Chapter 12 Biopharmaceuticals
Introduction
The term ‘biopharmaceutical’ was originally coined to define therapeutic proteins produced by genetic engineering, rather than by extraction from normal biological sources. Its meaning has broadened with time, and the term now encompasses nucleic acids as well as proteins, vaccines as well as therapeutic agents, and even cell-based therapies. In this chapter we describe the nature of biopharmaceuticals, and the similarities and differences in discovery and development between biopharmaceuticals and conventional small-molecule therapeutic agents. The usual starting point for biopharmaceuticals is a naturally occurring peptide, protein or nucleic acid. The ‘target’ is thus identified at the outset, and the process of target identification and validation, which is a major and often difficult step in the discovery of conventional therapeutics (see Chapter 6), is much less of an issue for biopharmaceuticals. Equally, the process of lead finding and optimization (Chapters 7, 8, 9) is generally unnecessary, or at least streamlined, because nature has already done the job. Even if it is desirable to alter the properties of the naturally occurring biomolecule, the chemical options will be much more limited than they are for purely synthetic compounds. In general, then, biopharmaceuticals require less investment in discovery technologies than do conventional drugs. Toxicity associated with reactive metabolites – a common cause of development failure with synthetic compounds – is uncommon with biopharmaceuticals. On the other hand, they generally require greater investment in two main areas, namely production methods and formulation. Production methods rely on harnessing biological systems to do the work of synthesis, and the problems of yield, consistency and quality control are more complex than they are for organic synthesis. Formulation problems arise commonly because biomolecules tend to be large and unstable, and considerable ingenuity is often needed to improve their pharmacokinetic properties, and to target their distribution in the body to where their actions are required.
It is beyond the scope of this book to give more than a brief account of the very diverse and rapidly developing field of biopharmaceuticals. More detail can be found in textbooks (Buckel, 2001; Ho and Gibaldi, 2003; Walsh, 2003). As the field of biopharmaceuticals moves on from being mainly concerned with making key hormones, antibodies and other signalling molecules available as therapeutic agents, efforts – many of them highly ingenious – are being made to produce therapeutic effects in other ways. These include, for example, using antisense nucleic acids, ribozymes or RNAi (see below and Chapter 6) to reduce gene expression, the use of catalytic antibodies to control chemical reactions in specific cells or tissues, and the development of ‘DNA vaccines’. So far, very few of these more complex ‘second-generation’ biopharmaceutical ideas have moved beyond the experimental stage, but there is little doubt that the therapeutic strategies of the future will be based on more sophisticated ways of affecting biological control mechanisms than the simple ‘ligand → target → effect’ pharmacological principle on which most conventional drugs are based.
Recombinant DNA technology: the engine driving biotechnology
The discovery of enzymes for manipulating and engineering DNA – the bacterial restriction endonucleases, polynucleotide ligase and DNA polymerase – and the invention of the enabling technologies of DNA sequencing and copying of DNA sequences by using the polymerase chain reaction (PCR) allowed rapid determination of the amino acid sequence of a protein from its mRNA message. Versatile systems for introducing nucleic acids into target cells or tissues and for the control of host nucleic acid metabolism brought the potential for correction of genetic defects and for new therapeutic products for disorders poorly served by conventional small-molecule drugs. The importance of these discoveries for the biological sciences was indicated by the many Nobel Prizes awarded for related work. No less critical was the impact that these reagents and technologies had on applied science, especially in the pharmaceutical industry. The business opportunities afforded by biotechnology spawned thousands of startup companies and profoundly changed the relationship between academia and industry. The mainstream pharmaceutical industry, with its 20th-century focus on small-molecule therapeutic agents, did not immediately embrace the new methodologies except as research tools in the hands of some discovery scientists. Entrepreneurs in biotechnology startup firms eventually brought technology platforms and products to large pharmaceutical companies as services or as products for full-scale development. This alliance allowed each party to concentrate on the part they did best.
Biotechnology products were naturally attractive to the small startup companies. Because the protein or nucleic acid itself was the product, it was unnecessary to have medicinal chemists synthesize large collections of organic small-molecule compounds to screen for activity. A small energetic company with the right molecule could come up with a profitable and very useful product. The niche markets available for many of the initial protein or nucleic acid products were sufficient to support a small research-based company with a high-profit-margin therapeutic agent.
The early days of protein therapeutics
Along with plant-derived natural products, proteins and peptides were some of the first therapeutic agents produced by the fledgling pharmaceutical industry in the latter half of the 19th century, before synthetic chemistry became established as a means of making drugs. Long before antibiotics were discovered, serum from immune animals or humans was successfully used to treat a variety of infectious diseases. Serotherapy was the accepted treatment for Haemophilus influenzae meningitis, measles, diphtheria, tetanus, hepatitis A and B, poliovirus, cytomegalovirus and lobar pneumonia. Antisera raised in animals were used to provide passive protection from diphtheria and tetanus infection.
Extracts of tissues provided hormones, many of which were polypeptides. After its discovery in 1921, insulin extracted from animal pancreas replaced a starvation regimen for treating diabetes. The size of the diabetic population, and the activity in humans of the hormone isolated from pancreas of pigs and cows, permitted early commercial success. Generally, however, the low yield of many hormones and growth factors from human or animal sources made industrial scale isolation difficult and often uneconomic. Nevertheless, several such hormones were developed commercially, including follicle-stimulating hormone (FSH) extracted from human urine to treat infertility, glucagon extracted from pig pancreas to treat hypoglycaemia, and growth hormone, extracted from human pituitary to treat growth disorders. Some enzymes, such as glucocerebrosidase, extracted from human placenta and used to treat an inherited lipid storage disease (Gaucher’s disease), and urokinase, a thrombolytic agent extracted from human urine, were also developed as commercial products.
Some serious problems emerged when proteins extracted from human or animal tissues were developed for therapeutic use. In particular:
• Repeated dosage of non-human proteins generated an immune response in some patients against the foreign sequences, which differed by several amino acids from the human sequence. Such immune responses could cause illnesses such as serum sickness, or loss of efficacy of the protein.
• Human tissue was in short supply and was subject to potential contamination with infectious agents. Growth hormone extracted from human cadaver pituitary glands was contaminated with prions that cause Creutzfeld–Jakob disease, a dementing brain-wasting disease similar to bovine spongiform encephalitis (BSE) and sheep scrapie. Human blood plasma-derived products have been tainted with hepatitis B virus and HIV.
• Many agents (e.g. cytokines) cannot be extracted in sufficient quantities to be used therapeutically.
• Batch-to-batch variability was considerable, requiring standardization by bioassay in many cases.
The recombinant DNA revolution and the subsequent development of biotechnology resolved many of these issues. Many vaccines and antisera, however, are still prepared from blood products or infectious organisms, rather than by recombinant DNA methods.
Currently available classes of biopharmaceuticals
The major classes of biopharmaceuticals currently on the market include hormones, cytokines, growth factors, antibodies, enzymes, vaccines and nucleotide-based agents. Examples of therapeutic proteins, including antibodies, enzymes and other proteins approved for clinical use, are presented in Table 12.1. Others not included in the compilation include therapeutic preparations such as serum albumin, haemoglobin and collagen, which are not drugs in the conventional sense.
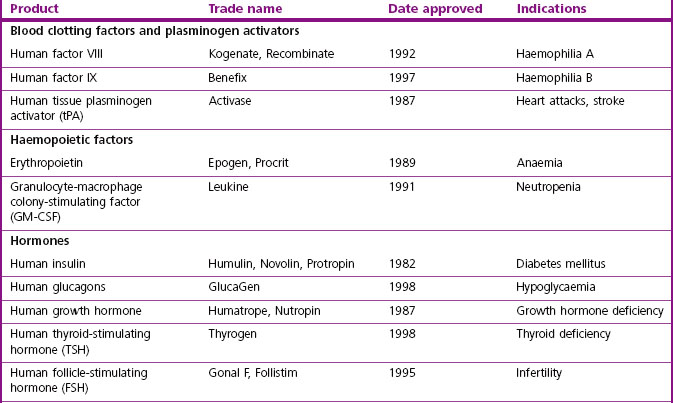
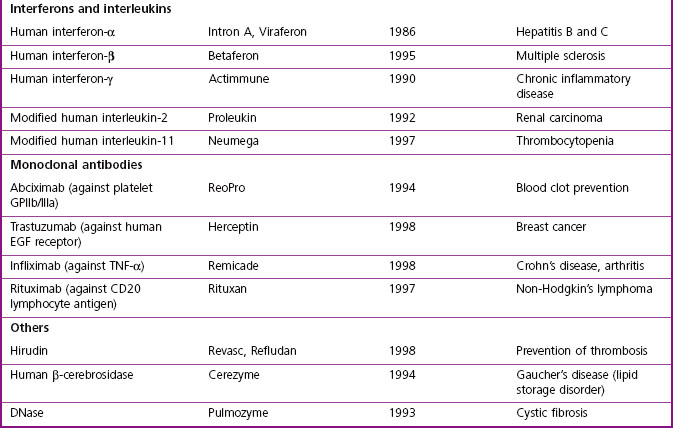
In addition to the ‘mainstream’ biopharmaceuticals considered here are numerous speciality products for niche markets that are under investigation or in development by small ‘boutique’ companies.
Growth factors and cytokines
The production, differentiation and survival of the various types of blood cell are tightly regulated by an interacting network of hormones, cytokines and growth factors. Species-specific activities of many of these chemical mediators, and their very low abundance, highlighted the need for biopharmaceutical products.
The most common uses of haemopoietic factors are for the treatment of various types of neutropenia, where specific white cell levels are depressed as a result of infection, immune disorders, recovery from chemotherapy or reaction to various drug regimens. They are especially useful in aiding recovery from the dose-limiting side effects of cancer chemotherapy. Granulocyte colony-stimulating factor (G-CSF) and granulocyte-macrophage colony-stimulating factor (GM-CSF) are used for this purpose to improve patient quality of life and allow the continuation of chemotherapy.
Erythropoietin (EPO), normally secreted by the kidney to stimulate the production of red blood cells, is the most successful biotechnology product so far marketed. EPO boosts red cell counts and reduces transfusion requirements for patients rendered anaemic by cancer chemotherapy or renal disease. Various forms of EPO with clearance profiles – and hence duration of action – modified by linkage with polyethylene glycol (PEGylation) or alteration of its glycosylation (see below and Chapter 17) are also available. Off-label use of EPO by athletes to improve performance has caused controversy.
Interferons are a complex group of proteins that augment immune effector cell function. Interferon-α was the first recombinant biotherapeutic agent approved by the FDA for cancer treatment. Recombinant interferons have been approved for melanoma, hepatitis C, Karposi’s sarcoma, T-cell lymphoma, chronic myelogenous leukaemia, multiple sclerosis and severe malignant osteopetrosis. Mechanism-based side effects have thus far restricted their utility to these severe disorders.
Hormones
Hormone replacement or augmentation is a commonly accepted medical practice in certain diseases of deficiency or misregulation. Insulin isolated from biological sources is administered to diabetics to control blood glucose levels. Immunological reaction to non-human (porcine or bovine) insulin preparations, which occur in a significant number of patients, are avoided by recombinant products incorporating parts of the human insulin sequence. A variety of human insulin and glucagon preparations are now on the market.
Human growth hormone (somatotropin) was originally developed for treating paediatric growth failure and Turner’s syndrome. Originally, growth hormone was extracted from human pituitary tissue post mortem, but this material carried a significant risk of transmitting Creutzfeld-Jakob disease, a fatal neurodegenerative condition now known to be transmitted by a prion, an abnormal protein found in affected nervous tissue, whose existence was unsuspected when human-derived growth hormone was introduced as a therapeutic agent. The production of human growth hormone by recombinant methods rather than extraction avoids this serious problem as well as providing a much more abundant source. Growth hormone has acquired notoriety since the potential for misuse to produce taller and stronger athletes was realized.
Human gonadotropin-releasing hormones have been extensively used in fertility management as well as in treating endometriosis and precocious puberty. Originally isolated from urine, there are now numerous recombinant products on the market.
Coagulation factors
Coagulation factors are a group of plasma proteins required for proper haemostasis. Deficiencies are associated with genetic lesions and occur as complications of viral infections such as hepatitis C and HIV. They were initially treated with plasma-derived concentrates, which carried a significant risk of viral and prion contamination. Recombinant factor VIII (Recombinate, Kogenate) and factor IX (BeneFix) avoid this risk and are now widely used.
Antithrombotic factors
To reduce blood coagulation, when conventional heparin or warfarin therapy is contraindicated, recombinant thrombin inhibitors (Leprudin, Bivalirudin) and antiplatelet gpIIb/IIIa antagonists (Eptifibatide) are now available. In conditions such as stroke, coronary thrombosis and pulmonary embolism early treatment with thrombolytic agents to relieve the vascular block is highly beneficial. Streptokinase and urokinase are proteases that process plasminogen to plasmin, activating its thrombolytic activity to dissolve fibrin clots. Newer products include tissue plasminogen activator (tPA) and TNKase, which catalyse the same reaction but in a fibrin-dependent fashion, so that plasmin production is concentrated in the region of the clot. These proteins are also less immunogenic than the bacterial streptokinase, which is important in cases where repeated administration of the thrombolytic agent is required.
Another regulator of coagulation is the serine protease protein C. The activated form of protein C breaks down the clotting factors Va and VIIIa and plasminogen activator inhibitor-1, tipping the balance in the favour of thrombolysis. These enzymes are important bio-pharmaceutical products that have no small-molecule counterpart.
Therapeutic antibodies
Monoclonal antibodies
A breakthrough in high-quality reproducible and scalable production of antibodies came with the development by Kohler and Milstein in 1975 of monoclonal antibodies. Fusion of primed T cells from an immunized mouse with an immortalized mouse myeloma (B-cell) line that secretes immunoglobulin light chains provided a cell-culture system that could produce unlimited quantities of antibody with defined specificity. Single-cell clones secreted antibody against a single epitope of the antigen. The initial immunization could, for toxic or scarce immunogens, be replaced by in vitro stimulation of isolated mouse thymocytes for fusion with the myeloma cells.
The technology for antibody production has now gone mouseless. It has moved into the realm of molecular biology, in which bacteriophages, gene libraries in plasmids and bacterial hosts are engineered to produce either whole antibodies or derivatives of antibodies with desired properties. ‘Humanizing’ the antibodies, or replacing the rodent constant domains with human sequences (chimerization), limits hypersensitivity reactions to foreign protein. Chimerization increases the half-life of the antibodies in human plasma up to six-fold and improves their function within the human immune network. The human Fc domain reacts with Fc receptors on human cells more avidly. Chimeric antibodies with human constant regions also interact optimally with human complement proteins, and are thus more effective in destroying target cells in patients than are their rodent counterparts.
Antibody selection by phage display
Phage display technology (Benhar, 2001) is a useful way to identify antigen combining regions to produce monoclonal antibodies that bind to therapeutically relevant antigens. Bacteriophages (or phages) are viruses that replicate in bacteria, Escherichia coli being the organism of choice in most cases. For antibody selection (Figure 12.1) a large DNA library, encoding millions of different putative antigen-binding domains, is incorporated into phage DNA, so that each phage particle encodes a single antigen combining region. The mixed phage population is added to E. coli cultures, where the phage replicate, each phage particle expressing copies of a single antigen combining region on its surface. The phage suspension is applied to plates coated with the antigen of interest (‘panning’) and those phage particles expressing antigen combining regions recognizing the antigen stick to the plates. The adherent phages are isolated, allowing the antigen combining region-encoding DNA to be identified and inserted into the DNA sequence encoding an appropriate full-size antibody scaffold to produce a specific antibody, or into a reduced size, simplified monomeric framework to produce a single-chain (sFv) antibody.
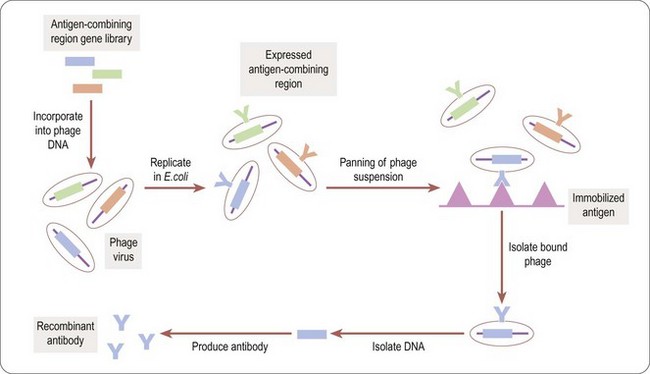
Uses of antibodies as therapeutic agents
Cancer immunotherapy
Although high-affinity mouse monoclonal antibodies to target antigens can be reproducibly produced in industrial quantities, and bind to specific human targets, they generally function poorly in recruiting human effector functions. The therapeutic potency of these antibodies can be enhanced by taking advantage of the targeting selectivity of antibodies (see Chapter 17) for the purposes of drug delivery. Linking other agents, such as cytotoxic drugs, biological toxins, radioisotopes or enzymes to activate prodrugs to targeting antibodies enhances their delivery to the target cells and reduces side effects by directing the toxic agents to the tumour and minimizing clearance. Bispecific antibodies with one H-L chain pair directed against a target cell antigen and the other against a soluble effector such as a complement component, or against a cell surface marker of an effector cell type, have also been developed to bring the components of the reaction together. A number of these are in clinical trials for a variety of different malignancies, but have in general proved less successful than had been expected on the basis of animal studies.
Antibody use in transplantation and immunomodulation
The major obstacle in the transplantation of cells, tissues or organs is the body’s recognition of foreign material. A strong immune response to rid the body of the non-self antigens leads to rejection of the transplant. Selective immunological suppression can ablate components of the antitransplant response. Fab fragments of antibodies to a number of surface antigens on T-cells are used to partially block T-cell responses to donor cells. Fab fragments are required because the Fc portion of the complete antibody targets the cell for destruction, resulting in unwanted complete immunosuppression.
Even human antibodies face disadvantages as therapeutics. Because of their size (MW ~150 kDa), their ability to penetrate into tissues, such as solid tumours, is limited. Engineered versions lacking the Fc region, which is largely responsible for hypersensitivity responses of patients treated with monoclonal antibodies, have replaced the (Fab)2 fragments generated by proteolysis from the intact antibody. Single sFv chains containing the H and L variable regions are the smallest antibodies containing a high-affinity antigen-combining site. Still in the experimental stage, ‘di-antibodies’ with two H-L units connected by a 15-amino acid linker (Gly4Ser)3 peptide have greatly increased affinity for antigen (see also Chapter 13).
Catalytic antibodies
Antibodies can be used to enhance the chemical reactivity of molecules to which they bind. Such catalytic antibodies (‘abzymes’) created with transition state analogs as immunogens specifically enhance substrate hydrolysis by factors of 102–105 over the rate in their absence, and this principle has been applied to the development of therapeutic agents (Tellier, 2002). Both esterase and amidase activities have been reported. Catalytic turnover of substrates by abzymes is low in comparison to true enzymes, as high-affinity binding impedes the release of products. Attempts to improve catalytic efficiency and to identify therapeutic uses for catalytic antibodies have engrossed both academic and biotech startup laboratories. Targets being approached with these antibodies include cocaine overdose and drug addiction, bacterial endotoxin, and anticancer monoclonal antibody conjugated with a catalytic antibody designed to activate a cytotoxic prodrug. Attempts are also being made to develop proteolytic antibodies containing a catalytic triad analogous to that of serine proteases, designed to cleave gp120 (for treatment of HIV), IgE (for treatment of allergy), or epidermal growth factor receptor (for treatment of cancer).
Therapeutic enzymes
Enzymes can be useful therapeutic agents as replacements for endogenous sources of activity that are deficient as a result of disease or genetic mutation. Because enzymes are large proteins they generally do not pass through cellular membranes, and do not penetrate into tissues from the bloodstream unless assisted by some delivery system. Lysosomal hydrolases such as cerebrosidase and glucosidase have targeting signals that allow them to be taken up by cells and delivered to the lysosome. Genetic lysosomal storage diseases (Gaucher’s, Tay–Sachs) are treated by enzyme replacement therapy. However, penetration of the enzymes into the nervous system, where the most severe effects of the disease are expressed, is poor. Cystic fibrosis, a genetic disorder characterized by deficits in salt secretion, is treated with digestive enzyme supplements and an inhaled DNase preparation (dornase-α) to reduce the extracellular viscosity of the mucous layer in the lung to ease breathing. All of these are produced as recombinant proteins.
Vaccines
Using the immune system to protect the body against certain organisms or conditions is a powerful way to provide long-term protection against disease. Unlike with small-molecule pharmaceuticals, which are administered when needed, once immunity is present subsequent exposure to the stimulus automatically activates the response. Most current vaccines are against disease-causing organisms such as bacteria, viruses and parasites. More complex conditions where the antigens are not so well defined are also being addressed by immunization. Vaccines are being tested for cancer, neurodegenerative diseases, contraception, heart disease, autoimmune diseases, and alcohol and drug addiction (Rousseau et al., 2001; Biaggi et al., 2002; BSI Vaccine Immunology Group, 2002; Kantak, 2003). Immune induction is complex. Pioneering experiments with attenuation of disease organisms showed that illness was not required for immunity. The goal in immunization is to retain enough of the disease-causing trait of an antigen to confer protection without causing the disease. For infectious agents, various methods of killing or weakening the organism by drying or exposure to inactivating agents still dominate manufacturing processes. Isolation of antigens from the organisms, or modifying their toxins, can be used in some cases. Vaccine production of isolated antigen ‘subunit’ vaccines benefits from protein engineering.
Novel approaches to presenting antigens are expected to have an impact on vaccination. An example is the phage display technique (Benhar, 2001), described earlier as a technique for antibody selection. The same approach can be used to provide the protein antigen in a display framework that enhances its immunogenicity and reduces the requirement for immune adjuvants (of which there are few approved for human use). Viruses encoding multiple antigens (‘vaccinomes’) can also be employed.
Genetic vaccination (Liu, 2003) employs a DNA plasmid containing the antigen-encoding gene, which is delivered to the host tissue by direct injection of DNA, or by more exotic techniques such as attaching the DNA to microparticles which are shot into tissues at high speed by a ‘gene gun’, or introduced by other transfection methods (Capecchi et al., 2004; Locher et al., 2004; Manoj et al., 2004). When the DNA is transcribed, the mRNA translated and the protein expressed in the host tissue, eukaryotic sequences will undergo appropriate post-translational modification, which does not occur with conventional protein vaccines. In general, partial sequences of pathogen-derived proteins are fully immunogenic, despite lacking the toxicity of full-length transcripts. The first human trial for a genetic vaccine against HIV took place in 1995, and others quickly followed, including hepatitis, influenza, melanoma, malaria, cytomegalovirus, non-Hodgkin’s lymphoma, and breast, prostate and colorectal tumours. Initial results have been promising. At the time of writing, recombinant vaccines against hepatitis A and B (e.g. Twinrix), papillomavirus virus for genital warts and cervical cancer (Gardisil), and against Haemophilus b/meningococcal protein for meningitis (Comvax) are approved. Single or multiple proteins from an organism can be included, and proteins that interfere with the immune response (common in many infectious agents) excluded.
Gene therapy
The development and present status of gene therapy are discussed in Chapter 3. Here we focus on the technical problems of gene delivery and achieving expression levels sufficient to produce clinical benefit, which are still major obstacles.
Introduction of genetic material into cells
Nucleic acid polymers are large, highly charged poly-anions at physiologic pH. The mechanism by which such cumbersome hydrophilic molecules traverse multiple membrane lipid bilayers in cells is poorly understood, although a variety of empirical techniques for getting DNA into cells have been devised by molecular biologists. Eukaryotic cells use many strategies to distinguish between self and foreign DNA to which they are continually exposed. Some mechanisms apparently interfere with the incorporation and expression of engineered genes in gene therapy and other DNA transfer applications. Despite advances in technology, expression of foreign genes in cells or animals in the right place, at the right moment, in the right amount, for a long enough time remains difficult. Doing so in a clinical situation where so many more of the variables are uncontrollable is yet more challenging. The high expectations of gene therapies, which conceptually seemed so straightforward when the first trials were started, have run aground on the shoals of cellular genetic regulatory mechanisms. Effective gene therapy awaits the selective circumventing of these biological controls to deliver precise genetic corrections.
Delivery of nucleic acids
A potential gene therapy requires three major components: the payload to accomplish the mission, a targeting system to direct the vehicle and its payload to the correct body compartment in the correct cell population, and finally gene regulatory element(s), such as promoters/enhancers, to control the time and place of expression of the payload sequence(s). Table 12.2 compares the properties of some gene therapy vectors that have been used in clinical trials.
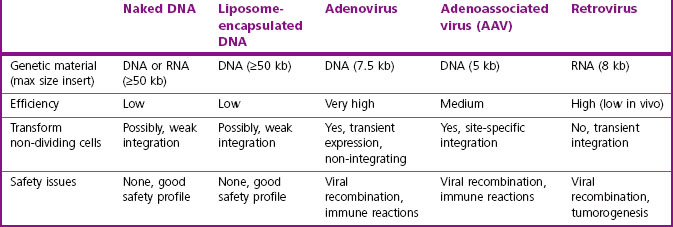
To be clinically useful, a vehicle must elude host immunological or toxicological responses, must persist in the body, and must avoid sequestration in non-target organs such as the liver or kidney. Ideally, for clinical use one would want the ability to modulate or remove the genetic modification in case of adverse effects – the equivalent of discontinuing administration of a traditional drug. In practice, major difficulties are experienced in obtaining sufficient expression of the desired product for long enough to measure a positive clinical outcome.
Strategies for nucleic acid-mediated intervention
There are several points of attack for nucleic acid therapeutics. Most diseases are not due to an identified gene mutation, but rather reflect secondary cellular malfunction, often in response to events entirely external to the cell being targeted. Overexpressing proteins or fragments, either normal or mutated so as to compete with the normal protein for participation in cellular functions – known as the ‘dominant negative’ strategy – is a commonly used approach.
RNA metabolism can be modulated in many different ways, including:
• Small interfering mRNA (siRNA)
• RNA decoys for viral RNA-binding proteins
• Specific mRNA-stabilizing proteins
• Interference with mRNA splicing to induce exon skipping or to correct abnormal splicing
• Sequence-specific cleavage by catalytic RNAs such as hammerhead and hairpin ribozymes. Bacterial introns that code for a multifunctional reverse transcriptase/RNA splicing/DNA endonuclease/integrase protein can be altered to target specific DNA sequences
Transcription of DNA into mRNA can also be controlled either through transcription factors or through antisense oligonucleotide triplex formation. Direct interference with genomic DNA through quadruplex formation is another strategy. These oligonucleotide sequences can be provided by biologically derived vector systems or by chemical synthesis. Short double- and single-strand oligonucleotide sequences are readily taken up by cells. Produced as single strands by automated solid-phase chemical synthesis, these short, single-stranded 8–20-nucleotide sequences can be chemically modified on the base, sugar, or phosphate linker to enhance their stability against nuclease degradation and cellular penetration (Figure 12.2).
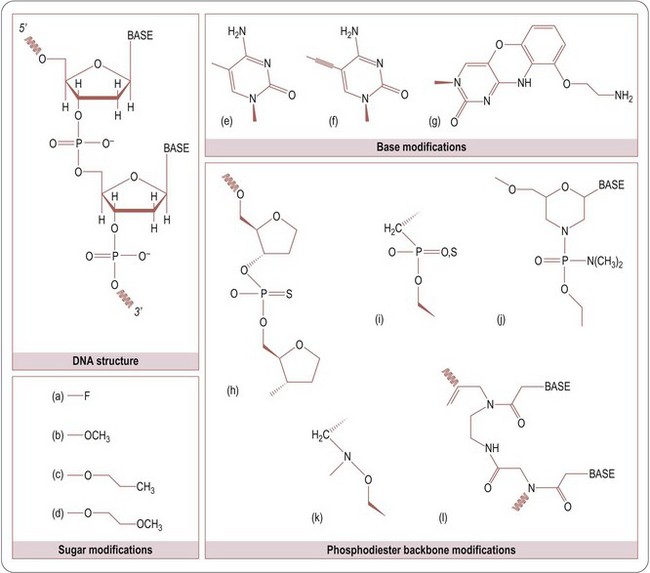
Fig. 12.2 Chemical modification of nucleotides. Key for modifications: Sugar modifications: (a) fluoro-, (b) methoxy-, (c) methoxyethyl-, (d) propoxy-. Base modifications: (e) 5-methyl cytosine, (f) 5-propyne cytosine, (g) tricyclic cytosine. Phosphodiester backbone modifications: (h) phosphorothioate, (i) mopholino-, (j) methylene- (on PDF), (k) methylene-methylimino, (l) peptide nucleic acid (PNA).
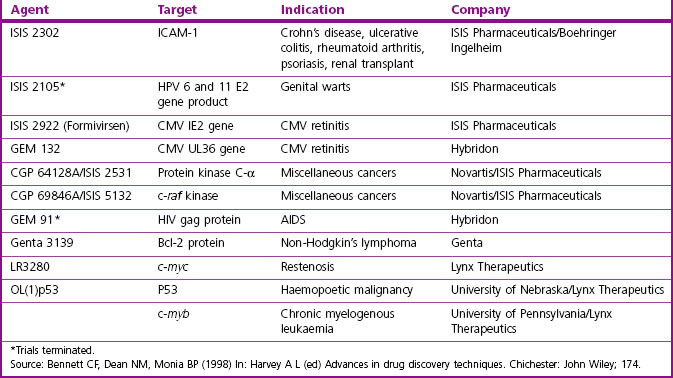
Table 12.3 lists a number of clinical trials with nucleic acid-based therapeutics that are at different stages of completion.
RNA interference – gene silencing by RNAi
RNAi is a technique for reducing the expression of or silencing specific genes. Its usefulness as an experimental tool, particularly for identifying potential drug targets, is discussed in Chapter 6, and it also has considerable potential as a means of silencing genes for therapeutic purposes. It is similar to antisense modulation of mRNA, but extends further into the command and control of cellular nucleic acids. Short double-stranded RNA (21–23 nucleotides) is used to silence homologous gene activity. Specific RNA-induced silencing complexes (RISC) guide multiple activities that target mRNA for degradation, block translation, or block gene transcription by methylation of chromatin (Denli and Hannon, 2003).
Examples of genes that have been targeted by RNAi include:
• Viruses such as HIV-1, poliovirus, respiratory syncytial virus and hepatitis C
• Oncogenes and tumour suppressors, such as Ras, bcl-abl, p53, p53bp and p73Dn
• Cell surface receptors for HIV-1-CD4, CCR5 and CXCR4, and the IL2 receptor α CD25 (Dykxhoorn et al., 2003).
Although RNA interference is highly efficient as a gene-silencing technique under laboratory conditions, most of the difficulties with antisense or expression technology still remain to be overcome. Unlike single-stranded antisense oligonucleotides, the double-stranded siRNAs are unable to penetrate cell membranes effectively without assistance. They are also highly susceptible to plasma degradation. Unfortunately, the same chemical modifications in the phosphodiester backbone used to stabilize antisense oligonucleotides reduces or eliminates silencing activity of interfering RNA constructs. Modification of the 2’ position of uridines and cytosines with fluorine increases plasma half-life and preserves their inhibitory capacity.
Although initial results suggested a high degree of specificity for siRNA in down-regulating a target molecule and the technique is widely used in cell culture studies, there is a surprising tolerance of mismatching leading to effects on unintended targets (Lin et al., 2005). Even if microarray analysis indicates that two siRNAs to the same protein give similar expression patterns, siRNAs with multiple mismatches in the centre of the sequence can operate as microRNAs (miRNA) that can inhibit expression of off-target proteins by arresting their translation without altering cellular mRNA levels, or affect mRNA stability (Saxena et al., 2003). Despite these challenges, several companies are moving ahead with siRNA candidates in clinical trials (Table 12.3), and other products in this class are in the pipeline for approval for testing. RNAi will probably dominate the next wave of gene therapeutics moving into clinical trials.
miRNAs are naturally occurring small RNA species that have been shown to have profound regulatory effects on the genome. Multiple genes may be regulated by a single miRNA and each gene may also be connected to multiple miRNAs, creating a vast network of interactions that may provide fine control to multiple processes from tissue development to a variety of disease states. They operate analogously to siRNA, but with a twist. The RISC complex differs in that noncomplementary nucleotides create bulges in the double-stranded nucleotide preventing the miRNA : RISC complex from continuing along the siRNA pathway. Instead, mRNA translation is affected and the mRNA is destabilized. Targeting microRNAs as a therapeutic strategy is in the very early stages of evaluation. Successful development faces both the known challenges of chemical matter and delivery as well as additional safety and efficacy hurdles because of the web of interactions among the genome and individual miRNAs (Seto, 2010).
Comparing the discovery processes for biopharmaceuticals and small molecule therapeutics
The approach to discovering new protein/peptide therapeutics differs in many ways from the drug discovery approach for synthetic compounds described in other chapters in this section.
Protein/peptide drug discovery usually starts with known biomolecules, which it may be desirable to trim down to smaller domains with desired activities. Alternatively, native proteins are often optimized for desirable properties by mutation or other kinds of modification. These modifications can also provide the basis for patent protection. Unlike small-molecule synthesis, where the possibilities are virtually limitless, there is a finite number of natural proteins and a limited range of feasible modifications that may be introduced to improve their biological properties. The race to protect important proteins as starting points for therapeutics is critical for the future application of biotechnology.
The production of biotechnology-based therapeutics also involves technologies and facilities quite different from those used for producing synthetic compounds.
Manufacture of biopharmaceuticals
Expression systems
Full-size proteins, including a number of hormones, growth factors and mediators, are produced recombinantly by inserting the cDNA version of the mRNA sequence coding for the protein of interest into specially designed plasmid vectors that orchestrate the expression of the desired protein product in the cell type of choice (Figure 12.3). The characteristics of different expression systems are compared in Table 12.4.
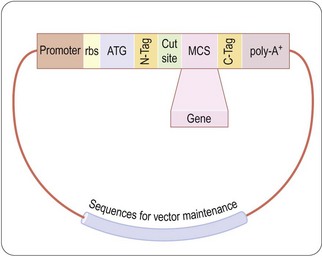
Fig. 12.3 Generic vector for protein expression. The cDNA for the protein to be expressed is cloned into restriction sites in the multiple cloning site (MCS) of an expression vector which contains appropriate sequences for maintenance in the chosen host cell. Expression of the gene is controlled by inducible promoter. A ribosome-binding site (rbs) optimized for maximum expression directs translation, beginning at the ATG initiation codon. Either N-terminal or C-terminal tags (N-, C-Tag) can be used to aid in purification or stabilization of the protein, and a protease cut site may be used to remove the tag after purification. A translation terminator codon is followed (in eukaryotic cells) by a poly-A+ tail to stabilize the mRNA.
These vectors contain a selection marker to maintain the vector in the host cell along with regulatory DNA sequences that direct the host cell transcriptional machinery to produce mRNA from the inserted cDNA sequence, with appropriate initiation and termination codons and a ribosome-binding site. A polyA+ tail is added to eukaryotic expression systems to stabilize the mRNA. mRNA transcription, and hence protein production, is controlled by an induction element, so that protein is produced only when required. This is important, as some expressed proteins are toxic to growing and dividing host cells. Other sequences, such as protein export signal peptides, hexahistidine tags, immunologic epitopes, or fusion with protein partners such as glutathione S-transferase, thioredoxin, maltose-binding protein or IgG Fc, facilitate secretion or provide handles for the detection/purification of the expressed protein. Purification tags are particularly useful in eukaryotic expression systems, which express exogenous proteins at lower levels than do bacterial systems, where the expressed protein can be 10–50% of the total cellular protein. The tags are designed to be removed from the protein once it has been purified.
Bacterial protein expression, usually in Escherichia coli, is the system of choice for high expression levels of many proteins, and because scale-up technology is well developed. However, not every protein is amenable to bacterial expression. Proteins that contain disulfide bonds or are comprised of multiple subunits are problematic, as are proteins containing critical carbohydrate, lipid or other post-translational modifications. In some cases the enzymes responsible for post-translational modification, such as fatty acylation, can be co-transfected with the desired protein. Highly expressed proteins are frequently produced as insoluble inclusion bodies in the bacterial cytoplasm or periplasmic space. Active proteins must be recovered from these inclusion bodies after denaturation and in vitro refolding. Intrinsic membrane-bound proteins can pose additional problems.
Eukaryotic microbial systems include Saccharomycetes cerevisiae and Picha pastoralis – yeasts that perform many mammalian post-translational modifications, including partial glycosylation. Insect cells, notably from Spodoptera frugiperda (Sf9), are infected with engineered baculovirus expression vectors carrying the protein of interest. This is a particularly efficient system for membrane protein expression. Many G-protein-coupled receptors, a large class of integral membrane proteins that includes many targets for currently marketed drugs, are often produced in these cells for use in screening assays for receptor ligands.
Cultured mammalian cells are used to express proteins on a research or production scale where faithful post-translational modification is required. Favourites include fibroblasts, anchorage-independent lymphocytes, Chinese hamster ovary (CHO) cells, and human embryonic kidney (HEK) cells. Mammalian cells grow more slowly and are more fastidious than the bacteria, yeasts or insect cells, and are therefore more expensive for production. Human cells are used in situations where there is some mammalian species dependence of bound carbohydrate. This can be particularly important for growth factors and the large glycoprotein hormones, as their glycosylation modulates the activity and stability of the protein product in vivo. Potential contaminating human pathogens in the human cell lines used for expression must be controlled. So must non-primate mammalian pathogens, as the possibility of transfer to humans cannot be ignored.
The use of transgenic animals and plants as ‘factories’ for genetically engineered proteins is discussed below.
Engineering proteins
Nature has devoted millions of years to optimizing protein structures for their biological roles. The natural configuration may not, however, be optimal for pharmaceutical applications. Recombinant DNA methods can be used to re-engineer the protein structure so as to better fulfil the requirements for a pharmaceutical agent.
Site-directed mutagenesis
Changes in the sequence of a protein at the level of a single amino acid can change the biochemical activity or the stability of the protein. It can also affect interactions with other proteins, and the coupling of those interactions to biological function. Stabilizing proteins to better withstand the rigours of industrial production and formulation is commercially important. Microorganisms adapted to extreme environments have evolved modified forms of enzymes and other functional proteins, such as increased disulfide (cystine) content and more extensive core interactions, to withstand high temperatures. Similar tricks are used to stabilize enzymes for use at elevated temperatures, such as in industrial reactors, or in the home washing machine with enzyme-containing detergents. Other mutations alter the response of the protein to environmental conditions such as pH and ionic strength. Mutations to add or remove glycosylation signal sequences or to increase resistance to proteolytic enzymes may be introduced to alter immunogenicity or improve stability in body fluids. Protein activation by conformational changes due to phosphorylation, binding of protein partners or other signals may sometimes be mimicked by amino acid changes in the protein. The solubility of a protein can often be improved, as can the ability to form well-ordered crystals suitable for X-ray structure determination.
Fused or truncated proteins
Sometimes the full-size form of a protein or peptide is too difficult or expensive to produce or deliver to the target. Domains of proteins can often be stripped to their essentials, so that they retain the required characteristics of activity or targeting specificity. They may require fusion with a partner for proper folding or stability, the partner being removed as part of purification.
Ligands and receptor-binding domains can be attached to the active biomolecule in order to target it to specific sites. Pathogens and infectious agents, in addition to appropriating intracellular targeting motifs, have evolved protein sequences that enable them to penetrate the cell membrane to access the machinery for their propagation. Some of these sequences can be incorporated into biopharmaceuticals in order to deliver peptides and proteins into cells. Hydrophobic portions of Kaposi fibroblast growth factor, Grb2 SH2 domain, and human integrin α and β subunits have been employed in this way as cell-penetrating agents. The fusion sequence (1–23) of HIV-1 gp41 has been used to deliver synthetic antisense oligo-nucleotides and plasmid DNA into cells. Amphipathic sequences containing periodic arrangements of polar and hydrophobic residues, such as those found in the fusion peptide of the influenza haemagglutinin-2 protein, promote fusion with cell membranes and the delivery of associated agents. Polycationic sequences in penetratin (residues 43–58 from the Antp protein), HIV-1 tat protein (47–57), and the HIV-1 transcription factor VP22 (267–300), permeabilize membranes and increase the uptake of associated material.
Protein production
Although relatively small quantities of highly potent biologicals are normally required, production often presents problems. Only synthetic peptides and oligonucleotides and their derivatives can be produced by a scalable chemical synthesis. The others require large-scale fermentation or cell culture. The proteins and nucleic acids are then isolated and purified from the cells or media. Biotechnological process development is a complex field (Sofer and Zabriskie, 2000; Buckel, 2001; Gad, 2007). High-level production of recombinant proteins can result in heterogeneity because of the misincorporation of amino acids such as norvaline into the product, depending on the culture conditions, the nutrient composition of the medium and the producing organism. Post-translational modifications depend on the cultured organism and the subcellular localization of the protein. In cultured eukaryotic cells proteins undergo many kinds of post-translational modification, including glycosylation, sulfation, phosphorylation, lipidation, acetylation, γ-carboxylation and proteolytic processing. These and other modifications increase the heterogeneity of the product, affecting its biological half-life, immunogenicity and activity. The most homogeneous preparation consistent with retention of the important biological properties is standardized for production and a reproducible analytical profile is established. Nevertheless, protein products are invariably less homogeneous in composition than synthetic compounds, and the quality criteria for approval of clinical materials have to be adjusted to take account of this.
Formulation and storage conditions are critical because the protein primary amino acid sequences naturally display a variety of chemical instabilities. A major degradation route of protein products is through unfolding of the polypeptide chain and aggregation. Proteins have a finite half-life in an organism, governed partly by built-in chemical and conformational instabilities. Adsorption to surfaces of glass or plastic containers, particulates and air–liquid interfaces is often a cause of product loss.
The chemical reactivity of the various amino acid residues contributes to protein loss and heterogeneity. Succinimide formation, isomerization and racemization, as well as peptide bond cleavage, tend to occur at aspartate residues, to an extent that depends somewhat on the surrounding amino acid sequence. Examples include hACTH, soluble CD4, OKT-3 monoclonal antibody, hGH-releasing factor, IL-1β and hEGF. Oxidation, particularly at methionine residues, is catalysed by transition metal ions, pH, light, and various oxygen free radicals. Examples are relaxin A, hIGF-I and enkephalin analogs. Disulfide exchange (cysteine/cystine) can generate inappropriate folding or intermolecular bond formation, thereby promoting aggregation and precipitation of protein. Examples are interferon, and the acidic and basic forms of FGF. Beta-elimination at cystine, cysteine, serine and threonine leads to dehydroalanine formation and reaction with nucleophilic side chains. Anhydride formation between the α amino group on a protein and the C terminus is a major degradation pathway for insulin stored between pH3 and pH5. Sometimes peroxides contaminate the excipients and stabilizing agents, such as the polyethylene glycols or the surfactants Polysorbate 20 and Polysorbate 80 used in product formulation (see Chapter 16).
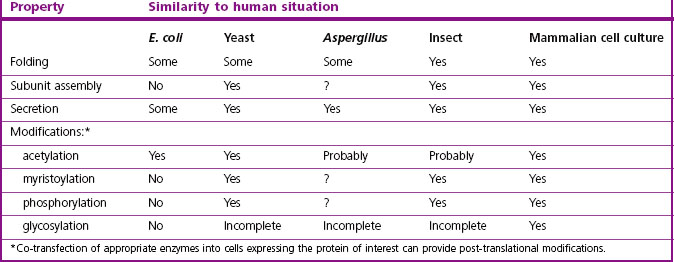
The first therapeutic proteins produced on an industrial scale were antibodies, which initially relied on immunization of large animals such as horses. Monoclonal technology paved the way for the production of murine antibodies in cell culture. With the ability to produce human antibody proteins and derivatives through molecular biological manipulation, the organism used to manufacture the protein is chosen on the basis of economics and requirements for post-translational modification. The characteristics of some commonly used organisms for protein production are summarized in Table 12.4.
Eukaryotic and prokaryotic microorganisms and eukaryotic cells in culture are the most common source of therapeutic proteins. Prokaryotic expression is generally the first choice, whereas higher organisms produce protein products that are most similar to human material and thus are more likely to be biologically active and pharmacokinetically stable. The production of proteins on a commercial scale by eukaryotic cell culture is, however, extremely expensive. The cells grow slowly (18–24 hours doubling time, compared with about 20 minutes for bacteria) and most require a serum source or expensive growth factor/hormone cocktails in the culture medium.
‘Pharming’ of protein expression in whole animals or plants is used for large-scale production of proteins for some therapeutics and for industrial applications. Transgenic farm animals – goats, cows, sheep and rabbits – have been engineered to secrete human proteins into their milk (van Berkel et al., 2002). Annual milk yields range from about 8 L (rabbit) to 8–10 000 L (cow). Expression levels of from 1 to 20 g of product per litre of milk have been achieved in favourable instances, allowing small herds of transgenic animals to do the work of large fermentation facilities. Many biopharmaceuticals are under development by specialized biotechnology companies (Table 12.5). The first drug produced in genetically engineered livestock (Atryn in goat milk) was approved by the FDA in February 2009. Thirty or more such products are expected to be registered in the next decade.
Table 12.5 Biopharmaceuticals produced in transgenic farm animals. (These products are in development; none have yet reached the market)
| Protein | Indication | Host |
|---|---|---|
| Antithrombin III | Inflammation | Goat |
| α1-Antitrypsin | Inflammation, inherited deficiency | Cow, goat, sheep |
| α-Glucosidase | Glycogen storage disease type II | Rabbit |
| h-Chorionic gonadotropin | Infertility | Cow, goat |
| Factor VIII | Haemophilia | Pig |
| Factor IX | Haemophilia | Pig |
| Factor XI | Haemophilia | Sheep |
| Fibrinogen | Burns, surgery | Pig, sheep |
| Lactoferrin | GI bacterial infection | Cow |
| Monoclonal antibody | Colon cancer | Goat |
| Protein C | Deficiency, adjunct to tPA | Pig, sheep |
| Serum albumin | Surgery, burns, shock | Cow, goat |
| Tissue plasminogen activator (tPA) | Infarction, stroke | Goat |
Plants can be directed to concentrate antibodies and other proteins in their leaves, seeds or fruit (Giddings et al., 2000; Daniell et al., 2001; Powledge, 2001; Streatfield and Howard, 2003), and are beginning to be used for the commercial production of antibodies and vaccines. Monoclonal antibodies expressed in plants – ‘plantibodies’ – have been produced in maize, tobacco and soybeans. Antibodies produced against Streptococcus mutans, the main cause of tooth decay in humans, have demonstrated efficacy against dental caries. CaroRX is in Phase II clinical trials for topical application for tooth decay. Other proteins, such as mouse and human interferon, human growth hormone, haemoglobin, human serum albumin and human epidermal growth factor, are also being targeted. Merispace, a recombinant mammalian gastric lipase, is being produced in corn for oral administration to counteract lipid maladsorption in cystic fibrosis and chronic pancreatitis Vaccine production for both animals and humans appears to be a most promising application of ‘pharming’. By splicing the gene for the antigen into a modified plant virus (cowpea mosaic virus), companies such as Agricultural Genetics (Cambridge, UK) have produced effective vaccines for animals against mink enterovirus, HIV-1 and foot and mouth disease, obtaining up to 200 doses of vaccine from a single cowpea (black-eyed pea) leaf. Other companies are pursuing vaccines for hepatitis A and B, cold and wart viruses and Plasmodium (malaria). Splicing the antigen genes directly into the plant genome has been successful for hepatitis B, Norwalk, cholera and rabies virus vaccines.
Feeding antigen-producing plants to animals has shown promise for inducing immune protection (Judge et al., 2004), despite the differences in the type of immune response from oral exposure and the standard humoral administration. The oral route would be ideal for human vaccines, particularly in countries where refrigeration is uncertain. Although antigen expressed in potato for use in humans was effective in animals, the cooking required for human consumption of the vegetable destroyed the immunogenicity of the antigen. Current efforts are developing the technology for human foods that are eaten raw and which are suitable for tropical climates, such as tomato and banana (Korban et al., 2002).
There are many developments in plant-derived biopharmaceuticals for human use, but none has yet become commercially available. Widespread environmental and other concerns among the general public about transgenic crop production are a significant impediment, especially outide of the United States.
Pharmacokinetic, toxicological and drug-delivery issues with proteins and peptides
The main source of the bias against biologicals (proteins) as therapeutic agents in major pharmaceutical companies comes from the difficulties in delivering these expensive, large, highly charged molecules to the target in sufficient concentrations for long enough to achieve pharmacologic effects. The complexities faced in taking small molecules through preclinical development into clinical trials are compounded with biologicals. More detailed expositions of pharmacokinetics, toxicology and the delivery of therapeutic proteins are available (Frokjaer and Hovgaard, 2000; Sofer and Zabriskie, 2000; Ho and Gibaldi, 2003; Walsh, 2003; Steffansen et al., 2010).
Figure 12.4 illustrates the pathways that govern the distribution and elimination of a pharmacological agent. Factors that assume particular importance for biopharmaceuticals are (a) the choice of formulation and route of administration so as to achieve consistent absorption, (b) control of distribution by targeting to the required site of action, and (c) protection against rapid inactivation or elimination.
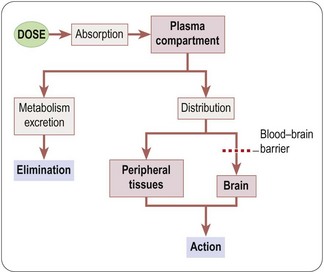
Fig. 12.4 Simplified scheme showing the processes involved in drug absorption, distribution and elimination. Pink boxes, body compartments; grey boxes, transfer processes.
Absorption of protein/peptide therapeutics
Mucosal delivery
Peptide and protein drugs are in general poorly absorbed by mouth because of their high molecular weight, charge, and susceptibility to proteolytic enzymes. The main barriers to absorption of proteins are their size and charge (Table 12.6).
Table 12.6 Physical properties of some therapeutic proteins
| Protein | Molecular wt (Da) | Isoelectric point |
|---|---|---|
| Somatostatin | 1600 | 10.4 |
| Insulin | 6000 | 5.6 |
| Human growth hormone | 20 000 | 5.3 |
| t-PA | 60 000 | 9.1 |
| Human serum albumin | 66 000 | 5.3 |
| IgG | 150 000 | Variable |
| Factor VIII | 270 000 | 7.4 |
| Typical small-molecule drug | <500 | Variable |
Large hydrophilic and charged molecules do not readily pass the cellular lipid bilayer membrane. Small peptides can be absorbed if they are relatively hydrophobic, or if specific transporters exist. Proteins can be transported by transcytosis, in which proteins enter cells at the basal surface via receptor-mediated endocytosis or fluid-phase pinocytosis into vesicles, are transported across the cell through the cytosol, and then released by exocytosis at the apical membrane. However, much of the protein is degraded by the lysosomal system during transit.
Formulations have been devised to optimize the stability and delivery of protein and peptide therapeutics (Frokjaer and Hovgaard, 2000). Protection from proteolysis is a major concern for many routes of administration. Various entrapment and encapsulation technologies have been applied to the problem (see Chapter 16). The greater the protection, though, the less rapidly the protein is released from the preparation. Hydrogels, hydrophilic polymers formed around the protein, avoid proteolysis but limit absorption. Microcapsule and microsphere formulations are made by enclosing the active compound within a cavity surrounded by a semipermeable membrane or within a solid matrix. The surface area for absorption is much greater than that provided by hydrogel or solid matrix formulations.
A variety of penetration-enhancing protocols have been applied to increase the rate and amount of protein absorption. These include surfactants (sodium dodecyl sulfate, polyoxyethylene fatty acyl ethers), polymers (polyacrylic acid, chitosan derivatives), certain synthetic peptides (occulin-related peptides), bile salts (sodium deoxycholate, sodium glycocholate, sodium taurocholate), fatty acids (oleic acid, caprylic acid, acyl carnitines, acylcholines, diglycerides), and chelating agents (EDTA, citric acid, salicylates, N-amino acyl β-diketones).
The nasal mucosal route of administration is emerging as an acceptable and reasonably effective method of drug delivery. Some peptide drugs, such as nafarelin, oxytocin, lypressin, calcitonin and desmopressin, are effective as nasal sprays, although the fraction absorbed in humans is generally low (≤10%). Pulmonary administration is surprisingly effective when –3 µm particles of the therapeutic are dispersed deep in the lung. The vast alveolar surface area and blood supply allow even macromolecules to be absorbed.
Transdermal delivery across the stratum corneum, the outermost, least-permeable skin layer, is being increasingly used for proteins and other drugs. Transport of drug molecules across the skin is facilitated by transient disruption of epithelial barrier function with sonic energy (sonophoresis), electric current (iontophoresis) or electric field (electroporation). Table 12.7 compares different routes of non-parenteral dosing for the relative proteolytic activity to which a protein drug would be exposed.
Table 12.7 Exposure of protein/peptide drugs to protease activity by different routes of administration
| Oral | Very high |
| Rectal | High |
| Buccal | Medium |
| Nasal | Medium |
| Vaginal | Medium |
| Transdermal | Low |
| Pulmonary | Low |
| Ocular | Low |
Parenteral delivery
Oral or mucosal absorption of native, full-length proteins is inefficient even with enhancing strategies. Thus, many protein therapeutics are delivered parenterally by intravenous, intramuscular or subcutaneous injection. With intramuscular and subcutaneous administration sustained-release formulations can be used to increase the duration of action.
The blood–brain barrier is impermeable to most proteins and peptides, but possesses transporters that facilitate the entry of some, such as apolipoprotein E, transferrin and insulin. Linking active peptides to an antibody directed against the transferrin receptor has been shown in experimental animals to allow neurotrophic factors such as nerve growth factor (NGF) to enter the brain and produce neurotrophic effects when given systemically and this strategy may prove applicable for human use.
Elimination of protein/peptide therapeutics
Most therapeutic proteins are inactivated by proteolysis, producing short peptides and amino acids which then enter dietary pathways. Unlike small-molecule drugs, metabolites of protein therapeutics are not considered to be a safety issue. The rate and extent of degradation of protein therapeutics before excretion depend on the route of administration. Breakdown occurs in both plasma and tissues. Attempts to minimize these losses include the use of glycosylated proteins, which more closely resemble the native protein, or chemical modification of the protein with polyethylene glycol (PEG) chains or other entities. PEGylation can increase the plasma half-life of a protein between three- and several hundredfold. Besides reducing proteolysis, PEGylation can shield antigenic sites, enhance protein solubility and stability, and prevent rapid uptake by organs. With small peptides, cyclization or the inclusion of D-amino acid residues, particularly at the N terminus, can be used to protect against exoproteolysis, though this approach is of course applicable only to synthetic peptides and not to biopharmaceuticals.
The kidney is an important organ for the catabolism and elimination of small proteins and peptides. Proteins of 5–6 kDa, such as insulin, are able freely to pass the glomerular filter of the kidney. Passage through the glomerulus decreases to about 30% for proteins of 30 kDa, and to less than 1% for 69 kDa proteins. Some proteins, such as calcitonin, glucagon, insulin, growth hormone, oxytocin, vasopressin and lysozyme, are reabsorbed by the proximal tubules via luminal endocytosis, and then are hydrolysed in lysosomes. Linear peptides less than 10 amino acids long are hydrolysed by proteases on the surface of brush border membranes of the proximal tubules.
Liver metabolism is highly dependent on specific amino acid sequences in proteins. Cyclic peptides, small (<1.4 kDa) and hydrophobic peptides are taken up by carrier-mediated transport and degraded. Large proteins use energy-dependent carrier-mediated transport, including receptor-mediated endocytosis and transcytotic pathways (polymeric IgA), and are targeted to lysosomes.
Summary
In the late 1970s recombinant DNA technology boosted biotechnology into a prominence it has not relinquished. Apart from fears about human genetic modification, the outlook has been positive for the production of new and better biopharmaceuticals. Whereas the traditional pharmaceutical companies were slow to embrace the new technology, a number of small biotech companies sprang up which have provided the innovative driving force for the protein and gene product industry. The genomic revolution is the latest manifestation of the technology, stimulating efforts to mine the information coming out of the various species genome projects for new products and therapies.
Recombinant DNA technology allows the production of individual proteins and peptides on a large scale regardless of their natural abundance. There are many advantages to recombinant production. Human sequence proteins reduce potential immunological problems and avoid potential infectious agents in materials isolated from natural sources. Proteins are expressed in a variety of cellular systems, determined by the properties of the molecule required. Bacterial systems are used wherever possible because of the high level of expression that can be obtained and the simplicity and availability of production. Bacteria, however, do not provide many of the post-translational modifications required for human protein stability and function, such as glycosylation, lipid modification, phosphorylation, sulfation and disulfide bond formation. Protein aggregation often occurs, and even though denaturation and refolding are successful for many proteins, some are problematic. In these cases eukaryotic systems, including various yeast species and cultured insect and mammalian cells, are used. Transgenic farm animals and plants are also coming into their own as protein ‘factories’.
Antibodies in the form of animal sera were the first widely used protein therapeutics. Although immunization is still used as a prophylactic and as a therapeutic, recombinant DNA technology is used to produce both immunogen and antibodies. Hypersensitivity reactions are reduced by producing altered antibodies with the required epitope specificity. They are ‘chimerized’ (human constant domain) or ‘humanized’ (rodent complementarity-determining region (CDR)), with human Fc portions for optimal interaction with the human complement system. Single-chain and multichain antibodies can be expressed intracellularly to attack previously sequestered antigens. These immunological agents are also used to target infectious organisms, deliver radioisotopes or cytotoxic agents to cancer cells, and to moderate tissue rejection in transplantation.
Proteins and enzymes have been engineered to hone their therapeutic usefulness. Protein structure can be stabilized or modified to slow degradation or increase uptake, and multiple functions can be built into the same molecule. Recombinant vaccine production of cloned protein domains uses these modifications to increase immunogenicity and avoid exposure to infectious agents. Delivery of synthetic vectors coding for antigenic epitopes into host tissues promotes endogenous antigen expression.
The biggest obstacle to the use of proteins and peptides as therapeutics is the delivery to the site of action of sufficient agent to create a biological effect. Formulating proteins and peptides to penetrate epithelial barriers in a biologically active form is difficult. Because of their size, charge and instability in the gastrointestinal tract, special delivery systems often must be used to achieve efficacious blood levels. Penetration through the blood–brain barrier can be even more challenging. Small-molecule drugs of <500 Da molecular weight have a much better record in this regard. Once in the blood, proteins are often rapidly eliminated by a variety of mechanisms, although there are modifications that can slow degradation.
Even though protein and peptide therapeutics faces significant pharmacokinetic and pharmacodynamic liabilities, much work is still being done on agents for which small molecules are not available to perform the same function.
Nucleic acids can be delivered into cells in a variety of ways. Complexation of the highly charged DNA with lipophilic cations, or derivitization of nucleic acid bases, sugars or the phosphodiester backbone can be efficient in cell culture, but less so in whole organisms. Ingenious schemes of antisense, RNAi, ribozyme, decoy sequences and RNA splicing inhibition are used to attain therapeutic effects. Viruses, nature’s DNA delivery machines, modified to eliminate their pathogenic capacity, are used in organisms to introduce the therapeutic gene into target cells. The inserted DNA directs the synthesis of a protein in the host cell, in the case of a growth factor deficiency in the brain providing a secreted product for the support of surrounding cells. Numerous gene therapy clinical trials are in progress, but the need to avoid side effects and host suppression of viral-based therapeutics has slowed progress.
Benhar I. Biotechnological applications of phage and cell display. Biotechnology Advances. 2001;19:1–33.
Biaggi E, Rousseau RF, Yvon E, et al. Cancer vaccines: dream, reality, or nightmare? Clinical and Experimental Medicine. 2002;2:109–118.
BSI Vaccine Immunology Group. Vaccination against non infectious disease (BSI Vaccine Immunology Group/BSACI session). Immunology. 2002;107(Suppl 1):67–70.
Buckel P, ed. Recombinant protein drugs. Basel: Birkhauser Verlag, 2001.
Capecchi B, Serruto D, Adu-Bobie J, et al. The genome revolution in vaccine research. Current Issues in Molecular Biology. 2004;6:17–27.
Daniell H, Streatfield SJ, Wycoff K. Medical molecular farming: production of antibodies, biopharmaceuticals and edible vaccines in plants. Trends in Plant Science. 2001;6:219–226.
Denli AM, Hannon GJ. RNAi: an ever growing puzzle. Trends in Biochemical Sciences. 2003;28:196–201.
Dykxhoorn DM, Novina CD, Sharp PA. Killing the messenger: short RNAs that silence gene expression. Nature Reviews Molecular Cell Biology. 2003;4:457–467.
Frokjaer S, Hovgaard L. Pharmaceutical formulation: development of peptides and proteins. Philadelphia, PA: Taylor & Francis; 2000.
Gad SC, ed. Handbook of Pharmaceutical Biotechnology. Hoboken, NJ: John Wiley and Sons, 2007.
Giddings G, Allison G, Brooks D, et al. Transgenic plants as factories for biopharmaceuticals. Nature Biotechnology. 2000;18:1151–1155.
Ho RJY, Gibaldi M. Biotechnology and biopharmaceuticals. Transforming proteins and genes into drugs. Hoboken, NJ: Wiley-Liss; 2003.
Judge NA, Mason HS, O’Brien AD. Plant cell-based intimin vaccine given orally to mice primed with intimin reduces time of Escherichia coli O157:H7 shedding in feces. Infection and Immunity. 2004;72:168–175.
Kantak KM. Anti-cocaine vaccines: antibody protection against relapse. Expert Opinion in Pharmacotherapy. 2003;4:213–218.
Korban SS, Krasnyanski SF, Buetow DE. Foods as production and delivery vehicles for human vaccines. Journal of the American College of Nutrition. 2002;21:212S–217S.
Lin X, Ruan X, Anderson MG, et al. SiRNA-mediated off-target gene silencing triggered by a 7 nt complementation. Nucleic Acids Res. 2005;33:4527–4535.
Liu MA. DNA vaccines: a review. Journal of Internal Medicine. 2003;253:402–410.
Locher CP, Soong NW, Whalen RG, et al. Development of novel vaccines using DNA shuffling and screening strategies. Current Opinion in Molecular Therapy. 2004;6:34–39.
Manoj S, Babiuk LA, van Drunen Littel-van den Hurk S. Approaches to enhance the efficacy of DNA vaccines. Critical Reviews in Clinical Laboratory. Science. 2004;41:1–39.
Powledge TM. Tobacco pharming. A quest to turn the killer crop into a treatment for cancer. Scientific American. 2001;285:25–26.
Rousseau RF, Hirschmann-Jax C, Takahashi S, et al. Cancer vaccines. Hematology/Oncology Clinics of North America. 2001;15:741–773.
Saxena S, Jonsson ZO, Dutta A. Small RNAs with imperfect match to endogenous mRNA repress translation. Implications for off-target activity of small inhibitory RNA in mammalian cells. J Biol Chem. 2003;278:44312–44319.
Seto AG. The road toward microRNA therapeutics. Int J Biochem Cell Biol. 2010;42:1298–1305.
Sofer G, Zabriskie DW. Biopharmaceutical process validation, vol. 25. New York: Marcel Dekker; 2000.
Steffansen B, Brodin B, Nielson CU. Molecular biopharmaceuticals: aspects of drug characterization, drug delivery, and dosage form evaluation. Chicago: Pharmaceutical Press; 2010.
Streatfield SJ, Howard JA. Plant production systems for vaccines. Expert Review on Vaccines. 2003;2:763–775.
Tellier C. Exploiting antibodies as catalysts: potential therapeutic applications. Transfusion Clinique et Biologique. 2002;9:1–8.
van Berkel PH, Welling MM, Geerts M, et al. Large scale production of recombinant human lactoferrin in the milk of transgenic cows. Nature Biotechnology. 2002;20:484–487.
Walsh G. Biopharmaceuticals, biochemistry, and biotechnology, 2nd ed. Chichester: John Wiley; 2003.