Lipoprotein Metabolism and Atherogenesis
Learning objectives
After reading this chapter you should be able to:
 Describe the composition and functions of lipoproteins present in plasma: the chylomicrons, very low-density lipoproteins, remnant particles, low-density lipoproteins and high-density lipoproteins.
Describe the composition and functions of lipoproteins present in plasma: the chylomicrons, very low-density lipoproteins, remnant particles, low-density lipoproteins and high-density lipoproteins.
Describe the fuel transport pathway and the overflow pathway of lipoprotein metabolism.
Describe the reverse cholesterol transport and its links with other pathways of lipoprotein metabolism.
Outline mechanisms and regulation of intracellular cholesterol concentration, including the role of relevant transcription factors, receptors and enzymes.
Comment on laboratory tests that assess lipid metabolism and cardiovascular risk.
Discuss the main component processes of atherogenesis – endothelial dysfunction, arterial deposition of lipids, chronic low-grade inflammation and thrombosis – as well as their relationships to atherosclerotic plaque growth, destabilization and rupture.
Introduction
Lipoproteins distribute cholesterol and triacylglycerols (esters of glycerol and fatty acids, synonymously called triglycerides; we use both terms), from the intestine and the liver, to peripheral tissues
These processes link closely with the energy metabolism. Abnormalities of lipoprotein metabolism are key factors in the development of atherosclerosis, a process affecting arterial walls and causing coronary heart disease, stroke and peripheral vascular disease. Atherosclerosis-related cardiovascular disease is presently the most frequent cause of death in the world: ischemic heart disease and cerebrovascular disease are together responsible for 23.6% of all deaths worldwide (WHO data 2011).
Free fatty acids and triacylglycerols are transported between organs and tissues
Fatty acids are, together with glucose, the key energy substrates. However, in contrast to glucose, the fatty acids can be stored long term in the adipose tissue (as triacylglycerols, Chapter 16) to provide energy during periods of fasting. They are absorbed from the gastrointestinal tract as components of ingested food but are also synthesized endogenously, primarily in the liver and intestine. Fatty acids need to be transported from their places of absorption or synthesis to peripheral tissues. While the free (nonesterified) short- and medium-chain fatty acids ‘travel’ in plasma bound to albumin, the long-chain fatty acids are too hydrophobic to be transported in this manner. Instead, they are transported as triacylglycerols packaged into particles known as lipoproteins.
Lipoproteins
Lipoproteins are composed of hydrophilic, hydrophobic and amphipathic molecules
Lipoprotein particles contain triacylglycerols, cholesterol, phospholipids, and proteins (apolipoproteins). They also transport fat-soluble vitamins such as vitamin A and vitamin E. The hydrophobic cholesteryl esters and triacylglycerols reside in the core of the lipoprotein particles and amphipathic phospholipids and free cholesterol, together with apolipoproteins, form their outer layer (Fig. 18.1). Some apolipoproteins, such as apolipoprotein B (apoB), are embedded in the particle surface while others, such as apoC, are only loosely bound and can be exchanged between different particles.
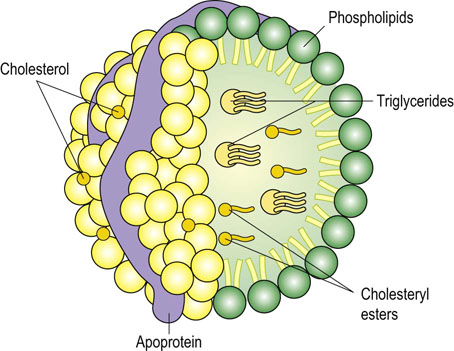
Plasma lipoproteins are particles of different size and density
Lipoproteins are classified on the basis of either their density or constituent set of apolipoproteins: in plasma they form a continuum of size and density (Table 18.1). The main lipoprotein classes are the chylomicrons, the very low-density lipoproteins (VLDL), the remnant particles (which are nearly identical to the intermediate-density lipoproteins, IDL), the low-density lipoproteins (LDL), and the high-density lipoproteins (HDL). The VLDL and remnant particles are triacylglycerol-rich, whereas the LDL are triacylglycerol-poor and cholesterol-rich. With a decreasing triacylglycerol content, the density of a particle increases and its size decreases. Thus the density increases from the chylomicrons (the lightest), through VLDL, IDL, LDL, to the HDL (the heaviest).
Table 18.1
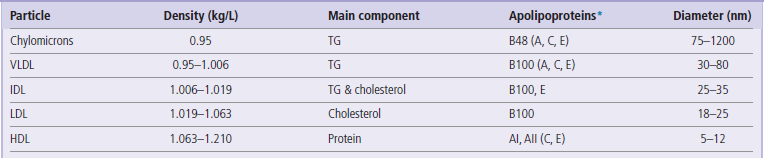
TG, triacylglycerol (triglyceride); VLDL, very low-density lipoproteins; IDL, intermediate-density lipoproteins; HDL, high-density lipoproteins. When separated by electrophoresis VLDL are called pre-β-lipoproteins, LDL, β-lipoproteins and HDL α-lipoproteins. *The most abundant apoproteins present in a given lipoprotein particle are indicated first, with those that are exchanged with other particles in parentheses.
 Advanced concept box Lipoproteins can be separated by ultracentrifugation
Advanced concept box Lipoproteins can be separated by ultracentrifugation
Every clinical laboratory uses centrifuges to separate red blood cells from serum or plasma. These machines develop a moderate centrifugal force, 2000–3000 g. However, in the specialist lipid, protein and nucleic acid biochemistry much larger centrifugal forces (40,000–100,000 g) are applied to plasma to separate particles and molecules. This technique is called ultracentrifugation and is extensively used in lipid research. When centrifugal force is applied to a solution, particles that are heavier than the surrounding solvent sediment, and those lighter than the solvent, float to the surface at a rate proportional to the applied centrifugal force and to the particle size. The formula below shows factors that affect particle movement:
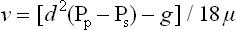
where v = sedimentation rate; d = diameter; Pp = particle density; Ps = solvent density; µ = viscosity of the solvent, and g = gravitational force.
In a technique known as flotation ultracentrifugation, the plasma is over-layered with a solution of defined density, e.g. 1.063 kg/L, the density of the VLDL. After several hours of centrifugation (with the rotor speeds around 40,000 rev/min), the VLDL float to the surface, where they can be harvested. Solutions of different density can be used to separate other lipoproteins. Modifications of the ultracentrifugation technique, such as density gradient centrifugation, can be applied to separate plasma into several ‘bands’ containing different lipoprotein fractions.
Apolipoproteins
Apolipoproteins are protein components of lipoprotein particles. Their role is both structural and metabolic
Apolipoproteins that are embedded into the surface of lipoprotein particles determine their metabolic fate through interactions with cellular receptors. They also regulate the activity of enzymes involved in lipid transport and distribution. Each class of lipoproteins contains a characteristic set of apolipoproteins. The main apolipoproteins are listed in Table 18.2. The most important are apoA, apoB, apoC, apoE, and apo(a).
Table 18.2
Structure and function of apolipoproteins
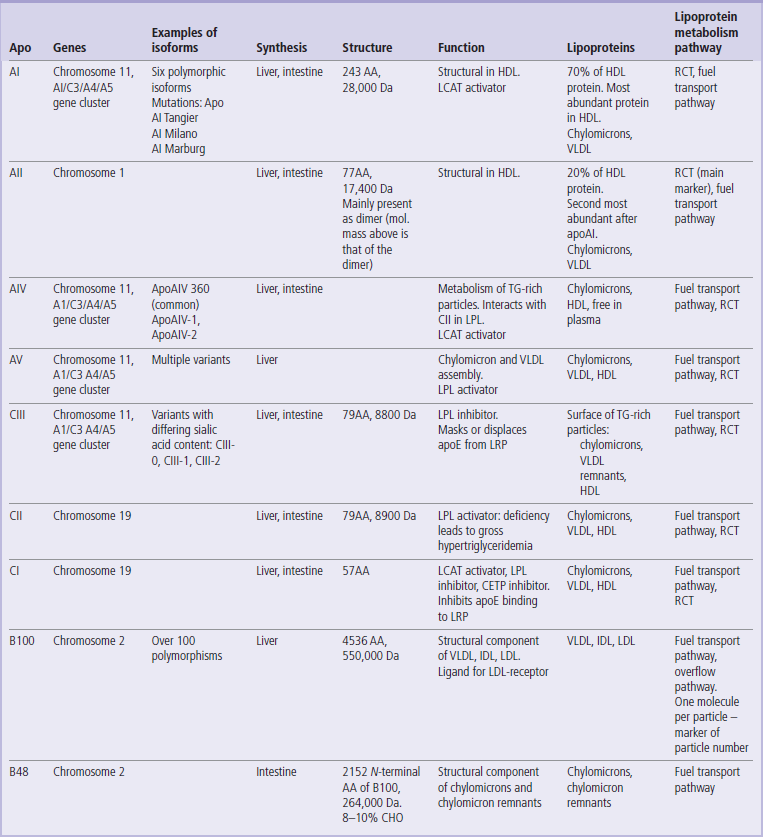
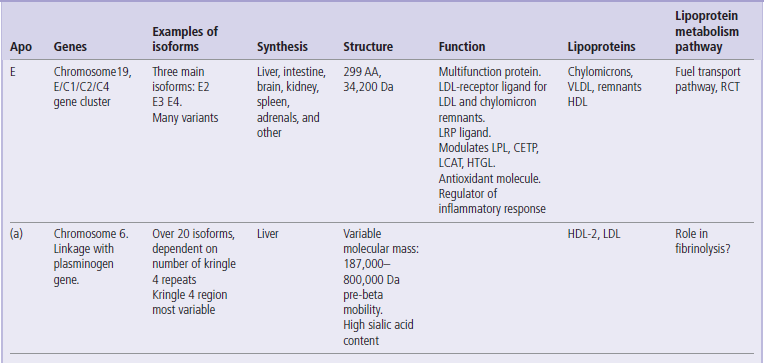
CHO, carbohydrates; AA, aminoacids; RCT, reverse cholesterol transport; LRP, LDL-receptor-like protein. LPL,lipoprotein lipase; LCAT, lecithin: cholesterol acyltransferase; CETP, cholesteryl ester transfer protein; HTGL, hepatic triglyceride lipase.
Reproduced, with permission, from Dominiczak MH, Caslake MJ. Apolipoproteins: metabolic role and clinical biochemistry applications. Ann Clin Biochem 2011; 48: 498–515.
Apolipoproteins A (AI and AII) are present in the HDL. Apo AI is a small protein of 243 amino acids and is synthesized in the liver and in the intestine. The APOA1 gene is part of the APOA1/C3/A4/A5 complex. Apo A1 is the main apolipoprotein in HDL particles. It activates the lecithin: cholesterol acyltransferase (LCAT; the cholesterol-esterifying enzyme) and has anti-inflammatory and antioxidant properties. Clinically, it is a marker of HDL concentration.
ApoAII is an even smaller protein (77 amino acids) and is mostly synthesized in the liver. It is also present primarily in the HDL. It is a cofactor for the LCAT and the cholesterol ester transfer protein (CETP), and inhibits lipoprotein lipase (LPL). It is not routinely measured in plasma. Apolipoprotein A binds to the scavenger receptor BI.
Apolipoprotein B exists in two common variants, apoB100 and apoB48. The apoB100 controls the metabolism of the LDL, whereas its truncated form, apoB48 (see Fig. 34.7) is present in the chylomicrons. ApoB100 is a relatively large protein with a molecular mass of 513,000 kDa, comprising 4509 amino acids. It is synthesized in the liver. There are over 100 variants of the APOB gene. Mutation at the amino acid residue 3500 decreases its binding to the LDL receptor and is the cause of a condition known as familial defective apoB (FDB). Because there is only 1 molecule of apoB per lipoprotein particle, the measurement of the apoB in plasma is a good marker of the sum of the VLDL, remnants, and the LDL particles. ApoB100 binds to the LDL receptor.
ApoB48 is a truncated form of apoB100. ApoB100 and apoB48 are synthesized from the same gene. A stop codon is introduced into the gene during editing of apoB100 mRNA (the ‘48’ designation signifies that it comprises the amino-terminal 48% of the apoB sequence). It is synthesized in the enterocytes and is secreted from the intestine in the chylomicrons. ApoB48 does not bind to the LDL receptor. Its measurement is a marker of the number of chylomicrons and chylomicron remnant particles.
Apolipoprotein E has a molecular mass of 34,200 Da and comprises 299 amino acids. It is present in all lipoprotein classes. It binds to the LDL receptor with a higher affinity than apoB100. It also binds to the LDL-receptor-related protein (LRP), driving the cellular uptake of the remnant particles. ApoE stimulates the LPL, the hepatic triglyceride lipase (HTGL) and the LCAT. ApoE exists in 3 isoforms, E2, E3 and E4. Its synthesis is controlled by three major alleles, ε2, ε3, and ε4. The E2 isoform results from cysteine-for-arginine substitution at position 158 (compared to E3) and has lower affinity to receptors. This, in homozygotes, slows down the uptake of the remnant particles and results in familial dyslipidemia (also known as type III hyperlipidemia). Isoforms E3 and E2 are also associated with higher plasma insulin and glucose concentrations than E4. In the HDL, apoE contributes, together with apoAI, to cholesterol removal from cells. Interestingly, in mice, inactivation of the LRP resulted in a reduction of body weight and a decreased lipid clearance. Thus, although apoE is atheroprotective, it may be associated with weight gain by promoting lipid transport to peripheral tissues.
ApoE is also synthesized in the brain by astrocytes and microglia: it affects growth and repair of the CNS cells and also is anti-inflammatory and antioxidant. Individuals with E4 phenotype were shown to be at an increased risk of the sporadic form of Alzheimer disease. ApoE is not routinely measured in plasma in clinical laboratories, but phenotyping and genotyping apoE isoforms is used in the diagnosis of familial dyslipidemia.
Apolipoproteins C (CI, CII and CIII) act as enzyme activators and inhibitors and they are extensively exchanged between different lipoprotein classes.
Apolipoprotein (a) is a component of lipoprotein (a) (Lp(a) ). Lp(a) is a hybrid particle that comprises apo(a) attached to apoB100 by a disulfide linkage. It is highly polymorphic and its molecular mass may vary from 187,000 to 800,000 Da. Apo(a) possesses a protease domain and a number of repeating sequences of approximately 80–90 amino acids in length, stabilized by disulfide bridges into a triple-loop structure. These structures are called kringles (the name of a Danish pastry of a similar shape). One of the kringles, kringle IV, is repeated 35 times within the apo(a) sequence. The number of kringle IV repeats determines the size of the lipoprotein (a) isoforms.
Apo(a) is synthesized in the liver and it binds to the LDL receptor. It is structurally related to plasminogen. Lp(a) concentration in plasma is almost entirely genetically determined, and is little influenced by lifestyle factors. Lp(a) is modestly associated with cardiovascular risk. Lp(a) is measured in plasma during specialist assessments of cardiovascular risk.
Clinical studies show that the measurements of plasma apolipoproteins predict the risk of CVD better than the lipid testing i.e. measurements of total cholesterol and LDL-cholesterol. However, since most of the large epidemiologic studies and treatment algorithms are referenced to lipid measurements, most laboratories still measure lipids for the assessment of cardiovascular risk.
Lipoprotein receptors
LDL receptor is regulated by the intracellular cholesterol concentration
Cellular uptake of the lipoproteins is mediated by apolipoprotein binding to receptors present on cell membranes. This allows cells to acquire cholesterol and other lipids. The key lipoprotein receptor is the LDL receptor (the apoB/E receptor). It was discovered by Joseph Goldstein and Michael Brown, who jointly received the Nobel Prize for this work in 1985. The receptor can bind either apoB100 or apoE. The mature receptor protein contains 839 amino acids and spans the cell membrane (Fig. 18.2). The receptor gene is located on chromosome 19 and its expression is regulated by the intracellular cholesterol concentration.
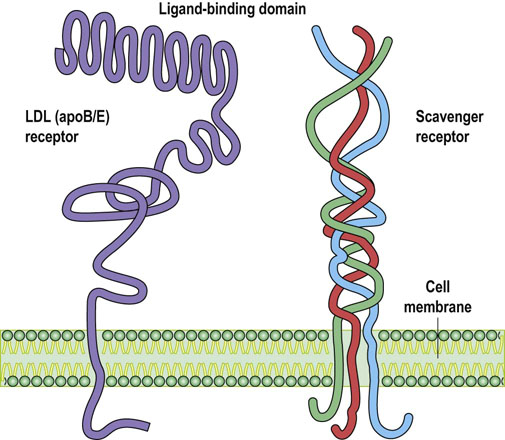
Fig. 18.2 Lipoprotein receptors.
The LDL receptor, also known as the apoB/E receptor, mediates cellular uptake of intact LDL particles. The scavenger receptor internalizes chemically modified (e.g. oxidized) LDL. Both types of receptor span cell membranes. The expression of LDL receptor is regulated by the intracellular cholesterol concentration, while the scavenger receptor remains unregulated. The scavenger receptor type A, illustrated here, is present on macrophages and has a collagen-like structure. Scavenger receptor type BI participates in the metabolism of the HDL particles.
Scavenger receptors are nonspecific and nonregulated
While the apoB/E receptor binds specific ligands, the scavenger receptors can bind many different molecules. These receptors are present on phagocytic cells such as macrophages. Importantly, they are not subject to feedback regulation, and therefore they may overload the cell with the ligand they bind. Scavenger receptors are designated as class A and class B, and CD36. Class A receptors have a collagen-like triple helical structure. They do not bind intact LDL but readily bind chemically modified (e.g. acetylated or oxidized) LDL (see Fig. 37.5). Class B receptor takes up HDL particles in the liver.
Enzymes and LIPID transfer proteins
Two hydrolases, lipoprotein lipase (LPL) and hepatic triglyceride lipase (HTGL), remove triacylglycerols from lipoprotein particles. LPL is bound to heparan sulfate proteoglycans on the surface of vascular endothelial cells, and HTGL is associated with plasma membranes in the liver. LPL digests triacylglycerols in chylomicrons and VLDL, and releases fatty acids and glycerol to cells. HTGL acts on particles already partially digested by LPL, and facilitates the conversion of IDL into LDL (see below).
Lecithin:cholesterol acyltransferase (LCAT) is a glycoprotein enzyme synthesized in the liver, which is associated with the HDL. LCAT esterifies cholesterol that HDL acquire from cells. LCAT is activated by apoAI. Within cells, however, cholesterol is esterified by a different enzyme – the acylCoA:acylcholesterol transferase (ACAT). There are two isoforms of ACAT: ACAT1 is present in macrophages and ACAT2 is present in the intestine and liver. Another protein, the cholesterol ester transfer protein (CETP), facilitates the exchange of cholesteryl esters for triacylglycerols between the HDL on the one hand, and the VLDL and IDL on the other, contributing to reverse cholesterol transport.
Pathways of lipoprotein metabolism
The fuel transport pathway and the overflow pathway reflect the fuel transport function of lipoprotein particles
The transport function of the lipoproteins is essential for the tissue distribution of two essential classes of compounds: triacylglycerols and cholesterol. Triacylglycerols and fatty acids are part of the body energy metabolism, whereas cholesterol transported by lipoproteins forms an extracellular pool available for cellular uptake, which ‘backs up’ the capacity of cells to synthesize cholesterol. The main stages of lipoprotein metabolism are:
The assembly of lipoprotein particles. The chylomicrons are assembled in the intestine, the VLDL in the liver, and the HDL are synthesized both in the liver and the intestine.
The transfer of lipoproteins to peripheral cells and the release of triacylglycerols/fatty acids from lipoproteins to cells. This is facilitated by the LPL and HTGL. As a result, chylomicrons and VLDL decrease in size and become remnant particles.
The binding of remnant particles to the liver receptors and their uptake.
Generation of LDL particles from remaining remnants by HTGL-mediated hydrolysis, their binding to the apoB/E receptor and cellular uptake.
The reverse cholesterol transport, i.e. removal of cholesterol from cells by the HDL particles.
The pathway of metabolism of the chylomicrons and the VLDL, called here the fuel transport pathway, is closely related to the feed–fast cycle (Chapter 21) and thus to energy metabolism (Fig. 18.3). The fuel transport pathway also links closely to the reverse cholesterol transport through triglyceride and cholesteryl ester exchanges with the HDL particles. The completion of the fuel transport stage (i.e. the delivery of triglycerides to the peripheral cells) is linked to generation of the remnants and then the LDL particles, which form an extracellular cholesterol pool. Because the LDL generation depends on the activity of the fuel transport pathway, further LDL metabolism is termed the overflow pathway.
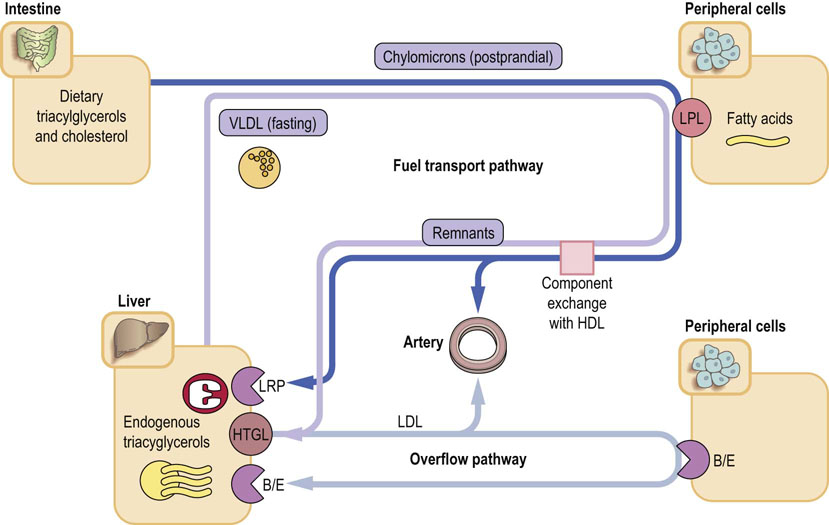
Fig. 18.3 Lipoprotein metabolism: the fuel transport pathway and the overflow pathway.
The fuel transport pathway is linked to energy metabolism and to the feed-fast cycle. In the fed state, chylomicrons transport triglycerides to the periphery, where the LPL hydrolyzes the them, liberating the fatty acids into cells. Chylomicron remnants are metabolized in the liver, after binding through the apoE to the LDL receptor, and also to the LRP. The VLDL particles transport fuel from the liver to peripheral tissues. The VLDL remnants, similarly to the chylomicron remnants, return to the liver. Approximately 65% are taken up after binding to the apoB/E receptor, and the remaining ones are hydrolyzed by HTGL, yielding the LDL particles. The triglyceride-rich particles (chylomicrons, VLDL and the remnant particles) acquire additional cholesteryl esters from the HDL in exchange for triglycerides. In clinical practice, the marker of the activity of the fuel transport pathway is the measurement of plasma triglycerides. The remnant particles can also be measured but this has been done mostly for research purposes. The overflow pathway is the pathway of LDL metabolism. The LDL particles are generated from remnants in the fuel transport pathway, and are cholesterol-rich. The LDL are taken up by the apoB/E receptor in response to a decrease in intracellular cholesterol concentration. In clinical practice, the markers of activity of the overflow pathway are the measurements of plasma total cholesterol and LDL-C LPL, lipoprotein lipase, LRP, LDL-receptor-related protein, HTGL, hepatic triglyceride lipase.
The fuel transport pathway of lipoprotein metabolism
Chylomicrons transport dietary lipids
The pathway involving the assembly of chylomicrons is active after a fat-containing meal. Triacylglycerols present in food are acted upon by pancreatic lipases and are absorbed as monoacylglycerols, free fatty acids, and free glycerol (see Chapter 10). The cells lining the intestine (enterocytes) resynthesize triacylglycerols and, together with phospholipids and cholesterol, assemble them on the apoB48 into chylomicron particles. These are secreted into the lymph and reach plasma through the thoracic duct.
The main apolipoprotein of the chylomicrons is the apoB48. Chylomicrons also contain apoA, C, and E. Once they reach peripheral tissues, their triacylglycerols are hydrolyzed by the LPL, and the fatty acids enter cells. What is now left of the chylomicrons are smaller and denser particles known as the chylomicron remnants. The remnants acquire some cholesteryl esters from the HDL (see below). Importantly, the change in particle size uncovers the apoE on their surface, which mediates the remnant binding to the apoB/E receptor and to the LRP in the liver. The half-life of chylomicrons in plasma is less than 1 h. Chylomicrons normally appear in plasma only after fat-containing meals, giving plasma a milky appearance (Fig. 18.3).
There is now substantial evidence that the postprandial lipemia may play a role in atherogenesis. Nonfasting plasma triglyceride concentration (reflecting an increase in atherogenic lipoprotein remnants) has been linked to the risk of cardiovascular disease (CVD). The postprandial elevation of triglycerides is particularly important in persons with diabetes mellitus and insulin resistance.
VLDL particles transport triacylglycerols synthesized in the liver
In contrast to the chylomicron transport of dietary fat, triacylglycerols that are synthesized in the liver are transported by the VLDL. The VLDL are assembled both during fasting and after meals. They are built up around apoB100 molecules. The lipidation of apoB is facilitated by the microsomal triglyceride transfer protein (MTP). Unused apoB100 is degraded by an ubiquitin-dependent protease (Fig. 34.10). After being secreted into plasma, VLDL acquire cholesteryl esters and apolipoproteins (apoC and apoE) from the HDL. In the peripheral tissues, their triacylglycerols are hydrolyzed by the LPL in a way analogous to chylomicrons; this yields VLDL remnants (also called IDL). The conformations of apoB100 and apoE in the VLDL do not allow binding to the apoB/E receptor. However, in the remnants, apoE assumes a conformation that allows such binding. The remnants are thus either taken up by the liver, or are further hydrolyzed by another enzyme, the HTGL, which, by removing practically all triacylglycerols, transforms them into LDL. Because of the previous triacylglycerol loss, the remnant particles are relatively cholesterol-rich. Their small size facilitates penetration of the vascular endothelium, and both these characteristics make them atherogenic.
VLDL enriched with cholesteryl esters give rise to highly atherogenic small-dense LDL particles
The VLDL are enriched by CETP in cholesteryl esters derived from HDL, in exchange for triglycerides. When such cholesteryl-ester-enriched particles are acted upon by HTGL, the removal of triglycerides decreases their size to an even greater extent creating the small-dense (sd) LDL, which are -not surprisingly- strongly atherogenic. It is thought that their presence might be responsible for an increased CVD risk in some patients who present with seemingly ‘normal’ plasma lipid concentrations – as happens in diabetes mellitus. Since only 1 molecule of apoB100 is present in each LDL particle, the presence of sd-LDL can manifest itself as hyperapobetalipoproteinemia, i.e an increased plasma apoB100 concentration with relatively normal cholesterol concentration. This condition is associated with increased CVD risk.
The overflow pathway of lipid metabolism
LDL particles are taken up by cells by the same route as remnant particles
The LDL are thus triacylglycerol-poor and relatively cholesterol-rich. They are generated from the VLDL remnants by the removal of almost all triglycerides by the HTGL. They are thus the ‘overflow’ products of the fuel transport pathway.
The LDL are small enough to penetrate the vascular wall. They contain only one apolipoprotein (the apoB100) and they are the main carrier of cholesterol in plasma. They remain in the circulation longer than the remnants, and are taken up through the apoB/E receptor, either in the liver (approximately 80% of particles) or in the peripheral cells (see Fig. 18.3). Their affinity for the receptor is lower than that of the apoE-containing remnant particles.
Intracellular cholesterol synthesis and its cellular uptake are interdependent
Most cells synthesize cholesterol for their own needs. However, when the concentration of intracellular cholesterol decreases, the cells can acquire it from the outside – and the lipoproteins constitute a pool of extracellular cholesterol upon which the cells draw. After internalization, the LDL–receptor complex is digested by the lysosomal enzymes. The released free cholesterol is esterified within the cell and the receptor protein recycles back to the membrane. The free cholesterol is a negative feedback regulator of its own synthesis. This is mediated by a family of transcription factors called sterol regulatory element-binding proteins (SREBPs). SREBPs regulate transcription of genes coding for enzymes in the pathway of cholesterol synthesis: the 3-hydroxy-3-methylglutaryl coenzyme A synthase and HMG-CoA reductase, but also the gene coding for the apoB/E receptor (Chapter 17). Depletion of hepatic sterols increases the SREBPs level and consequently both cholesterol synthesis and the expression of the apoB/E receptor. On the other hand, increased intracellular cholesterol concentration inhibits the SREBP pathway, decreasing cholesterol synthesis and receptor expression. This is dioscussed in more detail in Chapter 18 (Fig. 18.4; see also Fig. 17.7).
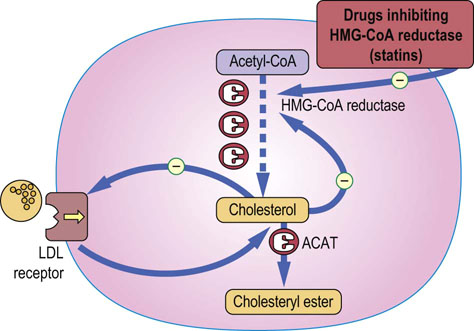
Fig. 18.4 Regulation of intracellular cholesterol concentration.
Intracellular cholesterol regulates the activity of the HMG-CoA reductase, the rate-limiting enzyme in cholesterol synthesis, and also the expression of LDL receptors on the cell membrane. The expression of relevant genes is controlled by the SREBP transcription factors. ACAT: acyl CoA:acylcholesterol transferase. See also Figure 17.7
The reverse cholesterol transport
HDL particles transport cholesterol from the peripheral cells to the liver – this makes them anti-atherogenic
The HDL particles transport cholesterol from the periphery to the liver – thus the ‘reverse’ transport, Fig. 18.5. This relieves the cholesterol burden of cells and makes the HDL anti-atherogenic: a high plasma concentration of HDL-cholesterol (HDL-C) is associated with longevity, and a decreased concentration of HDL-C (and apoAI) is associated with an increased CVD risk.
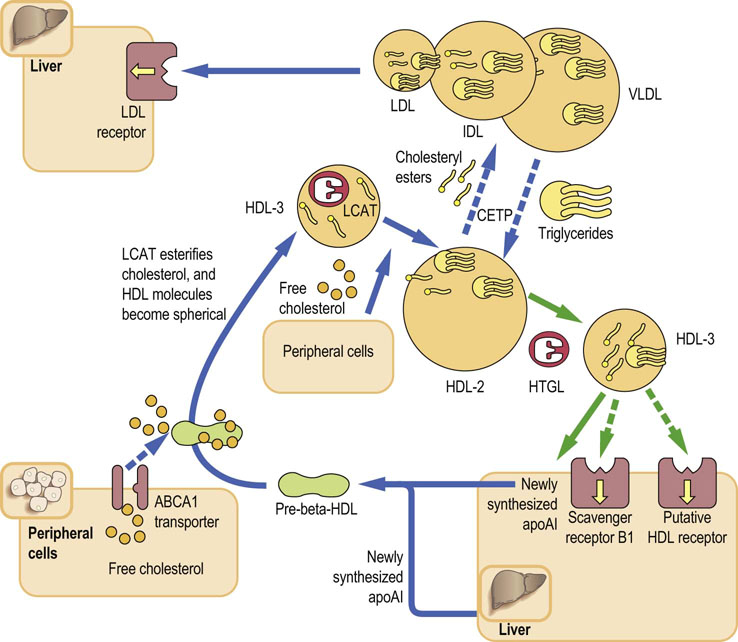
Fig. 18.5 Reverse cholesterol transport.
HDL are assembled in the liver and intestine as discoid particles. They acquire cholesterol from cell membranes through the ABCA1 transporter. The LCAT associated with HDL esterifies the acquired cholesterol. The cholesteryl esters move to the inside of the particle, making it spherical (HDL-2). The CETP facilitates exchange of apolipoproteins and cholesteryl esters between HDL and triglyceride-rich lipoproteins: this inserts the cholesteryl esters into the fuel transport pathway and is the main route of reverse cholesterol transport in humans. The HDL-2 particles which acquire triacylglycerols during the CETP-mediated exchange, increase in size, becoming HDL-3. They bind to the scavenger receptor BI on the hepatocyte membrane and transfer cholesteryl esters to the liver. When the transfer is completed, the size of the HDL particle decreases again. Some of the redundant surface material is released, forming apoAI-rich, lipid-poor pre-β-HDL, which re-enter the cholesterol removal cycle. LCAT, lecithin:cholesterol acyltransferase; CETP, cholesterol ester transfer protein; HTGL, hepatic triglyceride lipase.
The HDL particles are synthesized in the liver and the intestine. Their main apolipoproteins are apoAI and apoAII. They also contain apoC and apoE. Importantly, the HDL exchange their components (apolipoproteins, phospholipids, triacylglycerols and cholesteryl esters) with the triglyceride-rich particles: the chylomicrons, VLDL, and the remnant particles.
Advanced concept box Peroxisome proliferator-activated receptors control carbohydrate and lipid metabolism
Peroxisome proliferator-activated receptors (PPARs) belong to the superfamily of nuclear receptors that function as transcription factors. They regulate genes controlling carbohydrate and lipid homeostasis – and thus play a role in controlling energy metabolism. PPARs form dimers with retinoid X receptor (RXR); and the dimer subsequently binds to response elements in the promoter regions of target genes. The majority of their activating ligands derive from the metabolism of fatty acids.
The action of PPARα is associated also with catabolism of fatty acids. It stimulates fatty acid catabolism, ketogenesis and gluconeogenesis. It is also involved in the assembly of lipoproteins and in cholesterol metabolism. PPARα regulates expression of the AI-CIII-AIV gene cluster; this increases the expression of apoAI and apoAII and reduces the expression of apoCIII gene. It also increases the expression of the LPL.
PPARβ/δ is involved in the control of cell proliferation and differentiation, and also in fatty acid catabolism. PPARγ influences energy homeostasis and adipose tissue differentiation. Its action improves insulin sensitivity. The actions of both PPARα and PPARγ are anti-inflammatory.
The PPARs are important targets of drug action. The commonly used lipid-lowering drugs, derivatives of fibric acid (fibrates) activate PPARα (see also Chapter 20). The anti-diabetic drugs thiazolidinediones activate PPAR γ.
Cholesterol is removed from cells to the HDL by specific transporter molecules
The HDL are formed as discoid, lipid-poor particles (pre-β-HDL) which contain mainly apoAI; they are partly constructed from the excess phospholipids shed from the VLDL during their hydrolysis by LPL. These nascent HDL particles accept cholesterol from cells through the action of a membrane protein known as the ATP-binding cassette transporter A1 (ABCA1; Chapter 8). ABCA1 uses ATP as a source of energy and is rate-limiting for the efflux of free cholesterol to apoAI. Another ATP-binding cassette transporter, ABCG1, transfers cholesterol from cells to mature HDL particles. Others still, known as ABCG5 and ABCG8, reside in the apical membranes of hepatocytes where they control the transfer of cholesterol to bile (Chapter 17).
Cholesteryl ester exchange between HDL and triglyceride-rich particles is important in humans
The free cholesterol acquired by the nascent HDL is esterified by LCAT. The cholesteryl esters move into the interior of the particle, which enlarges and becomes spherical – it is known as the HDL3. Then, aided by the CETP, it transfers some cholesteryl esters to triglyceride-rich lipoproteins in exchange for triglycerides. The acquisition of triglycerides enlarges the particle further – it now becomes HDL-2. Note that the CETP-mediated exchange re-inserts the cholesterol into the fuel transport pathway, and channels it back to the liver. This is important, because the exchange route seems to be the main pathway of the reverse cholesterol transport in humans.
The cholesterol remaining in the HDL-2 is also carried to the liver. There, the HDL-2 binds to the class B scavenger receptor, transferring cholesterol to the cell membrane. Note that the particle binds to the receptor but it is not taken up by cells. The transfer of cholesterol returns it to a smaller size. The now redundant parts of its ‘shell’ become the nascent HDL ready for the next cycle of transport. The HDL-2 can also be further digested by the HTGL, yielding a subclass of small HDL-3.
HDL also exert anti-atherogenic actions other than reverse cholesterol transport
Apart from the reverse cholesterol transport, the HDL has other atheroprotective properties. For instance, they increase the production of nitric oxide (NO) by activating the endothelial nitric oxide synthase (eNOS). They also exert anti-inflammatory and free radical (reactive oxygen species, ROS)-scavenging actions, promote the integrity of the endothelial layer, and prevent endothelial adhesion of cells, platelet aggregation, and thrombosis.
Dyslipidemias
Defects in the lipoprotein metabolism lead to disorders known as dyslipidemias synonymously but less accurately called hyperlipidemias. Their original, now outdated, classification into types I to V had been based on the electrophoretic behavior of lipoproteins (Table 18.3). The electrophoretic classification was superseded by the genetic one (Table 18.4). Another clinically commonly used phenotypic classification divides the dyslipidemias into hypercholesterolemia, hypertriglyceridemia and mixed dyslipidemia. Figure 18.6 presents an overview of the abnormalities of lipoprotein metabolism.
Table 18.3
Phenotypic classification of dyslipidemias

On electrophoresis the α-lipoproteins (HDL) migrate furthest towards the anode ( (+) electrode), followed by the pre-β-lipoproteins (VLDL) and the β-lipoproteins (LDL). The chylomicrons remain at the cathodic end, at the origin of the electrophoretic strip.
This is the classification developed by Fredrickson and adopted by the WHO; it is based on the electrophoretic separation of serum lipoproteins. It has been largely superseded by the genetic classification. Dyslipidemias are also simply classified as hypercholesterolemia, hypertriglyceridemia or mixed dyslipidemia.
Table 18.4
The most important genetic dyslipidemias
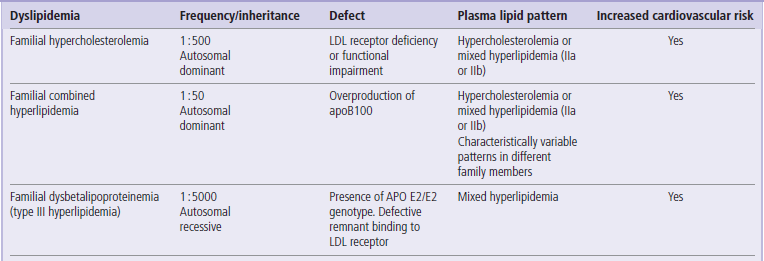
Mixed hyperlipidemia: increased both plasma cholesterol and plasma triglyceride concentration.

Fig. 18.6 An overview of the abnormalities of lipoprotein metabolism.
Compare this figure to Figure 18.3.
Conditions that primarily affect the fuel transport pathway: The fuel transport pathway is affected by excessive dietary intake of fats, obesity and diabetes. The LPL deficiency causes extreme elevation of chylomicrons and VLDL. Importantly, in this pathway the lipoprotein concentrations change with fast-feed cycle. The concentrations of atherogenic remnants increase postprandially and may be important in atherogenesis. Familial dysbetalipoproteinemia leads to an increased remnant concentration because of impaired uptake caused by the mutation in apoE.
Conditions that primarily affect the overflow pathway: The plasma LDL concentration can be affected by its increased generation ( a HTGL-mediated digestion of VLDL remnants) from the fuel transport pathway, or by their impaired cellular uptake. The most important defect is the impaired uptake caused by mutations in the apoB/E receptor gene, which causes familial hypercholesterolemia (FH). In FH the overflow pathway is primarily affected, and plasma LDL concentration is elevated. Note, however, that the remnant uptake is also impaired.
Conditions that affect both pathways: Familial combined hyperlipidemia affects both pathways because of the increased synthesis of the VLDL and consequent increase in LDL generation. Similarly, a high-fat diet affects both pathways.
 Clinical box Genetically determined dyslipidemias
Clinical box Genetically determined dyslipidemias
In the industrialized countries, approximately 30% of people have undesirably high plasma cholesterol concentration. The most frequent dyslipidemia (known as the common hypercholesterolemia) is polygenic and is a result of combined genetic and environmental factors. Then there are rarer disorders with defined genetic background. The most important ones are mentioned below.
Familial hypercholesterolemia is a monogenic disorder caused by a mutation in the gene coding for the apoB/E receptor and it affects the overflow pathway. Cellular uptake of the remnant particles and the LDL is either impaired (heterozygous FH) or completely inhibited (a very rare homozygous FH). Other mutations disrupt LDL receptor recycling to plasma membrane. Patients with FH have very high plasma cholesterol and LDL-C concentrations. The mode of inheritance is autosomal dominant, so there usually is a prominent family history of early heart disease. Some patients develop lipid deposits on hand and knee tendons, and particularly on the Achilles tendons: these are known as xanthomas, and are diagnostic for the disorder. Familial hypercholesterolemia carries a high risk of early cardiovascular disease (note: premature infarction is defined as one occurring in a man aged below 55 years or a woman below 65 years).
A mutation in the gene coding for the LDL-receptor-related protein 6 has also been linked to premature coronary heart disease, showing autosomal dominant inheritance.
Familial combined hyperlipidemia is characterized by an overproduction of apoB100 rather than the impairment of the receptor-mediated clearance. There is an increased production of the VLDL and, consequently, increased generation of the LDL: again, both the fuel transport and the overflow pathways are affected. This dyslipidemia presents with variable plasma lipid patterns (either with hypercholesterolemia alone or hypercholesterolemia with hypertriglyceridemia). Familial combined hyperlipidemia is a relatively common cause of premature myocardial infarctions.
Familial dysbetalipoproteinemia affects primarily fuel transport pathway and is caused by a mutation in the apoE gene, yielding apoE isoform with low affinity for the apoB/E receptor. In this condition, the remnants accumulate, and there is an increase in both cholesterol and triglyceride concentration in plasma. Characteristic palmar xanthomas are present. Familial dyslipidemia is associated with early coronary disease.
Lipoprotein lipase deficiency is a very rare dyslipidemia that affects the fuel transport pathway and is caused by the deficiency of LPL. Chylomicron and VLDL accumulation leads to very high plasma triacylglycerol concentrations. Clinical signs include characteristic rash-like skin xanthomas. The risk associated with the LPL deficiency is primarily that of pancreatic inflammation (pancreatitis, see Chapter 9) caused by the very high triacylglycerol concentrations.
Abetalipoproteinemia. Mutations in the gene coding for apoB can also lead to low VLDL, and consequently low LDL concentrations. A very rare dyslipidemia known as abetalipoproteinemia is associated with the mutation of the gene coding for the microsomal transfer protein (MTP), involved in the cellular assembly of the VLDL.
Clinical box The diagnosis of familial hypercholesterolemia
The UK Simon Broome criteria for the diagnosis of familial hypercholesterolemia used in Britain are:
Plasma total cholesterol above 7.5 mmol/L (290mg/dL) or LDL-cholesterol above 4.9 mmol/L (189 mg/dL) in an adult.
Tendon xanthomas in a patient or first-degree relative (parent, sibling, child) or in a second-degree relative (grandparent, uncle, aunt).
Currently, the basic genetic screening to support the diagnosis of FH involves searching for several mutations of the LDL-receptor gene: one is the sequence1637G>A, which results in the substitution of glycine by aspartic acid (Gly546Asp). It results in a reduced activity of the LDL receptor. Another mutation of the APOB gene, Arg3527Gln, has been found in 5-7% of the FH patients, as well as a less frequent mutation of the PCSK9 gene, Asp374Tyr.
Clinical box Dyslipidemia is common in diabetes mellitus
Mr B is 67 years old, overweight (BMI 28 kg/m2) and has type 2 diabetes and mild hypertension. When he visited the outpatient clinic, his cholesterol concentration was 6.9 mmol/L (265 mg/dL), triglycerides were 1.9 mmol/L (173 mg/dL), and HDL-C was 0.9 mmol/L (35 mg/dL). Fasting blood glucose was 8.5 mmol/L (153 mg/dL) and hemoglobin A1c (HbA11c) 31 mmol/mol (desirable value below 48 mmol/mol). He was being treated with diet and metformin, which improves insulin sensitivity and lowers blood glucose concentration.
Diabetes carries a 2–3 times increased risk of coronary heart disease. This patient's diabetes was well controlled (judged by the HbA1C concentration) but his cholesterol level remained high, so that he required lipid-lowering drug treatment. Low HDL-C concentration is relatively common in type 2 diabetes. The patient was prescribed a statin in addition to metformin. Blood pressure responded to treatment with an angiotensin-converting enzyme inhibitor (see Chapter 20).
Clinical box Familial hypercholesterolemia is a cause of early myocardial infarctions
A 32-year-old man who smoked heavily developed a sudden crushing chest pain. He was admitted to the casualty department. Myocardial infarction was confirmed by EKG changes and by high cardiac troponin concentration. On examination the patient had tendon xanthomas on hands and thickened Achilles tendons. There was a strong family history of coronary heart disease (his father had had a coronary bypass graft at the age of 40 and his paternal grandfather died of myocardial infarction in his early fifties). His cholesterol was 10.0 mmol/L (390 mg/dL), triglycerides 2 mmol/L (182 mg/dL) and HDL-C 1.0 mmol/L (38 mg/dL).
This patient has familial hypercholesterolemia (FH), an autosomal dominant disorder characterized by a decreased number of LDL receptors. FH carries a very high risk of premature coronary disease, and heterozygotes may suffer heart attacks as early as the 3rd or 4th decade of life. The frequency of FH homozygotes in Western populations is approximately 1 : 500. This patient was immediately given an intravenous thrombolytic treatment. Subsequently he underwent coronary artery bypass graft and was treated with lipid-lowering drugs. His cholesterol concentration decreased to 4.8 mmol/L (185 mg/dL) and triglyceride to 1.7 mmol/L, with HDL-C increasing to 1.1 mmol/L (42 mg/dL).
Note: An early myocardial infarction is the one occurring in a man aged below 55 or a woman younger than 65 years.
Conditions affecting the fuel transport pathway
Increased flux through the fuel transport pathway occurs in obesity and diabetes mellitus
The increased flux through the fuel transport pathway is most commonly due to increased synthesis of the VLDL. This happens in two common conditions, obesity and diabetes mellitus. Diabetic dyslipidemia also affects the reverse cholesterol transport; a frequent pattern seen in diabetes is an increase in plasma triglyceride concentration combined with a decrease in HDL-C. In diabetes, the overflow pathway remains relatively unaffected: patients often have normal LDL-cholesterol (LDL-C) concentration. However, diabetic patients seem to generate the small dense LDL, and therefore diabetic LDL, although not increased, may be more atherogenic than the nondiabetic particles. Importantly, weight loss decreases the activity of this pathway. The combination of increased VLDL remnants (resulting in mild hypertriglyceridemia), increased sd-LDL and low HDL is sometimes referred to as ‘the atherogenic triad’.
Another common condition causing increase in VLDL production is alcohol abuse. However, in contrast to diabetes, alcohol, while raising VLDL, also raises the HDL concentration.
LPL deficiency leads to extreme hypertriglyceridemia
The fuel transport pathway also becomes overloaded if the hydrolysis of chylomicrons or VLDL is inefficient. In diabetes, the lack of insulin suppresses LPL, and contributes to the development of hypertriglyceridemia. A very rare condition, the LPL deficiency, results in extremely high plasma triglyceride concentrations, which may exceed 100 mmol/L (8850 mg/dL).
Impaired metabolism of remnant particles leads to familial dysbetalipoproteinemia
The rare but important condition affecting the very last step of the fuel transport pathway, the uptake of remnants, is an inherited condition known as familial dysbetalipoproteinemia. It is caused by the mutation of the remnants ‘driver’ apolipoprotein, apoE (the presence of E2/E2 phenotype), which decreases its binding to the apoB/E receptor. Familial dysbetalipoproteinemia is linked to premature CVD and was previously known as Type III hyperlipidemia.
Conditions affecting the overflow pathway
High LDL-C concentration can be a result of its overproduction or impaired cellular uptake
Abnormalities of the overflow pathway lead to hypercholesterolemias, the most important of which is the familial hypercholesterolemia (FH). FH is characterized by defective cellular uptake of the LDL due to abnormalities (or the lack) of the apoB/E receptor caused by mutations in the relevant genes (see Box). Patients with FH have a high LDL concentration and some present with tendon xanthomas – deposits of cholesteryl esters in the Achilles tendons and tendons of the hands. FH is associated with premature cardiovascular disease and patients frequently present with a prominent family history of early coronary disease. Familial defective apolipoprotein B (FDB) is, on the other hand, caused by a mutation of the apoB100 molecule that impairs its binding to the receptor.
An increase in LDL can also be secondary to increased VLDL synthesis (according to the ‘overflow’ concept) and the consequent increased transformation of remnants into LDL. This happens in a condition known as the familial combined dyslipidemia. Finally, dietary intake of saturated fats affects the LDL concentration. A low-fat diet can decrease the LDL, and plasma cholesterol concentration, by 10–15%.
Note that the majority of mild-to-moderate hypercholesterolemias seen in clinical practice are polygenic (see Box on p. 225).
Conditions affecting the reverse cholesterol transport
Several rare mutations lead to a decreased HDL cholesterol concentration
Low HDL-cholesterol can result from mutations in genes coding for the apoA1, the ABCA1 transporter and the LCAT. Patients with apoAI deficiency present with low plasma HDL-C accompanied by xanthelasmas, corneal clouding, and arteriosclerosis. Heterozygotes occur in 1% of the population. They also develop amyloidosis.
Those with ABCA1 mutations, apart from low plasma HDL-C have large, orange-colored tonsils, hepatosplenomegaly, peripheral neuropathy and thrombocytopenia. It is known as Tangier disease.
The deficiency of the LCAT is known as fish-eye disease. It is characterized by HDL deficiency and also by corneal clouding, nephropathy and hemolytic anemia. In contrast, the deficiency of CETP leads to high HDL concentration.
Atherosclerosis, atherogenesis and atherothrombosis
Atherosclerosis is a process that leads to the narrowing, or a sudden complete occlusion, of the arterial lumen. The narrowing is due to slow-growing atherosclerotic plaques, whereas a sudden complete occlusion is caused by the thrombus that forms over a ruptured plaque. The complete occlusion may cause myocardial infarction (if the blockage is in a coronary artery), stroke (blockage in an artery supplying the brain) or peripheral vascular disease (blockage in leg arteries; this leads to a characteristic pain that occurs during walking, with fast relief when the person stops, known as intermittent claudication). The development of atherosclerotic plaque is termed atherogenesis. The term atherothrombosis is sometimes used to emphasize links between plaque growth and blood coagulation processes.
Atherogenesis involves lipid transport and deposition in the subendothelial layer of the arterial wall (intima). This occurs on the background of endothelial damage and is accompanied by inflammatory reaction that affects the intima, and involves elements of innate and adaptive immunity (Chapter 38). It leads to remodeling of the arterial wall, including new vessel formation (angiogenesis). Thrombosis (Chapter 7) is important in plaque maturation and destabilization (Fig.18.7).
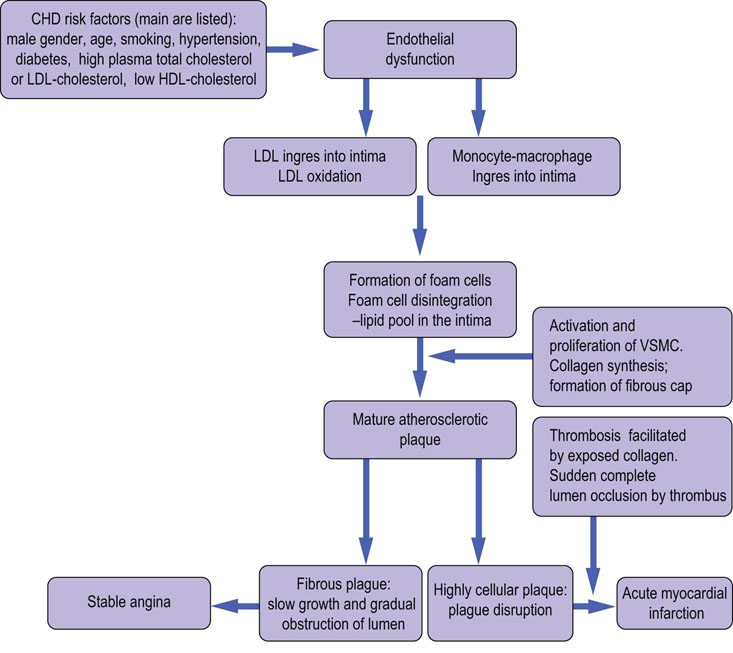
Fig. 18.7 The development of the atherosclerotic plaque.
The cardiovascular risk factors damage the endothelium. This leads to penetration of lipids and cells into the intima. Activation of macrophages leads to cytokine response and leads to activation and proliferation of smooth muscle cells. The lipid pool which results from disintegrating lipid-filled macrophages (the foam cells) becomes covered by a fibrous cap, forming the mature plaque. While stable fibrous plaques cause a slowly-progressing angina, the disruption of an unstable, highly cellular plaque leads to acute clinical events such as a myocardial infarction. VSMC, vascular smooth muscle cells.
Advanced concept box Plasma C-reactive protein concentration reflects chronic low-grade inflammation associated with atherogenesis
The inflammatory reaction associated with infection can be detected by measuring the concentration of plasma C-reactive protein (CRP), a protein synthesized in the liver (but also in VSMC and in the endothelium) in response to stimulation by pro-inflammatory cytokines. Its name comes from its binding to the capsular (C) polysaccharide of bacteria such as S. pneumoniae, by which way it mediates their clearance.
Minute increases in CRP concentration, which require a highly sensitive (hs) analytical method able to detect CRP concentrations below 10 mg/L, may reflect chronic inflammatory processes in the vascular walls. Epidemiologic studies demonstrated an association between the hsCRP concentration and cardiovascular events. Importantly, this association is independent of the link between plasma cholesterol concentration and coronary disease. Increased plasma concentrations of other pro-inflammatory molecules, such as interleukin-6 (IL-6) and serum amyloid A, have also been linked to coronary heart disease. hsCRP measurement enhances the predictive value of the LDL-C and its suggested use is to fine-tune cardiovascular risk assessment. It has been recently observed that adding either CRP or fibrinogen to the panel of conventional risk factors improves risk assessment.
It is suggested that CRP <1 mg/dL signifies low CVD risk, while CRP >3 mg/L is associated with high risk of coronary disease.
The role of vascular endothelium
Normal endothelium has anticoagulant and anti-adhesion properties
The lumen of a healthy artery is lined by a confluent layer of endothelial cells. Normal endothelial surface is strongly antithrombotic and anti-adhesive: it repels cells floating in plasma. The arterial wall itself consists of three layers: the subendothelial layer (the intima), the middle one (the media, which contains vascular smooth muscle cells, VSMC), and the outer layer (the adventitia, composed of looser connective tissue and containing relevant nerves). Substances can penetrate the endothelium either through junctions between the endothelial cells or by transgressing the cells themselves. Particles with a diameter greater than approximately 60–80 nm can lodge in the vascular wall.
Endothelium controls vasodilatation by secreting nitric oxide known as the endothelium-derived relaxing factor (EDRF)
The endothelium controls the ability of blood vessels to dilate (vasodilatation) and to constrict (vasoconstriction) and thus to regulate tissue and organ blood flow. Nitric oxide is the most important vasodilatory substance. It is synthesized from L-arginine by the endothelial NO synthase (eNOS). Activity of eNOS is controlled by the intracellular calcium concentration. The eNOS is constitutively (constantly) expressed in the endothelium, while another isoenzyme, the inducible NOS (iNOS), is found in VSMC and in macrophages. NO signals via guanylate cyclase and cyclic GMP (see Chapters 37 and 40). A decrease in NO production contributes to arterial hypertension. The drug glyceryl trinitrate, commonly used to relieve chest pain caused by inadequate oxygen supply to the heart muscle (causing the characteristic chest pain known as angina pectoris), dilates coronary arteries by stimulating NO release.
Endothelial dysfunction, lipid deposition, and inflammatory reaction in the vascular wall are the key processes in atherogenesis
Crucially, there is cross-talk between endothelial cells, VSMC, plasma-derived inflammatory cells (monocytes, macrophages and lymphocytes) and blood platelets, all of which secrete an array of chemokines, cytokines, and growth factors (Chapter 42). This attracts cells to the atherosclerotic lesions, induces cell migration, proliferation, apoptosis, and production of collagenous extracellular matrix (Chapter 27, Fig. 27.9).
Endothelial dysfunction precedes formation of atherosclerotic lesions
Atherogenesis is initiated by endothelial damage (Figs 18.8 and 18.9). Initially, the damage is functional rather than structural. The endothelium switches from anti-atherogenic to pro-atherogenic, inflammatory phenotype by losing its cell-repellent quality, and admits inflammatory cells into the vascular wall. It also becomes more permeable to lipoproteins, which subsequently deposit in the intima. Later, there is structural damage or a complete destruction of endothelial cells.

Fig.18.8 Atherogenesis: the process.
Atherogenesis involves endothelial dysfunction, deposition of lipids in the arterial intima, low-grade inflammatory reaction, migration and proliferation of the vascular smooth muscle cells, and thrombosis. Note the role of oxidized lipids in the formation of lipid-laden cells and subsequently the lipid pool that becomes the center of the atherosclerotic plaque. The sequence of events is described in the text.

Fig. 18.9 Atherogenesis: the role of growth factors and cytokines.
Atherogenesis is driven by signals mediated by cytokines and growth factors generated by endothelial cells, macrophages, T lymphocytes and vascular smooth muscle cells (VSMC). There are multiple activation paths: for instance, the expression of MCP-1 and VCAM-1 may be stimulated by signals generated by the macrophages as well as by the oxidized LDL. VSMC may be stimulated by the dysfunctional endothelial cells, by macrophages, and by T lymphocytes (note also the autocrine activation). A hormone, angiotensin II, also participates in these processes. MCP-1, monocyte chemoattractant protein 1; VCAM-1, vascular cell adhesion molecule 1; ICAM-1, intracellular cell adhesion molecule 1; TNFβ, tumor necrosis factor-β; TNFα, tumor necrosis factor-α; IFNγ, interferon-γ; NO, nitric oxide; PDGF, platelet-derived growth factor; bFGF, basic fibroblast growth factor; IGF-1, insulin-like growth factor 1; EGF, epidermal growth factor; TGFβ, transforming growth factor-β; IL-1, interleukin 1.
Cell adhesion to endothelium is mediated by the adhesion molecules present on its surface. Selectins (P-selectin and E-selectin) mediate the initial interactions with circulating cells. The vascular cell adhesion molecule 1 (VCAM-1) promotes adhesion of monocytes and T lymphocytes. In experimental animals, VCAM-1 deficiency decreases formation of atherosclerotic plaques. Adhering cells are subsequently stimulated by the monocyte chemoattractant protein 1 (MCP-1) to cross the endothelium and lodge in the intima. This is further facilitated by a protease (matrix metalloproteinase 9, MMP-9), secreted by the monocytes. Production of NO in the damaged endothelium decreases and this promotes vasoconstriction. Since NO normally reduces monocyte adhesion and VSMC migration and proliferation, in its absence these processes intensify.
Importantly, endothelial expression of the adhesion molecules is stimulated by most cardiovascular risk factors, including hypercholesterolemia, hypertension, components of the cigarette smoke, high saturated fat diet and also diabetes mellitus and obesity. Activation of the renin–angiotensin–aldosterone system, apart from its effect on blood pressure, also has pro-atherogenic effects. Angiotensin II (Chapter 23) increases expression of VCAM-1 and MCP-1. Clinical studies show that drugs inhibiting that axis (the ACE inhibitors) are beneficial in cardiovascular prevention.
Advanced concept box Genetics of atherosclerosis
Genes coding for the LDL receptors, apolipoproteins and LRP6 are currently the only ones that have been directly linked with atherosclerotic disorders. Deep sequencing (sequencing the genome a large number of times to minimize error rate) identified two variants of the PCSK9 gene (in patients of African descent) that are responsible for low lipid levels and a decreased risk of myocardial infarction. Altogether, approximately 95 loci have been associated with lipids LDL, HDL and triglycerides – their combined effect was responsible for approx 25% of variation in the LDL and HDL concentrations.
However, the majority of cardiovascular diseases are polygenic. The current working hypothesis (the common disease variant hypothesis) is that the common variants, occurring with a frequency below 5%, play a role in pathophysiology of polygenic diseases. The genome-wide association studies (GWAS) test associations between a disease and common variants across the entire genome. There are presently about 30 loci associated with myocardial infarction and coronary artery disease.
The technical ability to perform genome-wide scans changed the way this type of research is conducted. Formerly, accumulation of results from many single studies would lead to elucidation of mechanisms and then perhaps a search for their role in disease. In contrast, genome-wide studies often provide evidence of association between a pathologic condition and a gene before anything is known about the underlying mechanisms. Only later investigators would search for processes that underpin such association.
Monocytes migrate into the intima and transform into resident macrophages
Monocytes are attracted to the developing plaques by the chemoattractant cytokine CCL2, which binds to their receptors. At the next stage, monocytes transform into macrophages, under the influence of interferon-γ, tumor necrosis factor-α (TNFα), granulocyte-macrophage colony-stimulating factor, and the monocyte colony-stimulating factor 1 (MCSF-1), secreted by the endothelial cells and the VSMC. Macrophages also generate reactive oxygen species, which oxidize LDL in the intima. Some macrophages produce cytokines such as interleukin 1β (IL-1β), IL-6 and TNFα. Other express scavenger receptors (class A receptors, and CD36), which become active in endocytosis of the oxidized LDL.
Lipids enter the intima
The smaller lipoproteins, i.e. the remnants and the LDL, enter the vascular wall more easily than other particles, and thus are most atherogenic. While in the plasma, the LDL particles are protected against oxidation by antioxidants such as vitamin C and β-carotene. In the intima, however, they are deposited in association with proteoglycans. This removes their access to antioxidants, and the fatty acids and phospholipids in the LDL become prone to oxidation mediated by lipoxygenases, myeloperoxidase, and NADPH oxidases expressed by the macrophages. Oxidized LDL further stimulates expression of VCAM-1 and MCP-1, maintaining the ingress of cells into intima; they are also mitogenic for macrophages. The apoB100, once oxidized, binds to the scavenger receptors rather than to the apoB/E receptor on cells. Because the scavenger receptors are not feedback-regulated by intracellular cholesterol, the macrophages overload themselves with oxidized lipids and take the appearance of foam cells; conglomerates of such cells are visible in the arterial walls as the so-called fatty streaks. Dying foam cells release lipids, which form pools within the intima. These become centers of mature atherosclerotic plaques.
Migrating vascular smooth muscle cells change the structure of the vascular wall
Growth factors secreted by the endothelial cells and macrophages (the platelet-derived growth factor PDGF, the epidermal growth factor EGF, and the insulin-like growth factor 1, IGF-1) activate the VSMC present in the arterial media. Under the influence of PDGF and TGFβ, the VSMC proliferate and migrate into the intima. Migration is further facilitated by the MMP-9. The cells secrete adhesion molecules, as well as inflammatory cytokines IL-1 and TNFα. Activated VSMC also synthesize extracellular matrix, in particular collagen, which deposits in the growing plaque. All this disrupts the structure of the arterial wall: a newly formed plaque may protrude into the lumen of the artery, obstructing the blood flow.
Inflammation is fundamental to atherogenesis
The exit of the monocytes and T leukocytes from plasma and their activation in the intima are parts of the inflammatory response. Normally, such response is initiated by an antigen or trauma. Intriguingly, no specific antigen capable of initiating atherogenesis has been identified. There could be a molecular mimicry between such putative antigen(s) and the exogenous pathogens (Chapter 38). The antigen(s) might be infectious agents or modified molecules generated by reactive oxygen species. For instance, the phosphorylcholine group found in the oxidized LDL is also a component of the capsular polysaccharide of bacteria. The oxidized LDL remains a candidate antigen that could be responsible for the stimulation of inflammatory reaction in atherogenesis.
Atherogenesis involves both innate and adaptive immunity. Innate immunity includes recognition of molecules by scavenger receptors A and the membrane glycoprotein CD36. When molecules that possess patterns encoded in immune memory bind to these receptors, they activate cells through, for instance, the pathway involving the transcription factor NFκB. With regard to adaptive immunity, the T cells predominantly of CD4 subtype are present in atherosclerotic lesions, and circulating IgG and IgM-type antibodies against modified LDL have been identified in plasma.
Macrophages (and foam cells) present in the plaque continue to secrete cytokines, growth factors and adhesion molecules (IL-1β and TNFα, VCAM-1, IL8, IL-6) and the MMPs. IL-18 stimulates the secretion of the interferon-γ, which in turn stimulates chemokine-inducible protein 10, and the T-cell-α chemoattractant, which further facilitate T-cell ingress. Interferon also facilitates activation of T-helper 1 cells to effector cells that then secrete CD40 ligand (CD40L), a cytokine that is a member of the TNF family.
Lastly, there is new vessel formation: the newly formed vessels facilitate intraplaque hemorrhages. The thrombin generated there activates monocytes, macrophages, endothelial cells, VSMC and platelets to secrete inflammatory mediators such as CD40L (CD40L binding to CD40 receptor further increases secretion of MMPs, cytokines and adhesion molecules).
The adipokines secreted by the adipose tissue may also contribute to the atherogenic milieu. Adiponectin (Chapter 22) is an insulin-sensitizing adipokine. It has anti-inflammatory actions. It stimulates maturation of preadipocytes and decreases adipocyte mass. Its effects on plasma lipids include a decrease in plasma triglyceride concentration, and an increase in HDL-C. It also increases the activity of HTGL and LPL. Besides, it decreases the expression of VCAM-1, the scavenger receptor and TNFα. Obesity decreases adiponectin concentration, de-inhibiting pro-atherogenic processes.
The role of platelets
Platelets mediate leukocyte adhesion and secrete CD40L. Initial adhesion of platelets occurs through platelet glycoprotein receptors to von Willebrand factor and fibrinogen. Adhesion is further mediated by β3-integrins. The adherent platelets contribute to inflammation by secreting cytokines, chemokines, growth factors and adhesion molecules. There is binding between platelets and circulating cells, and such aggregates increase leukocyte activation, adhesion and cell migration.
The role of prostaglandins and leukotrienes
Prostaglandin synthesis from arachidonic acid is catalyzed by the enzyme cyclooxygenase (COX). COX-1 in platelets is inhibited by aspirin; this decreases production of pro-thrombotic thromboxane A2 and is responsible for cardioprotective effect of the drug. The pro-inflammatory prostaglandins such as prostaglandin E2 (PGE2) contribute to inflammatory process. Leukotrienes (leukotriene A4 and B4: lipids derived from the arachidonic acid) are generated by the enzyme 5-lipoxygenase and contribute to the recruitment of lymphocytes to atherosclerotic lesions.
Inflammatory activity destabilizes the plaque, making it prone to rupture
In the mature plaque (Fig. 18.10) the lipid pool is surrounded by foam cells, lymphocytes and VSMC that have migrated into the intima. The plaque ‘cap’ contains collagenous matrix synthesized by VSMC. The cap also contains active macrophages and T lymphocytes. The advanced lesions may calcify.
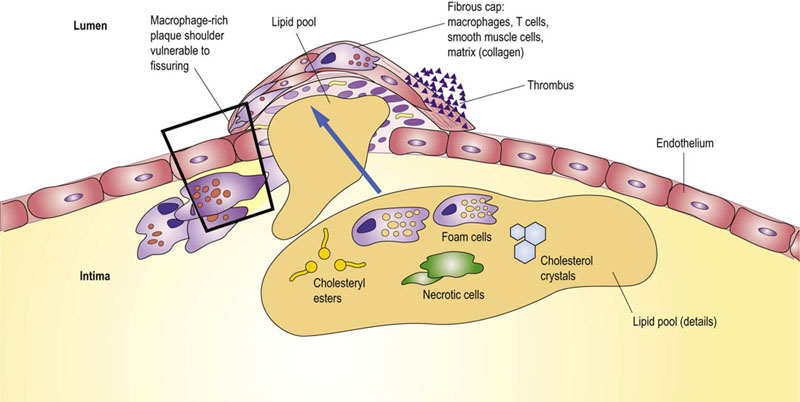
Fig. 18.10 The atherosclerotic plaque.
The lipid center and fibrous cap are the main parts of the mature atherosclerotic plaque which emerges from the structurally remodeled vascular wall. The plaque which is cell-poor and collagen-rich is relatively stable, and grows slowly over the years. On the other hand, the plaque which is cell-rich and collagen-poor becomes unstable and may easily rupture. The key process leading to plaque rupture is digestion of the collagenous matrix of the plaque cap by the metalloproteinase enzymes (MMPs). The figure illustrates areas vulnerable to breakage and shows the obstructing thrombus formed at the rupture site.
Growth of the plaque is accelerated by cycles of plaque mini-ruptures and thrombosis. Active macrophages and T lymphocytes reside preferentially at plaque edges. The unstable plaque has a decreased VSMC content and contains an increased number of macrophages. The macrophages continue to secrete MMPs (collagenases, gelatinases and stromyelysin) that degrade matrix. In addition, lysosomal proteases (cathepsins) degrade collagen and elastin. The activated T cells secrete IFNγ and pro-inflammatory cytokines that induce macrophages to release MMPs and inhibit VSMC collagen synthesis, further weakening the plaque cap. VSMC present in the most vulnerable edge regions of the plaque may undergo apoptosis.
The mature plaque also contains elements of the coagulation system. Tissue factor, a transmembrane cytokine receptor and the primary physiologic trigger of coagulation cascade (Chapter 7), is normally expressed in the VSMC and fibroblasts in the adventitia. In the plaque, it may be expressed in the VSMC and in macrophages. The MMP-9 activity in the plaque stimulates its expression. Apart from this, the tissue factor complexes with the coagulation factor VII (FVII) and this induces cell signaling through the protease activated receptor 2 (PAR2), stimulating a range of events from monocyte, chemotaxis, through VSMC migration and proliferation to angiogenesis and apoptosis.
Finally, thrombin continues to be generated in both the early and advanced plaques, and formation of small thrombi within plaques contributes to their instability. These minute thrombi accelerate plaque growth. On the other hand, after a major rupture, a thrombus forming on the plaque surface may completely occlude the lumen of the affected artery. Such an occlusion cuts off oxygen supply and causes tissue necrosis, precipitating sudden – and sometimes catastrophic – clinical events.
THE Assessment of cardiovascular risk
Cardiovascular risk means the probability of a clinical event occurring within a defined time frame
Cardiovascular risk is the probability that a person will suffer (or die of) heart attack or stroke in a defined period in the future. The main cardiovascular risk factors are listed in Table 18.5. Epidemiologic studies show that the risk of cardiovascular disease is strongly related to plasma concentrations of total cholesterol and LDL-C. It is also inversely related to the concentration of HDL-C. Recent research makes it quite clear that the plasma concentration of triglycerides also contributes to risk, albeit to a lesser extent.
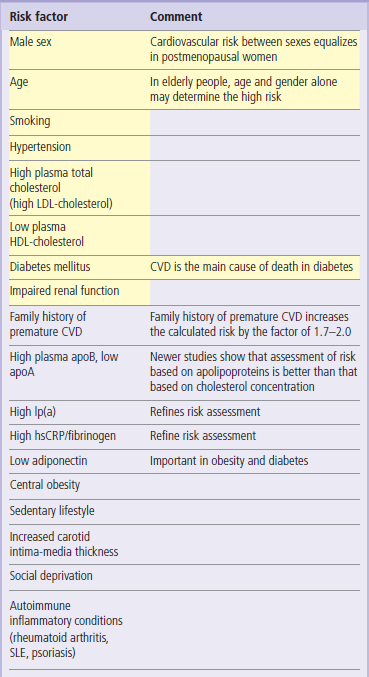
According to the US National Cholesterol Education Program Adult Treatment Panel III (ATP III), the desirable level of total cholesterol is below 5.2 mmol/L (200 mg/dL) and the optimal level of LDL-C is below 2.6 mmol/L (100 mg/dL). The risk steeply increases when total plasma cholesterol concentration increases above 5.2 mmol/L (200 mg/dL). It seems that there is no lower threshold of cholesterol concentration at which the risk would plateau (in other words, the lower the better).
Plasma lipid concentrations are an essential component of cardiovascular risk assessment
It is now accepted that cholesterol concentration that should be achieved with treatment needs to be lowest in people who are at the greatest risk of cardiovascular events, such as individuals with several risk factors, or those who already have atherosclerosis-related disease, diabetes or renal disease. In such persons the ATP III recommends lowering the LDL-C to below 2.6 mmol/L (100 mg/dL) and even 1.8 mmol/L (70 mg/dL).
The Joint British Societies' Guidelines (JBS2) published in 2005 recommend that the optimal total cholesterol concentration in persons who have an increased cardiovascular risk is 4 mmol/L (155 mg/dL) and optimal LDL-C concentration is 2.0 mmol/L (77 mg/dL). Triglyceride concentration should be no higher than 1.7 mmol/L.
A low concentration of HDL-C also signifies increased risk. HDL-C concentration below 1 mmol/L (40 g/dL) in men or 1.2 mmol/L (47 mg/dL) in women is regarded as low. Conversely, it seems that a concentration above 1.6 mmol/L (60 mg/dL) provides some protection against coronary disease. The principles of lipid testing in clinical practice are summarized in Figure 18.11.
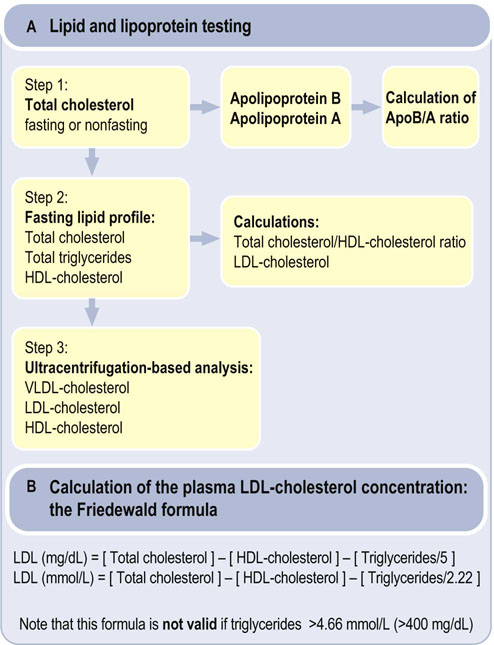
Fig. 18.11 Laboratory diagnosis of dyslipidemias.
(A) The measurement of plasma lipids and apolipoproteins. Several studies show that apolipoprotein B and apolipoprotein A measurements provide better assessment of the lipid-associated cardiovascular risk than total cholesterol and HDL-cholesterol, respectively. (B) Calculation of plasma LDL-cholesterol concentration. Note that the commonly used laboratory methods do not measure the concentration of lipoprotein particles such as LDL or HDL. These methods only measure the fraction of total cholesterol which is present in these particles: therefore we talk about plasma LDL-cholesterol (LDL-C), HDL-cholesterol (HDL-C) or VLDL- cholesterol (VLDL-C). Apo, apolipoprotein.
One should remember that the risk associated with plasma concentrations of the ‘main’ lipids can be modified by other factors, such as increased plasma concentration of Lp(a), fibrinogen or C-reactive protein (hsCRP, see Box). A sobering thought is that about 50% of persons who suffer from myocardial infarction have ‘average’ cholesterol and LDL-C concentrations. The search for new biomarkers continues.
Clinical box Lifestyle change improves plasma lipid profile
A 57-year-old man was referred to the lipid clinic because of hypertriglyceridemia. His triglycerides were 6 mmol/L (545 mg/dL), cholesterol was 5 mmol/L (192 mg/dL), and HDL-C was 1 mmol/L (39 mg/dL). He was obese, took 30 units of alcohol per week, and led a sedentary lifestyle. After initial difficulties, he eventually managed to lose 7 kg of weight over 6 months, cut drinking to below 20 units per week, and started to exercise regularly. Twelve months later, his triglycerides were 2.5 mmol/L (227 mg/dL), cholesterol 4.8 mmol/L (186 mg/dL), and HDL-C 1.2 mmol/L (46 mg/dL).
Lifestyle change can result in appreciable improvements in the lipid profile. To achieve this, individuals need to become committed to changing lifestyle and to maintaining the change over a prolonged period of time.
Note: 1 unit of alcohol is one measure (60 mL) of liquor, one glass (170 mL) of wine, or a half-pint (300 mL) of beer.
The overall CVD risk is calculated using population-based risk calculators
There is a defined risk of a cardiac event associated with every risk factor. However, what interests the clinician most is the overall risk. Calculations of such risk are the basis for preventive measures and drug treatment. The risk algorithms have been developed on the basis of data from long-term epidemiologic studies. The most widely used algorithm has been derived from the Framingham Study in the US. It is based on age, the presence of diabetes, smoking habits, systolic blood pressure, and total cholesterol and HDL-C concentrations. It relates to a population aged 34–74 years without CVD at baseline, and involves up to 12 years follow-up. Importantly, this algorithm does not take into account the family history of early cardiovascular disease.
In Europe, there is the Assign score, the Systematic Coronary Risk Evaluation (SCORE), as well as the Prospective Cardiovascular Munster Study (PROCAM) algorithm. The SCORE algorithm assesses a10-year risk of a first fatal event. The definition of a high risk there is the 10-year risk of CVD death equal or above 5%. This translates roughly into a risk of any CVD event (nonfatal or fatal: the basis of the Framingham algorithm) of about 15%. SCORE also differentiates between high- and low-risk populations in Europe. Yet another algorithm has been developed by the World Health Organization (WHO).
Drugs used in the treatment of dyslipidemias
Clinical box The presence of xanthelasma does not necessarily indicate dyslipidemia
A 28-year-old woman developed unsightly yellow marks around both eyes (xanthelasma). She was asymptomatic and had a good exercise tolerance. Her cholesterol was 5.0 mmol/L (192 mg/dL), triglycerides 0.7 mmol/L (64 mg/dL), and her HDL-C was 1.4 mmol/L (53 mg/dL). There was no family history of early coronary disease.
Clinical box Defining cardiovascular risk
The concept of cardiovascular risk is based on the probability of a cardiovascular event occurring within 10 years from the point of assessment. Different risk calculations use different endpoints: the Framingham Study uses the probability of any cardiovascular disease (CVD), whereas the European SCORE system focuses on the probability of a fatal event decision. Here are some examples of the criteria used to assess the risk as low, moderate or high:
The SCORE risk classification developed by the European Society of Cardiology/European Atherosclerosis Society (ESC/EAS) assesses the 10-year risk of fatal CVD:
To convert the risk of fatal CVD to risk of total (fatal + nonfatal) CVD, one needs to multiply the SCORE-calculated risk by 3 in men and 4 in women.
The risk stratification used in the US National Cholesterol Education Program Adult Treatment Panel III guidelines (ATP III) is based on data from the Framingham Study, and uses three levels of 10-year risk of fatal or nonfatal CHD: >20%, 10–20%, and <10%.
Note that diagnostic cutoff points and recommended treatment targets change, and are periodically updated by the relevant bodies. For instance, ATP IV guidelines are now under development. For current recommendations consult the websites of organizations listed at the end of the chapter.
Clinical box Cholesterol, triglycerides and glucose concentrations: conventional and si units
These are approximate equivalent values for cholesterol:
| mmol/L | mg/dL |
| 4 | 150 |
| 5 | 190 |
| 6 | 230 |
| 7 | 270 |
| 8 | 310 |
The conversion factors to use for obtaining exact values are:
Management of dyslipidemias combines lifestyle measures and drug treatment
Effective cardiovascular prevention needs an approach that combines lifestyle modification (smoking cessation, diet and regular exercise) with drug treatment of dyslipidemia, hypertension and diabetes (Table 18.5). The concentration of plasma LDL (and total plasma cholesterol) can decrease by approximately 15% when a person consistently follows a low-cholesterol diet. When lifestyle measures fail to correct the abnormalities, one resorts to drug treatment. There are several classes of drugs that lower plasma cholesterol concentration.
Statins inhibit HMG-CoA reductase
Statins such as simvastatin, pravastatin, atorvastatin and rosuvastatin are competitive inhibitors of HMG-CoA reductase, the rate-limiting enzyme in cholesterol synthesis. They primarily lower plasma LDL. The inhibition of this enzyme results in a decrease in intracellular cholesterol concentration. This decrease, through SREBP transcription factors (Chapter 17, Fig. 17.7), increases the expression of LDL receptors on the cell membrane. This leads to increased cellular uptake of LDL and, consequently, to a lower plasma cholesterol concentration. The treatment with statins decreases plasma cholesterol concentration by 30–60%, and decreases the risk of future cardiovascular events by 20–30%. Statins also seem to decrease the inflammatory phenomena in the arterial wall.
Fibrates act through PPARα transcription factor
Derivatives of fibric acid (fibrates) are agonists of the transcription factor PPARα. They stimulate the activity of LPL, decrease plasma triglyceride concentrations, and increase the concentration of HDL-C. Their effect on the LDL and total cholesterol is less pronounced than that of the statins.
Inhibitors of intestinal absorption bind bile acids and inhibit cholesterol transporter
Inhibitors of the intestinal absorption of cholesterol include older drugs, the bile acid-binding resins, that are now rarely used. They decrease plasma cholesterol concentration by interrupting the recirculation of cholesterol from the intestine and increasing its excretion. A newer drug, ezetimibe, inhibits the intestinal cholesterol transporter, the Niemann–Pick C1-like 1 (NPC1L1) protein in the intestinal brush border, and lowers total cholesterol by approximately 20%. Longer-term studies of its clinical benefit are in progress.
Omega-3 fatty acids lower plasma triglyceride concentration
A substantial decrease in plasma triglyceride concentration can be achieved by treatment with omega-3 fatty acids (fish oil). Interestingly, fish oil preparations are also anti-arrhythmic, particularly in patients who have already suffered myocardial infarction.
Role of antioxidants continues to be studied
In animals, antioxidants such as probucol or vitamin E inhibit development of atherosclerosis. Epidemiologic studies have shown that those who take antioxidants such as vitamin E and C or β-carotene have a decreased risk of cardiovascular disease. However, prospective clinical trials of antioxidant treatment failed to confirm such preventive benefit. One tentative explanation is that it is the natural antioxidants (such as those contained in fruits) or their combinations that are protective, rather than pure substances.
Summary
Lipoproteins transport hydrophobic lipids between organs and tissues.
Chylomicrons mediate the transport of dietary fat.
VLDL mediate the transport of endogenously synthesized fat.
Chylomicrons, VLDL and remnant lipoproteins are part of the organism's fuel distribution network: the fuel transport pathway.
LDL are cholesterol-rich lipoproteins which emerge from the fuel transport pathway. When present in excess they may enter the arterial wall.
HDL mediate reverse cholesterol transport, e.g. removal of cholesterol from the peripheral cells to the liver.
Atherogenesis involves endothelial dysfunction, lipid deposition, inflammatory reaction in the arterial wall, activation and proliferation of the arterial smooth muscle cells, and thrombosis. The major atherosclerosis-related diseases are coronary heart disease, stroke and peripheral vascular disease.
Interactions between cells participating in atherogenesis are mediated by an array of cytokines, growth factors and adhesion molecules.
Atherogenesis disrupts the structure of the arterial wall and results in the formation of atherosclerotic plaque, which narrows the lumen of the affected artery. However, the immediate cause of a heart attacks is not the slow growth of the plaque, but its sudden rupture.
Assessment of cardiovascular risk involves measurements of several lipid parameters and identification other risk factors such as hypertension, smoking and the presence of diabetes. The intensity of treatment is geared to the degree of calculated risk.
Borissoff, JI, Spronk, HMH, ten Cate, H. The hemostatic system as a modulator of atherosclerosis. N Engl J Med. 2011; 364(18):1746–1760.
Dominiczak, MH. Risk factors for coronary disease: the time for a paradigm shift? Clin Chem Lab Med. 2001; 39:907–919.
Dominiczak, MH, Caslake, MJ. Apolipoproteins: metabolic role and clinical biochemistry applications. Ann Clin Biochem. 2011; 48:498–515.
Durrington, P. Dyslipidaemia. Lancet. 2003; 362:717–731.
Duval, C, Muller, M, Kersten, S. PPAR alpha and dyslipidemia (review). Biochim Biophys Acta Mol Biol Cell Biol Lipids. 2007; 1771:961–971.
Libby, P, Aikawa, M. Stabilization of atherosclerotic plaques: New mechanisms and clinical targets. Nature Medicine. 2002; 8:1257–1262.
Michalik, L, Auwerx, J, Berger, JP, et al. International Union of Pharmacology. LXI. Peroxisome proliferator-activated receptors. Pharmacol Rev. 2006; 58:726–741.
O'Donnell, CJ, Nabel, EG. Genomics of cardiovascular disease. In: Genomic medicine. Feero WG, Guttmacher, AE, editors. N Engl J Med. 2011; 365:2098–2109.
Packard, RRS, Libby, P. Inflammation in atherosclerosis: from vascular biology to biomarker discovery and risk prediction. Clin Chem. 2008; 54:24–38.
Rader, DJ, Daugherty, A. Translating molecular discoveries into new therapies for atherosclerosis. Nature. 2008; 451:904–913.
Familial hypercholesterolemia. Simon Broome Diagnostic criteria for index individuals and relatives. http://www.nice.org.uk/nicemedia/live/12048/41707/41707.pdf. [(Accessed October 2012)].
General Cardiovascular disease. Framingham Heart Study. www.framinghamheartstudy.org/risk/gencardio.html. [(Accessed May 2013)].
The Task Force for the Management of Dyslipidaemias of the European Society of Cardiology (ESC) and the European Atherosclerosis Society (EAS). ESC/EAS Guidelines for the management of dyslipidaemias. www.escardio.org/guidelines-surveys/esc-guidelines/guidelinesdocuments/guidelines-dyslipidemias-ft.pdf. [(Accessed May 2013)].
Marks, D, Thorogood, M, Neil, HA, et al. A review on the diagnosis, natural history, and treatment of familial hypercholesterolaemia. Atherosclerosis. 2003; 168:1–14.
Morris, PB. ATP IV, CVD risk assessment and dyslipidemia. www.acli.com/Events/Documents/Tue22812%20-%20Lipidology%20-%20Pamela%20Morris.pdf. [(Accessed May 2013)].
National Cholesterol Education Program Adult Treatment Panel III (ATP III. www.nhlbi.nih.gov/guidelines/cholesterol/index.htm [(Accessed May 2013)].
National High Blood Pressure Education Program. Joint National Committee on the Prevention, Detection, Evaluation, and Treatment of High Blood Pressure (7th Report: known as JNC7) JNC8 is in development www.nhlbi.nih.gov/guidelines/hypertension/. [(Accessed May 2013)].
National Institute for Health and Clinical Excellence (NICE), United Kingdom. http://publications.nice.org.uk/lipid-modification-cg67 [(Accessed May 2013)].
Von Eckardstein, A. HDL in motion: molecular dynamics and disease pathophysiology. www.medscape.org/noscan/slideshow. [(Accessed May 2013)].