Amino acid and protein metabolism
Amino acids are the building block molecules of body proteins. In addition, they serve as precursors of a variety of biologically important compounds including peptides, polyamines, purines, pyrimidines, etc. The neurotransmitters such as dopamine, epinephrine and norepinephrine are also synthesized from amino acids. Catabolism of amino acids provides carbon skeleton for gluconeogenesis, ketogenesis, as also for the energy yielding pathways.
In this chapter, various aspects of amino acid and protein metabolism are described. After going through this chapter, the student should be able to understand:
Nitrogen metabolism; essential, non-essential and semi-essential amino acids, protein turnover and amino acid pool.
Amino group metabolism; transamination, oxidative deamination, transport of ammonia, ammonia toxicity and urea cycle.
Metabolism of carbon skeleton of amino acids; outline pathways of degradation of amino acids.
Disorders of amino acid metabolism; phenylketonuria, alkaptonuria, maple syrup urine disease, isovaleric acidaemia, methyl malonic aciduria, histidinaemia, homocystinuria, cystinuria, cystinosis. Hartnup disease, blue diaper syndrome and familial renal iminoglycinuria.
Amino acids as precursors of specialized products.
There are 20 primary amino acids which are classified into three categories: non-essential, essential and semi-essential. There are eight primary amino acids (viz., lysine, leucine, isoleucine, methionine, phenylalanine, trypto-phan, valine and threonine) which cannot be endoge-nously synthesized, and therefore, need to be obtained from the diet: they are classified as essential amino acids. The non-essential amino acids can be synthesized in body and hence their dietary intake is not essential. These amino acids can be produced from the essential amino acids, as also from intermediates of certain metabolic pathways, or even by interconversion among themselves. For example, tyrosine, a non-essential amino acid, is produced by hydroxylation of an essential amino acid, phenylalanine. Similarly glutamate can be formed by amination of α-ketoglutarate.
Two amino acids, arginine and histidine, are classified as semi-essential amino acids. Arginine can be synthesized in adequate amounts in adults, but in growing children additional dietary supplementation is required. This is because the body requirement for this amino acid is higher in the growing stage. Histidine is not synthesized in the human body and yet it is not essential in diet. This is because of its relatively lower requirement and a fairly high reserve in the body, especially in form of carnosine in muscles.
The lower living forms like microorganisms and plants are capable of synthesizing the essential amino acids. During evolution, animals lost the genes that encoded the enzymes necessary for the synthesis of these amino acids. This implies that the essential amino acids are relatively less important for the body, since animals could survive without synthesizing them.
I Nitrogen Metabolism
A Protein Turnover
Most proteins in the body are constantly and repetitively synthesized and degraded. In adults, the rate of synthesis is just sufficient to replace the protein that is degraded, so that the total amount of proteins in the body remains nearly the same. This process, called protein turnover, involves about 1-2% of body proteins per day (about 12 kg proteins are present in an adult of 70 kg body weight).
The rate of protein turnover shows wide variation for individual proteins. For example:
• Collagen and lens crystalline are degraded slowly, having very long half-life, whereas
• HMG CoA reductase has half-life of just about 2 hours only.
Protein turnover results in degradation of proteins to amino acids. About 80% of the amino acids so liberated are recycled, whereas the rest are catabolized. The average lifespan of an amino acid in plasma is only about 5 minutes.
B Amino Acid Pool
The amino acids released as a result of protein degradation join the pre-existing amino acids distributed throughout the body. Together, they constitute the amino acid pool. The ingested (dietary) proteins, as also the endogenously synthesized (non-essential) amino acids, contribute to this pool (Fig. 13.1 ). Although this pool of about 100 g of amino acids appears trivial compared to the total amount of proteins in the body, it is of vital significance because of its dynamic nature. It can be channeled in various pathways depending on body requirements.
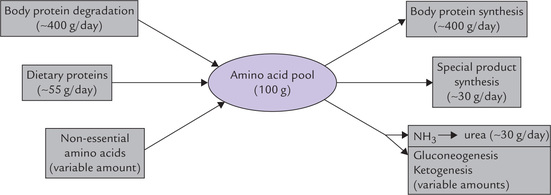
• A major proportion of the amino acids that join this pool every day are recaptured for the synthesis of body proteins and a variety of biologically important compounds (i.e. special products), as discussed later in this chapter.
• Amino acids corresponding to about 30 g of proteins are catabolized every day and their amino group nitrogen converted to urea for excretion.
Since the amino acids of this pool are readily available, deficiency of amino acids (especially the non-essential amino acids) does not manifest immediately. This is because amino acids are constantly replenished from the pool.
Nitrogen Balance
Nitrogen balance is the difference between ingested nitrogen and excreted nitrogen. Because dietary proteins are an important source of nitrogen (100 g proteins contain 16 g of nitrogen), nitrogen balance is an important index of protein and amino acid metabolism. It is determined by comparing the nitrogen entering the body with that leaving it.
In healthy adults, a state of nitrogen equilibrium exists, meaning that nitrogen intake equals nitrogen excretion. In growing children and pregnant women in whom nitrogen intake exceeds excretion, positive nitrogen balance occurs. Negative nitrogen balance (excretion exceeds intake) is observed in dietary protein deficiency. In an adult, even if protein starved, at least 30–40 g of amino acids are degraded each day; this amount defines the dietary requirement. If the dietary supply drops below this limit, a negative nitrogen balance occurs and the body protein is lost. Essential amino acid deficiency has the same effect because relative deficiency of even a single essential amino acid results in corresponding decrease in protein synthesis.
Negative nitrogen balance is also seen in a variety of non-physiological conditions including severe infections, metastatic carcinoma, burns, trauma and postsurgical stress. The negative nitrogen balance in these patients is related in part to increased secretion of stress hormones cortisol and epinephrine, which favour protein degradation over protein synthesis, and also stimulate the use of the liberated amino acids for gluconeogenesis by the liver. Cytokines, the biologically active products released by the leucocytes in a variety of disease states, also elicit metabolic effects similar to those of the stress hormones.
 Nitrogen balance, determined by comparing the nitrogen entering the body with that leaving it, is an important index of amino acid and protein metabolism.
Nitrogen balance, determined by comparing the nitrogen entering the body with that leaving it, is an important index of amino acid and protein metabolism.
Nitrogen balance from nutritional viewpoint is discussed in Chapter 28.
II Catabolism of Amino Group Nitrogen
The amino acids in excess of those needed by the body cannot be stored. They are catabolized to yield energy; about 5-10% of the total energy requirement of the body comes from catabolism of amino acids. Thus, amino acids are secondarily used as fuel molecules, while their primary role is in synthesis of body proteins and special products such as amines, porphyrins, nitrogenous bases of phospholipids, polyamines, etc. (Fig. 13.1). This is in contrast with carbohydrates and lipids whose primary role is to provide energy.
During catabolism, the fate of α-amino group is different from that of the rest of the carbon skeleton (Fig. 13.2 ). The α-amino group is released as ammonia and carried to liver where it is incorporated in urea by way of a cyclic pathway, called urea cycle. Urea is a waste product which is eliminated in urine. In this section, formation of urea by α-amino group catabolism has been described. Metabolism of the remaining carbon skeleton will be discussed later.
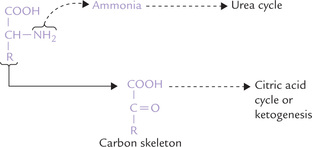
Amino acids are degraded by the removal of the α-amino group and its conversion to urea. The remaining carbon skeleton is converted to one or more metabolic intermediates.
The catabolism of α-amino group nitrogen occurs in the four stages: transamination, oxidative deamination, ammonia transport, and ureagenesis in a cyclic pathway of urea cycle.
A Transamination
Catabolism of all α-amino acids begins with removal of the α-amino group by a process called transamination; the only exceptions are lysine, threonine, glycine, proline and histidine. The amino group thus removed is transferred usually to α-ketoglutarate forming glutamate. Other reaction product is a keto acid corresponding to the amino acid substrate.
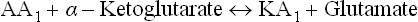
[KA1 = Keto acid corresponding to the amino acid substrate (AA1).]
The enzymes catalyzing the transamination reactions are aminotransferases (transaminases). Liver, kidney, muscle and brain contain appreciable amounts of these enzymes. At least a dozen different transaminases have been identified in these tissues, each one being specific for different amino acid substrates. They are named after the amino acid that serves as the amino group donor. For example, the enzymes catalyzing the transfer of the amino group from alanine and aspartate are termed ala-nine aminotransferase (ALT) and aspartate aminotransferase (AST) respectively
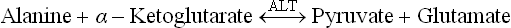

AST and ALT have considerable diagnostic significance in cardiac and hepatic disorders (Chapter 6).
The coenzyme, pyridoxal-5-phosphate (PLP), is required for the transamination reactions. PLP is derived from pyridoxine, which is vitamin B6. PLP is bound to the active site of the enzyme both by electrostatic interactions and by a Schiff base bond with a lysine side chain of the apoprotein. The α-amine group of the original amino acid is firstly transferred to PLP to form pyridoxamine phosphate, and then transferred from pyridoxamine phosphate to α-ketoglutarate (Fig. 13.3 ). Thus, the reaction follows a ping-pong mechanism: the prosthetic group is modified chemically during the reaction, and the first product is released before the second substrate binds (Chapter 6).
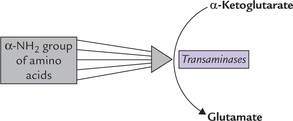
Fig. 13.3 Pyridoxal-5-phosphate (PLP), the coenzyme for all trasamination reactions, is transiently converted to pyridoxamine phosphate during transamination. Interconversion of pyridoxal phosphate and pyridoxamine phosphate is shown here.
Transamination involves removal of α-amino groups from amino acid to the α-keto acid acceptor, usually α-keto-glutarate, which results in formation of glutamate and the corresponding α-keto acid. The coenzymes for transaminases is pyridoxal phosphate (derived from vitamin B6).
α-Ketoglutarate is the acceptor of the transferred amino groups of various amino acids. In the process, it is converted to glutamate which acts as a collecting point for the α-amino groups (Fig. 13.4 ).
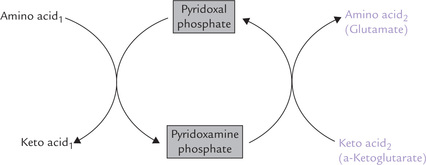
Fig. 13.4 α-Ketoglutarate serving as common acceptor of α-amino groups of various amino acids to form glutamate.
The biological advantage of such an arrangement is that the subsequent steps are common, since it is only the glutamate nitrogen which has to be handled. Glutamate is deaminated and the ammonia released is funneled into ureagenesis, as discussed in the following sections.
B Oxidative Deamination
The α-amino group of glutamate is released as ammonia by the following reaction:
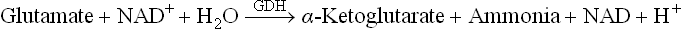
The enzyme catalyzing this reaction is glutamate dehydrogenase (GDH). Thus, successive actions by transaminases and glutamate dehydrogenase effectively cause release of the α-amino group as ammonia (Fig. 13.5 ).
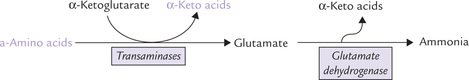
Fig. 13.5 The α-amino groups of various amino acids incorporated into glutamate during transamination reactions are liberated as ammonia by glutamate dehydrogenase.
Glutamate dehydrogenase is a complex allosteric enzyme consisting of six subunits. It is unusual in being able to utilize both NAD+ and NADP+ as co-substrates. Its activity is allosterically regulated by several modulators. Guanosine triphosphate (GTP) and adenosine triphosphate (ATP) are allosteric inhibitors, whereas guanosine diphosphate (GDP) and adenosine diphosphate (ADP) are allosteric activators. Thus, lowering of the cellular energy (i.e. increased ADP or GDP) activates GDH and therefore, increases catabolism of amino acids. Conversely, increased cellular energy (increased ATP and GTP) inhibits this enzyme.
GDH can catalyze the reverse reaction also, i.e. amina-tion of α-ketoglutarate to form glutamate. This reaction requires NADPH as coenzyme (cosubstrate).
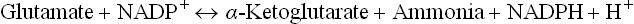
The reverse reaction is activated by ATP and GTP and inactivated by ADP and GDP. Thus, when the cellular energy charge is high amination of α-ketoglutarate to glutamate is favoured and deamination of glutamate to α-ketoglutarate is impeded.
ATP and GTP are allosteric inhibitors of glutamate dehydrogenase, and GDP and ADP are allosteric activators. Therefore, the enzyme is activated with lowering of cellular energy. The reverse reaction—amination of α-ketoglutarate to glutamate, is activated with increased cellular energy.
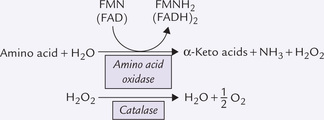
Box 13.1 Alternate Pathways of Deamination
Two other enzymes, besides glutamate dehydrogenase, provide alternative route for releasing nitrogen of α-amino groups as ammonia. They are L-amino acid oxidase (cofactor: FMN) and D-amino acid oxidase (cofactor: FAD). Both enzymes are present in liver and kidneys, localized in peroxisomes where they generate hydrogen peroxide. The latter is decomposed by catalase.
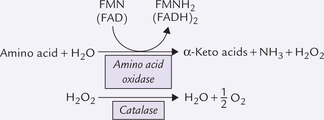
The L-amino acid oxidase can act on all amino acids except hydroxy amino acids and dicarboxylic amino acids. Activity of the D form is higher than that of the L form. It is not important for the degradation of common L-amino acids. Its main function is to degrade D-amino acids present in bacterial cell walls. Thus, D-amino acid oxidase functions during the degradation of D-amino acids of bacterial origin that are absorbed from the gut.
The utilization of two different coenzymes by glutamate dehydrogenase—NAD+ for release of ammonia and NADP+ for incorporation of ammonia—permits independent regulation of these two reactions. It is noteworthy that independent regulation is possible although both the reactions are catalyzed by the same enzyme.
In addition to glutamate dehydrogenase, amino acid oxidase provides an alternative pathway for deamination (Box 13.1).
To sum up, successive transamination and oxidative deam-ination reactions effectively release the α-amino group as ammonia. Because glutamate is the only amino acid that can undergo rapid oxidative deamination, most other amino acids form glutamate by transferring their amino group to α-ketoglutarate.
The ammonia so released is neurotoxic, and so safely transported, as discussed below.
C Transport of Ammonia
Release of the α-amino group as ammonia, discussed above, is potentially a hazardous event because ammonia is neurotoxic even in low concentrations (above 4 × 10-5 M) in blood. Therefore, it is vital to deal efficiently with the ammonia generated. Aquatic animals can release ammonia as such through their gills (ammonotelic), while birds, reptiles and insects convert it to uric acid (uricotelic). In terrestrial vertebrates, including humans, it is transported to liver chiefly as glutamine and alanine, where it is detoxified to urea (humans are ureotelic). Urea is then transported to the kidneys and excreted in the urine. Kidneys also form some urea.
Ammonia is produced in most tissues and must be transported to liver without causing ammonia toxicity. Glutamate, glutamine and alanine are the transport forms of ammonia from peripheral tissues to liver.
1. Glutamate (α-ketoglutarate plus ammonia) may be considered as the major participant in such interor-gan transport of ammonia. Concentration of glutamate in blood is about tenfold higher than other amino acids.
2. Glutamine is the transport form of ammonia from brain. Since brain is extremely sensitive to ammonia, it possesses a special mechanism for its immediate detoxification by combining it with glutamate to form glutamine (Fig. 13.6 ). Synthesis of glutamine is catalyzed by the enzyme glutamine synthetase
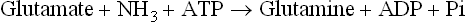
Glutamine is released into the blood circulation and carried to liver, where it enters hepatocytes (because of its neutral nature, glutamine can readily move across the cell membrane by facilitated diffusion). Glutaminase converts it into glutamate and ammonia, and the latter is channeled into the urea cycle. Thus, glutamine serves as the major transport form of ammonia from brain, based on successive actions of glutamine synthetase and glutaminases.
Asparagine may also similarly transport ammonia (by successive action of asparagine synthetase and asparagi-nase). However, asparaginase is quantitatively of much less significance.
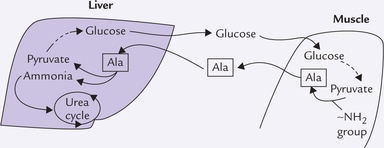
3. Alanine serves as the transport form of ammonia from muscles. The amine group of various amino acids is transferred to pyruvate in muscles to form alanine. The latter is transported to liver where the amine group is removed to reform pyruvate, which is used for glucose synthesis. This is glucose alanine cycle (or Cahill cycle), a major participant in interor-gan transport of ammonia (see Box 13.2).
Ammonia Toxicity
Ammonia is a highly toxic substance, particularly to the brain. The glial cells surrounding the neurons are highly susceptible to ammonia. So high is its toxicity that injection of extremely dilute solutions of ammonia into the blood stream is capable of rendering the experimental animal comatose. Ammonia can readily diffuse through cell membranes and enter tissues so that little is left in blood circulation (its concentration in peripheral blood is 30-60 μg/dL).
Most of the ammonia is converted to ammonium ion because of its high pK' value (less than 1% is left as ammonia) and even such small amount is capable of causing serious toxic effects. Biochemical basis of ammonia toxicity is not clearly understood. The following mechanisms have been proposed:
1. Decreased glucose utilization and ATP generation: In the brain cell mitochondria, excess ammonia may cause the reductive amination of α-ketoglutarate by glutamate dehydrogenase, forming glutamate. This reaction depletes α-ketoglutarate, a key intermediate of the TCA cycle, and leads to its impairment. As a result, there is severe inhibition of glucose utilization and fall of ATP generation.
This theory is, however, not universally accepted.
2. Glutamate depletion: Ammonia exerts an inhibitory effect on activity of glutaminase, resulting in depletion of glutamate, an excitatory neurotransmitter, in the neuronal cells. Glutamine, synthesized and stored in glial cells, is the most likely precursor of glutamate (it is transported into the neurons and hydrolyzed by glutaminase). Ammonia inhibits glutaminase and depletes the glutamate concentration.
Moreover, intracellular accumulation of glutamine causes osmotic shifts of water into the cell, resulting in oedema and swelling of astrocytes. This may aggravate the encephalopathy of hyperammonaemia.
3. Neuronal dysfunction: Hyperammonia increases permeability of the neuronal membrane to K+ and Cl-ions to cause neuronal dysfunction.
4. Accumulation of excito-toxins: Increased transport of tryptophan across the blood brain barrier and accumulation of its metabolites, which are excito-toxins, are also implicated in ammonia toxicity. Two of such tryptophan-derived metabolites are serotonin and quinolinic acid.
Other Sources of Ammonia
Although major source of ammonia is amino acids, additional sources are also known. These are:
1. Bacterial degradation of urea in the intestinal lumen.
2. Action of renal glutaminase on glutamine in renal tubular cells.
3. Action of intestinal glutaminase on glutamine in intestinal mucosal cells.
4. Release of amino groups of purines and pyrimidines as ammonia during catabolism of these nitrogen bases.
Box 13.2 Glucose—Alanine Cycle
In muscles, pyruvate accepts the ~NH2 group from various amino acids to form alanine. The latter is also released into blood circulation, from where it is taken up by the liver. The following transformations occur in the liver cell.
1. The α-amino group of alanine is removed by transamination and channeled into urea cycle.
2. The remaining 3-carbon skeleton (i.e. pyruvate) is converted to glucose by gluconeogenesis.
The glucose formed by gluconeogenesis is circulated back to muscles where it again forms pyruvate by way of glycolysis. Pyruvate then accepts the ~NH2 group to form alanine which again moves to liver, thus repeating the cycle. In this way, the glucose-alanine cycle plays dual role:
D Urea Cycle
The only organ where urea synthesis occurs is liver. Urea is the major excretory product in humans, accounting for an average of 86% of nitrogen eliminated. The rest of the nitrogen is eliminated as follows: 4.5% by creatinine, 2.8% as ammonium ions, 1.7% as uric acid, and 5.0% as other compounds. About 30 g urea is excreted per day; the amount excreted is dependent on protein intake. Higher the protein intake, more is the urea synthesis and excretion.
Reactions of Urea Cycle
The sequence of reactions leading to urea synthesis was first proposed by Krebs and Henseleit in 1932, five years before the elucidation of TCA cycle. Urea cycle was the first cyclic pathway to be identified. All the reactions of this pathway are shown in Figure 13.7 . The first two reactions take place in the mitochondria, and the rest occur in cytosol.
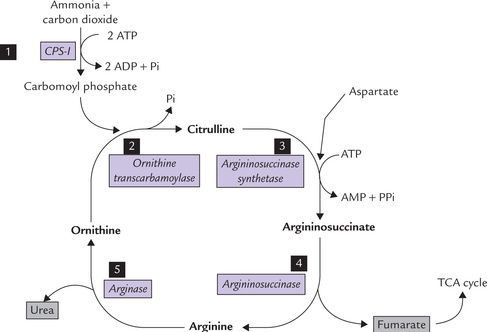
Reaction 1: Formation of Carbamoyl Phosphate
Carbamoyl phosphate is formed from ammonia and carbon dioxide in an energy-requiring reaction. Two ATP molecules are required to drive this reaction forward. The enzyme catalyzing this step, carbamoyl phosphate synthetase-I (CPS-I) is rate-limiting for the pathway. It is present in very high concentration in liver mitochondria and its Km for ammonia (250 μM) is not much higher than the physiological ammonia concentration. These properties enable the enzyme to effectively remove ammonia quantitatively from its environment.
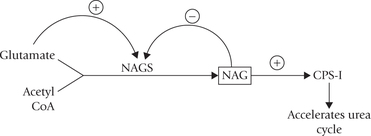
N-Acetyl glutamate (NAG) is an obligatory positive effector of CPS-I, as discussed later.
Formation of carbamoyl phosphate is the rate-limiting step of urea synthesis. Increased dietary consumption of proteins raises the level of N-acetylglutamate in liver, which stimulates carbamoyl phosphate synthesis and hence urea production.
In humans there are two immunologically distinct carbamoyl phosphate synthetases, one mitochondrial (CPS-I) and the other cytosolic (CPS-II).
• CPS-I is involved in ureagenesis, uses NH3 exclusively as the nitrogen donor and requires binding of NAG for its activity.
• CPS-II is used in pyrimidine synthesis, does not depend on NAG and uses glutamine as a substrate (Chapter 20).
These characteristics make possible compartmental-ization of the two pathways, one of which is degradative to remove nitrogen from the body, and the other is synthetic to build the bases for DNA or RNA.
Reaction 2: Formation of Citrulline
Carbamoyl phosphate, a high energy mixed anhydride, condenses with ornithine to form citrulline. The reaction is catalyzed by a mitochondrial enzyme ornithine trans-carbamoylase.
Citrulline diffuses out of the inner mitochondrial membrane so that the subsequent reactions of the urea cycle take place in the cytosol.
Reaction 3: Condensation of Citrulline with Aspartate
This is a complex condensation reaction between citrul-line and aspartate to form argininosuccinate. The reaction is catalyzed by the enzyme argininosuccinate synthetase. Free energy is required in this reaction which is provided by pyrophosphate cleavage of ATP. The pyrophosphate cleavage ensures irreversibility of the reaction.
Reaction 4: Cleavage of Argininosuccinate
The carbon skeleton of aspartate is released as fumarate while its nitrogen remains in the cycle, forming one of the nitrogen side chains of arginine. Reaction is catalyzed by the enzyme argininosuccinate lyase (also known as argininosuccinase).
Reaction 5: Formation of Urea
In the last step, arginine is hydrolyzed by the enzyme arginase to form urea. The other product of this reaction, ornithine, enters the mitochondrial matrix to participate in the urea cycle again.
Urea cycle is linked to TCA cycle
Synthesis of fumarate (reaction 4) is important because it links urea cycle to the citric acid cycle. Fumarate is converted to malate which is in turn oxidized to oxaloacetate. Oxaloacetate can condense with acetyl CoA to form citrate, the first intermediate of TCA cycle.
Energetics of Ureagenesis
The stoichiometry of urea synthesis is as below:
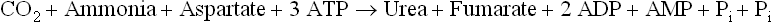
As shown, formation of one molecule of urea is powered by three ATPs and requires one molecule each of ammonia, carbon dioxide and aspartate. In all, hydrolysis of four high energy phosphate groups is required in each cycle: two are needed to drive the formation of carbamoyl phosphate and two for the formation of argininosuccinate.
However, the net energy expenditure may fall to only one ATP if fumarate (formed in the fourth step) is converted to malate. When this malate is oxidized to oxaloacetate, one NADH molecule is generated that can give rise to three ATP molecules through the electron transport chain. Thus, the energy expenditure becomes one (4-3) ATP molecule for each molecule of urea formed.
Control of Urea Cycle
Coarse Regulation
It occurs by induction-repression mechanism. The urea cycle enzymes are induced or repressed depending on the metabolic needs of the body. In starvation, urea cycle enzymes are induced. Their activities are elevated by 10-20-fold. It permits increased formation of urea in response to increased catabolism of the proteins that occurs in starvation. Moreover, cellular energy falls low in starvation, which activates the glutamate dehydrogenase. This results in increased production of ammonia which is channeled into urea cycle.
A protein rich diet also accelerates urea cycle through the activation of rate limiting enzyme, carbamoyl-phosphate synthetase.
Fine Regulation
It occurs by allosteric-modulation. The major regulatory enzyme of the urea cycle is carbamoyl phosphate synthetase-I, which is subject to allosteric activation by N-acetyl glutamate (NAG). Transfer of the acetyl group from acetyl CoA to glutamate by the enzyme NAG synthase (NAGS) forms this compound.
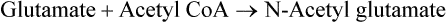
NAG synthase is under positive allosteric modulation by arginine and product inhibition by NAG. A high glutamate level also leads to increased NAG synthesis; this situation can occur when more amino acids are degraded. High glutamate level leads to increased NAG, hence enhanced activity of CPS-I, and thereby increased rate of ureagenesis.
Defects of Urea Cycle
Synthesis of urea provides the major route for the removal of toxic ammonia from body. Blockage of any of the steps of urea synthesis, therefore, results in accumulation of ammonia in the blood; the condition is known as hyperammonaemias. The blockage may result due to genetic deficiency of an enzyme of the urea cycle (i.e. familial hyperammonaemia) or due to some acquired defect (i.e. acquired hyperammonaemia).
Familial Hyperammonaemia
Genetic deficiency of each of the five enzymes of urea cycle have been described with an overall prevalence of about 1 in 30,000 live births. In addition, deficiency of NAG synthase has also been described. Inheritance pattern of the latter is not known, but the four of the urea cycle defects are autosomal recessive and ornithine car-bamoyltransferase deficiency is X-linked. They are enlisted below, with name of the enzyme in parenthesis:
• Hyperammonaemia type I (carbamoyl phosphate synthetase).
• Hyperammonaemia type II (ornithine transcarbamo-ylase) - X-linked. (All others are autosomal recessive).
• Citrullinaemia (argininosuccinate synthetase).
Feeding difficulties, lethargy or irritability, vomiting and poor intellectual development, and tendency for coma and death are common to all these disorders, though they vary in severity.
Earlier blocks: When the block is in one of the earlier enzymes, ammonia itself accumulates and the condition is more severe.
Later blocks: When the block is in later enzymes, accumulation of other intermediates occurs, which are less toxic, and therefore the condition is relatively mild.
Dietary proteins aggravate the symptoms and many patients develop spontaneous distaste for protein-rich foods.
Acquired Hyperammonaemia
Impaired detoxification of ammonia in advanced liver cirrhosis is a far more important cause of hyperam-monaemia. This condition is end point of several disease processes, such as alcoholism, hepatitis or biliary cirrhosis, and is characterized by progressive loss of hepatocytes. The lost hepatocytes are replaced by fibrous connective tissue, which impairs blood flow through the liver, and so the portal blood is shunted directly into the systemic circulation without having access to liver. The detoxification of ammonia in liver is, therefore, impaired and its concentration in systemic blood rises.
Portal hypertension also develops, which is dangerous because it can result in fatal haemorrhage from the dilated lower oesophageal veins. These derangements lead to CNS involvement, i.e. hepatic encephalopathy, also known as portal-systemic encephalopathy (because the shunting of blood around the cirrhotic liver is important in its pathogenesis). It has poor prognosis, frequently progressing to coma and death. Although various other biochemical and pathophysiological abnormalities have also been implicated, hyperammonaemia is a major cause of the CNS disorder.
Diagnosis of hyperammonaemias calls for detection of increased ammonia levels in blood and/or urine. Besides ammonia, glutamine level is also elevated because excess ammonia is diverted into glutamine synthesis. The immediate substrate of the deficient enzyme also is elevated in blood and urine in familial hyperammonaemia. Paper chromatography, paper electrophoresis, and ion-exchange chromatography are available for the quantitative determination of these compound amino acids in serum and urine. Final confirmation requires an assay-proven enzyme defect.
Treatment of hyperammonaemia involves a twofold strategy: dietary manipulations and activation of alternative routes of nitrogen excretion:
1. Dietary protein restriction is considered a mainstay of the long-term management of hyperammonaemia cases. The replacement of essential amino acids by their corresponding α-keto acids minimizes the requirement for nitrogen disposal, without precipitating the essential amino acid deficiencies. This is because transamination reactions generate most of the essential amino acids from their corresponding α-keto acids in diet.
2. The second treatment strategy promotes nitrogen excretion in forms other than urea. This is accomplished by activation of some latent biochemical pathway to bypass a genetic defect. The following examples illustrate this strategy:
(a) The metabolic block resulting due to argininosuc-cinase deficiency can be partially bypassed by supplementing the diet with arginine (Fig. 13.8 ). The surplus arginine is metabolized into argininosuc-cinate (via steps 5, 2 and 3), in the process utilizing ammonia. Argininosuccinate is subsequently disposed. In this way argininosuccinate substitutes urea for eliminating nitrogen from the body.
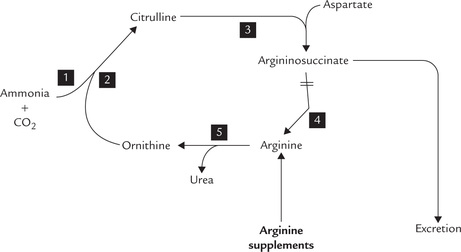
Fig. 13.8 Bypassing the metabolic block in argininosuccinase deficiency. Numbers 1 to 5 are steps of reactions discussed in text.
(b) A different approach is employed for the treatment of carbamoyl phosphate synthetase deficiency or of ornithine transcarbamoylase deficiency. In these conditions, excessive ammonia accumulates in glutamine (and glycine). The aim of treatment is to get rid of these two amino acids. This is accomplished by supplementing the diet with large amount of benzoate and phenylacetate. Benzoate is activated to benzoyl CoA which reacts with glycine to form hippurate. Likewise, phenylacetate is activated to phenylacetyl CoA which reacts with glutamine to form phenylacetylglutamine. These two conjugates act as substitutes for urea in the disposal of ammonia.
Case 13.1 describes clinical presentation and treatment strategy of a urea cycle disorder.
3. Other treatment strategies for management of hyper-ammonaemia may require aggressive approach. For instance, any neonatal hyperammonaemia, irrespective of the cause, is medical emergency and requires a rapid lowering of ammonia levels to prevent serious effects on the brain. These include haemodialysis, exchange transfusion, peritoneal dialysis, etc. Gastrointestinal haemorrhage from dilated oesophageal veins in portal-systemic encephalopathy needs to be controlled. Sterilization of gut in hepatic encephalopathy is helpful, although there is a risk of over growth by drug-resistant bacteria, resulting in enterocolitis.
III Catabolism of Carbon Skeleton of Amino Acids
Excess amino acids are metabolized by oxidative pathways to generate energy. As discussed earlier, the metabolism occurs in two stages:
• Metabolism of the amino group
• Metabolism of the carbon skeleton (Fig. 13.2).
One or more of the following compounds are generated as catabolic (end) products from the carbon skeleton: acetyl CoA, acetoacetyl CoA, pyruvate, or some intermediates of TCA cycle (e.g. oxaloacetate, α-ketoglu-tarate, succinyl CoA or fumarate). Thus, carbon skeletons of different amino acids are converted into a limited number of compounds, which are further metabolized in TCA cycle. This provides an example of the economy of metabolic conversions (Fig. 13.9 ).
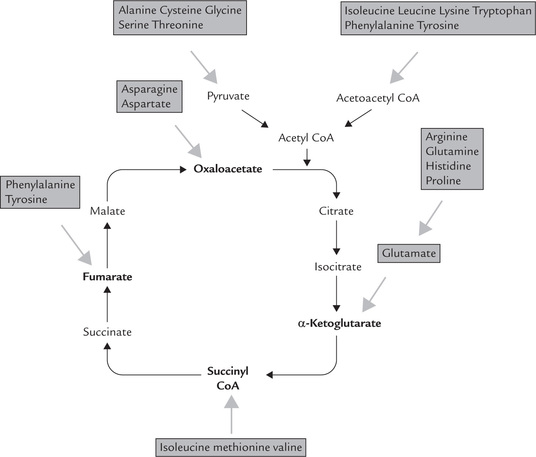
Further fate of these seven products depends on the cellular energy requirements. When the cell needs energy, these products undergo further degradation via TCA cycle to provide energy. On the other hand, when the cell is already supplied with adequate energy, they enter pathways of intermediary metabolism to yield glucose or ketone bodies. Evidently, strategy of amino acid catabolism is to form major metabolic intermediates that can be oxidized via TCA cycle, or converted to either glucose or ketone bodies.
Depending on the nature of the end product formed, the amino acids are classified in three categories (Table 13.1 ). They are designated as glycogenic if they can be converted to glucose, ketogenic if they can be converted to ketone bodies, and both glycogenic and ketogenic if they are convertible to both types of compounds. This classification is based on experiments performed by administration of each amino acid to experimental animals and determining whether there was an increase in glucose in the urine (glycogenic amino acid), an increase in ketone bodies in the urine (ketogenic amino acid), or both. Glycogenic amino acids are catabolized to pyruvate or a TCA intermediate, which are potential precursors for glucose and glycogen. Ketogenic amino acids yield acetyl CoA or acetoacetyl CoA, or both. Some of the carbon atoms of the amino acids, which are both glycogenic and ketogenic, emerge in acetyl CoA and acetoacetate, whereas others appear in potential precursors of glucose. Leucine and lysine are the only amino acids that are regarded as being exclusively ketogenic. The aromatic amino acids and isoleucine (four amino acids) are both glycogenic and ketogenic, and the remainder (14 amino acids) can be considered as purely glycogenic. This classification is, however, not universally accepted.
Table 13.1
Classification of amino acids on the basis of catabolic end products
| Convertible to glycogen (Glycogenic) | Convertible to fat (Ketogenic) | Convertible to glycogen and fat (Glycogenic and ketogenic) |
| Alanine | Leucine | Isoleucine |
| Glycine | Lysine | Phenylalanine |
| Histidine | Tyrosine | |
| Glutamine | Tryptophan | |
| Asparagine | ||
| Cysteine | ||
| Glutamate | ||
| Aspartate | ||
| Serine | ||
| Threonine | ||
| Valine | ||
| Methionine | ||
| Arginine | ||
| Proline |
Amino acids are designated as glycogenic if catabolized to pyruvate or a TCA cycle intermediate, ketogenic if convertible to acetyl CoA or acetoacetyl CoA, and both glycogenic and ketogenic if they can be converted to either type of compounds.
Catabolism of the primary amino acids, as discussed below, has been divided on the basis of nature of the cat-abolic end product. Generally, the oxidative metabolism of non-essential amino acids is simple, and that of the essential amino acids is relatively complicated.
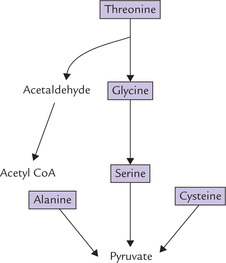
A Amino Acids that Form Pyruvate (Fig. 13.10)
The 3-carbon amino acids, alanine, serine and cysteine are directly convertible to the 3-carbon keto acid, i.e. pyruvate. The carbon skeletons of two other amino acids, glycine (2-C) and threonine (4-C), also enter the metabolic mainstream through pyruvate.
1. Alanine can be directly converted to pyruvate by transamination catalyzed by the enzyme ALT.
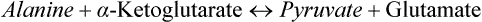
2. Serine undergoes dehydration followed by loss of α-amino group to yield pyruvate as depicted in Figure 13.11 (see Reactions 1 and 2). Both the reactions are catalyzed by serine dehydratase, a pyridoxal phosphate-dependent enzyme.
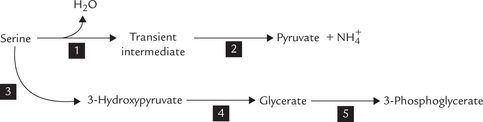
Alternatively, serine can undergo a transamination reaction (3) to produce 3-hydroxypyruvate, which is reduced by an NADH-dependent enzyme glycerate dehydrogenase (4) to yield D-glycerate. The latter is phosphorylated to 3-phosphoglycerate (5), a glyco-lytic intermediate.
Both these pathways produce a gluconeogenic intermediate that can also be metabolized by mainstream reactions.
3. Cysteine can be converted to pyruvate by loss of sulphur atom, which is released as H2S or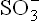 The metabolism is considered intricate because of the multiple pathways cysteine can enter during sulphur metabolism:
The metabolism is considered intricate because of the multiple pathways cysteine can enter during sulphur metabolism:
(a) Loss of 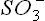 : Cysteine reacts with oxygen to yield cysteine sulphinate, which undergoes transamina-tion to yield β-sulphinylpyruvate. The latter then undergoes an exergonic hydrolysis to yield pyruvate and sulphite (
: Cysteine reacts with oxygen to yield cysteine sulphinate, which undergoes transamina-tion to yield β-sulphinylpyruvate. The latter then undergoes an exergonic hydrolysis to yield pyruvate and sulphite (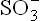 ) which is oxidized to produce sulphate by the enzyme sulphite oxidase. Cysteine sulphinate can be converted to taurine in a quantitatively minor pathway, which combines with cholyl-CoA to produce taurocholate, a bile salt.
) which is oxidized to produce sulphate by the enzyme sulphite oxidase. Cysteine sulphinate can be converted to taurine in a quantitatively minor pathway, which combines with cholyl-CoA to produce taurocholate, a bile salt.
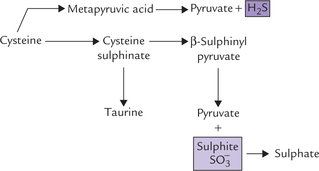
(b) Loss of H2S: Cysteine may undergo isoergonic transamination to mercaptopyruvic acid, which liberates H2S to form pyruvate.
The conversion of cysteine to pyruvate by either pathway accounts for the glycogenic nature of this amino acid.
4. Glycine metabolism is intimately linked with tetra-hydrofolate (THF).
The following pathways are involved:
(a) The major pathway is via the glycine cleavage enzyme, which catalyzes degradation of glycine to carbon dioxide and a THF-bound one carbon unit. The enzyme is also known as the glycine synthase for the reverse reaction
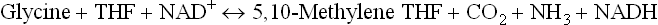
(b) Hydroxymethylene transfer reaction is another significant way to metabolize glycine. It bring about glycine (2-c) to serine (3-c) conversion by addition of one carbon (hydroxymethylene) group. The donor of one carbon group is 5,10-methylene tetrahydrofolate and the enzyme is hydroxymethyltransferase.
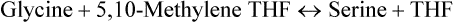
Serine is further metabolized as discussed.
(c) A minor pathway of glycine metabolism is its conversion to glyoxylate via glycine oxidase or D-amino acid oxidase. Glyoxylate can be oxidized to oxalate and excreted in urine; the precipitation of soluble calcium oxalate is the most common cause of renal calculi.
5. Threonine, the 4-C amino acid, is cleaved to 2-C products, glycine and acetaldehyde (Fig. 13.10 ). Glycine is then metabolized as above; and the other two carbons of threonine (that appear in acetalde-hyde) form acetyl CoA. Threonine metabolism can also produce another TCA cycle intermediate, succi-nyl CoA, as discussed later.
B Amino Acids That Form TCA Cycle Intermediates (Fig. 13.9)
Carbon skeleton of certain amino acids enter the metabolic mainstream via some intermediates of TCA cycle such as oxaloacetate, α-ketoglutarate, fumarate or succinyl CoA.
Oxaloacetate producing
The C-4 family of amino acids, i.e. aspartate and asparagine form oxaloacetate (Fig. 13.9). Asparagine degradation involves:
• An exergonic hydrolysis at the amide nitrogen by the enzyme asparaginase to produce aspartate and ammonia.
• Aspartate undergoes an isoergonic transamination reaction to yield oxaloacetate.
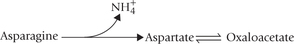
Asparaginase in treatment of cancer: Parenteral administration of the enzyme asparaginase has been used in various malignancies, in particular leukaemias in adults. The leukaemic cells lose ability to synthesize asparagine and depend for its supply from blood circulation. The injected asparaginase sufficiently lowers the plasma level of asparagine to decrease its uptake by these cells, thereby inhibiting their growth.
α-Ketoglutarate producing
End point of several amino acids of C-5 family, such as glutamate, glutamine, arginine, proline and histidine is α-ketoglutarate (Fig. 13.9). These pathways for α-ketoglutarate are discussed below:
1. Glutamate is converted to the corresponding 5-C keto-acid, α-ketoglutarate, by action of the enzyme glutamate dehydrogenase, as described earlier in detail.
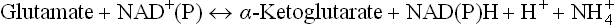
The other four amino acids of the C-5 family, i.e. glutamine, arginine, proline and histidine are first converted to glutamate which then yields α-ketoglutarate.
2. Glutamine loses amino group by action of the enzyme glutaminase. Glutamate so formed is then converted to α-ketoglutarate.
3. Proline undergoes a flavoprotein-dependent oxidation to form Δ1-pyrroline 5-carboxylate. Water adds to this compound and the ring opens non-enzymatically to form glutamate γ-semialdehyde (Fig. 13.12 ). The semialdehyde is oxidized to the carboxylate state in an NAD+-dependent reaction to produce glutamate. The conversion of aldehyde to a carboxylate is exergonic and irreversible because the carboxylate is stabilized by resonance.
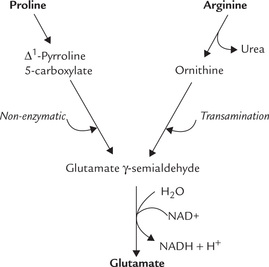
4. Arginine is hydrolyzed by the enzyme arginase to form ornithine; urea is the other reaction product (Fig. 13.12). Ornithine is a five-carbon amino acid that undergoes transamination reaction to form glutamate semialdehyde. The latter is oxidized to glutamate.
5. Histidine: One carbon of the histidine molecule is transferred to the one-carbon pool of tetrahydrofolate derivatives and the other five are converted to glutamate. The first step, catalyzed by the enzyme histidase involves a lyase reaction with the elimination of ammonia to form urocanate (Fig. 13.13 ). Urocanate undergoes a hydration and isomerization to form imidazolone pro-pionate, which then undergoes an exergonic hydrolytic reaction to form N-formimino-glutamate (FIGLU). The latter donates its formimino group to tetrahydro-folate, leaving behind glutamate. This reaction exemplifies the role of tetrahydrofolate as one carbon unit pool (Box 13.3).
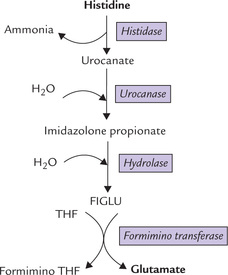
Folic acid is required for amino acid degradation. One-carbon fragments (formyl formimino, methylene, etc.) get attached to the reactive portions of this vitamin, from where they are transferred to several acceptor compounds.
As folic acid is essential for metabolism of histidine, in cases of folic acid deficiency, metabolism of histidine stops at FIGLU. If histidine is orally administered to such patients, plasma concentration of FIGLU and its urinary excretion rises. Thus, folic acid deficiency is diagnosed by giving oral load of histidine and measuring urinary excretion of FIGLU; in case of deficiency, marked rise in the urinary excretion of this compound is observed. The test is termed the FIGLU test.
Fumarate producing
Phenylalanine and tyrosine produce fumarate (and acetyl CoA; Fig. 13.9) They share a common pathway of degradation, which occurs in liver, and ultimately yields fumarate (and acetyl CoA).
The pathway starts with oxidation of phenylalanine to tyrosine. Phenylalanine is an essential amino acid, but tyrosine is not. Tyrosine in the diet, however, decreases the requirement for phenylalanine, a phenomenon called sparing.
Conversion of phenylalanine to tyrosine: The enzyme phenylalanine hydroxylase (E1) catalyzes the reaction between phe-nylalanine, oxygen and tetrahydrobiopterin (THB) to form tyrosine. Addition of a hydroxyl group to the aromatic ring of phenylalanine occurs in this reaction. The enzyme is a monooxygenase (also called oxidase) because in this reaction one atom of oxygen is incorporated in the substrate and the second atom is reduced to water. Tyrosine cannot be converted back to phenylalanine, thereby accounting for the essential nature of phenylalanine.
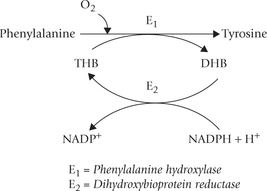
During the reaction, THB is converted to its oxidized form, the dihydrobiopterin (DHB). Reconversion of DHB to THB requires reducing power of NADPH; and the reaction is catalyzed by the enzyme DHB reductase (E2). Because the cellular [NADPH+]/[NADP+] ratio is greater than [NADH]/[NAD+], NADP+ is probably the physiologically important reductant.
Several biosynthetic reactions involve transfer of 1-C unit to a metabolic precursor. The 1-C units exist in a variety of oxidation states, including methane, methanol, formaldehyde, formate, formimino and methylene. The carriers of these compounds are folic acid and S-adenosyl methionine (SAM); the 1-C pool refers to these carrier compounds.
The 1-C units can be transferred from these carrier compounds to specific acceptors that are consequently modified. SAM carries methyl groups and can act as a methylating agent. Folic acid is a more important carrier because it can carry 1-C units in various oxidation states.

As described in Chapter 18 its active form is tetrahydrofolate (THF), and 1-C units are covalently attached to THF at positions N5, N10, or both. These 1-C units, in different oxidation states, are interconvertible by enzymatic redox reactions.
THF acquires 1-C unit in conversion of serine to glycine by serine hydroxymethyl transferase (page 292), in cleavage of glycine (page 292) and in histidine breakdown (Fig. 13.13). The 1-C units carried by THF are used in the synthesis of methionine from homocysteine (Fig. 18.7) and in the synthesis of purine nucleotides (Fig. 18.11) as described in Chapter 18.
Biotin, a water-soluble vitamin, is a carrier of carbon dioxide, which is transferred to certain acceptor molecules. These reactions are referred to as the carboxylation reactions; for example, conversion of pyruvate (3-C) to oxaloacetate (4-C). However, biotin is not considered as a member of the 1-C pool.
Degradation of tyrosine (phenylalanine): Further metabolism of tyrosine and phenylalanine is considered together. Because phenylalanine is converted to tyrosine, a single pathway, depicted in Figure 13.14 is responsible for the degradation of both amino acids. The sequence of reactions from tyrosine onwards is described below:
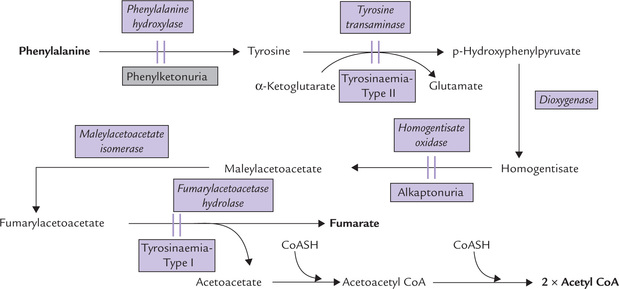
1. Tyrosine undergoes transamination to form p-hydroxyphenyl pyruvate. The reaction is catalyzed by a PLP-dependent enzyme, tyrosine transaminase.
2. p-Hydroxyphenyl pyruvate is a substrate for an irreversible dioxygenase reaction in which both atoms of oxygen are incorporated into the substrate, forming homogentisate (Fig. 13.14). The enzyme catalyzing this reaction, p-hydroxyphenyl pyruvate hydroxylase is a dioxygenase. It contains iron and requires ascorbic acid for keeping the ferrous iron in the reduced state.
3. Cleaving of the benzene ring of homogentisate occur next to form 4-maleylacetoacetate. Molecular oxygen is required for opening the aromatic ring, and the enzyme, homogentisate oxidase (iron metalloenzyme) catalyzes this reaction.
4. Maleylacetoacetate undergoes an isoergonic isomeriza-tion to form fumarylacetoacetate (maleate is the cis iso-mer of the 4-C dicarboxylate, and fumarate is the trans isomer). The enzyme catalyzing this reaction, maleylace-toacetate isomerase, requires glutathione as a cofactor.
5. Hydrolysis of fumarylacetoacetate occurs next by the enzyme fumarylacetoacetase. The reaction produces fuma-rate (glycogenic) and acetoacetate (ketogenic), accounting for the glycogenic as well as ketogenic nature of these amino acids.
Inherited deficiencies of the enzymes of this pathway lead to the diseases such as phenylketonuria, alkaptonuria and albinism, discussed later in this chapter.
Succinyl CoA producing
Methionine, valine, isoleucine and threonine yield succinyl CoA as end product (Fig. 13.9).
1. Methionine: Catabolism of methionine is divided into two parts. The first part comprises six steps that lead to the formation of propionyl CoA (Fig. 13.15 ); the second part consists of three steps that brings about conversion of propionyl CoA to succinyl CoA.
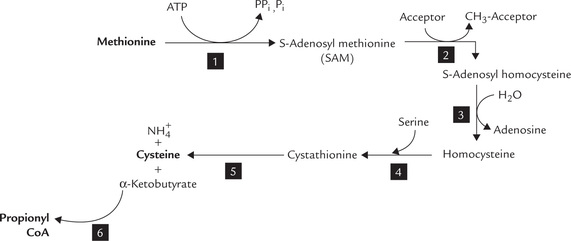
Formation of propionyl CoA: Methionine is activated by the enzyme methionine adenosyltransferase to form S-adenosyl methionine (SAM). It is an unusual reaction in which all three phosphates of ATP are released (Reaction 1). SAM is the most important methyl group donor in biological methylations; its methyl group can be transferred to a variety of acceptor molecules (Reaction 2).
By donating its methyl group, the S-adenosylmethionine is converted to S-adenosylhomocysteine. Displacement of adenosine from the latter by water produces homocysteine (Reaction 3). Homocysteine combines with serine to form cystathionine as catalyzed by cystathionine synthase, a pyridoxal phosphate-dependent enzyme (Reaction 4). Cystathionine lyase, also a pyridoxal phosphate-dependent enzyme, catalyzes hydrolysis of cystathionine to produce α-ketobutyrate, cysteine and ammonia (Reaction 5).
The α-ketobutyrate is then converted to propionyl CoA (Reaction 6) by a reaction sequence that parallels the conversion of pyruvate to acetyl CoA.
The first two reactions of this reaction-sequence illustrate role of SAM in transmethylation reaction. For more examples of transmethylation, refer to Box 13.4.
Conversion of propionyl CoA to succinyl CoA has been described earlier in Chapter 11. It should be noted that propionyl CoA derived from either amino acid degradation or odd chain fatty acid catabolism is converted to succinyl CoA by the same reaction sequence.
Reconversion of homocysteine to methionine: In reaction 3 homocysteine is produced, which must be reconverted to methionine. There exist two reactions for this purpose:
• The first involves transfer of a methyl group from betaine, a metabolite derived from choline (Chapter 12).
• The second reaction requires both folate (as methyl-tetrahydrofolate) and vitamin B12 (as methylcobala-min), as described in Chapter 18.
Methionine is an essential amino acid because humans cannot synthesize methionine from metabolites other than homocysteine.
During breakdown of methionine, serine is converted to cysteine by transfer of the sulphydryl group. Methionine is activated to SAM, which serves as a methyl group donor for synthesis of several biologically important compounds (choline, epinephrine, melatonin, creatine, etc.).
2. Valine and isoleucine: Valine and isoleucine are branched chain amino acids. Both yield succinyl CoA upon degradation (Fig. 13.16 ). Whereas valine yields only succinyl CoA, isoleucine yields acetyl CoA also (in addition to succinyl CoA).
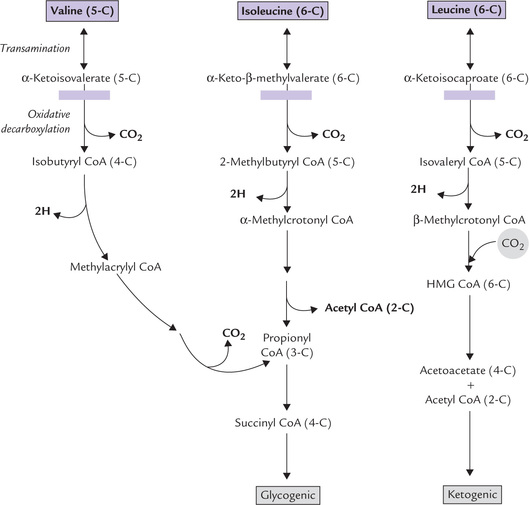
Fig. 13.16 Degradation of the branched amino acids. Blockage in the oxidative-decarboxylation step ( ) results in maple syrup urine disease.
) results in maple syrup urine disease.
Metabolism of these two amino acids, as also of leu-cine, shows a striking similarity. Therefore, the three branched chain amino acids (Valine, isoleucine and leucine) are considered together (Fig. 13.16), although leu-cine is purely ketogenic.
• The first reaction is an isoergonic transamination, occurring in muscle and other extrahepatic tissues. The resulting keto acids are transported to the liver, where they are metabolized further.
• The keto acids undergo oxidative decarboxylation by a single enzyme complex, the branched-chain α-keto acid dehydrogenase. This second reaction is analogous to pyruvate dehydrogenase and α-ketobutyr-ate dehydrogenase reactions. Similar to the pyruvate dehydrogenase reaction, five cofactors and three protein-activities participate in this reaction. The reaction products are: isobutyryl CoA from valine, 2-methylbutyryl CoA from isoleucine and isovaleryl CoA from leucine.
• The third reaction is a FAD-dependent dehydrogena-tion in which the bond between the a and the ( carbons of the CoA-thioester is oxidized to a double bond by a flavoprotein. This reaction resembles the first step of (β-oxidation (Fig. 11.3). This produces methylacrylyl CoA from valine, α-methylcrotonyl CoA from isoleucine and ( -methylcrotonyl CoA from leucine.
The remaining reactions are intricate and complex, differing for the three amino acids, and it is unnecessary to commit them to memory. An outline of these is presented in Figure 13.16. Evidently:
3. Threonine: Removal of a water molecule from threo-nine yields α-ketobutyrate. The latter is converted to pro-pionyl CoA which then forms succinyl CoA. Threonine metabolism can produce pyruvate also, as discussed earlier.
C Amino Acids that Form Acetyl CoA or Acetoacetyl CoA (Fig. 13.17)
There are six amino acids that yield acetyl CoA or acetoace-tyl CoA, making them ketogenic. These are: two branched chain-amino acids (isoleucine and leucine), aromatic amino acids (phenylalanine, tyrosine and tryptophan), and lysine. Figure 13.17 presents a simplified overview of their metabolism, which is otherwise quite intricate. For example, eight steps are required for conversion of tryp-tophan to α-ketoadipate and five more steps are required for conversion of α-ketoadipate to acetoacetyl CoA.
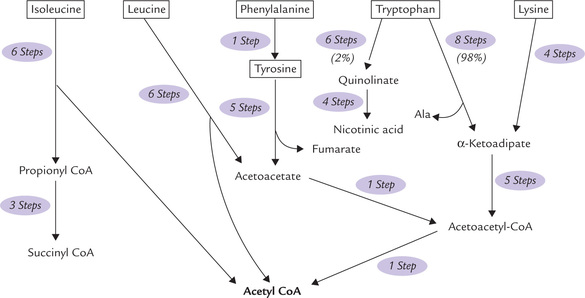
Fig. 13.17 Overview of the metabolism of ketogenic amino acids: the branched-chain amino acids (isoleucine and leucine), the aromatic amino acids (phenylalanine, tyrosine, and tryptophan), and lysine. Only leucine and lysine are exclusively ketogenic.
1. Leucine: Its metabolism has been discussed earlier (Fig. 13.16).
2. Tryptophan: Metabolism of tryptophan is highly complex, and as mentioned, tryptophan is converted to acetyl CoA (ketogenic) and alanine (glycogenic).
The process is initiated by tryptophan pyrrolase, a haem protein, which oxidatively cleaves the pyrrole ring to form N-formylkynurenine (Fig. 13.18 ). The further pathway may branch off as follows:
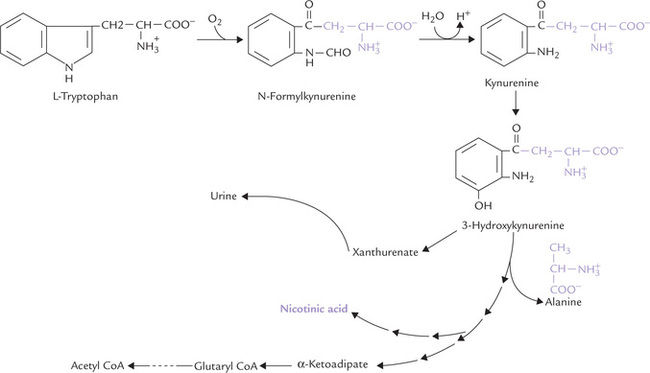
Fig. 13.18 Pathway of tryptophan catabolism. The portion of the molecule outside indole ring forms alanine (a glucogenic precursor), and the balance of carbons are ultimately converted to α-ketoadipate (a ketogenic precursor). This major pathway is diverted to form nicotinic acid, and vitamins.
(a) Glycogenic branch: The portion of tryptophan molecule outside the indole ring (shown in colour) becomes alanine a glucogenic precursor.
(b) Ketogenic branch: The indole ring itself is converted to a variety of other products, of which the α-keto-adipate is quantitatively the most important (98%). After four more sequential reactions, it is finally converted to acetoacetyl CoA, and then acetyl COA.
(c) Kynurenine branch: While bulk of tryptophan in metabolized as above, a minor fraction (less than 2%) is converted into nicotinic acid, a vitamin, by the kyn-urenine pathway. About 60 mg tryptophan is equivalent of 1 mg of nicotinic acid by this pathway, also called the nicotinic acid pathway of tryptophan (Chapter 18). It is diverted from the major pathway at the level shown in Figure 13.18.
Some tryptophan derived isoquinolines such as xanthurenate are not degraded further, but are excreted in urine. They are in part responsible for the yellow colour in urine.
3. Lysine: Catabolism of lysine is similar to that of tryptophan in that it comprises a lengthy pathway that forms α-ketoadipate, which finally yields acetyl CoA. This makes lysine a ketogenic amino acid. However, reports from animal studies indicate that lysine is both glycogenic and ketogenic. This discrepancy indicates that we lack complete information on the metabolism of this essential amino acid.
4. Phenylalanine and tyrosine: Metabolism of these two amino acids produce fumarylacetoacetate, which undergoes an exergonic ydrolysis to form fumarate (glycogenic) and acetoacetate (ketogenic), thereby accounting for the glycogenic and ketogenic nature of these amino acids (Fig. 13.14).
5. Isoleucine: This is, likewise, both glycogenic and ketogenic since it produces both succinyl CoA and acetyl CoA.
IV Disorders of Amino Acid Metabolism
A number of inborn errors of amino acid metabolism are known. They occur due to genetically determined deficiency or absence or modification of a specific protein. The affected protein may be an enzyme or a transport protein.
• Enzyme: In majority of the cases, the deficient protein is an enzyme of a metabolic pathway. As a result, the metabolic pathway is blocked (i.e. metabolic block), which causes abnormalities in the normal metabolism. For example, when the enzyme phenyl-alanine hydroxylase that causes conversion of phenylalanine to tyrosine, is deficient, normal metabolism of phenylalanine is disrupted; the condition is called phenylketonuria
• Transport protein: In some other cases, the transport proteins responsible for the renal or the intestinal absorption of amino acids, is defective. For example, renal tubular reabsorption of cystine is impaired in cystinuria; renal tubular and intestinal transport of the neutral amino acids is impaired in Hartnup disease.
Such disorders are rare, yet they are of considerable significance. Unless early diagnosis is made and appropriate treatment is initiated, the consequences of many of such disorders are disastrous. However, if timely measures are taken, the patient can lead a nearly normal life. Most of these disorders are inherited in an autosomal recessive manner. Phenotypically, the heterozygotes are usually normal.
For a better understanding of such disorders, it would be worthwhile to review the fundamental design of the normal metabolic pathways, effects of metabolic aberrations and the treatment strategies. Consider the following pathways consisting of three sequential reactions, catalyzed by enzymes designated as X, Y and Z.
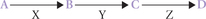
A metabolic block, where the enzyme Z is defective, produces the following effects:
• Decreased concentration of the reaction product, D
• Increased concentration of substrate, C
• Increased production of alternate metabolites from the accumulated substrate.
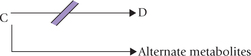
The treatment strategies of these disorders primarily aim at (i) restricting the substrate intake, and (ii) increasing the supply of the missing product.
For example, in phenylketonuria following measures are recommended:
• A restricted dietary intake of phenylalanine and generous intake of tyrosine.
• Providing the coenzyme of the defective enzyme or replacing the gene coding for the enzyme protein may also be helpful.
Finally, elimination of the accumulated amino acids or alternate metabolites by appropriate drugs ameliorates the patient’s symptoms in some of these disorders. For example, in treatment of cystinuria (an inherited disorder characterized by defective renal tubular reabsorption of cystine and the dibasic amino acids, i.e. lysine, ornithine and arginine) penicillamine is used. This drug complexes with the accumulated cysteine and thus prevents formation of renal stones from cystine.
Some most common errors of amino acid metabolism are discussed in this section.
A Disorders of Aromatic Amino Acid Metabolism
Phenylketonuria (PKU)
This autosomal recessive trait is the most common inborn error of metabolism. In its classical form (type I) PKU is caused by complete deficiency of the enzyme phenylalanine hydroxylase. In type II, the enzyme deficiency is partial. Many mutations of the phenylalanine hydroxylase gene (located on chromosome 12q) have been identified, such as missense, nonsense, insertions, deletions and duplications. The incidence of classical PKU is 1 in 10,000-20,000 live births, but shows considerable geographic variation: the incidence in Ireland is 1 in 4000, whereas the condition is rare among Asians.
Variant forms
Variant forms of PKU are also known, e.g. type III and type IV, which account for 2% of cases of hyperphenylalaninaemia.
• Type III is due to deficiency of dihydropteridine reductase or the reductant coenzyme, NADPH (page 15).
• Type IV is due to deficiency of one of the enzymes that catalyzes tetrahydrobiopterin synthesis from GTP.
Because tetrahydrobiopterin is required for hydrox-ylation of tyrosine and tryptophan also, synthesis of catecholamines from tyrosine and synthesis of serotonin and melatonin from tryptophan are also hampered. Therefore, these variant forms result in more severe clinical manifestations.
Biochemical Abnormalities
1. Hyperphenylalaninaemia: The effect of blocking of the phenylalanine hydroxylase reaction is accumulation of phenylalanine in blood.
2. Phenylpyruvate production: When the concentration of phenylalanine exceeds a certain limit (above 1200 μM; normal is 30-20 μM) conversion to phenylpyruvate by transamination reaction becomes the major metabolic fate of the accumulated amino acid (Fig. 13.19 ). Among unaffected individuals, such direct transamination is a very minor pathway of phenylalanine metabolism.
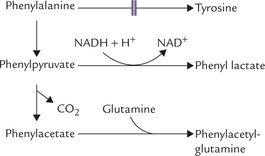
Fig. 13.19 Formation of alternate metabolites—phenylpyruvate, phenyllactate, phenylacetate and phenylacetylglutamine—from the accumulated phenylalanine in phenylketonuria.
3. Urinary elimination of phenylpyruvate: Phenylpyruvate is excreted in the urine and accounts for the name "phenylketonuria"
4. Other urinary metabolites: In addition to phenylalanine and phenylpyruvate, other major urinary metabolites are formed in these patients due to opening of alternate pathways. These include phenyllactate, phenylacetate and phenylacetylglutamine, a conjugation product of phenylacetate.
Diagnostic tests
The affected infants are normal at birth, so diagnosis of PKU is missed unless screening tests are carried out. These tests in the newborn period are mandatory in the developed Western nations.
Ferric chloride test for urinary phenylpyruvate is widely used: transient blue colour is given by the phenylketonuric urine with this reagent.
Guthrie bacterial inhibition assay: In this test, the blood of phenylketonuric patient—but not the blood of normal individuals—supports the growth of phenylalanine-dependent bacterial strain (Bacillus subtilis). Ideally, the test should be done not less than 2 days after birth because false negatives are common within the first 24 or even 48 hours after birth.
Detection of hyperphenylalaninaemia: Measurement of blood phenylalanine level is reliable test; in PKU the level is 1200 μM or even more.
Amino acid analysis of blood by paper chromatogra-phy, showing elevated phenylalanine and normal tyro-sine is highly diagnostic.
Prenatal diagnosis by the enzyme assay is difficult because phenylalanine hydroxylase is expressed only in the liver; but the amniotic cells, which are used for the prenatal diagnosis, do not express the enzyme. DNA-based diagnostic procedures are under study, but are mostly inconclusive because of the allelic heterogeneity in PKU. It means that different patients have different mutations in the gene for phenylalanine hydroxylase.
Clinical presentation
The most important clinical presentation is mental retardation. Phenylalanine and its metabolites are transferred from the mother to the fetus and impair the fetal brain development. However, PKU is not evident at birth and clinical manifestations appear a few days or weeks later. The child develops mental retardation, with IQ values typically between 25 and 50. The reasons for the development of mental retardation are not clear, some possible mechanisms are discussed in Case 13.2.
Neurological signs, such as hyperreactive deep tendon reflexes, hyperactivity and seizures are also present sometimes.
Carrier state
Because PKU is recessive, it manifests clinically only in homozygous state. Heterozygotes, which have approximately half of the normal enzyme activity, can metabolize the normal load of phenylalanine, but not when this amino acid is injected in a dose of 4 g. The transient increase in serum phenylalanine level, seen in unaffected individuals with this dose, is exaggerated in the heterozygotes (phenylalanine tolerance test).
Treatment
1. Low phenylalanine diet: PKU is treated effectively with a synthetic diet that is low in phenylalanine but which maintains normal nutrition (as an essential amino acid, phenylalanine cannot be omitted entirely from the diet). Since the developing brain is damaged by the biochemical abnormality, prompt initiation of the dietary restrictions is important. These restrictions can be tapered off in older children. If initiated within the first few weeks after birth, rigorously controlled and continued until 5 or 6 years of age, mental development is essentially normal. In some cases, however, continuation of this diet throughout the first decade, or for life, may be necessary.
Tyrosine becomes an essential amino acid for the affected (phenylketonuric) children, but dietary supplements of this amino acid are mostly not required.
2. Oral tetrahydrobiopterin supplement: In the variant disorders (biopterin and biopterin reductase deficiency) treatment consists not only of regulating the blood levels of phenylalanine but of supplying the missing form of coenzyme, mostly oral tetrahydrobiopterin.
3. Gene replacement therapy is under study with some encouraging results.
4. Successfully treated females who have reached reproductive age and become pregnant may expose their offsprings to excess phenylalanine. This will affect the brain development of the fetus and may cause congenital disorders or spontaneous abortions. An extremely strict dietary control throughout pregnancy may be required.
Case 13.2 gives relevant aspects regarding biochemical abnormalities, clinical features and management of a phenylketonuric infant.
Alkaptonuria
The enzyme deficient in this disorder is homogentisate oxidase (Fig. 13.14). The disorder is inherited as autosomal recessive trait. Deficiency of the enzyme prevents conversion of homogentisate to maleylacetoacetate, resulting in plasma accumulation and excretion of homogentisate, an uncoloured hydroquinone, in urine. The urine darkens upon exposure to air owing to oxidation of homogentis-ate to benzoquinone acetate, which is polymerized to black alkapton bodies.
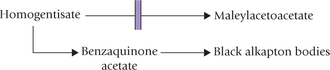
Alkaptonuria was the first to be described as an inherited disease, caused due to an enzyme deficiency (Archibald Garrod,1902). Along with cytinuria, albinism and pentosuria, it was included in Gerrod’s tetrad. The condition causes much anxiety to mother who notices dark nappies which become darker on washing in alkaline soaps and detergents. However, alkaptonuria is a harmless condition and as such does not need any treatment. The problem that the patient may face is development of arthritis in the middle age or later life. This is due to deposition of alkapton in cartilage; the condition is called ochronosis.
However, the relationship between pigment deposition and arthritis is not understood.
Tyrosinaemia
Two different types of tyrosinaemia have been identified: type I and type II, caused by deficiencies of the cytoplas-mic fumarylacetoacetate hydrolase and tyrosine transaminase respectively (Fig. 13.14).
Type I tyrosinaemia, also called tyrosinosis or hepato-renal tyrosinaemia, is more common, with an incidence of 1.5 per 1000 live births. Accumulation of fumarylaceto-acetate and related organic acids causes a cabbage-like odour, abnormal liver function and renal tubular dysfunction. Anaemia and vitamin D resistant rickets are also observed to develop. An abnormal metabolite, suc-cinyl acetone, derived from fumaryl acetoacetate, inhibits haem synthesis, resulting in porphyria-like neurological symptoms.
Type II tyrosinaemia, also referred to as oculo-cutaneous tyrosinaemia, manifests as painful corneal erosions and plaques, inflammation (from intracellular crystallization of tyrosine), keratosis of palmar surface and mental retardation. Low-tyrosine and low-phenylalanine diets are beneficial.
Transient tyrosinaemia of the newborn, particularly in premature infants, is the most common form of tyros-inaemia in infancy. It is caused by absence of the enzyme p-hydroxyphenylpyruvate dioxygenase (Fig. 13.14). It is mostly a benign condition and responds well to ascorbic acid.
B Disorders of Branched Chain Amino Acids
Maple Syrup Urine Disease
The condition results due to deficiency of the branched-chain α-ketoacid dehydrogenase, the second enzyme of the pathway, as shown in Figure 13.16. The enzyme is rather non-selective, acting on all three branched-chain α-keto-acids. The condition is termed maple syrup urine disease, the name derived from the fact that odor of urine resembles that of maple syrup. The prevalence in newborn infants is on the order of 1 in 200,000.
Diagnosis is confirmed by raised plasma levels and enhanced urinary excretion of the branched chain amino acids and their keto acids. If the diagnosis is made in the first week of life, and a diet low in branched chain amino acids is started, normal development is possible. Failure to initiate such measures results in serious consequences— severe neurological lesions develop with death occurring in a few weeks or months.
Isovaleric Acidaemia
The deficient enzyme in this condition is isovaleryl CoA dehydrogenase, the third enzyme of leucine catabolic pathway (Fig. 13.16). It results in plasma accumulation of isovalerate. The children suffering from this disorder emit a characteristic body odour. They have frequent episodes of vomiting, acidosis and coma, during which the unusual body odour is more noticeable. Treatment involves restriction of leucine in diet.
Methyl Malonic Aciduria
This disorder is due to inadequate metabolism of meth-ylmalonyl CoA. This intermediate is obtained during metabolism of propionyl CoA, which is produced during catabolism of some amino acids (isolucine, methionine) or from odd chain fatty acids (Chapter 11).
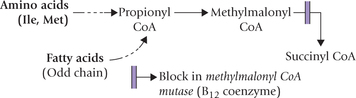
The defective enzyme is methylmalonyl CoA mutase which converts the methylmalonyl CoA to succinyl CoA. This enzyme requires deoxyadenosyl cobalamin, a vitamin B12 derivative, as a coenzyme.
Inadequate metabolism of methylmalonyl CoA may occur due to: (i) inborn error involving the enzyme protein, or (ii) to inadequate supply of the B12 coenzyme (Case 13.3).
C Inborn Errors of Histidine Metabolism
Histidinaemia
It is inherited as an autosomal recessive trait. It results due to deficiency of the enzyme histidase which is required for the normal metabolism of histidine (Fig. 13.13). Elevation of blood levels of histidine and an alternate metabolite, imidazole pyruvic acid results.
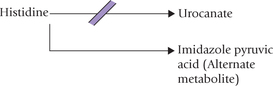
Increased urinary excretion of these metabolites also follows. Incidence of this disorder in newborns is on the order of 1 in 200,000. Most reported cases have shown mental retardation and speech defects, but some remained symptom-free.
Diagnosis is readily made with ferric chloride test since the imidazole pyruvic acid gives blue-green colour with this reagent. Other diagnostic procedures include determination of serum histidine, enzyme determination in skin biopsy, and the urocanate concentration in sweat. (Histidase is present only in the skin and liver, and urocanate is a normal constituent of sweat).
D Inborn Errors of Sulphur-Containing Amino Acids
Homocystinurias
These are autosomal recessive disorders of methionine metabolism. The metabolic defects known to lead to this group of heritable defects are deficiencies of methionine adenosyltransferase, cystathionine synthase and cystathionine lyase, that catalyze reactions 1, 4 and 5 respectively of the pathway shown in Figure 13.15. The first enzyme defect leads to hypermethioninaemia, and the third defect to cysta-thionuria, but both are benign conditions, not associated with any clinical abnormality.
The second enzyme defect (deficiency of cystathionine synthase) leads to plasma accumulation of methionine, homocysteine and homocystine (a dimer of two homocysteines), and urinary elimination of homocystine. In fact, this condition is the best known cause of homocystinuria in humans.
Homocystinurias lead to several clinical manifestations, discussed in following paragraphs, that appear to be initiated by the plasma accumulation of homocysteine, which gradually starts depositing in various tissues. The tissue accumulation interferes with the maturation of collagen and elastin, probably by binding copper, which is required for the activity of the enzyme lysyl oxidase. This results in skeletal deformities. The patient may also suffer from mental retardation, increased susceptibility to thrombosis and posterior dislocation of lens.
Restriction of dietary methionine is the mainstay of treatment of homocystinurias. Supplements of vitamin B12 and folic acid also are often given for enhancing the homocysteine to methionine reaction. Betaine (N,N,N-trimethylglycine), which is a methyl group donor in an alternative reaction for the synthesis of methionine from homocysteine, can be employed with the same aim of boosting the conversion of homocysteine to methionine.
Homocysteine as risk factor for CAD
Several studies have shown the relationship between homocysteine and altered endothelial cell function leading to thrombosis. Thus, elevated homocysteine in blood appears to be an independent risk factor for occlusive vascular disease. A rise of merely 6 μmol/L of homocysteine in plasma enhances the risk for occlusive vascular diseases as much as cholesterol increase of 20 mg/dL does. Five to ten per cent of the general population has mild hyperhomocysteinaemia.
Cystinuria
It is a disorder of renal and gastrointestinal tract transport of cystine, that also affects lysine, ornithine and arginine. The four amino acids share a common transport mechanism.
Because cystine is relatively insoluble, in cystinuric patients it may precipitate in the renal tubules and form cystine calculi. This is the major complication of the disease. To avoid this, penicillamine is used for the treatment. The cysteine-penicillamine complex being relatively more soluble, tends to be rapidly excreted.
Cystinosis
It is a familial condition, characterized by deposition of cysteine crystals in various tissues and organs. The crystals are deposited in lysosomes, because of an abnormality in transport of cysteine across cell membrane. Thus, cystinosis appears to be a lysosomal disorder. There is generalized amino-aciduria and the renal functions are seriously affected. Many patients die of renal failure at an early age.
Hereditary Sulphite Oxidase Deficiency
This may occur alone or along with xanthine oxidase deficiency. Both enzymes contain molybdenum. Patients with sulphite oxidase deficiency exhibit mental retardation, motor seizures, cerebral atrophy, and lens dislocation. Dietary deficiency of molybdenum can cause deficient activities of both xanthine oxidase and sulphite oxidase.
F Other Inborn Errors
Hartnup Disease
Impairment of transport of neutral amino acids in the intestinal mucosa and renal tubules is the cause of this rare autosomal recessive disease. Amino aciduria involving large neutral amino acids (valine, isoleucine, leucine, tyrosine, phenylalanine and tryptophan) is a prominent feature. Tryptophan is the most remarkably affected amino acid. Its depletion results in features of pellagra (dermatitis and dementia) because part of the niacin requirement normally is covered by endogenous synthesis from tryptophan.
In Hartnup disease, since the intestinal absorption of amino acids is decreased, the amino acids tend to accumulate in the intestinal lumen where they are degraded by the intestinal bacteria. Degradation of tryptophan in this way yields indole compounds, namely indolyl acetic acid and indolyl acetyl glutamine. These compounds have neurotoxic action, which accounts for the neurological symptoms of these patients.
Blue Diaper Syndrome and Familial Renal Iminoglycinuria
These are the other disorders resulting due to defective amino acid transport.
Hyperprolinaemias
Deficiencies of the proline oxidase and Δ1-pyrroline 5-carboxylate dehydrogenase, the first and the second enzyme, respectively of the metabolism of proline (Fig. 13.12) results in hyperprolinaemia type I and type II respectively. Both are clinically harmless autosomal recessive traits.
Non-ketotic Hyperglycinaemia
It is a rare recessively inherited disease (approximately 1 in 125,000 live births) of glycine metabolism. Glycine synthase is apparently absent, with the result that glycine levels in the blood are increased and glycine is excreted in the urine. Most infants die shortly after birth and some survive with profound mental retardation.
Primary Hyperoxaluria
It is a genetic disorder due to a deficiency of the enzyme glycine transaminase (converts glyoxylate to glycine coupled with impaired oxidation of glyoxylate to formate). Because of deficiency of glycine transaminase, there is accumulation of glyoxylate. The accumulated glyoxalate is channeled into production of oxalates which is excreted in urine. Enhanced urinary excretion of oxalate carries risk of developing calcium oxalate stone in the genitourinary tract.
V Biosynthesis of Amino Acids
The bacterial cell is capable of synthesizing all the 20 primary amino acids. The human cell can synthesize 11 of them (i.e. non-essential amino acids) but lacks capacity to make the other nine (i.e. essential amino acids). It is important to synthesize amino acids in adequate quantities and desired proportions so as to meet the body requirements. Failure to synthesize even a single amino acid results in negative nitrogen balance. In this state, more proteins are degraded than are synthesized and excessive excretion of nitrogen occurs.
This section deals with biosynthesis of the non-essential amino acids in the human cell and the control devices that coordinate the biosynthetic pathways.
A Nitrogen Fixation
Assimilation of nitrogen into amino acids starts with nitrogen fixation by certain microorganisms. These organisms bring about reduction of nitrogen to ammonia by the enzyme nitrogenase. The reaction is one of the most important reactions in cellular biosynthesis because it traps atmospheric nitrogen which can then be used for the synthesis of biomolecules. Thus, it serves as a link between the living and the non-living. Some of such nitrogen-fixing microorganisms, namely blue-green algae and certain soil-bacteria, invade roots of leguminous plants and form root nodules. It is in these nodules that nitrogen fixation occurs.
The next step in assimilation of the trapped nitrogen into biomolecules is its incorporation into amino acids. Glutamate and glutamine play a major role in this regard. Glutamate is synthesized by incorporation of ammonia into a keto acid (i.e. α-ketoglutarate); the reaction is catalyzed by glutamate dehydrogenase.

Another ammonia molecule is then assimilated into glutamate to form glutamine; the reaction is catalyzed by the enzyme glutamine synthetase, and is driven forward by ATP hydrolysis. Thus, nitrogen gets entry into the metabolic mainstream in the form of glutamate and glutamine.
B Biosynthesis of Non-essential Amino Acids
The carbon skeletons of the non-essential amino acids can be derived from glycolytic and Krebs cycle intermediates (Fig. 13.20 ). Alanine and serine can be derived from gly-colytic intermediates, pyruvate and 3-phosphoglycerate respectively, and glycine can be obtained from serine (discussed in the following sections). The carbon atoms of cysteine can be derived from serine via cystathionine as previously noted (Fig. 13.15). The carbon atoms of aspartate and asparagine can be derived from oxaloace-tate, and the carbon atoms of glutamate, glutamine and proline are derived from α-ketoglutarate. Tyrosine is produced from phenylalanine, as discussed.
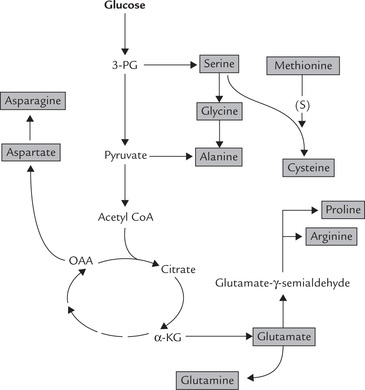
Fig. 13.20 Synthesis of non-essential amino acids (OAA = oxaloacetate, α-KG = α-ketoglutarate, 3-PG = 3-phospho-glycerate, S = sulfhydryl group).
Carbon skeletons for the non-essential amino acids can be derived from glycolytic and Krebs cycle intermediates.
Synthesis by Transamination: Pyruvate, oxaloacetate and α-ketoglutarate gain an amino group by participating in the transamination reaction to form the corresponding amino acids. In this way, pyruvate (3-C) forms alanine (3-C), oxaloacetate (4-C) forms aspartate (4-C) and α-ketoglutarate (5-C) yields glutamate (5-C). Glutamate can also be synthesized by amination of α-ketoglutarate by the enzyme glutamate dehydrogenase, as discussed earlier.
Synthesis by Amidation: Glutamine and asparagine can be synthesized from glutamate and aspartate respectively, by formation of an amide linkage. This linkage is formed between ammonia and the carboxyl group(s) present in R group(s) of these dicarboxylic amino acids.
Synthesis of glutamine is catalyzed by the enzyme glutamine synthetase, as discussed earlier. Synthesis of aspar-agine, catalyzed by asparagine synthetase, takes place by a similar reaction. However, the donor of amide group in this instance is glutamine. The reaction is driven forward by pyrophosphate cleavage of ATP.
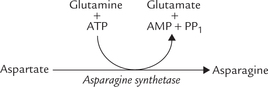
Synthesis of Proline and Arginine
Proline can be obtained from glutamate via glutamate γ-semialdehyde (refer Fig. 13.12). The conversion of glutamate to the semialdehyde is intricate because free car-boxylate groups are difficult to reduce. Formation of an energy rich intermediate (γ-glutamylphosphate) has been reported.
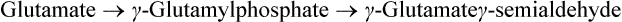
Spontaneous cyclization of the semialdehyde with removal of a water molecule occurs next to yield Δ1-pyrroline 5-carboxylate. The final reaction is to add hydrogen across a double bond, to form proline. This synthetic pathway occurring in intestinal mucosal cells is irreversible and different from the hepatic and renal pathway that catabolizes proline back to glutamate.
Glutamate-γ-semialdehyde can produce arginine also. First, this compound forms ornithine by transamination (Fig. 13.12), which then produces arginine via the last reactions (2, 3 and 4) of urea cycle (Fig. 13.7). The capacity for arginine synthesis is however limited, making this a semi-essential amino acid.
Synthesis of Serine, Glycine and Cysteine
Serine is synthesized from the glycolytic intermediate 3-phosphoglycerate (Fig. 13.20). The reactions are as below:
Reaction 1: An NAD+-dependent dehydrogenase catalyzes the oxidation of the alcohol of 3-phosphoglycerate to form 3-phosphopyruvate. This is a simple oxidation-reduction reaction similar to the lactate dehydrogenase reaction and is bidirectional.
Reaction 2: 3-Phosphopyruvate undergoes a transami-nation reaction with glutamate to form 3-phosphoserine.
Reaction 3: A phosphatase catalyzes removal of the phosphate group to yield serine.
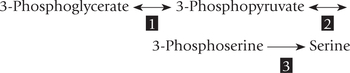
Glycine is formed from serine in one step by the serine hydroxymethyl transferase reaction, as noted earlier.
Cysteine is synthesized during catabolism of methionine (Fig. 13.15) obtaining its carbon atoms from serine and sulphur group from methionine.
C Biosynthesis of Semi-essential Amino Acids
Arginine can be synthesized in a limited amount, but his-tidine, the other semi-essential amino acid cannot be synthesized in humans. Nevertheless, histidine-free diet for several weeks does not produce any ill effects. This is probably due to its release from carnosine (α-alanyl-histidine), a dipeptide that is present in large quantities in muscle tissue. Carnosine also prevents alteration of pH during severe muscle contractions when large amounts of lactic acid are formed, thereby acting as a pH buffer.
VI Amino Acids as Precursors of Specialized Products
Amino acids serve as precursors of a large number of biologically important compounds; for example, amines, por-phyrins, nitrogenous bases of phospholipids, creatine phosphate, polyamines, etc. Compounds like purines and pyrimidines are derived in part from amino acids (Fig. 13.21 ).
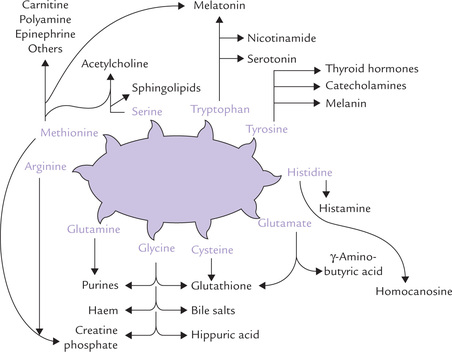
A number of useful products are obtained from amino acids, e.g. creatine, haem, glutathione and purines (from glycine), catecholamines, melanin and thyroxine (from tyrosine), histamine (from histidine), GABA (from glutamate), polyamines (from arginine), etc.
A Histamine
Histamine is a biogenic amine produced from histidine. It is a major mediator of anaphylaxis and several allergic processes. It is released by circulating basophils and mast cells typically in response to antigenic stimulation of surface-bound IgE.
Biosynthesis
Histamine is produced from histidine by removal of its α-carboxyl group; the reaction is catalyzed by histidine decarboxylase, a pyridoxal phosphate (PLP) dependent enzyme.

Another enzyme, aromatic α-amino acid decarboxylase, can also bring about decarboxylation of histidine. However, specificity of this enzyme is broader since it can cause decarboxylation of aromatic amino acids as well.
Functions: Histamine dilates small blood vessels, increases capillary permeability, contracts bronchial and intestinal smooth muscles, stimulates gastric acid secretion and nasal fluid discharge, and regulates cells of the immune system. These effects are elicited following its interaction with two different types of receptors: the H1 receptor and the H2 receptor.
• H1 receptors are located on some smooth muscle cells. Interaction of histamine with these receptors accounts for the above stated effects on blood vessels and smooth muscles. The anti-allergic drugs like diphen-hydramine and chlorophenaramine oppose action of histamine by occupying these receptors, hence blocking them from action of histamine.
• H2 receptors are located on the gastric mucosa. Binding of histamine to these receptors results in increased secretion of hydrochloric acid. Cimetidine, a common drug used for the treatment of hyperacidity, occupies the H2 receptor. This decreases the histamine-induced acid secretion.
B Gamma (γ) Aminobutyric Acid
γ-Aminobutyric acid (GABA) is an inhibitory neurotransmitter in the brain.
Biosynthesis
GABA is produced by removal of the γ-carboxyl group of glutamate by the enzyme glutamate decarboxylase. The enzyme is present in the GABAergic nerve terminals in the brain, especially the grey matter. The insulin secreting beta cells of pancreas also form this enzyme and this has a bearing on the occurrence of diabetes mellitus.

Further metabolism of GABA occurs by a transamina-tion reaction to form succinate semialdehyde which is then oxidized to succinic acid (Fig. 13.22 ).
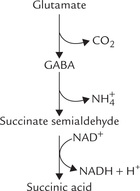
The first two enzymes of this pathway require pyri-doxal phosphate (PLP), but glutamate decarboxylase is more susceptible to drugs that interact with pyridoxal phosphate (probably because its Km for the PLP is higher). Example of such antipyridoxal drugs are the hydrazides, e.g. hydralazine, that are used for their vasodilatory effect on arteriolar smooth muscle. But overdosage with such antipyridoxals can cause convulsions as formation of the inhibitory GABA is prevented.
Functions
GABA is concentrated in the synaptic vesicles where it enhances passage of chloride ions through the post-synaptic membrane. This action of GABA may account for its role as an inhibitory neurotransmitter. It is noteworthy, that glutamate (the precursor of GABA) acts as an excitatory neurotransmitter.
GABA and Huntington’s disease
Various movement disorders arise as a result of defects in the basal ganglia of the brain, e.g. parkinsonism and Huntington’s disease. (The former is discussed in a subsequent section, while consideration of the latter is relevant here because it involves degeneration of GABAergic neurons.) The disease is named after the scientist George Huntington, who described it first in 1872. Loss of the inhibitory pathways results, which leads to uncontrolled (choreic) movements that characterize this condition.
C Catecholamines
Epinephrine, norepinephrine and dopamine are collectively referred to as catecholamines. They are synthesized from the same precursor amino acid, i.e. tyrosine. In common with the other compounds containing amino groups such as histamine or serotonin, catecholamines are also known as biogenic amines.
Biosynthesis
The reaction sequence for the synthesis of catecholamines is shown in Figure 13.23 . Synthesis of epinephrine requires four steps, whereas only three and two steps suffice for norepinephrine and dopamine respectively. The initial reactions up to dopamine take place in the cytoplasm, whereas the formation of norepi-nephrine is catalyzed by enzymes within the synaptic vesicles in neurons. Conversion of norepinephrine to epi-nephrine occurs in cytosol of the chromaffin granules of the adrenal medulla.
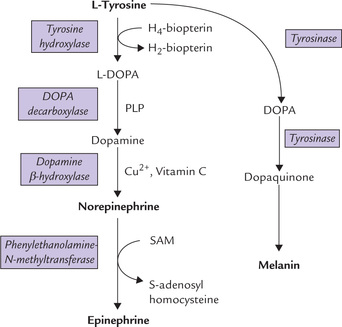
Fig. 13.23 Conversion of tyrosine to epinephrine, norepinephrine or melanin (DOPA = dehydroxy phenylalanine, PLP = pyri-doxal phosphate).
(a) The first step involves hydroxylation of tyrosine to dihydroxyphenylalanine (L-DOPA) by the enzyme, tyrosine hydroxylase, found in catecholaminergic nerve terminals. This is a monooxygenase that requires molecular oxygen, ferric ions and tetrahydrobiopterin as a reducing agent. The latter is formed from dihydrobi-opterin at the expense of NADPH.
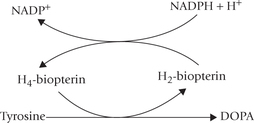
(b) In the second reaction, DOPA loses its α-carboxy group to form dopoamine. The enzyme catalyzing this step is DOPA decarboxylase. Dopamine is then taken up into synaptic vesicles where conversion of dopamine to norepinephrine would occur. This process is dependent on a vesicular ATPase.
(c) Within synaptic vesicles (of adrenergic neurons), the vesicular dopamine β-hydroxylase catalyzes the formation of noradrenaline. The enzyme is a copper containing monooxygenase that requires ascorbic acid and molecular oxygen.
(d) The final step involves formation of epinephrine. Though in majority of CNS, norepinephrine is the final product, but a few neurons in the brain, and chromaffin cells of adrenal medulla, epinephrine is produced by action of the cytosolic enzyme, N-methyl transferase. The enzyme catalyzes the transfer of a methyl group from S-adenosylmethionine (SAM) to form adrenaline.
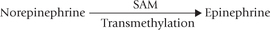
Although the pathway to three catecholamines is common, the end product formed in a given cell depends on its enzymatic outfit. Some cells (e.g. dopaminergic neurons of nigrostriatal system) having only the first two enzymes synthesize dopamine, whereas the cells of adrenal medulla having all the four enzymes of the pathway synthesize both epinephrine (80%) and norepinephrine (20%).
Synthesis of three catecholamines: dopamine, norepinephrine, epinephrine, occurs by a comman biosynthetic pathway starting from tyrosine. The pathway operates partly in cytosol and partly in synaptic vesicles of neurons. Chromaffin cells of adrenal medulla and few neurons of brain can synthesize epinephrine (from nor-epinephrine) also.
Regulation of biosynthesis
The committed step of the biosynthetic pathway is the tyrosine hydroxylase reaction, which is allosterically inhibited by dopamine and norepinephrine when the adrenal medulla is unstimulated. Continuous stimulation of the adrenal medulla (as during prolonged stress) promotes tyrosine hydroxylase activity, primarily because the turnover of dopamine and norepi-nephrine is rapid.
Functions
• Norepinephrine is a major transmitter in the sympathetic nervous system; stimulation of these nerves is responsible for various features of the ’fight or flight response, such as stimulation of the heart rate, sweating, vasoconstriction in the skin and bronchodilatation. There are also norepinephrine containing neurons in the CNS, largely in the brain stem.
• Epinephrine is produced in the adrenal medulla; it is more active than norepinephrine on the heart and lungs.
• Dopamine is both an intermediate in the synthesis of norepinephrine and a neurotransmitter. It is a major transmitter in nerves that interconnect the nuclei of the basal ganglia in the brain and control voluntary movements. Damage to these nerves causes Parkinson’s disease.
Parkinson’s disease
This second type of movement disorder, Parkinsonism (the first is Huntington’s disease, discussed earlier), was first described by James Parkinson in 1817. It involves loss of dopamine neurons resulting in deficiency of dopamine synthesis. The condition is relatively common after the fifth decade of life, with an incidence of 1 in 1000. Nerve transmission is affected in substantia nigra of the upper brain stem (because of dopamine depletion), which accounts for the clinical features of this disease, e.g. muscular rigidity, tremors, expressionless face and slow rhythmical movements.
Treatment involves administration of DOPA. This compound enters the brain cells where it is converted to dopamine. Administration of dopamine for the treatment is ineffective because dopamine is incapable of entering the brain cells.
D Serotonin and Melatonin
Both these compounds are derived from tryptophan (Fig. 13.24 ).
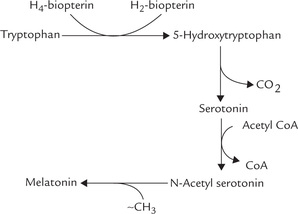
Biosynthesis
Serotonin is found in enterochromaffin cells, brain and platelets. In the former two it is synthesized from tryptophan, whereas in platelets, serotonin is taken up from plasma. Synthesis involves the following steps: (i) Hydroxylation of tryptophan by tryptophan 5-hydroxylase followed by (ii) decarboxylation by aromatic L-amino acid decarboxylase (Fig. 13.24). Hydrox-ylation, the rate-limiting reaction, is analogous to that of phenylalanine, and requires molecular oxygen and tetra-hydrobiopterin.
Synthesis of melatonin from serotonin occurs in pineal gland by N-acetylation followed by methylation.
Functions
Serotonin is a neurotransmitter in the brain and serves as a precursor of melatonin in the pineal gland. The serotoninergic neurons are concentrated in the upper brain stem, but project up to the cerebral cortex and down to the spinal cord. They are more active when the subject is awake than when he is asleep, and serotonin may control the degree of responsiveness of motor neurons in the spinal cord. In addition, it regulates various vegetative body functions, such as appetite, sleep, sexual activity and aggression. It is a potent smooth muscle constrictor and a vasoconstrictor. It may act as a transmitter in gastrointestinal tract to evoke release of peptide hormones. It might play a role in intestinal motility also.
Melatonin: is a biologically active compound present in pineal gland. It exerts an inhibitory effect on gonads. It blocks actions of melanocyte stimulating hormone (MSH) and adrenocorticotrophin. Synthesis of melato-nin is sensitive to light and darkness. In light, the synthesis is stimulated whereas in dark it is diminished. These effects are thought to be mediated via cAMP.
Under normal circumstances, only about 1% of tryptophan is converted to serotonin; the rest either undergoes degradation or enters kynurenine pathway to form nicotinamide (Chapter 18). However, about 60% or more of tryptophan is channeled towards serotonin formation in a condition called malignant carcinoid (argentaffinoma). Widespread tumour cells in the argentaffin tissue of the abdominal cavity characterize this condition. Symptoms of nicotin-amide deficiency (i.e. pellagra) and negative nitrogen balance develop in this condition.
E Melanin
Melanins are the dark pigment of skin, hair, iris and the retinal epithelial cells, derived from tyrosine. They are formed in organelles called melanosomes. The latter occur in pigment producing cells called melanocytes. Two types of melanins occur in humans: the brown-black eumelanins and the yellow-red pheomelanins. The latter are derived, in addition to tyrosine, from cyste-ine also.
Biosynthesis
A series of enzymatic and non-enzymatic oxidation and coupling reactions are required to form melanins (Fig. 13.23).
• Tyrosine is first hydroxylated by the enzyme tyrosinase to form dihydroxyphenylalanine (DOPA).
• The same enzyme then converts the DOPA to dopa-quinone.
• Depaquinone then undergoes a series of enzymatic and non-enzymatic reactions, some of which are catalyzed by tyrosinase to form polymeric melanin molecules.
It should be noted that the enzyme that catalyzes formation of DOPA from tyrosine in nerve and adrenal medulla (tyrosine hydroxylase) is different from tyrosinase, although both form the same product.
Functions
Melanin is derived from a Greek world, melan, meaning black. It is a polymeric product with heteroge-nous molecular weight and poorly defined structure. It is present in skin, hair, choroid plexus, substantia nigra, and in retina and ciliary body in eye, imparting them their characteristic brown colour. Melanin protects our skin because it absorbs not only the visible light but also the UV radiation. The latter is a natural component of sunlight (wavelength 280-320 nm) and is dangerous because it induces DNA damage, a common cause of both sunburn and skin cancer (Chapter 21).
In the eye, melanin is present in pigment epithelium underlying the sensor cells of the retina where it absorbs stray light. This enhances visual acuity and prevents over-stimulation of the photoreceptors.
Albinism
It refers to a variety of recessively inherited conditions that exhibit hypomelanosis based on metabolic defects in the melanocytes of the eye and skin. They occur with frequencies of approximately 1 in 50,000 in most parts of the world. The classical form of albinism arises due to deficiency of the enzyme tyrosinase. Such tyrosinase negative individuals lack detectable pigment in the skin, hair, or eyes. For this reason, this disorder is sometimes called complete perfect albinism.
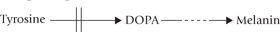
Other forms of albinism called complete imperfect albinism occur in tyrosinase-positive individuals. These individuals have some detectable tyrosinase activity but fail to synthesize melanin in adequate amounts. This may because of a transport defect-decreased ability to transport tyrosine to a site where it will form melanin (Case 13.4).
Various clinical forms of albinism have been recognized. These are: ocular, oculocutaneous, and cutaneous, depending on the tissue involved.
The albinos are abnormally sensitive to light; even a slight exposure to light causes burns and can lead to skin cancer. Photophobia is a common feature.
F Thyroid Hormones
The generic term "thyroid hormones" refers to the iodin-ated amino acid derivatives T3 (triiodo-L-thyronine) and T4 (tetraiodo-L-thyronine). They are synthesized from iodine and the tyrosyl residues present in thyroglobulin, a homodimeric glycoprotein (MW 669,000). It contains about 134 tyrosyl residues. Up to 25-30 of the tyrosine side chains become iodinated, but not more than 8-10 of these are processed to active hormones (Chapter 30).
G Creatine Phosphate and Creatinine
Creatine (N-methylguanidinoacetate) is an amino acid-derived product that is present in muscle tissue, and to a lesser extent in nervous tissue. It plays a pivotal role in the metabolism of high-energy phosphates. Creatinine is a dead end metabolite formed by spontaneous cycliza-tion of creatine and creatine phosphate.
Biosynthesis
Creatine phosphate is synthesized from three amino acids: glycine, arginine and methionine. The biosynthetic pathway consists of three sequential reactions, one reaction each occurs in kidney, liver and muscle (Fig. 13.25 ).
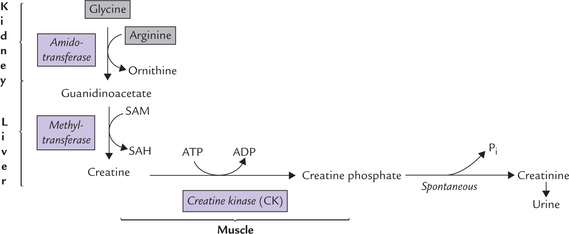
Functions
In vertebrate muscles, creatine phosphate occurs as a reservoir of high-energy phosphate groups. It helps generation of ATP in exercising muscles by substrate-level phosphorylation. Nearly 1% of the weight of skeletal muscle is accounted for by this compound. Small amount is present in smooth muscles, testes, liver and kidneys as well.
Creatine phosphate possess an energy rich phosphate bond, with a standard free energy of hydrolysis of -10.3 kcal/mole.
This value is much higher than the standard free energy of ATP hydrolysis ( — 7.3 kcal/mole). Therefore, hydrolysis of creatine phosphate can be coupled with concomitant generation of ATP (i.e. substrate level phosphorylation).

During muscle contraction, when the ATP: ADP ratio declines, ATP is regenerated rapidly by the above reaction. This reaction, called the reversible creatine kinase reaction (Fig. 13.25) is the most important source of ATP during the first few seconds of muscle contraction. For more sustained muscular activity, however, ATP has to be regenerated by (anaerobic) glycolysis or oxidative metabolism.
The compound corresponding to creatine phosphate in invertebrates is arginine phosphate. These high-energy compounds of muscles are termed phosphogens.
Clinical implications
Creatine phosphate is relatively unstable at the pH prevailing in the sarcoplasma, and is non-enzymatically converted to creatinine in this organ-elle. This conversion takes place continuously in healthy muscle. The creatinine so produced is released in circulation transported to kidneys and excreted in the urine.
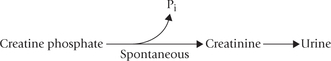
This has two important implications in clinical medicine:
1. The amount of creatinine excreted in the urine over a 24-hour period, which correlates with the muscle mass, is constant for a given individual (about 15 mg/kg of body weight). The quantity of creatinine is measured in the 24-hour urine specimens to validate that the collection was complete.
2. It is a useful kidney function test because its blood level is remarkably constant. It is neither secreted nor reabsorbed in the tubular system, and so its excretion during a specified time in a creatinine clearance test serves as a measure of glomerular filtration rate.
Small quantity of creatine is also excreted in urine. Muscle wasting due to any cause, such as muscular dystrophy, starvation, diabetes, fever and thyrotoxicosis results in increased excretion of creatine in urine.
H Carnitine
Carnitine is a coenzyme that carries long-chain fatty acids into mitochondrion, thereby playing an important role in β-oxidation (Chapter 11). It is synthesized from trimethyllysine, which is obtained during normal turnover of proteins like myosin and histone.
I Nicotinamide
Nicotinamide is a water-soluble vitamin synthesized from tryptophan (Fig. 13.21). In humans, about 3% of tryptophan is converted to nicotinamide derivatives. The pathway of its formation, termed the kynurenine pathway, is discussed in Chapter 18. Nicotinate, a related compound, is also obtained from tryptophan by the kyn-urenine pathway. Its administration decreases the serum cholesterol levels. However, larger doses are toxic. Nicotinamide is free from these side effects.
J Polyamines
There are aliphatic amines possessing multiple amino groups. Polyamines were originally identified in sperms, but now they are known to exist in a number of other tissues. Biologically important polyamines are putrescine, sperm-ine and spermidine.
Biosynthesis
Polyamines are synthesized from ornithine, which is derived from arginine. The following steps are involved:
1. Ornithine decarboxylase, a PLP-dependent enzyme, catalyzes decarboxylation of ornithine to form putrescine (Fig. 13.26 ).
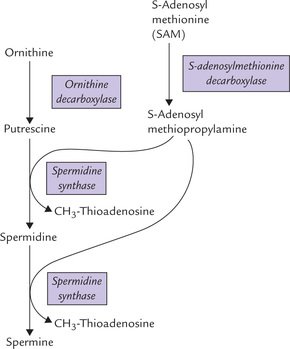
2. Another PLP-dependent enzyme, S-adenosylmethionine decarboxylase, catalyzes decarboxylation of S-adenosyl-methionine to form S-adenosylmethiopropylamine, an activated donor of propylamine group.
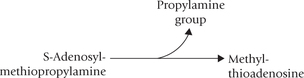
3. The S-adenosylmethiopropylamine donates the propylamine group first to putrescine to form spermi-dine, and then to spermidine to form spermine.
Functions
Polyamines are basic in nature and possess multiple positive changes. Hence, they readily associate with negatively charged cellular macromolecules such as nucleic acids. They also interact with the membrane phospholipids and negatively charged residues of membrane bound proteins. Because of these interactions, polyamines carry out a variety of biological functions:
1. The interaction of polyamines with DNA and RNA may stimulate synthesis of proteins and nucleic acids and enhance cellular proliferation.
2. The polyamines may open certain channels because of their binding with anionic sites on the cell membranes. This may lead to enhanced transport of specific substances.
3. Polyamines may play a role in hormone mediated actions and in membrane fusion during exocytosis and endocytosis due to their association with membrane phospholipids.
4. Certain enzymes are inhibited by polyamines, e.g. protein kinase.
Biogenic Amines
These are a group of basic compounds of diverse origin and functions. They are synthesized by decarboxylation of amino acids (and their derivatives), e.g. histamine from histidine and taurine cysteine. Examples are listed below:
| Histidine | → | Histamine |
| Tryptophan | → | Tryptamine |
| Tyrosine | → | Tyramine |
| Lysine | → | Cadoverine |
| Serine | → | Ethanolamine, choline |
| DOPA | → | Dopamine |
K Glutathione
Glutathione is a tripeptide of glutamic acid, cysteine and glycine with the sequence L-γ-glutamyl-L-cysteinylglycine. It plays a major role in the maintenance of proteins in their reduced forms (Chapter 27).
L Acetylcholine
Acetylcholine is a neurotransmitter of the parasympathetic nervous system and sympathetic ganglia. Stimulation of the parasympathetic system produces slowing of heart rate, bronchoconstriction, and stimulation of intestinal smooth muscle; these effects are broadly opposite to those of the sympathetic system.
Acetylcholine also serves as the neurotransmitter of the neu-romuscular junctions where motor nerves contact skeletal muscle cells (i.e. motor end plate) and cause them to contract. Neurons containing this transmitter also exist in the brain, and they are thought to be involved in learning and memory.
Synthesis of acetylcholine occurs by the cytoplasmic enzyme choline acetyl transferase. The initial precursor is the amino acid, serine (Fig. 13.21), which undergoes decar-boxylation to form ethanolamine. The latter receives three methyl groups from SAM molecules to form choline which combines with an acetyl group to form acetylcholine.
After acetylcholine is secreted into the synaptic cleft, it is largely degraded by the enzyme acetylcholinesterase. The breakdown products, choline and acetate, are taken up rapidly into the nerve terminal, where they are used for the resynthesis of acetylcholine.
M Others
A number of other compounds, in addition to those just mentioned, are also obtained from amino acids. Glycine, the smallest of all amino acids, is a precursor for the largest number of such products (Fig. 13.21).
Special products of glycine
Glycine participates in generation of several important products:
1. Hippuric acid: It is formed by an amide linkage between the carboxyl group of benzoic acid and the amino group of glycine.
2. Bile salts: Glycine becomes conjugated with bile acids to form conjugated bile salts.
3. Haem: Glycine combines with succinyl CoA in the first step of the haem biosynthetic pathway.
4. Creatine, discussed earlier.
5. Purine nucleotides (Chapter 19).
Exercises
Essay type questions
1. Describe the transport of ammonia, and highlight how ammonia detoxication in brain differs from that in liver. Explain biochemical basis of ammonia toxicity.
2. Describe reactions in the synthesis of urea.
3. Draw a thumbnail sketch of the TCA cycle and indicate where each of the amino acids may enter this cycle.
4. Outline metabolism of phenylalanine. Name special products obtained from tyrosine and describe synthesis of any one of them.
Write short notes on
Case 13.1 Lapsing into coma 3 days after birth
A full term child, born after normal pregnancy, developed grunting respiration 36 hours after birth. After three days he had severe bouts of vomiting and became lethargic and unresponsive to stimuli. He was admitted in the hospital nursery, where a blood sample was obtained for analysis.
Chromatography of the plasma sample was carried out and compared with that of a normal child. The results indicated that plasma levels of citrulline, glutamine and alanine were grossly elevated, but the argininosuccinate level was decreased. Treatment was started immediately.
Case 13.2 A 2-week old infant having convulsions
An infant of 2 weeks had convulsions. He was born after a normal pregnancy and had taken his feeds normally. His mother had observed a peculiar mousy odor in the child’s urine. The urine was tested by ferric chloride test: characteristic green colour was observed which indicated the presence of phenylpyruvic acid.
Quantitative analysis of the blood and urine yielded values for phenylalanine and its metabolites.
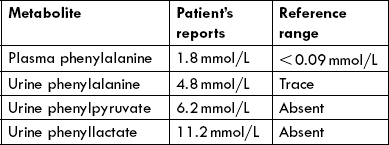
Similar results were obtained on repeating tests after few days. Liver biopsy was performed and cell-free extract was prepared from the hepatocytes. Radioactive phenyl-alnaine was added to the extract and amount of radioactive tyrosine produced from it was measured. It was found to be less than 1% of that in a normal subject. However, addition of tetrahydrobiopterin (THB) to the reaction mixture enhanced the tyrosine production. Addition of dihy-drobiopterin (DHB) did not make any difference.
Case 13.3 Recurrent vomiting in an infant
A 4-month-old-female infant was referred to the hospital OPD with recurrent vomiting, convulsions, respiratory distress and failure of adequate growth. Biochemical examination showed decreased plasma bicarbonate concentration. Urinary ammonium excretion was enhanced. Specialized analysis in the hospital laboratory found excessive methyl malonic acid in the urine sample. Injection of vitamin B12 markedly decreased the urinary excretion of this compound.
Q.1. Identify the metabolic block in this infant?
Q.2. Urine chromatography of the infant revealed presence of homocystine and cystathionine and plasma level of methionine was decreased. Comment on these observations.
Q.3. If an infant with similar clinical features and biochemical results does not respond to vitamin B12 injection, what could be the biochemical defect?
Q.4. Comment on the other biochemical test results, i.e. decreased plasma bicarbonate and enhanced urinary ammonium excretion.
Case 13.4 A 3-year-old child with white hair and abnormally fair skin
A 3-year-old child was exceedingly fair skinned, had white hair, and red pupils with bluish iris. On examination, visual impairment (astigmatism) was detected. A sample of melano-cytes was obtained by skin biopsy and a cell free extract was prepared from these cells. Radioactive tyrosine was added to the extract and formation of radioactive dihydroxyphe-nylalanine was measured. It was found to be markedly decreased as compared to that expected in a normal subject. Production of radioactive melanin was also found to be greatly diminished, being less than 1% of that expected in a normal person. Addition of terahydrobiopterin had no effect on production of either dihydroxyphenylalanine or melanin.
Q.1. Identify the biochemical defect in this child.
Q.2. Given that tyrosine to DOPA conversion is an important step in biosynthesis of catecholamines, do you expect any abnormality of catecholamine metabolism in this child?
Q.3. In an albino, addition of radioactive tyrosine to the cell free extract resulted in normal synthesis of melanin. However, when melanocytes were cultured and radioactive tyrosine was added to them, decreased synthesis of radioactive melanin occurred. Identify the biochemical defect in this patient.