Role of Kidneys in Metabolism
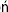 ska-Konkel
ska-KonkelLearning objectives
After reading this chapter you should be able to:
 Describe the handling of sodium and water in the nephron.
Describe the handling of sodium and water in the nephron.
Discuss links between renal oxygen consumption and sodium reabsorption.
Explain the role of sodium/potassium ATPase in the nephron.
Explain why urine analysis can provide clinically important information.
Describe clinical assessment of the glomerular filtration rate.
Introduction
The human kidneys weigh about 300 g. They have a rich blood supply, receiving about 25% of the cardiac output. About 80% of the blood is distributed in the renal cortex and 20% in the renal medulla. Almost all the blood passes through the glomerular capillaries, which act as high-pressure filters. Every day about 180 L of plasma containing several kilograms of plasma proteins, sodium chloride and other electrolytes, and metabolites, are filtered through the glomerular filtration area of 0.5–2 m2. More than 99.9% of the plasma proteins are retained by the filter, whereas almost all filtered water and sodium chloride and other solutes are retained by transport systems in the renal tubules. Under normal conditions, the kidneys form 1–2 liters of urine per day. Urine composition is summarized in Table 23.1.
Table 23.1
Daily excretion of nitrogen compounds and main ions in urine (mmol/24h)
Urea is a major contributor of the urine nitrogen excretion. It is the final product of protein catabolism in humans. Daily urea excretion also reflects nutrition status and strongly depends on protein intake.
Uric acid excretion depends mainly on endogenous purine degradation, but might be elevated on a purine-rich diet.
Creatinine is derived from the skeletal muscle phosphocreatine.
At metabolic steady state, urinary excretion of nitrogen compounds strictly depends on kidney function.
In renal failure urine output falls and this leads to an increase in plasma urea and creatinine concentrations.
Urinary excretion of sodium, potassium and chloride reflect their intake. Excessive sodium intake or impaired elimination might lead to hypertension.
Ammonia is generated in the kidney by deamination of glutamine and glutamate, and is excreted as the ammonium ion. Daily excretion of ammonia and phosphate depends on hydrogen ion excretion in urine (Chapter 25).
The key functions of the kidney are filtration of plasma, and subsequent tubular excretion and reabsorption of ions, low-molecular-weight substances and water
The excretory function of the kidneys involves filtration of the plasma in the glomeruli, transport of water and solutes from the tubular lumen back to the blood (tubular reabsorption), and secretion of different substances from the tubular cells into the lumen.
The kidneys remove, in urine, products of metabolism such as urea, uric acid and creatinine, and retain substances such as glucose, amino acids, and proteins. They also metabolize and remove drugs and toxins. They play a critical role in the regulation of extracellular fluid volume and composition and in maintaining the acid–base balance (Chapters 24 and 25). These functions are regulated hormonally by vasopressin (antidiuretic hormone, ADH) produced in the posterior pituitary, and by the renin–angiotensin system. Kidney function is also controlled by norepinephrine and dopamine released from nerve terminals in the kidney itself.
The kidneys also produce calcitriol (1,25-dihydroxycholecalciferol, 1,25(OH)2D3), a vitamin involved in calcium homeostasis (Chapter 26) and erythropoietin, which controls the production of erythrocytes.
The functional unit of the kidney is the nephron
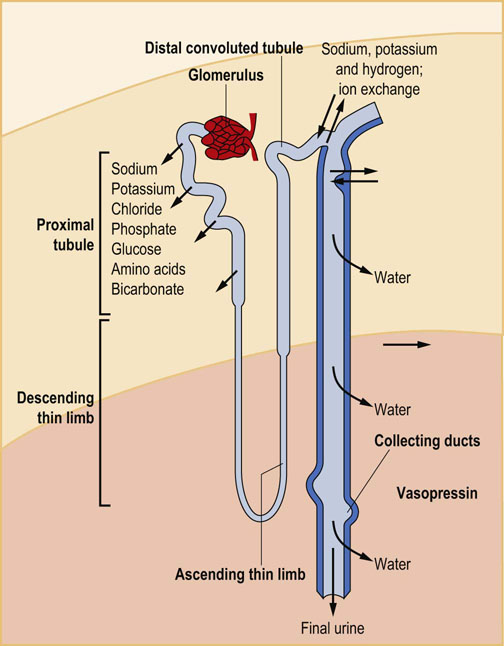
Each kidney consists of approximately 1 million nephrons composed of glomerulus and an excretory tubule (Fig. 23.1). Glomeruli, located in the kidney cortex, are biological filters, interfacing the plasma with the tubules that reabsorb and excrete substances. The segments of each tubule (starting from the glomerulus end) are known as the proximal tubule, the loop of Henle, the distal tubule, and the collecting duct.
The glomerular filtration barrier
Plasma from the glomerular capillaries is filtered into the interior of the glomerulus known as the Bowman's space. The filtration barrier includes a layer of endothelial cells that line glomerular blood vessels, the basement membrane, and the epithelial cells (podocytes) with characteristic foot processes (Fig. 23.2). Glomerular filtration depends on the filtration surface and on the permeability of the barrier.
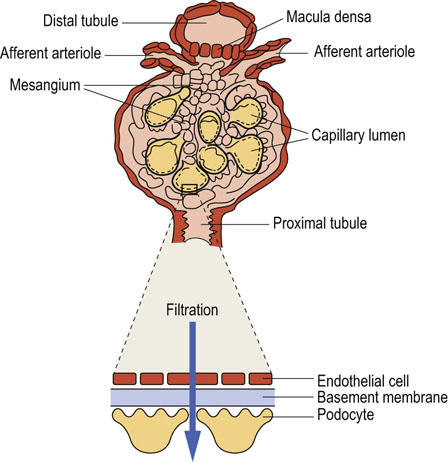
Fig. 23.2 Glomerular filtration barrier.
The barrier consists of the endothelial cells, the basement membrane, and the podocytes. The macula densa cells are part of the juxtaglomerular apparatus: they sense chloride concentration in the distal tubule and adjust the diameter of afferent arterioles, regulating the glomerular blood flow.
The main component of the glomerular basement membrane is type IV collagen, which forms a network of filaments providing elasticity and resistance to hydrostatic pressure. The membrane also contains laminin, fibronectin and proteoglycans with negatively charged heparan sulfate groups, which form an electrostatic barrier for proteins filtered from plasma.
The podocytes and mesangial cells possess receptors for a range of vasoactive substances such as angiotensin II, vasopressin, bradykinin, ATP, endothelin, prostaglandins, dopamine, natriuretic peptides and adenine nucleotides.
The fenestrations in the endothelial layer and the spaces between foot processes of the podocytes form a sieve that filters water and small molecules. Filtration of larger molecules is limited by their size, shape, and electric charge. For instance, at pH 7.4, most plasma proteins are negatively charged, and so is the filtration barrier; this hinders filtration of even the smallest proteins, such as myoglobin (molecular mass 17 kDa), and almost completely prevents filtration of the larger (69 kDa) albumin.
Glomerular filtration is driven by the hydrostatic pressure in the glomerular capillaries, which is approximately 50 mmHg. The hydrostatic pressure is counteracted by the oncotic pressure of the plasma and the back-pressure (approximately 10 mmHg) of the filtrate in the glomerular capsule. Changes in the glomerular filtration rate alter the total amount of filtered water and solute, but not the composition of the filtrate. A decrease in the blood pressure in the afferent arteriole of the glomerulus is sensed by the group of cells known as the juxtaglomerular apparatus. This stimulates renin secretion and activates the renin–angiotensin system. An increase in the intracapillary blood pressure and/or hyperfiltration may induce glomerular cells' injury and may lead to severe kidney damage.
 Advanced concept box Hyperglycemia and mechanical stress resulting from glomerular hypertension contribute to the development of diabetic nephropathy
Advanced concept box Hyperglycemia and mechanical stress resulting from glomerular hypertension contribute to the development of diabetic nephropathy
Mechanical stress and high glucose concentration trigger multiple signaling pathways that may accelerate glomerular cells' damage. Podocytes, the cells covering the outer aspect of glomerular basement membrane, are subjected not only to the load of filtered glucose but also to diverse mechanical forces. Both high glucose and mechanical stress may impair the protein systems anchoring the podocyte foot processes in the glomerular basement membrane (integrin, agrin), therefore blunting resistance of these cells to mechanical forces, and causing their detachment from the membrane (and appearance in urine). Modulation by these factors of expression and activity of numerous structural and functional proteins such as actin, nephrin, podocin, and other compounds such as phospholipase C, growth factors, cytokines, and chemokines results in the inflammatory responses, dysfunction, and apoptosis or necrosis of the podocytes. Loss of the podocytes is irreversible due to their inability to proliferate and to replenish damaged cells. Podocytes are injured early in the course of diabetic nephropathy.
Glomerular filtrate: formation of urine
Kidneys consume large amounts of oxygen, mainly to support active sodium transport
Most of the metabolic processes in the kidneys are aerobic, and consequently their oxygen consumption is high: it approximately equals that of the cardiac muscle, and is three times greater than that of the brain. Such a high metabolic activity is required to maintain tubular reabsorption; about 70% of the oxygen consumed by the kidney is used to support active sodium transport, which in turn determines reabsorption of glucose and amino acids.
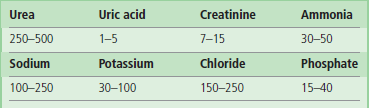
Reabsorption of sodium. Approximately 80% of the filtrate is reabsorbed in the proximal tubule. Sodium is reabsorbed by several mechanisms: through specific ion channels, in exchange for the hydrogen ion, and in co-transport with glucose, amino acids, phosphate, and other anions. The movement of sodium causes reabsorption of water (Fig. 23.3). The entry of sodium into the proximal tubular cells is passive. This is possible because the Na+/K+-ATPase maintains low sodium concentration in the cytoplasm of the tubular cells. More sodium is reabsorbed in the distal tubule and in the collecting duct in exchange for potassium or hydrogen ion. This process is controlled by aldosterone (Fig. 23.3 and 23.4).
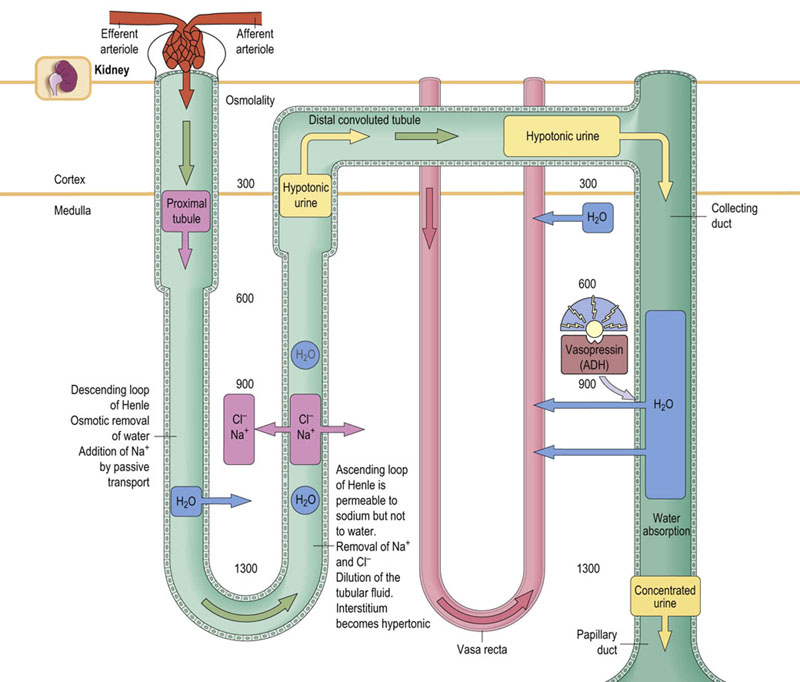
Fig. 23.3 Counter-current exchange and multiplication in the renal tubules.
The counter-current mechanism is essential for the formation of urine, and for reabsorption of water in the distal tubule. In the ascending arm of the loop of Henle, sodium and chloride ions are pumped into the interstitial fluid. They then diffuse freely into the lumen of the descending limb, creating a functional loop, which perpetuates the increase in osmolality of the filtrate reaching the ascending limb. This is known as counter-current multiplication. As a result of this, while the osmolality of the renal cortex is similar to that of plasma (300 mmol/L), in the medulla it might be as high as 1300 mmol/L. High osmolality of the medulla later facilitates reabsorption of water in the collecting ducts. This is known as counter-current exchange. The amount of reabsorbed water is controlled by vasopressin.
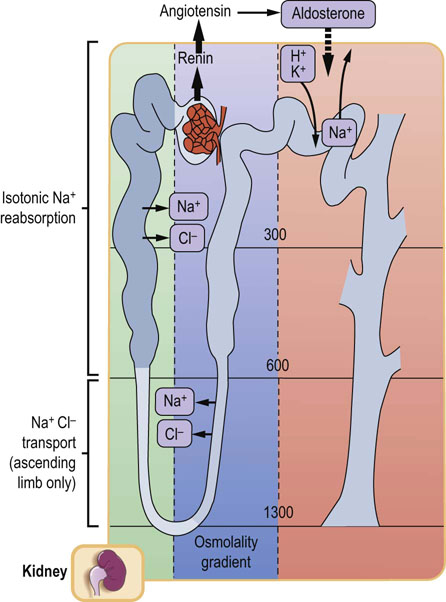
Fig. 23.4 Sodium reabsorption in the renal tubules.
More than 80% of filtered sodium is actively reabsorbed in the proximal tubule. Sodium and chloride ions are also reabsorbed in the ascending limb of the loop of Henle. A different mechanism operates in the distal tubule, where sodium reabsorption is stimulated by aldosterone and is coupled with the secretion of hydrogen and potassium ions. Aldosterone causes sodium retention and an increase in potassium excretion.
Reabsorption of water. The fluid leaving the proximal tubule is isotonic. Different permeability of the ascending and descending limbs of the loop of Henle maintains high osmolality of the medulla. This is essential for subsequent reabsorption of water in the collecting duct (Fig. 23.5). The fluid that leaves the loop of Henle is diluted (hypotonic). Water reabsorption in the collecting duct is controlled by vasopressin (Fig. 23.3).
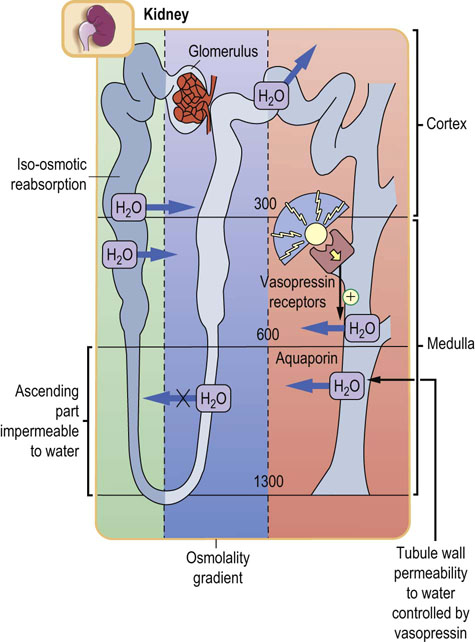
Fig. 23.5 Renal handling of water.
Permeability of the tubular walls to water differs along the nephron. About 80% of filtered water is reabsorbed in the proximal tubule, by iso-osmotic reabsorption. The ascending loop of Henle is impermeable to water. In the collecting duct, vasopressin controls the water reabsorption through aquaporin water channels (Chapter 24).
Role of kidneys in glucose homeostasis
Kidneys contribute to glucose homeostasis via three different mechanisms: the release of glucose into the circulation via gluconeogenesis, the uptake of glucose from the circulation to satisfy its own energy needs, and the reabsorption of glucose from the glomerular filtrate.
Renal gluconeogenesis
In humans, only the liver and kidney are capable of gluconeogenesis (Chapter 13). After an overnight fast, 75–80% of glucose released into the circulation derives from the liver and the remaining 20–25% derives from the kidney. Surprisingly, after a meal, renal gluconeogenesis increases approximately twofold and accounts for ~60% of endogenous glucose release in the postprandial period (hepatic gluconeogenesis decreases by ~80% at that time). Glutamine and lactate are preferential gluconeogenic precursors in the kidney.
The key enzymes of gluconeogenesis, pyruvate carboxylase, phosphoenol pyruvate carboxykinase, fructose-1,6-bisphosphatase, and glucose-6-phosphatase, are found mainly in the renal cortical cells. The rate of gluconeogenesis depends on the glucose concentration, substrate availability and hormonal control. Insulin suppresses, and epinephrine increases, glucose release by the kidney. Glucagon has no effect on renal glucose release. The produced glucose provides the brain and other organs with a needed energy substrate, particularly when liver gluconeogenesis is impaired: e.g. after liver transplantation, in hepatic failure, and during prolonged fasting, hypoglycemia and acidosis.
Renal glucose utilization
The metabolic fate of glucose is different in different regions of the kidney. Glucose is an essential energy substrate for the renal medulla, because of low oxygen tension and low levels of oxidative enzymes there. Consequently, in the medulla, the lactate is the main metabolic end product of glucose metabolism.
In contrast, the renal cortex contains high levels of oxidative enzymes. The main energy substrates in the renal cortex are thus fatty acids, lactate, glutamate, citrate, and ketone bodies. Of these, the fatty acids are the main source of energy.
Renal tubular cells with a high Na+/K+-ATPase activity possess multiple mitochondria situated close to the plasma membrane, so that the released ATP is easily accessible.
Neuronal control of kidney function
The kidneys communicate with the central nervous system via the sensory (afferent) nerves. An increase in renal afferent activity directly influences sympathetic outflow to the kidneys via efferent nerves. Norepinephrine released from the nerve terminals, together with co-transmitters (such as ATP, other adenine mono- and dinucleotides, and neuropeptide Y) modify kidney function (Fig. 23.6). Norepinephrine increases sodium reabsorption directly via tubular Na+/K+-ATPase activation, and indirectly via the stimulation of the renin–angiotensin system (Fig. 23.7). Increased renal sympathetic activity has been identified as a major contributor to the complex pathophysiology of hypertension. On the other hand, kidney denervation leads to blood pressure lowering.
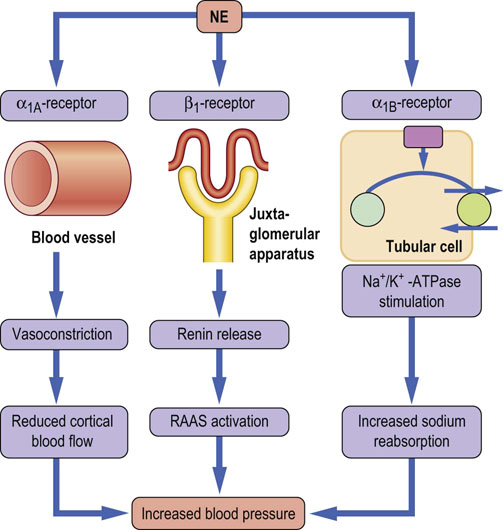
Fig. 23.6 Renal effects of norepinephrine.
Kidneys have dense sympathetic innervation, which terminates in the blood vessels, the juxtaglomerular apparatus, and the renal tubules. Stimulation of the renal sympathetic nerves causes norepinephrine release, which increases renin secretion rate by activation of β1-adrenoceptors, and increases the renal sodium reabsorption by activation of α1B receptors and tubular Na+/K+-ATPase, and decreases the renal blood flow by activation of α1A-adrenoceptors. Overall, the activation of renal adrenergic receptors leads to increased blood pressure. NE, norepinephrine; RAAS, renin-angiotessin-aldosterone system.
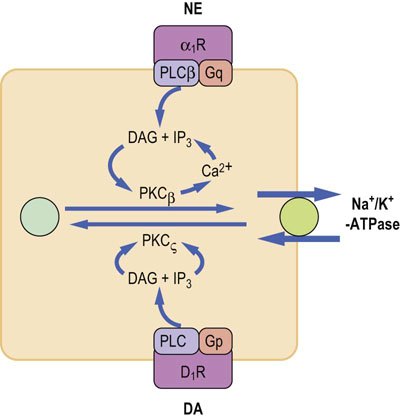
Fig.23.7 Norepinephrine and dopamine regulate sodium reabsorption in the renal proximal tubule.
Norepinephrine and dopamine control Na+/K+-ATPase. Norepinephrine stimulates Na+/K+-ATPase via α1B-adrenergic receptors, and through G-proteins stimulates phospholipase Cβ and protein kinase Cβ, leading to enzyme phosphorylation and its translocation to the cell membrane. On the other hand, dopamine inhibits Na+/K+-ATPase. Dopamine binds to its receptors and through G-proteins stimulates phospholipase C, which in turn activates protein kinase Cζ. PKCζ phosphorylates membrane Na+/K+-ATPase and induces its translocation to cytoplasm. NE, norepinephrine; DA, dopamine; PKCβ,protein kinase Cβ; PKCζ, protein kinase Cζ; α1R, α1B-adrenergic receptors; D1R, Dopamine receptor 1.
Membrane transport systems in the kidney
Na+/K+-ATPase
Na+/K+-ATPase activity in the kidney is several thousand times higher than in other tissues. In the kidney, its major function is sodium reabsorption by catalyzing the extrusion of sodium to the interstitial fluid. There is a close relationship between the amount of Na+/K+-ATPase and sodium reabsorptive capacity of the different segments of the nephron. In the renal tubular cells, as in all sodium-reabsorbing epithelial cells, Na+/K+-ATPase is present in the basolateral membrane. Its activity in the nephron is controlled by a number of hormones, mainly by aldosterone, angiotensin II, and by neurotransmitters norephinephrine and dopamine (Chapter 24). In humans, the kidney reabsorbs about 18 moles of sodium per day and utilizes about 6 moles of ATP for this process. Thus, Na+/K+-ATPase is an energy transducer that converts metabolic energy into ionic gradients.
Sodium transport system in the renal tubules
Sodium reabsorption occurs along the nephron, with the exception of the descending limb of the loop of Henle. The driving force for the reabsorption of sodium and other solutes is the electrochemical gradient generated by the Na+/K+-ATPase in the basolateral membrane of tubular cells. The transport of sodium ions across cellular membrane, similarly to other polar molecules, requires the involvement of specific membrane proteins.
Understanding the renal transport systems elucidates the action of diuretics
In the proximal tubule the sodium ions enter into the cell from the luminal side via the sodium–hydrogen exchanger (NHE3), through ion channels and via co-transporters with glucose, phosphates and amino acids. The sodium–bicarbonate co-transporter (known as NBC1) is located in the basolateral membrane.
In the thin ascending limb of the loop of Henle the sodium ions move into the cell via sodium–potassium–chloride co-transporter (known as NKCC2), which is inhibited by furosemide. In this segment, potassium ions are secreted into the lumen by the ATP-sensitive rectifier potassium channel.
In the distal tubule the reabsorption of sodium ions involves sodium–chloride co-transporter (NCC), which is thiazide-sensitive.
In the collecting duct the sodium ions are reabsorbed by amiloride-sensitive epithelial sodium channel (ENaC). Aldosterone, through the mineralocorticoid receptor stimulates both ENaC expression and the activity of the Na+/K+-ATPase. The pharmacologic antagonist of aldosterone is the diuretic spironolactone.
Reabsorption of sodium in turn drives the movement of water. The inhibitors of sodium reabsorption, e.g. thiazide diuretics (hydrochlorothiazide), loop diuretics (e.g. furosemide, torasemide), amiloride and spironolactone, are extensively used in clinical practice to induce natriuresis and diuresis.
 Clinical box Diuretics are used for treatment of edema, cardiac failure and hypertension
Clinical box Diuretics are used for treatment of edema, cardiac failure and hypertension
Diuretics are drugs that stimulate water and sodium excretion. Thiazide diuretics, e.g. bendrofluazide, decrease sodium reabsorption in the distal tubules by blocking sodium and chloride co-transport. Loop diuretics, such as frusemide, inhibit sodium reabsorption in the ascending loop of Henle. Spironolactone, a potassium-sparing diuretic, is a competitive inhibitor of aldosterone: it inhibits sodium–potassium exchange in the distal tubules, and decreases potassium excretion. An osmotic diuresis may be induced by the administration of the sugar alcohol, mannitol.
The net effect of treatment with diuretics is increased urine volume and loss of sodium and water. Diuretics are important in the treatment of edema associated with circulatory problems such as heart failure, in which impaired cardiac function may lead to a severe breathlessness caused by pulmonary edema. They are also essential in the treatment of hypertension.
Glucose reabsorption
Normally, at the average plasma glucose concentration around 5 mmol/L (~100 mg/dL), approximately 200 g of glucose are filtered by the kidney each day. In healthy individuals, all filtered glucose is reabsorbed into the circulation and the urine is glucose-free. Reabsorption of glucose from the filtrate in the proximal convoluted tubules occurs with the participation of sodium–glucose co-transporters (SGLTs). There are two sodium-dependent–glucose co-transporter proteins: SGLT2 is a low-affinity, high-capacity transporter reabsorbing 90% of filtered glucose, and SGLT1 is a high-affinity low-capacity transporter. Glucose transport mediated by the SGLTs is an active process, utilizing energy derived from the sodium electrochemical gradient maintained by the Na+/K+-ATPase. Glucose reabsorbed into the tubular epithelial cells via SGLTs is released into the circulation by the specific glucose transporters (GLUT-1 and GLUT-2) located in the basolateral membranes.
Reabsorption of filtered glucose increases linearly until the maximal capacity of the tubules (Tmax) is exceeded
The point of saturation of the transport system, the so-called renal threshold, is reached at the plasma glucose concentration of 11.0 mmol/L (198 mg/dL) in healthy adults. Above this concentration, the percentage of filtered glucose that is reabsorbed decreases and glucosuria appears. The threshold is decreased in individuals with a rare condition known as familial renal glucosuria, caused by a range of mutations to the SLC5A2 gene, which encodes the SGLT2. Depending on the nature of the mutations, affected individuals have varying degrees of glucosuria. In the most severe form they can lose more than 100 g of glucose per day in the urine.
Renal handling of amino acids
In humans, the total plasma concentration of amino acids is approximately 2.5 mmol/L (~25 mg/dL). Free plasma amino acids are filtered in the renal glomeruli (approximately 50 g per day). In parallel, amino acids are transported into the kidney cells and are metabolized. For example glutamine, transported through both luminal and basolateral membranes, is deaminated by glutaminase. The product of this reaction, ammonia, is secreted into the tubular lumen where it buffers the hydrogen ion (Chapter 25). In the kidney glutamine is also a substrate for gluconeogenesis.
The near-complete reabsorption of filtered amino acids is a fundamental transport function of the proximal tubule. Their transepithelial transport from the tubular lumen, across the cell and into the circulation, is driven by sodium electrochemical gradient. On the luminal membrane of tubular cells, there are three types of sodium-dependent amino acid co-transporters (belonging to solute carrier transporters family; SLC): one for neutral amino acids (glycine, alanine, proline), one for acidic amino acids (glutamate, aspartate) and one for basic amino acids (cystine, arginine, ornithine, lysine). The transport out of the cell involves both co-transporters and antiporters (the latter also belonging to the SLC family). Individual amino acids can be transported by more than one transporter, providing backup capacity if the transport is partially impaired. Elevated urine amino acid excretion may be caused by mutation in the gene(s) encoding for a particular amino acid transporter, as happens in cystinosis.
Only small amounts of amino acids (0.7 g/24 h) are normally present in the urine. Each substance, which is reabsorbed in the renal tubules, has its own renal transport maximum (Tmax). Tmax can be exceeded either when the amount of filtered substance becomes too large to handle or when the tubular cells do not function properly. Thus, aminoaciduria may result from the accumulation of amino acids such as phenylalanine, leucine, isoleucine, and valine in the plasma, or from an impaired tubular function. Aminoaciduria may also be symptom of proximal tubule damage (the Fanconi syndrome).
Renal handling of phosphate
Homeostasis of inorganic phosphate (Pi) depends mainly on the balance between the intestinal absorption of Pi and its urinary excretion. Adaptation of renal Pi handling to changing dietary Pi content is also well documented. The renal handling of Pi is regulated by hormonal and nonhormonal factors. Changes in urinary excretion of Pi are usually due to changes in the activity of the renal tubular transport system.
Approximately 90% of plasma phosphates are ultrafiltrable in the glomeruli (10% of Pi is bound to plasma albumin). Reabsorption of the filtered Pi occurs in the proximal, and also to some extent in the distal, tubules. The transport of Pi across tubular cells depends on the magnitude of sodium gradient formed by Na+/K+-ATPase in the basolateral membrane. However, the rate-limiting factor for Pi transport is the abundance of specific membrane transport proteins.
The transport of Pi across brush border membrane of the renal proximal tubules is mediated by the two Na+-dependent Pi co-transporter proteins, NaPi-IIa and NaPi-IIc (NaPi-IIb is also highly abundant in the brush border membrane of the small intestine). Na-Pi IIa is electrogenic, because the transport of one divalent Pi ion is coupled with three sodium ions (3Na+:HPO42–). NaPi-IIc, in contrast, is electroneutral and couples two sodium ions with one divalent Pi ion (2Na+:HPO42–). The expression of NaPi-IIa and NaPi-IIc adjusts the renal reabsorption of Pi to the organism's needs. For example, dietary restriction of Pi intake increases the quantity of NaPi-IIa and NaPi-IIc proteins in the luminal membrane. In contrast, high Pi diet leads to a decrease in the transporter expression. The membrane expression of NaPi-IIa is regulated by parathyroid hormone (PTH). PTH receptors are located on both the basolateral and luminal membranes of the tubular cells. PTH activates adenylate cyclase via receptors located on basolateral membrane and increases the intracellular cyclic AMP. cAMP, in turn, activates protein kinase A, which phosphorylates NaPi-IIa transporter, leading to its internalization (a shift into the cytosol). Also, through the binding to luminal receptors, PTH causes phosphorylation of NaPi-IIa by phospholipase C and protein kinase C, and, again, the subsequent internalization of NaPi-IIa transporter. All this leads to a decreased availability of transporters for the filtered Pi, and its urinary excretion increases.
Clinical box Inherited nephron transport disorders
Gitelman's syndrome is a result of inactivating mutations in the gene encoding the thiazide-sensitive sodium–chloride co-transporter (gene SLC12A3). It is an autosomal-recessive disorder. Homozygous individuals are generally normotensive. The observed biochemical abnormalities are similar to thiazide-induced side effects (e.g. hypochloremic metabolic alkalosis, hypokalemia, hypocalciuria and sometimes hypomagnesemia).
Bartter's syndrome is a group of inherited defects in ion transport along the thick ascending limb of the loop of Henle. The neonatal Bartter's syndrome is linked to mutation in the furosemide-sensitive sodium–potassium–chloride co-transporter gene (SLC12A2) or the thick ascending limb potassium channel gene (ROMK/KCNJ1). The classic Bartter's syndrome results from chloride channel gene (CLCNKB) mutation. Clinical symptoms include polyuria and polydypsia, and there also is hypokalemia and alkalosis.
Cystinosis is a result of autosomal-recessive inactivating mutations in the genes SLC3A1 or SLC79A related to the transport of dibasic amino acids. SLC3A1 encodes dibasic amino acids transporter (rBAT), which forms a heterodimer with SLC7A9 gene product (B0, +AT1). In effect, there is a liposomal storage of cystine, and impairment of proximal tubule function, which lead to a decreased glucose and phosphate reabsorption.
Drug transport by organic anion transporters
The kidney eliminates waste products through filtration and secretion into the tubular fluid. It also eliminates water-soluble xenobiotics, e.g. drugs and their metabolites. The transport takes place in the proximal tubules, and is facilitated by substrate-specific tubular transporters. The most important in this are the organic anion transporters (OATs) which belong to the families of ATP-binding cassette (ABC) transporters or the solute carrier transporters (SLC).
The ABC transporters are transmembrane proteins that utilize the energy of ATP hydrolysis to carry various substrates across membranes, e.g. metabolites, inorganic ions, lipids, sterols, peptides and drugs. Classification of the ABC transporters is based on their amino acids sequence and the organization of the ABC domain (Chapter 17 and 18). The ABC transporters are expressed in various organs – e.g. the kidneys, liver and the intestine – and play a role in tumor resistance, cystic fibrosis (Chapter 10) and other inherited diseases, as well as in the development of resistance to multiple drugs (such as colchicine). Competition of drugs for the transporters may lead to toxic effects.
The SLC superfamily includes more than 300 proteins, which possess the ability to exchange the extracellular organic ions for intracellular ones. The SLC transporters are subclassified into organic cation transporters (OCTs – e.g. nicotine, quinine), organic anion transporters (OATs – transporting, e.g., lactate and succinate), and organic zwitterion/cation transporters (OCTNs). These proteins are expressed on basolateral and luminal membranes of various epithelial tissues, including the kidney and the liver.
Urine
The kidneys excrete from 0.5 L to more than 10 L of urine daily, the average daily volume being 1–2 L. The minimum volume necessary to remove the products of metabolism (mainly nitrogen, excreted as urea) is approximately 0.5 L/24 h. The osmolality of the glomerular filtrate is about 300 mmol/L and the osmolality of urine varies from about 80 to 1200 mmol/L. Thus, the maximal urine concentration, is approximately fourfold. Conversely, to excrete excess water, urine may be diluted to below the osmolality of plasma.
Urine analysis provides clinically important information
Urine analysis (urinalysis) in clinical laboratories includes testing for the presence of protein, glucose, ketone bodies, bilirubin, and urobilinogen, and for traces of blood. Measuring urinary osmolality assesses the concentrating capacity of the kidney. The urine is also tested for the presence of leukocytes and various crystals and deposits. Specialist investigations include analysis of urinary amino acids, hormones and other metabolites.
Only traces of protein are normally detectable in the urine. This increases when the glomeruli are damaged: the presence of significant amounts of protein in urine is an important sign of renal disease. Even a minimal amount of albumin in the urine (microalbuminuria) predicts the development of diabetic nephropathy (Chapter 21). Larger proteins such as immunoglobulins appear in the urine when the damage is more extensive: the immunoglobulin light chains (the Bence Jones protein) are present in urine in multiple myeloma (Chapter 4). In a hemolytic anemia, urine may contain free hemoglobin and urobilinogen. The presence of myoglobin is a marker of muscle damage (rhabdomyolysis). The measurement of urine glucose and ketones is important in the assessment of glycemic control in diabetic patients (Chapter 21). Measurements of urobilinogen and bilirubin help to assess liver function (Chapter 29).
Assessment of renal function
Glomerular filtration rate is the most important characteristic describing kidney function
The renal clearance is the volume of plasma (in milliliters) that the kidney clears of a given substance every minute. The glomerular filtration rate (GFR) is the most important characteristic describing kidney function. GFR could be estimated by measuring the clearance of a substance, such as the polysaccharide inulin, that is neither secreted nor reabsorbed in the renal tubules. The amount of inulin filtered from plasma (i.e. its plasma concentration, Pin, multiplied by GFR) equals the amount recovered in the urine (i.e. its urinary concentration, Uin, multiplied by the urine formation rate, V):
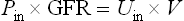
From this we calculate the GFR:
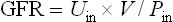
The average GFR is 120 mL/min in men and 100 mL/min in women. The renal clearance of inulin equals the GFR.
Serum creatinine and urea are first-line tests in the diagnosis of renal disease
To administer inulin intravenously every time one would want to assess the GFR is impractical. In clinical practice, we use the creatinine clearance instead.
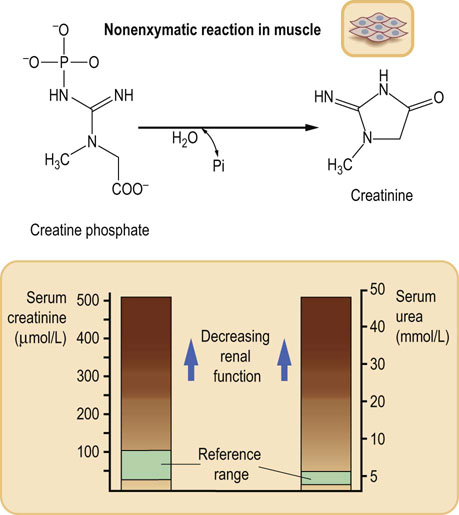
Creatinine is derived from skeletal muscle phosphocreatine
The clearance of creatinine is similar to that of inulin. Although some creatinine is reabsorbed in the renal tubules, this is compensated by an equivalent tubular secretion. To calculate creatinine clearance, one needs a sample of blood, and urine collected over 24 hours. The concentrations of creatinine in serum (PCre) and urine (UCre) are measured first. Urine excretion rate is calculated by dividing the urine volume by the collection time (V, above). The creatinine clearance is then calculated according to the formula:
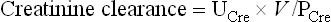
Serum concentration of creatinine is 20–80 mmol/L (0.28–0.90 mg/dL). An increase in serum creatinine concentration reflects the decrease in GFR: serum creatinine concentration doubles when the GFR decreases by 50%. Another test used to assess kidney function is measurement of serum urea concentration. However, because urea is the end product of protein catabolism, its level in plasma is also dependent on factors such as the dietary protein intake and the rate of tissue breakdown.
In clinical practice, serum urea and creatinine are first-line tests in the diagnosis of renal failure (Fig. 23.8). Renal failure leads to a decrease in urine volume and creatinine clearance, and to an increase in serum urea and creatinine. The refinements in laboratory testing include standardization of creatinine measurements by making methods used in clinical laboratories traceable to the reference method, which is isotope dilution mass spectroscopy.
Serum cystatin C concentration is another marker of the GFR
Cystatin C is a 122-amino acid, 13-kDa protein belonging to the family of cysteine proteinase inhibitors. It is a product of the housekeeping gene expressed in all nucleated cells, and is produced at a constant rate. Because of its small size and basic isoelectric point, cystatin C is freely filtered through the glomerulus. It is not secreted by the tubules and although it is reabsorbed, it is subsequently catabolized and therefore does not return to plasma. Its serum concentration is not significantly affected by age, and therefore it is a preferential marker of GFR in children. However, other factors, independent of the GFR, such as the inflammatory phenomena, may affect serum cystatin C concentration.
Neutrophil gelatinase-associated lipocalin is a novel biomarker of acute kidney injury
Neutrophil gelatinase-associated lipocalin (NGAL) is a 178 amino acid, 25-kDa protein belonging to lipocalin family (Greek lipos, ‘fat, grease’ and calyx, ‘cup’). This protein is produced in various epithelial cells (kidney, liver, lung, intestine) and in myelocytes, and is a component of innate immunity. It also plays a role in tissue remodeling, especially after heterodimerization with the metalloproteinase 9 (MMP-9, collagenase IV). NGAL synthesis and secretion into the bloodstream is immediately increased after tissue damage.
Serum NGAL is freely filtered through the glomeruli and is reabsorbed in the proximal tubules. The rising serum NGAL during acute kidney injury may reflect reduced GFR. Though the major source of the urinary NGAL is the distal nephron, a small fraction may come from the filtered pool, escaping tubular reabsorption, due to proximal injury.
Recent studies demonstrated a quantitative link between cellular stress, expression of NGAL in the kidney and excretion of NGAL in urine. The cells in the distal nephron are activated to express NGAL within a few hours following damage (e.g. by ischemia, hypoxia, drug toxicity), before another nephron segment may be affected. Urine NGAL is also being evaluated as a sign of the earliest stage of acute kidney injury, before there are evident clinical abnormalities. The limitations of urine NGAL measurements as biomarker of an acute renal injury are that it may be affected by coexisting chronic kidney disease, renal inflammation and urinary tract infections.
Clinical box A 25-year-old man admitted after a motorcycle accident: acute renal failure
A 25-year-old man was admitted to hospital unconscious after a motorcycle accident. He had evidence of shock with hypotension and tachycardia, a fractured skull and multiple injuries to his limbs. Despite treatment with intravenous colloid and blood, he showed persistent oliguria (urine output 5–10 mL/h; oliguria is <20 mL/h).
On the third day, his serum creatinine concentration had risen to 300 µmol/L (3.9 mg/dL) and his urea concentration to 21.9 mmol/L (132 mg/dL). eGFR was 22 mL/min/1.73m2. Reference values are:
Serum creatinine: 20–80 µmol/L (0.23–0.90 mg/dL)
Serum urea: 2.5–6.5 mmol/L (16.2–39 mg/dL).
eGFR: See table 23.2
Clinical box Diabetes often leads to impairment of renal function
A 37-year-old woman with a 12-year history of type 1 diabetes came for a routine visit to the diabetic clinic. Her glycemic control was poor and glycated hemoglobin (HbA1c) was 8% (64 mmol/mol). Blood pressure was mildly raised at 145/88 mmHg. A quantitative measurement of albumin in urine revealed protein concentration of 5 mg/mmol creatinine, indicating microalbuminuria. Reference values are:
Estimated GFR (eGFR)
Creatinine clearance changes with age, body surface and gender, and also varies with race. Also, the relationship between GFR and creatinine concentration may differ between healthy population and patients with renal disease. In current practice, the GFR values are being adjusted using formulas that include, apart from serum creatinine concentration, factors such as age, gender, weight and race.
Different equations provide useful estimates of GFR: in adults, the equations used are the one developed by the Modification of Diet in Renal Disease Study Group (MDRD) and the Cockcroft–Gault equation; in children, the Schwartz and Counahan–Barratt equations are used. The so-called “abbreviated MDRD formula” includes four variables: serum creatinine, age, sex and race. The original MDRD six-variable equation included serum albumin and serum urea in addition to these. A detailed discussion of these formulas is beyond the scope of this text – the reader is referred to the Further Reading section.
The eGFR equations are useful in the detection of renal impairment. Calculation of the estimated GFR is recommended for the identification and classification, as well as for screening and monitoring, of chronic kidney disease. The severity of chronic kidney disease is classified into six stages (Table 23.2).
Table 23.2
The staging of chronic kidney disease
| Stage | Description | eGFR (mL/min/1.73 m2) |
| 1 | Normal kidney function but urine findings or structural abnormalities of the kidney* | ≥90 |
| 2 | Slightly decreased GFR | 60–89 |
| 3 | Moderate decrease in GFR | 30–59 |
| 4 | Severe decrease in GFR | 15–29 |
| 5 | End-stage kidney failure or dialysis | <15 |
The severity of chronic kidney disease is categorized in six stages. The eGFRs used in this classification has been derived from the abbreviated MDRD equation. *Proteinuria, albuminuria, hematuria lasting for at least 3 months and/or structural abnormalities
Reference: KDOQI Clinical Practice Guidelines for Chronic Kidney Disease: Evaluation, Classification, and Stratification. (see Further Reading for details)
Kidney Disease Outcome Quality Initiative, Am J Kidney Dis 39(suppl. 1):S1–S266, 2002.
One should note though, that in individuals with exceptional dietary intake (vegan, vegetarian diet, creatinine supplements) or abnormal muscle mass (resulting from amputation, malnutrition, muscle wasting), the renal function should still be assessed by measuring creatinine clearance.
Summary
The kidneys maintain electrolyte and water homeostasis and thus play a critical role in the regulation of the composition and volume of the extracellular fluid. Sodium handling by the kidney is also a major determinant of the arterial blood pressure.
The kidneys are exposed to mechanical stress induced by hypertension, to hyperglycemia, and to various nephrotoxins, which may damage renal cells and impair kidney function. Therefore, assessment of renal function is an important part of clinical examination.
Serum concentrations of urea and creatinine are key tests in the assessment of renal function. For more accurate assessment of renal function, the clearance of creatinine is assessed.
Proteinuria is a symptom of renal filtration barrier damage. Microalbuminuria is an early marker of nephropathy; NGAL in urine is an early marker of acute kidney injury.
Broer, S. Amino acid transport across mammalian intestinal and renal epithelia. Physiol Rev. 2008; 88:249–286.
Burckhardt, G. Drug transport by Organic Anion Transporters (OATs). Pharmacol Thera. 2012; 136:106–130.
Chadha, V, Alon, US. Hereditary renal tubular disorders. Semin Nephrol. 2009; 29:399–411.
Devarajan, P. Neutrophil gelatinase-associated lipocalin: a promising biomarker for human acute kidney injury. Biomark Med. 2010; 4:265–280.
Haraldsson, B, Nystrom, J, Deen, WM. Properties of the glomerular barrier and mechanisms of proteinuria. Physiol Rev. 2008; 88:451–487.
Mather, A, Pollock, C. Glucose handling by the kidney. Kidney Int. 2011; 79(suppl 120):S1–S6.
Murer, H, Biber, J. Phosphate transport in the kidney. J Nephrol. 2010; 23(Suppl 16):S145–S151.
Schlaich, MP, Sobotka, PA, Krum, H, et al. Renal denervation as a therapeutic approach for hypertension. Hypertension. 2009; 54:1195–1201.
KDOQI Clinical practice guidelines for chronic kidney disease. evaluation, classification, and stratification. Part 5. Evaluation of laboratory measurements for clinical assessment of kidney disease. Guideline 4. Estimation of GFR www.kidney.org/professionals/kdoqi/guidelines_ckd/p5_lab_g4.htm. Kidney Disease Outcome Quality Initiative. Am J Kidney Dis. 2002; 39(suppl. 1):S1–S266.
KDOQI Clinical practice guidelines for chronic kidney disease: evaluation, classification, and stratification. Part 5. Evaluation of laboratory measurements for clinical assessment of kidney disease. Guideline 4. Estimation of GFR www.kidney.org/professionals/kdoqi/guidelines_ckd/p5_lab_g4.htm.
MDRD eGFR calculator. www.nephron.com/MDRD_GFR.cgi.
UniProt Protein knowledge base. www.uniprot.org/.