Eugene J. Barrett
The thyroid gland is located in the anterior neck, lying like a small bow tie across the front of the trachea. In adults, the normal thyroid weighs ~20 g. It is composed of left and right lobes and a small connecting branch, or isthmus.
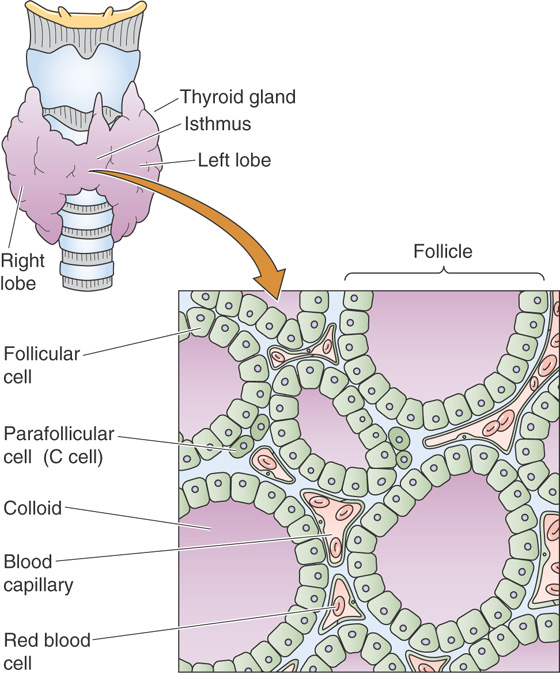
The thyroid gland possesses many features unique among endocrine glands, not the least of which is that it is the only endocrine gland that can be easily seen and palpated in the course of a routine clinical examination. At the biochemical level, the thyroid hormones are the only ones that require an essential trace element, iodine, for the production of active hormone. One of the rather unusual features of thyroid hormone physiology is that the hormone is stored in an extracellular site within a highly proteinaceous material called thyroid colloid. The major protein within this material is thyroglobulin, which contains—as part of its primary structure—the thyroid hormones thyroxine (tetraiodothyronine or T4) and triiodothyronine (T3). These sequestered hormones are entirely surrounded by thyroid follicular cells, which are responsible for the synthesis of thyroid hormones (Fig. 49-1).
Figure 49-1 Structure of the thyroid gland. The thyroid gland is located anterior to the cricoid cartilage in the anterior neck. The gland comprises numerous follicles, which are filled with colloid and lined by follicular cells. These follicular cells are responsible for the trapping of iodine, which they secrete along with thyroglobulin—the major protein of the thyroid colloid—into the lumen of the follicle.
The physiological actions of thyroid hormones also display several unique aspects. Although, like most peptide hormones, T4 and T3 are made as part of a larger protein, unlike peptide hormones, no cell-membrane receptors exist for these hormones. Instead, like the steroid hormones, thyroid hormones act by binding to nuclear receptors (see Chapter 3) and regulate the transcription of cell proteins. The hormones secreted by the thyroid act on multiple tissues and are essential for normal development, growth, and metabolism. The thyroid makes another hormone, calcitonin, which is synthesized by thyroid C cells (parafollicular cells); these C cells are not part of the follicular unit (Fig. 49-1). Calcitonin may play a role in Ca2+ and phosphate homeostasis. The physiology of calcitonin is discussed along with that of parathyroid hormone in Chapter 52.
The structures of T4 and T3, the two active thyroid hormones, are shown in Figure 49-2. T3 is far more active than T4. Also shown is reverse T3 (rT3), which has no known biological activity. It has two iodines on its outer benzyl ring, rather than two on its inner ring, as is the case for T3. All three compounds derive from the ether linkage of a tyrosine molecule to the benzyl group of a second tyrosine molecule; one or two iodine atoms are attached to each benzyl group. The bottom panel of Figure 49-2 shows T4 as part of the thyroglobulin molecule.
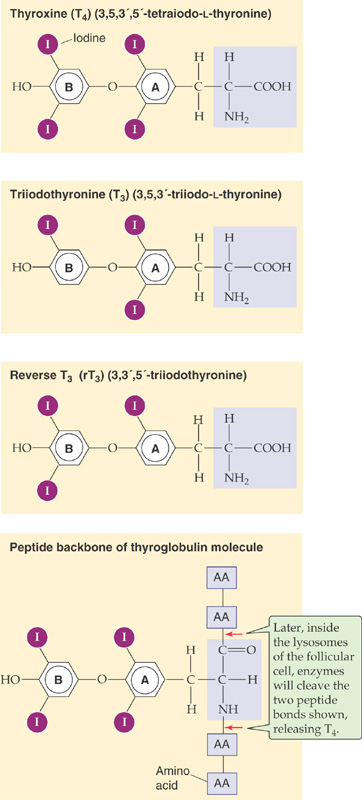
Figure 49-2 The structure of T4, T3, and rT3. T4, T3, and rT3 all are products of the coupling of two iodinated tyrosine derivatives. Only T4 and T3 are biologically active, and T3 is far more active than T4 because of a higher affinity for TRs. rT3 forms as an iodine is removed from the inner benzyl ring (labeled A) of T4; rT3 is present in approximately equal molar amounts with T3. However, rT3 is essentially devoid of biological activity. As shown in the bottom panel, T4 is part of the peptide backbone of the thyroglobulin molecule, as are T3 and rT3. Cleavage of the two indicated peptide bonds would release T4.
The synthesis of thyroid hormones begins with the trapping of iodide by the thyroid gland. Iodine is essential for the formation of thyroid hormones. It exists in nature as a trace element in soil and is incorporated into many foods. The iodide anion (I−) is rapidly absorbed by the gastrointestinal tract and is actively taken up by the thyroid gland. A specialized Na/I cotransporter (NIS) is located at the basolateral membrane (i.e., facing the blood) of the thyroid follicular cell (Fig. 49-3). NIS (for Na Iodide Symporter) is a 65-kDa integral membrane protein that is believed to have 12 membrane-spanning segments. NIS moves I− into the follicular cell against the I− electrochemical gradient, fueled by the energy of the Na+ electrochemical gradient (see Chapter 5). Several other anions (e.g., perchlorate, pertechnetate, and thiocyanate) can compete with I− for uptake by the thyroid. Iodide leaves the follicular cell and enters the lumen of the follicle across the apical membrane. Pendrin, a member of the SLC26 family of anion exchangers (see Chapter 5), is present on the apical membrane and may contribute to I− secretion. Mutations in this protein can lead to a congenital syndrome typically characterized by a large thyroid gland (goiter) and hearing loss. The thyroid enlarges because of deficient I− uptake, just as it would with an I−-deficient diet (see the box titled Iodine Deficiency). (See Note: Role of Pendrin in Apical Iodide Secretion by Thyroid Follicular Cells)
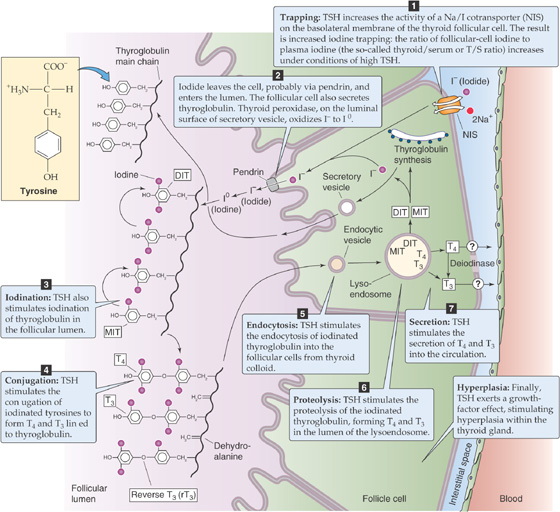
Figure 49-3 The follicular cell and its role in the synthesis of T4 and T3. The synthesis and release of T4 and T3 occurs in seven steps. Inside the follicular cell, a deiodinase converts some of the T4 to T3. Thyrotropin (or TSH) stimulates each of these steps except step 2. In addition, TSH exerts a growth factor or hyperplastic effect on the follicular cells.
In parallel with the secretion of I− into the follicle lumen, the follicular cell secretes thyroglobulin into the lumen; thyroglobulin contains the tyrosyl groups to which the I− will ultimately attach. The thyroglobulin molecule is a glycoprotein synthesized inside the follicular cell, following the secretory pathway (see Chapter 2). Thyroglobulin is a very large protein (>600 kDa), and it accounts for approximately half of the protein content of the thyroid gland. It has relatively few tyrosyl residues (~100/molecule of thyroglobulin), and only a few of these (<20) are subject to iodination. The secretory vesicles that contain thyroglobulin also carry the enzyme thyroid peroxidase on their intravesicular surfaces. As the secretory vesicles fuse with the apical membrane, this enzyme faces the follicular lumen and catalyzes the oxidation of I− to I0. As the thyroglobulin is entering the lumen of the thyroid follicle by the process of exocytosis, its tyrosyl groups react with I0.
One or two oxidized iodine atoms incorporate selectively into specific tyrosyl residues of thyroglobulin. Within the thyroglobulin molecule, an internal rearrangement occurs, resulting in the conjugation of two iodinated tyrosyl residues to form a single iodothyronine, as well as a remnant dehydroalanine. Both remain as part of the primary structure of the iodinated thyroglobulin until it is later degraded inside the follicular cell. This coupling of two tyrosines, catalyzed by thyroid peroxidase, does not occur unless they are iodinated. Because only a few tyrosyl groups become iodinated, something specific about the structure of the protein near these residues probably facilitates both iodination and conjugation. The thyroid hormones, although still part of the thyroglobulin molecule, are stored as colloid in the thyroid follicle.
While they are attached to thyroglobulin in the thyroid follicular lumen (Fig. 49-1), thyroid hormones remain inactive until the iodinated thyroglobulin is hydrolyzed. Before this proteolysis can begin, the follicular cells must resorb thyroglobulin from the follicular lumen by fluid-phase endocytosis (see Chapter 2). As the endocytic vesicle containing the colloid droplet moves from the apical toward the basolateral membrane, it fuses with lysosomes to form a lysoendosome. Inside this vesicle, lysosomal enzymes hydrolyze the thyroglobulin and form T4 and T3, as well as diiodothyronine (DIT) and monoiodothyronine (MIT). The vesicle releases both T4 and T3 near the basolateral membrane, and these substances exit the cell into the blood by an unknown mechanism. Approximately 90% of the thyroid hormone secreted by the thyroid is released as T4, and 10% is released as T3. The thyroid releases very little reverse T3 into the blood. As discussed in the next section, nonthyroidal tissues metabolize the T4 released by the thyroid into T3 and rT3. Approximately three fourths of circulating T3 arises from the peripheral conversion of T4, which occurs principally in the liver and kidneys.
In the circulation, both T4 and T3 are highly bound to plasma proteins. Thyroid-binding globulin (TBG), albumin, and transthyretin (TTR) account for most of this binding. The affinity of these binding proteins is sufficiently high that, for T4, more than 99.98% of the hormone circulates tightly bound to protein. T3 is bound only slightly less: ~99.5% is protein bound. Because the free or unbound hormone in the circulation is responsible for the actions of the thyroid hormones on their target tissues, the large amount of bound hormone has considerably confounded our ability to use simple measurements of the total amount of either T4 or T3 in the plasma to provide a reliable index of the adequacy of thyroid hormone secretion. For example, the amount of TBG in the serum can change substantially in different physiological states. Pregnancy, oral estrogen therapy, hepatitis, and chronic heroin abuse can all elevate the amount of TBG and hence the total concentration of T4 and T3. Decreased levels of TBG, associated with diminished concentration of total T4 and T3, can accompany steroid usage and the nephrotic syndrome. However, despite the marked increases or decreases in the amounts of circulating TBG, the concentrations of free T4 and T3 do not change in the aforementioned examples. The box titled Free Versus Bound Thyroxine indicates how one can calculate levels of free T4 or T3, knowing the concentration of TBG and the concentration of total T4 or total T3.
Iodine Deficiency
In areas where soil is relatively iodine deficient, human iodine deficiency is common. Because seawater and seafood contain large amounts of iodide, iodine deficiency is more common in inland areas, particularly in locales that rely on locally grown foods. For example, in inland areas of South America along the Andes Mountains, in central Africa, and in highland regions of Southeast Asia, iodine deficiency is common. In the early 1900s, investigators first recognized that iodide was present in high concentrations in the thyroid and that iodine deficiency promoted goiter formation. These observations led to efforts to supplement dietary iodine. Iodine deficiency causes thyroid hormone deficiency. The pituitary responds to this deficit by increasing the synthesis of thyrotropin (or TSH), which, in turn, increases the activity of the iodine-trapping mechanism in the follicular cell in an effort to overcome the deficiency. The increased TSH also exerts a trophic effect that increases the size of the thyroid gland. If this trophic effect persists for sufficient time, the result is an iodine-deficient goiter. The word goiter is simply a generic term for an enlarged thyroid. If this effort at compensation is not successful (i.e., if insufficient thyroid hormone levels persist), the person will develop signs and symptoms of goitrous hypothyroidism. When iodine deficiency occurs at critical developmental times in infancy, the effects on the CNS are particularly devastating and produce the syndrome known as cretinism. Persons so affected have a characteristic facial appearance and body habitus, as well as severe mental retardation. Dietary supplementation of iodine in salt and bread has all but eliminated iodine deficiency from North America. In many nations, especially in mountainous and landlocked regions of developing nations, iodine deficiency remains a major cause of preventable illness.
The liver makes each of the thyroid-binding proteins. TBG is a 54-kDa glycoprotein consisting of 450 amino acids. It has the highest affinity for T4 and T3 and is responsible for most of the thyroid-binding capacity in the plasma. The extensive binding of thyroid hormones to plasma proteins serves several functions. It provides a large buffer pool of thyroid hormones in the circulation, so that the active concentrations of hormone in the circulation change very little on a minute-to-minute basis. The binding to plasma proteins markedly prolongs the half-lives of both T4 and T3. T4 has a half-life of 8 days, and T3 has a half-life of ~24 hours; each is longer than the half-life of the steroid or peptide hormones. Finally, because much of the T3 in the circulation is formed by the conversion of T4 to T3 in extrathyroidal tissues, the presence of a large pool of T4 in the plasma provides a reserve of prohormone available for synthesis of T3. This reserve may be of particular importance because T3 is responsible for most of the biological activity of thyroid hormones.
Free Versus Bound Thyroxine
Most of the T4 and T3 in the serum is bound to proteins, the most important of which is TBG. For the binding of T4 to TBG, the reaction is as follows:
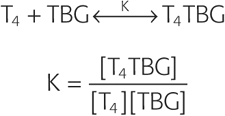
The binding constant K is ~2 × 1010 M−1 for T4. The comparable binding constant for T3 is ~5 × 108 M−1. Approximately one third of TBG’s binding sites are occupied by T4. Therefore, we have all the information we need to compute the concentration of free T4:
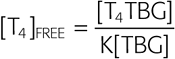
A reasonable value for [T4TBG] would be 100 nM, and for [TBG], 250 nM. Thus,
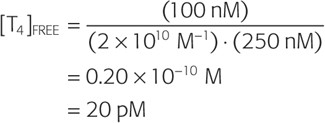
Because the bound T4 in this example is 100 nM, and the free T4 is only 20 pM, we can conclude that only ~0.02% of the total T4 in the plasma is free. Because 99.98% of the total T4 in the plasma is bound, moderate fluctuations in the rate of T4 release from the thyroid have only tiny effects on the level of free T4. To simplify, we have not included the minor contribution of albumin and TTR in this sample calculation.
The thyroid synthesizes and stores much more T4 than T3, and this is reflected by the ~10:1 ratio of T4/T3 secreted by the thyroid. However, certain tissues in the body have the capacity to selectively deiodinate T4, thereby producing either T3 or rT3. T3 and rT3 can each be further deiodinated to various DITs and MITs (Fig. 49-4); both DITs and MITs are biologically inactive. Both iodine atoms on the inner ring, and at least one iodine atom on the outer ring, appear essential for biological activity. Similarly, the loss of the amino group renders T4 or T3 inactive. The importance of the peripheral deiodination of T4 to T3 can be readily appreciated from the observation that persons whose thyroids have been removed have normal circulating concentrations of T3 when they receive oral T4 supplementation.
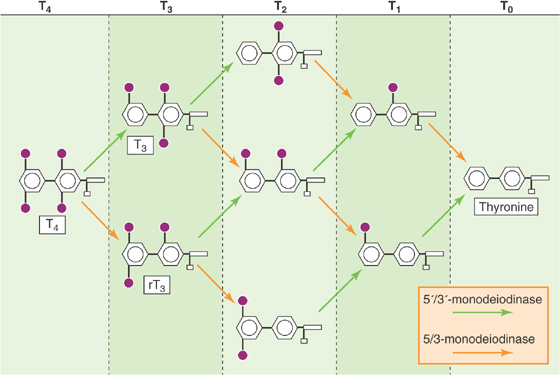
Figure 49-4 Peripheral metabolism of T4. A 5′/3′-monodeiodinase (green arrows) removes I from the outer benzyl ring, whereas a 5/3-monodeiodinase (orange arrows) removes I from the inner benzyl ring. Thus, the action of the 5′/3′-monodeiodinase on T4 yields T3, whereas the action of the 5/3-monodeiodinase yields rT3. Sequential deiodination yields T0 (thyronine).
Inasmuch as T3 is biologically much more active than the far more abundant T4, the regulation of the conversion of T4 to T3 in peripheral tissues assumes considerable importance. Two distinct deiodinases convert T4 to T3 (Fig. 49-4): The 5′/3′-deiodinase removes an I from the outer ring, thus producing T3, whereas the 5/3-deiodinase removes an I from the inner ring, thereby producing the inactive rT3. Because the 3′ and 5′ positions in T4 are equivalent stereochemically, removal of either of these yields T3. Similarly, removing the I from either the 3 or the 5 position of the inner ring of T4 yields rT3. Further deiodination by these two enzymes ultimately yields T0 (i.e., thyronine).
The 5′/3′-deiodinase, which acts on the outer ring, comes in two forms. Type 1 is present in high concentrations in the liver, kidneys, and thyroid. It appears to be responsible for generating most of the T3 that reaches the circulation. Type 2 is found predominantly in the pituitary, central nervous system (CNS), and placenta and is involved in supplying those tissues with T3 by local generation from plasma-derived T4. As shown later, the type 2 enzyme in the pituitary is of particular importance because the T3 that is generated there is responsible for the feedback inhibition of the release of thyrotropin (or thyroid-stimulating hormone, TSH).
The relative activity of the deiodinases changes in response to physiological and pathologic stimuli. Caloric restriction or severe stress inhibits the type 1 outer ring deiodinase; this process decreases the conversion of T4 to T3—and thus reduces the levels of T3. In contrast, levels of rT3 rise by default in these situations, in part because of reduced conversion to DITs. These decreases in T3 levels are accompanied by a decline in metabolic rate. You may think that because plasma levels of T3 fall, there would be a compensatory rise in TSH, the secretion of which is inhibited by T3. However, because type 2 deiodinase mediates the conversion of T4 to T3 within the pituitary and CNS, and because caloric restriction does not affect this enzyme, local T3 levels in the pituitary are normal. Thus, the thyrotrophs in the pituitary continue to have adequate amounts of T3, and no compensatory rise in TSH occurs. Teleologically, the rationale to restrain calorie expenditure in settings of decreased caloric intake is appealing. (See Note: Effect of Calorie Restriction on Type-1 Deiodinase)
Thyroid hormones act on many body tissues to exert both metabolic and developmental effects. Most, if not all, of the actions of thyroid hormones occur as thyroid hormones bind to and activate nuclear receptors (see Chapter 3). These receptors, in turn, are bound to chromatin and alter the transcription of specific genes. The multitude of thyroid hormone actions is mirrored by the ubiquitous expression of thyroid hormone receptors (TRs) throughout the body’s tissues. Once T4 and T3 leave the plasma, they enter the cell either by diffusing through the lipid of the cell membrane or by carrier-mediated transport (Fig. 49-5). Once in the cytoplasm, T4 and T3 bind to sites in the cytosol, microsomes, and mitochondria, as well as the nucleus. This observation has prompted speculation that thyroid hormones may exert actions through mechanisms not involving transcriptional regulation. However, current evidence suggests that if these nongenomic mechanisms are operative, they are exceptions to the general schema shown in Figure 49-5, which appears to account for most of the actions of thyroid hormones.

Figure 49-5 Action of thyroid hormones on target cells. Free extracellular T4 and T3 enter the target cell. Once T4 is inside the cell, a cytoplasmic 5′/3′-monodeiodinase converts much of the T4 to T3, so cytoplasmic levels of T4 and T3 are about equal. TRs bind to nuclear DNA at thyroid response elements in the promoter region of genes regulated by thyroid hormones. The binding of T3 or T4 to the receptor regulates the transcription of these genes. Of the total thyroid hormone bound to receptor, ~90% is T3. The receptor that binds to the DNA is preferentially a heterodimer of the TR and RXR.
Biologically, T3 is much more important than T4. This statement may be surprising inasmuch as the total concentration of T4 in the circulation is 50-fold or higher than that of T3. T3 has greater biological activity for three reasons. First, T4 is bound (only 0.02% is free) more tightly to plasma proteins than is T3 (0.50% is free; i.e., a 25-fold higher ratio of free to bound). The net effect is that the amount of free T4 in the circulation is only ~2-fold greater than the amount of free T3. Second, because the target cell converts T4—once it has entered the cell—to T3, it turns out that T4 and T3 are present at similar concentrations in the cytoplasm of target cells. Finally, the TR in the nucleus has ~10-fold greater affinity for T3 than T4, so that T3 is more potent on a molar basis. As a result, T3 is responsible for ~90% of the occupancy of TRs in the euthyroid state. (See Note: The Sick Euthyroid Syndrome)
When T3 or T4 binds to the TR in the nucleus, the hormone-bound receptors either activate or repress the transcription of specific genes. As discussed earlier in Chapter 4, TR preferentially binds to DNA as a heterodimer of TR and the retinoid X receptor (RXR). TR belongs to the superfamily of nuclear receptors that contains domains A through F (see Chapter 3). Three regions are especially important for TR: (1) The amino-terminal A/B region contains the first of two transactivation domains, which are responsible for transducing receptor binding into a conformational change in the DNA, thereby initiating transcription; (2) the middle or C region is responsible for DNA binding through two zinc fingers (see Chapter 4), as well as dimerization between receptors; and (3) the E region, toward the carboxyl terminus, is responsible for binding the ligand (T3 or T4), and also for dimerization.
Actually, two TR genes—α and β—are present on chromosomes 17 and 3, respectively. The expression of these receptor genes is tissue specific and varies with time of development. The liver expresses principally the β isoform, whereas the α isoform predominates in the brain. During development, the amount of α expressed may vary 10-fold or more.
Nongenomic actions of thyroid hormone have been observed in several tissues, including heart, muscle, fat, and pituitary. Early on, it was thought that thyroid hormone could act through nongenomic pathways to enhance mitochondrial oxidative phosphorylation—or at least energy expenditure as measured by O2 consumption. More recent data support this hypothesis as well as actions on ion channels, second messengers, and protein kinases. It is less clear whether these actions are mediated through the classical TR, as they are for nongenomic actions of the estradiol receptor (see Chapter 47), or another high-affinity thyroid-binding protein.
Investigators have long observed that excess thyroid hormone raises the basal metabolic rate (BMR) as measured by either heat production (direct calorimetry) or O2 consumption (indirect calorimetry). Conversely, thyroid hormone deficiency is accompanied by a decreased BMR. Figure 49-6 illustrates the effect of thyroid hormone levels on BMR, and Table 49-1 summarizes the effect of the thyroid hormones on several parameters. Thyroid hormones increase the BMR by stimulating both catabolic and anabolic reactions in pathways affecting fats, carbohydrates, and proteins.
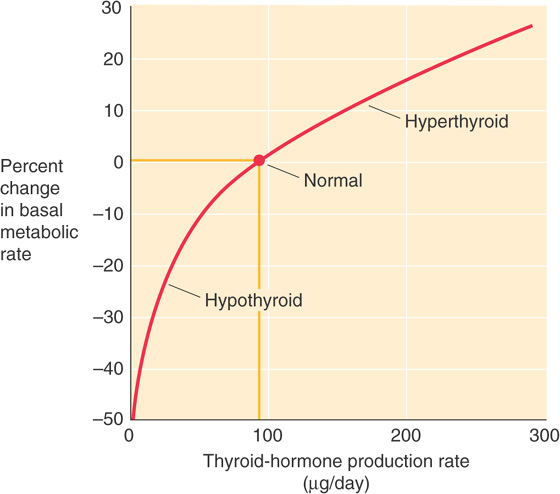
Figure 49-6 Effect of thyroid hormone on BMR. This graph shows the dependence of BMR on the daily rate of thyroid hormone secretion (T4 and T3). We use the secretion rate because it is difficult to know whether to use free T4 or free T3. Thus, the secretion rate is a crude measure of effective thyroid hormone levels. (Data from Guyton AC, Hall JE: Textbook of Medical Physiology, 9th ed. Philadelphia: WB Saunders, 1996.)
Table 49-1 Physiological Effects of the Thyroid Hormones (T3 and T4)
Parameter |
Low Level of Thyroid Hormones (Hypothyroid) |
High Level of Thyroid Hormones (Hyperthyroid) |
Basal metabolic rate |
↓ |
↑ |
Carbohydrate metabolism |
↓ Gluconeogenesis |
↑ Gluconeogenesis |
|
↓ Glycogenolysis |
↑ Glycogenolysis |
|
Normal serum [glucose] |
Normal serum [glucose] |
Protein metabolism |
↓ Synthesis |
↑ Synthesis |
|
↓ Proteolysis |
↑ Proteolysis |
|
|
Muscle wasting |
Lipid metabolism |
↓ Lipogenesis |
↑ Lipogenesis |
|
↓ Lipolysis |
↑ Lipolysis |
|
↑ Serum [cholesterol] |
↓ Serum [cholesterol] |
Thermogenesis |
↓ |
↑ |
Autonomic nervous system |
Normal levels of serum catecholamines |
↑ Expression of β adrenoceptors (increased sensitivity to catecholamines, which remain at normal levels) |
Carbohydrate Metabolism Thyroid hormones raise the rate of hepatic glucose production, principally by increasing hepatic gluconeogenic activity. This effect generally does not result in increases in plasma [glucose], provided the pancreas responds by augmenting insulin secretion. Thyroid hormones also enhance the availability of the starting materials required for increased gluconeogenic activity (i.e., amino acids and glycerol), and they specifically induce the expression of several key gluconeogenic enzymes, including phosphoenolpyruvate carboxykinase, pyruvate carboxylase, and glucose-6-phosphatase.
Protein Metabolism The amino acids required for increased hepatic gluconeogenesis, stimulated by thyroid hormones, come from increased proteolysis, predominantly in muscle. Thyroid hormones also increase protein synthesis. Because the increases in protein degradation usually outweigh the increases in synthesis, a net loss of muscle protein occurs. The catabolic effect is exaggerated when T3 is present in great excess, so that muscle wasting and weakness, as well as increased nitrogen loss in the urine as urea (see Chapters 36 and 46), can be prominent features of clinical thyrotoxicosis (hyperthyroidism).
Lipid Metabolism The glycerol required for increased hepatic gluconeogenesis, stimulated by thyroid hormones, comes from increased degradation of stored triglycerides in adipose tissue. The fatty acids released along with the glycerol provide fuel for the liver to support the energy demand of gluconeogenesis. Thyroid hormones not only increase lipolysis but also enhance lipogenesis. Indeed, modest amounts of thyroid hormones are needed for the normal synthesis of fatty acid by liver. Very high levels of T3 shift the balance in favor of lipolysis, with resulting generalized fat mobilization and loss of body fat stores.
By accelerating the rates of glucose production, protein synthesis, and degradation, as well as of lipogenesis and lipolysis, the thyroid hormones stimulate energy consumption. Therefore, to the extent that thyroid hormones stimulate both synthesis and degradation, they promote futile cycles that contribute significantly to the increased O2 consumption seen in thyrotoxicosis (hyperthyroidism).
How, at the molecular level, thyroid hormones affect the BMR in states of both spontaneous and experimentally induced thyroid hormone excess or deficiency has been a difficult question to answer. The changes in metabolic rate do not appear to be determined by changes in the expression of a single gene. Several specific examples of the effects of thyroid hormones on target tissues serve to illustrate their general mechanism of action.
Na-K Pump Activity In muscle, liver, and kidney, thyroid hormone–induced increases in O2 consumption are paralleled by increases in the activity of the Na-K pump in the plasma membrane (see Chapter 5). This increase in transport is the result, at least in part, of an increase in the synthesis of new transporter units that are inserted into the plasma membrane. At least in some tissues, blockade of the increases in Na-K pump activity with ouabain also blocks the increase in O2 consumption. T3 stimulates the transcription of the genes for both the α and β subunits of the Na-K pump. In addition, T3 increases translation by stabilizing the mRNA that encodes the Na-K pump. Increases in the activity of this transporter use additional ATP and thereby consume O2 and generate heat. Inasmuch as states of thyroid hormone excess are not accompanied by any noticeable derangement of plasma electrolyte levels, presumably the increase in Na-K pump activity is compensated in some manner by a leak of Na+ and K+, although such pathways have not yet been defined. Overall, the increased activity of the Na-K pump (with an accompanying cation leak) would result in a futile cycle whereby energy was consumed without useful work.
Thermogenesis In rodents, thyroid hormones may affect metabolic rate and thermogenesis through another futile cycle mechanism. Brown fat in these animals expresses a mitochondrial uncoupling protein (UCP), or thermogenin, that dissociates oxidative phosphorylation from ATP generation. Thus, mitochondria consume O2 and produce heat without generating ATP. Both T3 and β-adrenergic stimulation (acting through the β3 receptor) enhance respiration in brown adipose tissue by stimulating this uncoupling mechanism. We discuss thermogenin—and the vital role it plays in helping to keep newborn humans warm—in Chapter 57.
Thyroid hormones also increase the BMR by increasing the thermogenic effects of other processes. An example is the effect of adrenergic stimulation on thermogenesis, discussed earlier. In humans, plasma concentrations of catecholamines are normal in states of both excess and deficient T3 and T4. However, excess thyroid hormone raises the sensitivity of tissues to the action of adrenergic hormones. In heart, skeletal muscle, and adipose tissue, this effect is the result, at least in part, of increased expression of β-adrenergic receptors by these tissues. In patients who are acutely thyrotoxic, treatment with β-receptor antagonists is one of the first priorities. This treatment blunts the hypersympathetic state induced by the excess of thyroid hormones. Thyroid hormones may also exert postreceptor effects that enhance adrenergic tone. In the heart, thyroid hormones also regulate the expression of specific forms of myosin heavy chain. Specifically, in rodents, thyroid hormone increases the expression of the myosin α chain, thereby favoring the α/α isoform of myosin heavy chain (see Chapter 9). This isoform is associated with greater activity of both actin and Ca2+-activated ATPase, faster fiber shortening, and greater contractility.
In amphibians, thyroid hormone regulates the process of metamorphosis. Removing the thyroid gland from tadpoles causes development to arrest at the tadpole stage. Early administration of excess thyroid hormone can initiate premature metamorphosis. Iodothyronines are present even farther down the phylogenetic tree, at least as far as primitive chordates, although these animals lack a thyroid gland per se. However, the biological actions of iodothyronines in many species are not known.
Thyroid hormones are essential for normal human development as well, as starkly illustrated by the unfortunate condition of cretinism in regions of endemic iodine deficiency. Cretinism is characterized by profound mental retardation, short stature, delay in motor development, coarse hair, and a protuberant abdomen. Correction of iodine deficiency has essentially eliminated endemic cretinism in developed nations. Sporadic cases continue to occur, however, as a result of congenital defects in thyroid hormone synthesis. If hypothyroidism is recognized and corrected within a few days of birth, development—including mental development—can proceed almost normally. For this reason, many localities have initiated laboratory screening of newborns for hypothyroidism because clinical recognition of the full syndrome often occurs only after the developmental abnormalities in the CNS are irreversible.
Typically overshadowed by the impaired cognitive development that occurs in cretinism is the dwarfism that results from the effects of thyroid hormone deficiency on human growth (Fig. 49-7). In children with normal thyroid function at birth, development of hypothyroidism at any time before the fusion of the epiphyses of the long bones leads to growth retardation or arrest. Much of the loss in height that occurs can be recovered after thyroid hormone treatment is begun, a phenomenon called catch-up growth. If the diagnosis and treatment of hypothyroidism are delayed, loss of potential growth may occur. As indicated in Figure 49-7, catch-up growth may not apply to mental development unless the treatment is begun very early, certainly within a few days of birth. In general, the longer the duration of congenital hypothyroidism, the more profound is the mental retardation. The growth curve (i.e., a plot of the child’s height and weight versus age) can provide a particularly sensitive early indicator of hypothyroidism. An overactive thyroid is much less a problem than is an underactive thyroid with regard to its effect on growth; other signs and symptoms of an overactive thyroid predominate.

Figure 49-7 Effect of thyroid hormone on growth and development. This is a graph of developmental age—that is, the age that the child appears based on height, bone radiograph, and mental function—versus chronological age. For a normal child, the relationship is the straight line in red, for which developmental and chronological age are identical. The three green curves are growth curves for a child with thyroid hormone deficiency. At age 4.5 years, before initiation of therapy, height age, bone age, and mental age are all substantially lower than normal. Initiating replacement therapy with thyroid hormone at age 4.5 years causes a rapid increase in both height age and bone age (catch-up growth) but has no effect on mental age, which remains infantile. Treatment can help mental development only if the therapy is begun within a few days of birth. (Data from Wilkins L: The Diagnosis and Treatment of Endocrine Disorders of Childhood and Adolescence. Springfield, IL: Charles C Thomas, 1965.)
Cellular explanations of the effects of thyroid hormones on human development are incomplete. In rats, thyroid hormone induces the secretion of pituitary growth hormone (GH); thus, the growth retardation in thyroid-deficient rats may be partly the result of decreased GH secretion. However, in humans, who have no thyroid hormone response element in the promoter region of the GH gene, plasma [GH] is normal in hypothyroidism. Thus, the growth failure of hypothyroid human infants is not as readily explained. In humans, changes in the growth of long bones are more or less characteristic of thyroid hormone deficiency. These changes include a delay in formation of centers of ossification at the growth plate, followed by the appearance of several ossification centers, which eventually merge. Short stature in human juvenile or infantile hypothyroidism may be in part related to these abnormalities of cartilage growth and development as well as to resistance to the normal action of GH to promote growth. In rodents, thyroid hormone regulates the induction of expression of several neural proteins, including myelin basic protein. How deficiencies in these proteins may result in the generalized cortical atrophy seen in infantile hypothyroidism is not clear.
The pituitary regulates the synthesis and secretion of thyroid hormones through the release of thyrotropin—also known as thyroid stimulating hormone (TSH)—from the anterior pituitary. The hypothalamus, in turn, stimulates the release of TSH through thyrotropin-releasing hormone (TRH). Finally, circulating thyroid hormones exert feedback control on both TRH and TSH secretion.
Thyrotropin-Releasing Hormone TRH is a tripeptide pyro-Glu-His-Pro containing the modified amino acid pyro-Glu. It is found in many tissues, including the cerebral cortex, multiple areas of the gastrointestinal tract, and the β cells of the pancreas. However, the arcuate nucleus and the median eminence of the hypothalamus appear to be the major sources of the TRH that stimulates TSH synthesis and secretion (Fig. 49-8). TRH released by neurons in the hypothalamus travels to the anterior pituitary through the hypophyseal portal system (see Chapter 47). Hypothalamic lesions that interrupt TRH release or delivery cause a fall in basal TSH levels. Conversely, administering TRH intravenously can cause a rapid, dose-dependent release of TSH from the anterior pituitary. However, it is not clear that such bursts of TRH release and TSH secretion occur physiologically.
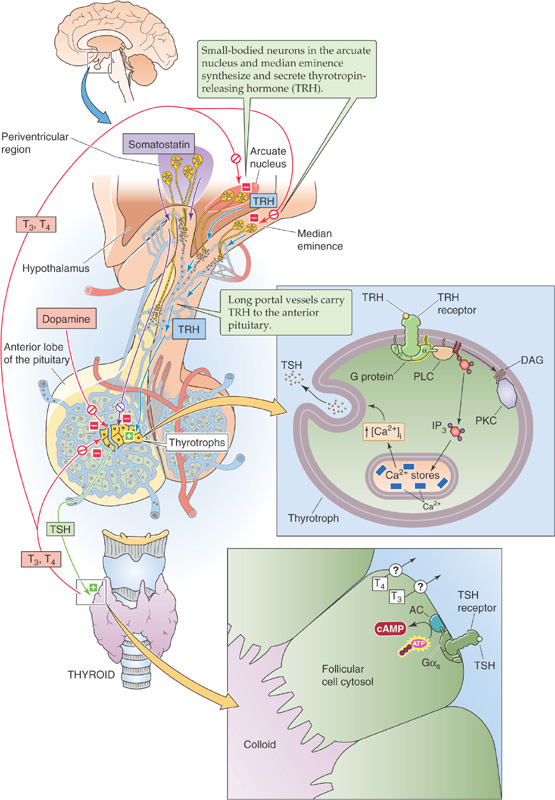
Figure 49-8 The hypothalamic-pituitary-thyroid axis. Small-bodied neurons in the arcuate nucleus and median eminence of the hypothalamus secrete TRH, a tripeptide that reaches the thyrotrophs in the anterior pituitary through the long portal veins. TRH binds to a G protein–coupled receptor on the thyrotroph membrane, thus triggering the DAG/IP3 pathway, leading to protein phosphorylation, and raising [Ca2+]i. These pathways stimulate the thyrotrophs to synthesize and release thyrotropin (or TSH), which is a 28-kDa glycoprotein stored in secretory granules. The TSH binds to receptors on the basolateral membrane of thyroid follicular cells, thereby stimulating Gαs. This process, in turn, activates adenylyl cyclase (AC) and raises [cAMP]i. As outlined in Figure 49-3, TSH stimulates certain steps in the synthesis and release of T4 and T3. Inside the pituitary, the type 2 form of 5′/3′-monodeiodinase converts T4 to T3, which negatively feeds back on the thyrotrophs as well as on the TRH-secreting neurons. Somatostatin and dopamine—released by hypothalamic neurons—inhibit TSH release and thus can influence the set point at which TSH is released in response to a given amount of T3 in the pituitary. PKC, protein kinase C; PLC, phospholipase C.
Thyrotropin-Releasing Hormone Receptor Once it reaches the thyrotrophs in the anterior pituitary, TRH binds to the TRH receptor, a G protein–coupled receptor on the cell membranes of the thyrotrophs. TRH binding triggers the phospholipase C pathway (see Chapter 3). The formation of diacylglycerols (DAGs) stimulates protein kinase C and leads to protein phosphorylation. The simultaneous release of inositol triphosphate (IP3) triggers Ca2+ release from internal stores, thus raising [Ca2+]i. The result is an increase in both the synthesis and release of TSH, which is stored in secretory granules. TRH produces some of its effects by activating phospholipase A2, a process leading to the release of arachidonic acid and the formation of a variety of biologically active eicosanoids (see Chapter 3). In healthy persons, administering TRH also raises plasma [prolactin] by stimulating lactotrophs in the anterior pituitary (see Chapter 52). However, no evidence indicates a regulatory role for TRH in prolactin secretion or action.
Thyrotropin The thyrotrophs represent a relatively small number of cells in the anterior pituitary. The TSH that they release is a 28-kDa glycoprotein with α and β chains. The α chain of TSH is identical to that of the other glycoprotein hormones: the gonadotropins luteinizing hormone (LH), follicle-stimulating hormone (FSH), and human chorionic gonadotropin (hCG). The β chain is unique to TSH and confers the specificity of the hormone. Once secreted, TSH acts on the thyroid follicular cell through a specific receptor.
Graves Disease
Surprisingly, it is not uncommon for B lymphocytes to synthesize immune globulins that bind to and activate the TSH receptor, thereby reproducing all the actions of TSH on the thyroid. Unfortunately, these errant lymphocytes do not regulate the production of these immunoglobulins in a manner analogous to the regulated secretion of TSH by the pituitary. As a result, iodide trapping by the thyroid increases, the synthesis and secretion of both T3 and T4 increase, and the thyroid enlarges to produce a goiter. Untreated, the affected individual becomes increasingly hyperthyroid. The clinical manifestations of hyperthyroidism include an increased metabolic rate with associated weight loss, sweating and heat intolerance, a rapid and more forceful heartbeat, muscle weakness and wasting, tremulousness, difficulty concentrating, and changes in hair growth and skin texture. Because TSH stimulates all areas of the thyroid, the thyroid is symmetrically enlarged, and even the isthmus is frequently palpable on clinical examination. (See Note: Thyroid Storm)
The abnormal immunoglobulin is designated TSI for thyroid-stimulating immunoglobulin. The constellation of symptoms noted previously, together with a symmetrically enlarged goiter, is called Graves disease, after Robert Graves, who provided one of the first detailed descriptions of the disorder in the early 19th century. These antibodies are also able to stimulate connective tissue in the extraocular muscles and in the dermis of the lower extremity to synthesize mucopolysaccharides, thus leading to thickening of both the muscle and the dermis. Therefore, in addition to the abnormalities of thyroid growth and hyperfunction, a few individuals with Graves disease develop a peculiar infiltrative abnormality in the extraocular muscles. When severe, this infiltrative ophthalmopathy impairs muscle function and causes diplopia (double vision) and forward protrusion of the eyes (exophthalmos). Even less frequently, patients with Graves disease develop infiltrating dermopathy in the skin over the lower legs called pretibial myxedema. This thickening of the skin occurs in localized patches and is pathologically distinct from the generalized thickening and coarsening of the skin seen in hypothyroidism (generalized myxedema).
Thyrotropin Receptor The TSH receptor on the thyroid follicular cells is a member of the family of G protein–coupled receptors. Like receptors for the other glycoprotein hormones (LH, FSH, and hCG), the TSH receptor activates adenylyl cyclase through Gαs (see Chapter 3). This activation of the TSH receptor stimulates a diverse range of physiological processes or events, summarized in Figure 49-3:
1. Iodide uptake by the NIS on the basolateral membrane of the thyroid follicular cell. Stimulation of this cotransporter allows for trapping of dietary iodine within the thyroid gland. The ratio of follicular cell iodine to serum iodine (the so-called thyroid/serum or T/S ratio) is 30 : 1 in euthyroid individuals. The T/S ratio decreases under conditions of low TSH (e.g., hypophysectomy) and increases under conditions of high TSH (e.g., a TSH-secreting pituitary adenoma);
2. Iodination of thyroglobulin in the follicular lumen;
3. Conjugation of iodinated tyrosines to form T4 and T3 linked to thyroglobulin;
4. Endocytosis of iodinated thyroglobulin into the follicular cells from thyroid colloid;
5. Proteolysis of the iodinated thyroglobulin in the follicular cell;
6. Secretion of T4 and T3 into the circulation; and
7. Hyperplasia of the thyroid gland because of the growth factor effects of TSH.
Figure 49-9 illustrates the goiter that occurs when TSH concentrations are elevated for a prolonged period of time and stimulates an otherwise normal thyroid gland (see the box on Iodine Deficiency). Hyperplasia of the thyroid gland also occurs in Graves disease because of stimulation of the TSH receptor by a thyroid-stimulating immunoglobulin. In contrast, the chronic elevation of TSH typically seen when the thyroid gland undergoes autoimmune injury (Hashimoto thyroiditis) does not respond by hypertrophy.
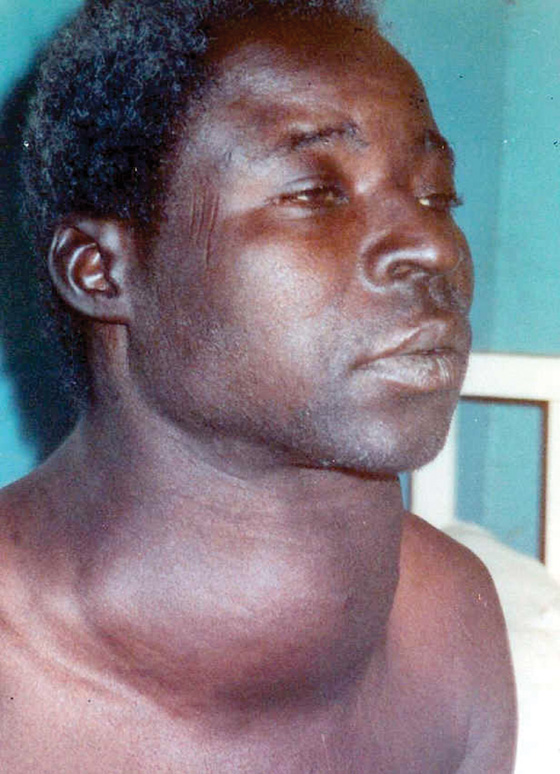
Figure 49-9 Goiter in iodine deficiency. A young woman from a region in Central Africa where iodine deficiency is prevalent exhibits a large goiter secondary to iodine deficiency and the growth-promoting effects of TSH, the levels of which as part of a feedback mechanism for achieving a sufficient amount of thyroid hormone.
Circulating free T4 and T3 inhibit both the synthesis of TRH by hypothalamic neurons and the release of TSH by the thyrotrophs in the anterior pituitary. Plasma [TSH] is very sensitive to alteration in the levels of free T4 and T3; a 50% decline in free T4 levels can cause plasma [TSH] to increase 50- to 100-fold. Conversely, as may be expected of a well-functioning feedback system, an excess of thyroid hormone leads to a decrease in plasma [TSH].
Hypothyroidism
Hypothyroidism is one of the most common of all endocrine illnesses—even more common than Graves disease. Hypothyroidism affects between 1% and 2% of all adults at some time in their lives. Women are much more commonly affected than men. Although hypothyroidism has several causes, the most common cause worldwide is iodine deficiency. In the United States, by far the most common cause is an autoimmune disorder called Hashimoto thyroiditis. Like Graves disease, Hashimoto thyroiditis is caused by an abnormal immune response that includes the production of antithyroid antibodies, in this case, antibodies against the thyroid follicular cells, microsomes, and TSH receptors. Unlike in Graves disease, the antibodies in Hashimoto thyroiditis are not stimulatory, but rather are part of an immune process that blocks and destroys thyroid function. The titers of these autoantibodies can reach colossal proportions.
Typically, hypothyroidism in Hashimoto thyroiditis is an insidious process that develops slowly; indeed, many patients are diagnosed, long before striking clinical manifestations are apparent, by routine blood tests revealing an elevated TSH despite normal levels of T3 and T4. These individuals, although not yet clinically hypothyroid, are sometimes treated with thyroid hormone replacement, so the clinical manifestations of hypothyroidism are never given a chance to develop.
In patients in whom the disease does evolve, the classical presentation consists of painless goiter, skin changes, peripheral edema, constipation, headache, joint aches, fatigue, and, in women, anovulation. The TSH level should be checked in any female patient with secondary amenorrhea. In time, some of these patients also develop other autoimmune disorders, such as pernicious anemia, myasthenia gravis, Addison disease, diabetes mellitus, and ovarian failure.
Like patients with hyperthyroidism, who may be threatened by thyroid storm, those with hypothyroidism also have their severe, life-threatening variant, in this case called myxedema coma. This malady is quite rare and occurs most commonly in elderly patients with established hypothyroidism. Hypothermia and coma evolve slowly in these patients, and the usual causes are failure to take prescribed thyroid hormone replacement, cold exposure, sepsis, heart failure, and alcohol abuse.
At the level of the thyrotroph, the sensor in this feedback system monitors the concentration of T3 inside the thyrotroph (Fig. 49-8). As noted earlier, T3 either can enter directly from the blood plasma or can form inside the thyrotroph by deiodination of T4. The negative feedback of T4 and T3 on TSH release occurs at the level of the pituitary thyrotroph by both indirect and direct mechanisms. In the indirect feedback pathway, intracellular T3 decreases the number of TRH receptors on the surface of the thyrotroph. As a result, thyroid hormones indirectly inhibit TSH release by reducing the sensitivity of the thyrotrophs to TRH. In the direct feedback pathway, intracellular T3 inhibits the synthesis of both the α and the β chains of TSH. Indeed, both the α and β TSH genes have T3 response elements in their promoter regions. These response elements, which are inhibitory, differ from those found in genes that are positively regulated by T3 (e.g., Na-K pump).
Clinical Assessment of Thyroid Function
Plasma thyrotropin levels. Direct measurements of T4/T3 provide a measure of total circulating hormone (i.e., the sum of free T4 and T3, as well as T4 and T3 bound to TBG, TTR, and albumin). However, these direct measurements do not allow one to distinguish between bound and free T4/T3. The sensitive response of TSH to changes in thyroid hormone levels provides an extremely valuable tool for assessing whether the free T4/T3 levels in the circulation are deficient, sufficient, or excessive. Indeed, the level of TSH reflects the amount of free, biologically active thyroid hormone in the target tissue. As a result, in recent years, measurements of plasma TSH using very sensitive immune assay methods have come to be regarded as the single best determinants of thyroid hormone status. Obviously, this approach is valid only if the thyrotrophs themselves are able to respond to T3/T4—that is, if patients have no evidence of pituitary dysfunction.
The health of the thyrotrophs themselves can be tested by injecting a bolus of synthetic TRH and monitoring changes in plasma [TSH]. In hypothyroid patients, the subsequent rise in plasma [TSH] is more dramatic than in physiologically normal individuals. This test was of great value in confirming the diagnosis of hypothyroidism before the advent of today’s sensitive assays, but it has largely been abandoned.
Radioactive iodine uptake. Measuring the amount of a standard bolus of radioactive iodine that the thyroid can take up was also once widespread as a measure of thyroid function. A hyperactive gland would take up increased amounts of the tracer, whereas an underactive gland would take up subnormal amounts. Today, the test is mostly used for three other purposes. First, radioactive iodine uptake can show whether a solitary thyroid nodule, detected on physical examination, is “hot” (functioning) or “cold” (nonfunctioning). Cold nodules are more likely than hot ones to harbor a malignancy. Second, radioactive iodine uptake can show whether hyperthyroidism is the result of thyroid inflammation (i.e., thyroiditis), in which tracer uptake is minimal, or Graves disease, in which tracer uptake is increased. Third, high doses of radioactive iodine are commonly used to treat patients with hyperthyroidism.
Free T4 and T3 concentrations in the plasma, which determine intracellular T3 levels in the thyrotroph, are relatively constant over the course of 24 hours, a finding reflecting the long half-lives of both T4 and T3. Given that the levels of T4 and T3 are the primary triggers in the afferent limb of the negative feedback for the hypothalamic-pituitary-thyroid axis, the feedback regulation of TSH secretion by thyroid hormones appears to be a slow process—essentially integrating thyroid hormone levels over time. Indeed, T3 feeds back on the thyrotroph by modulating gene transcription, which by its very nature is a slow process.
The feedback of T4 and T3 on the release of TSH may also be under the control of somatostatin and dopamine, which travel from the hypothalamus to the thyrotrophs through the portal vessels (Fig. 49-8). Somatostatin and dopamine both inhibit TSH secretion, apparently by making the thyrotroph more sensitive to inhibition by intracellular T3—that is, shifting the set point for T3. Thus, somatostatin and dopamine appear to counterbalance the stimulatory effect of TRH. Although these inhibitory effects are readily demonstrated with pharmacological infusion of these agents, their physiological role in the regulation of TSH secretion appears small. In particular, with long-term administration of somatostatin or dopamine, compensatory mechanisms appear to override any inhibition.
A special example of feedback between T3 and TSH is seen in neonates of mothers with abnormal levels of T3. If the mother is hyperthyroid, both she and the fetus will have low TSH levels because T3 crosses the placenta. After birth, the newborn rapidly metabolizes T3, but TSH remains suppressed, so the infant temporarily becomes hypothyroid. Conversely, if the mother’s thyroid gland has been removed and she is hypothyroid because she is not receiving sufficient thyroid hormone replacement therapy, both she and the fetus will have high levels of circulating TSH. Immediately after birth, the newborn will be temporarily hyperthyroid.
Books and Reviews
Bassett JH, Harvey CB, Williams GR: Mechanisms of thyroid hormone receptor–specific nuclear and extra nuclear actions. Mol Cell Endocrinol 2003; 213:1-11.
Cavalieri RR: Iodine metabolism and thyroid physiology: Current concepts. Thyroid 1997; 7:177-181.
Dumont JE, Lamy F, Roger P, Maenhaut C: Physiological and pathological regulation of thyroid cell proliferation and differentiation by thyrotropin and other factors. Physiol Rev 1992; 72:667-697.
Gershengorn MC, Osman R: Molecular and cellular biology of thyrotropin-releasing hormone receptors. Physiol Rev 1996; 76:175-191.
Guyton AC, Hall JE: Textbook of Medical Physiology, 9th ed. Philadelphia: WB Saunders, 1996.
Larsen PR: Update on the human iodothyronine selenodeiodinases, the enzymes regulating the activation and inactivation of thyroid hormone. Biochem Soc Trans 1997; 25:588-592.
Orban Z, Bornstein SR, Chrousos GP: The interaction between leptin and the hypothalamic-pituitary-thyroid axis. Horm Metab Res 1998; 30:231-235.
Samuels HH, Forman BM, Horowitz ZD, Ye Z-S: Regulation of gene expression by thyroid hormone. Annu Rev Physiol 1989; 51:623-639.
Wilkins L: The Diagnosis and Treatment of Endocrine Disorders of Childhood and Adolescence. Springfield, IL, Charles C Thomas, 1965.
Journal Articles
Arvan P, Kim PS, Kuliawat R, et al: Intracellular protein transport to the thyrocyte plasma membrane: Potential implications for thyroid physiology. Thyroid 1997; 7:89-105.
Dai G, Levy O, Carrasco N: Cloning and characterization of the thyroid iodide transporter. Nature 1996; 379:458-460.
Koenig RJ: Thyroid hormone receptor coactivators and corepressors. Thyroid 1998; 8:703-713.