembly to form collagen fibrils. The dominance of type 1 collagen in bone explains why bones are predominantly affected. However, there is remarkable clinical variability characterized by bone fragility, osteopenia, variable degrees of short stature, and progressive skeletal deformities. The most common form of OI, with a presentation that is sometimes mistaken for child abuse, has a good prognosis, with fractures decreasing after puberty, though the general reduction in bone mass means that lifetime risk remains high. Patients frequently develop deafness due to osteosclerosis, partly from recurrent fractures of the stapes. Bisphosphonate drugs (see Chapter 26), which inhibit osteoclast activity and thereby inhibit normal bone turnover, have reduced the incidence of fractures. Long-term follow-up studies are under way.
Nonfibrillar collagens are a heterogeneous group containing triple-helical segments of variable length, interrupted by one or more intervening nonhelical (noncollagenous) segments. This group includes basement membrane collagens (the type IV family), fibril-associated collagens with interrupted triple helices (FACITs), and collagens with multiple triple-helical domains with interruptions, known as multiplexins. Nonfibrillar collagens associate with the fibrillar collagens, forming microfibrils and network or mesh-like structures.
Basement membranes are relatively thin layers of ECM found on the basal aspect of epithelial cells and surrounding some other cell types including myocytes, Schwann cells and adipocytes. The basement membrane has a number of functions including anchorage of cells to surrounding connective tissue and filtration.
Type IV collagen assembles into a flexible mesh-like network. This collagen contains a long triple-helical domain interrupted by short noncollagenous sequences. These interruptions in the helical domain block continued association of two triple helices, oblige them to find another partner, and thus contribute to formation of a lattice-type structure. In the kidney, the thickened basement membrane (100–200 nm thick) on the basal aspect of the glomuerular capillary endothelial cells plays an essential role as a macromolecular filter (see Chapter 23). The meshwork of ECM proteins in the basement membrane restricts the passage of large molecules from the blood into the urine. In addition, the inclusion of negatively charged proteoglycans (described later in this chapter) in the glomerular basement membrane restricts the passage of charged molecules. Anomalies in type IV collagen in the glomerular basement membrane result in several glomerular diseases including Goodpasture's syndrome and Alport syndrome. Goodpasture's syndrome is a rare autoimmune disease caused by the production of antibodies that specifically bind to type IV collagen of basement membranes. This inflammatory condition leads to progressive worsening of basement membrane function in the kidney and sometimes in the lung. Alport syndrome results from mutations in the type IV collagen chains which cause defective collagen scaffold assembly within the basement membrane. The symptoms of both of these syndromes progress from blood in the urine (hematuria) to urine containing excessive protein (proteinuria) and eventually to kidney failure.
Synthesis and post-translational modification of collagens
Collagen synthesis begins in the rough endoplasmic reticulum (RER)
After synthesis in the RER, the nascent collagen polypeptide undergoes extensive modification, first in the RER, then in the Golgi apparatus, and finally in the extracellular space, where it is modified to a mature extracellular collagen fibril (Fig. 29.3). A nascent polypeptide chain, preprocollagen, is synthesized initially with a hydrophobic signal sequence that facilitates binding of ribosomes to the endoplasmic reticulum (ER) and directs the growing polypeptide chain into the lumen of the ER. Post-translational modification of the protein begins with removal of the signal peptide in the ER, yielding procollagen. Three different hydroxylases then add hydroxyl groups to proline and lysine residues, forming 3- and 4-hydroxyprolines and δ-hydroxylysine. These hydroxylases require ascorbate (vitamin C) as a cofactor (Fig. 29.3, step 1). Vitamin C deficiency leads to scurvy as a result of alterations in collagen synthesis and crosslinking (see Chapter 11).
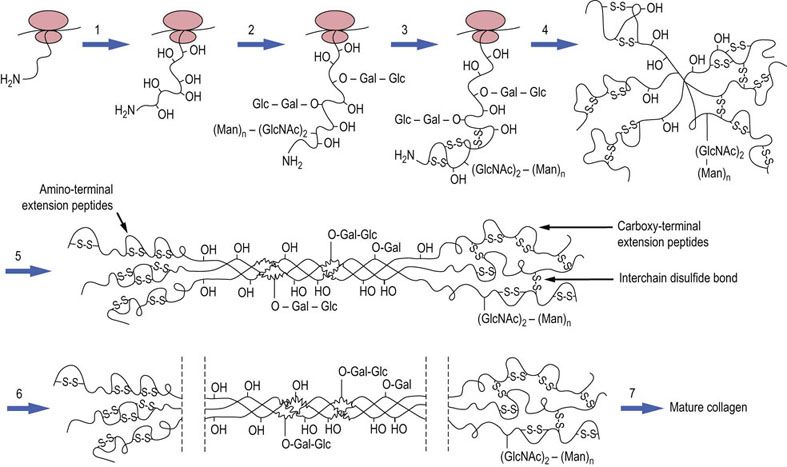
Fig. 29.3 Biosynthesis and post-translational processing of collagen in the endoplasmic reticulum.
Collagen is synthesized in the RER, post-translationally modified in the Golgi apparatus, then secreted, trimmed of extension peptides, and finally assembled into fibrils in the extracellular space. (1) Hydroxylation of proline and lysine residues. (2) Addition of O-linked and N-linked oligosaccharides. (3) Formation of intrachain disulfide bonds at the N-terminal of the nascent polypeptide chain. (4) Formation of interchain disulfides in the C-terminal domains, which assist in alignment of chains. (5) Formation of triple-stranded, soluble tropocollagen, and transport to Golgi vesicles. (6) Exocytosis and removal of N- and C-terminal propeptides. (7) Final stages of processing, including lateral association of triple helices, covalent crosslinking and collagen fiber formation. Gal, galactose; Glc, glucose; GlcNAc, N-acetylglucosamine; Man, mannose.
O-linked glycosylation occurs by the addition of galactosyl residues to hydroxylysine by galactosyl transferase; a disaccharide is formed by addition of glucose to galactosyl hydroxylysine by a glucosyl transferase (Fig. 29.3, step 2). These enzymes have strict substrate specificity for hydroxylysine or galactosyl hydroxylysine, and they glycosylate only those peptide sequences that are in noncollagenous domains. N-linked glycosylation also occurs on specific asparagine residues in nonfibrillar domains. The nonfibrillar collagens, with a greater extent of nonhelical domains, are more highly glycosylated than fibrillar collagens. Thus, the extent of glycosylation may influence fibril structure, interrupting fibril formation and promoting interchain interactions required for a meshwork structure. Intra- and interchain disulfide bonds are formed in the C-terminal domains by a protein disulfide isomerase, facilitating the association and folding of peptide chains into a triple helix (Fig. 28.3, steps 3–5). At this stage, the procollagen is still soluble and contains additional, nonhelical extensions at its N- and C-terminals.
Procollagen is finally modified to collagen in the Golgi apparatus
Clinical box Lathyrism: the result of lysyl oxidase inhibition
Lathyrism is a diet-induced disease characterized by deformation of the spine, dislocation of joints, demineralization of bones, aortic aneurysms, and joint hemorrhages. These problems develop as a result of inhibition of lysyl oxidase, an enzyme required for the crosslinking of collagen chains. Lathyrism can be caused by chronic ingestion of the sweet pea Lathyrus odoratus, the seeds of which contain β-aminopropionitrile, an irreversible inhibitor of lysyl oxidase. Penicillamine, a sulfhydryl agent used for chelation therapy in heavy-metal toxicity, also causes lathyrism, because of either chelation of copper required for lysyl oxidase activity or reaction with aldehyde groups of (hydroxy)allysine, inhibiting collagen crosslinking reactions.
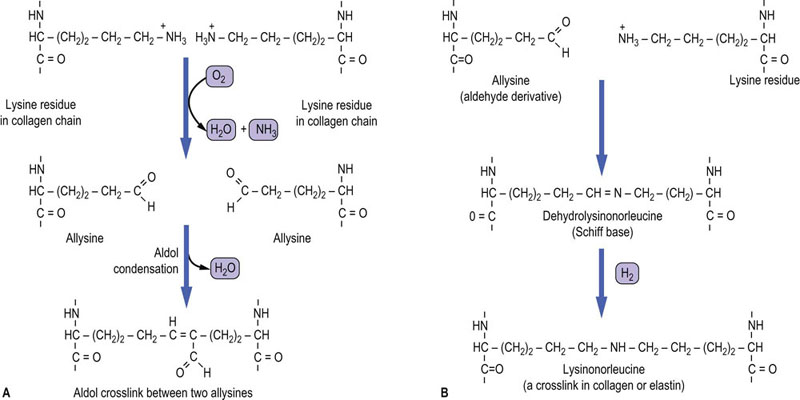
After assembly into the triple helix, the procollagen is transported from the RER to the Golgi apparatus, where it is packaged into cylindrical aggregates in secretory vesicles, then exported to the extracellular space by exocytosis. The nonhelical extensions of the procollagen are now removed in the extracellular space, by specific N- and C-terminal procollagen proteinases (Fig. 29.3, step 6). The ‘tropocollagen’ molecules then self-assemble into insoluble collagen fibrils, which are further stabilized by the formation of aldehyde-derived intermolecular crosslinks. Lysyl oxidase (not to be confused with lysyl hydroxylase involved in formation of hydroxylysine) oxidatively deaminates the amino group from the side chains of some lysine and hydroxylysine residues, producing reactive aldehyde derivatives, known as allysine and hydroxyallysine. The aldehyde groups now form aldol condensation products with neighboring aldehyde groups, generating crosslinks both within and between triple-helical molecules. They may also react with the amino groups of unoxidized lysine and hydroxylysine residues to form Schiff base (imine) crosslinks (Fig. 29.4). The initial products may rearrange, or be dehydrated, or reduced to form stable crosslinks, such as lysinonorleucine. Studies with β-aminopropionitrile, which inhibits the enzyme lysyl oxidase, have illustrated that collagen crosslink formation is a major determinant of tissue mechanical properties and strength.
Noncollagenous proteins in the extracellular matrix
Elastin
Weak hydrophobic interactions between valine residues permit the flexibility and extensibility of elastin
The flexibility required for function of blood vessels, lungs, ligaments and skin is imparted by a network of elastic fibers in the ECM of these tissues. The predominant protein of elastic fibers is elastin. Unlike the multigene collagen family, there is only one gene for elastin, coding for a polypeptide about 750 amino acids long. In common with collagens, it is rich in glycine and proline residues but elastin is more hydrophobic: one in seven of its amino acids is a valine. Unlike collagens, elastin contains little hydroxyproline and no hydroxylysine or carbohydrate chains, and does not have a regular secondary structure. Its primary structure consists of alternating hydrophilic and hydrophobic lysine and valine-rich domains. The lysines are involved in intermolecular crosslinking, while the weak interactions between valine residues in the hydrophobic domains impart elasticity to the molecule.
Clinical box Marfan syndrome: result from mutations in fibrillin gene
The ultrastructure of elastic fibers reveals elastin as an insoluble, polymeric, amorphous core covered with a sheath of microfibrils that contribute to the stability of the elastic fiber. The predominant constituent of microfibrils is the glycoprotein fibrillin. Marfan syndrome is a relatively rare genetic disease of connective tissues caused by mutations in the fibrillin gene (frequency: 1 in 10,000 births). People with this disease have typically tall stature, long arms and legs, and arachnodactyly (long, ‘spidery’ fingers). The disease in a mild form causes loose joints, deformed spine, floppy mitral valves (leading to cardiac regurgitation), and eye problems such as lens dislocation. In severely affected individuals, the aortic wall is prone to rupture because of defects in elastic fiber formation.
The soluble monomeric form of elastin initially synthesized on the RER is called tropoelastin. Except for some hydroxylation of proline, tropoelastin does not undergo post-translational modification. During the assembly process in the extracellular space, lysyl oxidase generates allysine in specific sequences: -Lys-Ala-Ala-Lys- and -Lys-Ala-Ala-Ala-Lys-. As with collagen, the reactive aldehyde of allysine condenses with other allysines or with unmodified lysines. Allysine and dehydrolysinonorleucine on different tropoelastin chains also condense to form pyridinium crosslinks – heterocyclic structures known as desmosine or isodesmosine (Fig. 29.5). Because of the way in which elastin monomers are cross-linked in polymers, elastin can stretch in two dimensions.
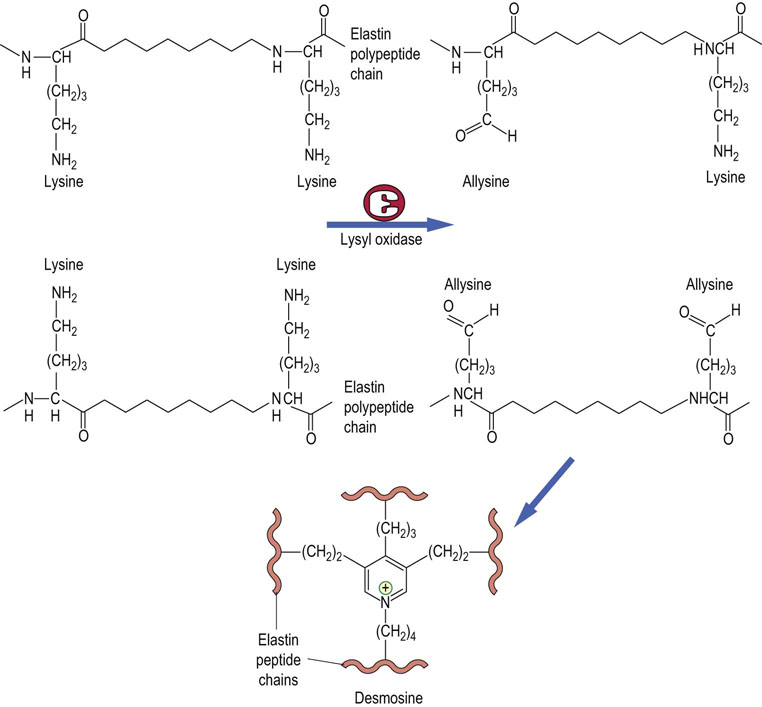
Fibronectin
Fibronectin and laminin have multiple binding sites for ECM proteins and proteoglycans
Fibronectin is a glycoprotein present as a structural component of the ECM and also in plasma as a soluble protein. Fibronectin is a dimer of two identical subunits, each of 230 kDa, joined by a pair of disulfide bonds at their C-terminals. Each subunit is organized into domains, known as type I, II, and III domains, and each of these has several homologous repeating units or modules in its primary structure: there are 12 type I repeats, two type II repeats, and 15–17 type III repeats. Each module is independently folded, forming a ‘string of beads’ type of structure. At least 20 different tissue-specific isoforms of fibronectin have been identified, all produced by alternative splicing of a single precursor messenger ribonucleic acid (mRNA). The alternative splicing is regulated not only in a tissue-specific manner but also during embryogenesis, wound healing, and oncogenesis. Plasma fibronectin, secreted mainly by liver cells, lacks two of the type III repeats that are found in cell- and matrix-associated forms of fibronectin. Because of its multidomain structure and its ability to interact with cells and with other ECM components, alterations in fibronectin expression affect cell adhesion and migration, embryonic morphogenesis, and cytoskeletal and ECM organization.
Functional domains in fibronectin have been identified by their binding affinity for other ECM components, including collagen, heparin, fibrin, and the cell surface. The type I modules interact with fibrin, heparin and collagen, type II modules have collagen-binding domains, and type III modules are involved in binding to heparin and the cell surface. The specific interactions have been further mapped to short stretches of amino acids. A short peptide containing Arg-Gly-Asp (RGD), present in the tenth type III repeat of fibronectin, binds to the integrin family of proteins present on cell surfaces; this sequence is not unique to fibronectin but is also found in other proteins in the ECM. Another sequence, Pro-X-Ser-Arg-Asn (PXSRN), present in the ninth type III repeat, is implicated in integrin-mediated cell attachment. The loss of fibronectin from the surface of many tumor cells may contribute to their release into the circulation and penetration through the ECM, one of the first steps in tumor metastasis.
Laminins
Laminins are a family of noncollagenous glycoproteins found in basement membranes and expressed in variant forms in different tissues. They are large (850 kDa), heterotrimeric molecules, composed of α, β and γ chains. To date, five α, four β and three γ chains have been identified which can associate to produce at least 15 different laminin variants. The three interacting chains in a heterotrimer are arranged in an asymmetric cruciform or cross-shaped molecule, held together by disulfide linkages. Laminins undergo reversible self-assembly in the presence of calcium to form polymers, contributing to the elaborate mesh-like network in the basement membrane. Biochemical and electron microscopic studies indicate that all full-length short arms of laminin are required for self-assembly and that the polymer is formed by joining the ends of the short arms. Like fibronectin, laminins interact with cells through multiple binding sites in several domains of the molecule. The α chains have binding sites for integrins and heparan sulfate (below). Laminin polymers are also connected to type IV collagen by a single-chain protein, nidogen/entactin, which has a binding site for collagen and, in common with fibronectin, also has an RGD sequence for integrin binding. Nidogen also binds to the core proteins of proteoglycans (below). It has a central role in formation of crosslinks between laminin and type IV collagen, generating a scaffold for anchoring of cells and ECM molecules in the basement membrane.
Clinical box Muscular dystrophies
Muscular dystrophies are a heterogeneous group of genetic disorders that result in progressive decline in muscle strength and structure. To date, mutations have been identified in more than 30 genes that result in muscular dystrophies. Many of the identified gene products are components of the ECM–cell surface–cytoskeletal complex of muscle cells. In particular, one class of muscular dystrophy is caused by mutations in the α2 chain of laminin-2. These mutations prevent normal polymer formation of laminin-2 and result in abnormal basement membrane organization surrounding skeletal muscle fibers of patients with this muscular dystrophy.
Clinical box Epidermolysis bullosa
Epidermolysis bullosa is a rare heritable disorder characterized by severe blistering of the skin and epithelial tissue. Three kinds are known:
simplex: blistering in the epidermis, caused by defects in keratin filaments
junctional: blistering in the dermal–epidermal junction, caused by defects in laminin
dystrophic: blistering in the dermis, caused by mutations in the gene encoding type VII collagen.
Epidermolysis bullosa illustrates the multifactorial nature of connective tissue diseases that have similar clinical features.
Proteoglycans
Proteoglycans are gel-forming components of the ECM and comprise what has classically been called the ‘ground substance’. Some proteoglycans are located on the cell surface, where they bind growth factors and other ECM components. They are composed of peptide chains containing covalently bound sugars. However, the peptide chains of proteoglycans are usually more rigid and extended than the protein portion of the glycoproteins, and the proteoglycans contain much larger amounts of carbohydrate – typically >95% carbohydrate. The sugar chains are linear, unbranched oligosaccharides that are much longer than those of the glycoproteins, and may contain more than 100 sugar residues in a chain. Furthermore, the oligosaccharide chains of proteoglycans have a repeating disaccharide unit, usually composed of a uronic acid and an amino sugar. Proteoglycan oligosaccharide chains are polyanionic because of the many negative charges of the carboxyl groups of the uronic acids, and from sulfate groups attached to some of the hydroxyl or amino groups of the sugars.
Structure of proteoglycans
Glycosaminoglycans are the polysaccharide components of proteoglycans
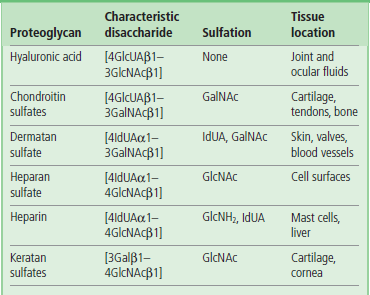
The general structures of the glycosaminoglycans (GAGs) are shown in Table 29.2. The disaccharide repeat is different for each type of GAG, but is usually composed of a hexosamine and a uronic acid residue, except in the case of keratan sulfate, in which the uronic acid is replaced by galactose. The amino sugar in GAGs is either glucosamine (GlcNH2) or galactosamine (GalNH2), both of which are present mostly in their N-acetylated forms (GlcNAc and GalNAc), although in some of the GAGs (heparin, heparan sulfate) the amino group is sulfated rather than acetylated. The uronic acid is usually D-glucuronic acid (GlcUA) but in some cases (dermatan sulfate, heparin) it may be L-iduronic acid (IdUA). With the exception of hyaluronic acid and keratan sulfate, all the GAGs are attached to protein by a core trisaccharide, Gal-Gal-Xyl; the xylose is linked to a serine or threonine residue of a core protein. Keratan sulfate is also attached to protein, but in that case the linkage is either through an N-linked oligosaccharide (keratan sulfate I) or an O-linked oligosaccharide (keratan sulfate II). Hyaluronic acid, which has the longest polysaccharide chains, is the only GAG that does not appear to be attached to a core protein.
Hyaluronic acid
Hyaluronic acid, the only nonsulfated glycosaminoglycan, has a unique role in proteoglycan assembly
Hyaluronic acid is composed of repeating units of GlcUA and GlcNAc. This polysaccharide chain is the longest of the GAGs, with molecular weight of 105–107 Da (250–25,000 repeating disaccharide units), and is the only nonsulfated GAG.
The chondroitin sulfates
The chondroitin sulfates are major components of cartilage. They contain GalNAc rather than GlcNAc as the amino sugar, and their polysaccharide chains are shorter: 2–5 × 105 Da. The chondroitin chains are attached to protein via the trisaccharide linkage region (Gal-Gal-Xyl), and they contain sulfate residues linked to either the 4- or 6-hydroxyl groups of GalNAc.
Dermatan sulfate
Dermatan sulfate was originally isolated from skin but is also found in blood vessels, tendon and heart valves. This GAG is similar in structure to chondroitin sulfate but has a variable amount of L-iduronic acid (IdUA), the C-5-epimer of D-GlcUA, formed in an unusual reaction by epimerization of GlcUA after it has been incorporated into the polymer. Dermatan sulfate has a higher charge density than the chondroitin sulfates, as it contains sulfate residues on the C-2 position of some IdUA residues, and on the 4-hydroxyl groups of GalNAc.
Heparin and heparan sulfate
Heparin is a small, highly charged GAG with strong anticoagulant activity
Heparin and heparan sulfate consist primarily of repeating disaccharide units of GlcNH2 with IdUA or GlcUA, respectively. The linkage between the amino sugar and the uronic acid is uniformly 1–4, rather than the alternating 1–4/1–3 linkages seen in other GAGs. Most of the GlcNH2 units of heparin are N-sulfated, whereas many of the IdUA residues are sulfated at the C-2 hydroxyl group, and the GlcNH2 residues at the C-6 hydroxyl group. Heparin and heparan sulfate are the most highly charged of the GAGs. Although the structures of these two polymers are closely related, their distribution in the body and their functions are quite different: heparin is a small microheterogeneous molecule (~3000–30,000 Da), found intracellularly as a proteoglycan. It is released into the extracellular space as a free polysaccharide (GAG) and has strong anticoagulant activity (Chapter 7). In contrast, heparan sulfate is bound in the ECM or on the surface of cells, and has only weak anticoagulant activity.
Keratan sulfate
The final GAG structure shown in Table 29.2 is keratan sulfate (KS). This is a rather unusual GAG because it is linked to protein by either an N-linked (KS I) or an O-linked (KS II) oligosaccharide. Thus it has features common to both proteoglycans and glycoproteins. It is considered to be a proteoglycan, however, because the glycan portion has a repeating disaccharide unit and a long, linear chain. The repeating unit is composed of GlcNAc and galactose, instead of the uronic acid. Both the GlcNAc and the galactose are generally sulfated on the C-6 hydroxyl groups.
Synthesis and degradation of proteoglycans
The structure of glycosaminoglycans is determined by the cell's complement of glycosyl and sulfotransferases
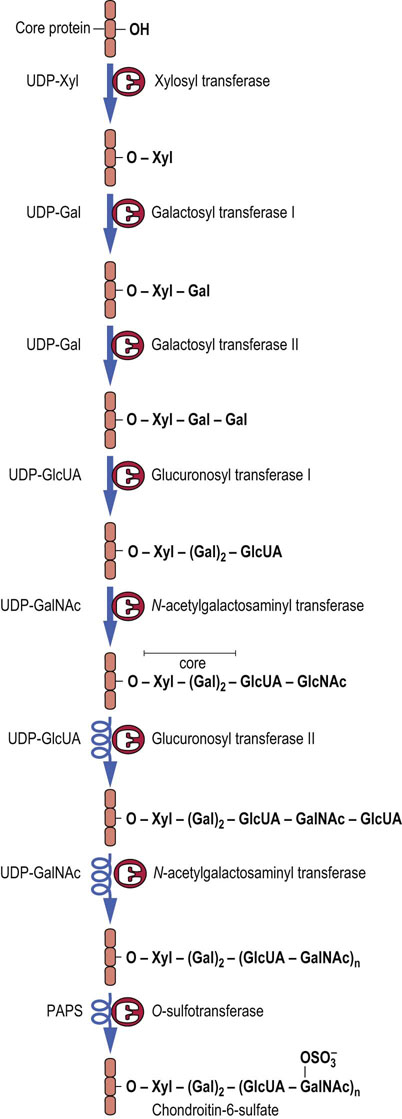
Proteoglycans are synthesized by a series of glycosyl transferases, epimerases and sulfotransferases, beginning with the synthesis of the core trisaccharide (Xyl→Gal→Gal) while the protein is still in the RER. Synthesis of the repeating oligosaccharide and other modifications take place in the Golgi apparatus. As with the synthesis of glycoproteins and glycolipids, separate enzymes are involved in individual steps. For example, there are separate galactosyl transferases for each of the galactose units in the core, a separate GlcUA transferase for the core and repeating disaccharides, and separate sulfotransferases for the C-4 and C-6 positions of the GalNAc residues of chondroitin sulfates. Phosphoadenosine phosphosulfate (PAPS) is the sulfate donor for the sulfotransferases. These pathways are illustrated in Figure 29.6, for chondroitin-6-sulfate.
Defects of proteoglycan degradation lead to mucopolysaccharidoses
The degradation of proteoglycans occurs in lysosomes. The protein portion is degraded by lysosomal proteases and the GAG chains are degraded by the sequential action of a number of different lysosomal acid hydrolases. The stepwise degradation of GAGs involves exoglycosidases and sulfatases, beginning from the external end of the glycan chain. This may involve the removal of sulfate by a sulfatase, then removal of the terminal sugar by a specific glycosidase, and so on. Figure 29.7 shows the steps in the degradation of heparan sulfate. As with degradation of glycosphingolipids, if one of the enzymes involved in the stepwise pathway is missing, the entire degradation process is halted at that point and the undegraded molecules accumulate in the lysosome. The lysosomal storage diseases resulting from accumulation of GAGs are known as mucopolysaccharidoses (Table 29.3), because of the original designation of GAGs as mucopolysaccharides. There are more than a dozen such mucopolysaccharidoses, resulting from defects in degradation of GAGs. In general, these diseases can be diagnosed by the identification of specific GAG chains in the urine, followed by assay of the specific hydrolases in leukocytes or fibroblasts.
Table 29.3
Enzymatic defects characteristic of various mucopolysaccharidoses
| Syndrome | Deficient enzyme | Product accumulated in lysosomes and secreted in urine |
| Hunter's | Iduronate sulfatase | Heparan and dermatan sulfate |
| Hurler's | α-Iduronidase | Heparan and dermatan sulfate |
| Morquio's A | Galactose-6-sulfatase | Keratan sulfate |
| Morquio's B | β-Galactosidase | Keratan sulfate |
| Sanfilippo's A | Heparan sulfamidase | Heparan sulfate |
| Sanfilippo's B | N-Acetylglucosaminidase | |
| Sanfilippo's C | N-Acetylglucosamine-6-sulfatase |
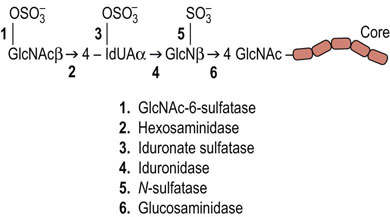
Fig. 29.7 Degradation of heparan sulfate.
This proceeds by a defined sequence of lysosomal hydrolase activities.
 Advanced concept box Mechanisms of the anticoagulant effect of heparin
Advanced concept box Mechanisms of the anticoagulant effect of heparin
Heparin is a heterogeneous (3000–30,000 kDa), polyanionic oligosaccharide activator of antithrombin III (AT) (Chapter 7). AT is a slow but quantitatively important inhibitor of thrombin (factor X) and other factors (IX, XI, XII) in the blood-clotting cascade. When heparin binds to AT, it converts AT from a slow inhibitor to a rapid inhibitor of coagulating enzymes. Heparin interacts with a lysine residue in AT and induces a conformational change that promotes covalent binding of AT to the active serine centers of coagulating enzymes, inhibiting their procoagulant activity. Heparin then dissociates from the ternary complex and can be recycled for anticoagulation.
The smallest, most active component of heparin is a pentasaccharide [GlcN-(N-sulfate-6-O-sulfate)-α1,4-GlcUA-β1,4-GlcN-(N-sulfate-3,6-di-O-sulfate)-α1,4-IdUA-(2-O-sulfate)-α-1,4-GlcN-(N-sulfate-6-O-sulfate], which has a Kd of ~10 μM for binding to ATlll. Heparin has an average half-life of 30 min in the circulation, so that it is commonly administered by infusion. Heparin does not have fibrinolytic activity; therefore, it will not lyse existing clots. In addition to its anticoagulant activity, heparin also releases several enzymes from proteoglycan binding sites on the vascular wall, including lipoprotein lipase, which is often assayed as heparin-releasable plasma lipoprotein lipase activity or postheparin lipase. Lipoprotein lipase is inducible by insulin, and decreased activity of this enzyme delays plasma clearance of chylomicrons and VLDL, contributing to hypertriglyceridemia in diabetes (Chapter 18).
Functions of the proteoglycans
Bottlebrushes, silly putty and reinforced concrete
Proteoglycans are found in association with most tissues and cells. One of their major roles is to provide structural support to tissues, especially cartilage and connective tissue. In cartilage, large aggregates, composed of chondroitin sulfate and keratan sulfate chains linked to their core proteins, are noncovalently associated with hyaluronic acid via link proteins, forming a jelly-like matrix in which the collagen fibers are embedded. This macromolecule, a ‘bottlebrush’ structure known as aggrecan (Fig. 29.8), provides both rigidity and stability to connective tissue. Because of their negative charge, the GAGs bind large amounts of monovalent and divalent cations: a cartilage proteoglycan molecule of 2 × 106 Da would have an aggregate negative charge of about 10,000. The maintenance of electrical neutrality consequently requires a high concentration of counter-ions. These ions draw water into the ECM, causing swelling and stiffening of the matrix, the result of tension between osmotic forces and binding interactions between proteoglycans and collagen. The structure and hydration of the ECM allow for a degree of rigidity, combined with flexibility and compressibility, enabling the tissue to withstand torsion and shock. The hyaluronic acid–proteoglycan–collagen aggregates in vertebral and articular disks have some of the viscoelastic properties of ‘silly putty’, bounce plus resilience, cushioning the impact between bones. These disks compress during the course of the day, expand elastically during the night, and deform gradually with age.
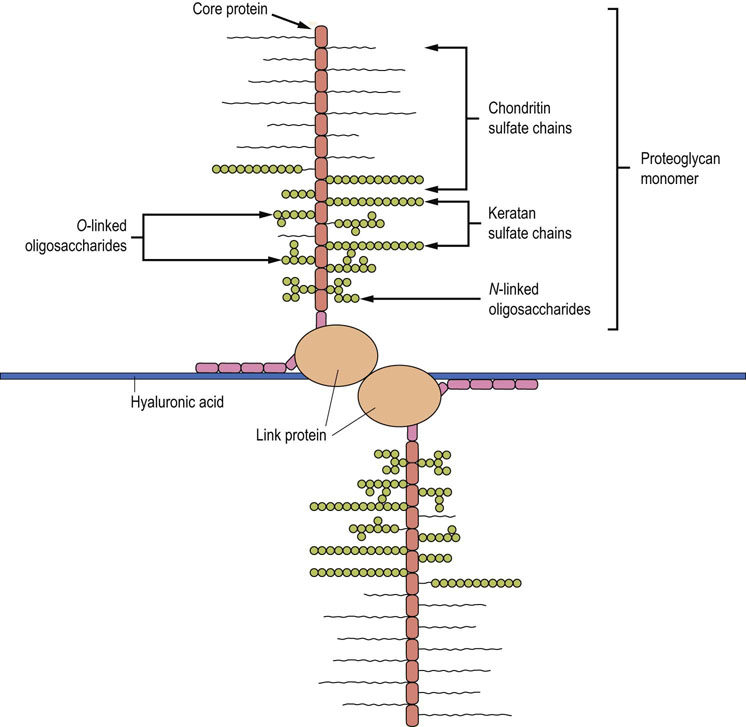
Fig. 29.8 Structure of aggrecan.
Associations between proteoglycans and hyaluronic acid form an aggrecan structure in the extracellular matrix (ECM). The extension of this structure yields a three-dimensional array of proteoglycans bound to hyaluronic acid, which creates a stiff matrix or ‘bottlebrush’ structure in which collagen and other ECM components are embedded.
The overall structure of cartilage can be likened to that of the vertical reinforced concrete slabs poured during the construction of large buildings, in which steel rods (collagen fibers) are embedded in an amorphous layer of cement (the proteoglycan aggregates). Collagen stabilizes the network of proteoglycans in cartilage in much the same way that the reinforcing rods in the concrete provide structural strength for the cement walls. The structure of earthquake-resistant buildings, like the ECM, provides a balance between integrity and flexibility.
Although the amounts involved are low compared with those in skin and cartilage, organs such as the liver, brain, or kidney also contain a variety of proteoglycans:
Liver: heparan sulfate is the principal GAG; it is present both intracellularly and on the cell surface of the hepatocyte, and the attachment of hepatocytes to their substratum in cell culture is mediated, in part, by this proteoglycan.
Kidney: changes in both the collagen and proteoglycan content of the renal basement membrane are associated with diabetic renal disease. In this case, the change in structure and charge of the proteoglycan aggregate, known as perlecan, is associated with a change in the filtration selectivity of the glomerulus (Chapter 23).
Cornea: two populations of proteoglycans have been identified in the cornea, one containing keratan sulfate and the other dermatan sulfate. These molecules have a much smaller hydrodynamic size than the large cartilage proteoglycans, which may be required for interaction of the corneal proteoglycans with the tightly packed and oriented collagen fibers in this transparent tissue. Corneal clouding in macular corneal dystrophy is associated with undersulfation of keratan sulfate I proteoglycan.
Some proteoglycans or GAGs, especially heparin and heparan sulfate, have important physiologic roles in binding proteins or other macromolecules. Heparin serves as an intracellular binding site for proteinases located in secretory granules of mast cells. Several proteoglycans are involved in binding of proteins and enzymes to the vascular wall. They may also function in the vascular wall to inhibit clot formation by activation of antithrombin III (Chapter 7).
Communication of cells with the extracellular matrix
Integrins are plasma membrane proteins that bind to and transmit mechanical signals between the ECM and intracellular proteins
Interactions between cells and the ECM regulate a wide variety of cellular processes including proliferation, migration, differentiation and even survival. Several cell surface receptors have been indentified that mediate these interactions including integrins, discoidin domain receptors, dystroglycan and others. Of these, the integrins appear to be the most ubiquitous form of ECM receptors. Integrins are widely expressed throughout the animal kingdom from sponges to humans. They are heterodimers of α and β chains that have been loosely grouped into subfamilies based upon the component β chain. To date, 18 α and eight β chains have been indentified in mammals. Through various combinations of α and β chains, over 20 different functional integrin heterodimers have been described. The specific combination of α and β chains dictates the specific ECM ligand for a particular integrin heterodimer. However, multiple integrin heterodimers can bind to some ECM components. For instance, α4β1, α5β1 and αvβ3 all interact with fibronectin. Adding to this complexity, several integrin heterodimers bind to multiple ECM components. For instance, αvβ3, which was originally described as a vitronectin receptor, can interact with not only vitronectin but also with fibronectin, fibrinogen and osteopontin as well.
In a functional integrin, both the α and β chains span the cell membrane (Fig. 29.9). Typically, each chain has a large extracellular domain, a single transmembrane domain and a short cytoplasmic tail. An exception to this is the β4 protein, which has an exceptionally long cytoplasmic tail of over 1000 amino acids. The extracellular region of the integrin heterodimer interacts with ECM components in a divalent cation-dependent manner. The integrins are in an optimal position to transmit physical or mechanical signals from the ECM to the interior of the cell. These physical signals can be further distributed through the cell via the actin-containing cytoskeleton and ultimately modulate gene expression in the nucleus. This transmission of physical signals via the ECM–integrin–cytoskeletal axis has been extensively investigated and termed ‘tensegrity’. Physical signals from the ECM can also be transduced into biochemical events in the cytoplasm of the cell via integrins. Unlike some other types of receptors, integrins do not themselves possess enzymatic activity. However, integrins associate with a number of cytoplasmic protein kinases including focal adhesion kinase (FAK) and Src. Activation of integrins initiates enzymatic cascades via these associated kinases that ultimately leads to changes in cell behavior and gene expression.
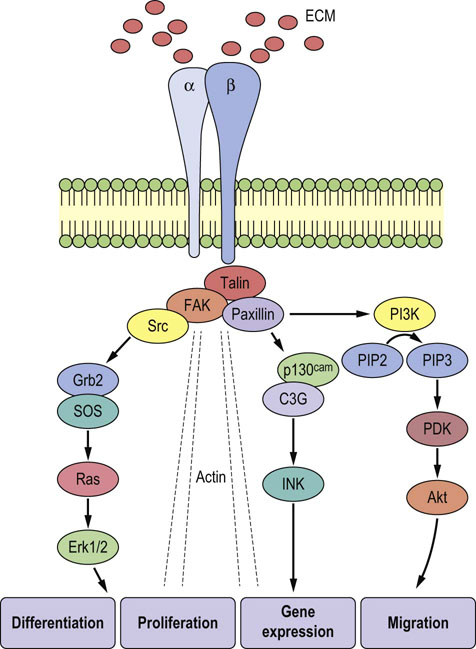
Fig. 29.9 Organization of integrins.
The α and β chains span the cell membrane, interacting with the ECM outside the cell and the cytoskeleton and signaling molecules inside. In this manner, integrins can transduce signals from the ECM into biochemical and mechanical events in the cytoplasm that ultimately lead to alterations in cell morphology and function. The ovals contain abbreviations for components of the complex signaling cascade that conveys information from the integrin molecule to the nucleus of the cell.
Advanced concept box Matrix remodeling
The ECM is in a constant state of synthesis and degradation, repair and remodeling: for example, during cell migration, morphogenesis, angiogenesis, and in response to inflammation and injury. ECM turnover is mediated by a family of matrix metalloproteinases (MMPs), about 30 zinc endoproteinases with specificity for different components of the matrix. The MMP family includes collagenases, stromelysins, matrilysins and elastases; these enzymes, with broad substrate specificities, catalyze degradation of collagen, aggrecan and accessory ECM proteins, such as fibronectin and laminin.
MMPs may be integral plasma membrane proteins, may be bound to the plasma membrane by a glycosylphosphatidylinositol (GPI) glycan anchor (Chapter 28), or secreted into the extracellular space; they exist as zymogens until activated locally by proteolytic cleavage in response to cellular signals or extracellular enzymes, such as thrombin and plasmin, activated during blood clotting and fibrinolysis. As with the cascade of protease reactions involved in blood coagulation, there are also tissue inhibitors of MMPs, known as TIMPs, a family of four proteins that inactivate MMPs and limit the spread of damage. The balance between the activation and inhibition of MMPs is critical to the integrity and function of the ECM; alterations in MMP activity are associated with skeletal dysplasias, coronary artery disease, arthritis and metastasis.
Advanced concept box Extracellular matrix and tissue engineering
Over the past decade, the interest in producing replacement tissues through tissue engineering has grown considerably. The ultimate goal of tissue engineering is to combine appropriate cells and biomaterials to produce tissue equivalents that mimic normal tissues and organs and can replace damaged or diseased tissues. As the biological and mechanical properties of tissues are determined in part by the heterogeneous composition and organization of the ECM, the successful generation of tissue equivalents will require the development of appropriate three-dimensional ECM scaffolds.
An attractive therapeutic approach is to combine undifferentiated stem cells with appropriate scaffolds and biochemical factors to promote differentiation of the cells along particular lines, depending upon the specific replacement tissue desired. Properties of the ECM scaffold, including ECM composition, porosity and mechanical properties, have important effects on stem cell differentiation. Culture of mesenchymal stem cells in scaffolds of relatively high stiffness tends to promote the formation of bone-like tissue and the formation of osteoblasts, while culture of the same stem cells in less stiff scaffolds results in formation of cartilage cells or chondroblasts. These and other studies illustrate that physical and mechanical cues from the ECM are important in regulating differentiation of stem cells. Advances in tissue engineering and production of replacement tissues will require a thorough understanding of the normal and pathologic ECM.
Summary
The ECM contains a complex array of fibrillar and network-forming collagens, elastin fibers, a stiff gelatinous matrix of proteoglycans, and a number of glycoproteins that mediate the interaction of these molecules with one another and with the cell surface.
Interactions between ECM components afford structure, stability, and elasticity to the ECM, and provide a route for communication between the intra- and extracellular environments in tissues.
The heterogeneity of both the protein and the carbohydrate components of the ECM provides for great diversity in the structure and function of the ECM in various tissues.
Couchman, JR, Pataki, CA. An introduction to proteoglycans and their localization. J Histochem Cytochem. 2012; 60:885–897.
Hanein, D, Horwitz, AR. The structure of cell-matrix adhesions: the new frontier. Curr Opin Cell Biol. 2012; 24:134–140.
Ingber, DE. Tensegrity-based mechanosensing from macro to micro. Prog Biophys Mol Biol. 2008; 97:163–179.
Kadler, KE, Baldock, C, Bella, J, Boot-Handford, RP. Collagens at a glance. J Cell Science. 2007; 120:1955–1958.
Kock, L, van Donkelaar, CC, Ito, K. Tissue engineering of functional articular cartilage: the current status. Cell Tissue Res. 2012; 347:613–627.
Murphy, G. Tissue inhibitors of metalloproteinases. Genome Biol. 2011; 12:233.
Stupack, DG. The biology of integrins. Oncology. 2007; 21:6–12.
Yamauchi, M, Sricholpech, M. Lysine post-translational modifications of collagen. Essays Biochem. 2012; 52:113–133.
Collagen in disease. www.dupuytrenfoundation.org.
Marfan's syndrome. www.marfan.org.
Mucopolysaccharidoses. www.ninds.nih.gov/health.