Nucleic Acid Chemistry And Nucleotide Metabolism
Nucleotides are structural units of nucleic acids consisting of a pentose sugar, a nitrogenous base and one or more phosphate groups. They are widely distributed and perform a variety of functions of vital significance. Covalently linked nucleotide monomers form the nucleic acids, DNA and RNA. Most nucleic acids are very large. They often contain thousands (in case of DNA, millions) of nucleotide units held together by phosphodiester bonds. Nucleotides play an important role in biosynthetic reactions and in energy metabolism.
This chapter deals with important structural features and biological significance of nucleotides and nucleic acids. After going through this chapter, the student should be able to understand:
• Chemistry, properties and functions of purine and pyrimidine bases, nucleosides and nucleotides.
• The de novo synthesis and salvage pathways of purine nucleotides, and the purine degradation pathway; regulation of these pathways, the positive and negative effectors, enzymes controlling rate of metabolism, and the attendant pathologies.
• Pyrimidine synthesis and degradation; aetiology of various disorders related to these pathways; steps (of the above stated purine and pyrimidine pathways) inhibited by antineoplastic drugs and the sulpha drugs, and the mechanisms of inhibition.
I Nucleotides: Chemistry and Biological Significance
A Basic Structure
A nucleotide has three structural components: a heterohmpound termed nitrogenous base, a five carbon (pentose) sugar, and at least one phosphate group. The nitrogenous bases found in nucleic acids belong to one of the heterocyclic groups, either purines or pyrimidines. When the nitrogenous bases are combined with a pentose sugar, they are known as nucleosides. Phosphate group can be attached either at the 5′-position or the 3′-position of the pentose, and the nucleoside-phosphate thus formed is known as nucleotide.
Nucleic acids are polymers of several nucleotide units, hold together by covalent linkages.
Nitrogenous Bases
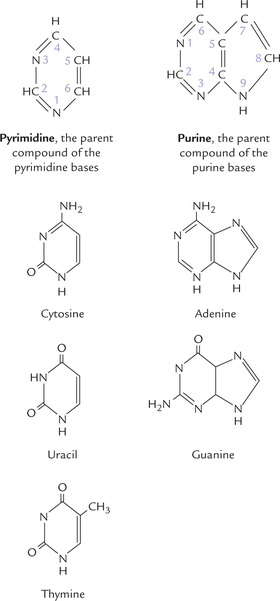
The bases are derivatives of purines or pyrimidines. Structures of the parent compounds of purine and pyrimidine bases (which do not occur in nature) are shown in Figure 20.1, with the atoms numbered. Purines are numbered in anti-clockwise direction, and the pyrimi-dines are numbered in the clockwise direction.
Major Bases
The major purines of both DNA and RNA are guanine (G) and adenine (A). In DNA, the major pyrimidines are thymine (T) and cytosine (C), while in RNA the major pyrimidines are uracil (U) and cytosine (C). Presence of T instead of U in DNA is important for preventing mutations (Chapter 21). Structures of the major bases are depicted in Figure 20.1.
Other Purine and Pyrimidine Bases
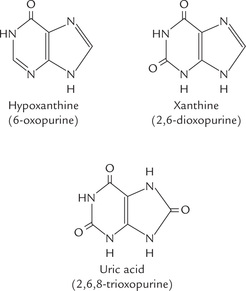
In addition to the five major purine and pyrimidine bases in nucleic acids, some minor or unusual bases are also found in nature. Hypoxanthine, xanthine and uric acid (Fig. 20.2 ) are present in free state intracellularly. The former two are the metabolites of adenine and guanine, which are ultimately converted to uric acid—the end product of purine catabolism.
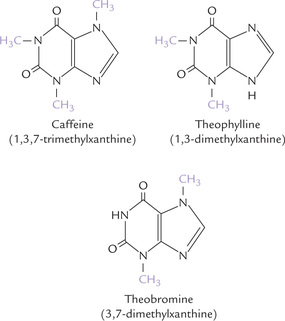
Some minor or unusual bases are found in nucleic acids, mostly in RNA. These include 5′-methylcytosine, pseudouracil, N 6-methyladenine and N7-methylguanine (Fig. 22.5, Chapter 22). Finally, some methylated purine bases are found in plants. These include alkaloids like theophylline (1,3-dimethylxanthine), caffeine (1,3,5-trimethylxanthine) and theobromine (3,7-dimethylxanthine) (Fig. 20.3 ). Theophylline is found in tea, and is used therapeutically in asthma; caffeine is a component of coffee beans and tea, that elevates intracellular cAMP level; and theobromine is found in chocolate, and is a diuretic, heart stimulant and vasodilator.
Chemical Properties of Bases
Nitrogenous bases include highly conjugated double bond systems within the ring structures. For this reason, nucleic acids have a very strong absorption maximum at about 260 nm, which is used for nucleic acid quantitation. Moreover, the heterocyclic rings of both purines and pyrimidines have oxo (—C = O—) functional group. Therefore, a given base can exist in two tautomeric forms, the lactam or keto form and the lactim or enol form.
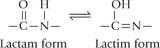
In DNA and RNA, the keto forms are by far more predominant, and this property makes it possible for the bases to form intermolecular hydrogen bonds (Chapter 21).
Sugars
The 5-carbon sugar found in nucleotides is either D-ribose or D-2′-deoxyribose. Each occurs in furanose form, the configuration at C-1 being β (see Chapter 2 for nomenclature). D-Ribose is present in RNA, while D-2′-deoxyribose is present in DNA. Deoxyribose, as the name suggests, differs from ribose in having one oxygen less (at C-2).
Nucleosides
A nucleoside is a compound of base and sugar; the latter two are linked by an N-glycosidic linkage (a carbon-nitrogen bond). The bond is formed between C-1 of sugar and nitrogen atom 1 of pyrimidine base or nitrogen atom 9 of purine base.
If the sugar is ribose, the compound is ribonucleoside and if the sugar is deoxyribose, the compound is deoxyribonucleoside. Adenosine (adenine + ribose), guanosine (guanine + ribose), cytidine (cytosine + ribose) and uridine (uracil + ribose) are the ribonucleosides of A, G, C and U, respectively. Similarly, deoxyadenosine, deoxyguanosine, deoxycytidine and deoxythymidine are the deoxyribonucleosides of A, G, C and T, respectively.
| Base | Ribonucleoside | Deoxyribonucleoside |
| Adenine (A) | Adenosine | Deoxyadenosine |
| Guanine (G) | Guanosine | Deoxyguanosine |
| Cytosine (C) | Cytidine | Deoxycytidine |
| Uracil (U) | Uridine | – |
| Thymine (T) | – | Deoxythymidine |
Nucleotides
As already stated, nucleotide is nucleoside plus phosphate. The phosphate group is esterified to one of the hydroxyl groups of the sugar component of the nucleoside. Mostly it is the hydroxyl group at the C-5 (less commonly C-3) that is phosphorylated.
To avoid confusion between the numbering of various atoms in the base and carbon atoms of the sugar, the latter are represented with an associated prime. Thus, carbons of sugar are numbered 1′, 2′, 3′, etc.
B Nomenclature
The nucleotides are named as phosphate derivatives of nucleosides. Thus, adenosine linked covalently with phosphate produces adenosine monophosphate (AMP). It is a mononucleotide. Likewise, other mononucleotides, e.g. guanosine monophosphate (GMP), cytidine monophosphate (CMP), and uridine monophosphate (UMP) are formed by phosphorylation of guanosine, cytidine and uridine, respectively.
The phosphate group can be attached either at the 5′-position or the 3′-position of the pentose, and accordingly the mononucleotides formed are designated as 5′ or 3′-monophosphates, respectively. However, attachment at the 5′-position is far more common, and therefore 5′ is usually omitted in the abbreviation. Thus, AMP means adenosine 5′-monophosphate, CMP means cytidine 5′-monophosphate, and so on. However, for adenosine 3′-monophosphate or cytidine 3′-monophosphate, the abbreviations 3′-AMP or 3′-CMP respectively are used.
A mononucleotide is also called nucleoside monophosphate.
Nucleoside-Diphosphate and Triphosphate
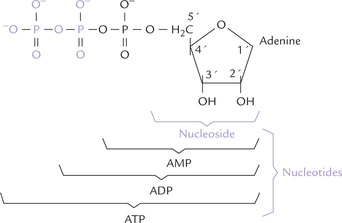
Additional phosphate groups may be attached to a mononucleotide. Addition of a second and a third phosphate to a nucleoside monophosphate yields a nucleoside-diphosphate, and triphosphate, respectively. AMP thus yields adenosine diphosphate (ADP) or adenosine triphosphate (ATP), respectively (Fig. 20.4 ). The subsequent phosphate groups are attached to the preceding ones by acid anhydride bonds. These bonds possess high free energy of hydrolysis, yielding more than 7 kcal/ mole of free energy (Chapter 8).
C Functions
Nucleotides are central to maintenance and propagation of life. They are most versatile of all biomolecules, being involved in a number of reactions including the energy transfer reactions.
Some important functions of nucleotides are:
1. The ribonucleotides such as ATP, GTP, CTP and UTP are important coenzymes and provide the basic unit of RNA. The deoxyribonucleotides, dATP, dGTP, dCTP and dTTP are required for DNA replication and DNA repair. The ribonucleotides are present in millimolar concentrations in the cell, and the deoxyribonucleotides are present in micromolar concentrations intracellularly.
2. Nucleoside sugars are activated precursors which are used in biosynthetic reactions. For example:
3. Nucleotides are the basis of high-energy compounds. The best known example is adenosine triphosphate (ATP), referred to as the currency of free energy in the body, which provides energy for all types of cellular activities. GTP is used as an energy source in protein synthesis.
4. Regulation of enzyme activity involves certain nucleotides, such as cAMP and cGMP. They are signal-conducting molecules, acting as intracellular messengers for certain hormones (Chapter 29).
5. The nucleic acids, DNA and RNA, are involved in storage and decoding of genetic information.
6. The coenzymes, NAD +, FAD and coenzyme A have nucleotides as essential components of their structures.
Although dietary nucleotides and nucleic acids are digested to nucleosides and free bases, the degradation products are poorly absorbed. The small amounts of absorbed products are not delivered to tissues but rather degraded within intestinal mucosa to form uric acid. Thus, dietary nucleic acids are not used for the synthesis of tissue nucleic acids.
II Purine Metabolism
A Synthesis of Purine Nucleotides
Human body is capable of synthesizing the purine and the pyrimidine rings de novo (anew) and also by salvaging (recycling) the nitrogenous bases arising from degradation products of nucleic acids.
• In de novo synthesis the elements of purine ring system are added step by step, using C-1 of ribose 5-phosphate as a primer. It provides the cell with the capacity to construct the purine ring afresh, with carbon and nitrogen atoms coming from various sources.
• In the salvage pathways, the free purine bases or nucleosides (arising from degradation of preexisting nucleic acid bases) are reutilized for the synthesis of new nucleotides. The salvage reactions reduce the requirement for the energetically expensive de novo biosynthesis and prevent wastage of raw materials. They are the principal source of nucleotides for many tissues, most notably brain, in which the de novo pathway is either poorly developed or absent.
The nucleotides arising from endogenous synthesis are sufficient to meet the body’s requirements. Therefore, humans are said to be prototrophic for purines and pyrimidines for not being dependent upon dietary sources.
The de novo Pathway
The de novo synthesis of purines is most active in liver, though it may occur in several other tissues. Liver also exports the surplus bases and nucleosides to other tissues. All reactions of the de novo pathway are cytosolic.
The first clue to the de novo synthesis was provided in 1948 by John Buchman. By feeding of a variety of isotopically labelled compounds to pigeons and chemically determining the positions of the labelled atoms in their excreted uric acid (a purine), it was demonstrated that various atoms of purine ring arise from different sources. Many compounds contribute to purine ring (Fig. 20.5 ).
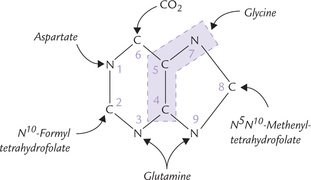
• N-1 arises from the amino group of aspartate;
• C-2 and C-8 originate from a single carbon moiety carried by tetrahydrofolate;
• N-3 and N-9 are contributed by the amide group of glutamine;
• C-6 comes directly from CO 2.
• C-4; C-5 and N-7 are derived from glycine (suggesting that glycine is wholly incorporated into the purine ring).
 Purines are built on foundation of ribose 5′-phosphate. The heterocyclic ring systems are assembled from simple precursors: glycine, glutamine, aspartate, formate and carbon dioxide.
Purines are built on foundation of ribose 5′-phosphate. The heterocyclic ring systems are assembled from simple precursors: glycine, glutamine, aspartate, formate and carbon dioxide.
The initially synthesized purine derivative is inosine monophosphate (IMP), the nucleotide of the base hypoxanthine. It is loosely referred to as the parent nucleotide. It can then serve as precursor for both AMP and GMP.
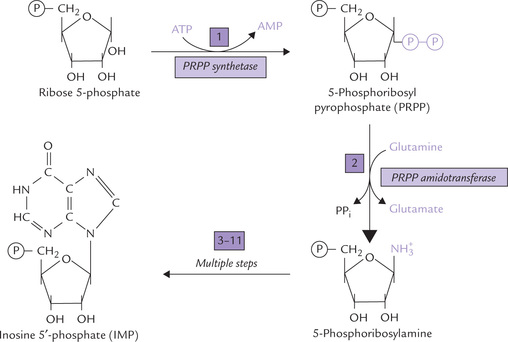
Reaction 1
Purine ring is built directly onto ribose 5′-phosphate, which is first converted to 5′-phosphoribosyl pyrophosphate (PRPP) by the enzyme PRPP synthetase (Fig. 20.6). The pyrophosphate group that gets attached to the ribose phosphate is provided by ATP. PRPP is also a precursor in the biosynthesis of pyrimidine nucleotides and the amino acids, histidine and tryptophan. Thus, PRPP lies at crucial biosynthetic crossroads and so its intracellular concentration is subject to meticulous regulation.
Reaction 2
Displacement of the pyrophosphate group (of PRPP) by amide nitrogen of glutamine occurs next. To form 5-phosphoribosylamine; the reaction is catalyzed by the enzyme PRPP amidotransferase. This is the first reaction unique to purine biosynthesis (i.e. committed step) and is also rate-limiting for the pathway.
Reactions 3—11
A series of nine reactions that follow construct the parent nucleotide, inosine monophosphate (IMP); these are shown in Figures 20.7 and 20.8 . Some of these reactions are energy demanding: ATP is required at each of the synthetase and ring closure steps. A summary of these reactions is given here.
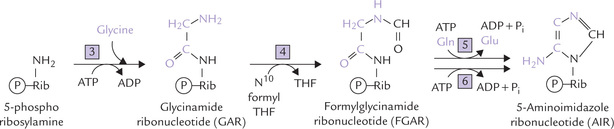
Fig. 20.7 Expansion of reactions 3–6 of IMP biosynthesis. The numbers also correspond to the enzymes for various reactions (3 = GAR synthetase, 4 = GAR transformylase, 5 = FGAM synthetase, 6 = AIR synthetase).
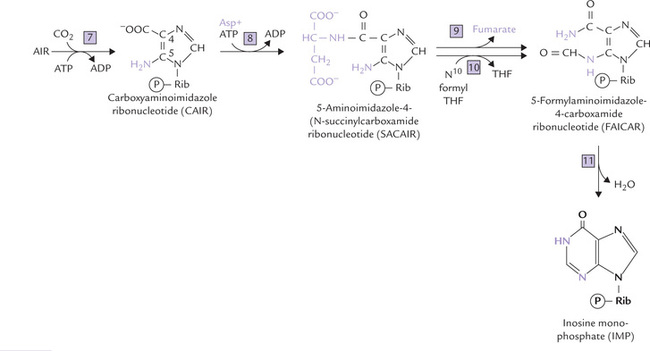
Fig. 20.8 Expansion of reactions 7–11 of IMP biosynthesis. The numbers also correspond to the enzymes for various reactions (7 = AIR carboxylase, 8 = SACAIR synthetase, 9 = Adenylosuccinate lyase, 10 = AICAR transformyltase, 11 = IMP synthase).
• Two carbons and a nitrogen are added in a single step, when glycine is attached to amine group of 5-phosphoribosylamine (Reaction 3). The reaction, catalyzed by the ATP-dependent enzyme, GAR synthetase, yields glycinamide ribonucleotide (GAR).
• A formyl (-CHO) group is then attached to the GAR to form formylglycinamide ribonucleotide (FGAR); the formyl group comes from N 10-formyl tetrahydro-folate (Reaction 4).
• In Reaction 5, another amino group is added onto FGAR to form formylglycinamidine ribonucleotide (FGAM). As for other amino group additions, the donor is glutamine.
• The imidazole ring of the purine is then closed in an ATP-dependent reaction to yield 5-aminoimidazole ribonucleotide (AIR) by action of AIR synthetase (Reaction 6).
The second ring is now built by stepwise addition of the final three atoms (Fig. 20.8).
• The first atom to be added is the C-6 carbon (Reaction 7). It comes from CO2 which is incorporated to form carboxyaminoimidazole ribonucleotide (CAIR). The reaction is an unusual one because the carbon atom arises directly from CO2, without involvement of biotin.
• The nitrogen atom at position 1 of the mature purine ring is now added in two steps (Reactions 8 and 9). The first step is the formation of an aspartate-CAIR covalent intermediate, termed succinylcarboxamide ribonucleotide (SACAIR), catalyzed by SACAIR synthetase (Reaction 8). The four-carbon dicarboxylic acid, fumarate is then cleaved off by the action of adenylosuccinate lyase and only the amino group of aspartate is retained to yield 5-aminoimidazole-4-carboxamide ribonucleotide (AICAR).
• The last purine ring carbon is added as a formyl group from N10-formyl tetrahydrofolate, by AICAR transformylase (Reaction 10); the product is 5-formylamino-imidazole-4-carboxamide ribonucleotide (FAICAR). With this reaction, all the carbon and nitrogen atoms of the purine ring have been contributed by respective sources.
• In the final step (Reaction 11), IMP synthase catalyzes closure of the second purine ring to form IMP
Finally, several reactions of the de novo pathway require input of ATP energy. One ATP each being required in reactions 1, 3, 5, 6, 7 and 8, a total of 6 ATPs are used up in synthesis of IMP.
In animals, as Mary Ellen Jones demonstrated, a single polypeptide catalyzes reactions 3, 4 and 6. Likewise, a different polyprotein is required for each set of reactions 7 and 8 and 10 and 11. This arrangement increases the efficiency of the pathway.
The de novo pathway of purine synthesis is complex, consisting of 11 steps, and requiring six ATPs till the parent nucleotide, IMP. Transfer of an amide from glutamine to phosphoribosylpyrophosphate is the committed and rate-limiting step in purine synthesis.
Stage II: Conversion of IMP to AMP and GMP
IMP is readily converted to AMP and GMP by a two-step process in each case (Fig. 20.9 ).
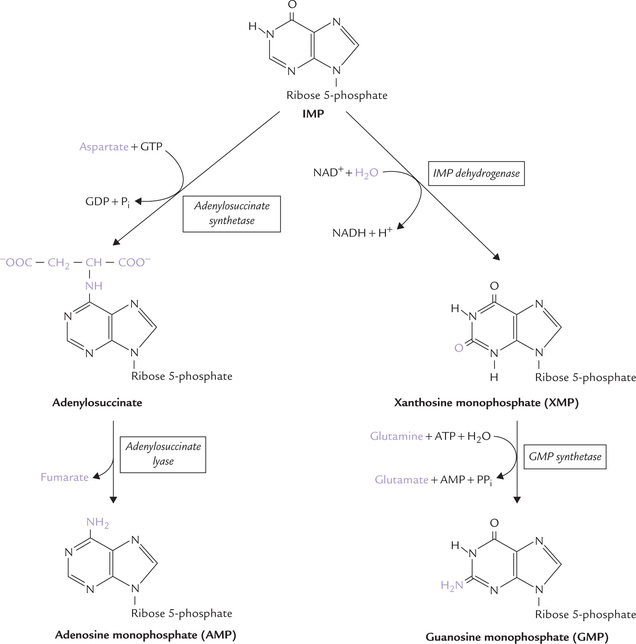
For IMP to AMP conversion, aspartate’s amino group is first linked to IMP to yield adenylosuccinate. This step is driven by hydrolysis of GTP. In the second step, adenylosuccinate lyase eliminates fumarate from adenylosuccinate to form AMP.
For IMP to GMP conversion, the C-2 of IMP is oxidized by IMP dehydrogenase (an NAD+-dependent enzyme) to form xanthosine monophosphate (XMP). XMP is then converted to GMP by GMP synthetase using glutamine as the nitrogen donor. The reaction is powered by ATP hydrolysis.
6-Mercaptopurine inhibits conversion of IMP to AMP by inhibiting the enzyme adenylsuccinate lyase and to GMP by inhibiting IMP dehydrogenase.
Conversion of Nucleoside Monophosphate to Nucleoside Diphosphate and Triphosphate
The monophosphates are the forms synthesized de novo, but the di- and triphosphates are the most commonly used forms. Therefore, monophosphates must be converted to the corresponding di- and triphosphates, which is brought about by sequential action of the following enzymes:
(a) The nucleoside monophosphate kinases convert nucleoside monophosphates to the corresponding diphosphates. ATP is generally the source of energy, being present in relatively higher concentration intracellularly than the other nucleoside triphosphates. These enzymes are specific to nitrogenous base; for example, adenylate kinase converts AMP to ADP while GMP is converted to GDP by the action of guanylate kinase.
(b) Conversion of the nucleoside diphosphates to the corresponding triphosphates is catalyzed by a single enzyme called nucleoside diphosphate kinase. Evidently, this enzyme has broad specificity.
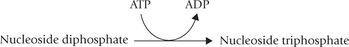
Conversion of Ribonucleotides to Deoxyribonucleotides
The compounds synthesized by the de novo pathway discussed so far are ribonucleotides, containing the ribose sugar. These can be used for RNA synthesis, but not for DNA synthesis. For synthesizing DNA, the deoxyribonucleotides are required, which differ from the ribonucleotides in having deoxyribose sugar instead of the ribose sugar. These compounds are synthesized by the enzyme ribonucleotide reductase (Fig. 20.10 ).
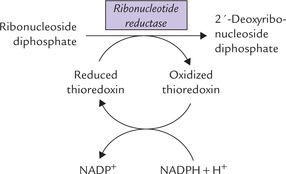
Fig. 20.10 Role of various components of the reaction system involved in reduction of ribonucleoside diphosphate to 2′-deoxyribonucleoside diphosphate.
The ribonucleotide reductase is specific for a variety of nucleoside diphosphates: purines (ADP, GDP) as well as the pyrimidines (CDP and UDP), converting them to the corresponding deoxyribonucleoside diphosphates (dADP, dGDP, dCDP and dUDP). The enzyme requires a peptide coenzyme, thioredoxin, which is the donor of the reducing equivalents; the latter are required for reducing the ribonucleoside diphosphates (Fig. 20.10). During the process, the thioredoxin (reduced) is converted to the corresponding oxidized form. Subsequently, it is reconverted to the reduced form by the reducing equivalents of NADPH. Thus, in this series of reactions, NADPH is the ultimate donor of reducing equivalent.
Regulation of the de novo Pathway
There are two important levels of regulation: First is the IMP pathway up to synthesis of IMP from ribose 5-phos-phate, and the second is immediately below the branch point leading from IMP to ATP and GTP.
The first level regulation controls the total amount of purine nucleotides synthesized for nucleic acid synthesis, while the second one controls relative amount of adenine and guanine nucleotides synthesized. Thus, an important feature of the regulatory network, Figure 20.11, is that the pathways synthesizing IMP, ATP and GTP are individually regulated (Fig. 20.11).
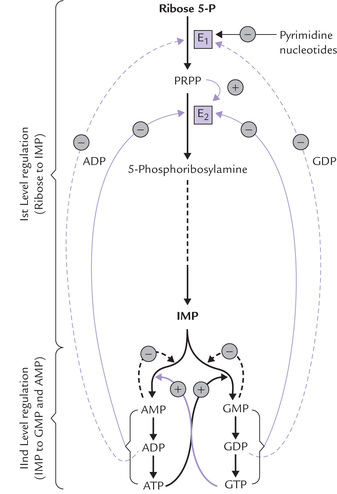
Fig. 20.11 Regulation of de novo purine biosynthesis (E 1 = PRPP synthetase, E 2 = PRPP amidotransferase, (-) = negative feedback, ( + ) = feed forward activation).
The first level regulation
The principal regulatory enzymes of the IMP pathway up to IMP synthesis are the first two enzyme, i.e. PRPP synthetase and PRPP amidotransferase:
• PRPP synthetase (E1) is inhibited by both ADP and GDP.
• PRPP-amidotransferase (E2), the committed and the rate-controlling enzyme, is likewise subject to feedback inhibition. It is a dimeric protein to which ATP, ADP and AMP bind at one inhibitory site, and GTP, GDP and GMP at the other.
The rate of IMP production is, therefore, independently but synergistically controlled by the levels of ade-nine nucleotides and guanine nucleotides. Moreover, the enzyme activity is allosterically stimulated by PRPP (feed-forward activation). It has been reported that the enzyme is apparently inactive in dimeric form. Increased amounts of PRPP stimulate its dissociation into two sub-units, which acquire catalytic activity.
The second level of regulation
• Feedback inhibitory mechanism is involved in conversion of IMP to AMP or GMP (Fig. 20.11).
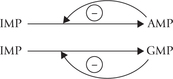
Both AMP and GMP are competitive inhibitors of IMP in their own synthesis, which balances the production of each of these nucleotides (they are roughly required in equal amounts).
• The IMP to GMP conversion is stimulated by ATP, and of IMP to AMP conversion gets stimulated by GTP.
The Salvage Pathways
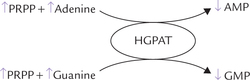
Purine bases that are obtained from the normal turnover of cellular nucleic acids can be used to resynthesize the corresponding nucleotides. Pathways of base to nucleotide conversion are referred to as the salvage pathways. A given base reacts with PRPP to yield the corresponding nucleotide; for example, guanine forms guanosine monophosphate (GMP).
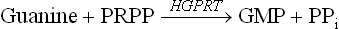
The enzyme catalyzing this step is hypoxanthine guanine phosphoribosyl transferase (HGPRT). This enzyme also converts another base, hypoxanthine, to its corresponding nucleotide, inosine monophosphate (IMP).
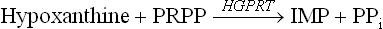
Adenine phosphoribosyl transferase (APRT), the other important enzyme of salvage pathway, is specific for adenine.
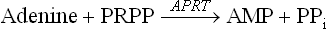
HGPRT is quantitatively a more important salvage enzyme. It is competitively inhibited by IMP and GMP. APRT is inhibited by AMP.
Compared to the de novo pathway, the salvage pathways account for production of smaller fraction of the total purine nucleotides in the human body. But they do have certain advantages for being:
• Simpler and more cost-efficient way of producing purine nucleotides.
• Preventing wastage of raw materials and are particularly important in brain and other such tissues where de novo pathway is slow or absent.
• Finally, in contrast to the de novo pathway, which is virtually identical in all cells, salvage pathways are diverse in character and distribution.
Inhibitors of Purine Synthesis
The inhibitors of purine biosynthesis can be divided in the following categories:
1. The PABA analogues, which structurally resemble para-amino-benzoic acid (PABA), inhibit synthesis of folic acid from PABA in bacterial cells.

This decreases availability of tetrahydrofolate (THF), a derivative of folic acid. Since the de novo purine synthesis requires THF (Reactions 4 and 10) decreased THF level inhibits this pathway. Diminished synthesis of purine nucleotides in the bacterial cell results, which in turn impairs bacterial cell growth and proliferation.
The above mechanism accounts for the bacteriostatic effect of sulphonamides, a PABA analogue (Fig. 18.8).
Humans lack capability to synthesize folic acid from PABA, but obtain this vitamin from dietary sources. Therefore, sulpha drugs have no deleterious effect on the human cell growth, so this drug can be safely used in humans.
2. Antifolates, such as methotrexate amethopterin, aminopterin, and related compounds interfere with the action of folate cofactors, and are widely used in the treatment of cancer. They competitively (and irreversibly) bind to dihydrofolate reductase (DHFR) with about 1000-fold more avidity than dihydrofolate, the natural substrate of this enzyme. This causes inhibition of the enzyme DHFR activity, thus blocking the synthesis of tetrahydrofolate.
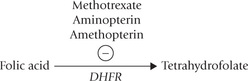
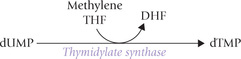
Decreased availability of tetrahydrofolate blocks synthesis of dTMP from dUMP by thymidylate synthase (because methylene THF is required in this reaction).
Further, these compounds (antifolates) are competitive inhibitors of several other THF-dependent reactions (4 and 10) used in the biosynthesis of purines.
These events interfere with the synthesis of new nucleotides and, therefore, of DNA and RNA. Consequently, cell proliferation is inhibited. This accounts for antineo-plastic effect of methotrexate and other antifolates. A number of other purine analogues are also known to act as antineoplastic agents, as discussed below.
Development of nucleic acid analogues as antineoplastic drugs
An understanding of metabolism of nucleotides has helped development of several antineoplastic drugs. The toxic effects of these drugs are accounted by their ability to interfere with nucleotide metabolism. However, they suffer from a serious limitation, i.e. development of drugs with selective toxicity for tumour cells is difficult because most biological activities of the neoplastic cells are same as those of normal cells from which they are derived. Drugs that are toxic to tumour cells are, therefore, likely to be nearly as toxic to normal cells.
The tumour cells, however, do have a higher mitotic rate, and therefore, a higher requirement for DNA synthesis than normal cells. Therefore, they are more likely to be affected by antagonists of nucleotide synthesis. With this in mind, several drugs have been developed as antagonists of nucleotide synthesis. Some commonly used drugs and their modes of action are as here:
• Glutamine antagonists, e.g. azaserine, are useful as antineoplastic drugs. They inhibit those steps in purine and pyrimidine metabolism in which glutamine donates a nitrogen: the incorporation of N-3 and N-9 into the purine ring, and conversion reactions—IMP to GMP and UTP to CTP.
• Structural analogues of bases of nucleosides act by inhibiting individual reactions in nucleotide metabolism or through their incorporation into DNA or RNA. But such DNA or RNA is functionally inactive. This arrests cell division, and hence useful in treatment of cancers. A uracil analogue, 5-fluorouracil, exemplifies this type (discussed in Case 20.4).
• Antifolates are best exemplified by methotrexate, as discussed earlier.
• 6-Mercaptopurine inhibits conversion of IMP to AMP and GM, as mentioned earlier.
B Catabolism of Purine Nucleotides
Catabolism of purine nucleotides is a continuous process, balancing the biosynthesis of these compounds. As a result, the nucleotides of every cell are constantly revised. Both adenine and guanine nucleotides are catabolized in a similar fashion (Fig. 20.12 ).
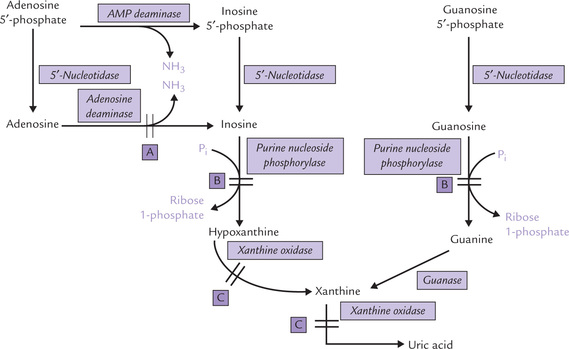
Fig. 20.12 Catabolism of AMP and GMP to uric acid (A = defect in severe combined immunodeficiency disease, B = defect in purine nucleoside phosphorylase deficiency, C = defect in xanthinuria. P i = phosphate).
Catabolism of Adenosine 5′-Phosphate
First, AMP loses an amino group through the action of enzyme AMP deaminase to produce inosine 5′-phosphate. The phosphate group of the latter is then removed by the enzyme 5′-nucleotidase to produce the corresponding nucleoside, inosine. Through phosphorolytic cleavage, ribose phosphate is liberated from inosine, leaving behind the hypoxanthine. The reaction is catalyzed by the enzyme purine nucleoside phosphorylase. Hypoxanthine is then converted to xanthine by the enzyme xanthine oxidase. Further conversion of xanthine to uric acid is catalyzed by the same enzyme.
Inosine can be produced by an alternate pathway as well, in which the phosphate group of AMP is removed by 5′-nucleotidase first to form adenosine. The latter then loses its amino group through action of adenosine deaminase to form inosine. (Adenosine, being a poor substrate for purine nucleoside phosphorylase, is not converted to adenine by this enzyme.)
Catabolism of Guanosine Monophosphate
It proceeds along the same lines. Sequential actions of 5′-nucleotidase and purine nucleoside phosphorylase produces guanine. The latter is deaminated by the enzyme guanase to form xanthine which is converted to uric acid.
Thus, xanthine is the common product in both adenine and guanine catabolic pathways, which is finally converted to uric acid by xanthine oxidase and excreted in urine. Thus uric acid is the final excretory product of purine catabolism. In addition, it can serve as an important antioxidant in primates by getting converted to allantoin. In animals other than primates an enzyme uricase converts uric acid to allantoin.
C Diseases of Purine Metabolism
Abnormalities in purine metabolism can lead to a number of disorders; the situation is akin to the disorders of amino acid metabolism. These disorders arise due to enzymatic defects in the pathway of purine degradation or biosynthesis. They may lead to altered serum uric acid concentration, i.e. hyperuricaemia or hypouricaemia (Table 20.1 ).

Hyperuricaemia and Gout
Hyperuricaemia refers to increased serum uric acid concentration. Gout is a clinical syndrome, associated with hyperuricaemia. Interestingly, all patients who develop gout would have had hyperuricaemia at some point in the development of the disease. However, not all patients with hyperuricaemia develop gout: only a minority of them do so. The reasons for this are not known.
Gout is a clinical syndrome which is characterized by hyperuricaemia and recurrent acute arthritis. Renal disease (nephropathy) is also a common complication.
History
Gout has a remarkable history: it has been known to humans since ancient times and was defined by Hippocrates, the “father of medicine” in fifth century bc. Some of the eminent people including Newton, Martin Luther, Charles Darwin, and Benjamin Franklin suffered from gout. The word gout comes from the Latin word gutta which means drop, for it was earlier thought that a poison dripping into the affected joints was the cause of this disorder. Subsequently, hyperuricaemia was shown to be the underlying cause.
Causes and Consequences of Hyperuricaemia
Normal concentration of uric acid in serum is 3–7 mg/dL (lower in women by about 1 mg/dL). Hyperuricaemia may result from increased production or decreased excretion of uric acid.
Some unrecognized defects may also lead to hyper-uricaemia (Table 20.1).
In about 25% cases over-production of uric acid causes hyperuricaemia, and in the rest, decreased renal excretion is mostly the underlying cause.
Uric acid is barely soluble in plasma, so even a moderate rise in its concentration leads to precipitation of uric acid crystals. This accounts for the development of characteristic features of gout, which include subcutaneous deposits of sodium urate crystals called tophi, arthritis and renal impairment (Case 20.1).
Gout is of two types: primary and secondary.
• In primary gout an inborn enzymatic defect leads to hyperuricaemia.
• Secondary gout refers to a state where some other primary disorder leads to the increase of uric acid level.
Over 90% of the hyperuricaemia patients are males; women suffering from this disorder are usually post-menopausal.
Primary Gout
Any disorder in which purine (de novo) synthesis is increased, causes enhanced production of purine nucleotides. Since purine nucleotides cannot be stored in human body, increased production is regulated by increased degradation. This results in enhanced uric acid production.
Some of the enzymatic defects leading to increased purine synthesis are as below:
Increased activity of PRPP synthetase
Some variants of this enzyme have altered kinetic properties, such as increased Vmax or low Km for ribose 5-phosphate, or resistance to feedback inhibition. These properties permit this enzyme to bring about increased transformation of ribose phosphate to PRPP, leading to enhanced purine biosynthesis.
Glucose 6-phosphatase deficiency
The condition is called von Gierke’s disease. Deficiency of this enzyme leads to intracellular accumulation of glucose 6-phosphate that forms pentose (ribose) phosphate through the stage II of hexose monophosphate pathway. Increased intracellular concentration of ribose phosphate leads to increased rate of purine synthesis.
Deficiency of hypoxanthine guanine phosphoribosyl transferase (HGPRT)
It is a purine salvage enzyme; its deficiency is inherited as an X-linked trait causing Lesch-Nyhan syndrome. It is associated with increased purine synthesis by a 3-fold mechanism.
• Firstly, recycling of the substrate bases (hypoxanthine and guanine) is impaired, they are metabolized to uric acid.
• Secondly, accumulation of the PRPP, the other substrate for HGPRT, occurs, which is channeled into the de novo purine biosynthetic pathway. The pathway is stimulated because of feed-forward activation effect of PRPP.
• Finally, the defect in the salvage pathway leads to decreased levels of AMP and GMP, which decreases the feedback inhibition of the rate-limiting enzyme, PRPP amidotransferase (Fig. 20.11). This further speeds up purine biosynthesis.
Most cases of primary gout, however, fall into a less defined category in which the specific defect and the mode of inheritance is not known.
Secondary Gout
It refers to a state where hyperuricaemia is consequence of some other primary disorder as noted earlier. Haematologic maliganancies such as leukaemia and multiple myeloma lead to increased cell turnover, which is accompanied by increased degradation of excess nucleic acids to uric acid. Chronic renal failure is associated with an impaired excretion of uric acid with consequent retention of abnormally large amounts of this compound. Various drugs, particularly diuretics and alcohol also minimize the excretion of uric acid in urine, thus leading to hyperuricaemia (Case 20.1). Urinary excretion of uric acid falls in starvation as well.
Uric Acid Pool in Gout
The miscible uric acid pool has been estimated to be around 1200 mg in normal subjects. It is increased to 3000 mg or more in patients suffering from gout. Uric acid may exist in the dissociated form (i.e. ionized form called urate) or the undissociated form (called uric acid). At pH of 5.75, only about half of the total molecules are in urate form (since pK’ of the most acidic group of uric acid is 5.75). At body pH of 7.4, almost all molecules are in the urate form because the body pH exceeds the pK’ value. Therefore, in all body fluids and blood, the urate predominates while uric acid accounts for a small proportion. In urine, where pH may fall below 5.75, the predominant form is uric acid. The solubility of these two forms differs vastly: uric acid is about 13 times less soluble than urate. Solubility of the sodium salt of the latter is still lower.
Solubility of uric acid = 15 mg/dL
Low Solubility: The Offending Factor in Gout
The low solubility and consequent precipitation of uric acid crystals is the cause of gouty arthritis and uric acid nephropathy. When concentration of serum urate is increased, its sodium salt (sodium urate) readily forms in the body. Its precipitation is a constant risk since limits of solubility (7 mg/dL) are exceeded with even moderate rise of serum urate level (normal serum urate is 3–7 mg/dL). Focal deposits of these precipitates, called tophi, are asymptomatic initially, but may lead to arthritis or nephropathy.
Gauty arthritis
Accumulation of trophi in joints triggers the inflammatory response of gouty arthritis by the following mechanism: the crystals are first ingested by leukocytes. These cells subsequently rupture, releasing the lysosomal enzymes that degrade articular tissue and induce the inflammatory response.
The crystals can be deposited in subcutaneous tissues also, most commonly in the external ear, over the knees and elbows, and along the tendons. However, a sustained hyperuricaemia for several years is required to bring about such deposition of tophi.
Nephropathy
Sodium urate or uric acid may precipitate in kidneys and ureters also to cause renal damage, termed uric acid nephropathy. As noted, formation of uric acid from urate is favoured as the pH falls. Therefore, with acidification of urine in renal tubules uric acid is generated. Being of limited solubility, it tends to crystallize as urinary stones. Sharp abdominal pain, obstruction to urine flow and damage to organ are the consequences.
Treatment of Gout
Is aimed at reducing joint inflammation (by anti-inflammatory drugs), increasing uric acid excretion (by uricosuric drugs), and decreasing uric acid production (by allopurinol). The most important drug treatments are:
• The anti-inflammatory drug, colchicine, is the classical treatment for the acute attack, though it is not effective in other forms of arthritis.
• Probenacid is a typical example of uricosuric agent.
• Allopurinol, an inhibitor of the xanthine oxidase, is a hypoxanthine analogue with interchanged N-7 and C-8 positions (Fig. 20.13 ). Xanthine oxidase hydroxylates allopurinol (as it does to hypoxanthine), yielding alloxanthine, which remains tightly bound to the enzyme, thereby inactivating it. Allopurinol is therefore classified as a suicide inhibitor of xanthine oxidase (Case 6.1).
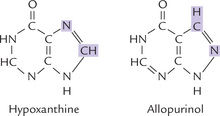
Decreased production of uric acid is accompanied by accumulation of hypoxanthine and xanthine. They are more soluble than uric acid, and hence readily excreted in urine.
Gout is a metabolic disease caused by the overproduction or under-excretion of uric acid. Due to its poor solubility, excess urate in the blood crystallizes and forms deposits in soft tissues such as kidneys (causing nephropathy) and in the toes and joints (causing gouty arthritis). Allopurinol, a purine analogue, is used for the treatment of gout due to its suicide inhibition effect on xanthine oxidase.
Besides drug therapy, low protein diet and restriction in dietary intake of purines is found beneficial in gout. Low protein diet is advised because amino acids increase de novo purine synthesis. Plenty of oral fluids and abstinence from alcohol is also recommended; the latter is harmful because of the associated dehydration and because the increased lactate levels during alcohol intoxication may impair renal excretion of uric acid. Acute attacks are precipitated by an alcoholic binge (Case 20.1).
Pseudogout
Deposition of calcium pyrophosphate crystals in joints leads to pseudogout. Clinical manifestations of this condition are similiar to those in gout but serum uric acid is not elevated.
Lesch—Nyhan Syndrome
The biochemical defect in this disorder is deficiency of the salvage enzyme HGPRT, as explained earlier. Partial deficiency of this enzyme causes only hyperuricaemia, but a complete deficiency results in Lesch-Nyhan syndrome. The disorder is named after the investigators who first described it in 1948. Since it is an X-linked trait, only males are affected.
Lesch-Nyhan syndrome (deficient HGPRT) presents as a vast array of neurological manifestations, superimposed on a strong tendency to develop gout.
Neurological abnormalities characterize Lesch-Nyhan syndrome and these appear after several months of life. Usually the initial presenting feature is delayed motor-development. Mental retardation is a predominant abnormality. The brain has an extremely low capacity for de novo biosynthesis, therefore, a complete deficiency of salvage enzymes damages the brain by depriving it of purine nucleotides. A unique feature of this disorder is the self-mutilating behaviour of the affected children who frequently bite their lips and fingertips, often violently; or jam their hands into spokes of their wheel-chair. Most patients die at an early age of uric acid nephropathy.
Hypouricaemia
Decreased concentration of serum uric acid (below 2 mg/dL) represents hypouricaemia. This is mostly due to enzymatic defects (in adenosine deaminase or xanthine oxidase), and less commonly due to some renal defect, e.g. Fanconi syndrome (Table 20.1).
Adenosine Deaminase Deficiency
Adenosine deaminase catalyzes deamination of adenosine to inosine, and of deoxyadenosine to deoxyinosine (Fig. 20.12). Deficiency of this enzyme results in accumulation of adenosine and deoxyadenosine (obtained from breakdown of DNA). It is believed that increased amount of deoxyadenosine is toxic to lymphocytes and suppresses immune function. This is because:
• The accumulated deoxyadenosine is converted to dAMP, dADP and dATP.
• The last one (dATP) is a powerful inhibitor of ribonucleotide reductase.
• This deprives the cell of precursor deoxyribonucleotide molecules for DNA synthesis. The selective impairment of lymphocytes but not of other cell types remains unexplained.
The enzyme depletion is associated with severe combined immunodeficiency disease (SCID). In this inherited disease, the B-cells as well as the T-cells are defective.
Xanthinuria
Deficiency of xanthine oxidase is the underlying cause of this rare disorder (Fig. 20.12). Since this enzyme is involved in the production of urate, its deficiency causes the purine metabolism to stop at xanthine and hypoxanthine. As a result, urate levels in blood and urine are exceedingly low, while xanthine and hypoxanthine become the major end products of purine metabolism. In most cases this condition is not associated with any clinical symptoms and goes unnoticed. Diagnosis is usually made after the incidental finding of low serum urate (Case 20.2).
Purine Nucleoside Phosphorylase Deficiency
Purine nucleoside phosphorylase converts inosine to hypoxanthine and guanosine to guanine. Deficiency of this enzyme impedes conversion of the above nucleosides to their respective bases (Fig. 20.12). Plasma accumulation of inosine and guanosine occurs, followed by increased urinary excretion of these compounds. Plasma concentration of uric acid is decreased because uric acid is the ultimate catabolic end product of inosine and guanosine.
Purine nucleoside phosphorylase is responsible for degradation of purine deoxyribonucleosides also. Therefore, accumulation of deoxyribonucleosides also occurs in this disorder.
Adenine Phosphoribosyltransferase Deficiency
Adenine phosphoribosyl transferase (APRT) is a salvage enzyme that converts adenine to AMP. Deficiency of this enzyme impedes the salvage pathway resulting in plasma accumulation of adenine. The series of events that follow ultimately result in development of renal stones (Case 20.3).
III Pyrimidine Metabolism
A Synthesis of Pyrimidine Nucleotides
Pyrimidine synthesis differs from purine synthesis in one important aspect. The pyrimidine ring is synthesized before being attached to ribose 5-phosphate; in contrast, the purine ring is constructed on a pre-existing ribose 5-phosphate. The constituent carbon and nitrogen atoms of pyrimidine are derived from various sources, namely aspartate, glutamine and carbon dioxide (Fig. 20.14 ).
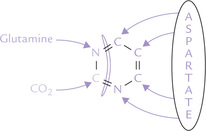
Reactions of Pyrimidine Synthesis
The de novo reactions can be divided in two stages:
• Synthesis of the parent pyrimidine nucleotide (UMP).
• Conversion of UMP to cytidine and thymidine nucleotides (Fig. 20.15 ).
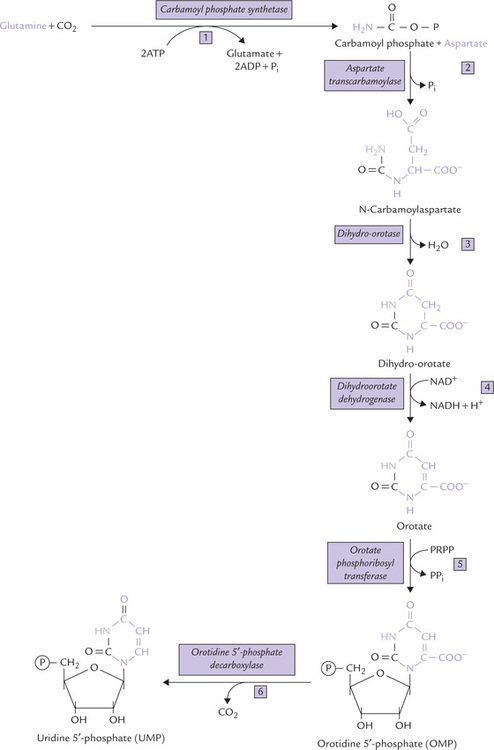
Fig. 20.15 Biosynthesis of pyrimidine nucleotides: the de novo pathway. Dihydro-orotate dehydrogenase is the only mitochondrial enzyme; all the others are cytosolic. Note that the pyrimidine ring is fully synthesized and then attached to ribose phosphate (in contrast purine ring is constructed on pre-existing ribose phosphate).
Stage I: Synthesis of UMP
The first reaction, the committed step of the pathway, involves synthesis of carbamoyl phosphate from glutamine and carbon dioxide. The enzyme catalyzing this step is carbamoyl phosphate synthetase II (CPS-II); 2 ATPs are required to drive forward the reaction.
It may be recalled that production of carbamoyl phosphate occurs by the mitochondrial enzyme, carbamoyl phosphate synthetase I (CPS I) during urea synthesis. Differences between the CPS I and CPS II are summarized in Table 20.2 .
Table 20.2
Differences between carbamoyl phosphate synthetase (CPS-I and CPS-II)
| CPS I | CPS II | |
| Pathway involved | Urea cycle | Pyrimidine synthesis |
| Cellular location | Mitochondrial matrix | Cytosol |
| Source of nitrogen | Ammonia | Glutamine |
In contrast to the different enzymes in eukaryotes, prokaryotes contain only one carbamoyl phosphate synthetase, which is responsible for the biosynthesis of arginine and pyrimidines.
In the second step, carbamoyl phosphate and aspartate condense to form N-carbamoyl aspartate, catalyzed by the enzyme aspartate transcarbamoylase (ATCase). The third reaction is an intramolecular condensation leading to pyrimidine ring closure by the enzyme dihydro-orotase. The reaction product is dihydro-orotate, which is then oxidized to orotate in the fourth reaction, catalyzed by dihydro-orotate dehydrogenase in which NAD+ serves as the coenzyme.
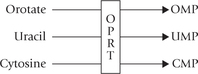
Orotate may be visualized as the parent pyrimidine base. Orotate is converted to the corresponding nucleotide, orotidine 5′-phosphate (OMP) by coupling with a phosphoribosyl group donated by PRPP. Thus, attachment with phosphoribosyl group is postponed till the penultimate step of the pathway. The final reaction involves decarboxylation of OMP to yield uridine 5′-monophosphate (UMP).
Stage II: Conversion of UMP to Cytidine and Thymidine Nucleotides
It occurs by a series of reactions shown in Figure 20.16 .
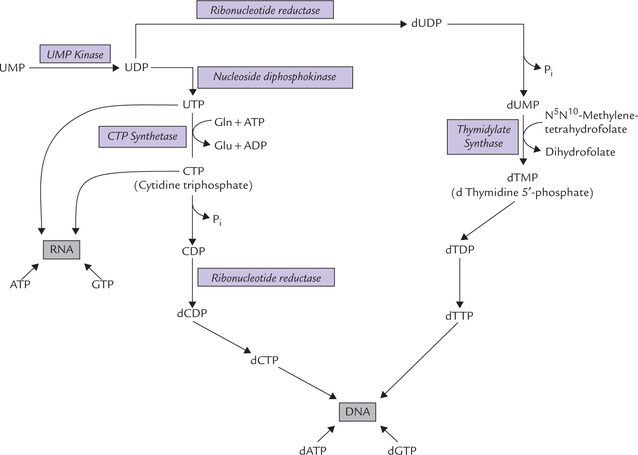
Fig. 20.16 Synthesis of cytidine and thymidine nucleotides from uridine nucleotides and their incorporation in nucleic acids.
Conversion of UMP to cytidine nucleotides
UMP is phosphorylated by two consecutive reactions, catalyzed by the enzymes UMP kinase and nucleoside diphosphokinase to produce UTP. The latter serves as a substrate for the cytidine nucleotide, cytidine 5′-triphosphate (CTP). The enzyme, CTP synthetase catalyzes this conversion by bringing about amination of the UTP; the source of amino group is glutamine.
Formation of thymidine nucleotides
The UMP is first converted to dUMP by the reaction sequence shown in Figure 20.16. The dUMP is then converted to dTMP by the enzyme thymidylate synthase which utilizes N 5N 10-methylenetetrahydrofolate as the source of the methyl group.
This is an unusual reaction where tetrahydrofolate donates not only a carbon unit but also undergoes change in its own oxidation state (from THF to DHF). Inhibitor of this reaction enzyme, 5′-fluorouracil (a thymine analogue), serves as an anti-tumour agent (Case 20.4).
Regulation of pyrimidine synthesis
1. In mammalian cells, the main regulatory enzyme of the de novo biosynthetic pathway is carbamoyl phosphate synthetase II, which exists in a polyprotein (Box 20.1). The enzyme is inhibited by UTP and activated by ATP and PRPP, thus providing good examples of feedback inhibition and feed-forward stimulation, respectively.
2. In prokaryotic cells, the second reaction catalyzed by the enzyme aspartate transcarbamoylase is a regulatedstep. It is inhibited by CTP and stimulated by ATP. In certain bacteria, UTP also inhibits ATCase.
3. OMP decarboxylase is inhibited by UMP and CMP. This also keeps pyrimidine formation under tight feedback control.
B Catabolism of Pyrimidine Nucleotides
Animal cells degrade pyrimidine nucleotides to their component bases, which are then broken down in the liver to amino acids, namely (β-alanine and (β-aminoiso-butyrate. The latter is further metabolized to certain intermediates of fatty acid metabolism; thus, catabolism of pyrimidine nucleotides contributes, to a limited extent, to the energy metabolism of the cell.
1. The major enzyme involved in pyrimidine degradation is pyrimidine specific 5′-nucleotidase. It brings about liberation of phosphate group from the pyrimidine nucleotides to form the corresponding nucleosides.
2. The next enzyme of the pathway, nucleoside phosphorylase, releases ribose 1-phosphate, leaving behind the corresponding free base. Conversion of uridine to uracil and of deoxythymidine to thymine takes place in this manner (Fig. 20.17 ).
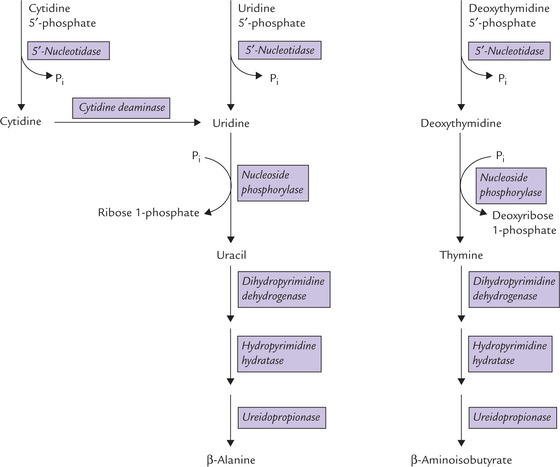
3. The terminal steps involve a series of reactions (such as ring splitting, reduction of carbon bond, and hydrolytic cleavage), which ultimately result in the formation of (β-alanine or (β-aminoisobutyrate. Both compounds are transaminated and eventually metabolized to carbon dioxide although the exact pathway is not known.
Figure 20.17 summarizes the reactions involved in catabolism of cytidine 5′-phosphate and uridine 5′-phosphate. Degradation of the deoxythymidine 5′-phosphate to ( β-aminoisobutyrate by parallel pathway is also shown in this Figure.
A pyrimidine nucleotide is synthesized by firstly forming the pyrimidine ring and then attaching it to ribosyl phosphate. It is degraded to amino acids, namely ( β-alanine and ( β-aminoisobutyrate, which are then further metabolized.
The patients having certain defects in the catabolic pathway of pyrimidines need to be evaluated carefully. Though asymptomatic normally, they are at increased risk of developing severe neurologic reactions following administration of certain drugs (Case 20.4).
Orotic Aciduria
Orotic aciduria is an autosomal recessive disorder, characterized by the urinary excretion of large amounts of orotic acid, retarded growth and severe anaemia.
Biochemical Defect
Orotic aciduria results from deficiency of either or both of the last two enzymes: orotate phosphoribosyl transferase and OMP decarboxylase (Fig. 20.15).
Clinical manifestations
Orotic acid is not inherently toxic, but excessive urinary level of this substance (more than 1 g/day; normal, 1.4 mg/day) leads to its crystallization and consequent obstruction to urine flow. Moreover, block in pyrimidine synthesis occurs in orotic aciduria, which leads to shortage of pyrimidine nucleotides for incorporation into DNA and RNA. Insufficient genetic material decreases generation of new cells. The rapidly proliferating cells such as red cells are most prominently affected, which results in anaemia. The erythrocyte precursors in the bone marrow become both abundant and unusually large. The diagnosis is confirmed by assay of the above enzymes in erythrocytes, or fibroblasts.
Treatment
Intake of diet rich in uridine (or cytidine) is an effective treatment for orotic aciduria. Uridine provides enough UMP through phosphorylation, and also inhibits CPS II so as to decrease the rate of orotic acid synthesis.
Orotic aciduria is an autosomal recessive defect in pyrimidine synthesis, treated by diet rich in uridine or cytidine, or by replacement therapy.
(Enzyme activities of both orotate phosphoribosyl transferase and OMP decarboxylase are present on a single protein as domains (bifunctional enzyme). Mutation in the phosphoribosyl transferase impairs its association with the other (decarboxylase) domain, resulting in loss of activity of both. This produces the type 1 situation (i.e. orotic aciduria type 1). In type 2, mutation in the decarboxylase occurs, but it does not affect its aggregation with phosphoribosyl transferase. In this case, only decarboxylase is inactive, whereas activity of phosphoribosyl transferase is not affected.)
Rey’s Syndrome
Excessive production of orotic acid may also occur in deficiency of ornithine transcarbamoylase, a urea cycle enzyme. It leads to accumulation of carbamoyl phosphate, which is diverted for enhanced synthesis of orotic acid. Increased urinary excretion of this compound follows, hence this defect is considered as a secondary orotic aciduria, also termed Rey’s syndrome.
No other genetic deficiency in pyrimidine nucleotide biosynthesis is known because such defects are lethal in utero.
Exercises
Essay type questions
1. Review the nomenclature of bases, nucleosides, and nucleotides.
2. Compare the de novo pathways of purine and pyrimidine nucleotide synthesis with respect to (a) precursors, (b) energy cost, (c) acquisition of the ribose moiety, and (d) number of enzymatic steps.
3. Describe the salvage pathways for purines and pyrimidines. How do PRPP levels influence these? Describe an inherited disorder caused by deficiency of a purine salvage enzyme.
4. Explain biochemical defects in orotic aciduria, gout and Lesch-Nyhan syndrome.
5. How are folate cofactors involved in nucleotide metabolism? What antifolates serve as effective drugs?
6. What are the various mechanisms for regulating de novo purine synthesis? How does the cell balance the production of (a) purine and pyrimidine nucleotides, and (b) ribonucleotides and deoxynucleotides.
7. Differentiate between the primary and the secondary gout. Explain the biochemical basis of clinical abnormalities in primary gout and their treatment.