The Newborn with a Perinatal Injury or Congenital Malformation
1 Define each key term listed.
2 List and define the more common disorders of the newborn.
3 Describe the classifications of birth defects.
4 Outline the nursing care for the newborn with hydrocephalus.
5 Describe the symptoms of increased intracranial pressure.
6 Discuss the prevention of neural tube anomalies.
7 Outline the preoperative and postoperative nursing care of a newborn with spina bifida cystica.
8 Differentiate between cleft lip and cleft palate.
9 Discuss the dietary needs of a newborn with phenylketonuria.
10 Discuss the early signs of developmental hip dysplasia.
11 Discuss the care of the newborn with Down syndrome.
12 Outline the causes and treatment of hemolytic disease of the newborn (erythroblastosis fetalis).
13 Devise a plan of care for a newborn receiving phototherapy.
14 Describe home phototherapy.
15 Discuss the assessment and nursing care of a newborn with macrosomia.
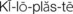 , p. 328)
, p. 328) , p. 337)
, p. 337)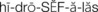 , p. 322)
, p. 322) , p. 339)
, p. 339)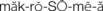 , p. 344)
, p. 344) , p. 325)
, p. 325) , p. 326)
, p. 326) , p. 325)
, p. 325) , p. 325)
, p. 325) http://evolve.elsevier.com/Leifer
http://evolve.elsevier.com/Leifer
Birth defects, abnormalities that are apparent at birth, occur in 3% to 4% of all live births. The rate is even higher if the defects that become evident later in life are counted. An abnormality of structure, function, or metabolism may result in a physical or mental disability, may shorten life, or may be fatal. Box 14-1 shows the system of classification of birth defects. Because these disorders include so many conditions, it is necessary to limit the number discussed in this chapter and to place others in relevant areas of the text (see the Index for specific conditions). Fetal alcohol syndrome and environmental influences on fetal growth are discussed in Chapter 5. Congenital heart disease is discussed in Chapter 26.
Defects present at birth often involve the skeletal system; limbs may be missing, malformed, or duplicated. Some abnormalities (e.g., congenital hip dysplasia) are more subtle, and the nurse must be alert to detect them. Inborn errors of metabolism include a number of inherited diseases that affect body chemistry. There may be an absence or a deficiency of a substance necessary for cell metabolism. The deficient substance is usually an enzyme. Almost any organ of the body may be damaged. Examples of inborn errors of metabolism include cystic fibrosis and phenylketonuria (PKU). In disorders of the blood, there is a reduced or missing blood component or an inability of a component to function adequately. Sickle cell disease, thalassemia, and hemophilia fall into this category. Chromosomal abnormalities number in the thousands. Most involve some type of mental retardation, and others are incompatible with life. The newborn with Turner’s syndrome or Klinefelter’s syndrome may have impaired physical growth and sexual development. Perinatal injuries have many causes and are seen in various forms, the most common of which is premature birth.
As the March of Dimes Birth Defect Foundation (2006) points out, “Few birth defects can be attributed to a single cause. The majority are thought to result from an interplay between environment and heredity, depending on inherited susceptibility, stage of pregnancy, and degree of environmental hazard.” Newborns with birth defects may need to remain in the neonatal unit for an extended period of time for intensive care and treatment.
Malformations Present at Birth
The following sections discuss some congenital malformations, or those defects present at birth, according to body systems.
Nervous System
Neural tube defects are most often caused by failure of neural tube closure at either the cranial (top) or the caudal (lower) end of the spinal cord. These defects include hydrocephalus and spina bifida.
Hydrocephalus
Pathophysiology: Hydrocephalus (hydro, “water,” and cephalo, “head”) in the newborn is a condition characterized by an increase of cerebrospinal fluid (CSF) within the ventricles of the brain, which causes pressure changes in the brain and an increase in head size. It results from an imbalance between the production and absorption of CSF or improper formation of the ventricles. Hydrocephalus may be congenital or acquired. It is most commonly acquired by an obstruction, such as a tumor, or as a sequela of infections (encephalitis or meningitis) or perinatal hemorrhage. The symptoms depend on the site of obstruction and the age at which it develops.
Hydrocephalus is classified as noncommunicating (obstructive) or communicating. Noncommunicating hydrocephalus results from the obstruction of CSF flow from the ventricles of the brain to the subarachnoid space. Communicating hydrocephalus results when CSF is not obstructed in the ventricles but is inadequately reabsorbed in the subarachnoid space (Figure 14-1).
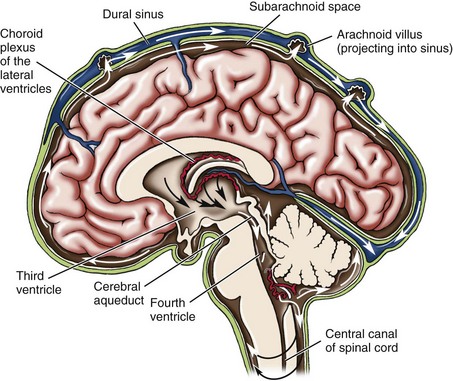
FIGURE 14-1 Cerebrospinal fluid circulation. Cerebrospinal fluid (CSF) is formed in the choroid plexus. The total volume of CSF is approximately 50 mL in the infant and 150 mL in the adult. It flows from the lateral ventricles through the foramen of Monro to the third ventricle. From the third ventricle, the CSF fluid flows through the aqueduct of Sylvius to the fourth ventricle, and then through the foramen of Luschka and the foramen of Magendie into the cisterns at the base of the brain. Flow continues to the spinal canal. The CSF is then absorbed by the arachnoid villi (which are also known as pacchionian bodies). Communicating or nonobstructive hydrocephalus occurs when the arachnoid villi are malformed or malfunction. Noncommunicating or obstructive hydrocephalus results when the tiny aqueduct of Sylvius is obstructed within the ventricles. When hydrocephalus occurs, excessive CSF causes the ventricles to enlarge and press the brain tissue against the bony skull.
Manifestations: The signs and symptoms of hydrocephalus depend on the time of onset and the severity of the imbalance. The classic sign is an increase in head size. If hydrocephalus occurs in utero, the enlarged head will necessitate a cesarean section delivery. At birth the head enlarges rapidly and the fontanelles bulge. The cranial sutures separate to accommodate the enlarging mass. The scalp is shiny, and the veins are dilated. In advanced cases the pupils of the eyes may appear to be looking downward and the sclera may be seen above the pupils, much like the look of a setting sun (Figure 14-2). A foreshortened occiput suggests pathology of the fourth ventricle, with the brain stem protruding through the cervical canal. This is called the Chiari malformation (Kliegman et al., 2007). When the enlarged head involves a prominent occiput, the condition usually involves an atresia of the foramen of Lushka and the foramen of Magendie and is known as the Dandy-Walker syndrome. The infant is helpless and lethargic. The body becomes thin, and the muscle tone of the extremities is often poor. The cry is shrill and high pitched. Irritability, vomiting, and anorexia are present, and convulsions may occur.
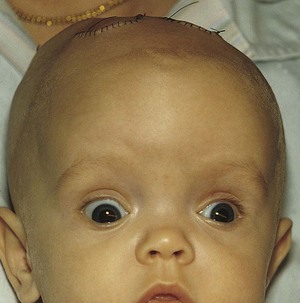
FIGURE 14-2 Marked hydrocephalus with “setting sun” sign of the eyes. Note the characteristic large head, distended scalp veins, and full fontanelle.
When hydrocephalus occurs in the older child, the head cannot enlarge because the cranial sutures are fused; therefore headache is the predominant symptom, with cognitive slowing, personality changes, spasticity, and other neurological signs.
Diagnosis: Transillumination (trans, “across,” and illuminare, “to enlighten”)—the inspection of a cavity or organ by passing a light through its walls—is a simple diagnostic procedure useful in visualizing fluid. A flashlight with a sponge-rubber collar is held tightly against the infant’s head in a dark room. The examiner observes for areas of increased luminosity. A small ring of light is normal, but a large halo effect is not. The child’s head is measured daily. Echoencephalography, computed tomography (CT) scanning, and magnetic resonance imaging (MRI) are used to visualize the enlarged ventricles and to identify the area of obstruction. A ventricular tap or puncture may be performed using sterile technique to determine pressure and drain CSF. The equipment needed is the same as that for a lumbar puncture. A specimen is labeled and sent to the laboratory for analysis.
Treatment: The use of acetazolamide and furosemide reduces the production of CSF and may provide some relief, but most often surgery is indicated (Kliegman et al., 2007). The surgeon attempts to bypass, or shunt, the point of obstruction. The CSF may thus be carried to another area of the body, where it is absorbed and finally excreted. This is accomplished by inserting special tubing, which is replaced at intervals as the child grows. The procedure, known as a ventriculoperitoneal shunt (Figure 14-3), allows the excess fluid to drain into the peritoneal cavity, where it is absorbed.
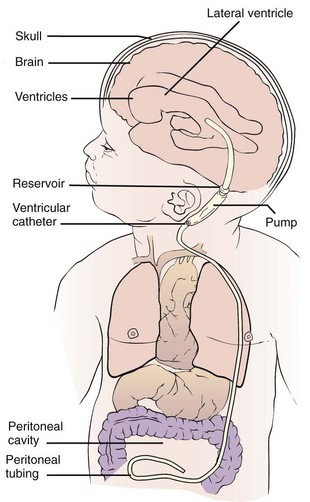
FIGURE 14-3 Shunting procedure for hydrocephalus, in which a catheter drains the ventricular system into the peritoneal cavity. Note the pump behind the ear, which can be depressed intermittently to clear obstructions.
The prognosis for the child with hydrocephalus has improved with modern drugs and surgical techniques. If the brain is not seriously damaged before the operation, mental function may be preserved. Motor development is sometimes slower if the child cannot lift the head, normally because of its weight. Complications of shunts are usually mechanical (kinking or plugging of tubing) or infectious. The shunt acts as a focal spot for infection and may need to be removed if infection persists.
Preoperative Nursing Care: The general nursing care of an infant with hydrocephalus who has not undergone surgery presents several challenges. The child may be barely able to raise the head. Mental development is delayed. Lack of appetite, a tendency to vomit easily, and poor resistance to infections complicate the management of these infants.
The position of the infant must be changed frequently to prevent hypostatic pneumonia and pressure sores. Hypostatic pneumonia occurs when the circulation of the blood in the lungs is poor and the infant remains in one position too long. It is particularly prevalent in infants who are poorly nourished or weak or who have a debilitating disease. When the nurse turns an infant who has hydrocephalus, the head must always be supported. To turn the infant in bed, the weight of the head is borne in the palm of one hand, and the head and body are rotated together to prevent a strain on the neck. When the infant is lifted from the crib, the head must be supported by the nurse’s arm and chest.
The tissues of the head, ears, and bony prominences tend to break down. A pad of lamb’s wool or sponge rubber placed under the head may help to prevent these lesions. If the skin becomes broken, it is given immediate attention to prevent infection. The infant must be kept dry, especially around the creases of the neck, where perspiration may collect.
In most cases the nurse may hold the infant for feedings. The nurse sits with the arm supported because the infant’s head is heavy. A calm, unhurried manner is necessary. The room should be as quiet as possible. After the feeding the infant is placed in a side-lying position. The infant is not disturbed once settled, because vomiting occurs easily. The nurse must organize daily care so it does not interfere with meals.
Observations to be recorded and reported include the type and amounts of food taken, any vomiting, the condition of the skin, motor abilities, restlessness, irritability, and changes in vital signs. Fontanelles are inspected for size and signs of bulging. Head circumference is measured around the occipitofrontal area and is recorded on the chart.
Symptoms of increased pressure within the head are an increase in blood pressure and a decrease in pulse rate and respirations. Signs of a cold or other infection are immediately reported to the nurse in charge and are recorded.
Postoperative Nursing Care: In addition to routine postoperative care and observations, the nurse observes the patient for signs of increased intracranial pressure (ICP) and of infection at the operative site or along the shunt line. As with any postoperative care, pain control management is essential.
Bacterial infection is a life-threatening complication that sometimes necessitates shunt removal. Signs of infection include an increase in vital signs, poor feeding, vomiting, pupil dilation, decreased levels of consciousness, and seizures. The operative area is observed for signs of inflammation. An internal flushing device may be used to ensure patency of the shunt tube when increased ICP is suspected. The surgeon may order the pump to be routinely depressed a certain number of times each day to facilitate drainage. This is accomplished by compressing the antechamber or reservoir that is under the skin behind the ear (see Figure 14-3).
Positioning of the infant depends on several factors and may vary with the infant’s progress. If the fontanelles are sunken, the infant is kept flat because too rapid a reduction of fluid may lead to seizures or cortical bleeding. If the fontanelles are bulging, the infant is usually placed in the semi-Fowler’s position to promote drainage of the ventricles through the shunt. The infant is always positioned in a way that prevents pressure on the operative site. The surgeon leaves orders for the patient’s position and activity. Assessment of skin remains a priority. Head and chest measurements are recorded. In patients with peritoneal shunts, the abdomen is measured or observed to detect malabsorption of fluid.
The infant should be observed for signs of increased ICP. The development of a high-pitched cry, unequal pupil size or response to light, bulging fontanelles, irritability or lethargy, poor feeding, or abnormal vital signs should be reported and recorded. (Evidence of increased ICP in the older child may be manifested by a change in personality, a change in level of consciousness, and complaints of headache that is unrelieved by over-the-counter medications.) The need for pain control should be assessed and medications given as needed. Intake and output are carefully recorded, and the infant is observed closely for signs of fluid overload. The infant is usually fed after active bowel sounds are heard. The surgical suture lines should be kept clean and dry and the infant’s diaper kept well below the abdominal suture line to prevent contamination.
Parent education, support, and guidance are essential. Parents are taught signs that indicate shunt malfunction, how and when to “pump” the shunt by pressing against the valve behind the ear, and the need for multidisciplinary follow-up care. Signs of tube malfunction in the older child involve signs of increasing ICP such as headache, lethargy, and changes in level of consciousness. Community resources, such as the National Hydrocephalus Foundation and information concerning special car seats for children with special needs, should be made known to the parents. There is approximately an 80% survival rate for infants treated early, and approximately one third of the cases result in normal physical and neurological functioning. Other survivors may have varying degrees of developmental disabilities.
Spina Bifida
Spina bifida, also known as myelodysplasia, refers to a group of central nervous system disorders characterized by malformation of the spinal cord.
Pathophysiology: Spina bifida (divided spine) is a congenital embryonic neural tube defect in which there is an imperfect closure of the spinal vertebrae. There are two forms: occulta (hidden) and cystica (sac or cyst) (Figure 14-4).
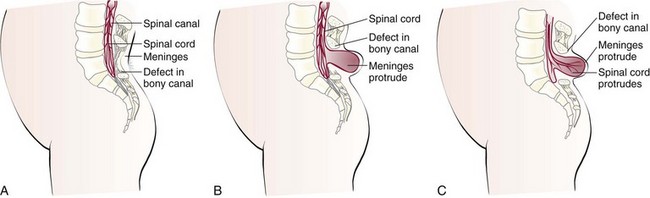
FIGURE 14-4 Types of spina bifida. A, Spina bifida occulta. There is a defect in the bony canal. The meninges and spinal cord are normal. B, Spina bifida cystica meningocele. The spinal cord is normal, but there is a defect in the bony canal. The meninges protrude through this defect. C, Spina bifida cystica meningomyelocele. There is a defect in the bony canal. The meninges protrude, and the spinal cord protrudes through the defect.
Spina bifida occulta is a relatively minor variation of the disorder in which the opening is small and there is no associated protrusion of structures. It often goes undetected and occurs most commonly at the L5 and S1 levels. There may be a tuft of hair (Figure 14-5), dimple, lipoma, or discoloration at the site. In general, treatment is not necessary unless neuromuscular symptoms appear. These symptoms consist of progressive disturbances of gait, such as footdrop, or disturbances of bowel and bladder sphincter function.
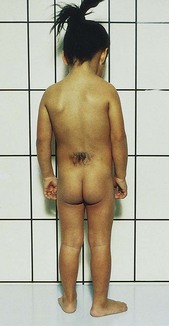
FIGURE 14-5 A child with a hairy patch in the lumbosacral region indicating the site of a spina bifida occulta.
Spina bifida cystica consists of the development of a cystic mass in the midline of the opening in the spine. Meningocele and meningomyelocele are two types of spina bifida cystica. A meningocele (meningo, “membrane,” and cele, “tumor”) contains portions of the membranes and CSF. The size varies from that of a walnut to that of a newborn’s head.
More serious is a protrusion of the membranes and spinal cord through this opening, or a meningomyelocele. Although it resembles a meningocele, there may be associated paralysis of the legs and poor control of bowel and bladder functions. Hydrocephalus is a common complication. Prenatal detection is possible through ultrasonography and testing for increased alpha-fetoprotein (AFP) in the amniotic fluid of the mother (see Chapter 5).
Prevention: The specific cause of meningomyelocele is unknown. The use of drugs during early pregnancy and poor nutrition may contribute to the development of a neural tube defect. The American Academy of Pediatrics (AAP) recommends that all women of childbearing age take a daily multivitamin that contains 0.4 mg of folic acid and continue the intake of folic acid until the twelfth week of pregnancy, when basic neural tube development is completed. Studies have shown that the intake of folic acid before conception dramatically decreases the occurrence of neural tube defects such as spina bifida.
Treatment: The treatment for spina bifida is surgical closure. The prognosis for patients with these conditions depends on the extent of involvement. When a patient with meningocele has no weakness of the legs or sphincter involvement, surgical correction is performed with excellent results. Surgery is also indicated for a patient with meningomyelocele for cosmetic purposes and to help prevent infection. A multidisciplinary approach is necessary because, depending on the extent of the defect, the child may have difficulties associated with hydrocephalus, orthopedic problems, and problems relating to urinary and bowel function.
Habilitation is necessary after the operation because the legs remain paralyzed and the patient is incontinent of urine and feces. Habilitation, rather than rehabilitation, is the term used to describe this treatment because the patient is disabled from birth and therefore is learning, not relearning. The aim of habilitation is to minimize the child’s disability and put to constructive use the unaffected parts of the body. Every effort is made to help the child develop a healthy personality so that he or she may experience a happy and productive life.
Eventually the child can be taught to use a wheelchair and possibly to walk with braces, crutches, or other walking devices. The implantation of an artificial urinary sphincter in early childhood can help some children to become continent and prevent the complications associated with constant urinary dribble. Medications such as oxybutynin chloride (Ditropan) are available to increase bladder storage. Children can also be “bowel trained” with the use of suppositories that promote timed bowel movements, helping the child avoid the social rejection that can be caused by bowel incontinence.
Nursing Care: The main objectives of nursing care include prevention of infection of or injury to the sac, correct positioning to prevent pressure on the sac and development of contractures, good skin care (particularly if the infant is incontinent of urine and feces), adequate nutrition, accurate observations and charting, education of the parents, continued medical supervision, and habilitation.
Immediate care of the sac is essentially the same regardless of whether the cord is involved. On delivery the newborn is placed in an incubator. Moist, sterile dressings of saline or an antibiotic solution may be ordered to prevent drying of the sac. Some method of protecting the mass is necessary if surgery is to be delayed. Protection from injury and maintenance of a sterile environment for the open lesion are essential.
Along with routine observations made for every newborn, other pertinent nursing observations must be made and recorded:
• The size and area of the sac are checked for any tears or leakage.
• The extremities are observed for deformities and movement. (There may be spasticity or paralysis of the limbs, or they may be normal depending on the type and location of the cyst.)
• The head circumference is measured to determine the possibility of associated hydrocephalus.
• Fontanelles are observed to provide baseline data.
• The lack of anal sphincter control and dribbling of urine are significant in the differential diagnosis (Figure 14-6). In general, the higher the defect on the spine, the greater the neurological deficit.
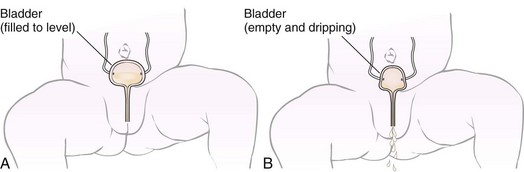
FIGURE 14-6 Incontinence in the newborn. A, Normally when the bladder fills to a certain level, a sensor stimulates contraction of the bladder, and expulsion of a volume of urine into the diaper occurs. A normal newborn has about six wet diapers a day. B, A newborn is considered incontinent when the sensor does not function and the bladder does not fill to its capacity before emptying. There is a constant dribble of urine into the diaper. The diaper is always wet.
Positioning of the infant is of importance. The goal is to prevent pressure on the sac and prevent postural deformities. When positioning infants with multiple deformities, the nurse must try to guard against aggravating existing problems. The infant is usually placed prone with a pad between the legs to maintain abduction and to counteract hip subluxation. A small roll is placed under the ankles to maintain foot position. Some infants may be supported in a side-lying posture to provide periods of relief. The disadvantage of this position is that it reduces movement of the arms and flexes the hips. The physical therapy staff may provide a helpful consultation. Surgery is generally done early.
Postoperative nursing care involves neurological assessment and prevention of infection. The status of the fontanelles and any signs of increased ICP, such as irritability or vomiting, are significant. Sometimes a shunt is performed shortly after closure of the spine, if hydrocephalus is present. Complications that can be life threatening include meningitis, pneumonia, and urinary tract infection.
Urological monitoring is essential, because many of these infants have urinary incontinence (see Figure 14-6). Medication to prevent urinary tract infections is routinely given. The Credé method of bladder emptying (applying pressure above the symphysis pubis) may be used for infants. Older children may be taught intermittent, clean self-catheterization. This technique can be performed by parents and learned by children.
Skin care is a challenge. Constant dribbling of feces and urine irritates the perineal area and can infect the sac or the incision. Meticulous cleanliness is necessary. The bedding must be dry and free of wrinkles. Frequent cleansing, application of a prescribed ointment or lotion, and light massage help to maintain skin integrity. If range of motion exercises are ordered, they are performed gently.
Feeding is facilitated by early closure of the defect. In delayed cases, gavage may be used. These patients need cuddling and sensory stimulation. An infant who cannot be held can be soothed by touch. The nurse talks to the infant and, when possible, provides face-to-face (en face) communication. Mobiles are placed appropriately. Periodically moving the incubator or crib provides diversity of view. Soft music is also soothing.
Many infants with spina bifida develop a latex allergy. A latex-free environment should be adopted whenever possible. Parents should be informed that latex products such as balloons, “koosh” balls, tennis balls, and adhesive strips can cause allergic reactions that may include rashes and wheezing. The parents should be informed about food sensitivities that are common to children with latex allergies. Foods to avoid include bananas, avocados, and kiwi. Other commonly used items to avoid include latex-based pacifiers, feeding nipples, and water toys. The child should wear a medical identification tag indicating the latex allergy. In some cases, antihistamines and steroids may be prescribed before and after surgery. Nurses should wear nitrile gloves instead of latex gloves while caring for these patients.
Special consideration must be given to the establishment of parent-infant relationships. This problem is complicated if the infant is transferred to a large medical center. Understanding and support are given to the parents, who may be overwhelmed. It is not unusual for them to be repulsed by the cyst. Most experience a sense of loss for what was to have been their “perfect baby.” Steps of the grieving process may be recognized by the astute nurse. Information and education about this disorder can be obtained from the Spina Bifida Association of America.
Gastrointestinal System
Pathophysiology: A cleft lip is characterized by a fissure or opening in the upper lip (Figure 14-7). It is a result of the failure of the maxillary and median nasal processes to unite during embryonic development, usually between the seventh and eighth weeks of gestation. In many cases it seems to be caused by hereditary predisposition, but occasionally it can be caused by environmental influences during the stage of oral development. This disorder appears more frequently in boys than in girls and may occur on one or both sides of the lip. The extent of the defect may vary from slight to severe. Sometimes it is accompanied by a cleft palate—a fissure in the midline of the roof of the mouth. Cleft lip and cleft palate are common congenital anomalies and occur in about 1 in 750 births. Transculturally, they occur more often in Asian Americans and Native Americans and less commonly in African Americans (Kliegman et al., 2007).
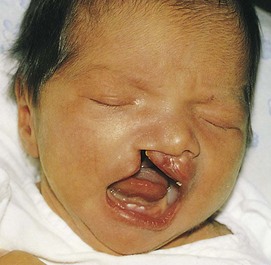
Treatment and Nursing Care: The initial treatment for cleft lip is a surgical repair known as cheiloplasty. The cleft lip is repaired by age 3 months when weight gain is established and the infant is free of infection. Surgery not only improves the infant’s sucking ability but also greatly improves appearance. Indirectly, this influences bonding and the amount of affection the infant receives because some parents refrain from cuddling an infant who is obviously disfigured.
A complete physical examination is done and routine blood tests are ordered before surgery. Photographs may also be taken. Any signs of oral, respiratory, or systemic infection are reported to the registered nurse. The physician may order elbow restraints to prevent the infant from scratching the lip and to acquaint the infant with them because they are necessary postoperatively. A syringe with a rubber tip, a long nipple with a large hole attached to a squeeze bottle, or a medicine dropper can be used to feed the infant before and after surgery, because sucking motions must be avoided to keep from applying tension on the suture line.
Postoperative Nursing Care: Postoperative nursing goals for the infant undergoing a cheiloplasty include the following:
• Preventing the infant from sucking and crying, which could cause tension on the suture line.
• Careful positioning (never on the abdomen) to prevent injury to the operative site.
• Preventing infection and scarring by gentle cleansing of the suture line to prevent crusts from forming.
• Preventing injury to the operative site by using elbow restraints. A Logan bow (a device used to immobilize the upper lip) may be applied for a short time postoperatively.
• Providing for the infant’s emotional needs by cuddling and other forms of affection. This is of particular importance because the infant cannot obtain the usual satisfactions from sucking.
• Providing appropriate pain relief and sedation, which may be required for active infants.
Feeding: The infant receives feedings by dropper until the wound is completely healed (1 to 2 weeks). The infant is usually fed as soon as clear liquids are tolerated postoperatively. Care should be taken to avoid touching the suture line when inserting the medicine dropper. Sucking is prevented as much as possible until the suture line is healed. Placing a small amount of formula into the infant’s mouth and allowing time for swallowing will prevent aspiration. Offering small amounts of sterile water will cleanse the mouth after feeding. Formula or drainage is gently cleaned from the suture line with saline solution, and an ointment may be applied to the skin as prescribed. Holding the infant during feedings, burping frequently, and placing the infant in an infant seat after feeding or on the right side propped with a rolled blanket will aid in a positive outcome for this infant. The mother who has fed her infant preoperatively and has been allowed to assist with feedings during hospitalization will feel more confident after discharge. The immediate improvement as a result of surgery is encouraging to the parents, particularly if the child must have further surgery for cleft palate repair.
Cleft Palate
Pathophysiology: A cleft palate is a failure of the hard palate to fuse at the midline during the seventh to twelfth weeks of gestation. This separation forms a passageway between the nasopharynx and the nose, which not only complicates feeding but also easily leads to infections of the respiratory tract and middle ear that can result in hearing loss. It is generally responsible for speech difficulties that occur in later life. The cleft may not be readily apparent at birth, and for this reason careful examination of the oral cavity and upper palate at birth is essential. Feeding is a problem because the cleft prevents negative pressure from being formed within the mouth, which is necessary for successful sucking.
Treatment: The goals of therapy are union of the cleft, improved feeding, improved speech, improved dental development, and the nurturing of a positive self-image. Some surgeons prefer to operate between 1 year and 18 months of age if at all possible so that speech patterns are minimally affected. If surgery has been deferred, a dental speech appliance may be used to facilitate communication. This appliance must be changed periodically as the child grows.
Treatment of the child with a cleft lip and palate requires multidisciplinary teamwork with a surgeon, pediatrician, pediatric dentist, orthodontist, nurse, psychologist, speech therapist, and social worker. The public health nurse should be responsible for coordinating parental counseling and referral as needed. The emotional problems that sometimes occur with this condition require more extensive attention than does the repair itself. A child born with a facial deformity encounters many problems. Feedings are difficult and are not relaxed in the initial period. As the child grows, irregular tooth eruptions, drooling, delayed speech, and the need for intermittent hospitalization and frequent clinic appointments can be frustrating. High-resolution ultrasound can detect cleft palate by 13 weeks of gestation, and therefore its correction (without scarring) by fetal surgery appears promising for the near future.
 Safety Alert!
Safety Alert!Psychosocial Adjustment of the Family: A mother’s first reaction to a disfigured newborn is one of shock, hurt, disappointment, and guilt. Some parents regard the deformity as a result of their inadequacies. They may desire to hide the child from relatives and friends. The developing child senses the parents’ feelings and acquires either a positive or a negative self-image. The patient and family need understanding, a concrete basis for hope, and practical advice. Family stress often occurs because of the multiple surgeries that may be required throughout childhood.
Follow-up Care and Home Care: In large cities, special cleft palate clinics are available in which several specialists can work together in convenient consultation. The parents are instructed about the resources available in the state in which they live. The American Cleft Palate–Craniofacial Association, the Cleft Palate Foundation, the March of Dimes Birth Defect Foundation, and state programs for children with special needs are examples of community referrals that should be offered to parents.
Postoperative Treatment and Nursing Care:
Nutrition: Fluids are taken by a cup, although a gravity feeder may be desirable in some cases. The method varies with the plastic surgeon. The diet is progressive, at first consisting of clear fluids and then full fluids. By the time of discharge, a soft diet can generally be taken. Hot foods and liquids are avoided to prevent injury to the operative site. The patient must not suck on a straw. When feeding with a spoon, the nurse should place the spoon into the side of the mouth. The spoon must not touch the roof of the mouth. The nurse teaches parents to keep objects such as the child’s thumb, tongue blades, toast, cookies, forks, and pacifiers out of the mouth. Elbow restraints are used to prevent the child from placing his or her fingers or objects in the mouth. The diet is advanced only on consultation with the physician.
Oral hygiene: The mouth is kept clean at all times. Feedings are followed by a little water. The physician may prescribe a mild antiseptic mouthwash.
Speech: It is helpful to speak slowly and distinctly to the child. The child is encouraged to pronounce words correctly. Children who have undergone extensive repairs or have associated deafness need the help of a speech therapist. The speech therapist evaluates the child and assists the parents in specific activities that facilitate speech development.
Diversion: Crying is to be prevented as much as possible. Play should be quiet, particularly in the immediate postoperative period. The nurse reads, draws, or colors with the child.
Complications: Ear infections and dental decay may accompany cleft palate. Parents are instructed to take the child to the health care provider at the first sign of earache. Regular visits to the dentist are scheduled. Throughout the long-term care, a stable goal in the care of this infant is to promote optimal growth and development and to establish positive self-esteem.
Musculoskeletal System
Pathophysiology: Clubfoot, one of the most common deformities of the skeletal system, is a congenital anomaly characterized by a foot that has been twisted inward or outward. The incidence is about 1 in 1000 live births. Many mild forms are caused by improper position in the uterus, and these clear up with manipulative exercises. In contrast, true clubfoot does not respond to simple exercise. Several types are recognized. Talipes (talus, “heel,” and pes, “foot”) equinovarus (equinus, “extension,” and varus, “bent inward”) is seen in 95% of patients. The feet are turned inward, and the child walks on the toes and the outer borders of the feet. It generally involves both feet (Figure 14-8).
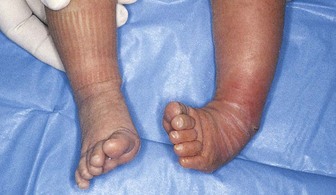
Treatment and Nursing Care: The treatment of clubfoot is started as early as possible or the bones and muscles will continue to develop abnormally. Conservative treatment that consists of splinting or casting to hold the foot in the right position is carried out during infancy. Passive stretching exercises may also be recommended. If these methods are not effective by age 3 months, surgery may be indicated. The infant with a clubfoot is under medical supervision for a long time. Parents must be instructed in developmental behaviors of the infant as well as the clinical aspects of care. Ongoing support is paramount.
Cast care: Casts are made of plaster or synthetic materials such as fiberglass or polyurethane. The plaster cast consists of crinoline that has powdered plaster in its meshwork. It is placed in warm water before being applied over cotton wadding or a stockinette. The wet plaster of Paris hardens as it dries. This type of cast dries from the inside out and takes 24 to 48 hours to dry.
If the patient returns to the unit before the cast is dry, the cast must be left uncovered and protected from pressures that could cause a depression in it. If the physician orders that the leg and foot be elevated on pillows to prevent swelling, the nurse who assists must use the palms of the hands, not the fingers, to lift the cast. Indentations made in a wet cast by fingers can press on the underlying skin and cause damage. This precaution is also explained to parents. Lighter synthetic casts dry in less than 30 minutes; however, they are more expensive and not as strong.
The toes are left exposed for observation. The nurse checks them for capillary refill and signs of poor circulation, pallor, cyanosis, swelling, coldness, numbness, pain, or burning. If circulation is impaired, the physician may split the cast to relieve the pressure, or the cast may need to be removed and reapplied. The nurse also reports irritation of the skin around the edges of the cast and lack of movement of the toes. Adhesive petals may be placed around the edges of the cast to prevent skin irritation.
As the infant grows the cast may need to be removed and reapplied. Because an infant grows rapidly, parents should be taught how to check for circulation impairments, which could be caused by a tight cast.
If surgery on tendons and bones has been performed, the nurse also observes the cast for evidence of bleeding. If a discolored area appears on the cast, it is circled and the time is recorded. Further bleeding can then be estimated. If bleeding is noted, the patient’s vital signs are also checked and compared with preoperative readings. After surgery the cast is changed about every 3 weeks to bring the foot gradually into position. When the cast is removed for the final time, exercise and use of special shoes may be indicated.
Emotional support: The nurse is an important figure in the care of the long-term patient with clubfoot. Nurses review the normal growth and development of children in the patient’s age range to anticipate problems and to educate caretakers in parenting.
Children in a cast may be slow in developing certain motor abilities. Education concerning the therapy and referral for follow-up care is an important nursing responsibility. The financial burdens of hospitalization, surgery, special shoes, and continued medical supervision may pose a serious problem. If the nurse suspects that the parents need financial help, a social service referral is made.
 Nursing Tip
Nursing Tip
In the long-term care of orthopedic patients, educating the parents about orthopedic devices, cast care, exercise, hygiene, and treatment goals is necessary. The nurse explains the importance of frequent clinic visits, reinforces physicians’ information, and clarifies directions as necessary.
Pathophysiology: Developmental hip dysplasia, formerly known as congenital hip dysplasia, is a common orthopedic deformity. The incidence is about 1 in 1000 births. The term hip dysplasia is a broad description applied to various degrees of deformity: subluxation or dislocation, either partial or complete. The head of the femur is partly or completely displaced as a result of a shallow hip socket (acetabulum). Hereditary and environmental factors appear to be causal factors. Hip malformation, joint laxity, breech position, and maternal hormones may all contribute. Developmental hip dysplasia is seven times more common in girls than in boys. Newborn infants seldom have complete dislocation. However, the child beginning to walk exerts pressure on the hip, which can cause complete dislocation. Therefore early detection and treatment are of particular importance.
There is a high risk for developmental hip dysplasia in cultures in which the newborn is wrapped snugly with the hips in adduction and extension. There is a lower risk for developmental hip dysplasia in cultures in which the infant is carried straddled on the mother’s waist with the infant’s hips flexed and widely abducted.
Manifestations: A dislocation of the hip is commonly discovered at the periodic health examination of the infant during the first or second month of life. One of the most reliable signs is a limited abduction of the leg on the affected side. When the infant is placed on the back with knees and hips flexed, the physician can press the thigh of the normal hip backward until it almost touches the examining table. This can be accomplished only partially on the affected side. The knee on the side of the dislocation is lower, and the skin folds of the thigh are deeper and often asymmetrical (Figure 14-9). When the infant is in a prone position, one buttock appears higher than the other.
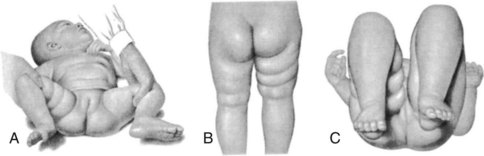
FIGURE 14-9 Early signs of dislocation of the right hip. A, Limitation of abduction. B, Asymmetry of skin folds. C, Shortening of femur.
Barlow’s test is performed by a physician to detect an unstable hip in the newborn. The physician adducts and extends the hips while stabilizing the pelvis and may “feel” the dislocation occur as the femur leaves the acetabulum.
In infants aged 1 to 2 months with developmental dislocation of the hip, the physician can actually feel and hear the femoral head slip back into the acetabulum under gentle pressure. This is called Ortolani’s sign or Ortolani’s click and is also considered diagnostic of the disorder. The child who is walking and has had no treatment displays a characteristic limp. Bilateral (bi, “two,” and latus, “side”) dislocation may occur; however, unilateral (uni, “one,” and latus, “side”) dislocation is more common. Radiographic studies confirm the diagnosis.
Treatment: Treatment begins immediately on detection of the dislocation. The hips are maintained in constant flexion and abduction for 4 to 8 weeks to keep the head of the femur within the hip socket. This constant pressure enlarges and deepens the acetabulum; thus, it can correct the dislocation.
A triple-thick diaper may be used in the newborn to maintain a “froglike” abduction of the legs until a Pavlik harness can be fitted properly (Kliegman et al., 2007). In infants more than 2 months of age, soft tissue contractures prevent stabilization of the hip, and longer-term immobilization with a Pavlik harness may be required (Figure 14-10). Traction may be necessary if the dislocation is severe or is not detected until the child begins to walk. This pulls the head of the femur down to the correct position opposite the acetabulum and helps to overcome muscle spasm. Casting in a froglike position is then done. This type of cast, known as a body spica cast, is shown in Figure 14-11. (See Chapter 24 for care of the orthopedic patient.)
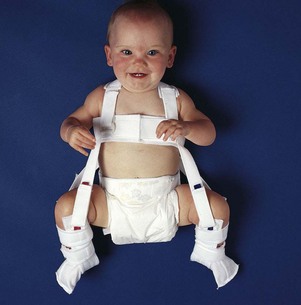
FIGURE 14-10 The Pavlik harness is used with infants aged 1 to 6 months to maintain hips in a position of flexion and abduction.
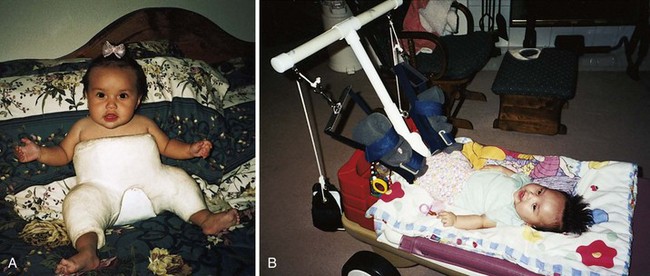
FIGURE 14-11 A, Infant in a spica body cast. This cast maintains the legs in a froglike position and is used to treat developmental hip dysplasia. Note that the infant is able to move her toes freely. A diaper tucked inside prevents the cast from becoming soiled with urine or feces. B, Traction is sometimes necessary before surgery or casting. Home care enables the child to be in familiar surroundings that will nurture growth and development.
The length of time spent in a cast varies according to the patient’s progress and growth and the condition of the cast; however, it is usually 5 to 9 months. During this time the cast may be changed about every 6 weeks. Surgery may be required in infants more than 18 months of age. In such cases, open reduction of the dislocation or repair of the shelf of the hip bone is performed. After surgery a cast is applied to keep the femur in the correct position.
Nursing care: The nursery nurse carefully observes each infant during the bath to detect signs of a hip dysplasia:
• When the infant is prone, the nurse observes the buttocks for variation in size.
• The legs of the infant should be equal in length.
• The infant should be kicking both legs, not just one leg.
• The depth and number of skin folds of the infant’s upper thighs should be symmetrical.
• In the well-baby clinic the nurse notes the posture and gait of older children and records observations.
Infants who progress well with the Pavlik harness or Frejka splint (or a similar brace) remain at home with regular visits to the physician. The parents need assurance that the infant may be held and may sit in a chair. They should be encouraged to ask questions of the clinic nurse and physician.
The child who is admitted to the hospital with a diagnosis of developmental hip dysplasia is given as much personal attention as possible. The first admission sets the pattern for future hospitalization; therefore, the child must make a satisfactory adjustment. The nurse becomes familiar with the child’s habits and plan of care. Every effort is made to provide a homelike environment for children who are hospitalized for many weeks.
The spica cast: The body spica cast encircles the waist and extends to the ankles or toes. Neurovascular assessment, discussed in Chapter 24, should be reviewed at this point. Nursing Care Plan 14-1 gives selected nursing diagnoses and interventions for the patient with a spica cast.
14-1  Nursing Care Plan
Nursing Care Plan
The Infant or Child with a Spica Cast
An infant returns from the cast room after having a spica cast applied. The infant is scheduled for discharge after the cast is dry.

Risk for injury, related to awkwardness and weight of cast
| Goals | Nursing Interventions | Rationales |
| Patient will remain safe and as independent as possible. | Inform older child when turning as to how and when you are going to proceed (e.g., “ready, set, go”). | Involving child in procedure, as age appropriate, gives him or her a sense of control; procedure will go more smoothly. |
| Leave articles and toys within reach. | Child will not need to strain or move awkwardly to reach articles; patient will feel greater control if articles can be obtained independently. | |
| Some car seats are adapted to accommodate a small child in a spica cast. | These children need protection in a car. |
1. A mother brings her 2-year-old child to the clinic complaining that the spica cast has a strong, unpleasant odor. The child has a temperature of 37.8° C (100° F). He appears to be in no acute distress and is playing with a small toy car in one hand and has a half-eaten cracker in the other hand. What nursing intervention is indicated?
Firm, plastic-covered pillows are required. These are placed beneath the curvatures of the cast for support. Older children may benefit from an overhead bar and trapeze. The room should be adequately ventilated. A fracture pan should be available at the bedside for toileting as developmentally appropriate.
The head of the patient’s bed is slightly elevated so that urine or feces drain away from the body of the cast. One should not use pillows to elevate the head or shoulders of a child in a body cast, because this thrusts the patient’s chest against the cast and causes discomfort or respiratory difficulty. The child who is not toilet trained may be placed on a Bradford frame to facilitate nursing care. Frequent changes of position are important; immobilized patients must be turned often. Infants may be held in the nurse’s lap after the cast has dried. A ride on a wagon or gurney to the playroom or around the hospital provides changes of position and scenery.
The supporting bar between the legs should not be used as a lever when turning the child (Skill 14-1). All body curvatures are supported with pillows or sheet rolls. Whenever possible, the older child should be on the abdomen during mealtime to facilitate swallowing and self-feeding. When placing a child in a body cast on a fracture pan, the upper back and legs are supported with pillows so that body alignment is maintained.
Skill 14-1 Technique for Turning the Child in a Body Cast








Two people, one on each side of the bed, are needed to turn a child in a body cast, as follows:
1. Move the child to the edge of the bed as far as possible so that the nurse who will receive the child is farther away from him or her.
2. The nurse nearer to the child places one hand under the head and back and one hand under the leg part of the cast and turns the child to the midway point on the side.
3. The nurse farther away then accepts the support of the child and cast as turning is completed.
Itching is a problem for the patient in a body cast. If at all possible, a strip of gauze is placed beneath the cast before it is applied; this gauze extends through the opened area required for toilet needs. It is gently moved back and forth to relieve itching. When the strip becomes soiled, a clean one is tied to one end of the soiled gauze and pulled through the cast; this soiled portion is then removed. Other methods to relieve itch that might cause injury to the skin beneath the cast are discouraged because any break in the skin under a cast is difficult to heal.
Toys small enough to be “hidden” inside the cast should not be given to the child. Toys that can be used when the child is in a prone position are best.
The child with this long-term disability requires help in meeting his or her everyday needs. This child is growing and developing rapidly, and therefore frequent adjustments in home and clinic care are necessary. Dressing and clothing are a problem. The child cannot fit into regular furniture and much of the play equipment enjoyed by other children. Transportation is difficult. A special wagon built up with pillows may be used (see Figure 14-11). The child should be included in everyday family and play activities to encourage normal growth and development. A referral for home health care should be made on discharge.
Metabolic Defects
The infant with an inborn error of metabolism has a genetic defect that may not be apparent before birth. As the infant adjusts to the birth process and begins to ingest nourishment, symptoms can rapidly emerge that quickly become life threatening. Symptoms such as lethargy, poor feeding, hypotonia, a unique odor to the body or urine, tachypnea, and vomiting must be reported by the nurse in the newborn nursery to prevent long-term or life-threatening sequelae. The nurse must also be prepared to offer psychological support and help parents deal with the impact of having an infant with a genetic problem.
Phenylketonuria
Pathophysiology: Classic phenylketonuria (PKU) is a genetic disorder caused by the faulty metabolism of phenylalanine, an amino acid that is essential to life and is found in all protein foods. This inborn error of metabolism, which is transmitted by an autosomal recessive gene, is termed classic PKU and is associated with blood phenylalanine levels above 20 mg/dL. The hepatic enzyme phenylalanine hydrolase, which is normally needed to convert phenylalanine into tyrosine, is missing. When the infant is fed formula, phenylalanine begins to accumulate in the blood. It can increase to as high as 20 times the normal amount. Its by-product, phenylpyruvic acid, appears in the urine within the first weeks of life.
Classic PKU results in severe retardation that is evidenced in infancy. Early detection and treatment are paramount. By the time the urine test is positive, brain damage has already occurred. The infant appears normal at birth but begins to show delayed development at about 4 to 6 months of age. The child may show evidence of failure to thrive, have eczema or other skin conditions, have a peculiar musty odor, or have personality disorders. About one third of the children have seizures. PKU occurs mainly in blonde and blue-eyed children; these features result from a lack of tyrosine, a necessary component of the pigment melanin. Less severe forms of the disorder are now recognized. They are designated as “atypical PKU” and “mild hyperphenylalaninemia.”
Diagnosis: The Guthrie blood test is widely used and is currently considered the most reliable test for PKU. Blood is obtained from a simple heel stick. A few drops of capillary blood are placed on filter paper and mailed to the laboratory for screening. It is recommended that the blood be obtained after 48 to 72 hours of life, preferably after the ingestion of proteins, to reduce the possibility of false-negative results. Many states require that the test be done on all newborns before they leave the nursery, but because of early discharge the test may be repeated within 2 weeks. The infant can be tested at home by a public health nurse or at the clinic or physician’s office. Confirmation of the diagnosis requires quantitative elevations of phenylalanine compound in the blood (Kliegman et al., 2007). Screening programs for pregnant women have also been advocated to detect elevated phenylalanine levels that could have an effect on the newborn.
Treatment and Nursing Care: Treatment of PKU consists of close dietary management and frequent evaluation of blood phenylalanine levels. Because phenylalanine is found in all natural protein foods, a food that provides enough protein for growth and tissue repair but little phenylalanine must be substituted. The most commonly used formulas are Lofenalac or Phenex-1 for infants, Phenyl-Free for children and Phenex-2 for adolescents. The goals of the diet are to provide enough essential proteins to support growth and development while maintaining phenylalanine blood levels between 2 and 10 mg/dL. A phenylalanine level below 2 mg/dL may result in growth retardation, whereas levels above 10 mg/dL can result in significant brain damage.
There is a low phenylalanine content in breast milk, and infants can be partially breastfed and supplemented with Lofenalac while phenylalanine blood levels are monitored. Solid foods that are low in phenylalanine are added at the same age that solid foods are added for infants without PKU. Phenyl-Free is introduced between ages 3 and 8 years. Cookbooks and family recipes provide ideas for variety. Eventually the child learns to assume full management of the diet.
A dietitian may be consulted concerning parental guidance and support in maintaining the dietary regimen, especially for the school-aged child and adolescent. Many foods that contain a high level of phenylalanine are clearly labeled and provide easier choices for parents at the supermarket. A single 12-oz can of diet cola containing NutraSweet or Equal (aspartame) will not significantly raise blood levels of phenylalanine, but the intake of most meat, dairy products, and diet drinks must be restricted. An exchange list for food selection can aid the child in participating in and monitoring his or her progress. Flavoring the milk substitute with a fruit-flavored powder or chocolate flavoring can increase the child’s compliance. Sapropterin dihydrochloride (Kuvan), a new drug approved in 2008, is the first on the market to treat this inherited disorder but there is little research data concerning its success (Hussar, 2008).
Genetic counseling is important for the affected child for future family planning. Women of childbearing age who have PKU must follow a low-phenylalanine diet before conception to prevent brain damage of the fetus during development. Phenylalanine levels greater than 6 mg/dL in pregnant women can affect development of the embryo.
Maple Syrup Urine Disease
Pathophysiology: Maple syrup urine disease is caused by a defect in the metabolism of branched-chain amino acids. It causes marked serum elevations of leucine, isoleucine, and valine. This results in acidosis, cerebral degeneration, and death within 2 weeks if left untreated.
Manifestations: The infant with maple syrup urine disease appears healthy at birth but soon develops feeding difficulties, loss of the Moro reflex, hypotonia, irregular respirations, and convulsions. The infant’s urine, sweat, and cerumen (earwax) have a characteristic sweet or maple syrup odor. This is caused by ketoacidosis, a process similar to that which may occur in diabetic children, and causes a fruity odor of the breath. However, the condition does not resolve with the correction of blood glucose levels. The urine contains high levels of leucine, isoleucine, and valine. Diagnosis is confirmed by blood and urine tests.
Treatment and Nursing Care: Early detection in the newborn period is extremely important. The nursery nurse should report any newborn whose urine has a sweet aroma. Initial treatment consists of removing these amino acids and their metabolites from the tissues of the body. This is accomplished by hydration and peritoneal dialysis to decrease serum levels. The patient is placed on a lifelong diet low in the amino acids leucine, isoleucine, and valine. Several formulas specifically for this disease are available. Exacerbations are most often related to the degree to which the leucine level is abnormal. These exacerbations are frequently related to infection and can be life threatening. The nurse must frequently assess the patient and instruct parents about the need to prevent infections.
Galactosemia
Pathophysiology: In galactosemia the body is unable to use the carbohydrates galactose and lactose. In the healthy person the liver converts galactose to glucose. In the patient with galactosemia, an enzyme is defective or missing and therefore there is a disturbance in a normally occurring chemical reaction. The result is an increase in the amount of galactose in the blood (galactosemia) and in the urine (galactosuria). This can cause cirrhosis of the liver, cataracts, and mental retardation if left untreated. Because galactose is present in milk sugar, early diagnosis is necessary so that a milk substitute can be used.
Manifestations: The symptoms of galactosemia begin abruptly and worsen gradually. Early signs consist of lethargy, vomiting, hypotonia, diarrhea, and failure to thrive. These commence as the newborn begins breastfeeding or ingesting formula. Jaundice may be present. Diagnosis is made by observing galactosuria, galactosemia, and evidence of decreased enzyme activity in the red blood cells. Screening tests are available.
Treatment and Nursing Care: Milk and lactose-containing products are eliminated from the diet of the patient with galactosemia. The nursing mother must discontinue breastfeeding. Lactose-free formulas and those with a soy-protein base are often substituted. The nurse must realize the frustration and anxiety that this diagnosis creates. Parents experience periods of feeling overwhelmed and inadequate. They can also become totally absorbed in the dietary program. A rare disease creates feelings of isolation and uncertainty. Because surveillance is ongoing, some of the emotional characteristics of the family with a child who has a chronic disease are pertinent.
Chromosomal Abnormalities
Pathophysiology: Down syndrome is one of the most common chromosomal abnormalities. Its incidence is approximately 9 in 10,000 live births. It may increase to 1 in 365 live births among children of mothers age 35 years or older (Kliegman et al., 2007). Paternal age is also a factor, particularly when the father is age 55 years or older. Sometimes the first infant of a young mother has Down syndrome, but subsequent children are usually born free of the defect. Children born with this birth defect have mild to severe mental retardation and generally some physical abnormalities. In the past, children with this condition were called “mongoloid” because of the Oriental (“Mongolian”) appearance of their faces, but this term is now considered inappropriate.
There are three phenotypes (genetic makeups) of Down syndrome: trisomy 21, mosaicism, and translocation of a chromosome. The most common type, trisomy 21 syndrome, accounts for 95% of patients. In this instance there are three number 21 chromosomes rather than the normal two. This is a result of nondisjunction, the failure of a chromosome to follow the normal separation process into daughter cells. The earlier in the embryo’s development this occurs, the greater the number of cells affected. When nondisjunction occurs late in development, both normal and abnormal cells are present in the newborn. This condition is mosaicism, and patients tend to be less severely affected in physical appearance and intelligence. The third condition is translocation. In translocation, a piece of chromosome in pair 21 breaks away and attaches itself to another chromosome.
Screening for Down syndrome is offered during the first trimester of pregnancy and includes an ultrasound assessment of the thickness of the fetal nuchal fold (called nuchal translucency) and absence of the nasal bone. This early screening allows parents to discuss options of terminating or continuing the pregnancy with preparation for the outcome. A second trimester “quad test” involves testing of the blood for levels of AFP, unconjugated estriol (UE), inhibin-A (a placental hormone), and human chorionic gonadotropin (hCG) (see Chapter 5). A low AFP or a high hCG and inhibin-A with a low UE may indicate a high risk for Down syndrome in the developing fetus. A test of pregnancy-associated plasma protein A (PAPP-A) may also indicate a risk for Down syndrome. Positive tests in the first or second trimester may indicate the need for amniocentesis to confirm the diagnosis.
Manifestations: Down syndrome can be diagnosed by the clinical manifestations, but a chromosomal analysis will confirm the specific type. The signs of Down syndrome, which are apparent at birth, are close-set and upward-slanting eyes, small head, round face, flat nose, protruding tongue that interferes with sucking, and mouth breathing (Figure 14-12, A). There is a deep straight line across the palm, which is called the simian crease (Figure 14-12, B). The hands of the infant are short and thick, and the little finger is curved (Figure 14-12, C). There is also a wide space between the first and the second toes. The undeveloped muscles and loose joints enable the child to assume unusual positions. Physical growth and development may be slower than normal (Tables 14-1 and 14-2). The child is limited intellectually. Some children have been found to have intelligence quotients (IQs) in the borderline to low-average range. Congenital heart deformities are also associated with this condition.
Table 14-1
Time of Occurrence of Developmental Milestones in Normal Children and Those with Down Syndrome (in Months)
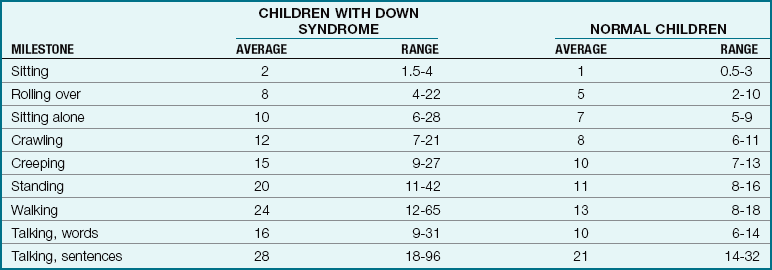
From Levine, M.D., Carey, W.B., & Crocker, A.C. (2003). Developmental-behavioral pediatrics (4th ed.). Philadelphia: Saunders.
Table 14-2
Time of Occurrence of Self-Help Skills in Normal Children and Those with Down Syndrome (in Months)
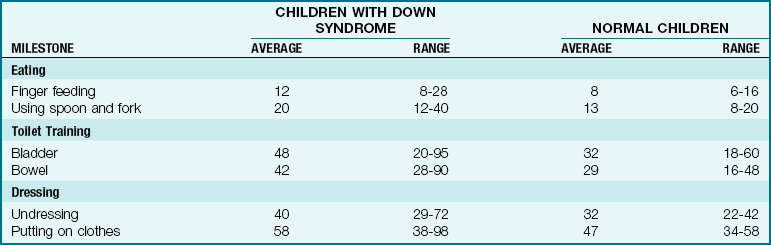
From Levine, M.D., Carey, W.B., & Crocker, A.C. (2003). Developmental-behavioral pediatrics (4th ed.). Philadelphia: Saunders.
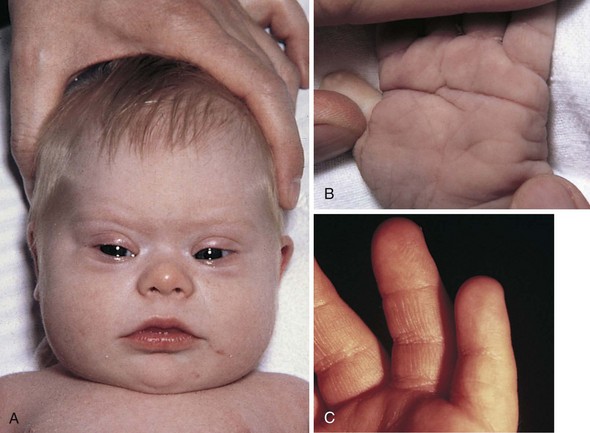
FIGURE 14-12 Down syndrome. A, The typical facial appearance of an infant with Down syndrome shows the upward slant of the canthal folds of the eyes, protruding tongue, and short, thick neck. B, The straight simian crease in the palm of the hand is a typical finding in children with Down syndrome. C, The short fifth finger is a typical finding in children with Down syndrome. The tip of the fifth finger does not extend to the distal joint of the adjoining finger.
Children with Down syndrome are very lovable. They may be restless and somewhat more difficult to train than the normal youngster. Their resistance to infection is poor, and they are prone to respiratory and ear infections as well as speech and hearing problems. The life span of children with Down syndrome has increased with the widespread use of antibiotics. The incidence of acute leukemia is higher in these children than in the normal population, and Alzheimer’s disease is common to those who reach middle adult life (Jackson et al., 2009).
The limp, flaccid posture of the infant is caused by hypotonicity of the muscles; it makes positioning and holding more difficult and contributes to heat loss from the exposed surface areas. The infant should be warmly wrapped to prevent chilling. The hypotonicity of muscles also causes respiratory problems and excess mucus accumulation. Bulb suctioning may be necessary before feedings. In addition, the hypotonicity of muscles contributes to the development of constipation, which can be controlled by dietary intervention.
Counseling parents: The counseling of families of Down syndrome children is ongoing. Maternity nurses must be aware of their own feelings before they can effectively support parents. They, too, will feel saddened at the birth of an imperfect child. They may identify with the parents. It is appropriate to express one’s feelings of initial helplessness, and it may encourage the parents to verbalize their concerns. The nurse must listen and provide honest, tactful, and compassionate support.
Empathy from the nurse is particularly important. Involving parents in the care and planning for the infant from the start facilitates bonding. The need for the staff’s warm concern cannot be overestimated. Pampering the infant by putting a little curl in the hair, for example, shows that others care.
Counseling family: Siblings of the patient must be informed and included in discussions about the newborn. Even very young children are aware of parental distress, and what they imagine can be more frightening than the reality. Open communications early on will prevent isolation and misconceptions and promote an easier transition period. The nurse should connect the family with a Down syndrome support group in their area if there is one. Other parents with a Down syndrome child are an important resource. The National Association for Down Syndrome is one organization that provides education and support to families. (See Chapter 23 for further discussion of nursing care of the cognitively impaired child.)
Perinatal Injuries
Hemolytic Disease of the Newborn: Erythroblastosis Fetalis
Pathophysiology: Erythroblastosis fetalis (erythro, “red,” blast, “a formative cell,” and osis, “disease condition”) is a disorder that becomes apparent in fetal life or soon after birth. It is one of many congenital hemolytic diseases found in the newborn. It is caused when an Rh-negative mother and an Rh-positive father produce an Rh-positive fetus. Although fetal and maternal blood does not mix during pregnancy, small leaks may allow fetal blood to enter the maternal circulation and sensitize the mother. The mother’s body responds by producing antibodies that cross the placenta and destroy the blood cells of the fetus, causing anemia and possible heart failure (see Chapter 5). The terms isoimmunization and sensitization refer to this process (Box 14-2). The incidence of erythroblastosis fetalis has greatly decreased as a result of the protective administration of an Rh immune globulin (RhoGAM) to women at risk (see following discussion of prevention). Incompatibility of ABO factors is now more common and generally less severe than Rh incompatibility (Box 14-3).
The process of maternal sensitization is depicted in Figure 14-13. The mother accumulates antibodies with each pregnancy; therefore the chance that complications may occur increases with each gestation. If large numbers of antibodies are present, the infant may be severely anemic. In the gravest form, hydrops fetalis, the progressive hemolysis causes anemia, heart failure, fetal hypoxia, and anasarca (generalized edema). This is rare today because of early detection methods.
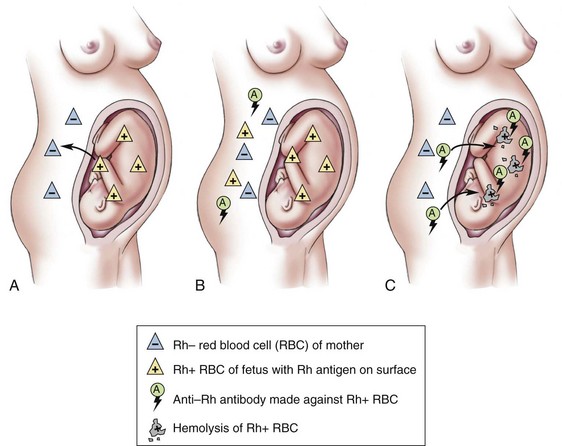
FIGURE 14-13 Maternal sensitization producing erythroblastosis in the newborn. A, During the first pregnancy, the mother is sensitized to the Rh-positive antigen from the fetus. B, The mother produces Rh antibodies to the Rh antigen to which she was exposed. C, During a second pregnancy, these Rh antibodies cross the placenta to the fetus and destroy the fetal Rh-positive blood cells.
Diagnosis and Prevention: An extensive maternal health history is obtained. Of particular interest are previous Rh sensitizations, an ectopic pregnancy, abortion, blood transfusions, or children who developed jaundice or anemia during the neonatal period. The mother’s blood titer is carefully monitored. An indirect Coombs’ test on the mother’s blood will indicate previous exposure to Rh-positive antigens.
Diagnosis of the disease in the prenatal period is confirmed by amniocentesis and monitoring of bilirubin levels in the amniotic fluid. Information gained from these tests helps to determine the necessity of early interventions such as induction of labor or intrauterine fetal transfusions that allow the fetus to remain in utero until the lungs mature. Repeated transfusions may be required (Kliegman et al., 2007). Fetal Rho(D) status can be determined noninvasively via free DNA in maternal plasma, and this new technique can be used in other areas of genetic testing (Moise, 2005a).
Prevention of erythroblastosis by the use of Rho(D) immune globulin (RhoGAM) is now routine. An intramuscular injection is given to the mother within 72 hours of delivery of an Rh-positive infant. RhoGAM may also be given to the pregnant woman at 28 weeks of gestation. It is also administered, when appropriate, after a spontaneous or therapeutic abortion, after amniocentesis, and to women who have bleeding during pregnancy, because fetal blood may leak into the mother’s circulation at these times.
Manifestations: At the time of delivery, a sample of the infant’s cord blood is sent to the laboratory. The direct Coombs’ test detects damaging antibodies. The symptoms of erythroblastosis fetalis vary with the intensity of the disease. Anemia and jaundice are present. The anemia is caused by hemolysis of large numbers of erythrocytes. This pathological jaundice differs from physiological jaundice in that it becomes evident within 24 hours after delivery. The liver is unable to handle the massive hemolysis, and bilirubin levels rise rapidly, causing hyperbilirubinemia (hyper, “excess,” bilis, “bile,” rubor, “red,” and emia, “blood”). Early jaundice is immediately reported to the physician. Techniques of assessing for jaundice are discussed in Chapter 12.
Enlargement of the liver and spleen and extensive edema may develop. The circulating blood usually contains an excess of immature nucleated red blood cells (erythroblasts) caused by the infant’s attempts to compensate for the destruction of cells. The oxygen-carrying power of the blood is diminished, as is the blood volume, and therefore shock or heart failure may result. Jaundice (with symptoms such as irritability, lethargy, poor feeding, and high-pitched shrill cry), muscle weakness progressing to opisthotonos positioning (arched back), and seizures are indications of bilirubin toxicity that could lead to kernicterus. Kernicterus (accumulation of bilirubin in the brain tissues) may cause serious brain damage and permanent disability.
Treatment and Nursing Care: Treatment includes prompt identification, laboratory tests, drug therapy, phototherapy, and exchange transfusion if indicated. Phototherapy may be used to reduce serum bilirubin levels. It may be used alone or in conjunction with an exchange transfusion. The newborn is placed in an incubator under a bank of fluorescent lights (Figure 14-14, A). The eyes are protected from the lights (Figure 14-14, B), and specific protocols are carried out. Intensive phototherapy may be provided by placing the fluorescent lights within 10 cm of a naked term infant in a bassinet rather than an incubator. (Light sources from tungsten, halogen, or spot lamps involve high heat and can burn the skin at close range.) Intensive phototherapy for a premature infant may involve standard light sources above an incubator and a fiberoptic pad under the infant. A term infant may require two or three fiberoptic pads under his or her body (Maisels, 2005b). Lining the sides of the incubator with aluminum foil or a white sheet may increase the effectiveness of intensive phototherapy treatment. Phototherapy is usually discontinued when the bilirubin level declines to 14 mg/dL.
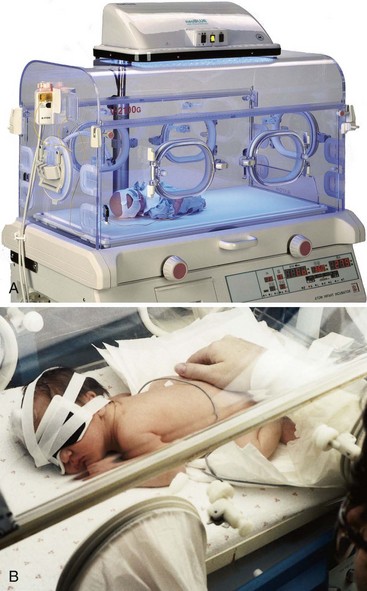
FIGURE 14-14 Phototherapy. A, The neoBLUE provides a high-intensity, narrow band of blue light that helps break down excess bilirubin. A flip of a switch allows change from conventional to intense phototherapy treatment. Note: In the clinical setting, the infant would be diapered only, not fully clothed. B, When an infant receives phototherapy in an incubator, the eyes are protected from the fluorescent lights. The infant is turned frequently so that all skin surfaces are exposed to the lights. (A, Photo courtesy Natus Medical Inc., San Carlos, Calif.)
Phototherapy is contraindicated in infants with a history of congenital porphyria (a metabolic disorder involving sensitivity to light) or the use of photosensitizing drugs. Although phototherapy may prevent an increase in the level of bilirubin, it has no effect on the underlying cause of jaundice. The nursing care of the infant receiving phototherapy is presented in Nursing Care Plan 14-2. If phototherapy fails to keep the total serum bilirubin at acceptable levels, an exchange transfusion may be indicated.
14-2 Nursing Care Plan
The Infant Receiving Phototherapy
A newborn, 12 hours of age, is diagnosed with hyperbilirubinemia and placed in an incubator for phototherapy.







During an exchange transfusion, a plastic catheter is inserted into the umbilical vein of the newborn, small amounts of blood (10 to 20 mL) are withdrawn, and equal amounts of Rh-negative blood are injected. The amount of donor blood used is about twice the infant blood volume to a limit of 500 mL. In this way, healthy cells are added to the infant’s blood, and antibodies are removed. Additional small transfusions may be necessary later. After a second exchange transfusion, approximately 85% of the infant’s blood will have been replaced. Antibiotics may be given to prevent infection.
Nursing Tip
An infant born with cardiac failure and edema as a result of hemolytic disease is a candidate for immediate exchange transfusion with fresh whole blood.
The nurse is usually responsible for the following: observing the newborn’s color and reporting any evidence of jaundice during the first and second days; stressing to mothers the importance of good prenatal care for subsequent pregnancies; helping to interpret the treatment to parents by giving reassurance as needed; and observing and assisting the physician with the exchange transfusion. A drug, TIN-mesoporphyrin, is under study for approval by the U.S. Food and Drug Administration (FDA). This drug is designed to treat hyperbilirubinemia, eliminating the need for exchange transfusions and perhaps even phototherapy. At present, the drug is reserved for use in extremely low-birth-weight infants who may experience neurological complications during phototherapy (Wong et al., 2007).
Home Phototherapy: Home phototherapy programs are being used for newborns with mild to moderate physiological (normal) jaundice. These programs are advocated because bilirubin levels generally begin to increase on the third day after birth, when the mother and newborn are discharged. An increase in bilirubin levels may necessitate the newborn’s return to the hospital and possible separation of mother and infant. Home therapy is less costly. A referral for home care is made by the infant’s pediatrician on the basis of the newborn’s health, bilirubin levels (generally between 10 and 14 mg/dL), evidence of jaundice, and suitability of the family for complying with the home program.
A phototherapy blanket in a bassinet (Figure 14-15) or a fiberoptic pad (Figure 14-16) can be used. These allow for holding the infant and decrease the risk of eye damage. Written instructions are given to the parents. Parents keep a daily record of their infant’s temperature, weight, intake and output, stools, and feedings (see Nursing Care Plan 14-2). Phototherapy tips are listed in Box 14-4.
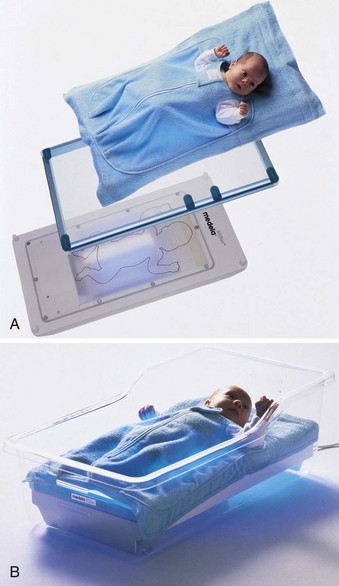
FIGURE 14-15 Phototherapy Bilibed. A, The infant is diapered and placed in the therapy blanket, which fits on a light-permeable infant support. This plastic support is placed over the irradiation unit, which fits into the standard bassinet instead of the mattress. B, The infant in the Bilibed can room-in with the mother and requires no eye-patch protection. The therapeutic light focuses directly on the infant’s skin through the underside of the phototherapy blanket. (Photos courtesy Medela.)
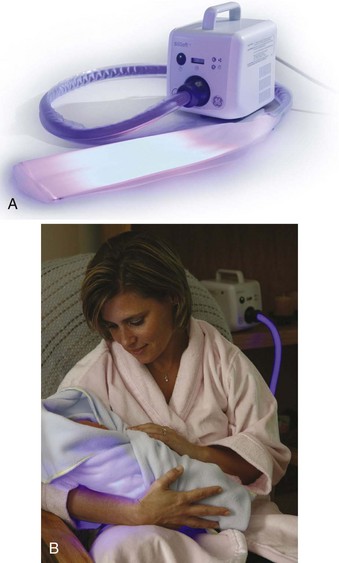
FIGURE 14-16 Biliblanket Plus High Output Phototherapy System. A, A pad of woven fibers is used to transport light from a light source to the infant. This fiberoptic pad is wrapped directly on the infant’s skin to bathe the skin in light. B, The infant can then be diapered, clothed, held, and nursed during treatment at home. (Photos courtesy Medela.)
Intracranial Hemorrhage
Pathophysiology: Intracranial hemorrhage, the most common type of birth injury, may result from trauma or anoxia. It occurs more often in the preterm infant, whose blood vessels are fragile. Blood vessels within the skull are broken, and bleeding into the brain occurs. When the diagnosis is made, the specific location of the hemorrhage may be noted (i.e., subdural, subarachnoid, or intraventricular). This injury may also occur during precipitate delivery or prolonged labor or when the newborn’s head is large in comparison with the mother’s pelvis.
Manifestations: The signs of intracranial hemorrhage may occur suddenly or gradually. Some signs or all signs may be present depending on the severity of the hemorrhage, and can include poor muscle tone, lethargy, poor sucking reflex, respiratory distress, cyanosis, twitching, forceful vomiting, a high-pitched shrill cry, and convulsions. Opisthotonic posturing may be observed (see Figure 23-8). The fontanelle may be tense and under pressure rather than soft and compressible. The pupil of one eye is likely to be small (constricted) and the other large (dilated).
If the symptoms are mild, most patients have a good chance of complete recovery. Death results if there is a massive hemorrhage. The infant who survives an extensive hemorrhage may suffer residual effects such as mental retardation or cerebral palsy. The diagnosis is established by the history of the delivery, CT scan, MRI, evidence of an increase in ICP, and the symptoms and course of the disease.
Treatment and Nursing Care: The newborn is placed in an incubator, which allows proper temperature control, ease in administering oxygen, and continuous observation (see Chapter 13). The infant is handled gently and as little as possible. The head is elevated. The physician may prescribe vitamin K to control bleeding and phenobarbital if twitching or convulsions are apparent. Prophylactic antibiotics and vitamins may be used. The infant is fed carefully because the sucking reflex may be affected. The infant vomits easily. The nurse observes the infant for signs of increased ICP (see Chapter 23) and convulsions and assists the physician with procedures such as lumbar punctures and aspiration of subdural hemorrhage. Performing neurochecks, monitoring vital signs and head circumference, and palpating fontanelles are essential.
If a convulsion occurs, observation of its character aids the physician in diagnosing the exact location of the bleeding. The following are of particular importance: Were the arms, legs, or face involved? Was the right or left side of the body involved? Was the convulsion mild or severe? How long did it last? What was the condition of the infant before and after the seizure? The nurse records observations and notifies the team leader or health care provider.
Transient Tachypnea of the Newborn
Transient tachypnea of the newborn (TTN) usually occurs after a cesarean section birth or an uneventful vaginal delivery of a term infant. It is characterized by tachypnea (rapid respirations) and may also include chest retractions, grunting, and mild cyanosis. The condition is often referred to as respiratory distress syndrome, type II (type I is discussed in Chapter 13). The distinctive feature of this condition is that it typically resolves suddenly after 3 days. TTN is thought to be caused by slow absorption of the fluid in the lungs after birth. Treatment is supportive, providing warmth, energy conservation, and supplemental oxygen.
Meconium Aspiration Syndrome
Meconium aspiration syndrome (MAS) is a group of symptoms that occur when the fetus or newborn aspirates meconium-stained amniotic fluid into the lungs.
In utero, the fetus often expels some meconium into the amniotic fluid during the prolonged labor process, especially if there is cord compression or another condition that temporarily interrupts fetal circulation. If asphyxia and acidosis occur in utero, the fetus may make gasping movements that draw meconium-stained amniotic fluid into the lungs. This condition can also occur when the infant takes its first breath before the nose and mouth are suctioned and meconium-stained amniotic fluid present in the upper airway passages is drawn into the lungs. Meconium aspiration can be prevented by giving the mother amnioinfusion (see Chapter 8) when fetal distress occurs with loss of amniotic fluid and by suctioning the nose and mouth of the infant as soon as the head is delivered, before the infant takes its first breath. Respiratory distress is the primary symptom of MAS, including nasal flaring, retractions, cyanosis, grunting, rales, and rhonchi. The tachypnea may persist for several weeks. Treatment includes supportive care with warmth, supplemental oxygen, and energy-conserving plans of care. Intubation and mechanical ventilation may be necessary, and the infant is transferred to the neonatal intensive care unit (NICU).
Neonatal Abstinence Syndrome
Neonatal abstinence syndrome occurs when the fetus has prenatal exposure to drugs such as opiates, amphetamines, or tranquilizers or multiple illicit drugs while in utero. Because these drugs cross the placenta, the infant born to an addicted mother is physiologically dependent on the drugs and suffers withdrawal symptoms after birth and possible long-term developmental and neurological deficits. Body tremors and hyperirritability are the principal signs of this condition in the newborn. Wakefulness, diarrhea, poor feeding, sneezing, and yawning may also be present. Meconium testing may be more accurate than neonatal urine testing for the presence of drugs (Kliegman et al., 2007). Treatment includes providing a quiet environment with swaddling (see Skill 12-3), reduction of external stimuli, and close observation for seizures. Drugs such as phenobarbital, paregoric, and tincture of opium and intravenous fluids are often prescribed. Fetal alcohol syndrome (FAS) and parent teaching are discussed in Chapter 5.
Infant of a Diabetic Mother
Diabetes in the mother presents various problems for the newborn. These are determined by the severity and duration of the disease in the mother, the degree of control of her condition, and the gestational age of the infant. Diabetes in pregnancy is discussed in Chapter 5. When diabetes of the mother is under good control from conception and throughout pregnancy, the adverse effects on the newborn infant are minimal.
Many newborn infants of diabetic mothers have serious complications. When the mother is hyperglycemic, large amounts of glucose are transferred to the fetus. This makes the fetus hyperglycemic. In response, the fetal pancreas (islet cells) produces large amounts of fetal insulin. Hyperinsulinism, along with excess production of protein and fatty acids, often results in a newborn infant who weighs more than 4082 g (9 lb). Such an infant is designated “large for gestational age” (LGA), and this condition is termed macrosomia (macro, “large,” and soma, “body”). This infant is prone to injuries at birth because of his or her size (Figure 14-17).
FIGURE 14-17 Macrosomia. A newborn with macrosomia caused by maternal diabetes mellitus during pregnancy. This infant weighed 5.5 kg (11 lb) at birth. Macrosomic infants often have respiratory disorders and other problems. (Courtesy Pat Spier, RN-C.)
After delivery the infant often has low blood glucose levels because of the abrupt loss of maternal glucose and hypertrophy of the pancreatic islet cells, which results in a temporary overproduction of insulin. The infant has a characteristic cushingoid appearance because of increased subcutaneous fat. The face is round and appears puffy, and the infant appears lethargic. The size of these newborn infants makes them appear healthy, but this is deceptive because they often have developmental deficits and may suffer complications of respiratory distress syndrome (RDS) or congenital anomalies. In contrast, some infants born to a mother with severe diabetes may be small for gestational age (SGA) because of poor placental perfusion. These infants suffer from hypoglycemia, hypocalcemia, and hyperbilirubinemia.
The nursing care of the infant of a diabetic mother includes close monitoring of vital signs, early feeding, and frequent assessment of blood glucose levels for the first 2 days of life. Hypoglycemia in the first days of life is defined as a blood glucose level that falls below 40 mg/dL. It can result in rapid and permanent brain damage. The infant should be closely watched for signs of irritability, tremors, and respiratory distress.
Get Ready for the NCLEX® Examination!
Key Points
• The nurse manages communication between parents and the multidisciplinary health care team to meet the needs of the newborn with congenital problems and of their families.
• Measuring head size is important in infants with hydrocephalus.
• Spina bifida is a congenital embryonic neural tube defect in which there is an imperfect closure of the spinal vertebrae.
• Folic acid supplementation during the early weeks of pregnancy can prevent neural tube anomalies.
• Postoperative nursing care of the infant with a cleft lip includes preventing the infant from sucking and crying, which could impair healing of the suture line.
• Newborns feel pain, and adequate pain control after invasive procedures and surgery is essential.
• The body spica cast encircles the waist and extends to the ankles or toes. It is used to treat developmental hip dysplasia.
• A positive Barlow’s test and Ortolani’s sign are indicative of developmental hip dysplasia.
• Newborn infants are routinely screened for phenylketonuria.
• Lofenalac is a formula used for infants with phenylketonuria.
• RhoGAM is given to an Rh-negative mother after delivering an Rh-positive fetus to prevent maternal Rh sensitization.
• Hyperbilirubinemia results from rapid destruction of red blood cells.
• Jaundice that occurs in the first 24 hours of life is considered pathological.
• Distinguishing pathological jaundice from physiological jaundice can facilitate early intervention and prevent serious complications.
• Macrosomia is a condition in which the infant is large for gestational age (LGA) and usually occurs in infants of diabetic mothers.
• In intracranial hemorrhage, blood vessels within the skull are broken and there is bleeding into the brain.
Additional Learning Resources
 Go to your Study Guide for additional learning activities to help you master this chapter content.
Go to your Study Guide for additional learning activities to help you master this chapter content.
Go to your Evolve website (http://evolve.elsevier.com/Leifer) for the following FREE learning resources:
• Answer Guidelines for Critical Thinking Questions
• Answers and Rationales for Review Questions for the NCLEX® Examination
• Glossary with English and Spanish pronunciations
• Interactive Review Questions for the NCLEX® Examination
• Patient Teaching Plans in English and Spanish
 Online Resources
Online Resources• Centers for Disease Control and Prevention: www.cdc.gov
• Cleft Palate Foundation: www.cleftline.org
• March of Dimes: www.marchofdimes.com
• National Association for Down Syndrome: www.nads.org
• Spina Bifida Association of America: www.spinabifidaassociation.org
Review Questions for the NCLEX® Examination
1. The practice of suctioning the nose and mouth of the newborn when the head is born and before the rest of the infant’s body is delivered is advisable because:
1. it prevents aspiration of amniotic fluid in the newborn.
2. there is more time to suction because many procedures must be done after delivery.
3. the physician is available to suction rather than the nurse.
4. the newborn will have a cleaner appearance when first seen by the mother.
2. A congenital defect that results in enlargement of the infant’s head and pressure changes within the brain is:
1. a protrusion of the meninges through an opening in the spine.
2. primarily a disorder of the muscular tissue of the body.
3. a protrusion of the membranes and cord through an opening in the spine.
4. A Pavlik harness is often used to correct:
5. When bathing an infant, the nurse observes the hips for dislocation. What observation may indicate developmental hip dysplasia?