Chapter 20 Drugs for Neurodegenerative Disorders and Headache
This chapter covers drugs used in treating various neurodegenerative disorders, such as Parkinson’s disease, myasthenia gravis, multiple sclerosis and dementias, including Alzheimer’s disease. Drugs with centrally-mediated actions on skeletal muscle are also discussed; these medications are used to treat muscle spasm and spasticity. The actions of the drugs are related to proposed neurotransmitter imbalances, especially of dopamine and acetylcholine in motor function and balance. Drugs used in the treatment of headache are considered, along with the role of 5-hydroxytryptamine (5-HT, serotonin) in the pathogenesis of migraine, and the use of 5-HT agonists and antagonists in treatment and prophylaxis.
Many of these conditions are progressive and incapacitating, and will become more common as our population ages, hence understanding the disease processes and appropriate pharmacological interventions is important.
Key abbreviations
COMT catechol-O-methyltransferase
DDCI dopa decarboxylase inhibitor
Key background: motor nervous system pathologies
The motor nervous system
CENTRAL and peripheral control of motor function and skeletal muscles are discussed in Chapters 13 and 14 (sections on CNS functional systems and neurotransmitters). These areas should be reviewed as background to drugs used in neurodegenerative conditions. Drugs that affect transmission at the neuromuscular junction (NMJ) are used in many different clinical contexts, e.g. as skeletal muscle relaxants during surgical operations, to stimulate acetylcholine receptors in muscle weakness, to relieve spasticity and spasms in skeletal muscle and to treat ocular disorders such as glaucoma. Drugs that have motor effects via actions on central DA receptors are also considered in Chapter 19 (on psychotropic agents). These clinical uses are discussed in the relevant chapters.
Neurodegenerative pathologies
The neurodegenerative disorders include a loose grouping of conditions such as Parkinson’s disease, myasthenia gravis, multiple sclerosis and other movement disorders and the dementias, including Alzheimer’s disease and stroke-related cognitive impairments. The pathological processes occurring in these nervous system dysfunctions are not completely understood, and good animal models of the diseases and specific drug therapies are not always available.
Spasticity of the skeletal muscles can also be debilitating; these muscles are affected by many drugs, with effects at different levels in the CNS, i.e. in the brain or the spinal cord or at the neuromuscular junction. Drugs used in treating migraine and other headaches include specific analgesics and vasodilators, as well as simple non-steroidal anti-inflammatory agents.
Currently there are no cures for these conditions, so drug therapies are the primary methods used to minimise the symptoms. In some of the conditions, novel techniques involving transplantation of neurons and gene therapy are being trialled.
Skeletal muscle spasm and spasticity
Skeletal muscle spasms, or cramps, result when there is an involuntary contraction of a muscle or group of muscles, accompanied by pain or limited function. Most skeletal muscle spasms are caused by local injuries, but some result from low calcium or sodium levels, epileptic myoclonic seizures or disease of the spinal nerves and their roots as a result of degenerative osteoarthritis, herniated discs or spondylosis. Each type of spasm is treated according to its cause. Skeletal muscle injuries and strains are usually selflimiting and can be treated with rest, physiotherapy or immobilisation by use of casts, neck collars, crutches or arm slings. When tissue damage and oedema are present, however, anti-inflammatory drugs may be used.
Spasticity (a form of muscular hypertonicity with increased resistance to stretch) occurs when gamma motor neurons, which tonically control muscle spindle contractile activity, become hyperactive as the result of stroke, closed head injuries, cerebral palsy, multiple sclerosis, spinal cord trauma and other neurological disorders. Spinal spasticity can be identified by a marked loss of inhibitory influences with hyperactive tendon stretch reflexes, clonus (alternate contraction and relaxation of muscles), primitive flexion withdrawal reflexes and a flexed posture. Varying degrees of spasticity of the bladder and bowel can also occur. Cerebral spasticity has less reflex excitability, increased or impaired muscle tone, and no primitive flexion withdrawal reflexes or flexed posture. Spasticity may require long-term use of muscle-relaxing agents.
Drug treatment of movement disorders
Drugs affecting skeletal muscles
In this chapter we discuss drugs affecting central control of skeletal muscle and some drugs that affect skeletal muscle actions in movement disorders with a central component, such as cerebral palsy and multiple sclerosis. These drugs affect central control of motor activity via gamma-aminobutyric acid (GABA) receptors or affect neurotransmission at the NMJ via acetylcholine receptors.
Anticholinesterases
The anticholinesterase agents (cholinesterase inhibitors) enhance cholinergic actions by inhibiting the effect of the cholinesterase enzymes that inactivate acetylcholine at cholinergic nerve terminals (see Figures 11-7 and 13-4; and Drug Monograph 13-3 on neostigmine). This permits the accumulation of acetylcholine and enhanced effects at autonomic ganglia, parasympathetic neuroeffector junctions and neuromuscular junctions. In myasthenia gravis (see Figure 20-3), the increased amount of acetylcholine (ACh) at the NMJ competes more successfully with antibodies against the ACh receptors for receptor binding sites and thus reduces fatigue in muscle. Anticholinesterases that are lipid-soluble and thus cross the blood–brain barrier are used for their central effects on cholinergic transmission, especially in dementias (see later section, and Clinical Interest Box 20-5).
The anticholinesterase agents are generally divided into three groups based on their duration of action, which is determined largely by the type of binding to the enzyme. Of the medium-acting agents, pyridostigmine has better oral bioavailability than neostigmine, a longer half-life and fewer GIT adverse reactions, so it is the first-line drug for myasthenia gravis. An old drug that has come back into use is edrophonium; it may be available in Australia through the Special Access Scheme for use in diagnosis and monitoring of myasthenia gravis.
 Skeletal muscle relaxants
Skeletal muscle relaxants
Centrally acting skeletal muscle relaxants
Centrally acting and directly acting skeletal muscle relaxants are the drugs of choice in treating muscle spasticity and spasms that do not quickly respond to other forms of therapy. These drugs include baclofen, diazepam and dantrolene. They are more effective in the treatment of spinal spasticity than cerebral spasticity; concurrent physiotherapy is always required for optimal treatment. Excessive muscle relaxation can cause the serious adverse reactions of dysphagia (difficulty in swallowing) and choking. The exact mechanism of action of the central skeletal muscle relaxants is not known. The drugs cause CNS depression in the brain (brainstem, thalamus and basal ganglia) and spinal cord that results in relaxation of striated muscle spasm; thus CNS depression accompanies the muscle relaxation. Consequently, these drugs create the adverse reactions of drowsiness, blurred vision, light-headedness, headache and feelings of weakness, lassitude and lethargy that make their long-term use undesirable.
Baclofen and benzodiazepines
The main centrally acting drugs used primarily as antispastic agents are baclofen (see Drug Monograph 20-1) and diazepam (Drug Monograph 16-1), both of which act via enhancing GABA inhibitory transmission. Baclofen is a selective agonist at presynaptic GABAB receptors; by inhibition of adenylyl cyclase it blocks calcium channels and thus has an antispasticity action, inhibiting motor neurons mainly in the spinal cord. A related compound, gamma-hydroxybutyric acid, previously used as an anaesthetic agent, has been subject to abuse as a street drug (see under ‘Hallucinogens’ in Chapter 21).
Tetrabenazine
Tetrabenazine is a centrally acting skeletal muscle relaxant that acts via effects on dopamine pathways. It releases monoamine neurotransmitters and depletes brain DA levels, and thus causes sedation and muscle relaxation. It was formerly used as a neuroleptic agent, but causes parkinsonism, extrapyramidal effects and depression, so is now used only occasionally in treatment of movement disorders.
Peripherally acting skeletal muscle relaxants
Neuromuscular blocking agents are clinically the most important skeletal muscle relaxants; they are mainly used during surgical operations, so are discussed in more detail in Chapter 14, under ‘Adjuncts to anaesthesia: muscle relaxants’. The two groups are:
Botulinum toxin A
The type A toxin from the bacteria Clostridium botulinum has long been known to be poisonous and is implicated in food poisoning in which the anaerobic organism multiplies in poorly preserved or refrigerated food. The toxin blocks release of ACh from cholinergic nerves and thus causes a chemical denervation. It has a permanent toxic effect, decreasing muscle tone and contractility, leading to flaccid paralysis and atrophy of the affected muscles. It is a protein toxin and extraordinarily potent: it is estimated that less than a 10−12 g (1 picogram) dose will kill a mouse.
These effects have been put to good clinical use in the parenteral administration of the toxin to specific muscle groups undergoing involuntary spasm, e.g. in blepharospasm (uncontrollable winking or sustained tight closure of the eyes due to spasm of the eyelid muscles; see Drug Monograph 31-3), equinus foot deformity or other focal muscle dystonias. Botulinum toxin is also used to paralyse superficial facial muscles to (apparently) reduce wrinkles (see Clinical Interest Box 13-1). The toxin is injected SC to the muscle and relieves muscle spasm for several months until new motor end-plates sprout and reinnervation occurs.
Botulinum toxin A is contraindicated in myasthenia gravis, which it exacerbates, and has adverse interactions with aminoglycoside antibiotics and other drugs that impair ACh release and cause neuromuscular blockade. Adverse reactions include muscle weakness in muscle groups adjacent to the site of injection.
Dantrolene
Dantrolene acts directly on skeletal muscles to produce skeletal muscle relaxation by inhibiting the release of calcium from the sarcoplasmic reticulum to the myoplasm. This results in a decreased muscle response to the action potential and decreased muscle contraction. As an antispastic agent, the direct effect of dantrolene on skeletal muscle dissociates the excitation–contraction coupling. Dantrolene reduces both monosynaptic- and polysynapticinduced muscle contractions (see Drug Monograph 20-2).
Parkinson’s disease
Pathology: dopamine-deficiency
Parkinson’s disease is a progressively debilitating disorder of the basal ganglia, a series of paired nuclei in the cerebral hemispheres including the corpus striatum (caudate nucleus and putamen) and globus pallidus, which regulate the tone and characteristics of all voluntary movements. It is characterised by tremors at rest, bradykinesia (abnormal slowing of all voluntary move ments and speech), forward flexion of the trunk, muscle rigidity, loss of postural reflexes and muscle weakness. It occurs usually between the ages of 50 and 80 years, affecting both sexes equally. The prevalence is about 110 per 100,000 in the Australian population; however, about 1% of the population over the age of 55, and 1.5% of people in the 70–79 age group, have Parkinson’s disease. Although the cause is unknown, genetic factors, viral influences and environmental contaminants have been suspected. Parkinsonism is classified as idiopathic (no known cause), postencephalitic (particularly after viral encephalitis), degenerative (e.g. due to arteriosclerosis) or drug-induced (especially by chronic administration of the antidopamine neuroleptic agents for schizophrenia).
The CNS has five major types of DA receptors (see Table 14-1). The exact roles of some DA receptors are not currently known (although D1 receptor activation is necessary for maximal expression of D2 receptor activity), but D2 and especially D2A receptors are particularly involved with the motor effects of dopamine agonists effective in Parkinson’s disease.
In the biosynthetic pathways for the formation of the catecholamine neurotransmitters (see Figure 12-1), the starting point is the absorption of the essential amino acids phenylalanine and tyrosine from proteins in the diet. These are converted in adrenergic nerves to dopa (dihydroxyphenylalanine), which is rapidly converted to DA by action of the enzyme dopa decarboxylase (DDC, a general aromatic amino acid decarboxylase). If the required enzymes are also present, DA may then be metabolised to noradrenaline and thence to adrenaline.
The signs and symptoms of Parkinson’s disease are caused by a dopamine-deficiency state of the extrapyramidal motor system (see Figure 14-5), particularly in the nigrostriatal tracts. In affected patients, the levels of DA, an inhibitory transmitter, in the basal ganglia fall to as low as 20% of normal levels. This produces DA/ACh imbalance, with a relative increase in ACh (excitatory neurotransmitter). The correct balance of DA and ACh is important in regulating posture, muscle tone and voluntary movement (Figures 14-6 and 20-1). The amounts of other monoamine neurotransmitters (e.g. noradrenaline and 5-hydroxytryptamine [5-HT, serotonin]) and of some other transmitters (e.g. somatostatin, substance P and enkephalins) are also decreased in the brain of a person with Parkinson’s disease. The condition has even been induced by designer drugs (Clinical Interest Box 20-1). Parkinsonian symptoms are present in other disorders, including dementia with Lewy bodies (see later section).
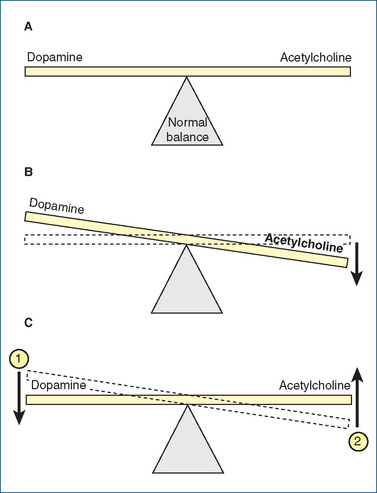
Figure 20-1 Central acetylcholine/dopamine balance. A Normal ‘balance’ of acetylcholine and dopamine. B In Parkinson’s disease, a decrease in dopamine results in an acetylcholine/dopamine imbalance: cholinergic effects outweigh dopaminergic. C Drug therapy for Parkinson’s disease focuses on (1) increasing the dopamine level, which restores the acetylcholine/dopamine balance towards normal by increasing the supply of dopamine or stimulating dopamine receptors, and/or (2) blocking acetylcholine receptors or reducing acetylcholine levels.
Clinical interest Box 20-1 Parkinsonism induced by drugs
Not surprisingly, movement disorders including parkinsonian symptoms can be induced by chronic administration of dopamine receptor antagonists. Thus conventional antischizophrenic (neuroleptic) agents, such as the phenothiazines (chlorpromazine, prochlorperazine), have been implicated in drug-induced parkinsonism; the atypical antipsychotics are less likely to produce extrapyramidal adverse effects.
Other drugs implicated in iatrogenic parkinsonism are the ‘designer drugs’ (i.e. chemical variations of illegal or controlled substances). Such products are usually not yet illegal but are produced to mimic the psychoactive effects of various illegal products. One such drug is 1-methyl–4-phenyl-1,2,3,6- tetrahydropyridine (MPTP), initially a contaminant of a preparation produced as an analogue of pethidine in clandestine laboratories, which has been sold on the streets as heroin, cocaine or a contaminant of other ‘street drugs’. MPTP in some users induces a severe degenerative CNS disorder characterised by tremors and muscle paralysis similar to the symptoms of Parkinson’s disease; in some patients the paralysis has been permanent.
MPTP causes irreversible destruction selectively of the nigrostriatal dopaminergic pathways in various species, and has been used to induce a parkinsonian syndrome in primates, as a useful animal model in which to study the actions of drugs and other methods potentially useful in treating Parkinson’s disease.
Pharmacotherapy
No agents have yet been found that cure the condition or slow its progression. Drug therapy is focused on correcting the DA/Ach imbalance by raising DA levels or blocking ACh effects. The classes of drugs used in treatment include: (1) drugs that raise brain DA levels or stimulate DA receptors to enhance dopaminergic mech anisms; (2) drugs with central anticholinergic activity (anticholinergics and antihistamines); (3) other drugs with varying actions as adjuncts and for symptomatic relief, e.g. anti-inflammatory agents, decongestants, laxatives and antipsychotics.
Drug treatment is usually not started until the symptoms have become disturbing to the patient. Choice of drugs is individualised, depending on which symptoms are most troublesome; severe tremor requires use of anticholinergic agents. Non-drug therapies that have been tried include physiotherapy, surgery to specific tracts in the CNS and transplantation of dopaminergic neuronal tissue from fetal CNS. This is an exciting and busy area of pharmacological research, with a wide range of new molecules under going clinical trials: esterified forms of levodopa, MAO-B inhibitors, DA agonists and reuptake inhibitors, and new formulations of some older drugs. In addition, efforts are being made to treat dyskinesias with drugs acting at various receptors and to halt or reverse disease progression with neuroprotective agents.
Drugs enhancing brain dopamine activity
Three classes of drugs mimic brain DA: those that raise brain levels of DA, those that release DA and directly acting dopaminergic agonists. The drugs of choice in the treatment of Parkinson’s disease are those that raise the brain levels of DA, either by increasing its synthesis or decreasing its metabolism. The other two groups are used as adjuncts or when normal therapy is contraindicated.
Drugs enhancing brain DA have their major effects on the bradykinesia (slowed movements), akinesia (difficulty in initiating or the lack of ability to initiate muscle movement) and rigidity caused in Parkinson’s disease by lowered levels of brain DA. The person with akinesia exhibits a mask-like facial expression, impairment of postural reflexes and eventually an inability for self-care. Dopamine enhancers are less effective in relieving the tremor associated with the condition. Tachyphylaxis (gradual reduction in efficacy) develops for all the DA-enhancing agents, making long-term treatment difficult.
Levodopa: a prodrug
Dopamine itself cannot be given as a drug to ‘top up’ the stores in CNS dopaminergic pathways, as it would be metabolised too rapidly, and would not pass the blood–brain barrier, so its precursor, levodopa, is administered. Levodopa is the first-line treatment for most Parkinson’s patients. A large proportion (99%) of orally-administered levodopa is metabolised in the liver and other cells by the enzyme dopa decarboxylase. Hence only a small proportion (1%) survives to cross the blood–brain barrier and be converted in dopaminergic neurons to DA (see Figure 20-2). For this reason large doses of levodopa used to be given, leading to major peripheral adverse reactions, including constipation, difficult urination, orthostatic hypotension, irregular heart rate and severe nausea or vomiting.
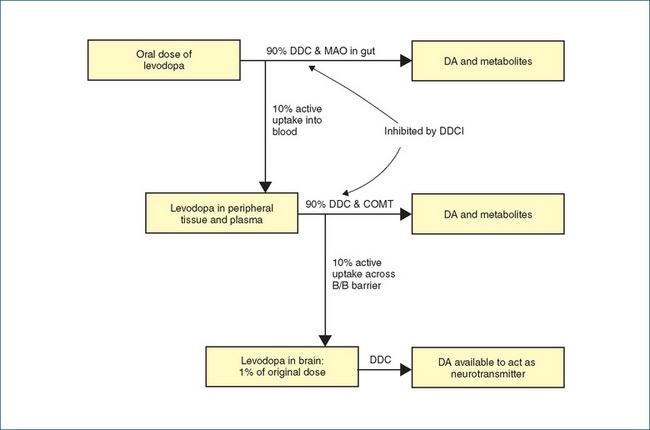
Figure 20-2 Levodopa in Parkinson’s disease. Of the orally administered dose, about 99% is metabolised in the periphery by the enzymes dopa decarboxylase (DDC), monoamine oxidase (MAO) and catechol-O-methyltransferase (COMT), allowing only 1% to cross the blood– brain barrier (B/B barrier) and be converted to dopamine (DA). In the presence of a DDC inhibitor (DDCI), the enzyme in the periphery is inhibited, thus allowing a much greater proportion of administered dose to reach the CNS and be converted to active dopamine.
In a fine piece of pharmacological detective work and synthetic organic chemistry research, compounds were developed that inhibit the DDC enzyme in the peripheral nervous system, thus allowing a greater proportion of the levodopa dose to enter the CNS. Because the DDC inhibitors (DDCI) themselves were designed so as not to pass the blood–brain barrier, the enzyme is not inhibited in the CNS, where DA can still be synthesised from dopa. Thus the stores in the remaining functional CNS dopaminergic pathways are replenished, DA release is facilitated and the remaining functional receptors are ‘flooded’ with DA. The dose of levodopa required when administered together with a DDCI is only 1/5 to 1/4 of that previously needed. The doses are taken orally immediately after meals.
The DDCI drugs used clinically, administered orally in conjunction with levodopa, are carbidopa (see Drug Monograph 20-3) and benserazide; both are structural analogues of DA and competitive inhibitors of the decarboxylase enzyme. So successful has this strategy been in reducing the dose of levodopa required and the peripheral adverse reactions, that levodopa is no longer available in Australia for use without a DDC inhibitor.
Adverse effects occur in both peripheral and central nervous systems (see Drug Monograph 20-3); some tolerance develops to the adverse effects in the GIT. CNS effects are a greater risk with the combination because more levodopa reaches the brain to be converted to DA. The effectiveness of the combination often declines over years of chronic administration, resulting in the ‘on–off syndrome’, with increasing fluctuations in motor control (see Clinical Interest Box 20-2), so levodopa plus DDCI may have only a few years usefulness in a patient. Doses of levodopa and the frequency of administration eventually need to be increased to maintain therapeutic effect, and taking the dose without food can increase the rapidity of onset of action. Controlled-release formulations are available. Due to the many difficulties in long-term management of patients with Parkinson’s disease, specialist neurologists are usually involved in patient care.
Clinical interest Box 20-2 Levodopa ‘on–off’ syndrome
On–off syndrome refers to a complication following prolonged levodopa therapy (2 years or more). The patient fluctuates from being symptom-free (‘on’) to demonstrating full-blown Parkinson’s symptoms (‘off’) during therapy. These effects can last from minutes to hours, and may be due to a decrease in delivery of dopamine centrally, an alteration in sensitivity of the dopamine receptors, a variation in the amount and rate of drug absorption, a dopamine metabolite interference or a combination of effects.
Treatment may require more frequent administration of levodopa or levodopa–DDC inhibitor, and perhaps the addition of a direct-acting dopamine agonist, bromocriptine. Concurrent administration of a COMT inhibitor (entacapone) helps reduce ‘off’ times. After a drug holiday (drug withdrawal) of several days, some people demonstrate an improved response to the drug therapy. This might be because of the re-establishment of dopamine receptor sensitivity to levodopa, which is usually only temporary. Because symptoms can worsen during the drug-free period, this approach should be instituted in a hospital setting.
Dopamine agonists
Adverse effects
Just as DA antagonists (used in schizophrenia) can cause parkinson-like (extrapyramidal) symptoms as adverse effects, so DA agonists used to treat Parkinson’s disease can tilt the DA–ACh ‘see-saw’ in the opposite direction and cause psychosis-like effects. Thus DA agonists can cause GIT disorders, hallucinations and impulse control disorders such as pathological gambling, overspending and hypersexuality; patients and their carers need to be warned of these possibilities.
Ergot derivatives
The ergot alkaloids are derivatives of a fungus, Claviceps purpurea, which grows on damp rye grains and can cause outbreaks of poisoning (ergotism). These compounds are renowned for having different effects on a variety of receptors, and thus can cause intense vasoconstriction, ischaemia and hypertension (α-agonist sympathomi metic effects), or vasodilation and flushing (by blocking α-adrenoceptors), uterine contractions (an oxytocic effect on uterine smooth muscle) and agonistic or antagonistic actions on 5-HT receptors. Ergot derivatives include ergometrine (used as an oxytocic agent: see Drug Monograph 38-4); ergotamine, dihydroergotamine and methysergide (used in migraine); bromocriptine (a central DA agonist, used to inhibit lactation); and lysergic acid diethylamide (LSD, the classic hallucinogenic agent, see Chapter 21 and Figure 21-4).
The ergot derivatives used in Parkinson’s disease are bromocriptine, pergolide and cabergoline. They stimulate central DA receptors and thus improve bradykinesia and rigidity, but are less effective than levodopa. The ergot derivatives, however, used together with a levodopa–DDC inhibitor combination allow lower doses of levodopa, and hence can delay the onset of motor fluctuations (on–off syndrome) and dyskinesias. A peripheral DA antagonist (domperidone, an antiemetic acting on the chemoreceptor trigger zone which is peripheral to the blood–brain barrier) is sometimes given concurrently to minimise peripheral DA effects such as nausea and hypotension (use of a centrally-acting DA antagonist antiemetic such as prochlorperazine or metoclopramide would be counterproductive as it would block the effects of DA produced in the CNS from levodopa).
Pergolide is more potent and longer-acting (for 4–6 hours) than bromocriptine and directly stimulates both D1 and D2 receptors. Many patients who did not respond to levodopa alone have improved with the addition of pergolide.
Drug interactions can be expected with DA antagonists, such as the phenothiazines, thioxanthines, haloperidol and metoclopramide. In addition, drugs that produce hypotension can have an additive hypotensive effect when administered concurrently with ergot alkaloids.
Adverse reactions are similar to those for levodopa, and include effects in the gastrointestinal, cardiovascular and central nervous systems. In addition, ergot derivatives especially cabergoline, pergolide and methysergide (a drug used to prevent migraines) can cause fibrotic effects in pleuropulmonary and retroperitoneal areas; patients on long-term therapy should be monitored for symptoms of fibrotic disorders. The drugs should be used with caution in patients with arrhythmias and psychosis. Because of their DA-agonist actions, these ergot derivatives inhibit lactation, so are contraindicated in breastfeeding women. Cabergoline is used as a lactation inhibitor and to treat hyperprolactinaemia (see Chapter 38).
Other dopaminergic agonists
Apomorphine is a DA agonist not related chemically to ergot derivatives; it is, as its name implies, a morphine derivative but has very little analgesic activity; it is strongly emetogenic (see Clinical Interest Box 20-3).
Clinical interest Box 20-3 Apomorphine, the archetypal emetic agent
Apomorphine is a morphine derivative with a four-ring structure that can be imagined to contain the dopamine (DA) backbone. It acts as a DA analogue and stimulates central DA receptors. Its pharmacology is interesting:
To prevent its powerful emetic actions, the antiemetic domperidone is given prophylactically before apomorphine; domperidone is a DA antagonist that does not cross the blood– brain barrier, so does not antagonise the actions of apomorphine or DA in the CNS. The combination of domperidone (peripheral DA antagonist), apomorphine (central DA agonist), carbidopa (peripheral dopa-decarboxylase inhibitor) plus levodopa (central DA precursor) is a powerful one.
In Parkinson’s disease, apomorphine is used in people severely disabled by motor fluctuations in levodopa response that are non-responsive to other treatment. Adverse reactions are similar to dopaminergic effects of levodopa or the ergot alkaloids, especially vomiting, hypotension and psychiatric disturbances. Apomorphine is contraindicated in many conditions, including cardiovascular diseases, respiratory or CNS depression, dyskinesias and psychiatric disorders. Because of its low therapeutic index and the need to determine an effective dose range during an ‘off’ motor period, the drug is best administered in a hospital setting under specialist supervision. An antiemetic such as domperidone must be given prophylactically before apomorphine.
Pramipexole, a new non-ergot DA agonist, acts at D2 and D3 receptors. When added to levodopa therapy, it has been shown to improve patient’s motor function and reduce ‘off’ time from levodopa. Adverse dopaminergic effects include hallucinations, nausea, insomnia and/or somnolence, and dyskinesia. A similar drug, rotigotine, is formulated as a skin patch; it also improves motor functions and activities of daily living, with similar adverse effects. Both drugs can cause sudden sleepiness and compulsive behaviours.
Drugs raising brain dopamine levels
Amantadine, a dopamine-releasing drug
Amantadine is a synthetic antiviral compound used occasionally to treat influenza. Although its mechanism of action is not completely known, it is postulated that amantadine releases DA and other catecholamines from neuronal storage sites. It also blocks the uptake of DA into presynaptic neurons, thus permitting peripheral and central accumulation of DA. It has useful antimuscarinic activity, and may induce a sense of wellbeing and elevation of mood. It is less effective than levodopa but produces more rapid clinical improvement and causes fewer adverse reactions.
Amantadine is indicated for use as an antidyskinetic agent in treatment of mild Parkinson’s disease, and as an antiviral drug used in treatment of influenza. It is well absorbed orally and is excreted by the kidneys unchanged, so doses need to be reduced in patients with kidney impairment. There are clinically significant drug interactions with dopamine antagonists (which oppose its effects) and other drugs with anticholinergic actions (which are additive).
Adverse reactions are typical anticholinergic (atropinic) effects and effects of DA agonists (GIT, mood and cardiovascular changes). In addition, amantadine can cause unusual purple-red skin spots (livedo reticularis), seen with chronic therapy.
Selegiline, a monoamine oxidase inhibitor
Monoamine oxidase A is relatively specific to metabolism of noradrenaline and 5-HT; monoamine oxidase B metabolises mainly DA. (Reversible inhibitors of MAO-A are used as antidepressants: see moclobemide in Chapter 18.) Selegiline irreversibly inhibits MAO-B, thus preventing the breakdown of DA, and blocks DA reuptake (see Drug Monograph 20-4). As a result it will enhance or prolong the antiparkinson effect of levodopa, which might allow a lowering of the daily dose of levodopa. Another MAO-B inhibitor, rasagiline, has been approved in some countries but is not marketed in Australia.
Entacapone, a catechol-O-methyltransferase inhibitor
The other main enzyme involved in metabolism of the catecholamines, including noradrenaline and DA, is catechol-O-methyltransferase (COMT). Thus a COMT inhibitor, analogous to an MAO inhibitor (MAOI), will also inhibit the inactivation of DA and prolong the clinical response to levodopa, increasing the ‘on’ time for motor response. Entacapone, a reversible and specific COMT inhibitor, is always used with a levodopa–DDC inhibitor combination.
As entacapone increases the clinical effects of and adverse reactions to levodopa, the dose of levodopa needs to be decreased by 10%–30%. Drug interactions are similar to those for other drugs that increase dopaminergic activity, i.e. with MAOIs, catecholamines and tricyclic antidepressants. Currently drug trials are underway to test whether addition of entacapone to levodopa–DDCI therapy early in the course of treatment will delay development of motor complications. Adverse effects include GIT, CNS and skin disorders. Entacapone is contraindicated in hepatic impairment, and liver functions are monitored as a similar drug was withdrawn from use due to severe hepatic reactions.
Drugs with central anticholinergic activity
Symptoms of Parkinson’s disease caused by an excess of cholinergic activity include muscle rigidity and muscle tremor (see Figure 20-1). The muscle rigidity or increased tone appears as ‘ratchet resistance’ or ‘cogwheel rigidity’, in which the affected muscle moves easily, then meets resistance or remains fixed in the new position. The muscle tremors appear to have a to-and-fro movement caused by the sequence of contractions of agonistic and antagonistic muscles involved. The tremors are usually worse at rest and are commonly manifested as a pill-rolling motion of the hands and a bobbing of the head. Anticholinergics are more useful early in the course of the disease because the adverse reactions to DA depletion are not prominent at this stage (see, however, geriatric implications of use of anticholinergics in Clinical Interest Box 20-4).
Clinical interest Box 20-4 Geriatric implications of anticholinergic drugs
Elderly people are highly susceptible to adverse effects of anticholinergic drugs, especially constipation, dry mouth and urinary retention (usually in men).
These agents should be avoided in people with narrow-angle glaucoma or a history of urinary retention.
Memory impairment has been reported with continuous administration of these agents, especially in older people.
When usual adult doses are administered, some elderly people may have a paradoxical reaction: hyperexcitability, agitation, confusion and sedation.
Chronic use decreases or inhibits the flow of saliva, which can contribute to oral discomfort, periodontal disease and candidiasis.
Overheating resulting in heat stroke has been reported in people receiving anticholinergic drugs during vigorous exercise or periods of hot weather.
Blurred vision and/or increased sensitivity to light can occur.
Anticholinergic dosing in the elderly should begin at the lowest dose, with gradual increases until maximum improvement is noted or intolerable adverse effects occur.
Anticholinergics which readily cross the blood–brain barrier can block central cholinergic excitatory pathways, returning the DA–ACh balance in the brain (especially in the basal ganglia) to normal and producing some improvement in functional capacity and relief of tremor. There is less effect on the rigidity and akinesia.
The belladonna alkaloids atropine (see Drug Monograph 11-2) and hyoscine were for many years the only agents available to treat parkinsonism. These drugs have been supplanted by synthetic anticholinergics, which were developed to have fewer adverse reactions. In this group benztropine is the key drug; other anticholinergics are benzhexol, biperiden and orphenadrine. The usefulness of these drugs is limited because of their peripheral anticholinergic (atropinic) adverse reactions and their tendency to be less effective with continued use. Some anticholinergics are also used to control extrapyramidal reactions, such as rigidity, akinesia (difficulty in or lack of ability to initiate muscle movement), tremor and akathisia, which are caused by antipsychotic drugs such as the phenothiazines. (Other anticholinergics are used mainly for their peripheral actions in the GIT [atropine, hyoscine], eye [cyclopentolate, tropicamide], NMJ [glycopyrrolate], urinary tract [darifenacin, oxybutynin, solifenacin, propantheline] or respiratory tract for asthma [ipratropium, tiotropium]; or for central actions in motion sickness [hyoscine].)
Drug treatment of other movement disorders
Multiple sclerosis
Multiple sclerosis (MS) is the most common cause of progressive neurological disability in the 20–50 age group. Its incidence varies with latitude, being higher further from the equator; thus in Australia the incidence in Tasmania is about seven times that in north Queensland. There is widespread demyelination of neurons in the brain (white matter) and spinal cord, leading to muscle weak ness and sensory and visual disturbances, urinary and gastrointestinal dysfunctions and anxiety and depression. Environmental and genetic factors have been implicated in triggering the autoimmune reaction against CNS myelin. There is usually a relapsing–remitting course of progressive disability over a period of about 40 years.
Centrally acting antispasticity drugs such as baclofen and diazepam are used, as well as dantrolene (Drug Monograph 20-2); immunosuppressants (such as corticosteroids, methotrexate, azathioprine and mitozantrone) and immunomodulators, anticonvulsants and antidepressants (for sensory disturbances, pain and depression) and autonomic drugs (for relief of urinary problems).
Immunomodulators
First-line therapy of MS is directed at damping-down the excessive immune response against components of the nervous system, thus reducing the frequency and severity of attacks and the number and size of lesions. Immunomodulators used include beta-interferons (see Chapter 47). Glatiramer is a new unusual drug specifically for use in MS, to reduce the frequency of relapses. A simple synthetic tetrapeptide, it appears to block T-lymphocyte action against myelin antigens. As a peptide, it must be injected daily, and reactions at the injection site are common. Other adverse effects include chest pain, dyspnoea, anxiety, oedema, tremor and delayed development of antibodies to the drug.
Another new immunomodulator is natalizumab, a monoclonal antibody that binds to integrins on the surface of leucocytes and thus slows entry of T-cells through cerebral capillaries into the CNS, and reduces inflammation and demyelination. Administered IV every 4 weeks, the drug reduces relapses and lesions in patients with relapsingremitting MS. There is a risk of progressive multifocal leukoencephalopathy, a serious viral infection of the brain; the drug is contraindicated in people who are immunosuppressed, and should only be prescribed by a specialist neurologist.
Restless legs syndrome
Restless legs syndrome affects from 5%–15% of the population; it can begin at any age, and may have a hereditary aetiology or be secondary to various metabolic, neurological or drug-induced conditions. Patients complain of limb discomfort (usually leg, possibly also arm) after lying quietly, plus an urge to move the affected part (which temporarily relieves the discomfort) and unpleasant sensory sensations deep in muscle or bone. The condition is usually chronic and progressive.
Some people do not require treatment, and good ‘sleep hygiene’ (described in Chapter 16) may be adequate to relieve mild symptoms. For more severe symptoms, dopaminergic drugs are first-line therapy, and initially 90% of patients obtain relief from levodopa or DA agonists such as cabergoline, pramipexole or ropinirole. Adverse reactions are typical of dopaminergic agonists, plus risk of augmentation of symptoms, and rebound after drug effects have worn off. Other drugs tried include benzodiazepines (clonazepam), opioid analgesics and neuropathic pain relievers (gabapentin); drugs with anticholinergic actions may worsen the condition by enhancing skeletal muscle activity.
Myasthenia gravis
Although this condition is uncommon—occurring with a worldwide prevalence of about 200–400 cases per million population—it has long been of great interest to pharmacologists because of the interesting pharmacological concepts that it exemplifies.
Pathology
Myasthenia gravis (MG) is a progressive, incurable disease char acterised by the loss of or decrease in ACh nicotinic motor end-plate receptors (or more rarely musclespecific tyrosine kinase), which is caused by an autoimmune process and results in skeletal muscle weakness and fatigue. The thymus gland is believed to be involved in the causation of MG, through the production of antibodies directed against the antigenic proteins. Nearly 15% of all MG patients have a thymoma, or tumour of the thymus gland. Clinical signs and symptoms, and implications, are shown in Figure 20-3.
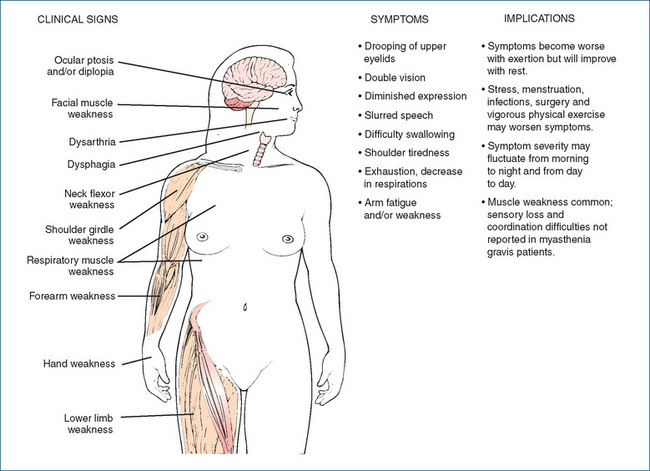
The most common early reported symptoms are ptosis and diplopia. The person might complain of shoulder fatigue after lifting the arm, or of hand weakness, finding it difficult to perform repetitive tasks, such as playing the piano. The most serious consequences of MG are dysphagia and respiratory muscle weakness, since these can result in aspiration pneumonia or respiratory failure.
The condition is exacerbated by many drugs that impair neuromuscular transmission or unmask auto-immune disorders. D-penicillamine is contraindicated, and the following drugs exacerbate weakness in most patients with MG: curare and other neuromuscular blockers; botulinum toxin; aminoglycoside, fluoroquinolone and macrolide antibiotics; quinine, quinidine and procainamide; interferons; magnesium and lithium; calcium channel blockers and beta-blockers; statins; and iodinated contrast agents.
Pharmacotherapy
It is a fluctuating but treatable condition, until the number of functioning ACh receptors drops too low; treatment may include thymectomy, which brings partial or complete remission in two-thirds of patients, plasmapheresis to remove antibodies and drug therapy, including anticholinesterases and immunosuppressants such as corticosteroids and azathioprine, cyclosporin or cyclophosphamide. Anticholinesterases allow increased levels of ACh at cholinergic nerve endings, thus competing with antibodies for binding sites on remaining functional nicotinic receptors. The drug of choice is pyridostigmine, a reversible inhibitor of acetylcholinesterase, as described earlier. Newer immunosuppressants used include tacrolimus and rituximab (see Chapter 47, and review by Meriggioli and Sanders [2009])
Motor neurone disease (amyotrophic lateral sclerosis)
Motor neurone disease is a progressive neuromuscular disorder, leading to bulbar palsy and atrophy of skeletal muscles, and is eventually fatal due to respiratory failure and/or choking. It is possibly due to accumulation of glutamate in affected neurons. Muscle cramps may respond to baclofen; physiotherapy and speech therapy are helpful.
Riluzole, a new drug that specifically inhibits the release of glutamate, has been trialled as a neuroprotective agent in this condition. It significantly slows the deterioration in muscle strength and prolongs life by a few months. Common adverse effects are weakness, nausea and decreased lung function; liver function and white cell counts require monitoring.
Other movement disorders
Hyperkinetic movement disorders, characterised by excessive movements, include tremor, chorea, tics and myoclonus. The term ‘Parkinson plus disorders’ has been coined to refer to conditions including multiple system atrophy, supranuclear palsy and corticobasilar ganglionic degeneration; some patients with these diseases respond to antiparkinson therapies plus symptomatic treatment of autonomic dysfunctions.
Tremor (muscle contractions in the frequency range 4–12 Hz) may be reduced after drinking alcohol; other drugs tried include beta-blockers, benzodiazepines, barbiturates and gabapentin. Choreas (abnormal involuntary movement disorders characterised by brief, irregular muscle contractions) may resolve spontaneously; drugs used include DA antagonists (neuroleptics) and anticonvulsants. Dystonias (movement disorders in which involuntary sustained muscle contractions cause twisting and repetitive movements or abnormal postures) may respond to botulinum toxin (see Chapter 31 and Drug Monograph 31-3 for treatment of blepharospasm in the eye) or anticholinergics, GABA agonists, and DA agonists or antagonists.
Drug-induced movement disorders
As discussed in the previous chapter, antipsychotic drugs commonly cause movement disorders (dystonias, akathisia, tardive dyskinesia) due to their DA-blocking actions. Anticholinergics are the first-line drugs for treatment of acute syndromes; whereas for tardive dyskinesia, withdrawal of the offending drug is the most important treatment.
 Drug treatment of dementias and stroke
Drug treatment of dementias and stroke
Dementia
Prevalence
Dementia is described as a progressive mental disorder characterised by chronic personality disintegration, confusion and deterioration of intellectual capacity and impulse control. Confusional states affect about 1% of the general population, but the incidence is about 16% in hospital admissions and up to 80% in geriatric or aged care units. Alzheimer’s disease accounts for about 50%–60% of dementia, while vascular dementia (including multi-infarct dementia, formerly known as cerebrovascu lar arteriosclerosis), Pick’s disease, Parkinson’s disease dementia and other forms comprise the balance. In Australia, dementia is the sixth leading cause of disease burden in the community, accounting for 3.5% of total disability-adjusted life years; it ranks ahead of diabetes and asthma in terms of disease burden.
Delirium, defined as an acute, transient disturbance of consciousness accompanied by a change in cognition and having a fluctuating course, can be considered as a type of acute, reversible dementia. It can be induced by drugs or by withdrawal of drugs (see below, ‘Causes’).
Causes
Reversible dementias can be caused by many factors, including
Pathogenesis
The syndrome of dementia usually develops slowly; early signs include depression, loss of ability to concentrate and increased anxiety, irritability and agitation. Intellectual (cognitive) ability is usually the first to decline, especially affecting memory, language and decision-making ability, with loss of orientation to time, place and person. Personal habits change; the person may become loud or obscene, or some personality characteristics that were present might become magnified. Helplessness, total dependency and loss of manual skills may occur next. In the final stages, the person may be bedridden, with loss of sphincter control, and eventually will die, usually of bronchopneumonia.
Management
All the possible reversible causes of dementia should be considered and managed first, and the patient’s general health and other conditions monitored and treated. Then treatment should be instituted to try to prevent or reduce the ongoing damage and to support the patient and family in managing this disease process. Drug treatment is indicated only for symptom control, i.e. the use of low-dose antipsychotic agents for treating severe agitation, delusions and hallucinations, or antidepressants for severe depression. Supportive care should include occupational therapy and assistance with activities of daily living, proper nutrition, moderate exercise if permitted, vitamins if indicated, plus help for the patient’s carers.
Alzheimer’s disease
Alzheimer’s disease is a dementia of insidious onset and gradually progressive course, characterised by confusion, memory failure, disorientation, restlessness, speech disturbances and impaired cognitive abilities. It is currently incurable, and accounts for more than half of cases of dementia. There is a linear incidence with age, so that about 50% of the population over 85 years shows some evidence of Alzheimer’s disease. It has been estimated to be the major underlying reason for over 50% of all nursing home admissions. Clinically, a progressive decline in intellectual functions is noted (see Table 20-1).
Table 20-1 Staging of cognitive decline in alzheimer’s disease
| STAGE | CLINICAL PHASE | SYMPTOMS |
| 1 | Normal | No change in cognition |
| 2 | Very mild | Forgets object location, some deficit in word finding |
| 3 | Mild (early confusion) | Early cognitive decline in one or more areas: memory loss, decreased ability to function in work situations, name-finding deficit, some decrease in social functioning, recall difficulties and anxiety |
| 4 | Moderate | Unable to perform complex tasks such as managing personal finances, planning a dinner party, concentrating and recalling knowledge of current events |
| 5 | Moderately severe (early dementia) | Usually needs assistance for survival: reminders to bathe, help in selecting clothes and other daily functions; may be disoriented; may become tearful |
| 6 | Severe (dementia) | Needs assistance with dressing, bathing and toilet functions; may forget names and details of personal life, and be unaware of surroundings; may have incontinence of urine and faeces; shows an increase in CNS disturbances such as agitation, delusions, paranoia, obsessive anxiety and violent behaviour |
| 7 | Very severe (late dementia) | Unable to speak (speech limited to 5 words or less); may scream or make other sounds; unable to ambulate, sit up, smile or feed self; unable to hold head erect, will ultimately slip into stupor or coma |
While researchers are still searching for the cause of Alzheimer’s disease, many aetiologies have been proposed. Some theories currently under study include:
Accumulation of amyloid plaques is due to inappropriate activity of some metalloproteinase enzymes in the CNS. Amyloid precursor protein (APP) is a transmembrane cellsurface protein formed in central neurons (its function is not known); it is cleaved by secretase enzymes to small proteins including a 42-amino-acid residue called A(42. This peptide spontaneously forms oligomers and then larger amyloid plaques, which accumulate and cause death of neurons. Loss of cholinergic neurons in the CNS, particularly the cholinergic input from the basal forebrain to the hippocampus and cerebral cortex, causes the impairment in memory and learning. There is also reactive gliosis (glial scarring), and formation of dendritic plaques and tangles, especially in the grey matter.
Current pharmacotherapy is focused on improving cognitive functioning or limiting disease progression and symptom control. Unfortunately, no known medication cures or prevents Alzheimer’s disease. The medications demonstrated to show cognitive improvement are centrally acting reversible anticholinesterases (cholinesterase inhibitors; see Clinical Interest Box 20-5), which act to raise and prolong ACh levels in cholinergic pathways and in some patients have been demonstrated to enhance cognitive functioning and slow the decline in functions. Other drugs may be used to relieve behavioural disturbances and mood changes, especially antipsychotics. New therapies currently under trial include muscarinic M1 agonists, nicotinic agonists, molecules that mimic nitric oxide, metal chelators and factors influencing amyloid beta-precursor protein, including antioxidants, vitamin E and the curry spice curcumin.
Clinical interest Box 20-5 Trial by anticholinesterases
In primitive societies, a person accused of a crime or of witchcraft was often subjected to trial by ordeal, involving dunking in deep water or administration of a potentially poisonous plant extract; if the person survived he or she was presumed innocent. The trick with poisons was to swallow the dose rapidly in the hope that the powerful emetic effect of the poison would cause severe vomiting and thus remove the toxin. An innocent person might do this, whereas a guilty person might be more hesitant and hence absorb more of the toxin.
One of the plants used this way was Physostigma venenosa, which grew on the Calabar Coast of western Africa. Its fruit, known as Calabar bean, contains an active alkaloid named physostigmine (or eserine). British missionaries described its use as an ordeal drug in about 1840. The active ingredient of the Calabar beans, eserine, was studied in Edinburgh by botanists and pharmacologists who grew the plants from seeds supplied by missionaries. After showing that the toxin caused death of animals by paralysis of heart and respiratory muscles, one valiant researcher (Robert Christison) tried an extract of seeds on himself. He described the effects as numbness, giddiness and, even after forced vomiting, feeble pulse and extreme pallor.
The major constituent of Calabar bean was purified and isolated in 1864, and named physostigmine. In 1875 it was used for the treatment of glaucoma, as it had been observed to reduce intraocular pressure and cause copious tears and a distinct contraction of the pupil. Its chemical structure was elucidated in 1925. Further pharmacological studies showed that physostigmine mimics the actions of ACh, and eventually its mechanism was shown to be inhibition of breakdown of ACh by the acetylcholinesterase enzyme. Since then, longer-acting anticholinesterases have been developed, so physostigmine is now rarely used.
Source: Mann 1992.
Centrally acting anticholinesterases
These drugs such as donepezil, galantamine (available as a prolonged-release formulation; see Clinical Interest Box 20-6) and rivastigmine (as a patch for transcutaneous administration) act to enhance neurotransmitter actions of ACh in CNS pathways; the mechanism is discussed in the previous section on drugs affecting skeletal muscle. As ACh is also the neurotransmitter at neuroeffector junctions in the parasympathetic nervous system, these drugs can be expected to have many adverse reactions, especially in the GIT and heart (see Drug Monograph 13-3 for neostigmine and Table 13-2). They should be used only cautiously in patients with asthma, chronic obstructive pulmonary disease, peptic ulcers, cardiac conduction disorders and cardiac arrhythmias. The enhanced vagal effect on the heart (bradycardia) can lead to arrhythmias and syncope; some patients have required hospitalisation and insertion of a pacemaker.
Clinical interest Box 20-6 Bulgarian snowdrops for alzheimer’s disease
Dementia is a CNS disorder developing over months to years, with cognitive, emotional and behavioural abnormalities leading to a decline in social and occupational functioning. The only drugs currently approved by the TGA in Australia for use in dementias are the cholinesterase inhibitors. These drugs do not cure Alzheimer’s disease, but may slow the progression and improve alertness and cognition for 1–4 years. Examples are donepezil, rivastigmine and, more recently, galantamine; the latter drug has been used for hundreds of years in a traditional European herbal remedy from snowdrops (Galanthus nivalis).
There are reports that in the 1950s a Bulgarian pharmacologist noticed people rubbing the common snow drop on their foreheads to ease nerve pain and giving an infusion of the bulbs to relieve poliomyelitis-associated paralysis. Russian pharmacologists identified anticholinesterase activity in extracts of Galanthus species in 1951 and determined the chemical structure of the active ingredient galantamine in 1952; it is a complex polycyclic alkaloid. It was soon introduced into Russian medicine as an antidote to neuromuscular blockade and for many neurological conditions, such as myasthenia gravis.
Perhaps because its pharmacology was originally studied in Russia during the Cold War period, it was some decades before galantamine made its appearance in the West. In Australia, it is now marketed in tablets and capsules (8, 16 or 24 mg), and is subsidised for use in mild-to-moderate Alzheimer’s disease. Galantamine has dual mechanisms of action: both by inhibition of acetylcholinesterase and enhancement of the binding of acetylcholine to nicotinic receptors. Clinically, galantamine improves cognitive performance (memory, attention, reasoning and language) and performance in activities of daily living. Its adverse reactions, as with other cholinomimetic agents, include gastrointestinal stimulation, depression and weakness.
Sources: Neurology Expert Group 2007, inter alia.
The clinical benefits have proven to be small, and are considered not cost-effective for all sufferers to be treated (see review by Pepeu and Giovannini [2009]). Currently in Australia there are strict guidelines as to provision of authority for a doctor to prescribe the drugs subsidised by the PBS.
Memantine, an NMDA antagonist
Memantine, a new non-competitive antagonist at glutamate N-methyl-D-aspartate (NMDA)-type receptors, is thought to reduce neuronal degradation due to excess glutamate present in Alzheimer’s disease; glutamate is an excitatory amino acid that can cause excitotoxicity and neuronal degradation (see Table 14-1). Memantine is approved for use in moderate-to-severe dementia and can be used in combination with an anticholinesterase; adverse CNS effects are common. Functional status should be reviewed regularly and the drug discontinued unless deterioration is slowed.
Trials are studying the efficacy of memantine in other CNS disorders; it appears safe and effective in some other dementias, alcohol dependence, post-traumatic stress disorder, headache and obesity; however, larger scale clinical trials are needed (see review by Kavirajan [2009]).
Symptom management
Due to the widespread prevalence of dementias, and the limited benefits from anticholinesterase agents, the search is on for better drugs. Many other drugs are under clinical investigation, including:
Clinical interest Box 20-7 Complementary and alternative therapies in neurological disorders
Many CAM methods have been tried in neurological disorders (stroke, brain injury, multiple sclerosis, Parkinson’s disease, dementias, epilepsy and spinal cord injury), possibly reflecting the inadequate success of conventional medicine in treating these distressing chronic/degenerative conditions. The best validated research is on acupuncture and Ginkgo biloba.
Acupuncture, in varied techniques, has provided some improvement in stroke patients and tension-type headache.
Extracts of the herb Ginkgo biloba have been used for 5000 years in traditional Chinese medicine; in experimental studies, Ginkgo biloba extracts have demonstrated antiplatelet, anticoagulant and free-radical scavenger activities and ‘vascular regulatory activity’. There is conflicting data as to its efficacy: some evidence for producing cognitive improvements in patients with stroke, Alzheimer’s disease and cerebral ischaemia; however, a large randomised trial has failed to demonstrate prevention of dementia, or any slowing of cognitive decline in elderly volunteers with mild cognitive impairment.
The herb feverfew (Tanacetum parthenium), which is proposed to have actions inhibiting serotonin release from platelets, has a long history of use in migraine patients.
Fish oil supplementation has been shown to alleviate dementia, and a diet high in fish reduces the risk of Alzheimer’s disease.
Galantamine, an alkaloid from the bulbs of the Russian snowdrop Galanthus woronowii, has anticholinesterase activity, with actions similar to those of neostigmine; it has been used effectively in neuromuscular disorders and is now available in tablet form (Clinical Interest Box 20-6).
Choline precursors are being trialled as neuroprotective agents in ischaemic and haemorrhagic stroke; one such agent, citicoline, is used as a dietary supplement in many countries.
Other CAM therapies shown to be useful are phyto-oestrogens, hypnotherapy and music therapy in improving mood and motivation, and massage therapy in improving circulation.
From: Spencer & Jacobs 1999; Australian Prescriber 24(4):100–101, 2001; Braun & Cohen 2007; Linde et al 2009.
Other dementias
Dementia with Lewy bodies is another common dementia, often difficult to distinguish from Alzheimer’s; post-mortem autopsy shows the presence of Lewy bodies (accumulated bits of alpha-synuclein protein) inside the nuclei of neurons from brain areas associated with memory and motor control. Two of the following symptoms must be present: visual hallucinations, parkinsonian tremors and stiffness and fluctuation in mental state; other symptoms may include impaired concentration, extreme confusion and difficulty judging distances. Progression is usually fairly rapid, leading to death within about seven years. Anticholinesterases may be helpful; antipsychotics can help reduce hallucinations, but will worsen parkinsonian symptoms.
Treatment of stroke
Neurological damage as a consequence of a cerebrovascular accident, or stroke, may lead to progressive mental dysfunction resembling dementia. A stroke is characterised by sudden onset of neurological dysfunction due to insufficient blood supply to the brain; the major types are ischaemic, due to embolism or infarction, and haemorrhagic, due to intracerebral or subarachnoid haemorrhage. Stroke is the third most common cause of death in Western countries; there are about 40,000 strokes per year in Australia, leading to long-term neurological disability in adults.
Prevention of stroke
Since stroke is primarily due to vascular disorders, the drugs involved in prevention are covered in the chapters on cardiovascular drugs. The main risk factors are: atrial fibrillation, hypertension, smoking, diabetes, cardiovascular disease and hypercholesterolaemia. Thus the drugs used to reduce these risk factors are low-dose aspirin, warfarin and other antiplatelet agents; antihypertensive agents, antidiabetes drugs and statins; these drugs are all covered in other chapters. Cessation of smoking is the most important modifiable risk factor, and surgery for carotid artery stenosis reduces the risk of stroke in suitable patients.
 Treatment of acute stroke
Treatment of acute stroke
Aspirin administered within 48 hours of onset of stroke (100–300 mg orally daily, except in patients with haemorrhagic stroke) has been shown to be life-saving. Anticoagulant (warfarin) or thrombolytic (alteplase) therapy is indicated only in specific patients. Thereafter, treatment is for the underlying predisposing conditions; e.g., recent research has shown that, in hypertensive patients who have had a stroke, aggressive lowering of blood pressure with an angiotensin-converting enzyme inhibitor and a diuretic significantly reduces the risk of another stroke. Other drugs tried include reperfusion therapies, neuroprotective agents and various complementary and alternative therapies (see Clinical Interest Box 20-7). After haemorrhagic stroke, urgent neurosurgery may be required to drain blood and evacuate haematomas. Nimodipine is the recommended antihypertensive agent.
Secondary prevention after a stroke may include warfarin, antiplatelet agents (aspirin, dipyridamole or clopidogrel), antihypertensive therapy and a statin. Carotid surgery and early rehabilitation are also important.
 Drugs used in migraine and other headaches
Drugs used in migraine and other headaches
Migraine
Pathology and prevalence
Migraine is a severe intermittent headache sometimes preceded or accompanied by flashing lights or other disturbances of brain function. More specifically, migraine attacks have been defined as recurrent headache with at least two of the following four features: pain affecting one side of the head only, pulsating in quality, moderate or severe in intensity, aggravated by exertion; plus nausea with or without vomiting, and sensitivity to light and sound. The nature of the attacks varies between patients, and within a patient at different times.
Migraine has been known and described for thousands of years. At various times, it has been considered a bad headache, a spontaneous ‘concussion’, an inflammatory disorder, a vascular disorder, a form of epilepsy or a platelet disorder. The fact that dilation of cerebral blood vessels is involved was proved by showing that if patients are (gently) centrifuged feet-outwards, the head pain is relieved, by taking the blood to the periphery. It is now generally considered that migraine is a syndrome of unstable cerebral blood vessels, probably mediated by 5-HT, with the first, prodromal, stage involving vasoconstriction of intracranial vessels, followed by reflex vasodilation with severe unilateral pulsating pain. Calcitonin gene-related peptide (CGRP) is also implicated in aetiology of migraine, with CGRP receptors found in trigeminal nerve pathways involved in migraine headaches.
The prevalence of migraine in the community is about 7% in males and 16% in females (during reproductive non-pregnant years). Migraine can occur in young children, and prevalence rises from about 3% in 7-year-olds to 9% in 15-year-olds. In Australia, it is estimated that a quarter of people suffering migraines require medical attention, and about 70% have some positive family history of migraines. While the exact aetiology is unknown, many factors are known to trigger a migraine attack; these are summarised in Clinical Interest Box 20-8.
Clinical interest Box 20-8 Complementary and alternative therapies in neurological disorders
While the specific factor(s) causing migraines are not known, many patients come to realise that particular events or factors seem to trigger an attack. Some of the common ‘triggers’ are:
The effects of baclofen are mediated via GABA, an inhibitory neurotransmitter in many pathways, especially in the grey matter in the brain, and at about 30% of CNS synapses. Baclofen is a GABA agonist, stimulating GABAB receptors and thus inhibiting release of transmitters from many types of nerve terminals; it depresses the CNS and has an antispastic action in the spinal cord, inhibiting activation of motor neurons.
INDICATIONS It is used orally in the treatment of spasticity resulting from multiple sclerosis or from injuries to the spinal cord or head injury; it may be effective in the chronic spasticity associated with cerebral palsy, but not in epilepsy. Baclofen may also reduce pain in spastic patients by inhibiting substance P release in the spinal cord.
PHARMACOKINETICS Absorption after oral administration (with meals) is generally good (bioavailability 70%–80%), but can vary among individuals. Baclofen crosses the blood–brain barrier and acts centrally. The time to peak plasma concentration is 2–3 hours. The onset of action is variable and can occur in hours or may take weeks. Baclofen has a half-life of 2.5–6 hours. Intrathecal administration minimises GIT adverse effects. Baclofen is partly metabolised in the liver and is excreted in the kidneys 70% unchanged.
DRUG INTERACTIONS Enhanced CNS-depressant and hypotensive effects can occur when baclofen is given concurrently with other CNS-depressant medications, antihypertensive agents, or with MAO inhibitors or tricyclic antidepressants. With levodopa, there is increased risk of psychotic reactions.
ADVERSE REACTIONS These include transient drowsiness, headache, vertigo, confusion, muscle weakness, nausea, hallucinations, respiratory and cardiovascular depression, urinary disorders, tinnitus and GIT upset.
WARNINGS AND CONTRAINDICATIONS Use with caution in patients with cerebral lesions, cerebrovascular accident, diabetes mellitus, seizure disorders, kidney impairment, respiratory disease or a history of psychiatric problems and in the elderly. Not recommended for cerebral palsy. Use with extreme caution in children <16 years. Avoid use in people with peptic ulcer or known baclofen hypersensitivity
DOSAGE AND ADMINISTRATION Dosage begins low to minimise adverse effects. The adult dose is 5 mg orally three times daily, increased by 5 mg per dose every 3 days until the desired response is achieved, not to exceed 80 mg/day.
Drug monograph 20-2 Dantrolene
INDICATIONS Dantrolene is used in the treatment of spasticity, especially upper motor neuron disorders such as multiple sclerosis, cerebral palsy, spinal cord injury and cerebrovascular accident, and in prophylaxis and treatment of malignant hyperthermia that occurs during surgery.
PHARMACOKINETICS Dantrolene is available orally and parenterally. Oral absorption is incomplete and slow; the onset of action when dantrolene is used to treat the spasticity of upper motor neurons can take 1 week or more. The drug has a half-life (orally) of about 9 hours. It is metabolised in the liver and excreted in the kidneys and bile.
DRUG INTERACTIONS When dantrolene is given concurrently with other CNS depressants, an increase in CNS-depressant effects can result. Monitor closely, as dosage reduction of one or both drugs may be necessary. When dantrolene is used chronically, concurrent use of hepatotoxic medications increases the potential risk for hepatotoxicity.
ADVERSE REACTIONS These include diarrhoea, dizziness, sleepiness, unusual fatigue, muscle weakness, nausea, vomiting, respiratory depression and cardiovascular, haematological and CNS changes.
WARNINGS AND CONTRAINDICATIONS Potentially fatal hepatitis can occur, so liver function should be monitored. Use with caution in patients with myopathy, liver or pulmonary function impairment, or neuromuscular diseases and in individuals older than 35 years (especially women), as they have an increased potential for hepatotoxicity. Patients should be cautioned against driving or other hazardous occupations.
Avoid use in people with dantrolene hypersensitivity and active liver diseases, such as hepatitis or cirrhosis.
Dosage begins low (25 mg orally daily), with gradual increases to a limit of 50 mg 4 times daily to monitor effects and minimise toxicity.
Drug monograph 20-3 Levodopa–carbidopa
This combination formulation consists of levodopa plus carbidopa in a 4:1 or 10:1 ratio.
INDICATIONS Levodopa–carbidopa is indicated for the treatment of idiopathic, postencephalitic and symptomatic Parkinson’s disease. The combination is particularly effective against rigidity and bradykinesia; tremor is less well treated.
PHARMACOKINETICS Levodopa is absorbed by active trans port; much is metabolised in the gut by monoamine oxidase (MAO) and dopa decarboxylase (DDC), so only about 30%–50% reaches the systemic circulation. Food reduces absorption of levodopa; it is recommended that the drug be given with food initially to minimise GIT discomfort, but later doses be given on an empty stomach to minimise fluctuations in plasma level and effects. The drug is distributed to most body tissues; the CNS receives less than 1% of the dose. Levodopa has a half-life of 1–3 hours. The duration of action is up to 5 hours per dose. Metabolites, principally homovanillic acid, are excreted by the kidneys.
After an oral dose of carbidopa, 40%–70% of the carbidopa dose is absorbed. Carbidopa is distributed widely to many body tissues, with the exception of the CNS. Peak plasma levels of carbidopa appear in 2–5 hours. The drug’s metabolism is insignificant; it is excreted by the kidneys.
Usually, improvement is seen within 2–3 weeks, although some patients require levodopa for up to 6 months to obtain maximal therapeutic effect.
DRUG INTERACTIONS See Drug Interactions 20-1.
ADVERSE REACTIONS Adverse reactions include peripheral dopaminergic effects (nausea and vomiting, postural hypotension, irregular heart rate, difficult urination, aggravation of peptic ulcers and dark discolouration of urine and sweat) and CNS effects: confusion, especially in the elderly, anxiety, nightmares, mood changes and choreiform and involuntary movements of the body, and sudden loss of mobility (the ‘off’ stage). A withdrawal syndrome can occur, resembling the ‘neuroleptic malignant syndrome’ related to decreased dopaminergic transmission. Eyelid spasms or closing (blepharospasm) may be an early sign of drug overdose.
Adverse reactions due to levodopa–carbidopa are similar to those for levodopa.
WARNINGS AND CONTRAINDICATIONS Caution is required in severe cardiovascular, pulmonary, renal, hepatic, psychiatric and endocrine diseases, in glaucoma and peptic ulcer, and during pregnancy (Category B3). The combination is contraindicated in wide-angle glaucoma. Levodopa can activate melanomas. Monitoring should be carried out for mental, behavioural and intraocular pressure changes and for arrhythmias.
Dopamine agonists are contraindicated during lactation, as dopamine inhibits secretion of prolactin (see Clinical Interest Box 33-5), and during treatment with typical antipsychotic agents.
DOSAGE AND ADMINISTRATION Levodopa dosage for adults is initiated with 50 mg twice daily, increasing by 100–125 mg until a therapeutic response is achieved, to a maximum of 2 g/day. The tablets should be taken before food.
Available levodopa–carbidopa combination dosage forms include 100/25 (100 mg levodopa and 25 mg carbidopa), 250/25 (250 mg levodopa and 25 mg carbidopa), a controlled-release formulation 200/50 (200 mg levodopa and 50 mg carbidopa) and an intestinal gel formulation containing 20 mg levodopa and 5 mg carbidopa per mL, to be administered via a percutaneous endoscopic gastrostomy (PEG), used only for uncontrolled advanced disease with severe motor fluctuations.
These combination dosage forms permit greater flexibility in prescribing sufficient amounts of both levodopa and carbidopa, and in titrating the dose–response relationship for the individual patient. The controlled-release formulations can minimise the on–off swings, but they have lower oral bioavailability, so higher levodopa doses may be required. There are also formulations containing three drugs: levodopa, carbidopa and entacapone (a COMT inhibitor).
Levodopa should not be stopped abruptly, due to risk of sudden drop in DA levels and a withdrawal syndrome resembling neuroleptic malignant syndrome.
Drug interactions 20-1 Levodopa plus a DDC inhibitor
| Other drug or drug group | Likely effects and management |
| Anticonvulsants, antipsychotics including phenothiazines, metoclopramide | Can result in decreased levodopa effects, because hydantoin anticonvulsants increase levodopa metabolism, and metoclopramide and antipsychotic neuroleptic agents such as phenothiazines block dopamine receptors in the brain. If possible, avoid the combination with levodopa |
| Antihypertensives | Increased risk of postural hypotension; decrease dose |
| Monoamine oxidase (MAO) inhibitors (type A) | This combination can result in a hypertensive crisis. MAO inhibitors should be discontinued 2–4 weeks before starting levodopa therapy |
| Monoamine oxidase (MAO) inhibitors (type B) (selegiline) | This combination may be used (see later), but can result in increased levodopa-induced nausea and CNS effects; levodopa dose should be reduced |
| Iron (ferrous sulfate) may decrease absorption of levodopa and carbidopa | Control of Parkinson’s disease may be impaired; administration times should be separated by as long an interval as possible |
| Other drugs with dopaminergic activity, including methyldopa | Increased adverse effects; monitor and reduce levodopa dose if necessary |
| Phenytoin, tetrabenazine | May reduce action of levodopa and worsen symptoms; reduce dose or avoid |
Drug monograph 20-4 Selegiline
Selegiline is used as adjunctive therapy in late-stage Parkinson’s disease, in combination with levodopa or levodopa–carbidopa.
PHARMACOKINETICS Selegiline, a phenylethylamine derivative, is well absorbed orally, reaching its peak plasma level in 30 minutes to 2 hours. It is rapidly metabolised and has three active metabolites, including L-amphetamine and methamphetamine (with half-lives of 2–20 hours), so has low bioavailability. It readily crosses the blood–brain barrier; metabolites are excreted slowly via the kidneys.
DRUG INTERACTIONS In usual doses (<10 mg/day), selegiline can be given with levodopa, and without dietary tyramine restrictions. Adverse effects are enhanced by other dopamine agonists or oral contraceptives. When used with serotonergic drugs, including other MAO inhibitors, selective serotonin reuptake inhibitors (SSRIs, including fluoxetine and sertraline), sumatriptan or pethidine, a reaction similar to the serotonin syndrome (confusion, restlessness, hyperreflexia, sweating, shivering, tremors, diarrhoea, ataxia and fever) can occur; these combinations should be avoided.
ADVERSE REACTIONS These are typical dopaminergic effects, including nausea, vomiting, insomnia, dizziness, stomach distress or pain, dyskinesias and mood alterations.
WARNINGS Use with caution in patients with move ment or cardiovascular disorders, psychoses, history of peptic ulcer disease or selegiline hypersensitivity.
DOSAGE AND ADMINISTRATION The usual adult dose of selegiline is 2.5–5 mg twice daily.
Neurotransmitters involved
The course and pathogenesis of a typical migraine attack begins with the prodrome or aura phase, during which there is vasoconstriction of the intracranial vessels. This is thought to lead to impairment of blood flow to the brain, starting in the visual cortex and causing the sensation of flashing lights, pin-pricking, impaired speech and weak ness. Blood flow is reduced in the parts of the brain subject to the symptoms.
The second phase of the attack is that of headache, thought to be due to a protective reflex vasodilation, during which blood flow to the brain, face and head is increased by about 20%. The nervous system is over-reactive, responding rapidly to intense stimuli. There are sensations of flashing lights, spectra, double vision, and increased sensitivity to light (photophobia), smells and noise. Autonomic effects include nausea and vomiting, diar rhoea, fluid retention and afterwards diuresis, and CNS effects of vertigo, ataxia, incoordination and impaired consciousness.
During the early phase, platelet levels of 5-HT drop due to release of 5-HT, which causes the vasoconstriction, aura and pain. Later, 5-HT levels are low, allowing vasodilation. GIT symptoms are due to effects of 5-HT on receptors in the gut. The headache is thought to be due both to arterial dilation and sensitisation to pain by released 5-HT and bradykinin. Spontaneous discharge along trigeminal nerve pathways releases other neurotransmitters (substance P, CGRP), which contribute to pain, vasodilation and visual and GIT disturbances.
Thus the main neurotransmitter involved in migraine appears to be 5-HT, and the specific drugs involved in treatment are 5-HT agonists in the acute attack and, apparently paradoxically, 5-HT antagonists in prophylaxis against attacks.
Non-pharmacological treatment
Before pharmacological treatment of migraine is commenced, it is important that the diagnosis be confirmed and other causes of severe headache excluded. Trigger factors need to be managed; often severe attacks can be pre-empted if the person can remove to a quiet dark room, avoid movement or any activity even sedentary, take mild analgesics (aspirin or paracetamol) and sleep off the attack. Behavioural therapies such as attention to sleep and rest habits, relaxation and assertiveness training, cognitive behavioural therapy and avoidance of trigger factors (including caffeine) and stress are useful.
Treatment of the acute attack
Analgesics and antiemetics
Mild analgesics should be tried early in the attack and in sufficient doses, with stronger drugs tried as necessary, moving up the analgesic ladder (see Figure 15-5). Suitable analgesics are paracetamol in children and in adults aspirin (600–900 mg every 4 hours) or paracetamol (1–1.5 g every 4 hours, to a maximum of 4 g/day). An NSAID (naproxen or ibuprofen, or rectal ketoprofen) can be added. Opioids including codeine combinations, especially pethidine, should be avoided as they exacerbate the GIT symptoms of the migraine attack and cause potential problems with dependence.
To enhance absorption of the anti-migraine drug, and reduce vomiting if nausea is severe, an antiemetic such as metoclopramide, domperidone or prochlorperazine may be given; parenteral administration may be necessary due to vomiting.
Ergot alkaloids
Ergotamine, a powerful vasoconstrictor, acts as a partial agonist at both (-adrenoceptors and 5-HT1 receptors. A dose of 1–2 mg can be given at the onset of the attack, either orally or rectally. Caffeine or diphenhydramine is sometimes given concurrently with ergotamine (with doubtful pharmacological rationale). If parenteral administration is necessary due to nausea or gastric stasis that occurs early in an attack, the dihydroergotamine derivative can be given SC or IM. Ergot alkaloids have many adverse reactions (see Clinical Interest Box 38-4) so are used cautiously, and rebound headaches can occur after cessation of administration.
Triptans
If the above therapies have been ineffective in relieving previous attacks, one of the ‘triptan’ drugs may be prescribed: sumatriptan (see Drug Monograph 20-5), zolmitriptan, rizatriptan or naratriptan. They are relatively new drugs that came into use in the 1990s and revolutionised treatment of migraine for many patients. Triptans are structural analogues of 5-HT and are selective agonists at 5-HT1D receptors and effective vasoconstrictors, especially on cerebral arteries, relieving migraine headache in 50%–75% of cases within 2–4 hours of oral administration. They should be given when the headache is beginning to develop and can be administered orally (but have low bioavailability), parenterally or as a nasal spray. Serotonin syndrome is a risk as an adverse reaction; hence triptans should be used only cautiously in patients concurrently taking lithium, MAOIs or SSRIs. Triptans and ergotamines should not be taken concurrently, due to additive vasoconstrictor effects.
Drug monograph 20-5 Sumatriptan
Sumatriptan selectively constricts cranial vessels by agonist actions at 5-HT receptors.
INDICATIONS It is indicated for treatment of an acute migraine attack, in patients unresponsive to or intolerant of other therapies. It is also administered by injection for acute relief of cluster headache pain.
PHARMACOKINETICS After oral administration, absorption of sumatriptan is rapid but incomplete; a high first-pass metabolism leads to low bioavailability. After SC administration, the peak plasma concentration is reached in about 30 minutes, and there is much higher bioavailability, so doses are considerably lower (6 mg compared with 50–100 mg orally). Intranasal administration as a spray has a quicker onset of action than orally (hence it is useful to relieve pain and slow development of the migraine attack, especially in people with severe nausea and vomiting) but shorter duration of action.
DRUG INTERACTIONS The triptan drugs interact significantly with other drugs that raise serotonin levels, especially monoamine oxidase inhibitors including moclobemide and selective serotonin reuptake inhibitors; there is increased risk of adverse drug reactions including serotonin syndrome and ischaemia, so effects should be monitored.
ADVERSE REACTIONS Minor reactions include dizziness, fatigue, drowsiness, chest pain, nausea and vomiting, feelings of heaviness and rash. Rarely, there have been reports of arrhythmias, stroke, anaphylactic reactions, seizures and even acute myocardial infarction and death.
WARNINGS AND CONTRAINDICATIONS Sumatriptan should be taken as monotherapy, i.e. other antimigraine preparations should be avoided. It should not be taken within 24 hours of ergotamine preparations. Caution is advised in the elderly and during pregnancy. The drug is contraindicated in ischaemic diseases (ischaemic heart disease, myocardial infarction, coronary vasospasm), hypertension and in a history of cerebroor peripheral vascular disease. It should not be used during lactation or in severe liver disease, and is not licensed for use in children.
DOSAGE AND ADMINISTRATION The oral dose is 50–100 mg as soon as possible in the attack, repeating after 2 hours if necessary to a maximum of 300 mg/day. The SC dose regimen is 6 mg, repeating after 1 hour to maximum of 12 mg/day. For intranasal administration the dose is 10–20 mg into one nostril, repeating after 2 hours to a maximum of 40 mg/day. Note: Sumatriptan must not be taken less than 24 hours after ergot, and ergot not for 6 hours after sumatriptan.
New therapies being trialled include telcagepant, an orally available antagonist of CGRP receptors; early studies show this new type of antimigraine therapy to be safe and effective.
Prevention of migraine attacks
Triggering factors need to be identified and avoided if possible, and any underlying conditions, such as hypertension or anxiety, treated first. Increasing frequency of attacks can be due to overuse of ergotamine, as the with drawal effect causes rebound headache.
If the patient suffers more than one severe attack per month, the following prophylactic agents are tried:
Only one preventive drug should be used at a time. It may take 1–3 months for a prophylactic effect to be evident; during this time treatment of acute attacks is still required (see review by Stark and Stark [2008]).
Methysergide
Methysergide is an ergot derivative that is a potent 5-HT2antagonist (compare to ergotamine, above, a 5-HT agonist). It suppresses migraine headache in about 25% of patients, but is ineffective in treatment of acute attacks. An initial prophylactic dose of 1 mg/day orally is given, increasing gradually to 3–4 times/day, then reducing over 2–3 weeks. The drug must be discontinued after 4 months due to the risk of fibrosis (retroperitoneal, or in the heart valves or pleura). The drug may then be reintroduced after a drug holiday of 1–2 months if no fibrosis has occurred.
There is a high incidence of adverse reactions, espec ially in the CNS, GIT and cardiovascular system, with behavioural changes and peripheral vascular disease. Methysergide is contraindicated in many conditions: CVS disease, hyperthyroidism, collagen disease, urinary tract disorders, renal or liver disease and in children, pregnancy or lactation.
Feverfew
The herb feverfew (Tanacetum parthenium), which is proposed to have actions inhibiting 5-HT release from platelets, has a long history of use in migraine patients. Recent clinical trials have confirmed that it has some prophylactic efficacy; however, the content of active ingredients varies widely among plants and at different times (see Clinical Interest Box 20-7).
Drugs used in other headaches
Normal headaches
There is a wide variety of conditions in which headaches can occur, with accompanying triggering factors:
In most of these types of headache, simple analgesics provide adequate pain relief. Appropriate doses are aspirin 600 mg orally every 4 hours or paracetamol 0.5–1 g orally every 3–6 hours, with a maximum of 4 g/day. In medication overuse headache, the causative drug must be withdrawn.
Although most headaches are mild and self-limiting, there are potentially serious causes of headaches that need to be checked: a space-occupying lesion (e.g. intracranial haemorrhage, inflammation due to meningitis or encephalitis, cerebral oedema or tumour), vascular insufficiency, temporal arteritis, severe hypertension or headache after head injury.
Tension headache
Tension headache is the commonest form of headache, without aura or vomiting, affecting both sides of the head, with a feeling of heaviness or tightness, often with depression or anxiety. It affects women more frequently than men. It has been suggested that it is a syndrome of low 5-HT levels; lack of sleep is a common trigger.
Physical management includes relaxation, exercises and massage, and avoidance or reduction of caffeine intake. Drug treatments tried (after simple analgesics) are NSAIDs (ibuprofen, diclofenac or naproxen), then amitriptyline (an antidepressant) and diazepam (an antianxiety agent).
Cluster headache
Cluster headache is a severe one-sided pain, centred on one eye with tearing and redness of the eye, often with one blocked or runny nostril, which recurs within 24 hours; multiple attacks may occur each day. There is decreased sympathetic function on the affected side; it has been called ‘migrainous neuralgia’. Men are affected more frequently than women; it is triggered particularly by vasodilators, including alcohol.
Treatment is with inhaled oxygen (100% for up to 15 minutes), and sumatriptan SC is effective. Prevention is with prophylactic use of ergotamine, methysergide, corticosteroids, lithium or calcium channel blockers.
Key points


 This chapter reviews the pathology and treatment of Parkinson’s disease, myasthenia gravis, movement disorders and muscle spasticity, dementia and Alzheimer’s disease, and migraine and other headaches. Drugs that act as skeletal muscle relaxants can have central actions (e.g. baclofen, diazepam) or peripheral actions (botulinum toxin A, dantrolene). They are usually the drugs of choice in the treatment of muscle spasticity. The neuromuscular blocking agents used in surgery (such as curare and suxamethonium) also act at the neuromuscular junction. Myasthenia gravis, which is characterised by skeletal muscle weakness and fatigue, is a debilitating disease. The drugs of choice for treatment are the anticholinesterase drugs acting at the neuromuscular junction, such as pyridostigmine; these increase acetylcholine activity at the motor end-plate. People with Parkinson’s disease usually require correction of the disorder’s imbalance of dopamine and acetylcholine neurotransmitters. They are therefore treated with dopaminergic agents, to raise brain levels of dopamine, and/or with drugs that have central anticholinergic effects. The main dopaminergic agents are levodopa with a decarboxylase inhibitor, the ergot derivatives, and apomorphine, amantadine and selegiline. These all tend to have adverse reactions on dopamine receptors in the CNS and GIT. Centrally acting anticholinergic drugs (such as benztropine) inhibit acetylcholine-mediated motor activity but have adverse reactions on other acetylcholine receptors in the motor and autonomic nervous systems. There is no current medication to cure or prevent Alzheimer’s disease and dementia. Centrally acting anticholinesterases (e.g. donepezil) slow cognitive decline by enhancing acetylcholine functions. Other pharmacotherapy is used for symptom control and to manage agitation, delusions and hallucinations. Migraine is a severe unilateral headache; it is thought to be due to fluctuating levels of 5-HT in the brain, causing vasoconstriction then vasodilation of cerebral blood vessels. For treatment of acute attacks, analgesics and 5-HT agonists (ergotamine, sumatriptan) are used.
This chapter reviews the pathology and treatment of Parkinson’s disease, myasthenia gravis, movement disorders and muscle spasticity, dementia and Alzheimer’s disease, and migraine and other headaches. Drugs that act as skeletal muscle relaxants can have central actions (e.g. baclofen, diazepam) or peripheral actions (botulinum toxin A, dantrolene). They are usually the drugs of choice in the treatment of muscle spasticity. The neuromuscular blocking agents used in surgery (such as curare and suxamethonium) also act at the neuromuscular junction. Myasthenia gravis, which is characterised by skeletal muscle weakness and fatigue, is a debilitating disease. The drugs of choice for treatment are the anticholinesterase drugs acting at the neuromuscular junction, such as pyridostigmine; these increase acetylcholine activity at the motor end-plate. People with Parkinson’s disease usually require correction of the disorder’s imbalance of dopamine and acetylcholine neurotransmitters. They are therefore treated with dopaminergic agents, to raise brain levels of dopamine, and/or with drugs that have central anticholinergic effects. The main dopaminergic agents are levodopa with a decarboxylase inhibitor, the ergot derivatives, and apomorphine, amantadine and selegiline. These all tend to have adverse reactions on dopamine receptors in the CNS and GIT. Centrally acting anticholinergic drugs (such as benztropine) inhibit acetylcholine-mediated motor activity but have adverse reactions on other acetylcholine receptors in the motor and autonomic nervous systems. There is no current medication to cure or prevent Alzheimer’s disease and dementia. Centrally acting anticholinesterases (e.g. donepezil) slow cognitive decline by enhancing acetylcholine functions. Other pharmacotherapy is used for symptom control and to manage agitation, delusions and hallucinations. Migraine is a severe unilateral headache; it is thought to be due to fluctuating levels of 5-HT in the brain, causing vasoconstriction then vasodilation of cerebral blood vessels. For treatment of acute attacks, analgesics and 5-HT agonists (ergotamine, sumatriptan) are used.Review exercises
References and further reading
Arulmozhi D.K., Veeranjaneyulu A., Bodhankar S.L. Migraine: current concepts and emerging therapies. Vascular Pharmacology. 2005;43(3):176-187.
Attia J., Schofield P. What now for Alzheimer’s disease? An epidemiological evaluation of the AD2000 trial. Australian Prescriber. 2005;28(6):134-135.
Australian Medicines Handbook 2010. Adelaide: AMH, 2010.
Beal M.F., Lang A.E., Ludolph A.C., editors. Neurodegenerative Diseases: Neurobiology, Pathogenesis and Therapeutics. Cambridge: Cambridge University Press, 2005.
Braun L., Cohen M. Herbs and Natural Supplements: An Evidence-Based Guide, 2nd edn. Sydney: Elsevier Mosby; 2007.
Byrne G.J. Pharmacological treatment of behavioural problems in dementia. Australian Prescriber. 2005;28(3):67-70.
Cooper J.R., Bloom F.E., Roth R.H. The Biochemical Basis of Neuropharmacology, 8th edn. New York: Oxford University Press; 2003.
Crouch A. Treating dementia. Australian Prescriber. 2009;32(1):9-12.
Johnston T.H., Brotchie J.M. Drugs in development for Parkinson’s disease. Current Opinion in Investigational Drugs. 2004;5(7):720-726.
Kavirajan H. Memantine: a comprehensive review of safety and efficacy. Expert Opinion on Drug Safety. 2009;8(1):89-109.
Lance J.W. Headache and face pain. Medical Journal of Australia. 2000;172:450-455.
Linde K., Allais G., Brinkhaus B., et al. Acupuncture for tensiontype headache. Cochrane Database of Systematic Reviews. 21(1), 2009. Jan CD007587
Lindley R.I., Landau P.B. Early management of acute stroke. Australian Prescriber. 2004;27(5):120-123.
Mann J. Murder, Magic and Medicine. Oxford: Oxford University Press; 1992.
Meriggioli M.N., Sanders D.B. Autoimmune myasthenia gravis: emerging clinical and biological heterogeneity. Lancet Neurology. 2009;8:475-490.
Neurology Expert Group. Therapeutic Guidelines: Neurology, version 3. Melbourne: Therapeutic Guidelines Limited; 2007.
Pepeu G., Giovannini M.G. Cholinesterase inhibitors and beyond. Current Alzheimer Research. 2009;6(2):86-96.
Psychotropic Expert Group. Therapeutic Guidelines: Psychotropic, version 6. Melbourne: Therapeutic Guidelines Limited; 2008.
Reddel S. Treatment of myasthenia gravis. Australian Prescriber. 2007;30(6):156-160.
Schrag A. Entacapone in the treatment of Parkinson’s disease. Lancet Neurology. 2005;4(6):366-370.
Spencer J.W., Jacobs J.J. Complementary/Alternative Medicine: An Evidence-Based Approach. St Louis: Mosby; 1999.
Stark R.J., Stark C.D. Migraine prophylaxis. Medical Journal of Australia. 2008;189(5):283-288.
Thyagarajan D. Restless legs syndrome. Australian Prescriber. 2008;31(4):90-93.
Williams D. Medication overuse headache. Australian Prescriber. 2005;28(6):143-145.
Alzheimer’s Australia website: www.alzheimers.org.au
New Zealand Medicines and Medical Devices Safety Authority: www.medsafe.govt.nz
More weblinks at: http://evolve.elsevier.com/AU/Bryant/pharmacology