CHAPTER 8 Lymphocyte Development and Antigen Receptor Gene Rearrangement
Lymphocytes express highly diverse antigen receptors capable of recognizing a wide variety of foreign substances. This diversity is generated during the development of mature B and T lymphocytes from precursor cells that do not express antigen receptors and cannot recognize and respond to antigens. The process by which lymphocyte progenitors in the thymus and bone marrow differentiate into mature lymphocytes that populate peripheral lymphoid tissues is called lymphocyte development or lymphocyte maturation. The immune repertoire is made up of the collection of antigen receptors—and therefore specificities—expressed by B and T lymphocytes that are produced during the maturation of these cells. Maturation is initiated by signals from cell surface receptors that have two main roles—they promote the proliferation of progenitors and also induce the expression of transcription factors that work together to initiate the rearrangement of specific antigen receptor genes and commit developing cells to a B or a T cell fate. The rearrangement of antigen receptor genes is a key event in the commitment of a progenitor cell to a lymphoid fate.
We begin this chapter by considering the process of commitment to the B and T lymphocyte lineages and discuss some common principles and mechanisms of B and T cell development. This is followed by a description of the processes that are unique to the maturation of B cells and then of those unique to the development of cells of the T lymphocyte lineage.
Overview of Lymphocyte Development
The maturation of B and T lymphocytes involves a series of events that occur in the generative lymphoid organs (Fig. 8-1). These events include the following:
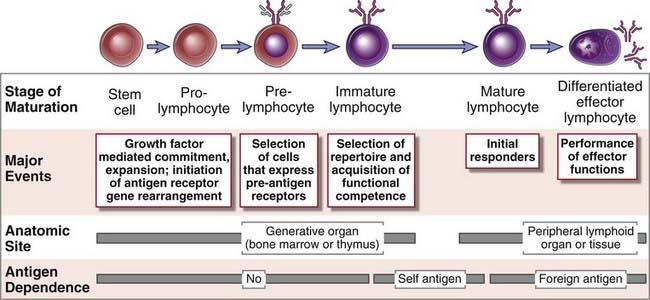
FIGURE 8–1 Stages of lymphocyte maturation.
Development of both B and T lymphocytes involves the sequence of maturational stages shown. B cell maturation is illustrated, but the basic stages of T cell maturation are similar.
We next describe features of each of these events that are common to both B and T lymphocyte lineages.
Commitment to the B and T Cell Lineages and Proliferation of Progenitors
Pluripotent stem cells in the bone marrow (and fetal liver), referred to as hematopoietic stem cells (HSCs), give rise to all lineages of blood cells, including lymphocytes. HSCs mature into common lymphoid progenitors that can give rise to B cells, T cells, NK cells, and some dendritic cells (Fig. 8-2). The maturation of B cells from progenitors committed to this lineage occurs mostly in the bone marrow and before birth in the fetal liver. Fetal liver–derived stem cells give rise mainly to a type of B cell called a B-1 B cell (best defined in rodents), whereas bone marrow–derived HSCs give rise to the majority of circulating B cells (follicular B cells). Precursors of T lymphocytes leave the fetal liver before birth and the bone marrow later in life and circulate to the thymus, where they complete their maturation. The majority of T cells, which are αβ T cells, develop from bone marrow–derived HSCs; most γδ T cells arise from fetal liver HSCs. In general, the B and T cells that are generated early in fetal life have less diverse antigen receptors. Despite their different anatomic locations, the early maturation events of both B and T lymphocytes are fundamentally similar.
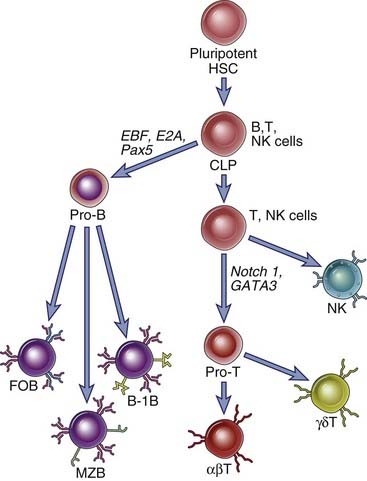
FIGURE 8–2 Pluripotent stem cells give rise to distinct B and T lineages.
Hematopoietic stem cells (HSCs) give rise to distinct progenitors for various types of blood cells. One of these progenitor populations (shown here) is called a common lymphoid progenitor (CLP). CLPs give rise mainly to B and T cells but may also contribute to NK cells and some dendritic cells (not depicted here). Pro-B cells can eventually differentiate into follicular (FO) B cells, marginal zone (MZ) B cells, and B-1 B cells. Pro-T cells may commit to either the αβ or γδ T cell lineages. Commitment to the T lineage depends on signals delivered by Notch-1, whose intracellular domain mediates transcriptional activation of T lineage genes in collaboration with other transcription factors such as GATA-3. Commitment to the B lineage is mediated initially by the EBF and E2A transcription factors and subsequently by Pax-5. These transcription factors work together to induce the transcription of B cell–specific genes and of genes of the recombination machinery. (Transcription factors are indicated by italics.)
Commitment to the B or T lineage depends on instructions received from several cell surface receptors, which signal to induce specific transcriptional regulators that drive a common lymphoid progenitor to specifically assume a B cell or a T cell fate. The cell surface receptors and transcription factors that contribute to commitment function to induce the proteins involved in antigen receptor gene rearrangement and make particular antigen receptor gene loci accessible to these proteins. In the case of developing B cells, the immunoglobulin (Ig) heavy chain locus, originally in a “closed” chromatin configuration, is opened up so that it becomes accessible to the proteins that will mediate antigen receptor gene rearrangement and expression. In developing αβ T cells, the T cell receptor (TCR) β gene locus is made available first. In addition to genes involved in the process of antigen receptor gene rearrangement, genes that drive the further differentiation of T and B cells are expressed at this stage.
Different sets of transcription factors drive the development of the B and T cell lineages from uncommitted precursors (see Fig. 8-2). The Notch family of proteins are cell surface molecules that are proteolytically cleaved when they interact with specific ligands on neighboring cells. The cleaved intracellular portions of Notch proteins migrate to the nucleus and modulate the expression of specific target genes. Notch-1, a member of the Notch family, is activated in lymphoid progenitor cells and collaborates with a transcription factor called GATA-3 to commit developing lymphocytes to the T lineage. These transcriptional regulators contribute to the induction of a number of genes that are required for the further development of αβ T cells. Downstream target genes include components of the pre-TCR and of the machinery for V(D)J recombination, described later. In B cells, the EBF and E2A transcription factors contribute to the induction of another transcription factor called Pax-5, and these three proteins collaborate to induce the process of commitment to the B lineage by facilitating the expression of a number of genes. These genes, which are described in more detail later, include those encoding the Rag-1 and Rag-2 proteins, surrogate light chains, and the Igα and Igβ proteins that contribute to signaling through the pre-B cell receptor and the B cell receptor. The role of these receptors in B cell development will be considered later in the chapter.
Early B and T cell development is characterized by the proliferation of committed progenitors induced by cytokine-derived signals. Proliferation ensures that a large enough pool of progenitor cells will be generated to eventually provide a highly diverse repertoire of mature, antigen-specific lymphocytes. In rodents, the cytokine interleukin-7 (IL-7) drives proliferation of both T and B cell progenitors; in humans, IL-7 is required for the proliferation of T cell progenitors but not of progenitors in the B lineage. IL-7 is produced by stromal cells in the bone marrow and by epithelial and other cells in the thymus. Mice with targeted mutations in either the IL-7 gene or the IL-7 receptor gene show defective maturation of lymphocyte precursors beyond the earliest stages and, as a result, profound deficiencies in mature T and B cells. Mutations in a chain of the IL-7 receptor, called the common γ chain because it is shared by several cytokine receptors, give rise to an immunodeficiency disorder in humans called X-linked severe combined immunodeficiency disease (X-SCID). This disease is characterized by a block in T cell and NK cell development, but normal B cell development, reflecting the role of IL-7 in humans (see Chapter 20).
The proliferative activity in early lymphocyte development, driven mainly by IL-7, ceases just before gene rearrangement for one chain of the antigen receptor is completed, and subsequent proliferation occurs only in cells that have successfully rearranged and expressed the gene encoding the first antigen receptor chain in a developing B or T cell. Once this receptor chain is produced, it forms the pre-B cell receptor (pre-BCR) or pre-T cell receptor (pre-TCR), described later, that selects cells for survival, proliferation, and further differentiation but is incapable of recognizing antigen. Pre-antigen receptor–based selection of the cells that have successfully made an Ig heavy chain protein in the B cell lineage and the TCR β chain in the αβ T cell lineage supports the greatest expansion of lymphocyte progenitors during development. This is an important developmental checkpoint that cells must successfully negotiate because only cells that express the first component of antigen receptors can survive, expand, and proceed to the next stage of maturation.
MicroRNAs and Lymphocyte Development
Although gene expression during lymphocyte development is driven primarily by transcription factors, an additional level of regulation is mediated by microRNAs (miRNAs). miRNAs are small endogenous noncoding RNAs that are initially generated in the nucleus as longer primary miRNA transcripts that are processed at this site by an endoribonuclease called Drosha into shorter pre-miRNAs that have a stem loop structure and can be exported into the cytosol. In the cytosol, the pre-miRNA is processed by another endoribonuclease called Dicer into a short double-stranded miRNA about 21 to 22 base pairs in length, one strand of which can be used to pair with a complementary sequence in a number of cellular mRNAs. These mRNAs associate with miRNAs and proteins called Argonaute proteins to form complexes known as RISC (RNA-induced silencing complex). If the 6– to 8–base pair miRNA seed sequence is not perfectly complementary to the mRNA, the mRNA is prevented from being translated efficiently. mRNAs may be targeted for degradation when complementarity is perfect. In either case, the result is a reduction in the abundance of proteins encoded by genes targeted by miRNAs. Gene ablation studies have revealed that miRNAs are involved in both B and T cell development. Numerous miRNAs have been reported to regulate lymphocyte activation as well.
Antigen Receptor Gene Rearrangement and Expression
The rearrangement of antigen receptor genes is the key event in lymphocyte development that is responsible for the generation of a diverse repertoire. Antigen receptor gene products also provide signals that ensure the selective survival of lymphocytes with useful specificities. As we discussed in Chapter 7, each clone of B or T lymphocytes produces an antigen receptor with a unique antigen-binding structure. In any individual, there may be 107 or more different B and T lymphocyte clones, each with a unique receptor. The ability of each individual to generate these enormously diverse lymphocyte repertoires has evolved in a way that does not require an equally large number of distinct antigen receptor genes; otherwise, a large proportion of the genome would be devoted to encoding the vast number of Ig and TCR molecules. Functional antigen receptor genes are produced in immature B cells in the bone marrow and in immature T cells in the thymus by a process of gene rearrangement, which is designed to generate a large number of variable region–encoding exons using a relatively small fraction of the genome. In any given developing lymphocyte, one of many variable region gene segments is randomly selected and joined to a downstream DNA segment. The DNA rearrangement events that lead to the production of antigen receptors are not dependent on or influenced by the presence of antigens. In other words, as the clonal selection hypothesis had proposed, diverse antigen receptors are generated and expressed before encounter with antigens (see Fig. 1-7, Chapter 1). We will discuss the molecular details of antigen receptor gene rearrangement later in the chapter.
Selection Processes That Shape the B and T Lymphocyte Repertoires
The process of lymphocyte development contains numerous intrinsic steps, called checkpoints, at which the developing cells are “tested” and continue to mature only if a preceding step in the process has been successfully completed. One of these checkpoints is based on the successful production of one of the polypeptide chains of the two chain antigen receptor protein and a second checkpoint requires the assembly of a complete receptor. The requirement for traversing these checkpoints ensures that only lymphocytes that have successfully completed antigen receptor gene rearrangement processes, and are therefore likely to be functional, are selected to mature. Additional selection processes operate after antigen receptors are expressed and serve to eliminate potentially harmful, self-reactive lymphocytes and to commit developing cells to particular lineages. We next summarize the general principles of these selection events.
Pre-antigen receptors and antigen receptors deliver signals to developing lymphocytes that are required for the survival of these cells and for their proliferation and continued maturation (Fig. 8-3). Antigen receptor gene rearrangement involves the opening of a particular receptor gene locus (such as a TCR gene locus in T cells and an Ig locus in B cells) and the joining of DNA segments in this locus to generate a functional antigen receptor gene. During this process, bases are randomly added or removed between the gene segments being joined together, thus maximizing variability among receptors. In developing B cells, the first antigen receptor gene to be completely rearranged is the Ig heavy chain or Ig H gene. In αβ T cells, the β chain of the TCR is rearranged first. Cells of the B lymphocyte lineage that successfully rearrange their Ig heavy chain genes express the Ig H chain protein and assemble a pre-antigen receptor known as the pre-BCR. In an analogous fashion, developing T cells that make a productive TCR β chain gene rearrangement synthesize the TCR β chain protein and assemble a pre-antigen receptor known as the pre-TCR. Only about one in three developing B and T cells that rearranges an antigen receptor gene makes an in-frame rearrangement and is therefore capable of generating a proper full-length protein. If cells make out-of-frame rearrangements at the Ig µ or TCR β chain loci, the pre-antigen receptors are not expressed, the cells do not receive necessary survival signals, and they undergo programmed cell death. The assembled pre-BCR and pre-TCR complexes provide signals for survival, for proliferation, for the phenomenon of allelic exclusion discussed later, and for the further development of early B and T lineage cells. Thus, expression of the pre-antigen receptor is the first checkpoint during lymphocyte development.
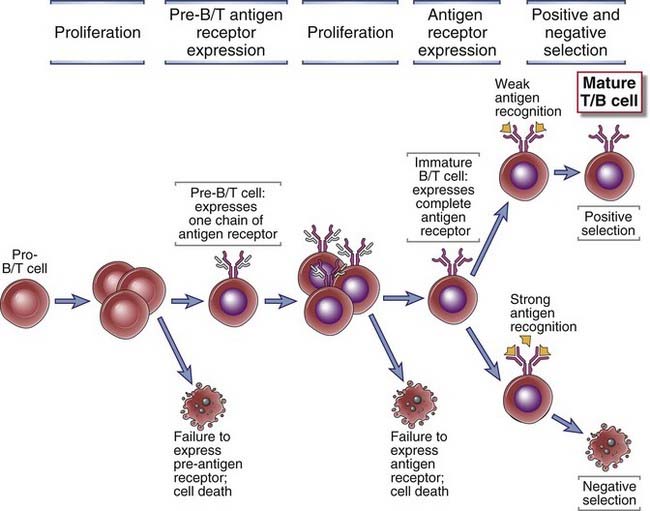
FIGURE 8–3 Checkpoints in lymphocyte maturation.
During development, the lymphocytes that express receptors required for continued proliferation and maturation are selected to survive, and cells that do not express functional receptors die by apoptosis. Positive selection and negative selection further preserve cells with useful specificities. The presence of multiple checkpoints ensures that only cells with useful receptors complete their maturation.
Lymphocytes that have successfully navigated this checkpoint continue to develop in the generative lymphoid organs and express the complete antigen receptor while they are still immature. At this immature stage, potentially harmful cells that strongly recognize self structures may be eliminated or induced to alter their antigen receptors, and cells that express useful antigen receptors may be preserved (Fig. 8-4). A process called positive selection facilitates the survival of potentially useful lymphocytes, and this developmental event is linked to lineage commitment, the process by which lymphocyte subsets are generated. In the T cell lineage, positive selection ensures the maturation of T cells whose receptors recognize self MHC molecules and also that the expression of the appropriate coreceptor on a T cell (CD8 or CD4) is matched to the recognition of the appropriate type of MHC molecule (MHC class I or MHC class II). Mature T cells whose precursors were positively selected by self MHC molecules in the thymus are able to recognize foreign peptide antigens displayed by the same self MHC molecules on antigen-presenting cells in peripheral tissues. In the B cell lineage, positive selection preserves receptor-expressing cells and is coupled to the generation of different subsets, discussed later.
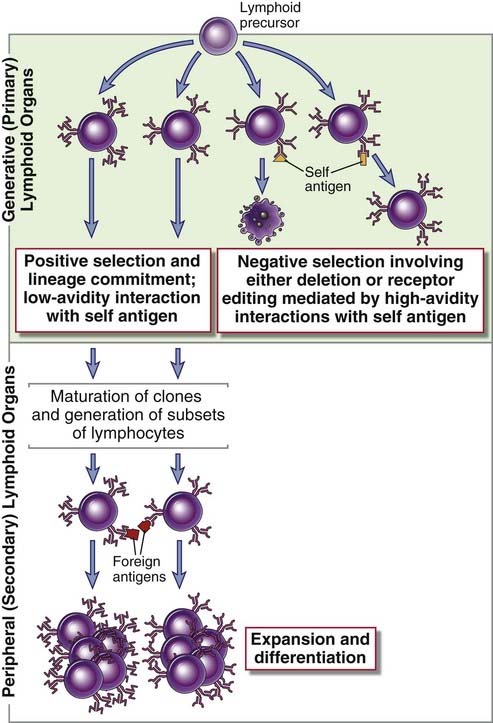
FIGURE 8–4 Positive and negative selection during lymphocyte maturation.
After immature clones of lymphocytes in generative lymphoid organs express antigen receptors, they are subject to both positive and negative selection processes. In positive selection, lymphocyte precursors with antigen receptors that bind some self ligand with low avidity are selected to survive and mature further. Developing B cells receive survival signals simply because of expression of complete antigen receptors, without recognition of a self antigen. However, as in T cells, self antigen of different affinities can drive the differentiation of different B cell subsets. Positive selection in both B and T lineages is therefore tightly linked to the process of generating lymphocyte subsets, also referred to as lineage commitment. Positively selected lymphocytes enter peripheral lymphoid tissues, where they respond to foreign antigens. In negative selection, cells that bind antigens present within the generative organs, with high avidity, receive signals that either lead to cell death or induce further rearrangement of antigen receptor genes, a process known as receptor editing. As a result, the repertoire of mature lymphocytes lacks cells capable of responding to these self antigens. The diagram illustrates selection of B cells; the principles are the same for T lymphocytes, except that there is no receptor editing in the T lineage.
Negative selection is the process that eliminates or alters developing lymphocytes whose antigen receptors bind strongly to self antigens present in the generative lymphoid organs. Both developing B and T cells are susceptible to negative selection during a short period after antigen receptors are first expressed. Developing T cells with a high affinity for self antigens are eliminated by apoptosis, a phenomenon known as clonal deletion. Strongly self-reactive immature B cells may be induced to make further Ig gene rearrangements and thus evade self-reactivity. This phenomenon is called receptor editing. If editing fails, the self-reactive B cells die, also called clonal deletion. Negative selection of immature lymphocytes is an important mechanism for maintaining tolerance to many self antigens; this is also called central tolerance because it develops in the central (generative) lymphoid organs (see Chapter 14).
The Generation of Lymphocyte Subsets
Another important feature of lymphocyte development that is linked to positive selection is the generation of functionally distinct subsets in both the T and the B cell lineages. Precursors that express both CD4 and CD8 differentiate into either CD4+ class II MHC–restricted T cells or CD8+ class I MHC–restricted T cells. The naive CD4+ T cells that leave the thymus can be activated by antigens to differentiate into helper T cells whose effector functions are mediated by specific membrane proteins and by secreted cytokines. The CD8+ cells can differentiate into cytotoxic T lymphocytes whose major effector function is to kill infected target cells. A similar lineage commitment process during positive selection of B cells drives the development of these cells into distinct peripheral B cell subsets. Bone marrow–derived developing B cells may differentiate into follicular B cells, which recirculate and mediate T cell–dependent immune responses in secondary lymphoid organs, or marginal zone B cells, which reside in the vicinity of the marginal sinus in the spleen and mediate largely T cell–independent responses to blood-borne antigens.
With this introduction, we proceed to a more detailed discussion of lymphocyte maturation, starting with the key event in the process, the rearrangement and expression of antigen receptor genes.
Rearrangement of Antigen Receptor Genes in B and T Lymphocytes
The genes that encode diverse antigen receptors of B and T lymphocytes are generated by the rearrangement in individual lymphocytes of different variable (V) region gene segments with diversity (D) and joining (J) gene segments. A novel rearranged exon for each antigen receptor gene is generated by fusing a specific distant upstream V gene segment to a downstream segment on the same chromosome. This specialized process of site-specific gene rearrangement is called V(D)J recombination. (The terms recombination and rearrangement are used interchangeably.) Elucidation of the mechanisms of antigen receptor gene rearrangement, and therefore of the underlying basis for the generation of immune diversity, represents one of the landmark achievements of modern immunology.
The first insights into how millions of different antigen receptors could be generated from a limited amount of coding DNA in the genome came from analyses of the amino acid sequences of Ig molecules. These analyses showed that the polypeptide chains of many different antibodies of the same isotype shared identical sequences at their C-terminal ends (corresponding to the constant domains of antibody heavy and light chains) but differed considerably in the sequences at their N-terminal ends that correspond to the variable domains of immunoglobulins (see Chapter 5). Contrary to one of the central tenets of molecular genetics, enunciated as the “one gene–one polypeptide hypothesis” by Beadle and Tatum in 1941, Dreyer and Bennett postulated in 1965 that each antibody chain is actually encoded by at least two genes, one variable and the other constant, and that the two are physically combined at the level of DNA or of messenger RNA (mRNA) to eventually give rise to functional Ig proteins. Formal proof of this hypothesis came more than a decade later when Susumu Tonegawa demonstrated that the structure of Ig genes in the cells of an antibody-producing tumor, called a myeloma or plasmacytoma, is different from that in embryonic tissues or in nonlymphoid tissues not committed to Ig production. These differences arise because DNA segments within the loci encoding Ig heavy and light chains are specifically ligated together only in developing B cells but not in other tissues or cell types. Similar rearrangements were found to occur during T cell development in the loci encoding the polypeptide chains of TCRs. Antigen receptor gene rearrangement is best understood by first describing the unrearranged, or germline, organization of Ig and TCR genes and then describing their rearrangement during lymphocyte maturation.
Germline Organization of Ig and TCR Genes
The germline organizations of Ig and TCR genetic loci are fundamentally similar and are characterized by spatial segregation of multiple sequences that encode variable and constant domains of receptor proteins; distinct variable region sequences are joined to constant region sequences in different lymphocytes. We first describe the Ig loci and then the TCR loci.
Organization of Ig Gene Loci
Three separate loci encode, respectively, all the Ig heavy chains, the Ig κ light chain, and the Ig λ light chain. Each locus is on a different chromosome. The organization of human Ig genes is illustrated in Figure 8-5, and the relationship of gene segments after rearrangement to the domains of the Ig heavy and light chain proteins is shown in Figure 8-6A. Ig genes are organized in essentially the same way in all mammals, although their chromosomal locations and the number and sequence of different gene segments in each locus may vary.
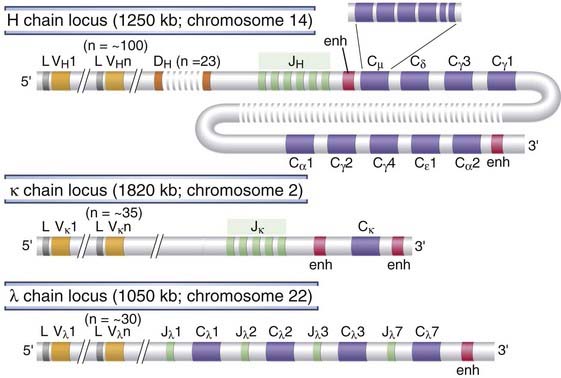
FIGURE 8–5 Germline organization of human Ig loci.
The human heavy chain, κ light chain, and λ light chain loci are shown. Only functional genes are shown; pseudogenes have been omitted for simplicity. Exons and introns are not drawn to scale. Each CH gene is shown as a single box but is composed of several exons, as illustrated for Cµ. Gene segments are indicated as follows: L, leader (often called signal sequence); V, variable; D, diversity; J, joining; C, constant; enh, enhancer.
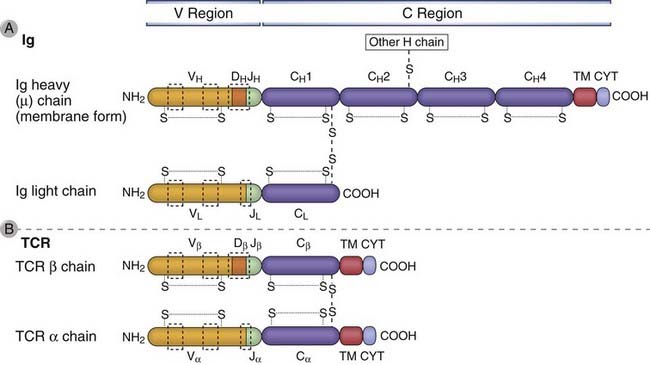
FIGURE 8–6 Domains of Ig and TCR proteins.
The domains of Ig heavy and light chains are shown in A, and the domains of TCR α and β chains are shown in B. The relationships between the Ig and TCR gene segments and the domain structure of the antigen receptor polypeptide chains are indicated. The V and C regions of each polypeptide are encoded by different gene segments. The locations of intrachain and interchain disulfide bonds (S-S) are approximate. Areas in the dashed boxes are the hypervariable (complementarity-determining) regions. In the Ig µ chain and the TCR α and β chains, transmembrane (TM) and cytoplasmic (CYT) domains are encoded by separate exons.
At the 5′ end of each of the Ig loci, there is a cluster of V gene segments, each V gene in the cluster being about 300 base pairs long. The numbers of V gene segments vary considerably among the different Ig loci and among different species. For example, there are about 35 V genes in the human κ light chain locus, about 30 in the λ locus, and about 100 functional V genes in the human heavy chain locus, whereas the mouse λ light chain locus has only two V genes and the mouse heavy chain locus has more than 1000 V genes. The V gene segments for each locus are spaced over large stretches of DNA, up to 2000 kilobases long. Located 5′ of each V segment is a leader exon that encodes the 20 to 30 N-terminal residues of the translated protein. These residues are moderately hydrophobic and make up the leader (or signal) peptide. Signal sequences are found in all newly synthesized secreted and transmembrane proteins and are involved in guiding nascent polypeptides being translated on membrane-bound ribosomes into the lumen of the endoplasmic reticulum. Here, the signal sequences are rapidly cleaved, and they are not present in the mature proteins. Upstream of each leader exon is a V gene promoter at which transcription can be initiated, but, as discussed later, this occurs most efficiently after rearrangement.
At varying distances 3′ of the V genes are several J segments that are closely linked to downstream constant region exons. J segments are typically 30 to 50 base pairs long and are separated by noncoding sequences. Between the V and J segments in the Ig H locus, there are additional segments known as D segments. As for V genes, the numbers of J and D genes vary in different Ig loci and different species.
Each Ig locus has a distinct arrangement and number of C region genes. In humans, the Ig κ light chain locus has a single C gene (Cκ) and the λ light chain locus has four functional C genes (Cλ). The Ig heavy chain locus has nine C genes (CH), arranged in a tandem array, that encode the C regions of the nine different Ig isotypes and subtypes (see Chapter 5). The Cκ and Cλ genes are each composed of a single exon that encodes the entire C domain of the light chains. In contrast, each CH gene is composed of five or six exons. Three or four exons (each similar in size to a V gene segment) each encode a CH domain of the Ig heavy chain, and two smaller exons code for the carboxyl-terminal ends of the membrane form of each Ig heavy chain, including the transmembrane and cytoplasmic domains of the heavy chains (see Fig. 8-6A).
In an Ig light chain protein (κ or λ), the V domain is encoded by the V and J gene segments; in the Ig heavy chain protein, the V domain is encoded by the V, D, and J gene segments (see Fig. 8-6A). All junctional residues between the rearranged V and D segments and the D and J segments as well as the sequences of the D and J segments themselves make up the third hypervariable region (also known as complementarity-determining region 3 or CDR3) in the case of the Ig H and TCR β V domains. The junctional sequences between the rearranged V and J segments as well as the J segment itself make up the third hypervariable region of Ig light chains. CDR1 and CDR2 are encoded in the germline V gene segment itself. The V and C domains of Ig molecules share structural features, including a tertiary structure called the Ig fold. As we discussed in Chapter 5, proteins that include this structure are members of the Ig superfamily.
Noncoding sequences in the Ig loci play important roles in recombination and gene expression. As we shall see later, sequences that dictate recombination of different gene segments are found adjacent to each coding segment in Ig genes. Also present are V gene promoters and other cis-acting regulatory elements, such as locus control regions, enhancers, and silencers, that regulate gene expression at the level of transcription.
Organization of TCR Gene Loci
The genes encoding the TCR α chain, the TCR β chain, and the TCR γ chain map to three separate loci on three different chromosomes, and the TCR δ chain locus is contained within the TCR α locus (Fig. 8-7). Each germline TCR locus includes V and J gene segments, the latter being just upstream of C region exons in each locus. In addition, TCR β and TCR δ loci also have D segments, like the Ig heavy chain locus. At the 5′ end of each of the TCR loci, there is a cluster of several V gene segments, arranged in a very similar way to the Ig V gene segments. Upstream of each TCR V gene is an exon that encodes a leader peptide, and upstream of each leader exon is a promoter for each V gene.
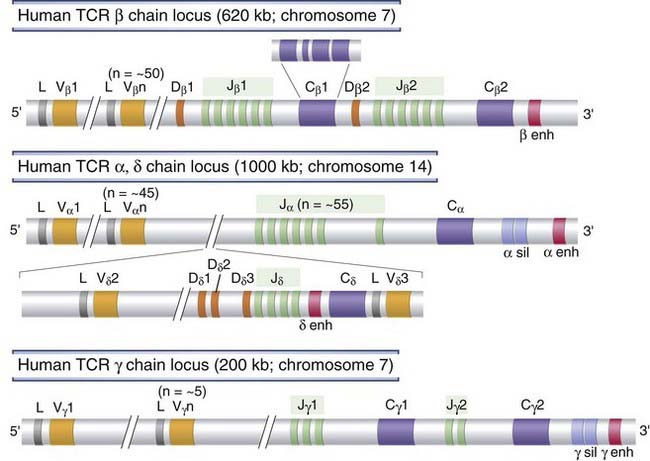
FIGURE 8–7 Germline organization of human TCR loci.
The human TCR β, α, γ, and δ chain loci are shown, as indicated. Exons and introns are not drawn to scale, and nonfunctional pseudogenes are not shown. Each C gene is shown as a single box but is composed of several exons, as illustrated for Cβ1. Gene segments are indicated as follows: L, leader (usually called signal sequence); V, variable; D, diversity; J, joining; C, constant; enh, enhancer; sil, silencer (sequences that regulate TCR gene transcription).
In each TCR locus, similar to the Ig loci, C region genes are located just 3′ of the J segments. There are two C genes in each of the human TCR β (Cβ) and TCR γ (Cγ) loci and only one C gene each in the TCR α (Cα) and TCR δ (Cδ) loci. Each TCR C region gene is composed of four exons encoding the extracellular C region Ig-like domain, a short hinge region, the transmembrane segment, and the cytoplasmic tail. The TCR β and TCR δ chain loci resemble the Ig heavy chain locus and contain D segments between V genes and J segments. Each human TCR C gene has its own associated 5′ cluster of J segments. In the TCR α or γ chains (which are analogous to Ig light chains), the V domain is encoded by the V and J exons; and in the TCR β and δ proteins, the V domain is encoded by the V, D, and J gene segments.
The relationship of the TCR gene segments and the corresponding portions of TCR proteins that they encode is shown in Figure 8-6B. As in Ig molecules, the TCR V and C domains assume an Ig fold tertiary structure, and thus the TCR is a member of the Ig superfamily of proteins.
V(D)J Recombination
The germline organization of Ig and TCR loci described in the preceding section exists in all cell types in the body. The germline genes cannot be transcribed into mRNAs that encode functional antigen receptor proteins. Functional antigen receptor genes are created only in developing B and T lymphocytes after DNA rearrangement events that bring randomly chosen V, (D), and J gene segments into contiguity. Figure 8-8 schematically depicts a Southern blot, including an Ig light chain locus in germline configuration in a nonlymphoid cell. The V segment shown in this configuration is a considerable distance away from the J segments depicted. A hypothetical rearrangement event in a B cell clone is also shown. In this clone, a specific upstream V segment has been joined to one of the J segments during the rearrangement process.
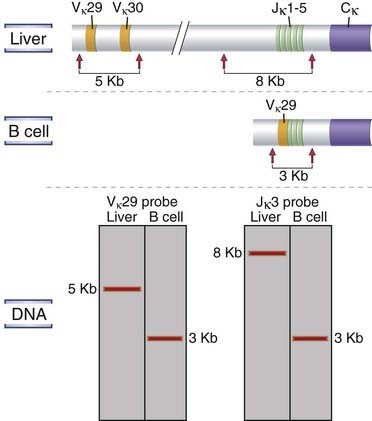
FIGURE 8–8 Antigen receptor gene rearrangements.
Southern blot analysis of DNA from nonlymphoid (liver) cells and from a monoclonal population of B lymphocyte lineage origin (e.g., a B cell tumor) is shown in schematic fashion. The DNA is digested with a restriction enzyme (EcoRI as depicted), different-sized fragments are separated by electrophoresis, and the fragments are transferred onto a filter. The sites at which the EcoRI restriction enzyme cleaves the DNA are indicated by arrows. The size of the fragments containing the Jκ3 segment of the Ig κ light chain gene or the Vκ29 V region gene was determined by use of a radioactive probe that specifically binds to Jκ3 segment DNA or to Vκ29 DNA. In the hypothetical example shown, Vκ29 is part of a 5-kb EcoRI fragment in liver cells but is on a 3-kb fragment in the B cell clone studied. Similarly, the Jκ3 fragment is 8 kb in liver cells but 3 kb in the B cell clone.
The process of V(D)J recombination at any Ig or TCR locus involves selection of one V gene, one J segment, and one D segment (when present) in each lymphocyte and rearrangement of these gene segments together to form a single V(D)J exon that will code for the variable region of an antigen receptor protein (Fig. 8-9). In the Ig light chain and TCR α and γ loci, which lack D segments, a single rearrangement event joins a randomly selected V gene to an equally randomly selected J segment. The IgH and TCR β and δ loci contain D segments, and at these loci two distinct rearrangement events must be separately initiated, first joining a D to a J and then a V segment to the fused DJ segment. Each rearrangement event involves a number of sequential steps. First, the chromatin must be opened in specific regions of the antigen receptor chromosome to make gene segments accessible to the enzymes that mediate recombination. Next, two selected gene segments must be brought next to one another across a considerable chromosomal distance. Double-stranded breaks are then introduced at the coding ends of these two segments, nucleotides are added or removed at the broken ends, and finally the processed ends are ligated to produce clonally unique but diverse antigen receptor genes that can be efficiently transcribed. The C regions lie downstream of the rearranged V(D)J exon separated by the germline J-C intron. This rearranged exon is transcribed to form a primary (nuclear) RNA transcript. Subsequent RNA splicing brings together the leader exon, the V(D)J exon, and the C region exons, forming an mRNA that can be translated on membrane-bound ribosomes to produce one of the chains of the antigen receptor. The use of different combinations of V, D, and J gene segments and the addition and removal of nucleotides at the joints contribute to the tremendous diversity of antigen receptors, as we will discuss in more detail later.
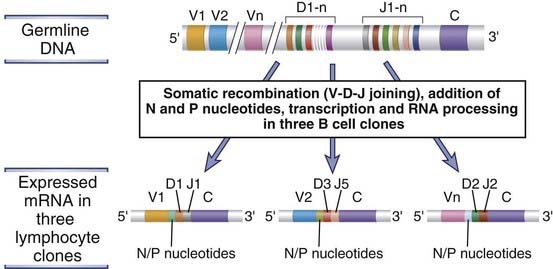
FIGURE 8–9 Diversity of antigen receptor genes.
From the same germline DNA, it is possible to generate recombined DNA sequences and mRNAs that differ in their V-D-J junctions. In the example shown, three distinct antigen receptor mRNAs are produced from the same germline DNA by the use of different gene segments and the addition of nucleotides to the junctions.
Recognition Signals That Drive V(D)J Recombination
Critical lymphocyte-specific factors that mediate V(D)J recombination recognize certain DNA sequences called recombination signal sequences (RSSs), located 3′ of each V gene segment, 5′ of each J segment, and flanking each D segment on both sides (Fig. 8-10A). The RSSs consist of a highly conserved stretch of 7 nucleotides, called the heptamer, usually CACAGTG, located adjacent to the coding sequence, followed by a spacer of exactly 12 or 23 nonconserved nucleotides, followed by a highly conserved AT-rich stretch of 9 nucleotides, called the nonamer. The 12- and 23-nucleotide spacers roughly correspond to one or two turns of a DNA helix, respectively, and they presumably bring two distinct heptamers into positions that are simultaneously accessible to the enzymes that catalyze the recombination process.
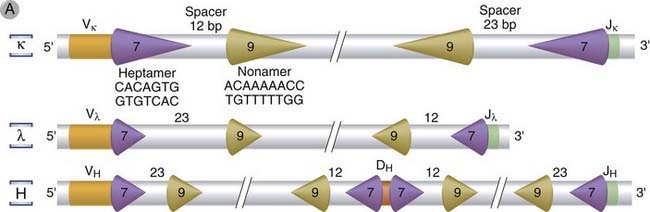
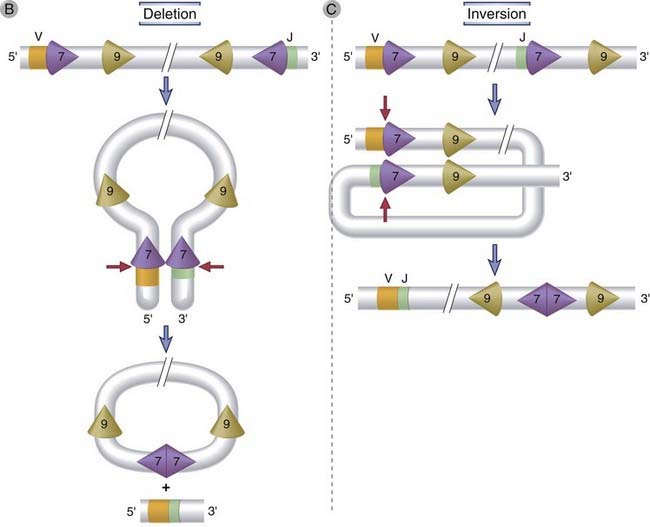
FIGURE 8–10 V(D)J recombination.
The DNA sequences and mechanisms involved in recombination in the Ig gene loci are depicted. The same sequences and mechanisms apply to recombinations in the TCR loci. A, Conserved heptamer (7 bp) and nonamer (9 bp) sequences, separated by 12- or 23-bp spacers, are located adjacent to V and J exons (for κ and λ loci) or to V, D, and J exons (in the H chain locus). The V(D)J recombinase recognizes these recombination signal sequences and brings the exons together. B, C, Recombination of V and J exons may occur by deletion of intervening DNA and ligation of the V and J segments (B) or, if the V gene is in the opposite orientation, by inversion of the DNA followed by ligation of adjacent gene segments (C). Red arrows indicate the sites where germline sequences are cleaved before their ligation to other Ig or TCR gene segments.
During V(D)J recombination, double-stranded breaks are generated between the heptamer of the RSS and the adjacent V, D, or J coding sequence. In Ig light chain V-to-J recombination, for example, breaks will be made 3′ of a V segment and 5′ of a J segment. The intervening double-stranded DNA, containing signal ends (the ends that contain the heptamer and the rest of the RSS), is removed in the form of a circle, and this is accompanied by the joining of the V and J coding ends (Fig. 8-10B). Some V genes, especially in the Ig κ locus, are in the same orientation as the J segments, such that the RSSs 5′ of these V segments and 3′ of the J segments do not “face” each other. In these cases, the intervening DNA is inverted and the V and J exons are properly aligned, and the fused RSSs are not deleted but retained in the chromosome (Fig. 8-10C). Most Ig and TCR gene rearrangements occur by deletion; rearrangement by inversion occurs in up to 50% of rearrangements in the Ig κ locus. Recombination occurs between two segments only if one of the segments is flanked by a 12-nucleotide spacer and the other is flanked by a 23-nucleotide spacer; this is called the 12/23 rule. A coding segment with a “one-turn” RSS therefore always recombines with a coding segment with a “two-turn” RSS. The type of the flanking RSSs (one turn or two turn) ensures that the appropriate gene segments will recombine. For example, in the Ig heavy chain locus, the RSSs flanking both V and J segments have 23-nucleotide spacers (two turns) and therefore cannot join directly; D-to-J recombination occurs first, followed by V-to-DJ recombination, and this is possible because the D segments are flanked on both sides by 12-nucleotide spacers, allowing D-J and then V-DJ joining. The RSSs described here are unique to Ig and TCR genes. Therefore, V(D)J recombination can occur in antigen receptor genes but not in other genes.
One of the consequences of V(D)J recombination is that the process brings promoters located immediately 5′ of V genes close to downstream enhancers that are located in the J-C introns and also 3′ of the C region genes (Fig. 8-11). These enhancers maximize the transcriptional activity of the V gene promoters and are thus important for high-level transcription of rearranged V genes in lymphocytes. Because Ig and TCR genes are sites for multiple DNA recombination events in B and T cells, and because these sites become transcriptionally active after recombination, genes from other loci can be abnormally translocated to these loci and, as a result, may be aberrantly transcribed. In tumors of B and T lymphocytes, oncogenes are often translocated to Ig or TCR gene loci. Such chromosomal translocations are frequently accompanied by enhanced transcription of the oncogenes and are believed to be one of the factors causing the development of lymphoid tumors.
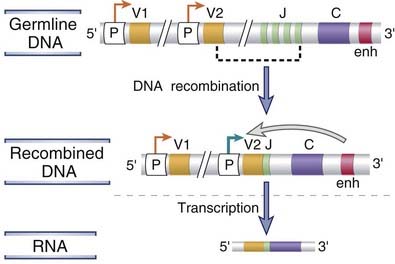
FIGURE 8–11 Transcriptional regulation of Ig genes.
V-D-J recombination brings promoter sequences (shown as P) close to the enhancer (enh). The enhancer promotes transcription of the rearranged V gene (V2, whose active promoter is indicated by a bold green arrow). Many receptor genes have an enhancer in the J-C intron and another 3′ of the C region. Only the 3′ enhancer is depicted here.
The Mechanism of V(D)J Recombination
Rearrangement of Ig and TCR genes represents a special kind of nonhomologous DNA recombination event, mediated by the coordinated activities of several enzymes, some of which are found only in developing lymphocytes, whereas others are ubiquitous DNA double-stranded break repair (DSBR) enzymes. Although the mechanism of V(D)J recombination is fairly well understood and will be described here, how exactly specific loci are made accessible to the machinery involved in recombination remains to be determined. It is likely that the accessibility of the Ig and TCR loci to the enzymes that mediate recombination is regulated in developing B and T cells by several mechanisms, including alterations in chromatin structure, DNA methylation, and basal transcriptional activity in the gene loci.
The process of V(D)J recombination can be divided into four distinct events that flow sequentially from one to the next (Fig. 8-12):
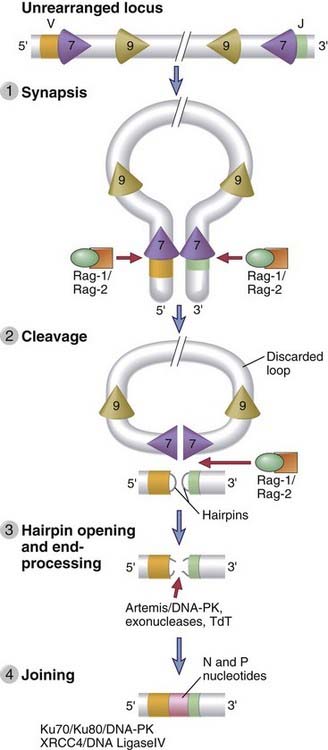
FIGURE 8–12 Sequential events during V(D)J recombination.
Synapsis and cleavage of DNA at the heptamer/coding segment boundary are mediated by Rag-1 and Rag-2. The coding end hairpin is opened by the Artemis endonuclease, and broken ends are repaired by the NHEJ machinery.
Rag genes are lymphoid specific and are expressed only in developing B and T cells. Rag proteins are expressed mainly in the G0 and G1 stages of the cell cycle and are inactivated in proliferating cells. It is thought that limiting DNA cleavage and recombination to these stages minimizes the risk of generating inappropriate DNA breaks during DNA replication or during mitosis.
Generation of Diversity in B and T Cells
The enormous diversity of the B and T cell repertoires is created not only by random combinations of germline gene segments being brought together but also by random addition or deletion of sequences at the junctions between segments that have been united. Several genetic mechanisms contribute to this diversity, and the relative importance of each mechanism varies among the different antigen receptor loci (Table 8-1). Not discussed here is somatic hypermutation, a mechanism that involves point mutations and other changes in DNA in activated B cells, which will be considered in Chapter 11.
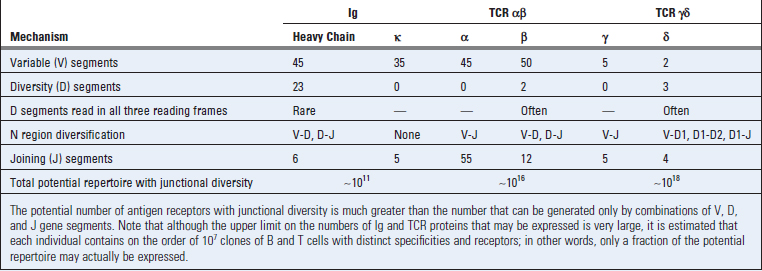
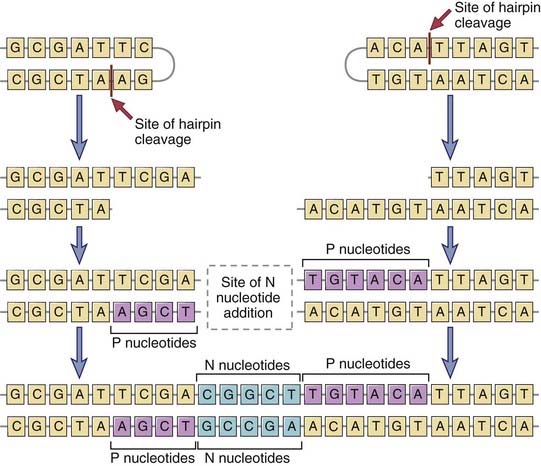
FIGURE 8–13 Junctional diversity.
During the joining of different gene segments, addition or removal of nucleotides may lead to the generation of novel nucleotide and amino acid sequences at the junction. Nucleotides (P sequences) may be added to asymmetrically cleaved hairpins in a templated manner. Other nucleotides (N regions) may be added to the sites of VD, VJ, or DJ junctions in a nontemplated manner by the action of the enzyme TdT. These additions generate new sequences that are not present in the germline.
Because of junctional diversity, antibody and TCR molecules show the greatest variability at the junctions of V and C regions, which form the third hypervariable region, or CDR3 (see Fig. 8-6). In fact, because of junctional diversity, the numbers of different amino acid sequences that are present in the CDR3 regions of Ig and TCR molecules are much greater than the numbers that can be encoded by germline gene segments. The CDR3 regions of Ig and TCR molecules are also the most important portions of these molecules for determining the specificity of antigen binding (see Chapters 5 and 7). Thus, the greatest diversity in antigen receptors is concentrated in the regions of the receptors that are the most important for antigen binding.
Although the theoretical limit of the number of Ig and TCR proteins that can be produced is enormous (see Table 8-1), the actual number of antigen receptors on B or T cells expressed in each individual is probably on the order of only 107. This may reflect the fact that most receptors, which are generated by random DNA recombination, do not pass the selection processes needed for maturation.
A practical application of our knowledge of junctional diversity is the determination of the clonality of lymphoid tumors that have arisen from B or T cells. This laboratory test is commonly used to distinguish monoclonal tumors from polyclonal proliferations of lymphocytes in response to some external stimulus. Because every lymphocyte clone expresses a unique antigen receptor CDR3 region, the sequence of nucleotides at the V(D)J recombination site serves as a specific marker for each clone. Thus, by measuring the length of the junctional regions of Ig or TCR genes in different B or T cell lesions by polymerase chain reaction assays, one can establish whether these lesions arose from a single clone (indicating a tumor) or independently from different clones (implying non-neoplastic proliferation of lymphocytes). The same method may be used to identify small numbers of tumor cells in the blood or tissues.
With this background, we proceed to a discussion of B lymphocyte development and then the maturation of T cells.
B Lymphocyte Development
The principal events during the maturation of B lymphocytes are the rearrangement and expression of Ig genes in a precise order, selection and proliferation of developing B cells at the pre-antigen receptor checkpoint, and selection of the mature B cell repertoire. Before birth, B lymphocytes develop from committed precursors in the fetal liver, and after birth, B cells are generated in the bone marrow. The majority of B lymphocytes arise from adult bone marrow progenitors that are initially Ig negative, develop into immature B cells that express membrane-bound IgM molecules, and then leave the bone marrow to mature further primarily in the spleen. In the spleen, cells that develop into follicular B cells express IgM and IgD on the cell surface and acquire the ability to recirculate and populate all peripheral lymphoid organs. These follicular B cells home to lymphoid follicles and are able to recognize foreign antigens and to respond to them. The development of a mature B cell from a lymphoid progenitor is estimated to take 2 to 3 days in humans.
Stages of B Lymphocyte Development
During their maturation, cells of the B lymphocyte lineage go through distinguishable stages, each characterized by distinct cell surface markers and a specific pattern of Ig gene expression (Fig. 8-14). The major stages and the events in each are described next.
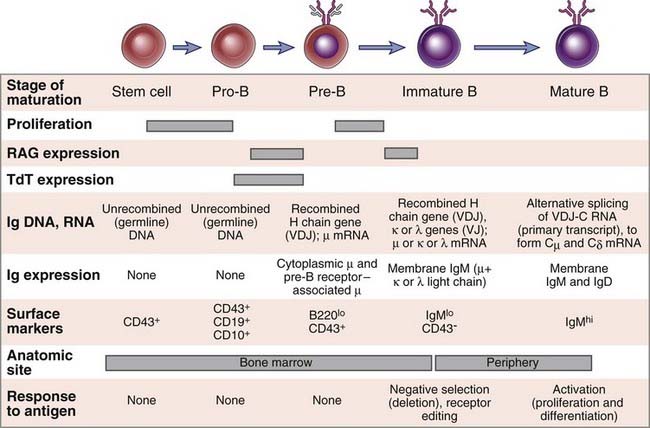
FIGURE 8–14 Stages of B cell maturation.
Events corresponding to each stage of B cell maturation from a bone marrow stem cell to a mature B lymphocyte are illustrated. Several surface markers in addition to those shown have been used to define distinct stages of B cell maturation.
The Pro-B and Pre-B Stages of B Cell Development
The earliest bone marrow cell committed to the B cell lineage is called a pro-B cell. Pro-B cells do not produce Ig, but they can be distinguished from other immature cells by the expression of B lineage–restricted surface molecules such as CD19 and CD10. Rag proteins are first expressed at this stage, and the first recombination of Ig genes occurs at the heavy chain locus. This recombination brings together one D and one J gene segment, with deletion of the intervening DNA (Fig. 8-15A). The D segments that are 5′ of the rearranged D segment and the J segments that are 3′ of the rearranged J segment are not affected by this recombination (e.g., D1 and J2 to J6 in Fig. 8-15A). After the D-J recombination event, one of the many 5′ V genes is joined to the DJ unit, giving rise to a rearranged VDJ exon. At this stage, all V and D segments between the rearranged V and D genes are also deleted. V-to-DJ recombination at the Ig H chain locus occurs only in committed B lymphocyte precursors and is a critical event in Ig expression because only the rearranged V gene is subsequently transcribed. The TdT enzyme, which catalyzes the nontemplated addition of junctional N nucleotides, is expressed most abundantly during the pro-B stage when VDJ recombination occurs at the Ig H locus and levels of TdT decrease before light chain gene V-J recombination is complete. Therefore, junctional diversity attributed to addition of N nucleotides is more prominent in rearranged heavy chain genes than in light chain genes. The heavy chain C region exons remain separated from the VDJ complex by DNA containing the distal J segments and the J-C intron. The rearranged Ig heavy chain gene is transcribed to produce a primary transcript that includes the rearranged VDJ complex and the Cµ exons. The Cµ nuclear RNA is cleaved downstream of one of two consensus polyadenylation sites, and multiple adenine nucleotides, called poly-A tails, are added to the 3′ end. This nuclear RNA undergoes splicing, an RNA processing event in which the introns are removed and exons joined together. In the case of the µ RNA, introns between the leader exon and the VDJ exon, between the VDJ exon and the first exon of the Cµ locus, and between each of the subsequent constant region exons of Cµ are removed, thus giving rise to a spliced mRNA for the µ heavy chain. If the mRNA is derived from an Ig locus at which rearrangement was productive, translation of the rearranged µ heavy chain mRNA leads to synthesis of the µ protein. For a rearrangement to be productive (in the correct reading frame), bases must be added or removed at junctions in multiples of three. This ensures that the rearranged Ig gene will be able to correctly encode an Ig protein. Approximately half of all pro-B cells make productive rearrangements at the Ig H locus on at least one chromosome and can thus go on to synthesize the µ heavy chain protein. Only cells that make productive rearrangements survive and differentiate further.
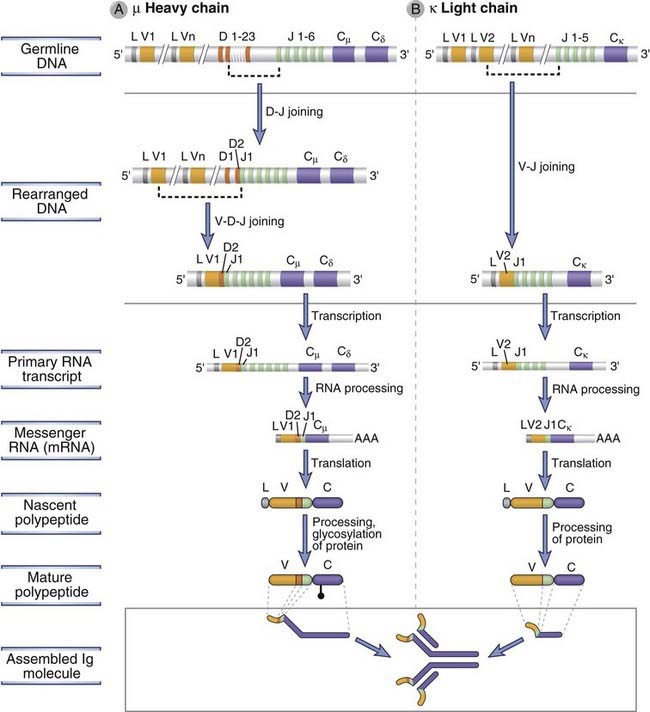
FIGURE 8–15 Ig heavy and light chain gene recombination and expression.
The sequence of DNA recombination and gene expression events is shown for the Ig µ heavy chain (A) and the Ig κ light chain (B). In the example shown in A, the V region of the µ heavy chain is encoded by the exons V1, D2, and J1. In the example shown in B, the V region of the κ chain is encoded by the exons V2 and J1.
Once a productive Ig µ rearrangement is made, a cell ceases to be called a pro-B cell and has differentiated into the pre-B stage. Pre-B cells are developing B lineage cells that express the Ig µ protein but have yet to rearrange their light chain loci. The pre-B cell expresses the µ heavy chain on the cell surface, in association with other proteins, as the pre-B cell receptor, which has several important roles in B cell maturation.
The Pre-B Cell Receptor
Complexes of µ, surrogate light chains, and signal-transducing proteins called Igα and Igβ form the pre-antigen receptor of the B lineage, known as the pre-B cell receptor (pre-BCR). The µ heavy chain associates with the λ5 and V pre-B proteins, also called surrogate light chains because they are structurally homologous to κ and λ light chains but are invariant (i.e., they are identical in all pre-B cells) and are synthesized only in pro-B and pre-B cells (Fig. 8-16A). Igα and Igβ also form part of the B cell receptor in mature B cells (see Chapter 7). Signals from the pre-BCR drive the pro-B to pre-B transition and are responsible for the largest proliferative expansion of B lineage cells in the bone marrow. It is not known what the pre-BCR recognizes; the consensus view at present is that this receptor functions in a ligand-independent manner and that it is activated by the process of assembly (i.e., the fully assembled receptor is in the “on” mode). The importance of pre-BCRs is illustrated by studies of knockout mice and rare cases of human deficiencies of these receptors. For instance, in mice, knockout of the gene encoding the µ chain or one of the surrogate light chains results in markedly reduced numbers of mature B cells because development is blocked at the pro-B stage.
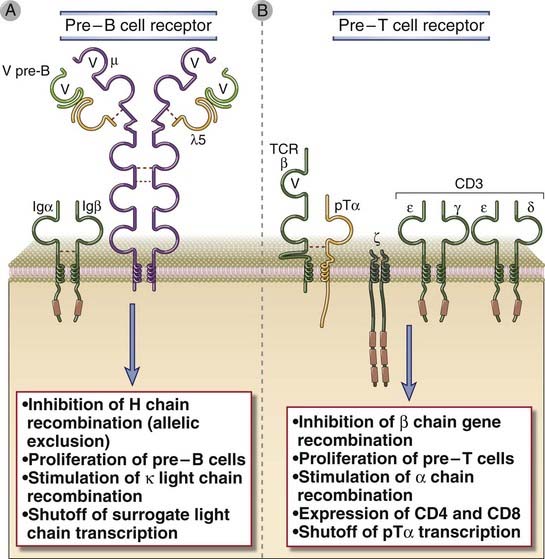
FIGURE 8–16 Pre-B cell and pre-T cell receptors.
The pre-B cell receptor (A) and the pre-T cell receptor (B) are expressed during the pre-B and pre-T cell stages of maturation, respectively, and both receptors share similar structures and functions. The pre-B cell receptor is composed of the µ heavy chain and an invariant surrogate light chain. The surrogate light chain is composed of two proteins, the V pre-B protein, which is homologous to a light chain V domain, and a λ5 protein that is covalently attached to the µ heavy chain by a disulfide bond. The pre-T cell receptor is composed of the TCR β chain and the invariant pre-T α (pTα)chain. The pre-B cell receptor is associated with the Igα and Igβ signaling molecules that are part of the BCR complex in mature B cells (see Chapter 9), and the pre-T cell receptor associates with the CD3 and ζ proteins that are part of the TCR complex in mature T cells (see Chapter 7).
The expression of the pre-BCR is the first checkpoint in B cell maturation. Numerous signaling molecules linked to both the pre-BCR and the BCR are required for cells to successfully negotiate the pre-BCR–mediated checkpoint at the pro-B to pre-B cell transition. A kinase called Bruton’s tyrosine kinase (Btk) is activated downstream of the pre-BCR and is required for delivery of signals from this receptor that mediate survival, proliferation, and maturation at and beyond the pre-B cell stage. In humans, mutations in the BTK gene result in the disease called X-linked agammaglobulinemia (XLA), which is characterized by a failure of B cell maturation (see Chapter 20). In mice, mutations in btk result in a less severe B cell defect in a mouse strain called Xid (for X-linked immunodeficiency). The defect is less severe than in XLA because murine pre-B cells express a second Btk-like kinase called Tec that compensates for the defective Btk.
The pre-BCR regulates further rearrangement of Ig genes in two ways. First, if a µ protein is produced from the recombined heavy chain locus on one chromosome and forms a pre-BCR, this receptor signals to irreversibly inhibit rearrangement of the Ig heavy chain locus on the other chromosome. If the first rearrangement is nonproductive, the heavy chain allele on the other chromosome can complete VDJ rearrangement at the Ig H locus. Thus, in any B cell clone, one heavy chain allele is productively rearranged and expressed, and the other is either retained in the germline configuration or nonproductively rearranged. As a result, an individual B cell can express Ig heavy chain proteins encoded by only one of the two inherited alleles. This phenomenon is called allelic exclusion, and it ensures that every B cell will express a single receptor, thus maintaining clonal specificity. If both alleles undergo nonproductive Ig H gene rearrangements, the developing cell cannot produce Ig heavy chains, cannot generate a pre-BCR–dependent survival signal, and thus undergoes programmed cell death. Ig heavy chain allelic exclusion involves changes in chromatin structure in the heavy chain locus that limit accessibility to the V(D)J recombinase.
The second way in which the pre-BCR regulates the production of the antigen receptor is by stimulating κ light chain gene rearrangement. However, µ chain expression is not absolutely required for light chain gene recombination, as shown by the finding that knockout mice lacking the µ gene do initiate light chain gene rearrangements in some developing B cells (which, of course, cannot express functional antigen receptors and proceed to maturity). The pre-BCR also contributes to the inactivation of surrogate light chain gene expression as pre-B cells mature.
Immature B Cells
Following the pre-B cell stage, each developing B cell initially rearranges a κ light chain gene, and if the rearrangement is in-frame, it will produce a κ light chain protein, which associates with the previously synthesized µ chain to produce a complete IgM protein. If the κ locus is not productively rearranged, the cell can rearrange the λ locus and again produce a complete IgM molecule. (Induction of λ light chain gene rearrangement occurs mainly when Ig κ-expressing B cell receptors are self-reactive, as will be discussed later). The IgM-expressing B cell is called an immature B cell. DNA recombination in the κ light chain locus occurs in a similar manner as in the Ig heavy chain locus (see Fig. 8-15B). There are no D segments in the light chain loci, and therefore recombination involves only the joining of one V segment to one J segment, forming a VJ exon. This VJ exon remains separated from the C region by an intron, and this separation is retained in the primary RNA transcript. Splicing of the primary transcript results in the removal of the intron between the VJ and C exons and generates an mRNA that is translated to produce the κ or λ protein. In the λ locus, alternative RNA splicing may lead to the use of any one of the four functional Cλ exons, but there is no known functional difference between the resulting types of λ light chains. Production of a κ protein prevents λ rearrangement, and, as stated before, λ rearrangement occurs only if the κ rearrangement was nonproductive or if a self-reactive rearranged κ light chain is deleted. As a result, an individual B cell clone can express only one of the two types of light chains; this phenomenon is called light chain isotype exclusion. As in the heavy chain locus, expression of κ or λ is allelically excluded and is initiated from only one of the two parental chromosomes at any given time. Also, as for heavy chains, if both alleles of both κ and λ chains are nonfunctional in a developing B cell, that cell fails to receive survival signals that are normally generated by the BCR and dies.
The assembled IgM molecules are expressed on the cell surface in association with Igα and Igβ, where they function as specific receptors for antigens. In cells that are not strongly self-reactive, the BCR provides ligand-independent tonic signals that keep the B cell alive and also mediate the shutoff of Rag gene expression, thus preventing further Ig gene rearrangement. Immature B cells do not proliferate and differentiate in response to antigens. In fact, if they recognize antigens in the bone marrow with high avidity, which may occur if the B cells express receptors for multivalent self antigens that are present in the bone marrow, the B cells may undergo receptor editing or cell death, as described later. These processes are important for the negative selection of strongly self-reactive B cells. Immature B cells that are not strongly self-reactive leave the bone marrow and complete their maturation in the spleen before migrating to other peripheral lymphoid organs.
Subsets of Mature B Cells
Distinct subsets of B cells develop from different progenitors (Fig. 8-17). Fetal liver–derived HSCs are the precursors of B-1 B cells, described later. Bone marrow–derived HSCs give rise to the majority of B cells, which are sometimes called B-2 B cells. These cells rapidly pass through two transitional stages and can commit to development either into marginal zone B cells, also described later, or into follicular B cells. The affinity of the B cell receptor for self antigens contributes to whether a maturing B-2 B cell will differentiate into a follicular or a marginal zone B cell. This cell fate decision represents a positive selection event in B lymphocytes that is linked to lineage commitment.
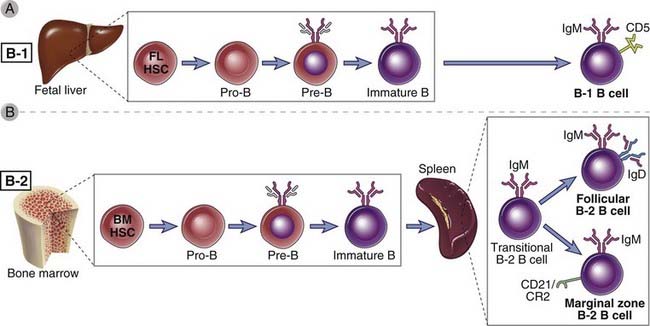
FIGURE 8–17 B lymphocyte subsets.
A, Most B cells that develop from fetal liver–derived stem cells differentiate into the B-1 lineage. B, B lymphocytes that arise from bone marrow precursors after birth give rise to the B-2 lineage. Two major subsets of B lymphocytes are derived from B-2 B cell precursors. Follicular B cells are recirculating lymphocytes; marginal zone B cells are abundant in the spleen in rodents but can also be found in lymph nodes in humans.
Follicular B Cells
Most mature B cells belong to the follicular B cell subset and produce IgD in addition to IgM. Each of these B cells coexpresses µ and δ heavy chains using the same VDJ exon to generate the V domain and in association with the same κ or λ light chain to produce two membrane receptors with the same antigen specificity. Simultaneous expression in a single B cell of the same rearranged VDJ exon on two transcripts, one including Cµ exons and the other Cδ exons, is achieved by alternative RNA splicing (Fig. 8-18). A long primary RNA transcript is produced containing the rearranged VDJ unit as well as the Cµ and Cδ genes. If the primary transcript is cleaved and polyadenylated after the µ exons, introns are spliced out such that the VDJ exon is contiguous with Cµ exons; this results in the generation of a µ mRNA. If, however, the VDJ complex is not linked to Cµ exons but is spliced to Cδ exons, a δ mRNA is produced. Subsequent translation results in the synthesis of a complete µ or δ heavy chain protein. Thus, selective polyadenylation and alternative splicing allow a B cell to simultaneously produce mature mRNAs and proteins of two different heavy chain isotypes. The precise mechanisms that regulate the choice of polyadenylation or splice acceptor sites by which the rearranged VDJ is joined to either Cµ or Cδ are poorly understood, as are the signals that determine when and why a B cell expresses both IgM and IgD rather than IgM alone. The coexpression of IgM and IgD is accompanied by the ability to recirculate and the acquisition of functional competence, and this is why IgM+IgD+ B cells are also called mature B cells. This correlation between expression of IgD and acquisition of functional competence has led to the suggestion that IgD is the essential activating receptor of mature B cells. However, there is no evidence for a functional difference between membrane IgM and membrane IgD. Moreover, knockout of the Ig δ gene in mice does not have a significant impact on the maturation or antigen-induced responses of B cells. Follicular B cells are also often called recirculating B cells because they migrate from one lymphoid organ to the next, residing in specialized niches known as B cell follicles (see Chapter 2). In these niches, these B cells are maintained, in part, by survival signals delivered by a cytokine of the tumor necrosis factor (TNF) family called BAFF or BlyS (see Chapter 11).
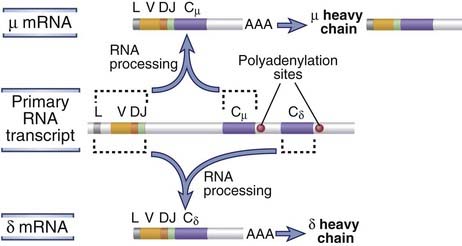
FIGURE 8–18 Coexpression of IgM and IgD.
Alternative processing of a primary RNA transcript results in the formation of a µ or δ mRNA. Dashed lines indicate the H chain segments that are joined by RNA splicing.
Mature, naive B cells are responsive to antigens, and unless the cells encounter antigens that they recognize with high affinity and respond to, they die in a few months. In Chapter 11, we will discuss how these cells respond to antigens and how the pattern of Ig gene expression changes during antigen-induced B cell differentiation.
B-1 and Marginal Zone B Cells
A subset of B lymphocytes, called B-1 B cells, differs from the majority of B lymphocytes and develops in a unique manner. These cells develop from fetal liver–derived HSCs and are best defined in rodents. Most murine B-1 cells express the CD5 (Ly-1) molecule. In the adult, large numbers of B-1 cells are found as a self-renewing population in the peritoneum and mucosal sites. B-1 cells develop earlier during ontogeny than conventional B cells do, express a relatively limited repertoire of V genes, and exhibit far less junctional diversity than conventional B cells do (because TdT is not expressed in the fetal liver). B-1 cells as well as marginal zone B cells spontaneously secrete IgM antibodies that often react with microbial polysaccharides and lipids. These antibodies are sometimes called natural antibodies because they are present in individuals without overt immunization, although it is possible that microbial flora in the gut are the source of antigens that stimulate their production. B-1 cells contribute to rapid antibody production against microbes in particular tissues, such as the peritoneum. At mucosal sites, as many as half the IgA-secreting cells in the lamina propria may be derived from B-1 cells. B-1 B cells are analogous to γδ T cells in that they both have antigen receptor repertoires of limited diversity, and they are both presumed to respond to commonly encountered microbial antigens early in immune responses.
The major marker used to delineate murine B-1 cells is CD5. In humans, B-1–like cells have been described but these cells do not express CD5. In humans, CD5 is found on transitional B cells and some activated B cell populations.
Marginal zone B cells are located primarily in the vicinity of the marginal sinus in the spleen and are similar to B-1 cells in terms of their limited diversity and their ability to respond to polysaccharide antigens and to generate natural antibodies. Marginal zone B cells exist in both mice and humans and express IgM and the CD21 coreceptor. In mice, marginal zone B cells exist only in the spleen, whereas in humans, they can be found in the spleen as well as in lymph nodes. Marginal zone B cells respond very rapidly to blood-borne microbes and differentiate into short-lived IgM-secreting plasma cells. Although they generally mediate T cell–independent humoral immune responses to circulating pathogens, marginal zone B cells also appear capable of mediating some T cell–dependent immune responses.
Selection of the Mature B Cell Repertoire
The repertoire of mature B cells is positively selected from the pool of immature B cells. As we shall see later, positive selection is well defined in T lymphocytes and is responsible for matching the TCRs on newly generated CD8+ and CD4+ T cells with their ability to recognize self MHC class I and MHC class II molecules, respectively. There is no comparable restriction for B cell antigen recognition. Nevertheless, positive selection appears to be a general phenomenon primarily geared to identification of lymphocytes that have completed their antigen receptor gene rearrangement program successfully. It is believed that only B cells that express functional membrane Ig molecules receive constitutive BCR-derived survival signals, also known as “tonic” BCR signals. Tonic BCR signals mediate the shutoff of Rag gene expression in immature B cells and activate cell survival pathways in all B cells. Self antigens may influence the strength of the BCR signal and thereby the subsequent choice of peripheral B cell lineage during B cell maturation.
Immature B cells that recognize self antigens with high avidity may be induced to change their specificities by a process called receptor editing. In this process, antigen recognition leads to reactivation of Rag genes, additional light chain V-J recombination events, and production of a new Ig light chain, allowing the cell to express a different B cell receptor that is not self-reactive. Receptor editing generally is targeted at self-reactive κ light chain genes. VJκ exons encoding the variable domains of autoreactive light chains are deleted and replaced by new VJκ exons or by newly rearranged λ light chain genes. The new VJκ exon may be generated by the rearrangement of a Vκ gene upstream of the original Vκ gene that produced an autoreactive light chain to a Jκ segment downstream of the originally rearranged Jκ segment.
If receptor editing fails, the immature B cells that express high-affinity receptors for self antigens and encounter these antigens in the bone marrow or the spleen may die by apoptosis. This process is also called negative selection. The antigens mediating negative selection—usually abundant or polyvalent (e.g., membrane-bound) self antigens—deliver strong signals to IgM-expressing immature B lymphocytes that happen to express receptors specific for these self antigens. Both receptor editing and deletion are responsible for maintaining B cell tolerance to self antigens that are present in the bone marrow (see Chapter 14).
Once the transition is made to the IgD+IgM+ mature B cell stage, antigen recognition leads to proliferation and differentiation, not to receptor editing or apoptosis. As a result, mature B cells that recognize antigens with high affinity in peripheral lymphoid tissues are activated, and this process leads to humoral immune responses. Follicular B cells make most of the helper T cell–dependent antibody responses to protein antigens (see Chapter 11).
Maturation of T Lymphocytes
The maturation of T lymphocytes from committed progenitors involves the sequential rearrangement and expression of TCR genes, cell proliferation, antigen-induced selection, and commitment to phenotypically and functionally distinct subsets (Fig. 8-19). In many ways, this is similar to B cell maturation. However, T cell maturation has some unique features that reflect the specificity of the majority of T lymphocytes for self MHC–associated peptide antigens and the need for a special microenvironment for selecting cells with this specificity.
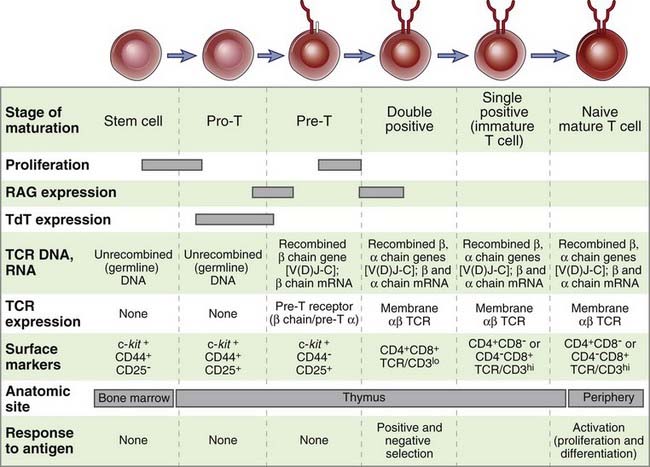
FIGURE 8–19 Stages of T cell maturation.
Events corresponding to each stage of T cell maturation from a bone marrow stem cell to a mature T lymphocyte are illustrated. Several surface markers in addition to those shown have been used to define distinct stages of T cell maturation.
Role of the Thymus in T Cell Maturation
The thymus is the major site of maturation of T cells. This function of the thymus was first suspected because of immunologic deficiencies associated with the lack of a thymus. The congenital absence of the thymus, as occurs in the DiGeorge syndrome in humans or in the nude mouse strain, is characterized by low numbers of mature T cells in the circulation and peripheral lymphoid tissues and severe deficiencies in T cell–mediated immunity (see Chapter 20). If the thymus is removed from a neonatal mouse, this animal fails to develop mature T cells. The thymic anlage develops from the endoderm of the third pharyngeal pouch and the underlying neural crest–derived mesenchyme and is subsequently populated by bone marrow–derived precursors. The thymus involutes with age and is virtually undetectable in postpubertal humans, resulting in a somewhat reduced output of mature T cells. However, maturation of T cells continues throughout adult life, as indicated by the successful reconstitution of the immune system in adult recipients of bone marrow transplants. It may be that the remnant of the involuted thymus is adequate for some T cell maturation. Because memory T cells have a long life span (perhaps longer than 20 years in humans) and accumulate with age, the need to generate new T cells decreases as individuals age.
T lymphocytes originate from precursors that arise in the fetal liver and adult bone marrow and seed the thymus. These precursors are multipotent progenitors that enter the thymus from the blood stream, crossing the endothelium of a postcapillary venule in the corticomedullary junction region of the thymus. In mice, immature lymphocytes are first detected in the thymus on the eleventh day of the normal 21-day gestation. This corresponds to about week 7 or 8 of gestation in humans. Developing T cells in the thymus are called thymocytes. The most immature thymocytes are found in the subcapsular sinus and outer cortical region of the thymus. From here, the thymocytes migrate into and through the cortex, where most of the subsequent maturation events occur. It is in the cortex that the thymocytes first express γδ and αβ TCRs. The αβ T cells mature into CD4+ class II MHC–restricted or CD8+ class I MHC–restricted T cells as they leave the cortex and enter the medulla. From the medulla, CD4+ and CD8+ single-positive thymocytes exit the thymus through the circulation. In the following sections, we discuss the maturation of αβ T cells; γδ T cells are discussed later in the chapter.
The thymic environment provides stimuli that are required for the proliferation and maturation of thymocytes. Many of these stimuli come from thymic cells other than the maturing T cells. Within the cortex, thymic cortical epithelial cells form a meshwork of long cytoplasmic processes, around which thymocytes must pass to reach the medulla. Epithelial cells of a distinct type known as thymic medullary epithelial cells are also present in the medulla. Bone marrow–derived dendritic cells are present at the corticomedullary junction and within the medulla, and macrophages are present primarily within the medulla. The migration of thymocytes through this anatomic arrangement allows physical interactions between the thymocytes and these other cells that are necessary for the maturation and selection of the T lymphocytes.
Two types of molecules produced by the nonlymphoid thymic cells are important for T cell maturation. The first are class I and class II MHC molecules, which are expressed on epithelial cells and dendritic cells in the thymus. The interactions of maturing thymocytes with these MHC molecules within the thymus are essential for the selection of the mature T cell repertoire, as we will discuss later. Second, thymic stromal cells, including epithelial cells, secrete cytokines and chemokines, which respectively stimulate the proliferation of immature T cells and orchestrate the cortical to medullary transit of developing αβ lineage thymocytes. The best defined of these cytokines is IL-7, which was mentioned earlier as a critical lymphopoietic growth factor. The movement of cells into and through the thymus is driven by chemokines. The progenitors of thymocytes express the chemokine receptor CCR9, which binds to the chemokine CCL25, which is produced in the thymic cortex. Entry of precursors into the thymus is dependent on CCL25 and CCR9. Chemokines such as CCL21 and CCL19, which are recognized by the CCR7 chemokine receptor on thymocytes, mediate the guided movement of developing T cells from the cortex to the medulla.
The rates of cell proliferation and apoptotic death are extremely high in cortical thymocytes. A single precursor gives rise to many progeny, and 95% of these cells die by apoptosis before reaching the medulla. The cell death is due to a combination of factors including a failure to productively rearrange the TCR β chain gene and thus to negotiate the pre-TCR/β selection checkpoint described later, a failure to be positively selected by MHC molecules in the thymus, and self antigen–induced negative selection (see Figs. 8-3 and 8-4). Cortical thymocytes are also sensitive to irradiation and glucocorticoids. In vivo, high doses of glucocorticoids induce apoptotic death of immature cortical thymocytes.
Stages of T Cell Maturation
During T cell maturation, there is a precise order in which TCR genes are rearranged and in which the TCR and CD4 and CD8 coreceptors are expressed (Fig. 8-20; see also Fig. 8-19). In the mouse, surface expression of the γδ TCR occurs first, 3 to 4 days after precursor cells first arrive in the thymus, and the αβ TCR is expressed 2 or 3 days later. In human fetal thymuses, γδ TCR expression begins at about 9 weeks of gestation, followed by expression of the αβ TCR at 10 weeks.
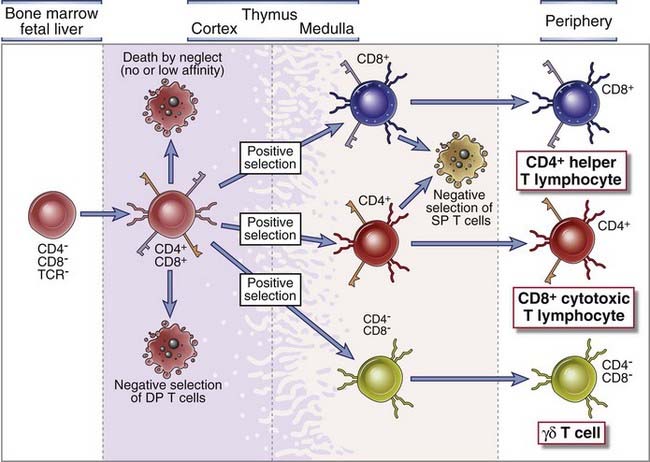
FIGURE 8–20 Maturation of T cells in the thymus.
Precursors of T cells travel from the bone marrow through the blood to the thymus. In the thymic cortex, progenitors of αβ T cells express TCRs and CD4 and CD8 coreceptors. Selection processes eliminate self-reactive T cells in the cortex at the double-positive (DP) stage and also single-positive (SP) medullary thymocytes. They promote survival of thymocytes whose TCRs bind self MHC molecules with low affinity. Functional and phenotypic differentiation into CD4+CD8− or CD8+CD4− T cells occurs in the medulla, and mature T cells are released into the circulation.
Double-Negative Thymocytes
The most immature cortical thymocytes, which are recent arrivals from the bone marrow, contain TCR genes in their germline configuration and do not express TCR, CD3, ζ chains, CD4, or CD8; these cells are called double-negative thymocytes. Thymocytes at this stage are also considered to be at the pro-T cell stage of maturation. The majority (>90%) of the double-negative thymocytes that survive thymic selection processes will ultimately give rise to αβ TCR–expressing, MHC-restricted CD4+ and CD8+ T cells, and the remainder of these thymocytes will give rise to γδ T cells. Rag-1 and Rag-2 proteins are first expressed at the pro-T stage of development and are required for the rearrangement of TCR genes. Dβ-to-Jβ rearrangements at the TCR β chain locus occur first; these involve either joining of the Dβ1 gene segment to one of the six Jβ1 segments or joining of the Dβ2 segment to one of the six Jβ2 segments (Fig. 8-21A). Vβ-to-DJβ rearrangements occur at the transition between the pro-T stage and the subsequent pre-T stage during αβ T cell development. The DNA sequences between the segments undergoing rearrangement, including D, J, and possibly Cβ1 genes (if Dβ2 and Jβ2 segments are used), are deleted during this rearrangement process. The primary nuclear transcripts of the TCR β genes contain the intron between the recombined VDJβ exon and the relevant Cβ gene. Poly-A tails are added after cleavage of the primary transcript downstream of consensus polyadenylation sites located 3′ of the Cβ region, and the sequences between the VDJ exon and Cβ are spliced out to form a mature mRNA in which VDJ segments are juxtaposed to either of the two Cβ genes (depending on which J segment was selected during the rearrangement process). Translation of this mRNA gives rise to a full-length TCR β chain protein. The two Cβ genes appear to be functionally interchangeable, and there is no evidence that an individual T cell ever switches from one C gene to another. Furthermore, the use of either Cβ gene segment does not influence the function or specificity of the TCR. The promoters in the 5′ flanking regions of Vβ genes function together with a powerful enhancer that is located 3′ of the Cβ2 gene once V genes are brought close to the C gene by VDJ recombination. This proximity of the promoter to the enhancer is responsible for high-level T cell–specific transcription of the rearranged TCR β chain gene.
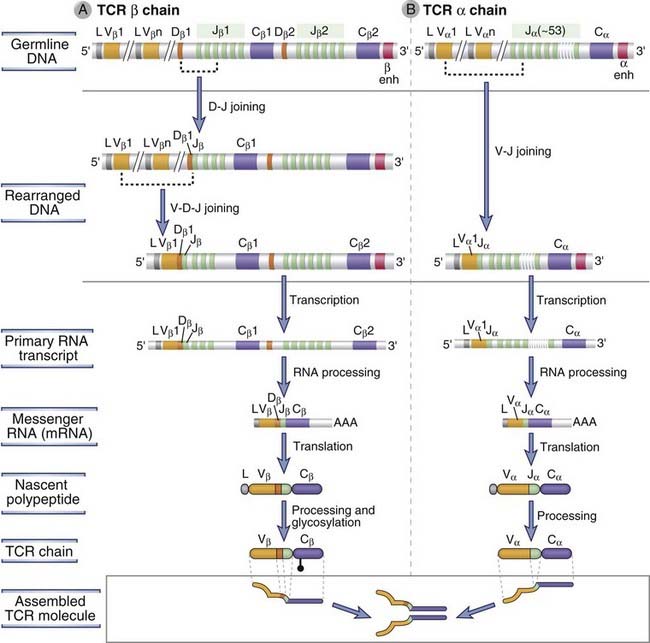
FIGURE 8–21 TCR α and β chain gene recombination and expression.
The sequence of recombination and gene expression events is shown for the TCR β chain (A) and the TCR α chain (B). In the example shown in A, the variable (V) region of the rearranged TCR β chain includes the Vβ1 and Dβ1 gene segments and the third J segment in the Jβ1 cluster. The constant (C) region is encoded by the Cβ1 exon. Note that at the TCR β chain locus, rearrangement begins with D-to-J joining followed by V-to-DJ joining. In humans, 14 Jβ segments have been identified, and not all are shown in the figure. In the example shown in B, the V region of the TCR α chain includes the Vα1 gene and the second J segment in the Jα cluster (this cluster is made up of at least 61 Jα segments in humans; not all are shown here).
Pre-T Cell Receptor
If a productive (i.e., in-frame) rearrangement of the TCR β chain gene occurs in a given pro-T cell, the TCR β chain protein is expressed on the cell surface in association with an invariant protein called pre-Tα and with CD3 and ζ proteins to form the pre-T cell receptor (pre-TCR) (see Fig. 8-16B). The pre-TCR mediates the selection of the developing pre-T cells that productively rearrange the β chain of the TCR. Roughly half of all developing pre-T cells add or remove bases at rearrangement junctions that are multiples of three (on at least one TCR β chromosome), and therefore approximately half of all developing pre-T cells fail to express a TCR β protein. The function of the pre-TCR complex in T cell development is similar to that of the surrogate light chain–containing pre-BCR in B cell development. Signals from the pre-TCR mediate the survival of pre-T cells and contribute to the largest proliferative expansion during T cell development. Pre-TCR signals also initiate recombination at the TCR α chain locus and drive the transition from the double-negative to the double-positive stage of thymocyte development (discussed later). These signals also inhibit further rearrangement of the TCR β chain locus largely by limiting accessibility of the other allele to the recombination machinery. This results in β chain allelic exclusion (i.e., mature T cells express only one of the two inherited β chain alleles). As in pre-B cells, it is not known what, if any, ligand the pre-TCR recognizes. Pre-TCR signaling, like pre-BCR signaling, is generally believed to be initiated in a ligand-independent manner, dependent on the successful assembly of the pre-TCR complex. Pre-TCR signaling is mediated by a number of cytosolic kinases and adaptor proteins that are known also to be linked to TCR signaling (see Chapter 7). The essential function of the pre-TCR in T cell maturation has been demonstrated by numerous studies with genetically mutated mice, in which lack of any component of the pre-TCR complex (i.e., the TCR β chain, pre-Tα, CD3, ζ, or Lck) results in a block in the maturation of T cells at the double-negative stage.
Double-Positive Thymocytes
At the next stage of T cell maturation, thymocytes express both CD4 and CD8 and are called double-positive thymocytes. The expression of CD4 and CD8 is essential for subsequent selection events, discussed later. The rearrangement of the TCR α chain genes and the expression of TCR αβ heterodimers occur in the CD4+CD8+ double-positive population soon after cells cross the pre-TCR checkpoint (see Figs. 8-19 and 8-20). A second wave of Rag gene expression late in the pre-T stage promotes TCR α gene recombination. Because there are no D segments in the TCR α locus, rearrangement consists solely of the joining of V and J segments (see Fig. 8-21B). The large number of Jα segments permits multiple attempts at productive V-J joining on each chromosome, thereby increasing the probability that a functional αβ TCR will be produced. In contrast to the TCR β chain locus, where production of the protein and formation of the pre-TCR suppress further rearrangement, there is little or no allelic exclusion in the α chain locus. Therefore, productive TCR α rearrangements may occur on both chromosomes, and if this happens, the T cell will express two α chains. In fact, up to 30% of mature peripheral T cells do express two different TCRs, with different α chains but the same β chain. It is possible that only one of the two α chains participates in the formation of the functional antigen-specific TCR. Transcriptional regulation of the α chain gene occurs in a similar manner to that of the β chain. There are promoters 5′ of each Vα gene that have low-level activity and are responsible for high-level T cell–specific transcription when brought close to an α chain enhancer located 3′ of the Cα gene. The inability to successfully rearrange the TCR α chain on either chromosome leads to a failure of positive selection (discussed later). Thymocytes that fail to make a productive rearrangement of the TCR α chain gene will die by apoptosis.
TCR α gene expression in the double-positive stage leads to the formation of the complete αβ TCR, which is expressed on the cell surface in association with CD3 and ζ proteins. The coordinate expression of CD3 and ζ proteins and the assembly of intact TCR complexes are required for surface expression. Rearrangement of the TCR α gene results in deletion of the TCR δ locus that lies between V segments (common to both α and δ loci) and Jα segments (see Fig. 8-7). As a result, this T cell is no longer capable of becoming a γδ T cell and is completely committed to the αβ T cell lineage. The expression of Rag genes and further TCR gene recombination cease after this stage of maturation.
Double-positive cells that successfully undergo selection processes go on to mature into CD4+ or CD8+ T cells, which are called single-positive thymocytes. Thus, the stages of T cell maturation in the thymus can readily be distinguished by the expression of CD4 and CD8 (Fig. 8-22A). This phenotypic maturation is accompanied by functional maturation. CD4+ cells acquire the ability to produce cytokines in response to subsequent antigen stimulation and to express effector molecules (such as CD40 ligand) that “help” B lymphocytes, dendritic cells, and macrophages, whereas CD8+ cells become capable of producing molecules that kill other cells. Mature single-positive thymocytes enter the thymic medulla and then leave the thymus to populate peripheral lymphoid tissues.
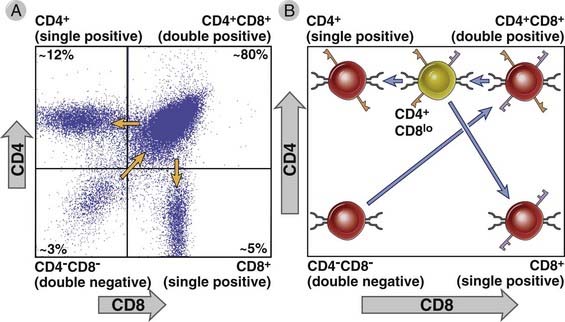
FIGURE 8–22 CD4 and CD8 expression on thymocytes and positive selection of T cells in the thymus.
A, The maturation of thymocytes can be followed by changes in expression of the CD4 and CD8 coreceptors. A two-color flow cytometric analysis of thymocytes using anti-CD4 and anti-CD8 antibodies, each tagged with a different fluorochrome, is illustrated. The percentages of all thymocytes contributed by each major population are shown in the four quadrants. The least mature subset is the CD4−CD8− (double-negative) cells. Arrows indicate the sequence of maturation. B, Positive selection of T cells. Double-positive T cells differentiate into a CD4+CD8low stage and are instructed to become CD4+ cells if the TCR on a double-positive T cell recognizes self class II MHC with moderate avidity and therefore receives adequate coreceptor signals. A CD4+CD8low T cell whose TCR recognizes MHC class I molecules fails to receive strong coreceptor signals and differentiates into a CD8+ T cell, silencing CD4 expression.
Selection Processes in the Maturation of MHC-Restricted αβ T Cells
The selection of developing T cells is dependent on recognition of antigen (peptide-MHC complexes) in the thymus and is responsible for preserving useful cells and eliminating potentially harmful ones. The immature, or unselected, repertoire of T lymphocytes consists of cells whose receptors may recognize any peptide antigen (self or foreign) displayed by any MHC molecule (also self or foreign). In addition, receptors may theoretically be expressed that do not recognize any peptide–MHC molecule complex. In every individual, the only useful T cells are the ones specific for foreign peptides presented by that individual’s MHC molecules, that is, self MHC molecules. When double-positive thymocytes first express αβ TCRs, these receptors encounter self peptides (the only peptides normally present in the thymus) displayed by self MHC molecules (the only MHC molecules available to display peptides) mainly on thymic epithelial cells in the cortex. The outcome of this recognition is determined primarily by the strength of the encounter between TCRs and self antigen–MHC complexes. Positive selection is the process that preserves T cells that recognize self MHC (with self peptides) with low avidity. This recognition preserves cells that can see antigens displayed by that individual’s MHC molecules. At the same time, the cells become committed to the CD4 or CD8 lineage based on whether the TCR on an individual cell respectively recognizes MHC class II or MHC class I molecules. Also, in every individual, T cells that recognize self antigens with high avidity are potentially dangerous because such recognition may trigger autoimmunity. Negative selection is the process in which thymocytes whose TCRs bind strongly to self peptide antigens in association with self MHC molecules are deleted (see Fig. 8-20). The net result of these selection processes is that the repertoire of mature T cells that leaves the thymus is self MHC restricted and tolerant to many self antigens, and only the useful cells complete their maturation. In the following sections, we discuss the details of positive and negative selection.
Positive Selection of Thymocytes: Development of the Self MHC–Restricted T Cell Repertoire
Positive selection is the process in which thymocytes whose TCRs bind with low avidity (i.e., weakly) to self peptide–self MHC complexes are stimulated to survive (see Figs. 8-20 and 8-22). Double-positive thymocytes are produced without antigenic stimulation and begin to express αβ TCRs with randomly generated specificities, which may be biased toward recognition of MHC-like structures. In the thymic cortex, these immature cells encounter epithelial cells that are displaying a variety of self peptides bound to class I and class II MHC molecules. Weak recognition of these self peptide–self MHC complexes promotes the survival of the T cells. Thymocytes whose receptors do not recognize self MHC molecules are permitted to die by a default pathway of apoptosis; this phenomenon is called death by neglect (see Fig. 8-20). Thus, positive selection ensures that T cells are self MHC restricted.
During the transition from double-positive to single-positive cells, thymocytes with class I–restricted TCRs become CD8+CD4−, and cells with class II–restricted TCRs become CD4+CD8−. Immature, double-positive T cells express TCRs that may recognize either self MHC class I or self MHC class II. Two models have been proposed to explain the process of lineage commitment, as a result of which coreceptors are correctly matched with the TCRs that recognize a specific class of MHC molecules. The “stochastic” or “probabilistic” model suggests that the commitment of immature T cells toward either lineage depends on the random probability of a double-positive cell differentiating into a CD4+ or a CD8+ T cell. In this model, a cell that recognizes self MHC class I may randomly differentiate into a CD8+ T cell (with the appropriate coreceptor) and survive or into a CD4+ T cell (with the “wrong” coreceptor) that may fail to receive survival signals. In this process of random differentiation into single-positive cells, the coreceptor would fail to be matched with recognition of the right class of MHC molecules approximately half the time. A more widely accepted view is that the process of lineage commitment linked to positive selection is not a random process but is “instructional.” Instructional models suggest that class I– and class II–restricted TCRs deliver different signals that actively induce expression of the correct coreceptor and shut off expression of the other coreceptor. It is known that double-positive cells go through a stage at which they express high CD4 and low CD8. If the TCR on such a cell is MHC class I restricted, when it sees the appropriate MHC class I and self peptide, it will receive a weak signal because levels of the CD8 coreceptor are low and, in addition, CD8 associates less well with the Lck tyrosine kinase than CD4. These weak signals activate transcription factors (such as Runx3) that stimulate CD8 expression, generating a class I–restricted CD8+ T cell. Conversely, if the TCR on the cell is class II restricted, when it sees class II, it will receive a stronger signal because CD4 levels are high and CD4 associates relatively well with Lck. These strong signals activate a different set of transcription factors (including ThPok), which stimulate CD4 expression and shut off CD8.
Peptides bound to MHC molecules on thymic epithelial cells play an essential role in positive selection. In Chapter 6, we described how cell surface MHC class I and class II molecules always contain bound peptides. These MHC-associated peptides on thymic antigen-presenting cells probably serve two roles in positive selection—first, they promote stable cell surface expression of MHC molecules, and second, they may influence the specificities of the T cells that are selected. It is also clear from a variety of experimental studies that some peptides are better than others in supporting positive selection, and different peptides differ in the repertoires of T cells they select. These results suggest that specific antigen recognition, and not just MHC recognition, has some role in positive selection. One consequence of self peptide–induced positive selection is that the T cells that mature have the capacity to recognize self peptides. We mentioned in Chapter 2 that the survival of naive lymphocytes before encounter with foreign antigens requires survival signals that are apparently generated by recognition of self antigens in peripheral lymphoid organs. The same self peptides that mediate positive selection of double-positive thymocytes in the thymus may be involved in keeping naive, mature (single-positive) T cells alive in peripheral organs, such as the lymph nodes and spleen.
The model of positive selection based on weak recognition of self antigens raises a fundamental question: How does positive selection driven by self antigens produce a repertoire of mature T cells specific for foreign antigens? The likely answer is that positive selection allows many different T cell clones to survive and differentiate, and many of these T cells that recognize self peptides with low affinity will, after maturing, fortuitously recognize foreign peptides with a high enough affinity to be activated and to generate immune responses.
Negative Selection of Thymocytes: Central Tolerance
Thymocytes whose receptors recognize peptide-MHC complexes in the thymus with high avidity undergo apoptosis (called negative selection) or differentiate into regulatory T cells (see Fig. 8-20). Among the double-positive T cells that are generated in the thymus, some may express TCRs that recognize self antigens with high affinity. The peptides present in the thymus are self peptides derived from widely expressed protein antigens as well as from some proteins believed to be restricted to particular tissues. (Recall that microbes that enter through the common routes, i.e., epithelia, are captured and transported to lymph nodes and tend to not enter the thymus.) In immature T cells, a major consequence of high-avidity antigen recognition is the triggering of apoptosis, leading to death, or deletion, of the cells. Therefore, many of the immature thymocytes that express high-affinity receptors for self antigens in the thymus are eliminated, resulting in negative selection of the T cell repertoire. This process eliminates the potentially most harmful self-reactive T cells and is one of the mechanisms ensuring that the immune system does not respond to many self antigens, called self-tolerance. Tolerance induced in immature lymphocytes by recognition of self antigens in the generative (or central) lymphoid organs is also called central tolerance, to be contrasted with peripheral tolerance induced in mature lymphocytes by self antigens in peripheral tissues. We will discuss the mechanisms and physiologic importance of immunologic tolerance in more detail in Chapter 14.
The deletion of immature self-reactive T cells may occur both at the double-positive stage in the cortex and in newly generated single-positive T cells in the medulla. The thymic antigen-presenting cells that mediate negative selection are primarily bone marrow–derived dendritic cells and macrophages, both abundant in the medulla, and thymic medullary epithelial cells, whereas cortical epithelial cells are especially (and perhaps uniquely) effective at inducing positive selection. Double-positive T cells are drawn to the thymic medulla by the CCR7-specific chemokines CCL21 and CCL19. In the medulla, thymic medullary epithelial cells express a nuclear protein called AIRE (autoimmune regulator) that induces the expression of a number of tissue-specific genes in the thymus. These genes are otherwise normally expressed only in specific peripheral organs, such as the pancreas and the thyroid. Their AIRE-dependent expression in the thymus makes many tissue-specific peptides available for presentation to developing T cells, facilitating the deletion (negative selection) of these cells. A mutation in the gene that encodes AIRE results in an autoimmune polyendocrine syndrome, underscoring the importance of AIRE in mediating central tolerance to tissue-specific antigens (see Chapter 14).
The mechanism of negative selection in the thymus is the induction of death by apoptosis. Unlike the phenomenon of death by neglect, which occurs in the absence of positive selection, in negative selection, active death-promoting signals are generated when the TCR of immature thymocytes binds with high affinity to antigen. The induction by TCR signaling of a proapoptotic protein called Bim probably plays a crucial role in the induction of mitochondrial leakiness and thymocyte apoptosis during negative selection (see Chapter 14). It is also clear that high-avidity antigen recognition by immature T cells triggers apoptosis, but the same recognition by mature lymphocytes (in concert with other signals, see Chapter 9) initiates T cell responses. The biochemical basis of this fundamental difference is not defined.
Recognition of self antigens in the thymus can generate a population of regulatory T cells that function to prevent autoimmune reactions (see Chapter 14). It is not clear what factors determine the choice between the two alternative fates of immature T cells that recognize self antigens with high avidity, namely, deletion of immature T cells and the development of regulatory T cells. It is possible that slightly lower avidity interactions than those required for deletion may lead to the development of natural regulatory T cells, but clear evidence for this kind of fine discrimination still needs to be obtained.
γδ T Lymphocytes
TCR αβ- and γδ-expressing thymocytes are separate lineages with a common precursor. In fetal thymuses, the first TCR gene rearrangements involve the γ and δ loci. Recombination of TCR γ and δ loci proceeds in a fashion similar to that of other antigen receptor gene rearrangements, although the order of rearrangement appears to be less rigid than in other loci. In a developing double-negative T cell, rearrangement of TCR β, γ, or δ loci is initially possible. If a cell succeeds in productively rearranging its TCR γ as well as its TCR δ loci before it makes a productive TCR β rearrangement, it is selected into the γδ T cell lineage. This happens in about 10% of developing double-negative T cells. About 90% of the time, a productive TCR β gene rearrangement is made first. In this situation, pre-TCR signaling selects these cells to mature into the αβ T cell lineage, and eventual deletion of TCR δ when TCR α is rearranged (the TCR δ locus is embedded in the TCR α locus) results in irreversible commitment to the αβ lineage.
The diversity of the γδ T cell repertoire is theoretically even greater than that of the αβ T cell repertoire, in part because the heptamer-nonamer recognition sequences adjacent to D segments permit D-to-D joining. Paradoxically, however, the actual diversity of expressed γδ TCRs is limited because only a few of the available V, D, and J segments are used in mature γδ T cells, for unknown reasons. This limited diversity is reminiscent of the limited diversity of the B-1 subset of B lymphocytes and is in keeping with the concept that γδ T cells serve as an early defense against a limited number of commonly encountered microbes at epithelial barriers.
NKT Cells
NKT cells are not MHC restricted and do not recognize peptides displayed by antigen-presenting cells. These cells express αβ TCRs that are CD1 restricted and also bear a surface marker found on NK cells, hence their name. The TCRs of NKT cells recognize lipid antigens presented by CD1 molecules. CD1 molecules are class I MHC–like molecules made up of a heavy chain and β2-microglobulin. The heavy chain has a groove made up of hydrophobic residues that can bind and present lipid antigens. These lipid antigens may be derived from endocytosed microbes or they may be self lipids (see Chapter 6). In the thymic cortex, double positive αβ T cells that express T cell receptors that recognize CD1 molecules expressed on neighboring double positive thymocytes are induced to differentiate into NKT cells. A large number of CD1-restricted NKT cells have an “invariant” TCR resulting from a unique and stereotypic TCR α chain gene rearrangement event. NKT cells secrete cytokines and participate in host defense and may also serve to regulate a variety of immune responses. The functions of NKT cells are described in Chapter 10.
Summary
Early B Cell Development and V(D)J Recombination
Cobaleda C, Busslinger M. Developmental plasticity of lymphocytes. Current Opinion in Immunology. 2008;20:139-148.
Jenkinson EJ, Jenkinson WE, Rossi SW, Anderson G. The thymus and T-cell commitment: the right choice for Notch? Nature Reviews Immunology. 2006;6:551-555.
Johnson K, Reddy KL, Singh H. Molecular pathways and mechanisms regulating the recombination of immunoglobulin genes during B-lymphocyte development. Advances in Experimental Medicine and Biology. 2009;650:133-147.
Jung D, Giallourakis C, Mostoslavsky R, Alt FW. Mechanism and control of V(D)J recombination at the immunoglobulin heavy chain locus. Annual Review of Immunology. 2006;24:541-570.
Nemazee D. Receptor editing in lymphocyte development and central tolerance. Nature Reviews Immunology. 2006;6:728-740.
Perlot T, Alt FW. Cis-regulatory elements and epigenetic changes control genomic rearrangements of the IgH locus. Advances in Immunology. 2008;99:1-32.
Schatz DG, JI Y. Recombination centers and the orchestration of V(D)J recombination. Nature Reviews Immunology. 11, March 2011.
Boehm T. Thymus development and function. Current Opinion in Immunology. 2008;20:178-184.
Carpenter AC, Bosselut R. Decision checkpoints in the thymus. Nature Immunology. 2010;11:666-673.
Godfrey DI, Stankovic S, Baxter AG. Raising the NKT cell family. Nature Immunology. 2010;11:197-206.
He X, Park K, Kappes DJ. The role of ThPOK in control of CD4/CD8 lineage commitment. Annual Review of Immunology. 2010;28:295-320.
Kyewski B, Klein L. A central role for central tolerance. Annual Review of Immunology. 2006;24:571-606.
Maillard I, Fang T, Pear WS. Regulation of lymphoid development, differentiation, and function by the Notch pathway. Annual Review of Immunology. 2005;23:945-974.
Rodewald HR. Thymus organogenesis. Annual Review of Immunology. 2008;26:355-388.
Rothenberg EV, Moore JE, Yui MA. Launching the T-cell-lineage developmental programme. Nature Reviews Immunology. 2008;8:9-21.
Singer A, Adoro S, Park JH. Lineage fate and intense debate: myths, models and mechanisms of CD4 versus CD8 lineage choice. Nature Reviews Immunology. 2008;8:788-801.
von Boehmer H, Aifantis I, Gounari F, Azugui O, Haughn L, Apostolou I, Jaeckel E, Grassi F, Klein L. Thymic selection revisited: how essential is it? Immunological Reviews. 2003;191:62-78.
MicroRNAs and Lymphocyte Development
Hoefig KP, Heissmeyer V. MicroRNAs grow up in the immune system. Current Opinion in Immunology. 2008;20:281-287.
Lodish HF, Zhou B, Liu G, Chen CZ. Micromanagement of the immune system by microRNAs. Nature Reviews Immunology. 2008;8:120-130.
Xiao C, Rajewsky K. MicroRNA control in the immune system: basic principles. Cell. 2009;136:26-36.