Hemostasis and Thrombosis
Learning objectives
After reading this chapter you should be able to:
 Outline the sequential mechanisms involved in normal hemostasis.
Outline the sequential mechanisms involved in normal hemostasis.
Summarize the processes through which the vessel wall regulates hemostasis and thrombosis.
Describe the role of platelets in hemostasis and thrombosis.
Outline pathways through which antiplatelet drugs act.
Describe the pathways of blood coagulation, and how these are tested in the clinical hemostasis laboratory to identify coagulation disorders.
Describe the physiologic inhibitors of blood coagulation.
Outline pathways through which anticoagulant drugs act.
Introduction
It is essential that blood should not leak excessively from blood vessels when they are injured by the traumas of daily life
Circulation of the blood within the cardiovascular system is essential for transportation of gases, nutrients, minerals, metabolic products, and hormones between different organs. It is also essential that blood should not leak excessively from blood vessels when they are injured by the traumas of daily life. Animal evolution has therefore resulted in the development of an efficient but complex series of hemodynamic, cellular, and biochemical mechanisms that limit such blood loss by forming platelet–fibrin plugs at sites of vessel injury (hemostasis). Genetic disorders that result in loss of individual protein functions, and therefore in excessive bleeding (e.g. hemophilia), have played an important part in the identification of many of the biochemical mechanisms in hemostasis.
It is essential also that these hemostatic mechanisms are appropriately controlled by inhibitory mechanisms, otherwise an exaggerated platelet–fibrin plug may produce local occlusion of a major blood vessel (artery or vein) at its site of origin (thrombosis), or may break off and block a blood vessel downstream (embolism).
Arterial thrombosis is the major cause of heart attacks, stroke, and non-traumatic limb amputations in developed countries (atherothrombosis is discussed in Chapter 18). Venous thrombosis and embolism are also major causes of death and disability. Clinical use of antithrombotic drugs (antiplatelet, anticoagulant, and thrombolytic agents) is now widespread in developed countries, and requires an understanding of how they interfere with hemostatic mechanisms to exert their antithrombotic effects.
Hemostasis
Hemostasis means ‘the arrest of bleeding’
After tissue injury that ruptures smaller vessels (including everyday trauma, injections, surgical incisions, and tooth extractions), a series of interactions between the vessel wall and the circulating blood normally occurs, resulting in cessation of blood loss from injured vessels within a few minutes (hemostasis). Hemostasis results from effective sealing of the ruptured vessels by a hemostatic plug composed of blood platelets and fibrin. Fibrin is derived from circulating fibrinogen, whereas platelets are small cell fragments that circulate in the blood and have an important role in the initiation of hemostasis.
Hemostasis requires the coordinated function of blood vessels, platelets, coagulation factors and the fibrinolytic system
Figure 7.1 provides an overview of hemostatic mechanisms and illustrates some of the interactions between blood vessels, platelets, and the coagulation system in hemostasis; each of these components of hemostasis also interacts with the fibrinolytic system. The initial response of small blood vessels to injury is arteriolar vasoconstriction, which temporarily reduces local blood flow. Flow reduction transiently reduces blood loss, and may also promote formation of the platelet–fibrin plug. Activation of blood platelets is followed by their adhesion to the vessel wall at the site of injury, and their subsequent aggregation to each other, building up an occlusive platelet mass that forms the initial (primary) hemostatic plug. This platelet plug is friable and, unless subsequently stabilized by fibrin, will be washed away by local blood pressure when vasoconstriction reverses.
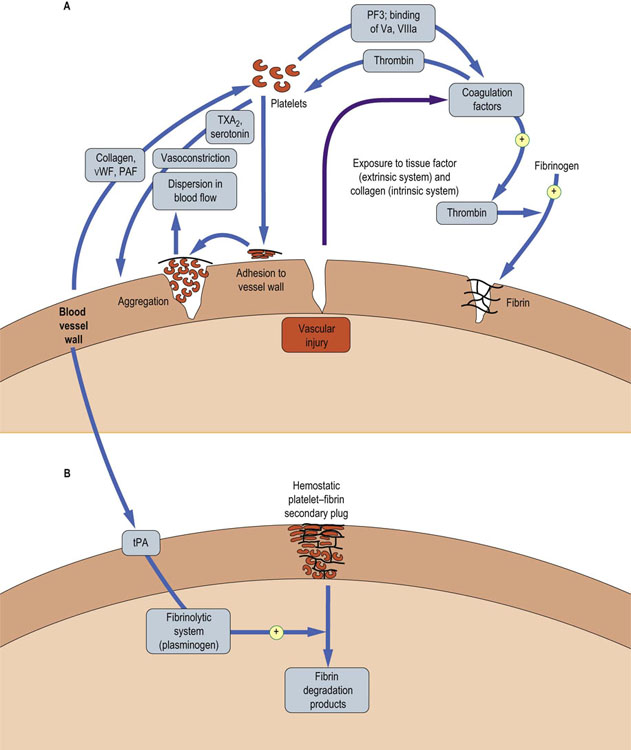
Fig. 7.1 Overview of hemostatic mechanisms.
(A) Vascular injury sets in motion a series of events that culminate in formation of a primary plug of platelets. This can be dispersed by blood flowing through the vessel unless the plug is stabilized. (B) The primary plug is stabilized by a network of fibrin (formed from crosslinked fibrinogen). The secondary plug is stable and is degraded only when the fibrinolytic system has been activated. PAF, platelet-activating factor; PS, phosphatidylserine; tPA, tissue-type plasminogen activator; TXA2, thromboxane A2; Va, activated coagulation factor V; VIIIa, activated coagulation factor VIII; vWF, von Willebrand factor. Reproduced from Dominiczak MH. Medical Biochemistry Flash Cards. London: Elsevier, 2012.
Vascular injury also activates coagulation factors, which interact sequentially to form thrombin, which converts circulating soluble plasma fibrinogen to insoluble, crosslinked fibrin. This forms the subsequent (secondary) hemostatic plug, which is relatively resistant to dispersal by blood flow or fibrinolysis. There are two pathways of the activation of coagulation factors: the extrinsic pathway, which is initiated by the exposure of the flowing blood to tissue factor, released from subendothelial tissue, and the intrinsic pathway, which has an important amplification role in generating thrombin and fibrin.
The lysis of fibrin is as important to health as its formation
Hemostasis is a continuous process throughout life, and would result in excessive fibrin formation and vascular occlusion if unchecked. Evolution has therefore produced a fibrinolytic system; this is activated by local fibrin formation, resulting in local generation of plasmin, an enzyme which digests fibrin plugs (in parallel with tissue repair processes), thus maintaining vascular patency. Digestion of fibrin results in generation of circulating fibrin degradation products (FDPs). These are detectable in plasma of healthy individuals at low concentration, which illustrates that fibrin formation and lysis are continuing processes in health.
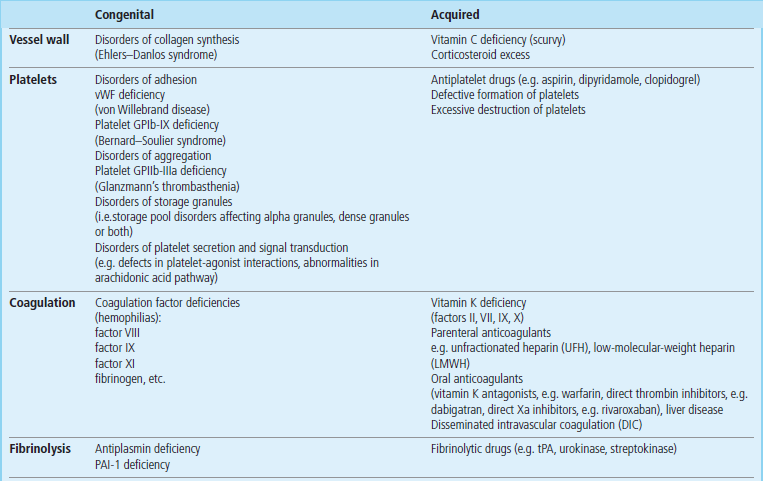
Excessive bleeding may result from defects in each of the components of hemostasis, which may be caused by disease (congenital or acquired) or by antithrombotic drugs (Table 7.1). The vascular, platelet, coagulation and fibrinolytic components of hemostasis will now be discussed in turn.
The vessel wall
Vascular injury has a key role in initiating local formation of the platelet–fibrin plug and in its subsequent removal by the fibrinolytic system
All blood vessels are lined by a flat sheet of endothelial cells, which have important roles in the interchange of chemicals, cells, and microbes between the blood and the body tissues. Endothelial cells in the smallest blood vessels (capillaries) are supported by a thin layer of connective tissue, rich in collagen fibers, called the intima. In veins, a thin layer (the media) of contractile smooth muscle cells allows some venoconstriction: for example, superficial veins under the skin constrict in response to surface cooling. In arteries and arterioles, a well-developed muscle layer allows powerful vasoconstriction, including the vasoconstriction after local injury that forms part of the hemostatic response. Larger vessels also have a supportive connective tissue outer layer (the adventitia).
Normal endothelium has an antithrombotic surface
Intact normal endothelium does not initiate or support platelet adhesion or blood coagulation. Its surface is antithrombotic. This thrombo-resistance is partly due to endothelial production of two potent vasodilators and inhibitors of platelet function: prostacyclin (prostaglandin I2, PGI2) and nitric oxide, otherwise known as endothelium-derived relaxing factor (EDRF).
 Advanced concept box Prostacyclin and nitric oxide: biochemical mediators of vasoconstriction and vasodilatation
Advanced concept box Prostacyclin and nitric oxide: biochemical mediators of vasoconstriction and vasodilatation
The diameters of arteries and arterioles throughout the body continuously alter to regulate blood flow according to local and general metabolic and cardiovascular requirements. Control mechanisms include neurogenic (sympathetic/adrenergic; Chapter 41.1) and myogenic pathways, and local biochemical mediators, including prostacyclin (PGI2) and nitric oxide.
Prostacyclin is the major arachidonic acid metabolite formed by vascular cells. It is a potent vasodilator, and also a potent inhibitor of platelet aggregation. It has a short half-life in plasma (3 min).
Nitric oxide is also a potent vasodilator formed by vascular endothelial cells, also with a short half-life. It was initially termed endothelium-derived relaxing factor (EDRF). In common with that of prostacyclin, its generation by endothelial cells is enhanced by many compounds, and also by blood flow and shear stress (the tangential force applied to the cells by the flow of blood). In the normal circulation, nitric oxide appears to have a key role in flow-mediated vasodilatation. It is synthesized by two distinct forms of endothelial nitric oxide synthase (eNOS): constitutive and inducible. Constitutive eNOS rapidly provides relatively small amounts of nitric oxide for short periods, related to vascular flow regulation. The beneficial effects of nitrate drugs in hypertension and angina may partly reflect their effects on this pathway (Chapter 18). Inducible eNOS is stimulated by cytokines in inflammatory reactions, and releases large amounts of nitric oxide for long periods. Its suppression by glucocorticoids may partly account for their antiinflammatory effects.
Both prostacyclin and nitric oxide appear to exert their vasodilator actions by diffusing locally from endothelial cells to vascular smooth muscle cells, where they stimulate guanylate cyclase, resulting in increased formation of cyclic guanosine 3′5′-monophosphate (cGMP) and relaxation of vascular smooth muscle via alteration of the intracellular calcium concentration (Chapter 40).
Endothelial damage exposes blood to the tissue factor and to collagen
The vasoconstriction that occurs after vascular injury is partly mediated by two platelet activation products: serotonin (5-hydroxytryptamine), and thromboxane A2 (TXA2), a product of platelet prostaglandin metabolism. The endothelial cell damage also exposes flowing blood to subendothelial tissue factor, which activates the extrinsic pathway of blood coagulation (Fig. 7.1). In addition, after a vascular injury that disrupts the endothelial cell lining, flowing blood is exposed to subendothelial collagen, which activates the intrinsic pathway of blood coagulation.
Advanced concept box Thromboxane A2 and aspirin
It has already been noted that prostacyclin, PGI2, the major arachidonic acid metabolite formed by vascular cells, is a potent vasodilator and inhibitor of platelet aggregation. In contrast, the major arachidonic acid metabolite formed by platelets is thromboxane A2 (TXA2), which is a potent vasoconstrictor and stimulates platelet aggregation. In common with prostacyclin, TXA2 has a short half-life. In the late 1970s, Salvador Moncada and John Vane contrasted the effects of PGI2 and TXA2 on blood vessels and platelets, and hypothesized that a balance between these two compounds was important in the regulation of hemostasis and thrombosis.
Congenital deficiencies of cyclooxygenase or thromboxane synthase (the enzymes involved in TXA2 synthesis) result in a mild bleeding tendency. Ingestion of even low doses of acetylsalicylic acid (aspirin) irreversibly acetylates cyclooxygenase and suppresses TXA2 synthesis and platelet aggregation for several days, resulting in an antithrombotic effect and a mild bleeding tendency. Bleeding is especially likely from the stomach, as a result of the formation of stomach ulcers secondary to the inhibition of cytoprotective gastric mucosal prostaglandins by aspirin. Although in persons at high risk of arterial thrombosis, e.g. previous myocardial infarction, this bleeding tendency is outweighed by a reduction in risk of thrombosis, aspirin is contraindicated in individuals with a history of bleeding disorders, or existing stomach or duodenal ulcers.
Exposure of flowing blood to collagen as a result of endothelial damage also stimulates platelet activation
Platelets bind to collagen via von Willebrand factor (vWF), which is released from the endothelial cells. vWF in turn binds both to collagen fibers and to platelets (via a platelet membrane glycoprotein receptor, GPIb-IX). Platelet-activating factor (PAF) from the vessel wall may also activate platelets in hemostasis (Fig. 7.1; see also box on p. 27).
Collagen has a key role in the structure and hemostatic function of small blood vessels
Because collagen has a key role in the structure and hemostatic function of small blood vessels, vascular causes of excessive bleeding include congenital or acquired deficiencies of collagen synthesis (Table 7.1). Congenital disorders include the rare Ehlers–Danlos syndrome. Acquired disorders include the relatively common vitamin C deficiency, scurvy (Chapter 11), and excessive exogenous or endogenous corticosteroids.
Platelets and platelet-related bleeding disorders
Blood platelets form the initial hemostatic plug in small vessels, and the initial thrombus in arteries and veins
Platelets are circulating, anuclear microcells of mean diameter 2–3 µm. They are fragments of bone marrow megakaryocytes, and circulate for approximately 10 days in the blood. The concentration of platelets in normal blood is 150–400 × 109/L
Advanced concept box Platelet activation exposes glycoprotein receptors
Platelets can be activated by several chemical agents, including adenosine diphosphate (ADP, released by platelets, erythrocytes, and endothelial cells), epinephrine, collagen, thrombin, and PAF; by infection e.g. HIV, Helicobacter pylori; and by high physical shear stresses. Most of the chemical agents appear to act by binding to specific receptors on the platelet surface membrane. After receptor stimulation, several pathways of platelet activation can be initiated, resulting in several phenomena:
Change in platelet shape from a disk to a sphere with extended pseudopodia, which facilitates aggregation and coagulant activity.
Release of several compounds involved in hemostasis from intracellular granules, e.g. ADP, serotonin, fibronectin and vWF.
Aggregation, via exposure of GPIb-IX membrane receptor and linking by vWF (under high shear conditions), and via exposure of another membrane glycoprotein receptor, GPIIb-IIIa, and linking by fibrinogen (under low shear conditions).
Adhesion to the vessel wall via exposure of the GPIb-IX membrane receptor, through which vWF binds platelets to subendothelial collagen.
Finally, stimulation of the platelet membrane receptor triggers the activation of platelet membrane phospholipases, which hydrolyze membrane phospholipids, releasing arachidonic acid. Arachidonic acid is metabolized by cyclooxygenase and thromboxane synthase to TXA2, a potent but labile (half-life 30 seconds) mediator of platelet activation and vasoconstriction.
Congenital defects in platelet adhesion/aggregation can cause lifelong excessive bleeding
A simple screening test – measurement of the skin bleeding time (reference range, 2–9 minutes) – can frequently detect congenital defects of platelet adhesion/aggregation, in which the time is characteristically prolonged. The most common such defect is von Willebrand disease (Table 7.1), a group of both autosomal dominant and autosomal recessive disorders that result in either quantitative or qualitative defects of vWF multimers. These multimers are composed of subunits (molecular weight 220–240 kDa) that are released from storage granules known as the Weibel–Palade bodies in endothelial cells and alpha granules in platelets. Not only does vWF have an important role in platelet hemostatic function but it also transports coagulation factor VIII (antihemophilic factor) in the circulation and delivers it to sites of vascular injury. Hence, plasma concentrations of factor VIII may also be low in von Willebrand disease. Treatment of this disease is to increase the low plasma vWF activity, usually by means of either desmopressin (a synthetic analogue of vasopressin (Chapter 24), which releases vWF from endothelial cells into plasma), or administering VWF concentrates derived from human plasma.
Less common congenital platelet-related bleeding disorders include GPIb-IX deficiency (Bernard–Soulier syndrome), GPIIb-IIIa deficiency (Glanzmann's thrombasthenia), and fibrinogen deficiency (because fibrinogen bridges GPIIb-IIIa receptors of adjacent platelets).
Acquired disorders may be caused by defective formation, and excessive destruction or consumption of platelets
Acquired disorders of platelets include a low platelet count (thrombocytopenia), which may be the result of either defective formation of platelets by bone marrow megakaryocytes, e.g. in myelodysplasia or acute myeloid leukaemia, excessive destruction of platelets, e.g. by antiplatelet antibodies, and excessive consumption of platelets, e.g. in disseminated intravascular coagulation or by sequestration in an enlarged spleen.
Antiplatelet drugs are used in the prevention or treatment of arterial thrombosis
Antiplatelet drugs are used in the prevention or treatment of arterial thrombosis; their sites of action are illustrated in Figure 7.2. Aspirin inhibits cyclooxygenase and hence reduces the formation of TXA2. Because it also has the effect of reducing the formation of PGI2, which itself has antiplatelet activity, agents acting more specifically as thromboxane synthase inhibitors, e.g. picotamide, or thromboxane receptor antagonists, such as ifetroban, have also been investigated as potential antiplatelet agents. However, these do not appear to be more effective than aspirin. Dipyridamole acts by both reducing the availability of ADP and inhibiting thromboxane synthase, and ticlopidine and clopidogrel inhibit the ADP receptor (Fig. 7.2). These drugs have antithrombotic effects similar to those of aspirin, but cause less gastric bleeding because they do not interfere with synthesis of prostaglandins in the stomach. GPIIb-IIIa antagonists, e.g. tirofiban or abciximab, can also be used in acute coronary thrombosis. Each of these antiplatelet drugs adds to the antithrombotic efficacy of aspirin but also increases the risk of bleeding when used in combination.
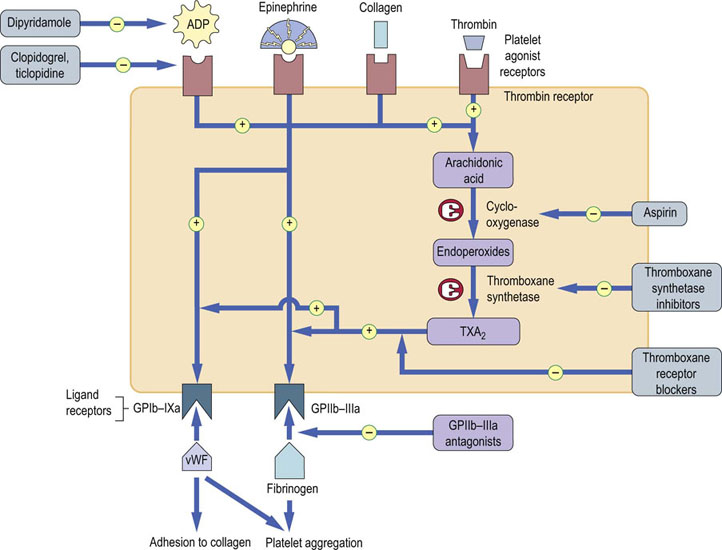
Fig. 7.2 Pathways of platelet activation and mechanisms of action of antiplatelet drugs.
Stimulation of platelet agonist receptors results in exposure of platelet ligand receptors, partly through the platelet prostaglandin (cyclooxygenase) pathway. Ligand receptors bind vWF and fibrinogen in platelet adhesion/aggregation. vWF, von Willebrand factor; TXA2, thromboxane A2.
Advanced concept box Platelet membrane receptors: their ligands vWF and fibrinogen
Platelets have a key role in hemostasis and thrombosis, through adhesion to the vessel wall and subsequent aggregation to form a platelet-rich hemostatic plug or thrombus. These processes involve exposure of specific membrane glycoprotein receptors after platelet activation by several compounds.
Platelet receptor GPIb-IX plays a key part in the adhesion of platelets to subendothelium. It binds vWF, which also interacts with specific subendothelial receptors, including those on subendothelial collagen. Congenital deficiencies of GPIb-IX (Bernard–Soulier syndrome) or, more commonly, of vWF, result in a bleeding tendency.
Another receptor, GPIIb-IIIa, has a key role in platelet aggregation. After platelet activation, hundreds of thousands of GPIIb-IIIa receptors can be exposed in a single platelet. These receptors interact with fibrinogen or vWF, which bind platelets together, forming a hemostatic or thrombotic plug. Congenital deficiency of GPIIb-IIIa (the rare Glanzmann's thrombasthenia) causes a severe bleeding disorder; in contrast, deficiencies of either fibrinogen or vWF cause a milder bleeding disorder, because these two ligands can substitute for each other. GPIIb-IIIa inhibitors (e.g. tirofiban, abciximab) have been developed for patients undergoing angioplasty for coronary artery disease to prevent further coronary events.
Coagulation
Blood coagulation factors interact to form the secondary, fibrin-rich, hemostatic plug in small vessels, and the secondary fibrin thrombus in arteries and veins
Plasma coagulation factors are identified by Roman numerals: they are listed in Table 7.2, together with some of their properties. Tissue factor was formerly known as factor III, calcium ion as factor IV; factor VI does not exist.
Table 7.2
Coagulation factors and their properties
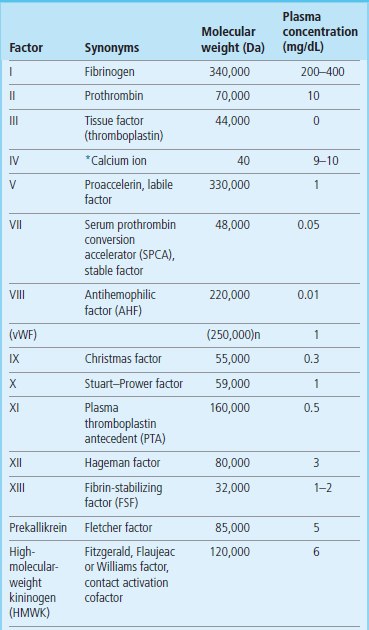
n indicates number of subunits.
*To convert calcium ion to mmol/L divide by 4.
The coagulation cascade
Figure 7.3 illustrates the currently accepted scheme of blood coagulation. Since the early 1960s, this has been accepted as a ‘waterfall’ or ‘cascade’ sequence of interactive proenzyme to enzyme conversions, each enzyme activating the next proenzyme in the sequence(s). Activated factor enzymes are designated by the letter ‘a’: for example, factor XIa. Although the process of blood coagulation is complex and nonlinear, traditionally, the scheme has been divided into three parts:
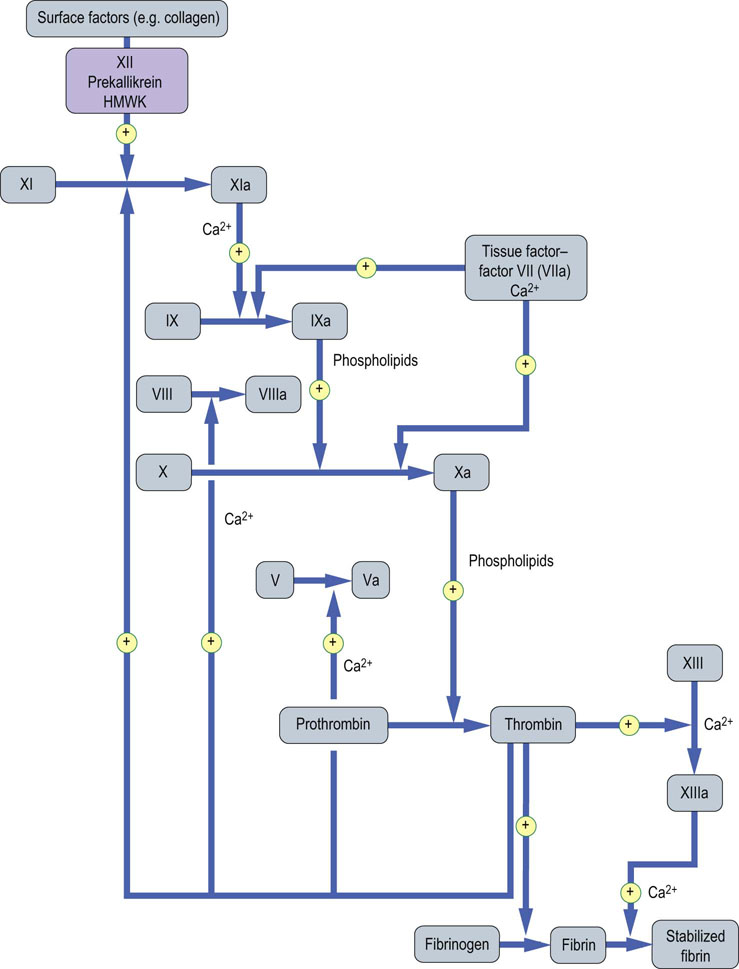
Fig. 7.3 Blood coagulation: activation of coagulation factors.
After the initiation of blood coagulation, the coagulation factor proenzymes are activated sequentially: activated factor enzymes are designated by the letter ‘a’. The purple box indicates contact factors that have no apparent function in in vivo hemostasis. Phospholipids are supplied in vivo by platelets. The intrinsic pathway: blue arrows. The extrinsic pathway: red arrows. The common pathway: green arrows. HMWK, high-molecular-weight kininogen. Reproduced from Dominiczak MH. Medical Biochemistry Flash Cards. London: Elsevier, 2012.
The status of the intrinsic, extrinsic and final common pathway is assessed by specific laboratory tests
The three components of the coagulation system are distinguished on the basis of the nature of the initiating factor and its corresponding test in the clinical hemostasis laboratory; hence, three tests of coagulation are performed in clinical laboratories on citrated, platelet-poor plasma:
activated partial thromboplastin time (APTT), testing the intrinsic pathway
prothrombin time (PT) – the extrinsic pathway, and
thrombin clotting time (TCT) testing the final common pathway.
Platelet-poor plasma is used in these tests because the platelet count influences clotting time results. To obtain platelet-poor plasma, blood is collected in tubes containing citrate anticoagulant to sequester calcium ions reversibly, and the blood is centrifuged at 2000 g for 15 minutes. The coagulation time tests are initiated by adding calcium and appropriate initiating agents.
However, these tests have their limitations in describing the in vivo phenotype of a patient's blood to coagulate effectively. The so-called global assays of coagulation have therefore been developed, and they are thought to better reflect an individual's ability to clot. These include thromboelastography and thrombin generation.
 Clinical test boxGlobal coagulation assays
Clinical test boxGlobal coagulation assays
Thromboelastography (TEG) and rotational thromboelastometry (ROTEM) assess the ability of whole blood to clot in response to a mechanical stimulus, allowing an assessment of all aspects of hemostasis: platelet function, fibrin cross-linking and fibrinolysis.
Thrombin generation assay is a global coagulation assay thought better able to assess an individual's ability to coagulate than standard coagulation assays. Tests such as the PT and APTT described above measure only 5% of the total thrombin generated: i.e. at the time of first clot generation (Fig. 7.4).
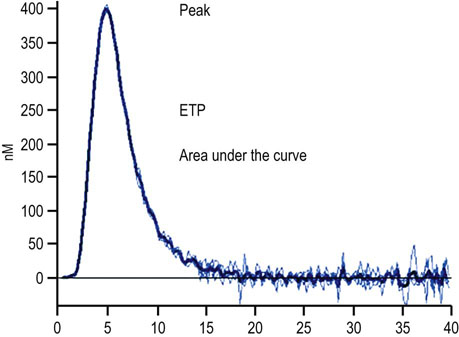
Fig. 7.4 Thrombin generation curve.
The curve is a measurement of thrombin concentration over time with the parameters lag time, time to the first development of thrombin, peak thrombin, the maximum amount of thrombin generated, time to peak, and endogenous thrombin potential (ETP) or the area under the curve.
Thrombin is central to the coagulation cascade in that it converts fibrinogen to fibrin and also has numerous positive and negative feedback roles. The measurement of thrombin generation enables quantification over time of all thrombin generated in a plasma sample by its ability to ‘cut’ either a chromophore or fluorochrome and measure the resultant chromogenic or fluorescent activity.
Despite promising results from ROTEM and thrombin generation, both assays are limited by numerous preanalytical and analytical variables, which make comparisons between laboratories difficult. There is still no reliable standardization of these assays with robust internal or external quality control. For this reason, both remain research tools.
Congenital deficiencies of coagulation factors (I–XIII) result in excessive bleeding
Congenital deficiencies of coagulation factors (I–XIII) result in excessive bleeding, which illustrates their physiologic importance in hemostasis. The exception is factor XII deficiency, which does not increase the bleeding tendency, despite prolonging blood clotting times in vitro; the same is true for its cofactors, prekallikrein or high-molecular-weight kininogen (HMWK). A possible explanation for this is given below.
Activated partial thromboplastin time (APTT) assesses the intrinsic pathway
The term ‘intrinsic’ implies that no extrinsic factor such as tissue factor or thrombin is added to the blood, besides a contact with nonendothelial ‘surface’. The clinical test of this pathway is the activated partial thromboplastin time (APTT), also known as the kaolin–cephalin clotting time (KCCT) because kaolin (microparticulated clay) is added as a standard ‘surface’, and cephalin (brain phospholipid extract) as a substitute for platelet phospholipid. The reference range of the APTT is about 30–40 seconds; prolongations are observed in deficiencies of factors XII (or its cofactors, prekallikrein or HMWK), XI, IX (or its cofactor, factor VIII), X (or its cofactor, factor V), or prothrombin (factor II) (see Table 7.1 and 7.2).
The test is used to exclude the common congenital hemophilias (deficiencies of factors VIII, IX or XI), and to monitor unfractionated heparin treatment. Hemophilias caused by factor VIII or IX deficiency occur in approximately 1 in 5000 and 1 in 30,000 males, respectively; inheritance is X-linked recessive, transmitted by carrier females. Treatment is usually with recombinant factor VIII or IX concentrates.
 Clinical boxA boy with extensive bruising: classic Hemophilia (Congenital Factor VIII Deficiency)
Clinical boxA boy with extensive bruising: classic Hemophilia (Congenital Factor VIII Deficiency)
A 3-year-old boy was admitted from the emergency room of his local hospital because of extensive bruising after a fall down a few stairs. A routine coagulation screen test showed a greatly prolonged APTT of more than 150 seconds (normal range 30–40 seconds). Assay of coagulation factor VIII showed a very low level; the vWF level was normal. His mother recollected a family history of excessive bleeding, which had affected her brother and father.
Because of this typical history of an X-linked recessive bleeding disorder, a low coagulation factor VIII level, and a normal vWF level, the diagnosis of congenital factor VIII deficiency was made. The family were referred to the local hemophilia center and counseled about the risks of further affected sons and carrier daughters. The child was treated with intravenous recombinant factor VIII concentrate for the present bleed, and prophylactically in the future to prevent further bleeding episodes.
Prothrombin time assesses the extrinsic pathway
The term ‘extrinsic’ refers to the effect of tissue factor, which (after combining with coagulation factor VII) greatly accelerates coagulation, by activating both factor IX and factor X (Fig. 7.3). Tissue factor is a polypeptide that is expressed in all cells other than endothelial cells. The clinical test of this pathway is the prothrombin time (PT), in which tissue factor is added to plasma. The reference range is approximately 10–15 seconds; prolongations are observed in deficiencies of factors VII, X, V, or II. In clinical practice, the test is used to diagnose both the rare congenital defects of these factors and, much more commonly, acquired bleeding disorders, resulting from:
Vitamin K deficiency, e.g. in malabsorption or obstructive jaundice (Chapter 11), which reduces hepatic synthesis of factors II, VII, IX and X. Treatment is by oral or intravenous administration of vitamin K.
Administration of Oral vitamin K antagonists, e.g. warfarin, which reduce hepatic synthesis of these factors. Excessive bleeding in patients taking warfarin can be treated by stopping the drug, giving vitamin K or replacing factors II, VII, IX and X with prothrombin complex concentrates containing only the relevant factors, e.g. Beriplex or fresh frozen plasma.
Liver disease, which reduces hepatic synthesis of these factors. For example, the prothrombin time is a prognostic marker of liver failure after acetaminophen (paracetamol) overdose (Chapter 30). Treatment is by replacing factors II, VII, IX and X with fresh frozen plasma.
Clinical test boxMonitoring oral anticoagulant therapy
Oral anticoagulant therapy with vitamin K antagonists, e.g. warfarin, is given long term to patients at risk of thrombosis within the chambers of the heart, e.g. patients with atrial fibrillation or heart valve prostheses, which may embolize to the brain, causing a stroke.
Monitoring of an internationally standardized prothrombin time, i.e. the International Normalized Ratio (INR), every few weeks is essential to minimize the risk not only of thromboembolism but also of excessive bleeding. Up to 1% of the adult population in developed countries now receive long-term anticoagulants; hence traditional monitoring by doctors and nurses (taking blood samples, sending them to the laboratory, getting results and giving dosage instructions to patients) has created an unsustainable workload.
In recent years, near-patient or point-of-care INR testing has become available for warfarin monitoring with a ‘finger prick’ capillary sample being drawn into a portable INR analyzer. With this technique some patients can self-test and occasionally self-monitor, similar to blood glucose self-monitoring by persons with diabetes. Computerized algorithms for dosing warfarin have also been developed to assist health care workers to alter dosing accurately. New oral anticoagulants, e.g. dabigatran and rivaroxaban, for patients with atrial fibrillation have recently been introduced, which require no monitoring of anticoagulation levels.
Thrombin clotting time assesses the final common pathway
The term ‘final common pathway’ refers to the conversion of prothrombin to thrombin via Xa, with Va acting as a cofactor
This in turn allows the conversion of fibrinogen to fibrin. This final stage of fibrin production in the common pathway is tested clinically by the thrombin clotting time (TCT), in which exogenous thrombin is added to plasma. The reference range of values is approximately 10–15 seconds; prolongations are observed in fibrinogen deficiency and in the presence of inhibitors, e.g. heparin, fibrin degradation products. Fibrinogen deficiency may be congenital or due to acquired consumption of fibrinogen in disseminated intravascular coagulation (DIC), or may occur after administration of fibrinolytic drugs (see below). Treatment is with cryoprecipitate or fibrinogen concentrates.
Several assays assess platelet function
Apart from assessing platelet number, size and morphology through the ‘complete blood count’ analysis and blood film review, platelet function can also be assessed in other ways.
One method of platelet function assessment is the Platelet Function Analyzer (PFA-100, Siemens). Whole blood is passed over a cartridge containing an aperture coated with a combination of two platelet agonists: e.g. collagen/epinephrine, or collagen/ADP. The time to aperture closure as a result of platelet aggregation is measured. It cannot define specific disorders but an abnormal result is suggestive of a platelet disorder and can be used as a screening test.
Light transmission aggregometry (LTA) is considered the gold standard for investigating specific disorders of platelet function. Platelet-rich plasma is exposed to various platelet agonists, e.g. collagen, ADP and epinephrine, and light transmission is monitored to produce standard curves. The pattern of curves obtained with a combination of agonists can help determine which platelet function defect is present.
The production and release of platelet nucleotides, i.e. ATP and ADP, can be measured to assess nucleotide production and nucleotide release from granules. Flow cytometry analysis of various platelet receptors can also be performed.
Such an array of tests should mean that platelet function disorders should be easy to diagnose. However, pre-analytical and analytical variables mean that results can often be unreliable and the results are often difficult to interpret.
Thrombin
Thrombin converts circulating fibrinogen to fibrin and activates factor XIII, which crosslinks the fibrin, forming a clot
It is currently believed that activation of blood coagulation is usually initiated by vascular injury, causing exposure of flowing blood to tissue factor, which results in activation of factors VII and IX. Subsequently, activation of factors X and II (prothrombin) occurs preferentially at sites of vascular injury, alongside activated platelets: the latter provide procoagulant activity as a result of exposure of negatively charged platelet surface membrane phospholipids, such as phosphatidylserine, and high affinity binding sites for several activated coagulation factors, allowing the formation of the prothrombinase complex (Va, Xa and II) and tenase complex (VIIIa, IXa and Xa), which both greatly enhance the production of thrombin. As a result of these biochemical interactions, thrombin and fibrin formation are efficiently localized at sites of vascular injury.
Thrombin has a central role in hemostasis
Not only does thrombin convert circulating fibrinogen to fibrin at sites of vascular injury, producing the secondary, fibrin-rich hemostatic plug, It also activates factor XIII, a transglutaminase, which crosslinks fibrin, rendering it resistant to dispersion by local blood pressure or by fibrinolysis (Figs 7.1 and 7.3). Furthermore, thrombin stimulates its own generation in a positive feedback cycle in three ways:
Thrombin inhibitors have been developed as anticoagulant drugs
Now that the central role of thrombin in hemostasis and thrombosis has been recognized, a number of direct thrombin inhibitors (DTIs) have been developed as anticoagulant drugs. Dabigatran, an oral direct thrombin inhibitor (DTI), has been demonstrated in large randomized controlled clinical trials to be as effective as warfarin in the treatment of acute venous thrombosis and in the prevention of stroke in patients with atrial fibrillation. The major advantage of this drug for these indications is that dose monitoring is not required. Recently it has obtained a licence in the UK for stroke prevention in atrial fibrillation. However, to date there is no effective antidote and for this reason, it is being introduced into clinical practice with caution.
Argatroban is another oral DTI which is an effective alterative to heparin when the latter is contraindicated following an episode of heparin-induced thrombocytopenia (HIT). It has recently been granted a licence in the UK for this indication. Bivalirudin, a derivative of hirudin, originally obtained from the medicinal leech, Hirudo medicinalis, is a parenteral DTI which has been shown to be effective for the treatment of acute coronary syndromes. It is also an alternative to heparin for patients with acute coronary syndromes who have HIT.
This central role of thrombin is also the rationale for the intensive research that is now being performed to refine the thrombin generation assay and apply it to both bleeding and thrombotic clinical pathologies.
Coagulation inhibitors are essential to prevent excessive thrombin formation and thrombosis
Three systems of naturally occurring coagulation inhibitors have been identified (Fig. 7.5 and Table 7.3).
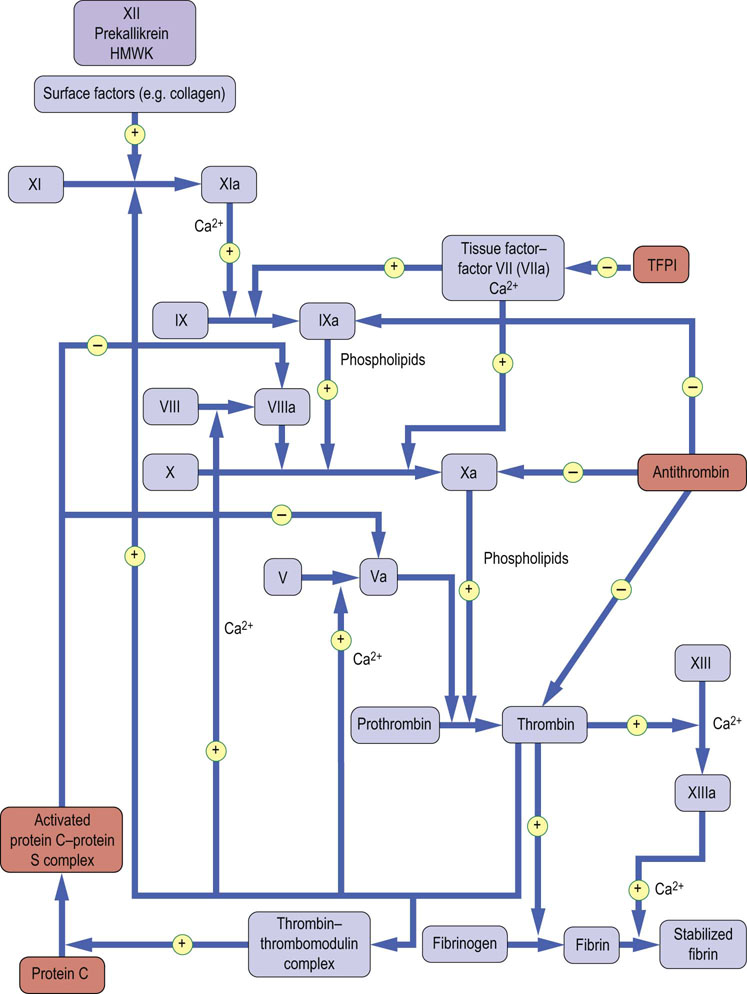
Fig. 7.5 Sites of action of blood coagulation inhibitors.
Antithrombin, protein C and protein S, and tissue factor pathway inhibitor (TFPI).
Antithrombin: this is a protein synthesized in the liver. Its activity is catalyzed by the antithrombotic drug heparin (unfractionated and low-molecular-weight heparins), and by heparin-like endogenous glycosaminoglycans (GAGs) that are present on the surface of vascular endothelial cells. It inactivates not only thrombin, but also factors IXa and Xa (Fig. 7.5). As a result, congenital antithrombin deficiency results in an increased risk of venous thromboembolism.
Heparins are referred to as indirect Xa inhibitors due to their augmentation of antithrombin activity. Heparins are used both in the treatment and prevention of acute venous thrombosis, usually in the form of a low-molecular-weight heparin (LMWH), e.g. enoxaparin or dalteparin. They are usually replaced by oral anticoagulants such as warfarin for longer-term anticoagulation. LMWHs also have a role in the management of acute arterial thrombosis as it pertains to acute coronary syndromes, e.g. enoxaparin or fondaparinux.
Direct Xa inhibitors have now been developed as anticoagulants. Rivaroxaban has been shown to be an effective alternative to warfarin for the treatment and prevention of venous thrombosis and for the prevention of stroke in patients with atrial fibrillation. Apixaban is licenced for the prevention of venous thrombosis in patients undergoing total knee or hip arthroplasties. Trials have also shown it to be effective in preventing thromboembolic stroke in patients with atrial fibrillation. Both medications are in an oral form and no monitoring is required of their anticoagulation effect. However, there is currently no effective antidote.
Protein C and its cofactor, protein S: these are vitamin K-dependent proteins, synthesized in the liver. When thrombin is generated, it binds to thrombomodulin (molecular weight 74 kDa), which is present on the surface of vascular endothelial cells. The thrombin–thrombomodulin complex activates protein C, which forms a complex with its cofactor, protein S. This complex selectively degrades factors Va and VIIIa by limited proteolysis (Fig. 7.5). Hence, this pathway forms a negative feedback upon thrombin generation. Congenital deficiencies of protein C or protein S result in an increased risk of venous thromboembolism. A further cause of increased risk of venous thromboembolism is a mutation in coagulation factor V (factor V Leiden), which confers resistance to its inactivation by activated protein C. This mutation is common, occurring in approximately 5% of the population in Western countries.
Tissue factor pathway inhibitor (TFPI): this protein is synthesized in endothelium and the liver; it circulates bound to lipoproteins. It inhibits the tissue factor–VIIa complex (Fig. 7.5). However, deficiency of TFPI does not appear to increase the risk of thrombosis.
Clinical boxA 40-year-old man with pain and leg swelling: antithrombin Deficiency
A 40-year-old man was admitted from the emergency room of his local hospital because of acute pain and swelling of his left leg 10 days after recent major surgery. Ultrasound imaging of the leg confirmed occlusion of the left femoral vein by thrombus.
He was prescribed anticoagulant therapy with low-molecular-weight heparin at standard doses. The patient volunteered a strong family history of ‘clots on the legs’ at a young age. He was commenced on warfarin and LMWH, the latter being discontinued when his INR was >2. He was followed up at the anticoagulation and specialist thrombophilia clinic for long-term management.
Fibrinolysis
Fibrinolytic system acts to limit excessive formation of fibrin through plasmin-mediated fibrinolysis
The coagulation system acts to form fibrin; the fibrinolytic system acts to limit excessive formation of fibrin (both intra- and extravascular) through plasmin-mediated fibrinolysis. Circulating plasminogen binds to fibrin via lysine-binding sites; it is converted to active plasmin by plasminogen activators. Tissue-type plasminogen activator (tPA) is synthesized by endothelial cells; it normally circulates in plasma in low basal concentrations (5 ng/mL), but is released into plasma by stimuli that include venous occlusion, exercise, and epinephrine. Together with plasminogen, it binds strongly to fibrin, which stimulates its activity (the Km for plasminogen decreases from 65 to 0.15 µmol/L in the presence of fibrin), thereby localizing plasmin activity to fibrin deposits.
Clinical test boxMeasurement of fibrin D-dimer in the diagnosis of suspected deep vein thrombosis
Fibrin D-dimer (a degradation product of crosslinked fibrin and a marker of fibrin turnover) is normally present in blood at concentrations of <250 µg/L. In deep vein thrombosis of the leg (DVT), deposition of a large mass of crosslinked fibrin within the leg veins, followed by partial lysis by the body's fibrinolytic system, increases fibrin turnover and blood D-dimer levels are elevated. Many patients attend accident and emergency departments with a swollen and/or painful leg, which may be due to a DVT.
Rapid immunoassays for blood D-dimer can be performed in the emergency department and are now widely used as an adjunct to clinical diagnosis. About one-third of patients with clinically suspected DVT have normal D-dimer levels, which in combination with a low clinical score usually excludes the diagnosis and may allow early discharge of such patients without the need for further investigation or treatment. In patients with raised D-dimer levels, heparin treatment is started and imaging of the leg performed (usually by ultrasound) to confirm the presence and extent of a DVT.
Clinical boxAntithrombotic treatment in acute coronary syndrome
Occlusion of a coronary artery by thrombus causes the features of acute coronary syndrome which include electrocardiographic and biochemical changes. Myocardial infarction refers to permanent death of that part of the heart muscle which is supplied by the artery. In acute coronary syndromes, including myocardial infarction, the patient typically experiences severe chest pain.
Aspirin and heparin are usually given in acute myocardial infarction and other acute coronary syndromes, to inhibit the platelet and fibrin components of the developing coronary artery thrombus. Some patients may require the addition of clopidogrel and/or GPIIb-IIIa inhibitors.
Many patients with evolving acute myocardial infarction are candidates for thrombolytic treatment with a plasminogen activator drug, given intravenously. Prompt thrombolysis dissolves the coronary artery thrombus, reduces the size of the infarct, and reduces the risk of complications, including death and heart failure. However, in recent years direct removal of the thrombus (percutaneous coronary intervention (PCI)) is given instead of thrombolytic therapy as it appears to give favorable outcomes to thrombolysis and does not increase the risk of bleeding, for example into the brain. Patients undergoing PCI should in addition receive a GPIIb-IIIa inhibitor.
Plasmin inhibitors prevent excessive fibrinolytic activity
Excessive tPA activity in plasma is normally prevented by an excess of its major inhibitor, plasminogen activator inhibitor type 1 (PAI-1), which is synthesized by both endothelial cells and hepatocytes. Urinary-type plasminogen activator (uPA) circulates in plasma both as an active single-chain precursor form (scuPA, pro-urokinase) and as a more active two-chain form (tcuPA, urokinase). One activator of scuPA is surface-activated coagulation factor XII, which therefore links the coagulation and fibrinolytic systems. The major components of the fibrinolytic system are illustrated in Table 7.4 and Figure 7.6. Excessive formation of plasmin is normally prevented by:
Table 7.4
The components of fibrinolytic system
| Component (synonym) | Molecular weight (Da) | Plasma concentration (mg/dL) |
| Plasminogen | 92,000 | 0.2 |
| Tissue-type plasminogen activator, tPA | 65,000 | 5 (basal) |
| Urinary-type plasminogen activator type 1, uPA | 54,000 | 20 |
| Plasminogen activator inhibitor type 1, PAI-1 | 48,000 | 200 |
| Antiplasmin (α2-antiplasmin) | 70,000 | 700 |

Fig. 7.6 The fibrinolytic system.
Plasminogen can be activated to plasmin by uPA (urokinase), tPA or streptokinase. uPA and tPA are inhibited by plasminogen activator inhibitor type 1 (PAI-1). Plasmin is inhibited by antiplasmin. Plasmin degrades fibrin to fibrin degradation products (FDPs). HMWK, high-molecular-weight kininogen; scuPA, pro-urokinase; tcuPA, two-chain urokinase; tPA, tissue-type plasminogen activator. Reproduced from Dominiczak MH. Medical Biochemistry Flash Cards. London: Elsevier, 2012.
binding of 50% of plasminogen to histidine-rich glycoprotein (HRG), and
rapid inactivation of free plasmin by its major inhibitor, α2-antiplasmin.
The physiologic importance of PAI-1 and α2-antiplasmin is illustrated by the increased bleeding tendency that is associated with the rare cases of their congenital deficiencies (Table 7.1); the excessive plasma plasmin activity that results from these deficiencies has the effect of lysing hemostatic plugs.
Summary
Hemostasis constitutes a number of processes which guard the body against blood loss.
Injury to the blood vessel wall sets in motion complex phenomena which involve blood platelets (activation, adhesion, aggregation) and a cascade of coagulation factors, classified into intrinsic, extrinsic and final common pathways.
The integrity of these three systems may be tested by simple laboratory tests. Global coagulation assays such as thrombin generation and thromboelastography, currently used in research, may be more effective in assessing an individual's coagulation phenotype.
Deficiencies of factors participating in the coagulation cascade, and/or disordered platelet function, result in bleeding disorders.
Eventually, blood clots are degraded by the fibrinolytic system. The process of fibrinolysis prevents thrombotic phenomena and there is normally a balance between hemostasis and thrombosis.
Aspirin and heparin are used in patients with acute myocardial infarction or other acute coronary syndromes.
Aspirin (or other antiplatelet agents) are also used to reduce risk of recurrent myocardial infarction and stroke.
Anticoagulant drugs, e.g. heparin, warfarin, or rivaroxaban, are used in the treatment of acute venous thrombosis or embolism.
Anticoagulant drugs, e.g. warfarin, dabigatran and rivaroxaban, are used long term to prevent thromboembolism arising from the heart (atrial fibrillation, heart valve prostheses).
Active learning
1. When a patient presents with excessive bleeding from multiple sites, what laboratory tests are available to identify the likely cause of their hemostatic defect?
2. When a patient presents with a painful swollen leg, possibly due to acute deep venous thrombosis (DVT), what laboratory tests can be performed to help the clinician to:
Establish or exclude this diagnosis?
Monitor anticoagulant treatment, after the diagnosis has been confirmed?
3. When a patient presents with acute coronary artery thrombosis (evolving to myocardial infarction), what antithrombotic drugs should be urgently considered to reduce the risk of complications?
Anaesthesia, UK. Tests of coagulation. www.frca.co.uk/article.aspx?articleid=100101.
International Society on Thrombosis and Haemostasis. www.isth.org.
Walker HK, Hall WD, Hurst JW, eds. Clinical Methods: The History, Physical, and Laboratory Examinations. ed 3. Butterworths, Boston, 1990. www.ncbi.nlm.nih.gov/books/NBK265
Wright, IS, The nomenclature of blood clotting factors. Can Med Assoc J 1962; 86:373–37. www.ncbi.nlm.nih.gov/pmc/articles/PMC1848865/?page=1