CHAPTER 57
Ervin E. Jones
Growth of the fetus begins soon after fertilization, when the first cell division occurs. Cell division, hypertrophy, and differentiation are highly coordinated events that result in the growth and development of specialized organ systems. The fetus, fetal membranes, and placenta develop and function as a unit throughout pregnancy, and their development is interdependent. The growth trajectory of fetal mass is relatively flat during the first trimester, increases linearly at the beginning of the second trimester, and rises rapidly during the third trimester.
The growth of an organ occurs as a result of an increase in cell number (hyperplasia), an increase in cell size (hypertrophy), or both. We can define three sequential phases of growth: (1) pure hyperplasia, (2) hyperplasia and concomitant hypertrophy, and (3) hypertrophy alone. The time courses of the three phases of growth are organ specific. For example, the placenta goes through all three phases of growth, but these phases are compressed because the placental life span is relatively short. Moreover, simple hypertrophy is the primary form of placental growth. Thus, the weight, RNA content, and protein content of the human placenta increase linearly until term, but cell number does not increase during the third trimester.
In contrast to placental growth and development, growth of the fetus occurs almost entirely by hyperplasia. Thus, DNA content increases linearly in all fetal organs beginning early in the second trimester. Stimuli that either increase or decrease cell number, cell size, or both may accelerate or retard the growth of the whole fetus or of individual organs. The phase of growth during which the stimulus acts determines the response of the organ. For example, malnutrition occurring during the period of hyperplasia retards cell division and causes a deficiency in cell number. Therefore, adequacy of nutrition early in life may determine the number of cells in any organ. This effect on cell number may be irreversible, even if normal nutrition is restored later. Conversely, malnutrition occurring during the period of hypertrophy causes a reduction in cell size. However, this effect can be reversed, and normal cell size can be achieved if adequate nutrition is restored. Thus, reversibility depends on the timing of the stimulus.
The fertilized egg contains the genetic material that directs cell multiplication and differentiation and guides development of the human phenotype. For specific developmental events to occur at precise times (Table 57-1), a programmed sequence of gene activation and suppression is necessary. Ignoring apoptosis, the fertilized egg must undergo an average of ~42 divisions to reach newborn size. A fertilized ovum, weighing less than 1 ng, gives rise to a newborn weighing slightly more than 3 kg (an increase of more than 1012 fold). Not only must the total cell number in a term fetus lie within relatively narrow limits, but also the developmental program must trigger cell differentiation after a specified number of cell divisions. After birth, only approximately five additional divisions are necessary for the net increase in mass that is necessary to achieve adult size. Obviously, many tissues (e.g., gastrointestinal tract, skin, blood cells) must continually undergo cell division to replenish cells lost by apoptosis.
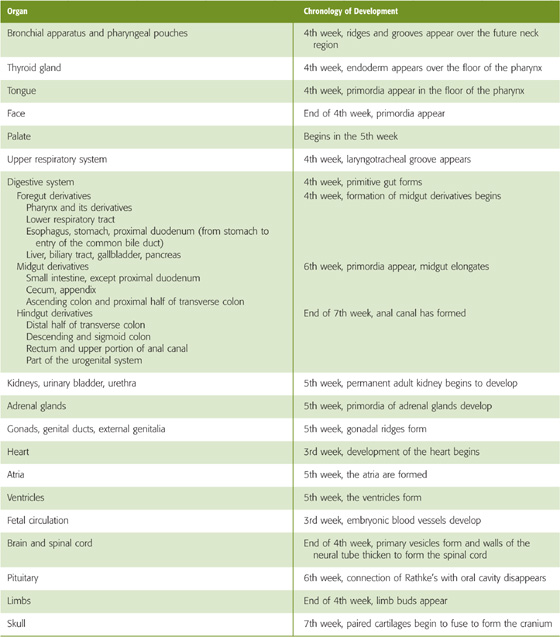
Table 57-1 Chronologic Development of Organs, Systems, and Body Form
Although the genetic makeup of the fetus principally determines its growth and development, other influences—both stimulatory and inhibitory—are superimposed on the genetic program. During the first half of pregnancy, the fetus’ own genetic program is the primary determinant of growth, thus constraining patterns of growth. During the second half of pregnancy, the patterns of growth and development are more variable. The four primary epigenetic factors at work during the second half of pregnancy are placental, hormonal, environmental (e.g., maternal nutrition, disease, drugs, altitude), and metabolic (e.g., diabetes). We discuss the first two factors (placental and hormonal) in the next two sections.
Studies of birth weights in families reveal that both parental and fetal genotypes affect birth weight (Table 57-2). The mother contributes to birth weight both through her influence on the environment that she provides for the fetus (~30%) and through the genes that she passes on to the fetus (~20%). The mother’s contribution to the fetal environment includes maternal health and nutritional status, environment, lifestyle, age (e.g., adolescents and older women have infants with lower birth weight), parity, prepregnancy weight and prenatal weight gain, early fat deposition, height, chronic diseases, infection, and stress. The father contributes to birth weight only through the genes that he passes on to his child (~20%). The unique fetal genotype—the interaction of the alleles provided by the parents (e.g., dominant versus recessive genes) considered apart from the individual contributions of the two parents—contributes ~15%. The gender of the fetus contributes ~2%. The remaining ~13% of the contribution to birth weight is multifactorial and may include variations in such factors as gestational age at delivery and multiple gestation (e.g., twinning).
Table 57-2 Determinants of Birth Weight
Factor |
Contribution to Final Birth Weight |
Maternal environment |
30% |
Maternal genotype |
20% |
Paternal genotype |
20% |
Fetal genotype (excluding gender) |
15% |
Fetal gender |
2% |
Multifactorial (e.g., gestational age at delivery, multiple gestation) |
13% |
The placenta plays several important roles in fetal growth and development. In addition to its transport and storage functions, the placenta is involved in numerous biosynthetic activities. These include the synthesis of steroids, such as estrogen and progesterone, and protein hormones, such as human chorionic gonadotropin (hCG) and the human chorionic somatomammotropins (hCSs) (see Chapter 56).
Fetal growth closely correlates with placental weight. During periods of rapid fetal growth, placental weight increases. As the placental mass increases, the total surface area of the placental villi (see Chapter 56) increases to sustain gas transport and fetal nutrition. Moreover, maternal blood flow to the uterus and fetal blood flow to the placenta also increase in parallel with the increase in placental mass. Placental growth increases linearly until ~4 weeks before birth. Intrauterine growth restriction (IUGR; see the box titled Growth Restriction) may occur as a result of decreased placental reserve caused by any insult. Adequate placental reserve is particularly important during the third trimester, when fetal growth is very rapid. For example, mothers who smoke during pregnancy tend to have small placentas and are at high risk of delivering a low birth weight baby.
Chapter 48 includes a discussion of several hormones—including glucocorticoids, insulin, growth hormone (GH), the insulin-like growth factors (IGFs), and thyroid hormones—that are important for achieving final adult mass.
Glucocorticoids and Insulin As its major energy source, the growing fetus uses glucose, which moves across the placenta by facilitated diffusion. Unlike the adult, who uses sophisticated hormonal systems to control blood glucose levels (see Chapter 51), the fetus is passive: the exchange of glucose across the placenta controls fetal blood glucose levels. The fetus normally has little need for gluconeogenesis, and the levels of gluconeogenic enzymes in the fetal liver are low. Glucocorticoids in the fetus promote the storage of glucose as glycogen in the fetal liver, a process that increases greatly during the final month of gestation in preparation for the increased glycolytic activity required during and immediately after delivery. Near term, when fetal glucose metabolism becomes sensitive to insulin, this hormone contributes to the storage of glucose as glycogen, as well as to the uptake and utilization of amino acids, and lipogenesis (see Chapter 51). Transient increases in maternal blood glucose levels after meals are closely mirrored by increases in fetal blood glucose levels. This transient fetal hyperglycemia leads to increased fetal production of insulin. Maternal insulin cannot cross the placenta.
In a mother with poorly controlled diabetes (see the box on diabetes mellitus in Chapter 51), sustained maternal hyperglycemia leads to sustained fetal hyperglycemia and therefore fetal hyperinsulinemia. The resulting high levels of fetal insulin, which is a growth factor (see Chapter 48), increase both the size of fetal organs (organomegaly) and fetal body mass (macrosomia). During the last half of the third trimester, fetal weight in poorly controlled diabetic pregnancies generally exceeds that in normal pregnancies. In some cases, large fetal size leads to problems at delivery. Indeed, the frequency of cesarean section is much higher in deliveries of fetuses born to diabetic mothers.
Insulin-Like Growth Factors Postnatally, GH acts by binding to GH receptors, primarily in the liver, and triggering the production of somatomedin or IGF-1. IGF-2 is not so much under the control of GH. The IGF-1 receptor is similar, but not identical, to the insulin receptor and can bind both IGF-1 and IGF-2, as well as insulin (see Chapter 48). In the fetus, both IGF-1 and IGF-2, which are mitogenic peptides, are extremely important for growth. IGF-1 and IGF-2 are present in the fetal circulation from the end of the first trimester, and their levels increase thereafter in both mother and fetus. Birth weight correlates positively with IGF levels. However, both relative levels of the IGFs and control of the IGFs are very different in the fetal stage than they are postnatally. First, fetal IGF-2 levels are much higher than IGF-1 levels; IGF-1 and IGF-2 levels resemble those in adults soon after birth. Second, in the fetus, IGF-1 and IGF-2 levels correlate poorly with GH levels. Indeed, it appears that GH may have only a minimal effect on fetal growth. For example, anencephalic fetuses (see Chapter 10 for the box on abnormalities of neural tube closure), which have low GH levels, generally grow normally. Moreover, unlike the adult liver, the fetal liver has relatively few GH receptors.
Epidermal Growth Factor The fetus has abundant epidermal growth factor (EGF) receptors (see Chapter 3), and EGF is well known for its mitogenic properties, especially with regard to development of ectodermal and mesodermal structures. However, the fetus has no detectable mRNA encoding EGF. Thus, transforming growth factor α (TGF-α), another potent mitogen, which binds to EGF receptors on target cells, may act as a ligand for the EGF receptor.
Thyroid Hormones The thyroid hormones are obligatory for normal growth and development (see Chapter 49). Before the second trimester, most of the thyroxine (T4) in the fetus is maternal. Fetal production of thyrotropin (thyroid-stimulating hormone [TSH]) and the thyroid hormone T4 begin to increase in the second trimester, concurrent with development of the hypothalamic-pituitary portal system. Hypothyroidism has adverse effects on fetal growth, generally reflected as a reduction in the size of organs such as the heart, kidney, liver, muscle, and spleen.
Peptide Hormones Peptide hormones secreted by the placenta (see Table 56-4) can act through endocrine, paracrine, and autocrine mechanisms to stimulate growth and differentiation in several organ systems.
Early during gestation, production of red blood cells (erythropoiesis) occurs in many tissues not normally thought of as erythropoietic in the adult. Erythropoiesis begins during the third week of fetal development in the yolk sac and placenta. At approximately the fourth week of gestation, the endothelium of blood vessels and the mesenchyme also begin to contribute to the erythrocyte pool, shortly followed by the liver. The bone marrow, spleen, and other lymphoid tissues begin to produce red blood cells only near the end of the first trimester. All these organ systems except bone marrow gradually lose their ability to manufacture blood cells, and by the third trimester, the bone marrow becomes the dominant source of blood cells.
Growth Restriction
IUGR is an abnormality of fetal growth and development. IUGR has been variously defined as a birth weight lower than the 3rd, 5th, or 10th percentile for gestational age or a birth weight that is more than two standard deviations lower than the mean for gestational age.
The growth-restricted fetus is at substantial risk of morbidity and mortality. Specific risks include birth asphyxia, neonatal hypoglycemia, hypocalcemia, meconium aspiration, persistent pulmonary hypertension of the newborn, pulmonary hemorrhage, thrombocytopenia, polycythemia, delayed neurologic development, and hypothermia. Ultrasound methods offer objective, reliable means for identifying IUGR. Intrauterine measurements of biparietal diameter (distance between the two parietal eminences of the head) and abdominal circumference predict IUGR in as many as 90% of the cases.
The three recognized categories of IUGR are related to the time of onset of the pathologic process, as follows:
Type I or symmetrical IUGR refers to the infant with decreased growth potential. Type I IUGR accounts for 20% to 30% of growth-restricted fetuses. The entire fetus is small for gestational age. Length, weight, and abdominal and head circumferences are all less than the 10th percentile for gestational age. Type I IUGR results from growth inhibition during early fetal development (4 to 20 weeks’ gestation), a period referred to as the hyperplastic stage of fetal development. Thus, the pathologic result is fewer cells in the fetus. Causes include intrauterine infections (e.g., rubella, cytomegalovirus), chromosomal disorders, congenital malformations, maternal drug ingestion, and maternal smoking. Of fetuses with severe, early onset of growth retardation, ~25% have aneuploidy (i.e., abnormal number of chromosomes). Uniformly (or symmetrically) diminished growth of these fetuses may result from inhibition of mitosis during early development.
Type II or asymmetric IUGR refers to the infant with restricted growth, most frequently caused by uteroplacental insufficiency. This type accounts for 70% to 80% of growth-restricted fetuses. This type of growth restriction results from an insult that occurs later in gestation than type I IUGR, usually after 28 weeks’ gestation. Late in the second trimester, hypertrophy dominates. A rapid increase in cell size and increases in the formation of fat, muscle, bone, and other tissues occur. Fetuses with type II IUGR have a normal total number of cells, but these cells are smaller than normal. The distinguishing feature of the fetus with asymmetric IUGR is that the fetus has a normal length and head circumference (brain-sparing effect), but abdominal growth slows during the late second and early third trimesters. Redistribution of fetal CO occurs, with increased flow to the brain, heart, and adrenals and decreased glycogen storage and liver mass. This form of IUGR is most often associated with maternal disease such as kidney disease, chronic hypertension, and severe diabetes mellitus, among others.
Intermediate IUGR is a combination of types I and II IUGR and accounts for 5% to 10% of all growth-restricted fetuses. It probably occurs during the middle phase of fetal growth (20 to 28 weeks’ gestation), between the hyperplastic and hypertrophic phases. During this middle period, mitotic rate decreases and overall cell size increases progressively. Chronic hypertension, lupus nephritis, or other maternal vascular diseases that are severe and begin early in the second trimester may result in intermediate IUGR, with symmetric growth and no significant brain-sparing effect.
The erythrocytes formed early in gestation are nucleated, but as fetal development progresses, more and more of the circulatory erythrocytes are non-nucleated. The blood volume in the common circulation of the fetoplacental unit increases as the fetus grows. The fraction of total erythrocytes that are reticulocytes (immature, non-nucleated erythrocytes with residual polyribosomes) is high in the young fetus, but it decreases to only ~5% at term. In the adult, the reticulocyte count is normally less than 1%. The life span of fetal erythrocytes depends on the age of the fetus; in a term fetus, it is ~80 days, or two thirds that in an adult. The life span of erythrocytes of less mature fetuses is much shorter. (See Note: Reticulocytes)
The hemoglobin (Hb) content of the fetal blood rises to ~15 g/dL by midgestation, equivalent to the level in normal men. The Hb concentration of fetal blood at term is higher than the Hb concentration of maternal blood, which may be only ~12 g/dL. Embryonic Hb with different combinations of α-type and β-type chains (see Table 29-1) is present very early in gestation. A genetic program of development governs the eventual transition to fetal Hb (HbF), which predominates at birth. HbA and a small amount of HbA2 gradually replace HbF during the first 12 months of life, thus culminating in the adult pattern of Hb expression (see Table 29-2).
The fetus imbibes considerable quantities of amniotic fluid by 20 weeks’ gestation. However, not until the final 12 weeks of gestation is fetal gastrointestinal function similar to that of the normal infant at term. The fetal gastrointestinal tract continuously excretes small amounts of meconium into the amniotic fluid. Meconium consists of excretory products from the gastrointestinal mucosa and glands, along with unabsorbed residua from the imbibed amniotic fluid.
By the beginning of the second trimester, the fetus also begins to urinate. Fetal urine constitutes ~75% of amniotic fluid production (see Chapter 56). The fetal renal system does not acquire the capacity to regulate fluid, electrolyte, and acid-base balance until the beginning of the third trimester. Full development of the renal system does not occur until several months following delivery.
Fetal tissues constantly synthesize and break down proteins. Protein synthesis predominates throughout gestation, especially during the third trimester, when fetal protein synthesis—primarily in muscle and liver—increases 3- to 4-fold. The number of ribosomes per cell increases throughout gestation and early postnatal life. The efficiency of ribosomes at translating mRNA may also improve during gestation. Substrate availability (i.e., amino acids) and modulation of the synthetic apparatus by endocrine and other factors play important roles in regulating protein synthesis during gestation.
The formation of each peptide bond requires four molecules of ATP, so the energy cost of protein synthesis is 0.86 kcal/g. Protein synthesis comprises 15% to 20% of fetal metabolic expenditure in the third trimester. At equivalent phases of development, fetuses across several species invest similar fractions of total energy in protein synthesis. Because glucose is the major metabolic fuel, a shortfall of oxidized metabolic substrates (e.g., glucose and lactate) has a direct, negative impact on protein synthesis.
Increases in skeletal muscle mass account for 25% to 50% of fetal weight gain during the second half of gestation, when the number of muscle cells increases 8-fold and cell volume increases ~2.6-fold. Although skeletal muscle fibers are not differentiated in the first half of gestation, distinct type I and type II muscle fibers (see Chapter 9) appear in equal amounts between 20 and 26 weeks of gestation.
Fetal fat stores account for only 1% of fetal body weight during the first trimester. By the third trimester, as much as 15% of fetal body weight is fat. At birth, humans have more fat than other warm-blooded animals (e.g., the newborn cat has 2%; the guinea pig, 9.5%; the rat, 11%), with the exception of hibernating mammals and migratory birds.
Approximately half the increase in body fat reflects increased lipid transport across the placenta, and the other half reflects increased fatty acid (FA) synthesis in the fetal liver. Blood levels of fetal lipids (i.e., triglycerides, FAs, and ketone bodies) remain low before 32 weeks’ gestation. In the last 2 months, the fetus increases its lipid storage as triglycerides in white and brown adipose tissue as well as in liver. During this period, both subcutaneous fat (i.e., white fat) and deep fat (i.e., white and brown) increase exponentially. The stored fat ensures adequate fuel stores for postnatal survival, and it also provides thermal insulation to the newborn. In addition, brown fat is important for thermogenesis in the postnatal period.
Several factors are responsible for increased lipid stores in the near-term fetus. Increases in fetal albumin facilitate FA transfer across the placenta. Insulin acts on fetal hepatocytes to stimulate lipogenesis. Insulin also promotes the availability of substrates, including glucose and lactate, which, in turn, increase the synthesis of fat (see Chapter 51).
The fetal lung begins as an outpouching of the foregut at ~24 days’ gestation. Several days later, this lung bud branches into two tubular structures, the precursors of the main bronchi. At 4 to 6 weeks’ gestation, the bronchial tree begins to branch repetitively. The further maturation of the lungs occurs in four overlapping phases: (1) the pseudoglandular period, (2) the canalicular period, (3) the terminal sac period, and (4) the alveolar period.
During the pseudoglandular period (5 to 17 weeks), the lung “airways” resemble branching exocrine glands. The canalicular period is characterized by canalization of the airways (16 to 25 weeks) and is complete when ~17 generations of airways have formed, including the respiratory bronchioles. Each respiratory bronchiole gives rise to as many as six alveolar ducts, which give rise to the primitive alveoli during the second trimester. The branching of the pulmonary arterial tree parallels, both temporally and spatially, the branching of the bronchial tree. However, at ~24 weeks’ gestation, considerable interstitial tissue separates the capillaries from the respiratory epithelium. Thus, if the fetus were born at this stage of its development, the premature infant would have a very low diffusing capacity (see Chapter 30), owing to the great distance between the edge of the alveolar lumen and the edge of the capillary lumen.
During the terminal sac period (24 weeks’ gestation to birth), the respiratory epithelium thins greatly, and the capillaries push into the alveolar sacs. The potential for gas exchange improves after ~24 weeks’ gestation, when capillaries proliferate and come into closer proximity to the thin type I alveolar pneumocytes (see Chapter 26). During this period, surfactant synthesis and storage begin (although not extensively) in the differentiated type II cells.
In the alveolar period of lung development (late fetal life to 8 years of age), final alveolar growth occurs. Alveolar-like structures are present at ~32 weeks’ gestation, and at 34 to 36 weeks’ gestation, 10% to 15% of the adult number of alveoli will be present. Alveolar number continues to increase until as late as 8 years of age.
Hormones play a major role in controlling fetal lung growth and development in preparation for ex utero function. A key target is surfactant (see Chapter 27), which increases lung compliance. Numerous hormones stimulate surfactant biosynthesis, including glucocorticoids, thyroid hormones, thyrotropin-releasing hormone, and prolactin, as well as growth factors such as EGF. Glucocorticoids in particular play an essential role in stimulating fetal lung maturation by increasing the number of both type II alveolar pneumocytes and lamellar bodies (see Chapter 27) within these cells. Glucocorticoid receptors are probably present in lung tissue at midterm. Fetal cortisol levels rise steadily during the third trimester and surge just before birth. Two thirds of this cortisol is of fetal origin; the rest crosses the placenta from the mother.
The predominant phospholipid in surfactant is dipalmitoylphosphatidylcholine (DPPC). Glycogen serves as a primary energy and carbon source for the FAs involved in phospholipid synthesis (Fig. 57-1). The FAs used in the synthesis of surfactant enter the type II cells directly from the bloodstream. The condensation of diacylglycerol with cytosine diphosphate choline ultimately leads to the production of DPPC. At ~32 weeks’ gestation, increases in cortisol and in the other hormones mentioned previously stimulate several regulatory enzymes, including FA synthase and phosphocholine transferase. Thus, the net effect is vastly increased production of pulmonary surfactant late in gestation. Coincident with increased surfactant synthesis are large increases in lung distensibility and stability on inflation.
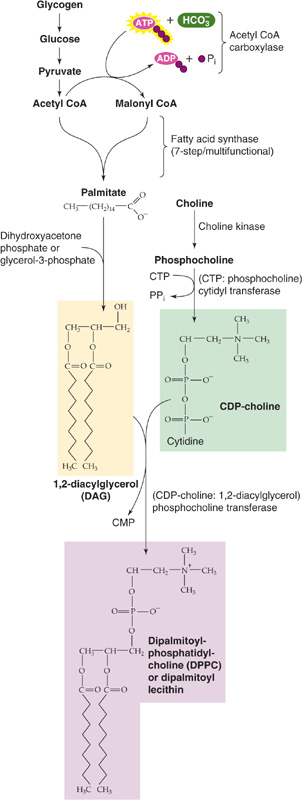
Figure 57-1 Synthesis of DPPC. Before birth, cortisol upregulates several enzymes that are important for the synthesis of surfactant, including FA synthase and phosphocholine transferase. CoA, coenzyme A.
Respiratory Distress Syndrome
Respiratory distress syndrome (RDS) affects 10% to 15% of infants born prematurely. In very immature infants, delivered before 30 weeks of gestation, cyanosis, tachypnea, nasal flaring, intercostal and subcostal retractions, the use of accessory musculature, and grunting may be immediately apparent in the delivery room. In more mature preterm infants, these symptoms may evolve over several hours. A chest radiograph reveals atelectasis with air bronchograms (i.e., air-filled bronchi standing out against the white background of collapsed lung tissue). Infants with severe RDS may develop edema and respiratory failure that requires mechanical ventilation. Uncomplicated cases usually resolve spontaneously. Because RDS occurs in premature infants, the course is often confounded by the coexistence of a patent ductus arteriosus. This combination of problems raises the risk for short- and long-term complications, such as alveolar rupture with pneumothorax and pulmonary interstitial emphysema, necrotizing enterocolitis, intraventricular hemorrhage, and bronchopulmonary dysplasia. (See Note: Respiratory Distress Syndrome of the Newborn)
RDS is caused by a deficiency of pulmonary surfactant. Although prematurity is by far the single most important risk factor for developing RDS, others include male sex, cesarean section, perinatal asphyxia, second twin pregnancy, and maternal diabetes. Surfactant insufficiency can result from abnormalities of surfactant synthesis, secretion, or reutilization. Decreased lung compliance and atelectasis result from both structural immaturity and surfactant deficiency, thus promoting airway collapse. The consequent right-to-left shunting of blood past poorly ventilated alveoli results in hypoxemia, which—at the level of the alveoli—causes capillary damage and leakage of plasma proteins into the alveolar space. These proteins may inactivate surfactant, thus exacerbating the underlying condition.
The discovery that a deficiency of surfactant is the underlying problem in infants with RDS led investigators to look for ways of assessing fetal lung maturity and adequacy of surfactant production before delivery, so elective induction or cesarean section could be timed successfully in infants who need to be delivered prematurely. Clinical tests for assessing lung maturity exploit the knowledge that the major surfactant lipids are phosphatidyl cholines (i.e., lecithins) and that phosphatidylglycerol (PG) is also overrepresented (see Chapter 27). A ratio of lecithin to sphingomyelin (L/S ratio) greater than 2.0 in the amniotic fluid is consistent with mature lungs, as is a positive PG assay. The 2000 National Institutes of Health Antenatal Steroid Consensus Conferences recommended antenatal steroid therapy for pregnant women with fetuses between 24 and 34 weeks’ gestational ages who are at risk of preterm delivery within 7 days. This treatment accelerates lung maturation and surfactant production.
In the newborn who develops signs of RDS soon after birth, surfactant is instilled into the trachea, preferably within the first hour after delivery. The administration of antenatal steroids and postnatal surfactant has markedly reduced the mortality from RDS and has improved the clinical course described earlier.
Fetal breathing movements have been confirmed in humans by both Doppler ultrasound and tocodynamometer (an external device that records uterine movements) studies, commencing near the end of the first trimester. It appears that hypoxia and tactile stimulation of the fetus promote these breathing movements, which occupy less than half of any 24-hour period. Near term, breathing movements are regular, similar to those found after birth. However, just before labor, fetal breathing decreases.
The fetal lung undergoes many changes in preparation for birth. In utero, the alveoli and airways of the fetal lung are filled with a volume of fluid approximating the functional residual capacity (see Chapter 27) of the neonatal lung. The onset of labor is accompanied by increases in catecholamines and arginine vasopressin, which decrease fluid production by the fetal lung and initiate its active reabsorption. The pulmonary circulation absorbs the majority of the fluid, and the pulmonary lymphatics absorb some as well. A small portion of the lung fluid is forced out of the trachea as the fetus passes through the birth canal.
The circulatory system differentiates from the mesoderm of the embryo. The fetal heart begins to beat in the fourth week of gestation. The major difference between the circulatory system of the fetus and that of the adult is the presence of the placenta. The placenta performs for the fetus functions that—at least in part—are performed by four organ systems in extrauterine life: (1) the lungs (gas exchange), (2) the gastrointestinal tract (nutrition), (3) the liver (nutrition, waste removal), and (4) the kidneys (fluid and electrolyte balance, waste removal). Thus, the fetal heart pumps large quantities of blood through the placenta and smaller amounts of blood through the other four organ systems. The key principle governing the unique pattern of blood flow in the fetus is the presence of four shunts, that is, pathways that allow blood to bypass the future postnatal route.
These shunts are illustrated in Figure 57-2. Because, to a large extent, the right and left sides of the fetal heart pump in parallel rather than in series, and because the inputs and outputs of these two sides mix, we define the combined cardiac output (CCO) as the sum of the outputs of the right and left ventricles. Figure 57-2A shows the fraction of the CCO that flows through the fetal circulatory system at important checkpoints. Figure 57-2B shows values for PO2 and the percentage of saturation of HbF at these same checkpoints. The numeric values in Figure 57-2 are reasonable values for a healthy fetus.
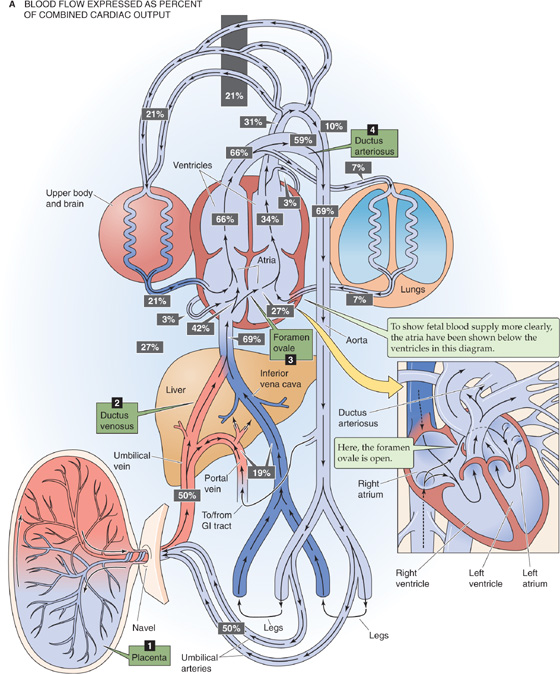
Figure 57-2 Fetal circulation. A, This schematic drawing shows the major elements of the fetal circulation. Note that—in the main drawing—the heart is upside down, for the sake of presenting the blood flow as simply as possible. The heart is right side up in the inset. Because the inputs and outputs of the right and left hearts mix, we define the CCO as the sum of the outputs of the right and left ventricles. The percentage of the CCO that passes various checkpoints is represented as a number in a black box. The CCOs of the right ventricle (66%) and the left ventricle (34%) add up to 100%. The fetal circulation has four major shunts: the placenta, the ductus venosus, the foramen ovale, and the ductus arteriosus.
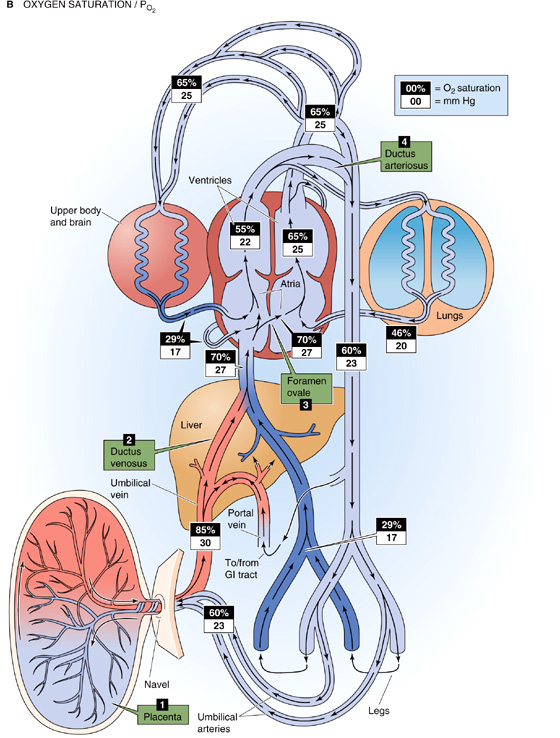
Figure 57-2 cont’d B, The schematic drawing is the same as in A, except—at each checkpoint—we show the O2 saturation of HbF against a black background and the PO2 (in mm Hg) against a white background. The relationship between the O2 saturation and PO2 figures is based on the O2 saturation curve for HbF (similar to the curve labeled Hb + CO2 in Fig. 29-7).
Fetal Asphyxia
Any insult that interferes with the ability of the placenta to exchange O2 and CO2 between the maternal and fetal circulations may lead to fetal asphyxia. Common causes include maternal hypotension, abruptio placentae (i.e., breaking away of a portion of the placenta from the uterine wall), and a prolapsed umbilical cord (i.e., the umbilical cord falls into the birth canal in front of the head or other part of the fetus). The results are low fetal PO2, high PCO2, and acidosis. These effects can decrease myocardial function, lead to lowering of CO and further compromise of O2 delivery to the tissues, and thereby create a vicious cycle.
In the brain, asphyxia produces substantial alterations in cerebral intracellular metabolism. As the brain is forced to shift from aerobic to anaerobic metabolism, high-energy phosphate compounds (e.g., ATP) decrease in concentration, and their breakdown products (e.g., ADP and inorganic phosphate) increase. Lactic acid accumulates. Changes in PO2, PCO2, pH, and metabolism can profoundly affect neurotransmitter release and re-uptake and hence concentrations of various ions in neurons, glia, and brain extracellular fluid (see Chapter 11). Asphyxia also may lead to accumulation of prostaglandins and leukotrienes, vasoactive compounds that can dilate microvessels in critical areas of the brain, thus permitting the generation of free radicals that, in turn, lead to cell damage.
Fetuses experiencing chronic O2 deficiency in utero are at increased risk of delayed breathing immediately after birth, in part because their energy reserves are already low. Therefore, fetal hypoxia can lead to neonatal hypoxia. The metabolic derangements of asphyxia remain evident for as long as 24 hours after birth.
Placenta The first of these shunts is the placenta itself. Of the CCO late in gestation, ~69% reaches the thoracic aorta (Fig. 57-2A). Half of the CCO enters the placenta as deoxygenated blood through the paired umbilical arteries, which arise from the two common iliac arteries. This massive blood flow to the placenta not only shunts blood away from the lower trunk, but also lowers effective blood flow to all abdominal viscera, including the kidneys. The umbilical arteries branch repeatedly under the amnion and ultimately form dense capillary networks within the terminal villi (see Chapter 56). The single umbilical vein returns oxygenated blood (which has a PO2 of 30 to 35 mm Hg) back to the fetus from the placenta. This blood enters the ductus venosus, which then merges with the inferior vena cava.
Ductus Venosus This second shunt bypasses the liver, which is largely nonfunctional. The ductus venosus allows blood from the umbilical vein, ~50% of CCO, to enter the inferior vena cava directly, without ever entering the liver. In addition, some blood from the portal circulation may enter the ductus venosus. Blood from the ductus venosus then combines with blood from the inferior vena cava, ~19% of CCO, which drains the lower body and liver. Thus, ~69% of the CCO (PO2 ≅ 27 mm Hg) enters the right atrium.
Foramen Ovale The third major shunt is blood entering the right atrium and then crossing the foramen ovale to enter the left atrium. The foramen ovale is an oval hole in the septum dividing the atria, located in the posterior aspect of the right atrium. Of the 69% of the CCO that enters the right atrium through the inferior vena cava, ~27% shunts through the foramen ovale directly into the left atrium. This movement represents a right-to-left shunt. Therefore, the left side of the heart receives relatively well oxygenated blood (PO2 ≅ 27 mm Hg) from the inferior vena cava. In addition, the left atrium receives 7% of the CCO as poorly oxygenated blood (PO2 ≅ 20 mm Hg) from the nonfunctional lungs. Thus, the left ventricle pumps a total of 27% + 7% = 34% of the CCO (PO2 ≅ 25 mm Hg). Because this blood enters the aorta upstream from the ductus arteriosus, it primarily flows to the head and forelimbs.
Returning now to the right atrium, 69% − 27% = 42% of the CCO entering the right atrium from the inferior vena cava does not shunt through the foramen ovale. This relatively well-oxygenated blood (PO2 ≅ 27 mm Hg) joins the relatively poorly oxygenated 21% of the CCO (PO2 ≅ 17 mm Hg) that enters the right atrium from the superior vena cava and another 3% from the coronary vessels—a total of 24% of CCO. Because of the valve-like nature of the septum surrounding the foramen ovale, none of the incoming blood from the superior vena cava or coronary vessels shunts through the foramen ovale. Rather, it goes through the tricuspid valve to the right ventricle. Thus, the right ventricle receives 42% + 21% + 3% = 66% of the CCO (PO2 = 18 to 22 mm Hg). The PO2 in the fetal right ventricle is somewhat lower than that in the left ventricle. The blood from the right ventricle then enters the trunk of the pulmonary artery.
Ductus Arteriosus The fourth major shunt, also a right-to-left shunt, directs blood from the pulmonary artery to the aorta through the ductus arteriosus. The ductus arteriosus contains substantial smooth muscle in its vessel wall. The patency of this vessel is due to active relaxation of this smooth muscle, mediated by prostaglandins, particularly prostaglandin E2 (PGE2). Fetal PGE2 levels are as much as 5-fold higher than adult levels are. Administering prostaglandin inhibitors to an experimental fetal animal causes the ductus arteriosus to vasoconstrict.
Although 66% of the CCO enters the pulmonary artery, only 7% of the CCO perfuses the unventilated fetal lungs, reflecting the high resistance of the pulmonary vasculature in the fetus. This high resistance is the result of hypoxic vasoconstriction and acidosis (see Chapter 31), the collapsed state of the airways, and perhaps leukotrienes (particularly leukotriene D4 [LTD4]). The rest of the blood entering the pulmonary artery, 66% − 7% = 59% of the CCO (PO2 ≅ 22 mm Hg), enters the descending aorta through the ductus arteriosus and mixes with the blood from the aortic arch, 10% of CCO, that did not perfuse the head and upper body (PO2 ≅ 25 mm Hg). Thus, the descending aorta receives 59% + 10% = 69% of the CCO (PO2 ≅ 23 mm Hg). The placenta receives blood with a PO2 of 23 mm Hg, and returns blood to the fetus with a PO2 of 30 to 35 mm Hg.
As the newborn exits the birth canal, it takes its first breath, which not only expands the lungs, but also triggers a series of changes in the circulatory system. At the same time, the newborn loses its nutritional connection to the mother and apprehends a cold new world. Three major changes in metabolism accompany birth: hypoxia, hypoglycemia, and hypothermia. We discuss the adaptations of the respiratory and cardiovascular systems in this major section and adjustments of other organ systems in the next.
Although separation of the placenta does not occur until several minutes after birth, vasoconstriction in the umbilical arteries terminates the ability of the placenta to deliver oxygenated blood to the newborn immediately upon birth. Thus, even though the newborn may remain attached to its placenta during the first few moments of life, it is essential that the baby begins to breathe immediately. Umbilical vasoconstriction has two origins. First, stretching the umbilical arteries during delivery stimulates them to constrict. Second, the sudden rise in the systemic arterial PO2 in the newborn also stimulates and maintains vasoconstriction in the umbilical arteries. Birth may also be associated with an “autotransfusion” as blood in the placental circulation preferentially moves into the body of the emerging baby. Because the umbilical veins do not constrict, as do the umbilical arteries, blood flows from placenta to newborn if the newborn is below the level of the placenta, and if the umbilical cord is not clamped. This autotransfusion may constitute 75 to 100 mL, which is a substantial fraction of the newborn’s total blood volume of ~300 mL.
At birth, the newborn must transform its circulatory system from one that supports gas exchange in the placenta to one that supports O2 and CO2 exchange in the lungs. In addition, other circulatory adjustments must occur as the gastrointestinal tract, liver, and kidneys assume their normal roles. As the lungs become functional at birth, the pulmonary and systemic circulations shift from interconnected and parallel systems to separate entities that function in series.
The first breath is the defining event for the newborn. Not only does it inflate the lungs, but also—as discussed later—it triggers circulatory changes that convert the fetal pattern of blood flow to the adult pattern. The functional capabilities of the lungs depend on their surface area available for gas exchange, the ability of surfactant to maximize lung compliance, neural mechanisms that control breathing, and the aforementioned circulatory changes.
The first breath is normally also the most difficult inspiration of a lifetime. A considerable negative pressure within the intrapleural space is necessary to overcome the effects of surface tension. The infant’s first inspiratory effort requires a transpulmonary pressure (PTP)—the pressure difference between the intrapleural space and alveolar air spaces—of 60 cm H2O to increase the lung volume by ~40 mL. In contrast, a typical adult only needs to change PTP by ~2.5 cm H2O during a typical tidal volume of 500 mL (see Chapter 27). The newborn’s first ventilatory effort creates an air-water interface for the first time, opening the alveoli. Breathing becomes far easier once the alveoli are open and the type II alveolar pneumocytes deliver surfactant to the air-water interface. Thus, the second inspiration may require a PTP of only 40 cm H2O. The newborn may not achieve the adult level of relative lung compliance until 1 hour after birth. Very immature neonates, who lack adequate surfactant (see the box titled Respiratory Distress Syndrome), may have difficulty expanding the lungs.
The rapid onset of breathing immediately after delivery appears to be induced by a temporary state of hypoxia and hypercapnia. In most normal deliveries, these changes in PO2 and PCO2 result from the partial occlusion of the umbilical cord. Tactile stimulation and decreased skin temperature also promote the onset of breathing. When newborns do not begin to breathe immediately, hypercapnia and hypoxia increase and provide further simulation for the infant to breathe.
The peripheral and central chemoreceptors are responsible for sensing the blood gas parameters (i.e., low PO2, high PCO2, and low pH) characteristic of the asphyxia that accompanies birth. In addition, increased sympathetic tone may stimulate breathing at the time of birth by constricting vessels to the peripheral chemoreceptors, thereby lowering the local PO2 in the microenvironment of the glomus cells and mimicking even more severe hypoxia (see Chapter 32). Finally, independent of the initial stimuli that trigger breathing, other central nervous system mechanisms may help to sustain breathing in the newborn.
The neonate’s ability to control the blood gas parameters depends on the sensitivity of the lung’s mechanical (i.e., stretch) reflexes, the sensitivity of the central and peripheral chemoreceptors, the gestational and postnatal age, the ability of the respiratory muscles to resist fatigue, and the effects of the sleep state.
Sleeping newborn infants, especially premature newborns, tend to have increased respiratory variability from breath to breath. For example, they exhibit periodic breathing, which consists of breaths with intermittent respiratory pauses (generally of a few to several seconds’ duration) and varying tidal volumes. Periodic breathing and increased respiratory variability, including periodic breathing, occur more frequently in rapid eye movement sleep than during quiet sleep, a state characterized by regular breathing. In human adults and in adult experimental animals, periodic breathing may reflect an exaggerated ventilatory response to CO2—which causes arterial PCO2 to fall, thus lowering respiratory drive. However, the mechanisms underlying periodic breathing in the newborn may not be the same. (See Note: Apnea in the Newborn)
As noted earlier, the fetal circulation has four unique shunts absent in the adult: the placental circulation, the ductus venosus, the foramen ovale, and the ductus arteriosus. At or around birth, these shunts disappear. In addition, the pulmonary circulation, which received only ~7% of the CCO in the fetus, now accepts the entire cardiac output (CO). In this and the next three sections, closure of each of these four shunts is discussed.
Closure of the Placental Circulation In the fetus, the placental circulation receives ~50% of the CCO (Fig. 57-2A). Thus, the placental circulation represents a major parallel path in the systemic circulation and accounts for the low vascular resistance of the fetal systemic circulation. As the placental circulation disappears at birth, the total peripheral resistance doubles. Because blood flow through the descending aorta is essentially unchanged, aortic pressure must increase, thereby causing upstream pressure in the left ventricle to increase as well.
Opening of the Pulmonary Circulation As noted earlier, during fetal life, pulmonary vascular resistance is high as the result of hypoxic vasoconstriction, acidosis, the collapsed state of the airways, and perhaps agents such as LTD4 (see earlier). As a result, only ~7% of the CCO of the term fetus flows through the lungs, a figure corresponding to ~11% of the right ventricular output. At birth, expansion of the lungs by itself markedly decreases pulmonary vascular resistance, perhaps by triggering the release of prostaglandin I2 (PGI2 or prostacyclin). In addition, the increase in PO2 and pH that occurs with breathing leads to pulmonary vasodilation. Together, these changes reduce pulmonary vascular resistance more than 5-fold (Fig. 57-3). Because the blood flow through the pulmonary vasculature increases by a slightly smaller factor, pressure in the pulmonary artery decreases. As a result, upstream pressure in the right ventricle also falls.
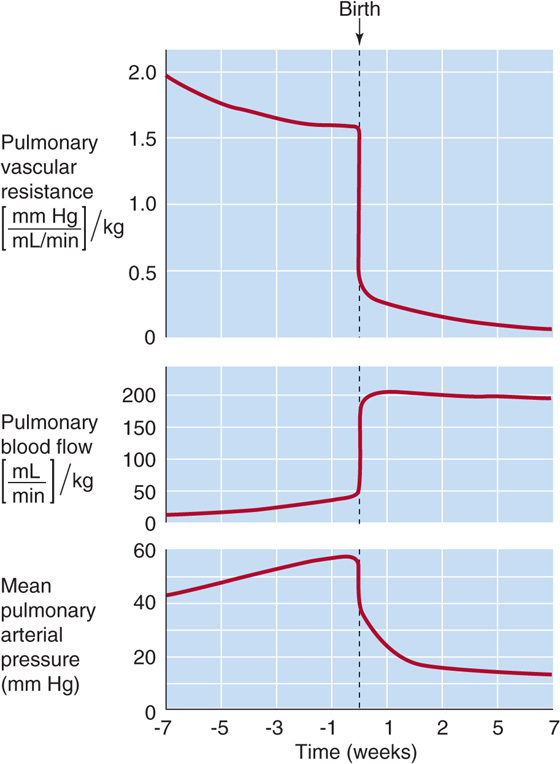
Figure 57-3 Effect of birth on pulmonary vascular resistance, blood flow, and mean arterial pressure. In the fetus, pulmonary vascular resistance is high, pulmonary blood flow is low, and mean pulmonary arterial pressure is high. At birth, each of these three situations rapidly reverses. The primary event is the fall in resistance, which occurs because of the following: (1) the pulmonary blood vessels are no longer being crushed; (2) breathing causes increased PO2, which, in turn, causes vasodilation; and (3) local prostaglandins cause vasodilation. The reason that pressure falls after birth is that the fall in pulmonary vascular resistance is greater than the rise in blood flow. (Data from Rudolf AM: Congenital Diseases of the Heart: Clinical-Physiological Considerations. Armonk, NY: Futura, 2001.)
During fetal life, a large fraction of the blood in the portal vein bypasses the liver by entering the ductus venosus and merging with blood from the umbilical vein (Fig. 57-2A). Although blood flow through the umbilical vein ceases soon after birth, the majority of the portal blood continues to flow through the ductus venosus. Thus, immediately after birth, portal flow through the liver remains low. Within ~3 hours after term birth, however, constriction of the vascular smooth muscle within the ductus venosus completely occludes this shunt pathway. As a result, pressure in the portal vein increases markedly, thereby diverting blood into the liver. The mechanisms underlying the contraction of the muscular walls of the ductus venosus remain unknown.
In the fetus, blood from the inferior vena cava moves preferentially from the right atrium across the foramen ovale into the left atrium (Fig. 57-2A). After entering the left ventricle, this well-oxygenated blood moves into the ascending aorta and primarily perfuses the head, neck, and coronary arteries. At birth, the decrease in the pulmonary vascular resistance increases blood flow through the lungs; the results are increased venous return to the left atrium and elevated left atrial pressure. At the same time, the venous return to the right atrium falls from 69% + 21% + 3% = 93% of the CCO of both ventricles in the fetus to 100% of the CO of a single ventricle at birth. Thus, right atrial pressure falls. The net effect is a reversal of the pressure gradient across the atrial septum, pushing the foramen ovale’s “valve”—situated on the “left” side of the septum—against the opening of the foramen ovale (Fig. 57-4A). Closing this valve usually prevents what would otherwise be movement of blood from the left to the right atrium of the newborn. The left side of the heart now receives blood only from the lungs.
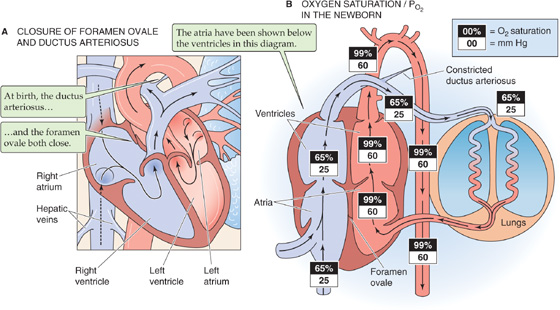
Figure 57-4 Changes in the circulation at and around birth. A, Closure of these two shunts establishes separate right and left circulatory systems. As the pressure in the left atrium rises higher than the pressure in the right atrium—owing to the large decrease in pulmonary vascular resistance—the flap of the foramen ovale pushes against the septum, thus preventing blood flow from the left to the right atrium. Eventually, this flap seals shut. As aortic pressure exceeds the pressure of the pulmonary artery, blood flow through the ductus arteriosus reverses. Well-oxygenated aortic blood now flows through the ductus arteriosus. This high PO2 causes vasoconstriction, which functionally closes the ductus arteriosus within a few hours. Falling prostaglandin levels also contribute to the rapid closure. Eventually, the lumen of the ductus becomes anatomically obliterated. B, The elimination of the fetal shunts and the oxygenation of blood in the lungs lead to major increases in the O2 saturation and PO2 in the circulation.
Gradually, a permanent seal forms between the valve and the wall of the septum, a process that can take as little as a few months or as long as a few years. In some newborns, the valve does not completely seal to the septum, thus leaving a remnant potential pathway between the two atria. However, the 15% to 20% of adults with this condition do not have left-to-right shunting, because the valve of the foramen ovale is effective even if it is not completely sealed. However, if right atrial pressure should increase to more than left atrial pressure for some pathologic reason (e.g., pulmonary hypertension), then a right-to-left shunt between the atria would occur.
During fetal life, blood flows from the pulmonary artery, through the ductus arteriosus, and into the aorta (Fig. 57-2A). Prostaglandins maintain the patency of the ductus arteriosus during fetal life. Immediately after birth, the ductus arteriosus remains open. However, it now conducts blood in the direction opposite from that of the fetus: from the aorta to the pulmonary artery. This reversal of blood flow is the result of the increased systemic resistance (which elevates aortic pressure) and decreased pulmonary vascular resistance (which lowers pulmonary arterial pressure).
Obviously, the open ductus arteriosus during the first few hours of postnatal life constitutes an undesirable left-to-right shunt. Fortunately, within a few hours after term birth, the ductus arteriosus closes functionally because its muscular wall constricts (Fig. 57-4A). Usually, all blood flow through the ductus arteriosus ceases within 1 week after birth. Within a month or so, the lumen becomes obliterated anatomically because of thrombosis (i.e., blood clot within the lumen), proliferation of the vessel’s intimal layer, and growth of fibrous tissue. Occasionally, the ductus arteriosus fails to close. The incidence of patent ductus arteriosus is one in several thousand.
The relatively rapid functional closure of the ductus arteriosus is primarily the result of the increased PO2 of the blood perfusing this vessel immediately after birth. As the PO2 of blood flowing through the ductus arteriosus rises from 18 to 22 mm Hg in utero to ~60 mm Hg a few hours after birth, the smooth muscle in the wall of the ductus arteriosus contracts. In newborns who are hypoxemic, the low PO2 has three effects: (1) pulmonary vascular resistance and pressure remain high, (2) the ductus arteriosus remains patent, and (3) the patent ductus arteriosus maintains a right-to-left shunt. In these infants, raising the inspired PO2 closes the ductus arteriosus. If these infants are allowed to breathe room air again too quickly, the ductus arteriosus will reopen.
Other factors, in addition to a high PO2, contribute to the rapid functional closure of the ductus arteriosus. Shortly after birth, circulating levels of prostaglandins fall quickly, thus relieving the ductus arteriosus of the vasodilating influence of these substances. Preterm infants tend to maintain high circulating prostaglandin levels, a feature that may account for their tendency to patent ductus arteriosus. Treating such infants with indomethacin (a nonsteroidal anti-inflammatory drug that inhibits cyclooxygenase and thereby reduces prostaglandin synthesis) (see Chapter 3 for the box on inhibition of cyclooxygenase isoforms by aspirin) induces closure of the ductus arteriosus, even at a low PO2. Norepinephrine, acetylcholine, and bradykinin also produce constrictor responses.
The closure of the ductus arteriosus completes the separation of the right and left circulations initiated with closure of the foramen ovale. Whereas the ventricles functioned in parallel in the fetus, now they function in series in the neonate. As a result, the O2 saturation of the newborn’s Hb is similar to the adult’s (Fig. 57-4B). However, because the O2 saturation curve of HbF is shifted relatively leftward (see Chapter 29), the newborn achieves these O2 saturations at lower PO2 values. In the neonate, the sum of the ventricular outputs of the two ventricles (i.e., twice the CO) is larger than the CCO in the fetus, a result primarily of a marked rise in the output of the left ventricle, which doubles its stroke volume. Compared with the adult, the newborn has a markedly lower systemic vascular resistance and thus can achieve a relatively high blood flow with a relatively low perfusion pressure.
In humans, the neonatal period is defined as the first 4 weeks of life. The newborn’s ability to survive during this period depends on the adequate development and maturation of various fetal organ systems, as well as on adaptations of these organ systems to extrauterine life. As the newborn loses the nutritional link with the placenta, the infant must now rely on his or her own gastrointestinal tract. Moreover, other functions normally carried out by the placenta are now entrusted to the liver and kidneys. Finally, on exiting its uterine “incubator,” the newborn must stabilize his or her body temperature.
The body loses heat to the environment by radiation, conduction, convection, and evaporation (see Chapter 59). The relative importance of these processes depends on the circumstances. For instance, at birth, the infant moves from a warm and liquid environment to cool and dry surroundings. Hence, evaporation is the main source of heat loss immediately after delivery. However, even after the newborn’s skin is dry, the infant is at risk for losing body heat by each of the previously discussed mechanisms. The major reasons are as follows: (1) the large skin surface area of the newborn relative to the small body mass (i.e., large surface-to-volume ratio), (2) the limited ability of the newborn to generate heat through muscle contraction (e.g., shivering thermogenesis), (3) the newborn’s poor thermal insulation from the environment by adipose tissue, and (4) the inability of the newborn to adjust his or her own protection (e.g., put on warmer clothes) from the thermal stress of the environment or to modify that environment (e.g., turn up the thermostat). Premature and growth-retarded infants are at an even higher risk for heat loss and hypothermia.
Fortunately, the newborn has one important asset for fighting hypothermia: nonshivering thermogenesis, a process that occurs primarily in liver, brain, and brown fat (Fig. 57-5). Cold stress triggers an increase in the levels of epinephrine and TSH. TSH stimulates the release of the thyroid hormones, predominantly T4 (see Chapter 49). Working in parallel, epinephrine activates, particularly in brown fat, the 5′/3′-monodeiodinase responsible for the peripheral conversion of circulating T4 to the far more active triiodothyronine (T3). T3 acts locally in brown fat to uncouple mitochondrial oxidation from phosphorylation and thereby to increase heat production.
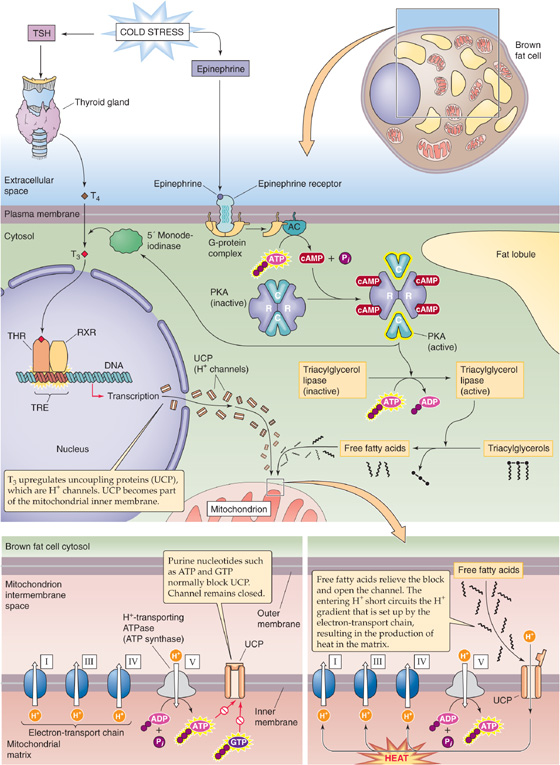
Figure 57-5 Nonshivering thermogenesis in brown fat. AC, adenylyl cyclase; RXR, retinoid X receptor; THR, thyroid hormone receptor; TRE, thyroid response element.
Brown fat differs from white fat in having a high density of mitochondria; the cytochromes in these mitochondria give the brown fat cells their color. Newborns have particularly high levels of brown fat in the neck and midline of the upper back. In brown fat, the locally generated T3 upregulates a protein called uncoupling protein UCP1, originally called thermogenin. This protein is an H+ channel located in the inner mitochondrial membrane. Normally, intracellular purine nucleotides (e.g., ATP, GDP) inhibit UCP1. However, epinephrine, acting through a cAMP pathway, activates the lipase that liberates FAs from triglycerides. These FAs relieve the inhibition of the H+ channel and increase its conduction of protons. Consequently, the protons generated by electron transport enter the mitochondrion through UCP1, which dissipates the H+ gradient needed by the H+-translocating ATP synthase (see Chapter 5). Thus, the mitochondria in brown fat can produce heat without producing useful energy in the form of ATP. The oxidation of FAs in brown fat generates ~27 kcal/kg of body weight each day and contributes a large fraction of the neonate’s high metabolic rate.
Carbohydrate Metabolism Elimination of the placental circulation at birth means that the newborn now has to forage for his or her own food. However, the newborn may not start suckling for ~6 hours. During late fetal life, glucocorticoids promote rapid accumulation of glycogen through their action on glycogen synthase. In its first few hours, the neonate uses glycogenolysis to mobilize hepatic glycogen stores, thereby releasing glucose into the bloodstream. The two enzymes needed for breaking down hepatic glycogen, phosphorylase, and glucose-6-phosphatase (G6Pase; see Chapter 58) are present in the fetus but do not become active until soon after birth.
The newborn depletes his or her hepatic glycogen stores in the first 12 hours of life. Stores of glycogen in cardiac muscle are 10 times those in the adult, and those in the skeletal muscle are 3 to 5 times those in the adult, but the fetus mainly uses the glycogen stored in these tissues to provide glucose for local use. The net effect is that, during the first day of life, blood glucose levels may decline to 40 to 50 mg/dL, although they soon rise to near adult values once nutrition becomes adequate.
Infants born of diabetic mothers run a very high risk of having pathologic hypoglycemia (i.e., <35 mg/dL). The high maternal blood glucose levels translate to high fetal blood glucose levels and cause hyperplasia of β cells in the fetal pancreatic islets and hyperinsulinism in the fetus. Insulin levels remain high after birth, thereby producing extreme symptomatic hypoglycemia. Blood glucose levels tend to reach their low point within a few hours after birth and begin to recover spontaneously within 6 hours.
Even in the physiologically normal newborn, low levels of blood glucose in the immediate postnatal period lead to a decrease in blood levels of insulin and a reciprocal increase of glucagon (see Chapter 51). This hormonal milieu promotes the net release of glucose by the liver. In the liver, glucagon acts through cAMP to stimulate glycogenolysis, inhibit glycogen synthesis, stimulate gluconeogenesis, and initiate synthesis of some gluconeogenic enzymes. All these actions promote formation of glucose for release into the bloodstream (see Fig. 51-12). In contrast to the adult, in whom phosphorylation is the main mechanism for regulating the enzymes involved in glycogen metabolism, in the fetus, the relative concentration of the enzymes may be more important.
Epinephrine also promotes glucose mobilization (see Chapter 3) in the immediate postnatal period, and it uses the same cAMP pathway as glucagon. Hypoxia, hypoglycemia, and hypothermia all stimulate the release of epinephrine from the adrenal medulla.
Fat Metabolism During the final 2 months of gestation, the fetus stores ~500 g of fat (i.e., ~15% of body weight), an important source of energy for the neonate. Decreased blood glucose just after birth raises levels of glucagon and epinephrine, which stimulate hormone-sensitive lipase in adipose tissue through cAMP (see Chapter 58). This lipase breaks down triglycerides into glycerol and FAs, which enter the bloodstream. The liver can take up glycerol and can ultimately synthesize glucose (i.e., gluconeogenesis). The liver can also take up FAs and can generate ketone bodies, which are important as the newborn deals with glycogen depletion. (See Note: Gluconeogenesis with Glycerol as the Starting Material)
Metabolic Rate The neonate’s metabolic rate—expressed per kilogram of body weight—is much higher than that of the adult. Through the first year of life, the growing infant has a daily resting metabolic rate of ~55 kcal/kg, which is nearly double the value of ~30 kcal/kg for a healthy young adult. At birth, the growing infant has a daily caloric requirement of 100 to 120 kcal/kg, a requirement that falls to 90 to 100 kcal/kg by the end of the first year. The difference between the caloric requirement and resting metabolic rate represents physical activity and growth.
Provided the mother’s diet is adequate during pregnancy, the newborn is in complete nutritional balance at birth. The newborn’s natural nutrition for the first few days of life is colostrum (a milk-like substance secreted by the mammary glands), and thereafter it is breast milk (see Table 56-8 for compositions), both of which the newborn’s gastrointestinal tract is prepared to digest. The American Academy of Pediatrics recommends breast-feeding until an infant is 1 year old. If the infant is breast-fed, and if the mother’s nutritional status is good, colostrum and breast milk will meet all the newborn’s nutritional needs. Moreover, colostrum and breast milk make important contributions to the newborn’s immune status. Both contain high concentrations of immunoglobulin A (or secretory) antibodies directed against bacteria and viruses, and they also contain macrophages. Breast milk contains factors that promote the growth of lactobacilli, which colonize the colon and may protect the infant from some virulent strains of Escherichia coli.
If the mother’s dietary intake of iron was adequate during pregnancy, the infant’s hepatic stores of iron will be adequate for hematopoiesis for 6 to 9 months following delivery. Administering medicinal iron or providing iron-fortified milk can prevent iron-deficiency anemia.
The calcium in breast milk can meet the infant’s needs for the rapid calcification of bones and teeth. However, fluoride for minimizing tooth decay must be provided as a supplement. Although the supply of calcium per se is unlikely to be a problem, vitamin D is necessary for the proper absorption of calcium by the intestines. Vitamin D supplementation may be necessary if the newborn or the mother is not exposed to sufficient sunlight to generate vitamin D in the skin (see Chapter 52). Supplementation may also be required in formula-fed infants and in infants born prematurely. Rickets (also see Chapter 52 for the box on rickets and osteomalacia) can develop rapidly in a vitamin D–deficient infant.
Vitamin C (ascorbic acid) is necessary for the synthesis of hydroxyproline in collagen and chondroitin sulfate (see Table 45-3) in cartilage, bone, and other connective tissues. The neonate is normally born with sufficient stores of vitamin C and receives adequate amounts of this vitamin in breast milk. If the breast milk or formula is vitamin C deficient, the infant may develop scurvy, which can be prevented with vitamin C supplements for the mother or infant.
The newborn normally loses 6% to 10% of his or her body weight during the first week of life, a finding reflecting a decrease in interstitial and intravascular volume. After ~1 week, the rate of fluid intake begins to exceed that rate of loss, and term infants return close to birth weight by 1½ weeks.
The newborn’s total body water is ~75% of body weight compared with 60% for men and 50% for women (see Table 5-1). Once the newborn is in a steady state, daily turnover of body water is 100 to 120 mL/kg, a rate that is 3-to 4-fold higher than in adults. Thus, even small changes in the balance between fluid intake and loss can lead to rapid and profound disturbances in body fluid compartments. The sick newborn can have a very difficult time maintaining fluid status and requires very careful management.
The immaturity of the neonate’s kidneys further complicates matters. For example, the glomerular filtration rate (GFR; see Chapter 35) is extremely low at birth and increases rapidly during the first 2 weeks of life. However, even when normalized for body weight, the GFR does not reach adult levels until ~1 year of age. Moreover, the concentrating ability of the newborn kidney (i.e., a maximal urine osmolality of ~450 mOsm) is substantially less than that of an adult (~1200 mOsm).
Because of its relatively high metabolic rate, the neonate’s CO and ventilation are also correspondingly higher than those of the adult. Metabolism generates fixed acids (see Chapter 39), and the newborn’s relative acid load is also greater. The immature state of neonatal kidneys with respect to acid excretion puts the neonate at risk for developing metabolic acidosis (see Chapter 28).
Maternal antibodies play a key role in protecting the infant from infection both in utero and during the first several months after birth.
Fetus The placenta actively transports the small IgG immunoglobulins from mother to fetus, so that fetal IgG levels are even higher than those in the mother. These maternal IgG antibodies ward off infection by viruses and some bacteria. However, maternal IgA (which is primarily present in secretions), IgE, and IgM antibodies generally do not cross the placenta in appreciable amounts, and the baby is generally born with very low levels of these other immunoglobulins.
The fetus begins to develop its own immune capabilities at very early stages of development. However, because the fetus is isolated from antigens, the fetal immune system normally does not make large amounts of antibodies. Nevertheless, the fetus can respond to intrauterine infections by generating IgM antibodies. In addition, the fetus also begins to produce other proteins that help to protect against bacterial and viral infections. Among these are the following: (1) the components of the complement pathway; (2) lysozyme, an enzyme found in secretions such as tears and mucus, which digests the cell walls of bacteria and fungi; and (3) interferon γ, which is produced by T lymphocytes and which activates B lymphocytes, macrophages, and endothelial cells.
Neonate In addition to high prenatal levels of maternal IgG, the newborn receives copious amounts of secretory IgA antibodies in colostrum and breast milk. However, the blood levels of maternal IgG antibodies progressively fall, and IgG levels in the infant’s blood reach a nadir at ~3 months of age. After that time, the infant’s own production of IgG antibodies causes total IgG levels to increase gradually. However, even at 1 year of age, IgG levels—as well as the levels of IgA, IgM, and IgE—are still only half of adult levels.
Antibodies obtained in utero from the mother protect against most childhood diseases, including diphtheria, measles, and poliomyelitis. The persistence of antibodies—at levels high enough for protection—varies considerably from one disease to another. For example, maternal measles antibodies are so persistent that vaccinations against measles often fail if they are attempted before 15 months of age. In contrast, maternal antibodies against whooping cough (pertussis) are generally inadequate to protect the infant beyond 1 to 2 months. The infant normally receives a first DTP immunization (diphtheria, tetanus, and pertussis), as well as a first poliomyelitis immunization, at 2 months of age.
According to the World Health Organization, a premature infant is defined as one born sooner than 37 weeks after the mother’s last menstrual period, compared with the normal 40 weeks (see Chapter 50). For the premature infant whose birth weight is appropriate for gestational age, the only problem is that the uterus was unable to retain the fetus for the appropriate term for any of a variety of reasons (e.g., premature rupture of the membranes, incompetent cervix). The concept of IUGR (see the box titled Growth Restriction) describes a newborn who not only has a low birth weight, but also is small for gestational age. Virtually all the challenges to the health of the neonate are made more severe in prematurity or IUGR, conditions that are therefore associated with reduced chances of survival. These problems generally reflect immaturity either of certain organ systems (e.g., lungs, intestines, liver, kidneys) or of homeostatic mechanisms (e.g., thermoregulation). (See Note: Problems of Prematurity)
Books and Reviews
Ballard PL: Hormonal regulation of pulmonary surfactant. Endocr Rev 1989; 10:165-181.
Benirschke K: Normal embryologic development. In Moore TR, Reiter RC, Rebar RW, Baker VV (eds): Gynecology and Obstetrics: A Longitudinal Approach. New York: Churchill Livingstone; 1993: 37-53.
Furon J-C, Skoll A: Fetal cardiovascular physiology and response to stress conditions. In: Reece A, Hobbins J, eds: Medicine of the Fetus and Mother. Philadelphia: WB Saunders; 1998: 115-130.
Polin RA, Fox WW, Abman SH: Fetal and Neonatal Physiology, 3rd ed. Philadelphia: WB Saunders; 2004.
Strang LB: Fetal lung liquid: Secretion and reabsorption. Physiol Rev 1991; 71:991-1016.
Journal Articles
Avery ME, Mead J: Surface properties in relation to atelectasis and hyaline membrane disease. Am J Dis Child 1959; 97:517.
Gluck K, Kulovich M: Lecithin/sphingomyelin ratios in amniotic fluid in normal and abnormal pregnancy. Am J Obstet Gynecol 1973; 115-539.
Goldenberg RL, Cutter GR, Hoffman HJ, et al: Intrauterine growth retardation: Standards for diagnosis. Am J Obstet Gynecol 1989; 161:271-277.
Gross I: Regulation of fetal lung maturation. Am J Physiol 1990; 259:L337-L344.
Kari MA, Hallman M, Eronen M, et al: Prenatal dexamethasone treatment in conjunction with human surfactant therapy: A randomized placebo-controlled multicenter study. Pediatrics 1994; 94:730-736.
Wilkening RB, Meschia G: Fetal oxygen uptake, oxygenation, and acid-base balance as a function of uterine blood flow. Am J Physiol 1983; 244:H749-H755.