5 Regulation of the Heartbeat
1. Describe the neural control of heart rate.
2. Explain the role of preload in the regulation of myocardial contraction.
3. Describe the neural regulation of myocardial contraction.
4. Explain the effects of hormones on myocardial contraction.
5. Explain the effects of blood gases on myocardial contraction.
The quantity of blood pumped by the heart each minute (i.e., cardiac output, CO) may be varied by changing the frequency of its beats (i.e., heart rate, HR) or the volume ejected per stroke (i.e., stroke volume, SV). Cardiac output is the product of heart rate and stroke volume; that is,
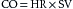
A discussion of the control of cardiac activity may therefore be subdivided into considerations of the regulation of pacemaker activity and the regulation of myocardial contraction. However, in the intact organism, a change in the function of one of these features of cardiac activity almost invariably alters the other.
Certain local factors, such as temperature changes and tissue stretch, can affect the discharge frequency of the sinoatrial (SA) node. However, the principal control of heart rate is relegated to the autonomic nervous system, and the discussion is restricted to this aspect of heart rate control. Also considered are the intrinsic and extrinsic factors that regulate myocardial performance.
Heart Rate is Controlled Mainly by the Autonomic Nerves
In normal adults the average heart rate at rest is approximately 70 beats per minute (beats/min); the rate is significantly higher in children. During sleep the heart rate decelerates by 10 to 20 beats/min, but during emotional excitement or muscular activity it may accelerate to rates considerably above 100 beats/min. In well-trained athletes at rest, the rate is usually only about 50 beats/min.
The SA node is usually under the tonic influence of both divisions of the autonomic nervous system. The sympathetic system enhances automaticity, whereas the parasympathetic system inhibits it. Changes in heart rate usually involve a reciprocal action of the two divisions of the autonomic nervous system. Thus, an increased heart rate is produced by a diminution of parasympathetic activity and a concomitant increase in sympathetic activity; deceleration is usually achieved by the opposite mechanisms. Under certain conditions, the heart rate may be changed by selective action of just one division of the autonomic nervous system rather than by reciprocal changes in both divisions.
Ordinarily, parasympathetic tone predominates in healthy, resting individuals. Blockade of parasympathetic effects by administration of atropine (a muscarinic receptor antagonist) usually increases heart rate substantially, whereas blockade of sympathetic effects by administration of propranolol (a β-adrenergic receptor antagonist) usually decreases heart rate only slightly (Figure 5-1). When both divisions of the autonomic nervous system are blocked, the heart rate of young adults averages about 100 beats/min. The rate that prevails after complete autonomic blockade is called the intrinsic heart rate.
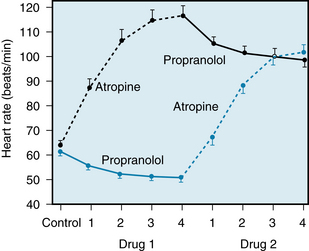
FIGURE 5-1 Effects of four equal doses of atropine (0.04 mg/kg total) and of propranolol (0.2 mg/kg total) on the heart rate of 10 healthy young men (mean age, 21.9 years). In half of the trials, atropine was given first (top curve); in the other half, propranolol was given first (bottom curve).
(Redrawn from Katona PG, McLean M, Dighton DH, et al: Sympathetic and parasympathetic cardiac control in athletes and nonathletes at rest. J Appl Physiol 52:1652, 1982.)
Parasympathetic Pathways
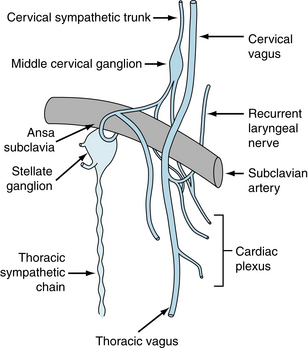
Cardiac parasympathetic fibers originate in the medulla oblongata, in cells that lie in the dorsal motor nucleus of the vagus or in the nucleus ambiguus. The precise location varies from species to species. Efferent vagal fibers pass inferiorly through the neck as the cervical vagus nerves (Figure 5-2), which lie close to the common carotid arteries. They then pass through the mediastinum to synapse with postganglionic cells on the epicardial surface or within the walls of the heart itself. Most of the cardiac ganglion cells are found in plexuses and are located near the SA node and atrioventricular (AV) conduction tissue.
The right and left vagi are distributed differentially to the various cardiac structures. The right vagus nerve affects the SA node predominantly; its stimulation slows SA nodal firing or may even stop it for several seconds. The left vagus nerve mainly inhibits AV conduction tissue to produce various degrees of AV block. However, the distributions of the efferent vagal fibers overlap, such that left vagal stimulation also depresses the SA node and right vagal stimulation impedes AV conduction.
The SA and AV nodes are rich in cholinesterase. Hence the effects of any given vagal impulse are brief because cholinesterase rapidly hydrolyzes the neurally released acetylcholine. The effects of vagal activity on SA and AV nodal function also display a very short latency (about 50 to 100 milliseconds), because the released acetylcholine activates special K+ channels (IK,ACh, an inwardly rectifying potassium channel) in the cardiac cells. The opening of these channels is so prompt because it does not require the operation of a second messenger, such as cyclic adenosine monophosphate (cAMP).
When the vagus nerves are stimulated at a constant frequency for several seconds, the heart rate decreases abruptly and attains a steady-state value within one or two cardiac cycles (Figure 5-3A). Also, when stimulation is discontinued, the heart rate returns very quickly to its basal level. The combination of the brief latency and rapid decay of the response (because of the abundance of cholinesterase) allows the vagus nerves to exert a beat-by-beat control of SA and AV nodal function.
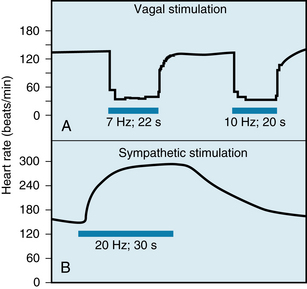
FIGURE 5-3 Changes in heart rate evoked by stimulation (horizontal bars) of the vagus (A) and sympathetic (B) nerves in an anesthetized dog.
(Modified from Warner HR, Cox AJ: A mathematical model of heart rate control by sympathetic and vagus efferent information. J Appl Physiol 17:349, 1962.)
Parasympathetic influences predominate over sympathetic effects at the SA node, as shown in Figure 5-4. This vagal predominance in the regulation of heart rate is mediated mainly by suppression of the release of norepinephrine from the sympathetic nerve endings by the acetylcholine released from neighboring vagus nerve endings. This nerve-nerve interaction between the two divisions of the autonomic nervous system is discussed more fully in association with Figure 5-5.
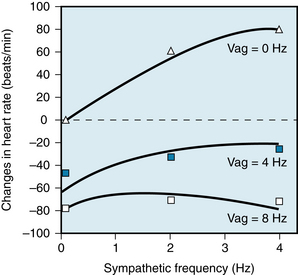
FIGURE 5-4 Changes in heart rate in an anesthetized dog when the vagus and cardiac sympathetic nerves were stimulated simultaneously. The sympathetic nerves were stimulated at 0, 2, and 4 Hz; the vagus nerves at 0, 4, and 8 Hz. As the frequency of sympathetic stimulation increased from 0 to 4 Hz, the heart rate rose by about 80 beats/min in the absence of vagal stimulation (Vag = 0 Hz). However, when the vagi were stimulated at 8 Hz, increasing the sympathetic stimulation frequency from 0 Hz to 4 Hz had a negligible influence on heart rate. The symbols represent the observed changes in heart rate; the curves were derived from a computed regression equation.
(Modified from Levy MN, Zieske H: Autonomic control of cardiac pacemaker activity and atrioventricular transmission. J Appl Physiol 27:465, 1969.)
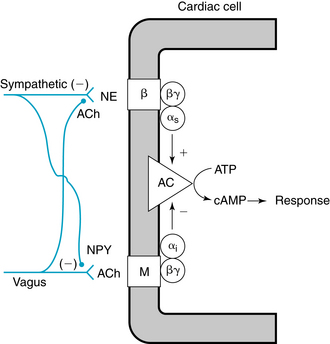
FIGURE 5-5 Interneuronal and intracellular mechanisms responsible for the interactions between the sympathetic and parasympathetic systems in the neural control of cardiac function. Acetylcholine (ACh) and norepinephrine (NE) act on muscarinic (M) and β-adrenergic (β) receptors, respectively. Inhibitory (αi) and stimulatory (αs) components of guanine nucleotide–binding proteins transduce the neurotransmitter signals to adenylyl cyclase (AC) to regulate synthesis of cyclic adenosine monophosphate (cAMP). ATP, adenosine triphosphate; βγ, βγ subunit of G protein; NPY, neuropeptide Y.
(Modified from Levy MN: in Kulbertus HE, Franck G, editors: Neurocardiology, Mt Kisco, NY, 1988, Futura.)
Sympathetic Pathways
The cardiac sympathetic fibers originate in the intermediolateral columns of the upper five or six thoracic and lower one or two cervical segments of the spinal cord. They emerge from the spinal column through the white communicating branches and enter the paravertebral chains of ganglia. Preganglionic and postganglionic neurons synapse mainly in the stellate and middle cervical ganglia (see Figure 5-2). The middle cervical ganglia lie close to the vagus nerves in the superior portion of the mediastinum. Sympathetic and parasympathetic fibers then join to form a plexus of mixed efferent nerves to the heart (see Figure 5-2).
Postganglionic cardiac sympathetic fibers approach the base of the heart along the adventitial surface of the great vessels. On reaching the base of the heart, these fibers are distributed to the various chambers as an extensive epicardial plexus. They then penetrate the myocardium, usually along the coronary vessels. The adrenergic receptors in the nodal regions and in the myocardium are predominantly of the β type; that is, they are activated by β-adrenergic agonists, such as isoproterenol, and are inhibited by β-adrenergic blocking agents, such as propranolol.
Like the vagus nerves, the left and right sympathetic fibers are distributed differentially. In the dog, for example, fibers on the left side have more pronounced effects on myocardial contractility than do fibers on the right side, whereas the fibers on the left side have much less effect on heart rate than do the fibers on the right side (Figure 5-6). In some dogs, left cardiac sympathetic nerve stimulation may not affect the heart rate at all. This bilateral asymmetry probably also exists in humans. In one group of patients, block of the right stellate ganglion caused a mean reduction in heart rate of 14 beats/min, whereas left-sided blockade decreased heart rate by only 2 beats/min.
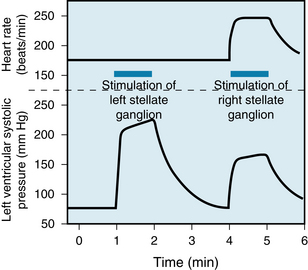
FIGURE 5-6 In the dog, stimulation of the left stellate ganglion has a greater effect on ventricular contractility than does right-sided stimulation, but a lesser effect on heart rate. In this example, traced from an original record, left stellate ganglion stimulation had no detectable effect at all on heart rate but had a considerable effect on ventricular performance in an isovolumic left ventricle preparation.
(From Levy MN: Unpublished tracing.)
Figures 5-3B and 5-6 show that the effects of sympathetic stimulation decay very gradually after the cessation of stimulation, in contrast to the abrupt termination of the response after vagal activity (see Figure 5-3A). Most of the norepinephrine released during sympathetic stimulation is taken up again by the nerve terminals, and much of the remainder is carried away by the bloodstream. These processes are relatively slow. Furthermore, at the beginning of sympathetic stimulation, the facilitatory effects on the heart attain steady-state values much more slowly than do the inhibitory effects of vagal stimulation (see Figure 5-3).
At least two factors are responsible for the more gradual onset of the heart rate response to sympathetic activity than to vagal activity. First, the response to sympathetic activity depends mainly on the intracellular production of second messengers, mainly cAMP, in the automatic cells in the SA node (see Figure 5-5). This is a slower process than the process that transduces the response to vagal activity. The muscarinic receptors that respond to the acetylcholine released from the vagal terminals are coupled directly to the acetylcholine-regulated K+ channels by a G protein; this direct coupling allows a prompt response. Second, the postganglionic nerve endings of each of the two autonomic divisions release neurotransmitters at different rates. Intense vagal activity releases enough acetylcholine during a brief period (e.g., 1 s) to arrest the heartbeat. Conversely, even during intense sympathetic activity, enough norepinephrine is released during each cardiac cycle to change cardiac behavior by only a small increment. Thus the vagus nerves are able to exert beat-by-beat control of heart rate, whereas the sympathetic nerves are not able to alter cardiac function very much within one cardiac cycle.
The predominance of parasympathetic over sympathetic effects on SA and AV node activities depends on the nature of their interactions. There are prejunctional (nerve-nerve) and postjunctional (within the cardiac cell) interactions between vagal and sympathetic neurons (see Figure 5-5). Within the terminal autonomic innervation, parasympathetic and sympathetic fibers are interlaced within micrometer distances of each other.
The experiment illustrated in Figure 5-7 demonstrates that stimulation of the cardiac sympathetic nerves results in the overflow of substantial amounts of norepinephrine into the coronary sinus blood. The amount of norepinephrine that overflows into the coronary sinus blood parallels the amount of norepinephrine released at cardiac sympathetic terminals. Concomitant vagal stimulation reduces the overflow of norepinephrine by about 30%. The sympathetic adrenergic terminal membrane has muscarinic receptors activated by acetylcholine that causes inhibition of norepinephrine release. Thus, vagal activity decreases cardiac frequency (see Figure 5-4) and contractility (see Figure 5-31) partly by antagonizing the facilitatory effects of any concomitant sympathetic activity. Physiologically, neuropeptide Y (NPY), a cotransmitter with norepinephrine in sympathetic adrenergic nerves, can inhibit acetylcholine from neighboring vagal fibers (see Figure 5-5).
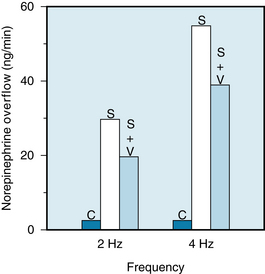
FIGURE 5-7 Mean rates of overflow of norepinephrine into the coronary sinus blood in a group of seven dogs under control conditions (C), during cardiac sympathetic stimulation (S) at 2 or 4 Hz, and during combined sympathetic and vagal stimulation (S + V). The combined stimulus consisted of sympathetic stimulation at 2 or 4 Hz and vagal stimulation at 15 Hz.
(Redrawn from Levy MN, Blattberg B: Effect of vagal stimulation on the overflow of norepinephrine into the coronary sinus during cardiac sympathetic nerve stimulation in the dog. Circ Res 38:81, 1976.)
Postjunctional interaction of parasympathetic and sympathetic transmitter occurs largely, but not exclusively, through regulation of cAMP formation (see Figure 5-5). Postjunctional targeting of transmitter action begins at guanine nucleotide–coupled receptors (GPCRs), namely muscarinic and β-adrenergic receptors. Inhibitory G proteins (Gi) for muscarinic receptor and stimulatory G proteins (Gs) for β-adrenergic receptors provide another means of targeting transmitter action. Figure 5-8 illustrates the general features of postjunctional action of a transmitter. Transmitter occupancy of its GPCR facilitates the exchange of GTP for GDP and the G protein α subunit dissociates from the βγ subunits. The α subunit regulates adenylyl cyclase activity (inhibition via Gi, stimulatory via Ga) and thereby the concentration of cAMP. In the case of Gi, the βγ subunits serve as a transducer to activate IK,ACh channels that cause membrane hyperpolarization in SA (see Figure 3-5) and AV (see Figure 3-10) nodes. Cyclic AMP is a water-soluble substance whose message is targeted by intracellular compartments of its synthetic enzyme, adenylyl cyclase, its hydrolytic enzymes, phosphodiesterases, and its anchoring peptides, cyclic AMP-dependent protein kinase anchoring proteins (AKAPs). Thus, norepinephrine raises intracellular cAMP, which interacts with its cognate protein kinase to regulate cardiac function by phosphorylation of strategic proteins (troponin I, phospholamban, various ionic channels, glycolytic enzymes). Acetylcholine, by reducing cAMP formation, exerts opposite actions on these intracellular effectors. Thus, the postjunctional interaction between these autonomic transmitters adds another level of integration to their regulation of heart function (see Figure 5-8).
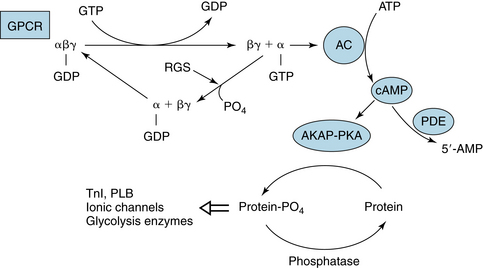
FIGURE 5-8 General reaction scheme for regulation of adenylyl cyclase (AC) by a G protein–coupled receptor (GPCR). Agonist occupancy of GPCR promotes exchange of guanosine triphosphate (GTP) for guanosine diphosphate (GDP) on the α subunit; this exchange stimulates (αs-GTP) or inhibits (αi-GTP) AC activity and synthesis of cyclic adenine monophosphate (cAMP). The βγ subunit of Gi can activate muscarinic potassium (KACh) channels in heart. The deactivation of α subunits is effected by regulators of G proteins (RGSs). Phosphodiesterases (PDEs) regulate cAMP concentration by hydrolysis to 5′-AMP. cAMP activates cAMP-dependent protein kinase (PKA) to phosphorylate proteins in compartments determined by the presence of PKA-kinase–anchoring proteins (AKAPs). The state (phosphorylated/dephosphorylated) of various proteins—troponin I (TnI), phospholamban (PLB), ionic channels, enzymes—regulates their function.
There are many targets at which signals initiated at G protein-coupled receptors (GPCRs) can be modified (see Figure 5-8). Among these are regulators of G protein signaling (RGSs), proteins that inactivate G proteins by augmenting their intrinsic GTPase activity. Thus, the signal from GPCR is reduced when α-GTP is degraded to α-GDP. RGS6 is a protein found in heart muscle, where it interacts with the β subunit to limit signals through Gi. Knockout of RGS6 removes this restraint on inhibition by Gi-mediated signaling. Subsequently, mice displayed a resting bradycardia (reduced heart rate). In atrial cells from these animals, activation of KACh channels by agonist persisted for a longer time. Also, muscarinic agonist caused greater reduction of beating rate of the SA node and prolongation of AV node conduction time. Thus, parasympathetic regulation of nodal function is critically regulated by modifying the transducer function of Gi.
Higher Centers Also Influence Cardiac Performance
Stimulation of various regions of the brain induces dramatic alterations in cardiac rate, rhythm, and contractility. In the cerebral cortex, the centers that regulate cardiac function are mostly in the anterior half of the brain—principally in the frontal lobe, the orbital cortex, the motor and premotor cortex, the anterior part of the temporal lobe, the insula, and the cingulate gyrus. In the thalamus, tachycardia may be induced by stimulation of the midline, ventral, and medial groups of nuclei. Variations in heart rate also may be evoked by stimulating the posterior and posterolateral regions of the hypothalamus.
Stimuli applied to the H2 fields of Forel in the diencephalon elicit various cardiovascular responses, including tachycardia; such changes closely resemble those observed during muscular exercise. Undoubtedly the cortical and diencephalic centers are responsible for initiating the cardiac reactions that occur during excitement, anxiety, and other emotional states. Hypothalamic centers are also involved in the cardiac response to alterations in environmental temperature. Localized temperature changes in the preoptic anterior hypothalamus alter heart rate and peripheral resistance.
Stimulation of the parahypoglossal area of the medulla activates cardiac sympathetic and inhibits cardiac parasympathetic pathways. In certain dorsal regions of the medulla, distinct cardiac accelerator and augmentor sites have been detected in animals with transected vagi. Stimulation of accelerator sites raises heart rate, whereas stimulation of augmentor sites increases cardiac contractility. The accelerator regions were found to be more abundant on the right and the augmentor sites more prevalent on the left. A similar distribution also exists in the hypothalamus. Therefore it appears that for the most part the sympathetic fibers descend the brainstem ipsilaterally.
Heart Rate Can Be Regulated via the Baroreceptor Reflex
Acute changes in arterial blood pressure reflexly elicit inverse changes in heart rate (Figure 5-9) via the baroreceptors located in the aortic arch and carotid sinuses (see also Chapter 9). The inverse relation between heart rate and arterial blood pressure is usually most pronounced over an intermediate range of arterial blood pressures. In the experiment shown in Figure 5-9, this range varied between about 70 and 160 mm Hg. Below the intermediate range of pressures, the heart rate maintains a constant, high value, whereas above this pressure range, it maintains a constant, low value.
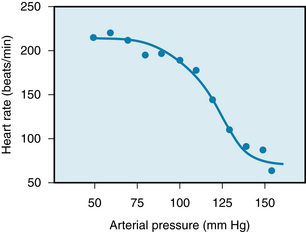
FIGURE 5-9 Heart rate as a function of mean arterial pressure in a group of five conscious, chronically instrumented monkeys. The mean control arterial pressure was 114 mm Hg. Pressure was increased above the control value by infusion of phenylephrine and was decreased below the control value by infusion of nitroprusside.
(Adapted from Cornish KG, Barazanji MW, Yong T, et al: Volume expansion attenuates baroreflex sensitivity in the conscious nonhuman primate. Am J Physiol 257:r595, 1989.)
The effects of changes in carotid sinus pressure on the activity in the cardiac autonomic nerves of an anesthetized dog are shown in Figure 5-10. Alterations in heart rate are achieved by reciprocal changes in vagal and sympathetic neural activity over an intermediate range of arterial pressures (from about 100 to 200 mm Hg). Below this range of arterial blood pressures, the high heart rate is achieved by intense sympathetic activity and the virtual absence of vagal activity. Conversely, above the intermediate range of arterial blood pressures, the low heart rate is achieved by intense vagal activity and a constant low level of sympathetic activity.
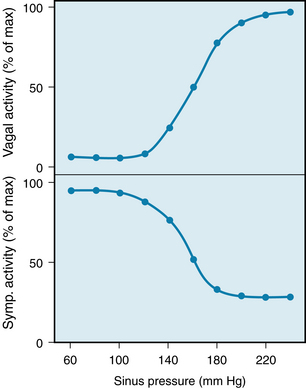
FIGURE 5-10 Changes in neural activity in cardiac vagal and sympathetic (Symp.) nerve fibers induced by changes in pressure in the isolated carotid sinuses in an anesthetized dog.
(Adapted from Kollai M, Koizumi K: Cardiac vagal and sympathetic nerve responses to baroreceptor stimulation in the dog. Pflugers Arch 413:365, 1989.)
The Bainbridge Reflex and Atrial Receptors Regulate Heart Rate
In 1915 Bainbridge reported that infusions of blood or saline accelerated the heart rate in dogs. This increase in heart rate occurred whether arterial blood pressure did or did not rise. Tachycardia was observed whenever central venous pressure rose sufficiently to distend the right side of the heart, and the effect was abolished by bilateral transection of the vagi.
The magnitude and direction of the heart rate changes evoked by the Bainbridge reflex depend on the prevailing heart rate. When the heart rate is slow, intravenous infusions usually accelerate the heart. At more rapid heart rates, however, infusions ordinarily slow the heart. Increases in blood volume not only evoke the Bainbridge reflex but also activate other reflexes (notably the baroreceptor reflex) that tend to change the heart rate in the opposite direction. The actual change in heart rate evoked by an alteration of blood volume is, therefore, the result of these antagonistic reflex effects (Figure 5-11).
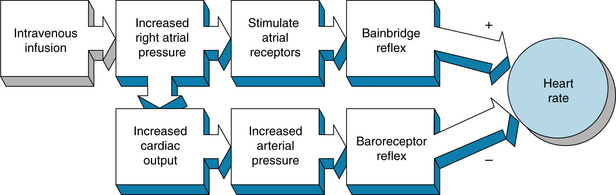
FIGURE 5-11 Intravenous infusions of blood or electrolyte solutions tend to increase heart rate via the Bainbridge reflex and to decrease heart rate via the baroreceptor reflex. The actual change in heart rate induced by such infusions is the result of these two opposing effects.
In unanesthetized dogs, infusions of blood increased heart rate and cardiac output proportionately (Figure 5-12); consequently, stroke volume remained virtually constant. Conversely, reductions in blood volume diminished the cardiac output but increased heart rate. Undoubtedly, the Bainbridge reflex prevailed over the baroreceptor reflex when the blood volume was raised, but the baroreceptor reflex prevailed over the Bainbridge reflex when the blood volume was diminished.
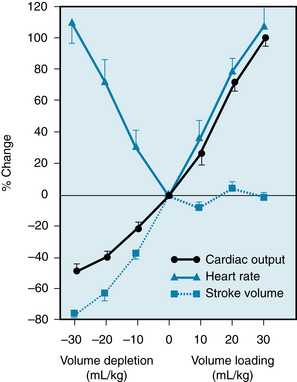
FIGURE 5-12 Effects of blood transfusion and of bleeding on cardiac output, heart rate, and stroke volume in unanesthetized dogs.
(From Vatner SF, Boettcher DH: Regulation of cardiac output by stroke volume and heart rate in conscious dogs. Circ Res 42:557, 1978.)
Both atria have receptors that influence heart rate. They are located principally in the venoatrial junctions—in the right atrium at its junctions with the venae cavae and in the left atrium at its junctions with the pulmonary veins. Distention of these atrial receptors sends impulses centrally in the vagi. The efferent impulses are carried by fibers from both autonomic divisions to the SA node. The cardiac response is highly selective. Even when the reflex increase in heart rate is large, changes in ventricular contractility are negligible. Furthermore, the increase in sympathetic activity is restricted to the heart rate; there is no increase of sympathetic activity to the peripheral arterioles.
Stimulation of the atrial receptors also increases the urine volume. Reduced activity in the renal sympathetic nerve fibers might be partially responsible for this diuresis. However, the principal mechanism appears to be a neurally mediated reduction in the secretion of vasopressin (antidiuretic hormone) by the posterior pituitary gland.
Atrial natriuretic peptide (ANP) is released from storage granules within atrial myocytes in response to increases in blood volume by virtue of the resulting stretch of the atrial walls. ANP, a 28–amino acid peptide, has potent diuretic and natriuretic effects on the kidneys, and it dilates the resistance and capacitance of blood vessels. Thus, ANP is an important regulator of blood volume and blood pressure.
CLINICAL BOX
In congestive heart failure, sodium chloride (NaCl) and water are retained, mainly because of the increased release of aldosterone from the adrenal cortex consequent to stimulation by the renin-angiotensin system. The plasma level of ANP is also increased in congestive heart failure. This peptide acts to enhance the renal excretion of NaCl and water and thereby attenuates the fluid retention and consequent elevations of central venous pressure and cardiac preload.
Respiration Induces a Common Cardiac Dysrhythmia
Rhythmic variations in heart rate, occurring at the frequency of respiration, are detectable in most individuals and tend to be more pronounced in children. Typically, the cardiac rate accelerates during inspiration and decelerates during expiration (see Figure 5-13).
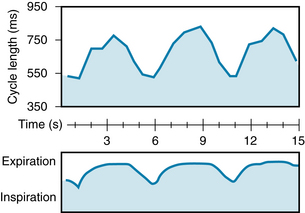
FIGURE 5-13 Respiratory sinus dysrhythmia in a resting, unanesthetized dog. Note that the cardiac cycle length increases during expiration and decreases during inspiration.
(Modified from Warner MR, de Tarnowsky JM, Whitson CC, et al: Beat-by-beat modulation of AV conduction. II. Autonomic neural mechanisms. Am J Physiol 251:H1134, 1986.)
Recordings from cardiac autonomic nerves reveal that activity increases in the sympathetic nerve fibers during inspiration, whereas activity in the vagal nerve fibers increases during expiration (Figure 5-14). Acetylcholine released at the vagal endings is hydrolyzed so rapidly that the rhythmic changes in activity are able to elicit rhythmic variations in heart rate. Conversely, norepinephrine released at the sympathetic endings is removed more slowly, thus damping out the effects of rhythmic variations in norepinephrine release on heart rate. Hence rhythmic changes in heart rate arise almost entirely from oscillations in vagal activity. Respiratory sinus dysrhythmia is exaggerated when vagal tone is enhanced.
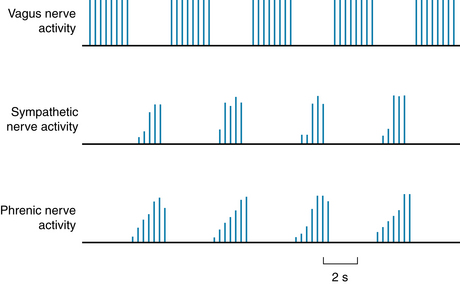
FIGURE 5-14 Respiratory fluctuations in efferent neural activity in the cardiac nerves of an anesthetized dog. Note that the sympathetic nerve activity occurs synchronously with the phrenic nerve discharges (which initiate diaphragmatic contraction), whereas the vagus nerve activity occurs between the phrenic nerve discharges.
(From Kollai M, Koizumi K: Reciprocal and non-reciprocal action of the vagal and sympathetic nerves innervating the heart. J Auton Nerv Syst 1:33, 1979.)
Both reflex and central factors contribute to the genesis of the respiratory cardiac dysrhythmia (Figure 5-15). During inspiration, intrathoracic pressure decreases, and therefore venous return to the right side of the heart is accelerated and right atrial pressure increases (see also Chapter 10) and elicits the Bainbridge reflex (see Figure 5-15). After the time delay required for the increased venous return to reach the left side of the heart, left ventricular output increases and raises arterial blood pressure. This greater pressure, in turn, reduces heart rate reflexly through baroreceptor stimulation (see Figure 5-15).
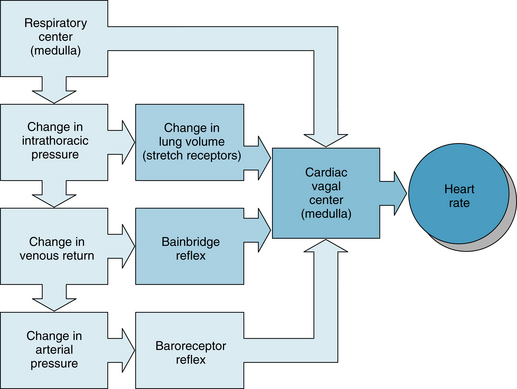
FIGURE 5-15 Respiratory sinus dysrhythmia is generated by a direct interaction between the respiratory and cardiac centers in the medulla as well as by reflexes originating from stretch receptors in the lungs, stretch receptors in the right atrium (Bainbridge reflex), and baroreceptors in the carotid sinuses and aortic arch.
Fluctuations in sympathetic activity to the arterioles cause peripheral resistance to vary at the respiratory frequency. Consequently, arterial blood pressure fluctuates rhythmically, affecting heart rate via the baroreceptor reflex. Stretch receptors in the lungs may also affect heart rate (see Figure 5-15). Moderate pulmonary inflation may increase heart rate reflexly. The afferent and efferent limbs of this reflex are located in the vagus nerves.
Central factors are also responsible for respiratory cardiac dysrhythmia (see Figure 5-15). The medullary respiratory center influences cardiac autonomic centers. In heart-lung bypass experiments conducted on animals, the chest is opened, the lungs are collapsed, venous return is diverted to a pump-oxygenator, and arterial blood pressure is maintained at a constant level. In such experiments, rhythmic movements of the rib cage attest to the activity of the medullary respiratory centers, and the movements of the rib cage are often accompanied by rhythmic changes in heart rate at the respiratory frequency. This respiratory cardiac dysrhythmia is almost certainly induced by an interaction between the respiratory and cardiac centers in the medulla (see Figure 5-15).
Activation of the Chemoreceptor Reflex Affects Heart Rate
The cardiac response to peripheral (or arterial) chemoreceptor stimulation merits special consideration because it illustrates the complexity that may be introduced when one stimulus excites two organ systems simultaneously. In intact animals, stimulation of the arterial chemoreceptors consistently increases ventilatory rate and depth, but heart rate usually changes only slightly. The directional change in heart rate evoked by the peripheral chemoreceptors is related to the enhancement of pulmonary ventilation (Figure 5-16). When respiratory stimulation is mild, heart rate usually diminishes; when the increase in pulmonary ventilation is more pronounced, heart rate usually accelerates.
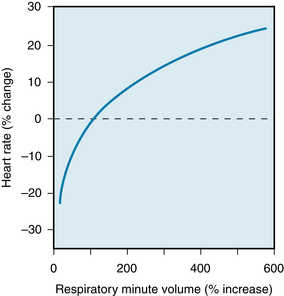
FIGURE 5-16 Relationship between the change in heart rate and the change in respiratory minute volume during carotid chemoreceptor stimulation in spontaneously breathing cats and dogs. When respiratory stimulation was relatively slight, heart rate usually diminished; when respiratory stimulation was more pronounced, heart rate usually increased.
(Modified from Daly MD, Scott MJ: An analysis of the reflex systemic vasodilator response elicited by lung inflation in the dog. J Physiol 144:148, 958.)
The cardiac response to arterial chemoreceptor stimulation is the result of primary and secondary reflex mechanisms (Figure 5-17). The primary reflex effect of arterial chemoreceptor excitation is to stimulate the medullary vagal center and thereby to decrease heart rate. Secondary effects are mediated by the respiratory system. Respiratory stimulation by the arterial chemoreceptors tends to inhibit the medullary vagal center. This inhibitory effect varies with concomitant stimulation of respiration.
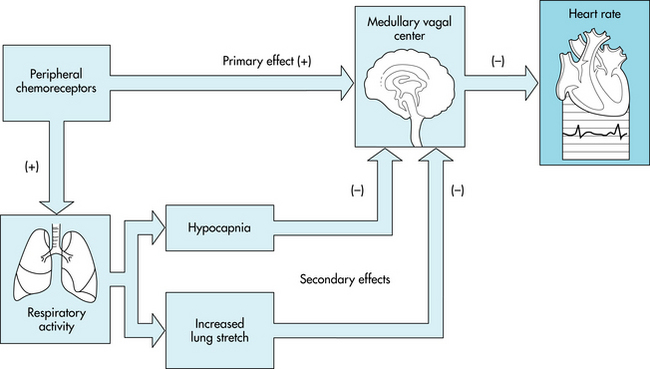
FIGURE 5-17 The primary effect of stimulation of the peripheral chemoreceptors on heart rate is to excite the cardiac vagal center in the medulla and thus to decrease heart rate. Peripheral chemoreceptor stimulation also excites the respiratory center in the medulla. This effect produces hypocapnia and increases lung inflation, both of which secondarily inhibit the medullary vagal center. Thus, these secondary influences attenuate the primary reflex effect of peripheral chemoreceptor stimulation of the heart rate.
An example of the primary inhibitory influence of arterial chemoreceptor stimulation is displayed in Figure 5-18. In this experiment, the lungs were completely collapsed and blood oxygenation was accomplished with an artificial oxygenator. When the carotid chemoreceptors were stimulated, an intense bradycardia and some degree of AV block ensued. These effects are mediated primarily by efferent vagal fibers.
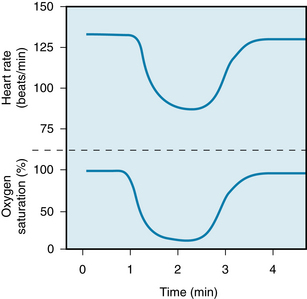
FIGURE 5-18 Changes in heart rate during carotid chemoreceptor stimulation in an anesthetized dog on total heart bypass (upper tracing). The lungs remain deflated, and respiratory gas exchange is accomplished by an artificial oxygenator. The lower tracing represents the oxygen saturation of the blood perfusing the carotid chemoreceptors. The blood perfusing the remainder of the animal, including the myocardium, was fully saturated with oxygen throughout the experiment.
(Modified from Levy MN, Degeest H, Zieske H: Cardiac response to cephalic ischemia. Circ Res 18:67, 1966.)
The hyperventilation that is ordinarily evoked by carotid chemoreceptor stimulation influences heart rate secondarily, both by initiating more pronounced pulmonary inflation reflexes and by producing hypocapnia (see Figure 5-17). Each of these influences tends to depress the primary cardiac response to chemoreceptor stimulation and thereby to accelerate the heart. Hence, when pulmonary hyperventilation is not prevented, the primary and secondary effects tend to neutralize each other, and carotid chemoreceptor stimulation affects heart rate only minimally.
The identical primary vagal inhibitory effect also operates in humans. The electrocardiogram shown in Figure 5-19 was recorded from a quadriplegic patient who could not breathe spontaneously but required tracheal intubation and artificial respiration. When the tracheal catheter was briefly disconnected to permit nursing care, profound bradycardia quickly developed. The heart rate was 65 beats/min just before the tracheal catheter was disconnected. In less than 10 s after cessation of artificial respiration, the heart rate fell to about 20 beats/min. This bradycardia could be prevented by blocking of the effects of efferent vagal activity with atropine, and its onset could be delayed considerably by hyperventilation of the patient before disconnection of the tracheal catheter.
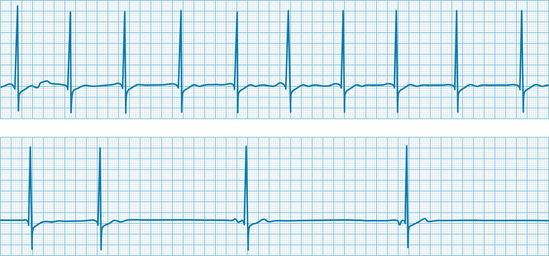
FIGURE 5-19 Electrocardiogram of a 30-year-old quadriplegic man who could not breathe spontaneously and required tracheal intubation and artificial respiration. The two strips are continuous. The tracheal catheter was temporarily disconnected from the respirator at the beginning of the top strip.
(Modified from Berk JL, Levy MN: Profound reflex bradycardia produced by transient hypoxia or hypercapnia in man. Eur Surg Res 9:75, 1977.)
Ventricular Receptor Reflexes Play a Minor Role in the Regulation of Heart Rate
Sensory receptors near the endocardial surfaces of the ventricular walls initiate reflexes similar to those elicited by the arterial baroreceptors. Excitation of these endocardial receptors diminishes heart rate and peripheral resistance. Other sensory receptors have been identified in the epicardial regions of the ventricles. Ventricular receptors are excited by a variety of mechanical and chemical stimuli, but their physiological functions are not clear.
Ventricular receptors are suspected of being involved in the initiation of vasovagal syncope, which is light-headedness or brief loss of consciousness that may be triggered by psychological or orthostatic stress. The ventricular receptors are thought to be stimulated by a reduced ventricular filling volume combined with a vigorous ventricular contraction. In a person standing quietly, ventricular filling is diminished because blood tends to pool in the veins in the abdomen and legs, as explained in Chapter 10. Consequently, the reduction in cardiac output and arterial blood pressure leads to a generalized increase in sympathetic neural activity via the baroreceptor reflex (see Figure 5-10). The enhanced sympathetic activity to the heart evokes a vigorous ventricular contraction, which thereby stimulates the ventricular receptors. Excitation of the ventricular receptors appears to initiate the autonomic neural changes that evoke vasovagal syncope, namely, a combination of a profound, vagally mediated bradycardia and a generalized arteriolar vasodilation mediated by a diminution in sympathetic neural activity.
Myocardial Performance is Regulated by Intrinsic Mechanisms
Just as the heart can initiate its own beat in the absence of any nervous or hormonal control, the myocardium can adapt to changing hemodynamic conditions by mechanisms that are intrinsic to cardiac muscle. Experiments on denervated hearts reveal that this organ adjusts remarkably well to stress. For example, racing greyhounds with denervated hearts perform almost as well as those with intact innervation. Their maximal running speed was found to be only 5% less after complete cardiac denervation. In these dogs the threefold to fourfold increase in cardiac output during exertion was achieved principally by an increase in stroke volume. In normal dogs the increase of cardiac output with exercise is accompanied by a proportionate increase of heart rate; stroke volume does not change much (see Chapter 13). The cardiac adaptation in the denervated animals is not achieved entirely by intrinsic mechanisms; circulating catecholamines undoubtedly contribute. If the β-adrenergic receptors are blocked in greyhounds with denervated hearts, their racing performance is severely impaired.
The intrinsic cardiac adaptation that has received the greatest attention involves changes in the resting length of the myocardial fibers. This adaptation is designated Starling’s law of the heart or the Frank-Starling mechanism. The mechanical, ultrastructural, and physiological bases for this mechanism are explained in Chapter 4.
The Frank-Starling Mechanism Is an Important Regulator of Myocardial Contraction Force
Isolated Hearts
In 1895, Otto Frank described the response of the isolated heart of the frog to alterations in the load on the myocardial fibers just before ventricular contraction. He noted that as the load was increased, the heart responded with a more forceful contraction.
In 1914, Ernest Starling described the intrinsic response of the heart to changes in right atrial and aortic pressure in the canine isolated heart-lung preparation. In this preparation the right ventricular filling pressure is varied by altering the height of a reservoir connected to the right atrium. The ventricular filling pressure just before ventricular contraction constitutes the preload for the myocardial fibers in the ventricular wall (see also Chapter 4). The right ventricle then pumps this blood through the pulmonary vessels to the left atrium. The lungs are artificially ventilated. Blood is pumped by the left ventricle into the aortic arch and then through some external tubing back to the right atrial reservoir. A resistance device in the external tubing allows the investigator to control the aortic pressure; this pressure constitutes the afterload for left ventricular ejection (see also Chapter 4).
One of Starling’s recordings of the changes in ventricular volume evoked by a sudden increase in right atrial pressure is shown in Figure 5-20. Aortic pressure in this experiment was permitted to increase only slightly when the right atrial pressure (preload) was increased. In the top tracing, the upper border of the tracing represents the systolic ventricular volume, the lower border indicates the diastolic ventricular volume, and the width of the tracing reflects the stroke volume (i.e., the volume of blood ejected by a ventricle during each heartbeat).
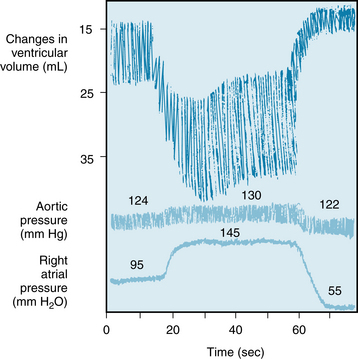
FIGURE 5-20 Changes in ventricular volume in a canine heart-lung preparation when the right atrial pressure was suddenly increased from 95 to 145 mm H2O and subsequently lowered to 55 mm H2O. Note that an increase in ventricular volume is registered as a downward shift in the volume tracing.
(Redrawn from Patterson SW, Piper H, Starling EH: The regulation of the heart beat. J Physiol (Lond) 48:465, 1914.)
For several beats after the rise in preload, the ventricular volume progressively increased. This indicates that a disparity must have existed between ventricular inflow during diastole and ventricular outflow during systole. During a given systole, the volume of blood expelled by the ventricles was not as great as the volume that had entered the ventricles during the preceding diastole. This progressive accumulation of blood dilated the ventricles and lengthened the individual myocardial fibers in the walls of the ventricles. The increased diastolic fiber length somehow facilitates ventricular contraction and enables the ventricles to pump a greater stroke volume, so that cardiac output exactly matches the augmented venous return at steady state. Increased fiber length alters the structure of cardiac constituents. In particular, the structural protein titin may serve as a mechanotransducer because it forms a scaffold around and binds to actin and myosin. As cardiac muscle is stretched, actin and myosin are brought into closer apposition. Thus, length dependence of cardiac performance is achieved mainly by changing the Ca++ sensitivity of the myofilaments and, in part, by changing the number of myofilament crossbridges that can interact (see also Chapter 4). Beyond an optimal fiber length, contraction is actually impaired. Therefore, excessively high filling pressures may depress rather than enhance the pumping capacity of the ventricles by overstretching the myocardial fibers (see Figure 4-4). Such excessive degrees of stretch are rarely encountered under normal conditions.
Changes in diastolic fiber length also permit the isolated heart to compensate for an increase in peripheral resistance. In a Starling preparation, when the arterial resistance is abruptly increased but ventricular filling rate is held constant, the stroke volume diminishes initially in response to the sudden increase in arterial resistance. However, after a brief period, the stroke volume enlarges to equal the constant ventricular filling volume. Concomitantly, the arterial pressure and end-diastolic ventricular volume are greater than the values that prevailed before the experimentally induced increase in afterload. Thus, when the afterload is first increased, the stroke volume ejected by the ventricles during systole is less than the filling volume that enters the ventricles during diastole. The consequent excess of volume in the ventricles stretches the myocardial fibers in the ventricular walls. This increase in myocardial fiber length enables the ventricles to eject a given stroke volume against a greater afterload.
Changes in ventricular volume are also involved in the cardiac adaptation to alterations in heart rate. During bradycardia, for example, the longer duration of diastole permits greater ventricular filling. The consequent augmentation of myocardial fiber length increases stroke volume. Therefore, the reduction in heart rate may be fully compensated by the increase in stroke volume, such that cardiac output may remain constant (see Figure 10-22).
The curves that relate cardiac output to mean atrial pressure for the two ventricles are not coincident; the curve for the left ventricle usually lies below that of the right, as shown in Figure 5-21. At equal right and left atrial pressures (points A and B), right ventricular output would exceed left ventricular output. Hence venous return to the left ventricle (a function of right ventricular output) would exceed left ventricular output, and therefore left ventricular diastolic volume and pressure would rise. By the Frank-Starling mechanism, left ventricular output would increase (from point B toward point C). Only when the outputs of both ventricles are identical (points A and C) would the equilibrium be stable. Under such conditions, however, left atrial pressure (point C) would exceed right atrial pressure (point A), and this is precisely the relationship that ordinarily prevails in normal subjects.
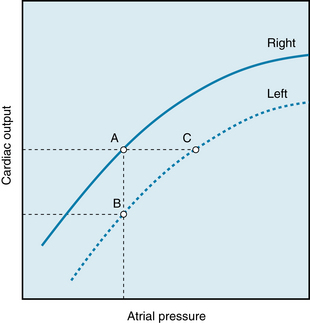
FIGURE 5-21 Outputs of the right and left ventricles as functions of the mean right and left atrial pressure, respectively. At a given level of cardiac output, mean left atrial pressure (e.g., point C) exceeds mean right atrial pressure (point A).
When cardiac compensation involves ventricular dilation, the force required by each myocardial fiber to generate a given intraventricular systolic pressure must be appreciably greater than that developed by the fibers in a nondilated ventricle. The relationship between wall tension and cavity pressure resembles the Laplace relationship for cylindric tubes (see Chapter 8), in that, for a constant internal pressure, wall tension varies directly with the radius. As a consequence, the dilated heart requires considerably more O2 to perform a given amount of external work than does the normal heart.
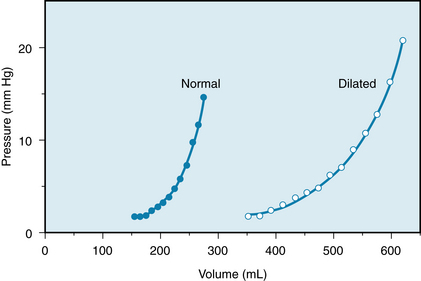
The relatively rigid pericardium that encloses the heart determines the pressure-volume relationship at high levels of pressure and volume. The pericardium exerts this limitation of volume even under normal conditions—when an individual is at rest and the heart rate is slow (see also Chapter 4).
In the cardiac dilation and hypertrophy that accompanies chronic heart failure, the pericardium is stretched considerably (Figure 5-22). The pericardial limitation of cardiac filling is exerted at pressures and volumes entirely different from those in normal individuals.
Intact Preparations
The major problem in assessing the role of the Frank-Starling mechanism in intact animals and humans is the difficulty of measuring end-diastolic myocardial fiber length. The Frank-Starling mechanism has been represented graphically by plotting an index of ventricular performance along the ordinate and an index of fiber length along the abscissa. The most commonly used indexes of ventricular performance are cardiac output, stroke volume, and stroke work. The indexes of fiber length include ventricular end-diastolic volume, ventricular end-diastolic pressure, ventricular circumference, and mean atrial pressure.
The Frank-Starling mechanism is better represented by a family of so-called ventricular function curves than by a single curve. To construct a control ventricular function curve, one must alter the blood volume of an experimental animal over a range of values, and then measure stroke work and end-diastolic pressure at each step. One then makes similar observations during the desired experimental intervention. For example, the ventricular function curve obtained during a norepinephrine infusion lies above and to the left of a control ventricular function curve (Figure 5-23). For a given level of left ventricular end-diastolic pressure, the left ventricle performs more work during a norepinephrine infusion than under control conditions. Hence a shift of the ventricular function curve to the left usually signifies an increase of ventricular contractility (see Chapter 4); a shift to the right usually indicates a reduction of contractility and a consequent tendency toward cardiac failure. Contractility is a measure of cardiac performance at a given level of preload and afterload. The end-diastolic pressure is ordinarily used as an index of preload; the aortic systolic pressure is used as an index of afterload.
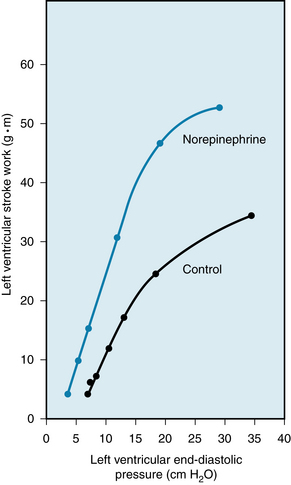
FIGURE 5-23 A constant infusion of norepinephrine in a dog shifts the ventricular function curve to the left. This shift signifies an enhancement of ventricular contractility.
(Redrawn from Sarnoff SJ, Brockman SK, Gilmore JP, et al: Regulation of ventricular contraction: Influence of cardiac sympathetic and vagal nerve stimulation on atrial and ventricular dynamics. Circ Res 8:1108, 1960.)
The Frank-Starling mechanism is ideally suited for matching cardiac output to venous return. Any sudden, excessive output by one ventricle soon increases the venous return to the other ventricle. The consequent increase in diastolic fiber length in the second ventricle augments the output of that ventricle to correspond with the output of its mate. Therefore, it is the Frank-Starling mechanism that maintains a precise balance between the outputs of the right and left ventricles. Because the two ventricles are arranged in series in a closed circuit, even a small, maintained imbalance in the outputs of the two ventricles would be catastrophic.
Changes in Heart Rate Affect Contractile Force
The effects of the frequency of contraction on the force developed in an isometrically contracting cat papillary muscle are shown in Figure 5-24. Initially, the strip of cardiac muscle was stimulated to contract once every 20 s (Figure 5-24A). When the muscle was suddenly made to contract once every 0.63 s, the developed force increased progressively over the next several beats. This progressive increase in developed force induced by a change in contraction frequency is known as the staircase, or Treppe, phenomenon. At the new steady-state level, the developed force was more than five times as great as it was during stimulation at the longer contraction interval. A return to the longer interval (20 s) had the opposite influence on developed force.
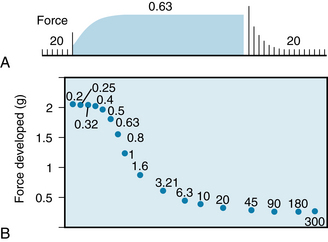
FIGURE 5-24 Changes in force development in an isolated papillary muscle from a cat as the interval between contractions is varied. The numbers in both panels denote the interval (in seconds) between beats.
(Redrawn from Koch-Weser J, Blinks JR: The influence of the interval between beats on myocardial contractility. Pharmacol Rev 15:601, 1963.)
The effect of the interval between contractions on the steady-state level of developed force is shown in Figure 5-24B for a wide range of intervals. As the interval is reduced from 300 s down to about 20 s, developed force changes only slightly. As the interval is reduced further, to a value of about 0.5 s, force increases sharply. Further reduction of the interval to 0.2 s has little additional effect on developed force.
The initial progressive rise in developed force when the interval between beats is suddenly decreased (e.g., from 20 to 0.63 s in Figure 5-24A) is achieved by a gradual increase in intracellular Ca++ content. Two mechanisms contribute to the rise in Ca++ content: (1)! an increase in the number of depolarizations per minute and (2) an increase in the inward Ca++ current per depolarization.
With respect to the first mechanism, Ca++ enters the myocardial cell during each action potential plateau (see Figure 2-20). As the interval between beats is diminished, the number of plateaus per minute increases. Even though the duration of each action potential (and of each plateau) decreases as the interval between beats is reduced (see Figure 2-20), the overriding effect of the higher number of plateaus per minute on the influx of Ca++ would increase the intracellular content of Ca++.
With respect to the second mechanism, as the interval between beats is suddenly diminished, the inward Ca++ current (ICa) progressively increases with each successive beat until a new steady-state level is attained at the new basic cycle length. Figure 5-25 shows that in an isolated ventricular myocyte that was subjected to repetitive depolarizations, the Ca++ current into the myocyte increased on successive beats. Thus, the maximal ICa was considerably greater during the seventh depolarization than it was during the first depolarization. Furthermore, the decay of current (i.e., its inactivation) was substantially slower during the seventh depolarization than during the first depolarization. Both of these characteristics of the ICa would result in a greater Ca++ influx of the myocyte during the seventh depolarization than during the first depolarization. The greater influx of Ca++ would strengthen the contraction by increasing the Ca++ content of the sarcoplasmic reticulum.
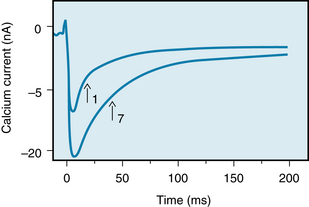
FIGURE 5-25 Calcium currents induced in a guinea pig myocyte during the first and seventh depolarizations in a sequence of depolarizations. Arrows indicate the half-times of inactivation. Note that during the seventh depolarization, the maximal inward Ca++ current and the half-time of inactivation were greater than the respective values for the first depolarization.
(Modified from Lee KS: Potentiation of the calcium-channel currents of internally perfused mammalian heart cells by repetitive depolarization. Proc Natl Acad Sci U S A 84:3941, 1987.)
Transient changes in the intervals between beats also affect the strength of contraction. When a premature ventricular systole (Figure 5-26, beat A) occurs, the premature contraction (extrasystole) itself is feeble, whereas the beat after the subsequent pause (B) is very strong. In intact animals, this response depends partly on the Frank-Starling mechanism. Inadequate ventricular filling just before the premature beat accounts partly for the weak premature contraction. The exaggerated degree of filling associated with the subsequent pause explains in part the vigorous postextrasystolic contraction.
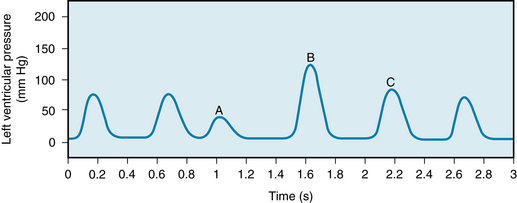
FIGURE 5-26 In an isovolumic canine left ventricle preparation, a premature ventricular systole (beat A) is typically feeble, whereas the postextrasystolic contraction (beat B) is characteristically strong, and the enhanced contractility may persist to a diminishing degree over a few beats (e.g., contraction C). In this preparation the animal is on total heart-lung bypass, and a balloon filled with saline is positioned in the left ventricle.
(From Levy MN: Unpublished tracing.)
The Frank-Starling mechanism is not the exclusive mechanism involved in the usual ventricular adaptation to a premature beat. Directionally similar results are obtained even from isovolumically contracting ventricles. In the experiment shown in Figure 5-26 the coronary circulation was perfused with oxygenated blood; then a balloon was inserted into the left ventricle and filled with enough saline to fill the entire ventricle. Thus, the left ventricle contracts against an incompressible fluid; that is, the ventricle contracts isovolumically. Figure 5-26 shows the changes in pressure that were recorded from this balloon during a series of contractions. Note that the premature beat (A) was much weaker than the preceding beat, and that the postextrasystolic beat (B) that immediately followed the premature beat was very strong. Such enhanced contractility in contraction B is an example of postextrasystolic potentiation, and it may persist for one or more additional beats (e.g., contraction C).
The weakness of the premature beat is directly related to the degree of prematurity. Conversely, as the time (coupling interval) between the premature beat and the preceding beat is increased, the strength of the premature beat is more nearly normal. The curve that relates the strength of contraction of a premature beat to the coupling interval is called a mechanical restitution curve. Figure 5-27 shows the restitution curve obtained by varying the coupling intervals of test beats in an isolated ventricular muscle preparation from a guinea pig.
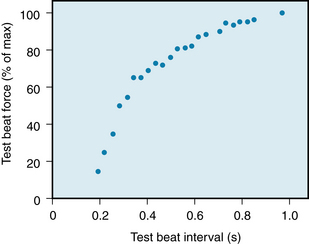
FIGURE 5-27 Force generated during premature contractions in a guinea pig isolated ventricular muscle preparation. The muscle was driven to contract once per second. Periodically, the muscle was stimulated to contract prematurely. The scale along the abscissa denotes the time between the driven and premature beat. The ordinate denotes the ratio of the contractile force of the premature beat to that of the driven beat.
(Modified from Seed WA, Walker JM: Review: Relation between beat interval and force of the heartbeat and its clinical implications. Cardiovasc Res 22:303, 1988.)
The restitution of contractile strength depends on the time course of intracellular Ca++ circulation during the contraction and relaxation process (see Figure 4-8). During relaxation, the Ca++ that dissociates from the contractile proteins is rapidly taken up by the sarcoplasmic reticulum for subsequent release. However, the ryanodine receptor recovers slowly from adaptation or inactivation such that about 500 to 800 ms are required before the Ca++ that had been taken up becomes available for release in response to the next depolarization.
The premature beat itself (Figure 5-26, beat A) is weak because not enough time has elapsed to allow much of the Ca++ taken up by the sarcoplasmic reticulum during the preceding relaxation to become available for release in response to the premature depolarization. Conversely, the postextrasystolic beat (Figure 5-26, beat B) is considerably stronger than normal. The reason is that after the pause between beats A and B, the sarcoplasmic reticulum had available for release the Ca++ that had been taken up during two heartbeats: the extrasystole (beat A) and the preceding normal beat.
Myocardial Performance is Regulated by Nervous and Humoral Factors
Although the completely isolated heart can adapt well to changes in preload and afterload, various extrinsic factors also influence the heart in the intact animal. Under many normal conditions, these extrinsic regulatory mechanisms may overwhelm the intrinsic mechanisms. These extrinsic regulatory factors may be subdivided into nervous and chemical components.
Nervous Control
Sympathetic Influences
Sympathetic nervous activity enhances atrial and ventricular contractility. The effects of increased cardiac sympathetic activity on the ventricular myocardium are asymmetrical. The cardiac sympathetic nerves on the left side of the body usually have a much greater effect on left ventricular contraction than do those on the right side (see Figure 5-6).
Electrical stimulation of the left stellate ganglion markedly raised both the peak pressure and the maximal rate of pressure rise (dP/dt) during systole in an isovolumic left ventricle preparation (Figure 5-28). Also, the duration of systole is reduced and the rate of ventricular relaxation is increased during the early phases of diastole. The shortening of systole and more rapid ventricular relaxation can enhance ventricular filling in the intact circulatory system. For a given cardiac cycle length, the briefer systole allows more time for diastole and hence for ventricular filling. In the experiment shown in Figure 5-29, for example, the animal’s heart was paced at a constant rapid rate. Sympathetic stimulation (right panel) shortened systole, allowing substantially more time for ventricular filling.
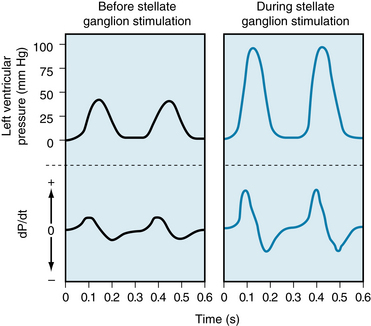
FIGURE 5-28 In a canine isovolumic left ventricle preparation, stimulation of cardiac sympathetic nerves evokes a substantial rise in peak left ventricular pressure and in the maximal rates of intraventricular pressure rise and fall (dP/dt).
(From Levy MN: Unpublished tracing.)
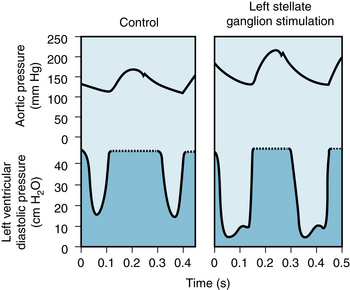
FIGURE 5-29 Stimulation of the left stellate ganglion of a dog increases arterial pressure, stroke volume, and stroke work despite a concomitant reduction in ventricular end-diastolic pressure. Systole is also abbreviated, thereby allowing more time for ventricular filling; the heart was paced at a constant rate. In the ventricular pressure tracings, the pen excursion is limited at 45 mm Hg; actual ventricular pressures during systole can be estimated from the aortic pressure tracings.
(Redrawn from Mitchell JH, Linden RJ, Sarnoff SJ: Influence of cardiac sympathetic and vagal nerve stimulation on the relation between left ventricular diastolic pressure and myocardial segment length. Circ Res 8:1100, 1960.)
Sympathetic nervous activity enhances myocardial performance. Neurally released norepinephrine or circulating catecholamines interact with β-adrenergic receptors on the cardiac cell membranes (see Figure 5-5). This reaction activates adenylyl cyclase, which raises the intracellular cAMP levels. Consequently, protein kinases are activated that promote the phosphorylation of various proteins within the myocyte. Phosphorylation of specific sarcolemmal proteins activates the calcium channels in the myocardial cell membranes. Phosphorylation of phospholamban facilitates the reuptake of systolic Ca++ by the sarcoplasmic reticulum, and phosphorylation of troponin I reduces Ca++ sensitivity. These effects accelerate the relaxation of myocardial cells (see also Chapter 4).
Activation of Ca++ channels increases Ca++ influx during the action potential plateau, and more Ca++ is released from the sarcoplasmic reticulum in response to each cardiac excitation. The contractile strength of the heart is thereby increased. Figure 5-30 shows the correlation between the contractile force developed by a thin strip of ventricular muscle and the Ca++ concentration (as reflected by the aequorin light signal) in the myocytes as the concentration of isoproterenol (a β-adrenergic agonist) was increased in the tissue bath.
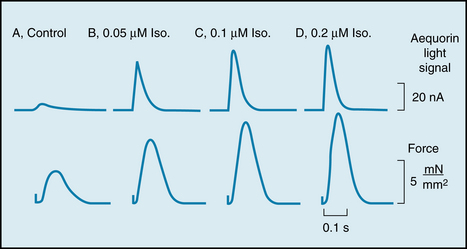
FIGURE 5-30 Effects of various concentrations of isoproterenol (Iso) on the aequorin light signal (in nA) and contractile force (in mN/mm2) in a rat ventricular muscle injected with aequorin. The aequorin light signal reflects the instantaneous changes in intracellular Ca++ concentration.
(Modified from Kurihara S, Konishi M: Effects of beta-adrenoceptor stimulation on intracellular Ca transients and tension in rat ventricular muscle. Pflugers Arch 409:427, 1987.)
The overall effect of increased cardiac sympathetic activity in intact animals can best be appreciated in terms of families of ventricular function curves. When stepwise increases in the frequency of electrical stimulation are applied to the left stellate ganglion, the ventricular function curves shift progressively to the left. The changes parallel those produced by catecholamine infusions (see Figure 5-23). Hence for any given left ventricular end-diastolic pressure, the ventricle is capable of performing more work as the level of sympathetic nervous activity is raised.
During cardiac sympathetic stimulation, the enhanced cardiac response is usually accompanied by a reduction in left ventricular end-diastolic pressure (see Figure 5-29). This reduction in end-diastolic pressure represents a decrease in the preload. The reason for the reduction in ventricular preload is explained in Chapter 10.
Parasympathetic Influences
The vagus nerves inhibit the cardiac pacemaker, atrial myocardium, and AV conduction tissue. The vagus nerves also depress the ventricular myocardium, albeit less markedly. In the isovolumic left ventricle preparation, vagal stimulation decreases the peak left ventricular pressure, maximal rate of pressure development (dP/dt), and maximal rate of pressure decline during diastole (Figure 5-31). In pumping heart preparations, vagal stimulation affects the relation between ventricular performance and preload such that the ventricular function curve shifts to the right.
The vagal effects on the ventricular myocardium are achieved by at least two mechanisms, as shown in Figure 5-5. The acetylcholine (ACh) released from the vagal endings can interact with muscarinic (M) receptors in the cardiac cell membrane to inhibit adenylyl cyclase and the cAMP/protein kinase A (PKA) cascade. This inhibition diminishes the Ca++ conductance of the cardiac cell membrane, reduces phosphorylation of the Ca++ channel, and hence decreases myocardial contractility. The ACh released from vagal endings can also inhibit norepinephrine release from neighboring sympathetic nerve endings (see Figures 5-5 and 5-7).
Baroreceptor Reflex
Stimulation of the carotid sinus and aortic arch baroreceptors changes the heart rate (see Figure 5-9) and also alters myocardial performance. Evidence of reflex alterations of ventricular contractility is presented in Figure 5-32. Ventricular function curves were obtained at four levels of carotid sinus pressure. With each successive rise in pressure, the ventricular function curves were displaced farther and farther to the right, denoting a progressively greater reflex depression of ventricular performance.
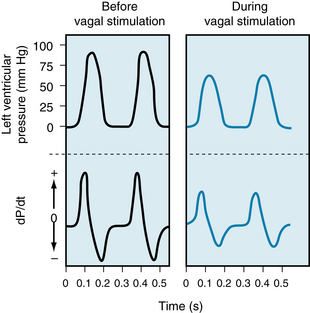
FIGURE 5-31 In an isovolumic left ventricle preparation, when the ventricle is paced at a constant frequency, vagal stimulation decreases the peak left ventricular pressure and diminishes the maximal rates of pressure rise and fall (dP/dt).
(From Levy MN: Unpublished Tracing.)
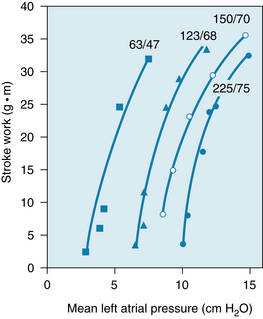
FIGURE 5-32 As the pressure in the isolated carotid sinus is progressively raised, the ventricular function curves shift to the right. The numbers at the tops of each curve represent the systolic/diastolic perfusion pressures (in millimeters of mercury) in the carotid sinus regions of the dog.
(Redrawn from Sarnoff SJ, Gilmore JP, Brockman SK, et al: Regulation of ventricular contraction by the carotid sinus: its effect on atrial and ventricular dynamics. Circ Res 8:1123, 1960.)
Cardiac Performance Is Also Regulated by Hormonal Substances
Hormones
Adrenomedullary Hormones
The adrenal medulla is essentially a component of the autonomic nervous system. Epinephrine is the principal hormone secreted by the adrenal medulla, although it also releases some norepinephrine. The rate of secretion of catecholamines by the adrenal medulla is largely regulated by the same mechanisms that control sympathetic nervous activity. The catecholamine concentrations in the blood rise under the same conditions that activate the sympathoadrenal system. However, the cardiovascular effects of circulating catecholamines are probably minimal under normal resting conditions.
The changes in myocardial contractility induced by norepinephrine infusions have been tested in resting, unanesthetized dogs. The maximal rate of rise of left ventricular pressure (dP/dt), an index of myocardial contractility, was proportional to the norepinephrine concentration in the blood (Figure 5-33). In these same animals, moderate exercise increased the maximal dP/dt by almost 100%, but it raised the circulating catecholamines by only 500 pg/mL. Such a rise in blood norepinephrine concentration, by itself, would have had only a negligible effect on left ventricular dP/dt (see Figure 5-33). Hence the pronounced change in dP/dt observed during exercise must have been achieved mainly by the norepinephrine released from cardiac sympathetic nerve fibers rather than by catecholamines released from the adrenal medulla.
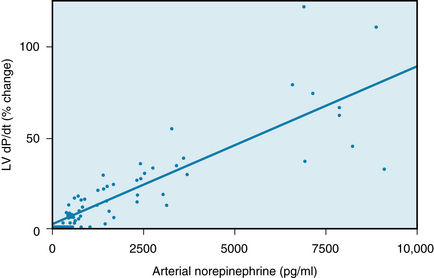
FIGURE 5-33 Effect of norepinephrine infusions on ventricular contractility in a group of resting, unanesthetized dogs. The plasma concentrations of norepinephrine (pg/mL) plotted along the abscissa are the increments above the control values. The maximal rate of rise of left ventricular pressure (Lv dP/dt) is plotted along the ordinate as percentage change from the control value; it is an index of contractility.
(Redrawn from Young MA, Hintze TH, Vatner SF: Correlation between cardiac performance and plasma catecholamine levels in conscious dogs. Am J Physiol 248:H82, 1985.)
Adrenocortical Hormones
The influence of adrenocortical steroids on cardiac function is not well understood. Cardiac muscle removed from adrenalectomized animals and placed in a tissue bath is more likely to fatigue than that obtained from normal animals. In some species the adrenocortical hormones enhance contractility. Furthermore, hydrocortisone potentiates the cardiotonic effects of the catecholamines. This potentiation may be mediated in part by an inhibition of the extraneuronal uptake mechanisms for the catecholamines by the adrenocortical steroids. The adrenocortical hormone aldosterone is synthesized locally in the heart. Aldosterone promotes cardiac hypertrophy in heart failure and, when applied over the long term in large doses, also augments the L-type Ca++ current. How it functions under physiological conditions is not clear.
Thyroid Hormones
Thyroid hormones (levothyroxine, T4; triiodothyronine, T3) enhance heart rate and myocardial contractility in humans. Triiodothyronine is the principal agent responsible for cardiac effects. Some of the effects of thyroid hormone arise from changes in gene expression via activation of transcription factors. Increased activity of sarcoplasmic reticulum Ca++-ATPase and of Na+,K+-ATPase are among these genomic effects. The rates of Ca++ uptake and of adenosine triphosphate (ATP) hydrolysis by the sarcoplasmic reticulum are increased in response to thyroid hormones (hyperthyroidism). Expressions of α-myosin heavy chain with high ATPase activity and of β-adrenergic receptor are also greater and contribute to the higher rate and greater contractility of the heartbeat. Systemically, these effects contribute to increased cardiac output. When in excess, thyroid hormone also reduces systemic vascular resistance and changes preload. Nongenomic effects on ion channels in vascular smooth muscle and heart contribute to this action. Opposite effects occur when thyroid hormones are present at inadequate levels (hypothyroidism). For example, preferential synthesis of β-myosin heavy chains and of phospholamban occurs in thyroid hormone deficiency. Thus, a balanced or euthyroid state is very important in the regulation of cardiovascular function.
CLINICAL BOX
The cardiovascular changes in thyroid dysfunction also depend on indirect mechanisms. Thyroid hyperactivity increases the body’s metabolic rate, and this in turn results in arteriolar vasodilation. The consequent reduction in total peripheral resistance increases cardiac output (see Chapter 10). Substantial evidence indicates that hyperthyroidism increases the density of β-adrenergic receptors in cardiac tissue and that it increases the responsiveness of the heart to sympathetic neural activity. Cardiac activity is sluggish in patients with inadequate thyroid function (hypothyroidism); that is, the heart rate is slow and cardiac output is diminished. The converse is true in patients with overactive thyroid glands (hyperthyroidism). Characteristically, hyperthyroid patients exhibit tachycardia, high cardiac output, palpitations, and dysrhythmias (such as atrial fibrillation).
Insulin
Insulin has a prominent, direct, positive inotropic effect on the human heart. This acute, nongenomic action of insulin appears, in part, to be due to increased Ca++ entry secondary to stimulation of Na+-Ca++ exchange. In addition to increased intracellular Ca++ transients, myofilament sensitivity to Ca++ increases when glucose is present. The effect of insulin is evident even when hypoglycemia is prevented by glucose infusions.
Glucagon
Glucagon has potent positive inotropic and chronotropic effects on the heart. This endogenous hormone probably plays no significant role in the normal regulation of the cardiovascular system, but it has been used to treat various cardiac conditions. The effects of glucagon on the heart closely resemble those of the catecholamines, and certain of their metabolic effects are similar. Both glucagon and catecholamines activate adenylyl cyclase to raise the myocardial tissue levels of cAMP. The catecholamines activate adenylyl cyclase by interacting with β-adrenergic receptors, but glucagon activates this enzyme through a different mechanism.
Blood Gases
Oxygen Changes in O2 tension (Pao2) of the blood perfusing the brain and the peripheral chemoreceptors affect the heart through nervous mechanisms, as described earlier in this chapter. These indirect cardiac effects of hypoxia are usually greater than those caused by the direct effect of hypoxia. Moderate degrees of systemic hypoxia characteristically increase heart rate, cardiac output, and myocardial contractility. These changes are largely mediated by the sympathetic nervous system, and they are effectively abolished by β-adrenergic receptor blockade.
The Pao2 of the blood perfusing the myocardium also influences myocardial performance directly. Moderate to severe degrees of hypoxia depress myocardial contractility. One mechanism for the diminished contractility is reduced sensitivity of the contractile proteins to Ca++. The impaired contractility has also been attributed to higher intracellular concentrations of inorganic phosphate and of H+. Indeed, it is difficult to separate the effect of reduced Pao2 from the acidosis caused by hypoxia.
Carbon Dioxide and AcidosisChanges in Paco2 may also affect the myocardium directly and indirectly. The indirect, neurally mediated effects produced by increased Paco2 are similar to those evoked by a decrease in Pao2.
With respect to the direct effects on the heart, alterations in myocardial performance elicited by changes of Paco2 in the coronary arterial blood are illustrated in Figure 5-34. In intact animals, systemic hypercapnia activates the sympathoadrenal system, which tends to compensate for the direct depressant effect of the greater Paco2 on the heart.
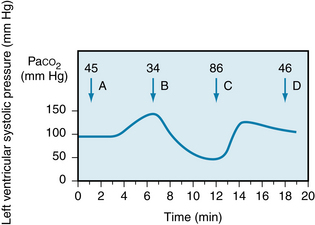
FIGURE 5-34 Decrease in Paco2 increases left ventricular systolic pressure (arrow B) in an isovolumic left ventricle preparation; a rise in Paco2 (arrow C) has the reverse effect. when Paco2 is returned to the control level (arrow D), left ventricular systolic pressure returns to its original value (arrow A).
(From Levy MN: Unpublished tracing.)
Neither the Paco2 nor the blood pH is a primary determinant of myocardial function. The associated change in intracellular pH is the critical factor. The reduced intracellular pH diminishes the the L-type Ca++ current and the amount of Ca++ released from the sarcoplasmic reticulum in response to excitation. The diminished pH also decreases myofilament sensitivity to Ca++ by reducing TnC affinity for Ca++. This effect of acidosis on sensitivity to Ca++ is reflected by a shift in the relationship between developed force and pCa (the negative logarithm of the intracellular Ca++ concentration). Figure 5-35 illustrates such a shift in an experiment on isolated ventricular fibers immersed in a tissue bath. When the pH of the bath was changed from 7.1 to 6.8, the curve of contractile force as a function of pCa was shifted substantially to the right (normal intracellular pH is about 7.1). Furthermore, in this same preparation, at high intracellular Ca++ concentrations (i.e., at values of pCa below about 4.6), a reduction in pH diminishes the maximal developed force. This reduction in maximal force suggests that the low pH depresses the actomyosin interactions by reducing crossbridge efficiency.
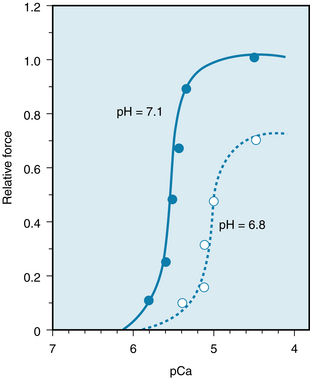
FIGURE 5-35 Effect of pH on the relationship between relative force and pCa (the negative logarithm of the intracellular Ca++ concentration) in a “skinned” ventricular fiber from a rat. Relative force is the force developed by the preparation under the various experimental conditions, expressed as a percentage of the maximal force developed by the preparation when intracellular pH was normal (i.e., 7.1) and pCa was less than 4.6. The “skinned” fiber was prepared by treating the preparation with a detergent to solubilize the cell membranes, thus exposing the contractile proteins in the fiber to the concentrations of H+ and Ca++ that prevailed in the bathing solution.
(Modified from Mayoux E, Coutry N, Lechêne P, et al: Effects of acidosis and alkalosis on mechanical properties of hypertrophied rat heart fiber bundles. Am J Physiol 266:H2051, 1994.)
Summary
 Cardiac function is regulated by intrinsic and extrinsic mechanisms.
Cardiac function is regulated by intrinsic and extrinsic mechanisms.
Sympathetic nervous activity increases heart rate, whereas parasympathetic (vagal) activity decreases heart rate. When both systems are active, the vagal effects usually dominate.
The baroreceptor, chemoreceptor, pulmonary inflation, atrial receptor (Bainbridge), and ventricular receptor reflexes regulate heart rate. The efferent path for the reflexes is the autonomic nervous system.
A change in the resting length of the muscle alters the distance between actin and myosin and thereby influences the subsequent contraction in part by altering the affinity of the myofilaments for Ca++; this is called the Frank-Starling mechanism.
A sustained change in contraction frequency affects the strength of contraction by altering the influx of Ca++ into the cell per minute. A transient change in contraction frequency alters contractile strength because an appreciable delay exists between the time that Ca++ is taken up by the sarcoplasmic reticulum and the time that it becomes available again for release.
The autonomic nervous system regulates myocardial performance mainly by varying the phosphorylation state of proteins (L-type Ca++ channel, phospholamban, troponin I) that regulate the entry and intracellular disposition of Ca++.
Various hormones, including epinephrine, adrenocortical steroids, thyroid hormones, insulin, glucagon, and anterior pituitary hormones, regulate myocardial performance.
Changes in the blood concentrations of O2, CO2, and H+ alter cardiac function directly (by interacting with ion channels and proteins associated with contraction/relaxation) and reflexly (via the central and peripheral chemoreceptors).
Bernardi L., Porta C., Gabutti A., Spicuzza L., Sleight P. Modulatory effects of respiration. Auton Neurosci. 2001;90:47.
Bers D.M. Excitation-Contraction Coupling and Cardiac Contractile Force, ed 2. Boston: Kluwer Academic Publishers; 2001.
Chapleau M.W., Li Z., Meyrelles S.S., Ma X., Abboud F.M. Mechanisms determining sensitivity of baroreceptor afferents in health and disease. Ann N Y Acad Sci. 2001;940:1.
Cini G., Carpi A., Mechanick J., et al. Thyroid hormones and the cardiovascular system: Pathophysiology and interventions. Biomed Pharmacother. 2009;63:742.
Dillmann W.H. Cellular action of thyroid hormone on the heart. Thyroid. 2002;12:447.
Fu Y., Huang X., Piao L., et al. Endogenous RGS proteins modulate SA and AV nodal functions in isolated heart: implications for sick sinus syndrome and AV block. Am J Physiol. 2007;292:H2532.
Iancu R.V., Jones S.W., Harvey R.D. Compartmentation of cAMP signaling in cardiac myocytes. Biophys J. 2007;92:3317.
Jiang C., Rojas A., Wang R., Wang X. CO2 central chemosensitivity: Why are there so many sensing molecules? Respir Physiol Neurobiol. 2005;145:115.
Klein I., Danzi S. Thyroid disease and the heart. Circ. 2007;116:1725.
Lewinski D., Rainer P.P., Gasser R., et al. Glucose-transporter-mediated positive inotropic effects in human myocardium of diabetic and nondiabetic patients. Metabolism. 2010;59:1020.
Posokova E., Wydeven N., Allen K.L. RGS6/Gβ5 complex accelerates IKACh gating kinetics in atrial myocytes and modulates parasympathetic regulation of heart rate. Circ Res. 2010;107:1350.
Potts J.T. Neural circuits controlling cardiorespiratory responses: Baroreceptor and somatic afferents in the nucleus tractus solitarius. Clin Exp Pharmacol Physiol. 2002;29:103.
Scott J.D., Santana L.F. A-kinase anchoring proteins. Circ. 2011;121:1264.
Shepherd J.T., Vatner S.F., eds. Nervous Control of the Heart. Amsterdam: Harwood Academic Press, 1996.
Shimizu J., Todaka K., Burkhoff D. Load dependence of ventricular performance explained by model of calcium-myofilament interactions. Am J Physiol Heart Circ Physiol. 2002;282:H1081.
Stangherlin A., Gesellchen F., Zoccarato A., et al. cGMP signals modulate cAMP levels in a compartment-specific manner to regulate catecholamine-dependent signaling in cardiac myocytes. Circ Res. 2011;108:929.
Taggart P., Critchley H., Lambiase P.D. Heart-brain interactions in cardiac arrhythmia. Br Med J. 2011;97:698.
Yang J., Huang J., Mait B., et al. GRS6, a modulator of parasympathetic activation in heart. Circ Res. 2010;107:1345.
Zhang P., Mende U. Regulators of G-protein signaling in the heart and their potential as therapeutic targets. Circ Res. 2011;109:320.
CASE 5-1
History
A 48-year-old woman was susceptible to occasional, usually brief, episodes of light-headedness. She noticed that her heart rate was very rapid during these episodes and that the light-headedness disappeared when the heart rate returned to normal. Her doctor noted no significant abnormalities on physical examination. Review of a 24-hour recording of the patient’s electrocardiogram revealed a 7-minute period during which the patient’s heart rate increased abruptly from a resting value of about 75 beats/min to a steady level of about 145 beats/min. At the end of the 7-minute period of tachycardia, the heart rate decreased within 1 minute to the resting value of about 75 beats/min. The patient’s problem was diagnosed as paroxysmal supraventricular tachycardia, which is a sudden, pronounced increase in heart rate. This problem is mediated usually by a reentry circuit in the atrioventricular junction.
During a subsequent visit to her doctor, the patient’s paroxysmal tachycardia appeared spontaneously. The doctor was able to terminate the tachycardia promptly with carotid sinus massage (i.e., massaging the patient’s neck just below the angles of the jaw, in the region of the bifurcations of the common carotid arteries). The physician noted that the patient’s arterial blood pressure during tachycardia was 95 mm Hg systolic/75 mm Hg diastolic and that it returned to a value of 130/85 mm Hg (the patient’s usual resting blood pressure) soon after the termination of the tachycardia.
Questions
1. A reduction in the patient’s mean arterial pressure during the paroxysmal tachycardia would cause a reflex:
2. A sudden, substantial increase in efferent vagal activity (induced by carotid sinus massage, for example) would:
3. When the subject was in a normal sinus rhythm, administration of a drug that antagonizes the muscarinic cholinergic receptors would:
4. If the patient had a prominent respiratory sinus dysrhythmia when she was not experiencing paroxysmal tachycardia, the following functional change would take place:
5. When neural activity in the vagus nerves suddenly ceases, the heart rate response to that vagal activity disappears rapidly because: