Vitamins
Vitamins are organic nutrients of low molecular mass (< 1500 Da) that play a central role in metabolism. Most vitamins are either not synthesized or are synthesized in inadequate amounts by the human organism; therefore, they must be obtained from an exogenous source, such as diet or bacterial flora in the gut.
Dietary vitamin deficiency may lead to diseases and even death. Vitamins and their utility for humans were known much before their structure and function were elucidated. It was observed that in some countries where the staple food of the people was rice, a disorder called “beriberi” was prevalent. In Sinhalese, beriberi means ‘I cannot’ (said twice), signifying that the patient is too ill to do anything. Diet of these patients was believed to be deficient in some essential organic nutrient which was subsequently purified by a Polish biochemist, Caimir Funk. Since this nutrient exhibited properties of an amine, it was called “vitamine”, denoting that it was like an amine. Later, a number of other essential nutrients were discovered and it was revealed that all of them were not amines. Therefore, the suffix ‘-e’ was dropped and the name “vitamin” was adopted. Presently 13 different vitamins are known to be required in the diet of the humans and many animal species for normal growth and function.
Vitamins are biosynthetic precursors of physiologically active forms called coenzymes. Coenzymes participate in many enzyme reactions. In addition, certain specialized functions in humans are performed by vitamins. For example, vitamin A plays an important role in visual process. The biology of vitamins may be examined from two viewpoints: nutritional and biochemical. The former is concerned with minimum daily requirements, dietary sources, bioavailability, and deficiency syndromes. The biochemist examines structure, conversion to coenzymes, mechanism of action, mode of transport and storage, metabolism and biochemical role. Both the aspects are covered in this chapter, though the emphasis will be on biochemical properties of vitamins.
After going through this chapter, the student should be able to understand:
• Sources, daily requirements and deficiency of vitamins.
• Water-soluble vitamins: structure, biochemical role and nutritional disorders of riboflavin, niacin, pyridoxine, thiamine, pantothenic acid, cobalamin, biotin and folic acid.
• Fat-soluble vitamins: structure, biochemical role and nutritional disorders of vitamins A, D, E, and K.
I Classification and Nomenclature
Vitamins are divided into fat-soluble and water-soluble groups (Table 18.1 ). The fat-soluble vitamins are A, D, I and K, whereas water-soluble vitamins include B group of vitamins (B1, B2, B3, B5, B6, B7, B12 and folate) and ascorbic and (vitamin C).
The classification in two broad groups—fat-soluble and water-soluble—has persisted, although the compounds within a given group have widely different structures and functions. For example, vitamin A, D, E, and K of the fat-soluble group have a limited structural resemblance with one another and perform vastly different roles. But all fat soluble vitamins have same common properties:
1. They are soluble in fats, and therefore handled in the same way with respect to their absorption from the gut, transport and distribution.
2. They may be stored in the body.
3. They may be toxic or even lethal if taken in excessive quantities.
Water soluble vitamins cannot accumulate to toxic levels in the body as the excessive intake results in their excretion in the urine. Except for vitamin B12 there is no storage capacity for the water-soluble vitamins, so their intake has to be more frequent than fat-soluble vitamins that are stored. A well-nourished adult, for example, may have three years’ supply of vitamin A but only three months’ supply of vitamin C.
Some vitamins of the B-complex group are known as the energy-releasing vitamins because they participate in the energy-yielding catabolic pathways. Examples include vitamins B1′, B2′, B3′, B5′, B7′, etc. Folic acid and vitamin B12 are loosely referred to as haematopoietic.
II Sources, Daily Requirements and Deficiency of Vitamins
Humans obtain vitamins from two sources:
1. Diet: Since vitamins cannot be manufactured by humans, they must be obtained from food (exception is vitamin D).
2. Intestinal microorganisms: Some vitamins can be synthesized by the intestinal microorganisms. However, the quantity synthesized in this manner may not be sufficient to meet the daily requirement. Biotin, however, is an exception; it is synthesized in larger quantities than required by the body.
Vitamins are referred to as micronutrients because their dietary requirements are in negligible quantities, amounting to few micrograms or milligrams per day. The dietary requirements for vitamins are specified in terms of a recommended daily allowance (RDA). The RDA defines not a minimal requirement but dietary intake that is considered optimal under ordinary conditions. Age, sex, body weight, diet and physiological status have a significant effect on the RDA. Thus, increased dietary intake of many vitamins are recommended during pregnancy and lactation. Table 18.1 summarizes the RDAs for most important vitamins for a 70 kg male.
 A number of water-soluble vitamins are grouped into B-complex because they occur together in the same food source.
A number of water-soluble vitamins are grouped into B-complex because they occur together in the same food source.
Dietary Vitamin Deficiencies
May result in pathology and even death, as noted earlier. Various causes of vitamin deficiency are as below:
1. Inadequate dietary intake due to faulty dietary habits or poverty.
2. Inadequate intestinal absorption, which may result from various gastrointestinal disorders, or from biliary obstruction. The latter may lead to deficiency of the fat-soluble vitamins (A, D, E, and K) since bile is required for their absorption. Lack of intrinsic factor, a factor required for the intestinal absorption of vitamin B12, results in deficiency of this vitamin which leads to pernicious anaemia. Some vitamins participate in enterohepatic circulation. Hence, deficiency of such vitamins is a natural consequence of impaired enterohepatic circulation.
3. Inadequate utilization by the target tissues may also lead to vitamin deficiency, even when dietary intake and absorption are normal. Causes of inadequate use include:
(i) Lack of transport proteins that carry the vitamins to the peripheral sites where they are utilized.
(ii) Defective uptake of the vitamins by the target tissues or impaired interaction of the (fat-soluble) vitamins with the receptors located on target tissues.
(iii) Failure to convert the vitamin precursor(s) to activated form(s).
4. Increased requirements which occur during pregnancy, lactation, growth, wound healing and convalescence. In some cases, an increased requirement precipitates a borderline deficiency into a frank deficiency.
5. Drug-induced deficiency such as loss of vitamin synthesis in the gastrointestinal tract due to elimination of the microorganisms by antibiotics. Pyridoxine deficiency develops in patients receiving isoniazid for the treatment of tuberculosis (isoniazid forms a hydrazone with pyridoxal; the pyridoxal-hydrazone is rapidly excreted in urine, and the vitamin deficiency results).
Absolute or relative deficiencies of individual vitamins cause characteristic diseases or syndromes, whose symptoms can be traced to specific metabolic functions of the missing nutrient. They are a major public health concern, especially for such at-risk groups as infants, pregnant women, alcoholics and the elderly.
III Water-soluble Vitamins
The water-soluble vitamins are the largest class of essential micronutrients, performing a broad range of biological functions. They are biosynthetic precursors of coenzymes for various enzymes, thereby playing pivotal role in cellular metabolism. Table 18.2 lists vitamins with their structures and coenzyme forms.
Table 18.2
Water-soluble vitamins: structures and coenzyme forms. The arrows indicate the portions which are important for bioloaical activity
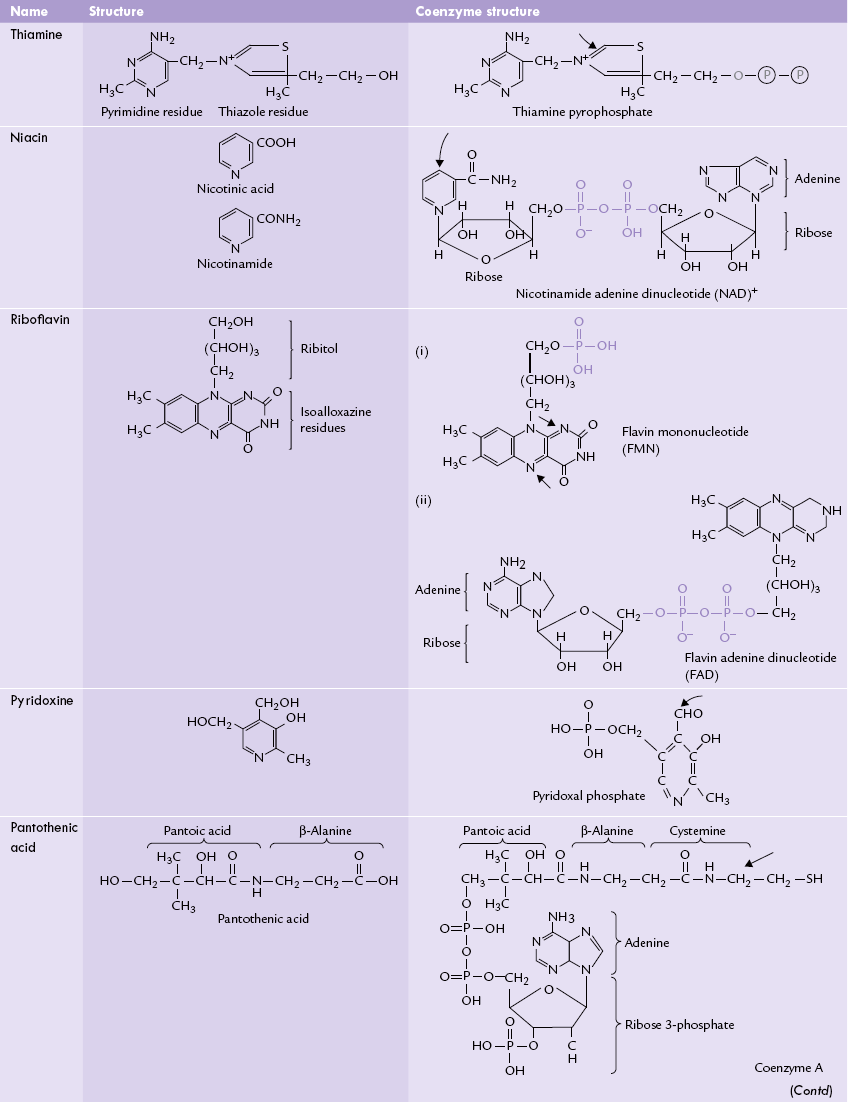
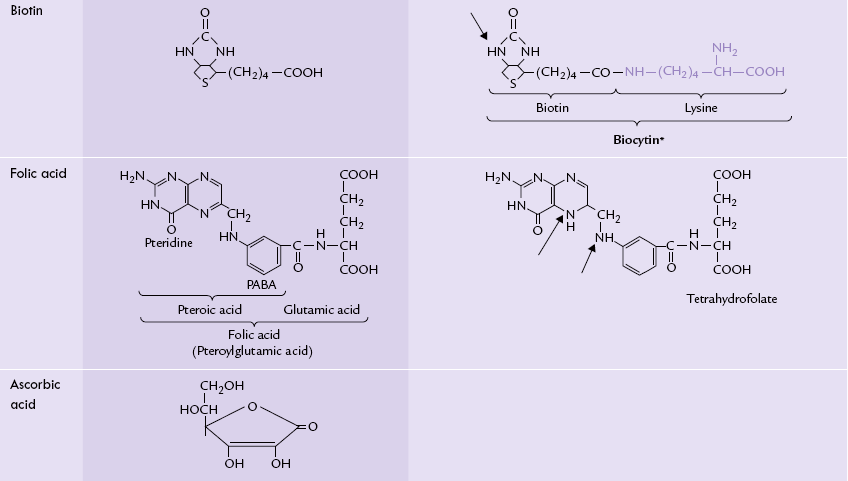
Most water-soluble vitamins are precursors of coenzymes: riboflavin, for example, is required for the synthesis of the flavin coenzymes, niacin for NAD+ and NADP+, thiamine for thiamine pyrophosphate, pantothenic acid for coenzyme A, and folic acid for tetrahydrofolate.
A Thiamine (Vitamin B1)
Thiamine, also called aneurine, was first recognized by a Dutch physician, Christian Eijkman in 1926 as the antiberiberi factor.
Structure
As elucidated by Adolf Windaus, thiamine consists of a substituted pyrimidine residue linked to a thiazole (sulphur and nitrogen) ring through a methylene bridge. Thiamine-dependent reactions are aldehyde transfers, in which the aldehyde is bound covalently to one of the carbons in the thiazole ring, shown by arrow in Table 18.2.
Absorption and Transport
Dietary thiamine is absorbed readily from jejunum and proximal ileum and is transported to tissues where it is converted to the active form, thiamine pyrophosphate. Two different mechanisms are involved in absorption depending on the level of intake:
1. At levels below 5 mg/day, the absorption occurs by an active, ATP-dependent process, which is saturable at concentrations of 0.5–1.0 µmol/L.
2. At levels higher than this, the absorption occurs by passive diffusion.
Ethanol inhibits the active transport of thiamine, and this could be one of the causes of thiamine deficiency in alcoholics, whose thiamine intakes are usually also low because of their penchant for drinking but not eating.
Coenzyme Functions
Following absorption, thiamine is transported to tissues bound to albumin and, to a small extent, other proteins.
After cellular uptake it is converted into its active form, thiamine pyrophosphate (TPP) in an ATP-dependent reaction catalyzed by thiamine pyrophosphokinase. Total amount of thiamine in the body is approximately 30 mg, about 80% of which is in the form of TPP.
TPP serves as a coenzyme in several enzyme catalyzed reactions, which involve aldehyde transfer. These include oxidative-decarboxylation, transketolase reaction, decarboxylation, etc.
1. Oxidative-decarboxylation: TPP is a component of mitochondrial multienzyme complexes which bring about oxidative-decarboxylation of α-keto acids, e.g. pyruvate and α-ketoglutarate.

Malfunction of TCA is thus an obvious consequence of thiamine deficiency, and results in defective energy metabolism.
Moreover, acetyl CoA is a precursor for the synthesis of the neurotransmitter acetylcholine, and also for the synthesis of lipids, including myelin, which may explain the importance of thiamine in the correct functioning of the nervous system.
2. Transketolase reactions: TPP is also a coenzyme for the transketolase, in the pentose phosphate pathway. The pathway generates ribose sugars and supplies NADPH necessary for a wide variety of redox and biosynthetic reactions.
3. Decarboxylation reactions: TPP serves as a coenzyme for decarboxylation reactions in the metabolism of branched chain amino acids. Patients with maple syrup disease suffer from a defect in the decarboxylation reaction during metabolism of these amino acids, and therefore, may be successfully treated with large doses of thiamine.
4. Others: Another thiamine derivative, thiamine trisphosphate, is known to be involved in nerve conduction, but the precise role of the vitamin in this respect is not clear.
Requirement and Dietary Sources
Thiamine is related to energy metabolism, therefore its dietary requirement depends on the caloric intake. The recommended daily allowance is 0.5 mg/1000 kcal for adults with the recommendation for a minimum absolute intake of 0.5 mg/day. Dietary sources are given in Table 18.1.
Deficiency
Thiamine deficiency leads to beriberi. In rice-consuming areas of Asia, where it is customary to polish this staple grain to remove its coarse, thiamine-containing outer layer, the disease is most prevalent. Patients with history of malnutrition, chronic inflammation and parenteral nutrition are especially prone to develop thiamine deficiency.
Thiamine deficiency, however, is not common in developed western nations, except in chronic alcoholics.
Mild thiamine deficiency is associated with weakness and gastrointestinal disturbances. Peripheral neuropathy, mental confusion and ataxia develop in moderate deficiency conditions; and, severe deficiency presents with severe neuromuscular and cardiovascular disorders.
The full blown deficiency, known as beriberi, is categorized into the following types.
1. Dry beriberi: The neuromuscular symptoms predominate in this type. In longstanding cases, there is degeneration and demyelination of sensory and motor nerves and severe wasting of muscles.
2. Wet beriberi: This type develops in more severe cases of thiamine deficiency. The cardiovascular manifestations appear in this type and oedema is a notable feature. These signs and symptoms are largely accounted by accumulation of pyruvic and lactic acids (Case 18.1).
3. Infantile beriberi: This type develops in breastfed infants, mostly between the second and the fifth month, in the areas where beriberi is endemic. Classical signs of beriberi, however, are present in only about 50% of the mothers.
4. Thiamine deficiency due to alcoholism: Chronic alcoholism does not result in beriberi but in Wernicke-Korsakoff syndrome. The acute stage of this disease, known as Wernicke’s encephalopathy, is characterized by mental derangement, delirium and ataxia. In the chronic stage, known as Korsakoff psychosis, the patient has anterograde amnesia.
5. Thiamine deficiency due to thiaminase: Thiamine can be destroyed if the diet contains thiaminases, which cleave the pyrimidine ring from the thiazole ring. These enzymes are present in some ferns, raw fishes and sea foods, and they are thought to contribute to the incidence of beriberi in areas in Japan where raw fish (Sushi) is a common dietary constituent.
Assessment of Thiamine Status
Thiamine status is evaluated by estimating urinary thiamine excretion and plasma levels of pyruvate and lactate, particularly after an oral glucose load (these acids accumulate because of the decreased activity of pyruvate dehydrogenase). Determination of erythrocyte transketolase activity, which requires TPP as a coenzyme, confirms the deficiency (Case 18.1).
Toxicity
Excess intake of thiamine, like other water soluble vitamins, is usually not toxic because it is promptly excreted in the urine. However, it has been recorded that chronic intake of thiamine in excess of 3 g/day are toxic to adults, causing headaches, irritability and dermatitis, and in extreme cases may even cause death.
B Redox Vitamins: Niacin (Vitamin B3) and Riboflavin (Vitamin B2)
Niacin and riboflavin give rise to coenzymes that participate in redox reactions.
Niacin is converted to the coenzymes, NAD+ and NADP+, and riboflavin to flavin mononucleotide (FMN) and flavin adenine dinucleotide (FAD).
Niacin
Niacin is a nutritional term used to refer to the vitamers (different structural forms of a vitamin with the same biological activity): nicotinic acid and nicotinamide. The word niacin is coined from letters of three words: nicotinic acid and vitamin. This was to avoid confusion with nicotine, a compound present in tobacco. Pellagra caused by niacin deficiency, was once endemic among poor peasants of Latin America; these people subsisted chiefly on maize (American corn), which is deficient in tryptophan.
Because nicotinic acid can be formed in the body from tryptophan (1 mg of the vitamin from 60 mg tryptophan), deficiency of this amino acid leads to deficiency of nicotinic acid as well.
Structure
Structures of the two vitamers, nicotinic acid and nicotinamide are given in Table 18.2. Nicotinic acid is a pyridine-3-carboxylic acid. Niacinamide, the form present in tissues, is the acid amide.
Coenzyme Forms of Niacin
Niacin, in the form of nicotinamide, is incorporated into the structure of two coenzymes: nicotinamide adenine dinucleotides (NAD+) and nicotinamide adenine dinucleotide phosphate (NADP+). The niacin is attached to a ribose phosphate to form a mononucleotide, which is then attached to AMP to form the nicotinate adenine dinucleotide (Fig. 18.1 ). The latter reacts with glutamine to form its amide, i.e., NAD+. The nitrogen atom of nicotinamide contains one positive charge, hence the structure is abbreviated with a positive sign, as NAD+.
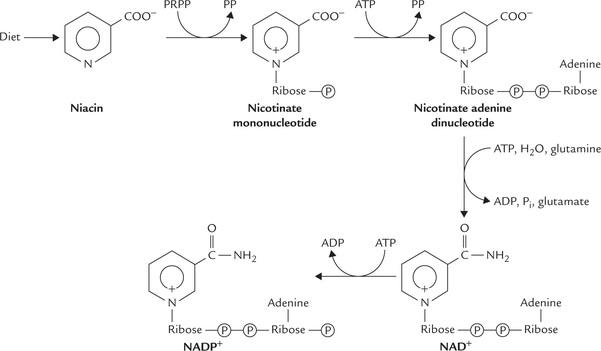
Fig. 18.1 Synthesis of nicotinamide adenine dinucleotide (NAD+). Attachment of a phosphate group to a 2’-OH in the NAD+ yields NADP+.
In the case of NADP+, a phosphate group is attached to the 2’-hydroxyl group of ribose (of the AMP).
Coenzyme Activities
1. Oxidation and reduction: Both the coenzymes, NAD+ and NADP+, participate in a vast variety of oxidation-reduction reactions. NAD+ is used largely in the catabolic redox reactions occurring in the mitochondrial matrix Table (14.1). NADP+ in its reduced form, NADPH, is used largely in the reductive biosynthesis reactions (e.g. cholesterogenesis and lipogenesis) in the extramitochondrial compartment of the cell. Thus, NAD+ and NADP are present in distinct cellular compartments and catalyze different reaction types. In line with this is the observation that in rat liver cytosol, NADPH : NADP+ ratio is about 80, whereas the NADH : NAD+ ratio is only 8 X 10 4.
2. ADP-Ribosylation: NAD+ functions as an ADP-ribose donor. Such reactions are called the ADP-ribosylation reactions, and they occur in the nucleus; for example, the eukaryotic post-translational modifications catalyzed by the enzyme poly ADP-ribose polymerase. The enzyme seems to play a role in DNA repair and other cellular responses to DNA damage.
Clinical Deficiency
The niacin deficiency disease is pellagra (derived from Italian words pelle = skin + agro = rough) involves the gastrointestinal tract, skin and central nervous system. The symptoms comprise three D’s: diarrhoea, dermatitis and dementia. The fourth D, i.e. death, follows in the untreated cases. The dermatitis, which resembles sunburn, is seen in areas of the skin exposed to the sunlight. It is probably related to the role of NAD+ in DNA repair reactions following damage caused by the UV light.
Pellagra is also seen in conditions where dietary amino acids, including tryptophan, are not properly absorbed, as in Hartnup’s disease, a genetic disorder of amino acid transport. Severe pellagra can be fatal.
Niacin (B3) is a generic name for nicotinic acid or nicotinamide, either of which is an essential nutrient. Severe niacin deficiency produces dermatitis, diarrhoea and dementia.
Most cases of pellagra respond to nicotinamide in a dose of 100 mg every 4 to 6 hours: response is so rapid that within 24 hours the erythema of skin diminishes, the diarrhoea ceases and there is striking improvement in the patient’s behaviour and mental attitude. Nicotinic acid is equally effective for the treatment.
Therapeutic Uses
Nicotinic acid is used in pharmacological doses (several grams daily) for the treatment of hyperlipidaemias. It is observed to lower total plasma cholesterol, LDL-cholesterol and VLDL-triacylglycerols in patients with hyperlipoproteinaemias. It also lowers the circulating free fatty acid levels by inhibiting hormone-sensitive lipase, thereby blocking adipose tissue lipolysis. Hypoglycaemic effects with large doses (1–2 g daily) have also been observed. These effects are unrelated to the vitamin activity of nicotinic acid.
The large doses necessary to produce the lipid- and glucose-lowering effects, have undesirable side effects, mainly vasodilatation and flushing. Cases of hepatic toxicity have also been reported in some patients.
Riboflavin
Structure
Chemically, riboflavin is 6,7-dimethyl-9D-ribitol isoalloxazine, consisting of an isoalloxazine ring with a ribitol side chain (Table 18.2). Ribitol is an alcohol of ribose sugar. The ring system of isoalloxazine accounts for the intense yellow colour of the vitamin and its emission of greenish-yellow fluorescence following exposure to UV light.
Coenzyme Activity
Riboflavin is a component of two coenzymes: flavin mononucleotide (FMN) and flavin adenine dinucleotide (FAD). They are known as flavin nucleotides. FMN is formed (in intestine) by attachment of a phosphate group to the ribitol side chain. FAD, which contains adenosine linked via phosphate group to FMN, is formed by adenylation of the latter in liver (Fig. 18.2 ).
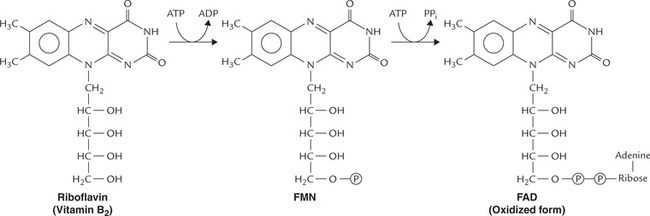
The flavin nucleotides serve as prosthetic groups for the enzymes called flavin-dependent (flavoprotein) enzymes. These enzymes remove a pair of hydrogen atoms from the substrate and thereby participate in a number of oxidation-reduction reactions in metabolism.
• FMN is a cofactor for L-amino acid oxidase.
• The enzyme NADH dehydrogenase, of respiratory chain contains FMN.
• FAD is a constituent of the Complex II of the respiratory chain.
• FAD is a constituent of the microsomal hydroxylase system.
• FAD is a cofactor for several enzymes, e.g. D-amino acid oxidase, succinate dehydrogenase, acyl CoA dehydrogenase, glycerol 3-phosphate dehydrogenase, xanthine oxidase, pyruvate dehydrogenase, α-ketoglutarate dehydrogenase, etc.
During oxidation process, FAD accepts two hydrogen atoms from substrate and gets reduced to FADH2. The two nitrogen atoms of the isoalloxazine ring accept the hydrogen atoms, as indicated by the arrow in Table 18.2. FMN is likewise reduced to FMNH2.
Absorption, Transport and Storage
Riboflavin is ingested in form of flavoproteins. The FAD and FMN components are released from the protein complex in the stomach, and free riboflavin is released in the intestine, from which it is absorbed by an active ATP-dependent process. The activation of riboflavin via an ATP-dependent enzyme system occurs next, resulting in the production of FMN and FAD. The main storage form of the vitamin, found mainly in the liver, is FAD.
Clinical Deficiency
Dietary requirement of riboflavin is low (Table 18.1), and the intestinal flora synthesizes it. Therefore, isolated deficiency of riboflavin is rare. It is generally combined with other deficiencies, such as beriberi, pellagra and kwashiorkor. Owing to light sensitivity of riboflavin, phototherapy for physiological jaundice may induce transient riboflavin deficiency in infants.
Deficiency Manifestations
Symptoms are confined to skin and mucous membranes. Angular stomatitis (inflammation of the mouth), glossitis (inflammation of the tongue), cheilosis (reddening of the mucous membrane of lips), seborrheic dermatitis (rough and scaly skin around nasolabial and scrotal areas), corneal vascularization, and a form of peripheral neuropathy are the prominent features. The condition is referred to as ariboflavinosis.
The symptoms of ariboflavinosis are relatively mild and certainly not life threatening even though flavoproteins are essential for life. There are two main reasons for this. One is that riboflavin is associated with proteins in the diet and any diet providing protein will also provide a fair amount of riboflavin. The other reason is that the recycling of riboflavin released from FAD and FMN is extremely efficient and, therefore, only small amounts need be ingested.
Like thiamine deficiency, riboflavin deficiency can be seen in conditions such as chronic alcoholism, malnutrition, anorexia and malabsorption. Drugs, such as barbiturates, may also cause riboflavin deficiency by inducing microsomal oxidation of the vitamin.
Laboratory diagnosis of riboflavin deficiency is difficult. Serum and urine riboflavin fall low only in severe deficiency. Erythrocyte enzyme activity measurement glutathione reductase (a riboflavin-dependent enzyme) used as index of riboflavin status.
C Pyridoxine (Vitamin B6)
Vitamin B6 plays a key role in amino acid metabolism, being required in several reactions for the synthesis, catabolism and interconversion of amino acids (Chapter 13). It consists of three different vitamers: pyridoxine, pyridoxal, and pyridoxamine, all of which can be phosphorylated and converted to the active form, pyridoxal phosphate (Fig. 18.3 ). Pyridoxine is the major form of vitamin B6 in diet, occurring widely in both animal and plant tissues.
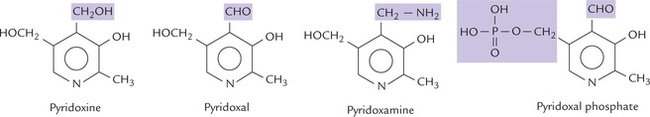
Fig. 18.3 Structures of B6 vitamins. Pyridoxine is primary alcohol, pyridoxal is an aldehyde, and pyridoxamine is an amine.
Structure
All forms of the B6 vitamin are pyridine derivatives, differing from one another only in nature of the functional group attached to the pyridine ring.
The aldehyde group of PLP forms aldimine derivatives with primary amino groups of amino acids.
Absorption, Transport and Excretion
Vitamin B6 is rapidly absorbed from the intestine by passive diffusion. The absorption is highly efficient: about 80% of the ingested vitamin is absorbed. Phosphorylated pyridoxine vitamers are hydrolyzed by intestinal membrane alkaline phosphatase and dephosphorylated forms are absorbed.
The major circulating form of vitamin B6 is pyridoxal phosphate (PLP). It is produced from the absorbed pyridoxine and pyridoxamine. Pyridoxic acid is the principal excretory form of the vitamin in urine. Its formation is catalyzed by aldehyde oxidase.
Limited storage of pyridoxine is possible: most of the body stores are associated with the enzyme glycogen phosphorylase, for which it is a coenzyme. In fact, about 70% of total PLP content of the body resides in muscles, in association with this enzyme.
Coenzyme Functions
Pyridoxal phosphate serves as a coenzyme for a broad range of reactions in intermediary metabolism, especially of amino acid metabolism where it seems to play a central role (Chapter 13). For this reason, requirement of B6 also increases with protein intake. Some of the more important PLP-dependent reactions are:
1. Transamination: PLP is a coenzyme for transaminases, where it acts as an amino group carrier (Chapter 13).
2. Decarboxylation: All decarboxylation reactions of amino acid metabolism require PLP. Particularly important in this respect are glutamate decarboxylase and DOPA decarboxylase. These enzymes are involved in the production of γ-aminobutyric acid (GABA) and catecholamine neurotransmitters (epinephrine, norepinephrine, dopamine) respectively in the nervous system.
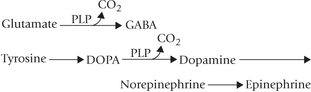
Synthesis of the serotonin neurotransmitter also requires B6’ as does that of histamine.
3. Condensation: The enzyme 8- aminolevulinic acid synthase that catalyzes joining of glycine and succinyl CoA in the haem biosynthetic pathway requires PLP as a cofactor (Chapter 16). This accounts for anaemia seen in B6 deficiency.
4. Transulfuration: PLP is a coenzyme for cystathionine synthase and cystathionine γ-lyase, which play important role in methionine and cysteine metabolism (Fig. 13.15). In these reactions transfer of sulphur from methione to serine occurs to produce cysteine. In PLP deficiency, blood level of homocysteine (substrate for cystathionine synthase) is increased, which is correlated with occlusive vascular diseases. Therefore, pyridoxine intake may be recommended in clinical practice to prevent CAD.
5. Tryptophan metabolism: PLP is a cofactor for the enzyme kynureninase, which catalyzes release of alanine from 3-hydroxykynurenine to form 3-hydroxyan-thranilic acid, during catabolism of tryptophan (Fig. 13.18).
In deficiency of PLP, 3-hydroxyanthranilic acid accumulates and is converted into an alternate metabolite, xanthurenic acid which is excreted in urine. The urinary excretion of xanthurenic acid serves as an indicator of pyridoxine deficiency: it increases in the deficiency state.
6. Others: PLP is a cofactor for glycogen phosphorylase in both liver and muscle, and therefore, favours glycogenolysis. PLP also serves as coenzyme for the enzymes responsible for the specific deamination of serine, threonine and cysteine. It is also required for the synthesis of sphingosine, a component of sphingomyelin and sphingolipids. Cellular uptake of L-amino acids also requires participation of PLP.
Clinical Deficiency
Deficiency of vitamin B6 is rare because of its widespread distribution in a variety of foodstuffs. Deficiency symptoms are most often seen in alcoholics, women taking oral contraceptives, and in infants fed with formula diet low in this vitamin. Probably the commonest cause of deficiency is drug antagonism. Isoniazid, an antitubercular drug, reacts with PLP to form a hydrazone that is biologically inactive, rapidly excreted in urine, and also inhibits pyridoxal kinase. Penicillamine, used in the treatment of Wilson’s disease and rheumatoid arthritis, also combines with PLP to render it unavailable.
The major symptoms of B6 deficiency include neural dysfunction and anaemia. The former is accounted by an impairment in the synthesis of neurotransmitters such as norepinephrine and serotonin. Anaemia is accounted by impairment of haem biosynthesis. Other features of B6 deficiency include lesions of the skin and mucosa, sideroblastic anaemia and personality changes. In rare instances, vitamin B6 can be toxic and cause convulsions at very high levels. Possible mechanism is by enhancing decarboxylation of L-DOPA in several tissues. In patients treated for parkinsonism, efficiency of DOPA is thereby reduced.
Assessment of pyridoxine status
As with other B-complex vitamins, assessment of pyridoxine status is based on the measurement of erythrocyte enzymes; the enzyme in this case is aspartate aminotransferase. Tryptophan load test, involving measurement of urinary excretion of xanthurenic acid following a load dose of tryptophan is the other important test: xanthurenic acid excretion is increased in B6 deficiency. Measurement of urinary homocysteine and cystathionine after a methionine load, and urinary pyridoxic acid and PLP level in blood are the other important tests.
D Pantothenic Acid
Pantothenic acid consists of pantoic acid and (β-alanine (Table 18.2). It is a building block of coenzyme A and of the phosphopantotheine group in the fatty acid synthase complex (Chapter 11). The CoA plays a significant role in several body reactions, especially the biosynthetic pathways where the thiol (SH) group of CoA forms thioester bonds with many organic acids. This results in activation of the organic acids. Conversion of succinate to succinyl CoA and of acetate to acetyl CoA are some examples. Succinyl CoA is a precursor in haem biosynthetic pathway, and acetyl CoA in lipogenesis.
Deficiency of pantothenic acid in humans is unknown. In experimental studies, the isolated deficiency can be induced only under rigorously controlled experimental conditions. The RDA of 5–10 mg is readily met by ordinary diets.
E Biotin (Vitamin B7 or H)
Biotin is a coenzyme for carbon dioxide fixation reactions. It serves as prosthetic group of ATP-dependent carboxylases, including pyruvate carboxylase, acetyl CoA carboxylase and propionyl CoA carboxylase. It was first isolated by Vincent du Vigneaud in 1942 (Nobel Prize 1955).
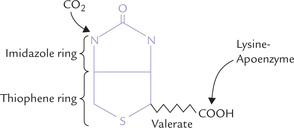
Structure
Biotin consists of an imidazole ring that is cis-fused to a thiophene ring bearing a valerate side chain (Fig. 18.4 ). The valeryl carboxyl group is attached to the e-amino group of a lysine residue in the apoenzyme.
Requirement, Sources and Causes of Deficiency
About 5µg/1000 kcal biotin is required daily. Biotin is widely distributed in foods. Liver, yeast, peanuts, kidney, and egg yolk are especially rich sources. Biotin is mostly bound to dietary protein by an amide linkage. The linkage is cleaved prior to absorption of biotin from intestine.
The intestinal flora synthesize biotin in quantities that far exceed the daily requirement. This is reflected in urinary and fecal excretion of biotin, which is 2–3 times its daily dietary intake. Thus, biotin deficiency is unknown, except in a small number of people with unusual dietary habit of consuming large amounts of uncooked eggs. Egg white contains the glycoprotein avidin, a homotetramer of four subunits (MW 70,000), which binds the imidazole group of biotin tightly, thereby preventing its absorption from the intestine. However, consumption of more than 20 raw egg whites per day only is likely to induce biotin deficiency.
Sterilization of the intestine by antibiotics or administration of biotin analogues can also induce biotin deficiency.
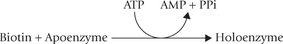
Coenzyme Functions
Biotin is covalently attached to the apoenzyme by an amide linkage with a lysyl ε-amino group, as noted earlier. This binding is catalyzed by the enzyme holocarboxylase synthetase:
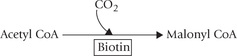
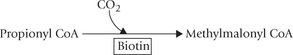
Biotin serves as a carrier of the activated carbon dioxide transferring it to various acceptor molecules, e.g. acetyl CoA, pyruvate and propionyl CoA, etc. Such reactions are termed as carboxylation reactions and the enzymes catalyzing them are called carboxylases.
The most important carboxylation reactions are:
1. Conversion of acetyl CoA to malonyl CoA, catalyzed by acetyl CoA carboxylase, in fatty acid synthesis.
2. Pyruvate to oxaloacetate conversion, catalyzed by pyruvate carboxylase, in gluconeogenesis. It is an important anaplerotic reaction also.

3. Conversion of propionyl CoA to methylmalonyl CoA, by propionyl CoA carboxylase, in catabolism of branched chain amino acids and odd chain fatty acids
Carboxylations not dependent on biotin: Not all carboxylation reactions, however, require biotin. Some examples are: (a) formation of carbamoyl phosphate by carbamoyl phosphate synthetase, (b) vitamin K-dependent γ-carboxylation of glutamyl residues of several of the clotting factors, (c) conversion of pyruvate to malate by malic enzyme, and (d) addition of C-6 to the purine ring.
Recycling: Proteolytic degradation of biotin-containing enzymes, both in the intestinal lumen and in the tissues, produces biotinyl-lysine or biocytin. It consists of biotin bound to the ε-group of lysine (Fig. 18.4). Biotinidase cleaves biocytin to biotin and lysine. Thus, biotin is recycled.
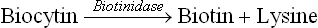
Biotinidase deficiency is an autosomal recessive disorder which disrupts the recycling. Thus, biotinidase deficiency causes non-dietary biotin deficiency.
Clinical Deficiency
Patients with biotin deficiency develop seborrhoeic dermatitis, anorexia, and alopecia with loss of hair follicles. The infants with biotinidase deficiency present with hypotonia, seizures, optic atrophy, dermatitis and conjunctivitis. In experimental biotin deficiency, produced by feeding excessive egg-white (at levels that provide 30% of the dietary energy), subjects suffered from muscle pain, hallucinations, depression, sleepiness as well as hair loss and dermatitis.
Toxicity due to excessive consumption of biotin is not known.
F Cobalamin (Vitamin B12)
Vitamin B12 is important haematopoietic vitamin (together with folic acid) deficiency of which causes megaloblastic anaemic—large sized RBCs. The anaemia is associated with neurological deterioration, and called pernicious anaemia.
Structure
Cobalamin is chemically the most complex of all vitamins (Fig. 18.5 ). It is unique among vitamins as it contains an essential trace element, cobalt. Cobalt lies at the centre of a corrin ring system which consists of four pyrrole rings. The tetrapyrrole structure of corrin is similar to that of porphyrin, except that it is more hydrogenated and two of the pyrrole rings are linked directly rather than through a methenyl bridge.
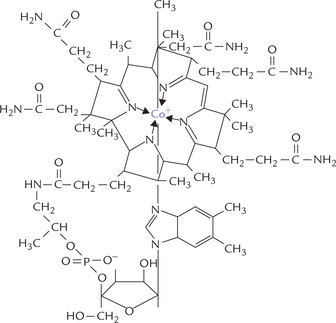
Synthesis
Cobalamin is the only vitamin that is synthesized by neither plants nor animals, but only by a few species of bacteria. It is, therefore, absent from all plants but is concentrated in the liver of animals in three forms: methylcobalamin, adenosylcobalamin and hydroxycobalamin. Liver is thus a useful source of this vitamin.
Vitamin B12 when isolated from natural sources, is called cyanocobalamin because it contains a cyano group, which is essential for chelation of the cobalt ion. The cyano group needs to be removed for converting the vitamin to its active form. Interestingly, the only known role of cobalt in mammalian systems is that it is a part of the vitamin B12.
Absorption, Transport and Distribution
Cobalamin is synthesized by microorganisms, but it enters the food chain only through foods of animal origin. It usually occurs in protein-bound forms and must be separated from these proteins by either acid hydrolysis in the stomach or trypsin digestion in the intestine. In stomach, dietary vitamin B12 is bound by a glycoprotein called the intrinsic factor which is produced by the stomach’s parietal cells. The vitamin-intrinsic factor complex travels to the ileum, where the vitamin is absorbed, via a receptor in the intestinal mucosa (Fig. 18.6 ). The rate-limiting factor in this process is the number of ileal receptor sites. The complex then dissociates and the liberated vitamin B12 is converted to methyl cobalamin and released into the bloodstream.
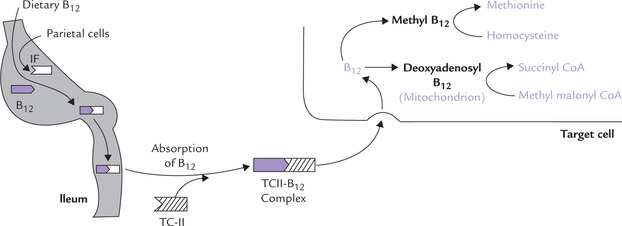
Fig. 18.6 Intestinal absorption of vitamin B12’ transport in bloodstream and utilization in the target cell (IF = intrinsic factor, TC-II = transcobalamin II).
Methyl cobalamin is the predominant circulating form. It is transported in a bound form with transcobalamin-II (TCII), a glycoprotein. The TCII-B12 complex enters virtually all cells of the body via a specific cell-surface receptor. The ligand receptor complex is internalized and appears in lysosomes, where TCII is degraded, thus freeing the cobalamin. The latter then passes into the cytoplasm through mediation of a specific transport mechanism.
Vitamin B12 Transport Proteins
1. Intrinsic factor (IF) is a highly specific glycoprotein, which accounts for absorption of about 97% of the ingested B12 (simple diffusion of the vitamin across intestinal membrane accounts for 3%).
2. Transcobalamin-I, transcobalamin-II, transcobalamin-III and R-proteins are the other transport proteins involved in the delivery or storage of the cobalamin. TC-II is a plasma transport protein, whereas TC-I and TC-III exist in liver also. Both provide storage forms for the B12 vitamin.
R-proteins are secreted by the salivary gland and gastric mucosa, and together with transcobalamin-I and -III they are termed cobalaphilins.
For more information on transport proteins, refer Box 18.1.
Vitamin B12 can be Stored
Water-soluble vitamins cannot be stored, but vitamin B12 is an important exception for it is stored in liver. The transcobalamin-I and -III provide excellent storage forms of the vitamin. Whole liver contains about 2 mg of the vitamin which is sufficient for the requirement of 2–3 years: a situation unique in terms of water soluble vitamins, more akin to that of vitamin A and E.
Coenzyme Functions
Cobalamin is required in two reactions that lead to:
1. Homocysteine to methionine conversion: Methylcobalamin donates a methyl group to homocysteine to form methionine; the reaction is catalyzed by the enzyme homocysteine methyltransferase (Fig. 18.6). After donating its methyl group, the methylcobalamin is reconverted to cobalamin. Methylcobalamin is re-formed from cobalamin by receiving methyl group of methyl-tetrahydrofolate (Fig. 18.7 ).
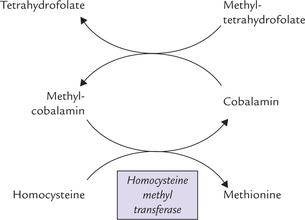
Fig. 18.7 Conversion of homocysteine to methionine requires vitamin B12 (as methyl cobalamin) along with folate (as methyl-THF).
Thus, cobalamin and folate are involved together in this methylation reaction. An absence of cobalamin leads to cessation of the reaction and build up of methyltetrahy-drofolate, known as the ‘folate trap’, discussed later.
2. Formation of succinyl CoA: Vitamin B12’ as deoxyadenosyl cobalamin, acts as a cofactor for the enzyme methylmalonyl CoA mutase (Case 18.2). This enzyme catalyzes conversion of methyl malonyl CoA to succinyl CoA.
As discussed earlier, methylmalonyl CoA is formed from (a) valine, isoleucine and methionine via propionyl CoA, and (b) fatty acids having odd number of carbon atoms. Succinyl CoA may be oxidized in Krebs cycle or converted to glucose.
Requirement and Dietary Sources
| Infants and children | 1–1.5 µg/day |
| Adults | 3.0 µg/day |
| Pregnancy/lactation | 4.5 µg/day |
Clinical deficiency manifests after a long time because normal requirement for cobalamin is very small (~3 µg/ day) and the liver stores a 3- to 5-year supply of this vitamin.
Apart from liver, which is the largest source of cobalamin, kidney, meat, eggs, milk and cheese are good sources.
Causes and Effects of B12 Deficiency
Vitamin B12 deficiency may arise due to decreased absorption or decreased dietary intake. The important causes are:
1. Pernicious anaemia: Cobalamin deficiency in developed countries is most commonly seen due to deficiency of intrinsic factor in the stomach and hence, decreased intestinal absorption. The condition, termed pernicious anaemia, is rare in India. It was named so because it was pernicious (fatal) when described first in 1849.
Pernicious anaemia is an autoimmune disease with a strong familial background affecting people over 40 years of age. Antibodies against IF are generated leading to deficiency of IF and decreased absorption of cobalamin.
2. Nutritional: B12 deficiency is very common in India, especially among vegetarians of low socio-economic groups who cannot usually afford milk and milk products which are the only vegetarian sources of the vitamin. Interestingly, bacteria of the large bowel contain cobalamin, but humans are unable to absorb it because the bacteria are distal to the stomach (source of intrinsic factors) and the ileum (site of absorption). Interestingly, rabbits overcome this problem by habitually contaminating food with fecal matter.
3. Gastrointestinal: A situation similar to pernicious anemia will arise upon surgical removal of the ileum. Individuals with stomach cancer may develop pernicious anaemia because the diseased stomach fails to produce adequate amounts of intrinsic factors.
4. Enterohepatic circulation: Cobalamin is also secreted in the bile and there is a marked enterohepatic circulation of the vitamin. Disturbances in this circulation can, therefore, have major effects on vitamin B12 status.
5. Others: These include iron deficiency anaemia (associated with gastric atrophy), infection with fish tapeworm (the parasite binds B12), or increased requirement in pregnancy.
Clinical Manifestations of B12 Deficiency
Major deficiency manifestations are of two types: haematologic abnormalities (megaloblastic anaemia, hypersegmentation of neutrophils), and demyelination in peripheral nerves and the spinal cord leading to neurological deficiency.
Haematological abnormalities: In initial stages, the predominant feature is macrocytic (“large cell”) anaemia: the red cells are 25–50% larger than normal with fragile membranes and a tendency to haemolyze. Macrocytosis is probably due to secondary deficiency of folate. As discussed later, it is a consequence of the accumulation of methyltetrahydrofolate (folate trap), and hence cannot be distinguished from anaemia of folate deficiency.
Neurological disorders: The neurological damage may sometimes occur in the absence of haematological abnormalities and may be mistaken for diabetic neuropathy or neuropsychiatric disorders. This is known as subacute combined degeneration of the cord and is unique to B12 deficiency (not associated with folate deficiency). The lateral and posterior columns of the cord are affected, as are neurons of cerebral cortex, resulting in sensory as well as motor disturbances.
The anaemia of B12 deficiency is haematologically indistinguishable from that of folate deficiency, but a similar neurological disorder is not seen in patients with folate deficiency.
Laboratory diagnosis: Since cobalamin is required in only two reactions, its deficiency results in accumulation of methylmalonic acid (methylmalonic aciduria) and homocysteine (homocystinuria); both may be measured in laboratory. The plasma levels of cobalamin can be determined by bacteriological methods or radioimmunoassay.
G Folic Acid
Folic acid is obtained from green leafy vegetables and its name reflects this—foliage, derived from the Latin word folium, meaning leaf. It serves as a carrier of one-carbon (C1) units during several biosynthetic processes. Two other co-factors are also known to be involved in the addition of C1 unit to a metabolic precursor, biotin in carboxylation reactions, and S-adenosylmethionine (SAM) as a methylating agent. However, folic acid is more versatile than either of these two because it can transfer the C1 units in several oxidation states.
Structure
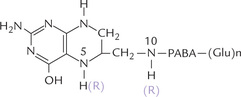
Folic acid consists of three components: a pteridine ring linked in sequence to para-aminobenzoic acid (PABA) and a glutamate residue (Table 18.2). Up to five additional glutamate residues are linked to the first glutamate via isopeptide bonds (between terminal carboxylate group and the amino group of the next glutamate residue) to form a polyglutamyl tail.
Synthesis
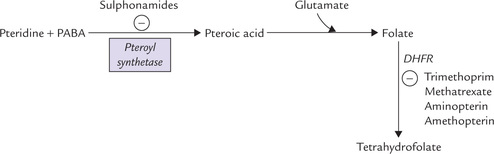
Pteridine and para-aminobenzoic acid are linked covalently by pteroyl synthetase to form pteroic acid. The latter is attached to a glutamate residue to form folic acid (Fig. 18.8 ). A polyglutamyl tail is built by addition of more glutamyl residues, which imparts multiple negative charges to the molecule and so it cannot traverse biological membranes by passive diffusion. Thus, polyglutamylation serves to sequester folate in the cells in which it is required.
Activation
Folate in the human organism must be doubly reduced to become an active coenzyme tetrahydrofolate (THF). The reduction reaction is a stepwise one: folate to dihydrofolate and then to THF. A single NADPH-dependent enzyme dihydrofolate reductase (DHFR) catalyzes both the steps.
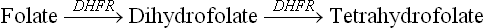
Folate antagonists: The DHFR reaction is inhibited by the antitumour agents (e.g. methotrexate, amithopterin, and amenopterin), which competitively inhibit the DHFR. This blocks the synthesis of tetrahydrofolate. Because THF is required for DNA biosynthesis and tumour cells have a very high level of DNA biosynthetic activity, even modest decrease in THF availability will inhibit tumour growth.
Mammals cannot synthesize folic acid, so it must be provided in the diet or by intestinal microorganisms. Many microorganisms can synthesize their own folate as long as PABA is present in the medium. Therefore, the PABA analogues (e.g. sulphonamides) can inhibit formation of folate (Fig. 18.8) in these organism, thereby inhibiting their growth. Sulpha drugs are effective in many infections, especially those of genitourinary tract. Trimethopterin inhibits DHFR, and hence acts synergistically with sulpha drugs.
Absorption and Distribution
Folic acid is absorbed in the jejunum. In intestinal lumen, all but one of the glutamyl residues are removed by hydrolysis prior to absorption. This is achieved by two γ-glutamyl hydrolases. Following absorption, folic acid is transported in blood by two (β-globulins. The major circulating form is methyltetrahydrofolate and the normal concentration range is 5–15 ng/mL. Once it arrives in the liver, the methyl derivatives are taken up by hepatocytes where various coenzyme forms are produced. Folic acid is not stored in tissues.
Coenzyme Functions
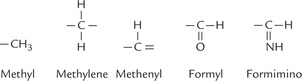
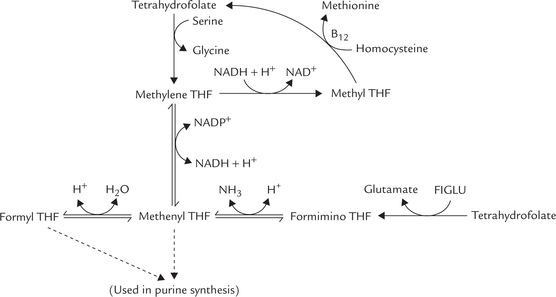
Tetrahydrofolate serves as carrier of one-carbon units at different oxidation levels (Fig. 18.9 ). These C1 units are bound to one or both of the two nitrogens in the molecule, N5 and N10 (Fig. 18.10 ).
THF receives the C1 units from various donor molecules during catabolic reactions and can transfers them to specific acceptors for the synthesis of various compounds. Role of THF is thus vital in those reactions that require either addition or removal of C1 units of various oxidation states.
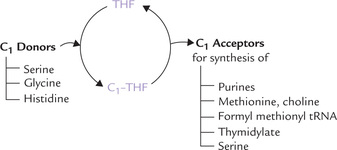
1. Donors of C1 units: THF acquires C1 units from various donors during the following reactions:
(a) Serine to glycine conversion by serine hydroxy-methyltransferase.
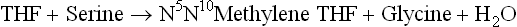
(b) Glycine breakdown by the glycine-cleavage enzyme

(c) Histidine breakdown, where a C1 unit from FIGLU is transferred to THF (Fig. 13.13).
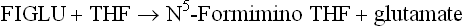
Once bound to THF, the C1 unit can be oxidized or reduced enzymatically. Thus, various oxidation states are interconvertible (Fig. 18.11 ).
2. Acceptors of C1 units: Some important one-carbon addition reactions in which C1 unit is transferred to an acceptor are as here.
(a) Synthesis of purine nucleotides: The C-2 and C-8 of purines are contributed by formyl THF and methenyl THF, respectively (Chapter 20).
(b) Conversion of homocysteine to methionine: The methyl group required for the synthesis of methionine from homocysteine is provided by methyl THF (Fig. 18.7).
(c) Synthesis of formylmethionyl-tRNA: This is required for initiation of protein synthesis in prokaryotes and in mitochondria (Chapter 22).
(d) Methylation of deoxyuridylate to thymidylate: Methylene THF donates a carbon unit (C1 unit) for the formation of deoxythymidylate (dTMP) (a nucleotide unit present in DNA) from deoxyuridylate (dUMP).
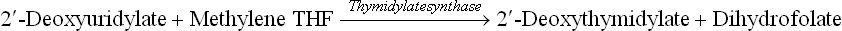
This reaction is commonly referred to as thymidylate synthase reaction. It is unique as the tetrahydrofolate changes to dihydrofolate in this reaction. This is the only 1-C transfer reaction in which the redox state of methylene (the transferred 1-C unit) changes as the methyl group is added to dUMP.
(e) Synthesis of serine: The hydroxymethyl group required for glycine to serine conversion is provided by N5, N10 Methylene THF.
(f) Synthesis of choline: Serine to choline conversion requires methyl group from N5, N10 methylene THF.
Causes and Effects of Folate Deficiency
Deficiency of this vitamin is very common in India, particularly during pregnancy when the requirement is increased. Common causes are dietary deficiency and defective intestinal absorption. Chronic alcoholism is also a known cause.
DNA synthesis indirectly requires folic acid because of its role in the synthesis of purines and in thymidylate synthesis. Hence, folic acid is needed in DNA replication and cell division. The cytoplasm of the folate-deficient cell grows at a normal rate, but DNA replication and cell division are delayed. This results in oversized cells that have an abnormally large amount of cytoplasm (i.e. megaloblasts). The cells most vulnerable to the deficiency state are the rapidly dividing cells of the bone marrow and intestinal mucosa, as they are most affected by slower than normal rates of cell division. The haematological changes may progress to a condition known as megaloblastic anaemia or macrocytic anaemia, where the red cells are large with diffuse nuclei (macrocytes; megaloblasts are oversized RBC precursors in the bone marrow). Formation of other blood cells, e.g. platelets and granulocytes are also compromised.
Megaloblastic anaemia seen in cobalamin deficiency is indistinguishable from the anaemia of folate deficiency. This is because in cobalamin deficiency, folate is trapped in the form of 5-methyl-THF. As a result, cobalamin deficiency leads to functional folate deficiency.
Folate Trap Hypothesis
Deficiency of cobalamin leads to functional folate deficiency by the following mechanism.
During the metabolism of one-carbon units, a small amount of methylene tetrahydrofolate is reduced irreversibly to methyltetrahydrofolate (Fig. 18.11). Since it cannot be used for the synthesis of purines or thymine, methyl-THF has to be converted back to one of the other coenzyme forms. The only reaction of methyl-THF is the methylation of homocysteine to methionine, which regenerates free THF. This reaction requires cobalamin, and therefore, methyl THF tends to accumulate in cobalamin deficiency.
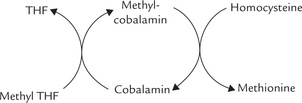
Accumulation of methyl THF leads to depletion of the other coenzyme forms that are needed for nucleotide synthesis. Thus, folate trap hypothesis explains the anaemia of cobalamin deficiency but it cannot account for the neurological manifestations of pernicious anaemia.
H Ascorbic Acid (Vitamin C)
King and Waugh in 1933, isolated from orange, an anti-scurvy substance having strong reducing nature. Its structure was established in 1938 by Howarth and it was named as ascorbic acid. Its reducing nature was due to its strong tendency to donate reducing equivalents.
Absorption and Storage
Vitamin C is absorbed from the small intestine by a carrier-mediated process at the luminal surface that requires a sodium gradient. The transport resembles the sodium-dependent transport of sugars and amino acids (Chapter 7). The efficiency of absorption is high (80–90%). Following absorption, the vitamin circulates in plasma, red cells and leukocytes. It is found in highest concentrations in the adrenals, the pituitary and the retina, in that order.
Structure and Synthesis
The structure of ascorbic acid resembles monosaccharides (hexoses) and it can exist as L- and D-isomers. Only the L form possesses the vitamin activity.
Vitamin C is synthesized by most of the plant and the animal kingdom in uronic acid pathway (Chapter 10). Only humans, higher primates, guinea pigs, and fruit-eating bats have lost the ability to synthesize the vitamin because of lack of the enzyme L-gluconolactone oxidase, that converts gluconolactone to ascorbic acid. Therefore, vitamin C is an essential nutrient in these animals.
Functions
Ascorbic acid functions as a reducing agent and a scavenger of free radicals (antioxidant).
1. As a reducing agent: Ascorbic acid is promptly oxidized to its biological equivalent dehydroascorbic acid, which can be readily reduced to re-form ascorbic acid. Mechanism of action of ascorbic acid relative to its many activities is explained by its ability to undergo such reversible oxidation and reduction reactions. In a large proportion of reactions, the prime function of this vitamin is to maintain metal co-factors in their lower valence state, e.g. Fe2+ and Cu+. Some of the important ascorbate-dependent reactions are as below:
(a) During collagen biosynthesis, the hydroxylases causing post-translational hydroxylation of prolyl and lysyl residues require ascorbate.

Thus, vitamin C plays a role in the formation of matrix of bones, cartilages, and connective tissue. In absence of vitamin C, newly synthesized collagen cannot form fibres properly, which accounts for the prominent connective tissue abnormalities of scurvy.
(b) Synthesis of norepinephrine from dopamine by the enzyme dopamine β-monooxygenase depends on vitamin C (Fig. 13.23).
(c) Carnitine synthesis requires two Fe2+-containing ascorbate-dependent dioxygenases. Carnitine deficiency decreases mitochondrial fatty acid oxidation and thereby contributes to the fatigue, characteristic of scurvy.
(d) During bile acid synthesis in liver mitochondria, the 7-α-hydroxylase reaction requires ascorbic acid.
(e) Absorption of iron is aided by vitamin C by converting ferric to ferrous ions (Fig. 19.2).
(f) During steroidogenesis, ascorbic acid is thought to participate in several oxidation-reduction reactions. This may explain highest tissue concentrations of ascorbate in the adrenal cortex.
(g) Ascorbate participates in tyrosine catabolism by serving as coenzyme for 4-hydroxyphenylpyruvate dioxygenase.
2. As an antioxidant: Ascorbic acid is not only one of the strongest naturally occurring reducing agents known, but it can also serve as an antioxidant in several non-enzymatic reactions. It decreases oxidation of DNA and arrests protein damage, reduces lipid peroxidation and oxidation of low-density lipoproteins, and decreases production of extracellular oxidants from neutrophils. Because of these actions it provides several health benefits, especially in prevention of atherosclerosis and coronary heart disease.
Vitamin C deficiency (scurvy) results in connective tissue problems because of impaired collagen synthesis.

An important area associated with its antioxidant properties is in the prevention and treatment of cancer. For example, it may suppress the formation of potentially carcinogenic nitrosamines from dietary nitrite and nitrate in the stomach, which may explain its protective effect in cancer. Epidemiological studies suggest that vitamin C exerts a synergistic effect with other dietary antioxidants, vitamin E and carotenoids, and this may have a significant role in the prevention of cancer, cardiovascular disease and cataract formation. The quantitative contributions of these components to the overall effect are not known.
Clinical Deficiency
Scurvy, the vitamin C deficiency disease, first became prominent during the 15th century among sailors on long voyages whose diets were devoid of fresh foods. The introduction of limes to the diet of the British navy alleviated scurvy and led to the nickname “limey” for the British sailors. The disease is characterized by reduced cross-linking of collagen fibres, resulting in fragile blood vessels and haemorrhagic diathesis, which manifests in various forms. There is tendency to bleeding, especially in joints and under the skin. Gums become soft and spongy, teeth become loose and there is poor wound healing. Bones become weakened and anaemia and infections develop. If untreated, these infections may prove fatal.
Ascorbic acid saturation test is a highly sensitive test that can detect even subclinical cases of scurvy. This test assumes that exogenously administered ascorbic acid is first taken up by tissues. The rest enters blood circulation from where it is eliminated in urine. In scurvy, the body stores of vitamin C are grossly depleted and therefore, any administered vitamin is promptly taken up by tissues; little or none is left for urinary excretion. Therefore, in a patient of scurvy, urinary excretion of ascorbic acid would be much lower compared to that in a normal subject.
IV Fat-soluble Vitamins
The fat-soluble vitamins (A, D, E, and K or their precursor provitamins) are assembled from isoprenoid units by plants or bacteria. Despite limited chemical similarity, they share many properties:
1. Absorption: They are absorbed into the intestinal lymphatics along with other dietary lipids (or their digestion products).
2. Transport: The absorbed vitamins are delivered to liver (by chylomicrons), from where they are transported to other organs; the interorgan transport is affected by either plasma lipoproteins or specific transport proteins.
3. Coenzyme role: Though they participate in a variety of biological processes, their coenzyme functions are unknown (with possible exception of vitamin K).
4. Storage: Because they are predominantly non-polar, they cannot be excreted by the kidneys, and so tend to be stored in the body. Deficiency disease (except in the case of vitamin K) is difficult to produce in adults because of the storage in liver (A, D, K) or adipose tissue (E).
5. Toxicity: Excessive intake leads to accumulation of these vitamins to toxic levels in lipid laden structures of the body, which may prove even lethal (as in case of vitamins A and D).
Ample reserves of fat-soluble vitamins are stored in the tissues as they are not readily absorbed from food. Excessive accumulation in lipid laden structures may lead to toxicity.
Table 18.3 represents structures and function of fat-soluble vitamins. Daily requirements, dietary sources and deficiency diseases are given in Table 18.1.
Table 18.3
The fat-soluble vitamins: structures and functions
* Vitamin D2 differs from vitamin D3 in having a double bond in the side chain and an extra methyl group (C28)
A Vitamin A
The term vitamin A refers to some polyisoprenoid compounds found only in animals. However, their precursors, carotenoids are found in plants.
Chemistry and Nomenclature
Vitamin A refers to three biologically active vitamers: retinol (an alcohol), retinal (an aldehyde) and retinoic acid (an acid), all of which are found only in animals. They are polyisoprenoid compounds comprising two distinct components: (a) a cyclohexenyl ring, and (b) a side-chain made up of several isoprene units, which is attached to the cyclohexenyl ring (Fig. 18.12 ).
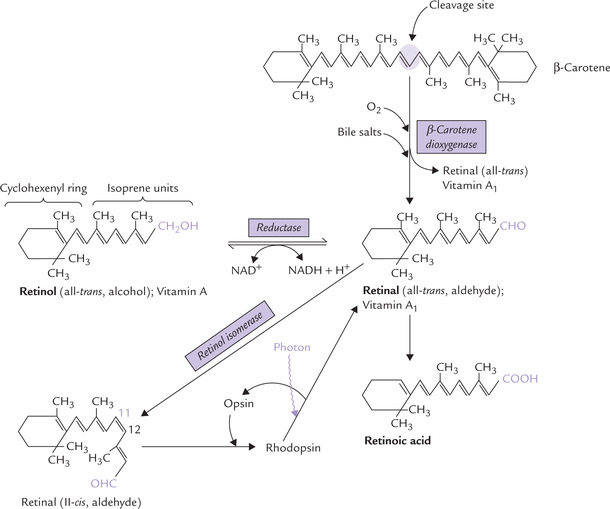
The term retinoids has been used to define these three compounds (and other associated synthetic compounds with vitamin A-like activity). The provitamin carotenoids are also included in the vitamin A family. They are present in a variety of plants. The most important, quantitatively, is β-carotene (Table 18.3) (responsible for the orange colour of carrots).
Absorption
β carotenes and retinol esters are handled differently.
(a) β-carotene is absorbed in small intestine and enters mucosal cells where it is cleaved into two molecules of trans-retinal (all-trans variety) by a dioxygenase and molecular oxygen; bile salts facilitate the reaction. Retinal is reduced to retinol (all-trans variety) by an NADH or NADPH-dependent retinal reductase. Retinol is esterified with a fatty acid, incorporated into chylomicrons together with dietary lipids, and secreted into lacteals (Fig. 18.13 ).
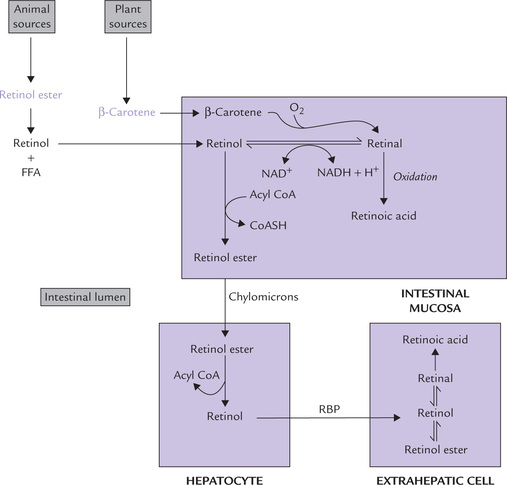
Fig. 18.13 Absorption, transport and metabolism of retinoids (RBP = retinol-binding protein, FFA = free fatty acid).
(b) The retinol esters of animal foods are handled differently. They are hydrolyzed in the intestinal lumen by a pancreatic enzyme (carboxylic ester hydrolase) and the free retinol generated is transferred across the intestinal mucosal cell. It is then esterified, incorporated into chylomicrons and secreted into lacteals.
The presence of lipids in the intestine ensures efficient absorption of retinol, up to 80% of the intake. Carotenoid absorption is less efficient, about 40% of the intake. Its activity is 1/6th that of retinol.
Transport
Chylomicrons carry the absorbed retinol into the circulation and then to liver parenchymal cells where the retinol esters are hydrolyzed. The retinol so released is reversibly bound to retinol-binding protein (RBP) in a one-to-one proportion and released into the circulation for transport to other organs. The target cells take up the retinol-RBP by a RBP-specific receptor-mediated process, and the retinol is bound to cellular retinol-binding protein (CRBP). Subsequently it may be oxidized to retinal or retinoic acid (Fig. 18.13).
RBP in human plasma is a monomeric polypeptide which has a single binding site. Being of low molecular weight (MW 20,000), RBP can be cleared by kidneys, so it circulates after being reversibly complexed with a plasma protein, transthyretin (T4-binding prealbumin).
Storage
The hepatocytes not only dispatch retinol with RBP, they can also store the surplus in the form of retinol esters. More than 90% of the body’s supply of vitamin A is usually stored in the liver cells, which contain a year’s supply of the vitamin.
Functions
The role of vitamin A in vision has been known since the 1940s mainly through the studies of G. Wald (Nobel Prize, 1967). Its importance in various other cellular functions, unrelated to visual process is also being recognized:
• Retinal is involved in vision.
• Retinoic acid is involved in growth and cellular differentiation.
• Retinol is necessary for the reproductive system.
• Carotenoids have an antioxidant role per se (not through their conversion into vitamin A).
Vitamin A and Vision
Vitamin A is the prosthetic group of rhodopsin, the light sensing protein in retinal rod cells. Rhodopsin is located in a membrane system in the outer segment of the rod cell.
(a) Rhodopsin synthesis: Rhodopsin is made up of the protein opsin and 11-cis-retinal. The 11-cis retinal serves as the light absorbing part of rhodopsin, and opsin is an integral membrane protein with seven transmembrane helices. In the pigment epithelium of retina, all trans-retinal is isomerized to 11-cis retinal. The aldehyde group of the 11-cis-retinal then spontaneously links with a lysyl residue in the apoprotein (by a protonated, schiff-base bond) to form rhodopsin. This rhodopsin reaction places the 11-cis-retinal at the centre of the molecule, interacting with the membrane spanning α-helices. (In the cone cells, opsin is somewhat different and the rhodopsin equivalent is iodopsin.)
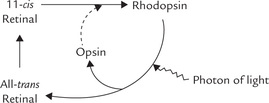
(b) Wald’s visual cycle: Rhodopsin is a photosensitive pigment, which plays a central role in the dim light vision. Exposure to light of wavelength centred around 500 nm on the rod cells induces isomerization of 11-cis-retinal (Fig. 18.14 ). This photo-isomerization switches rhodopsin to a series of unstable intermediates in the following sequence:
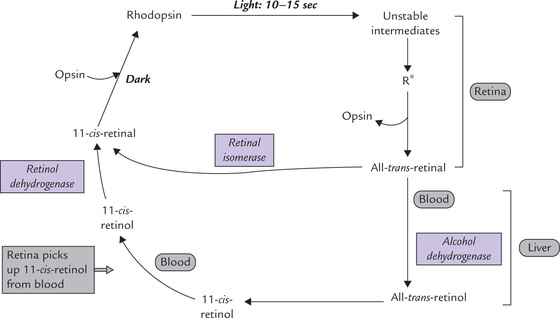
Opsin + 11-cis-Retinal: Rhodopsin → Bathorhodopsin → Lumirhodopsin →Metarhodopsin-I → Metarhodopsin-II γ and finally, all-trans-retinal + Opsin.
The last one (metarhodopsin-II) is an activated “photoexcited conformation” referred to as active rhodopsin (R*). The Schiff-base between the all-trans-retinal and the apoprotein of R* then hydrolyzes, and all-trans-retinal dissociates from the apoprotein. Before this bond is hydrolyzed, the photoexcited R* does something useful: it activates the G-protein (transducin)-based cascade. Activation of this cascade is implicated in generation of nerve impulse that is transmitted in optic nerve and is perceived by the brain as light.
A complex series of biochemical events are involved in generation of the nerve impulse, which are described later (see Mechanism of vision).
(c) Regeneration of 11-cis-retinal: Conversion of all-trans-retinal (released following dissociation of R*) back to 11- cis-retinal is the final event of the Wald’s visual cycle. This reaction can occur in the retina itself, or it may require participation of an enzyme system located in hepatocytes (Fig. 18.14).
• In retinal cells: The trans- to cis-isomerization reaction is catalyzed in the dark by the enzyme retinal isomerase. The 11 cis-isomer then combines with opsin to regenerate rhodopsin, as discussed earlier.
• In liver: The all-trans-retinal is released into blood circulation and transported to liver. Following uptake by hepatocytes, it is reduced to all-trans-retinol by alcohol dehydrogenase, an NADH-dependent, zinc containing, enzyme. The all-trans-retinol is isomerized to 11-cis-retinol and carried to retina by blood where it is oxidized to 11-cis-retinal. This completes the Wald’s visual cycle.
(d) Dark adaptation mechanism: The time taken for regeneration of rhodopsin (following light induced depletion of rhodopsin) is known as dark adaptation time. It is a common experience that when a person shifts from bright light to dark, there is difficulty in seeing (e.g. in cinema hall), and after a few minutes the vision improves. During these few minutes rhodopsin is resynthesized, and the time taken is referred to as the dark adaptation time. It depends on vitamin A status of the person. It is prolonged in the vitamin A deficiency state.
Mechanism of Vision
Rhodopsin is a transmembrane protein located in rod cells, bound to membranous structures in the outer segment.
Rod cell physiology: The rod cell consists of outer and inner segments connected to each other by a narrow cilium (Fig. 18.15 ). The outer segment is packed with membranous disc structures and the inner segment is rich in mitochondria. Next comes the cell body with its characteristic axon leading to the nerve ending and synapse. The discs in the outer segment are closed membranous structures, some 16 nm thick, and there are about 1000 packed into each segment. The light-sensitive rhodopsin is bound to these discs. The rod plasma membrane enclosing the outer segment contains sodium channels that admit Na+ into the cell. Sodium is later pumped out of the cell by an ATPase located in the inner segment. Such movement of sodium ions is called the dark current because it takes place in dark (absence of light). The sodium channels are kept open by the intracellular nucleotide, cGMP. When light strikes the rhodopsin, these sodium channels close, and the dark current ceases. This leads to polarization of the cell membrane and consequent generation of an electric current which results in a visual impulse. What causes closure of the sodium channel, and how is it related to light exposure? This intricate problem has been solved and is outlined here.
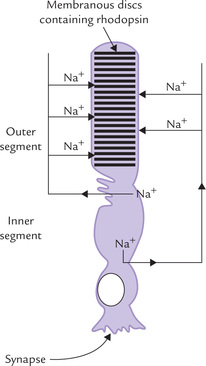
Fig. 18.15 A rod cell showing the dark current: the sodium ions enter the outer segment through cGMP-sensitive channels and are actively pumped out from the inner segment by an ATPase.
Active rhodopsin (formed on exposure of rhodopsin to light) plays a key role in the events that lead to closure of sodium channels. A G-type protein, transducin, also participates in this complex series of events. In general, the G-type proteins function by binding GTP or GDP, and by triggering the second messenger phenomenon (Chapter 29).
• Transducin consists of three subunits: a, (3 and y, and it binds with GDP.
• Active rhodopsin (R*) interacts with transducin and triggers the following changes in its composition: (a) GDP is replaced by GTP, and (b) the whole complex dissociates into the (βγ complex and the α-GTP complex (GTP-Tα).
• GTP-Tα activates a phosphodiesterase (PDE), which causes hydrolysis of cyclic GMP.
• This leads to depletion cGMP. Since cGMP is needed to keep the sodium channel open, its depletion leads to closure of these channels.
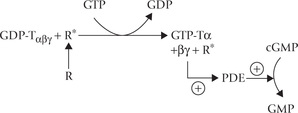
• Closure of Na+-channels prevents influx of Na+ resulting in hyperpolarization.
• A single photon can cause hydrolysis of up to 100,000 cGMP molecules and thereby prevent the influx of more than a million sodium ions to cause hyperpolarization of the cell by approximately 1mV. Thus, the transducin-based cascade has an extraordinary amplifying power.
The light absorbed by the rods causes the plasma membrane to hyperpolarize by causing closure of the Na+ channels. Photoexcited rhodopsin (R*) and a G-protein play key role in closing of the Na+ channels.
The final question that is yet to be resolved is how the hyperpolarization of the rod plasma membrane results in depolarization of the ganglion cells, the axons of which project in the optic tract. The student is advised to refer to a textbook of physiology for the same.
Other Functions of Vitamin A
1. Regulation of gene expression: Retinoic acid is an important regulator of gene expression. It is transported to the nucleus bound to intracellular proteins: cellular retinoic acid-binding protein (CRABP)-I and -II. In nucleus, retinoic acid binds to and activates two families of nuclear receptors, which regulates gene expression by binding to response elements on the DNA.
Action of retinoic acid resembles that of steroid hormones, and the retinoic acid receptors (like the thyroid and steroid hormone receptors) belong to a large super-family of ligand-regulated transcription factors (Chapter 29).
2. Growth and differentiation: Because retinoic acid regulates gene expression, several processes associated with growth and differentiation depend on retinoic acid. These include maintenance of healthy epithelial cells, cell differentiation in spermatogenesis, and the differentiation of epithelial cells, among others. Severe vitamin A deficiency leads to production of abnormal epithelial tissues (keratinization).
3. Glycoprotein synthesis: Retinyl phosphates, obtained from retinol play a role in glycoprotein synthesis. Its hydrophilic portion serves as an anchor for growing oli-gosaccharide chains. This function appears analogous to that of dolichol phosphate (Chapter 5).
4. Role in reproduction: Retinol is required in reproduction. This function is mediated by control of expression of certain genes by retinol bound to cellular retinol-binding protein.
Retinol, and to a lesser extent the retinal, support spermatogenesis in males and prevents fetal resorption in females.
5. Antioxidant role of vitamin A: The antioxidant properties of the carotenoids at low oxygen partial pressures in tissues have been reported. It is in this role that carotenoids are thought to prevent the development of diseases in which the action of free radicals is implicated, such as cancer and cardiovascular diseases.
6. Others: Retinoic acid exerts a number of metabolic effects on tissues, such as control of the biosynthesis of proteoglycans, particularly sulphation of the latter.
Requirements and Dietary Sources
Vitamin A requirement is difficult to calculate because oi the different forms in which it is present. It is usually expressed in terms of international units (IU: one IU is the activity present in 0.3 µg of retinol, 0.344 µg of retinol acetate or 0.6 µg of (β-carotene).
Only animal foods contain vitamin A and the provitamins are found in foods of plant origin (Table 18.1).
Clinical Deficiency
Vitamin A deficiency may be primary (dietary) or sec ondary. Causes of secondary deficiency include:
2. Failure to synthesize chylomicrons into which vita min A is normally incorporated after absorption (mostly due to inability to synthesize apoB48).
3. Failure to cleave (3-carotene because of an enzyme defect.
4. Impaired storage in hepatic cells in liver disease.
5. Failure to synthesize retinol-binding protein (RBP) thus impeding transport from liver to target tissues.
The deficiency leads to the following clinical manifestations:
1. Effect on vision: Retinal is an essential component of the pigment rhodopsin (visual purple) on which the dim light vision depends. Therefore, lack of retinal may result in impairment of ‘dark adaptation’. Inability to see in dim light (i.e. night blindness) results because of elevation in the visual threshold. Prolonged deficiency leads to an irreversible loss of visual cells. Pathological dryness of the conjunctiva and cornea also follow prolonged deficiency since retinoic acid is required for growth and maintenance of epithelium. This condition is known as xerophthalmia (“dry eyes”). If untreated, the condition progresses to corneal ulceration and consequent scarring. The ultimate result of these changes is blindness. This hazardous outcome is preventable with a timely supplementation of vitamin A. The ophthalmological manifestation of vitamin A deficiency is a major health problem in the developing countries. The worst sufferers are children.
Vitamin A (retinal) is necessary for vision mediated by the rod cells, so deficiency often presents as night blindness, dry eyes, and more serious consequences if untreated.
2. Failure of bone remodelling occurs leading to thick, solid bones in the skull with an increase in the cerebrospinal fluid pressure. This may lead to hydrocephalus.
3. Gonadal dysfunctions occur in deficiency of the vitamin A. Testicular degeneration in males and an increased incidence of miscarriage, or malformed offspring have also been reported.
4. Follicular hyperkeratosis: Transformation of columnar epithelium into heavily keratinized squamous epithelia, a process known as squamous metaplasia. Follicular hyperkeratosis (gooseflesh) is an important sign, developing early in the disease process.
5. Nerve lesions, often occurring with bone lesions, are frequently seen. Certain forms of skin diseases are also common in vitamin A deficiency.
Vitamin A Toxicity
The first recorded cases of vitamin A toxicity were among the arctic explorers because the polar bear liver, which they consumed, has high content of the vitamin. However, with normal foods it is virtually impossible to develop the vitamin A toxicity unless pharmacological amounts are prescribed or when the patient self-medicates. A large single dose of more than 300 mg, or more commonly, chronic ingestion of amounts grossly in excess of requirements, leads to toxic effects; the condition is called hypervitaminosis. It is characterized by dry and pruritic skin, hepatomegaly and raised intracranial pressure, which sometimes mimics the symptoms of a brain tumour. Excessive ingestion can cause congenital malformation in the growing fetus and is teratogenic. Pregnant women should be particularly cautioned against these risks.
B Vitamin D
Vitamin D is a group of sterol compounds that are required as accessory food factors in the individuals not exposed to sunlight. They are also required in the diet of growing children and pregnant women, but normal adults receiving even mild sunshine do not need a dietary source because they can photochemically synthesize these compounds in skin.
Historical Perspective
The vitamin deficiency disease, rickets, has been known in the European countries, notably England, for many hundreds of years. During Industrial revolution in the early 1900s, it was observed that the disease could be treated successfully by either incorporating fish, particularly fish oil, into the diet or by exposing the children to sunshine. An essential factor was later discovered to be missing in rickets, which was named vitamin D (the fourth vitamin to be discovered after A, B and C). Its structure was subsequently elucidated by Otto Diels and Kurt Alder (Nobel Prize 1950).
Synthesis
Vitamin D is synthesized by irradiation of 7-dehydrocho-lesterol, an intermediate of a minor pathway of cholesterol biosynthesis, present in all tissues. Largest amount of this steroid is present in malpighian layer of skin. It is photo-chemically cleaved by radiation in the ultraviolet range (290–315 nm), which causes scission of the B-ring of the steroid nucleus of 7-dehydrocholecalciferol. An unstable intermediate is produced which undergoes spontaneous, but slow, rearrangement to form cholecalciferol or vitamin D3 (Fig. 18.16 ). Thus 7-dehydrocholesterol serves as a provitamin.
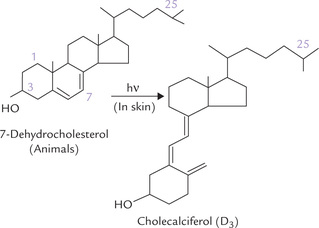
Fig. 18.16 Photochemical cleavage of B-ring of 7-dehydro-cholesterol by ultraviolet rays to form cholecalciferol.
In addition to the naturally produced form of the vitamin (cholecalciferol; D3), there is an artificially produced form D2, or ergocalciferol, differing slightly in the structure of its side chain (Table 18.3). It is produced in the laboratory by irradiation of the plant sterol, ergosterol, and is the form readily available for pharmaceutical use. The biological actions of the two forms are comparable.
Requirements, Dietary Sources, Absorption and Transport
Daily requirement of the vitamin is 5–10 µg (400 IU). It is higher (12 µg) in children, and during pregnancy and lactation.
Exposure to sunlight is the major source of vitamin D, but it can also be obtained from food sources. Fish liver oil, egg yolk and fish are good sources, and milk contains moderate amount.
Dietary vitamin D is absorbed in the duodenum and jejunum from bile salt micelles and appears in the circulation as a constituent of chylomicrons. It is transported to the liver in chylomicron remnants.
Transport of the cholecalciferol formed in the skin to liver occurs in tight non-covalent binding to a vitamin D-binding globulin.
Activation
Cholecalciferol is activated in two steps in liver and kidneys. It is initially transported to liver by cholecalciferol-binding globulin or a vitamin D-binding globulin. The hepatic enzyme 25-hydroxylase brings about hydroxylation at the 25th position (C-25) to form 25-hydroxycholecalciferol or calcidiol (Fig. 18.17 ).
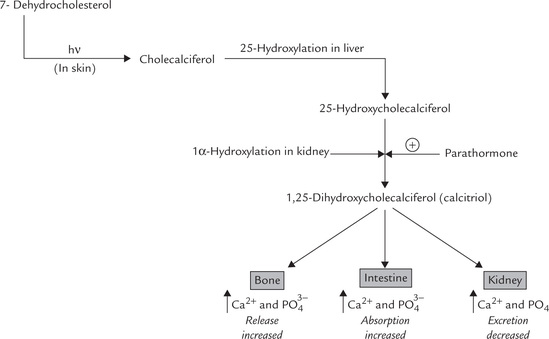
Fig. 18.17 Biosynthesis of biologically active vitamin D compound, calcitriol, from the pro-vitamin precursor.
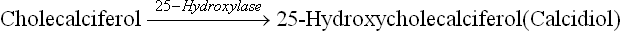
Calcidiol thus formed is the major transport form of vitamin D. It is transported to kidneys by an α2-globulin where it is further hydroxylated by 1-α-hydroxylase to form 1,25-dihy-droxycholecalciferol (DHCC or calcitriol). The enzyme is found in the inner mitochondrial membrane of the cells lining the proximal convoluted renal tubules. Note that DHCC is also termed calcitriol since it contains three hydroxyl groups at 1, 3 and 25 positions. It is the physiologically active form of the vitamin and is considered a hormone.
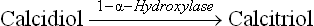
In kidneys, calcidiol can be hydroxylated at C-24 by 24-hydroxylase. Like the 1-α-hydroxylase, the 24-hydroxylase is also a mitochondrial mixed-function oxidase.
Calcitriol is a Hormone
Calcitriol is thought of as a hormone rather than a vitamin because of its several hormone-like properties:
• It can be synthesized in the body, is released in the circulation and has distinct target organs.
• Further, its mechanism of action resembles the Group II hormones (Chapter 29). It binds with a nuclear receptor, termed vitamin D receptor (VDR), which is a ligand-regulated transcription factor belonging to the same superfamily as the receptors for steroid hormones and thyroid hormones. The binding of calcitriol to VDR is analogous to initiation of biochemical action by steroid-thyroid hormones (Chapter 29).
• The ligand-VDR complex then binds to vitamin D response elements in the promoter regions of target genes. The binding results in either an increase or a decrease in gene expression, depending on the gene in question. It is known, for example, that the expression of genes coding for calcium binding proteins, such as calbindin and osteocalcin, is stimulated, whereas the expression of the gene coding for parathyroid hormone is inhibited.
Biochemical Effects
The predominant target organs for calcitriol are intestine, bone and kidney. It regulates serum calcium (normal = 9–11mg/dL) and phosphate (2.5–4.5 mg/dL) concentrations by stimulating:
• Absorption of calcium and phosphate from intestine.
• Reabsorption of calcium and phosphate from the renal tubules.
Evidently, calcitriol plays a major role in calcium homeostasis (Fig. 18.17).
In addition, pancreas, pituitary and thymus are also target organs for calcitriol.
(a) Effect on intestine: The intestinal effect is considered the most important effect of vitamin D on calcium homeostasis, as it allows the entry of calcium into the body and thereby increases the total amount of available calcium. Actions on bone and kidney, on the other hand, involve rearrangement of already existing calcium pools. As noted, calcitriol-receptor complex interacts with DNA and causes transcription of the genes that code for calbindins, the calcium-binding proteins, that increase intestinal absorption of calcium, thus elevating serum calcium levels.
Phosphate absorption is also stimulated by calcitriol.
(b) Effect on kidneys: In distal convoluted tubule, events similar to those described above take place to stimulate reabsorption of calcium and phosphate.
• Bone resorption: In bone, calcitriol facilitates the resorption of calcium and phosphate by stimulating osteoclasts. This is the best established action of vitamin D on bone, and in this instance, calcitriol acts cooperatively with parathormone to cause bone erosion.
• Bone mineralization: Vitamin D promotes bone mineralization as well, which seems opposite to the above stated effect. Increased mineralization may be
It had been generally believed that the latter was the case but some more recent studies on calcium-binding proteins in bone, especially calbindin 9K, indicate that vitamin D may have a more direct effect on bone mineralization. Synthesis of this protein is increased by calcitriol and its binding with calcium affects bone mineralization. Other effects of the vitamin that promote mineralization are:
• Synthesis of vitamin K-dependent calcium-binding protein, osteocalcin, is increased by calcitriol.
• Cross linking of collagen and synthesis of various other calcium binding proteins (osteopontin, and the third component of complement) are also known to be increased.
Since cytoplasmic receptors play a vital role in action of vitamin D, any defect in these receptors results in rickets. In addition to increasing the synthesis of the intestinal calcium-binding protein, calcitriol may also enhance conversion of this protein to its activated phosphorylated form. The intestinal absorption of phosphate is also stimulated by calcitriol, although the exact mechanism of action is not well defined.
Regulation of Calcitriol
Serum calcium and phosphate levels exert important influence on biosynthesis of calcitriol by controlling hydroxylation at position-1 (Fig. 18.18 ).
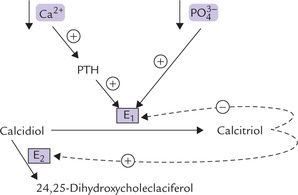
Fig. 18.18 Regulation of calcitriol production by serum calcium and phosphorus (E1 = 1-α-hydroxylase, E2 = 24-hydroxylase, PTH = parathormone).
1. Decreased serum calcium (hypocalcaemia) induces parathyroid glands to secrete the parathormone (PTH) which is a potent activator of 1-α-hydroxylase. This results in enhanced conversion of calcidiol to calcitriol (Fig. 18.17). Consequently the serum concentration of calcitriol is increased, which tends to replenish serum calcium through its effects on intestine, bones and renal tubules, described earlier in this chapter.
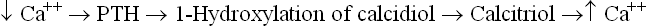
Thus, serum calcium participates in a self-regulatory loop: hypocalcaemia tends to elevate serum calcium levels, acting through parathormone and calcitriol.
2. Decreased serum phosphate (hypophosphataemia) directly stimulates the calcitriol formation by stimulating the 1-α-hydroxylase (Fig. 18.18). The calcitriol increases intestinal absorption of phosphate, and enhances reabsorption of this ion from renal tubules. These effects bring back the serum phosphate level to normal.
In addition to the above-stated action on serum calcium and phosphate levels, the calcitriol has following effects on hydroxylation reactions:
Other Vitamin D Metabolites
1. 24,25-dihydroxycholecalciferol is produced by the enzyme 24-hydroxylase from 25-hydroxycaldferol. It decreases the serum concentrations of both calcium and phosphate. It has pronounced mineralization promoting effect on bones. Its production is stimulated by decreased PTH levels.
2. A 26-hydroxylated metabolite is produced from calcitriol by hydroxylation of calcitriol at C-26 in course of its oxidative degradation. Hepatic microsomal mixed-oxidases are involved in these reactions.
3. Other polar metabolites (e.g. 23-hydroxylated derivatives) are also produced in course of the oxidative reactions. These metabolites possess little of vitamin D activity. The oxidases that produce these metabolites are induced by numerous pharmacological agents, including phenytoin and phenobarbital. This may account for development of rickets in children receiving anticonvul-sant therapy since the latter enhances formation of these metabolites (Case 18.3).
Causes and Manifestations of Vitamin D Deficiency
• Inadequate exposure to sunlight among people living in congested slums may result in vitamin D deficiency (i.e. rickets) in children. The adult counterpart of rickets is called osteomalacia. There is now evidence that with advancing age, capacity of the skin to synthesize cholecalciferol diminishes, resulting in the age-related disorders of calcium metabolism.
The immediate effect of the deficiency is a decrease of the plasma calcium concentration, which is promptly restored by parathormone (PTH) at the expense of bones (PTH mobilizes calcium from bones). Thus, bones are tapped as a calcium reservoir and are gradually depleted of their mineral content, resulting in soft, poorly mineralized and pliable bones that bend rather than break under stress (especially in weight bearing areas). The classical features of rickets, therefore, are bone deformities such as bow legs, knock-knee, Harrison’s sulcus, rickety rosary, bossing of frontal bones and pigeon chest. In osteomalacia, the patients suffer from bone pains and are likely to suffer fractures. Abnormalities of biochemical parameters in both conditions are low serum calcium and phosphate, and elevated serum alkaline phosphatase (bone isoenzyme) activity. Serum calcitriol level is low.
Note
Decreased responsiveness of the target tissues to the vitamin D may also result in signs and symptoms of the vitamin D deficiency. Decreased responsiveness results because of defect in the tissue receptor so that its affinity with calcitriol is decreased.
Study of Cases 18.5 and 31.1 will show that apart from classical vitamin D deficiency rickets, several other disorders affecting kidneys, liver and end organs may cause rickets.
C Vitamin E (α-Tocopherol)
Vitamin E is an important antioxidant. It was isolated from wheat germ oil and its structure was determined in 1936 by Paul Karrer (Nobel prize 1937). Vitamin E was earlier referred to as anti-sterility vitamin; in fact the word tocopherol is derived from Greek words, tokos, meaning childbirth and pherin, to bear, because it was believed that tocopherol is required for fertility. However, such anti-sterility effect has been observed only in some animals, not in human beings.
Chemical Nature
At least eight naturally occurring plant compounds with vitamin E activity are presently known, the most important being α-, β-, γ-, and δ-tocopherol. All are viscous, light yellow oils that are heat stable but readily degraded by oxygen or ultraviolet light. They have a substituted chromane nucleus with a polyisoprenoid side chain of variable length: usually three carbon atoms (Table 18.3). Of these vitamers, the most abundant and potent is α-tocopherol.
Absorption, Transport and Metabolism
The richest sources of the naturally occurring vitamin E are vegetable oils and nuts (Table 18.1). It is absorbed as free tocopherol along with other lipid components. The absorption requires bile salts: in their presence the vitamin is absorbed with efficiency of 20–40%. It has no specific binding proteins and is found in circulation in association with plasma lipoproteins and erythrocytes (plasma concentration is 0.5–1 mg/dL). Tissue distribution also shows little selectivity: vitamin E is enriched in all lipid-laden structures, with highest concentrations in fat depots of adipose tissue. It is mainly excreted in faeces via hepato-biliary route, after the chromane ring is oxidized followed by its conjugation with gluc-uronic acid.
Functions
1. Antioxidant role: Vitamin E is accepted as nature’s most potent and most abundant biological antioxidant, in particular a membrane antioxidant. In cellular and subcellular membranes, it acts as a first line of defense against free radicals by acting as chain-breaking antioxidants (Fig. 18.19 ) as described in Chapter 27. As such, it is associated with the membrane lipid structure and so can promptly protect the membranes from attack by endogenous and exogenous free radicals. Vitamin E may be located near enzyme complexes that produce free radicals, such as NADPH-dependent oxidase systems.
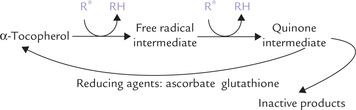
Vitamin E is enriched in fat depots and lipo-proteins also, where it scavenges free radicals that are formed during lipid peroxidation, thereby interrupting free radical chain reaction. Further details are presented in Chapter 27.
2. Antiatherogenic role: Several epidemiological studies suggest an inverse relationship between vitamin E intake and the incidence of morbidity and mortality from coronary artery disease. Supplements of 400 IU of vitamin E for about 2 years result in about 40% reduction in the incidence of heart attack (1 mg of α-tocopherol is equivalent to one IU of vitamin E). The following properties of the vitamin may contribute toward its antiatherogenic role:
(a) It retards oxidation of LDL, thereby decreasing production of the pro-atherogenic oxidized LDL.
(b) It impedes various cellular signalling pathways (e.g. protein kinase-C initiated pathways), which results in inhibition of proliferation of smooth muscle cells, platelet adhesion and aggregation, and function of adhesion molecules.
(c) Vitamin E may also decrease the synthesis of leukotrienes and increase synthesis of prostacyclins by upregulating phospholipase A2 and cyclooxygenase.
3. Interrelationship with selenium metabolism: Selenium alleviates some symptoms of vitamin E deficiency, probably through its role as a cofactor for glutathione peroxidase, an important enzyme that oxidizes and destroys the free radicals (Chapter 27). Vitamin E also decreases requirement of selenium, and vice versa, thus they appear to act synergistically.
4. Others: Vitamin E boosts immune response, protects RBC from haemolysis, keeps structural and functional integrity of all cells, slows ageing process and appears to offer protection against Alzheimer’s disease. These effects appear to be by virtue of its antioxidant role. Glutathione and ascorbate prolong action of vitamin E by (a) reducing the initially formed free radicals, and (b) reducing the quinone intermediates back to active form. Claims that high doses of the vitamin E are beneficial for treatment of skin disorders, fibrocystic breast disease, sexual dysfunction, cancer and baldness are yet to be substantiated.
Clinical Deficiency
Vitamin E deficiency is not uncommon in the premature infants but is rarely seen in full term infants (in spite of poor placental transport) because the breast milk is a good source (Table 18.1). It occurs rarely in adults because of its widespread distribution in foods and because the body vitamin stores can meet the requirement for several months. A daily intake of 10–30 mg is considered adequate and is provided by most diets. Defective lipid absorption or transport may sometimes cause the deficiency.
Vitamin E is often called a “vitamin in search of disease”; human deficiency does occur sometimes. It is seen in infants who are born with low tissue stores and who have poor intestinal absorption for several weeks after birth.
A genetic defect in the formation of hepatic α-tocopherol transfer protein (which facilitates incorporation of α-tocopherol in nascent VLDL) leading to the vitamin deficiency has been described. The deficiency may also be acquired—associated with defective lipid absorption or transport.
Haemolysis due to lack of protection for RBCs against peroxides, and creatinuria due to increased muscle breakdown are the features of vitamin E deficiency.
Hypervitaminosis E
High doses of vitamin E (more than 400 IU/day) depress coagulability. Patients with bleeding disorders and those receiving warfarin, should be cautioned against its use. Otherwise, vitamin E is least toxic of all fat soluble vitamins—even 50 times the recommended intake has been reported non-toxic.
D Vitamin K
Vitamin K was initially recognized as an antihaemorrhagic dietary factor. It is the only fat-soluble vitamin that acts as a coenzyme. Its major function is to synthesize γ-carboxyglutamate by incorporating carbon dioxide into specific glutamyl residues of certain proteins such as prothrombin, osteocalcin and certain clotting factors.
The designation “K” derived from the initial description of the vitamin as the “koagulation vitamin”

Chemistry
Vitamin K activity is present in a group of structurally related compounds. All have a 2-methyl-1,4-naphtho-quinone nucleus, but vary in the number of isoprenoid units in its side chain (Fig. 18.20 ). Two naturally occurring forms are menaquinone (K2) and phylloquinone (K1), present in animals and plants respectively. Menaquinone is synthesized by intestinal microorganisms and stored in liver. Some water soluble synthetic forms with vitamin K activity e.g., menadione have also been prepared for use in clinical practice. Sodium menadiol diphosphate and menadione sodium bisulphate are some other examples.
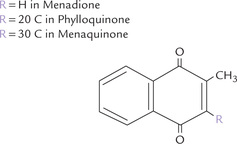
Absorption and Storage
The intestinal absorption of vitamin K is dependent on appropriate fat absorption and it requires bile salts. It may be obtained from diet or intestinal bacterial synthesis. But colonic bacteria do not make a significant contribution because vitamin K is absorbed only in the small intestine. Chylomicrons carry the absorbed vitamin to liver where it is stored. From liver, it is released into blood circulation where it is transported in association with β-lipoproteins. Unlike the other fat-soluble vitamins, the body stores of vitamin K are insignificant (50–100 µg), so vitamin K is the first fat-soluble vitamin to be deficient in acute fat malabsorption.
Functions
Vitamin K plays an important role in blood coagulation for it is required for the post-translational processing of several proteins required in the coagulation cascade (e.g. factor-II, -VII, -IX and -X) in the ER of liver cells. All these protein clotting-factors are initially synthesized as inactive precursors in the liver. Formation of mature clotting factors requires that the glutamyl residues of the precursor proteins be converted to γ-carboxyglutamate (Gla) residues by addition of carboxylate group. This reaction is dependent on vitamin K, which serves as a coenzyme (Fig. 18.21 ).
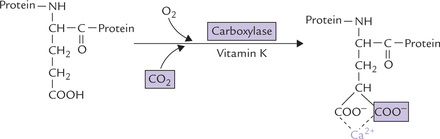
Role of Gla in clotting
The Gla residues so formed serve as binding site for calcium ions; each Gla contains two negative charges which chelate the positive calcium ion. In prothrombin molecule, for instance, there are 10 such carboxylated Gla residues and all of these are required for this protein’s specific chelation of calcium ions. The calcium then binds with the negatively charged phospholipids present on the platelet cell membrane. In this way, bridging of the phospholipids to the Gla residue of pro-thrombin occurs via calcium ion.
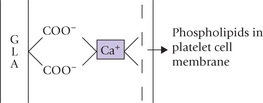
Thus, prothrombin becomes anchored to the surface of the activated platelets. This provides a high local concentration of the prothrombin, which accelerates its activation by about 50- to 100-fold, an essential requirement for blood clotting.
Functional activation of a number of other proteins also requires the vitamin K-dependent carboxylation reaction. Such proteins include osteocalcin, C-reactive protein and structural proteins ofkidneys, lungs and spleen. Osteocalcin is a small-sized protein (40–50 amino acids), synthesized by osteoblasts, which binds to hydroxyapatite crystals of bone. Vitamin K creates calcium binding sites on osteocalcin, which helps it to retain calcium.
Clinical Deficiency
Vitamin K deficiency is rare. It may be induced in following ways: (a) due to treatment with antibiotics that eliminates normal intestinal flora, (b) in fat malabsorption syndromes, (c) in liver diseases, and (d) by vitamin K antagonists, such as dicoumarin or warfarin. They act by competitively inhibiting the gamma carboxylation system due to structural similarity with vitamin K.
Deficiency of vitamin K increases the time for blood coagulation, and so bleeding tendency is the prominent deficiency manifestation. Even a minor cut may cause prolonged bleeding. The only important deficiency sign is increase in prothrombin time (PT), and it is the most important laboratory test for the evaluation of vitamin K status. Note that PT may rise in liver damage also due to inability of liver to synthesize prothrombin. To distinguish between these two conditions, vitamin K is administered parenterally. PT remains unaffected in vitamin K deficiency, but returns to normal in liver diseases.
Newborn infants with their inadequate vitamin K stores may suffer from haemorrhagic disease of the newborn. It is the most common nutritional deficiency in newborns.
Toxicity
Vitamin K is non-toxic even in large amounts. However, some special cases merit attention. Large doses (5 mg) of menadione and its water-soluble derivatives are known to cause haemolytic anaemia and kernicterus in infants; the premature infants are more at risk. Haemolytic reactions may also occur in adults having glucose 6-phosphate dehydrogenase deficiency.
Exercises
1. Explain the biochemical basis of anaemia due to deficiency to: vitamin B6’ vitamin C and vitamin B12.
2. Mention biologically active forms of folic acid. How is it involved in one carbon metabolism. Explain action and clinical significance of folate antagonists.
3. Why is vitamin D considered as a hormone? Explain the role of calcitriol in regulation of serum calcium level.
4. What is the active, form of vitamin D and how is it formed? What is renal rickets and hepatic rickets? Explain biochemical basis of the clinical manifestations seen in vitamin D deficiency.
5. How is cis-retinal synthesized in the body? Explain its role in vision.
6. What are the biochemical roles of vitamin C in human body? What the is biochemical basis of anti-cancer role of this vitamin?
Write short notes on
CASE 18.1 A malnourished man with warm extremities and swollen feet
A 35-year-old man reported in the hospital OPD with complaints of lack of appetite, listlessness, and weakness of legs. Often he had a feeling of heaviness and numbness of legs with “pin and needle” sensation. He appeared malnourished and came from poor socio-economic background.
Examination showed warm extremities and loss of sensation in the skin over tibia. There was moderate oedema, mainly involving legs and face. Tachycardia and marked distension of the neck was noticed. X-Ray chest showed cardiac enlargement. Laboratory investigations showed raised plasma pyruvic acid level. Transketolase activity in the erythrocytes was low, but it showed a rise by about 40% after administration of thiamine pyrophosphate (TPP).
CASE 18.2 A 6-month-old child with feeding problems and recurrent vomiting
A 6-month-old male infant was referred to the teaching hospital for investigation of feeding difficulties, recurrent vomiting, respiratory distress and seizures. On examination, his body weight and head circumference were very small for his age. Analysis of blood revealed that serum levels of both the D- and L-forms of methylmalonic acid were elevated. High urinary excretion of these two compounds and of other compounds related to methionine metabolism (e.g. cystathionine and homocysteine) were detected. Vitamin B12 deficiency was suspected since this vitamin is required by certain key enzymes in metabolism of the above-mentioned compounds. However, serum level of this vitamin was well within the normal range of 150–600 pg/mL. Analysis of mother’s milk was carried out since decreased B12 content of milk is a major cause of deficiency of this vitamin in infants. It was 1650 pg/mL, which is well within the normal range of 1000–3000 pg/mL. In view of these findings, deficiency of B12 vitamin as the likely cause was ruled out.
To further investigate the underlying defect, the following tests were conducted. Cell-free extract was obtained from the cultured fibroblasts. Addition of radioactive methylmalonyl CoA to it showed that conversion of this compound to succinyl CoA was hampered (experiment 1). However, addition of cobalt of +1 state (which means valency of cobalt is + 1) to the reaction mixture made this conversion possible (experiment 2).
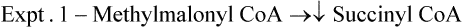
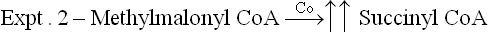
CASE 18.3 Listlessness and persistent bodyache in an 8-year-old boy
An 8-year-old boy was brought to the orthopedic OPD with back injury, following a fall. X-Ray spine revealed a fracture in the first lumbar vertebrae. The other significant radiological finding was diffuse haziness at the metaphyseal border and inadequate mineralization of primary spongiosa. He had been feeling mild fatigue, bodyache, upper abdominal discomfort and fatty food intolerance. During the past three months, he had developed slight abnormality of gait and found walking a tiring experience. Examination showed diffuse bony tenderness, especially over tibia, mild liver enlargement, and proximal muscle weakness.
| Investigations test | Patient’ report | Reference range |
| Serum bilirubin | 2.8 mg/dL | 0.1–1.0 mg/dL |
| Alanine transaminase | 88 U/L | 10–35 U/L |
| Aspartate transaminase | 102U/L | 10–40 U/L |
| Alkaline phosphatase | 128U/L | 40–100U/L |
| Serum calcium | 8.2 mg/dL | 8.5–10.5 mg/dL |
| Serum phosphate | 2.3 mg/dL | 2.5–4.5 mg/dL |
Serum parathormone level was moderately raised. Circulating level of 25-hydroxycholecalciferol (i.e. calcidiol) was 3 ng/mL (normal 8–55 ng/mL). Analysis of a bone biopsy sample revealed a marked fall in the mineral content.
Q.1. What is the probable diagnosis?
Q.2. Identify the underlying cause of vitamin D deficiency in this child.
Q.3. What would you expect the circulating calcitriol levels to be?
Q.4. Enumerate various factors that lead to development of rickets in hepatobiliary disorders.
Q.5. What accounts for the development of rickets in children receiving anticonvulsant therapy?