CHAPTER 36
Gerhard Giebisch and Erich Windhager
The kidney plays a central role in controlling the plasma levels of a wide range of solutes that are present at low concentrations in the body. The renal excretion of a solute depends on three processes—filtration, reabsorption, and secretion (see Chapter 33). The kidney filters and then totally reabsorbs some of the substances we discuss in this chapter (e.g., glucose). Others, it filters and also secretes (e.g., the organic anion p-aminohippurate [PAH]). Still others, the kidney filters, reabsorbs, and secretes (e.g., urea).
The liver generates urea from NH+4, the primary nitrogenous end product of amino acid catabolism (see Chapter 46). The primary route for urea excretion is the urine, although some urea exits the body through the stool and sweat. The normal plasma concentration of urea is 2.5 to 6 mM. Clinical laboratories report plasma urea levels as blood urea nitrogen (BUN) in the units “mg of elemental nitrogen per dL plasma”; normal values are 7 to 18 mg/dL. For a 70-kg person ingesting a typical Western diet, and producing 1.5 to 2 L/day of urine, the urinary excretion of urea is ~450 mmol/day.
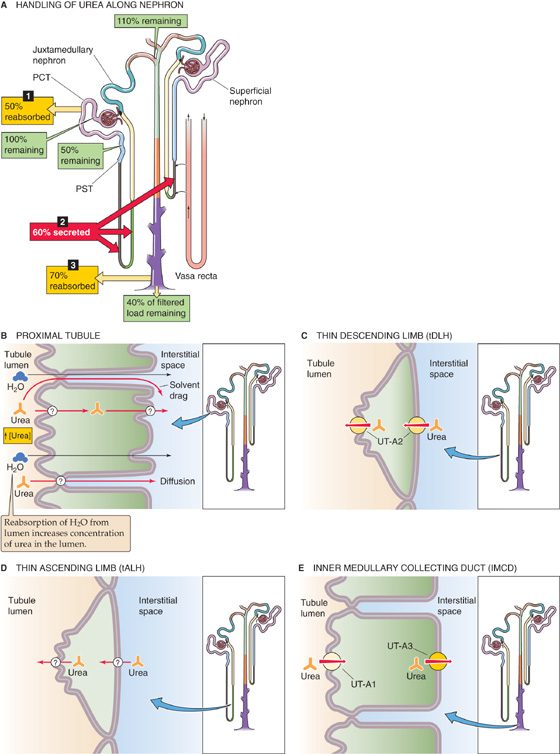
The kidney freely filters urea at the glomerulus, and then it both reabsorbs and secretes it. Because the tubules reabsorb more urea than they secrete, the amount of urea excreted in the urine is less than the quantity filtered. In the example shown in Figure 36-1A (i.e., average urine flow), the kidneys excrete ~40% of the filtered urea. The primary sites for urea reabsorption are the proximal tubule and the medullary collecting duct, whereas the primary sites for secretion are the thin limbs of the loop of Henle.
Figure 36-1 A to E, Urea handling by the kidney. In A, we assume a normal urine flow and thus a urea excretion of 40% of the filtered load. The numbered yellow boxes indicate the fraction of the filtered load that various nephron segments reabsorb. The tALH and the tip of the tDLH in juxtamedullary nephrons secrete urea. In superficial nephrons, the entire tDLH may secrete urea. The red box indicates the fraction of the filtered load jointly secreted by both nephron types. The green boxes indicate the fraction of the filtered load that remains in the lumen. The values in the boxes are approximations. UT-A1, UT-A2, and UT-A3 are urea transporters.
In the very early proximal tubule (Fig. 36-1B), [urea] in the lumen is the same as in blood plasma. However, paracellular fluid reabsorption along the proximal tubule sweeps some urea along with it through solvent drag (see Chapter 20). In addition, water reabsorption tends to increase [urea] in the lumen, thereby generating a favorable transepithelial gradient that drives urea reabsorption by diffusion through the transcellular or paracellular pathway. The greater the fluid reabsorption along the proximal tubule, the greater is the reabsorption of urea through both solvent drag and diffusion.
In juxtamedullary nephrons, as the tubule fluid in the thin descending limb (tDLH) approaches the tip of the loop of Henle, [urea] is higher in the medullary interstitium than in the lumen (see Chapter 38). Thus, the deepest portion of the tDLH secretes urea through facilitated diffusion (Fig. 36-1C) mediated by the urea transporter UT-A2, which is encoded by the SLC14A2 gene. As the fluid turns the corner to flow up the thin ascending limb (tALH), the tubule cells continue to secrete urea into the lumen, probably also by facilitated diffusion (Fig. 36-1D). (See Note: Urea Transporters)
The tDLH of superficial nephrons is located in the inner stripe of the outer medulla. Here, the interstitial [urea] is higher than the luminal [urea] because the vasa recta carry urea from the inner medulla. Because the tDLH cells of these superficial nephrons appear to have UT-A2 along their entire length, these cells secrete urea. Thus, both superficial and probably also juxtamedullary nephrons contribute to urea secretion, raising urea delivery to ~110% of the filtered load at the level of the cortical collecting ducts.
Finally, the inner medullary collecting duct (IMCD) reabsorbs urea by a transcellular route that is unusual in that both the apical and basolateral steps occur by facilitated diffusion (Fig. 36-1E). The UT-A1 urea transporter moves urea across the apical membrane of the IMCD cell, whereas UT-A3 probably mediates urea movement across the basolateral membrane. Arginine vasopressin (AVP)—also known as antidiuretic hormone (ADH)—stimulates UT-A1 but not UT-A3. In Chapter 38, we discuss the role of urea transport in the urinary concentrating mechanism.
UT-A2, the prototypical member of the UT family, is a glycosylated 55-kDa integral membrane protein with 10 putative membrane-spanning segments. UT-A2 is unusual among membrane proteins in that it is extremely hydrophobic; except for a large extracellular loop, almost the entire central portion of the protein may be embedded within the membrane. UT-A1, a splice variant of the same gene, is a 97-kDa protein with 20 membrane-spanning segments. UT-A1 is basically UT-A3 linked to UT-A2 by an intracellular loop. This loop has several putative phosphorylation sites for protein kinase A (PKA), a feature that explains why AVP—which acts through cAMP (see Chapter 3)—stimulates only UT-A1. As discussed in Chapter 38, UT-B1 and UT-B2, which are encoded by a different gene (SLC14A1), are present in the descending limb of the vasa recta. (See Note: Urea Transporters)
Because urea transport depends primarily on urea concentration differences across the tubule epithelium, changes in urine flow unavoidably affect renal urea handling (Fig. 36-2). At low urine flow, when the tubule reabsorbs considerable water and therefore much urea, the kidneys excrete only ~15% of filtered urea (see Fig. 38-6). However, the kidneys may excrete as much as 70% of filtered urea at high urine flow, when the tubules reabsorb relatively less water and urea. During the progression of renal disease, the decline of the glomerular filtration rate (GFR) leads to a low urine flow and urea retention, and thus an increase in BUN.
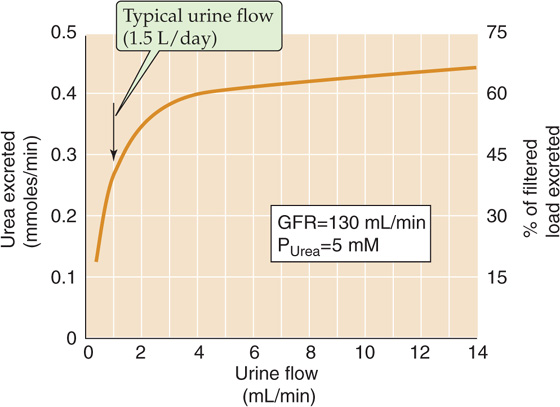
Figure 36-2 Urea excretion versus urine flow. (Data from Austin JH, Stillman E, Van Slyke DD: J Biol Chem 1921; 46:91-112.)
In clinical conditions such as volume depletion, in which the urine flow declines sharply (see Chapter 40), urea excretion decreases out of proportion to the fall in GFR. The resulting high BUN can thus serve as laboratory confirmation of dehydration. The flow dependence of urea clearance contrasts with the behavior of creatinine clearance, which, like inulin clearance (see Chapter 34), is largely independent of urine flow. Consequently, in patients with reduced effective circulating volume (see Chapter 23), and hence low urine flow, the plasma [BUN]/[creatinine] ratio increases from its normal value of ~10 (both concentrations in mg/dL).
The fasting plasma glucose concentration (see Chapter 51) is normally 4 to 5 mM (70 to 100 mg/dL) and is regulated by insulin and other hormones. The kidneys freely filter glucose at the glomerulus and then reabsorb it, so that only trace amounts normally appear in the urine (Fig. 36-3A). The proximal tubule reabsorbs nearly all the filtered load of glucose, mostly along the first third of this segment. More distal segments reabsorb almost all the remainder. In the proximal tubule, luminal [glucose] is initially equal to plasma [glucose]. As the early proximal tubule reabsorbs glucose, luminal [glucose] drops sharply, falling to levels far lower than those in the interstitium. Accordingly, glucose reabsorption occurs against a concentration gradient and must therefore be active.
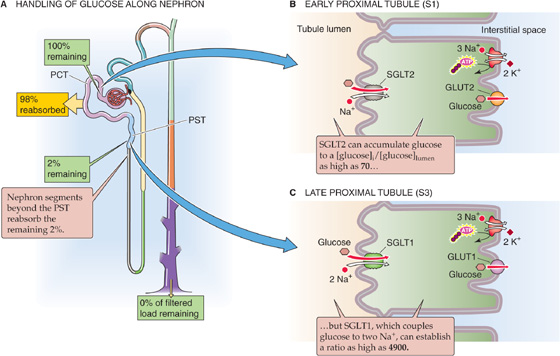
Figure 36-3 A to C, Glucose handling by the kidney. The yellow box indicates the fraction of the filtered load that the proximal tubule reabsorbs. The green boxes indicate the fraction of the filtered load that remains in the lumen at various sites. The values in the boxes are approximations. PCT, proximal convoluted tubule; PST, proximal straight tubule.
Glucose reabsorption is transcellular; glucose moves from the lumen to the proximal tubule cell through Na/glucose cotransport and from cytoplasm to blood by facilitated diffusion (Fig. 36-3B, C). At the apical membrane, a member of the Na/glucose cotransporter (SGLT) family (see Chapter 5) couples the movements of the electrically neutral D-glucose (but not L-glucose) and Na+. Phloridzin, extracted from the root bark of certain fruit trees (e.g., cherry, apple), inhibits the SGLTs. The basolateral Na-K pump maintains the intracellular Na+ concentration ([Na+]i) lower than that of the tubule fluid. Moreover, the electrically negative cell interior establishes a steep electrical gradient that favors the flux of Na+ from lumen to cell. Thus, the electrochemical gradient of Na+ drives the uphill transport of glucose into the cell (i.e., secondary active transport), thereby concentrating glucose in the cytoplasm (see Chapter 5). (See Note: Using Membrane Vesicles to Study Glucose Transport)
In the early part of the proximal tubule (S1 segment), a high-capacity/low-affinity transporter called SGLT2 mediates apical glucose uptake (Fig. 36-3B). This cotransporter has Na+/glucose stoichiometry of 1 : 1. In the later part of the proximal tubule (S3 segment), a high-affinity/low-capacity cotransporter called SGLT1 is responsible for apical glucose uptake (Fig. 36-3C). Because this cotransporter has Na+/glucose stoichiometry of 2:1 (i.e., far more electrochemical energy per glucose), it can generate a far larger glucose gradient across the apical membrane (see Chapter 5). Paracellular glucose permeability progressively diminishes along the proximal tubule, further contributing to the tubule’s ability to maintain high transepithelial glucose gradients and to generate near-zero glucose concentrations in the fluid emerging from the proximal tubule.
Once inside the cell, glucose exits across the basolateral membrane by a member of the GLUT family of glucose transporters (see Chapter 5). These transporters are Na+ independent and move glucose by facilitated diffusion. Thus, the GLUTs are quite distinct from the SGLTs. Like the apical SGLTs, the basolateral GLUTs differ between early and late proximal tubule segments, with GLUT2 in the early proximal tubule (Fig. 36-3B) and GLUT1 in the late proximal tubule (Fig. 36-3C). In contrast to the apical SGLTs, the basolateral GLUTs have a much lower sensitivity to phloridzin.
The relationship between plasma [glucose] and the rate of glucose reabsorption is the glucose titration curve. Figure 36-4A shows how rates of glucose filtration (orange curve), excretion (green), and reabsorption (red) vary when one increases plasma [glucose] by infusing intravenous glucose. As plasma [glucose] rises—at a constant GFR—from control levels to ~200 mg/dL, glucose excretion remains zero. It is only above a threshold of ~200 mg/dL (~11 mM) that glucose appears in the urine. Glucose excretion rises linearly as plasma [glucose] increases further. Because the threshold is considerably higher than the normal plasma [glucose] of ~100 mg/dL (~5.5 mM), and because the body effectively regulates plasma [glucose] (see Chapter 51), healthy people do not excrete any glucose in the urine, even after a meal. Likewise, patients with diabetes mellitus, who have chronically elevated plasma glucose concentrations, do not experience glucosuria until the blood sugar level exceeds this threshold value.
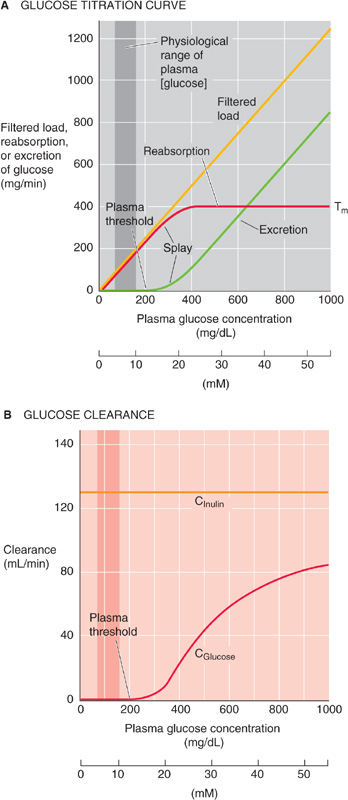
Figure 36-4 The effect of increasing plasma glucose concentrations on glucose excretion and clearance. In A, Tm is the transport maximum for reabsorption. In A and B, the darker vertical bands represent the physiological range of plasma [glucose], which spans from a fasting [glucose] to the peak value after a meal.
The glucose titration curve shows a second property—saturation. The rate of glucose reabsorption reaches a plateau (Tm) at ~400 mg/min. The reason for the Tm value is that the low-capacity SGLT (i.e., SGLT1) in the late proximal tubule—and perhaps even the high-capacity cotransporter (i.e., SGLT2) in the early proximal tubule—now become fully saturated. Therefore, these transporters cannot respond to further increases in filtered glucose.
Figure 36-4A also shows that the rate of glucose reabsorption reaches the Tm gradually, not abruptly. This splay in the titration curve probably reflects both anatomical and kinetic differences among nephrons. Therefore, a particular nephron’s filtered load of glucose may be mismatched to its capacity to reabsorb glucose. For example, a nephron with a larger glomerulus has a larger load of glucose to reabsorb. Different nephrons may have different distributions and densities of SGLT2 and SGLT1 along the proximal tubule. Accordingly, saturation in different nephrons may occur at different plasma levels. (See Note: Clinical Correlates of Splay in the Glucose Titration Curve)
At low filtered glucose loads, when no glucose appears in the urine (Fig. 36-4A), the clearance (see Chapter 33) of glucose is zero. As the filtered load increases beyond the threshold, and glucose excretion increases linearly with plasma [glucose], the clearance of glucose progressively increases. At extremely high glucose loads, when the amount of glucose reabsorbed becomes small compared with the filtered load, glucose behaves more like inulin (i.e., it remains in the tubule lumen). Thus, if we replot the glucose excretion data in Figure 36-4A as clearance (i.e., excretion divided by plasma [glucose]), we see that, as the filtered glucose load rises, glucose clearance (Fig. 36-4B, red curve) approaches inulin clearance (orange). (See Note: Glucose Clearance)
The four key characteristics of glucose transport—(1) threshold, (2) saturation (Tm), (3) splay, and (4) clearance approaching GFR at infinite plasma concentrations—apply to several other solutes as well, including amino acids, Krebs cycle intermediates (e.g., acetate, citrate, and α-ketoglutarate [α-KG]), PAH, and phosphate.
The total concentration of amino acids in the blood is ~2.4 mM. These L-amino acids are largely those absorbed by the gastrointestinal tract (see Chapter 45), although they also may be the products of protein catabolism or of the de novo synthesis of nonessential amino acids.
The glomeruli freely filter amino acids (Fig. 36-5A). Because amino acids are important nutrients, it is advantageous to retrieve them from the filtrate. The proximal tubule reabsorbs more than 98% of these amino acids by a transcellular route, using a wide variety of amino acid transporters, some of which have overlapping substrate specificity (Table 36-1). At the apical membrane, amino acids enter the cell by Na+-driven or H+-driven transporters as well as amino acid exchangers (Fig. 36-5B). At the basolateral membrane, amino acids exit the cell by amino acid exchangers—some of which are Na+ dependent—and also by facilitated diffusion (see Chapter 5). Particularly in the late proximal tubule and “postproximal” nephron segments, where the availability of luminal amino acids is low, SLC38A3 mediates the Na+-dependent uptake of amino acids across the basolateral membrane. This process is important for cellular nutrition or for metabolism. For example, in proximal tubule cells, SLC38A3 takes up glutamine—the precursor for NH+4 synthesis and gluconeogenesis (see Chapter 58 and Fig. 39-5A).
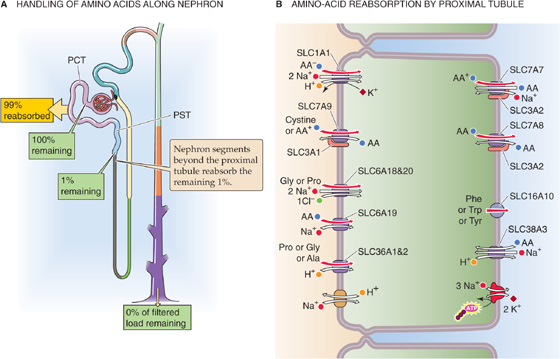
Figure 36-5 A and B, Amino acid handling by the kidney. In A, the yellow box indicates the fraction of the filtered load that the proximal tubule reabsorbs. The green boxes indicate the fraction of the filtered load that remains in the lumen at various sites. The values in the boxes are approximations. PCT, proximal convoluted tubule; PST, proximal straight tubule.
Table 36-1 Amino Acid Transporters*
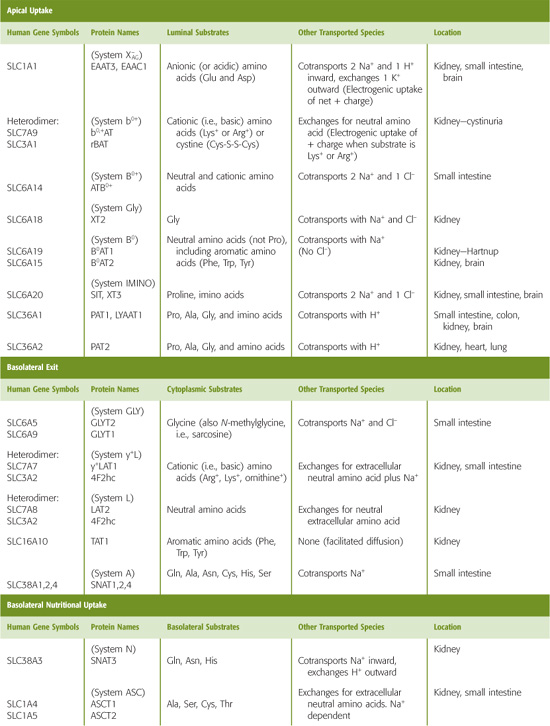
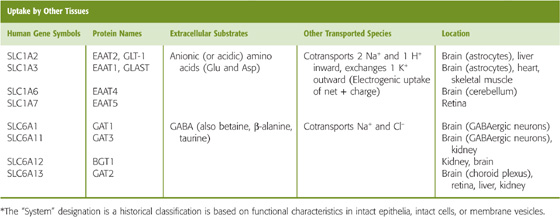
For an amino acid to cross the proximal tubule epithelium, it must move through both an apical and a basolateral transporter. For example, glutamate enters the cell across the apical membrane through SLC1A1. This transporter simultaneously takes up Na+ and H+ in exchange for K+ (Fig. 36-5B). Inside the cell, glutamate can be metabolized to α-KG in the synthesis of NH+4 and gluconeogenesis, or it can exit across the basolateral membrane, perhaps by SLC1A4 or SLC1A5. The positively charged lysine and arginine cross the apical membrane in exchange for neutral amino acids, mediated by the heterodimeric transporter SLC7A9/SLC3A1. This process is driven by the cell-negative voltage and by relatively high intracellular concentrations of the neutral amino acids. Lysine and arginine exit across the basolateral membrane through the electroneutral, heterodimeric transporter SLC7A7/SLC3A2, which simultaneously takes up Na+ and a neutral amino acid. Neutral amino acids other than proline can cross the apical membrane through SLC6A19, driven by Na+, and exit across the basolateral membrane through the heterodimeric SLC7A8/SLC3A2, which exchanges neutral amino acids. Neutral aromatic amino acids such as tyrosine, can exit by facilitated diffusion, mediated by SLC16A10. Proline enters across the apical membrane together with H+ through SLC36A1 and exits across the basolateral membrane through the neutral amino acid exchanger SLC7A8/SLC3A2.
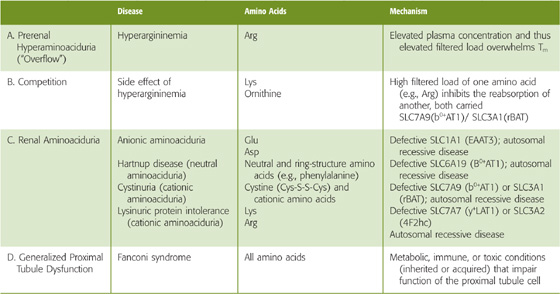
Because the same carrier can reabsorb structurally similar amino acids, competitive inhibition may occur in the presence of two related amino acids. This effect may explain why the tubules do not fully reabsorb some amino acids (e.g., glycine, histidine, and some nonproteogenic amino acids, such as L-methylhistidine and taurine), even though the transporter itself is normal. Competition can also occur in patients with hyperargininemia (Table 36-2; see also the box titled Hyperaminoacidurias).
Table 36-2 Patterns of Hyperaminoacidurias
Apparent competition between transported solutes occurs when they compete for the same energy source. Because the apical uptake of many organic and some inorganic solutes (e.g., phosphate, sulfate) depends on the electrochemical Na+ gradient, increasing the activity of one such transporter can slow others. For example, glucose uptake by electrogenic Na+/glucose cotransport may compromise the reabsorption of some amino acids for two reasons: (1) raising [Na+] diminishes the chemical Na+ gradient for other Na+-driven transporters; and (2) carrying net positive charge into the cell depolarizes the apical membrane, thus decreasing the electrical gradient.
With a few exceptions, the kinetics of amino acid reabsorption resembles that of glucose: The titration curves show saturation and transport maxima (Tm). In contrast to the case of glucose, in which the Tm is relatively high, the Tm values for amino acids are generally low. As a consequence, when plasma levels of amino acids increase, the kidneys excrete the amino acids in the urine, thus limiting the maximal plasma levels.
Oligopeptide The proximal tubules reabsorb ~99% of filtered oligopeptides (Fig. 36-6). Segments beyond the proximal tubule contribute little to peptide transport.
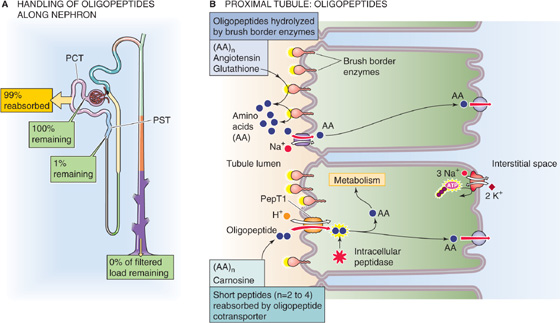
Figure 36-6 A and B, Oligopeptide handling by the kidney. In A, the yellow box indicates the fraction of the filtered load that the proximal tubule reabsorbs. The green boxes indicate the fraction of the filtered load that remains in the lumen at various sites. The values in the boxes are approximations. PCT, proximal convoluted tubule; PST, proximal straight tubule.
Several peptidases are present at the outer surface of the brush border membrane of proximal tubule cells (Fig. 36-6B), just as they are in the small intestine (see Chapter 45). These brush border enzymes (e.g., γ-glutamyltransferase, aminopeptidases, endopeptidases, and dipeptidases) hydrolyze many peptides, including angiotensin II (see Chapter 40), thereby releasing into the tubule lumen the free constituent amino acids and oligopeptides. Tubule cells reabsorb the resulting free amino acids as described in the previous section. The cell also absorbs the resulting oligopeptides (2 to 5 residues)—as well as other peptides (e.g., carnosine) that are resistant to brush border enzymes—using the apical H/oligopeptide cotransporters PepT1 and PepT2 (see Chapter 5). PepT1 is a low-affinity/high-capacity system in the early proximal tubule, whereas PepT2 is a high-affinity/low-capacity transporter in the late proximal segments, analogous to the properties of SGLT2 and SGLT1.
In general, an increase in the renal excretion of an amino acid (hyperaminoaciduria) may occur when the plasma concentration increases owing to any of several metabolic derangements, or when the carrier-mediated reabsorption of the amino acid decreases abnormally.
Prerenal (i.e., the Defect Is Before the Kidney) Hyperaminoacidurias
Hyperargininemia (Table 36-2A) is an inherited condition in which a metabolic defect leads to an increase in plasma arginine (Arg) levels that, in turn, increases the filtered load of Arg. Although the reabsorption of Arg increases, the filtered load exceeds the Tm, and the renal excretion increases.
Competition
Because the same transporter (the heterodimeric SLC7A9/SLC3A1 in Table 45-3) that carries Arg across the apical membrane also transports lysine (Lys) and ornithine, competition from Arg decreases the reabsorption of the other two (Table 36-2B). As a result, the urinary excretion of Lys and ornithine also increases. Because the metabolic production of Lys and ornithine does not change, plasma concentrations of these two amino acids, in contrast to that of Arg, usually fall.
Renal Aminoacidurias
This category of diseases results from an autosomal recessive defect in an amino acid transporter (Table 36-2C), and thus also affects absorption in the gastrointestinal tract (see Chapter 45 for the box on Defects in Apical Amino Acid Transport: Hartnup Disease and Cystinuria). In Hartnup disease, the defective apical transporter (SLC6A19) normally handles neutral amino acids (e.g., Ala, Ser), including those with rings (i.e., Phe, Trp, Tyr). In cystinuria, the affected apical transporter is the heterodimeric SLC7A9/SLC3A1 that carries cystine (Cys-S-S-Cys) and cationic amino acids (i.e., Arg, Lys, ornithine). An increased filtered load of one of these amino acids leads to increased excretion of all of them. Nephrolithiasis (i.e., kidney stones) may be a consequence of the increased excretion of the poorly soluble cystine.
Probably the most severe renal hyperaminoaciduria is lysinuric protein intolerance (LPI), resulting from the reduced reabsorption of Lys and Arg. The resulting low blood [Arg] impairs the urea cycle and detoxification of ammonium (hyperammonemia). Other features include alveolar proteinosis (the leading cause of death), hepatosplenomegaly, and—in severe cases—mental deterioration. The defective proximal tubule basolateral transporter (the heterodimeric SLC7A7/SLC3A2) normally mediates the efflux of Arg and Lys into blood in exchange for the uptake of Na+ and neutral amino acids.
Generalized Proximal Tubule Dysfunction
The Fanconi syndrome (Table 36-2D), which can be inherited or acquired, is characterized by a generalized loss of proximal tubule function. The kidney fails to reabsorb appropriately not only amino acids, but also Na+, Cl−, HCO−3, glucose, and water.
Once inside the cell, the oligopeptides undergo hydrolysis by cytosolic peptidases; this pathway is involved in the degradation of neurotensin and bradykinin. The distinction between which oligopeptides the cells fully digest in the lumen and which they take up through a PepT is not clear-cut. Oligopeptides that are more resistant to hydrolysis by peptidases are probably more likely to enter through a PepT.
Proteins Although the glomerular filtration barrier (see Chapter 33) generally prevents the filtration of large amounts of protein, this restriction is incomplete (see Chapter 34). For example, the albumin concentration in the filtrate is very low (4 to 20 mg/L), only 0.01% to 0.05% of the plasma albumin concentration. Nevertheless, given a GFR of 180 L/day, the filtered albumin amounts to 0.7 to 3.6 g/day. In contrast, albumin excretion in the urine normally is only ~30 mg/day. Thus, the tubules reabsorb some 96% to 99% of filtered albumin (Fig. 36-7A). In addition to albumin, the tubules extensively reabsorb other proteins (e.g., lysozyme, light chains of immunoglobulins, and β2-microglobulin), SH-containing peptides (e.g., insulin), and other polypeptide hormones (e.g., parathyroid hormone [PTH], atrial natriuretic peptide [ANP], and glucagon). It is not surprising that tubule injury can give rise to proteinuria even in the absence of glomerular injury.
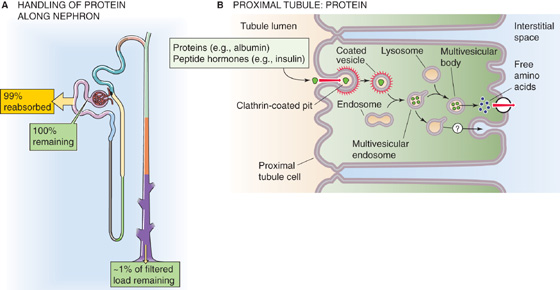
Figure 36-7 A and B, Protein handling by the kidney. In A, the yellow box indicates the fraction of the filtered load that the proximal tubule reabsorbs. The green boxes indicate the fraction of the filtered load that remains in the lumen at various sites. The values in the boxes are approximations.
Proximal tubule cells use receptor-mediated endocytosis (see Chapter 5) to reabsorb proteins and polypeptides (Fig. 36-7B). The first step is binding to receptors at the apical membrane (e.g., megalin, cubilin), followed by internalization into clathrin-coated endocytic vesicles. Factors that interfere with vesicle formation or internalization, such as metabolic inhibitors and cytochalasin B, inhibit this selective absorption. The vesicles fuse with endosomes; this fusion recycles the vesicle membrane to the apical surface and targets their content for delivery to lysosomes. At the lysosomes, acid-dependent proteases largely digest the contents over a period that is on the order of minutes for peptide hormones and many hours or even days for other proteins. The cells ultimately release the low-molecular-weight end products of digestion, largely amino acids, across the basolateral membrane into the peritubular circulation. Although the proximal tubule hardly reabsorbs any protein in an intact state, a small subset of proteins avoids the lysosomes and moves by transcytosis for release at the basolateral membrane.
In addition to the apical absorption and degradation pathway, the kidney has two other pathways for protein degradation. The first may be important for several bioactive proteins, particularly those for which receptors are present on the basolateral membrane (e.g., insulin, ANP, AVP, and PTH). After transcytosis, the proximal tubule cell partially hydrolyzes peptide hormones at the basolateral cell membrane. The resulting peptide fragments re-enter the circulation, where they are available for glomerular filtration and ultimate handling by the apical absorption/degradation pathway. The second alternative pathway for protein degradation involves receptor-mediated endocytosis by endothelial cells of the renal vascular and glomerular structures. This pathway participates in the catabolism of small peptides, such as ANP.
In conclusion, the kidney plays a major role in the metabolism of small proteins and peptide hormones. Renal extraction rates may be large, and they account for as much as 80% of the total metabolic clearance. Thus, it is not surprising that end-stage renal disease can lead to elevated levels of glucagon, PTH, gastrin, and ANP. Under physiological conditions, glomerular filtration represents the rate-limiting step for the removal of low-molecular-weight proteins from the circulation—apical absorption by the tubules, intracellular hydrolysis, and peritubular hydrolysis do not saturate over a wide range of filtered loads.
The combined concentration of carboxylates in the blood plasma is a few millimolars, of which lactate represents the largest fraction. The monocarboxylates pyruvate and lactate are products of anaerobic glucose metabolism (see Chapter 58). The dicarboxylates and tricarboxylates include intermediates of the citric acid cycle. Because these carboxylates are important for energy metabolism, their loss in the urine would be wasteful. Normally, the proximal tubule reabsorbs virtually all these substances (Fig. 36-8A).
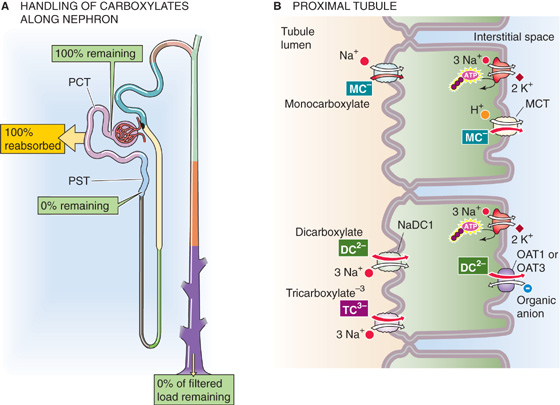
Figure 36-8 A and B, Monocarboxylate, dicarboxylate, and tricarboxylate handling by the kidney. In A, the yellow box indicates the fraction of the filtered load that the proximal tubule reabsorbs. The green boxes indicate the fraction of the filtered load that remains in the lumen at various sites. The values in the boxes are approximations. MCT, H+/monocarboxylate cotransporter; PCT, proximal convoluted tubule; PST, proximal straight tubule.
At least two distinct Na+-dependent cotransporters carry carboxylates across the apical membranes (Fig. 36-8B). One system is specific for monocarboxylates, including lactate, pyruvate, acetoacetate, and β-hydroxybutyrate. A second cotransporter (NaDC1 or SLC13A2) carries dicarboxylates and tricarboxylates, such as α-KG, malate, succinate, and citrate. Once inside the cell, the monocarboxylates exit across the basolateral membrane through an H+/monocarboxylate cotransporter 2 (MCT2, SLC16A7). Because monocarboxylates enter across the apical membrane coupled to Na+, and then exit across the basolateral membrane coupled to H+, monocarboxylate reabsorption leads to an accumulation of Na+ by the cell and a rise in intracellular pH.
Dicarboxylates exit the cell across the basolateral membrane through multiple organic anion-carboxylate exchangers (e.g., the renal organic anion transporter OAT1 and OAT3; see Chapter 5) that may overlap in substrate specificity or may even carry anions of different valence. Because the molecular identities and stoichiometries of some of these transporters are unknown, we present organic anion exchangers in a generic sense in this and the next three sections.
Although the proximal tubule normally reabsorbs carboxylates fully, carboxylates may appear in the urine when their plasma levels are elevated. Urinary excretion may occur when the filtered load of acetoacetate and β-hydroxybutyrate—ketone bodies (see Chapter 51) produced during starvation or during low-insulin states (diabetes mellitus)—exceeds the Tm in the proximal tubule.
The kidneys handle PAH (a monovalent anion), as well as many organic anions (e.g., many metabolites of endogenous compounds and administered drugs), by both filtration and secretion (Fig. 36-9A). The synthetic anion PAH is somewhat unusual in that ~20% of it binds to plasma proteins, largely albumin. Thus, only ~80% of PAH is available for filtration. Assuming a filtration fraction (see Chapter 34) of 20%, only 80% × 20%, or 16%, of the arterial load of PAH appears in Bowman’s space. Nevertheless, at low plasma [PAH], the kidneys excrete into the urine nearly all (~90%) of the PAH entering the renal arteries, so that very little PAH remains in the renal veins. Because the kidneys almost completely clear it from the blood in a single passage, PAH is useful for measuring renal plasma flow (see Chapter 34).
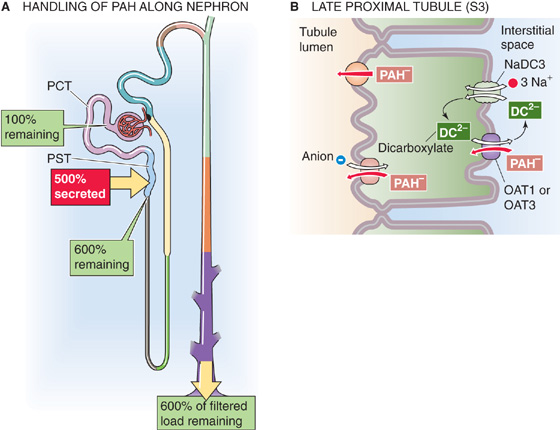
Figure 36-9 A and B, PAH handling by the kidney. In A, the red box indicates the fraction of the filtered load secreted by the proximal tubule. The green boxes indicate the fraction of the filtered load that remains in the lumen at various sites when plasma [PAH] is low (<12 mg/dL). The values in the boxes are approximations. PCT, proximal convoluted tubule; PST, proximal straight tubule.
The nephron secretes PAH mainly in the late proximal tubule (S3 segment), and it does so by the transcellular route, against a sizable electrochemical gradient. PAH uptake across the basolateral membrane occurs through the high-affinity OAT1 and the lower-affinity OAT3 transporters, driven by the outward gradient of the α-KG, which is a dicarboxylate (Fig. 36-9B). This uptake of PAH is an example of tertiary active transport because the basolateral Na/dicarboxylate cotransporter NaDC3—in a process of secondary active transport—elevates α-KG in the cell, thereby creating the outward α-KG gradient. NaDC3 carries three Na+ ions and one dicarboxylate into the cell. Finally, the basolateral Na-K pump—in a process of primary active transport—establishes the Na+ gradient used to drive the accumulation of α-KG. (See Note: Role of OAT1 and OAT3 in the Renal Transport of Organic Anions)
The apical step of PAH secretion probably occurs through exchange for luminal anions or through electrogenic facilitated diffusion, driving by the inside-negative membrane potential. Several anionic drugs (e.g., probenecid) that compete at the basolateral PAH-anion exchanger or the apical PAH-anion exchanger inhibit PAH secretion from blood to lumen.
Just as glucose reabsorption saturates at its Tm as one increases plasma [glucose], PAH secretion (Fig. 36-10A, red curve) saturates at a sufficiently high plasma [PAH]. Starting from an initially low value, increasing plasma [PAH] causes excretion (green) to rise much faster than filtration (orange). Subtracting the amount filtered from the amount excreted, we see that the rate of PAH secretion at first rises rapidly with plasma [PAH]. However, as plasma [PAH] approaches ~20 mg/dL, the amount of secreted PAH reaches a plateau (Tm), typically 60 to 80 mg/min, indicating saturation of secretory mechanisms.
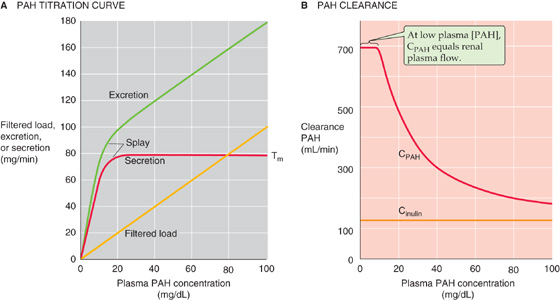
Figure 36-10 A and B, Effect of increasing plasma PAH concentrations on PAH excretion and clearance. In A, Tm represents the transport maximum for reabsorption.
After plasma [PAH] has increased enough to reach the Tm, further increases in plasma [PAH] increase urinary excretion, but only as a consequence of the increase in the filtered load, not because of increased tubule secretion. At these high plasma PAH levels, the kidneys can no longer fully remove PAH from the blood in a single pass, and therefore it would no longer be appropriate to use PAH secretion to estimate renal plasma flow. At low plasma [PAH] values (<12 mg/dL), PAH extraction from plasma flowing through the kidney is nearly complete (~90%), forming the basis for using PAH clearance as a measure of renal plasma flow.
Recall that as plasma [glucose] increases, glucose clearance rises to approach inulin clearance (Fig. 36-4B). In contrast, PAH clearance (Fig. 36-10B, red curve) decreases with increasing plasma [PAH] and falls to approach inulin clearance (orange). The reason is that, as plasma [PAH] increases, secreted PAH forms a progressively smaller fraction of the PAH appearing in the urine.
In addition to PAH, the late proximal tubule secretes a wide variety of organic anions. These anions include the following (Table 36-3): (1) endogenous anions, such as oxalate and bile salts; (2) exogenous anions, such as the drugs penicillin and furosemide; and (3) uncharged molecules conjugated to anionic groups, such as sulfate or glucuronate (see Chapter 46). The proximal tubule secretes these anions into the lumen by using basolateral and apical anion exchangers that are similar to those involved in PAH secretion (Fig. 36-9B). At the apical membrane, the secreted anion appears to exchange for luminal Cl−, urate, or OH−.
Table 36-3 Organic Anions and Cations Secreted by the Late Proximal Tubule
|
Anions |
Cations |
Endogenous |
cAMP and cGMP |
Epinephrine |
|
Bile salts |
Acetylcholine |
|
Hippurate |
Choline |
|
Oxalate |
Dopamine |
|
α-Ketoglutarate |
Histamine |
|
Short-chain fatty acids |
Serotonin |
|
Prostaglandins (e.g., PGE2) |
Thiamine |
|
Urate Creatinine (zwitterion) |
Guanidine |
|
Uremic organic anions* |
|
Exogenous |
Acetazolamide |
Atropine |
|
|
Quinine |
|
Chlorothiazide |
Amiloride |
|
Furosemide |
Cimetidine |
|
PAH |
NMN |
|
Penicillin G |
Morphine |
|
Probenecid |
Paraquat |
|
Saccharin |
Procainamide |
|
Salicylate |
Tetraethylammonium |
|
Indomethacin |
Chlorpromazine |
Conjugated (Endogenous and Exogenous) |
Glucuronate conjugates† |
|
|
Glutathione conjugates† |
|
|
Sulfate conjugates† |
|
* Hippurate-like aryl organic anions that interfere with PAH transport.
NMN, N-methylnicotinamide; PGE2, prostaglandin E2.
† See Chapter 46.
Urate, a monovalent anion, is the end product of purine catabolism (Fig. 36-11A). Plasma [urate] is typically 3 to 7 mg/dL (0.2 to 0.4 mM) and is elevated in gout. The glomeruli filter urate, and then the proximal tubule successively reabsorbs (S1 segment), secretes (S2 segment), and again reabsorbs (S3 segment) urate (Fig. 36-11B). Reabsorption is the more important transport pathway, so that the kidneys excrete only ~10% of filtered urate. (See Note: Tertiary Active Transport of Urate)
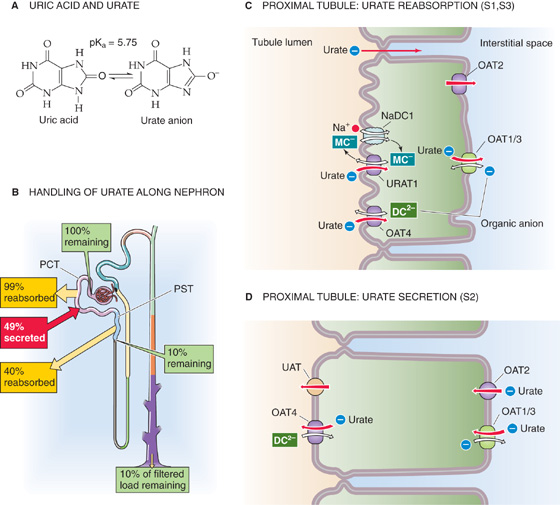
Figure 36-11 A and B, Urate handling by the kidney. In A, the yellow arrows indicate reabsorption, whereas the red arrow indicates secretion. The green boxes indicate the fraction of the filtered load remaining at various sites. The values in the boxes are approximations. In B, the molecular identities of the apical and basolateral urate transporters are unknown. PCT, proximal convoluted tubule; PST, proximal straight tubule.
Reabsorption Reabsorption of urate occurs by both a paracellular route involving passive diffusion and a transcellular route involving active transport (Fig. 36-11C). The contribution of the paracellular pathway can become apparent during extracellular volume depletion, which can lead to a compensatory enhancement of proximal-tubule fluid reabsorption. Solvent drag (p. 488) can then enhance paracellular urate reabsorption, thereby decreasing urate excretion and raising plasma levels of urate. The transcellular mechanism predominates in the early proximal tubule. The active apical step does not involve Na+ cotransport, but rather the exchange of luminal urate for any of a host of intracellular anions. URAT1 exchanges luminal urate for monocarboxylates (e.g., lactate), whereas OAT4 exchanges luminal urate for dicarboxylates. Once inside the cell, urate exits across the basolateral membrane either by facilitated diffusion via OAT2, or by exchange with an organic anion via OAT1 or OAT3.
Uricosuric agents such as probenecid, salicylate, and other non-steroidal anti-inflammatory drugs inhibit URAT1. Indeed, probenecid is useful in the treatment of gout.
Secretion The basolateral step of urate secretion (Fig. 36-11D) is mediated by organic anion exchange via reversal of OAT1 and OAT3 (which also mediates basolateral PAH uptake, see Fig. 36-9B), as well as by facilitated diffusion via OAT2. Urate exits the cell across the apical membrane by reversal of OAT4 (Fig. 36-11D), as well as by facilitated diffusion via UAT. Under certain conditions, renal urate excretion can exceed the quantity filtered, indicating net secretion.
The late proximal tubule (Fig. 36-12A) is also responsible for secreting a wide range of both endogenous and exogenous organic cations (Table 36-3). Some of the most important endogenous organic cations are the monoamine neurotransmitters such as dopamine, epinephrine, norepinephrine, and histamine (see Chapter 13). Exogenous secreted organic cations include morphine, quinine, and the diuretic amiloride (see Chapter 35).
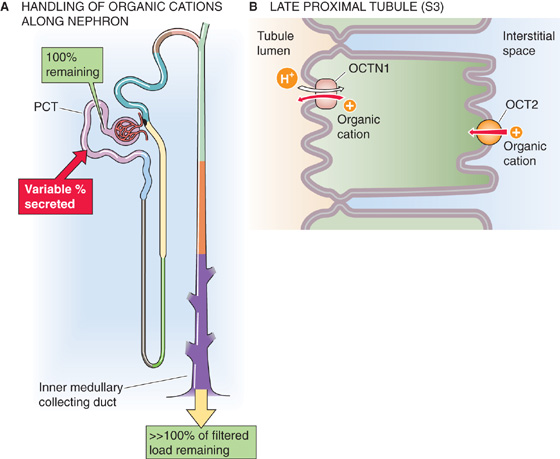
Figure 36-12 A and B, Organic cation handling by the kidney. In A, the red arrow indicates secretion. The green boxes indicate the fraction of the filtered load remaining at various sites. The values in the boxes are approximations. OCT, organic cation transporter; PCT, proximal convoluted tubule.
The polyspecific organic cation transporter OCT2 (see SLC22 family in Table 5-4 on p. 118) is responsible for the basolateral uptake of these organic cations (Fig. 36-12B). OCT2 mediates facilitated diffusion and is electrogenic.
At the apical membrane, an organic cation-H+ exchanger OCTN1 (Fig. 36-12B) moves these cations from the cell to the lumen. The energy for the extrusion of the organic cation is the H+ electrochemcal gradient across the apical membrane, from lumen to cell. Because the apical Na-H exchanger (a secondary active transporter) is largely responsible for establishing this H+ gradient, the cation-H exchange is an example of tertiary active transport.
An organic cation secreting mechanism is also responsible for secreting creatinine, the breakdown product of phosphocreatine. Despite this modest secretion, creatinine is a useful index of GFR (see Chapter 34).
Many cations or anions are, in fact, weak acids or bases in equilibrium with a neutral species (see Chapter 28), which generally diffuses across a membrane much faster than the corresponding cation or anion. This rapid diffusion usually occurs because the neutral species is far more soluble in the lipid bilayer of cell membranes than is the charged species.
Changing the luminal pH can substantially affect the overall transport of a buffer pair. For example, acidifying the tubule lumen promotes the reabsorption of a neutral weak acid and the secretion of a neutral weak base. In the case of a neutral weak acid such as salicylic acid (Fig. 36-13A), the glomerular filtrate contains both the neutral weak acid (HA) and its conjugate weak base, which is an anion (A−). The secretion of H+ into the lumen (see Chapter 39) titrates the A− to HA, thus raising luminal [HA] above intracellular [HA], so that HA rapidly diffuses across the apical membrane—by the process of nonionic diffusion—into the tubule cell and across the basolateral membrane into the blood. The more acidic the luminal fluid, the greater is the titration of A− to HA, and the greater is the nonionic diffusion of HA from lumen to blood.
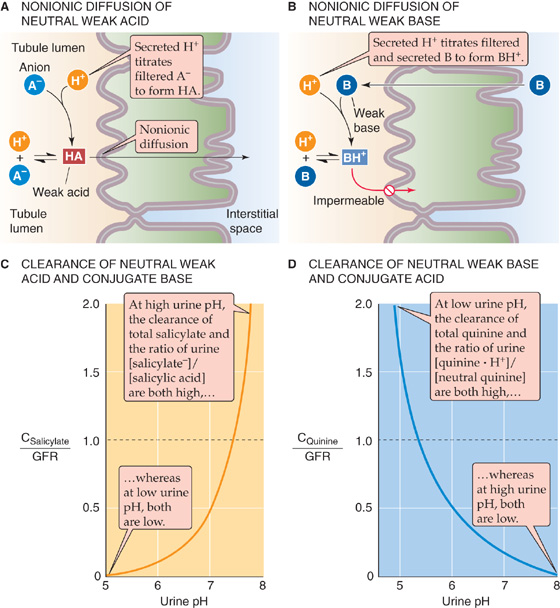
Figure 36-13 A to D, Nonionic diffusion.
In the case of a weak base such as ammonia (Fig. 36-13B), the glomerular filtrate contains both the neutral weak base (B) and the cationic species (BH+). The secretion of H+ into the lumen titrates B to BH+, thus lowering luminal [B] below intracellular [B], so that B rapidly diffuses from cell to lumen. The resulting fall in intracellular [B] sets up a gradient for B to diffuse from blood to cell. The more acidic the luminal fluid, the greater is the titration of B to BH+, and the greater is the nonionic diffusion of B from blood to lumen.
A striking example of how luminal pH affects the clearance of a weak acid is the renal handling of salicylic acid and its anion species, salicylate. At a urinary pH of ~7.5, the amount of total salicylate (salicylic acid + salicylate anion) excreted is the same as the filtered load (Fig. 36-13C); that is, the clearance of total salicylate (CSalicylate) equals GFR. Lower urinary pH values favor the titration of luminal salicylate anion to salicylic acid, which the tubule readily reabsorbs. Hence, lowering pH causes the fractional excretion of total salicylate (FESalicylate = CSalicylate/GFR; see Chapter 33) to fall to less than unity. At very low urinary pH values, the kidney reabsorbs virtually all salicylate, and FESalicylate approaches zero. However, at urinary pH values higher than ~7.5, FESalicylate increases markedly because the alkalinity keeps luminal levels of the salicylate anion high and levels of salicylic acid low, so that the neutral weak acid now diffuses from blood to lumen.
It is possible to treat overdoses of salicylate or acetylsalicylate (i.e., aspirin) by alkalinizing the urine with HCO−3 or by increasing urine flow with diuretics. The reason is that both high pH and high urine flow keep luminal levels of salicylic acid low, thereby maintaining a sink for salicylic acid in the lumen. Both treatments enhance the urinary excretion of the drug and lower plasma concentrations. The converse example of urinary pH dependence is seen in the excretion of a neutral weak base, such as ammonia or quinine (Fig. 36-13D). (See Note: Treatment of Salicylate Poisoning; Treatment of a PCP Overdose)
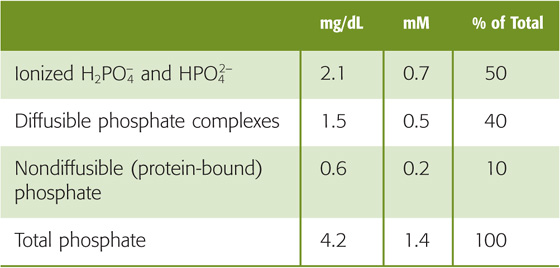
As discussed in Chapter 52, the metabolism of inorganic phosphate (Pi) depends on bone, the gastrointestinal tract, and kidneys. About half of total plasma phosphate (Table 36-4) is in an ionized form, and the rest is either complexed to small solutes (~40%) or bound to protein (10% to 15%). The plasma concentration of total Pi varies rather widely, between 0.8 and 1.5 mM (2.5 to 4.5 mg/dL of elemental phosphorus). Thus, the filterable phosphate (i.e., both the ionized and complexed) varies between ~0.7 and 1.3 mM. At a normal blood pH of 7.4, 80% of the ionized plasma phosphate is HPO2−4, and the rest is H2PO4−. Assuming that the total plasma phosphate concentration is 4.2 mg/dL, that only the free and complexed phosphate is filterable, and that the GFR is 180 L/day, each day the kidneys filter ~7000 mg of phosphate. Because this amount is more than an order of magnitude greater than the total extracellular pool of phosphate (see Chapter 52), it is clear that the kidney must reabsorb most of the phosphate filtered in the glomerulus.
Table 36-4 Components of Total Plasma Phosphate
The proximal tubule reabsorbs ~80% of the filtered phosphate (Fig. 36-14A). The loop of Henle and the collecting ducts reabsorb only negligible amounts of phosphate, but the classic distal tubule (see Chapter 33) reabsorbs ~10% of the filtered load. The kidneys excrete the remaining ~10% in the urine.
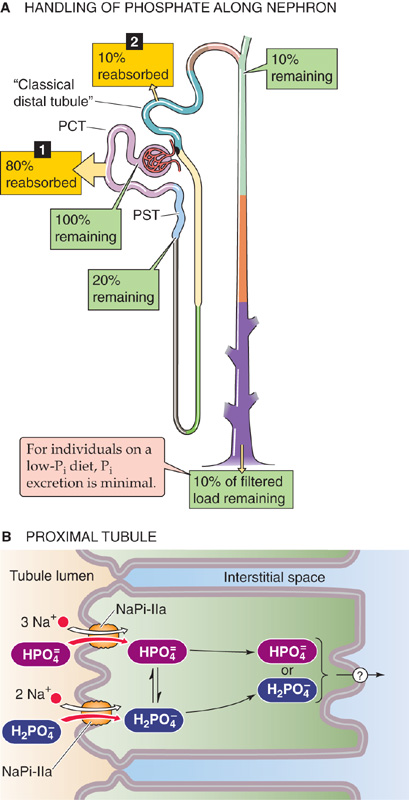
Figure 36-14 A and B, Phosphate handling by the kidney. In A, the numbered yellow boxes indicate the fraction of the filtered load that various nephron segments reabsorb. The green boxes indicate the fraction of the filtered load that remains in the lumen after these segments. The values in the boxes are approximations. In B, the top path illustrates reabsorption of divalent phosphate (HPO2−4), and the bottom path illustrates the reabsorption of monovalent phosphate (H2PO−4). PCT, proximal convoluted tubule; PST, proximal straight tubule.
The proximal tubule reabsorbs most of the filtered phosphate by the transcellular route (Fig. 36-14B). Phosphate ions enter the cell across the apical membrane by secondary active transport, energized by the apical electrochemical Na+ gradient. The apical Na/phosphate cotransporter NaPi-IIa (SLC34A1) translocates three Na+ ions and one divalent phosphate ion (HPO2−4). Thus, the process is electrogenic (net positive charge into the cell). In contrast, NaPi-IIc (SLC34A3)—which is in low abundance in adults—is electroneutral. As pH along the proximal tubule falls, the relative concentration of H2PO−4 increases, and overall phosphate uptake becomes even more electrogenic. (See Note: The Type II Na/Phosphate Cotransporter NaPi-IIa)
Apical phosphate uptake is under the control of intracellular pH as well as luminal pH and [Na+]. Intracellular H+ appears to stimulate NaPi allosterically. However, luminal H+ is a competitive inhibitor of Na+ binding to the extracellular face of NaPi, so that when luminal pH falls, it is more difficult for Na+ to bind. In addition, even when luminal [Na+] is saturating, an acidic luminal pH inhibits phosphate uptake, possibly by shifting the phosphate equilibrium toward H2PO−4, which NaPi may not transport as well as HPO2−4.
The passive exit of phosphate across the basolateral membrane occurs by a mechanism that is not well understood. The basolateral membrane also has a type III Na/Pi cotransporter that carries three Na+ with each phosphate. It thus mediates electrogenic uptake of phosphate—presumably for nutrition of the tubule cells—rather than exit across the basolateral membrane.
Although it is clear that the proximal tubule reabsorbs almost all filtered phosphate at low filtered loads, the role of the distal nephron in phosphate handling is controversial. When experimental animals are maintained on a low-phosphate diet, the classic distal tubule absorbs a significant fraction of the filtered load of phosphate, thus minimizing phosphate loss in the urine.
Phosphate handling by the kidney shares some of the properties of renal glucose handling, including threshold, saturation with Tm kinetics, and splay (Fig. 36-15). However, important differences exist. First, some phosphate excretion (green curve) occurs even at normal levels of plasma [phosphate]. Thus, a small increment in the plasma [phosphate] results in significant acceleration of phosphate excretion (i.e., to higher values along the green curve), whereas plasma [glucose] must double before glucose excretion even commences. Accordingly, the kidney plays an important role in the regulation of plasma [phosphate], but it does not normally modulate plasma [glucose]. Second, the kidney reaches the Tm for phosphate (red curve) at the high end of normal plasma [phosphate] values, whereas the kidney reaches the Tm for glucose at levels that are far higher than physiological plasma [glucose] levels. Third, the renal phosphate Tm is sensitive to a variety of stimuli, including hormones and acid-base balance, whereas the glucose Tm is insensitive to regulators of glucose metabolism, such as insulin.
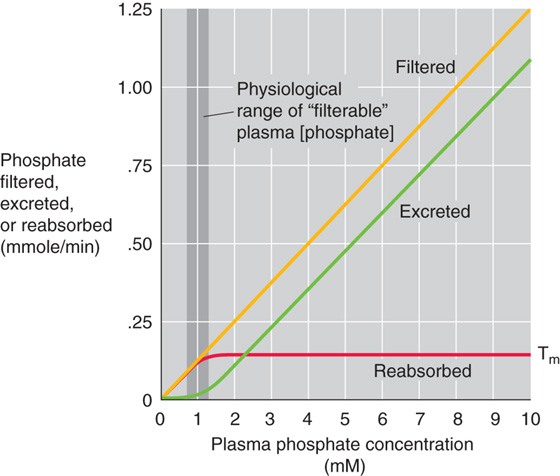
Figure 36-15 Phosphate titration curves. The plasma phosphate concentration is the filterable phosphate (i.e., ionized plus complexed to small solutes). The dark vertical band is the approximate range of normal filterable values.
Table 36-5 summarizes the major hormones and other factors that regulate phosphate transport by the proximal tubule. Although these factors affect phosphate transport through many different signaling pathways, they fall into two major categories. Some act rapidly and independently of protein synthesis. Others act more slowly, through mechanisms requiring protein synthesis.
Table 36-5 Factors That Modulate Phosphate Excretion
↓ Reabsorption |
↑ Reabsorption |
↑ Excretion |
↓ Excretion |
PTH |
1, 25-dihydroxy-vitamin D |
Extracellular volume expansion |
High Ca2+ intake |
High dietary phosphate intake |
Phosphate deprivation |
High plasma [phosphate] |
|
Glucocorticoids |
Insulin |
Acidosis |
Alkalosis |
Calcitonin |
Thyroid hormone |
ANP |
Growth hormone |
Dopamine |
|
Diuretics |
|
PTHrP |
|
Fasting |
|
Acute renal denervation |
|
Autosomal and X-linked hypophosphatemia |
|
The most important hormonal regulator is PTH, which inhibits phosphate reabsorption and thus promotes phosphate excretion. PTH is a classic example of a rapid regulator that does not require protein synthesis. PTH binds to PTH 1R receptors, which appear to couple to two heterotrimeric G proteins (see Chapter 52). The first (Gαs) activates adenylyl cyclase and, through cAMP, stimulates PKA. The second (Gαq), particularly at low PTH levels, activates phospholipase C (PLC), and then protein kinase C (PKC) (see Chapter 3). Once activated, PKA and PKC promote endocytotic removal of NaPi from the apical membrane, thus reducing phosphate reabsorption. Conversely, when PTH levels decrease, a net insertion of membrane vesicles containing NaPi occurs, thereby leading to an increase in phosphate reabsorption.
Another inhibitor of phosphate reabsorption, ANP (see Chapter 40), may also activate protein kinase, but it does so through cGMP rather than through cAMP. An additional example of a rapid effect on phosphate reabsorption is extracellular fluid expansion, which accelerates proximal phosphate excretion along with Na+ excretion (see Chapter 35).
Several modulators of phosphate transport are slow, acting by controlling protein synthesis and thus Na/Pi cotransporter mRNA levels. High dietary phosphate intake decreases phosphate reabsorption, even without changes in PTH levels. Changes in plasma [phosphate], such as those that occur in phosphate depletion or loading, also modulate the balance between retrieval and insertion of Na/Pi cotransporters. Both phosphate depletion and 1, 25-dihydroxy-vitamin D (see Chapter 52) upregulate the cotransporter. Glucocorticoid excess and metabolic acidosis downregulate the cotransporter; the latter effect is synergistic with the direct effects of H+ on the NaPi protein (see earlier). Because phosphate is the primary pH buffer in normal urine, factors that increase urinary [phosphate] (e.g., acidosis) increase the titratable acidity component of renal acid excretion (see Chapter 39).
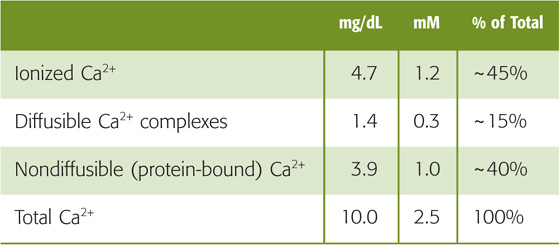
Whole-body Ca2+ balance—as well as the hormonal control of free, ionized plasma calcium (Ca2+) levels—is the subject of Chapter 52. Here we discuss the role of the kidney in Ca2+ balance. The total concentration of Ca2+ in plasma is normally 2.2 to 2.6 mM (8.8 to 10.6 mg/dL). Table 36-6 summarizes the forms of total Ca2+ in the plasma. Some 40% binds to plasma proteins, mainly albumin, and constitutes the nonfilterable fraction. The filterable portion, ~60% of total plasma Ca2+, consists of two moieties. The first, ~15% of the total, complexes with small anions such as carbonate, citrate, phosphate, and sulfate. The second, ~45% of total Ca2+, is the ionized Ca2+ that one may measure with Ca2+-sensitive electrodes or dyes. It is the concentration of this free, ionized Ca2+ that the body tightly regulates; plasma [Ca2+] normally is 1.0 to 1.3 mM (4.0 to 5.2 mg/dL).
Table 36-6 Components of Total Plasma Ca2+
The distribution among the various forms of total Ca2+ is not fixed. Because H+ competes with Ca2+ for binding to anionic sites on proteins or small molecules, acidosis generally increases plasma [Ca2+]. Thus, the decrease in luminal pH that occurs along the nephron (see Chapter 39) makes additional Ca2+ available for recovery. In addition, Ca2+ binding to proteins in the plasma depends on the concentration of proteins, particularly albumin. A change in the level of the anions with which Ca2+ can form complexes also affects the distribution of different Ca2+ entities. Tubule cells probably reabsorb Ca2+-anion complexes poorly. However, because the concentration of these anions declines along the nephron owing to anion reabsorption, free Ca2+ becomes available for transport.
The kidney reabsorbs ~99% of the filtered load of Ca2+, principally at the proximal tubule, the thick ascending limb (TAL), and the distal convoluted tubule (DCT) (Fig. 36-16A).
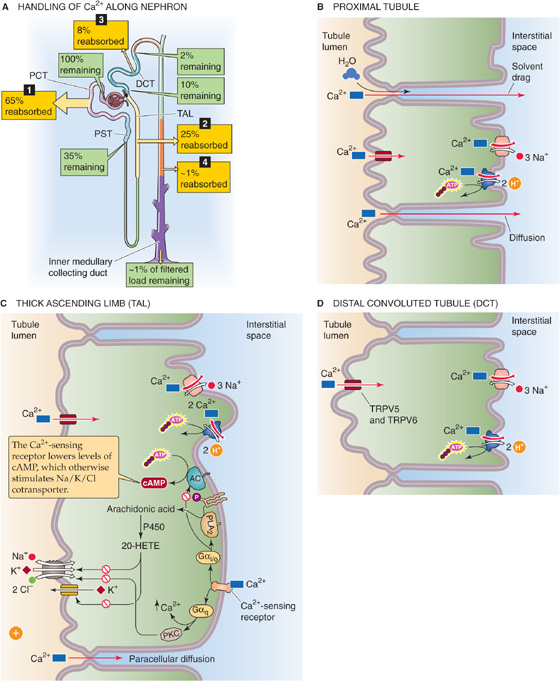
Figure 36-16 A to D, Calcium handling by the kidney. In A, the numbered yellow boxes indicate the approximate fraction of the filtered load that various nephron segments reabsorb. The green boxes indicate the fraction of the filtered load that remains in the lumen after these segments. The values in the boxes are approximations. AC, adenylyl cyclase; PCT, proximal convoluted tubule; PST, proximal straight tubule.
Proximal Tubule The proximal tubule reabsorbs ~65% of the filtered Ca2+, a process that is not subject to hormonal control. A small part of the Ca2+ reabsorbed by the proximal tubule (~20% of the 65%) moves by the transcellular route that we discuss in the next section. However, most proximal tubule Ca2+ reabsorption (~80% of the ~65%) occurs by the paracellular route (Fig. 36-16B). The evidence for a large component of paracellular Ca2+ transport is the high Ca2+ permeability of the tubule, as well as the sensitivity of net Ca2+ transport to changes in the transepithelial electrochemical gradient for Ca2+. For example, imposing a lumen-negative transepithelial voltage induces Ca2+ secretion, whereas a lumen-positive voltage induces reabsorption. Because the S2 and S3 segments of the proximal tubule normally have a small lumen-positive voltage, it is not surprising that they are responsible for reabsorbing a large fraction of filtered Ca2+ by the paracellular route. In addition to Ca2+ diffusion through tight junctions, proximal Ca2+ reabsorption parallels that of Na+ and water, a finding suggesting participation of solvent drag (see Chapter 20).
Thick Ascending Limb The TAL reabsorbs ~25% of the filtered Ca2+ (Fig. 36-16C). Under normal conditions, about half of the Ca2+ reabsorption in the TAL occurs passively by a paracellular route, driven by the lumen-positive voltage. Thus, it is not surprising that hormones such as AVP, which make the transepithelial voltage more positive, indirectly increase Ca2+ reabsorption. The other half of Ca2+ reabsorption by the TAL occurs by the transcellular pathway (see next section), which PTH stimulates.
Distal Convoluted Tubule This segment reabsorbs ~8% of the filtered Ca2+ load (Fig. 36-16D). Despite the relatively small amount of Ca2+ delivered, the DCT is a major regulatory site for Ca2+ excretion. In contrast to the proximal tubule and TAL, the DCT reabsorbs Ca2+ predominantly by an active, transcellular route (see next section). Evidence against a substantial component of paracellular Ca2+ reabsorption is that—under a variety of experimental maneuvers (see Chapter 36)—it is easy to dissociate Ca2+ transport in the DCT from Na+ and water reabsorption. The quantitative contribution of the collecting ducts and tubules to Ca2+ reabsorption is quite small (~1% of the filtered load), and their role in regulating renal Ca2+ excretion is not well defined.
We have seen that the contribution of the transcellular route to Ca2+ reabsorption increases steadily from the proximal tubule to the TAL and to the DCT. Regardless of the nephron segment, tubule cells apparently use the same general mechanism to move Ca2+ through the cell from lumen to blood (Fig. 36-16B–D).
Because [Ca2+]i is only ~100 nM (~10,000-fold less than in extracellular fluid), and because the membrane voltage across the apical membrane is ~−70 mV, a steep electrochemical gradient favors Ca2+ entry across the apical membrane. Work on the DCT shows that apical Ca2+ entry is passive and is mediated by epithelial Ca2+ channels TRP5 and TRP6 (ECaC1 and ECaC2), which are not gated by voltage. This Ca2+ channel has consensus phosphorylation sites for PKA, PKC, and cGMP-stimulated protein kinase (PKG), as well as interaction sites for ankyrin (see Chapter 2), a component of the cytoskeleton. Mg2+ competes with Ca2+ for entry into the cytoplasm, and extracellular H+ blocks channel activity. Whether similar Ca2+ channels play a role in transcellular Ca2+ reabsorption in the proximal tubule and TAL is unknown.(See Note: Regulation of Ca2+ and Mg2+ Reabsorption)
As Ca2+ enters the tubule cell across the apical membrane, Ca2+-binding proteins buffer the Ca2+, helping to keep [Ca2+]i low, and maintaining a favorable gradient for Ca2+ influx. In addition, Ca2+ may temporarily enter certain organelles, particularly the endoplasmic reticulum and mitochondria. In the steady state, however, the cell must extrude across the basolateral membrane all the Ca2+ that enters across the apical membrane. Both primary and secondary active transporters participate in this Ca2+ extrusion against a steep electrochemical gradient. The primary active transporter is an ATP-driven plasma-membrane Ca2+ ATPase or pump (PMCA; see Chapter 5). PMCA is present in several nephron segments, with its highest density in the basolateral membrane of the DCT. Increases in [Ca2+]i—such as those triggered by PTH through PLC and inositol triphosphate (IP3)—activate calmodulin and thus stimulate PMCA (see Chapter 3).
In addition to the Ca2+ pump, an Na-Ca exchanger (NCX; see Chapter 5) also extrudes Ca2+ across the basolateral membrane of tubule cells. At physiologically low [Ca2+]i levels, when the NCX is not very active, the major pathway for Ca2+ extrusion is the Ca2+ pump. Only when [Ca2+]i increases does the NCX begin to make a significant contribution. Conversely, if the [Ca2+]i is normal and the inward Na+ electrochemical gradient falls (e.g., [Na+]i rises), the NCX can reverse direction and load the cell with Ca2+.
Table 36-7 summarizes factors that modulate Ca2+ handling by various segments of the nephron.
Table 36-7 Factors Affecting Ca2+ Reabsorption Along the Nephron (See Note: Regulation of Ca2+ Reabsorption;)
Site |
Increase Reabsorption |
Decrease Reabsorption |
Proximal tubule |
Volume contraction |
Volume expansion |
Thick ascending limb |
PTH |
Furosemide and related diuretics |
|
Calcitonin |
|
Distal convoluted tubule |
PTH |
Phosphate depletion |
|
Vitamin D |
|
|
AVP |
|
|
Alkalosis |
|
|
Thiazide diuretics |
|
Collecting duct |
Amiloride |
— |
Parathyroid Hormone The most important regulator of renal Ca2+ reabsorption is PTH, which stimulates Ca2+ reabsorption in the TAL, the DCT, and the connecting tubule. PTH appears to increase the open probability of apical Ca2+ channels (Fig. 36-16C, D). Such an increase in Ca2+ permeability would increase [Ca2+]i, which, in turn, would stimulate basolateral Ca2+ extrusion mechanisms, increase Ca2+ reabsorption, and raise plasma [Ca2+].
As we saw in our discussion of phosphate handling, PTH acts by binding to the PTH 1R receptor, which apparently couples to two heterotrimeric G proteins and activates two kinases (see Chapter 52). Activation of Gαs raises [cAMP]i and stimulates PKA. Activation of Gαq ultimately stimulates a PKC that is unusual in being insensitive to phorbol esters (see Chapter 3). Both pathways are essential for the action of PTH on apical Ca2+ entry. Like PTH, calcitonin at low concentrations also increases Ca2+ reabsorption through cAMP in both the TAL and the DCT.
Vitamin D Acting on gene transcription, vitamin D (see Chapter 52) increases Ca2+ reabsorption in the distal nephron, thus complementing the major Ca2+-retaining action of vitamin D, Ca2+ absorption in the gastrointestinal tract (see Chapter 45). In renal tubule cells, vitamin D upregulates the Ca2+-binding protein, which contributes to enhanced Ca2+ reabsorption by keeping [Ca2+]i low during increased Ca2+ traffic through the cell.
Plasma Ca2+ Levels The kidney, similar to the parathyroid gland (see Chapter 52), responds directly to changes in extracellular [Ca2+]. In the cortical TAL, an increase in basolateral [Ca2+] inhibits both the Na/K/Cl cotransporter and K+ channels on the apical membrane (Fig. 36-16C), thereby decreasing the lumen-positive transepithelial potential and reducing paracellular Ca2+ reabsorption. The mechanism appears to be the following: Extracellular Ca2+ binds to a basolateral Ca2+-sensing receptor (CaSR), which couples to at least two G proteins. First, activation of Gαi decreases [cAMP]i, thus reducing stimulation of Na/K/Cl cotransport by cAMP (see Chapter 35). Second, activation of a member of the Gi/Go family stimulates phospholipase 2 (PLA2; see Chapter 3), thereby increasing levels of arachidonic acid and one of its cytochrome P450 metabolites, probably 20-hydroxyeicosatetraenoic acid (HETE). The latter inhibits apical Na/K/Cl cotransporters and K+ channels. Third, CaSR activates Gαq, elevating [Ca2+]i and stimulating PKC (see Chapter 3), which also inhibits Na/K/Cl cotransport. Regardless of the mechanism, inhibition of apical Na/K/Cl cotransport (1) lowers [Cl−]i and thus hyperpolarizes the basolateral membrane and (2) lowers [K+]i and thus depolarizes the apical membrane. Together, these two effects reduce the lumen-positive transepithelial voltage, thereby inhibiting the paracellular reabsorption of Ca2+ and Mg2+ (see later). The Ca2+-sensing receptor is also present in the proximal tubule, the medullary TAL, the DCT, and the collecting ducts.
An interesting consequence of reduced Na+ reabsorption induced by high plasma [Ca2+] is an inhibition of the kidney’s ability to generate concentrated urine (see Chapter 38). Indeed, chronic hypercalcemia results in dilute urine, a form of nephrogenic diabetes insipidus (see Chapter 38 for the box on this topic). By keeping urine [Ca2+] relatively low, this effect may help to avoid Ca2+ stones under conditions of high Ca2+ excretion.
Diuretics Among diuretics, those acting on the TAL, such as furosemide, decrease Ca2+ reabsorption, whereas those acting on the distal nephron, such as thiazides and amiloride, increase Ca2+ reabsorption. Thus, of these drugs, only the powerful loop diuretic furosemide is appropriate for treating hypercalcemic states. In the TAL, furosemide (see Chapter 35) reduces the lumen-positive voltage and thus the driving force for passive, paracellular Ca2+ reabsorption. Accordingly, urinary Ca2+ excretion increases in parallel with Na+ excretion.
In contrast, the effects of thiazides and amiloride are exceptions to the general rule that Ca2+ and Na+ excretion parallel each other. Thiazide diuretics inhibit Na/Cl cotransport and stimulate Ca2+ reabsorption in the DCT. Inhibiting apical Na/Cl cotransport lowers [Cl−]i in the DCT cell, thus hyperpolarizing the cell. The steeper electrical gradient increases apical Ca2+ entry, secondarily stimulating basolateral Ca2+ extrusion. Amiloride inhibits apical Na+ channels in the initial and cortical collecting tubules and hyperpolarizes the apical membrane. Thus, like the thiazides, amiloride augments Ca2+ reabsorption by enhancing the gradient for apical Ca2+ entry. The stimulatory effects of thiazides and of amiloride on apical Ca2+ uptake require physiological PTH levels to keep the apical Ca2+ channels open (see Chapter 35).
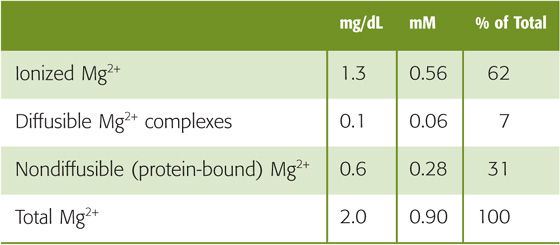
Approximately 99% of the total body stores of magnesium reside either within bone (~54%) or within the intracellular compartment (~45%), mostly muscle. Renal magnesium excretion plays an important role in maintaining physiological plasma magnesium levels. The body maintains the total magnesium concentration in blood plasma within narrow limits, 0.8 to 1.0 mM (1.8 to 2.2 mg/dL). Of this total, ~30% is protein-bound (Table 36-8). The remaining ~70% of total magnesium, which is filterable, is made up of two components. Less than 10% is complexed to anions such as phosphate, citrate, and oxalate, thus leaving ~60% of the total as free, ionized magnesium (Mg2+).
Table 36-8 Patterns of Hyperaminoacidurias
Disturbances of Mg2+ metabolism usually involve abnormal losses, and these occur most frequently during gastrointestinal malabsorption (see Chapter 45) and diarrhea, in the course of renal disease, and following the administration of diuretics. Clinical manifestations of Mg2+ depletion include neurologic disturbances such as tetany (especially when associated with hypocalcemia), cardiac arrhythmias, and increased peripheral vascular resistance. Conversely, increased Mg2+ intake may lower blood pressure and may reduce the incidence of hypertension. Severe hypermagnesemia, which may occur following excessive intake or renal failure, results in a toxic syndrome involving nausea, hyporeflexia, respiratory insufficiency, and cardiac arrest.
Normally, 5% or less of the filtered magnesium load appears in the urine (Fig. 36-17A). In contrast to the predominantly “proximal” reabsorption pattern of the major components of the glomerular filtrate, Mg2+ reabsorption occurs mainly along the TAL. In all segments, Mg2+ reabsorption is mainly paracellular.
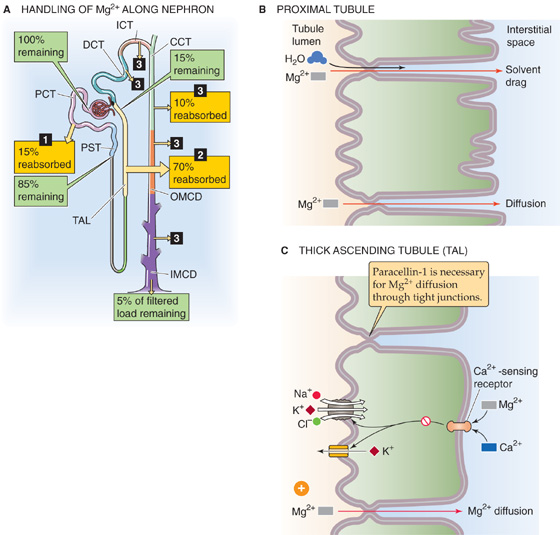
Figure 36-17 A to C, Magnesium handling by the kidney. In A, the numbered yellow boxes indicate the fraction of the filtered load that various nephron segments reabsorb. The DCT, initial collecting tubule (ICT), cortical collecting tubule (CCT), outer medullary collecting duct (OMCD), and IMCD together reabsorb 10% of the filtered load. The green boxes indicate the fraction of the filtered load that remains in the lumen after these segments. The values in the boxes are approximations. PCT, proximal convoluted tubule; PST, proximal straight tubule.
Normally, the proximal tubule reabsorbs only ~15% of the filtered magnesium load (Fig. 36-17B). Water reabsorption along the proximal tubule causes luminal [Mg2+] to double compared with the value in Bowman’s space, thereby establishing a favorable Mg2+ electrochemical gradient for passive, paracellular Mg2+ reabsorption. Solvent drag may also contribute to the paracellular reabsorption of Mg2+.
The TAL absorbs ~70% of the filtered magnesium load and is also the main site of control of Mg2+ transport (Fig. 36-17C). The driving force for paracellular Mg2+ reabsorption is the lumen-positive voltage of the TAL. In states of Mg2+ depletion (see later), the TAL may reabsorb some Mg2+ by a transcellular route.
In the TAL, a specific tight junction protein called claudin 16 or paracellin-1 (see Chapter 5) is necessary for paracellular Mg2+ reabsorption. A mutation of the paracellin-1 gene leads to an autosomal recessive disorder characterized by severe renal Mg2+ wasting. Mg2+ reabsorption along the DCT and the collecting tubules and ducts accounts for only ~10% of the filtered load.
Mg2+ Reabsorption Increases with Depletion of Mg2+ or Ca2+ or with Elevated PTH Levels Table 36-9 shows the site of action of factors modulating renal Mg2+ excretion.
Table 36-9 Factors Affecting Mg2+ Reabsorption Along the Nephron (See Note: Regulation of Mg2+Reabsorption)
Site |
Increase Reabsorption |
Decrease Reabsorption |
Proximal tubule |
Volume contraction |
Volume expansion |
Thick ascending limb |
PTH, calcitonin, glucagon, AVP |
Furosemide and related loop diuretics |
|
Low plasma [Mg2+] |
Mannitol |
|
Metabolic alkalosis |
High plasma [Mg2+] or [Ca2+] |
Distal convoluted tubule and collecting tubules/ducts |
PTH, calcitonin, glucagon, AVP, aldosterone, PGE2 |
High plasma [Mg2+] or [Ca2+] |
|
Low plasma [Mg2+] |
Metabolic acidosis |
|
Amiloride |
K+ or phosphate depletion |
Mg2+ Depletion In response to low plasma [Mg2+], the TAL increases its reabsorption of Mg2+, independent of Na+ and Ca2+ transport. This observation has been taken to suggest that a fraction of Mg2+ reabsorption may be transcellular and may involve an active transport step. Because [Mg2+]i in TAL cells is relatively low (0.5 to 0.6 mM) and the apical membrane voltage is cell negative, the apical electrochemical gradient favors passive Mg2+ uptake. It is likely that cation-selective channels, perhaps TRPV5/6, mediate apical Mg2+ uptake. The basolateral step for transcellular Mg2+ reabsorption is also uncertain, but it may include a unique ATP-dependent Mg2+ pump or an Na-Mg exchanger.
Hypermagnesemia and Hypercalcemia The kidney seems to discriminate poorly between increases in plasma [Mg2+] and [Ca2+]. Thus, each of these disturbances inhibits the reabsorption of both Mg2+ and Ca2+ in the TAL, thereby leading to increased urinary excretion of both Mg2+ and Ca2+. It is thought that high plasma [Mg2+] and [Ca2+] diminish Mg2+ reabsorption because each can bind to the Ca2+-sensing receptor.
Hormones PTH, the most important hormone for Mg2+ regulation, increases both proximal and distal Mg2+ reabsorption. When tested separately in hormone-depleted animals, AVP, glucagons, and calcitonin all stimulate Mg2+ reabsorption in the TAL, by acting through cAMP and PKA. Many of these hormones probably act by modulating passive Mg2+ movement through the paracellular pathway, either by changing NaCl transport and transepithelial voltage or by increasing paracellular permeability.
Diuretics In general, diuretics decrease Mg2+ reabsorption and thus enhance Mg2+ excretion. This statement is particularly true for diuretics targeting the TAL. Similar to their effects on Ca2+, loop diuretics act by depressing the lumen-positive voltage and thus the gradient for the passive, paracellular Mg2+ reabsorption. Osmotic diuretics, such as mannitol, also reduce Mg2+ reabsorption along the loop of Henle.
Books and Reviews
Chattopadhyay N, Brown EM (eds): Calcium-Sensing Receptor, pp 69-102. Boston: Kluwer Academic, 2002: 69-102.
Chillarón J, Roca R, Valencia A, et al: Heteromeric amino acid transporters: Biochemistry, genetics and physiology. Am J Physiol 2001; 281:F995-F1018.
Ciarimboli G, Schlatter E: Regulation of organic cation transport. Pflugers Arch Eur J Physiol 2005; 449:423-441.
Hoenderop JGJ, Bindels RJM: Epithelial Ca2+ and Mg2+ channels in health and disease. J Am Soc Nephrol 2005; 16:15-26.
Murer H, Hernando N, Forster I, Biber J: Proximal tubular phosphate reabsorption: Molecular mechanisms. Physiol Rev 2000; 80:1373-1409.
Sands JM: Mammalian urea transporters. Annu Rev Physiol 2003; 65:543-566.
Shayakul C, Hediger MA: The SLC14 gene family of urea transporters. Pflügers Arch 2004; 447:603-609.
Journal Articles
Austin JH, Stillman E, Van Slyke DD: Factors governing the excretion rate of urea. J Biol Chem 1921; 46:91-112.
Fei YJ, Kanai Y, Nussberger S, et al: Expression cloning of a mammalian proton-coupled oligopeptide transporter. Nature 1994; 368:563-566.
Forster IC, Wagner CA, Busch AE, et al: Electrophysiological characterization of the flounder type II Na/Pi cotransporter (NaPi-5) expressed in Xenopus laevis oocytes. J Membr Biol 1997; 160:9-25.
Hediger MA, Coady MJ, Ikeda IS, Wright EM: Expression cloning and cDNA sequencing of the Na+/glucose transporter. Nature 1987; 330:379-381.
Simon DB, Lu Y, Choate KA, et al: Paracellin-1, a renal tight junction protein required for paracellular Mg2+ resorption. Science 1999; 285:103-106.
You G, Smith CP, Kanai Y, et al: Expression cloning and characterization of the vasopressin-regulated urea transporter. Nature 1993; 365:844-847.
Zhao H, Shiue H, Palkon S, et al: Ezrin regulates NHE3 translocation and activation after Na+-glucose cotransport. Proc Natl Acad Sci U S A 2004; 101:9485-9490.