Chapter 13 Overview of the Somatic Nervous System and Drugs Affecting Neuromuscular Transmission
The somatic nervous system is the division of the peripheral nervous system that coordinates consciously controlled functions such as movement, posture and respiration. In this system a single motor neuron connects the central nervous system to the skeletal muscles, which are the effector organs. Blockade of neuromuscular transmission by drugs is used as an adjunct to anaesthesia for producing muscle relaxation. In clinical practice, anticholinesterase agents are used for Alzheimer’s disease and myasthenia gravis and to reverse neuromuscular blockade. Poisoning from organophosphate anticholinesterase agents can also occur as a result of their use as pesticides and chemical warfare agents.
Key abbreviations
Key background
THE second major division of the peripheral nervous system is the somatic nervous system (Figure 11-1), which coordinates consciously controlled functions, including movement, posture and respiration. In this system, a single motor neuron connects the central nervous system (CNS) to the skeletal muscles, which are the effector organs of the somatic nervous system. Often called the voluntary nervous system, this system allows us to consciously control our skeletal muscles and hence movement. Initiating and controlling both gross movements (such as jumping or walking) and precise movements (such as those done with our hands) involves the motor cortex, which initiates and controls movement, the basal ganglia, which integrate and establish our muscle tone, and the cerebellum, which ensures our movements are smooth and coordinated. Integration of these systems aids in the maintenance of normal posture and balance.
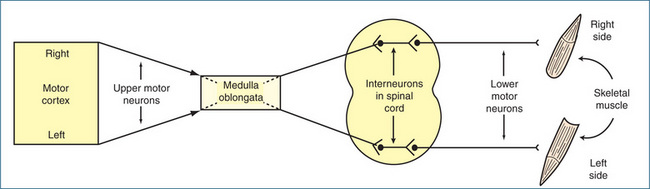
Figure 13-1 Diagrammatic representation of motor pathways from the right and left sides of the motor cortex innervating skeletal muscles on the opposite sides of the body.
Once the primary motor area of the cerebral cortex initiates a voluntary movement, nerve impulses propagate from the motor cortex through upper motor neurons that cross over in the medulla oblongata to the other side; thus, muscles on the right side of the body are controlled by the left motor cortex, and the right side of the brain controls the muscles on the left side of the body. The upper motor neurons terminate in the anterior grey horn of the spinal cord at each spinal segment. In many instances, the upper motor neurons synapse first with interneurons, which act as the connection with the lower motor neurons; they in turn innervate skeletal muscles of the trunk and limbs (Figure 13-1). The lower motor neurons are the final common pathway that connects the CNS to the skeletal muscles.
The neuromuscular junction and nicotinic receptors
The synapse between the lower motor (somatic) neuron and the skeletal muscle is called the neuromuscular junction (NMJ). At the NMJ, the motor neuron divides, forming a cluster of synaptic end bulbs that contain vesicles carrying the neurotransmitter acetylcholine (ACh). Following arrival of a nerve action potential (Figure 11-5), ACh is released from the vesicles and diffuses across the synaptic cleft to act on postsynaptic nicotinic receptors on the motor end-plate of the muscle fibre. As muscle fibres tend to be long, the NMJ is usually near the centre of the fibre. This allows the impulse to spread evenly towards the ends of the muscle fibres and ensures that contraction occurs simultaneously throughout the length of the muscle. As each nerve impulse produces only one muscle contraction, the action of ACh is rapidly terminated (within 1 ms) by acetylcholinesterase (AChE), which is attached to the collagen fibres. The release and metabolism of ACh occur by the same mechanisms as those described for the parasympathetic nervous system (Chapter 11). The difference is that in the somatic nervous system ACh acts on postsynaptic nicotinic receptors on the motor end-plate, whereas the postsynaptic receptors in the parasympathetic system are muscarinic receptors.
Motor end-plate nicotinic receptors
Nicotinic receptors mediate the effect of ACh on skeletal muscles, and they are the main biological targets of the tobacco alkaloid nicotine. The receptor is classed as an ion channel and is composed of five subunits arranged in a circular manner with the ion channel in the centre. The human adult skeletal muscle receptor subunits are designated using a Greek letter: there are two alpha (α) subunits and one beta (β), one delta (δ) and one epsilon (ε) subunit. The bulk of the receptor faces the extracellular surface. The density of the receptors is very high on the motor end-plate. When two molecules of ACh bind (one molecule to each of the α subunits), the channel opens immediately and sodium ions flow through, causing depolarisation of the motor end-plate. This triggers the muscle action potential, causing muscle contraction (Figure 13-2). Contraction occurs because of a sliding filament mechanism involving actin and myosin (see Chapter 22).
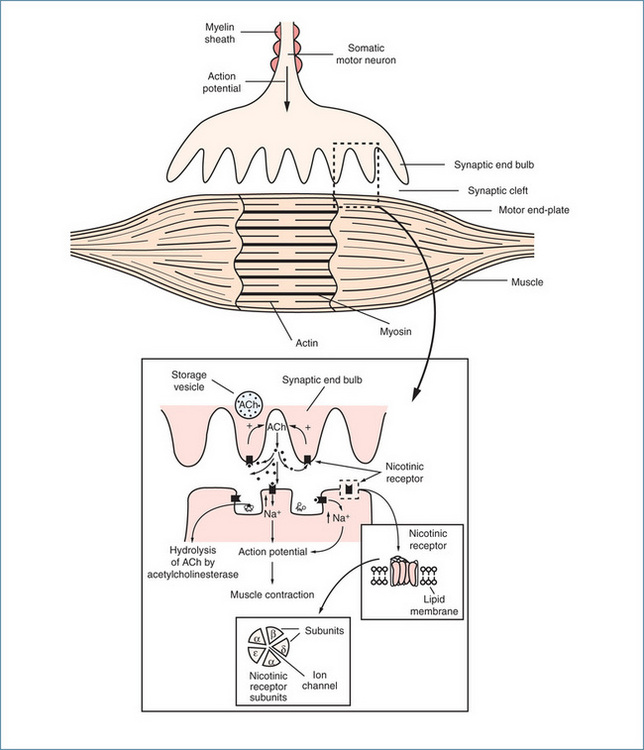
Figure 13-2 The neuromuscular junction, showing release of acetylcholine (ACh), which acts on both presynaptic and postsynaptic nicotinic receptors. The insets show enlargements of the relevant structures.
There are many sites at which drugs and toxins can interrupt neuromuscular transmission. These include blockade of action potential generation in the motor neuron, inhibition of release of ACh (Clinical Interest Box 13-1), inhibition of the breakdown of ACh and blockade of postsynaptic receptors (Figure 13-3). The pharmacological agents of clinical relevance are those used principally as adjuncts to anaesthesia, and include drugs acting at postsynaptic receptors, commonly referred to as neuromuscular blocking drugs, and anticholinesterase agents, which are also used for a variety of therapeutic purposes including the treatment of Alzheimer’s disease and myasthenia gravis. In addition, anticholinesterase agents are used as insecticides and chemical warfare agents, which can lead to situations of acute human exposure.
Clinical interest box 13-1 Cosmetic use of botulinum toxin, an inhibitor of acetylcholine release
Although botulinum toxin is commonly associated with outbreaks of lethal food poisoning and is a potential biowarfare/bioterror agent, it has been used since the 1970s for the treatment of facial dystonias (see Chapter 20 and Drug Monograph 31-3). Botulinum acts presynaptically, blocking the release of ACh and causing generalised muscle weakness. The muscle weakness produced slowly recovers over several months with the growth of new nerve terminals.
Different types of botulinum toxin exist and highly localised injections of small quantities of botulinum toxin type A (Botox) are widely used for cosmetic correction. The cosmetic use of botulinum toxin followed the observation by Jean Carruthers in 1987 that Botox reduced frown lines. Its use has now extended from frown lines to crow’s feet, horizontal forehead creases, eyebrow shaping and chin dimpling (Klein & Glogau 2000). The drug was approved in Australia in 1994 for the treatment of blepharospasm but it is also used widely in private cosmetic clinics for wrinkle treatment. In the USA in 2005 > 3.8 million Botox treatments at a cost of US$1.4 billion were performed. The popularity of Botox continues to grow with an everincreasing number of high profile individuals having Botox injections prior to public appearances and the proliferation of in-home Botox parties. Botox is often termed a ‘cosmeceutical’ and the aim is to use a sufficient dose at the right anatomical facial site to accomplish muscle weakening without muscle paralysis. Using the incorrect dose or imprecision in the injection site may result in a mask-like or frozen face or ptosis (eyelid drooping). Such is the social acceptability of Botox that it is now being used for vertical lip lines, flaring nostrils and to soften nasolabial folds. Unwanted paralysis is the biggest drawback and may not always be predictable. Of more concern is the sale from unreliable sources of impure and/or adulterated toxin via the internet, which increases the risk of adverse effects and the risk of botulism. Drug interactions can also occur and the action of Botox is prolonged by concomitant use of aminoglycoside or spectinomycin antibiotics.
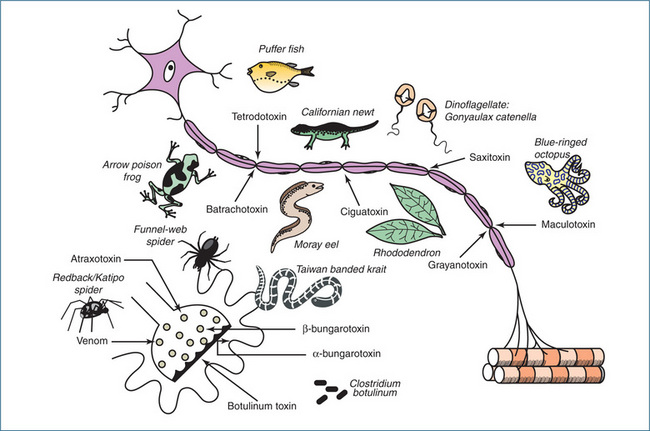
Figure 13-3 Summary diagram illustrating the sites of action of various toxins on somatic motor neurons and the motor end-plate. Tetrodotoxin, saxitoxin and maculotoxin prevent conduction in the axon by blocking the sodium channels; batrachotoxin, ciguatoxin and grayanotoxin block conduction by opening the sodium channels and thereby depolarising the axon membrane. Latrodectus spider spp (redback [AUST]/katipo [NZ]) venom, atraxotoxin and β-bungarotoxin disrupt the vesicles and deplete the nerve ending of acetylcholine. Botulinum toxin prevents the release of acetylcholine by acting on the axon terminal membrane. β-Bungarotoxin combines specifically with the acetylcholine receptors on the muscle side of the junction.
Source: Bowman 1973; published with permission from the Pharmaceutical Journal.
Neuromuscular blocking drugs
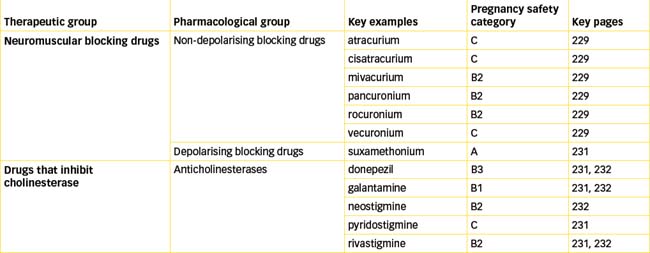
The neuromuscular blocking drugs are principally of two types:
Non-depolarising blocking drugs
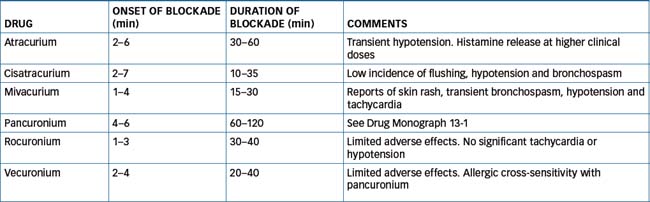
Curare is synonymous with the South American arrow-tip poisons that were used by indigenous people along the Amazon and Orinoco Rivers for killing animals. The pharmacologist Claude Bernard investigated the muscle paralysing effect of curare in 1856. He showed that the drug prevents response of skeletal muscle to nerve stimulation but does not inhibit contraction from a direct stimulus, nor does it block nerve conduction. These elegant experiments established the concept of nerve–muscle conduction, and in 1942 curare was introduced for promoting muscle relaxation during general anaesthesia. This heralded the search for other curare-like drugs. Although tubocurarine (the active constituent of curare) is no longer in clinical use, various synthetic drugs have been produced. These include atracurium, cisatracurium, mivacurium, pancuronium, rocuronium and vecuronium. As these drugs are quaternary ammonium compounds, they are poorly absorbed and do not readily cross the blood–brain barrier or placenta; the latter is an advantage when operating on pregnant women.
Effects on skeletal muscle
In general, the non-depolarising drugs produce rapid blockade characterised by motor weakness that progresses to total flaccid paralysis. Small muscles (e.g. those of the eyelid) are affected first, proceeding through to the limbs, neck, trunk and finally the diaphragm and intercostal muscles. With paralysis of the respiratory muscles, respiration ceases and mechanical ventilatory support is required. Return to normal muscle function varies markedly between individuals and between individual muscle groups. Normally function returns first to the respiratory system, the diaphragm and intercostal muscles; pharyngeal and facial muscles recover more slowly.
Effects on mast cells
Typically the non-depolarising neuromuscular blocking agents cause histamine release from mast cells. This often manifests as harmless cutaneous reactions (flushing and rash) but more severe symptoms can occur, including hypotension and bronchospasm. The effect is not related to an action at nicotinic receptors but is more likely due to the highly basic nature of these drugs. The tendency to cause histamine release varies among these drugs, with tubocurarine eliciting the greatest release and pancuronium, vecuronium and rocuronium showing lesser tendencies. The most frequently implicated drug is the depolarising blocking drug suxamethonium, and severe anaphylactoid reactions are more frequent in women.
Drug Monograph 13-1 describes pancuronium, and the main characteristics of the other non-depolarising NMJ blockers are summarised in Table 13-1.
Drug monograph 13-1 Pancuronium
Mechanism of action
Pancuronium is a potent competitive antagonist of acetylcholine at nicotinic receptors on the skeletal muscle motor end-plate (Figure 13-4). Interruption of neuro-muscular transmission requires occupancy of > 70% of the nicotinic receptors while blockade requires > 95% occupancy.
Indications
As an adjunct to general anaesthesia to provide muscle relaxation during surgery and during intensive care.
Pharmacokinetics
Pancuronium is widely distributed following IV administration and within 5 minutes high concentrations can be found in the kidney, liver and spleen. As the drug is highly water-soluble, urinary excretion begins almost immediately and up to 25% is excreted as unchanged drug. The remainder of the drug is cleared via hepatic metabolism and biliary excretion, both of which can be reduced in persons with liver disease. The half-life is > 30 minutes. In the presence of pre-existing renal disease clearance can be reduced and the half-life prolonged.
Drug interactions
Potentiation of effect can occur with inhalation anaesthetics, suxamethonium, antibiotics such as the aminoglycosides (which themselves cause blockade), diazepam, calcium channel blockers, lithium, propranolol and magnesium salts. A decrease in effect can occur with adrenaline, carbamazepine, anticholinesterase agents such as neostigmine, high-dose corticosteroids and the chloride salts of calcium, sodium and potassium.
Adverse reactions
These are uncommon but a slight increase in heart rate, cardiac output and blood pressure can occur. A life-threatening anaphylactoid reaction can occur but the incidence is less than 1 in 10,000 anaesthetics.
Depolarising blocking drugs
Currently the only depolarising neuromuscular blocking drug in clinical use is suxamethonium (also known as succinylcholine, Drug Monograph 13-2). In contrast to tubocurarine, which blocks nicotinic receptors and produces flaccid muscle paralysis, suxamethonium acts as an agonist at the nicotinic receptors on the motor end-plate. Binding to the receptor results in persistent stimulation and maintains the depolarised state of the motor end-plate. Loss of electrical excitability ensues because the sodium channels remain open and the motor end-plate can no longer respond to an electrical stimulus (Figure 13-4). Suxamethonium causes excessive salivation due to muscarinic-like actions, which can be prevented by the use of atropine. In addition, initial muscle fasciculations (twitching) occur because as each end-plate is depolarised it produces a localised action potential in the muscle fibre. As each fibre has only one motor end-plate, when they are depolarised individually it is not sufficient to produce complete muscle contraction. These fasciculations subside quickly and neuromuscular blockade follows.
Drug monograph 13-2 Suxamethonium
Mechanism of action
Suxamethonium, an analogue of acetylcholine, was introduced into clinical practice in 1951 and remains widely used. It is an agonist at muscle end-plate nicotinic receptors and maintains the depolarised state. It is the only truly short-acting muscle relaxant and reversal by an anticholinesterase drug is unnecessary because of the short duration of action of suxamethonium but also because use of an anticholinesterase agent will prolong the depolarisation blockade.
Indications
It is used when brief muscle relaxation is required (e.g. electroconvulsive therapy, tracheal intubation, short surgical procedures and orthopaedic manipulations). In addition to its muscle relaxant properties suxamethonium affects the cardiovascular system, causing bradycardia.
Pharmacokinetics
The onset of action of suxamethonium is rapid and the estimated half-life is in the order of 2–4 minutes. Blockade persists for about 10 minutes and the drug is rapidly hydrolysed by butyrylcholinesterase (also known as pseudocholines terase or plasma cholinesterase) to choline and succinyl monocholine; the latter is then hydrolysed to choline and succinic acid. In some individuals with atypical butyrylcholinesterase, blockade can persist for an extended period (refer to Chapter 7).
Drug interactions
Many drugs enhance the neuromuscular blocking activity of suxamethonium (e.g. lignocaine, nonpenicillin antibiotics, β-blockers, quinidine, lithium carbonate, high-dose corticosteroids and some cancer chemotherapy drugs). Current sources should be consulted for a more extensive list.
Adverse reactions
Suxamethonium can cause profound and complex effects on the cardiovascular system, including bradycardia, tachycardia, arrhythmias, hypertension and cardiac arrest. Because of loss of potassium from the motor end-plate, an increase in plasma potassium concentration can occur and this is important in situations of extensive burns and massive trauma and in people with muscular disorders. In rare situations suxamethonium can precipitate malignant hyperthermia, an often fatal condition characterised by intense muscle spasm and a rapid rise in body tempera ture (refer to Chapter 14). Although the action of suxamethonium is short, in some individuals prolonged apnoea occurs as a result of a butyrylcholinesterase deficiency, the use of antichol ines terase drugs that inhibit the action of butyrylcholinesterase or the presence of liver disease, which can cause a low butyrylcholinesterase concentration.
Warnings and contraindications
Care should be taken with the use of suxamethonium in people with electrolyte disturbances, low butyrylchol inesterase activity, renal disease and concomitant digitalis therapy. The drug is contraindicated in people with a known or suspected familial history of malignant hyperthermia and in cases of extensive burns or multiple trauma.
Dosage and administration
Dosage is individualised depending on the circumstances of use and the degree of relaxation required. The drug is usually administered IV but the IM route may be used when a suitable vein is not accessible. Under no circumstances should suxamethonium be administered to a conscious person.

Figure 13-4 Sites of action of neuromuscular blocking drugs and anticholinesterase agents. Schematic representation of postsynaptic membrane of motor end-plate showing nicotinic receptors and acetylcholinesterase (AChE). The enlargement shows acetylcholine (ACh) within the active site of acetylcholinesterase. The critical amino acids forming the catalytic site are indicated: Glu = glutamate, His = histidine, Ser = serine. The zigzag line indicates the site of hydrolysis of acetylcholine, yielding choline and acetic acid. NMJ = neuromuscular junction.
Anticholinesterase agents
Acetylcholinesterase (AChE) hydrolyses the neurotransmitter acetylcholine, forming choline and acetate (Figure 11-7). The enzyme is bound to the postsynaptic membrane and the active site, which resembles a deep gorge, contains within its structure three crucial amino acids: a serine and a histidine, which form the esteratic site, and a glutamate, termed the anionic site. Together, these three amino acids are crucial for hydrolysis of ACh and are the targets for the reversible and irreversible AChE inhibitors (Figure 13-4). Anticholinesterase agents are used for conditions such as Alzheimer’s disease and myasthenia gravis, and to reverse neuromuscular blockade after anaesthesia. In addition, AChE is the biological target of pesticides and chemical warfare agents.
Three broad categories of AChE agents exist:
Drug monograph 13-3 Neostigmine
Neostigmine is a reversible inhibitor of acetylcholin esterase, forming a carbamylated enzyme complex at the active site. This complex is hydrolysed slowly by AChE over the following 3–4 hours.
Indications
Neostigmine is most commonly used for the reversal of neuromuscular blockade induced by non-depolarising NMJ blockers such as pancuronium. In addition to its use as an adjunct to anaesthesia, it is used for the treatment of myasthenia gravis.
Pharmacokinetics
Neostigmine is a quaternary ammonium compound. It is poorly absorbed from the gastrointestinal tract and does not cross the blood–brain barrier. The plasma half-life is in the order of 0.5–1.5 hours and the drug is predominantly excreted in the faeces (>50%) and urine (about 30%). It is metabo lised principally by plasma cholinesterases and the kinetics of the drug are unlikely to be affected by liver disease.
Drug interactions
The anticholinesterase effect of neostigmine is diminished by corticosteroids. Many of the drug interactions are more relevant to the situation where the drug is used to treat myasthenia gravis as any drugs with anticholinergic activity may antagonise the effects of neostigmine, e.g. drugs for urinary incontinence. Similarly the efficacy of anticholinergic drugs (e.g. benzhexol, benztropine, biperiden and orphenadrine) used in the treatment of Parkinson’s disease will be substantially diminished by administration of an anticholinesterase drug.
Adverse reactions
These often relate to the overdose situation and resemble a cholinergic crisis, with many of the symptoms as listed in Table 13-3.
Donepezil, galantamine and rivastigmine
These three anticholinesterase drugs and the N-methyl-Daspartate (NMDA) antagonist, memantine (refer to Chapter 20), are approved for the treatment of Alzheimer’s disease but not other types of dementia. The use of donepezil, galantamine and rivastigmine increases the level of ACh in the brain and provides marginal improve ments in cognition and global assessment of dementia (Raina et al 2008). It is important to appreciate that the AChE drugs are classified on the basis of their duration of inhibition of AChE, which differs from the pharmacokinetics of the drug. For example donepezil forms a stable complex with AChE but is hydrolysed within minutes while its plasma half-life is 60 hours.
Donepezil is a synthetic reversible inhibitor of AChE that exhibits a relatively high degree of selectivity for neuronal AChE with little effect on intestinal or cardiac AChE. The drug is well absorbed after oral administration and is metabolised by oxidation (CYP3A4 and CYP2D6) and glucuronidation. The active metabolite 6-O-desmethy donepezil inhibits AChE and is present at about 20% of the donepezil concentration.
Galantamine is also a reversible inhibitor of AChE, while rivastigmine is classed as a reversible carbamoylating AChE inhibitor, which has high lipid solubility and readily crosses the blood–brain barrier. Table 13-2 provides a comparison of the pharmacokinetics of these three drugs.
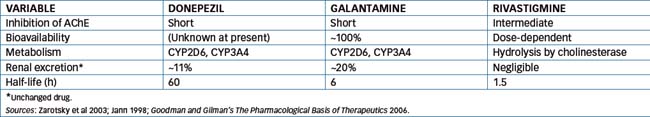
Donepezil and galantamine are subject to drug interactions with agents that are substrates, inducers or inhibitors of CYP3A4 and CYP2D6. For example, increased plasma concentration of donepezil and galantamine is likely to occur with co-administration of the CYP3A4 inhibitors ketoconazole or erythromycin. In contrast, as rivastigmine is hydrolysed by cholinesterase, interaction with other drugs metabolised by CYP is unlikely. As would be anticipated, combination with drugs with anticholinergic activity may antagonise the effect of anticholinesterases and worsen the dementia. Classes of drugs that may antagonise the effects of AChE inhibitors include drugs for urinary incontinence (refer to Chapter 25), antihistamines and some antipsychotics and antidepressants.
Adverse effects of these drugs are commonly cholinergic effects such as nausea, vomiting, diarrhoea, anorexia, headache, insomnia, dizziness, tremor and urinary incontinence. Infrequently bradycardia occurs and combination with drugs that also cause this may increase the risk of bradycardia and hypotension. The main pharmacological and toxicological effects of all the anticholinesterases are explained by enhanced levels of acetylcholine (Table 13-3).
Table 13-3 Therapeutic and toxicological effects of anticholinesterase agents
| SITE | EFFECT |
| NMJ | Inhibition of AChE leads to an increased synaptic concentration of ACh, which antagonises the action of the competitive non-depolarising NMJ blockers. This results in reversal of blockade. Toxicological effects include fasciculations, weakness, muscular paralysis, depressed ventilation |
| In myasthenia gravis, which is characterised by muscle weakness and profound fatigue, inhibition of AChE results in an increase in synaptic ACh, which increases the likelihood of postsynaptic action potential at the NMJ | |
| Postganglionic, parasympathetic synapses | Increased ACh leads to increased salivation, tears, gastrointestinal tract and bronchial secretions, augmentation of motor activity of bowel, bronchoconstriction, bradycardia, hypotension and constricted pupils, vomiting, diarrhoea and urination |
| CNS | Stimulation or depression (larger doses), headache, anxiety, irritability, ataxia, fatigue, amnesia, hypothermia, lethargy, unconsciousness, convulsions, coma, central respiratory depression |
| CVS | Actions on the CVS are complex, reflecting both ganglionic and postganglionic effects resulting from accumulation of ACh. This initially causes excitation but with increasing concentration of ACh ganglionic blockade occurs through persistent depolarisation. Augmentation of vagal action results in bradycardia, shortening of the effective refractory period of the atria and increases in the refractory period and conduction time at the SA and AV nodes |
Irreversible anticholinesterase agents
With the exception of ecothiopate, which was formerly used in the treatment of glaucoma, most irreversible inhibitors of AChE are pesticides of the organophosphate class or chemical warfare agents such as the nerve gases sarin, tabun and soman. The organophosphate pesticides (e.g. parathion and malathion) are widely used in agriculture, horticulture and urban gardening and are a common cause of poisoning in humans. The organophosphate pesticides inhibit AChE by forming a very stable complex principally with the esteratic site. This phosphorylated form of the enzyme is not degraded and return of AChE activity is dependent on synthesis of new enzyme. In addition some of these agents also inhibit plasma butyrylcholinesterase.
Nerve gases are also organophosphate anticholinesterase agents. A great deal of interest has been rekindled since the use of chemical nerve agents in various wars since the 1980s and in terrorist attacks, e.g. Japan and the Gulf War (see Clinical Interest Box 13-2). These agents are highly
Clinical interest box 13-2 Chemical warfare agents
Chemicals (chlorine and phosgene) hazardous to humans were first used as ‘weapons of mass destruction’ during World War I. Since that time refinement of chemical processes has resulted in the continued production of chemical weapons. Organophosphate nerve agents such as tabun, sarin, cyclosarin and soman were manufactured during WW II, and sarin and soman have been stockpiled in a number of countries including the USA. Another class of nerve gases is the V class, which are organophosphate esters of various 2-aminoethanethiols of which VX is the most lethal. It is estimated that the lethal dose in humans is 0.3 mg/person via the inhalational route and 5 mg/person via dermal absorption (Szinicz 2005). These agents are inhibitors of acetylcholinesterase and the antidote carried by military personnel is atropine, which antagonises the persistent stimulation of muscarinic receptors, and pralidoxime, an acetylcholinesterase-reactivating drug.
Sulfur mustard, which is classed as a vesicant, was explicitly developed as a chemical warfare agent and was first used in battle in 1917. The most recent use of sulfur mustard was in the Iran–Iraq War in the 1980s, where it is estimated that over 100,000 Iranians were injured by the chemical and approximately one-third are still suffering from late effects (Kehe & Szinicz 2005). Sulfur mustard has a different mechanism of action from the nerve gases. It is generally accepted that following exposure the sulfur mustard is metabo lised to reactive intermediates, which then alkylate DNA, RNA and proteins, resulting in subsequent failure of cellu lar functions. Clinical manifestations include respira tory tract damage, chronic obstructive lung disease, eye lesions, bone marrow depression and cancer. Currently there is no antidote to sulfur mustard.
volatile and pose a significant health problem. Toxicity occurs as a result of irreversible inactivation of AChE leading to an accumulation of acetylcholine. Persistent stimulation by ACh at presynaptic and postsynaptic receptors occurs initially, followed finally by paralysis of cholinergic neurotransmission. This ultimately affects the somatic, autonomic and central nervous systems (Table 13-3).
The signs and symptoms of poisoning from pesticides and nerve gases can be categorised according to whether excessive stimulation occurs at muscarinic or nicotinic receptors. The mnemonic ‘DUMBELS’ describes the muscarinic symptoms: Diarrhoea; Urination; Miosis; Bronchorrhoea, bronchoconstriction and bradycardia; Emesis; Lacrimation; Salivation (Geoghegan et al 2006). Nicotinic effects would occur more from stimulation of the somatic nervous system (e.g. skeletal muscle twitching, weakness and flaccid paralysis), and from the release of catecholamines from the adrenal medulla (Sidell & Borak 1992).
Treatment of organophosphate poisoning
This is a complex area and requires considerable expertise to recognise the effects of common chemical warfare agents and interpret the severity of nerve poisoning based on clinical symptoms. Drugs that are used include atropine (Chapter 11), which antagonises the muscarinic effects of excess acetycholine but is ineffective in antagonising the nicotinic effects of acetylcholine and hence muscle weakness and paralysis does not improve, and pralidoxime. Pralidoxime is a reactivating oxime that exerts a nucleophilic attack on the phosphorylated esteratic site (disrupts the covalent bond between the nerve agent and AChE) resulting in formation of a phosphoryloxime, which ‘regenerates’ the enzyme. This reverses nicotinic receptor dysfunction and reduces the paralysis. Early administration (usually IV) is necessary because, as the phosphorylated enzyme ‘ages’ (within hours), it becomes resistant to reactivation. Pralidoxime is excreted unchanged by the kidney and a continuous infusion is usually administered for 24 hours after symptoms resolve (Geoghegan et al 2006). As this drug has anticholinesterase activity adverse effects include cholinergic symptoms e.g. nausea, blurred vision.
Key points
 The somatic nervous system coordinates consciously controlled functions such as posture, movement and respiration. The synapse between the lower motor (somatic) neuron and the skeletal muscle is called the neuromuscular junction. The transmitter at the neuromuscular junction is acetylcholine, which acts on both presynaptic and postsynaptic nicotinic receptors. The neuromuscular blocking drugs are principally of two types: competitive non-depolarising drugs and depolarising nicotinic receptor agonists. Non-depolarising drugs such as pancuronium competitively block the action of ACh. They produce rapid blockade at the motor end-plate, which is characterised by initial motor weakness that progresses to flaccid paralysis. Non-depolarising blockers characteristically cause a release of histamine that may manifest as a rash or, in more severe cases, as hypotension and bronchoconstriction. Adverse effects of suxamethonium in clude bradycardia, tachycardia, arrhythmias, hypertension and cardiac arrest. In rare situations suxamethonium can precipitate malignant hyperthermia, an often fatal condition characterised by intense muscle spasm and a rapid rise in body tempera ture. Acetylcholinesterase is the biological target for anticholinesterase drugs, pesticides and chemical warfare agents such as nerve gases. The anticholinesterase drug neostigmine is commonly used to reverse neuromuscular blockade produced by non-depolarising blockers; it is also used as an adjunct to anaesthesia. Donepezil, galantamine and rivastigmine are approved for the treatment of Alzheimer’s disease but not other types of dementia. They increase the level of acetylcholine in the brain and provide marginal improvements in cognition and global assessment of dementia. Irreversible anticholinesterase agents are, in general, organophosphates. They are used as pesticides (e.g. parathion and malathion) and chemical warfare agents (e.g. tabun, sarin, soman). Drugs that are used to treat nerve agent poisoning include atropine (Chapter 11), which antagonises the muscarinic effects of excess acetylcholine, and pralidoxime, which ‘regenerates’ acetylcholinesterase.
The somatic nervous system coordinates consciously controlled functions such as posture, movement and respiration. The synapse between the lower motor (somatic) neuron and the skeletal muscle is called the neuromuscular junction. The transmitter at the neuromuscular junction is acetylcholine, which acts on both presynaptic and postsynaptic nicotinic receptors. The neuromuscular blocking drugs are principally of two types: competitive non-depolarising drugs and depolarising nicotinic receptor agonists. Non-depolarising drugs such as pancuronium competitively block the action of ACh. They produce rapid blockade at the motor end-plate, which is characterised by initial motor weakness that progresses to flaccid paralysis. Non-depolarising blockers characteristically cause a release of histamine that may manifest as a rash or, in more severe cases, as hypotension and bronchoconstriction. Adverse effects of suxamethonium in clude bradycardia, tachycardia, arrhythmias, hypertension and cardiac arrest. In rare situations suxamethonium can precipitate malignant hyperthermia, an often fatal condition characterised by intense muscle spasm and a rapid rise in body tempera ture. Acetylcholinesterase is the biological target for anticholinesterase drugs, pesticides and chemical warfare agents such as nerve gases. The anticholinesterase drug neostigmine is commonly used to reverse neuromuscular blockade produced by non-depolarising blockers; it is also used as an adjunct to anaesthesia. Donepezil, galantamine and rivastigmine are approved for the treatment of Alzheimer’s disease but not other types of dementia. They increase the level of acetylcholine in the brain and provide marginal improvements in cognition and global assessment of dementia. Irreversible anticholinesterase agents are, in general, organophosphates. They are used as pesticides (e.g. parathion and malathion) and chemical warfare agents (e.g. tabun, sarin, soman). Drugs that are used to treat nerve agent poisoning include atropine (Chapter 11), which antagonises the muscarinic effects of excess acetylcholine, and pralidoxime, which ‘regenerates’ acetylcholinesterase.Review exercises
References and further reading
Bowman W.C. Therapeutically useless drugs from unusual sources. Pharmaceutical Journal. 1973;211:219-223.
Geoghegan J., Tong J.L. Chemical warfare agents. Continuing Education in Anaesthesia, Critical Care and Pain. 2006;6:230-234.
Hogan D.B. Practical approach to the use of cholinesterase inhibitors in patients with early Alzheimer’s disease. Geriatrics and Aging. 2009;12:202-207.
Jann M.W. Pharmacology and clinical efficacy of cholinesterase inhibitors. American Journal of Health-System Pharmacy. 1998;55(Suppl 1):22S-25S.
Kehe K., Szinicz L. Medical aspects of sulphur mustard poisoning. Toxicology. 2005;214:198-209.
Klein A., Glogau R.G. Botulinum toxin: beyond cosmesis. Archives of Dermatology. 2000;136:539-541.
Lukas R.J., Changeux J.P., Le Novere N., et al. International union of pharmacology: current status of the nomenclature for nicotinic acetylcholine receptors and their subunits. Pharmacological Reviews. 1999;51:397-401.
Naguid M., Magboul M.A. Adverse effects of neuromuscular blockers and their antagonists. Drug Safety. 1998;18:99-116.
Raina P., Santaguida P., Ismaila A., et al. Effectiveness of cholinesterase inhibitors and memantine for treating dementia: evidence review for a clinical practice guideline. Annals of Internal Medicine. 2008;148:379-397.
Shintani E.Y., Uchida K.M. Donepezil: an anticholinesterase inhibitor for Alzheimer’s disease. American Journal of Health-System Pharmacy. 1997;54:2805-2810.
Sidell F.R., Borak J.B. Chemical warfare agents: II. Nerve gases. Annals of Emergency Medicine. 1992;21:865-871.
Szinicz L. History of chemical and biological warfare agents. Toxicology. 2005;214:167-181.
Taylor P. Anticholinesterase agents. In Brunton L.L., Lazo J.S., Parker K.L., editors: Goodman & Gilman’s The Pharmacological Basis of Therapeutics, 11th edn, New York: McGraw-Hill, 2006. [ch 8]
Taylor P. Agents acting at the neuromuscular junction and autonomic ganglia. In Brunton L.L., Lazo J.S., Parker K.L., editors: Goodman & Gilman’s The Pharmacological Basis of Therapeutics, 11th edn, New York: McGraw-Hill, 2006. [ch 9]
Zarotsky V., Sramek J.J., Cutler N.R. Galantamine hydrobromide; an agent for Alzheimer’s disease. American Journal of Health-System Pharmacy. 2003;60:446-452.