CHAPTER 20 Congenital and Acquired Immunodeficiencies
Integrity of the immune system is essential for defense against infectious organisms and their toxic products and therefore for the survival of all individuals. Defects in one or more components of the immune system can lead to serious and often fatal disorders, which are collectively called immunodeficiency diseases. These diseases are broadly classified into two groups. The congenital, or primary, immunodeficiencies are genetic defects that result in an increased susceptibility to infection that is frequently manifested early in infancy and childhood but is sometimes clinically detected later in life. It is estimated that in the United States, approximately 1 in 500 individuals is born with a defect in some component of the immune system, although only a small proportion are affected severely enough for development of life-threatening complications. Acquired, or secondary, immunodeficiencies are not inherited diseases but develop as a consequence of malnutrition, disseminated cancer, treatment with immunosuppressive drugs, or infection of cells of the immune system, most notably with the human immunodeficiency virus (HIV), the etiologic agent of acquired immunodeficiency syndrome (AIDS). This chapter describes the major types of congenital and acquired immunodeficiencies, with an emphasis on their pathogenesis and the components of the immune system that are involved in these disorders.
General Features of Immunodeficiency Diseases
Before beginning our discussion of individual diseases, it is important to summarize some general features of immunodeficiencies.
TABLE 20–1 Features of Immunodeficiencies Affecting T or B Lymphocytes
| Feature | B Cell Deficiency | T Cell Deficiency |
|---|---|---|
| Susceptibility to infection | Pyogenic bacteria (otitis, pneumonia, meningitis, osteomyelitis), enteric bacteria and viruses, some parasites | Pneumocystis jiroveci, many viruses, atypical mycobacteria, fungi |
| Diagnosis | ||
| Serum Ig levels DTH reactions to common antigens |
Reduced Normal |
Normal or reduced Reduced |
| Morphology of lymphoid tissues | Absent or reduced follicles and germinal centers (B cell zones) | Usually normal follicles, may be reduced parafollicular cortical regions (T cell zones) |
DTH, delayed-type hypersensitivity.
In this chapter, we first describe congenital immunodeficiencies, including defects in components of the innate immune system, and defects in the humoral and cell-mediated arms of the adaptive immune system. We conclude with a discussion of acquired immunodeficiencies, with an emphasis on AIDS.
Congenital (Primary) Immunodeficiencies
In different congenital immunodeficiencies, the causative abnormality may be in components of the innate causative system, at different stages of lymphocyte development, or in the responses of mature lymphocytes to antigenic stimulation. Inherited abnormalities affecting innate immunity most commonly affect the complement pathway or phagocytes. Abnormalities in lymphocyte development may be caused by mutations in genes encoding a variety of molecules, including enzymes, adaptors, transport proteins, and transcription factors. These inherited defects, and the corresponding targeted disruptions in mice, have been useful for elucidating the mechanisms of lymphocyte development (see Chapter 8). Abnormalities in B lymphocyte development and function result in deficient antibody production and are diagnosed by reduced levels of serum immunoglobulin (Ig), defective antibody responses to vaccination, and, in some cases, reduced numbers of B cells in the circulation or lymphoid tissues or absent plasma cells in tissues (see Table 20-1). Abnormalities in T lymphocyte maturation and function lead to deficient cell-mediated immunity and may also result in reduced antibody production. Primary T cell immunodeficiencies are diagnosed by reduced numbers of peripheral blood T cells, low proliferative responses of blood lymphocytes to polyclonal T cell activators such as phytohemagglutinin, and deficient cutaneous delayed-type hypersensitivity (DTH) reactions to ubiquitous microbial antigens, such as Candida antigens. Defects in both humoral and cell-mediated immunity are classified under severe combined immunodeficiencies. In the following sections, we describe immunodeficiencies caused by inherited mutations in genes encoding components of the innate immune system or in genes required for lymphocyte development and activation. We conclude with a brief discussion of therapeutic strategies for these diseases.
Defects in Innate Immunity
Innate immunity constitutes the first line of defense against infectious organisms. Two important mediators of innate immunity are phagocytes and complement, both of which also participate in the effector phases of adaptive immunity. Therefore, congenital disorders of phagocytes and the complement system result in recurrent infections. Complement deficiencies were described in Chapter 12. Deficiencies have been described in the classical and alternative complement pathways as well as in the lectin pathway. They typically present with recurrent bacterial infections, particularly by encapsulated bacteria and also Neisseria species, and often also contribute to susceptibility to autoimmune disorders, particularly systemic lupus erythematosus.
In this section of the chapter, we discuss some examples of congenital phagocyte disorders (Table 20-2) and inherited defects in Toll-like receptor (TLR) pathways and in the IL-12/IFN-γ pathway. Phagocyte defects generally result in infections of the skin and respiratory tract with bacteria or fungi, the latter predominantly involving Aspergillus and Candida species. Deep-seated abscesses and oral stomatitis are also common. Defects in TLR signaling and in type I interferon signaling may contribute to recurrent pyogenic infections as well as to severe viral infections; defects in IL-12 and the IFN-γ pathway are linked to susceptibility to intracellular pathogens, particularly mycobacterial infections.
TABLE 20–2 Congenital Disorders of Innate Immunity
| Disease | Functional Deficiencies | Mechanism of Defect |
|---|---|---|
| Chronic granulomatous disease | Defective production of reactive oxygen species by phagocytes; recurrent intracellular bacterial and fungal infections | Mutation in genes of phagocyte oxidase complex; phox-91 (cytochrome b588 α subunit) is mutated in X-linked form |
| Leukocyte adhesion deficiency type 1 | Defective leukocyte adhesion and migration linked to decreased or absent expression of β2 integrins; recurrent bacterial and fungal infections | Mutations in gene encoding the β chain (CD18) of β2 integrins |
| Leukocyte adhesion deficiency type 2 | Defective leukocyte rolling and migration linked to decreased or absent expression of leukocyte ligands for endothelial E- and P- selectins, causing failure of leukocyte migration into tissues; recurrent bacterial and fungal infections | Mutations in gene encoding a GDP-fucose transporter required for the synthesis of the sialyl Lewis X component of E- and P- selectin ligands |
| Leukocyte adhesion deficiency type 3 | Defective leukocyte adhesion and migration linked to defective inside-out signaling and therefore defective integrin activation | Mutations in gene encoding KINDLIN-3 |
| Chédiak-Higashi syndrome | Defective vesicle fusion and lysosomal function in neutrophils, macrophages, dendritic cells, natural killer cells, cytotoxic T cells, and many other cell types; recurrent infections by pyogenic bacteria | Mutation in LYST leading to defect in secretory granule exocytosis and lysosomal function |
| Toll-like receptor signaling defects | Recurrent infections because of defects in TLR and CD40 signaling and defective type I interferon production | Mutations in NEMO, UNC93B, MyD88, IκBα, and IRAK-4 compromise NF-κB activation downstream of Toll-like receptors |
IRAK-4, IL-1 receptor–associated kinase 4; LYST, lysosomal trafficking protein; NEMO, NF-κB essential modulator.
Defective Microbicidal Activities of Phagocytes: Chronic Granulomatous Disease
Chronic granulomatous disease (CGD) is caused by mutations in components of the phagocyte oxidase (phox) enzyme complex. It is a rare disease, estimated to affect about 1 in 1 million individuals in the United States. About two thirds of cases show an X-linked recessive pattern of inheritance, and the remainder are autosomal recessive. The most common X-linked form of the disease is caused by a mutation in the gene encoding the 91-kD α subunit of cytochrome b558, an integral membrane protein also known as phox-91. This mutation results in defective production of superoxide anion, one of several reactive oxygen species, which constitute a major microbicidal mechanism of phagocytes (see Chapter 4). Mutations in other components of the phox complex contribute to autosomal recessive variants of CGD. Defective production of reactive oxygen species results in a failure to kill phagocytosed microbes. The disease is characterized by recurrent infections with catalase-producing intracellular bacteria and fungi, usually from early childhood. Many of the organisms that are particularly troublesome in CGD patients produce catalase, which destroys the microbicidal hydrogen peroxide that may be produced by host cells from the residual reactive oxygen radical superoxide. Because the infections are not controlled by phagocytes, they stimulate chronic cell-mediated immune responses, resulting in T cell–mediated macrophage activation and the formation of granulomas composed of activated macrophages. Presumably, these activated macrophages try to limit or to eliminate the microbes despite defective production of reactive oxygen species. This histologic appearance is the basis for the name of the disorder. The disease is often fatal, even with aggressive antibiotic therapy.
The cytokine interferon-γ (IFN-γ) enhances transcription of the gene encoding phox-91 and also stimulates other components of the phagocyte oxidase enzyme complex. Therefore, IFN-γ stimulates the production of superoxide by normal neutrophils as well as by CGD neutrophils, especially in cases in which the coding portion of the phox-91 gene is intact but its transcription is reduced. Once neutrophil superoxide production is restored to about 10% of normal levels, resistance to infection is greatly improved. IFN-γ therapy is now commonly used for the treatment of X-linked CGD.
Leukocyte Adhesion Deficiencies
The leukocyte adhesion deficiencies are a group of autosomal recessive disorders caused by defects in leukocyte and endothelial adhesion molecules. These diseases are characterized by a failure of leukocyte, particularly neutrophil, recruitment to sites of infection, resulting in severe periodontitis and other recurrent infections starting early in life, and the inability to make pus. Different types of leukocyte adhesion deficiencies are caused by mutations in different genes.
Defects in NK Cells and Other Leukocytes: The Chédiak-Higashi Syndrome
The Chédiak-Higashi syndrome is a rare autosomal recessive disorder characterized by recurrent infections by pyogenic bacteria, partial oculocutaneous albinism, and infiltration of various organs by non-neoplastic lymphocytes. The neutrophils, monocytes, and lymphocytes of these patients contain giant lysosomes. This disease is caused by mutations in the gene encoding the lysosomal trafficking regulator protein LYST, resulting in defective phagosome-lysosome fusion in neutrophils and macrophages (causing reduced resistance to infection), defective melanosome formation in melanocytes (causing albinism), and lysosomal abnormalities in cells of the nervous system (causing nerve defects) and platelets (leading to bleeding disorders). Giant lysosomes form in neutrophils during the maturation of these cells from myeloid precursors. Some of these neutrophil precursors die prematurely, resulting in moderate leukopenia. Surviving neutrophils may contain reduced levels of the lysosomal enzymes that normally function in microbial killing. These cells are also defective in chemotaxis and phagocytosis, further contributing to their deficient microbicidal activity. NK cell function in these patients is impaired, probably because of an abnormality in the cytoplasmic granules that store proteins mediating cytotoxicity. The severity of the defect in cytotoxic T lymphocyte (CTL) function is variable among patients. A mutant mouse strain called the beige mouse is an animal model for the Chédiak-Higashi syndrome. This strain is characterized by deficient NK cell function and giant lysosomes in leukocytes. The beige mutation has been mapped to the mouse Lyst locus.
Other mutations that affect both CTL and NK cell function will be considered later when we discuss defects in T lymphocyte activation and function. A mutation in CD16/FcγRIII, the Fc receptor on NK cells that is required for antibody-dependent cellular cytotoxicity (see Chapter 12), has been described in a patient with recurrent viral infections.
Inherited Defects in TLR Pathways, Nuclear Factor κB Signaling, and Type I Interferons
Inherited defects in TLR-dependent responses are rare and have been recognized only recently. The major signaling pathway downstream of most TLRs as well as of the interleukin-1 receptor (IL-1R) involves the MyD88 adaptor and the IRAK-4 and IRAK-1 kinases (see Chapter 4), and this pathway results in the nuclear factor κB (NF-κB)–dependent induction of proinflammatory cytokines. TLR 3, 7, 8, and 9 recognize nucleic acids, are located in endosomes, and require a protein called UNC93B for their function. UNC93B is an endoplasmic reticulum membrane protein that interacts with endosomal TLRs when they are synthesized in the endoplasmic reticulum and helps deliver these TLRs to the endosomes. The UNC93B protein is also critical for signaling by nucleic acid–specific TLRS. Signaling downstream of the endosomal TLRs results in the synthesis and secretion of type I interferons. Defects in TLR signaling tend to have a fairly circumscribed clinical phenotype. Severe invasive bacterial infections early in life, especially pneumococcal disease, are observed in individuals with mutations in MYD88 and IRAK4. Later in life, infections tend to be less severe. Heterozygous mutations in TLR3 as well as homozygous mutations in UNC93B result in reduced type I interferon generation and susceptibility to herpes simplex encephalitis. Type I interferon receptors activate the STAT1 transcription factor. Loss-of-function STAT1 mutations (which interfere with interferon signaling) have also been linked to severe viral infections, notably herpes simplex encephalitis.
Some immune deficiencies are caused by defects in signaling pathways downstream of TLRs. Point mutations in the inhibitor of κB kinase γ (IKKγ), also known as nuclear factor κB essential modulator (NEMO), a component of the IκB kinase complex that is required for NF-κB activation, contribute to the X-linked recessive condition known as anhidrotic ectodermal dysplasia with immunodeficiency (EDA-ID). In this disorder, differentiation of ectoderm-derived structures is abnormal, and immune function is impaired in a number of ways. Responses to TLR signals as well as CD40 signals are compromised. These patients suffer from infections with encapsulated pyogenic bacteria as well as with intracellular bacterial pathogens including mycobacteria, viruses, and fungi such as Pneumocystis jiroveci (see also discussion later in the section on hyper-IgM syndromes). An autosomal recessive form of EDA-ID has been described in which a hypermorphic point mutation in IκBα prevents the phosphorylation, ubiquitination, and degradation of IκBα, thus leading to impaired NF-κB activation.
Defects in the IL-12/IFN-γ Pathway
IL-12 is secreted by dendritic cells and macrophages, and IL-12R signaling induces the synthesis of IFN-γ by helper T cells, cytotoxic T cells, and NK cells (see Chapter 4). Mutations in the genes encoding IL-12p40, the IL-12Rβ1 chain, and both chains of the IFN-γ receptor, as well as some hypomorphic mutations in STAT1, all result in susceptibility to environmental Mycobacterium species (often called atypical mycobacteria), such as Mycobacterium avium, Mycobacterium kansasii, and Mycobacterium fortuitum. IKKγ/NEMO mutations also lead to susceptibility to intracellular pathogens including mycobacteria, as discussed in the previous section.
Severe Combined Immunodeficiencies
Congenital immunodeficiencies that affect both humoral and cell-mediated immunity are called combined immunodeficiencies, and a subset of these in which most peripheral T cells are missing or defective are known as severe combined immunodeficiencies (SCIDs) (Table 20-3). These diseases are characterized by deficiencies of both B and T cells or only of T cells; in the latter cases, the defect in humoral immunity is due to the absence of T cell help. Children with SCID usually have infections during the first year of life, Pneumocystis jiroveci pneumonia being particularly common, and they succumb to these infections unless they are treated.
TABLE 20–3 Severe Combined Immunodeficiencies
| Disease | Functional Deficiencies | Mechanism of Defect |
|---|---|---|
| Defects in cytokine signaling | ||
| X-linked SCID | Marked decrease in T cells; normal or increased B cells; reduced serum Ig | Cytokine receptor common γ chain mutations; defective T cell development in the absence of IL-7–derived signals |
| Autosomal recessive forms | Marked decrease in T cells; normal or increased B cells; reduced serum Ig | Mutations in IL2RA, IL7RA, JAK3 |
| Defects in nucleotide salvage pathways | ||
| ADA deficiency | Progressive decrease in T, B, and NK cells; reduced serum Ig | ADA deficiency caused by mutations in the gene, leading to accumulation of toxic metabolites in lymphocytes |
| PNP deficiency | Progressive decrease in T, B, and NK cells; reduced serum Ig | PNP deficiency caused by mutations in the gene, leading to accumulation of toxic metabolites in lymphocytes |
| Defects in V(D)J recombination | ||
| RAG1 or RAG2 deficiency recombination* | Decreased T and B cells; reduced serum Ig; absence or deficiency of T and B cells | Cleavage defect during V(D)J recombination; mutations in RAG1 or RAG2 |
| Double-stranded break repair and checkpoint | Decreased T and B cells; reduced serum Ig; absence or deficiency of T and B cells | Failure to resolve hairpins during V(D)J recombination; mutations in ARTEMIS, DNA-PKcs, CERNUNNOS, LIG4, NBS1, MRE11, ATM |
| Defective thymus development | ||
| Defective pre-TCR checkpoint | Decreased T cells; normal or reduced B cells; reduced serum Ig | Mutations in CD45, CD3D, CD3E, ORAI1 (CRAC channel component), STIM1 |
| DiGeorge syndrome | Decreased T cells; normal B cells; normal or reduced serum Ig | 22ql 1 deletion; T-box 1 (TBX1) transcription factor mutations |
| FoxN1 deficiency | Thymic aplasia with defective thymic cell development | Recessive mutation in FOXN1 |
| Other defects | ||
| Reticular dysgenesis | Decreased T, B, and myeloid cells | Mutation in AK2 |
ADA, adenosine deaminase; AK2, adenylate kinase 2; ATM, ataxia-telangiectasia mutated; CRAC, calcium release activated channel; DNA-PKcs, DNA-dependent protein kinase catalytic subunit; LIG4, DNA ligase 4; MRE11, meiotic recombination homologue 11; NBS1, Nijmegen breakpoint syndrome 1; PNP, purine nucleoside phosphorylase.
* Hypomorphic mutations in RAG genes and in ARTEMIS can contribute to Omenn’s syndrome.
SCID results from impaired T lymphocyte development with or without defects in B cell maturation (Fig. 20-1). The thymic epithelium contributes in a major way to early T cell development. The process of T (and B) lymphocyte maturation from hematopoietic stem cells to functionally competent mature lymphocytes involves proliferation of early lymphocyte progenitors, rearrangement of the locus encoding one chain of the antigen receptor followed by selection of cells that have made in-frame productive rearrangements at a pre-antigen receptor checkpoint, expression of both chains of the antigen receptor, and selection of cells with useful specificities (see Chapter 8). Defects in many of these steps have been described in different forms of SCID. About 50% of SCIDs are autosomal recessive; the rest are X-linked. The most common cause of autosomal recessive SCID is deficiency of the enzyme adenosine deaminase, required for purine metabolism. X-linked SCID is caused by mutations in the gene encoding a cytokine receptor component called the common γ chain. The individual disorders are described here.
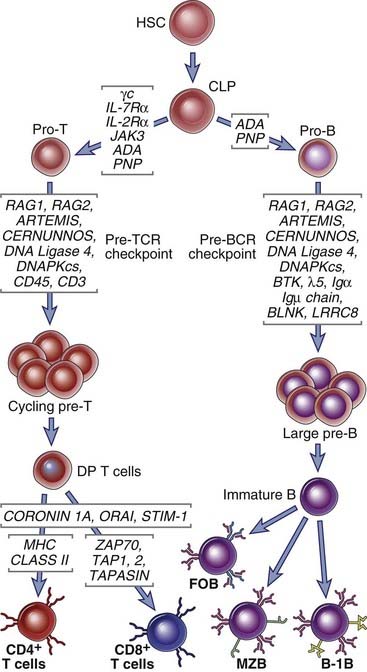
FIGURE 20–1 Immunodeficiency caused by defects in B and T cell maturation.
Primary immunodeficiencies caused by genetic defects in lymphocyte maturation are shown. These defects may affect T cell maturation alone, B cell maturation alone, or both. CLP, common lymphoid progenitor; DP, double-positive; FoB, follicular B cells; HSC, hematopoietic stem cell; MZB, marginal zone B cells.
The DiGeorge Syndrome and Other Forms of SCID due to Defective Thymic Epithelial Development
Failure or incomplete development of the thymic anlage can lead to defective T cell development. The most common defect in thymic development linked to SCID is seen in children with the DiGeorge syndrome. This selective T cell deficiency is due to a congenital malformation that results in defective development of the thymus and the parathyroid glands as well as other structures that develop from the third and fourth pharyngeal pouches during fetal life. The congenital defect is manifested by hypoplasia or agenesis of the thymus leading to deficient T cell maturation, absent parathyroid glands causing abnormal calcium homeostasis and muscle twitching (tetany), abnormal development of the great vessels, and facial deformities. Different patients may show varying degrees of these abnormalities. The disease is caused most frequently by a deletion in chromosome 22q11. Mutations in the murine homologue of a gene encoding a transcription factor called T-box 1 (TBX1), which lies within the region deleted in DiGeorge syndrome, also result in a similar defect in thymic development. It is likely that the immunodeficiency associated with DiGeorge syndrome can be explained, at least in part, by the deletion of the TBX1 gene. In this syndrome, peripheral blood T lymphocytes are absent or greatly reduced in number, and the cells do not respond to polyclonal T cell activators or in mixed leukocyte reactions. Antibody levels are usually normal but may be reduced in severely affected patients. As in other severe T cell deficiencies, patients are susceptible to mycobacterial, viral, and fungal infections.
The immunodeficiency associated with DiGeorge syndrome can be corrected by fetal thymic transplantation or by bone marrow transplantation. Such treatment is usually not necessary, however, because T cell function tends to improve with age in a large fraction of patients with this syndrome and is often normal by 5 years. Improvement with age probably occurs because of the presence of some thymic tissue or because some as yet undefined extrathymic sites assume the function of T cell maturation. It is also possible that as these patients grow older, thymus tissue develops at ectopic sites (i.e., other than the normal location).
An animal model of T cell immunodeficiency resulting from abnormal development of the thymus is the nude (athymic) mouse. These mice have an inherited defect of certain types of epithelial cells in the skin, leading to hairlessness, and in the lining of the third and fourth pharyngeal pouches, causing thymic hypoplasia. The disorder is due to a mutation in the FoxN1 gene encoding a Forkhead family transcription factor that is required for the normal development of certain ectoderm-derived cell types. Affected mice have rudimentary thymuses in which T cell maturation cannot occur normally. As a result, few or no mature T cells are present in peripheral lymphoid tissues, and cell-mediated immune reactions cannot occur. Autosomal recessive FOXN1 mutations have been described in a small number of patients who present with SCID, alopecia (hair loss), and nail dystrophy. An even rarer defect in the thymus has been described involving a mutation in CORONIN-1A, which encodes a protein that regulates the actin cytoskeleton. The absence of functional CORONIN-1A results in defective egress of mature T cells from the thymus.
ADA Deficiency and Other Forms of SCID Caused by Defects in Nucleotide Metabolism
The most common cause of autosomal recessive SCID is deficiency of an enzyme called adenosine deaminase (ADA) due to mutations in the ADA gene. ADA functions in the salvage pathway of purine synthesis and catalyzes the irreversible deamination of adenosine and 2′-deoxyadenosine to inosine and 2′-deoxyinosine, respectively. Deficiency of the enzyme leads to the accumulation of deoxyadenosine and its precursors S-adenosylhomocysteine and deoxyadenosine triphosphate (dATP). These byproducts have many toxic effects, including inhibition of DNA synthesis. Although ADA is present in most cells, developing lymphocytes are less efficient than most other cell types at degrading dATP into 2′-deoxyadenosine, and therefore lymphocyte maturation is particularly sensitive to ADA deficiency. Other features of the disease can include deafness, costochondral abnormalities, liver damage, and behavioral problems. ADA deficiency leads to reduced numbers of B and T cells; lymphocyte cell numbers are usually normal at birth but fall off precipitously during the first year of life. A few patients may have a nearly normal number of T cells, but these cells do not proliferate in response to antigenic stimulation.
A rarer autosomal recessive form of SCID is due to the deficiency of purine nucleoside phosphorylase (PNP), an enzyme that is also involved in purine catabolism. PNP catalyzes the conversion of inosine to hypoxanthine and guanosine to guanine, and deficiency of PNP leads to the accumulation of deoxyguanosine and deoxyguanosine triphosphate, with toxic effects on immature lymphocytes, mainly T cells. Autoimmune hemolytic anemia and progressive neurologic deterioration are also features of this disorder.
A particularly severe form of SCID is seen in a disease called reticular dysgenesis. This rare disorder is characterized by the absence of T and B lymphocytes and most myeloid cells, including granulocytes, and is due to a defect in the development of lymphoid and myeloid progenitors. This autosomal recessive disease is due to a mutation in the adenylate kinase 2 (AK2) gene. The AK2 protein regulates the level of adenosine diphosphate, and in the absence of AK2 there is increased apoptosis of lymphoid and myeloid precursors.
X-Linked SCID
X-linked SCID is caused by mutations in the gene encoding the common γ (γc) chain shared by the receptors for the interleukins IL-2, IL-4, IL-7, IL-9, and IL-15 (see Chapters 4 and 9). X-linked SCID is characterized by impaired maturation of T cells and NK cells and greatly reduced numbers of mature T cells and NK cells, but the number of B cells is usually normal or increased. The humoral immunodeficiency in this disease is due to a lack of T cell help for antibody production. This disease is a result of the inability of the lymphopoietic cytokine IL-7, whose receptor uses the γc chain for signaling, to stimulate the growth of immature thymocytes. In addition, the receptor for IL-15, which is a potent stimulus for the proliferation of NK cells, also uses the γc signaling chain, and the failure of IL-15 function accounts for the deficiency of NK cells.
Heterozygous females are usually phenotypically normal carriers, whereas males who inherit the abnormal X chromosome manifest the disease. Because developing cells in females randomly inactivate one of the two X chromosomes, the normal allele encoding a functional γc protein will not be expressed in half the lymphocyte precursors in a female carrier. These cells will fail to mature, and consequently, all the mature lymphocytes in a female carrier will have inactivated the same X chromosome (carrying the mutant allele). In contrast, half of all nonlymphoid cells will have inactivated one X chromosome, and half the other. A comparison of X chromosome inactivation in lymphoid cells versus nonlymphoid cells may be used to identify carriers of the mutant allele. The nonrandom use of X chromosomes in mature lymphocytes is also characteristic of female carriers of other X-linked mutations of genes that affect lymphocyte development, as discussed later.
Autosomal Recessive Mutations in Cytokine Signaling Components
Some patients with a disease identical to X-linked SCID show an autosomal recessive inheritance. These patients have mutations in the IL-7 receptor α chain or the JAK3 kinase, which associates with the γc chain and is required for signaling by this receptor (see Chapter 7). Patients with mutations in the gene encoding the IL-7Rα chain have a defect in T cell development but exhibit normal NK cell development, because IL-15 signaling is unaffected, and have normal numbers of B cells.
Severe Combined Immunodeficiency Caused by Defects in V(D)J Recombination and Pre-TCR Checkpoint Signaling
Absence of V(D)J recombination leads to a failure to express the pre-TCR and the pre-BCR and a block in T and B cell development. Mutations in the RAG1 or RAG2 genes (whose protein products mediate the cleavage step during V(D)J recombination) or the ARTEMIS gene, which encodes an endonuclease that resolves coding-end hairpins during V(D)J recombination, all result in a failure of V(D)J recombination. These diseases are rare, but they account for a large number of the autosomal recessive forms of SCID. The normal functions of these genes are discussed in Chapter 8. In children with these mutations, B and T lymphocytes are absent and immunity is severely compromised. Mutations in genes encoding proteins involved in double-stranded break repair/nonhomologous end joining of DNA also lead to SCID because of defects in V(D)J recombination. Homozygous mutations in the gene encoding the catalytic subunit of the DNA-dependent protein kinase (DNA-PK), CERNUNNOS/XLF, and DNA LIGASE 4 all lead to SCID. Among the many functions of DNA-PK is the phosphorylation and activation of ARTEMIS, and CERNUNNOS interacts with the XRCC4/DNA ligase 4 complex and presumably facilitates the ligation event that completes the nonhomologous end-joining process. Genetic defects in this end-joining process also result in increased cellular sensitivity to radiation and can result in other manifestations, such as microcephaly, facial dysmorphisms, and defective tooth development.
Hypomorphic mutations (that only partially reduce function) in the RAG genes, in ARTEMIS, or in the IL7RA gene are the cause of a disorder characterized by restricted generation of T and B cells, immunodeficiency, and immune dysregulation. This disorder is known as Omenn’s syndrome. It is phenotypically different from the diseases described above because in this disease immunodeficiency coexists with exaggerated immune activation and autoimmunity. This may be caused by relative absence of regulatory T cells, or in cases with decreased V(D)J recombination, defective receptor editing in immature B cells.
Although most autosomal recessive forms of SCID are linked to mutations in ADA, RAG1, RAG2, and ARTEMIS, rare forms of this syndrome are caused by mutations in the genes encoding the CD45 phosphatase (that is a positive regulator of Src family kinases, such as Fyn, Lck, and Lyn) and mutations in the CD3 δ or ε chains or in the CD3-associated ζ chain. These mutations contribute to defective pre-TCR signaling and result in a block in αβ T cell development.
The Bare Lymphocyte Syndrome and Other Defects in T Cell Positive Selection
The generation of single-positive CD4+ and CD8+ T cells from double-positive thymocytes depends on positive selection and lineage commitment events. Specific inherited mutations in genes that regulate the process of positive selection abrogate the development of CD4+ T cells or of CD8+ T cells.
Class II major histocompatibility complex (MHC) deficiency, also called bare lymphocyte syndrome, is a rare heterogeneous group of autosomal recessive diseases in which patients express little or no HLA-DP, HLA-DQ, or HLA-DR on B lymphocytes, macrophages, and dendritic cells and fail to express class II MHC molecules in response to IFN-γ. They express normal or only slightly reduced levels of class I MHC molecules and β2-microglobulin. Most cases of the bare lymphocyte syndrome are due to mutations in genes encoding proteins that regulate class II MHC gene transcription. For example, mutations affecting the constitutively expressed transcription factor RFX5 or the IFN-γ–inducible transcriptional activator CIITA lead to reduced class II MHC expression and a failure of APCs to activate CD4+ T lymphocytes. Failure of antigen presentation may result in defective positive selection of T cells in the thymus, with a reduction in the number of mature CD4+ T cells or defective activation of cells in the periphery. Affected individuals are deficient in DTH responses and in antibody responses to T cell–dependent protein antigens. The disease appears within the first year of life and is usually fatal unless it is treated by bone marrow transplantation.
Autosomal recessive class I MHC deficiencies have also been described and are characterized by decreased CD8+ T cell numbers and function. In some cases, the failure to express class I MHC molecules is due to mutations in the TAP-1 or TAP-2 genes, which encode the subunits of the TAP (transporter associated with antigen processing) complex, which normally transports peptides from the cytosol into the endoplasmic reticulum, where they are required for class I MHC assembly (see Chapter 6). These TAP-deficient patients express few cell surface class I MHC molecules, a phenotype similar to TAP gene knockout mice. Such patients suffer mainly from necrotizing granulomatous skin lesions and respiratory tract bacterial infections, but not viral infections, which is surprising considering that a principal function of CD8+ T cells is defense against viruses. A similar deficiency of class I MHC expression has been observed in patients with mutations in the gene encoding the tapasin protein (see Chapter 6).
Patients with ZAP-70 deficiency have a lineage commitment defect resulting in reduced CD8+ T cells but not CD4+ T cells; the reason for the selective loss is not clear. This specific tyrosine kinase defect does not compromise CD4+ T cell development or emigration to the periphery. However, these CD4+ T cells fail to proliferate normally when challenged with antigens.
SCID Caused by Defective T Cell Activation
Another rare form of SCID is caused by mutation in a gene encoding Orai1, a component of the CRAC channel (see Chapter 7). Antigen receptor signaling leads to the activation of the γ isoform of phospholipase C (PLCγ) and the inositol trisphosphate (IP3)–dependent release of calcium ions from the endoplasmic reticulum and mitochondria (see Chapter 7). The released calcium is replenished by store-operated CRAC channels that facilitate an influx of extracellular calcium. This process is crucial for lymphocyte activation, and it is defective in cells with mutant ORAI1. A similar phenotype is observed in patients with mutations in STIM1, which encodes an endoplasmic reticulum protein that senses the depletion of calcium stores and contributes to the opening of the CRAC channel. Patients with ORAI1 and STIM1 mutations do not exhibit a defect in T cell development, but their T cells cannot be properly activated.
Antibody Deficiencies: Defects in B Cell Development and Activation
Whereas defects in T cell development or in both T and B cell development contribute to the SCID phenotype, more circumscribed defects in B cells result in disorders in which the primary abnormality is in antibody synthesis (Table 20-4). Some of these disorders are caused by defects in B cell development (see Fig. 20-1) and others by abnormal B cell activation and antibody production (Fig. 20-2). However, in one subset of hyper-IgM syndromes discussed later, antibody deficiencies are also accompanied by defects in macrophage and APC activation, which in turn result in attenuated cell-mediated immunity.
TABLE 20–4 Antibody Deficiencies
| Disease | Functional Deficiencies | Mechanism of Defect |
|---|---|---|
| Agammaglobulinemias | ||
| X-linked | Decrease in all serum Ig isotypes; reduced B cell numbers | Pre-B receptor checkpoint defect; Btk mutation |
| Autosomal recessive forms | Decrease in all serum Ig isotypes; reduced B cell numbers | Pre-B receptor checkpoint defect; mutations in IgM heavy chain (µ), surrogate light chains (λ5), Igα, BLNK |
| Hypogammaglobulinemias/isotype defects | ||
| Selective IgA deficiency | Decreased IgA; may be associated with increased susceptibility to bacterial infections and protozoa such as Giardia lamblia | Mutations in TACI in some patients |
| Selective IgG2 deficiency | Increased susceptibility to bacterial infections | Small subset have deletion in IgH γ2 locus |
| Common variable immunodeficiency | Hypogammaglobulinemia; normal or decreased B cell numbers | Mutations in ICOS and TACI in some patients |
| ICF syndrome | Hypogammaglobulinemia, occasional mild T cell defects | Mutations in DNMT3B |
| Hyper-IgM syndromes | ||
| X-linked | Defects in T helper cell–mediated B cell, macrophage, and dendritic cell activation; defects in somatic mutation, class switching, and germinal center formation; defective cell-mediated immunity | Mutation in CD40L |
| Autosomal recessive with cell- mediated immune defects | Defects in T helper cell–mediated B cell, macrophage, and dendritic cell activation; defects in somatic mutation, class switching, and germinal center formation; defective cell-mediated immunity | Mutations in CD40, NEMO |
| Autosomal recessive with antibody defect only | Defects in somatic mutation and isotype switching | Mutations in AID, UNG |
AID, activation-induced cytidine deaminase; DNMT3B, DNA methyltransferase 3B; ICF, immunodeficiencies-centromeric instability-facial anomalies; ICOS, inducible costimulator; NEMO, NF-κB essential modulator; TACI, transmembrane activator and calcium modulator and cyclophilin ligand interactor; UNG, uracil N-glycosylase.
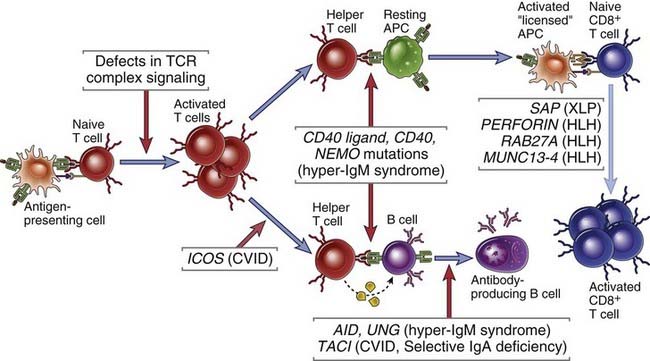
FIGURE 20–2 Immunodeficiency caused by defects in B and T cell activation.
Primary immunodeficiencies may be caused by genetic defects in molecules required for T or B lymphocyte antigen receptor signaling, for helper T cell–mediated activation of B cells and APCs, or for activation of cytotoxic T lymphocytes and NK cells. CVID, common variable immunodeficiency; HLH, hemophagocytic lymphohistiocytosis.
X-Linked Agammaglobulinemia: An X-linked Pre-BCR Signaling Defect
X-linked agammaglobulinemia, also called Bruton’s agammaglobulinemia, is caused by mutations or deletions in the gene encoding an enzyme called Bruton tyrosine kinase (Btk) that results in a failure of B cells to mature beyond the pre-B cell stage in the bone marrow (see Fig. 20-1). The disease is characterized by the absence of gamma globulin in the blood, as the name implies. It is one of the most common congenital immunodeficiencies and the prototype of a failure of B cell maturation. Btk is involved in transducing signals from the pre-B cell receptor (pre-BCR) that are required for the survival and differentiation of pre-B cells (see Chapter 8). In female carriers of this disease, only B cells that have inactivated the X chromosome carrying the mutant allele mature. Patients with X-linked agammaglobulinemia usually have low or undetectable serum Ig, reduced or absent B cells in peripheral blood and lymphoid tissues, no germinal centers in lymph nodes, and no plasma cells in tissues. The maturation, numbers, and functions of T cells are generally normal. Some studies have revealed reduced numbers of activated T cells in patients, which may be a consequence of reduced antigen presentation caused by the lack of B cells. Autoimmune disorders develop in almost 20% of patients, for unknown reasons. The infectious complications of X-linked agammaglobulinemia are greatly reduced by periodic (e.g., weekly or monthly) injections of pooled gamma globulin preparations. Such preparations contain preformed antibodies against common pathogens and provide effective passive immunity.
Knockout mice lacking Btk, as well as naturally Btk mutant Xid mice, show a less severe defect in B cell maturation than humans do because a Btk-like tyrosine kinase called Tec is active in mouse pre-B cells that lack Btk and partially compensates for the mutant Btk. The main abnormalities in Xid mice are defective antibody responses to some polysaccharide antigens and a deficiency in mature follicular and B-1 B cells.
Autosomal Recessive Pre-BCR Checkpoint Defects
Autosomal recessive forms of agammaglobulinemia have been described, most of which can be linked to defects in pre-BCR signaling. Mutant genes that have been identified in this context include genes encoding the µ (IgM) heavy chain, the λ5 surrogate light chain, Igα (a signaling component of the pre-BCR and BCR), and BLNK (an adaptor protein downstream of the pre-BCR and BCR).
Selective Immunoglobulin Isotype Deficiencies
Many immunodeficiencies that selectively involve one or a few Ig isotypes have been described. The most common is selective IgA deficiency, which affects about 1 in 700 Caucasians and is thus the most common primary immunodeficiency known. IgA deficiency usually occurs sporadically, but many familial cases with either autosomal dominant or recessive patterns of inheritance are also known. The clinical features are variable. Many patients are entirely normal; others have occasional respiratory infections and diarrhea; and rarely, patients have severe, recurrent infections leading to permanent intestinal and airway damage, with associated autoimmune disorders. IgA deficiency is characterized by low serum IgA, usually less than 50 µg/mL (normal, 2 to 4 mg/mL), with normal or elevated levels of IgM and IgG. The defect in these patients is a block in the differentiation of B cells to IgA antibody–secreting plasma cells. The α heavy chain genes and the expression of membrane-associated IgA are normal. No gross abnormalities in the numbers, phenotypes, or functional responses of T cells have been noted in these patients. In a small proportion of patients with selective IgA deficiency, mutations have been described in TACI (transmembrane activator and calcium modulator and cyclophilin ligand interactor), one of the three types of receptors for the cytokines BAFF (B cell–activating factor) and APRIL (a proliferation-inducing ligand). TACI mutations are also an important cause of common variable immunodeficiency, discussed later. IgA deficiency may represent a forme fruste of common variable immunodeficiency.
Selective IgG subclass deficiencies have been described in which total serum IgG levels are normal but concentrations of one or more subclasses are below normal. Deficiency of IgG3 is the most common subclass deficiency in adults, and IgG2 deficiency associated with IgA deficiency is the most common in children. Some individuals with these deficiencies have recurrent bacterial infections, but many do not have any clinical problems. Selective IgG subclass deficiencies are usually due to abnormal B cell differentiation and rarely to homozygous deletions of various constant region (Cγ) genes.
Defects in B Cell Differentiation: Common Variable Immunodeficiency
Common variable immunodeficiency is a group of heterogeneous disorders defined by reduced levels of serum Ig, impaired antibody responses to infection or vaccines, and increased incidence of infections. The diagnosis is usually one of exclusion when other primary immunodeficiency diseases are ruled out. The presentation and pathogenesis are, as the name implies, highly variable. Although Ig deficiency and associated pyogenic infections, typically with Haemophilus influenzae and Streptococcus pneumoniae, are major components of these disorders, autoimmune diseases, including pernicious anemia, hemolytic anemia, inflammatory bowel disease, and rheumatoid arthritis, may be just as clinically significant. A high incidence of malignant tumors, particularly lymphomas, is also associated with common variable immunodeficiency. These disorders may be diagnosed early in childhood or late in life. Both sporadic and familial cases occur, the latter with both autosomal dominant and recessive inheritance patterns. Mature B lymphocytes are present in these patients, but plasma cells are absent in lymphoid tissues, which suggests a block in B cell differentiation to antibody-secreting cells. The defective antibody production has been attributed to multiple abnormalities, including intrinsic B cell defects, deficient T cell help, and excessive “suppressor cell” activity. A small proportion of patients with common variable immunodeficiency have a mutation in the ICOS (inducible T cell costimulator) gene. ICOS is required for T follicular helper cell generation (see Chapter 11). A more common cause of this syndrome is the existence of mutations in TACI, described before in the context of selective IgA deficiency. A few cases of common variable immunodeficiency are linked to mutations in the CD19 gene. CD19 is a signaling component of the CR2 (CD21) coreceptor complex (see Chapter 7).
Defects in T Cell–Dependent B Cell Activation: Hyper-IgM Syndromes
The X-linked hyper-IgM syndrome is caused by mutations in the gene encoding the T cell effector molecule CD40 ligand (CD154). It is a rare disorder associated with defective switching of B cells to the IgG and IgA isotypes; these antibodies are therefore reduced, and the major isotype detected in the blood is IgM. The mutant forms of CD40 ligand produced in these patients do not bind to or transduce signals through CD40 and therefore do not stimulate B cells to undergo heavy chain isotype switching, which requires T cell help (see Chapter 11). Patients suffer from infections similar to those seen in other hypogammaglobulinemias. Patients with X-linked hyper-IgM syndrome also show defects in cell-mediated immunity, with an increased susceptibility to infection by the intracellular fungal microbe Pneumocystis jiroveci. This defective cell-mediated immunity occurs because CD40 ligand is also involved in T cell–dependent activation of macrophages and dendritic cells (see Chapter 10). Knockout mice lacking CD40 or CD40 ligand have a phenotype similar to that of the human disease.
Rare cases of hyper-IgM syndrome show an autosomal recessive inheritance pattern. In these patients, the genetic defects may be in CD40 or in the enzyme activation-induced deaminase (AID), which is involved in heavy chain isotype switching and somatic mutation (see Chapter 11). Mutations in AID are generally homozygous recessive. A small fraction of mutations in the region of the AID gene that corresponds to the C-terminal part of this enzyme exhibit an autosomal dominant inheritance pattern. One form of the hyper-IgM syndrome is caused by autosomal recessive mutations in uracil N-glycosylase (UNG; see Chapter 11), an enzyme that removes U residues from Ig genes during class switching and somatic mutation. An inherited disorder, EDA-ID, in which hypomorphic NEMO mutations contribute to a hyper-IgM state as well as defects in ectodermal structures, is described earlier in the section on TLR signaling defects.
AID and UNG mutations affect class switch recombination and somatic hypermutation in distinct ways. In the absence of AID, both switching and hypermutation are defective because AID is absolutely required for both processes. In the absence of UNG, isotype switching is defective but somatic hypermutation is largely preserved, although it exhibits less A : T mutations without the activity of UNG. The role of DNA repair gene mutations in class switching defects will be considered in the section on ataxia-telangiectasia later in this chapter.
Defects in T Lymphocyte Activation and Function
Congenital abnormalities in the activation of T lymphocytes are being increasingly recognized as our understanding of the molecular basis of lymphocyte activation improves (Table 20-5). Included in this broad category are some disorders of CTL and NK cell granule composition or exocytosis. Although we classify disorders linked to defective MHC expression with disorders of T cell development, these abnormalities also result in defective activation of T cells that do mature and emerge from the thymus.
TABLE 20–5 Defects in T Cell Activation
| Disease | Functional Deficiencies | Mechanism of Defect |
|---|---|---|
| Defects in MHC expression | ||
| Bare lymphocyte syndrome | Defective MHC class II expression and deficiency in CD4+ T cells; defective cell-mediated immunity and T-dependent humoral immune responses | Defects in transcription factors regulating MHC class II gene expression, including CIITA, RFXANK, RFX5, and RFXAP |
| MHC class I deficiency | Decreased MHC class I levels; reduced CD8+ T cells | Mutations in TAP1, TAP2, and TAPASIN |
| Defective T cell signaling | ||
| Proximal TCR signaling defects | Defects in cell-mediated immunity and T-dependent humoral immunity | Mutations in CD3 genes, CD45, STIM1, ORAI1 |
| Wiskott-Aldrich syndrome | Defective T cell activation, leukocyte mobility | TCR-dependent actin-cytoskeletal rearrangements are defective because of mutations in WASP |
| Familial hemophagocytic lymphohistiocytoses | ||
| X-linked lymphoproliferative syndrome | Uncontrolled EBV-induced B cell proliferation, uncontrolled macrophage and CTL activation, defective NK cell and CTL function | Mutations in SAP |
| Perforin deficiencies | Uncontrolled macrophage and CTL activation, defective NK cell and CTL function | Mutations in PERFORIN |
| Granule fusion | Uncontrolled macrophage and CTL activation, defective NK cell and CTL function | Defective cytotoxic granule exocytosis; mutations in RAB27A, MUNC13-4, SYNTAXIN, AP3 (and in LYST in Chédiak-Higashi syndrome—see Table 20-2) |
AP3, adaptor-related protein complex 3; LYST, lysosomal trafficking regulator protein; SAP, SLAM-associated protein; TAP, transporter associated with antigen processing; WASP, Wiskott-Aldrich syndrome protein.
Defects in TCR Signal Transduction
Many examples of rare immunodeficiency diseases caused by defects in the expression of molecules required for T cell activation and function have been identified, and some have already been discussed in the context of SCID. Biochemical and molecular analyses of affected individuals have revealed mutations in the genes encoding various T cell proteins (see Table 20-5). Examples include impaired TCR complex expression or function caused by mutations in the CD3 ε or γ genes, defective TCR-mediated signaling caused by mutations in the ZAP70 gene, reduced synthesis of cytokines such as IL-2 and IFN-γ (in some cases caused by defects in transcription factors), and lack of expression of IL-2 receptor chains. These defects are often found in only a few isolated cases or in a few families, and the clinical features and severity vary widely. Patients with these abnormalities may have deficiencies predominantly in T cell function or have mixed T cell and B cell immunodeficiencies despite normal or even elevated numbers of blood lymphocytes. We have previously considered the importance of the CD3 complex at the pre-TCR checkpoint, the role of ZAP70 mutations in CD8+ T cell development, and the relevance of ORAI1 and STIM1 in T cell activation, all in the clinical context of SCID. Other syndromes involving the defective activation of mature T cells are considered here.
Wiskott-Aldrich Syndrome
Variable degrees of T and B cell immunodeficiency occur in certain congenital diseases with a wide spectrum of abnormalities involving multiple organ systems. One such disorder is Wiskott-Aldrich syndrome, an X-linked disease characterized by eczema, thrombocytopenia (reduced blood platelets), and susceptibility to bacterial infection. Some of the abnormalities in this disorder can be traced to defective T cell activation, although intrinsic loss of B cell function also contributes to the pathogenesis. In the initial stages of the disease, lymphocyte numbers are normal, and the principal defect is an inability to produce antibodies in response to T cell–independent polysaccharide antigens, because of which these patients are especially susceptible to infections with encapsulated bacteria. The lymphocytes (and platelets) are smaller than normal. With increasing age, the patients show reduced numbers of lymphocytes and more severe immunodeficiency. The defective gene responsible for Wiskott-Aldrich syndrome encodes a cytoplasmic protein called WASP (Wiskott-Aldrich syndrome protein), expressed exclusively in bone marrow–derived cells, which interacts with several proteins, including adaptor molecules downstream of the antigen receptor, such as Grb-2 (see Chapter 7), the Arp2/3 complex involved in actin polymerization, and small G proteins of the Rho family that regulate actin cytoskeletal rearrangement. Defective activation and synapse formation in lymphocytes and defective mobility of all leukocytes may account for the immunodeficiency observed in this syndrome.
The X-Linked Lymphoproliferative Syndrome
X-linked lymphoproliferative (XLP) disease is a disorder characterized by an inability to eliminate Epstein-Barr virus (EBV), eventually leading to fulminant infectious mononucleosis and the development of B cell tumors and associated hypogammaglobulinemia. In about 80% of cases, the disease is due to mutations in the gene encoding an adaptor molecule called SAP (SLAM-associated protein) that binds to a family of cell surface molecules involved in the activation of NK cells and T and B lymphocytes, including the signaling lymphocyte activation molecule (SLAM). SAP links the membrane proteins SLAM and 2B4 (see Chapter 7) to the Src family kinase Fyn. Defects in SAP contribute to attenuated NK and T cell activation and result in increased susceptibility to viral infections. As discussed in Chapter 11, SAP is required for TFH cell development, and the inability of XLP patients to generate germinal centers and high-affinity antibodies also likely contributes to susceptibility to viral infection. In about 20% of cases of XLP, the genetic defect resides not in SAP but in the gene encoding XIAP (X-linked inhibitor of apoptosis). The resulting enhanced apoptosis of T cells and NKT cells leads to a marked depletion of these cell types. This immunodeficiency is most commonly manifested by severe EBV infections, which probably arise opportunistically because of the ubiquitous nature of EBV.
Defective CTL and NK Cell Activation: The Familial Hemophagocytic Lymphohistiocytosis Syndromes
The hemophagocytic lymphohistiocytosis (HLH) syndromes are a group of life-threatening immunodeficiency disorders in which NK cell and CTL granule secretion is defective. As a result, viral infections are not held in check, and uncontrolled macrophage activation is a feature of these syndromes. A late but striking feature of these disorders is the ingestion of red blood cells by activated macrophages (hemophagocytosis). Mutations in the perforin gene, as well as mutations in genes encoding the cellular machinery involved in granule exocytosis, can contribute to the phenotypes observed in this syndrome. Specifically, mutations in RAB27A, a small guanosine triphosphatase involved in vesicular fusion, and in MUNC13-4, which encodes an adaptor that participates in granule exocytosis, compromise the fusion of lytic granules with the plasma membrane and thus contribute to various subtypes of HLH. Similarly, mutations in the gene for one component of the AP-3 cytosolic adaptor protein complex can also disrupt intracellular transport and contribute to a form of HLH. It is believed that T cells and macrophages respond strongly to microbes to compensate for the CTL and NK cell defects, and these compensatory responses are manifested by hemophagocytosis and lymphadenopathy in the context of immunodeficiency.
Multisystem Disorders with Immunodeficiency: Ataxia-Telangiectasia
Immunodeficiency is often one of a constellation of symptoms in a number of inherited disorders. Examples of such syndromes discussed before include Chédiak-Higashi syndrome, Wiskott-Aldrich syndrome, and DiGeorge syndrome. Ataxia-telangiectasia is an autosomal recessive disorder characterized by abnormal gait (ataxia), vascular malformations (telangiectases), neurologic deficits, increased incidence of tumors, and immunodeficiency. The immunologic defects are of variable severity and may affect both B and T cells. The most common humoral immune defects are IgA and IgG2 deficiency, probably because of the crucial role the ATM protein plays in class switch recombination (discussed later). The T cell defects, which are usually less pronounced, are associated with thymic hypoplasia. Patients experience upper and lower respiratory tract bacterial infections, multiple autoimmune phenomena, and increasingly frequent cancers with advancing age. The gene responsible for this disorder is located on chromosome 11 and encodes a protein called ATM (ataxia-telangiectasia mutated) that is related structurally to phosphatidylinositol 3-kinase but is a protein kinase. The ATM protein can activate cell cycle checkpoints and apoptosis in response to double-stranded DNA breaks and has also been shown to contribute to the stability of DNA double-stranded break complexes during V(D)J recombination. Because of these abnormalities in DNA repair, the generation of antigen receptors may also be abnormal.
DNA repair during class switch recombination not only involves the nonhomologous end-joining pathway but also requires the ATM protein, the MRE11 (meiotic recombination 11) protein, and the NBS1 (Nijmegen breakpoint syndrome 1) protein. Patients with mutations in the genes encoding these proteins or ATM often exhibit decreased levels of IgG, IgA, and IgE.
Therapeutic Approaches for Congenital Immunodeficiencies
The current treatment of immunodeficiencies has two aims: to minimize and control infections and to replace the defective or absent components of the immune system by adoptive transfer or transplantation. Passive immunization with pooled gamma globulin is very beneficial for agammaglobulinemic patients and has been lifesaving for many boys with X-linked agammaglobulinemia. Hematopoietic stem cell transplantation is currently the treatment of choice for many immunodeficiency diseases and has been successful in the treatment of SCID with ADA deficiency, Wiskott-Aldrich syndrome, bare lymphocyte syndrome, and leukocyte adhesion deficiencies. It is most successful with careful T cell depletion from the marrow and HLA matching to prevent graft-versus-host disease (see Chapter 16). Enzyme replacement therapy for ADA and PNP deficiencies has been attempted, with red blood cell transfusions used as a source of the enzymes. This approach has produced temporary clinical improvement in several patients with autosomal SCID. Injection of bovine ADA conjugated to polyethylene glycol to prolong its serum half-life has proved successful in some cases, but the benefits are usually short-lived.
In theory, the therapy of choice for congenital disorders of lymphocytes is to replace the defective gene in self-renewing stem cells. Gene replacement remains a distant goal for most human immunodeficiencies at present, despite considerable effort. The main obstacles to this type of gene therapy are difficulties in purifying self-renewing stem cells, which are the ideal target for introduction of the replacement gene, and in introducing genes into cells to achieve stable, long-lived, and high-level expression. Some progress has been made in gene therapy for ADA deficiency by use of a milder conditioning approach to deplete host bone marrow cells, which facilitates the grafting and proliferation of modified stem cells introduced into the host. A small number of patients with X-linked SCID have been successfully treated by transplantation of autologous bone marrow cells engineered to express a normal γc gene. However, a few treated patients have developed leukemia, apparently because the introduced γc gene inserted adjacent to an oncogene and activated this gene. As a result, the future of gene therapy for this disease is uncertain.
Acquired (Secondary) Immunodeficiencies
Deficiencies of the immune system often develop because of abnormalities that are not genetic but acquired during life (Table 20-6). The most prominent of these abnormalities is HIV infection, and this is described in the next section. Acquired immunodeficiency diseases are caused by two main types of pathogenic mechanisms. First, immunosuppression may occur as a biologic complication of another disease process. Second, so-called iatrogenic immunodeficiencies may develop as complications of therapy for other diseases.
TABLE 20–6 Acquired Immunodeficiencies
| Cause | Mechanism |
|---|---|
| HIV infection | Depletion of CD4+ T cells |
| Protein-calorie malnutrition | Metabolic derangements inhibit lymphocyte maturation and function |
| Irradiation and chemotherapy for cancer | Decreased bone marrow lymphocyte precursors |
| Cancer metastases and leukemia involving bone marrow | Reduced site of leukocyte development |
| Immunosuppression for transplants, autoimmune diseases | Reduced lymphocyte activation |
| Removal of spleen | Decreased phagocytosis of microbes |
Diseases in which immunodeficiency is a common complicating element include malnutrition, neoplasms, and infections. Protein-calorie malnutrition is common in developing countries and is associated with impaired cellular and humoral immunity to microorganisms. Much of the morbidity and mortality that afflict malnourished people is due to infections. The basis for the immunodeficiency is not well defined, but it is reasonable to assume that the global metabolic disturbances in these individuals, caused by deficient intake of protein, fat, vitamins, and minerals, will adversely affect maturation and function of the cells of the immune system.
Patients with advanced widespread cancer are often susceptible to infection because of impaired cell-mediated and humoral immune responses to a variety of organisms. Bone marrow tumors, including cancers metastatic to marrow and leukemias that arise in the marrow, may interfere with the growth and development of normal lymphocytes and other leukocytes. In addition, tumors may produce substances that interfere with lymphocyte development or function. An example of malignancy-associated immunodeficiency is the impairment in T cell function commonly observed in patients with a type of lymphoma called Hodgkin’s disease. This defect was first characterized as an inability to mount a DTH reaction on intradermal injection of various common antigens to which the patients were previously exposed, such as Candida or tetanus toxoid. Other in vitro measures of T cell function, such as proliferative responses to polyclonal activators, are also impaired in patients with Hodgkin’s disease. Such a generalized deficiency in DTH responses is called anergy. The cause of these T cell abnormalities is unknown.
Various types of infections lead to immunosuppression. Viruses other than HIV are known to impair immune responses; examples include the measles virus and human T cell lymphotropic virus 1 (HTLV-1). Both viruses can infect lymphocytes, which may be a basis for their immunosuppressive effects. Like HIV, HTLV-1 is a retrovirus with tropism for CD4+ T cells; however, instead of killing helper T cells, it transforms them and produces an aggressive T cell malignant neoplasm called adult T cell leukemia/lymphoma (ATL). Patients with ATL typically have severe immunosuppression with multiple opportunistic infections. Chronic infections with Mycobacterium tuberculosis and various fungi frequently result in anergy to many antigens. Chronic parasitic infections may also lead to immunosuppression. For example, African children with chronic malarial infections have depressed T cell function, and this may be one reason why these children have an increased propensity to develop EBV-associated malignant tumors.
Iatrogenic immunosuppression is most often due to drug therapies that kill or functionally inactivate lymphocytes. Some drugs are given intentionally to immunosuppress patients, either for the treatment of inflammatory diseases or to prevent rejection of organ allografts. The most commonly used anti-inflammatory and immunosuppressive drugs are corticosteroids and cyclosporine, respectively. Various chemotherapeutic drugs are administered to patients with cancer, and these drugs are usually cytotoxic to mature and developing lymphocytes as well as to granulocyte and monocyte precursors. Thus, cancer chemotherapy is almost always accompanied by a period of immunosuppression and risk for infection. Iatrogenic immunosuppression and tumors involving the bone marrow are the most common causes of immunodeficiency in developed countries.
One other form of acquired immunosuppression results from the absence of a spleen caused by surgical removal of the organ after trauma and as treatment of certain hematologic diseases or by infarction in sickle cell disease. Patients without spleens are more susceptible to infection by some organisms, particularly encapsulated bacteria such as Streptococcus pneumoniae. This enhanced susceptibility is partly due to defective phagocytic clearance of opsonized blood-borne microbes, an important physiologic function of the spleen, and partly because of defective antibody responses resulting from the absence of marginal zone B cells.
Human Immunodeficiency Virus And The Acquired Immunodeficiency Syndrome
AIDS is the disease caused by infection with HIV and is characterized by profound immunosuppression with associated opportunistic infections and malignant tumors, wasting, and central nervous system (CNS) degeneration. HIV infects a variety of cells of the immune system, including CD4+ helper T cells, macrophages, and dendritic cells. HIV evolved as a human pathogen very recently relative to most other known human pathogens, and the HIV epidemic was first identified only in the 1980s. However, the degree of morbidity and mortality caused by HIV and the global impact of HIV infection on health care resources and economics are already enormous and continue to grow. HIV has infected 50 to 60 million people and has caused the death of more than 22 million adults and children. Approximately 35 million people are living with HIV infection and AIDS, of which approximately 70% are in Africa and 20% in Asia, and almost 2 million die of the disease every year. The disease is especially devastating because about half of the approximately 3 million new cases every year occur in young adults (15 to 24 years old). AIDS has left approximately 14 million orphans and resulted in the death of approximately 30 million people. Currently, there is no effective vaccine or cure for AIDS, but quite effective antiretroviral therapies have been developed. In this section of the chapter, we describe the molecular and biologic properties of HIV, the pathogenesis of HIV-induced immunodeficiency, and the clinical and epidemiologic features of HIV-related diseases.
Molecular and Biologic Features of HIV
HIV is a member of the lentivirus family of animal retroviruses. Lentiviruses, including visna virus of sheep and the bovine, feline, and simian immunodeficiency viruses, are capable of long-term latent infection of cells and short-term cytopathic effects, and they all produce slowly progressive, fatal diseases that include wasting syndromes and CNS degeneration. Two closely related types of HIV, designated HIV-1 and HIV-2, have been identified. HIV-1 is by far the most common cause of AIDS; HIV-2, which differs in genomic structure and antigenicity, causes a form of AIDS with slower progression than HIV-1–linked disease.
HIV Structure and Genes
An infectious HIV particle consists of two identical strands of RNA packaged within a core of viral proteins and surrounded by a phospholipid bilayer envelope derived from the host cell membrane but including virally encoded membrane proteins (Fig. 20-3). The RNA genome of HIV is approximately 9.2 kb long and has the basic arrangement of nucleic acid sequences characteristic of all known retroviruses (Fig. 20-4). Long terminal repeats (LTRs) at each end of the genome regulate viral gene expression, viral integration into the host genome, and viral replication. The gag sequence encodes core structural proteins. The env sequence encodes the envelope glycoproteins gp120 and gp41, which noncovalently associate with each other and are required for infection of cells. The pol sequence encodes reverse transcriptase, integrase, and viral protease enzymes required for viral replication. In addition to these typical retrovirus genes, HIV-1 also includes six other regulatory genes, namely, the tat, rev, vif, nef, vpr, and vpu genes, whose products regulate viral reproduction and host immune evasion in various ways. The functions of these genes are summarized in Figure 20-4.
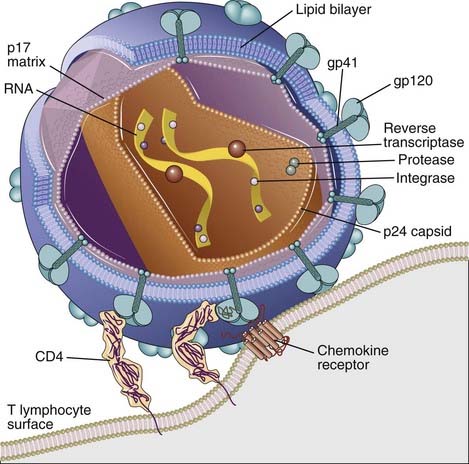
FIGURE 20–3 Structure of HIV-1.
An HIV-1 virion is shown next to a T cell surface. HIV-1 consists of two identical strands of RNA (the viral genome) and associated enzymes, including reverse transcriptase, integrase, and protease, packaged in a cone-shaped core composed of p24 capsid protein with a surrounding p17 protein matrix, all surrounded by a phospholipid membrane envelope derived from the host cell. Virally encoded membrane proteins (gp41 and gp120) are bound to the envelope. CD4 and chemokine receptors on the host cell surface function as HIV-1 receptors.
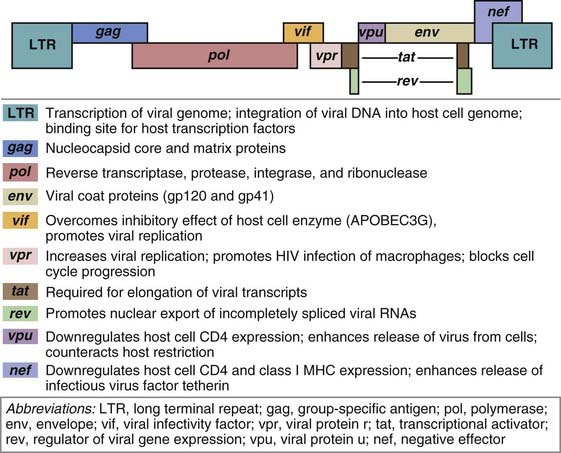
The genes along the linear genome are indicated as differently colored blocks. Some genes use some of the same sequences as other genes, as shown by overlapping blocks, but are read differently by host cell RNA polymerase. Similarly shaded blocks separated by lines indicate genes whose coding sequences are separated in the genome and require RNA splicing to produce functional mRNA.
(Modified from Greene W. AIDS and the immune system.
Viral Life Cycle
HIV infection of cells begins when the envelope glycoprotein (Env) of the virus binds to both CD4 and a coreceptor that is a member of the chemokine receptor family (Fig. 20-5). The viral particles that initiate infection are usually in the blood, semen, or other body fluids of one individual and are introduced into another individual by sexual contact, needle stick, or transplacental passage. Env is a complex composed of a transmembrane gp41 subunit and an external, noncovalently associated gp120 subunit. These subunits are produced by proteolytic cleavage of a gp160 precursor. The Env complex is expressed as a trimeric structure of three gp120/gp41 pairs. This complex mediates a multistep process of fusion of the virion envelope with the membrane of the target cell (Fig. 20-6). The first step of this process is the binding of gp120 subunits to CD4 molecules, which induces a conformational change that promotes secondary gp120 binding to a chemokine coreceptor. Coreceptor binding induces a conformational change in gp41 that exposes a hydrophobic region, called the fusion peptide, which inserts into the cell membrane and enables the viral membrane to fuse with the target cell membrane. After the virus completes its life cycle in the infected cell (described later), free viral particles are released from one infected cell and bind to an uninfected cell, thus propagating the infection. In addition, gp120 and gp41, which are expressed on the plasma membrane of infected cells before virus is released, can mediate cell-cell fusion with an uninfected cell that expresses CD4 and coreceptors, and HIV genomes can then be passed between the fused cells directly.
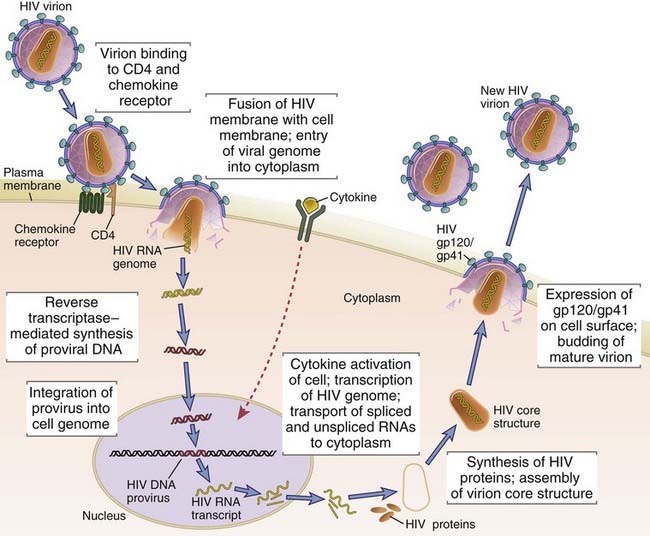
The sequential steps in the life cycle of HIV are shown, from initial infection of a host cell to viral replication and release of a new virion. For the sake of clarity, the production and release of only one new virion are shown. An infected cell actually produces many virions, each capable of infecting cells, thereby amplifying the infectious cycle.
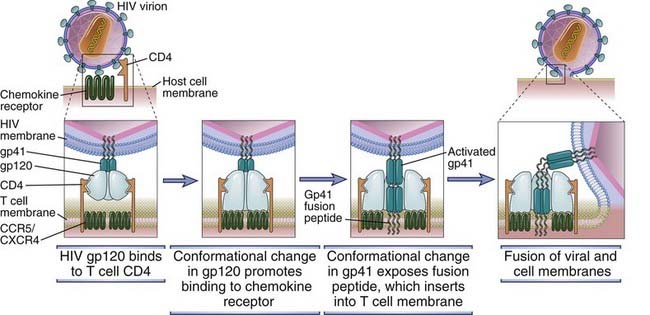
FIGURE 20–6 Mechanism of HIV entry into a cell.
In the model depicted, sequential conformational changes in gp120 and gp41 are induced by binding to CD4. These changes promote binding of the virus to the coreceptor (a chemokine receptor) and fusion of the HIV-1 and host cell membranes. The fusion peptide of activated gp41 contains hydrophobic amino acid residues that mediate insertion into the host cell plasma membrane.
The most important chemokine receptors that act as coreceptors for HIV are CXCR4 and CCR5. More than seven different chemokine receptors have been shown to serve as coreceptors for HIV entry into cells, and several other proteins belonging to the seven-transmembrane–spanning G protein–coupled receptor family, such as the leukotriene B4 receptor, can also mediate HIV infection of cells. Different isolates of HIV have distinct tropisms for different cell populations that are related to the specificity of gp120 variants for different chemokine receptors. All HIV strains can infect and replicate in freshly isolated human CD4+ T cells that are activated in vitro. In contrast, some strains will infect primary cultures of human macrophages but not continuous T cell lines (macrophage-tropic, or M-tropic, virus), whereas other strains will infect T cell lines but not macrophages (T-tropic virus). Some virus strains also infect both T cell lines and macrophages (dual-tropic virus). Macrophage-tropic virus isolates express a gp120 that binds to CCR5, which is expressed on macrophages (and some memory T cells), whereas T cell–tropic viruses bind to CXCR4, which is expressed on T cell lines. HIV variants are described as X4 for CXCR4 binding, R5 for CCR5 binding, or R5X4 for the ability to bind to both chemokine receptors. In many HIV-infected individuals, there is a change from the production of virus that uses CCR5 and is predominantly macrophage tropic early in the disease to virus that binds to CXCR4 and is T cell line tropic late in the disease. The T-tropic strains tend to be more virulent, presumably because they infect and deplete T cells more than do M-tropic strains. The importance of CCR5 in HIV infection in vivo is supported by the finding that individuals who do not express this receptor on the cell surface because of an inherited homozygous 32-bp deletion in the CCR5 gene are resistant to HIV infection.
Once an HIV virion enters a cell, the enzymes within the nucleoprotein complex become active and begin the viral reproductive cycle (see Fig. 20-5). The nucleoprotein core of the virus becomes disrupted, the RNA genome of HIV is reverse-transcribed into a double-stranded DNA form by viral reverse transcriptase, and the viral DNA enters the nucleus. The viral integrase also enters the nucleus and catalyzes the integration of viral DNA into the host cell genome. The integrated HIV DNA is called the provirus. The provirus may remain transcriptionally inactive for months or years, with little or no production of new viral proteins or virions, and in this way HIV infection of an individual cell can be latent.
Transcription of the genes of the integrated DNA provirus is regulated by the LTR upstream of the viral structural genes, and cytokines or other physiologic stimuli that trigger T cells and macrophages enhance viral gene transcription. The LTRs contain polyadenylation signal sequences, the TATA box promoter sequence, and binding sites for two host cell transcription factors, NF-κB and SP1. Initiation of HIV gene transcription in T cells is linked to activation of the T cells by antigen or cytokines. For example, polyclonal activators of T cells, such as phytohemagglutinin, and cytokines such as IL-2, tumor necrosis factor (TNF), and lymphotoxin stimulate HIV gene expression in infected T cells, and IL-1, IL-3, IL-6, TNF, lymphotoxin, IFN-γ, and granulocyte-macrophage colony-stimulating factor (GM-CSF) stimulate HIV gene expression and viral replication in infected monocytes and macrophages. TCR and cytokine stimulation of HIV gene transcription probably involves the activation of NF-κB and its binding to sequences in the LTR. This phenomenon is significant to the pathogenesis of AIDS because the normal response of a latently infected T cell to a microbe may be the way in which latency is ended and virus production begins. The multiple infections that AIDS patients acquire thus stimulate HIV production and infection of additional cells.
The Tat protein is required for HIV gene expression and acts by enhancing the production of complete viral mRNA transcripts. Even in the presence of optimal signals to initiate transcription, few if any HIV mRNA molecules are actually synthesized without the action of Tat because transcription of HIV genes by mammalian RNA polymerase is inefficient and the polymerase complex usually stops before the mRNA is completed. Tat protein binds to the nascent mRNA and increases the “processivity” of RNA polymerase by several hundred-fold, which allows transcription to be completed to produce a functional viral mRNA.
Synthesis of mature, infectious viral particles begins after full-length viral RNA transcripts are produced and the viral genes are expressed as proteins. The mRNAs encoding the various HIV proteins are derived from a single full-genome-length transcript by differential splicing events. HIV gene expression may be divided into an early stage, during which regulatory genes are expressed, and a late stage, during which structural genes are expressed and full-length viral genomes are packaged. The Rev, Tat, and Nef proteins are early gene products encoded by fully spliced mRNAs that are exported from the nucleus and translated into proteins in the cytoplasm soon after infection of a cell. Late genes include env, gag, and pol, which encode the structural components of the virus and are translated from singly spliced or unspliced RNA. The Rev protein initiates the switch from early to late gene expression by promoting the export of these incompletely spliced late gene RNAs out of the nucleus. The pol gene product is a precursor protein that is sequentially cleaved to form reverse transcriptase, protease, ribonuclease, and integrase enzymes. As mentioned before, reverse transcriptase and integrase proteins are required to produce a DNA copy of the viral RNA genome and to integrate it as a provirus into the host genome. The gag gene encodes a 55-kD protein that is proteolytically cleaved into p24, p17, and p15 polypeptides by the action of the viral protease encoded by the pol gene. These polypeptides are the core proteins that are required for assembly of infectious viral particles. The primary product of the env gene is a 160-kD glycoprotein (gp160) that is cleaved by cellular proteases within the endoplasmic reticulum into the gp120 and gp41 proteins required for HIV binding to cells, as discussed earlier. Current antiviral drug therapy for HIV disease includes inhibitors of the enzymes reverse transcriptase, protease, and integrase.
After transcription of various viral genes, viral proteins are synthesized in the cytoplasm. Assembly of infectious viral particles then begins by packaging full-length RNA transcripts of the proviral genome within a nucleoprotein complex that includes the gag core proteins and the pol-encoded enzymes required for the next cycle of integration. This nucleoprotein complex then buds from the plasma membrane, capturing Env and host glycoproteins as part of its envelope. The rate of virus production can reach sufficiently high levels to cause cell death, as discussed later.
A host factor that prevents virion release in certain cell types is a protein called tetherin. Tetherin prevents the pinching off of certain viruses including HIV, and its inhibition of the budding process can be antagonized by an HIV protein called Vpu.
Pathogenesis of HIV Infection and AIDS
HIV disease begins with acute infection, which is only partly controlled by the adaptive immune response, and advances to chronic progressive infection of peripheral lymphoid tissues (Fig. 20-7). The virus typically enters through mucosal epithelia. The subsequent events in the infection can be divided into several phases.
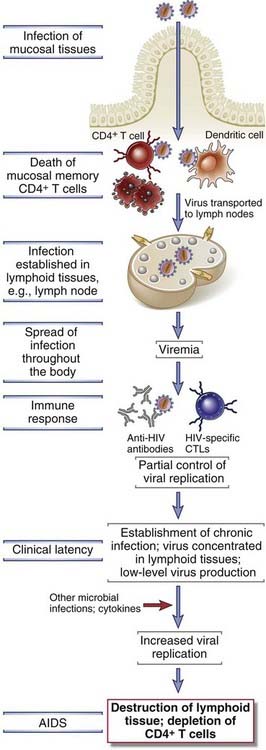
FIGURE 20–7 Progression of HIV infection.
The progression of HIV infection correlates with spread of the virus from the initial site of infection to lymphoid tissues throughout the body. The immune response of the host temporarily controls acute infection but does not prevent the establishment of chronic infection of cells in lymphoid tissues. Cytokine stimuli induced by other microbes serve to enhance HIV production and progression to AIDS.
Acute (early) infection is characterized by infection of memory CD4+ T cells in mucosal lymphoid tissues and death of many infected cells. Because the mucosal tissues are the largest reservoir of T cells in the body and the major site of residence of memory T cells, this local loss is reflected in considerable depletion of lymphocytes. In fact, within 2 weeks of infection, a large fraction of CD4+ T cells may be destroyed.
The transition from the acute phase to a chronic phase of infection is characterized by dissemination of the virus, viremia, and the development of host immune responses. Dendritic cells in epithelia at sites of virus entry capture the virus and then migrate into the lymph nodes. Dendritic cells express a protein with a mannose-binding lectin domain, called DC-SIGN, which may be particularly important in binding the HIV envelope and transporting the virus. Once in lymphoid tissues, dendritic cells may pass HIV on to CD4+ T cells through direct cell-cell contact. Within days after the first exposure to HIV, viral replication can be detected in the lymph nodes. This replication leads to viremia, during which large numbers of HIV particles are present in the patient’s blood, accompanied by an acute HIV syndrome that includes a variety of nonspecific signs and symptoms typical of many viral infections (described later). The viremia allows the virus to disseminate throughout the body and to infect helper T cells, macrophages, and dendritic cells in peripheral lymphoid tissues. As the HIV infection spreads, the adaptive immune system mounts both humoral and cell-mediated immune responses directed at viral antigens, which we will describe later. These immune responses partially control the infection and viral production, and such control is reflected by a drop in viremia to low but detectable levels by approximately 12 weeks after the primary exposure.
In the next, chronic phase of the disease, lymph nodes and the spleen are sites of continuous HIV replication and cell destruction (see Fig. 20-7). During this period of the disease, the immune system remains competent at handling most infections with opportunistic microbes, and few or no clinical manifestations of the HIV infection are present. Therefore, this phase of HIV disease is called the clinical latency period. Although the majority of peripheral blood T cells do not harbor the virus, destruction of CD4+ T cells within lymphoid tissues steadily progresses during the latent period, and the number of circulating blood CD4+ T cells steadily declines (Fig. 20-8). More than 90% of the body’s approximately 1012 T cells are normally found in peripheral and mucosal lymphoid tissues, and it is estimated that HIV destroys up to 1 to 2 × 109 CD4+ T cells every day. Early in the course of the disease, the body may continue to make new CD4+ T cells, and therefore these cells can be replaced almost as quickly as they are destroyed. At this stage, up to 10% of CD4+ T cells in lymphoid organs may be infected, but the number of circulating CD4+ T cells that are infected at any one time may be less than 0.1% of the total CD4+ T cells in an individual. Eventually, over a period of years, the continuous cycle of virus infection, T cell death, and new infection leads to a steady decline in the number of CD4+ T cells in the lymphoid tissues and the circulation.
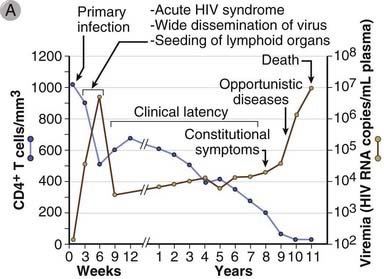
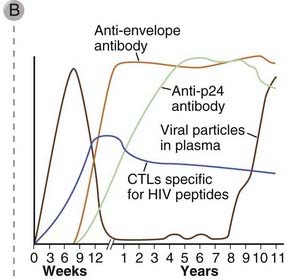
FIGURE 20–8 Clinical course of HIV disease.
A, Plasma viremia, blood CD4+ T cell counts, and clinical stages of disease. About 12 weeks after infection, blood-borne virus (plasma viremia) is reduced to very low levels (detectable only by sensitive reverse transcriptase–polymerase chain reaction assays) and stays this way for many years. Nonetheless, CD4+ T cell counts steadily decline during this clinical latency period because of active viral replication and T cell infection in lymph nodes. When CD4+ T cell counts drop below a critical level (about 200/mm3), the risk for infection and other clinical features of AIDS is high.
(From Pantaleo G, C Graziosi, and AS Fauci. New concepts in the immunopathogenesis of human immunodeficiency virus infection. New England Journal of Medicine 328:327-335, 1993.
B, Immune response to HIV infection. A CTL response to HIV is detectable by 2 to 3 weeks after the initial infection and peaks by 9 to 12 weeks. Marked expansion of virus-specific CD8+ T cells occurs during this time, and up to 10% of a patient’s CTLs may be HIV specific at 12 weeks. The humoral immune response to HIV peaks at about 12 weeks.
Mechanisms of Immunodeficiency Caused by HIV
HIV infection ultimately results in impaired function of both the adaptive and innate immune systems. The most prominent defects are in cell-mediated immunity, and they can be attributed to several mechanisms, including direct cytopathic effects of the virus and indirect effects.
An important cause of the loss of CD4+ T cells in HIV-infected people is the direct cytopathic effect of infection of these cells by HIV. Death of CD4+ T cells is associated with production of virus in infected cells and is a major cause of the decline in the numbers of these cells, especially in the early (acute) phase of the infection. Several direct toxic effects of HIV on infected CD4+ cells have been described.
Mechanisms in addition to direct lysis of infected CD4+ T cells by virus have been proposed for the depletion and loss of function of these cells in HIV-infected individuals. One mechanism that has been suggested is related to chronic activation of uninfected cells by the infections that are common in patients infected with HIV and also by cytokines produced in response to these infections. Chronic activation of the T cells may predispose the cells to apoptosis; the molecular pathway involved in this type of activation-induced cell death is not yet defined. Apoptotic death of activated lymphocytes may account for the observation that the loss of T cells greatly exceeds the numbers of HIV-infected cells. HIV-specific CTLs are present in many patients with AIDS, and these cells can kill infected CD4+ T cells. In addition, antibodies against HIV envelope proteins may bind to HIV-infected CD4+ T cells and target the cells for antibody-dependent cell-mediated cytotoxicity. Binding of gp120 to newly synthesized intracellular CD4 may interfere with normal protein processing in the endoplasmic reticulum and block cell surface expression of CD4, making the cells incapable of responding to antigenic stimulation. It has also been suggested that the maturation of CD4+ T cells in the thymus becomes defective in infected individuals. The relative importance of these indirect mechanisms of CD4+ T cell depletion in HIV-infected patients is uncertain and controversial.
Functional defects in the immune system of HIV-infected individuals exacerbate the immune deficiency caused by depletion of CD4+ T cells. These functional defects include a decrease in T cell responses to antigens and weak humoral immune responses even though total serum Ig levels may be elevated. The defects may be a result of the direct effects of HIV infection on CD4+ T cells, including the effects of soluble gp120 released from infected cells binding to uninfected cells. For example, CD4 that has bound gp120 may not be available to interact with class II MHC molecules on APCs, and thus T cell responses to antigens would be inhibited. Alternatively, gp120 binding to CD4 may deliver signals that downregulate helper T cell function. HIV-infected T cells are unable to form tight synapses with APCs, and this may also interfere with T cell activation. Some studies have demonstrated that patients with HIV infection have increased numbers of CD4+CD25+ regulatory T cells, but it is not yet clear if this is a consistent finding or if these cells actually contribute to defective immunity.
The Tat protein may play some role in the pathogenesis of immunodeficiency caused by HIV. Within T cells, Tat can interact with a variety of regulatory proteins, and these interactions can interfere with normal T cell functions such as cytokine synthesis. Remarkably, Tat not only enters the nucleus of infected T cells but can also escape across the plasma membrane and enter neighboring cells, thus interfering with activation of uninfected T cells in a paracrine fashion.
Macrophages, dendritic cells, and follicular dendritic cells are infected or injured by HIV, and their abnormalities also contribute to the progression of immunodeficiency.
HIV Reservoirs and Viral Turnover
The virus detected in patients’ blood is produced mostly by short-lived infected CD4+ T cells and in smaller amounts by other infected cells. Three phases of decay of plasma viremia have been observed in patients treated with antiretroviral drugs or predicted by mathematical modeling, and these decay curves have been used to surmise the distribution of HIV in different cellular reservoirs. More than 90% of plasma virus is believed to be produced by short-lived cells (half-lives of ~1 day), which are most likely activated CD4+ T cells that are major reservoirs and sources of the virus in infected patients. About 5% of plasma virus is produced by macrophages, which have a slower turnover (half-life of about 2 weeks). It is hypothesized that a small fraction of the virus, perhaps as little as 1%, is present in latently infected memory T cells. Because of the long life span of memory cells, it could take decades for this reservoir of virus to be eliminated even if all new rounds of infection were blocked.
Clinical Features of HIV Disease
A vast amount of information has accumulated about the epidemiology and clinical course of HIV infection. As antiretroviral drug therapy is improving, many of the clinical manifestations are changing. In the following section, we describe the “classical” features of HIV infection and refer to the changing pictures when relevant.
Transmission of HIV and Epidemiology of AIDS
HIV is transmitted from one individual to another by three major routes:
Clinical Course of HIV Infection
The course of HIV disease can be followed by measuring the amount of virus in the patient’s plasma and by the blood CD4+ T cell count (see Fig. 20-8).
TABLE 20–7 Clinical Features of HIV Infection
| Phase of Disease | Clinical Feature |
|---|---|
| Acute HIV disease | Fever, headaches, sore throat with pharyngitis, generalized lymphadenopathy, rashes |
| Clinical latency period | Declining blood CD4+ T cell count |
| AIDS |
Although this summary of the clinical course is true for the most severe cases, the rate of progression of the disease is highly variable, and some individuals are long-term nonprogressors. The immunologic correlates of variable progression remain unknown. Also, recent antiretroviral therapy has changed the course of the disease and greatly reduced the incidence of severe opportunistic infections (such as Pneumocystis) and tumors (such as Kaposi’s sarcoma).
Immune Responses to HIV
HIV-specific humoral and cell-mediated immune responses develop after infection but generally provide limited protection. The early response to HIV infection is, in fact, similar in many ways to the immune response to other viruses and serves to clear most of the virus present in the blood and in circulating T cells. Nonetheless, it is clear that these immune responses fail to eradicate all virus, and the infection eventually overwhelms the immune system in most individuals. Despite the poor effectiveness of immune responses to the virus, it is important to characterize them for three reasons. First, the immune responses may be detrimental to the host, for example, by stimulating the uptake of opsonized virus into uninfected cells by Fc receptor–mediated endocytosis or by eradication of CD4+ T cells expressing viral antigens by CD8+ CTLs. Second, antibodies against HIV are diagnostic markers of HIV infection that are widely used for screening purposes. Third, the design of effective vaccines for immunization against HIV requires knowledge of the types of immune responses that are most likely to be protective (the “correlates of protection”).
Many innate immune responses against HIV have been described. These include antimicrobial peptides (defensins), NK cells, dendritic cells (particularly plasmacytoid dendritic cells producing type I interferons), and the complement system. The role of these responses in combating the infection is not established.
The initial adaptive immune response to HIV infection is characterized by expansion of CD8+ T cells specific for HIV peptides. As many as 10% or more of circulating CD8+ T cells may be specific for HIV during the early stages of infection. These CTLs control infection in the acute phase (see Fig. 20-8) but ultimately prove ineffective because of the emergence of viral escape mutants (variants with mutated antigens). CD4+ T cells also respond to the virus, and these CD4+ T cells may contribute to viral control in a number of ways. An effective CD4+ T cell response is required as a source of help for the generation of CD8+ memory T cells, but CD4+ T cells have also been shown to mediate cytolytic responses against HIV-infected cells, perhaps using Fas ligand to target Fas on infected CD4+ T cells.
The importance of CTL responses in HIV control is underscored by the evolution of the virus under immune pressure, resulting in viral isolates that have lost their original CTL epitopes. The evolution of the virus also results in the loss of epitopes recognized by CD4+ T cells, indicating that both CD8+ and CD4+ cells contribute to host defense against the virus.
Antibody responses to a variety of HIV antigens are detectable within 6 to 9 weeks after infection. The most immunogenic HIV molecules that elicit antibody responses appear to be the envelope glycoproteins, and high titers of anti-gp120 and anti-gp41 antibodies are present in most HIV-infected individuals. Other anti-HIV antibodies found frequently in patients’ sera include antibodies to p24, reverse transcriptase, and gag and pol products (see Fig. 20-8). The effect of these antibodies on the clinical course of HIV infection is uncertain. The early antibodies are not neutralizing and are generally poor inhibitors of viral infectivity or cytopathic effects. Neutralizing antibodies against gp120 develop 2 to 3 months after primary infection, but even these antibodies cannot cope with a virus that is able to rapidly change the most immunodominant epitopes of its envelope glycoproteins.
Mechanisms of Immune Evasion by HIV
HIV is the prototype of an infectious pathogen that evades host defenses by destroying the immune system. In addition, several features of HIV may help the virus to evade host immunity.
HIV has an extremely high mutation rate because of error-prone reverse transcription, and in this way it may evade detection by antibodies or T cells generated in response to viral proteins. It has been estimated that in an infected person, every possible point mutation in the viral genome occurs every day. A region of the gp120 molecule, called the V3 loop, is one of the most antigenically variable components of the virus; it varies in HIV isolates taken from the same individual at different times. Furthermore, the regions of the V3 loop that are critical for viral entry and therefore are less frequently mutated are not readily exposed to the humoral immune system.
HIV-infected cells may evade CTLs through downregulation of class I MHC molecule expression. The HIV Nef protein inhibits expression of class I MHC molecules, mainly by promoting internalization of these molecules. Other mechanisms of inhibiting cell-mediated immunity have been demonstrated in some cases. These include a preferential inhibition of TH1 cytokines, activation of regulatory T cells, and suppression of dendritic cell functions. The mechanisms of these actions of the virus as well as their pathogenic significance are not established.
Elite Controllers and Long-term Nonprogressors: A Possible Role for Host Genes
Although most individuals infected with HIV eventually develop AIDS, approximately 1% of individuals who are infected do not develop disease. Such individuals have high CD4+ and CD8+ T cell counts, do not require therapy, and have persistent viremia but no disease for at least 10 to 15 years. On the basis of the degree of viremia, this group can be divided into two subsets: long-term nonprogressors have detectable viremia of around 5000 copies of HIV-1 RNA per milliliter of blood; and a much smaller subset of “elite controllers” present with viral loads of about 50 copies or less of HIV-1 RNA per milliliter of blood. There is considerable interest in understanding the genetic basis of HIV control by examining these cohorts of individuals in detail. So far, a strong role for the MHC locus in protecting individuals and preventing progression has been suggested by genetic association studies. Specific HLA class I and even some HLA class II loci have been linked to the absence of disease progression. We have previously mentioned the importance of the inheritance of the CCR5 homozygous 32-bp deletion in protection from infection, and other genetic factors contributing to resistance are likely to be revealed in the coming years.
Treatment and Prevention of AIDS and Vaccine Development
Active research efforts have been aimed at developing reagents that interfere with the viral life cycle. Treatment of HIV infection and AIDS now includes the administration of three classes of antiviral drugs, used in combination, that target viral molecules for which no human homologues exist. The first antiretroviral drugs to be widely used were nucleoside analogues that inhibit reverse transcriptase activity. These drugs include deoxythymidine nucleoside analogues such as 3′-azido-3′-deoxythymidine (AZT), deoxycytidine nucleoside analogues, and deoxyadenosine analogues. When these drugs are used alone, they are often effective in significantly reducing plasma HIV RNA levels for several months to years, but they usually do not halt progression of HIV-induced disease, largely because of the evolution of virus with mutated forms of reverse transcriptase that are resistant to the drugs. Non-nucleoside reverse transcriptase inhibitors directly bind to the enzyme and inhibit its function. Viral protease inhibitors have been developed that block the processing of precursor proteins into mature viral capsid and core proteins. When these protease inhibitors are used alone, mutant viruses resistant to their effects rapidly emerge. However, protease inhibitors are now being used in combination with two different reverse transcriptase inhibitors. This new triple-drug therapy, commonly referred to as HAART (highly active antiretroviral therapy) or ART (antiretroviral therapy), has proved to be effective in reducing plasma viral RNA to undetectable levels in most treated patients for up to 3 years. An integrase inhibitor is also now used as part of antiviral therapy. “Entry inhibitors,” which prevent viral entry by targeting either CD4 or CCR5 on the host cell or gp41 or gp120 on the virus, are another novel category of therapeutics. Maraviroc is an entry inhibitor that blocks CCR5 and thus prevents viral entry. Drugs that target gp41 include compounds such as enfuvirtide that prevent fusion of the viral envelope with the host cell plasma membrane. Although antiretroviral therapy has reduced viral titers to below detection for up to 10 years in some patients, it is unlikely that such treatment can eliminate the virus from all reservoirs (especially long-lived infected cells), and resistance to the drugs may ultimately develop. Other formidable problems associated with these new drug therapies, which will impair their effective use in many parts of the world, include high expense, complicated administration schedules, and significant adverse effects.
The individual infections experienced by patients with AIDS are treated with the appropriate prophylaxis, antibiotics, and supportive measures. More aggressive antibiotic therapy is often required than for similar infections in less compromised hosts.
Efforts at prevention of HIV infection are extremely important and potentially effective in controlling the HIV epidemic. In the United States, the routine screening of blood products for evidence of donor HIV infection has already reduced the risk of this mode of transmission to negligible levels. Various public health measures to increase condom use and to reduce the use of contaminated needles by intravenous drug users are now widespread. Perhaps the most effective efforts at prevention are campaigns to increase public awareness of HIV.
The development of an effective vaccine against HIV is a priority for biomedical research institutions worldwide. The task has been complicated by the ability of the virus to mutate and vary many of its immunogenic antigens. It is likely that an effective vaccine will have to stimulate both humoral and cell-mediated responses to viral antigens that are critical for the viral life cycle. To achieve this goal, several approaches are being tried for HIV vaccine development. Much of the preliminary work has involved simian immunodeficiency virus (SIV) infection of macaques, and effective vaccines against SIV have already been developed. This success is encouraging because SIV is molecularly closely related to HIV and causes a disease in macaques that is similar to AIDS in humans. Various live virus vaccines have been tested in the hope that they will induce strong CTL responses. Such vaccines include nonvirulent recombinant hybrid viruses composed of part SIV and part HIV sequences or viruses that have been attenuated by deletions in one or more parts of the viral genome, such as the nef gene. One concern with live virus vaccines is their potential to cause disease if they are not completely attenuated and possibly to recombine with wild-type HIV to produce a pathogenic variant. Another approach that avoids this safety concern but retains efficacy in inducing CTL-mediated immunity is the use of live recombinant non-HIV viral vectors carrying HIV genes. Preliminary trials in human volunteers have already shown that canarypox vaccines expressing several HIV-1 genes can induce strong CTL responses to the HIV antigens. Many DNA vaccines have also been studied; these vaccines are composed of combinations of structural and regulatory genes of SIV or HIV packaged in mammalian DNA expression vectors. Combinations of vaccines, such as initial immunization with a DNA vaccine followed by boosting with a canarypox vector expressing HIV genes, have yielded some of the most promising results to date. Recombinant protein or peptide subunit vaccines that elicit antibodies have so far been of limited value because the antibodies induced by these vaccines typically do not neutralize clinical isolates of HIV. Attempts are currently under way to generate vaccines that can induce neutralizing antibodies to HIV.
Summary
Congenital (Primary) Immunodeficiencies
Blackburn MR, Kellems RE. Adenosine deaminase deficiency: metabolic basis of immune deficiency and pulmonary inflammation. Advances in Immunology. 2005;86:1-4.
Bonilla FA, Geha RS. Update on primary immunodeficiency diseases. Journal of Allergy and Clinical Immunology. 2006;117(Suppl):435-441.
Buckley RH. Molecular defects in human severe combined immunodeficiency and approaches to immune reconstitution. Annual Review of Immunology. 2004;22:625-655.
Bustamante J, Boisson-Dupuis S, Jouanguy E, Picard C, Puel A, Abel L, Casanova JL. Novel primary immunodeficiencies revealed by the investigation of paediatric infectious diseases. Current Opinion in Immunology. 2008;20:39-48.
Castigli E, Geha RS. Molecular basis of common variable immunodeficiency. Journal of Allergy and Clinical Immunology. 2006;117:740-746.
Chen X, Jensen PE. MHC class II antigen presentation and immunological abnormalities due to deficiency of MHC class II and its associated genes. Experimental and Molecular Pathology. 2008;85:40-44.
Conley ME, Dobbs AK, Farmer DM, Kilic S, Paris K, Grigoriadou S, Coustan-Smith E, Howard V, Campana D. Primary B cell immunodeficiencies: comparisons and contrasts. Annual Review of Immunology. 2009;27:199-227.
Cunningham-Rundles C, Ponda PP. Molecular defects in T- and B-cell primary immunodeficiency diseases. Nature Reviews Immunology. 2005;5:880-892.
Fischer A. Human primary immunodeficiency diseases. Immunity. 2008;28:835-846.
Groneberg DA, Quarcoo D, Frossard N, Fischer A. Gene therapy for severe combined immunodeficiency. Annual Review of Medicine. 2005;56:585-602.
Janka GE. Hemophagocytic lymphohistiocytosis. Hematology. 2005;10(Suppl 1):104-107.
Lavin MF. Ataxia-telangiectasia: from a rare disorder to a paradigm for cell signalling and cancer. Nature Reviews Molecular and Cell Biology. 2008;9:759-769.
Notarangelo LD. Primary immunodeficiencies. Journal of Allergy and Clinical Immunology. 2010;125:S182-S194.
Ochs HD, Thrasher AJ. The Wiskott-Aldrich syndrome. Journal of Allergy and Clinical Immunology. 2006;117:725-738.
Puel A, Yang K, Ku CL, et al. Heritable defects of the human TLR signaling pathways. Journal of Endotoxin Research. 2005;11:220-224.
Barouch DH. Challenges in the development of an HIV-1 vaccine. Nature. 2008;455:613-619.
Brenchley JM, Price DA, Douek DC. HIV disease: fallout from a mucosal catastrophe? Nature Immunology. 2006;7:235-239.
Derdeyn CA, Silvestri G. Viral and host factors in the pathogenesis of HIV infection. Current Opinion in Immunology. 2005;17:366-373.
Haase AT. Perils at mucosal front lines for HIV and SIV and their hosts. Nature Reviews Immunology. 2005;5:783-792.
Hladik F, McElrath MJ. Setting the stage: host invasion by HIV. Nature Reviews Immunology. 2008;8:447-457.
Johnston MI, Fauci AS. An HIV vaccine—evolving concepts. New England Journal of Medicine. 2007;356:2073-2081.
Letvin NL. Progress and obstacles in the development of an AIDS vaccine. Nature Reviews Immunology. 2006;6:930-939.
Lusso P. HIV and the chemokine system: 10 years later. EMBO Journal. 2006;25:447-456.
Mascola JR, Montefiori DC. The role of antibodies in HIV vaccines. Annual Review of Immunology. 2010;28:413-444.
McMichael AJ, Borrow P, Tomaras GD, Goonetilleke N, Haynes BF. The immune response during acute HIV-1 infection: clues for vaccine development. Nature Reviews Immunology. 2010;10:11-23.
Nixon DF, Aandahl EM, Michaelsson J. CD4+CD25+ regulatory T cells in HIV infection. Microbes and Infection. 2005;7:1063-1065.
Walker BD, Burton DR. Towards an AIDS vaccine. Science. 2008;320:760-764.