Lipid Metabolism I: Metabolism Of Fatty Acids And Compound Lipids
Fatty acids are major sources of energy in humans. They are used as fuel by most tissues except red and white blood cells, nervous tissue, retina and adrenal medulla. They are stored as triacylglycerols (TAGs) in adipose tissue. When the need for energy arises, the fatty acids are mobilized from the TAG stores and released in circulation. They are transported to various parts of the body after being complexed with albumin; each molecule of albumin can bind 6 to 8 fatty acid molecules. Subsequently they are taken up by peripheral tissues where they are oxidized to release energy. The principal oxidative pathway is β-oxidation, though the other pathways (such as α-oxidation and ω-oxidation) play a supplementary role. The de novo synthesis of fatty acids (i.e. lipogenesis) occurs from acetyl CoA with the help of fatty acid synthase complex.
In this chapter, various aspects of metabolism of fatty acids and the compound lipids (e.g. triacylglycerol, phospholip-ids, and sphingolipids), particularly in adipose tissue and liver, have been described. After going through this chapter the student should be able to understand:
• The oxidative pathways (e.g. β-, α-, ω-, or peroxisomal oxidation) for degradation of fatty acids and catabolism of unsaturated and odd chain fatty acids.
• Mechanism of fatty acids synthesis; fatty acid synthase system; and desaturase and chain elongation systems.
• Metabolic processes occurring within adipocytes; mechanism of mobilization of depot fat from adipose tissue and the factors influencing it, particularly the role of hormones.
• Pathways of biosynthesis and catabolism of compound lipids such as phosphoglycerides, triacylglycerols and sphingolipids.
I β-Oxidation
β-Oxidation is the principal pathway for catabolism of fatty acid. The scheme of β-oxidation, whereby fatty acids are degraded by successive loss of two-carbon units, was first elucidated by a German scientist Fray Knoop in 1904. It consists of repeated cycles of a series of reactions. With each cycle, a two-carbon unit (i.e. acetyl CoA molecule) is removed from the carboxyl terminal of the fatty acid. Thus complete oxidation of the 16-carbon fatty acid (e.g. palmitic acid) requires seven such cycles and generates eight molecules of acetyl CoA.
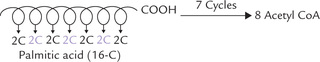
The pathway occurs in three stages:
 Oxidation of fatty acids occurs in the mitochondrial matrix of most cells by the pathway of β-oxidation. There occurs sequential removal of two-carbon acetyl CoA units from the end of the acyl chain.
Oxidation of fatty acids occurs in the mitochondrial matrix of most cells by the pathway of β-oxidation. There occurs sequential removal of two-carbon acetyl CoA units from the end of the acyl chain.
A Activation of Fatty Acid
As the priming step for catabolism, the fatty acids are activated through formation of a thioester linkage between a fatty acid molecule and coenzyme A. The product is an acyl coenzyme A, and therefore the name of the enzyme that catalyzes this reaction is acyl CoA synthetase, also called thiokinase. This reaction requires a great deal of energy and in the process ATP is converted to AMP
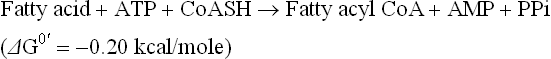
This was the first reaction in biochemistry found to yield pyrophosphate by cleavage of ATP. The reaction favours the formation of fatty acyl CoA, since the pyro-phosphate formed is hydrolyzed by the enzyme, pyrophosphatase: PPi 2→Pi. A large drop of free energy (-6.9 kcal/mol) accompanies the pyrophosphate cleavage, which ensures irreversibility of the overall reaction. Thus activation of fatty acid to fatty acyl CoA with an energy-rich thioester bond requires expenditure of two high-energy phosphate bonds.
The fatty acid is activated by forming a thioester link with coenzyme A before entering the mitochondria. The reaction uses a molecule of ATP, and is irreversible due to subsequent hydrolysis of PPi to two molecules of Pi.
The activation reaction actually takes place in three steps:
1. The carboxyl group is first activated to an enzymebound, high-energy, acyl-adenylate intermediate (fatty acyl-AMP).
2. The acyl group then reacts with coenzyme-A to give fatty acyl CoA.
3. The pyrophosphate is now hydrolyzed to ensures irreversibility of the overall reaction.
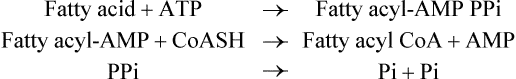
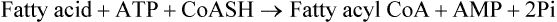
Types of Thiokinase Enzymes
At least four different thiokinase enzymes, one each for short chain, medium chain, long chain fatty acids, and one for arachidonate, have been identified (Table 11.1 ). The chemistry and bioenergetics of each of the enzyme-catalyzed reactions for the biosynthesis of variable length acyl CoA molecules are the same.
B Transport of Activated Fatty Acid into Mitochondria
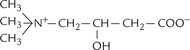
Formation of fatty acyl CoA takes place in the cytosol. Further degradation occurs in the mitochondrial matrix. This creates a problem because the inner mitochondrial membrane (IMM) is impermeable to fatty acyl CoA. To overcome this problem, participation of a carrier, carnitine (Fig. 11.1 ) is required. It is beta-hydroxyl-gamma-trimethyl ammonium butyrate, synthesized from lysine and methionine in liver and kidney.
The transport process, referred to as carnitine shuttle, comprises three steps:
Step 1: The acyl group of fatty acyl CoA is transferred to carnitine, resulting in formation of fatty acyl carnitine. The reaction is catalyzed by the rate-limiting enzyme of the pathway, acyl CoA carnitine transferase (E1), which is located on outer surface of the inner mitochondrial membrane.
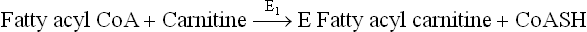
Step 2: Fatty acyl carnitine is translocated across the inner mitochondrial membrane into the mitochondrial matrix. Entry of the acyl-carnitine is linked to the exit of carnitine, both mediated by a translocase, called carnitine/acylcarnitine translocase (E2) (Fig. 11.2 ).
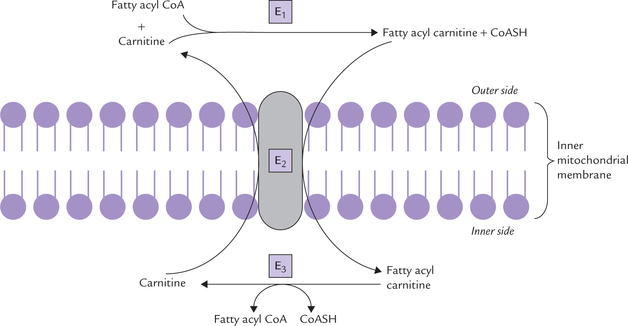
Fig. 11.2 Transport of fatty acids (16 ± 4 carbons) from cytosol into the mitochondrion (E1 = acyl CoA-carnitine transferase I, E2 = translocase, E3 = acyl CoA carnitine transferase II).
Step 3: Once inside the mitochondrial matrix, the fatty acyl carnitine is reconverted to fatty acyl CoA by the enzyme acyl CoA carnitine transferase II (E3). The enzyme is located on the inner surface of IMM.
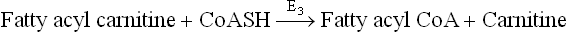
The inner mitochondrial membrane is not permeable to long chain fatty acids and so these are transported into the mitochondria as carnitine derivatives by carnitine/ acylcarnitine translocase.
Since carnitine is an essential component of the transport of fatty acyl CoA, its deficiency leads to decreased entry of fatty acids into mitochondrial matrix. This results in impaired fatty acid utilization, specially in muscles (Case 11.1).
The carnitine shuttle is primarily for the transport of the long chain fatty acids (16 ± 4 carbons).
• Short and medium-chain fatty acids can cross the mitochondrial membrane by passive diffusion, and are activated to their CoA derivative within the mitochondrion.
• Very long-chain fatty acids are shortened to long-chain fatty acids in peroxisomes and then transported by carnitine shuttle into the mitochondrion.
C Standard β-Oxidation Process of Activated Fatty Acid
The principal fate of fatty acyl CoA in the mitochondrion is β-oxidation.
Reactions of β-Oxidation
The standard β-oxidation process comprises a series of identical cycles. Each cycle shortens the fatty acid by two carbons and produces an acetyl residue in the form of acetyl CoA. Repeated cycles of β-oxidation generate several acetyl CoA molecules which enter TCA cycle and are further degraded to carbon dioxide and water. The reaction sequence is called β-oxidation because it is the β-carbon (C-3) that is oxidized.
In each cycle of β-oxidation, fatty acyl CoA is acted upon in a sequential manner by four enzymes—FAD-linked dehydrogenase, hydratase, NAD+-linked dehydrogenase, and thiolase. These reactions (Fig. 11.3 ) proceed as follows:
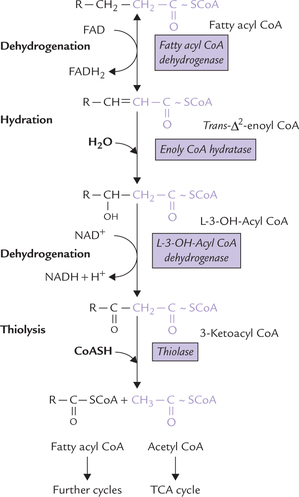
Fig. 11.3 Reactions of β-oxidation: dehydrogenation, hydra-tion, dehydrogenation and thiolysis. These four reactions form one ’round’ of degradation and their overall effect is to remove a 2-C (acetyl CoA) unit.
1. First dehydrogenation: A pair of hydrogen atoms is removed across the α- and β-carbons of the fatty acyl CoA by action of fatty acyl CoA dehydrogenase resulting in the formation of trans-Δ2-enoyl CoA. Four distinct dehydrogenases are known to exist, each one is specific for a given range of fatty acid chain length. All four are flavoproteins and contain a tightly bound molecule of flavin adenine dinucleotide (FAD).
2. Hydration: Addition of a water molecule converts trans-Δ2-enoyl CoA to L-3-OH acyl CoA. The reaction is catalyzed by enoyl CoA hydratase. In fact there are two hydratases: one preferring short chain fatty acids and the second long chain. Both are highly specific and act only on the trans isomer (i.e. trans-Δ2-enoyl CoA).
3. Second dehydrogenation: The L-3-OH-acyl CoA then loses a pair of hydrogen atoms to NAD+. The reaction is catalyzed by the enzyme L-3-OH-acyl CoA dehydro-genase. Two hydrogen atoms are removed from C-3 so that the hydroxyl group at that position is converted to a keto group. The reaction product is accordingly called 3-ketoacyl CoA.
Thus, by a sequential action of three enzymes, a keto group is introduced at C-3 position of the fatty acyl CoA molecule (compare structure of fatty acyl CoA with that of 3-ketoacyl CoA; Figure 11.3).
4. Thiolysis: Finally, a (thiolytic) cleavage occurs at the β-carbon atom of the 3-ketoacyl CoA, liberating an acetyl CoA molecule. The enzyme catalyzing this step is thiolase.
The active fatty acids (acyl CoA) are transported into mitochondrion by a carrier, carnitine, where they are oxidized at the β-carbon β-oxidation) by oxidation (removal of hydrogen) of the acyl group, followed by an hydration, and a second oxidation (removal of hydrogen) at the β-carbon, followed by thiolysis (cleavage).
Repeating Sequence of Reactions
In the reaction sequence discussed above, one NADH and one FADH2 are produced. At the end of the one cycle, the fatty acyl CoA is shortened by two carbons. The shortened fatty acyl CoA then undergoes a second cycle to liberate another acetyl CoA. These cycles are continued till whole of the acyl chain is degraded to acetyl CoA molecules. In case of palmitic acid there are seven such cycles, and the overall reaction can be represented by the following equation:
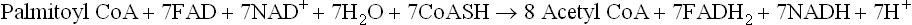
D Energy Production from β-Oxidation
For each round of degradation, one FADH2, one NADH and one acetyl CoA molecule are produced.
Energy is generated by oxidation of these products of β-oxidation: 2ATPs are produced by oxidation of FADH2; 3ATPs by NADH and 12 ATPs by degradation of acetyl CoA (through TCA cycle). Therefore, when a 16-C palmitic acid is fully oxidized a total of 131 ATPs (8 × 12 + 7 × 2 + 7 × 3) are generated:
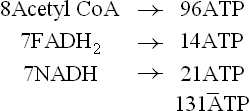
2 ATPs (i.e. two phosphodiester bonds) are used during initial activation of the fatty acid. Hence, net gain in terms of ATP molecules is 131-2, i.e. 129 ATPs.
Each cycle of β-oxidation produces an acetyl CoA and an acyl CoA with two carbons less, as also one FADH2 and NADH each. The cycles are repeated until complete degradation occurs. Complete oxidation of one mole of palmitate generates 129 ATPs.
Regulation of β-oxidation
The regulation occurs at the following two levels:
Supply of Fatty Acids
The β-oxidation is primarily regulated by the supply of fatty acids. Within rather wide limits, the use of fatty acids by tissues is proportional to the plasma-free fatty acid level, and therefore fatty acid oxidation is for the most part regulated at the level of adipose tissue metabolism. During fasting and uncontrolled diabetes, for example, a large amount of free fatty acids are released from adipose tissue (Chapter 12) and β-oxidized to acetyl CoA.
Mitochondrial Uptake of Fatty Acids
An additional regulatory step is the uptake of fatty acids into the mitochondrion. The enzyme, acyl CoA carnitine transferase I, which regulates entry of fatty acids into mitochondrial matrix, is allosterically inhibited by malonyl-CoA. This is important because malonyl-CoA is a substrate for fatty acid synthesis and its cytoplasmic concentration is high whenever fatty acid synthesis is stimulated. It would be futile to have fatty acid synthesis occurring at the same time as fatty acid degradation, and hence, a substrate for fatty acid synthesis inhibits fatty acid degradation (in this case by preventing the acyl group being transferred into the mitochondrial matrix).
β-Oxidation is regulated by the supply of fatty acids, and by the enzyme which regulates entry of these fatty acids into mitochondrial matrix.
Note
Non-esterified fatty acids in plasma are referred to as free fatty acids, but it should be noted that they are neither free (99% bound to albumin) nor acids (at pH 7.4 present as anions). Plasma concentration ranges from 0.2 to 0.6 mM depending on nutritional status, but may rise as high as 2.0 mM in severe stress.
E Defects of β-oxidation
The tissues such as muscle and liver, which derive most of their energy from fatty acids, at least during fasting are compromised by defects of β-oxidation. Among these common defects are discussed below:
Deficiency of Enzymes of β-Oxidation
Several defects of enzymes within the β-oxidation are known, the most common of which is medium-chain acyl-CoA dehydrogenase deficiency. The affected enzyme catalyzes the first dehydrogenation reaction of the β-oxidation of medium-chain (C4 to C 12) fatty acids. This defect presents with episodic non-ketotic hypoglycaemia initiated by fasting during the first 2 years of life. Many of such patients initially diagnosed with “Rey’s syndrome” or “sudden infant death syndrome” later turned out to suffer from medium chain acyl-CoA dehydrogenase deficiency. The hypoglycaemia is often lethal and at autopsy the patients who die of this disease show a typical fatty infiltration of the liver. These patients should be advised to avoid excessive fasting and recommended a low fat diet.
Deficiency of Translocase
A few cases of translocase deficiency have been reported, where the transport of long chain fatty acids across the mitochondrial membrane is impaired. Muscle cramps, precipitated by fasting, exercise and high fat diet, is the predominant clinical feature.
Carnitine Deficiency
Defects in carnitine biosynthesis, defective transport into the cells, or excessive renal excretion may result in deficiency of this carrier protein, which most commonly affects liver and muscles. When liver is affected, hypoke-totic hypoglycaemia during periods of extended fasting is the most obvious consequence. Muscle weakness and muscle cramps on exertion are the most typical complaints when muscle suffers in carnitine deficiency (Case 11.1). Histologically, many patients show an unusual abundance of fat droplets in muscle tissue and liver, and some develop fatty degeneration of the liver. This is to be expected, because excess fatty acyl CoA that cannot be transported into the mitochondrion is diverted into tri-glyceride synthesis.
II Other Oxidative Pathways
Mitochondrial β-oxidation can smoothly oxidize unbranched saturated fatty acids with an even number of carbons and a chain length up to 18 or 20 carbons. Fatty acids that do not fit this description require additional enzymatic reactions:
• Methylated fatty acids require α-oxidation initially, before they can be beta-oxidized.
• Very long chain fatty acids (VLCFA) are handled initially by peroxisomal oxidation pathway.
• Medium chain fatty acids are degraded by ω-oxidation.
• Unsaturated and odd chain fatty acids also require additional processing before they are beta-oxidized.
Oxidation of some fatty acids (e.g. unsaturated, branched, odd chain) requires specialized reactions.
A α-Oxidation of Fatty Acids
α-Oxidation is a minor pathway occurring in the endo-plasmic reticulum and mitochondria. It removes one carbon unit at a time from the carboxyl end. Unlike β-oxidation, the function of this pathway is not the production of any high-energy phosphate bonds, but rather the degradation of methylated fatty acids.
A methylated fatty acid cannot be β-oxidized without prior modification. Its degradation is initiated by oxidation of the carbon-2 (the α-carbon) and then release of carbon-1 as CO2, thus shortening the fatty acid by one carbon at a time. The shortened fatty acid then undergoes a round of β-oxidation yielding propionyl CoA rather than acetyl CoA (Fig. 11.4 ).
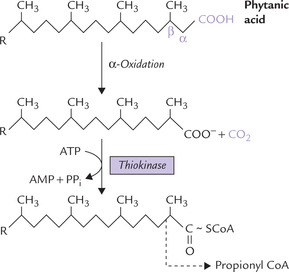
Fig. 11.4 Degradation of a methylated acid (phytanic acid) is initiated by α-oxidation, followed by a round of β-oxidation, which yields propionyl CoA rather than acetyl CoA.
Refsum’s Disease
It is a rare recessively inherited defect of α-oxidation. The disease is also called phytanic acid storage disease because the patients accumulate excessive quantities of phytanic acid (Fig. 11.4) in their body lipids. This unusual fatty acid is derived from the alcohol phytol, an isoprenoid, that occurs as a constituent of chlorophyll and in milk.
The β-carbon of phytanic acid is methylated, which blocks β-oxidation (Fig. 11.4). The phytanic acid levels in blood increase, reaching up to 100 mg/dl (normal value is around 0.3 mg/dl). Phytanic acid also accumulates in liver, sometimes to such an extent that nearly 50% of the total fatty acids in the liver is phytanic acid. Clinical features include abnormalities of the nervous system like cerebellar ataxia, motor weakness and distal sensory loss. Such abnormal neurological features probably result due to incorporation of the branched chain fatty acids in the membrane lipids. This increases fluidity of the neuronal membranes, which affects nerve conduction. The patients respond to dietary restriction of green vegetables and of ruminant milk and meat, which contain large amounts of phytanic acid.
α-Oxidation oxidizes carbon 2 (the α-carbon) and then releases carbon-1 as CO2, thereby shortening the fatty acid by one-carbon at a time. It is used for oxidation of methylated fatty acids, such as phytanic acid. The latter cannot be β-oxidized without prior modification because the β-carbon is methylated.
B Fatty Acid Oxidation in Peroxisomes
The peroxisomal β-oxidation handles very long chain fatty acids (VLCFA), of chain length > 20 carbons, which are poor substrates for mitochondrial β-oxidation. It uses essentially the same reaction sequence as the mitochondrial system, except for the very first reaction, which is catalyzed by an H2O2-producing flavoprotein oxidase (it is FAD-dehydrogenase in mitochondria).
The flavoprotein oxidase acts on the CoA derivatives of VLCFA, after the latter crosses the peroxisomal membrane (without involvement of carnitine). It catalyzes removal of a pair of hydrogen atoms from the hydrocarbon chain of the fatty acid molecule and transfers it directly to oxygen to form hydrogen peroxide. Removal of hydrogen atoms introduces a double bond in the hydrocarbon chain to form trans-Δ2-enoyl CoA. The latter is an intermediate of β-oxidation. It undergoes similar sequence of reactions as in mitochondrial β-oxidation to liberate an acetyl CoA molecule, leaving behind a fatty acyl CoA molecule shortened by two carbons. In this manner, action of the flavoprotein oxidase introduces a shortcut, whereby the substrate fatty acid directly enters the standard β-oxidation cycle.
The above cycle is repeated several times, each time an acetyl CoA is removed, the fatty acid chain is shortened by two carbon atoms. This cycle proceeds only to the stage of octanoyl-CoA (8-carbon); the latter moves to the cytosol and then into the mitochondrial matrix with the help of specific membrane transport proteins, where it undergoes further degradation by the standard β-oxidation process. Thus it may be said that main purpose of peroxisomal β-oxidation is to shorten very long chain fatty acids, thereby boosting β-oxidation.

High fat diet and the hypolipidaemic drugs like clofi-brate cause a marked increase in the number of peroxi-somes in a cell in order to increase efficiency of fatty acid utilization.
Zellweger syndrome
It is an inborn error of VLCFA oxidation because of absence of peroxisomes. Substrate accumulation in tissues, particularly brain, liver and kidneys, and excretion in urine (shown by chromatography) and non-ketotic hypo-glycaemia are the predominant features of this defect.
C Omega (ω)-Oxidation of Fatty Acids
Omega oxidation is a microsomal system for oxidation of the carbon atom most remote from the carboxyl group in a fatty acid to produce a dicarboxylic acid. The basic reaction, catalyzed by a monooxygenase that requires NADPH, O2, and cytochrome P-450, occurs in smooth endoplasmic retic-ulum. First, methyl group at the ω-carbon is converted to CH2OH and subsequently oxidized with the help of NAD+ to a COOH group to produce a dicarboxylic acid. The latter can be activated at either end, followed by β-oxidation (Fig. 11.5 ).
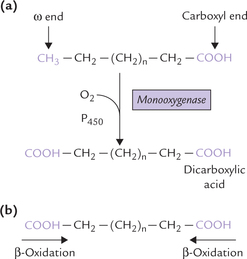
Fig. 11.5 The ω-oxidation of medium chain fatty acid. (a) The last carbon (ω) of a fatty acid is oxidized to a carboxyl group to produce a dicarboxylic acid. (b) The dicarboxylic acid is activated at either end, followed by β-oxidation.
Medium chain fatty acids mobilized from the adipocyte triacylglycerol stores are oxidized in liver by ω-oxidation. Moreover, ω-oxidation becomes important when (oxidation is defective. Dicarboxylic acids (C-6 and C-8 acids) are excreted in urine under these circumstances and the condition is termed as dicarboxylic aciduria.
D Oxidation of Odd Chain Fatty Acids
Even chain fatty acids, by far the most abundant fatty acids in human organism, are β-oxidized to acetyl CoA. Odd chain fatty acids are also β-oxidized normally but the last step produces a 3-carbon propionyl CoA instead of acetyl CoA. If C-17 fatty acid, for example, were cata-bolized, 7 acetyl CoA molecules and one propionyl CoA molecule would result after seven cycles of the β-oxidation process (Fig. 11.6 ).
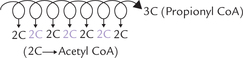
Fig. 11.6 Odd chain fatty acid produces several acetyl CoA and a single propionyl CoA (3-C) molecule in the final round of fatty acid degradation.
Propionyl CoA is catabolized to succinyl CoA, a Krebs cycle intermediate via methylmalonyl-CoA (Fig. 11.7 ). In this series of reactions, bicarbonate is incorporated into propionate by a biotin-dependent enzyme propionyl CoA carboxylase to yield D-methylmalonyl CoA. The latter undergoes intramolecular rearrangements in presence of a racemase to give L-methylmalonyl CoA, which is then converted to succinyl-CoA in the presence of a vitamin B12-containing mutase. The last step involves migration of the bulky COSCoA moiety to the methyl carbon.
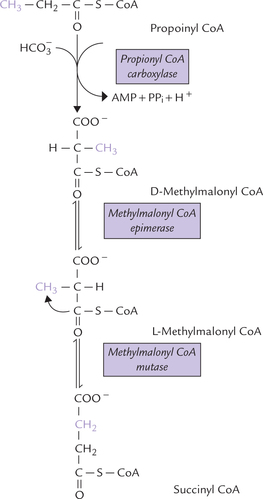
Odd chain fatty acids can therefore give net biosynthesis of glucose through propionyl CoA. Thus, three carbon units from odd chain fatty acids (comprising ω-1 to ω-3 carbons) are glucogenic.
Inborn Errors of Propionate Metabolism
Propionic Acidaemia
It results when propionyl-CoA carboxylase is deficient because of a genetic lesion. It is characterized by developmental abnormalities and ketoacidosis.
Methylmalonic Aciduria
This results when the B12-dependent methylmalonyl CoA mutase reaction is blocked. Methylmalonic acid accumulates in the blood and is excreted in urine. Methylmalonic aciduria can be caused by (a) inherited defect in the mutase enzyme, or (b) inherited defect in cobalamin (B12) metabolism. In the latter condition, there is inability to convert dietary B12 to the appropriate coenzyme form, i.e. adenosylcobalamin. (Lack of vitamin B12 from dietary sources may also lead to this condition.)
The inherited forms of methylmalonic aciduria are severe maladies (overall frequency 1 in 10,000), with life threatening acidosis in affected infants and children. The patients suffering from inherited defect in cobala-min metabolism respond to injected adenosylcobalamin (Case 13.3), whereas those with inherited defect in the mutase enzyme do not. In either case accumulation of methylmalonic acid causes severe metabolic acidosis, CNS damage and growth retardation. It is desirable to restrict intake of odd chain fatty acids. Restriction of dietary valine, isoleucine, threonine and methionine is also required because propionyl CoA is derived from the metabolism of these amino acids also (Chapter 13).
Note
The disorders of metabolism of fatty acids, such as methyl malonic aciduria, propionic acidaemia, medium chain acyl CoA dehydrogenase deficiency and flavoprotein oxidase deficiency are collectively referred to as organic acidurias. The accumulation of organic acids in body tissues and their excretion in urine characterize them all. Their collective incidence is about one in 3000 live births.
E Oxidation of Unsaturated Fatty Acids
Oxidation of unsaturated fatty acids initially proceeds by the β-oxidation process in the same way as described for saturated fatty acids. It begins at the carboxyl terminal, and successive removal of two carbon units occurs with each cycle of β-oxidation. But when a double bond is reached, the cycle cannot proceed any further because the intermediate formed at this stage is not a natural substrate for the enzymes of β-oxidation. Participation of additional enzymes is required at this stage. These enzymes convert the above mentioned intermediate to a natural intermediate of β-oxidation.
Monounsaturated Fatty Acids
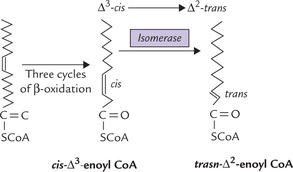
The monounsaturated fatty acids, for example, oleic acid (cis-Δ9-octadecanoic acid), has 18 carbon atoms with a cis double bond in position 9. After three β-oxidation cycles, the chain is shortened by six carbons, and now the oxidative mechanism encounters cis-Δ3-enoyl CoA.
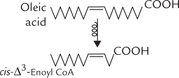
This compound is not a natural substrate for β-oxidation because there is a double bond between C-3 and C-4 in cis configuration. For β-oxidation to proceed, we need a double bond in trans-Δ2-configuration in enoyl CoA (Fig. 11.3). This problem is circumvented by the action of an isomerase which converts the cis-Δ3 bond into trans-Δ2 bond. As a result cis-Δ3-enoyl CoA is converted to trans-Δ2-enoyl CoA (Fig. 11.8 ). The latter being a normal substrate for β-oxidation, the reaction cycle can now proceed further.
Polyunsaturated Fatty Acids (PUFA)
The enzymatic difficulty in linolenic acid (cis cis 9, 12 octadecanoic acid) arises due to presence of two cis double bonds at position 9 and 12. The initial reaction sequence take place as in case of monosaturated fatty acid (Reactions I-III; Fig. 11.9 ). The oxidative process moves past the cis-Δ9 double bond through action of the same isomerase, which convert the cis-Δ3 bond into trans-Δ2 bond, leaving behind Δ4-cis -enoyl CoA. This compound, needs additional processing before it can be completely degraded by β-oxidation, as outlined below:
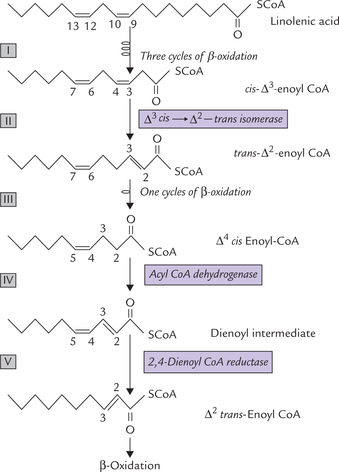
(a) Δ4-cis enoyl CoA is acted upon by an acyl CoA dehydrogenase which introduces a double bond at the C-2 (Reaction IV). This produces a 2,4-dienoyl intermediate (Δ2-trans-Δ4-cis-dienoyl CoA).
(b) The 2,4-dienoyl intermediate is a poor substrate for enoyl CoA hydratase, the next enzyme of the standard β-oxidation process. This difficulty is solved by an NADPH-dependent enzyme dienoyl CoA reductase (Reaction V), which reduces the double bond at C-4 to produce Δ2-trans-enoyl CoA.
(c) The A2-trans enoyl CoA is a normal substrate for the enoyl CoA hydratase, and hence for β-oxidation.
Unsaturated fatty acids require additional processing before they can be degraded by β-oxidation. Monoun-saturated fatty acids (e.g. oleic acid) requires an isomer-ase, and polyunsaturated fatty acids (e.g. linoleic acid) requires additional actions of an isomerase, a dehydroge-nase and a reductase.
III Ketone Body Production and Metabolism
Ketone bodies is a term that refers to the three biosyntheti-cally related compounds acetoacetate, β-hydroxybutyrate and acetone. They are produced in the liver from acetyl CoA substrate and are carried to extrahepatic tissues where acetoacetate and β-hydroxybutyrate are metabolized to produce energy. Being energy rich they serve as concentrated packets of metabolic energy. Acetone is however, an exception, since it cannot be metabolized and is readily exhaled. As diagrammed Figure 11.10 acetoacetate and acetone are true ketones, while β-hydroxybutyrate (or 3-hydroxybutyrate) does not posses a keto group.
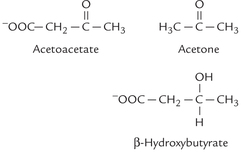
Fig. 11.10 Structures of ketone bodies. The ketone group (C = O) is present in acetoacetate and acetone, but not in β-hydroxybutyrate.
Because normally ketone bodies are produced in liver in such amount that can be rapidly extracted from circulation by various organs, their plasma level remains relatively low: 0.3 mM (1 mg/dl) and only traces are excreted in urine (daily excretion less than 1 mg). However, they are synthesized in inordinate amounts in some conditions (e.g. starvation and diabetes mellitus), which exceeds the ability of extrahepatic tissues to utilize them. Consequently, they get accumulated in blood leading to ketonaemia and excretion in urine (ketonuria). Ketonaemia and ketonuria together constitute a condition called ketosis.
In starvation and diabetes mellitus, the liver converts excess fatty acids to ketone bodies: acetoacetate, β-hydroxybutyrate and acetone, which are released into the blood and oxidized in extrahepatic tissues.
A Ketone Body Synthesis
Ketone bodies are synthesized exclusively in liver mitochondria from acetyl CoA or acetoacetyl CoA; the process is called ketogenesis (Fig. 11.11 ). Acetyl CoA is obtained by oxidation of pyruvate, fatty acids or the ketogenic amino acids (lysine, leucine, tyrosine, phenylalanine, etc.). The following steps are involved in ketogenesis:
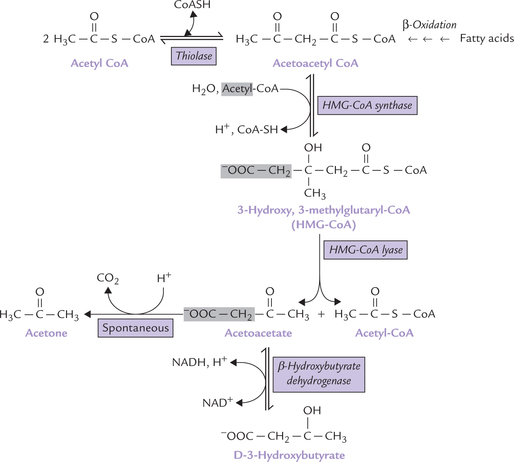
Fig. 11.11 Synthesis of ketone bodies in liver mitochondria. Only acetoacetate and β-hydroxybutyrate are present in plasma; the small amount of acetone normally formed by the non-enzymatic decarboxylation of acetoacetate is exhaled through lungs.
1. Acetoacetyl CoA is produced by condensation of two molecules of acetyl CoA. The condensation of acetyl CoA molecules is catalyzed by thiolase, the enzyme involved in final step of β-oxidation (where it cleaves acetoacetyl CoA). When, however, the concentration of acetyl CoA is high, the reaction occurs in the reverse direction, producing acetoacetyl CoA (Fig. 11.11).
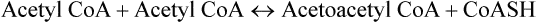
2. Acetoacetyl CoA combines with another acetyl CoA molecule to produce HMG-CoA (3-hydroxy-3-methylglutaryl CoA). The enzyme catalyzing this step, HMG CoA synthase, is rate-limiting for the pathway; it regulates the synthesis of ketone bodies.
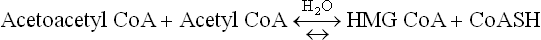
3. HMG CoA undergoes an aldose-lyase reaction, catalyzed by HMG CoA lyase. The cleaved products are acetoacetate and acetyl CoA.
4. Acetoacetate is reduced by NADH to form β-hydroxybutyrate by the enzyme β-hydroxybutyrate dehydrogenase, and a small fraction undergoes spontaneous decarboxylation to form acetone.
Note
Oxidation of some ketogenic amino acids directly yields acetoacetate, a ketone body (Chapter 13). Acetoacetyl CoA, an important intermediate in ketogenesis can be directly formed by β-oxidation of fatty acids.
B Ketone Body Oxidation
The two main ketone bodies, acetoacetate and β-hydroxybutyrate, are water soluble. They are readily transported from the liver to various tissues, which use them as important sources of energy, e.g. skeletal muscles, cardiac muscle, renal cortex, etc. Within mitochondria of these tissues, the ketone bodies are metabolized in following steps:
• First, oxidation of β-hydroxybutyrate produces acetoacetate, catalyzed by β-hydroxybutyrate dehydrogenase by reversal of the synthetic reaction (Fig. 11.12 ).
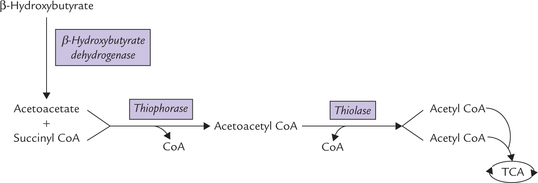
Fig. 11.12 Oxidation of ketone bodies: acetoacetate and β-hydroxybutyrate. Acetone cannot be thus oxidized and most is exhaled through lungs.
• Acetoacetate is activated to acetoacetyl CoA via the mitochondrial thiophorase. Donor of the coenzyme group in this reaction is succinyl CoA.
• Acetoacetyl CoA is cleaved by thiolase to yield two acetyl CoA molecules, which are oxidized in TCA cycle.
Acetone, however cannot be metabolized and most is exhaled through lungs.
C Excessive Production of Ketone Bodies
Normally, rates of ketone body synthesis and oxidation are balanced so that their serum concentration is maintained around 1 mg/dl and urinary excretion is negligible (1 mg/24 hours). When, however, rate of ketogenesis exceeds their utilization, their concentration in blood rises, and the condition is known as ketonaemia. Excess ketone bodies are eliminated in urine, this is known as ketonuria. Ketonaemia and ketonuria, as mentioned earlier, present an overall picture of ketosis, a risky and potentially lethal condition.
Any substrate, including glucose, that is degraded to acetyl CoA in the liver can be converted to ketone bodies, But ketogenesis is usually associated with excessive fatty acid oxidation, which occurs most notably in starvation and diabetes mellitus: 3-day-fast produces ketone bodies amounting to plasma level of 2-3 mM and in uncontrolled diabetes this may rise to 25 mM.
Starvation
It is a low insulin : high glucagon state, which causes flow of fatty acids from the adipose tissue and to the liver, where they become a major energy source (Chapter 12).
Excessive generation of acetyl CoA results, which cannot be fully handled by TCA cycle because of the following reasons:
Oxaloacetate depletion
In the liver, an enhanced β-oxidation results in depletion of oxaloacetate because the excess of NADH produced converts it to malate. This causes dearth of oxaloacetate, thereby limiting activity of the TCA cycle.
Exhanced gluconeogenesis
Oxaloacetate is directed towards glucose synthesis for meeting the essential requirements for tissues like brain, and this further exacerbates the dearth of oxaloacetate and retards TCA cycle.
TCA being virtually blocked, the acetyl CoA must find another outlet. One of the possible outlets, the fatty acid biosynthesis, is largely shut down under such circumstances (reasons given later), and acetyl CoA is instead channeled into ketone body production. Consequently, large amounts of acetyl CoA (produced by β-oxidation in liver) are diverted towards overproduction of ketone bodies (Fig. 11.13 ).
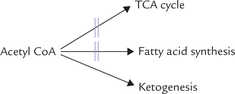
Fig. 11.13 Excessive amounts of acetyl CoA generated in starvation and diabetes mellitus are channeled into ketone body production; TCA cycle and fatty acid synthesis being retarded in these conditions.
Diabetes Mellitus
Severe and uncontrolled diabetes mellitus is the most common cause of ketosis. Persons with type 1 diabetes have little or no plasma insulin and the insulin : glucagon ratio falls very low in decompensated patients, resulting in overproduction of ketone bodies by the aforementioned series of events.
Acetoacetate and β-hydroxybutyrate decrease the pH of blood and cause metabolic acidosis; the condition called diabetic ketoacidosis is life threatening. In type 1 diabetes it develops very quickly, even after missing a single insulin dose, whereas in type 2 diabetes, ketoacidosis is relatively rare but may be precipitated by a major stress, such as myocardial infarction. Production of acetone being significantly enhanced, its smell in the patient’s breath is a valuable aid to diagnosis of diabetic ketoaci-dosis. The breathing is characteristically deep and fast (i.e. Kaussamal’s breathing).
Dangers of Ketosis
Ketosis causes metabolic acidosis and loss of sodium and potassium ions from the body, which are potentially lethal conditions. Large amounts of ketone bodies are excreted in urine, and because of their anionic character they also carry away Na+. The problem is aggravated because uncontrolled diabetes is accompanied by an osmotic diuresis. Moreover, insulin increases potassium uptake by cells, therefore lack of insulin in diabetes leads to release of potassium, which is then uncontrollably lost in urine.
Loss of circulating Na+ may aggravate acidosis, because the Na+ depletion often results in decrease in circulatory bicarbonate ion concentration. One may view this problem in terms of ketone bodies displacing bicarbonate ions even if Na+ concentration remains constant (Case 15.3).
D Regulation of Ketogenesis
Synthesis of ketone bodies is regulated at the following levels:
1. Availability of fatty acids: Fatty acids are precursors of ketone bodies, and the source of free fatty acids is depot fat adipose tissue. Therefore, factors influencing mobilization of fatty acids from adipose tissue also influence the rate of ketogenesis. These are described later in this chapter.
2. Entry of fatty acid into mitochondrial matrix: Carnitine acyl CoA transferase-I regulates entry of fatty acids into mitochondrial matrix for oxidation, so its activity regulates ketogenesis as well. Regulation of the enzyme activity by malonyl CoA has been discussed earlier.
3. Channelization of acetyl CoA into ketogenesis: Liver has a very high capacity for fatty acid oxidation, and the acetyl CoA formed by β-oxidation has a choice between progressing into TCA cycle, fatty acid biosynthesis, or ketogenesis or even cholesterol synthesis. However, TCA cycle is impeded and acetyl CoA is not used for biosynthetic activity during fasting. This results in channelization of acetyl CoA into ketogenesis.
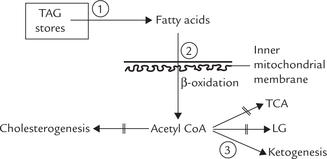
4. Effect of hormones: Insulin inhibits and glucagons stimulate ketogenesis, as discussed earlier.
IV De Novo Synthesis of Fatty Acids
Fatty acids are obtained from both diet and de novo (new) synthesis. Because most fatty acids have multiples of two carbon atoms, they are synthesized from successive addition of two carbon units, the donor of which is acetyl CoA. NADPH is required for the reductive reactions of the pathway which occurs in the cytosol of the cell. The elongation of the fatty acid chain stops upon formation of palmitate (16-C). Further elongation (and desaturation) are carried out by other enzyme systems.
In humans, liver and lactating mammary glands are the main organs for the endogenous synthesis of fatty acids, although adipose tissue, brain and kidneys are involved to a lesser extent. Liver convert excess dietary carbohydrates into fatty acids and triacylglycerols.
The pathway of synthesis of fatty acids involves condensation of two carbon units, in the form of acetyl CoA, to form long hydrocarbon chain a series of reaction. The reactions are carried out on fatty acid synthase complex using NADPH as reductant.
The biosynthetic pathway occurs in 3 stages:
(a) Transport of 2-carbon units (acetyl CoA) to cytosol.
A Transport of Two Carbon Units to Cytosol
Fatty acid synthesis occurs in the cytosolic compartment of the cell. However, acetyl CoA, the precursor, is generated in the mitochondrial matrix as end product of various catabolic processes. It must reach cytosol by crossing the mitochondrial membranes. But the inner mitochondrial membrane (IMM) is impermeable to most molecules and ions, including acetyl CoA. Transport of acetyl CoA across the inner mitochondrial membrane requires that it is first converted to citrate which is capable of moving across the IMM.
In the cytosol the citrate is cleaved to regenerate acetyl CoA (Fig. 11.14 ).
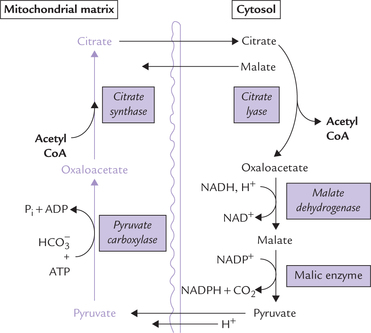
Fig. 11.14 Transport of acetyl CoA across inner mitochon-drial membrane (IMM). It combines with oxaloacetate to form citrate, which readily crosses the membrane. In the cytosol citrate is cleaved to regenerate oxaloacetate.
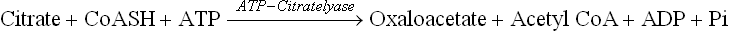
Thus, by the coordinated action of two enzymes-citrate-synthase and -lyase, the acetyl CoA is effectively transported from mitochondrial matrix to the cytosol. While it is used for fatty acid synthesis, oxaloacetate is shuttled back into the mitochondrion either as malate or as pyruvate. It is converted to malate via malate dehydrogenase that uses NADH as the proton donor. Malate is then converted to pyruvate by the NADP+-dependent malic enzyme. The reduced NADPH so formed becomes available for lipogenesis. Pyruvate is then translocated across the mitochondrial membrane to be cycled again.
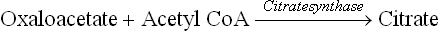
The malic enzyme reaction can theoretically supply half of the NADPH required for fatty acid synthesis. The remaining NADPH has to be obtained from the pentose phosphate pathway.
B Conversion of Acetyl CoA to Malonyl CoA
The key reactions of fatty acid synthesis are the carbon-to-carbon condensations. They require input of considerable energy and are, therefore, thermodynamically unfavourable. To overcome this energy barrier, acetyl CoA needs to be converted to activated form, which will serve as the donor of carbon units to the growing fatty acid chain. Malonyl CoA, a 3-carbon compound, is one such activated form. It is produced by carboxylation of acetyl CoA. The reaction is catalyzed by the enzyme acetyl CoA carboxylase, and requires free energy which is provided by ATP hydrolysis.

This is a typical ATP-dependent carboxylation reaction, with enzyme-bound biotin serving as a carrier of the car-boxyl group (Chapter 18). Since the malonyl CoA is not used in other metabolic pathways, this reaction is the committed step of fatty acid synthesis. It also serves as a control point, since activity of acetyl CoA carboxylase is allosterically modulated by citrate (positive modulator) and palmitoyl CoA (negative modulator).
C Reactions Catalyzed by Fatty Acid Synthase
Synthesis of fatty acid from acetyl CoA precursor occurs on a cytoplasmic polyprotein called fatty acid synthase (MW 400,000). A poly-protein is a single protein with more than one enzymatic activities. Fatty acid synthase is a dimer formed from two identical chains of such polyprotein (Fig. 11.15 ). Each one of these polyproteins exhibits multifunctionality, possessing seven enzyme activities (designated E1 to E7). Electron microscopic studies with this enzyme complex have shown that contiguous stretches of the polypeptide chain are folded to form seven autonomous domains, each with a different enzyme activity. (For additional information on fatty acyl synthase, refer to Box. 11.1). These domains are arranged in such a way that they catalyze the successive steps in the fatty acid synthesis cycle. The biosynthetic intermediates do not diffuse away from the polyprotein but remain attached to terminal sulphydryl (SH) group of an acyl carrier protein (ACP), which passes them from one enzyme active site to the next.
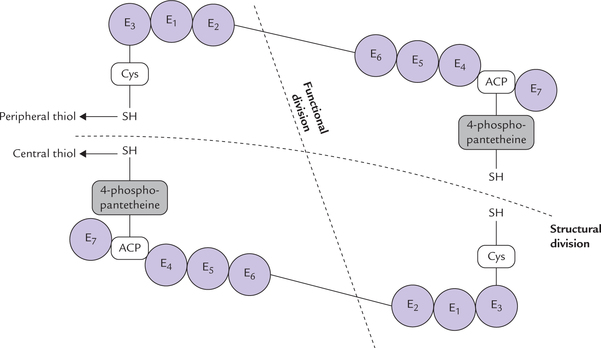
Fig. 11.15 The enzyme activities of fatty acid synthesis in higher organisms are present in a single, multifunctional polypeptide chain (as a diamer) called fatty acid synthase. The two subunits of this dirner interact in head-to-tail manner. Each subunit contains seven distinct enzyme activities (E1 to E7) and an acyl carrier protein (ACP). Cys = cysteine.
The growing fatty acid chain is bound covalently to two types of sulphydryl groups (SH) during its synthesis. These groups, two in each polyprotein chain, serve as carriers of acyl groups:
(a) One of these, called the central thiol, is contributed by ACP. The reactive unit of ACP is 4-phosphopantetheine (Pan) (Fig. 11.16 ) that is covalently bound to a swinging arm, 2.0 nm long, carrying the acyl intermediates from one enzyme active site to another. In other words, it acts as mobile carrier for shuttling the growing chain to successive catalytic domains.

Fig. 11.16 The 4-phosphopantetheine group of the acyl carrier protein (ACP). It contains pantothenic acid and cystamine (shown in colour) with a terminal SH group, and thus resembles coenzyme A.
(b) The second important thiol group, called the peripheral thiol, is contributed by a cysteine residue (Cys) of the enzyme 3-ketoacyl synthase (E3).
The two types of thiol groups lie in close proximity in the dimer, suggesting that they are present on separate (subunits) that interact in a head-to-tail manner.
Each subunit (or monomer) contains all the seven enzyme activities, but the actual functional unit consists of one-half of a monomer interacting with the complementary half of the other monomer. Thus, two fatty acid chains can be synthesized simultaneously.
Fatty acid synthesis occurs in cytosol through involvement of a polyprotein (fatty acid synthase) that has seven enzymatic sites (in which successive reactions take place). The intermediates are linked to the terminal sulphydryl group of the phosphopantetheine reactive unit in ACP.
The reactions catalyzed by the seven domains of fatty acid synthase are described below. The first two reactions (called priming reactions) ensure that both the sulphy-dryl groups are ’loaded’ with correct acyl groups, and the remaining reactions called elongation steps are involved in actual building of the fatty acid chain. The sequence of reactions is shown in Figure 11.17 .
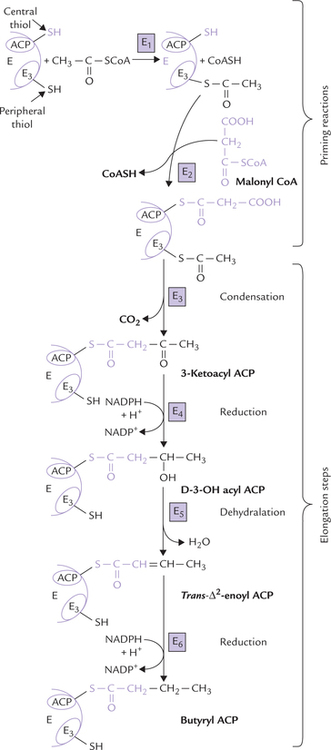
Fig. 11.17 Reactions in a single cycle of fatty acid synthesis (E1 = acetyl transferase, E2 = malonyl transferase, E3 = 3-ketoacyl ACP synthase, E4 = ketoacyl ACP reductase, E5 = D-3-OH-acyl ACP dehydratase, E6 = trans-Δ2-eonyl-ACP reductase).
Priming Reactions
The two priming reactions are catalyzed by enzymes acetyl transferase and malonyl transferase.
1. Acetyl transferase: This enzyme catalyzes transfer of an acetyl group from acetyl CoA to the cysteine - SH group, i.e. the peripheral thiol group furnished by E3
2. Malonyl transferase: This enzyme catalyzes transfer of a malonyl group from malonyl CoA to the central thiol group furnished by ACP.
At this state, both central and peripheral-thiol groups are charged by malonyl and an acetyl group respectively (Fig. 11.16). Since the two acyl groups are close to each other on the enzyme complex, the stage is now set for their condensation and further elongation steps.
Elongation Steps
Condensation, reduction, dehydration and reduction are the four elongation steps.
1. Condensation by 3-Ketoacyl ACP synthase: The condensation reaction between the previously attached acetyl group and the malonyl group forms 3-ketoacyl ACP. This frees the peripheral thiol group of E3 that had been occupied by acetyl CoA. A CO2 molecule is liberated from the malonyl group during condensation, and this (decarboxylation) provides the “pull” for the reaction.
2. Reduction by 3-Ketoacyl ACP reductase: The keto group is reduced by this enzyme to form D-3-OH-acyl ACP. The reducing power for this reaction is provided by NADPH.
3. Dehydration by D-3-OH-acyl ACP dehydratase: A water molecule is removed from the D-3-OH acyl ACP to form trans-Δ2-enoyl ACP.
4. Reduction by Trans-Δ2-enoyl-ACP reductase: The trans-Δ2-enoly-ACP, formed in the previous reaction, is reduced by this enzyme to form butyryl ACP. NADPH serves as a coenzyme in this reaction also.
This final reaction of elongation produces the four carbon butyryl-ACP; thus the acyl group has grown longer by two carbon atoms.
All the above enzymatic steps are carried out in different enzyme domains of fatty acid synthase with the flexible arm of the phosphopantetheine group moving the acyl group from one active site to the next. When it gets to the last part of the sequence, the new acyl group (here butyrate) is passed to the peripheral thiol-SH group of cysteine residue (that was previously occupied by acetyl CoA) on the other monomer. The central thiol group of ACP is now reloaded with malonyl group and the elongation cycle is repeated in the same way, but the acyl group is now longer by two carbon atoms.
Further cycles
The elongation steps described above, are repeated with malonyl ACP adding 2-carbon units in each cycle. It is noteworthy that pairs of carbons from the malonyl group are added at the carboxyl (C-1) end and not the methyl (ω) end of the growing chain. This continues until the acyl group is 16-carbon long (palmi-toyl-ACP). This is not a substrate for the condensing enzyme (E3), but for thiolase (deacylase; E7), which catalyzes the hydrolysis of palmitoyl-ACP. As a result, release of palmitic acid from the fatty acid synthase occurs and HS-ACP is regenerated. This completes synthesis of palmitic acid.
Cycle summary
Only two carbons in palmitic acid come directly from acetyl CoA. The remaining 14 are obtained from malonyl CoA, which in turn is produced from acetyl CoA. The overall stochiometry for the synthesis of palmitate therefore is
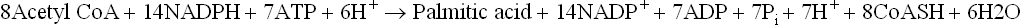
The coenzyme in lipogenesis is NADPH (not NAD+), which differs from NAD+ in being phosphorylated at C-2 of ribose. In general, NADP+ is associated with synthetic pathways and NAD+ is associated with degradative pathways.
Palmitic acid is produced in liver and adipose tissue; but in lactating mammary gland, medium chain fatty acids, 10-carbon capric acid, and 12-carbon lauric acid, are produced. Human milk contains both; cow’s milk by contrast contains odd numbered fatty acids.
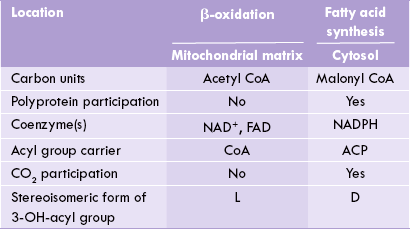
Comparison of Fatty Acid Synthesis and Oxidation
Fatty acid synthesis and β-oxidation are distinct pathways, and not a simple reversal of each other. Comparative features of the two are outlined in Table 11.2 .
Sources of NADPH
NADPH, which plays a key role in fatty acid synthesis, is generated from various sources.
1. Reactions of HMP shunt: Two of the reactions of this pathway yield an NADPH each. These reactions are catalyzed by the enzymes glucose 6-phosphate dehydrogenase (G6PD) and 6-phosphogluconate dehydrogenase, respectively.
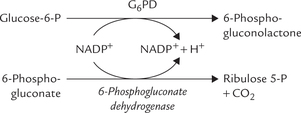
2. Malate to pyruvate conversion also yields a NADPH molecule. The reaction is catalyzed by the malic enzyme.
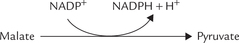
3. Isocitrate to α-ketoglutarate conversion by the cytosolic isocitrate dehydrogenase (IDH) is a minor source of NADPH. In fact, this enzyme can use both NAD+ and NADP+ as coenzymes.
D Regulation of Fatty Acid Biosynthesis
Availability of Citrate in Cytoplasm
The citrate formed in mitochondria has a choice between progressing further in the TCA cycle or diffusing into cytoplasm, where it can supply the acetyl CoA precursor for synthesis of fatty acids. As discussed in Chapter 9, in high-energy states, when NADH:NAD+ ratio is high and ADP low, the pathway for progression of citrate into the TCA cycle is blocked and hence its concentration builds up in the mitochondrial matrix. This excess citrate is then passed into the cytoplasm, where it supplies the acetyl CoA precursor. Citrate also allosterically stimulates the key enzyme of lipogenesis, acetyl CoA carboxylase.
Regulation of Enzyme Activities
Acetyl CoA carboxylase
Fatty acid synthesis is regulated at the acetyl CoA carboxylase step by allosteric mechanism, covalent modulation and induction-repression mechanism of enzyme synthesis. This provides a good example of control at committed step of a pathway.
(a) Allosteric modulation: Acetyl CoA carboxylase can exist in an inactive monomeric form (MW 400 kDa) and an active polymeric form (MW 6000-8000 kDa). The most important allosteric regulators are citrate, which stabilizes the active polymeric form, and palmitoyl-CoA, which dissociates the complex into inactive monomers. Thus, citrate has stimulatory effect and palmitoyl CoA has inhibitory effect. This is entirely logical: under conditions of high citrate concentration, energy storage is desirable, but decreased synthesis of fatty acids is appropriate when the product (palmitoyl CoA) accumulates.
(b) The covalent mechanism involves hormone-dependent protein kinase A/phosphatase: phos-phorylation inhibits the enzyme activity and dephosphorylation activates the enzyme activity. These effects are under influence of hormones— insulin promotes dephosphorylation and thereby stimulates the acetyl CoA carboxylase activity. Glucagon or epinephrine, on the other hand, inactivates the enzyme by cAMP-dependent phosphorylation. Thus, insulin promotes fatty acid synthesis, while epinephrine and glucagons inhibit it (Fig. 11.18 ).
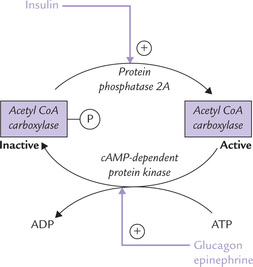
Fig. 11.18 Hormonal regulation of acetyl CoA carboxylase, the key lipogenic enzyme, by covalent mechanism.
(c) Induction-repression enzyme synthesis effected by diet: synthesis of acetyl CoA carboxylase is up regulated with high carbohydrate/low fat intake (insulin:glucagon ratio is high) and down regulated by high-fat low-carbohydrate diet and starvation (low insulin : glucagon ratio).
Finally, activity of acetyl CoA carboxylase is subject to control by energy charge of cell. Short-term regulation is effected by allosteric and covalent modulation, and a long term mechanism relates to induction or repression of enzyme activity.
Fatty acid synthase
In common with acetyl CoA carboxyl-ase system, fatty acid synthase activity also is similarly affected by regulatory mechanisms, such as, allosteric effect, and induction-repression of the enzyme. For instance, high carbohydrate low fat diet increases synthesis of fatty acid synthase and conversely low carbohydrate/ high fat diet or fasting decreases the synthesis.
These regulatory mechanisms ensure that the excess dietary carbohydrates are converted to fatty acids in liver. The latter are esterified to triacylglycerols, which are released as constituents of very low density lipoprotein. This carbohydrate-to-fat pathway is important only on a high carbohydrate diet, as the regulated enzymes are stimulated by insulin. This explains why people with carbohydrate-based diets are prone to develop obesity, even when they restrict fat intake.
V Chain Elongation and Desaturase Systems
Fatty acids of longer chain (above 16-carbon) length cannot be synthesized by fatty acid synthase system. But these fatty acids are important for the body: they are required in triacylglycerols and membrane lipids, which have fatty acids of chain lengths of up to 24 or 26 carbons, (Carbon-18 fatty acids being the commonest). Also only approximately 50% of the fatty acids of our body are saturated; another 40% are monounsaturated and approximately 10% are polyunsaturated. These long chain and unsaturated fatty acids are synthesized respectively by chain elongation system and desaturase systems.
A The Chain Elongation System
Chain elongation beyond C-16 palmitate is possible in endoplasmic reticulum (the microsomes) and in the mitochondrion.
The microsomal system elongates palmitate by the addition of 2-carbon fragments derived from malonyl CoA. The system of reactions during the chain elongation is similar to that involved in fatty acid synthesis, except that here the fatty acid is attached to CoA, instead of the pante-theine residue of acyl-carrier protein. Mitochondrial elongation system is less active and uses acetyl CoA as a donor of carbons. It is NADH-dependent and the substrates for chain elongation are short- and medium-chain fatty acids containing fewer than 16 carbon atoms. Elongation of fatty acids is greatly reduced during fasting and starvation.
B The Desaturase System
Monosaturated fatty acids can be obtained by introduction of double bond in the corresponding saturated fatty acids by a membrane bound desaturase system in the ER of the liver and other organs. The bond is most commonly introduced at position Δ9 (between C-9 and C-10) of palmitic or stearic acid, producing palmitoleic acid or oleic acid, respectively. The system for desaturation requires molecular oxygen, NADH, and cytochrome B5. The desaturases are monooxygenases: molecular oxygen is the oxidant and the process of desaturation results in the oxidation of both the fatty acid and the NADH.
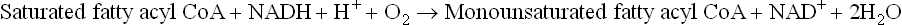
Polyunsaturated fatty acids may be formed by introduction of additional double bonds between the first double bond and the carboxyl end of the fatty acid, but not beyond Δ9. Hence, linoleic acid (having two double bonds at the positions Δ9 and Δ12) cannot be synthesized from the monounsaturated oleic acid, but the fatty acid having two double bonds at Δ6 and Δ9 (called γ-linoleic acid) can be synthesized.
Most fatty acids can be synthesized from palmitate by desaturase and elongase enzymes, which introduce double bonds and lengthen the existing acyl chains, respectively.
Some polyunsaturated fatty acids are nutritionally essential. Because of the inability of the human desaturase system to introduce double bonds beyond Δ9, linoleic acid (C-18, Δ9,12) and α-linolenic acid (C-18, Δ9,12,15 )—the parent compounds of the ω6 and the ω3 classes of poly-unsaturated fatty acids—cannot be synthesized in the human body. They must be present in the diet or synthesized from other fatty acids in the diet, hence called essential fatty acids. Arachidonic acid (ω6 20-C, Δ5,8,11,14) can be formed if the dietary supply of linoleic acid is adequate. The reaction sequence requires a combined action of the desaturase and chain elongation systems (Fig. 11.19 ).
Fig. 11.19 Arachidonic acid can be synthesized from linoleic acid in the human body by the elongation and the desaturase systems.
1. First linoleic acid is converted to γ-linolenic acid (C-18; Δ6,9,12) by introduction of a double bond at C-6.
2. Conversion of the γ-linolenic acid to arachidonic acid (C-20; Δ5,8,11,14) occurs by combined action of the elongation and desaturase systems, as shown here.
Thus, arachidonic acid, generally regarded as an essential fatty acid, can be synthesized provided linoleic acid is available. Therefore, strictly speaking arachidonic acid is not an essential fatty acid.
VI Metabolism of Triacylglycerol
A Synthesis of Triacylglycerols
Triacylglycerols are synthesized mainly in liver, adipose tissue and intestinal cells. There are two pathways: the general pathway, which is operational in liver, adipose and other organs where fatty acid biosynthesis occurs; and the intestinal pathway, which is responsible for resyn-thesis of triacylglycerols following digestion and absorption of dietary fats.
General Pathway
Glycerol-3-phosphate serves as the primary starting material and phosphatidic acid is the key intermediate of the pathway which proceeds in the following steps:
1. Synthesis of phosphatidic acid: Glycerol-3-phosphate (glycerol-3-P) is produced in most tissues, including adipose, by reduction of the glycolytic intermediate, dihy-droxyacetone phosphate (DHAP). In liver and kidneys, glycerol-3-P can also be formed directly via phosphorylation of glycerol by a specific kinase (glycerol kinase).
It is then acylated by transfer of two long chain fatty acids from fatty acyl CoA to the hydroxyl groups at C-1 and C-2 producing phosphatidic acid (Fig. 11.20 ). The first fatty acid, usually a saturated fatty acid, is linked to C-1 forming monoacyl-glycerol-P, also called lyso-phosphatidic acid; the prefix lyso indicates that one of the hydroxyl groups is not acylated. Then a second fatty acid, usually an unsaturated fatty acid, establishes an ester bond to C-2 to form phosphatidic acid. Two different enzymes catalyze these two acylation steps— glycerol phosphate acyltransferase (E) and monoacyl glycerol acyltransferase (E’):
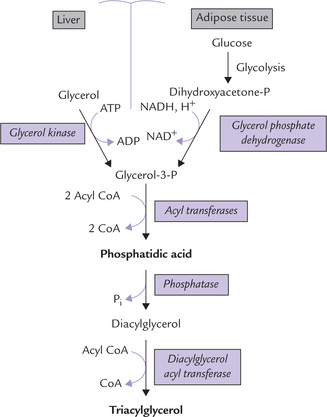
Fig. 11.20 Synthesis of TAG in liver and adipose tissue. The source of glycerol-3-P is different in the two tissues because there is no glycerol kinase in adipose tissue.
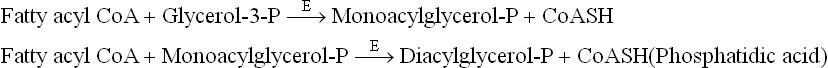
2. Synthesis of triacylglycerol from phosphatidic acid: Phosphatidic acid is first converted to diacyglycerol (DAG) by a specific cytosolic phosphatase, which remove a phosphate group. Transfer of an acyl group from fatty acyl CoA to DAG by the enzyme 1,2-diacylglycerol acyl transferase (E") finally generates triacylglycerol.
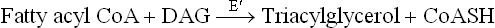
Note that the ester bonds of triacylglycerol are low energy in nature. Fatty acyl CoA, the acyl group donor, on the other hand, has a high energy thioester. Therefore, the acyl transfer reactions proceed with a decrease in the number of high energy bonds, and are therefore exergonic. This general principle provides rationale for understanding nearly all acyl transfer reactions in lipid metabolism.
Intestinal Pathway
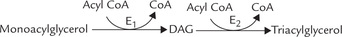
2-Monoacylglycerol, rather than glycerol-3-P is a precursor in intestinal epithelium. The pathway is simpler and more straightforward. An intestinal monoacylglycerol acyl transferase (E1) catalyzes the formation of DAG and 1,2-diacylglycerol acyl transferase (E2) catalyzes acylation of the remaining free carbon of DAG to yield a triacylglycerol.
B Hydrolysis of Triacylglycerols (Lipolysis)
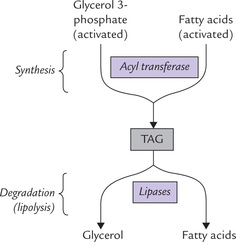
Lipases are the enzymes involved in degradation of triac-ylglycerols, catalyzing sequential removal of the fatty acids from glycerol backbone. Fatty acids are beta-oxidized to produce energy. The glycerol is converted to dihydroxy-acetone phosphate which enters glycolysis.
Various types of lipases are known, most important of which are hormone-sensitive lipase, lipoprotein lipase and hepatic lipase. Their actions are described below:
1. Hormone sensitive lipase (HSL) acts on triacylglycerol stores in adipose tissue, catalyzing the first cleavage to remove the fatty acid esterified to C-1 of glycerol. Lipases specific for monoacylglycerol and diacylglyc-erol then cleave the fatty acids linked to C-2 and C-3.

2. Lipoprotein lipase (LPL) causes hydrolysis of triacylg-lycerols present in some lipoproteins (very low density lipoproteins and chylomicrons) and is thus required for utilization of circulating triacylglycerols. This enzyme is synthesized in adipose tissue, skeletal muscle, the myocardium, the lactating mammary gland, spleen, lung, kidney and aorta (bound non-covalently to heparan-sulphate proteoglycans of the endothelial plasma membrane). As the chylomicrons or very low density lipoproteins (very low density lipoproteins and chylomiarons) pass through the capillaries of these organs, they bind to LPL, and their triacylglycerol is hydrolyzed to free fatty acids and 2-monoacylglycerol. Most of these breakdown products diffuse directly into the tissue cells.
3. Hepatic lipase hydrolyzes triacylglycerols present in other lipoprotein fractions, e.g. intermediate- and high-density lipoproteins (described in Chapter 12).
VII Adipose Tissue Metabolism
Metabolic processes in the adipose tissue have consequences that extend beyond the tissue itself. For instance, the plasma level of free fatty acids is determined by these processes. The circulating free fatty acids in turn have profound effect upon the metabolism of other tissues, particularly of liver and muscles.
Source and determinant of the circulating free fatty acids are the triacylglycerol (TAG) molecules that are stored in the adipose tissue. TAGs account for approximately 90% weight of the adipose tissue. Also termed depot fats, the stored triacylglycerols form major fuel reserve of the body. These stores exist in a dynamic steady state, being continually degraded and resynthesized (Fig. 11.21 ); life span of a stored triacylglycerol molecule is only 2-3 days. The degradation of TAG, known as lipolysis, is catalyzed by enzymes called lipases. Lipolysis results in production of a glycerol and three fatty acid molecules from each triacylglycerol molecule. The fatty acid molecules thus produced are re-used for synthesis of triacylglycerol.
A TAG Synthesis
Synthesis of TAG requires glycerol phosphate and three fatty acid molecules, activated to their CoA-thioesters.
• The fatty acids are mostly derived by the action of lipo-protein lipase on chylomicron TAGs, after a mixed meal.
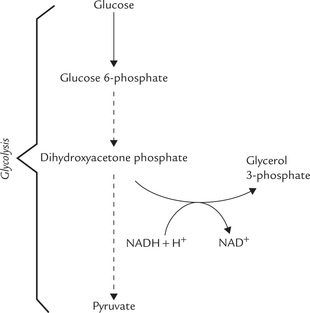
• The glycerol phosphate is derived from adipose tissue by the glycerol phosphate dehydrogenase reaction. In this reaction, dihydroxyacetone phosphate, a glycolytic intermediate, is reduced in an NADH-dependent step (Fig. 11.22 ). The alternative reaction for the generation of glycerol phosphate (the phosphorylation of glycerol by the enzyme glycerol kinase) is important in liver, but not in adipose tissue, because of extremely low concentration of this enzyme in adipocytes.
The acyl groups of the activated fatty acids are then sequentially transferred to glycerol 3-phosphate by acyl transferases to form triacylglycerol, as described earlier (Fig. 11.20).
• The first acyl group is linked to C-1 of glycerol phosphate by the enzyme glycerol phosphate acyl transferase (E1),
• The second acyl group is transfersed to C-2 of glycerol phosphate by monoacylglycerol acyl transferase (E2),
• The third acyl group is transfersed to C-3 by diacylg-lycerol acyl transferase (E3).
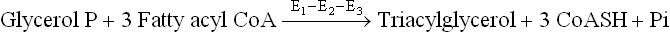
B TAG Hydrolysis (Lipolysis)
This process requires the sequential removal of three fatty acids from glycerol. The first cleavage which is rate-limiting is catalyzed by the enzyme hormone-sensitive lipase (HSL). Subsequent cleavages are brought about by other lipases which are specific for monoacylglycerol and diacylglycer-ols. A different fate awaits each of the two products of lipolysis: glycerol and fatty acids.
• Glycerol cannot be utilized within the adipocytes because low concentration of glycerol kinase in adipose tissue prevents its conversion to glycerol 3-phosphate in any substantial amount. Therefore, intracellular level of glycerol builds up. Subsequently, it diffuses into the plasma from where it is taken up by tissues, mainly liver and kidney. In these tissues, activity of glycerol kinase being high, the glycerol is phosphorylated to glycerol 3-phosphate which then forms dihydroxyacetone phosphate, a glycolytic intermediate. Thus, glycerol is ultimately used via the gly-colytic sequence in other tissues (Fig. 11.23 ).
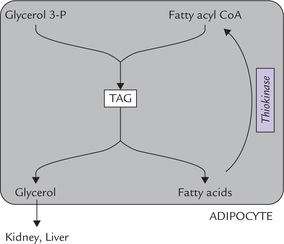
Fig. 11.23 Fatty acids are recycled for triacylglycerol (TAG) synthesis in adipose tissue but glycerol is metabolized in other tissues.
• Fatty acids, on the other hand, are used within the adipose tissue for TAG synthesis. Under normal circumstances, rates of TAG synthesis and hydrolysis are nearly balanced and most of the fatty acids obtained by lipolysis are activated to Co-thioesters which are recycled for TAG formation. However, imbalance between the two processes disturbs the dynamic steady state.
C The Dynamic Steady State
TAG production and hydrolysis are not forward and reverse phases of the same reaction. They are entirely different processes using different reactants and catalyzed by different enzymes. Under normal circumstances, a dynamic steady state exists where the rates of the two processes are balanced. Hormonal, nutritional and metabolic, factors regulate the metabolism of adipose tissue by influencing these two processes. Net balance of the two determines the amount of TAG stores in the adipose tissue, as also the levels of circulating free fatty acids in the plasma.
D Hormonal Regulation of Adipose Tissue Metabolism
The metabolic processes operative within adipocytes are regulated by a number of hormones which control activities of both the key enzymes of adipose tissue metabolism, viz. the hormone-sensitive lipase and the glycerophosphate acyl transferase.
The hormonal influences on the lipase are summarized in Figure 11.24 . The enzyme activity is regulated by the cAMP-dependent cascade; activation occurs when the enzyme is phosphorylated by the cAMP-dependent protein kinase-A (PKA). It is the same enzyme that operates in regulation of glycogen metabolism.
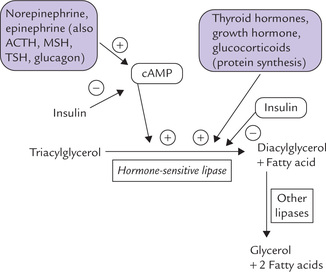
Fig. 11.24 Hormonal regulation of lipolysis in adipose tissue. Some lipolytic hormones activate HSL, others enhance synthesis of the enzyme-protein; and insulin antagonizes effects all lipolytic hormones.
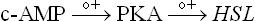
Thus, the hormones which increase intracellular level of cAMP activate the hormone sensitive lipase. Such hormones are lipolytic.
The most important lipolytic agents are catechol-amines which act through β-adrenergic receptors, cAMP and protein kinase A (PKA). Norepinephrine, released from sympathetic nerve terminals in adipose tissue, is a more potent lipolytic agent than epinephrine. The catechola-mines ensure that the TAGs are hydrolyzed during cold exposure, stress and physical exercise through stimulation of the lipase activity.
Catecholamines elevate intracellular level of cAMP, which allosterically activate PKA. The PKA in turn phos-phorylates hormone sensitive lipase, activating it.
Glucocorticoids, growth hormone and the thyroid hormones enhance lipolysis by inducing synthesis of lipolytic proteins. Thus, glucocorticoids induce the de novo synthesis of the hormone-sensitive lipase, and thereby accelerate the lipolytic response to catecholamines. This effect shows regional differences in action of glucocorticoids (For additional information on lipolytic action of corticoids refer to Box 11.2).
Lipolytic hormones facilitate lipolysis either by direct stimulation of cAMP formation or by inducing synthesis of lipolytic protein (HSL).
Role of Insulin
In contrast to the lipolytic hormones-action of insulin is antilipolytic, i.e. it antagonizes these hormones by causing inhibition of the hormone-sensitive lipase. The mechanism of action is poorly understood. Either insulin decreases the level of cAMP in the cell (by activating a phosphatase) or induces dephosphorylation of the HSL. Adipose tissue is highly sensitive to insulin: a moderate rise in insulin level (during feeding) is able to halt lipolysis.
Insulin enhances TAG synthesis: In addition to inhibiting lipolysis, insulin can also enhance TAG synthesis in adi-pocytes by several mechanisms:
1. By increasing production of glycerol 3-phosphate substrate: Insulin increases the rate of glycolysis, which enhances production of dihydroxy-acetone phosphate. The latter is then reduced to glycerol 3-phosphate.
In adipose tissue, the most important control site for glycolysis is tissue up-take of glucose (unlike in other tissues where it is the PFK reaction). Insulin enhances the glucose uptake by increasing the number of functional glucose carriers in plasma membrane.
2. By increasing synthesis of fatty acid substrate: Insulin enhances lipogenesis by bringing about the following metabolic alternations. It increases production of acetyl CoA from glucose (by augmenting glycolytic sequence and pyruvate dehydrogenase complex activity), and acetyl CoA is the substrate for lipogenesis. Further, it stimulates activity of acetyl CoA carboxylase, the key enzyme of lipogenesis. Insulin favors generation of NADPH, the coenzyme required in lipogenesis, by promoting the pentose phosphate pathway.
3. By increasing lipoprotein lipase activity: Stimulation of activity of this enzyme results in increased production of fatty acids from the TAG fraction of lipopro-tein (Chapter 12). This further increases the supply of fatty acid substrate.
4. By increasing esterification of fatty acid and glycerol substrates: Insulin induces the enzyme glycerophosphate-acyl transferase which adds the first fatty acid to glycerol phosphate in the bio-synthetic pathway of TAG.
Insulin ensures that triacylglycerols are synthesized after a meal, and degraded during fasting. Under its stimulatory influence, the adipose cells use the fatty acids (generated by lipoprotein lipase) for synthesis of storage triglycerides.
Thus, insulin has a dual role—inhibition of lipolysis and stimulation of triacylglycerol production. This helps to maintain a balance between these two pathways, under normal circumstances. In diabetes mellitus, loss of this balance occurs. Consequently, lipolysis is accelerated, but triacylglycerol synthesis slows. The net result is that the rate of lipolysis now exceeds that of triacylglycerol synthesis. Quantity of fatty acids produced is far in excess than that can be reutilized for re-esterification. The surplus flows out into the circulation to elevate the plasma levels of free fatty acids. This explains a marked elevation of plasma free fatty acid levels in diabetes mellitus.
E Effect of Nutritional States on Adipose Tissue Metabolism
The metabolic events in adipocytes show dramatic changes in different nutritional states. TAG synthesis depends on the supply of glycerol phosphate, which is synthesized from glucose via dihyroxyacetone phosphate (Fig. 11.22). The insulin-dependent uptake of glucose into the cell is the most important rate-limiting step in this pathway. Thus, in the fed state, increased availability of glucose and elevated insulin level in serum enhance TAG synthesis. Conversely, during starvation synthesis of TAG is impeded.
Utilization of chylomicron for providing fatty acids is also altered in different nutritional states. The utilization depends on the enzyme lipoprotein lipase which is induced by insulin in adipose tissue (but not in other tissues like cardiac and skeletal muscles). Therefore, the lipoprotein triacylglycerols are routed preferentially through adipose tissue in the well-fed state but not during fasting.
Insulin ensures that TAGs are synthesized after a meal and degraded during fasting. Fatty acids are major fuel for most tissues during fasting. Glycerol, which is released from adipose tissue together with fatty acids is a substrate for gluconeogenesis. This has a role in maintenance of normal glucose levels in blood during fasting and starvation, which is essential for survival.
VIII Metabolism of Complex Lipids
Complex lipids comprise a diverse group of saponifiable compounds found in biological membranes. They may be polar (phospholipids, sphingolipids) or non-polar (cholesterol esters, triacylglycerols). In this section metabolism of two major classes of the polar lipids, phospholipids and sphingolipids, are discussed; metabolism of non-polar lipids are discussed elsewhere.
A Metabolism of Phospholipids
The term phospholipids generally refers to two types of compounds: (a) glycerophospholipids, that contain a glycerol backbone, and (b) sphingomyelins, that contain an amino alcohol sphingosine instead of glycerol (the latter are discussed along with sphingolipids).
B Glycerophospholipid Metabolism
The commonest glycerophospholipids are phosphatidyl-choline, phosphatidylethanolamine, and phosphatidyl-serine. A number of more complicated structures, such as phosphatidylglycerol or cardiolipin, found in inner mito-chondrial membrane; and phosphatidylinositol, which has a role in signal transduction and in anchoring proteins in the plasma membranes, is also classed as glyc-erophospholipid (Chapter 3).
Biosynthesis of Glycerophospholipids
Glycerophospholipids are synthesized from phospha-tidic acid and diacylglycerol, the intermediates in production of triacylglycerol (Fig. 11.20). The same set of enzymes is used till diacylglycerol. These enzymes are located in the endoplasmic reticulum.
Synthesis of Lecithin and Cephalin
It involves transfer of choline/ethanolamine to the dia-cylglycerol, from an activated donor, e.g. CDP-choline or CDP-ethanolamine. How is the activated donor synthesized? Synthesis of CDP-choline occurs in a series of reactions given below (Fig. 11.25 ):
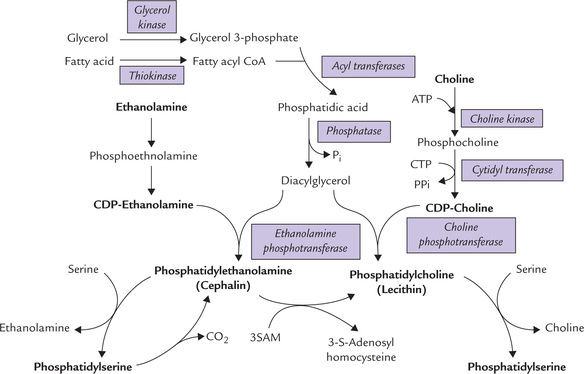
Fig. 11.25 Synthesis of phospholipids (SAM = S-adenosyl methionine, CTP = cytidine triphosphate, CDP = cytidine diphosphate).
• Choline is first converted to phosphocholine, and then activated to CDP-choline by a pyrophosphorylase reaction.
• The pyrophosphate bond is cleaved to drive forward the reaction.
The CDP-choline then transfers its phosphocholine group to diacylglycerol (DAG) to form phosphatidylcholine (lecithin). The last reaction is analogous to the transfer of glucose from UDP-glucose during glycogenesis.
Phosphatidylethanolamine (cephalin) is generated by a similar pathway using CTP and phosphoethanolamine to form CDP-ethanolamine; the latter transfers its phos-phoethanolamine group to DAG to form phosphatidyl-ethanolamine (Fig. 11.25).
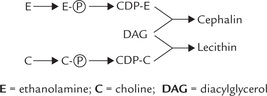
Synthesis of Phosphatidylserine
Phosphatidylethanolamine can react with free serine to form phosphatidylserine and the free base, ethanol-amine (Fig. 11.25). Likewise, phosphatidylcholine can form phosphatidylserine by such an exchange reaction.
(Liver has another route to phosphatidylethanolamine, involving decarboxylation of phosphatidylserine by a specific mitochondrial decarboxylase.)
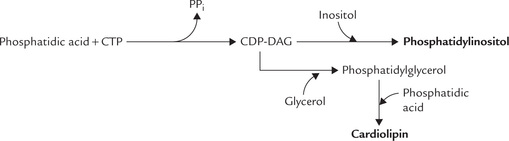
Synthesis of Cardiolipins or Phosphatidylinositol
In contrast to the pathways of lecithin and cephalin synthesis, where the head group (choline or ethanolamine) was activated by cytidine nucleotide, an alternate pathway exists for synthesis of other phospholipids (e.g. car-diolipin and phosphatidylinositol). In this pathway, phosphatidic acid is activated, rather than head group, yielding CDP-diacylglycerol (CDP-DAG).
The CDP-DAG is activated donor. It can readily transfer the phosphatidic acid group to free glycerol or inosi-tol to form phosphatidylglycerol or phosphatidylinositol, respectively. A second phosphatidic acid may also be likewise added to phosphatidylglycerol to form diphos-phatidylglycerol or cardiolipin (Fig. 11.27 ).
Others
In a secondary pathway, phosphatidylcholine is formed by transfer of methyl groups to phosphatidylethanolamine with the methyl donor, S-adenosylmethionine (SAM). The methylation pathway involves the sequential transfer of three activated methyl groups from SAM (Fig. 11.26 ).
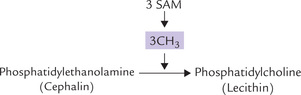
Fig. 11.26 Formation of lecithin by transfer of 3 methyl groups to cephalin, with the methyl donor S-adenosyl methionine (SAM).
De novo and salvage pathways
The above description makes it clear that there are two ways to synthesize a glyc-erophosphate. First, the head group is synthesized anew (this is de novo pathway). Second, the head group (choline or ethanolamine) is not synthesized anew, rather preformed choline and ethanolamine are reutilized or salvaged.
As depicted in Figure 11.25, when (i) phosphatidyl-ethanolamine is derived from phosphatidylserine by decarboxylation (Fig. 11.25), or (ii) when phosphatidyl-choline is derived from phosphatidylethanolamine by methylation pathway (Fig. 11.26), new ethanolamine and choline moieties, respectively are formed. Therefore, there pathways are the de novo pathways. However, eth-anolamine and choline per se are the starting substances in some reaction sequences depicted in the diagram on page 228, 11.25 and are called salvage pathways.
Degradation of Glycerophospholipids
The glycerophospholipids are in a continuous state of turnover in most membranes; the turnover especially increases in response to oxidative damage during inflammation, or in response to hormonal stimuli. As shown in Figure 11.28, a number of phospholipases are involved in degradation of phosphoglycerides. These enzymes are present in essentially all tissues and are classified according to their cleavage specificity: each one acts on a specific bond in the phospholipid structure.
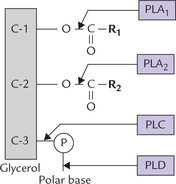
Fig. 11.28 Enzymatic degradation of phospholipid (PLA1 = phospholipase A1, PLA2 = phospholipase A2, PLC = phospholipase C, PLD = phospholipase D, P = phosphate group, and R2 = acyl groups).
Phospholipase A1 and -A2 remove the first and the second acyl group, respectively to produce the corresponding lyso-phospholipids. PLA2 acts on phosphatidylinositol to liberate arachidonic acid for the synthesis of prostaglandins. It is particularly active during the inflammatory response. Snake venom is especially a rich source of PLA2.
Phospholipase B (not shown) is a phospholipase that removes the second acyl group from lysophospholipid after the action of PLA1 or PLA2.
Phospholipase C (PLC) cleaves off the bond between the glycerol and phosphate to yield a phosphoryl base and a diacylglycerol molecule. It is especially active during signal transduction. Toxins isolated from Clostridium and other bacilli are rich sources of PLC.
Phospholipase D (PLD) liberates the polar base to form phosphatidic acid. It is found in plants, such as cabbage and cotton seeds.
The products obtained by degradation of glycerophos-pholipids enter the metabolic pool and are funneled into various important pathways.
Finally, phospholipases are used not only for the final degradation of the lipid molecule, but also for the remodelling of phosphoglycerides. The fatty acids in positions 1 and 2 for example, can be exchanged by hydrolysis and reacylation to form a new molecule. The cleavage specificities of phospholipases are largely responsible for the presence of specific fatty acids in positions 1 and 2 of phosphoglycerides : C-1 is usually occupied by a saturated fatty acid, and the fatty acid in position 2 is usually unsaturated.
C Sphingolipid Metabolism
Sphingolipids are a complex group of amphipathic polar lipids that are built on a core structure of the long chain (18-C) amino alcohol, sphingosine. They are divided into three general classes: sphingomyelins, cerebrosides and ganglio-sides, which differ with respect to the substituents attached to the C-1 hydroxyl group of sphingosine (Chapter 3).
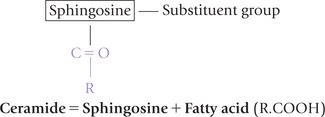
Sphingomyelins contain phosphoryl base, cerebrosides contain a monosaccharide, and gangliosides contain an oligosaccharide unit.
Biosynthesis of Sphingolipids
The pathway consists of two stages: firstly, synthesis of sphingosine/ceramide, and secondly, conversion of ceramide to any of the above classes of sphingolipids.
Synthesis of sphingosine and ceramide
Sphingosine is synthesized in the ER of most cells. The carbon atoms that contribute to sphingosine (C-18) synthesis come from palmitoyl CoA (C-16) and serine (which contributes two carbons). The condensation reaction requires reductive power of NADPH and is followed by a series of reactions to ultimately yield sphingosine.
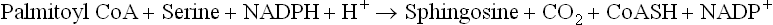
Ceramide, the next important intermediate, is formed by N-acylation of sphingosine:
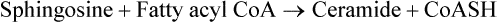
Formation of three classes of sphingolipids
Subsequent conversion of ceramide to sphingolipids occurs in ER and Golgi apparatus. It involves transfer of a substituent group to ceramide, usually from an activated donor, e.g. a nucleotide sugar (UDP glucose) or CDP choline (Fig. 11.29 ).
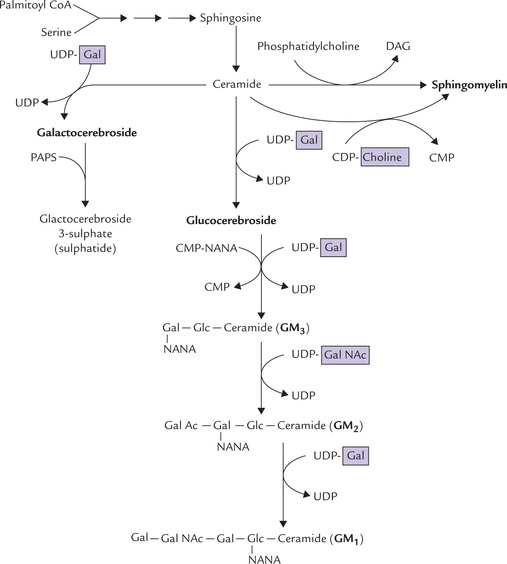
Fig. 11.29 Biosynthesis of sphingolipids from ceramide and nucleotide-activated monosaccharide or polar head group (e.g. choline) (PAPS = 3-phosphoadenosine-5-phosphosulphate, NANA = N-acetyl neuraminic acid or sialic acid, GM1, GM2, GM3 = gangliosides. Gal NAc = N-acetyl galactosamine).
(a) Conversion of ceramide to sphingomyelin: Transfer of phosphocholine to the terminal hydroxyl group of ceramide produces sphingomyelin. It is the only sphin-golipid that contains phosphorus. The formation of sphingomyelin from ceramide is unusual in that a high-energy precursor is not involved. The most important phosphocholine donor in this exchange reaction is phos-phatidylcholine. The reactants and products are low-energy compounds. Alternatively, CDP-choline may act as the phosphocholine donor.
(b) Conversion of ceramide to cerebrosides: The donor of carbohydrate(s) of cerebrosides are energy-rich uri-dine diphosphate sugars. The monosaccharide residues are introduced from these activated precursors; for example, formation of galactocerebrosides (a cerebroside) involves a reaction of ceramide with UDP-galactose, and formation of glucocerebroside involves UDP-glucose.
(c) Conversion of ceramide to gangliosides: The oligo-saccharide chains of these complex glycosphingolipids are constructed by the stepwise addition of monosaccha-ryl units, e.g. activated hexoses, hexosamine, and N-acetyl neuraminic acid (NANA). UDP serves as carrier of the sugar units in these reactions also. Each one of these exergonic reactions is catalyzed by an individual glyco-syltransferase. Various gangliosides, e.g. GM1, GM2 and GM3 are formed at different stages.
Sulphatides are the sulphate esters, formed by the combination of a sulphate with an alcohol, group present in glycosphingolipid. The donor of sulphate in biosynthetic reactions is 3’-phosphoadenosine-5’-phosphosulphate or PAPS. The standard free energy of hydrolysis of PAPS (to form 3’-phosphoadenosine monophosphate plus sulphate) is about -75 kJ/mol, which drives the biosynthetic reaction.
Globosides: When carbohydrate portion of the cere-broside contains two or more sugars plus an N-acetyl galactosamine group, it is referred to as globoside. The globosides are synthesized as gangliosides; UDP serving as carrier of the sugar units.
Degradation of Sphingolipids
Sphingolipids are degraded by lysosomal enzymes. In case of sphingomyelin, there are two enzymes: sphingomyelinase, which cleaves off the phosphocholine head group to form ceramide; and ceramidase, which further degrades the ceramide (Fig. 11.30 ).
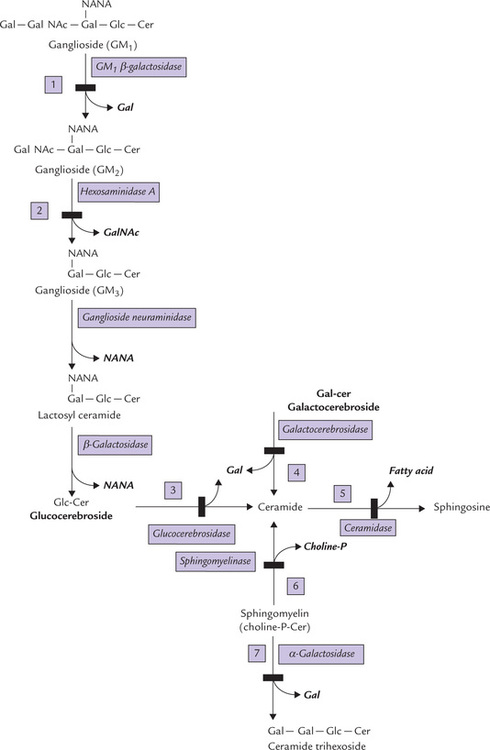
Fig. 11.30 Lysosomal degradation of sphingolipids, and the associated storage diseases ((1) generalized gangliosidosis, (2) Tay-Sachs disease, (3) Gaucher’s disease, (4) Krabbe’s disease, (5) Farber’s disease (6) Niemann-Pick disease, (7) Fabry’s disease).
In case of glycosphingolipids (cerebrosides and gan-gliosides), these degrading enzymes are specific lysosomal exoglycosidases, which remove terminal sugars from the oligosaccharide. They have narrow specificities for the monosaccharide and the type of bond they cleave. Figure 11.30 shows degradation of some important sphingolip-ids along with the associated inborn errors.
D Sphingolipidoses
Partial or complete deficiencies of these sphingolipid-degrading enzymes result in lysosomal accumulation of the missing enzyme substrate, which is insoluble, non-degradable, and non-excretable. This either causes lyso-somes to lyse and release their hydrolytic enzymes into a cell, or prevents the lysosome from functioning normally. In either case, the resulting disease is called a lipid storage disease lipid storage disease or sphingolipidosis. The enzyme deficiency is expressed in all tissues but clinical effect is most obvious in nervous tissue because of its high sphingolipid content and turnover. Enlarged liver and spleen (hepatosplenomegaly) is also common because phagocytic cells in these organs remove erythrocytes from the circulation, and non-degradable lipids from the RBC membrane accumulate in these tissues.
Deficiencies of individual sphingolipid-degrading lyso-somal enzymes result in lipid storage diseases.
Depending on the enzyme affected, several types of sphingolipidoses have been recognized (Fig. 11.30). For example, in Niemann-Pick disease, sphingomyelinase is defective, leading to sphingomyelin accumulation (Fig. 22.27; number 6).
General Features
Certain features are common to all types of sphingolipi-doses. These are:
1. Inheritance: Sphingolipidoses are autosomal recessive diseases. The only exception is Fabry’s disease, which is X-linked.
2. Severity: With exception of Fabry’s disease and Gaucher’s disease (adult form), all other types of sphingolipidoses are relentlessly progressive, causing death in early life.
Milder variants of lipid storage disease also occur, in which affected homozygotes have greatly decreased, but not completely absent enzyme activity. The severity and prognosis of disease depends largely on the residual enzyme activity.
3. Diagnosis: Sphingolipidoses rests on determination of enzyme activities in cultured leukocytes, cultured skin fibroblasts; or, for prenatal diagnosis, cultured amniotic cells are used for determination of the enzyme activities.
Heterozygotes can be identified because their enzyme activity is approximately one-half of normal; the homozygotes on the other hand have near-zero enzyme activity.
Major Types of Storage Diseases
Some major types of sphingolipidoses are depicted in Figure 11.30; numbered reactions refer to storage diseases in the Figure.
1. Generalized gangliosidosis is because of deficiency of β-galactosidase, so that there is impaired degradation of GM1 gangliosides and it accumulates in tissues. Mental retardation, skeletal deformities and hepatomegaly are the prominent features.
2. Tay-Sachs disease is a rare disease (except in Ashkenazi Jews) because of complete deficiency of hexosaminidase A. It leads to accumulation of the substrate, ganglioside GM2. The affected child appears normal at birth but within the first year of life develops signs of mental and neurological deterioration, and also hepato-splenomegaly. The lipid laden cells, the ganglion cells in retina (fovea centralis) cause visual impairment. Death occurs before the child reaches the age of 3 years.
3. Gaucher’s disease is a rare disease caused by deficiency of the enzyme glucocerebrosidase, so that tissue levels of glucocerebrosides elevate. The clinical course of the infantile form is similar to Tay-Sachs disease; and the adult onset form is without mental retardation.
4. Krabbe’s disease results because of deficiency of β-galactosidase, therefore there is accumulation of galactocerebrosides. There is near complete absence of myelin in the nervous tissue. The clinical features include mental retardation, blindness, deafness and hypertonia.
Some other disorders associated with abnormal sphingolipid metabolism are also shown in Table 11.3 .
Table 11.3
Some less commonly described sphingolipidoses

* The numbers refer to the numbered reactions of Figure 11.30.
Exercises
Essay type questions
1. Describe β-oxidation of a 16-carbon saturated fatty acid and its energetics. Explain the role of carnitine in transport of the fatty acid into mitochondrion.
2. Describe synthesis and oxidation of ketone bodies and discuss regulation of ketogenesis.
3. Describe the shuttle systems for transport of fatty acids into the mitochondria and acetyl CoA into the cytosol.
4. Describe the functions and metabolism of phos-pholipids.
5. Describe in detail the pathway of de novo synthesis of fatty acids and its regulation.
6. Describe the major mechanisms of regulating fatty acid metabolism in humans.
7. How are triacylglycerols, glycerophospholipids and sphingolipids synthesized?