Chapter 25 Diuretics and Drug Treatment of Urinary Incontinence
The kidneys play an important role in maintaining homeostasis by regulating the composition and volume of the extracellular fluid. In addition to this role, the kidneys are essential for eliminating metabolic byproducts such as creatinine, uric acid and urea, and they play a central role in acid–base balance. The functional unit of the kidney is the nephron. Diuretics are an important group of drugs that alter renal function, increasing urine volume and enhancing the excretion of sodium and chloride. They are widely prescribed for treating conditions such as heart failure and hypertension. Electrolyte imbalance and volume depletion are common adverse effects of diuretic administration and can be minimised by administering the lowest effective dose and monitoring both clinical response and plasma electrolytes.
Formed urine is stored in the bladder and urinary incontinence is a common embarrassing problem that afflicts the elderly population in particular. Drug therapy is an option but it is essential to eliminate possible contributing factors, such as urinary tract infection, metabolic disorders or the current use of drugs that may contribute to urinary incontinence.
Key abbreviations
GFR glomerular filtration rate
NKCC Na+–K+–2Cl− co-transporter
Key background
THE urinary system is composed of two kidneys, two ureters, the bladder and the urethra (Figure 25-1). Urine formed in the kidneys flows through the ureters to the bladder, where it is stored. When voluntary control of the muscles preventing voiding is removed, urine flows from the bladder into the urethra and is expelled from the body.
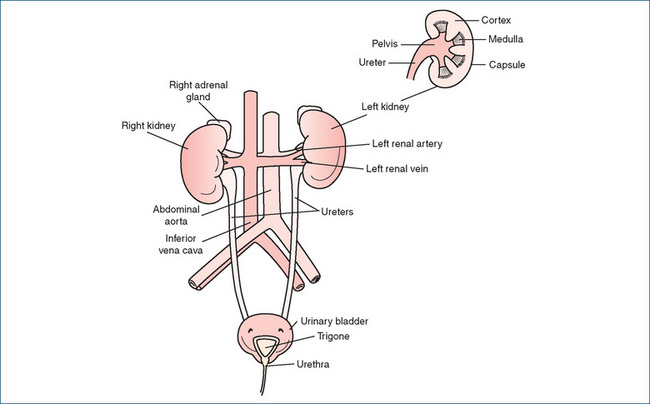
The kidneys, which are approximately the size of an individual’s closed fist, are essential for maintaining homeostasis. By processing salts and water and balancing excretion they control the volume and ionic composition of the extracellular fluid; regulate blood pH by excreting hydrogen ions (H+); excrete waste products such as urea, uric acid and creatinine; and modulate blood pressure via the release of renin and activation of the renin–angiotensin–aldosterone system. In addition to these roles, the kidneys synthesise calcitriol, an active vitamin D metabolite (see Chapter 37), and the growth factor erythropoietin, which stimulates red blood cell production (see Chapter 27).
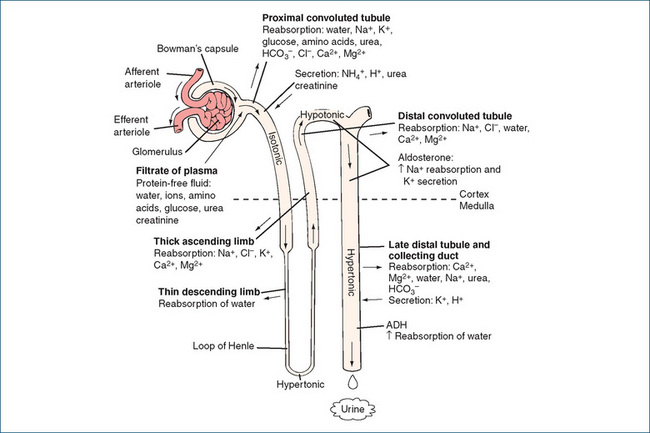
A cross-section of the kidney shows three distinct regions: the outer cortex, the inner medulla and the central hollow pelvis, which is continuous with the ureter (Figure 25-1). The functional units of the kidney are the nephrons, the number of which (about 1 million) is determined at birth. Each nephron consists of a glomerulus (the glomerular capillaries and the Bowman’s capsule) and the renal tubular network comprising, in order of the flow through them, the proximal convoluted tubule, the loop of Henle, the distal convoluted tubule and, finally, the collecting duct. Most nephrons (around 80%) have short loops of Henle that penetrate superficially into the medulla and are termed cortical nephrons. The remaining nephrons—with long loops of Henle that almost traverse the medullary region—are called juxtamedullary nephrons and provide the kidney with the ability to concentrate the urine.
The kidneys are highly vascularised and the complex network of arterioles, capillaries and venules gives rise to the glomerular capillaries, the peritubular capillaries that form a dense plexus surrounding the cortical aspects of the renal tubule and the vasa recta, the long hairpin loop-shaped capillaries that supply the renal medulla. The peritubular capillaries reunite to form the peritubular venules and blood finally leaves the kidney through a single renal vein. Control of renal blood flow is a complex process involving myogenic (intrinsic) responses, tubuloglomerular feedback via the juxtaglomerular apparatus, the renin–angiotensin system (refer to Chapter 23), the sympathetic nervous system and vasoactive agents including renal prostaglandins that play a role in vasodilation in compromised kidneys, leukotrienes and nitric oxide. The interface between the vascular network and the renal tubular system provides the basis for the three major renal processes: glomerular filtration, tubular reabsorption and tubular secretion.
Glomerular filtration
Glomerular filtration, the initial step in urine formation, occurs as a result of ultrafiltration of water and small solutes through pores of the glomerular capillaries into the capsular space of the Bowman’s capsule. In the absence of disease, the glomerular membrane does not allow molecules larger than 6–7 nm in diameter, including plasma proteins such as haemoglobin and albumin, to pass through. The heart works to create pressure in the blood vessels, which in turn provides the force necessary to accomplish glomerular filtration. Blood flow to the kidney is normally around 1200 mL/min, which is 20%–25% of cardiac output, and the glomerular filtration rate (GFR) is about 125 mL/min. This amounts to around 180 litres of filtrate formed per day in a healthy individual, of which 99% is ultimately reabsorbed throughout the length of the nephron.
Maintenance of glomerular hydrostatic pressure depends on systemic blood pressure and is aided by the ability of the afferent and efferent arterioles to constrict and dilate (Figure 25-2). Associated with the afferent arteriole are juxtaglomerular (JG) cells, which are the primary site of renin storage and release in the body. A reduction in afferent arteriole pressure causes the release of renin from JG cells into the general circulation. The renin released causes increased production of angiotensin II, a powerful vasoconstrictor that increases systemic blood pressure. Systemic blood pressure has to be significantly reduced before glomerular filtration is greatly altered. Usually, some degree of filtration will exist if the pressure in the glomerular capillaries remains above 50 mmHg. Renin release is also controlled by specialised tubular cells located in the cortical thick ascending limb of the loop of Henle where it makes contact with the afferent arteriole. These cells, known as the macula densa, along with the JG cells form the juxtaglomerular apparatus. The macula densa cells respond to changes in the flow of tubular fluid and sodium chloride concentration and influence the release of renin from JG cells.
Renal function changes with age and in various disease states and often an accurate estimation of renal function or GFR is used clinically to guide management or adjustment of drug dosage (refer to Chapter 8). Common measures of renal function include the use of serum creatinine and creatinine clearance. Serum creatinine represents the balance between production by muscle and excretion or clearance by the kidney. Creatinine clearance by the kidneys is a measure of the ‘volume’ of serum cleared of creatinine per unit time and has the units of mL/min or mL/second. It is considered a reasonable measure of GFR, hence renal function (http://www.australianprescriber.com/magazine/30/1/17/20/).
Direct determination of creatinine clearance requires simultaneous measurements of both serum creatinine and a timed urine creatinine, usually a 24-hour timed collection. The latter is often not convenient for the patient and an exact 24-hour collection is more frequently unreliable than reliable. The most widely recognised measure of calculating creatinine clearance is the Cockcroft–Gault formula that relies only on knowledge of the patient’s serum creatinine, age, gender and weight. This formula is used for estimating renal function when there is a need for adjustment of drug dosage in patients with renal impairment. However it has limitations, which include lack of established validity in some populations, lack of reliability in severe malnutrition and obesity, and in situations of rapidly changing renal function e.g. acute renal failure.
The Cockcroft–Gault formula is:
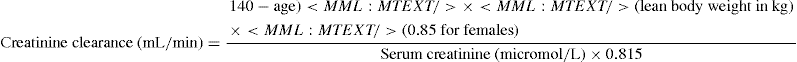
Clinical interest Box 25-1 Renal disease in indigenous populations
 The Australian Aboriginal and New Zealand Maori popula tions have been subject to rapid cultural changes and erosion of traditional lifestyles. One of the most concerning impacts of this on Indigenous health is the rise in prevalence of endstage renal disease (ESRD), now particularly prevalent in Australian Indigenous people. The crude incidence of ESRD was reported as six times higher for Aboriginals than non- Aboriginals in 1994, but by 1998 the incidence was 21 times that of non-Aboriginal Australians and doubling every four years (Hoy 1996; Hoy et al 1998).
The Australian Aboriginal and New Zealand Maori popula tions have been subject to rapid cultural changes and erosion of traditional lifestyles. One of the most concerning impacts of this on Indigenous health is the rise in prevalence of endstage renal disease (ESRD), now particularly prevalent in Australian Indigenous people. The crude incidence of ESRD was reported as six times higher for Aboriginals than non- Aboriginals in 1994, but by 1998 the incidence was 21 times that of non-Aboriginal Australians and doubling every four years (Hoy 1996; Hoy et al 1998).
The commonest conditions leading to ESRD are diabetes, hypertension and glomerulonephritis.
Haemodialysis is the most common in-hospital treatment for Indigenous people; of the total number of persons registered between 2002 and 2004 with the Australian and New Zealand Dialysis Transplant Registry, 9% were Indigenous and, of those, 66% were under 55 years of age. Over the past decade, the number of Indigenous people starting therapy for ESRD has more than tripled with 54 in 1991 and 188 in 2004. At the end of 2004, 87% of registered Indigenous people were reliant on dialysis and 13% had received a kidney transplant. By comparison, 54% of non-Indigenous Australians were reliant on dialysis and 46% had received a kidney transplant. This disproportion can be related to factors such as miscommunication between the health professionals and the patient, lack of understanding of the disease and treatment by the patient, and the lower rates of matched kidney donors for Indigenous patients (AIHW 2005).
Tubular reabsorption
Reabsorption of sodium and nutrients leads to the reabsorption of water by osmosis. Of the 180 L of glomerular filtrate delivered to the nephrons per day, about 99% is reabsorbed from the lumen of the proximal convoluted tubule into the peritubular capillaries, with the remainder excreted as urine. This tubular reabsorption is a selective process, and the main transport mechanisms that prevail throughout the nephron are:
Within the nephron there are also multiple renal tubular drug transporters. The main ones are the organic cation transporters and the organic anion transporters (Chapter 6). Competition for transport by a single transporter between ions and drugs often manifests as an adverse effect, e.g. hyperuricaemia with some diuretics. As ion transport within the nephron is complex, specific mechanisms will be discussed in those sections detailing the mechanism of action of the various classes of diuretics.
For almost every substance that is actively transported across the membrane, there is a maximum rate at which the transport mechanism can function. This is called the tubular transport maximum (TM). For example, the TM for glucose averages 320 mg/min for most adults. If the tubular concentration (mg) of glucose is very high the TM of 320 mg/min will be reached and the excess glucose remaining will not be reabsorbed but will appear in the urine. Every substance that has a tubular transport maximum also has a renal threshold concentration, which is the plasma concentration at which a substance begins to appear in the urine because of saturation of transporters, that is, the exceeding of TM.
Tubular secretion
The third major renal process is tubular secretion, which is the movement of substances from peritubular or interstitial capillaries into the renal tubular cells and then into the tubular lumen. The proximal convoluted tubule plays an important role in the secretion of hydrogen ions (H+) which, coupled with preferential absorption of bicarbonate (HCO3−), regulates acid–base balance and in the secretion of metabolic byproducts (e.g. ammonium ions, creatinine) and certain drugs (e.g. penicillin and radio-contrast agents). All of these secretory processes are saturable and exhibit a TM, just as the reabsorptive processes do.
The roles of various segments of the renal tubule in the movement of water and solutes are summarised in the following sections and in Figure 25-2. The final urinary excretion of a substance, which is influenced by glomerular filtration, tubular reabsorption and tubular filtration, can be summarised as:
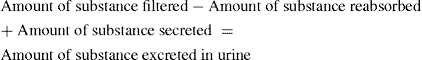
The proximal convoluted tubule
Most of the glomerular filtrate is reabsorbed in the proximal convoluted tubule and returned to the bloodstream. About 70% of the salt and water in the filtrate is reabsorbed rapidly, maintaining nearly the same osmolality between the tubular fluid and the interstitial fluid at the end of the proximal tubule (i.e. the solutions are isotonic). The secretion of H+ that occurs in the proximal tubule is linked to the reabsorption of HCO3− in the tubular filtrate. This process involves intracellular formation of carbonic acid (H2CO3) from carbon dioxide and water. The carbonic acid formed dissociates to give HCO3− and H+. This reversible reaction is catalysed by carbonic anhydrase. The hydrogen ions formed are secreted into the lumen and combine with bicarbonate in the glomerular filtrate to form carbonic acid in the lumen. This in turn dissociates into water and carbon dioxide, which diffuses into tubule cells and reforms H2CO3. Dissociation releases bicarbonate, which is then reabsorbed into the blood.
Acid–base balance is maintained in healthy humans by the action of the body’s buffer systems, changes in the rate and depth of breathing and excretion of hydrogen ions by the kidneys. As the blood becomes more acidic (decreasing pH), the kidneys will respond by increasing the renal tubule excretion of hydrogen and ammonia, which results in an increase in blood bicarbonate and an increase in pH (towards normal).
The loop of Henle
The loop of Henle is important in regulating urine osmolarity1 and osmolality2 of body fluids. The descending limb is highly permeable to water, and movement of water out of the tubule produces a hypertonic (more concentrated) filtrate at the tip of the loop of Henle (the papilla). Permeability to urea and sodium is low in this segment of the loop. In contrast, in the ascending limb of the loop of Henle water permeability is almost nil whereas sodium and chloride permeability is high. About 20%–25% of the sodium chloride in the filtrate is reabsorbed and this is not accompanied by water. Consequently, the tubular filtrate becomes very dilute, or hypotonic (this is often termed ‘free water production’), and the medullary interstitium becomes hypertonic, which is necessary for the concentrating capacity of the countercurrent between the renal tubules and the vasa recta. The concentration gradient established across the tubular epithelium becomes multiplied in a longitudinal direction, resulting in a large osmotic gradient between the isosmotic renal cortex and the hyperosmotic medulla and papilla. Potassium is also reabsorbed from the proximal tubules and loop of Henle in percentages equivalent to those for sodium; around 8% of the filtered potassium reaches the distal tubules.
The distal convoluted tubule
Between 5% and 10% of sodium reabsorption takes place actively in the distal convoluted tubule. The net loss of sodium from the filtrate is greater than the reabsorption of water; coupled with reabsorption of chloride, this makes the urine progressively more dilute. Uptake of sodium is largely determined by the presence of the mineralocorticoid aldosterone, produced by the adrenal cortex. When the extracellular fluid volume is decreased, the renin– angiotensin–aldosterone system is activated (see Chapter 23), stimulating the release of aldosterone, which acts to promote secretion of potassium and also the active reabsorption of sodium. The latter occurs by stimulating both Na+/H+ exchange via an action on aldosterone receptors and the insertion of more sodium pumps in the basolateral membrane. Parathyroid hormone and calcitriol also act on this segment of the nephron to increase reabsorption of calcium (see Chapter 37).
The collecting duct
Composition of the hypotonic fluid entering the collecting duct may be altered in the medullary portion by the action of antidiuretic hormone (ADH, also called vasopressin). ADH is a water-conserving hormone synthesised in the hypothalamus and stored in the posterior pituitary gland. When plasma osmolality increases as a result of dehydration or water deprivation, osmoreceptors in the supraoptic area of the hypothalamus stimulate the release of ADH.
Clinical interest Box 25-2 Renal damage from drugs, foods and plants
The kidneys are exposed to numerous chemicals every day, forming around 5 million litres of ultrafiltrate over a lifespan of 75 years. Exposure to nephrotoxins occurs from the ingestion of drugs, environmental chemicals, food and plants. Common drugs such as NSAIDs and ACE inhibitors can cause deterioration in renal function, and aminoglycoside antibiotics have been reported to cause an increase in plasma creatinine in about 30% of patients (Saker 2000).
Analgesic abuse was widespread in Australia in the 1970s and was related to extensive advertising and marketing of compound analgesics containing e.g. aspirin combined with phenacetin and caffeine or codeine. So successful was the advertising campaign that a significant proportion of the population started to regularly take large doses of these drugs. The renal function abnormalities included a reduction in glomerular filtration, haematuria and proteinuria and pathological changes typified by renal papillary necrosis. As a result of legislation introduced in Australia in 1979, restricting the advertising and sale of analgesics, the incidence of analgesic abuse and end-stage renal failure declined (Nanra 1993).
Heavy metals, such as cadmium and lead, and environmental chemicals, such as the pesticides paraquat, chlordane and diquat, all cause renal lesions if ingested, and many of these have been the offending chemical used for suicide. Nephrotoxicity from foods is rare and is more the exception than the rule, although many plants are incredibly toxic to the kidneys. These include the Amanita phalloides mushroom, Datura species (e.g. angel trumpets), autumn crocus and water hemlock. The damage produced varies enormously and can range from papillary necrosis to acute tubular necrosis to interstitial nephritis and glomerulonephritis.
The released ADH increases the expression of aquaporin (water channels) in the apical membrane thereby increasing permeability of the distal tubule and collecting duct to water, which is passively reabsorbed increasing plasma volume, thus lowering plasma osmolality. When ADH concentration is low, a large volume of hypotonic fluid passes from the distal tubule into the collecting ducts leading to the excretion of dilute urine. In the complete absence of ADH, a condition known as diabetes insipidus occurs and the individual affected can excrete as much as 20 L dilute urine daily.
Diuretics
Diuretics are among the most extensively used drugs. They are widely prescribed for the treatment of hypertension (Chapter 23) and are an integral part of drug therapies in oedematous conditions such as acute and chronic congestive heart failure (Chapter 22), chronic renal failure, nephrotic syndrome and cirrhosis.
Mechanism of action
Diuretics modify renal function and induce diuresis (increased rate of urine flow) and natriuresis (enhanced excretion of sodium chloride). The increase in urine volume is achieved primarily by inhibiting reabsorption of sodium and chloride in the nephron. The increased excretion of salt leads to an increase in the excretion of water. The three major classes of diuretics are:
Although carbonic anhydrase inhibitors were introduced as diuretics during the 1940s and 1950s, their diuretic action was weak and they were found to be ineffective over the long term. Acetazolamide, a carbonic anhydrase inhibitor introduced in 1950, is now reserved for the treatment of open-angle glaucoma (see Chapter 31) and is used as adjunct treatment with anticonvulsants to manage absence seizures (see Chapter 17).
Figure 25-3 shows the various sites of action of diuretic drugs on the nephron, the main mechanisms of ion absorption and the percentages of ions filtered.
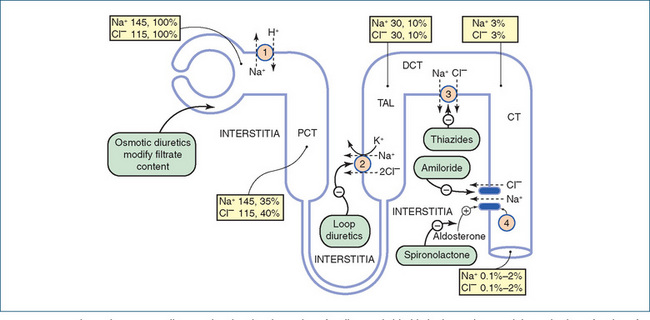
Figure 25-3 Schematic summary diagram showing the absorption of sodium and chloride in the nephron and the main sites of action of drugs. Mechanisms of ion absorption at the apical margin of the tubule cell (not of course shown to scale): 1 Na+/H+ exchange; 2 Na+/K+/2Cl– co-transport; 3 Na+/Cl− co-transport; 4 Na+ entry through sodium channels. Sodium is pumped out of the cells into the interstitia by the Na+/K+-ATPase in the basolateral margin of the tubular cells (not shown). Chloride ions may pass out of the tubule through the paracellular pathway. The numbers in the boxes give the concentrations of ions as millimoles per litre of filtrate and the percentages of ions filtered at the sites specified. No absolute concentrations are given for the DT and CT because they can vary considerably. CT = collecting tubule; DCT = distal convoluted tubule; PCT = proximal convoluted tubule; TAL = thick ascending loop. Data from: Greger 2000. Reproduced from: Rang et al 2007, with permission.
Loop diuretics
 The drugs commonly referred to as loop diuretics are bumetanide, ethacrynic acid and frusemide (Drug Monograph 25-1). The pharmacological effects of all the loop diuretics are similar—all produce a rapid and intense diuresis and in general have a short duration of action (4–6 hours). These powerful diuretics are actively secreted into the lumen of the nephron via the organic-base pump in the proximal tubule cells. On reaching the thick ascending limb of the loop of Henle, they inhibit the Na+–K+–2Cl− co-transporter (NKCC), thus preventing reabsorption of sodium and chloride from the lumen into the epithelial cells. As this site accounts for about 15%–25% of the reabsorption of sodium and chloride, their diuretic effect is greater than that reported with the other diuretics. The mechanism by which they inhibit the co-transporter is not known, but evidence suggests that they bind to the chloride-binding site on the transporter as these drugs also inhibit the reabsorption of calcium and magnesium.
The drugs commonly referred to as loop diuretics are bumetanide, ethacrynic acid and frusemide (Drug Monograph 25-1). The pharmacological effects of all the loop diuretics are similar—all produce a rapid and intense diuresis and in general have a short duration of action (4–6 hours). These powerful diuretics are actively secreted into the lumen of the nephron via the organic-base pump in the proximal tubule cells. On reaching the thick ascending limb of the loop of Henle, they inhibit the Na+–K+–2Cl− co-transporter (NKCC), thus preventing reabsorption of sodium and chloride from the lumen into the epithelial cells. As this site accounts for about 15%–25% of the reabsorption of sodium and chloride, their diuretic effect is greater than that reported with the other diuretics. The mechanism by which they inhibit the co-transporter is not known, but evidence suggests that they bind to the chloride-binding site on the transporter as these drugs also inhibit the reabsorption of calcium and magnesium.
In addition to diuresis, loop diuretics exert direct vascular effects. In particular, frusemide acutely causes venodilation, but the duration of this effect is short, occurring before the onset of diuresis. The mechanisms of the vascular actions are not fully understood but include reduced responsiveness to angiotensin II and noradrenaline, both vasoconstrictors.
Loop diuretics are indicated for the treatment of oedema associated with heart failure, cirrhosis, renal impairment and nephrotic syndrome. In addition, these agents are used as adjunct therapy in patients with acute pulmonary oedema, in people whose conditions are refractory to the other diuretics and, as they promote excretion of calcium, they are used in people with severe hypercalcaemia. These drugs are often included in multiple drug regimens and are subject to numerous drug interactions (see Drug Interactions 25-1).
Loop diuretics should be used with caution in people with diabetes mellitus, gout, hearing impairment, hepatic and renal impairment and in those in whom hypokalaemia might precipitate arrhythmias, such as people taking digoxin. These drugs should be avoided in people with known hypersensitivity to loop diuretics, anuria or severe kidney disease or significant renal impairment. As they are ADEC category C drugs, their use in pregnancy should be avoided. Refer to Clinical Interest Box 25-3 for a discussion of bone loss associated with loop diuretics.
Clinical interest Box 25-3 Loop diuretics and bone loss in men
The use of loop diuretics is common in older adults and as these drugs increase the urinary excretion of calcium loss of bone is not unexpected. Bone loss studies are principally associated with post-menopausal women but there have been studies that have identified a reduction in bone mineral density in women using diuretics. Frusemide is commonly prescribed to men but in general there are a limited number of studies that have investigated bone loss in older males.
A recent study over 4.6 years of 3269 men aged 65 years and older identified 84 men as chronic users of loop diuretics, 181 men as intermittent users and 3004 men as non-users of loop diuretics. The authors concluded that intermittent loop diuretic use was associated with a 2-fold greater rate of loss of hip bone and continuous use with a 2.5-fold greater rate of loss compared with nonusers of diuretics. This study highlights the importance of taking into account the use of loop diuretics when evaluating older men for fracture risk (Lim et al 2008).
Thiazide diuretics
The thiazide diuretics were synthesised during the 1950s and were the first real challengers to the use of mercurial diuretics. The current drugs are chemically related to the sulfonamides and include chlorthalidone, hydrochlorothiazide (Drug Monograph 25-2) and the thiazidelike drug indapamide.
Mechanism of action
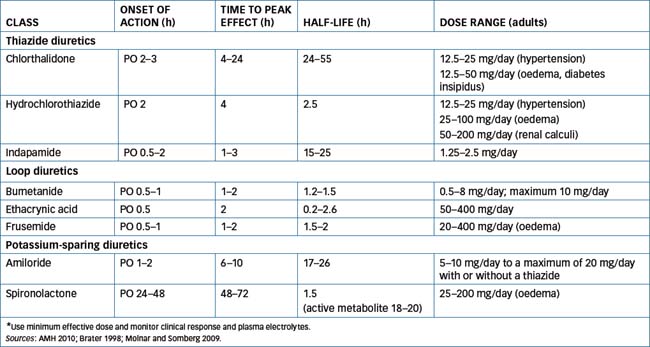
These drugs inhibit reabsorption of sodium and chloride in the proximal (diluting) segment of the distal convoluted tubule (Figure 25-3) by binding to the chloride-binding site of the Na+–Cl− symporter. This symporter is in the luminal membrane and, using the free energy in the electrochemical gradient of sodium, the Na+–Cl− symporter moves chloride into the epithelial cell against its electrochemical gradient. Inhibition of the Na+–Cl− symporter increases the excretion of sodium and chloride. However, because the maximum portion of the sodium load they can affect at the distal tubule is ∼5%, thiazides are considered only moderately potent diuretics in comparison with the loop diuretics. Like the loop diuretics inhibitors of the Na+–Cl− symporter also increase potassium excretion by the same mechanism discussed for frusemide. In general, thiazides are well absorbed orally and are usually excreted unchanged by the kidneys. The onset of action is usually within 12 hours, but the duration of action differs between the drugs. For pharmacokinetics and dosages, see Table 25-1.
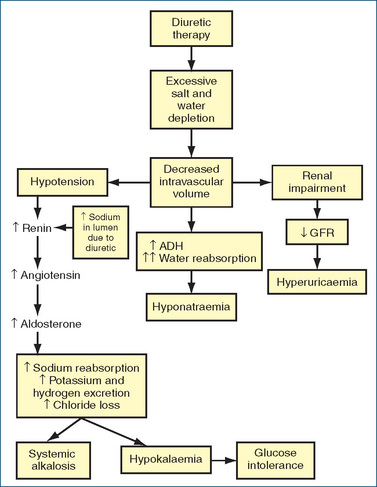
The thiazide diuretics promote the renal excretion of water, sodium, chloride, potassium and magnesium, whereas excretion of uric acid and calcium is decreased. When an increased sodium load is presented to the distal tubule, there is a corresponding increase in potassium secretion. In addition, as the extracellular fluid volume decreases, plasma renin activity and aldosterone levels increase, with resulting potassium loss (Figure 25-4). Potassium is one of the most common electrolytes lost, with loss occurring in 14%–60% of ambulatory hyper tensive patients. This loss is dose-related, occurring early in treatment (first month) and more frequently with larger diuretic doses or with the long-acting type of diuretics (e.g. chlorthalidone). Potassium loss can be a serious issue in people who are taking digitalis preparations, as it can precipitate serious arrhythmias as a result of digitalis toxicity. Hypokalaemia may also predispose people with cirrhosis to hepatic encephalopathy or coma. Potassium loss can be minimised by using the lowest possible dose of thiazide, by adding a potassium-sparing diuretic and, if necessary, using potassium supplements. Potassium replacement can be dangerous in the elderly, in patients with renal dysfunction or when used in combination with potassium-sparing diuretics, because high plasma potassium levels may occur. Dietary modification to include potassium-rich foods may in some circumstances be recommended by practitioners. A cup of dried fruit (e.g. apricots, figs, peaches, pears, prunes or raisins) provides approximately 1000 mg potassium.
Indications
The indications for the thiazide diuretics include the treatment of mild to moderate hypertension, oedema associated with heart failure or cirrhosis with ascites, the treatment of nephrogenic diabetes insipidus and the prevention of renal calculi formation. Although the initial diuresis produces a fall in blood pressure because of decreased blood volume, during chronic therapy a reduction in total peripheral resistance via an action on blood vessels appears to explain the continued antihypertensive effect.
Adverse reactions and drug interactions
Patients receiving thiazide diuretics may have an increase in plasma uric acid. This increase is persistent and may result from inhibition of tubular secretion of uric acid resulting from competition for the organic acid secretory pump in the proximal tubule or increased uric acid reabsorption. This effect is reversible when the drugs are discontinued. In the absence of gout, the hyperuricaemia is usually asymptomatic and requires no treatment; however, in a person with a history of gout, higher doses of thiazides can precipitate an attack that requires treatment (see Chapter 47).
Hyperglycaemia, or impaired glucose tolerance, has been reported with the thiazides and, rarely, with loop diuretics. This effect is reported most often in the elderly, and thiazides can unmask latent diabetes. The mechanism of the thiazide-induced hyperglycaemia is not known but may involve reduced insulin secretion and alterations in glucose metabolism. With use of low doses, effects on glucose tolerance are minimal (AMH 2010).
In higher doses the thiazide diuretics have been reported to increase plasma levels of LDL cholesterol, total cholesterol and triglycerides, and to reduce HDL cholesterol. The clinical relevance of changes in an individual’s lipid profile would need to be considered in the context of the overall health status of the person concerned.
In addition to electrolyte imbalances, common adverse reactions include dizziness, weakness, muscle cramps and hypotension. Infrequently rash, blurred vision and male impotence have been reported and, rarely, diarrhoea, photosensitivity, agranulocytosis, cholecystitis, jaundice, and haemolytic anaemia and thrombocytopenia.
Similar to loop diuretics, thiazides are also subject to a number of drug interactions (see Drug Interactions 25-2).
Warnings and contraindications
Thiazide diuretics should be used with caution in people with type 1 diabetes, gout, renal or hepatic impairment or dyslipidaemias, and in the elderly (see Clinical Interest Box 25-4). These drugs are contraindicated in severe renal impairment, anuria and Addison’s disease, and in people with known thiazide or sulfonamide hypersensitivity (refer to Chapter 44).
Clinical interest Box 25-4 Diuretics and the elderly
As kidneys age, their ability to concentrate and acidify urine and to retain potassium and sodium diminishes. Hence the elderly are more sensitive to diuretic-induced hypotension and electrolyte disturbances than younger adults. Diuretics are often referred to as ‘water pills’, and many people believe fluid intake should be restricted with this drug category. Fluid intake should be discussed with the individual.
For the elderly patient, start with the lowest dose possible, and titrate slowly to achieve the desired effect. Avoid or use extreme caution and close monitoring if concurrent potassium supplementation or a potassium chloride salt substitute is ordered for people receiving a potassiumsparing diuretic. Hyperkalaemia and death have been reported with this combination.
Be aware that diuretics can increase urinary incontinence and be alert to signs and symptoms of diuretic toxicity, such as anorexia, nausea, vomiting, confusion, increased weakness and paraesthesia of the extremities. When a diuretic is to be discontinued, reduce the drug gradually to avoid the development of fluid retention and oedema.
Diuretic combinations
A number of diuretic combination products are available, which are generally used in people whose hypertension is not controlled adequately by a single drug. Fixeddose combinations can provide additional diuretic activity and decrease the potassium depletion characteristic of the thiazide diuretics (e.g. triamterene 50 mg plus hydrochlorothia zide 25 mg, or amiloride 5 mg plus hydrochlorothiazide 50 mg). Additionally, combinations of hydrochlorothiazide with either an ACE inhibitor or an angiotensin-receptor antagonist are available (Table 25-2).
Table 25-2 Diuretic combinations
| COMBINATION | DRUGS/DOSES |
| Combination diuretics | Amiloride (5 mg)/hydrochlorothiazide (50 mg) |
| Triamterene (50 mg)/hydrochlorothiazide (25 mg) | |
| ACE inhibitor/diuretic combination | Enalapril (20 mg)/hydrochlorothiazide (6 mg) |
| Fosinopril (10 or 20 mg)/hydrochlorothiazide (12.5 mg) | |
| Perindopril (4 mg)/indapamide (1.25 mg) | |
| Quinapril (10 or 20 mg)/hydrochlorothiazide (12.5 mg) | |
| Angiotensin-receptor antagonist /diuretic combination | Candesartan (16 mg)/hydrochlorothiazide (12.5 mg) |
| Eprosartan (600 mg)/hydrochlorothiazide (12.5 mg) | |
| Irbesartan (150 or 300 mg)/hydrochlorothiazide (12.5 or 25 mg) | |
| Olmesartan (20 or 40 mg/hydrochlorothiazide (12.5 or 25 mg) | |
| Telmisartan (40 or 80 mg)/hydrochlorothiazide (12.5 mg) | |
| Valsartan (80 or 160 mg)/hydrochlorothiazide (12.5 mg) |
Potassium-sparing diuretics
The potassium-sparing diuretics are amiloride, triamterene and the aldosterone antagonist spironolactone. Amiloride is also available in combination with hydrochlorothiazide, and triamterene is available only as a combination with hydrochlorothiazide. All three are considered to have limited diuretic efficacy and are primarily considered useful when combined with potassium-depleting diuretics such as the thiazides. In addition to limited diuretic effects spironolactone is also a mineralocorticoid receptor antagonist. There is clinical evidence that low-dose spironolactone prolongs survival in some patients with severe heart failure by blocking the actions of aldosterone.
Amiloride and triamterene have similar mechanisms of action. They act on the late distal tubules and collecting ducts where they inhibit the reabsorption of sodium by blocking sodium channels in the luminal membrane. Blockade of sodium channels hyperpolarises the luminal membrane and the consequential reduction in the lumen-negative potential leads to a decrease in the excretion of potassium. Spironolactone, a synthetic steroidal compound, is a specific antagonist for the mineralocorticoid receptor and blocks the action of aldosterone (see Chapter 35). This action results in inhibition of the sodium-retaining property of aldosterone and a concomitant reduction in its potassium-secreting property. The effectiveness of spironolactone is directly related to the circulating plasma concentration of aldosterone: if the concentration is high, the effect of spironolactone is greater. It does not interfere with renal tubule transport of sodium and chloride, and does not inhibit carbonic anhydrase. When used alone, all of these drugs have the potential to cause life-threatening hyperkalaemia.
Amiloride has poor oral absorption (15%–25%), whereas triamterene and spironolactone are moderately well absorbed from the gastrointestinal tract (30%–70%). Amiloride is principally excreted as unchanged drug in urine while triamterene is partly metabolised in the liver and also partly excreted unchanged. Spironolactone is extensively metabolised to the active metabolite canrenone, which has a plasma half-life of 18–20 hours. The actions of spironolactone are largely attributable to canrenone. For pharmacokinetic and dosage information, see Table 25-1.
The potassium-sparing diuretics are indicated for the prevention and treatment of diuretic-induced hypokalaemia. They are also used as adjunct therapy in the treatment of oedema due to heart failure and hepatic cirrhosis. Spironolactone is used for the treatment of primary hyperaldosteronism, hirsutism in females, for refractory oedema associated with secondary hyperaldosteronism and severe heart failure.
Refer to Drug Interactions 25-3 for interactions with potassium-sparing diuretics. Common adverse reactions include electrolyte disturbances, particularly hyperkalaemia, hyponatraemia and hypochloraemia (worsened by the combination with hydrochlorothiazide), nausea, vomiting, dizziness, constipation, impotence and headache. As spironolactone is structurally similar to progesterone it binds to progesterone and androgen receptors and hence its use for prolonged periods or at high dose is associated with endocrine adverse effects. These endocrine adverse effects, which include gynaecomastia, decreased libido, impotence and menstrual irregularities, tend to limit the usefulness of spironolactone.
Potassium-sparing diuretics are contraindicated in situations of pre-existing hyperkalaemia (potassium >5 mmol/L) and renal failure. Caution should also be exercised in people with type 1 diabetes, renal or hepatic impairment and debilitating cardiopulmonary disease, and in the elderly, who are prone to hyperkalaemia and hypotension. Both amiloride and spironolactone should be avoided in pregnant women; amiloride can cause electrolyte disturbances in the fetus (ADEC Category C) and spironolactone can cause feminisation of the male fetus (ADEC Category B3).
Osmotic diuretics
Osmotic diuretics such as mannitol are pharmacologically inactive but cause diuresis by adding to the solutes already present in the tubular fluid; they are particularly effective in increasing osmotic pressure because they are not reabsorbed by the tubules. Passive water reabsorption is reduced in their presence; as more fluid remains in the lumen, this alters the electrochemical gradients so less sodium and chloride are reabsorbed in the proximal tubule. Urine volume increases but there is only a small increase in sodium excretion. The availability of other highly effective diuretics has resulted in relegation of these agents for use in non-diuretic indications such as cerebral oedema, reducing intraocular pressure before and after intraocular surgery and for acute closed-angle glaucoma (see Chapter 31).
For general information on signs and symptoms of fluid and electrolyte imbalances resulting from diuretic use, refer to Table 25-3.
Table 25-3 signs and symptoms of fluid and electrolyte imbalances associated with diuretic therapy*
| Hypovolaemia | Hypotension, weak pulse, tachycardia, clammy skin, rapid respirations and reduced urinary output |
| Hyponatraemia | Low sodium concentration (reference range 135–145 mmol/L*), lethargy, disorientation, muscle tenseness, seizures and coma |
| Hypokalaemia | Low serum potassium concentration (reference range 3.8–4.9 mmol/L*), weakness, abnormal ECG, postural hypotension and flaccid paralysis |
| Hypocalcaemia | Low total plasma calcium concentration (reference range 2.1–2.6 mmol/L*), irritability, vomiting, diarrhoea, twitching, hyperactive reflexes, cardiac dysrhythmias, tetany and seizures |
| Hypochloraemia | Low plasma chloride concentration (reference range 95–110 mmol/L*) |
| Hypomagnesaemia | Low plasma magnesium concentration (reference range 0.8–1.0 mmol/L*), nausea and vomiting, lethargy, muscle weakness, tremors and tetany |
| With potassium-sparing diuretics, be alert for: | |
| Hyperkalaemia | Above the upper limit of the reference range for plasma potassium; nausea, diarrhoea, muscle weakness, postural hypotension and ECG changes |
* Consult local/regional laboratories for the equivalent reference ranges.
Drugs for urinary incontinence
Once formed, urine is carried from the kidneys by the ureters to the urinary bladder, a hollow muscular organ the shape of which is determined by the volume of urine contained at the time. The ureters enter the bladder through the detrusor muscle in the floor of the bladder (the trigone area). The normal tone of the detrusor muscle prevents backflow of urine from the bladder to the ureters. The urethra exits from the bladder at the tip of the trigone, with the detrusor muscle forming the internal sphincter, and passes through the floor of the pelvis (Figure 25-1). In this region, the outer wall of the urethra contains a circular muscle band that forms the external urethral sphincter, which is under voluntary control and prevents urination until socially acceptable circumstances are achieved.
The micturition reflex
The storage of urine and the emptying of the bladder involve complex neural integration between the central nervous system, the spinal cord and peripheral nerves. The bladder has somatic, parasympathetic and sympathetic innervation. Sympathetic innervation via the release of noradrenaline acts on β3 receptors on the detrusor muscle, mediating smooth muscle relaxation and increasing bladder compliance. Stimulation of α1 adrenoceptors in the bladder neck and proximal urethra mediates smooth muscle contraction and increases bladder outlet resistance. The average capacity of the urinary bladder is about 500 mL in an adult. Volume expansion increases tension in the wall of the bladder, triggering stretch receptors in the detrusor muscle and the transmission of sensory impulses by parasympathetic afferent fibres. Reflex parasympathetic discharge via motor efferent fibres releases acetylcholine that acts on M3 muscarinic receptors causing contraction of the detrusor muscle and relaxation (opening) of the internal urethral sphincter. This reflex arc initiates a conscious desire to urinate and, when impulses from the cerebral cortex of the brain inhibit activity in motor neurons to the external sphincter, voluntary relaxation occurs and the bladder contents are expelled. When the bladder is empty the nerve signals reverse and the bladder is able to fill with urine again.
Micturition may be initiated and stopped voluntarily because of control exerted at the level of the cerebral cortex. The specific mechanisms within the CNS are not fully understood but may include neurotransmitters such as dopamine, serotonin and endorphins. A lack of voluntary control is referred to as incontinence, while failure to either completely or normally urinate may lead to urine retention. In children wetting the bed or nocturnal enuresis is of concern if it persists beyond the age when control of micturition is normally achieved. Following exclusion of structural or organic causes, desmopressin (refer to Chapter 33) may be used to treat nocturnal enuresis.
Urinary incontinence is a common and embarrassing problem that afflicts in particular the elderly population. Before instituting drug treatment, potential contributing factors should be eliminated. These include the possibility of a urinary tract infection, excessive fluid intake, high caffeine consumption, metabolic disorders (e.g. hyperglycaemia) and the administration of certain drugs (see Table 25-4). Incontinence can be categorised into a number of types including:
Table 25-4 Drug therapy that may contribute to urinary incontinence
| DRUG CLASS | MECHANISM | CONSEQUENCE |
| α-Adrenoceptor antagonists | Decreased urethral pressure | Stress incontinence |
| Anticholinergics | Incomplete bladder emptying | Overflow incontinence |
| Antidepressants | Detrusor overactivity | Urge incontinence |
| Antiparkinsonism agents | Incomplete bladder emptying | Overflow incontinence |
| Antipsychotics | Decreased urethral pressure | Stress incontinence |
| β-Adrenoceptor antagonists | Incomplete bladder emptying | Overflow incontinence |
| Benzodiazepines | Decreased urethral pressure | Stress incontinence |
| Diuretics | Excessive urine production | Urge incontinence |
| Hormone replacement | Detrusor overactivity | Urge incontinence |
Adapted from: Tsakiris et al 2008.
Clinical interest Box 25-5 Stress incontinence
Urinary incontinence is not normal in adults. Reports of incontinence vary from simply reporting a sensation of urgency (urge incontinence), to complaints of leakage of urine when laughing, coughing, sneezing, exercising etc (stress incontinence), to overflow incontinence (when the bladder fails to empty completely, often as a result of an obstruction) and functional incontinence arising as a result of either the inability to recognise the need to urinate (e.g. as a result of loss of memory) or a physical inability to get to the toilet. However, for younger women stress urinary incontinence (SUI) is frequently associated with pregnancy and childbirth. The majority of women with SUI may be managed conservatively, particularly when considering the ADEC category of risk in pregnancy for many of the drugs used to treat this condition. A comprehensive medical history is essential to exclude confounding factors (e.g. constipation, urinary tract infection, history of voiding difficulty, diabetes mellitus etc) and a gynaecological history including gravity, parity, number of vaginal deliveries and vaginal/ bladder surgery etc. Conservative management includes pelvic floor exercises, biofeedback, vaginal cones and pessaries. Restricting fluid intake to 1.5–2 L per day and reducing caffeine and alcohol intake are also recommended. As pelvic floor exercises take time to produce results, encouragement and support is critical to ensure adherence to the exercise regimen. When conservative or behavioural management alone is insufficient pharmacological therapy may be necessary (Herbruck 2008).
Frusemide, a sulfamoylbenzoic acid, is a commonly prescribed loop diuretic. The degree of diuresis is dependent on the amount of drug reaching the tubular lumen, not the plasma concentration, as frusemide is active from inside the lumen of the thick ascending limb of the loop of Henle (TALH). Hence, adequate urine concentration of frusemide via glomerular filtration and active secretion is essential for maximal diuresis. Frusemide is actively secreted by the renal organic anion transporter OAT3 and to a lesser extent by OAT1.
MECHANISM OF ACTION There are two isoforms of NKCC in mammalian kidney. NKCC1 is predominantly a ‘secretory’ transporter while NKCC2 is an ‘absorptive’ transporter specific to the luminal membrane of the TALH. The affinity of loop diuretics is greater for NKCC2 than NKCC1. In the TALH the movement of sodium, potassium and chloride from the lumen into the epithelial cells of the TALH is driven by NKCC2. Translocation of these ions depends on their simultaneous binding to all three ion-binding sites on the luminal side of the membrane. Frusemide inhibits NKCC2, blocking its function and virtually halting transport of sodium, potassium and chloride in the TALH. The mechanism of inhibition has not been fully elucidated but it is thought that frusemide binds to the chloride binding site of NKCC2. Inhibition of NKCC2 also alters the transepithelial electrochemical gradient between the luminal and the basolateral membranes of the epithelial cells. This change in potential difference reduces the driving force for the reabsorption of calcium and magnesium. Hence, frusemide increases the urinary excretion of sodium, chloride, potassium, calcium and magnesium. Acutely it enhances excretion of uric acid but chronic administration reduces uric acid excretion. This may be explained by increased uric acid reabsorption due to volume depletion or competition between uric acid and frusemide for active secretion in the proximal tubule.
Prolonged use of frusemide can lead to ‘loop diuretic resistance’. The underlying mechanism has not been fully established but may involve rebound sodium retention resulting from significant reabsorption of sodium in the distal nephron. Whatever the mechanism, diuretic resistance is thought to be a protective mechanism against profound sodium and intravascular volume depletion (Asare 2009). Strategies to deal with this problem include fluid and salt restriction, use of frusemide IV, increasing the dose and use in combination with a thiazide diuretic that blocks sodium reabsorption in the distal tubule.
PHARMACOKINETICS Frusemide is highly protein-bound (>95%) and around 50% of a dose of frusemide is excreted unchanged in urine; the remaining 50% is conjugated with glucuronic acid in the kidney. The oral bioavailability ranges from 10% to 100% (average about 50%) and the elimination half-life in normal subjects is 1.5–2 hours (Brater 1998). The peak effect occurs within 30 minutes when given intravenously and in approximately 1 hour following oral administration.
DRUG INTERACTIONS See comprehensive listing in Drug Interactions 25-1, which is relevant to all the loop diuretics.
ADVERSE REACTIONS The most common adverse reactions are electrolyte disturbances, including hyponatraemia, hypokalaemia, hypomagnesaemia and hyperuricaemia, and dizziness and postural hypotension (Figure 25–4). Increases in low-density lipoprotein (LDL) cholesterol and triglycerides with a fall in high-density lipoprotein (HDL) cholesterol plasma levels have been reported. High intravenous doses increase the risk of ototoxicity (e.g. tinnitus, vertigo and deafness). This risk is further increased if frusemide in used in combination with other drugs that also cause ototoxicity e.g. aminoglycosides. In addition, frusemide has been implicated in the ‘triple whammy’ (refer to Clinical Interest Box 23-4).
WARNINGS AND CONTRAINDICATIONS Frusemide is contraindicated in states of severe sodium and fluid depletion and where there is an existing history of allergy to frusemide and sulfonamides.
DOSAGE AND ADMINISTRATION This varies according to the condition being treated, and current information sources should be consulted.
Drug monograph 25-2 Hydrochlorothiazide
Hydrochlorothiazide is a diuretic used in the treatment of mild to moderate hypertension and oedema associated with hepatic cirrhosis or heart failure.
PHARMACOKINETICS Hydrochlorothiazide is absorbed after oral administration (about 70%) and the maximum plasma concentration occurs 2–4 hours after dosing. The plasma half-life is in the order of 2.5 hours. Hydrochlorothiazide is not hepatically metabolised and is excreted almost entirely (>95%) as unchanged drug in urine.
DRUG INTERACTIONS See comprehensive listing in Drug Interactions 25-2.
ADVERSE REACTIONS Common adverse reactions include dizziness, hypotension and electrolyte disturbances (hyponatraemia, hypokalaemia, hyperuricaemia and hypomagnesaemia). More serious reactions include intrahepatic cholestatic jaundice and a variety of haematological effects (agranulocytosis, aplastic anaemia and thrombocytopenia).
WARNINGS AND CONTRAINDICATIONS Hydrochlorothiazide is contraindicated in anuria and in people with known hypersensitivity to sulfonamides. The drug should be used with extreme caution in people with renal disease or cirrhosis.
DOSAGE AND ADMINISTRATION This varies according to the condition being treated, and current information sources should be consulted. Hydrochlorothiazide may be administered as a single agent or in combination with a potassium-sparing diuretic (amiloride or triamterene), an ACE inhibitor (fosinopril, enalapril or quinapril) or an angiotensin-receptor antagonist (candesartan, eprosartan, irbesartan, olmesartan, telmisartan or valsartan).
Sources: Dollery 1991; AMH 2010.
Drug interactions 25-1 Loop diuretics
| Drug | Possible effects and management |
| Amphotericin | Potentiates hypokalaemic effect of frusemide; monitor serum potassium level and use supplement if indicated |
| Angiotensin-receptor antagonists | Loop diuretics increase risk of severe first-dose hypotension. Begin with low dose of angiotensin receptor antagonist and withhold loop diuretic for at least 1 day if possible |
| Angiotensin-converting enzyme (ACE) inhibitors | In people on high-dose loop diuretics, increased risk of severe first-dose hypotension. Begin with low dose of ACE inhibitor and withhold loop diuretic for at least 1 day if possible |
| Aminoglycosides | Increased risk of ototoxicity and renal toxicity. Care required in dosing people with renal impairment.(krithiga)Combination with frusemide is not recommended |
| Cisplatin | Increased risk of nephrotoxicity in combination with frusemide |
| Digoxin | Increased risk of digoxin-induced arrhythmia in people with diuretic-induced hypokalaemia and hypomagnesaemia. Monitor serum potassium concentration and use supplement if indicated |
| Lithium | Increased risk of lithium toxicity because of reduced renal clearance. Monitor closely and adjust lithium dose if necessary |
| NSAIDs | Reduce the effect of loop diuretics; predispose to renal failure in presence of pre-existing hypovolaemia.(krithiga)Monitor BP and renal function |
| Sucralfate | Reduces the absorption of frusemide; avoid administration within 2 hours of each other |
| Thiazide diuretics | Combination with loop diuretics may cause profound diuresis and electrolyte disturbances. Monitor BP, renal function and electrolytes |
Drug interactions 25-2 Thiazide diuretics
| Drug | Possible effects and management |
| ACE inhibitors | Increased risk of severe first-dose hypotension. Commence therapy with a low dose of ACE inhibitor. Combination may increase risk of ACE inhibitor-induced renal impairment. Monitor renal function |
| Angiotensinreceptor antagonists | Thiazide diuretics increase risk of severe first-dose hypotension. Begin with low dose of angiotensin-receptor antagonist and withhold thiazide diuretic for at least 1 day if possible |
| Cholestyramine and colestipol | Concurrent administration can decrease gastrointestinal absorption of thiazide diuretics. Schedule administration of diuretics at least 1 hour before or 4–6 hours after administration of these drugs |
| Digitalis glycosides | Increases risk of digitalis toxicity in presence of hypokalaemia. Monitor serum potassium and ECG changes |
| Lithium | Increased risk of lithium toxicity because of decreased lithium excretion. Monitor plasma lithium concentration and adjust lithium dose if necessary |
| NSAIDs | Decreased natriuresis and reduced antihypertensive effect. In view of increased potential for nephrotoxicity, avoid concurrent use or adjust dose of diuretic |
Drug interactions 25-3 Potassium-sparing diuretics
| Drug | Possible effects and management |
| ACE inhibitors, angiotensin-receptor antagonists, potassium supplements | Increased risk of hyperkalaemia.(krithiga)Avoid combined use |
| Cyclosporin, NSAIDs | Increased risk of hyperkalaemia with both and increased risk of renal failure with NSAIDs. Use with caution and monitor serum potassium concentration |
| Digoxin | Spironolactone increases risk of digoxin toxicity. Monitor digoxin concentration and reduce dose if necessary |
| Lithium | Concurrent use increases the risk of lithium toxicity by reducing renal clearance. Monitor plasma lithium concentration |
Anticholinergics
Acetylcholine is the neurotransmitter that controls the detrusor muscle. Overactivity or spontaneous contraction of the detrusor muscle that leads to urge incontinence can be controlled by drugs that block the action of acetylcholine (muscarinic receptor antagonists) on the detrusor M3 receptor. This reduces contractility of the bladder muscle, which leads to an increase in bladder capacity. Drugs in this class include darifenacin, oxybutynin, propantheline, solifenacin and tolterodine. Oxybutynin is generally considered the first-line drug because of its safety, efficacy and tolerability (Kuteesa & Moore 2006). Imipramine, a tricyclic antidepressant (refer to Chapter 18), has significant anticholinergic effects but it also stimulates β3 receptors on the detrusor causing relaxation. As imipramine causes drowsiness it tends to be used for the treatment of nocturia or nocturnal enuresis.
Darifenacin and solifenacin are newer drugs with high affinity for the M3 receptor. This greater receptor selectivity tends to lessen impairment of cognitive and cardiac function. Although superior to placebo these drugs have adverse effects due to muscarinic receptor blockade and in several studies have been shown to be less well tolerated than tolterodine. Both drugs are well absorbed (90%–98%) and extensively metabolised in the liver by CYP3A4 (solifenacin) and CYP2D6/CYP3A4 (darifenacin) and excreted in urine (60%–70%) and faeces (20%–40%). Unchanged drug in urine accounts for only 3%–10% of the dose. Due to the involvement of CYP3A4 darifenacin and solifenacin are subject to a significant number of drug interactions involving inhibitors of CYP3A4, e.g. ketoconazole and itraconazole, which may inhibit the metabolism of both drugs. The dose of either darifenacin or solifenacin is reduced when administered with potent inhibitors of CYP3A4. The combination of darifenacin and imipramine (a CYP3A4 substrate) should be avoided as darifenacin may increase the concentration of imipramine and the active metabolite, desipramine, leading to the increased risk of adverse effects. Similarly, tolterodine is metabolised by CYP3A4 and CYP2D6 and the same precautions regarding dose reduction apply when administered concomitantly with inhibitors of CYP3A4, e.g. ketoconazole.
In addition to metabolic drug interactions the main synergistic interactions are with other drugs that have anticholinergic properties. These include tricyclic antidepressants, antihistamines, phenothiazines and butyrophenones. Common adverse reactions related to muscarinic receptor blockade include dry mouth, blurred vision, mydriasis, constipation, urinary hesitancy, orthostatic hypotension and tachycardia. These drugs should not be used in people with narrow-angle glaucoma, partial or complete gastrointestinal tract obstruction, severe colitis, urinary obstruction, myasthenia gravis or unstable cardiac rhythms. Caution should be exercised in patients with hepatic impairment if considering use of either darifenacin or solifenacin.
Key points


 The kidneys are essential for maintaining homeostasis and comprise the outer cortex, the inner medulla and the central hollow pelvis. The kidneys process salts and water, balance excretion, regulate the pH of blood via excretion of hydrogen ions, excrete metabolic waste products such as urea and creatinine, modulate blood pressure via release of renin and synthesise calcitriol and the growth factor erythropoietin. The functional unit of the kidney is the nephron, which consists of the glomerulus, the renal tubule and the collecting duct. The three major renal processes are glomerular filtration, tubular reabsorption and tubular secretion. The glomerular membrane filters water, ions, glucose, amino acids and urea. Plasma proteins are not filtered. Glomerular filtration rate (GFR) is 125 mL/min; about 180 L of filtrate are formed per day in healthy individuals. About 99% of the filtrate is reabsorbed and differential reabsorption of water and ions occurs along the length of the renal tubule. Reabsorbed substances include glucose, amino acids, water and bicarbonate, sodium, potassium and chloride ions. Tubular secretion involves movement of substances such as ammonium ions, creatinine and certain drugs from the blood into the lumen of the nephron. Antidiuretic hormone (ADH) and aldosterone regulate salt and water reabsorption in the distal convoluted tubule and collecting duct. Diuretics are important drugs used for the treatment of hypertension and for other conditions in which fluid volume excess is a problem, such as congestive heart failure, cirrhosis and nephrotic syndrome. Diuretics modify renal function and induce diuresis (increased rate of urine flow) and natriuresis (enhanced excretion of sodium chloride). There are three major classes of diuretics: the loop diuretics (e.g. frusemide), the thiazide diuretics (e.g. hydrochlorothiazide) and the potassium-sparing diuretics (e.g. amiloride). The loop diuretics are potent inhibitors of the reabsorption of sodium and chloride in the thick ascending limb of the loop of Henle. Drug interactions with the loop diuretics include ACE inhibitors, angiotensin-receptor antagonists, aminoglycosides, NSAIDs and thiazide diuretics. Thiazide diuretics inhibit absorption of sodium and chloride in the proximal (diluting) segment of the distal convoluted tubule and are considered less potent than the loop diuretics. Thiazide diuretics primarily promote the renal excretion of water, sodium, chloride, potassium and magnesium, whereas excretion of uric acid and calcium is decreased. Hyperglycaemia, or impaired glucose tolerance, has been reported with high-dose thiazide diuretics but rarely with loop diuretics. The potassium-sparing diuretics are amiloride, triamterene and the aldosterone antagonist spironolactone. All three are considered to have limited diuretic efficacy and are primarily considered useful when combined with potassium-depleting diuretics such as the thiazides. Common adverse reactions associated with loop and thiazide diuretics include electrolyte disturbances (e.g. hyponatraemia, hypokalaemia, hypomagnesaemia). Micturition (voiding of urine) can be initiated and stopped voluntarily through conscious control at the level of the cerebral cortex. A lack of voluntary control over micturition is referred to as incontinence.
The kidneys are essential for maintaining homeostasis and comprise the outer cortex, the inner medulla and the central hollow pelvis. The kidneys process salts and water, balance excretion, regulate the pH of blood via excretion of hydrogen ions, excrete metabolic waste products such as urea and creatinine, modulate blood pressure via release of renin and synthesise calcitriol and the growth factor erythropoietin. The functional unit of the kidney is the nephron, which consists of the glomerulus, the renal tubule and the collecting duct. The three major renal processes are glomerular filtration, tubular reabsorption and tubular secretion. The glomerular membrane filters water, ions, glucose, amino acids and urea. Plasma proteins are not filtered. Glomerular filtration rate (GFR) is 125 mL/min; about 180 L of filtrate are formed per day in healthy individuals. About 99% of the filtrate is reabsorbed and differential reabsorption of water and ions occurs along the length of the renal tubule. Reabsorbed substances include glucose, amino acids, water and bicarbonate, sodium, potassium and chloride ions. Tubular secretion involves movement of substances such as ammonium ions, creatinine and certain drugs from the blood into the lumen of the nephron. Antidiuretic hormone (ADH) and aldosterone regulate salt and water reabsorption in the distal convoluted tubule and collecting duct. Diuretics are important drugs used for the treatment of hypertension and for other conditions in which fluid volume excess is a problem, such as congestive heart failure, cirrhosis and nephrotic syndrome. Diuretics modify renal function and induce diuresis (increased rate of urine flow) and natriuresis (enhanced excretion of sodium chloride). There are three major classes of diuretics: the loop diuretics (e.g. frusemide), the thiazide diuretics (e.g. hydrochlorothiazide) and the potassium-sparing diuretics (e.g. amiloride). The loop diuretics are potent inhibitors of the reabsorption of sodium and chloride in the thick ascending limb of the loop of Henle. Drug interactions with the loop diuretics include ACE inhibitors, angiotensin-receptor antagonists, aminoglycosides, NSAIDs and thiazide diuretics. Thiazide diuretics inhibit absorption of sodium and chloride in the proximal (diluting) segment of the distal convoluted tubule and are considered less potent than the loop diuretics. Thiazide diuretics primarily promote the renal excretion of water, sodium, chloride, potassium and magnesium, whereas excretion of uric acid and calcium is decreased. Hyperglycaemia, or impaired glucose tolerance, has been reported with high-dose thiazide diuretics but rarely with loop diuretics. The potassium-sparing diuretics are amiloride, triamterene and the aldosterone antagonist spironolactone. All three are considered to have limited diuretic efficacy and are primarily considered useful when combined with potassium-depleting diuretics such as the thiazides. Common adverse reactions associated with loop and thiazide diuretics include electrolyte disturbances (e.g. hyponatraemia, hypokalaemia, hypomagnesaemia). Micturition (voiding of urine) can be initiated and stopped voluntarily through conscious control at the level of the cerebral cortex. A lack of voluntary control over micturition is referred to as incontinence.Review exercises
Abbott L.M., Kovacic J. The pharmacologic spectrum of furosemide. Journal of Veterinary Emergency and Critical Care. 2008;18:26-39.
Asare K. Management of loop diuretic resistance in the intensive care unit. American Journal of Health-System Pharmacy. 2009;66:1635-1640.
Australian Institute of Health Welfare. Australia’s Health 2005. Canberra: AIHW; 2005.
Australian Medicines Handbook 2010. Adelaide: AMH, 2010.
Brater D.C. Diuretic therapy. New England Journal of Medicine. 1998;339:387-395.
Dollery C., editor. Therapeutic Drugs, Vols1 and 2. Churchill Livingstone, London, 1991.
Greger R. Physiology of sodium transport. American Journal of Medical Science. 2000;319:51-62.
Herbruck L.F. Stress urinary incontinence: an overview of diagnosis and treatment options. Urologic Nursing. 2008;28:186-198.
Hoy W.E. Renal disease in Australian Aboriginals. Medical Journal of Australia. 1996;165:126-127.
Hoy W.E., Mathews J.D., McCredie D.A., et al. The multidimensional nature of renal disease: rates and associations of albuminuria in an Australian Aboriginal community. Kidney International. 1998;54(4):1296-1304.
Kuteesa W., Moore K.H. Anticholinergic drugs for overactive bladder. Australian Prescriber. 2006;29:22-24.
Lim L.S., Fink H.A., Kuskowski M.A., et al. Loop diuretic use and increased rates of hip bone loss in older men. The Osteoporotic Fractures in Men Study. Archives of Internal Medicine. 2008;168:735-740.
Molnar J., Somberg J. The clinical pharmacology of ethacrynic acid. American Journal of Therapeutics. 2009;16:86-92.
Nanra R.S. Analgesic nephropathy in the 1990s: an Australian perspective. Kidney International. 1993;44(Suppl 42):S86-S92.
Rang H.P., Dale M.M., Ritter J.M., Flower R.J. Pharmacology, 6th edn. Edinburgh: Churchill Livingstone; 2007. [ch 24]
Saker B.M. Everyday drug therapies affecting the kidneys. Australian Prescriber. 2000;23:17-19.
Tortora G.J., Grabowski S.R. Principles of Anatomy and Physiology, 9th edn. New York: Harper Collins; 2000. [ch 26]
Tsakiris P., Oelke M., Michel M.C. Drug-induced urinary incontinence. Drugs Aging. 2008;25:541-549.
Vogl A. The discovery of the organic mercurial diuretics. American Heart Journal. 1950;39:881-883.
Caring for Australasians with Renal Impairment: www.cari.org.au
Continence Foundation of Australia: www.continence.org.au
Kidney Health Australia: www.kidney.org.au
Faull R. Prescribing in renal disease: www.australianprescriber.com/magazine/30/1/17/20/