9 The Peripheral Circulation and its Control
1. Indicate the intrinsic and extrinsic (neural and humoral) factors that regulate peripheral blood flow.
2. Explain autoregulation of blood flow and the myogenic mechanism for local adjustments of blood flow.
3. Elucidate metabolic regulation of blood flow.
4. Explain the role of the sympathetic nerves in blood flow regulation.
5. Describe vascular reflexes in the control of blood flow.
6. Describe the role of humoral agents in the regulation of blood flow.
The Functions of the Heart and Large Blood Vessels
The principal function of the heart is to pump blood to the tissues of the body. However, the distribution of blood to the crucial regions of the body depends on the large and small arteries and arterioles. The regulation of peripheral blood flow is essentially under dual control: centrally, by the nervous system, and locally, in the tissues by the conditions in the immediate vicinity of the blood vessels. The relative importance of the central and local control mechanisms varies among tissues. In some areas of the body, such as the skin and the splanchnic regions, neural regulation of blood flow predominates; in other regions, such as the heart and the brain, local factors are dominant.
The small arteries and arterioles that regulate the blood flow throughout the body are called the resistance vessels. These vessels offer the greatest resistance to the flow of blood pumped to the tissues by the heart. As such, they are important in the maintenance of arterial blood pressure. Smooth muscle fibers are the main component of the walls of the resistance vessels (see Figure 1-2). Hence, the vessel lumen can vary from complete obliteration by strong contraction of the smooth muscle with infolding of the endothelial lining, to maximal dilation by full relaxation of the smooth muscle. At any given time, some resistance vessels are closed by partial contraction (tone) of the arteriolar smooth muscle. If all the resistance vessels in the body dilated simultaneously, blood pressure would fall precipitously. Passive stretch of the microvessels by an increase in intravascular pressure decreases vascular resistance, whereas a decrease in intravascular pressure increases vascular resistance by recoil of the stretched vascular muscle.
Contraction and Relaxation of Arteriolar Vascular Smooth Muscle Regulate Peripheral Blood Flow
Vascular smooth muscle is responsible for the control of total peripheral resistance, arterial and venous tone, and the distribution of blood flow throughout the body. The smooth muscle cells are small, mononucleate, and spindle shaped. They are usually arranged in helical or circular layers around the large blood vessels and in a single circular layer around arterioles (Figure 9-1A and B). Also, parts of endothelial cells project into the vascular smooth muscle layer (myoendothelial junctions) at various points along the arterioles (Figure 9-1C). These projections suggest a functional interaction between endothelium and adjacent vascular smooth muscle.
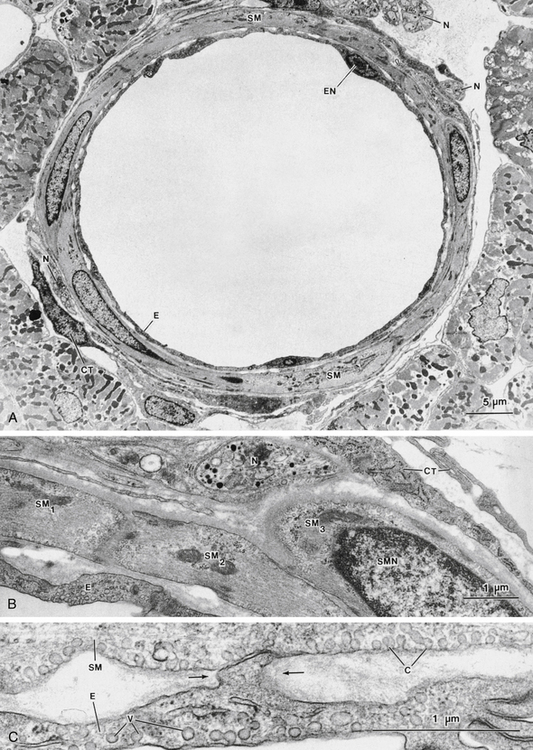
FIGURE 9-1 A, Low-magnification electron micrograph of an arteriole in cross section (inner diameter of approximately 40 µm) in cat ventricle. The wall of the blood vessel is composed largely of vascular smooth muscle cells (SM) whose long axes are directed approximately circularly around the vessel. A single layer of endothelial cells (E) forms the innermost portion of the blood vessel. Connective tissue elements (CT), such as fibroblasts and collagen, make up the adventitial layer at the periphery of the vessel; nerve bundles also appear in this layer (N). EN, endothelial cell nucleus. B, Detail of the wall of the blood vessel in A. This field contains a single endothelial layer (E), the medial smooth muscle layer (with three smooth muscle cell profiles: SM1, SM2, and SM3), and the adventitial layer, containing nerves (N) and connective tissue (CT). SMN, smooth muscle nucleus. C, Another region of the arteriole, showing the area in which the endothelial (E) and smooth muscle (SM) layers are apposed. A projection of an endothelial cell (between arrows) is closely applied to the surface of the overlying smooth muscle, forming a “myoendothelial junction.” Plasmalemmal vesicles (V) are prominent in both the endothelium and the smooth muscle cell (where such vesicles are known as “caveolae” [C]).
In general, the close association between action potentials and contraction observed in skeletal and cardiac muscle cells cannot be demonstrated in vascular smooth muscle. Also, vascular smooth muscle lacks transverse tubules. Graded changes in membrane potential are often associated with changes in force. Contractile activity is generally elicited by neural or humoral stimuli, and the activity of smooth muscle varies in different vessels. For example, some vessels, particularly in the portal or mesenteric circulation, contain longitudinally oriented smooth muscle. This muscle is spontaneously active, and it displays action potentials that are correlated with the contractions and the electrical coupling between cells.
Vascular smooth muscle cells contain large numbers of thin actin filaments and small numbers of thick myosin filaments. These filaments are aligned in the long axis of the cell, but they do not form visible sarcomeres with striations. Nevertheless, the sliding filament mechanism is believed to operate in this tissue, and phosphorylation of crossbridges regulates their rate of cycling. Compared with skeletal muscle, the smooth muscle contracts very slowly, develops high forces, and maintains force for long periods. The adenosine triphosphate (ATP) utilization is diminished, and it operates over a considerable range of lengths under physiological conditions. Cell-to-cell conduction occurs via gap junctions, as it does in cardiac muscle (see p. 57).
In smooth muscle, the interaction between myosin and actin, which leads to contraction, is controlled by the myoplasmic Ca++ concentration, as it is in cardiac and skeletal muscle. The molecular mechanism by which Ca++ regulates contraction in smooth muscle (Figure 9-2) is fundamentally different, however, because smooth muscle does not utilize the Ca++-binding regulatory protein troponin. For smooth muscle crossbridges to be activated to cycle, the 20-kDa regulatory light chain of myosin (MLC20, a protein subunit of myosin) must be phosphorylated. MLC20 is phosphorylated by myosin light-chain kinase (MLCK) and de-phosphorylated by myosin light-chain phosphatase (MLCP). This requirement for phosphorylation provides a means to regulate contraction in smooth muscle in addition to that in cardiac and skeletal muscles, because both MLCK and MLCP are themselves regulated by other kinases. MLCK is activated by a complex between 4 Ca++ and the Ca++-binding messenger protein calmodulin (CaM), which is present in high abundance in smooth muscle cells. The concentration of Ca++/calmodulin, and thus the activation of MLCK, is driven by the cytoplasmic Ca++ concentration (i.e., “‘Ca++ ’activation” of contraction). However, the level of phosphorylation of the MLC20 is also determined by MLCP. Inhibiting the activity of MLCP has the effect of increasing contraction, even when cytoplasmic Ca++ (and MLCK activity) does not change, because inhibition of MLCP results in increased phosphorylation of MLC20. Inhibition of MLCP activity thus increases the “‘Ca++ ’sensitivity” of contraction. Conversely, stimulation of MLCP activity decreases MLC20 phosphorylation and contraction, even at a constant Ca++, and thus decreases Ca++ sensitivity of contraction.
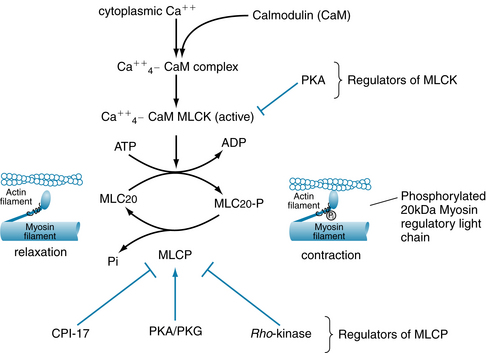
FIGURE 9-2 For smooth muscle actin-myosin crossbridges to cycle and generate force or shortening, the 20-kDa myosin regulatory light-chain subunit (MLC20) of myosin must be phosphorylated. Phosphorylation of MLC20 is controlled by myosin light-chain kinase (MLCK) and myosin light-chain phosphatase (MLCP). MLCK activity is inhibited by protein kinase A (PKA). MLCP activity is inhibited by Rho-kinase and CPI-17 (17-kDa C-kinase potentiated protein phosphatase-1 inhibitor). Pi, inorganic phosphate, released from MLC20-P by the phosphatase action of MLCP; PKA, cyclic adenosine monophosphate (AMP)–dependent protein kinase; PKG, cyclic guanosine monophosphate (GMP)–dependent protein kinase.
MLCP is inhibited primarily by rho-kinase (a regulator of the cytoskeleton in many types of cells). Rho-kinase is activated in a signaling cascade that begins with activation of certain G-protein–coupled receptors (GPCRs) on the surface membrane. Another protein, CPI-17 (17-kDa C-protein–potentiated inhibitor of protein phosphatase), which is activated by protein kinase C (PKC), also inhibits MLCP. Activity of MLCP may also be increased, particularly by nitric oxide (NO), through cyclic GMP and protein kinase G (PKG), and by cyclic adenosine monophosphate (AMP), acting through protein kinase A (PKA). The release of NO by endothelial cells, and subsequent stimulation of smooth muscle MLCP, constitutes a major mechanism by which endothelium may cause smooth muscle relaxation and arterial or venous dilation. In summary, regulation of smooth muscle contraction by neurotransmitters, circulating hormones, and autocoids often involves both changes in “‘Ca++ ’activation” of contraction (MLCK) and in Ca++ sensitivity of contraction (MLCP) (see later). The contractile state of smooth muscle is thus governed finally by the ratio of Ca++-activated MLCK activity to MLCP activity, because this ratio determines the level of phosphorylation of MLC20.
Cytoplasmic Ca++ is Regulated to Control Contraction, via MLCK
The cytoplasmic Ca++ concentration, and thus MLCK activity, is determined by the summation of the Ca++ that enters the cytosol (influx) and that leaving the cytosol (efflux) (Figure 9-3). Ca++ enters the cytosol in two ways: (1) from the extracellular space, via influx through voltage-operated calcium channels (typically L-type, activated by depolarization), receptor-operated calcium channels (ROCs, activated after the action of agonists on membrane receptors), and store-operated calcium channels (activated after depletion of sarcoplasmic reticulum Ca++ stores ), and (2) from the sarcoplasmic reticulum (SR) via activation of either ryanodine receptor channels, RyRs or inositol-1,4,5 triphosphate (IP3) receptor channels (IP3-stimulated) located on the SR. Ca++ ions leave the cytosol via ATP-driven calcium transporters (i.e., Ca++ pumps) located on both the SR (termed SERCAs) and plasma membrane (termed PMCAs) as well as activation of the Na/Ca exchangers on the plasma membrane as in cardiac muscle (see Figure 4-8).
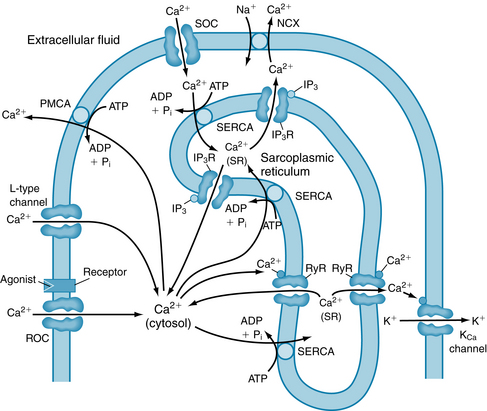
FIGURE 9-3 Control of cytoplasmic calcium ion ([Ca++], shown as Ca2+ here) in vascular smooth muscle. Calcium can enter the cell via electrically activated channels (e.g., voltage-operated Ca++ channel, L-type Ca++ channel), via receptor-operated channels (ROCs) in the sarcolemma, or via store-operated channels (SOCs). SOCs open when the sarcoplasmic reticulum (SR) becomes depleted of Ca++. Calcium is also released from the sarcoplasmic reticulum through ryanodine receptor channels (RyRs) and through inositol triphosphate receptors (IP3Rs) in response to inositol triphosphate (IP3) stimulation and is taken back into the SR by a calcium pump (SERCA). Calcium is extruded from the cell by a plasma membrane calcium pump (PMCA) and by the Na-Ca exchanger (NCX). Membrane potential is determined by KCa channels, which may be activated by local Ca++, and by ROCs, which are permeable to both Na+ and Ca++. ADP, adenosine diphosphate; ATP, adenosine triphosphate; MLCK, myosin light-chain kinase; MLCP, myosin light-chain phosphatase; Pi, inorganic phosphate.
(Modified from Blaustein MP, Kao JPY, Matteson DR: Cellular Physiology, Philadelphia, 2004, Mosby.)
Contraction is Controlled By Excitation-Contraction Coupling and/or Pharmacomechanical Coupling
Cellular responses to agonist vary among different blood vessels as well as smooth muscle types. This diversity arises partly from differences in ion channels that may be present (such as potassium and calcium channels important in excitation-contraction [E-C] coupling) and in agonist-specific receptors (such as those that bind angiotensin II, norepinephrine, serotonin, histamine, and acetylcholine) expressed on the plasma membrane of vascular smooth muscle.
Excitation-Contraction Coupling in Vascular Smooth Muscle
The potential across the plasma membrane of vascular smooth muscle is an important determinant of cytoplasmic Ca++, and thus of contractile state of vascular smooth muscle. The reason is that both entry of Ca++ into the cell and extrusion of Ca++ from the cell are voltage-dependent (involving voltage-dependent Ca++ channels and sodium/calcium exchange [NCX]). Depolarization increases Ca++ influx and decreases Ca++ efflux. In many types of arteries, membrane potential is strongly influenced by the transmural pressure (i.e., the blood pressure) through the “‘myogenic’” mechanism (discussed later in this chapter), with increases in pressure tending to cause depolarization and consequent entry of Ca++ through voltage-dependent Ca++ channels. Membrane potential is, however, always strongly influenced by K+ channels, with activation of K+ channels tending to hyperpolarize the membrane and inhibition of K+ channels tending to depolarize it. Some types of vascular smooth muscle produce action potentials in which the depolarizing current is carried by voltage-dependent (L-type) Ca++ channels. The resting membrane potential of vascular smooth muscle is determined primarily by K+ permeability because of the relative abundant expression of K+ channels. Stimuli that open K+ channels can alter membrane potential by altering K+ efflux across the plasma membrane (since resting potassium concentration inside the cell, [K+]i, is greater than that outside the cell, [K+]o). Opening of K+ channels causes hyperpolarization (due to increased K+ efflux) of the membrane potential, whereas closure of K+ channels causes depolarization (due to decreased K+ efflux). L-type calcium channels are voltage-sensitive and are activated (i.e., opened) by membrane depolarization, resulting in influx of extracellular Ca++ and elevating intracellular Ca++ concentration. If the increase in Ca++ levels is sufficient, then the Ca++/calmodulin complex activates MLCK, promoting actin-myosin interaction and contraction. Thus, changes in membrane potential alter extracellular Ca++ influx and efflux, modulate intracellular cytoplasmic Ca++, and affect vascular smooth muscle contraction, artery diameter, and vascular resistance.
Pharmacomechanical Coupling
Contraction of arteries and veins is very importantly modulated by hormones, neurotransmitters, and autocoids acting on receptors located in the plasma membrane (Figure 9-4). Most of these receptors are GPCRs. Pharmacomechanical (PM) coupling is a mechanism of contractile activation that causes little or no change in membrane potential (making it distinct from E-C coupling). Pharmacomechanical coupling can result either in contraction (Figure 9-4A) or relaxation (Figure 9-4B). The three major components of PM coupling are (1) the GPCR itself, (2) the coupling complex, and (3) the second messenger (e.g., cyclic AMP or IP3). G-protein coupled receptors are characterized by the ligands they bind (e.g., adrenergic [catecholamines], serotinergic [serotonin], cholinergic [acetylcholine]) and by the particular heterotrimeric G-proteins to which they are coupled. Important G-proteins coupled to GPCRs on vascular smooth muscle cells include Gαq/11, Gα12/13, Gαs, and Gαi/o. The G-protein α subunits, and sometimes the G-protein βγ subunits, activate specific effector molecules, such as kinases. In many cases, second messenger molecules are ultimately generated that stimulate downstream cellular mechanisms (ion influx, Ca++ release, enzyme activation or inhibition). G-protein–coupled receptors are important regulators of cardiovascular function, with many drugs acting on them. (It has been estimated that, overall, about 40% of currently used drugs act on GPCRs in the cardiovascular system and elsewhere in the body).
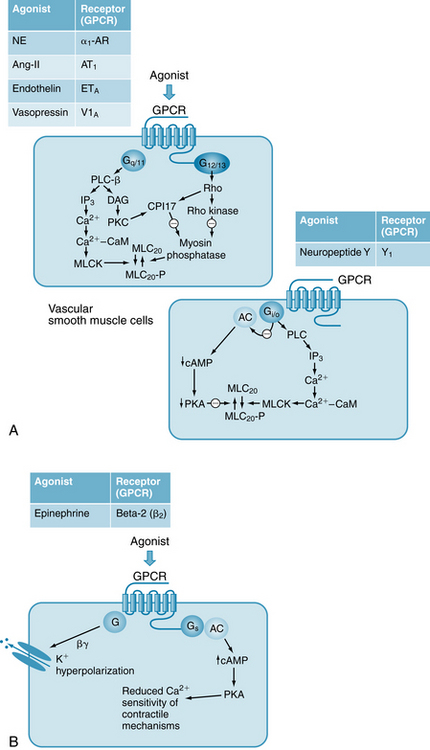
FIGURE 9-4 Key physiological neurotransmitters and hormones that control vascular tone, their receptors and intracellular mechanisms. A, Physiological agonists bind to G-protein–coupled receptors (GPCRs) that are coupled to different G proteins and to different effector molecules and second messengers. These pathways all result in vasoconstriction. The α subunits of the G-proteins are: Gαq/11, Gα12/13, and Gαi/o. AC, adenyl cyclase; CaM, calmodulin; CPI-17, 17-kDa C-protein–potentiated inhibitor of protein phosphatase; DAG, diacylglycerol; IP3, inositol triphosphate; MLC20, 20-kDa myosin regulatory light chain; MLCK, myosin light-chain kinase; MLCP, myosin light-chain phosphatase (also known as protein phosphatase); PKC, protein kinase C; PLC-β, phospholipase C-β. B, Epinephrine binds to the GPCR β2-AR (β2-adrenoceptor, AR) to promote vasodilation through increased cyclic adenosine monophosphate (cAMP) and activation of K+ channels. β2-AR is coupled to the G-protein GαS, which inhibits MLCK via phosphorylation by its effector, cAMP-dependent protein kinase A (PKA).
Control of Vascular Tone by Catecholamines
The catecholamines norepinephrine (NE) and epinephrine are key controllers of cardiovascular function. Norepinephrine is released from sympathetic nerve endings in the heart and blood vessels and acts locally, whereas epinephrine is released from the adrenal medulla and circulates in the blood to act widely throughout the body. In general, the GPCRs that bind catecholamines are known as “‘adrenoceptors’,” a term deriving from the fact that these receptors all bind the adrenal medullary hormones, epinephrine and NE, although with varying affinities. The adrenoceptors are large and diverse family of GPCRs, consisting of five major types (α1, α2, β1, β2, β3) and several subtypes, and a full discussion of their function and pharmacology is reserved for pharmacology texts. In arteries and veins, NE binds most strongly to α1-adrenoceptors (α1-ARs), which are GPCRs located in the sympathetic neuroeffector junctions, on the smooth muscle cell membrane. These receptors couple to both Gαq/11 and Gα12/13 (see Figure 9-4A), and their activation results in contraction (PM coupling). Gαq/11 activates phospholipase C (PLC), which catalyzes the production of diacylglycerol (DAG) and IP3 from PIP2 (phosphatidylinositol 4,5-bisphosphate). Diacylglycerol activates PKC, which in turn activates CPI-17, which inhibits MLCP. IP3 activates IP3 receptors (IP3Rs) on the SR membrane to release Ca++, which binds to CaM and activates MLCK. Thus, phosphorylation of MLC20 is increased as a result of increased “Ca++ activation” of contraction, and as a result of increased “Ca++ sensitivity” of contraction. Release of NE from sympathetic nerve endings, and the subsequent contraction of vascular smooth muscle, is a major way in which vascular resistance and vascular capacitance (through contraction of small veins) are regulated. (Note that heart tissue contains few α1-ARs, and there NE acts primarily on β1 receptors, which are coupled to Gas, increasing cAMP and strengthening cardiac contraction (see Figures 5-23 and 5-28). Many vascular tissues contain few or no β1-ARs). Most of the arteries and veins of the body are innervated solely by fibers of the sympathetic nervous system, and thus neurally released NE plays a major role in controlling vascular function, through sympathetic nerve activity directed by the central nervous system. The sympathetic nerve fibers release NE to exert a tonic vasoconstrictor effect on the blood vessels, as evidenced by the fact that cutting or freezing the sympathetic nerves to a vascular bed (such as muscle) increases the blood flow. Activation of the sympathetic nerves either directly or reflexly (see pp. 184 and 185) enhances vascular resistance. (In contrast to the sympathetic nerves, the parasympathetic nerves tend to decrease vascular resistance. However, these nerves innervate only a small fraction of the blood vessels in the body, mainly in certain viscera and pelvic organs.)
Epinephrine is released from the adrenal medulla and circulates in the blood. Epinephrine acts most potently on β2-ARs on vascular smooth muscle and causes vasodilation, through increases in cAMP (see Figure 9-4B) and reduced “Ca++ sensitivity” of contraction. At very high concentrations in the plasma, however, epinephrine also binds to a1-ARs to cause contraction and vasoconstriction, overriding its effects mediated by the vascular β2-ARs. In the heart, both epinephrine and NE bind equally well to β1-ARs.
Control of Vascular Contraction by Other Hormones, Other Neurotransmitters, and Autocoids
Angiotensin II has many actions, most acting to increase arterial blood pressure. It is a direct vasoconstrictor, acting primarily on AT1 receptors, which are coupled to both both Gαq/11 and Gα12/13 (see Figure 9-4A). Endothelin, a 21–amino acid peptide, acts primarily on ETA receptors on vascular smooth muscle to cause vasoconstriction (see Figure 9-4A). Endothelin is synthesized and released from endothelial cells. Vasopressin, or antidiuretic hormone (ADH), is a neurohypophysial hormone that is a potent constrictor that is called into play to elevate blood pressure (and conserve fluid volume) particularly as a compensatory mechanism in hemorrhage. Vasopressin binds primarily to the V1A receptor, which is coupled to Gαq/11 and Gα12/13. Neuropeptide Y is a sympathetic neurotransmitter that is co-released with NE from sympathetic nerve endings on vascular smooth muscle and binds primarily to the Y1 receptor, which is coupled to Gαi/o. Activation of Y1 thus can decrease cAMP and enhance contraction by decreasing the PKA-mediated inhibition of MLCP.
Intrinsic Control of Peripheral Blood Flow
Autoregulation and the Myogenic Mechanism Tend to Keep Blood Flow Constant
Blood flow is adjusted to the existing metabolic activity of the tissue. Furthermore, imposed changes in the arterial perfusion pressure at constant levels of tissue metabolism are met with vascular resistance changes that maintain a constant blood flow. This mechanism is commonly referred to as the autoregulation of blood flow, which is illustrated in Figure 9-5. In the skeletal muscle preparation from which these data were derived, the muscle was completely isolated from the rest of the animal and was in a resting state. From a control pressure of 100 mm Hg, the pressure was abruptly increased or decreased, and the blood flows observed immediately after changing the perfusion pressure are represented by the closed circles in the figure. Maintenance of the altered pressure at each new level was followed within 30 to 60 seconds by a return of flow toward the control levels; the open circles represent these steady-state flows. Over the pressure range of 20 to 120 mm Hg, the steady-state flow is relatively constant. Calculation of resistance across the vascular bed (pressure/flow) during steady-state conditions indicates that, with elevation of perfusion pressure, the resistance vessels have constricted, whereas with reduction of perfusion pressure, they have dilated.
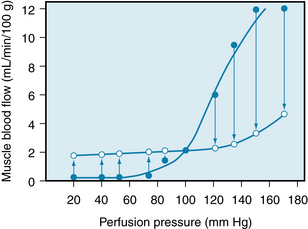
FIGURE 9-5 Pressure-flow relationship in the skeletal muscle vascular bed of the dog. Closed circles represent the flows obtained immediately after abrupt changes in perfusion pressure from the control level (point where lines cross). Open circles represent the steady-state flows obtained at the new perfusion pressure.
(Redrawn from Jones RD, Berne RM: Intrinsic regulation of skeletal muscle blood flow. Circ Res 14:126, 1964.)
The reason for this constancy of blood flow in the presence of an altered perfusion pressure is not known. It is explained best by the myogenic mechanism. According to this mechanism, the vascular smooth muscle contracts in response to an increase in transmural pressure, and it relaxes in response to a decrease in transmural pressure. Therefore, the initial flow increment is produced by an abrupt increase in perfusion pressure. This increase passively distends the blood vessels. The distention is followed by a return of flow to the previous control level by contraction of the vascular smooth muscles.
An example of a myogenic response is shown in Figure 9-6. Arterioles isolated from the hearts of young pigs were cannulated at each end, and the transmural pressure (intravascular pressure minus extravascular pressure) and flow through the arteriole could be adjusted to desired levels. With no flow through the arteriole, successive increases in transmural pressure elicited progressive decreases in the vessel diameter (Figure 9-6A). This effect was independent of the endothelium, because the responses were identical in intact vessels and in vessels that were denuded of endothelium (Figure 9-6B). Arterioles that were relaxed by direct action of nitroprusside on the vascular smooth muscle showed only a passive increase in diameter when transmural pressure was increased. In vascular smooth muscle having spontaneous action potentials (arterioles, portal vein), the action potential frequency increases with stretch to cause greater Ca++ influx via electromechanical coupling. In vascular smooth muscle lacking spontaneous action potentials, stretch activates plasma membrane Ca++ channels to elevate intracellular Ca++. The nature of the Ca++ sensor that allows this process is not completely understood, but it allows an increase in transmural pressure to activate membrane calcium channels. Indeed, membrane depolarization itself has been shown to activate phospholipase C and thereby generate second messengers.
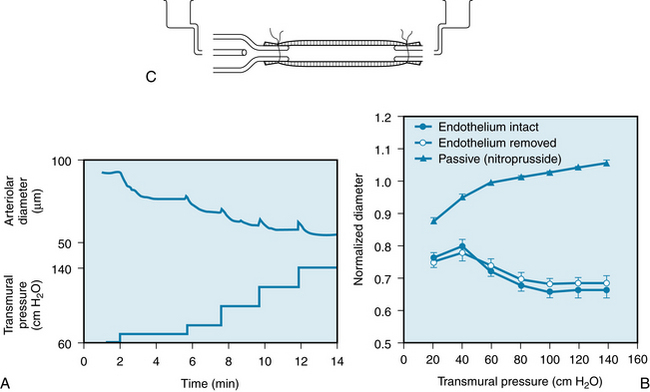
FIGURE 9-6 A, Constriction of an isolated cardiac arteriole in response to increases in transmural pressure without flow through the blood vessel. B, Constrictor response of the arteriole to an increase in transmural pressure is unaffected by removal of its endothelium. C, Diagram of cannulated arteriole. When the smooth muscle is relaxed by nitroprusside, the arteriole is passively distended by the increase in transmural pressure.
(Redrawn from Kuo L, Davis MJ, Chilian WM: Endothelium-dependent, flow-induced dilation of isolated coronary arterioles. Am J Physiol 259:H1063, 1990.)
It would be expected that operation of a myogenic mechanism would be minimized, because blood pressure is reflexly maintained at a fairly constant level under normal conditions. However, when a person changes position (e.g., from lying to standing), a large change in transmural pressure occurs in the lower extremities. The precapillary vessels constrict in response to this imposed stretch. Consequently, flow ceases in most capillaries. After flow stops, capillary filtration diminishes until the increase in plasma oncotic pressure and the increase in interstitial fluid pressure balance the elevated capillary hydrostatic pressure that is produced by changing from a horizontal to a vertical position (see Chapters 8 and 10).
The Endothelium Actively Regulates Blood Flow
Stimulation of the endothelium elicits a response of the underlying vascular smooth muscle. To demonstrate this response, transmural pressure is kept constant in an isolated arteriole. A perfusion fluid reservoir connected to one end of an arteriole is raised to increase intravascular pressure. A reservoir that is connected to the other end of the arteriole is lowered simultaneously by an equal distance. This maneuver increases the longitudinal pressure gradient along the vessel, and flow-mediated vasodilation occurs (Figure 9-7A). If the arteriole is denuded of endothelium, the dilation of the vessel in response to increased flow is abolished (Figure 9-7B). The mechanism of flow-mediated vasodilation is ascribed to endothelial-derived hyperpolarization factor (EDHF), which comprises several soluble components released from endothelial cells in response to shear stress secondary to the increase in velocity of flow.
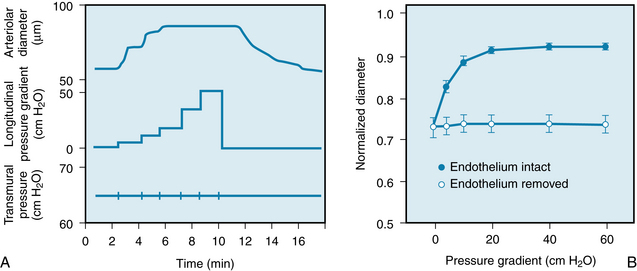
FIGURE 9-7 A, Flow-induced vasodilation in an isolated cardiac arteriole at constant transmural pressure. Flow was increased progressively by increasing the pressure gradient in the long axis of the arteriole (longitudinal pressure gradient). B, Flow-induced vasodilation is abolished by removal of the endothelium of the arteriole.
(Redrawn from Kuo L, Davis MJ, Chilian WM: Endothelium-dependent, flow-induced dilation of isolated coronary arterioles. Am J Physiol 259:H1063, 1990.)
The mechanism of EDHF’s action includes a hyperpolarization of the endothelial cell itself as well as of adjacent vascular smooth muscle. Hyperpolarization of the endothelial cell is initiated by activation of small (KCa 2.3) and intermediate (KCa 3.1) conductance K+ channels from which K+ leaves the cell. The rise of extracellular K+ also stimulates the Na+,K+-ATPase, an action that contributes to endothelial cell hyperpolarization. The hyperpolarization is transmitted to adjacent vascular smooth muscle cells via myoendothelial cell gap junctions. The endothelial cell also releases soluble factors (NO, prostacyclin, H2O2, and epoxyeicosatrienoic acid) that cause hyperpolarization of vascular smooth muscle by activation of large conductance KCa channels (BKCa). This action moves the membrane potential farther from the threshold at which Ca++ entry occurs. EDHF becomes an increasingly important as a vasodilator in the microcirculation as vessel radius decreases.
Tissue Metabolic Activity Is the Main Factor in the Local Regulation of Blood Flow
According to the metabolic mechanism, any intervention that results in an O2 supply that is inadequate for the requirements of the tissue gives rise to the formation of vasodilator metabolites. These metabolites are released from the tissue, and they act locally to dilate the resistance vessels. When the metabolic rate of the tissue increases or the O2 delivery to the tissue decreases, more vasodilator substance is released and the metabolite concentration in the tissue increases.
Many substances have been proposed as mediators of metabolic vasodilation. Some of the earliest substances suggested are lactic acid, CO2, and hydrogen ions. However, the decrease in vascular resistance induced by supernormal concentrations of these dilator agents falls considerably short of the dilation observed under conditions of increased metabolic activity.
Changes in O2 tension can evoke changes in the contractile state of vascular smooth muscle; an increase in Po2 elicits contraction, whereas a decrease in Po2 evokes relaxation. If significant reductions in the intravascular Po2 occur before the arterial blood reaches the resistance vessels (diffusion through the arterial and arteriolar walls; see p. 157), small changes in O2 supply or in consumption could elicit contraction or relaxation of the resistance vessels. However, direct measurements of Po2 at the resistance vessels indicate that over a wide range of Po2 (11 to 343 mm Hg), there is no correlation between O2 tension and arteriolar diameter. Furthermore, if Po2 were directly responsible for vascular smooth muscle tension, one would not expect to find a parallelism between the duration of arterial occlusion and the duration of the reactive hyperemia (Figure 9-8). In response to either short occlusions (5 to 10 seconds) or long occlusions (1 to 3 minutes), the venous blood becomes bright red (well-oxygenated) within 1 or 2 seconds after release of the arterial occlusion. Hence the smooth muscle of the resistance vessels must be exposed to a high Po2 in each instance. Nevertheless, the longer occlusions result in longer periods of reactive hyperemia. These observations are more compatible with the release of a vasodilator metabolite from the tissue than with a direct effect of Po2 on the vascular smooth muscle.
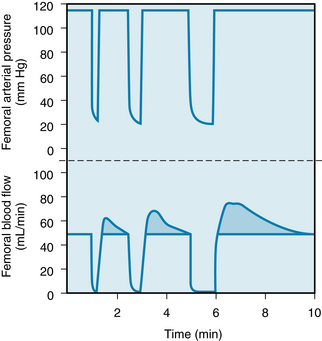
FIGURE 9-8 Reactive hyperemia in the hindlimb of the dog after 15-, 30-, and 60-second occlusions of the femoral artery.
(From Berne RM: Unpublished observations.)
Potassium ions, inorganic phosphate, and interstitial fluid osmolarity can also induce vasodilation. Because K+ and phosphate are released and osmolarity is increased during skeletal muscle contraction, these factors may contribute to active hyperemia (i.e., increased blood flow caused by enhanced tissue activity). However, significant increases of phosphate concentration and osmolarity are not consistently observed during muscle contraction, and they may produce only transient increases in blood flow. Therefore, they are not likely candidates as mediators of the vasodilation observed with muscular activity. Potassium release occurs with the onset of skeletal muscle contraction or with an increase in cardiac activity. This could be responsible for the initial decrease in vascular resistance observed with increased cardiac work. However, K+ release is not sustained, despite continued arteriolar dilation throughout the period of enhanced muscle activity. Therefore some other agent must mediate the vasodilation associated with the greater metabolic activity of the tissue. Reoxygenated venous blood obtained from active cardiac and skeletal muscles under steady-state conditions of exercise does not elicit vasodilation when infused into a test vascular bed. It is difficult to see how oxygenation of the venous blood could alter its K+ or phosphate content or its osmolarity and thereby destroy its vasodilator effect.
Adenosine, which is involved in coronary blood flow regulation, may also participate in the control of the resistance vessels in skeletal muscle. Furthermore, nitric oxide (NO) and prostaglandins have a role in flow-induced dilation of isolated perfused arterioles. In one study, wild-type (WT) mice and knockout (KO) mice (mice with deletion of the gene for encoding endothelial NO synthase) showed the same degree of arteriolar dilation to enhanced flow. In another, NG-nitro-l-arginine methyl ester, an inhibitor of NO synthase, partially reduced the increased flow response in WT mice, but it had no effect on the response in the KO mice. Indomethacin, an inhibitor of prostaglandin synthesis, elicited partial reduction of flow-induced dilation in WT mice, but it abolished the response in KO mice. Combined administration of l-arginine and indomethacin abolished flow-induced dilation in the WT and in the KO mice.
Thus, a number of candidates exist for the role of mediator of metabolic vasodilation. The relative contribution of each of the various factors remains the subject for future investigation. Several factors may be involved in any given vascular bed, and different factors predominate in different tissues.
Metabolic control of vascular resistance by the release of a vasodilator substance is predicated on the existence of basal vessel tone. This tonic activity, or basal tone, of the vascular smooth muscle is readily demonstrable. However, in contrast to tone in skeletal muscle, it is independent of the nervous system. The factors responsible for basal tone in blood vessels are not known, but one or more of the following factors may be involved: (1) an expression of myogenic activity in response to the stretch imposed by the blood pressure, (2) the high O2 tension of arterial blood, (3) the presence of Ca++, or (4) some unknown factor in plasma, because addition of plasma to the bathing solution of isolated vessel segments evokes partial contraction of the smooth muscle.
If arterial inflow to a vascular bed is discontinued briefly, the blood flow, on release of the occlusion, immediately exceeds the flow before occlusion. The period of reactive hyperemia only gradually returns to the control level. This response is illustrated in Figure 9-8: Blood flow to the leg was stopped by clamping the femoral artery for 15, 30, and 60 seconds. Release of the 60-second occlusion resulted in a peak blood flow that was 70% greater than the control flow, and the flow returned to the control level within about 110 seconds. When this same experiment is performed in humans by inflating a blood pressure cuff on the upper arm, dilation of the resistance vessels of the hand and forearm, immediately after release of the cuff, is evident from the bright red color of the skin and the fullness of the veins. Within limits, the peak flow and particularly the duration of the reactive hyperemia are proportional to the duration of the occlusion (see Figure 9-8). If the extremity is exercised during the occlusion period, reactive hyperemia increases. These observations, and the close relationship that exists between metabolic activity and blood flow in the unoccluded limb, are consonant with a metabolic mechanism in the local regulation of tissue blood flow.
When the vascular smooth muscle of the arterioles relaxes in response to vasodilator metabolites released by a decrease in the O2 supply/O2 demand ratio of the tissue, resistance may diminish in the arteries that feed these arterioles. This results in a greater blood flow than is produced by arteriolar dilation alone. Two possible mechanisms can account for this coordination of arterial and arteriolar dilation. First, vasodilation in the microvessels is propagated, and when initiated in the arterioles, it can propagate from arterioles back to arteries. Second, the metabolite-mediated dilation of the arterioles accelerates blood flow in the feeder arteries. This increases the shear stress on the arterial endothelium and thereby induces vasodilation.
Extrinsic Control of Peripheral Blood Flow is Mediated Mainly by the Sympathetic Nervous System
Impulses that Arise in the Medulla Descend in the Sympathetic Nerves to Increase Vascular Resistance
Several regions in the medulla influence cardiovascular activity. Some of the effects of stimulation of the dorsal lateral medulla (pressor region) are vasoconstriction, cardiac acceleration, and enhanced myocardial contractility. Caudal and ventromedial to the pressor region is a zone whose stimulation decreases the blood pressure. This depressor area exerts its effect by direct spinal inhibition and also by inhibition of the medullary pressor region. These areas constitute a center—not in an anatomic sense in that a discrete group of cells is discernible, but, in a physiologic sense, in that the stimulation of the pressor region produces the responses described previously. From the vasoconstrictor regions, fibers descend in the spinal cord and synapse at different levels of the thoracolumbar region (T1 to L2 or L3). Fibers from the intermediolateral gray matter of the cord emerge with the ventral roots, but they leave the motor fibers to join the paravertebral sympathetic chains through the white communicating branches. These preganglionic white (myelinated) fibers may pass up or down the sympathetic chains to synapse in the various ganglia. Postganglionic gray branches (unmyelinated) then join the corresponding segmental spinal nerves and accompany them to the periphery to innervate the arteries and veins. Postganglionic sympathetic fibers from the various ganglia join the large arteries and accompany them as an investing network of fibers to the resistance vessels (small arteries and arterioles) and capacitance vessels (veins).
The vasoconstrictor regions are tonically active, and reflexes or humoral stimuli that enhance this activity increase the frequency of impulses that reach the terminal branches to the vessels. At this location, a constrictor neurotransmitter (norepinephrine) is released and elicits constriction (α-adrenergic effect) of the resistance vessels. Inhibition of the vasoconstrictor areas reduces their tonic activity. Hence the frequency of impulses in the efferent nerve fibers diminishes, resulting in vasodilation. In this manner, neural regulation of the peripheral circulation is accomplished primarily by alteration of the frequency of impulses that pass down the vasoconstrictor fibers of the sympathetic nerves to the blood vessels. The vasomotor regions may show rhythmic changes in tonic activity that are manifested as oscillations of arterial pressure. Some oscillations occur at the frequency of respiration (Traube-Hering waves), and they are caused by an increase in sympathetic impulses to the resistance vessels coincident with inspiration. Other oscillations occur at a lower frequency than that of respiration (Mayer waves).
Sympathetic Nerves Regulate the Contractile State of the Resistance and Capacitance Vessels
The vasoconstrictor fibers of the sympathetic nervous system supply the arteries, arterioles, and veins. However, neural influence on the larger vessels is far less important than it is on the arterioles and small arteries. Capacitance vessels are more responsive to sympathetic nerve stimulation than are resistance vessels; they reach the maximal constriction at a lower frequency of stimulation than do the resistance vessels. However, capacitance vessels respond less to vasodilator metabolites. NE is the neurotransmitter released at the sympathetic nerve terminals in the blood vessels. Many factors, such as circulating hormones and in particular locally released substances, modify the liberation of norepinephrine from the vesicles of the nerve terminals.
The response of the resistance and capacitance vessels to stimulation of the sympathetic fibers is illustrated in Figure 9-9. At constant arterial pressure, sympathetic fiber stimulation reduces blood flow (constriction of the resistance vessels) and decreases blood volume of the tissues (constriction of the capacitance vessels). The abrupt decrease in tissue volume shown in the figure was caused by the movement of blood out of the capacitance vessels and out of the hindquarters of the cat, whereas the late, slow, progressive decline in volume (to the right of the arrow) was caused by the movement of extravascular fluid into the capillaries and, hence, away from the tissue. The loss of tissue fluid is a consequence of the lowered capillary hydrostatic pressure brought about by constriction of the resistance vessels. A new equilibrium of the forces responsible for filtration and absorption was established across the capillary wall (see p. 165).
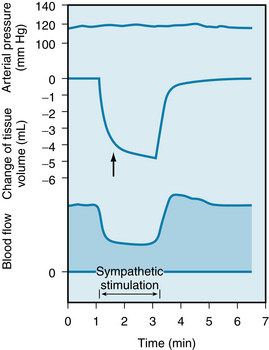
FIGURE 9-9 Effect of sympathetic nerve stimulation (2 Hz) on blood flow and tissue volume in the hindquarters of the cat. The arrow denotes the change in slope of the tissue volume curve where the volume decrease caused by emptying of capacitance vessels ceases and loss of extravascular fluid becomes evident.
(Redrawn from Mellander S: Comparative studies on the adrenergic neuro-hormonal control of resistance and capacitance blood vessels in the cat. Acta Physiol Scand 50[Suppl 176]:1, 1960.)
Section of the sympathetic nerves to an extremity, as might occur during peripheral arterial surgery, abolishes sympathetic vascular tone, thereby increasing limb flow. With time, vascular tone is regained by an increase in basal (intrinsic) tone.
Approximately one third of the blood volume of a tissue can be mobilized on stimulation of the sympathetic nerves at basal physiological frequencies. The basal tone is very low in capacitance vessels. When the veins are denervated, only small increases in volume are obtained with maximal doses of acetylcholine. Therefore, the blood volume at basal tone is close to the maximal blood volume of the tissue. More blood can be mobilized from the skin than from the muscle capacitance vessels. This action depends in part on the greater sensitivity of the skin vessels to sympathetic stimulation, but also because basal tone is lower in skin vessels than in muscle vessels. Therefore in the absence of neural influence, the skin capacitance vessels contain more blood than do the muscle capacitance vessels.
Blood is mobilized from capacitance vessels in response to physiological stimuli. In exercise, activation of the sympathetic nerve fibers constricts the veins and hence augments the cardiac filling pressure. In arterial hypotension, as induced by hemorrhage, the capacitance vessels constrict and thereby aid in overcoming the associated decrease in central venous pressure. Furthermore, the resistance vessels constrict in hemorrhage shock, and thereby help to restore the arterial pressure (see Chapter 13). Also, extravascular fluid is mobilized by a greater reabsorption of fluid from the tissues into the capillaries in response to the lowered capillary hydrostatic pressure that is caused by the lower arterial pressure.
Neural and humoral stimuli can exert similar or dissimilar effects on different segments of the vascular tree. In so doing, they can alter blood flow, tissue blood volume, and extravascular volume to meet the physiological requirements of the organism.
The Parasympathetic Nervous System Innervates Blood Vessels Only in the Cranial and Sacral Regions of the Body
The efferent fibers of the cranial division of the parasympathetic nervous system supply blood vessels of the head and viscera, whereas fibers of the sacral division supply blood vessels of the genitalia, bladder, and large bowel. Skeletal muscle and skin do not receive parasympathetic innervation. Because only a small proportion of the resistance vessels of the body receive parasympathetic fibers, the effect of these cholinergic fibers on total vascular resistance is small.
Epinephrine and Norepinephrine Are the Main Humoral Factors That Affect Vascular Resistance
Epinephrine and norepinephrine exert a profound effect on the peripheral blood vessels. In skeletal muscle, epinephrine in low concentrations dilates resistance vessels (β-adrenergic effect), and in high concentrations, it produces constriction (α-adrenergic effect). In skin, only vasoconstriction is obtained with epinephrine, whereas in all vascular beds the primary effect of norepinephrine is vasoconstriction. When stimulated, the adrenal gland can release epinephrine and norepinephrine into the systemic circulation. However, under physiologic conditions, the effect of catecholamine release from the adrenal medulla is less important than the norepinephrine released by sympathetic nerve activation.
The Vascular Reflexes Are Responsible for Rapid Adjustments of Blood Pressure
Areas of the medulla that mediate sympathetic and vagal effects are under the influence of neural impulses that arise in the baroreceptors, chemoreceptors, hypothalamus, cerebral cortex, and skin. These areas of the medulla are also affected by changes in the blood concentrations of CO2 and O2.
Arterial Baroreceptors
The baroreceptors (or pressoreceptors) are stretch receptors located in the carotid sinuses (slightly widened areas of the internal carotid arteries at their points of origin from the common carotid arteries) and in the aortic arch (Figure 9-10). Impulses that arise in the carotid sinus travel up the sinus nerve to the glossopharyngeal nerve and, via the latter, to the nucleus of the tractus solitarius (NTS) in the medulla. The NTS is the site of central projection of the chemoreceptors and baroreceptors. Stimulation of the NTS inhibits sympathetic nerve impulses to the peripheral blood vessels (depressor effect), whereas lesions of the NTS produce vasoconstriction (pressor effect). Impulses arising in the pressoreceptors of the aortic arch reach the NTS via afferent fibers in the vagus nerves. The pressoreceptor nerve terminals in the walls of the carotid sinus and aortic arch respond to the stretch and deformation of the vessel induced by the arterial pressure. The frequency of firing is enhanced by an increase in blood pressure and diminished by a reduction in blood pressure. An increase in impulse frequency, as occurs with a rise in arterial pressure, inhibits the vasoconstrictor regions. This inhibition results in peripheral vasodilation and a lowering of blood pressure. Bradycardia, elicited by stimulation of the vagal nuclei in the medulla, contributes to a lowering of the blood pressure.
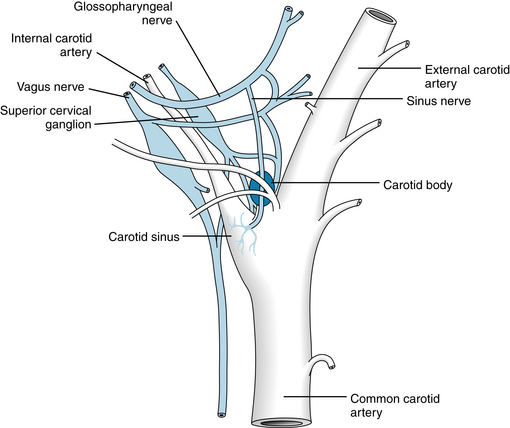
FIGURE 9-10 Diagrammatic representation of the carotid sinus and carotid body and their innervation in the dog.
(Redrawn from Adams WE: The comparative morphology of the carotid body and carotid sinus, Springfield, Ill, 1958, Charles C Thomas.)
The carotid sinus and aortic baroreceptors are not equipotent in their effects on peripheral resistance in response to nonpulsatile alterations in blood pressure. Baroreceptors in the carotid sinus are more sensitive than are those in the aortic arch. Changes in pressure in the carotid sinus evoke greater alterations in systemic arterial pressure than do equivalent changes in aortic arch pressure. However, with pulsatile changes in blood pressure, the two sets of baroreceptors respond similarly.
The carotid sinus, with the sinus nerve intact, can be isolated from the rest of the circulation and perfused by either a donor animal or an artificial perfusion system. Under these conditions, changes in the pressure within the carotid sinus are associated with reciprocal changes in the blood pressure of the experimental animal. The receptors in the walls of the carotid sinus display adaptation. Therefore, they respond more to constantly changing pressures than to sustained constant pressures. This is illustrated in Figure 9-11, which shows that at normal levels of mean blood pressure (about 100 mm Hg) a barrage of impulses from a single fiber of the sinus nerve is initiated in early systole by the pressure rise. Only a few spikes are observed during late systole and early diastole. At lower pressures, these phasic changes are even more evident, and the overall frequency of discharge is reduced. The blood pressure threshold for eliciting sinus nerve impulses is about 50 mm Hg. A maximal, sustained firing is reached at around 200 mm Hg.
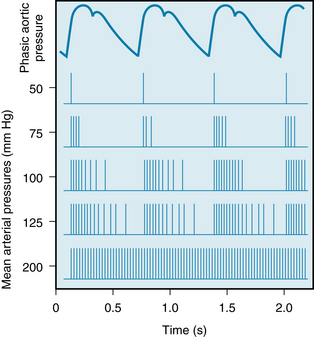
FIGURE 9-11 Relationship of phasic aortic blood pressure in the firing of a single afferent nerve fiber from the carotid sinus at different levels of mean arterial pressure.
Because the pressoreceptors show some degree of adaptation, their response at any level of mean arterial pressure is greater with a large than with a small pulse pressure. This is illustrated in Figure 9-12, which shows the effects of damping pulsations in the carotid sinus on the frequency of firing in a the sinus nerve fiber and on the systemic arterial pressure. When the pulse pressure in the carotid sinuses is reduced with an air chamber, but mean pressure remains constant, the rate of electrical impulses recorded from a sinus nerve fiber decreases and the systemic arterial pressure increases. Restoration of the pulse pressure in the carotid sinus restores the frequency of sinus nerve discharge and systemic arterial pressure to control levels (see Figure 9-12).
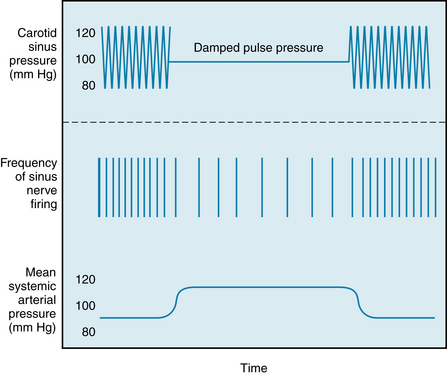
FIGURE 9-12 Effect of reducing pulse pressure in the vascularly isolated perfused carotid sinuses (top record) on impulses recorded from a fiber of a sinus nerve (middle record) and on mean systemic arterial pressure (bottom record). Mean pressure in the carotid sinuses (horizontal line, top record) is held constant when pulse pressure is damped.
The increases in resistance that occur in the peripheral vascular beds in response to reduced pressure in the carotid sinus vary from one vascular bed to another and thereby produce a redistribution of blood flow. For example, in the dog the resistance changes elicited by altering carotid sinus pressure around the normal operating sinus pressure are greatest in the femoral vessels, less in the renal, and least in the mesenteric and celiac vessels. Furthermore, the sensitivity of the carotid sinus reflex can be altered. Local application of norepinephrine, or stimulation of sympathetic nerve fibers to the carotid sinuses, enhances the sensitivity of the receptors in the sinus. Hence, a given increase in intrasinus pressure produces a greater depressor response. A decrease in baroreceptor sensitivity occurs in hypertension, when the carotid sinus becomes stiffer and less deformable as a result of the high intra-arterial pressure. Under these conditions, a given increase in carotid sinus pressure elicits a smaller decrement in systemic arterial pressure than it does at normal levels of blood pressure. In other words, the set-point of the baroreceptors is raised in hypertension, such that the threshold is increased and the receptors are less sensitive to change in transmural pressure.
As would be expected, denervation of the carotid sinuses can produce temporary, and in some instances prolonged, hypertension. The arterial baroreceptors play a key role in short-term adjustments of blood pressure, when relatively abrupt changes in blood volume, cardiac output, or peripheral resistance occur (as in exercise). However, long-term control of blood pressure—that is, over days, weeks, and longer—is determined by the fluid balance of the individual, namely, the balance between fluid intake and fluid output. By far the single most important organ in the control of body fluid volume, and hence of blood pressure, is the kidney. With overhydration, excessive fluid intake is excreted, whereas with dehydration, there is a marked reduction in urine output.
In some individuals the carotid sinus is quite sensitive to pressure. Hence tight collars or other forms of external pressure over the region of the carotid sinus may elicit marked hypotension and fainting.
CLINICAL BOX
Arterial blood pressure is maintained upon assuming an upright position because the tendency for blood to pool in peripheral vessels is opposed by the myogenic mechanism in arterioles (see Figure 9-6), by compression of veins by contracting skeletal muscle (see Figure 12-2), and by baroreceptor-mediated reflexes (see Figure 9-10) that modulate the output of the autonomic nervous system. Orthostatic or postural hypotension occurs when arterial blood pressure decreases (>10-20 mm Hg) when a subject moves from a supine to an upright position. Malfunction of the autonomic nervous system can be one of several various causes of this disorder. Thus, the increased heart rate (see Figure 5-9) and vasoconstriction (see Figure 9-9) arising from sympathetic nervous activity are less than optimal, and blood pressure falls in the upright position. Orthostatic hypotension can arise from primary autonomic failure in which either preganglionic or postganglionic sympathetic neurons degenerate. The reduction of transmitter release at the ganglionic synapse or at the neuroeffector junction impairs the reflex adjustments needed to sustain arterial pressure. Medical management of orthostatic hypotension due to primary autonomic failure includes increased salt and fluid consumption, physical exercises before rising, and elastic support stockings. In more severe cases, drugs that activate α-adrenergic receptors (midodrine) or cause fluid retention (fludrocortisone) are employed. Secondary autonomic failure can occur in subjects having peripheral neuropathies involving autonomic neurons, as in diabetes. Orthostatic hypotension is also caused by drugs used to treat hypertension (diuretics, drugs that block α-adrenergic receptors or Ca++ channels on vessels). Adjustment of dose can alleviate the condition.
Cardiopulmonary Baroreceptors
In addition to the carotid sinus and aortic baroreceptors, there are cardiopulmonary receptors with vagal and sympathetic afferent and efferent nerves. These cardiopulmonary reflexes are tonically active, and they can alter peripheral resistance with changes in intracardiac, venous, or pulmonary vascular pressures. The receptors are located in the atria, ventricles, and pulmonary vessels.
The atria contain two types of receptors: those activated by the tension developed during atrial contraction (A receptors) and those activated by the stretch of the atria during atrial filling (B receptors). Stimulation of these atrial receptors sends impulses up vagal fibers to the vagal center in the medulla. Consequently, the sympathetic activity is decreased to the kidney and increased to the sinoatrial (SA) node. These changes in sympathetic activity increase renal blood flow, urine flow, and heart rate.
Activation of the cardiopulmonary receptors can also lower blood pressure reflexly by inhibiting the vasoconstrictor center in the medulla. Stimulation of the receptors inhibits angiotensin, aldosterone, and vasopressin (ADH) release; and interruption of the reflex pathway has the opposite effects. Changes in urine volume elicited by changes in cardiopulmonary baroreceptor activation are important in the regulation of blood volume. For example, a decrease in blood volume (hypovolemia), as occurs in hemorrhage, enhances sympathetic vasoconstriction in the kidney and increases secretion of renin, angiotensin, aldosterone, and ADH. The renal vasoconstriction (primarily afferent arteriolar) reduces glomerular filtration and increases renin release from the kidney. Renin acts on a plasma substrate to form angiotensin I, a decapeptide. In turn, angiotensin I is metabolized to angiotensin II by angiotensin-converting enzyme. Angiotensin II increases aldosterone release from the adrenal cortex, which in turn enhances retention of NaCl. The enhanced release of ADH increases water reabsorption. The net result is retention of salt and water by the kidney and a sensation of thirst. Angiotensin II also causes vasoconstriction to raise systemic arteriolar tone.
The Peripheral Chemoreceptors Are Stimulated by Decreases in Blood Oxygen Tension and pH and by Increases in Carbon Dioxide Tension
The peripheral chemoreceptors consist of small, highly vascular bodies in the region of the aortic arch (aortic bodies) and in the carotid bodies just medial to the carotid sinuses (see Figure 9-10). Although they are primarily concerned with the regulation of respiration, the chemoreceptors reflexly influence the vasomotor regions to a minor degree. A reduction in arterial blood O2 tension (Pao2) stimulates the chemoreceptors. The consequent increase in the number of impulses in the afferent nerve fibers from the carotid and aortic bodies stimulates the vasoconstrictor regions. This action increases the tone of the resistance and capacitance vessels.
Stimulation of the peripheral chemoreceptors by increased arterial blood CO2 tension (Paco2) and reduced pH elicit a reflex response that is minimal compared with the direct effect of hypercapnia (high Paco2) and of H+ on the vasomotor regions in the medulla. When hypoxia and hypercapnia coexist (asphyxia), the stimulation of the chemoreceptors is greater than the sum of the two gas stimuli when each acts alone. The effects of asphyxia on blood pressure, heart rate, and respiration are shown in Figure 9-13 (see also Figures 5-14 and 5-15).
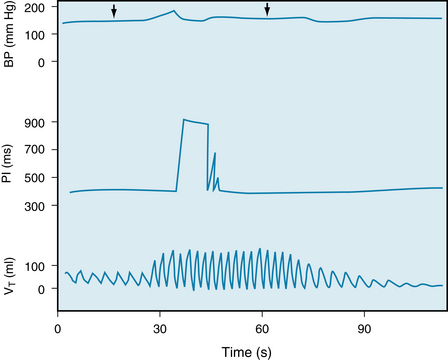
FIGURE 9-13 Effects of stimulation of the isolated perfused carotid body chemoreceptors at constant carotid sinus perfusion pressure, through substitution of hypoxic hypercapnic blood (Po2, 31.1 mm Hg; Pco2, 84.9 mm Hg; pH, 7.242) for arterial blood (Po2, 140.4 mm Hg; Pco2, 42.1 mm Hg; pH, 7.33) between arrows. Note that the bradycardia was transient. The increase in pulse interval (PI) indicates a decrease in heart rate. The enhanced respiratory response (bottom record) abolishes bradycardia and can produce tachycardia, especially with sustained stimulation of the carotid body receptors (see Figures 5-16 and 5-17). BP, mean arterial blood pressure; VT, tidal volume.
(Redrawn from de Burgh Daly M, Korner PI, Angell-James JE, et al: Cardiovascular and respiratory effects of carotid body stimulation in the monkey. Clin Exp Pharmacol Physiol 5:511, 1978.)
When the chemoreceptors are stimulated simultaneously with a pressure reduction in the baroreceptors, the chemoreceptors potentiate the vasoconstriction observed in the peripheral vessels. However, when the baroreceptors and chemoreceptors are stimulated together (e.g., high carotid sinus pressure and low Paco2), the effects of the baroreceptors predominate.
The Central Chemoreceptors Are Sensitive to Changes in Paco2
Increases of Paco2 stimulate chemosensitive regions of the medulla, thereby eliciting vasoconstriction and increased peripheral resistance. Reduction in Paco2 below normal levels (as with hyperventilation) decreases the degree of tonic activity of these areas in the medulla, thereby reducing peripheral resistance. The chemosensitive regions are also affected by changes in pH. A lowering of blood pH stimulates, and a rise in blood pH inhibits, these areas. These effects of changes in Paco2 and blood pH possibly operate through changes in cerebrospinal fluid pH, as appears to apply to the respiratory center.
Oxygen tension has relatively little direct effect on the medullary vasomotor region. The primary effect of hypoxia is reflexly mediated via the carotid and aortic chemoreceptors. Moderate reduction of Paco2 stimulates the vasomotor region, but severe reduction depresses vasomotor activity, in the same manner that other areas of the brain are depressed by very low O2 tensions.
Other Vascular Reflexes
Hypothalamus
Optimal function of the cardiovascular reflexes requires the integrity of the pontine and hypothalamic structures. Furthermore, these structures are responsible for behavioral and emotional control of the cardiovascular system. Stimulation of the anterior hypothalamus decreases the blood pressure and heart rate, whereas stimulation of the posterolateral region of the hypothalamus increases the blood pressure and heart rate. The hypothalamus also contains a temperature-regulating center that affects the skin vessels. Stimulation by cold applications to the skin or by cooling of the blood perfusing the hypothalamus results in constriction of the skin vessels and heat conservation, whereas warm stimuli result in cutaneous vasodilation and enhanced heat loss (see Chapter 12).
Cerebrum
The cerebral cortex can also exert a significant effect on blood flow distribution in the body. Stimulation of the motor and premotor areas can affect blood pressure; usually a pressor response is obtained. However, vasodilation and depressor responses may be evoked (e.g., blushing or fainting) in response to an emotional stimulus.
Cerebral ischemia, which may occur because of excessive pressure exerted by an expanding intracranial tumor, results in a marked increase in peripheral vasoconstriction. The stimulation is probably caused by a local accumulation of CO2 and reduction of O2, and possibly by excitation of intracranial baroreceptors. With prolonged, severe ischemia, central depression eventually supervenes and blood pressure falls.
Skin and Viscera
Painful stimuli can elicit either pressor or depressor responses, depending on the magnitude and location of the stimulus. Distention of the viscera often evokes a depressor response, whereas painful stimuli on the body surface usually evoke a pressor response.
Pulmonary Reflexes
Inflation of the lungs reflexly induces systemic vasodilation and a decrease in arterial blood pressure. Conversely, collapse of the lungs evokes systemic vasoconstriction. Afferent fibers that mediate this reflex are carried by the vagus nerves. Their stimulation, by stretch of the lungs, inhibits the vasomotor areas. The magnitude of the depressor response to lung inflation is directly related to the degree of inflation and to the existing level of vasoconstrictor tone; the greater the vascular tone, the greater the hypotension produced by lung inflation.
Balance Between Extrinsic and Intrinsic Factors in Regulation of Peripheral Blood Flow
Dual control of the peripheral vessels by intrinsic and extrinsic mechanisms makes possible a number of vascular adjustments. Such regulations enable the body to direct blood flow to areas where the need is greater and away from areas where the need is less. The relative potency of extrinsic and intrinsic mechanisms is constant in some tissues, whereas in other tissues the ratio is changeable, depending on the state of activity of that tissue.
In the brain and the heart, both vital structures with very limited tolerance for a reduced blood supply, intrinsic flow-regulating mechanisms are dominant.
Massive discharge of the vasoconstrictor region over the sympathetic nerves, which might occur in severe, acute hemorrhage, has negligible effects on the cerebral and cardiac resistance vessels, whereas skin, renal, and splanchnic blood vessels become greatly constricted.
In the skin the extrinsic vascular control is dominant. The cutaneous vessels not only participate strongly in a general vasoconstrictor discharge but also respond selectively through hypothalamic pathways to subserve the heat loss and heat conservation function required in body temperature regulation. However, intrinsic control can be demonstrated by local changes of temperature that can modify or override the central influence on resistance and capacitance vessels.
In skeletal muscle the interplay and changing balance between extrinsic and intrinsic mechanisms can be clearly seen. In resting skeletal muscle, neural control (vasoconstrictor tone) is dominant, as can be demonstrated by the large increment in blood flow that occurs immediately after section of the sympathetic nerves to the tissue. In anticipation of exercise, and at the start of exercise, blood flow increases in the leg muscles. After the onset of exercise, the intrinsic flow-regulating mechanisms elicit vasodilation in the active muscles because of the local increase in metabolites. Vasoconstriction occurs in the inactive muscles as a manifestation of the general sympathetic discharge associated with exercise. However, the constrictor impulses that reach the resistance vessels of the active muscles are overridden by the local metabolic effects that dilate them. Hence, operation of this dual-control mechanism provides increased blood flow where it is required and shunts it away from inactive areas.
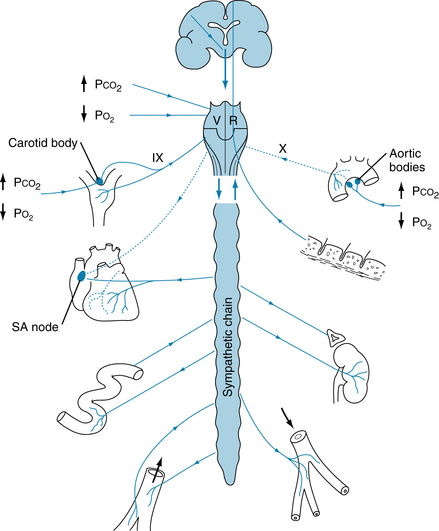
Similar effects may be achieved with an increase in Paco2. Normally, the hyperventilation associated with exercise keeps Paco2 at normal levels. However, were Paco2 to increase, a generalized vasoconstriction would occur, because of stimulation of the medullary vasoconstrictor region by CO2. In the active muscles, where the CO2 concentration is highest, the smooth muscle of the arterioles would relax in response to the local Pco2. Factors affecting and affected by the vasomotor region are summarized in Figure 9-14.
Summary
 The arterioles, often referred to as the resistance vessels, are important in the regulation of blood flow through their cognate capillaries. The smooth muscle, which makes up a major fraction of the walls of the arterioles, contracts and relaxes in response to neural and humoral stimuli.
The arterioles, often referred to as the resistance vessels, are important in the regulation of blood flow through their cognate capillaries. The smooth muscle, which makes up a major fraction of the walls of the arterioles, contracts and relaxes in response to neural and humoral stimuli.
Most tissues show autoregulation of blood flow, a phenomenon characterized by a constant blood flow in the face of a change in perfusion pressure. A logical explanation of autoregulation is the myogenic mechanism whereby an increase in transmural pressure elicits a contractile response, whereas a decrease in transmural pressure elicits relaxation.
The striking parallelism between tissue blood flow and tissue O2 consumption indicates that blood flow is largely regulated by a metabolic mechanism. A decrease in the O2 supply/O2 demand ratio of a tissue releases one or more vasodilator metabolites that dilate arterioles and thereby enhance the oxygen supply.
Neural regulation of blood flow is accomplished mainly by the sympathetic nervous system. Sympathetic nerves to blood vessels are tonically active; inhibition of the vasoconstrictor center in the medulla reduces peripheral vascular resistance. Stimulation of the sympathetic nerves constricts resistance and capacitance (veins) vessels.
Blood vessels in the head, viscera, and genitalia are supplied by the cranial and sacral divisions of the parasympathetic nervous system as well as by the sympathetic nervous system. Parasympathetic activity usually induces vasodilation, but the effect is generally weak.
The baroreceptors (pressoreceptors) in the internal carotid arteries and aorta are tonically active and regulate blood pressure on a moment-to-moment basis. Stretch of these receptors by an increase in arterial pressure reflexly inhibits the vasoconstrictor center in the medulla and induces vasodilation, whereas a decrease in arterial pressure disinhibits the vasoconstrictor center and induces vasoconstriction.
The carotid baroreceptors predominate over baroreceptors in the aorta and both respond more vigorously to pulsatile (stretch) than they do to steady (nonpulsatile) pressure; they adapt to an imposed constant pressure.
Baroreceptors are also present in the cardiac chambers and large pulmonary vessels (cardiopulmonary baroreceptors); they have less influence on blood pressure but they participate in blood volume regulation.
Peripheral chemoreceptors (carotid and aortic bodies) and central chemoreceptors in the medulla oblongata are stimulated by a decrease in blood oxygen tension (Pao2) and an increase in blood carbon dioxide tension (Paco2). Stimulation of these chemoreceptors increases the rate and depth of respiration but also produces peripheral vasoconstriction.
Peripheral resistance, and hence blood pressure, can be affected by stimuli arising in the skin, viscera, lungs, and brain.
The combined effect of neural and local metabolic factors is to distribute blood to active tissues and divert it from inactive tissues. In vital structures such as the heart and brain and in contracting skeletal muscle, the metabolic factors predominate over the neural factors.
Bellien J.M., Iacob M., Gutierres L., et al. Crucial role of NO and endothelium-derived hyperpolarizing factor in the radial artery in humans. Am J Physiol. 2006;290:H1347.
Berg B.R., Cohen K.D., Sarelius I.H. Direct coupling between blood flow and metabolism at the capillary level in striated muscle. Am J Physiol. 1997;272:H2693.
Bolton T.B., Prestwich S.A., Zholos A.V., Gordienko D.V. Excitation-contraction coupling in gastrointestinal and other smooth muscles. Annu Rev Physiol. 1999;61:85.
Dampney R.A., Horiuchi J., Tagawa T., et al. Medullary and supramedullary mechanisms regulating sympathetic vasomotor tone. Acta Physiol Scand. 2003;177:209.
Edwards G., Félétou M., Weston A.H. Endothelium-derived hyperpolarizing factors and associated pathways: a synopsis. Pflügers Arch. 2010;459:863.
Kara T., Narkiewicz K., Somers V.K. Chemoreflexes—physiology and clinical implications. Acta Physiol Scand. 2003;177:377.
Kohan D.E., Rossi N.F., Inscho E.W., Pollock D.M. Regulation of blood pressure and salt homeostasis by endothelin. Physiol Rev. 2011;91:1.
Liu Y., Bubolz A.H., Mendoza S., et al. H2O2 is the transferrable factor mediating flow-induced dilation in human coronary arterioles. Circ Res. 2011;108:566.
Maguire J.J., Davenport A.P. Regulation of vascular reactivity by established and emerging GPCRs. Trends Pharmacol Sci. 2005;26:448.
Mifflin S.W. What does the brain know about blood pressure? News Physiol Sci. 2001;16:266.
Monos E., Berczi V., Nadasy G. Local control of veins: Biomechanical, metabolic, and humoral aspects. Physiol Rev. 1995;75:611.
Moore A., Mangoni A.A., Lyons D., Jackson S.H. The cardiovascular system. Br J Clin Pharmacol. 2003;56:254.
Osborn J.W., Jacob F., Guzman P. A neural set point for the long-term control of arterial pressure: Beyond the arterial baroreceptor reflex. Am J Physiol. 2005;288:R846.
Persson P.B. Modulation of cardiovascular control mechanisms and their interaction. Physiol Rev. 1996;76:193.
Shamsuzzaman A.S., Somers V.K. Cardiorespiratory interactions in neural circulatory control in humans. Ann N Y Acad Sci. 2001;940:488.
Somlyo A.P., Somlyo A.V. Ca2+ sensitivity of smooth muscle and nonmuscle myosin II: modulated by G proteins, kinases, and myosin phosphatase. Physiol Rev.. 2003;83:1325.
Sun D., Huang A., Smith C.J., et al. Enhanced release of prostaglandins to flow-induced arteriolar dilation in eNOs knockout mice. Circ Res. 1999;85:288.
Thrasher T.N. Baroreceptors, baroreceptor unloading, and the long-term control of blood pressure. Am J Physiol. 2005;288:R819.
Timmers H.J., Wieling W., Karemaker J.M., Lenders J.W. Cardiovascular responses to stress after carotid baroreceptor denervation in humans. Ann N Y Acad Sci. 2004;1018:515.
CASE 9-1
History
A 40-year-old man sees his physician because of pain in the calves of both legs when he walks moderate distances; the pain is especially noticeable when he walks uphill or when he climbs stairs. The onset of the pain was insidious and has progressively increased in frequency and severity. He has had no other symptoms. He eats a normal diet, has two cocktails before dinner, and has smoked two packs of cigarettes per day for the past 22 years. Physical examination was essentially normal except for the presence of borderline hypertension with a brachial pressure of 140/90 mm Hg and for weak pulses in the dorsalis pedis and posterior tibial arteries in both legs. Measurement of blood pressure in the dorsalis pedis artery gave a systolic pressure of 112 mm Hg. Arteriography revealed a narrowing of the major arteries of both lower legs. He was diagnosed as having thromboangiitis obliterans, a severe progressive obstructive disease of large arteries.
Questions
1. At rest the arterioles in the lower legs show:
2. From the blood pressure measurements you obtain an ankle-brachial index of (see Chapter 7) of: