The nervous system
Nervous system examination
Anatomy
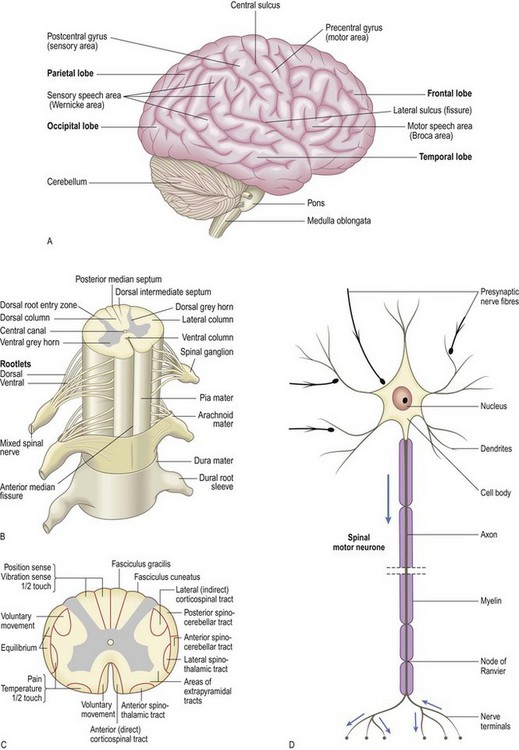
The nervous system consists of the brain and spinal cord (central nervous system, CNS) and peripheral nerves (peripheral nervous system, PNS). The PNS includes the autonomic nervous system, responsible for control of involuntary functions.
The neurone is the functioning unit of the nervous system. Each neurone has a cell body and axon terminating at a synapse, supported by astrocytes and microglial cells. Astrocytes provide the structural framework for the neurones, control their biochemical environment and form the blood–brain barrier. Microglial cells are blood-derived mononuclear macrophages with immune and scavenging functions. In the CNS, oligodendrocytes produce and maintain a myelin sheath around the axons. In the PNS myelin is produced by Schwann cells.
The brain consists of two cerebral hemispheres, each with four lobes (frontal, parietal, temporal and occipital), the brainstem and the cerebellum. The brainstem comprises the midbrain, pons and medulla. The cerebellum lies in the posterior fossa, with two hemispheres and a central vermis attached to the brainstem by three pairs of cerebellar peduncles. Between the brain and the skull are three membranous layers: dura mater next to the bone, arachnoid and pia mater next to the nervous tissue. The subarachnoid space between the arachnoid and pia is filled with cerebrospinal fluid (CSF).
The spinal cord contains afferent and efferent fibres arranged in discrete bundles which are responsible for the transmission of motor and sensory information. Peripheral nerves have myelinated and unmyelinated axons. The sensory cell bodies of peripheral nerves are situated in the dorsal root ganglia. The motor cell bodies are in the anterior horns of the spinal cord (Fig. 11.1).
Symptoms and definitions
Common neurological symptoms are headache, weakness, numbness, disturbance/loss of consciousness, imbalance, abnormal movements and memory loss. The history is crucial as many neurological diseases, e.g. migraine or epilepsy, have no clinical signs. Some symptoms, e.g. loss of consciousness or amnesia, demand an eye-witness history.
Headache
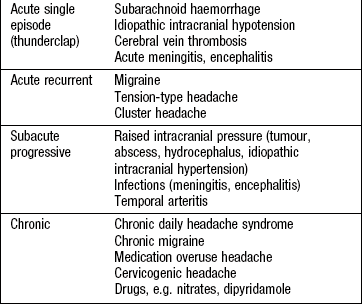
Headache is the most common neurological symptom and may be either primary or secondary to other pathology (Box 11.1). The most common causes of headache are migraine and tension-type headache (Box 11.2).
 11.1 Primary and secondary headache syndromes
11.1 Primary and secondary headache syndromes
Migraine
Trigeminal autonomic cephalalgias (including cluster headache)
Transient loss of consciousness (TLOC)
Syncope is loss of consciousness due to inadequate cerebral perfusion and is the commonest cause of TLOC. Vasovagal syncope (a ‘faint’) is the most common type and is usually precipitated by stimulation of the parasympathetic nervous system, e.g. pain, prolonged standing. Exercise-related syncope suggests a cardiac cause (Box 11.3). An epileptic seizure can cause TLOC. These are caused by paroxysmal electrical discharges from the brain involving the whole brain (generalised seizures: Box 11.4) or part of the brain (focal seizures: Box 11.5). The history from the patient and witnesses wherever possible helps distinguish syncope from epilepsy (Box 11.6).
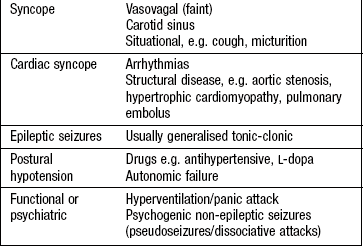
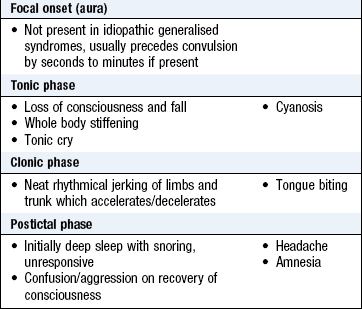

11.6 Features which help discriminate vasovagal syncope from epileptic seizure
| Vasovagal syncope | Seizure | |
| Triggers | Typically present (pain, illness, emotion) | Often none (sleep deprivation, alcohol, drugs) |
| Prodrome | Feeling faint, nausea, tinnitus, vision dimming | Focal onset (not always present) |
| Duration of unconsciousness | Less than 60 seconds | 1–2 minutes |
| Convulsion | May occur but brief myoclonic jerks | Usual, tonic-clonic 1–2 minutes |
| Colour | Pale/grey | Red/blue, may be pale |
| Lateral tongue biting | Very rare (may bite tip) | Common |
| Recovery | Rapid, no confusion | Gradual, over 30 minutes, often confused, amnesic |
Stroke and transient ischaemic attack (TIA)
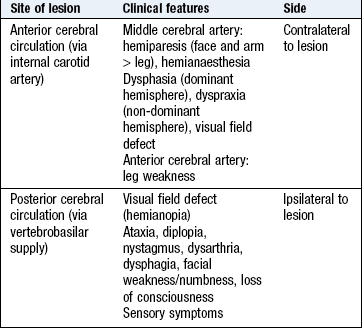
A stroke is a focal (occasionally global) neurological deficit of rapid onset due to a vascular cause. Hemiplegia following middle cerebral artery occlusion is a typical example, but symptoms are dictated by the vascular territory involved (Box 11.7). In industrialised countries, about 80% of strokes are ischaemic, the remainder haemorrhagic, but haemorrhagic stroke is much more prevalent in Asian populations. A TIA is the same, but with symptoms resolving within 24 hours; TIAs are an important risk factor for impending stroke, and demand urgent assessment and treatment. Spinal strokes are exceedingly rare.
Dizziness and vertigo
Patients use ‘dizziness’ to describe many sensations. Recurrent ‘dizzy spells’ affect ~30% of those >65 years and can be due to postural hypotension, cerebrovascular disease, cardiac arrhythmia or hyperventilation induced by anxiety and panic. Vertigo (the illusion of movement) specifically indicates a problem in the vestibular apparatus (peripheral) or, much less commonly, the brain (central). TIAs do not cause isolated vertigo.
The history
Neurological symptoms may be difficult for patients to describe, so clarify exactly what the patient tells you. Words such as ‘blackouts’, ‘dizziness’, ‘weakness’ and ‘numbness’ may indicate a different symptom from what you first imagined, so ensure you understand what the patient means. Clarifying or reviewing the history with the patient and/or witness is essential and provides diagnostic clues.
Time relationships: The onset, duration and pattern of symptoms over time often provide clues to the diagnosis, e.g. headache (Box 11.2) or vertigo (Box 13.5).
• When did the symptoms start (or when was the patient last well)?
• Are they persistent or intermittent?
• If persistent, are they getting better, worse, or staying the same?
• If intermittent, how long do they last?
• Was the onset sudden, e.g. subarachnoid haemorrhage, or gradual, e.g. migraine headache?
Associated symptoms: Associated symptoms might aid diagnosis, e.g. headache may be associated with other symptoms such as nausea, vomiting, photophobia (aversion to light), suggesting meningism, or phonophobia (aversion to sound), suggesting migraine.
Headache: Use SOCRATES to define the nature of the headache (Box 2.10); the onset and periodicity may provide aetiological clues (Box 11.2).
Transient loss of consciousness: If patients are unaware of their symptoms, obtain a witness account. This is more valuable than an unfocused neurological examination. Ask the witness about symptoms before, during and after the TLOC – were there any warning symptoms, any colour changes, did the patient lie still or move, what was the patient like immediately afterwards?
Past history
Forgotten symptoms may be important, e.g. a history of recovered visual loss (optic neuritis) in a patient now presenting with numbness suggests multiple sclerosis. Birth history and development may be important in some situations, e.g. epilepsy. Contact parents or family doctors to obtain such information. If considering a vascular cause for neurological symptoms, ask about important risk factors, e.g. other vascular disease, hypertension, family history and smoking.
Drug history
Always consider drugs, including prescribed, over-the-counter and complementary therapies, as they may cause many neurological symptoms (Box 11.8). Adverse reactions may be idiosyncratic, dose-related or caused by chronic use.
Family history
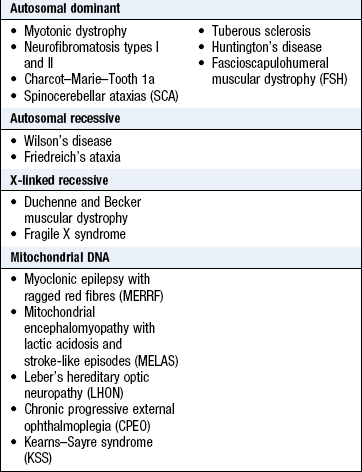
Many neurological disorders are caused by single-gene defects. Others have an important polygenic influence, e.g. multiple sclerosis. Some conditions have a variety of inheritance patterns, e.g. Charcot–Marie–Tooth disease. Neurological disease may also be caused by mitochondrial DNA abnormalities (Box 11.9).
Social history
Alcohol is the most common neurological toxin and damages both the CNS (ataxia, seizures, cognitive symptoms) and the PNS (neuropathy). Poor diet with vitamin deficiency compounds these problems. Other recreational drugs may damage the nervous system, e.g. cocaine and ecstasy can cause seizures and strokes, and smoking contributes to vascular and malignant disease. Always consider sexually transmitted or blood-borne infection, e.g. human immunodeficiency virus (HIV) or syphilis, especially in high-risk groups.
Social circumstances are relevant. How are patients coping with their symptoms? Do they drive? If so, should they? What are the physical and emotional support circumstances? Always ask what they think or fear might be wrong with them, as neurological symptoms cause much anxiety. Patients commonly research their symptoms on the internet; searches of common benign neurological symptoms, e.g. numbness, usually list the most alarming (and unlikely) diagnoses (multiple sclerosis, motor neurone disease, tumours) first.
The physical examination
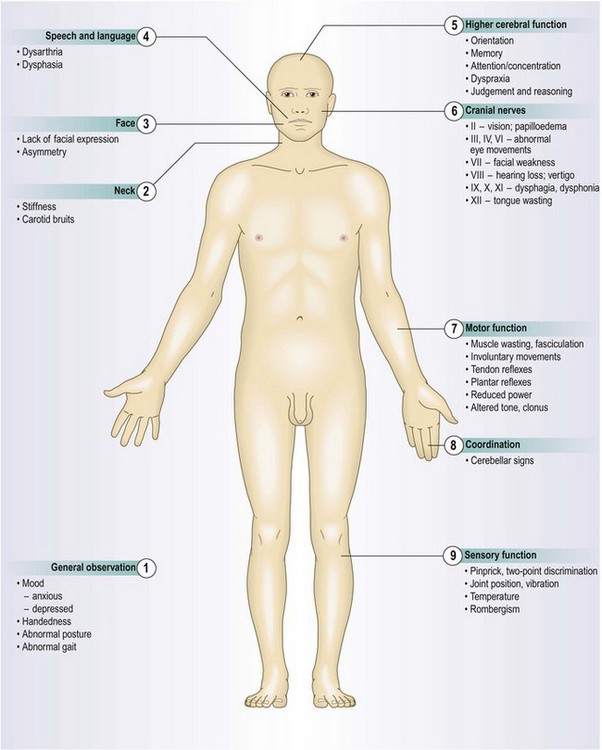
Neurological assessment begins with your first contact with the patient and continues during the history. Note facial expression, demeanour, dress, posture, gait and speech. Mental state examination (p. 21) and general examination (Ch. 3) are integral parts of the neurological examination.
Assessment of conscious level
Consciousness has two main components:
• The state of consciousness depends largely on integrity of the ascending reticular activating system, which extends from the brainstem to the thalamus.
• The content of consciousness refers to how aware the person is and depends on the cerebral cortex, the thalamus and their connections.
Do not use ill-defined terms such as stuporose or obtunded. Use the Glasgow Coma Scale (Box 19.14), a reliable and reproducible tool, to record conscious level.
Meningeal irritation
Meningism (inflammation or irritation of the meninges) can lead to increased resistance to passive flexion of the neck (neck stiffness) or the extended leg (Kernig’s sign). Patients may lie with flexed hips to ease their symptoms. Meningism suggests infection (meningitis) or blood within the subarachnoid space (subarachnoid haemorrhage), but can occur with non-neurological infections, e.g. urinary tract infection. Absence of meningism does not exclude pathology within the subarachnoid space. In meningitis, a finding of neck stiffness has relatively low sensitivity but higher specificity.
 Examination sequence
Examination sequence
 Support the patient’s head with the fingers of your hands at the occiput and the ulnar border of your hands against the paraspinal muscles of the patient’s neck (Fig. 11.2A).
Support the patient’s head with the fingers of your hands at the occiput and the ulnar border of your hands against the paraspinal muscles of the patient’s neck (Fig. 11.2A).
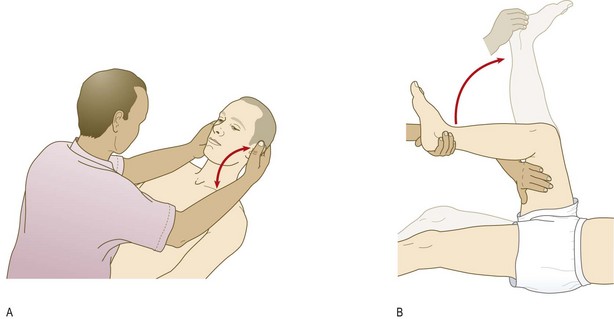
Flex the patient’s head gently until his chin touches his chest.
Ask the patient to hold that position for 10 seconds. If neck stiffness is present, the neck cannot be passively flexed and you may feel spasm in the neck muscles.
Flexion of the knees in response to neck flexion is Brudzinski’s sign.
Flex one of the patient’s legs at the hip and knee, with your left hand placed over the medial hamstrings.
Use your right hand to extend the knee while the hip is maintained in flexion. Look at the other leg for any reflex flexion (Fig. 11.2B). Kernig’s sign is positive when extension is resisted by spasm in the hamstrings. The other limb may flex at the hip and knee. Kernig’s sign is absent in local causes of neck stiffness, e.g. cervical spine disease or raised intracranial pressure (Boxes 11.10 and 11.11).
 11.10
11.10
Meningitis
The absence of all three signs of fever, neck stiffness and an altered mental state virtually eliminates the diagnosis of meningitis.
A positive Kernig’s or Brudzinski’s sign is highly specific for bacterial meningitis but absence of these signs cannot exclude meningitis. McGee S. Evidence based physical diagnosis. St Louis, MO: Saunders/Elsevier, 2007, p. 279.
11.11
Subarachnoid haemorrhage
In patients with acute headache, predictive features of subarachnoid haemorrhage are: age > 40 years, onset with exertion, neck stiffness or pain, raised blood pressure, loss of consciousness and vomiting.
Perry JJ, Stiell IG, Sivilotti MLA et al. High risk clinical characteristics for subarachnoid haemorrhage in patients with acute headache: prospective cohort study. BMJ 2010;341:1035.
Disorders of movement
The principal motor pathway has CNS (corticospinal or pyramidal tract – upper motor neurone) and PNS (anterior horn cell – lower motor neurone) components. Other parts of the nervous system, e.g. basal ganglia and cerebellum, have important modulating effects on movement. It is essential to distinguish upper from lower motor neurone signs (Box 11.12).
11.12 Features of motor neurone lesions
| Upper motor neurone lesion | Lower motor neurone lesion | |
| Inspection | Usually normal (wasting in longstanding lesions) | Wasting, fasciculation |
| Tone | Increased with clonus | Normal or decreased, no clonus |
| Weakness | Preferentially affects extensors in arms, flexors in leg | Usually more focal, in distribution of nerve root or peripheral nerve |
| Deep tendon reflexes | Increased | Decreased/absent |
| Plantar response | Extensor | Flexor |
Upper motor neurone lesions: If the lesion affects the CNS pathways, the lower motor neurones are under the uninhibited influence of the spinal reflex. The motor units then have an exaggerated response to stretch with increased tone (spasticity), clonus and brisk reflexes. There is weakness but not wasting (although atrophy may develop with longstanding lesions). Primitive reflexes, e.g. plantar extensor response (Babinski sign), may be present.
Lower motor neurone lesions: The group of muscle fibres innervated by a single anterior horn cell forms a ‘motor unit’. A lower motor neurone lesion causes weakness and wasting in these muscle fibres, reduced tone (flaccidity), fasciculation and reduced or absent reflexes.
Stance and gait: Stance and gait depend upon intact visual, sensory, corticospinal, extrapyramidal and cerebellar pathways, together with functioning lower motor neurones and spinal reflexes. Non-neurological gait disorders are discussed in Chapter 14. Certain abnormal gait patterns are recognisable, suggesting diagnoses (Box 11.13 and Fig. 3.2).
11.13 Common gait abnormalities
| Gait disturbance | Description | Causes |
| Parkinsonian | Stooped Shuffling (reduced stride length) Loss of arm swing Postural instability Freezing |
Parkinson’s disease Other parkinsonian syndromes |
| Gait apraxia | Small shuffling steps (marche à petit pas) Difficulty in starting to walk/freezing Better ‘cycling’ on bed than walking |
Cerebrovascular disease Hydrocephalus |
| Spastic paraparesis | Stiff ‘walking through mud’ or scissors gait | Spinal cord lesions |
| Myopathic | Waddling (proximal weakness) Bilateral Trendelenburg signs |
Muscular dystrophies Acquired myopathies |
| Foot drop | Foot slapping | Neuropathies L5 radiculopathy |
| Central ataxia | Wide based ‘drunken’ Tandem gait poor |
Cerebellar disease |
| Sensory ataxia | Wide-based Positive Romberg sign |
Neuropathies Spinal cord disorders |
| Functional gait | Variable, often bizarre, inconsistent Knees flexed, buckling Dragging immobile leg behind them |
Conversion disorder |
Examination sequence
Ask the patient to stand with his (preferably bare) feet close together and eyes open.
Swaying, lurching, or inability to stand with the feet together with the eyes open suggest a cerebellar ataxia.
Ask the patient to close his eyes (Romberg’s test) but be prepared to steady/catch the patient. Repeatedly falling is a positive result.
Time the patient walking a measured 10 metres, with a walking aid if needed, turning through 180° and returning.
Note stride length, arm swing, steadiness (including turning), limping or other difficulties.
Listen for the slapping sound of a foot drop gait.
Ask the patient to walk first on tip toes, then on the heels. Ankle dorsiflexion weakness (foot drop) is much more common than plantar flexion weakness, and makes walking on the heels difficult or impossible.
Ask the patient to walk heel to toe in a straight line (tandem gait). This emphasises any gait ataxia.
• Unsteadiness on standing with the eyes open is common in cerebellar disorders.
• Instability which only occurs, or is markedly worse, on eye closure (Romberg’s sign) indicates proprioceptive sensory loss in the feet (sensory ataxia).
• Hemiplegic gait (unilateral upper motor neurone lesion) is characterised by extension at the hip, knee and ankle and circumduction at the hip, such that the foot on the affected side is plantar flexed and describes a semicircle as the patient walks. The upper limb will be flexed.
• Bilateral upper motor neurone damage causes a scissor-like gait due to spasticity.
• Cerebellar dysfunction leads to a broad-based, unsteady (ataxic) gait, which usually makes walking heel to toe in a straight line impossible.
• In parkinsonism, initiation of walking may be delayed; the steps are short and shuffling with loss/reduction of arm swing. A pill-rolling tremor may be apparent. The stooped posture and impairment of postural reflexes can result in a festinant (rapid, short-stepped, hurrying) gait. As a doorway or other obstacle approaches, the person may freeze. Turning involves many short steps, with the risk of falls.
• Proximal muscle weakness may lead to a waddling gait with bilateral Trendelenburg signs (p. 346).
• Bizarre gaits, such as dragging a leg behind the patient, are often functional, but some diseases, e.g. Huntington’s disease, produce unusual gaits.
Speech
Symptoms and definitions: Dysarthria is slurred speech caused by articulation problems due to a motor deficit.
Dysphonia is loss of volume caused by laryngeal disorders.
Dysphasia is disturbance of language resulting in abnormalities of speech production and/or understanding and may also involve other language symptoms, e.g. writing and reading, unlike dysarthria and dysphonia.
Examination sequence
Listen to the patient’s spontaneous speech, noting volume, rhythm and clarity.
Ask the patient to repeat phrases such as ‘yellow lorry’ to test lingual (tongue) sounds and ‘baby hippopotamus’ for labial (lip) sounds, then a tongue twister, e.g. ‘the Leith police dismisseth us’.
Ask the patient to count steadily to 30 to assess fatigue.
Ask the patient to cough and to say ‘Ah’; observe the soft palate rising bilaterally.
Dysarthria: Disturbed articulation may result from lesions of the tongue, lips or mouth, ill-fitting dentures or disruption of the neuromuscular pathways.
Bilateral upper motor neurone lesions of the corticobulbar tracts cause a pseudobulbar dysarthria, characterised by a contracted, spastic tongue and difficulty pronouncing consonants, and may be accompanied by a brisk jaw jerk and emotional lability.
Bulbar palsy results from bilateral lower motor neurone lesions affecting the same group of cranial nerves. The nature of the speech disturbance is determined by the specific nerves and muscles involved. Weakness of the tongue results in difficulty with lingual sounds, while palatal weakness gives a nasal quality to the speech.
Cerebellar dysarthria may be slow and slurred, similar to alcohol intoxication.
Myasthenia gravis is the most common cause of fatiguing speech.
Parkinsonism may cause dysarthria and dysphonia, with a low-volume, monotonous voice in which the words run into each other.
Dysphonia: This usually results from either vocal cord pathology, as in laryngitis, or damage to the vagal (X) nerve supply to the vocal cords (recurrent laryngeal nerve). Inability to abduct one of the vocal cords leads to a ‘bovine’ (and ineffective) cough (p. 141).
Dysphasias
Anatomy: The language areas are located in the dominant cerebral hemisphere, which is the left in almost all right- and most left-handed people.
Broca’s area (inferior frontal region) is concerned with word production and language expression.
Wernicke’s area (superior posterior temporal lobe) is the principal area for comprehension of spoken language. Adjacent regions of the parietal lobe are involved in understanding written language and numbers.
The arcuate fasciculus connects Broca’s and Wernicke’s areas.
Examination sequence
During spontaneous speech, listen to the fluency and appropriateness of the content, particularly for paraphasias and neologisms.
Show the patient a common object, e.g. coin or pen, and ask its name.
Give a simple three-stage command, e.g. pick up this piece of paper, fold it in half and place it under the book.
Ask the patient to repeat a simple sentence, e.g. ‘Today is Tuesday’.
Ask the patient to read a passage from a newspaper.
Ask the patient to write a sentence; examine his handwriting.
Abnormal findings: Expressive (motor) dysphasia results from damage to Broca’s area. It is characterised by reduced verbal output with non-fluent speech and errors of grammar and syntax. Comprehension is intact.
Receptive (sensory) dysphasia occurs with dysfunction in Wernicke’s area. There is poor comprehension, and although speech is fluent, it may be meaningless and contain paraphasias (incorrect words) and neologisms (nonsense or meaningless new words).
Global dysphasia is a combination of expressive and receptive difficulties due to involvement of both areas.
Dysphasia (a focal sign) is frequently misdiagnosed as confusion (non-focal sign). Always consider dysphasia before assuming confusion, as this fundamentally alters the differential diagnosis and investigation plan.
Dominant parietal lobe lesions affecting the supramarginal gyrus may cause dyslexia (difficulty comprehending written language), dyscalculia (problems with simple addition and subtraction) and dysgraphia (impairment of writing).
Cortical function
Thinking, emotions, language, behaviour, planning and initiating movements, and perceiving sensory information are functions of the cerebral cortex and are central to awareness of, and interaction with, the environment. Certain cortical areas are associated with specific functions, so particular patterns of dysfunction can help localise the site of pathology (Fig. 11.3A). Assessment of higher cortical function is difficult and time-consuming. There are various tools. For the bedside, the Mini-Mental State Examination (p. 26) is quick to administer, whereas a global tool such as the Addenbrooke’s Cognitive Examination helps detect early cognitive changes but takes much longer to administer (Box 11.14).
11.14
Dementia screening
The revised Addenbrooke’s Cognitive Examination is a validated dementia screening test, sensitive to early cognitive dysfunction.
Mioshi E, Dawson K, Mitchell J et al. The Addenbrooke’s Cognitive Examination Revised (ACE-R): a brief cognitive test battery for dementia screening. Int J Geriatr Psychiatry 2006;21:1078–1085.
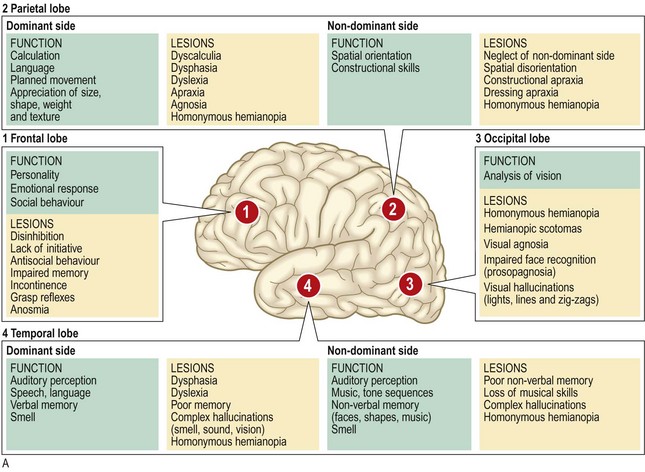
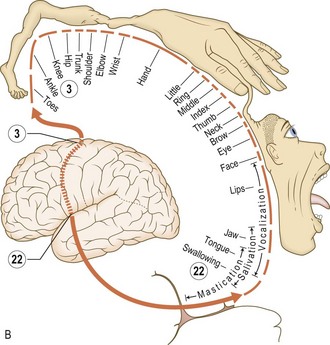
Fig. 11.3 Cortical function.
(A) Features of localised cerebral lesions. (B) Somatotropic homunculus.
Anatomy: The posterior part of the frontal lobe is the motor strip (precentral gyrus) which controls voluntary movement. The motor strip is organised somatotopically (Fig. 11.3B). The area anterior to the precentral gyrus is concerned with personality, social behaviour, emotions, cognition and expressive language, and contains the frontal eye fields and cortical centre for micturition (Fig. 11.4).
Abnormal findings: Frontal lobe damage may cause:
• personality and behaviour changes, e.g. apathy or disinhibition
• loss of emotional responsiveness or emotional lability
• cognitive impairments, e.g. memory, attention and concentration
• dysphasia (dominant hemisphere)
• conjugate gaze deviation to the side of the lesion
Anatomy: The temporal lobe contains the primary auditory cortex, Wernicke’s area and parts of the limbic system. The latter is crucially important in memory and smell appreciation. The temporal lobe also contains the lower fibres of the optic radiation and the area of auditory perception.
Abnormal findings: Temporal lobe dysfunction may cause:
• focal seizures with psychic symptoms (Box 11.5)
Anatomy: The postcentral gyrus (sensory strip) is the most anterior part of the parietal lobe and is the principal destination of conscious sensations. The upper fibres of the optic radiation pass through it. The dominant hemisphere contains aspects of language function and the non-dominant lobe is concerned with spatial awareness.
Abnormal findings: Damage to the parietal lobes is often associated with re-emergence of primitive reflexes. Features of parietal lobe dysfunction include:
• cortical sensory impairments
• contralateral lower quadrantanopia (Fig. 12.3 (part 5))
• dyslexia, dyscalculia, dysgraphia
• apraxia (an inability to carry out complex tasks despite having an intact sensory and motor system)
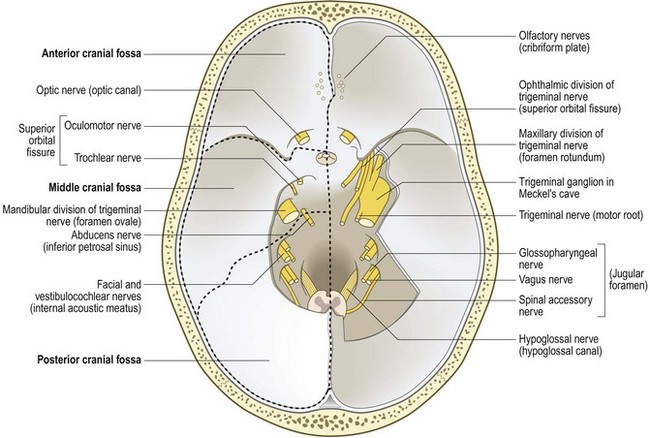
The cranial nerves
The 12 pairs of cranial nerves (with the exception of the olfactory (I) pair) arise from the brainstem (Fig. 11.4). Cranial nerves II, III, IV and VI relate to the eye (Ch. 12) and the VIII nerve to hearing and balance (Box 11.17, Ch. 13).
11.17 Summary of all 12 cranial nerves
| Nerve | Examination | Abnormalities/symptoms |
| I | Sense of smell, each nostril | Anosmia/parosmia |
| II | Visual acuity Visual fields Pupil size and shape Pupil light reflex Fundoscopy |
Partial sight/blindness Scotoma; hemianopia Anisocoria Impaired or lost Optic disc and retinal changes |
| III | Accommodation reflex | Impaired or lost |
| III, IV and VI | Eye position and movements | Strabismus, diplopia, nystagmus |
| V | Facial sensation Corneal reflex Muscles of mastication Jaw jerk |
Impaired, distorted or lost Impaired or lost Weakness of chewing movements Increased in upper motor neurone lesions |
| VII | Muscles of facial expression Taste over anterior two-thirds of tongue |
Facial weakness Ageusia |
| VIII | Whisper and tuning fork tests Vestibular tests |
Impaired hearing/deafness Nystagmus and vertigo |
| IX | Pharyngeal sensation | Not routinely tested |
| X | Palate movements | Impaired unilaterally or bilaterally |
| XI | Trapezius and sternomastoid | Weakness of neck movement |
| XII | Tongue appearance and movement | Dysarthria and chewing/swallowing problems |
The olfactory (I) nerve: The olfactory nerve conveys the sense of smell.
Anatomy: Bipolar cells in the olfactory bulb form olfactory filaments with small receptors projecting through the cribriform plate high in the nasal cavity. These cells synapse with second-order neurones, which project centrally via the olfactory tract to the medial temporal lobe and amygdala.
Abnormal findings: Hyposmia or anosmia (reduction or loss of the sense of smell) may result from ear, nose and throat disease, damage to the olfactory filaments after head injury or local compression or invasion by basal skull tumours. Disturbance of smell may also occur in the presymptomatic stages of Parkinson’s and Alzheimer’s diseases. Patients often also note hypogeusia/ageusia (altered taste) with anosmia.
Parosmia is when pleasant odours are perceived as unpleasant; it may occur with head trauma, sinus infection or as an adverse effect of drugs. Olfactory hallucinations may occur in Alzheimer’s disease and focal epilepsies.
The optic (II), oculomotor (III), trochlear (IV) and abducens (VI) nerves: See Chapter 12.
Anatomy: The V nerve provides sensation to the face, mouth and part of the dura, and motor supply to the muscles of mastication.
The cell bodies of the sensory fibres are located in the trigeminal (Gasserian) ganglion, which lies in a cavity (Meckel’s cave) in the petrous temporal dura (Fig. 11.4). There are three major branches of the nerve (Fig. 11.5):
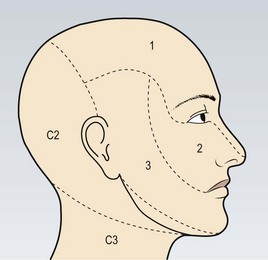
Fig. 11.5 The sensory distribution of the three divisions of the trigeminal nerve.
(1) Ophthalmic division. (2) Maxillary division. (3) Mandibular division.
The ophthalmic branch leaves the ganglion and passes forward to the superior orbital fissure via the wall of the cavernous sinus. In addition to the skin of the upper nose, upper eyelid, forehead and scalp, V1 supplies sensation to the eye (cornea and conjunctiva) and the mucous membranes of the sphenoidal and ethmoid sinuses and upper nasal cavity.
The maxillary branch (V2) passes from the ganglion via the cavernous sinus to leave the skull by the foramen rotundum. It contains sensory fibres from the mucous membranes of the upper mouth, roof of pharynx, gums, teeth and palate of the upper jaw and the maxillary, sphenoidal and ethmoid sinuses.
The mandibular branch (V3) exits the skull via the foramen ovale and supplies the floor of the mouth, common sensation, i.e. not taste, to the anterior two-thirds of the tongue, the gums and teeth of the lower jaw, mucosa of the cheek and the temporomandibular joint in addition to the skin of the lower lips and jaw area, but not the angle of the jaw (Fig. 11.5).
• From the trigeminal ganglion, the V nerve passes to the pons. From here, pain and temperature pathways descend to the C2 segment of the spinal cord, so ipsilateral facial numbness may occur with cervical cord lesions.
The motor fibres of V run in the mandibular branch (V3) and innervate the temporalis, masseter, medial and lateral pterygoids (muscles of mastication).
Examination sequence
There are four functions: sensory, motor and two reflexes.
Ask the patient to close his eyes and say ‘yes’ each time he feels you lightly touch them using a cotton wool tip. Do this in the areas of V1, V2 and V3.
Repeat using a fresh neurological pin, e.g. Neurotip, to test superficial pain.
Compare both sides. If you identify an area of reduced sensation, map it out. Does it conform to the distribution of the trigeminal nerve or branches? Remember the angle of the jaw is not served by the trigeminal nerve, but V1 does extend towards the vertex (Fig. 11.5).
‘Nasal tickle’ test: use a wisp of cotton wool to ‘tickle’ the inside of each nostril and ask the patient to compare: it is an unpleasant sensation easily appreciated by the patient.
Explain to the patient what you are going to do, and ask him to remove contact lenses, if relevant.
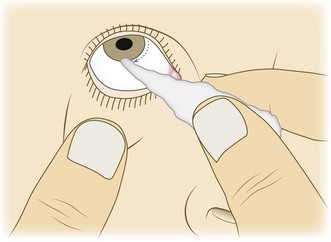
Gently depress the lower eyelid while the patient looks upwards.
Lightly touch the lateral edge of the cornea with a wisp of damp cotton wool (Fig. 11.6):
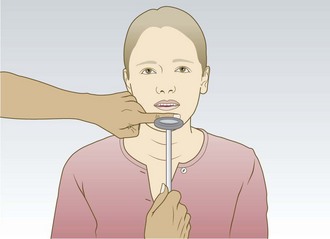
Ask the patient to let his mouth hang loosely open.
Place your forefinger in the midline between lower lip and chin.
Percuss your finger gently with the tendon hammer in a downwards direction (Fig. 11.7), noting any reflex closing of the jaw. An absent, or just present, reflex is normal.
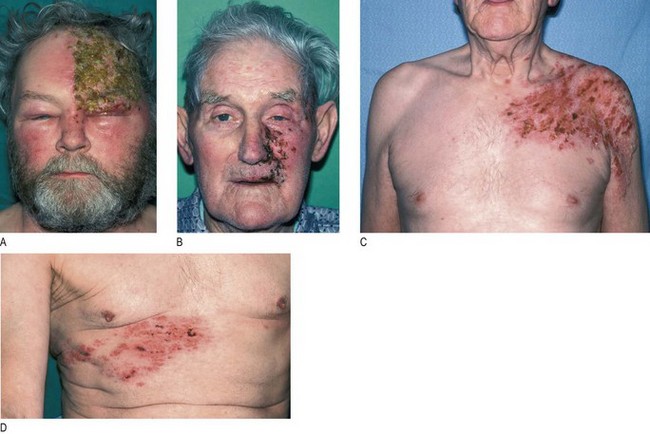
Abnormal findings: Sensory symptoms include facial numbness and pain (trigeminal neuralgia). Unilateral loss of sensation in one or more branches of the V nerve may result from direct injury in association with facial fractures (particularly V2) or local invasion by cancer. Lesions in the cavernous sinus often cause loss of the corneal reflex and V1 or V2 cutaneous sensory loss. Cranial nerves III, IV and VI may also be involved (Ch. 12). Trigeminal neuralgia causes severe, lancinating pain typically in distribution of V2 or V3, and is often due to neurovascular compression. Reactivation of herpes varicella zoster virus (chickenpox) can affect any sensory nerve, but typically either a thoracic dermatome or V1 (Fig. 11.8). Clinically significant weakness of the muscles of mastication is unusual, but may occur in myasthenia, with fatigable chewing. A brisk jaw jerk occurs in pseudobulbar palsy.
The facial (VII) nerve: The facial nerve supplies the muscles of facial expression, and carries parasympathetic secretomotor fibres to the lacrimal, submandibular and sublingual salivary glands (via nervus intermedius). It receives taste sensation from the anterior two-thirds of the tongue (via the chorda tympani branch), and also provides the efferent supply to several reflexes (Fig. 11.9).
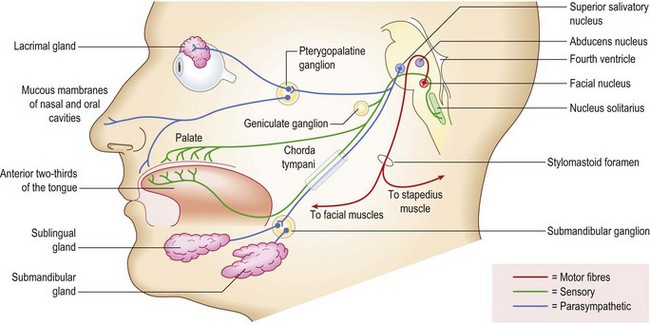
Anatomy: From its motor nucleus in the lower pons, fibres of the VII nerve pass back to loop around the VI nucleus before emerging from the lateral pontomedullary junction in close association with the VIII nerve; together they enter the internal acoustic meatus (Figs 11.4 and 11.9). At the lateral end of the meatus the VII nerve continues in the facial canal within the temporal bone, exiting the skull via the stylomastoid foramen. Passing through the parotid gland, it gives off its terminal branches. In its course in the facial canal it gives off branches to the stapedius muscle and its parasympathetic fibres, as well as being joined by the taste fibres of the chordae tympani (Fig. 11.10).
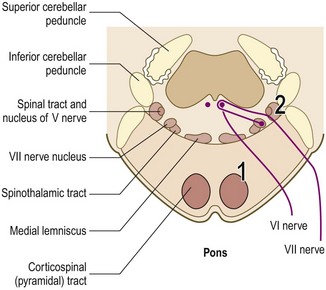
Fig. 11.10 Lesions of the pons.
Lesions at (1) may result in ipsilateral VI and VII nerve palsies and contralateral hemiplegia; at (2) ipsilateral cerebellar signs and impaired sensation on the ipsilateral side of the face and on the contralateral side of the body may occur.
Examination sequence
Examination is usually confined to motor function; taste is rarely tested.
Inspect the face for asymmetry or differences in blinking or eye closure on one side. Note that minor facial asymmetry is common and rarely pathological.
Watch for spontaneous or involuntary movement.
Ask the patient to raise the eyebrows and observe for symmetrical wrinkling of the forehead (Fig. 11.11A).
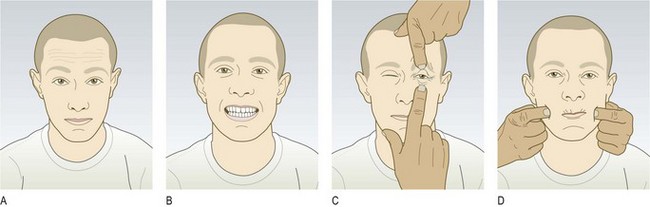
Fig. 11.11 Testing the motor function of the facial nerves.
(A) Ask the patient to raise his eyebrows. (B) Ask him to show his teeth. (C) Ask him to close eyes against resistance. (D) Ask him to blow out his cheeks.
Demonstrate baring your teeth and ask the patient to mimic you. Look for asymmetry (Fig. 11.11B).
Test power by saying: ‘Screw your eyes tightly shut and stop me from opening them,’ then ‘Blow out your cheeks with your mouth closed’ (Fig. 11.11C and D).
Abnormal findings: In a unilateral lower motor neurone VII nerve lesion, there is weakness of both upper and lower facial muscles. Bell’s palsy is a common condition presenting with acute lower motor neurone VII nerve paralysis, often preceded by mastoid pain. It may be associated with impairment of taste and hyperacusis (high-pitched sounds appearing unpleasantly louder than normal). Bell’s phenomenon occurs when the patient is unable to close his eye. As he tries, the eyeball rolls upwards, exposing the conjunctiva below the cornea (Fig. 11.12A). Ramsay Hunt syndrome occurs in herpes zoster infection of the geniculate (facial) ganglion. This produces a severe lower motor neurone facial palsy, ipsilateral loss of taste and buccal ulceration, and a painful vesicular eruption in the external auditory meatus. Other causes of a lower motor neurone VII lesion include cerebellopontine angle tumours, e.g. acoustic neuroma, trauma and parotid tumours. Synkinesis (most commonly twitching of the corner of the mouth on ipsilateral blinking) is a sign of aberrant reinnervation, and may be seen in recovering lower motor neurone VII lesions.
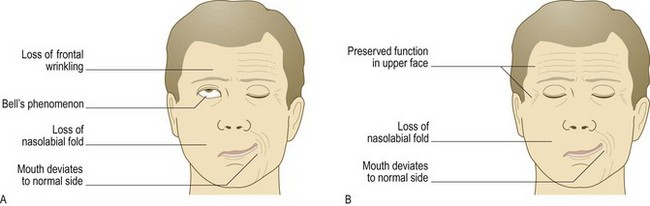
Fig. 11.12 Types of facial weakness.
(A) Right-sided lower motor neurone lesion (within facial nerve or nucleus); Bell’s phenomenon is also shown. (B) Right-sided upper motor neurone lesion.
In unilateral VII nerve upper motor neurone lesions, weakness is marked in the lower facial muscles with relative sparing of the upper face. This is because there is bilateral cortical innervation of the upper facial muscles. The nasolabial fold may be flattened and the corner of the mouth droop, but eye closure is usually preserved (Fig. 11.12B). Involuntary emotional movements, e.g. spontaneous smiling, have different pathways and may be preserved in the presence of paresis.
Bilateral facial palsies are less common, but occasionally occur, e.g. Guillain–Barré syndrome, sarcoidosis, Lyme disease and HIV infection. Distinct from VII nerve palsies, Parkinson’s disease can cause loss of spontaneous facial movements, including a slowed blink rate, and involuntary facial movements (levodopa-induced dyskinesias) may complicate advanced disease.
The vestibulocochlear (VIII) nerve: See Chapter 13.
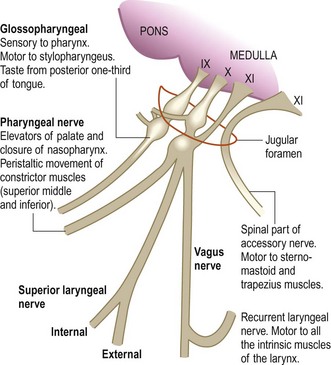
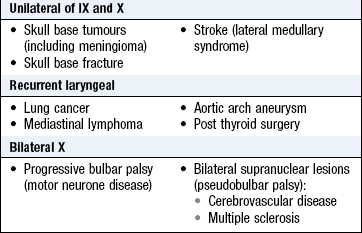
The glossopharyngeal (IX) and vagus (X) nerves: The IX and X nerves have an intimate anatomical relationship. Both contain sensory, motor and autonomic components. The glossopharyngeal (IX) nerve mainly carries sensation from the pharynx and tonsils, and sensation and taste from the posterior one-third of the tongue. The vagus (X) nerve carries important sensory information but also innervates upper pharyngeal and laryngeal muscles. The main functions of IX and X are swallowing, phonation/articulation and sensation from the pharynx/larynx.
Anatomy: Both nerves arise as several roots from the lateral medulla and leave the skull together via the jugular foramen (Fig. 11.4). The IX nerve passes down and forward to supply the stylopharyngeus muscle, the mucosa of the pharynx, the tonsils and the posterior one-third of the tongue, and sends parasympathetic fibres to the parotid gland. The X nerve courses down in the carotid sheath into the thorax, giving off several branches, including pharyngeal and recurrent laryngeal branches, which provide motor supply to the pharyngeal, soft palate and laryngeal muscles. The main nuclei of these nerves in the medulla are the nucleus ambiguus (motor), the dorsal motor vagal nucleus (parasympathetic) and the solitary nucleus (visceral sensation) (Fig. 11.13).
Examination sequence
Assess the patient’s speech for dysarthria or dysphonia.
Ask him to say ‘Ah’; look at the movements of the palate and uvula using a torch. Normally, both sides of the palate elevate symmetrically and the uvula remains in the midline.
Ask the patient to puff out his cheeks with the lips tightly closed. Listen for air escaping from the nose. For the cheeks to puff out, the palate must elevate and occlude the nasopharynx. If palatal movement is weak, air will escape audibly through the nose.
Ask the patient to cough; assess the strength of the cough.
Testing pharyngeal sensation and the gag reflex is unpleasant and has poor predictive value for aspiration. Instead, and in fully conscious patients only, use the swallow test. Administer 3 teaspoons of water and observe for absent swallow, cough or delayed cough, or change in voice quality after each teaspoon. If there are no problems, watch for the same reactions while the patient swallows a glass of water.
Abnormal findings: Isolated unilateral IX nerve lesions are rare. Unilateral X nerve damage leads to ipsilateral reduced elevation of the soft palate, which may cause deviation of the uvula (away from the side of the lesion) when the patient says ‘Ah’. Damage to the recurrent laryngeal branch of the X nerve due to lung cancer, thyroid surgery, mediastinal tumours and aortic arch aneurysm causes dysphonia and a ‘bovine’ cough (p. 141). Bilateral X nerve lesions cause dysphagia and dysarthria. Less severe cases can result in nasal regurgitation of fluids and nasal air escape when the cheeks are puffed out (dysarthria and nasal escape are often evident during history taking: Box 11.15).
The accessory (XI) nerve: The accessory nerve has two components:
• a cranial part closely related to the vagus nerve
• a spinal part which provides fibres to the upper trapezius and the sternocleidomastoid muscles, responsible for elevating (shrugging) the shoulders, and head turning/flexing.
The spinal component is discussed here.
Anatomy: The spinal nuclei arise from the anterior horn cells of C1–5. Fibres emerge from the spinal cord, ascend through the foramen magnum, and exit via the jugular foramen (Fig. 11.4), passing posteriorly.
Examination sequence
Face the patient and inspect the sternocleidomastoid muscles for wasting or hypertrophy; palpate them to assess their bulk.
Stand behind the patient to inspect the trapezius muscle for wasting or asymmetry.
Ask the patient to shrug the shoulders, then apply downward pressure with your hands to assess the power (Fig. 11.14A).
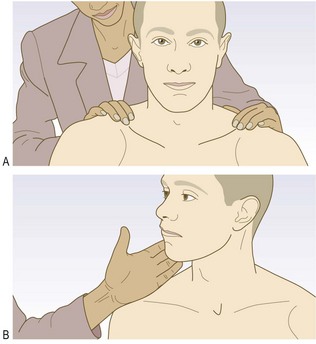
Fig. 11.14 Testing the trapezius and left sternocleidomastoid muscles.
(A) Trapezius. (B) Left sternocleidomastoid.
Test power in the left sternocleidomastoid by asking the patient to turn the head to the right while you provide resistance with your hand placed on the right side of the patient’s chin (Fig. 11.14B). Reverse the procedure to check the right sternocleidomastoid.
Abnormal findings: Isolated XI nerve lesions are uncommon but the nerve may be damaged during surgery in the posterior triangle of the neck, penetrating injuries or local invasion by tumour. Wasting of the upper fibres of trapezius may be associated with displacement of the upper vertebral border of the scapula away from the spine, while the lower border is displaced towards it. Wasting and weakness of the sternocleidomastoids are characteristic of myotonic dystrophy, and head drop may be seen in myasthenia, motor neurone disease and some myopathies.
The hypoglossal (XII) nerve: The XII nerve innervates the tongue muscles; the nucleus lies in the dorsal medulla beneath the floor of the fourth ventricle.
Anatomy: The nerve emerges anteriorly and exits the skull in the hypoglossal canal, passing to the root of the tongue (Fig. 11.4).
Abnormal findings: Unilateral lower motor XII nerve lesions lead to tongue wasting on the affected side and deviation to that side on protrusion (Fig. 11.15). Bilateral lower motor neurone damage results in global wasting, the tongue lies thin and shrunken and fasciculation may be evident. Normal rippling or undulating movements may be mistaken for fasciculation, especially if the tongue is protruded; these usually settle when the tongue is at rest in the mouth. When associated with lesions of IX, X and XI nerves, typically in motor neurone disease, these features are called bulbar palsy.
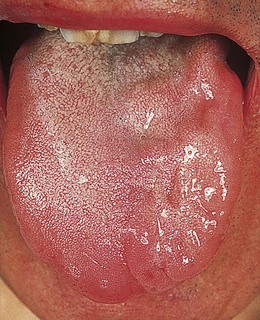
Unilateral upper motor XII nerve lesions are uncommon; bilateral lesions lead to a tongue with increased tone (spastic), and the patient has difficulty flicking the tongue from side to side. Bilateral upper motor lesions of IX–XII nerves may also affect the V and VII, and are called pseudobulbar palsy. They usually result from vascular disease, motor neurone disease or occasionally multiple sclerosis (Box 11.16). Tremor of the resting or protruded tongue may occur in Parkinson’s disease, although jaw tremor is more common. Other orolingual dyskinesias (involuntary movements of the mouth and tongue) are often drug-induced, e.g. tardive dyskinesias due to neuroleptics.
11.16 Comparison of bulbar and pseudobulbar palsy
| Bulbar palsy | Pseudobulbar palsy | |
| Motor lesion | Lower motor neurone | Upper motor neurone |
| Speech | Dysarthria | Dysarthria and dysphonia |
| Swallowing | Dysphagia | Dysphagia |
| Tongue | Weakness, wasting and fasciculation | Spastic, slow moving |
| Jaw jerk | Absent | Present/brisk |
| Emotional lability | Absent | May be present |
Examination sequence
Ask the patient to open his mouth. Look at the tongue at rest for wasting, fasciculation or involuntary movement.
Ask the patient to put out his tongue. Look for deviation or involuntary movement.
Ask the patient to move the tongue quickly from side to side.
Test power by asking the patient to press the tongue against the inside of each cheek in turn while you press from the outside with your finger.
Assess speech by asking the patient to say ‘yellow lorry’.
Assess swallowing with a water swallow test (p. 257).
The motor system
Assess the motor system under the following headings:
Inspection and palpation of the muscles
Anatomy: Motor fibres, together with input from other systems involved in the control of movement, including extrapyramidal, cerebellar, vestibular and proprioceptive afferents, converge on the cell bodies of lower motor neurones in the anterior horn of the grey matter in the spinal cord (Fig. 11.16).
Muscle bulk: Lower motor neurone lesions may cause muscle wasting. This is not seen in acute upper motor neurone lesions, although disuse atrophy may develop with longstanding lesions. A motor neurone lesion in childhood may impair growth (causing a smaller limb or hemiatrophy) or cause limb deformity, e.g. pes cavus. Muscle disorders usually result in proximal wasting (the notable exception is myotonic dystrophy, in which it is distal, often with associated temporalis wasting). Certain occupations, e.g. professional sports players, may lead to physiological muscle hypertrophy. Pseudohypertrophy may occur in muscular dystrophy but the muscles are weak. If you suspect wasting, ask the patient and/or partner whether they have also noticed this, as minor asymmetry in muscle bulk is often normal.
Fasciculation: Fasciculation is irregular twitches under the skin overlying resting muscles caused by individual motor units firing spontaneously. This occurs in lower motor neurone disease, usually in wasted muscles. Fasciculation is seen, not felt, and you may need to observe carefully for several minutes to be sure that this is not present. Physiological fasciculation is common, especially in the calves, but is not associated with weakness or wasting. Myokymia is rapid bursts of repetitive motor unit activity often occurring in an eyelid or first dorsal interosseus, and is rarely pathological.
Myoclonic jerks: These are sudden shock-like contractions of one or more muscles which may be focal or diffuse and occur singly or repetitively. Healthy people commonly experience these when falling asleep (hypnic jerks). They may also occur pathologically in association with epilepsy, diffuse brain damage and dementia.
Tremor: Tremor is an oscillatory movement about a joint or a group of joints resulting from alternating contraction and relaxation of muscles. Tremors are classified according to their frequency, amplitude, position (at rest, on posture or on movement) and body part affected.
Physiological tremor is a fine (low-amplitude), fast (high-frequency) postural tremor seen with anxiety. A similar tremor occurs in hyperthyroidism and with excess alcohol or caffeine intake, and is a common adverse effect of β-agonist bronchodilators.
Essential tremor is the most common pathological cause of an action tremor, typically affecting the upper limbs and head, with postural and action components. It may be improved by alcohol, and often demonstrates an autosomal dominant pattern of inheritance.
Parkinson’s disease causes a slow, coarse tremor, worse at rest but reduced with voluntary movement. It is more common in the upper limbs, usually asymmetrical, and does not affect the head.
Isolated head tremor is usually dystonic, and may be associated with abnormal neck postures such as torticollis (twisting to one side), anterocollis (neck flexion) or retrocollis (neck extension).
Intention tremor is absent at rest but maximal on movement, and is usually due to cerebellar damage. It is assessed with the finger-to-nose test (p. 266).
Functional tremors: movement disorders, including tremor, are common functional symptoms. They are often inconsistent, with varying frequencies and amplitudes, and may be associated with other signs.
Other involuntary movements: These are classified according to their appearance.
Dystonia is caused by sustained muscle contractions, leading to twisting, repetitive movements and sometimes tremor. It may be focal, e.g. torticollis, a twisting neck, or global.
Chorea describes brief, random, purposeless movements which may affect various body parts, but commonly the arms.
Athetosis is a slower, writhing movement, more similar to dystonia than chorea.
Ballism refers to violent flinging movements sometimes affecting only one side of the body (hemiballismus).
Tics are repetitive, stereotyped movements which can be briefly suppressed by the patient.
Tone
Tone is the resistance felt by the examiner when moving a joint passively.
Examination sequence
Ask the patient to lie supine on the examination couch, and to relax and ‘go floppy’. Enquire about any painful joints or limitations of movement before proceeding.
Passively move each joint tested through as full a range as possible, both slowly and quickly in all anatomically possible directions. Be unpredictable with these movements, both in direction and speed, to prevent the patient actively moving with you; you want to assess passive tone.
Hold the patient’s hand as if shaking hands, using your other hand to support his elbow. Assess tone at the wrist and elbow.
Activation is a technique used to exaggerate subtle increase in tone, and is particular useful for assessing extrapyramidal tone increase. Ask the patient to describe circles in the air with the contralateral limb while assessing tone. A transient increase in tone with this manoeuvre is normal.
Roll the leg from side to side, then briskly lift the knee into a flexed position, observing the movement of the foot (Fig. 11.17A and B). Typically the heel moves up the bed, but increased tone may cause it to lift off the bed due to failure of relaxation.
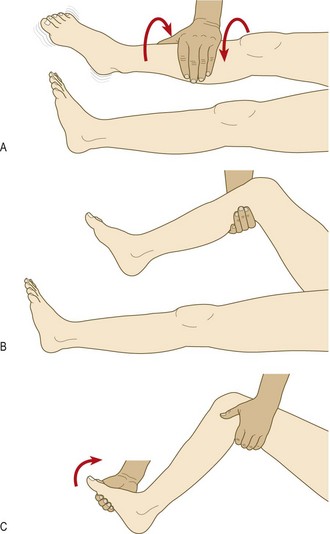
Support the patient’s leg, with both the knee and ankle resting in 90° flexion.
Briskly dorsiflex and partially evert the foot, sustaining the pressure (Fig. 11.17C). Clonus is felt as repeated beats of dorsiflexion/plantar flexion.
Abnormal findings: Hypotonia (decreased muscle tone) or hypertonia (increased) suggest a lower or upper motor neurone lesion respectively.
Hypotonia: This may occur in lower motor neurone lesions and is usually associated with muscle wasting, weakness and hyporeflexia. It may be a feature of cerebellar disease or in the early phases of cerebral or spinal shock, when the paralysed limbs are atonic prior to developing spasticity. Reduced tone can be difficult to elicit.
Hypertonia: There are two types of hypertonia: spasticity and rigidity.
Spasticity is velocity-dependent resistance to passive movement: it is detected with quick movements and is a feature of upper motor neurone lesions. It is usually accompanied by weakness, hyperreflexia, an extensor plantar response and sometimes clonus. In mild forms it is detected as a ‘catch’ at the beginning or end of passive movement. In severe cases it limits the range of movement and may be associated with contracture. In the upper limbs it may be more obvious on attempted extension; in the legs it is more evident on flexion.
Rigidity is a sustained resistance throughout the range of movement and is most easily detected when the limb is moved slowly. In parkinsonism this is classically described as ‘lead pipe rigidity’. In the presence of a parkinsonian tremor there may be a regular interruption to the movement, giving it a jerky feel (‘cog wheeling’).
Clonus is a rhythmic series of contractions evoked by sudden stretch of the muscle and tendon. Unsustained (<6 beats) clonus may be physiological. When sustained, it indicates upper motor neurone damage and is accompanied by spasticity. It is best elicited at the ankle; knee (patella) clonus is rare, and not routinely tested.
Power
Strength varies with age, occupation and fitness. Grade muscle power using the Medical Research Council scale (Box 11.18). In practice, most cases of weakness are grade 4. Plus or minus signs, e.g. 4+ or 4–, are helpful. Record what the patient can actually do in terms of daily activities, e.g. whether he can stand, walk, raise both arms above the head. Lesions at different sites may produce different clinical patterns of weakness (Boxes 11.19 and 11.20).
 11.18 Medical Research Council scale for muscle power
11.18 Medical Research Council scale for muscle power
| 0 | No muscle contraction visible |
| 1 | Flicker of contraction but no movement |
| 2 | Joint movement when effect of gravity eliminated |
| 3 | Movement against gravity but not against examiner’s resistance |
| 4 | Movement against resistance but weaker than normal |
| 5 | Normal power |
 11.19 Causes of muscle weakness
11.19 Causes of muscle weakness
| Anatomical aetiology | Associated features | Common causes |
| Lower motor neurone | Wasting | Peripheral neuropathies or mononeuropathies |
| Fasciculation | Radiculopathies | |
| Hypotonia | Anterior horn cell damage, e.g. poliomyelitis or motor neurone disease | |
| Reflexes absent or diminished | ||
| Upper motor neurone | ‘Patterned’ weakness (flexed arm, extended leg) | Stroke |
| No muscle wasting | Spinal cord pathology | |
| Hyperreflexia | Multiple sclerosis | |
| Hypertonia | Brain tumour | |
| Myopathies | Usually proximal weakness | Muscular dystrophies |
| Inflammatory myopathies | ||
| Corticosteroids | ||
| Alcohol | ||
| Functional weakness | Inconsistent weakness | Conversion disorders |
| Hoover’s sign | ||
| No ‘hard’ neurological signs |
11.20 Definitions of paralysis
| Term | Definition |
| Paresis | Partial paralysis |
| Plegia | Complete paralysis |
| Monoplegia | Involvement of a single limb |
| Hemiplegia | Involvement of one-half of the body |
| Paraplegia/diplegia | Paralysis of the legs |
| Tetraplegia | Paralysis of all four limbs |
Examination sequence
Do not test every muscle in most patients; the commonly tested muscles are listed in Box 11.21.
11.21 Nerve and muscle supplies of commonly tested movements
| Movement | Muscle | Nerve/root |
| Shoulder abduction | Deltoid | Axillary C5 |
| Elbow flexion | Biceps Brachioradialis |
Musculocutaneous C5, 6 Radial C6 |
| Elbow extension | Triceps | Radial C7 |
| Wrist extension | Extensor carpi radialis longus | Posterior interosseus nerve (radial) C6 |
| Finger extension | Extensor digitorum communis | Posterior interosseous (radial) C7 |
| Finger flexion | Flexor pollicis longus (thumb) Flexor digitorum profundus (index and middle fingers) |
Anterior interosseus (median) C8 |
| Flexor digitorum profundus (ring and little fingers) | Ulnar C8 | |
| Finger abduction | First dorsal interosseous | Ulnar T1 |
| Thumb abduction | Abductor pollicis brevis | Median T1 |
| Hip flexion | Iliopsoas | Iliofemoral nerve L1, 2 |
| Hip extension | Gluteus maximus | Sciatic L5/S1 |
| Knee flexion | Hamstrings | Sciatic S1 |
| Knee extension | Quadriceps | Femoral L3/4 |
| Ankle dorsiflexion | Tibialis anterior | Deep peroneal L4, L5 |
| Ankle plantar flexion | Gastrocnemius and soleus | Tibial S1/2 |
| Great toe extension (dorsiflexion) | Extensor hallucis longus | Deep peroneal L5 |
| Ankle eversion | Peronei | Superficial peroneal L5/S1 |
| Ankle inversion | Tibialis posterior | Tibial nerve L4, 5 |
Ask about pain which may interfere with testing.
Test upper limb power with the patient sitting on the edge of the couch. Test lower limb power with the patient reclining.
Ask the patient to undertake a movement. First assess whether he can overcome gravity, e.g. instruct the patient ‘Lift your right leg off the bed’ to test hip flexion. Then apply resistance to this movement testing across a single joint, e.g. apply resistance to the thigh in hip flexion, not the lower leg.
Ask the patient to lift his arms above his head.
Ask him to ‘play the piano’, checking movements of the outstretched arms (asymmetric loss of fine finger movement may be a very early sign of cortical or extrapyramidal disease).
Observe the patient getting up from a chair and walking. Assess individual muscles depending on the history.
Observe the patient with his arms outstretched and supinated (palms up) and eyes closed for ‘pronator drift’, when one arm starts to pronate (Box 11.22). Asking the patient to squeeze your fingers with his hand assesses the patient’s ability to obey commands, not power.
11.22
An early feature of upper motor neurone lesion
Pronator drift is an early feature of an upper motor neurone lesion and the test has good specificity and sensitivity.
Anderson NE. The forearm and finger rolling tests. Pract Neurol 2010;10:39–42.
To test truncal strength, ask the patient to sit up from the lying position, or rise from a chair, without using the arms.
Abnormal findings: Upper motor neurone lesions produce weakness of a relatively large group of muscles, e.g. a limb or more than one limb. Lower motor neurone damage can cause paresis of an individual and specific muscle so more detailed examination of individual muscles is required (Ch. 14). Look for patterns of weakness which may suggest a diagnosis (Box 11.20). Patients may find it difficult to sustain maximum power for reasons other than weakness, most commonly pain. You need only show that the patient can achieve maximum power briefly. Very few organic diseases cause power to fluctuate; the fatigable weakness of myasthenia is the chief exception. Wildly fluctuating or sudden ‘giveway’ weakness suggests a functional explanation. Hoover’s sign is often present in functional leg weakness, and is helpful diagnostically and therapeutically (you can show patients that the leg is not actually weak using this sign).
Deep tendon reflexes
Anatomy: A tendon reflex is the involuntary contraction of a muscle in response to stretch. It is mediated by a reflex arc consisting of an afferent (sensory) and an efferent (motor) neurone with one synapse between (a monosynaptic reflex). Muscle stretch activates the muscle spindles, which send a burst of afferent signals that lead to direct efferent impulses, causing muscle contraction. These stretch reflex arcs are served by a particular spinal cord segment which is modified by descending upper motor neurones.
Abnormal findings: Hyperreflexia (abnormally brisk reflexes) is a sign of upper motor neurone damage. Diminished or absent jerks are most commonly due to lower motor neurone lesions. In healthy elderly people the ankle jerks may be reduced or lost (Box 11.23), and in the Holmes–Adie syndrome, myotonic pupils (Fig. 12.26B and p. 292) are associated with loss of some deep tendon reflexes. Isolated loss of a reflex suggests a mononeuropathy or radiculopathy, e.g. loss of ankle jerk with L5/S1 lumbosacral disc prolapse compressing the S1 nerve root. Reflex patterns are helpful in localising neurological lesions, but you should know the nerve roots which serve the commonly tested reflexes (Box 11.24). There are several reflex-grading systems, but interobserver agreement is poor; record reflexes as present (and if so, whether normal, increased or decreased) or absent. Never conclude a reflex is absent until you have used reinforcement; this is a technique when concurrent motor activity in other muscles may augment (reinforce) the reflex tested.
11.23
Ankle jerks
Reduced or lost ankle jerks may be normal in elderly people.
Vrancken AFJE, Kalmijn S, Brugman F et al. The meaning of distal sensory loss and absent ankle reflexes in relation to age: a meta-analysis. J Neurol 2006;253:578–589.
11.24 Monosynaptic (deep tendon) reflexes and root innervation
| Reflex (muscle) | Nerve root |
| Biceps | C5 |
| Supinator (brachioradialis) | C6 |
| Triceps | C7 |
| Knee (quadriceps) | L3, 4 |
| Ankle (gastrocnemius, soleus) | S1 |
An ‘inverted’ biceps reflex is caused by combined spinal cord and root pathology localising to a specific spinal level. It is most common at the C5/6 level. When elicited, the biceps reflex is absent or reduced but finger flexion occurs. This is because the lesion at the C5/6 level affects the efferent arc of the biceps jerk (C5 nerve root), causing it to be reduced or lost, and also the spinal cord increasing reflexes below this level (including the finger jerks). It is most commonly seen in cervical spondylotic myeloradiculopathy.
A positive Hoffmann’s reflex (thumb flexion elicited by flicking the distal phalanx of the middle finger) and finger jerks suggest hypertonia, but can occur in healthy individuals, and are not useful signs in isolation. In cerebellar disease the reflexes may be pendular, and muscle contraction and relaxation tend to be slow, but these are not sensitive or specific cerebellar signs.
Examination sequence
Ask the patient to lie supine on the examination couch with the limbs exposed. He should be as relaxed and comfortable as possible, as anxiety and pain can cause an increased response.
Flex your wrist and allow the weight of the tendon hammer head to determine the strength of the blow. Strike the tendon, not the muscle or bone.
Ensure that both limbs are positioned identically with the same amount of stretch.
Compare each reflex with the other side; check for symmetry of response (Figs 11.18 and 11.19).
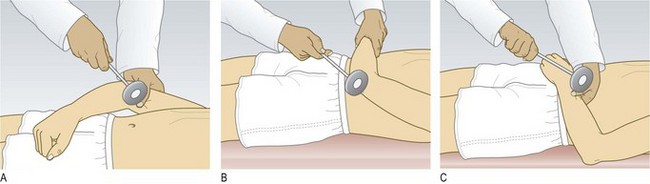
Fig. 11.18 Testing the deep tendon reflexes of the upper limb.
(A) Eliciting the biceps jerk, C5. (B) Triceps jerk, C7. (C) Supinator jerk, C6.
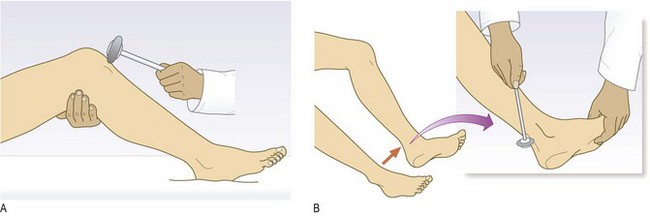
Fig. 11.19 Testing the deep tendon reflexes of the lower limb.
(A) Eliciting the knee jerk (note that the legs should not be in contact with each other), L3, L4. (B) Ankle jerk of recumbent patient, S1.
Use reinforcement whenever a reflex appears absent. For knee and ankle reflexes, ask the patient to interlock the fingers and pull one hand against the other on your command, immediately before you strike the tendon (Jendrassik’s manœuvre; Fig. 11.20).
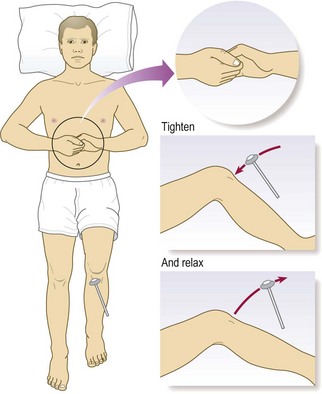
To reinforce upper limb reflexes, ask the patient to clench the teeth or to make a fist with the contralateral hand. The patient should relax between repeated attempts. Strike the tendon immediately after your command to the patient.
Place your right index finger under the distal interphalangeal joint of the patient’s middle finger.
Use your right thumb to flick the patient’s finger downwards.
Look for any reflex flexion of the patient’s thumb (Fig. 11.21A).
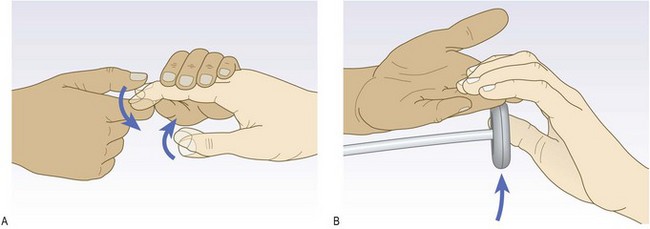
Place your middle and index fingers across the palmar surface of the patient’s proximal phalanges.
Tap your own fingers with the hammer.
Watch for flexion of the patient’s fingers (Fig. 11.21B).
Superficial reflexes
This group of reflexes is polysynaptic and elicited by cutaneous stimulation rather than stretch. With the exception of the plantar response, they are not part of the routine examination, and have poor sensitivity and specificity. The cremasteric reflex applies only in males.
Abnormal findings: An abnormal plantar response is extension of the large toe (extensor plantar or Babinski response). This is a sign of upper motor neurone damage and is usually associated with other upper motor neurone signs, e.g. spasticity, clonus and hyperreflexia. Fanning of the toes is normal and not pathological.
Superficial abdominal reflexes (T8–12) are lost in upper motor neurone lesions but are also affected by lower motor neurone damage affecting T8–12. They are usually absent in the obese, the elderly or after abdominal surgery.
The cremasteric reflex in males (L1 and L2) is rarely elicited, but typically is lost in spinal cord or root lesions.
Examination sequence
Run a blunt object (orange stick) along the lateral border of the sole of the foot towards the little toe (Fig. 11.22).
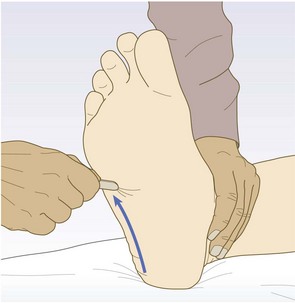
Watch both the first movement of the great toe and the other leg flexor muscles. The normal response is flexion of the great toe with flexion of the other toes.
The patient should be supine and relaxed.
Use an orange stick and briskly, but lightly, stroke the upper and lower quadrants of the abdomen in a medial direction (Fig. 11.23).
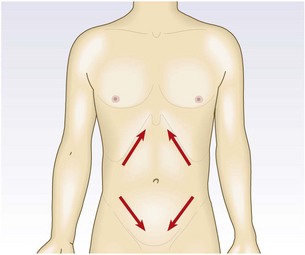
The normal response is contraction of the underlying muscle, with the umbilicus moving laterally and up or down depending upon the quadrant tested.
Primitive reflexes
These are present in normal neonates and young infants but disappear as the nervous system matures. People with congenital or hereditary cerebral lesions and a few healthy individuals retain these reflexes, but their return after early childhood is often associated with brain damage or degeneration. The primitive reflexes (snout, grasp, palmomental and glabellar tap) have little localising value and in isolation are of little significance, but in combination suggest diffuse or frontal cerebral damage (Box 11.25). Unilateral grasp and palmomental reflexes may occur with contralateral frontal lobe pathology. The glabellar tap is an unreliable sign of Parkinson’s disease.
Coordination
Performing complex movements smoothly and efficiently depends upon intact sensory and motor function and an intact cerebellum.
Anatomy: The cerebellum lies in the posterior fossa and consists of two hemispheres with a central vermis. Afferent and efferent pathways convey information to and from the cerebral motor cortex, basal ganglia, thalamus, vestibular and other brainstem nuclei and the spinal cord. In general, midline structures, e.g. vermis, influence body equilibrium, while each hemisphere controls ipsilateral coordination.
Examination sequence
Test cerebellar function by testing limb coordination, then for dysarthria (p. 250), nystagmus (p. 291), stance and gait (p. 249).
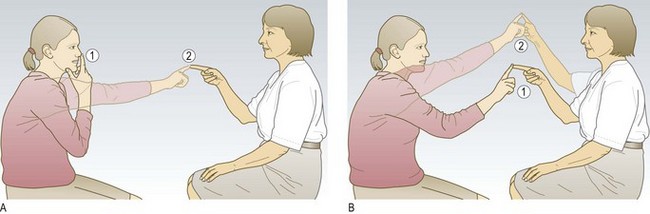
Ask the patient to touch her nose with the tip of her index finger and then touch your finger tip. Hold your finger just within the patient’s arm’s reach (you should make the patient use her arm outstretched).
Ask her to repeat the movement between nose and target finger as quickly as possible.
Make the test more sensitive by changing the position of your target finger. Timing is crucial – move your finger just as the patient’s finger is about to leave her nose, otherwise you will induce a false-positive finger-to-nose ataxia.
Some patients are so ataxic that they may injure their eye/face with this test. If so, use your two hands as the targets (Fig. 11.24).
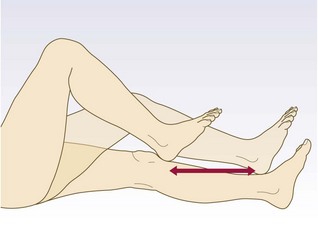
With the patient lying supine, ask him to place his heel on his opposite knee, and then slide his heel up and down the shin between knee and ankle (Fig. 11.25).
Demonstrate repeatedly patting the palm of your hand with the palm and back of your opposite hand as quickly and regularly as possible.
Ask the patient to copy your actions.
Repeat with the opposite hand.
Alternatively, ask the patient to tap a steady rhythm rapidly with his hand on the other hand or table, and ‘listen to the cerebellum’; ataxia makes this task difficult, with a slower, irregular rhythm than normal.
Abnormal findings: The finger-to-nose test may reveal a tendency to fall short or overshoot the examiner’s finger (dysmetria or past-pointing). In more severe cases there may be a tremor of the finger as it approaches the target finger and the patient’s own nose (intention tremor). The movement may be slow, disjointed and clumsy (dyssynergia). The heel-to-shin test is the equivalent test for the lower limbs. It is abnormal if the heel wavers away from the line of the shin. Weakness may produce false-positive finger-to-nose or heel-to-shin tests, so demonstrate that power is normal first.
Dysdiadochokinesis (impairment of rapid alternating movements) is evident as slowness, disorganisation and irregularity of movement. Dysarthria (p. 250) and nystagmus (p. 283) also occur with cerebellar disease. Much less reliable signs of cerebellar disease include: the rebound phenomenon, when the displaced outstretched arm may fly up past the original position (the normal response is to return to the original position); pendular reflexes; and hypotonia.
In disorders predominantly affecting midline cerebellar structures, e.g. tumours of the vermis and alcoholic cerebellar damage, the above tests may be normal, and truncal ataxia may be the only finding. In the most severe cases, this may mean the patient cannot sit unsupported. Tandem gait (heel–toe walking) may be impaired in less severe cases. Cerebellar dysfunction occurs in many conditions, and the differential diagnosis varies with age and speed of presentation (Box 11.26).
Apraxia: Dyspraxia or apraxia is difficulty or inability to perform a task, despite no impairment of the necessary individual functions. It is a sign of higher cortical dysfunction, usually localising to the non-dominant frontal or parietal lobes.
Examination sequence
Ask the patient to perform an imaginary act, e.g. drinking a cup of tea, combing the hair, folding a letter and placing it in an envelope. Ask the patient to copy movements you make with your fingers, e.g. pointing.
Ask the patient to copy a geometrical figure (interlocking pentagons or cube).
Ask the patient to put on a pyjama top or dressing gown, one sleeve of which has been pulled inside out.
Abnormal findings: The patient may be unable to initiate a task or perform it in an odd or bizarre fashion.
Constructional apraxia (difficulty drawing a figure) is a feature of parietal disturbance.
Dressing apraxia, often associated with spatial disorientation and neglect, is usually due to non-dominant hemisphere parietal lesions.
The sensory system
Detailed examination of sensation is time-consuming and unnecessary unless the patient volunteers sensory symptoms or you suspect a specific pathology, e.g. spinal cord compression or mononeuropathy.
Anatomy
Proprioception (joint position sense) and vibration are conveyed in large, myelinated fast-conducting fibres in the peripheral nerves and in the posterior (dorsal) columns of the spinal cord. Pain and temperature sensation are carried by small, slow-conducting fibres of the peripheral nerves and the spinothalamic tract of the spinal cord. The posterior column remains ipsilateral from the point of entry up to the medulla, but most pain and temperature fibres cross to the contralateral spinothalamic tract within one or two segments of entry to the spinal cord. All sensory fibres relay in the thalamus before sending information to the sensory cortex in the parietal lobe (Fig. 11.26).
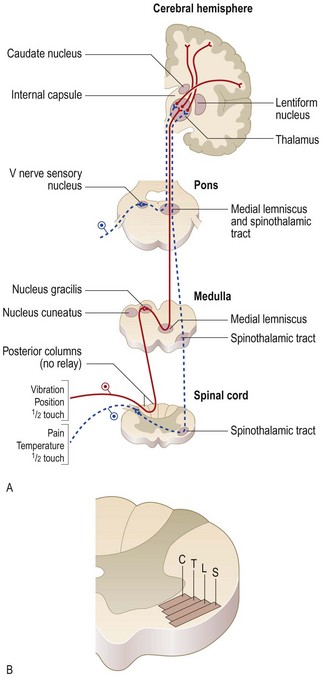
Fig. 11.26 The sensory system.
(A) Main sensory pathways. (B) Spinothalamic tract: layering of the spinothalamic tract in the cervical region. C represents fibres from cervical segments which lie centrally; fibres from thoracic, lumbar and sacral segments (labelled T, L and S respectively) lie progressively more laterally.
Symptoms and definitions
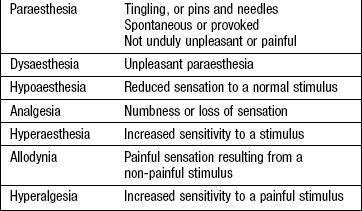
Sensory symptoms are numerous (Box 11.27), and it is important to discern what the patient is describing. Clarify that by ‘numbness’ the patient means lack of sensation rather than weakness or clumsiness. Neuropathic pain (pain due to disease or dysfunction of the PNS or CNS) is often severe and refractory to standard analgesia. Reduced ability to feel pain may be accompanied by scars from injuries or burns.
The sensory modalities: In addition to the modalities conveyed in the principal ascending pathways (touch, pain, temperature, vibration and joint position sense), sensory examination includes tests of discriminative aspects of sensation which may be impaired by lesions of the sensory cortex. Only assess these cortical sensory functions if the main pathway sensations are intact.
Consider abnormalities on sensory testing according to whether the lesion(s) is in the peripheral nerve(s), dorsal root(s), spinal cord, or intracranial.
Peripheral nerve and dorsal root: Many diseases affect peripheral nerves, generally resulting in peripheral neuropathies or polyneuropathies (Box 11.28). Peripheral neuropathies tend to affect the lower limbs first (length-dependent). Symptoms affecting the upper limbs first suggest a demyelinating rather than axonal neuropathy or a disease process in the spinal cord. In many cases, touch and pinprick sensation are lost in a ‘stocking and glove’ distribution (Fig. 11.27A).
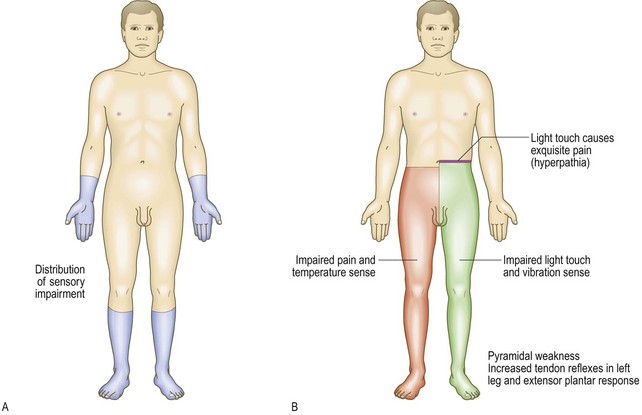
Fig. 11.27 Patterns of sensory loss.
(A) In length dependent peripheral neuropathy. (B) Brown-Séquard syndrome. Note the distribution of corticospinal, posterior column and lateral spinothalamic tract signs. The cord lesion is in the left half of the cord.
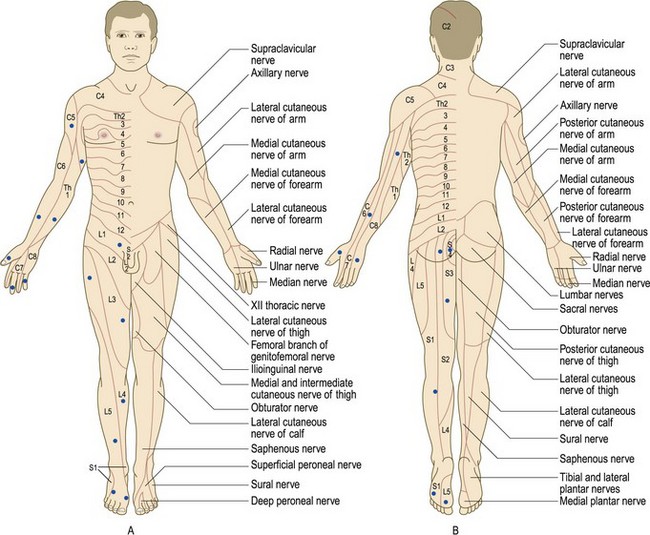
In large-fibre neuropathies, vibration and joint position sense are disproportionately affected. Patients may report staggering when they close their eyes during hair washing (Romberg’s sign: p. 249). When joint position sense is affected in the arms, pseudoathetosis may be demonstrated by asking the patient to close his eyes and hold his hands outstretched: the fingers will make involuntary, slow wandering movements, mimicking athetosis. Interpretation of sensory signs requires knowledge of the relevant anatomy of sensory nerves and dermatomes (Box 11.29, Fig. 11.28 and Fig. 11.32).
Spinal cord: Traumatic and compressive spinal cord lesions cause loss or impairment of sensation in a dermatomal distribution below the level of the lesion. A zone of hyperaesthesia may be found immediately above the level of sensory loss.
Anterior spinal artery syndrome usually results in loss of spinothalamic sensation and motor function, with sparing of dorsal column sensation. A similar dissociated pattern of pain and temperature loss and sparing of dorsal column sensation occurs in syringomyelia.
When one-half of the spinal cord is damaged, the Brown-Séquard syndrome may occur. This is characterised by ipsilateral motor weakness and loss of vibration and joint position sense, with contralateral loss of pain and temperature (Fig. 11.27B).
Intracranial: Brainstem lesions are often vascular, and you must understand the relevant anatomy to determine the site of the lesion (Fig. 11.29). Lower brainstem lesions may cause ipsilateral numbness on one side of the face (V nerve nucleus) and contralateral body numbness (spinothalamic tract).
Thalamic lesions may cause a patchy sensory impairment on the opposite side with unpleasant, poorly localised pain, often of a burning quality.
Cortical parietal lobe lesions typically cause sensory inattention but may also affect joint position sense, two-point discrimination, stereognosis (tactile recognition) and localisation of point touch. Two-point discrimination and touch localisation are not helpful signs and are not performed routinely.
Examination sequence
Use a fresh neurological pin, e.g. Neurotip, not a hypodermic needle. Dispose of the pin after each patient to avoid transmitting infection.
Explain and demonstrate that the ability to feel a sharp pinprick is being tested.
Map out the boundaries of any area of reduced, absent or increased sensation and compare with Figure 11.28. Move from reduced to higher sensibility: i.e. from hypoaesthesia to normal, or normal to hyperaesthesia.
Place a vibrating 128-Hz tuning fork over the sternum.
Ask the patient, ‘Do you feel it buzzing?’
Place it on the tip of the great toe (Fig. 11.30).
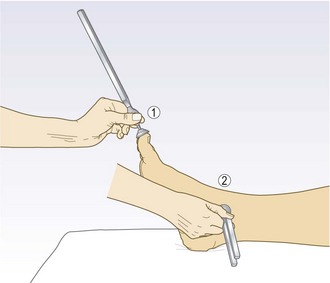
If sensation is impaired, place the fork on the interphalangeal joint and progress proximally, to the medial malleolus, tibial tuberosity and anterior iliac spine, depending upon the response.
Repeat the process in the upper limb. Start at the distal interphalangeal joint of the forefinger, and if sensation is impaired, proceed proximally.
If in doubt as to the accuracy of the response, ask the patient to close his eyes and to report when you stop the fork vibrating with your fingers.
With the patient’s eyes open, demonstrate the procedure.
Hold the distal phalanx of the patient’s great toe at the sides. Tell the patient you are going to move his toe up or down, demonstrating as you do so (Fig. 11.31).
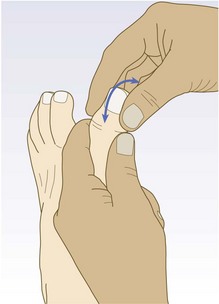
Ask the patient to close his eyes and to identify the direction of small movements in random order.
Test both great toes (or middle fingers). If impaired, move to more proximal joints in each limb.
The peripheral nerves
Peripheral nerves may be damaged individually (mononeuropathy) or multiply (peripheral neuropathy or mononeuritis multiplex). Certain nerves (median nerve at the wrist, common peroneal nerve at the knee) are prone to trauma or compression.
Median nerve
This may be compressed as it passes between the flexor retinaculum and the carpal bones at the wrist (carpal tunnel syndrome); it is the most common entrapment neuropathy and initially produces sensory symptoms (Box 11.30).
11.30 Common features of carpal tunnel syndrome
• Unpleasant tingling in the hand
• May not observe anatomical boundaries, radiating up the arm to the shoulder
• Weakness uncommon, but affects thumb abduction if occurs
• Symptoms commonly occur at night, wakening patient from sleep
• The patient may hang the hand and arm out of the bed for relief
Examination sequence
Test for altered sensation over the hand involving the thumb, index and middle fingers and the lateral half of the ring finger – splitting of the ring finger (Fig. 11.32A and Fig. 14.29).
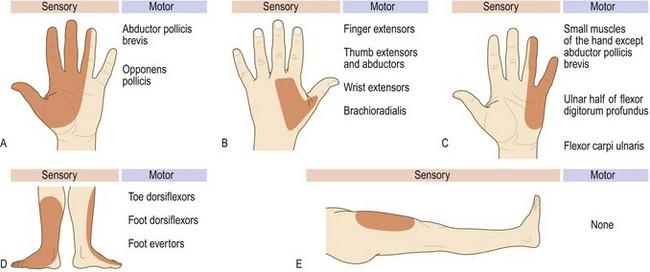
Fig. 11.32 Sensory and motor deficits in nerve lesions.
(A) Median. (B) Radial. (C) Ulnar. (D) Common peroneal. (E) Lateral cutaneous of the thigh.
Look for wasting of the thenar eminence.
Test thumb abduction with the patient’s hand held palm up on a flat surface. Ask the patient to move the thumb vertically against your resistance (abductor pollicis brevis).
Test opposition by asking the patient to touch the thumb and ring finger together while you attempt to pull them apart (opponens pollicis).
Radial nerve
This may be compressed as it runs through the axilla, or injured in fractures of the humerus. It typically causes wrist drop.
Examination sequence
Test for weakness of arm and forearm extensors (triceps and the wrist and fingers).
Look for sensory loss over the dorsum of the hand (Fig. 11.32B) and loss of triceps tendon jerk.
Ulnar nerve
This is most often affected at the elbow by external compression or injury, e.g. dislocation.
Examination sequence
Look for wasting of interossei (dorsal guttering).
Test for weakness of finger abduction with the patient’s fingers on a flat surface, and ask him to spread the fingers against resistance from your fingers.
Test adduction by placing a card between the patient’s fingers and pulling it out using your own fingers.
Assess for sensory loss on the ulnar side of the hand, splitting the ring finger (Fig. 11.32C).
Examine the elbow (the commonest place of entrapment). Note any scars or other signs of trauma.
Examine the range of movement and feel for the nerve in the ulnar groove.
Common peroneal nerve: This typically presents with foot drop. It may be damaged in fibular head fractures, or compressed particularly in immobile patients, or as a result of repetitive kneeling or squatting.
Examination sequence
Test for weakness of ankle dorsiflexion and eversion. Inversion will be preserved.
Test for sensory loss over the dorsum of the foot (Fig. 11.32D).
Lateral cutaneous nerve of thigh: This purely sensory nerve may be compressed as it passes under the inguinal ligament, producing paraesthesiae in the lateral thigh (meralgia paraesthetica) (Fig. 11.32E).
Putting it all together
Having completed the history and examination, decide whether the symptoms are due to neurological disease. Determine the site(s) of damage (where is the lesion?). Try to localise the lesion to a single area of the nervous system, although some conditions may cause multiple symptoms and signs due to several lesions, e.g. multiple sclerosis. Consider the likely underlying pathology: what is the lesion?
Draw up a differential diagnosis and then consider which (if any) investigations are pertinent.
Do not place undue emphasis on an isolated sign that fails to fit with the history, e.g. an apparently isolated extensor plantar response in a patient with typical migraine – it is likely this is a false-positive sign rather than indicating underlying pathology.
Remember that medically unexplained symptoms are common but distinguishing them from organic disease is difficult, even for experts (p. 27).
Investigations
See Box 11.31 and Figures 11.33-35. Lumbar puncture is a key investigation in a number of acute and chronic neurological conditions (Box 11.32). Always measure the CSF opening pressure (in a lying position, not sitting). CSF is routinely examined for cells, protein content, and glucose (in comparison to simultaneously taken blood glucose); it is also stained and cultured for bacteria. Other specific tests may be carried out, e.g. analysis for oligoclonal bands, meningococcal and pneumococcal antigens, polymerase chain reaction (PCR) for certain viruses or cytology for malignant cells.
 11.31
11.31
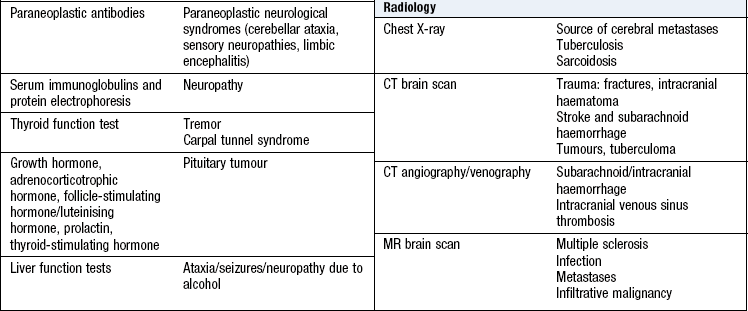
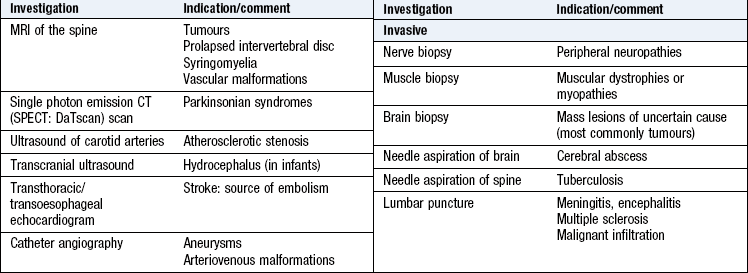
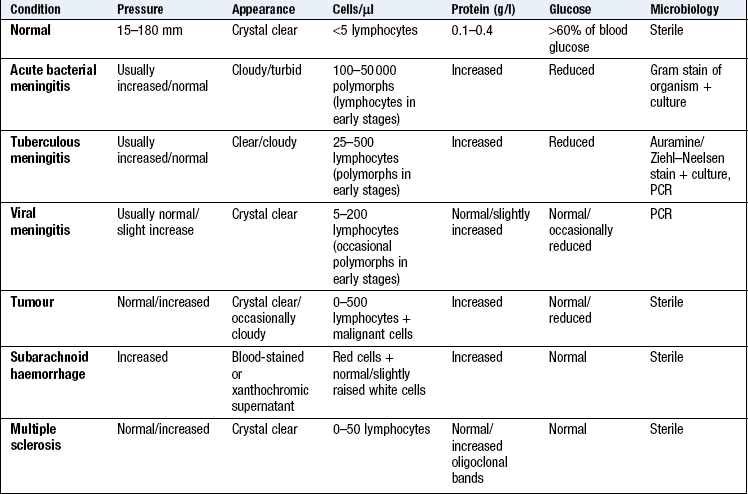
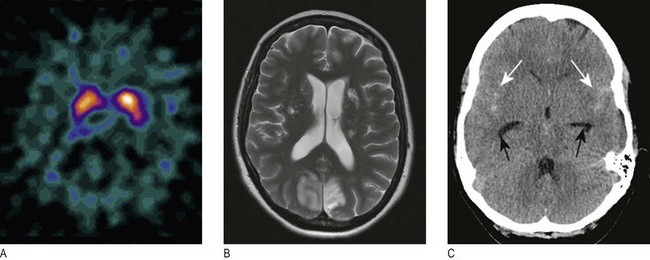
Fig. 11.33 Scanning of the head:
(A) DaTSCAN scan showing the distribution of blood flow on cross-section of the brain. (B) MR scan showing ischaemic stroke T2 imaging demonstrates bilateral occipital infarction and bilateral hemisphere lacunar infarction. (C) Unenhanced CT scan showing subarachnoid blood in both sylvian fissures (white arrows) and early hydrocephalus, with temporal horns of the lateral ventricles visible (black arrows).
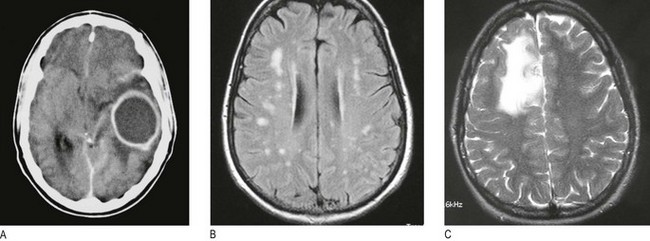
Neurophysiological tests
Electroencephalography (EEG) records the spontaneous electrical activity of the brain, using scalp electrodes. It is used in the investigation of epilepsy, encephalitis or dementia. Modifications to the standard EEG improve sensitivity, including sleep-deprived studies, prolonged video telemetry and invasive EEG monitoring.
Electromyography (EMG) involves needle electrodes inserted into muscle. Electrical activity is displayed on an oscilloscope and an audio monitor, allowing the neurophysiologist to see and hear the pattern of activity. Neurogenic and myogenic pathology may cause characteristic EMG abnormalities.
Nerve conduction studies involve applying electrical stimuli to nerves and measuring the speed of impulse conduction. They are used for both motor and sensory nerves, and are helpful in diagnosing peripheral nerve disorders such as nerve compressions or polyneuropathies.