Frederick J. Suchy
After the skin, the liver and the brain (see Chapter 11) are the largest organs in the human body. The liver weighs between 1200 and 1500 g, representing 2% to 5% of body weight in the adult and ~4% to 5% in the newborn. The liver is strategically situated in the circulatory system to receive the portal blood that drains the stomach, small intestine, large intestine, pancreas, and spleen (see Fig. 17-3). In this position, the liver plays a key role in handling foodstuffs assimilated by the small intestine. However, the liver’s role is far more diverse; it serves as a chemical factory, an excretory system, an exocrine gland, and an endocrine gland.
A major function of the liver is to metabolize, detoxify, and inactivate both endogenous compounds (e.g., steroids and other hormones) and exogenous substances (e.g., drugs and toxins). In addition, by virtue of its large vascular capacity and abundance of phagocytes (Kupffer’s cells), the liver provides an important filtering mechanism for the circulation by removing foreign particulate matter, including bacteria, endotoxins, parasites, and aging red blood cells. Kupffer’s cells constitute 80% to 90% of the fixed macrophages of the reticuloendothelial system.
The liver has the capacity to convert important hormones and vitamins into a more active form. Examples include the initial hydroxylation of vitamin D and the deiodination of the thyroid hormone thyroxine (T4) to triiodothyronine (T3). Moreover, numerous enzymes in the liver process lipophilic chemicals into more polar, water-soluble metabolites, which are more readily excreted into bile.
Bile is a complex secretory product produced by the liver. Biliary secretion has two principal functions: (1) elimination of many endogenous and exogenous waste products from the body, such as bilirubin and cholesterol; and (2) promotion of digestion and absorption of lipids from the intestine. The composition of bile is modified significantly as a result of the absorptive and secretory properties of epithelial cells that line the intrahepatic and extrahepatic bile ducts. Moreover, bile solutes are further concentrated as bile is stored in the gallbladder.
The products of digested food, including carbohydrates, peptides, vitamins, and some lipids, are avidly extracted from portal blood by the liver. Depending on the metabolic requirements of the body, these substrates may be stored by the hepatocytes or released into the bloodstream either unbound (e.g., glucose) or associated with a carrier molecule (e.g., a triglyceride molecule complexed to a lipoprotein).
The liver also synthesizes—in a highly regulated fashion—many substances that are essential to the metabolic demands of the body. These substances include albumin and other plasma proteins, glucose, cholesterol, fatty acids for triglyceride biosynthesis, and phospholipids. The liver must provide a supply of substrates as fuels for other organs, particularly in the fasted state. For example, the liver produces ketone bodies, which can be used by the central nervous system during periods of fasting, thereby sparing ~50% of the amount of glucose that would otherwise be used by this tissue. Thus, the liver has a critical and unique role in the energy metabolism of all nonhepatic organs.
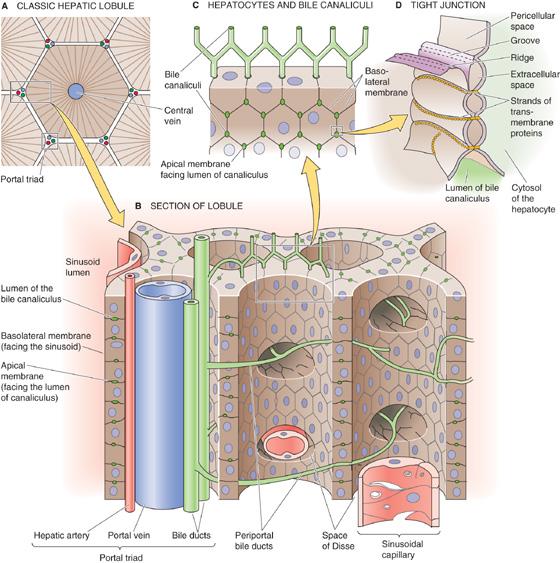
One way of looking at the organization of the liver is to imagine that a classic lobule is a hexagon in cross section (Fig. 46-1A), with a branch of the hepatic vein at its center and, at each of the six corners, triads composed of branches of the hepatic artery, portal vein, and bile duct. Hepatocytes account for ~80% of the parenchymal volume in human liver. Hepatocytes form an epithelium, one cell thick, that constitutes a functional barrier between two fluid compartments with differing ionic compositions: the tiny canalicular lumen containing bile and the much larger sinusoid containing blood (Fig. 46-1B). Moreover, hepatocytes significantly alter the composition of these fluids by vectorial transport of solutes across the hepatocyte. This vectorial transport depends critically on the polarized distribution of specific transport mechanisms and receptors that are localized to the apical membrane that faces the canalicular lumen and the basolateral membrane that faces the pericellular space between hepatocytes and the blood-filled sinusoid (Fig. 46-1B, C). As in other epithelia, the apical and basolateral membrane domains of hepatocytes are structurally, biochemically, and physiologically distinct.
Figure 46-1 Hepatocytes, sinusoids, and the intrahepatic bile system.
The space of Disse, or perisinusoidal space, is the extracellular gap between the endothelial cells lining the sinusoids and the basolateral membranes of the hepatocytes. These basolateral membranes have microvilli that project into the space of Disse to facilitate contact with the solutes in sinusoidal blood. The microvilli greatly amplify the basolateral membrane, which accounts for ~85% of the total surface area of the hepatocyte.
The bile canaliculi, into which bile is initially secreted, are formed by the apical membranes of adjoining hepatocytes. The apical membrane of the hepatocyte runs as a narrow belt that encircles and grooves into the polygonal hepatocyte (Fig. 46-1B, C). Two adjacent hepatocytes form a canaliculus that is ~1 μm in diameter by juxtaposing their groove-like apical membranes along their common face (i.e., one side of the polygon). Because a hepatocyte has many sides and a different neighbor on each side, the canaliculi form a chicken wire–like pattern along the contiguous surfaces of hepatocytes and communicate to form a three-dimensional tubular network. Although the apical membrane belt is very narrow (i.e., ~1 μm), its extensive microvillous structure amplifies its surface area so that the canalicular membrane constitutes as much as 15% of the total membrane surface area. Because of this high surface-to-volume ratio, the total apical surface area available for the movement of water and solutes in the human liver is in excess of 10.5 m2.
The seal that joins the apical membranes of two juxtaposed hepatocytes and that separates the canalicular lumen from the pericellular space—which is contiguous with the space of Disse—comprises several elements, including tight junctions (Fig. 2-25) and desmosomes (see Chapter 2). By virtue of their permeability and morphology, hepatic junctions can be classified as having an intermediate tightness, somewhat between tight epithelia (e.g., toad bladder) and leaky epithelia (e.g., proximal tubule). Specialized structures called gap junctions (see Chapter 4) allow functional communication between adjacent hepatocytes.
Hepatocytes do not have a true basement membrane, but rather they rest on complex scaffolding provided by the extracellular matrix in the space of Disse, which includes several types of collagens (I, III, IV, V, and VI), fibronectin, undulin, laminin, and proteoglycans. Cells are linked to the matrix through specific adhesion proteins on the cell surface. The extracellular matrix not only provides structural support for liver cells but also seems to influence and maintain the phenotypic expression of hepatocytes and sinusoidal lining cells.
Slightly more than 6% of the volume of the liver parenchyma is made up of cells other than hepatocytes, including endothelial cells (2.8%), Kupffer’s cells (2.1%), and stellate cells (fat-storing or Ito’s cells, 1.4%). The endothelial cells that line the vascular channels or sinusoids form a fenestrated structure with their bodies and cytoplasmic extensions. Plasma solutes, but not blood cells, can move freely into the space of Disse through pores, or fenestrae, in the endothelial cells. Some evidence indicates that the fenestrae may regulate access into the perisinusoidal space of Disse by means of their capacity to contract.
Kupffer’s cells are present within the sinusoidal vascular space. This population of fixed macrophages removes particulate matter from the circulation. Stellate cells are in the space of Disse and are characterized morphologically by the presence of large fat droplets in their cytoplasm. These cells play a central role in the storage of vitamin A, and evidence suggests that they can be transformed into proliferative, fibrogenic, and contractile myofibroblasts. On liver injury, these activated cells participate in fibrogenesis through remodeling of the extracellular matrix and deposition of type I collagen, which can lead to cirrhosis. (See Note: Kupffer Cells)
The blood supply to the liver has two sources. The portal vein contributes ~ 75% of the total circulation to the liver; the hepatic artery contributes the other 25% (Fig. 46-2A). Blood from portal venules and hepatic arterioles combines in a complex network of hepatic sinusoids (Fig. 46-2B). Blood from these sinusoids converges on terminal hepatic venules (or central veins), which, in turn, join to form the hepatic veins (Fig. 46-2C). Branches of the portal vein, hepatic artery, and a bile duct (i.e., the triad), as well as lymphatics and nerves, travel together as a portal tract.
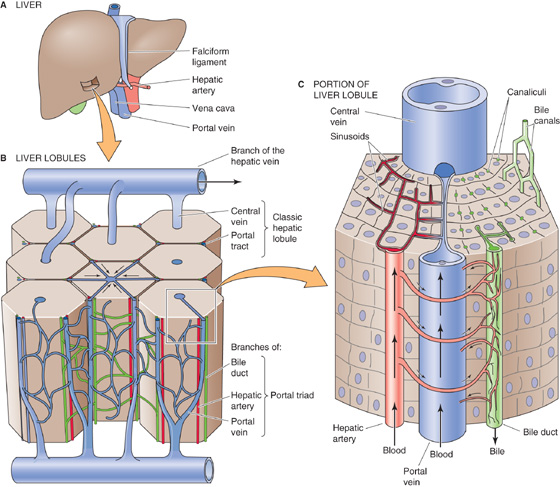
Figure 46-2 Blood supply to the liver.
The arterial supply for the bile ducts arises mainly from the right hepatic artery (Fig. 46-2C). These arterioles give rise to an extraordinarily rich plexus of capillaries that surround the bile ducts as they pass through the portal tracts. Blood flowing through this peribiliary plexus empties into the sinusoids by way of branches of the portal vein so that this blood may pick up solutes from the bile ducts and cycle them back to the hepatocytes. Thus, the peribiliary plexus may provide the means for modifying biliary secretions through the bidirectional exchange of compounds such as proteins, inorganic ions, and bile acids between the bile and blood within the portal tract.
The complex structure of the liver makes it difficult to define a single unit—something analogous to the nephron in the kidney—that is capable of performing the functions of the entire liver. One way of viewing the organization of the liver is depicted in Figures 46-1 and 46-2, in which we regard the central vein as the core of the classic hepatic lobule. Thus, the classic hepatic lobule (Fig. 46-3A) includes all hepatocytes drained by a single central vein, and it is bounded by two or more portal triads. Alternatively, we can view the liver as though the triad is the core of a portal lobule (Fig. 46-3B). Thus, the portal lobule includes all hepatocytes drained by a single bile ductule and is bounded by two or more central veins. A third way of viewing the liver is to group the hepatocytes according to their supply of arterial blood (Fig. 46-3C). Thus, the portal acinus is a small, three-dimensional mass of hepatocytes that are irregular in size and shape, with one axis formed by a line between two triads (i.e., high PO2) and another axis formed by a line between two central veins (i.e., low PO2).
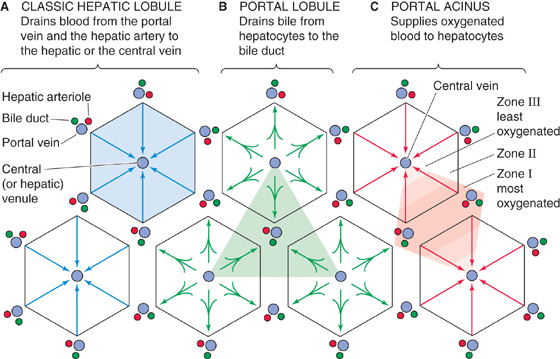
Figure 46-3 Zones in the acinus. A, The classic lobule includes all hepatocytes drained by a single central vein. At each corner of the hexagon are triads composed of branches of the hepatic artery, portal vein, and bile duct. B, The portal lobule includes all hepatocytes drained by a bile ductule. C, This organization emphasizes the arterial blood supply to the hepatocytes and thus the oxygenation gradient between a branch of the hepatic artery and branches of the hepatic vein (i.e., central vein). The periportal hepatocytes of zone I immediately surround the portal tract and thus have the highest PO2 and the most nutrients. They specialize in oxidative metabolism and certain other functions. The pericentral hepatocytes of zone III surround the central vein and have the lowest PO2. The zone III hepatocytes specialize in biotransformations and drug detoxification. Zone II is an intermediate zone between zones I and III.
Rappaport first proposed that a zonal relationship exists between cells that constitute the portal acini and their blood supply (Fig. 46-3C). Hepatocytes close to the vascular core formed by the terminal portal venule and terminal hepatic arteriole are perfused first and thus receive the highest concentrations of oxygen and solutes. These periportal hepatocytes are said to reside in zone I, and as a consequence of their location, they are the most resistant to the effects of circulatory compromise or nutritional deficiency. These cells are also more resistant to other forms of cellular injury and are the first to regenerate. Hepatocytes in the intermediate zone II and the most distal population of pericentral hepatocytes located near the terminal hepatic venule (central vein) in zone III are sequentially perfused with blood that is already modified by the preceding hepatocytes; thus, they are exposed to progressively lower concentrations of nutrients and oxygen. The exact boundaries of these zones are difficult to define.
The concept of zonal heterogeneity of liver function has evolved as a result of these differences in access to substrate. Because of the specialized microenvironments of cells in different zones, some enzymes are preferentially expressed in one zone or another (Table 46-1). For example, in zone I, oxidative energy metabolism with β oxidation, amino acid metabolism, ureagenesis, gluconeogenesis, cholesterol synthesis, and bile formation is particularly important. Localized in zone III are glycogen synthesis from glucose, glycolysis, liponeogenesis, ketogenesis, xenobiotic metabolism, and glutamine formation. Molecular techniques have allowed an even more precise definition of which hepatocytes express particular mRNA and proteins. For example, the enzyme glutamine synthetase is expressed exclusively in only one or two hepatocytes immediately adjacent to the hepatic venules. Hepatocytes of zone III also seem to be important for general detoxification mechanisms and the biotransformation of drugs. The zonal distribution of drug-induced toxicity manifested as cell necrosis may be attributed to zone III localization of the enzymatic pathways involved in the biotransformation of substrates by oxidation, reduction, or hydrolysis. Although it appears that each hepatocyte is potentially capable of multiple metabolic functions, the predominant enzymatic activity appears to result from adaptation to the microenvironment provided by the hepatic microcirculation. In some cases, it has been possible to reverse the zone I–to–zone III gradient of hepatocyte function by experimentally reversing the direction of blood supply (i.e., nutrient flow).
Table 46-1 Zonal Heterogeneity of Preferential Hepatocyte Function
Zone 1 |
Zone III |
Amino acid catabolism |
Glycolysis |
Gluconeogenesis |
Glycogen synthesis from glucose |
Glycogen degradation |
Liponeogenesis |
HMG-CoA reductase (cholesterol synthesis) |
Cholesterol 7α-hydroxylase (bile acid biosynthesis) |
Ureagenesis (all hepatocytes with exception of the last one or two rows encircling the hepatic venules) |
Ketogenesis |
Bile acid–dependent canalicular bile flow |
Glutamine synthesis |
Oxidative energy metabolism and probably β oxidation of fatty acids |
Bile acid–independent canalicular bile flow |
|
The adult human liver has more than 2 km of bile ductules and ducts, with a volume of ~20 cm3 and a macroscopic surface area of ~400 cm2. Microvilli at the apical surface magnify this area by ~5.5 fold.
As noted earlier, the canaliculi into which bile is secreted form a three-dimensional polygonal meshwork of tubes between hepatocytes, with many anastomotic interconnections (Fig. 46-1). From the canaliculi, the bile enters the small terminal bile ductules (i.e., canals of Hering), which have a basement membrane and in cross section are surrounded by three to six ductal epithelial cells or hepatocytes (Fig. 46-4A). The canals of Hering then empty into a system of perilobular ducts, which, in turn, drain into interlobular bile ducts. The interlobular bile ducts form a richly anastomosing network that closely surrounds the branches of the portal vein. These bile ducts are lined by a layer of cuboidal or columnar epithelium that has microvillous architecture on its luminal surface. The cells have a prominent Golgi apparatus and numerous vesicles, which probably participate in the exchange of substances among the cytoplasm, bile, and blood plasma through exocytosis and endocytosis.
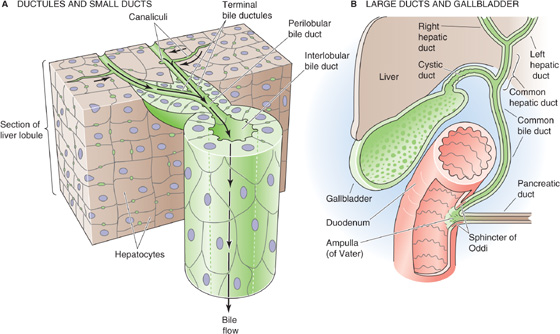
Figure 46-4 Structure of biliary tree. A, The bile canaliculi, which are formed by the apical membranes of adjacent hepatocytes, eventually merge with terminal bile ductules (canals of Hering). The ductules eventually merge into perilobular ducts, and then interlobular ducts. B, The interlobular ducts merge into septal ducts, lobar ducts, and eventually the right and left hepatic ducts, which combine as the common hepatic duct. The confluence of the common hepatic and the cystic ducts gives rise to the common bile duct. The common bile duct may merge with the pancreatic duct and form the ampulla of Vater before entering the duodenum, as shown in the figure, or it may have a completely independent lumen. In either case, a common sphincter—the sphincter of Oddi—simultaneously regulates flow out of the common bile duct and the pancreatic duct.
The interlobular bile ducts unite to form larger and larger ducts, first the septal ducts and then the lobar ducts, two hepatic ducts, and finally a common hepatic duct (Fig. 46-4B). Along the biliary tree, the biliary epithelial cells, or cholangiocytes, are similar in their fine structure except for size and height. However, emerging evidence suggests that they differ in their complement of transporters and receptors. Increasing emphasis has been placed on the absorptive and secretory properties of the biliary epithelial cells, properties that contribute significantly to the process of bile formation. As with other epithelial cells, cholangiocytes are highly cohesive, with the lateral plasma membranes of contiguous cells forming tortuous interdigitations. Tight junctions seal contacts between cells that are close to the luminal region and thus limit the exchange of water and solutes between plasma and bile.
The common hepatic duct emerges from the porta hepatis after the union of the right and left hepatic ducts. It merges with the cystic duct emanating from the gallbladder to form the common bile duct. In adults, the common bile duct is quite large, ~7 cm in length and ~0.5 to 1.5 cm in diameter. In most individuals, the common bile duct and the pancreatic duct merge before forming a common antrum known as the ampulla of Vater. At the point of transit through the duodenal wall, this common channel is surrounded by a thickening of both the longitudinal and the circular layers of smooth muscle, the so-called sphincter of Oddi. This sphincter constricts the lumen of the bile duct and thus regulates the flow of bile.
The gallbladder lies in a fossa beneath the right lobe of the liver. This distensible, pear-shaped structure has a capacity of 30 to 50 mL in adults. The absorptive surface of the gallbladder is enhanced by numerous prominent folds that are important for concentrative transport activity, as discussed later. The gallbladder is connected at its neck to the cystic duct, which empties into the common bile duct (Fig. 46-4B). The cystic duct maintains continuity with the surface columnar epithelium, lamina propria, muscularis, and serosa of the gallbladder. Instead of a sphincter, the gallbladder has, at its neck, a spiral valve (the valve of Heister) formed by the mucous membrane. This valve regulates flow into and out of the gallbladder.
The liver metabolizes an enormous variety of compounds that are brought to it by the portal and systemic circulations. These compounds include endogenous molecules (e.g., bile salts and bilirubin, which are key ingredients of bile) and exogenous molecules (e.g., drugs and toxins). The hepatocyte handles these molecules in four major steps (Fig. 46-5A): (1) the hepatocyte imports the compound from the blood across its basolateral (i.e., sinusoidal) membrane, (2) the hepatocyte transports the material within the cell, (3) the hepatocyte may chemically modify or degrade the compound intracellularly, and (4) the hepatocyte excretes the molecule or its product or products into the bile across the apical (i.e., canalicular) membrane. Thus, compounds are secreted in a vectorial manner through the hepatocyte.
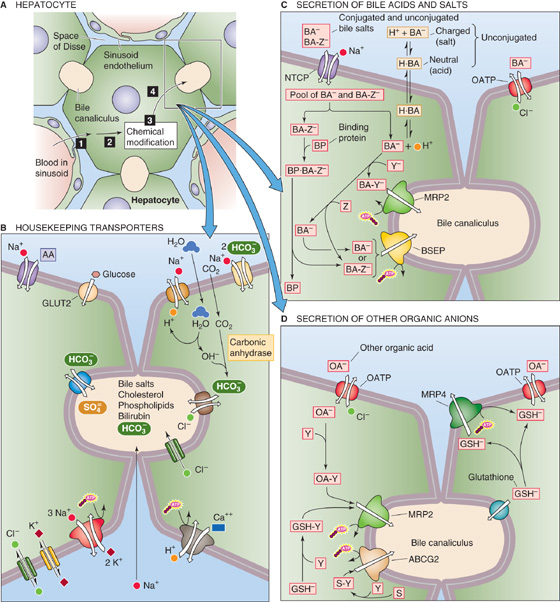
Figure 46-5 Transporters in hepatocyte. A, The hepatocyte can process compounds in four steps: (1) uptake from blood across the basolateral (i.e., sinusoidal) membrane, (2) transport within the cell, (3) control chemical modification or degradation, and (4) export into the bile across the apical (i.e., canalicular) membrane. B, The hepatocyte has a full complement of housekeeping transporters. C, Bile acids can enter the hepatocyte in any of several forms: the unconjugated salt (BA−); the neutral, protonated bile acid (H · BA); the bile salt conjugated to taurine or glycine (BA-Z−, where Z represents taurine or glycine). The three pathways for bile acid entry across the basolateral membrane are as follows: the Na+-driven transporter NTCP, which prefers BA-Z−, but also carries BA−; nonionic diffusion of H · BA; and an OATP. Binding proteins (BP) may ferry conjugated bile acids across the cytoplasm. Some bile acids are conjugated to sulfate or glucuronate (Y); these exit the cell across the canalicular membrane through the MRP2 transporter. Most bile acids are conjugated to glycine or taurine (Z) before their extrusion into the bile through the BSEP. D, Organic anions (including bile acids) may enter across the basolateral membrane through an OATP. After conjugation with sulfate or glucuronate (Y), these compounds may be extruded into the bile by MRP2. GSH synthesized in the hepatocyte, after conjugation to Y, can enter the canaliculus through MRP2. Unconjugated GSH can enter the canaliculus through an unidentified transporter. GSH can exit the hepatocyte across the basolateral membrane through an OATP. ABCG2, G2 member of ABC protein family.
Like other epithelial cells, the hepatocyte is endowed with a host of transporters that are necessary for basic housekeeping functions. To the extent that these transporters are restricted to either the apical or basolateral membrane, they have the potential of participating in net transepithelial transport. For example, the Na-K pump (see Chapter 3) at the basolateral membrane of hepatocytes maintains a low [Na+]i and high [K+]i (Fig. 46-5B). A basolateral, ATP-dependent Ca2+ pump maintains [Ca2+]i at an extremely low level, ~100 nM, as in other cells. The hepatocyte uses the inwardly directed Na+ gradient to fuel numerous active transporters, such as the Na-H exchanger, the Na/HCO3 cotransporter, and Na+-driven amino acid transporters. As discussed later, the Na+ gradient also drives one of the bile acid transporters. The hepatocyte takes up glucose through the GLUT2 facilitated diffusion mechanism (see Chapters 3 and 50), which is insensitive to regulation by insulin. (See Note: Hepatocyte Housekeeping Functions)
The resting basolateral membrane has a voltage (Vm) of −30 to −40 mV and is endowed with both K+ and Cl− channels. Basolateral K+ conductance helps to maintain a negative Vm; the resting Vm is considerably more positive than the equilibrium potential for K+ (EK) because of the presence of numerous “leak” pathways, such as the aforementioned electrogenic Na+-driven transporters, as well as channels for ions other than K+. For example, Cl− is passively distributed (i.e., ECl = Vm).
Bile Acids and Salts The primary bile acids are cholic acid and chenodeoxycholic acid, both of which are synthesized by hepatocytes, as described later in Figure 46-9. Other “secondary” bile acids form in the intestinal tract as bacteria dehydroxylate the primary bile acids. Because the pK values of the primary bile acids are near neutrality, most of the bile acid molecules are neutral; that is, they are bile acids (H · BA) and thus are not very water soluble. Of course, some of these molecules are deprotonated and hence are bile salts (BA−). The liver may conjugate the primary bile acids and salts to glycine or taurine (Z in Fig. 46-5C), as well as to sulfate or glucuronate (Y in Fig. 46-5C). Most of the bile acids that the liver secretes into the bile are conjugated, such as taurocholate (the result of conjugating cholic acid to taurine). These conjugated derivatives have a negative charge, and hence they, too, are bile salts (BA-Z− and BA-Y−). Bile salts are far more water soluble than the corresponding bile acids.
Because the small intestine absorbs some bile acids and salts, they appear in the blood plasma, mainly bound to albumin, and are presented to the hepatocytes for re-uptake. This recycling of bile acids, an example of enterohepatic circulation, is discussed later in Figure 46-13. Dissociation from albumin occurs before uptake. Surprisingly, the presence of albumin actually stimulates Na+-dependent taurocholate uptake, perhaps by increasing the affinity of the transporter for taurocholate.
Uptake of bile acids has been studied extensively and is mediated predominantly by an Na+-coupled transporter known as Na-taurocholate cotransporting polypeptide or NTCP (a member of the SLC10A1 family) (Fig. 46-5C). This transporter is a 50-kDa glycosylated protein, and it appears to have seven membrane-spanning segments. NTCP handles unconjugated bile acids, but it has a particularly high affinity for conjugated bile acids. In addition, NTCP can also transport other compounds, including neutral steroids (e.g., progesterone, 17β-estradiol sulfate), cyclic oligopeptides (e.g., amantadine and phalloidin), and a wide variety of drugs (e.g., verapamil, furosemide). As is the case for other transporters, NTCP activity is low in the fetus and neonate and increases with development. (See Note: Regulation of Na/Taurocholate Cotransport)
Although NTCP also carries unconjugated bile acids, as much as 50% of these unconjugated bile acids may enter the hepatocyte by passive nonionic diffusion (Fig. 46-5B). Because unconjugated bile acids are weak acids of the form
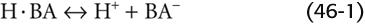
the neutral form can diffuse into the cell. Conjugation of bile acids enhances their hydrophilicity (taurine more so than glycine) and promotes dissociation of the proton from the side chain (i.e., lowering the pKa), thus raising the concentration of BA−. Both properties decrease the ability of bile acid to traverse membranes through passive nonionic diffusion.
Organic Anions The organic anion transporting polypeptides or OATPs (members of the SLC21 family) are a group of polyspecific membrane carriers (Fig. 46-5D) with partially overlapping substrate specificities for a wide range of amphipathic solutes, including bile salts, organic dyes, steroid conjugates, thyroid hormone, anionic oligopeptides, numerous drugs, toxins, and other xenobiotics. The driving force for OATP-mediated transport appears to be anion exchange for intracellular Cl−, glutathione, and possibly other substrates. Several human OATPs—including OATP-A, OATP-C, and OATP8—appear to be liver specific and transport bile acids and many other amphipathic substrates. Others—OATP-B, OATP-E, and OATP-F—are widely distributed and multispecific. Another ubiquitous OATP—PGT—transports prostanoids (e.g., prostaglandins E2 and F2α and thromboxane B2, but not arachidonic acid). Thus, basolateral uptake of bile acids into the hepatocyte is a complex process that involves both an Na+-dependent transporter (NTCP) and Na+-in dependent transporters (OATPs), as well as nonionic diffusion of unconjugated bile acids. (See Note: Organic Anion Transporters)
Bilirubin Senescent erythrocytes are taken up by macrophages in the reticuloendothelial system, where the degradation of hemoglobin leads to the release of bilirubin into the blood (Fig. 46-6A; see the box titled Jaundice). The mechanism by which hepatocytes take up unconjugated bilirubin remains controversial. As evidenced by yellow staining of the sclerae and skin in the jaundiced patient, bilirubin can leave the circulation and enter cells by diffusion. However, uptake of albumin-bound bilirubin by the isolated, perfused rat liver and isolated rat hepatocytes is faster than can occur by diffusion and is consistent with a carrier-mediated process. Electroneutral, electrogenic, and Cl−-dependent transport have been proposed (Fig. 46-6B). However, although one of the OATPs may account for a minor portion of uptake, the majority of transport occurs through proteins that have not been convincingly identified.
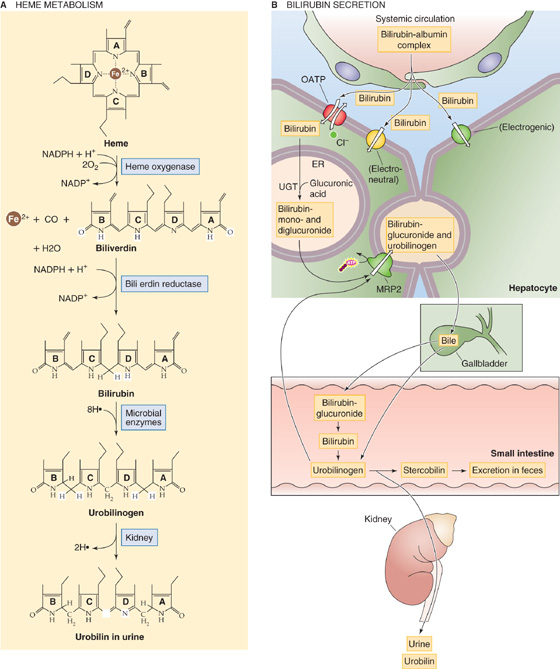
Figure 46-6 Excretion of bilirubin. A, Macrophages phagocytose senescent red blood cells and break the heme down to bilirubin, which travels in the blood, linked to albumin, to the liver. The conversion to the colorless urobilinogen occurs in the terminal ileum and colon, whereas the oxidation to the yellowish urobilin occurs in the urine. B, The hepatocyte takes up bilirubin across its basolateral membrane through an OATP and other unidentified mechanisms. The hepatocyte then conjugates the bilirubin with one or two glucuronic acid residues and exports this conjugated form of bilirubin into the bile. Bacteria in the terminal ileum and colon convert some of this bilirubin glucuronide back to bilirubin. This bilirubin is further converted to the colorless urobilinogen. If it remains in the colon, the compound is further converted to stercobilin, which is the main pigment of feces. If the urobilinogen enters the plasma and is filtered by the kidney, it is converted to urobilin and gives urine its characteristic yellow color.
Organic Cations The major organic cations transported by the liver are aromatic and aliphatic amines, including such important drugs as cholinergics, local anesthetics, and antibiotics, as well as endogenous solutes such as choline, thiamine, and nicotinamide (Fig. 46-7). The basolateral membrane of the hepatocyte contains several well-characterized transporters for organic cations. The polyspecific organic cation transporters OCT1 and OCT2 mediate electrogenic, facilitated diffusion (see Chapter 3) of small (type 1) organic cations, including many drugs, toxins, and endogenous compounds. These Na+-independent transporters, also expressed in the intestine and kidney, may reverse direction, depending on transmembrane concentration and voltage gradients. OATP-A mediates the uptake of bulky (type 2) organic cations. Physiological studies have identified proton-organic cation exchangers at both the basolateral and the canalicular membranes; however, the molecular identities of these transporters are currently unknown.
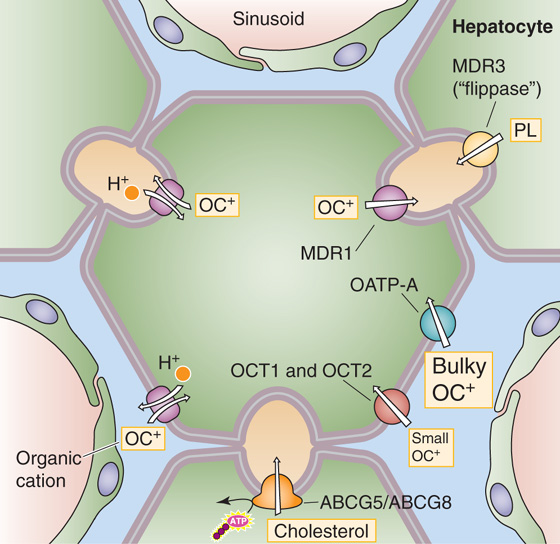
Figure 46-7 Excretion of organic cations and lipids. Small organic cations (type 1) enter the hepatocyte across the basolateral (i.e., sinusoidal) membrane through OCT1 and OCT2. Bulky organic cations (type 2) enter through OATP-A. Organic cations may also enter through a proton–organic cation exchanger. The organic cations exit the hepatocyte across the apical (i.e., canalicular) membrane through MDR1. MDR3 transports phospholipids (PL), and ABCG5/ABCG8 heterodimer transports cholesterol (C).
Neutral Organic Compounds This group of molecules is also taken up by an Na+-independent, energy-dependent process, but the nature of the driving force is not known. The best characterized substrate is ouabain, uptake of which is inhibited by other neutral steroids, such as cortisol, aldosterone, estradiol, and testosterone. OATP8 transports some of these compounds. We return to Figures 46-5, 46-6, and 46-7 later, when we discuss the movement of solutes into the bile canaliculus.
Bile Salts Some compounds traverse the cell while bound to intracellular “binding” proteins (Fig. 46-5C). The binding may serve to trap the molecule within the cell, or it may be involved in intracellular transport. For bile salts, three such proteins have been identified. In humans, the main bile acid–binding protein appears to be the hepatic dihydrodiol dehydrogenase, one of a large family of dehydrogenases, the catalytic and binding properties of which are organ and species specific. The two others are glutathione-S-transferase B and fatty acid–binding protein. Intracellular sequestration of bile salts by these proteins may serve an important role in bile acid transport or regulation of bile acid synthesis. Transcellular diffusion of bile salts bound to proteins can be detected within seconds after bile salts are applied to hepatocytes; this mechanism may be the primary mode of cytoplasmic transport under basal conditions. Free, unbound bile salts may also traverse the hepatocyte by rapid diffusion.
At high sinusoidal concentrations, hydrophobic bile acids may partition into membranes of intracellular vesicles. These conditions may also cause increased targeting of the vesicles to the canalicular membrane—that is, transcellular bile acid transport by a vesicular pathway.
Bilirubin After uptake at the basolateral membrane, unconjugated bilirubin is transported to the endoplasmic reticulum (ER), where it is conjugated to glucuronic acid (Fig. 46-6). Because the resulting bilirubin glucuronide is markedly hydrophobic, it was thought that intracellular transport was mediated by binding proteins such as glutathione transferase B. More recently, however, spontaneous transfer of bilirubin between phospholipid vesicles was observed to occur by rapid movement through the aqueous phase, in the absence of soluble proteins. Therefore, investigators have suggested that direct membrane-to-membrane transfer is the principal mode of bilirubin transport within the hepatocyte. In addition, the membrane-to-membrane flux of bilirubin is biased toward the membrane with the higher cholesterol/phospholipid ratio. Hence, the inherent gradient for cholesterol from the basolateral membrane to the ER membrane may direct the flux of bilirubin to the ER.
The liver is responsible for the metabolism and detoxification of many endogenous and exogenous compounds. Some compounds (e.g., proteins and other ligands) taken up by hepatocytes are completely digested within lysosomes. Specific carriers exist for the lysosomal uptake of sialic acid, cysteine, and vitamin B12. Clinical syndromes resulting from an absence of these carriers have also been identified. The lysosomal acid hydrolases cleave sulfates, fatty acids, and sugar moieties from larger molecules.
Hepatocytes handle other compounds by biotransformation reactions that usually occur in two phases. Phase I reactions represent oxidation or reduction reactions in large part catalyzed by the P-450 cytochromes. The diverse array of phase I reactions includes hydroxylation, dealkylation, and dehalogenation, among others. The common feature of all these reactions is that one atom of oxygen is inserted into the substrate. Hence these monooxygenases make the substrate a more polar compound, poised for further modification by a phase II reaction. For example, when the phase I reaction creates a hydroxyl group, the phase II reaction may increase the water solubility of ROH by conjugating it to a highly hydrophilic compound such as glucuronate, sulfate, or glutathione:
Finally, the conjugated compound is secreted into the blood or bile.
The major enzymes involved in phase I reactions are the P-450 cytochromes. Cytochromes are colored proteins that contain heme for use in the transfer of electrons. Some cytochromes—not the P-450 system—are essential for the electron transport events that culminate in oxidative phosphorylation in the mitochondria. The P-450 cytochromes, so named because they absorb light at 450 nm when bound to CO, are a diverse, but related group of enzymes that reside mainly in the ER and typically catalyze hydroxylation reactions. More than 150 P-450 isoforms have been characterized.
In this text, we encounter P-450 oxidases in two sets of organs. In cells that synthesize steroid hormones—the adrenal cortex (see Chapter 49), testes (see Chapter 53), and ovary and placenta (see Chapters 54 and 55)—P-450 oxidases are localized either in the mitochondria or in the ER, where they catalyze various steps in steroidogenesis. In the liver, these enzymes are located in the ER, where they catalyze a vast array of hydroxylation reactions involving the metabolism of drugs and chemical carcinogens, bile acid synthesis, and the activation and inactivation of vitamins. The same reactions occur in other tissues, such as the intestines and the lungs.
Hepatic microsomal P-450 enzymes have similar molecular weights (48 to 56 kDa). The functional protein is a holoenzyme that consists of an apoprotein and a heme prosthetic group. The apoprotein region confers substrate specificity, which differs among the many P-450 enzymes. These substrates include RH moieties that are as wide ranging as the terminal methyl group of fatty acids, carbons in the rings of steroid molecules, complex heterocyclic compounds, and phenobarbital. In general, phase I processes add or expose a functional group, a hydroxyl group in the case of the P-450 oxidases, which renders the molecule reactive with phase II enzymes. The metabolic products of phase I may be directly excreted, but more commonly, because of only a modest increment in solubility, further metabolism by phase II reactions is required.
In phase II, the hepatocyte conjugates the metabolites generated in phase I to produce more hydrophilic compounds, such as glucuronides, sulfates, and mercapturic acids. These phase II products are readily secreted into the blood or bile. Conjugation reactions are generally considered to be the critical step in detoxification. Either a defect in a particular enzyme, which may result from a genetic defect, or saturation of the enzyme with excess substrate may result in a decrease in the overall elimination of a compound. One example is the gray syndrome, a potentially fatal condition that occurs after the administration of chloramphenicol to newborns who have low glucuronidation capacity. Infants have an ashen gray appearance and become weak and apathetic, and complete circulatory collapse may ensue.
Jaundice
Jaundice denotes a yellowish discoloration of body tissues, most notable in the skin and sclera of the eyes. The condition is caused by an accumulation of bilirubin in extracellular fluid, either in free form or after conjugation. Bilirubin is a yellow-green pigment that is the principal degradation product of heme (Fig. 46-6A), the iron-binding portion of hemoglobin. The metabolism of hemoglobin of senescent red cells accounts for 65% to 80% of total bilirubin production. Because of avid extraction and conjugation by the liver, the normal plasma concentration of bilirubin, which is mostly of the unconjugated variety, is ~0.5 mg/dL or lower. The skin or eyes may begin to appear jaundiced when the bilirubin level rises to 1.5 to 3 mg/dL.
Hemoglobin released into the circulation is phagocytized by macrophages throughout the body and is split into globin and heme. Cleavage of the heme ring releases both free iron, which travels in the blood by transferrin, and a straight chain of 4-pyrrole nuclei called biliverdin (Fig. 46-6A), which the cell rapidly reduces to free bilirubin. This form of bilirubin is often referred to as free or unconjugated bilirubin (we discuss its conjugation later). After it enters the circulation, unconjugated bilirubin binds reversibly to albumin and travels to the liver, which avidly removes it from the plasma (Fig. 46-6B). The hepatocyte esterifies the bilirubin, which is extremely insoluble, with glucuronic acid to form the monoconjugated and diconjugated derivatives. Conjugated bilirubin is more soluble and thus is suitable for excretion into bile, but it cannot be absorbed by the biliary or intestinal epithelia.
Jaundice occurs under several circumstances. Increased destruction of red blood cells or hemolysis may occur with rapid release of free, unconjugated bilirubin into the circulation. Unconjugated hyperbilirubinemia occurs commonly in neonates not only because of increased production of heme but also because of the immaturity of the pathways for glucuronidation in the liver. Obstruction of the bile ducts or damage to the liver may also result in jaundice. In this setting, conjugated bilirubin produced by the liver cannot be excreted in bile and consequently refluxes back into the systemic circulation. Therefore, most of the bilirubin in plasma is the highly soluble conjugated bilirubin, a small amount of which can be filtered by the kidneys, rather than the poorly soluble free form of bilirubin, which is mostly bound to albumin and is not excreted by the kidneys. Thus, in obstructive jaundice, conjugated bilirubin imparts a dark yellow color to the urine. Measurement of free and conjugated bilirubin in serum serves as a sensitive test for detecting liver disease.
Under normal conditions, approximately half of the bilirubin reaching the intestinal lumen is metabolized by bacteria into the colorless urobilinogen (Fig. 46-6A). The intestinal mucosa reabsorbs ~20% of this soluble compound into the portal circulation. The liver then extracts most of the urobilinogen and re-excretes it into the gastrointestinal tract. The kidneys excrete a small fraction (~20% of daily urobilinogen production) into the urine. Urobilinogen may be detected in urine by using a clinical dipstick test. Oxidation of urobilinogen yields urobilin, which gives urine its yellow color. In the feces, metabolism of urobilinogen yields stercobilin, which contributes to the color of feces. In obstructive jaundice, no bilirubin reaches the intestine for conversion into urobilinogen, and therefore no urobilinogen appears in the blood for excretion by the kidney. As a result, tests for urobilinogen in urine are negative in obstructive jaundice. Because of the lack of stercobilin and other bile pigments in obstructive jaundice, the stool becomes clay colored.
Hepatocytes use three major conjugation reactions, as follows:
1. Conjugation to glucuronate. The uridine diphosphate glucuronosyl transferases (UGTs), which reside in the smooth ER (SER) of the liver, are divided into two families based on their substrate specificity. Family 1 consists of at least four members and is encoded by genes that are located on chromosome 2. These UGTs catalyze the conjugation of glucuronic acid with phenols or bilirubin (Fig. 46-6B). Family 2 contains at least five UGTs that are encoded by genes on chromosome 4. These UGTs catalyze the glucuronidation of steroids or bile acids. Because family 1 UGTs are essential for the dual conjugation of bilirubin (Fig. 46-6B) and because only conjugated bilirubin can be excreted in bile, congenital absence of bilirubin UGT activity results in jaundice from birth and bilirubin encephalopathy, as seen in patients with Crigler-Najjar type I syndrome.
2. Conjugation to sulfate. The sulfotransferases—which are located in the cytosol rather than in the SER—catalyze the sulfation of steroids, catechols, and foreign compounds such as alcohol and metabolites of carcinogenic hydrocarbons. Their substrate specificity is greater than that of the UGTs. The different cellular localization of these two groups of enzymes suggests that they act cooperatively rather than competitively. In general, sulfates are not toxic and are readily eliminated, with the exception of sulfate esters of certain carcinogens.
3. Conjugation to glutathione. Hepatocytes also conjugate a range of compounds to reduced glutathione (GSH) for excretion and later processing in either the bile ducts or kidney (Fig. 46-8). Glutathione is a tripeptide composed of glutamate γ-linked to cysteine, which, in turn, is α-linked to glycine. The liver has the highest concentration of glutathione (~5 mM), with ~90% found in the cytoplasm and 10% in the mitochondria. Glutathione-S-transferases, which are mainly cytosolic, catalyze the conjugation of certain substrates to the cysteine moiety of GSH. Substrates include the electrophilic metabolites of lipophilic compounds (e.g., epoxides of polycyclic aromatic hydrocarbons), products of lipid peroxidation, and alkyl and aryl halides. In some cases, the conjugates are then secreted into bile and are further modified by removing the glutamyl residue from the glutathione by γ-glutamyl transpeptidase on the bile duct epithelial cell. The fate of glutathione-S-conjugates in bile is largely unknown. Some (e.g., the leukotrienes) undergo enterohepatic circulation. In other cases, the glutathione conjugates are secreted into plasma and are filtered by the kidney, where a γ-glutamyl transpeptidase on the proximal tubule brush border again removes the glutamyl residue. Next, a dipeptidase removes the glycine residue to produce a cysteine-S-conjugate. The cysteine-S-conjugate is either excreted in the urine or is acetylated in the kidney or liver to form a mercapturic acid derivative, which is also excreted in the urine.
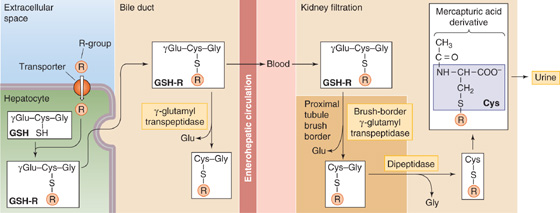
Figure 46-8 Conjugation to GSH and formation of mercapturic acids. The hepatocyte detoxifies compounds by chemically linking (conjugating) them to small molecules, such as GSH, which is a tripeptide. The first step is for glutathione-S-transferase to couple the target compound (R) to the S on the cysteine residue of GSH. After MRP2 transports this GSH conjugate into the canalicular lumen (see Fig. 46-5D), a γ-glutamyl transpeptidase may remove the terminal glutamate residue. Alternatively, the conjugate may reach the blood and may be filtered by the kidney, where a γ-glutamyl transpeptidase at the brush border and a dipeptidase generate a cysteine derivative of R. Acetylation yields the mercapturic acid derivative, which appears in the urine.
Although glutathione conjugation is generally considered a detoxification reaction, several such conjugates undergo activation into highly reactive intermediates.
Other Conjugations Other forms of conjugation include methylation (e.g., catechols, amines, and thiols), acetylation (e.g., amines and hydrazines), and conjugation (e.g., bile acids) with amino acids such as taurine, glycine, or glutamine. The involvement of multiple enzyme systems in these detoxification reactions facilitates the rapid removal of toxic species and provides alternative pathways in the event of failure of the preferred detoxification mechanism.
The liver and intestine express a nuclear receptor (see Chapter 4) called the steroid and xenobiotic receptor (SXR). A chemically diverse array of substances binds to this relatively “promiscuous” transcription factor, which then binds to response elements on DNA and alters the expression of multiple drug-metabolizing enzymes as well as transporters that excrete drug metabolites. Thus, SXR serves as a master regulator of xenobiotic metabolism. Enzymes upregulated by SXR include the phase I drug-metabolizing enzymes of the P-450 family, such as CYP3A, which metabolizes more than 50% of all drugs in humans. SXR also activates the phase II enzyme glutathione-S-transferase, which is critical for catalyzing conjugation of many substrates to glutathione. SXR also upregulates the multidrug resistance transporter MDR1 (see later). Although these pathways are for the most part hepatoprotective, a particular compound may elicit SXR-mediated alterations in CYP3A activity that may profoundly influence the metabolism of another drug—perhaps thereby compromising the therapeutic efficacy of that drug or enhancing the production of a toxic metabolite. Another nuclear receptor—the constitutive androstane receptor (CAR)—is also an important regulator of drug metabolism. CAR regulates all the components of bilirubin metabolism, including uptake (possibly through OATP), conjugation (UTG1A1), and excretion (through multidrug resistance-associated protein 2 [MRP2], as discussed later).
At the apical membrane, the transport of compounds is generally unidirectional, from cell to canalicular lumen. An exception is certain precious solutes, such as amino acids and adenosine, which are reabsorbed from bile by Na+-dependent secondary active transport systems.
Bile Salts Bile salt transport from hepatocyte to canalicular lumen (Fig. 46-5C) occurs through an ATP-dependent transporter called the bile salt export pump (BSEP). The ABCB11 member of the ABC (ATP-binding cassette) protein family (see Table 5-6 on p. 124), BSEP has a very high affinity for bile salts (taurochenodeoxycholate > taurocholate > tauroursodeoxycholate > glycocholate). The electrical charge of the side chain is an important determinant of canalicular transport inasmuch as only negatively charged bile salts are effectively excreted. Secretion of bile salts occurs against a significant cell-to-canaliculus concentration gradient, which may range from 1 : 100 to 1 : 1000. Mutations in the BSEP gene can, in children, cause a form of progressive intrahepatic cholestasis that is characterized by extremely low bile acid concentrations in the bile.
Organic Anions Organic anions that are not bile salts move from the cytoplasm of the hepatocyte to the canalicular lumen through MRP2, the ABCC2 member of the ABC protein family (see Table 5-6) (Fig. 46-5D). MRP2 is electrogenic, ATP dependent, and has a broad substrate specificity—particularly for divalent, amphipathic, phase II conjugates with glutathione, glucuronide, glucuronate, and sulfates. Its substrates include bilirubin diglucuronide, sulfated bile acids, glucuronidated bile acids, and several xenobiotics. In general, transported substrates must have a hydrophobic core and at least two negative charges separated by a specific distance. MRP2 is critical for the transport of GSH conjugates across the canalicular membrane into bile. Although MRP2 has a low affinity for GSH, functional studies suggest that other mechanisms for GSH transport exist. Animal models of defective MRP2 exhibit hyperbilirubinemia, which corresponds phenotypically to the Dubin-Johnson syndrome in humans. Another canalicular efflux pump for sulfated conjugates is the human ABC protein ABCG2, which transports estrone-3-sulfate (see Fig. 55-10) and dehydroepiandrosterone sulfate (see Fig. 54-5)—breakdown products of sex steroids. Other anions, such as HCO−3 and SO4−2, are excreted by anion exchangers. (See Note: ABCC2 (MRP2))
Organic Cations Biliary excretion of organic cations is poorly understood. With the exception of transport that is mediated by the MDR proteins such as BSEP (discussed earlier), the hepatic MDR proteins belong to the ABC family of transporters (see Table 5-6). MDR1 (ABCB1) is present in the canalicular membrane, where it mediates the excretion of some organic cations into the bile canaliculus (Fig. 46-7). The nomenclature of the MDRs is especially confusing because different and conflicting numbering systems have been used for different species; we use the human numbering system. MDR1 secretes bulky organic cations, including xenobiotics, cytotoxins, anticancer drugs, and other drugs (e.g., colchicine, quinidine, verapamil, cyclosporine).
Other organic cations appear to be secreted into the canaliculus by a transport process driven by a pH gradient (Fig. 46-7). The presence of an electroneutral H-organic cation exchanger has been demonstrated at the canalicular membrane. However, the importance of this process is uncertain because major H+ gradients probably do not exist in the bile canaliculus. In some cases, it appears that organic cations passively move across the apical membrane into the canaliculus, where they are sequestered by biliary micelles.
Biliary Lipids Phospholipid is a major component of bile. MDR3 (ABCB4) is a “flippase” that promotes the active translocation of phosphatidylcholine (PC) from the inner to the outer leaflet of the canalicular membrane. Bile salts then extract the PC from the outer leaflet so that the PC becomes a component of bile, where it participates in micelle formation. Indeed, in humans with an inherited deficiency of MDR3, progressive liver disease develops, characterized by extremely low concentrations of phospholipids in the bile.
Bile is also the main pathway for elimination of cholesterol. A heterodimer composed of the “half” ABC transporters ABCG5 and ABCG8 is located on the canalicular membrane. This transporter is responsible for the secretion of cholesterol into bile. Although the mechanism is uncertain, the ABCG5/ABCG8 complex may form a channel for cholesterol translocation or alternatively may undergo a conformational change following ATP hydrolysis, thus flipping a cholesterol molecule into the outer membrane leaflet in a configuration favoring release into the canalicular lumen. Mutations in the genes encoding either of the two ABC monomers lead to sitosterolemia, a disorder associated with defective secretion into the bile of dietary sterols, increased intestinal absorption of plant and dietary sterols, hypercholesterolemia, and early-onset atherosclerosis.
The hepatocyte takes up macromolecules, such as plasma proteins, from the blood plasma through endocytosis, transports these molecules across the cytoplasm, and then secretes them into the bile through exocytosis. Three forms of endocytosis have been identified in the basolateral (sinusoidal) membrane: fluid-phase endocytosis, adsorptive endocytosis, and receptor-mediated endocytosis. (See Note: Protein Transport by Hepatocytes)
Fluid-phase endocytosis involves the uptake of a small amount of extracellular fluid, with its solutes, and is a result of the constitutive process of membrane invagination and internalization (see Chapter 2). The process is nondiscriminatory and inefficient. Adsorptive endocytosis involves nonspecific binding of the protein to the plasma membrane before endocytosis, and it results in more efficient protein uptake. Receptor-mediated endocytosis is quantitatively the most important mechanism for the uptake of macromolecules (see Chapter 2). After endocytosis, the receptor recycles to the plasma membrane, and the ligand may be excreted directly into bile by exocytosis or delivered to lysosomes for degradation. Receptor-mediated endocytosis is involved in the hepatic removal from the blood of proteins such as insulin, polymeric immunoglobulin A (IgA), asialoglycoproteins, and epidermal growth factor.
The formation of bile occurs in three discrete steps. First, the hepatocytes actively secrete bile into the bile canaliculi. Second, intrahepatic and extrahepatic bile ducts not only transport this bile but also secrete into it a watery, HCO−3-rich fluid. These first two steps may produce ~900 mL/day of so-called hepatic bile (Table 46-2). Third, between meals, approximately half the hepatic bile—perhaps 450 mL/day—is diverted to the gallbladder, which stores the bile and isosmotically removes salts and water. The result is that the gallbladder concentrates the key remaining solutes in bile fluid—bile salts, bilirubin, cholesterol, and lecithin—by 10-to 20-fold. The 500 mL/day of bile that reaches the duodenum through the ampulla of Vater is thus a mixture of relatively “dilute” hepatic bile and “concentrated” gallbladder bile.
Table 46-2 Composition of Bile
Parameter |
Hepatic Bile |
Gallbladder Bile |
pH |
7.5 |
6.0 |
Na+ (mM) |
141-165 |
220 |
K+ (mM) |
2.7-6.7 |
14 |
Ca2+ (mM) |
1.2-3.2 |
15 |
Cl− (mM) |
77-117 |
31 |
HCO−3 (mM) |
12-55 |
19 |
Total phosphorus (g/L) |
0.15 |
1.4 |
Bile acids (g/L) |
3-45 |
32 |
Total fatty acids (g/L) |
2.7 |
24 |
Bilirubin (g/L) |
1-2 |
3 |
Phospholipids (g/L) |
1.4-8.1 |
34 |
Cholesterol (g/L) |
1-3.2 |
6.3 |
Proteins (g/L) |
2-20 |
4.5 |
Data from Boyer JL: In Andreoli TE, Hoffman JF, Fanestil DD, Schultz SG (eds): Physiology of Membrane Disorders. New York: Plenum, 1986.
The first step in bile formation cannot be ultrafiltration because the hydrostatic pressure in the canaliculi is significantly higher than the sinusoidal perfusion pressure. This situation is in marked contrast to glomerular filtration by the kidney (see Chapter 33), which relies predominantly on passive hydrostatic forces for producing the fluid in Bowman’s space. Instead, bile formation is an active process. It is sensitive to changes in temperature and to metabolic inhibitors. Bile formation by hepatocytes requires the active, energy-dependent secretion of inorganic and organic solutes into the canalicular lumen, followed by the passive movement of water. This movement of water through the tight junctions between hepatocytes carries with it other solutes by solvent drag (see Chapter 19). Canalicular bile is an isosmotic fluid; thus, the intercellular junctions allow the passage of water and small ions. The canalicular membrane expresses the water channel aquaporin 8 (AQP8). Under basal conditions, AQP8 is predominantly localized to intracellular vesicles but redistributes to the canalicular domain with stimulation by the secretagogue cAMP, thereby increasing apical water permeability. Thus, water transport into the bile canaliculus follows both paracellular and transcellular pathways. Further down the biliary tree (i.e., ducts and gallbladder), where the pore size of paracellular junctions is significantly smaller, solvent drag is not as important. Organic solutes do not readily enter bile distal to the canaliculi.
Bile has two important functions: (1) bile provides the sole excretory route for many solutes that are not excreted by the kidney, and (2) secreted bile salts and acids are required for normal lipid digestion and absorption (see Chapter 44).
Both hepatic bile and gallbladder bile are complex secretions that are isosmotic with plasma (~300 mosmol/kg) and consist of water, inorganic electrolytes, and a variety of organic solutes, including bilirubin, cholesterol, fatty acids, and phospholipid (Table 46-2). The predominant cation in bile is Na+, and the major inorganic anions are Cl− and HCO−3. Solutes whose presence in bile is functionally important include micelle-forming bile acids, phospholipids, and IgA.
Bile acids promote dietary lipid absorption through their micelle-forming properties (see Chapter 45). As shown in Figure 45-9, hepatocytes synthesize the so-called primary bile acids—cholic acid and chenodeoxycholic acid—from cholesterol. Indeed, biliary excretion of cholesterol and conversion of cholesterol to bile acids are the principal routes of cholesterol excretion and catabolism, thus making bile formation pivotal for total body cholesterol balance. The first step in this conversion is catalyzed by cholesterol 7α-hydroxylase (CYP7α1), a specific cytochrome P-450 enzyme located in the SER. As we see later, secondary bile acids are the products of bacterial dehydroxylation in the terminal ileum and colon. After being absorbed and returning to the liver (enterohepatic circulation, discussed later), these secondary bile acids may also undergo conjugation. Figure 46-9 shows typical examples of conjugation reactions. Phospholipids in bile help to solubilize cholesterol as well as diminish the cytotoxic effects of other bile acids on hepatocytes and bile duct cells. IgA inhibits bacterial growth in bile. (See Note: Cholesterol 7 α-Hydroxylase)
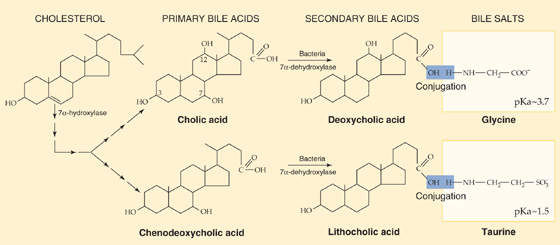
Figure 46-9 Synthesis of bile acids. The liver converts cholesterol to the primary bile acids—cholic acid and chenodeoxycholic acid—in a series of 14 reactions occurring in four different cellular organelles. The first reaction is the 7α-hydroxylation of cholesterol. In addition, the action of bacteria in the terminal ileum and colon may de hydroxylate bile acids, thus yielding the secondary bile acids deoxycholic acid and lithocholic acid. The hepatocytes conjugate most of the primary bile acids to small molecules such as glycine and taurine (not shown) before secreting them into the bile. In addition, those secondary bile acids that return to the liver through the enterohepatic circulation may also be conjugated to glycine or taurine, as shown in the figure. The liver may also conjugate some primary and secondary bile acids to sulfate or glucuronate (not shown).
Excretory or waste products found in bile include cholesterol, bile pigments, trace minerals, plant sterols, lipophilic drugs and metabolites, antigen-antibody complexes, and oxidized glutathione. Bile is also the excretory route for compounds that do not readily enter the renal glomerular filtrate, either because they are associated with proteins such as albumin or because they are associated with formed elements in blood. Although these compounds are generally lipophilic, they also include the heavy metals. Some bile acids (e.g., the trihydroxy bile acid cholic acid) are only partly bound to serum albumin and may therefore enter the glomerular filtrate. However, they are actively reabsorbed by the renal tubule. In health, bile acids are virtually absent from the urine.
Total bile flow is the sum of the bile flow from hepatocytes into the canaliculi (canalicular flow) and the additional flow from cholangiocytes into the bile ducts (ductular flow). In most species, the rate of canalicular bile secretion (i.e., milliliters per minute) increases more or less linearly with the rate of bile acid secretion (i.e., moles per minute). Canalicular bile flow is the sum of two components (Fig. 46-10): (1) a “constant” component that is independent of bile acid secretion (bile acid–independent flow) and (2) a rising component that increases linearly with bile acid secretion (bile acid–dependent flow). In humans, most of the canalicular bile flow is bile acid dependent. If we now add the ductular secretion, which is also “constant,” we have the total bile flow in Figure 46-10. We discuss the canalicular secretion in the remainder of this section and ductular secretion in the following section.
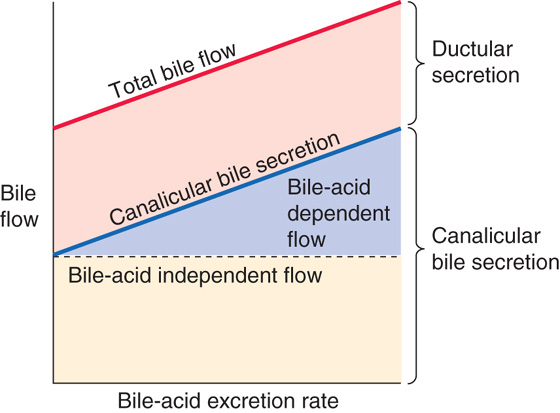
Figure 46-10 Components of bile flow.
Bile Acid–Independent Flow in the Canaliculi The secretion of organic compounds probably provides the major driving force for bile acid–independent flow. For example, glutathione, present in bile in high concentrations, may generate a potent osmotic driving force for canalicular bile formation. (See Note: Contribution of Inorganic Solutes to Bile Acid-Independent Flow)
Bile Acid–Dependent Flow in the Canaliculi The negatively charged bile salts in bile are in a micellar form and are—in a sense—large polyanions. Thus, they are effectively out of solution and have a low osmotic activity coefficient. However, the positively charged counter ions accompanying these micellar bile acids are still in aqueous solution and may thus represent the predominant osmotic driving force for water movement in bile acid–dependent flow. If one infuses an animal with a nonphysiological bile acid that does not form micelles or one that forms micelles only at a rather high concentration, the osmotic activity will be higher, and thus the exogenous bile acid will be more effective in producing bile acid–dependent flow. In other words, the slope of the blue bile acid–dependent line in Figure 46-10 would be steeper than for physiological bile acids.
Bile flow does not always correlate with the osmotic activity of the bile acid. In some cases, bile acids increase electrolyte and water flux by other mechanisms, such as by stimulating Na+-coupled cotransport mechanisms or by modulating the activity of other solute transporters. For example, the bile acid ursodeoxycholic acid produces a substantial increase in bile flow by markedly stimulating biliary HCO−3 excretion.
Bile acids in the lumen may also stimulate the secretion of other solutes by trapping them in the lumen. These solutes include bilirubin and other organic anions, as well as lipids such as cholesterol and phospholipids. The mixed micelles formed by the bile acids apparently sequester these other solutes, thus lowering their effective luminal concentration and favoring their entry. Therefore, excretion of cholesterol and phospholipid is negligible when bile acid output is low, but it increases and approaches maximum values as bile acid output increases.
As discussed in the previous section, biliary epithelial cells, or cholangiocytes, are the second major source of the fluid in hepatic bile. Experimentally, one can isolate cholangiocytes from normal liver or from the liver of experimental animals in which ductular hyperplasia has been induced by ligating the bile duct. These cholangiocytes have numerous transporters (Fig. 46-11), including the apical Cl-HCO3 exchanger AE2, 6 of the 11 known human aquaporins (AQPs), and several apical Cl− channels, including the cystic fibrosis transmembrane regulator (CFTR). In a mechanism that may be similar to that in pancreatic duct cells, the Cl-HCO3 exchanger, in parallel with the Cl− channels for Cl− recycling, can secrete an HCO−3-rich fluid (see Chapter 42). AQP1, CFTR, and AE2 co-localize to intracellular vesicles in cholangiocytes; secretory agonists cause all three to co-redistribute to the apical membrane. (See Note: Bicarbonate Secretion by Cholangiocytes)
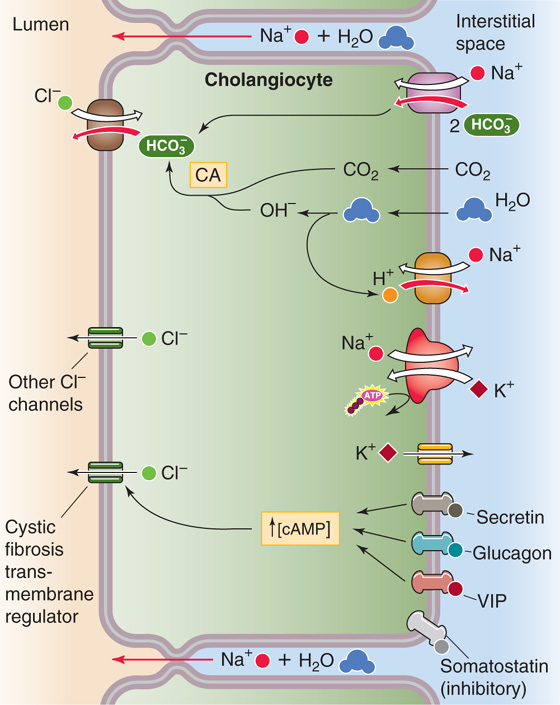
Figure 46-11 Secretion of an HCO−3-rich fluid by cholangiocytes. The apical step of HCO−3 secretion by the duct cell is mediated by a Cl-HCO3 exchanger. The Cl− recycles back to the lumen through Cl− channels, such as CFTR. The basolateral step of HCO−3 secretion probably is mediated in part by the uptake of HCO−3 through an electrogenic Na/HCO3 cotransporter. The uptake of CO2, combined with the extrusion of H+ through an Na-H exchanger and an H+ pump, generates the rest of the HCO−3 through carbonic anhydrase (CA). Secretin, glucagon, VIP, and gastrin-releasing peptide (GRP) all are choleretics. Somatostatin either enhances fluid absorption or inhibits secretion.
A complex network of hormones, mainly acting through cAMP, regulates cholangiocyte secretory function. Secretin receptors (see Chapter 42) are present on the basolateral membranes of cholangiocytes, a finding that explains why secretin produces water-rich choleresis—that is, bile rich in HCO−3 (i.e., alkaline) but diluted in bile acids. Similarly, the hormones glucagon (see Chapter 50) and vasoactive intestinal peptide (VIP; see Chapter 43) also produce HCO−3-rich choleresis at the level of the ducts. These hormones raise [cAMP]i and thus stimulate apical Cl− channels and the Cl-HCO3 exchanger. A Ca2+-activated Cl− channel is also present in the apical membrane. (See Note: Regulation of Cholangiocyte Secretion; Calcium-Activated Chloride Channels)
Cholangiocytes are also capable of reabsorbing fluid and electrolytes, as suggested by the adaptation that occurs after removal of the gallbladder (i.e., cholecystectomy). Bile found within the common bile duct of cholecystectomized, fasting animals is similar in composition to the concentrated bile typically found in the gallbladder. Thus, the ducts have partially taken over the function of the gallbladder (see later).
The hormone somatostatin inhibits bile flow by lowering [cAMP]i, an effect opposite that of secretin. This inhibition may be caused by enhancing fluid reabsorption by bile ducts or by inhibiting ductular secretion of the HCO−3-rich fluid discussed earlier.
Solutes reabsorbed from bile by cholangiocytes can be returned to the hepatocyte for repeat secretion. As shown earlier in Figure 46-2, the intralobular bile ducts are endowed with a rich peribiliary vascular plexus that is supplied by the hepatic artery. The blood draining this plexus finds its way into the hepatic sinusoids. This plexus is analogous to the capillaries of the gut, which, through the portal vein, also find their way into the hepatic sinusoids. Thus, some solutes, such as the hydrophilic bile acid ursodeoxycholic acid, may be absorbed by the cholangiocytes from bile and returned to the hepatocytes for repeat secretion, thus inducing significant choleresis.
The gallbladder is not an essential structure of bile secretion, but it does serve to concentrate bile acids up to 10-or even 20-fold during interdigestive periods. Tonic contraction of the sphincter of Oddi facilitates gallbladder filling by maintaining a positive pressure within the common bile duct. As we noted earlier, up to 50% of hepatic bile—or ~450 mL/day—is diverted to the gallbladder during fasting. The remaining ~450 mL/day passes directly into the duodenum. Periods of gallbladder filling between meals are interrupted by brief periods of partial emptying of concentrated bile and probably aspiration of dilute hepatic bile in a process analogous to the function of a bellows.
Bile salts and certain other components of bile are concentrated up to 20-fold within the gallbladder lumen because they are left behind during the isotonic reabsorption of NaCl and NaHCO3 by the leaky gallbladder epithelium (Fig. 46-12). The apical step of NaCl uptake and transport is electroneutral and is mediated by parallel Na-H and Cl-HCO3 exchangers. At the basolateral membrane, Na+ exits through the Na-K pump, whereas Cl− most likely exits by Cl− channels. Both water and HCO−3 move passively from lumen to blood through the tight junctions, which are rather leaky. Water can also move through the cell. The net transport is isotonic, which leaves behind gallbladder bile that is also isotonic but has a higher concentration of bile salts, K+, and Ca2+. Net fluid and electrolyte transport across the gallbladder epithelium is under hormonal regulation. Both VIP (released from neurons innervating the gallbladder) and serotonin inhibit net fluid and electrolyte absorption. Conversely, α-adrenergic blockade of neuronal VIP release increases fluid absorption.
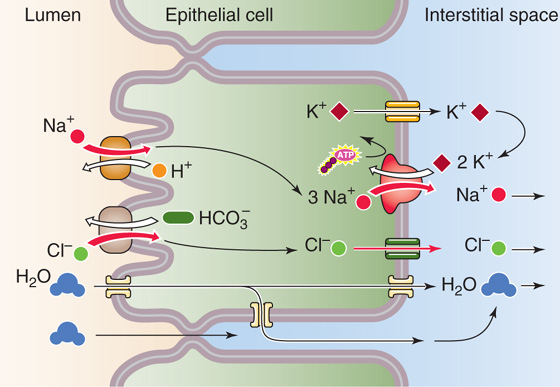
Figure 46-12 Isotonic fluid reabsorption by the gallbladder epithelium. The gallbladder epithelium performs the isotonic absorption of NaCl. The apical step is parallel Na-H exchange and Cl-HCO3 exchange. Because Na-H exchange is somewhat faster, net secretion of acid into the lumen occurs. The basolateral step of NaCl absorption is mediated by the Na-K pump and by Cl− channels. K+ channels provide a route for basolateral K+ recycling. Water follows passively through the tight junctions and through the basolateral membrane.
Although the gallbladder reabsorbs NaCl by parallel Na-H and Cl-HCO3 exchange at the apical membrane, Na-H exchange outstrips Cl-HCO3 exchange; the end result is net secretion of H+ ions. This action neutralizes the HCO−3 and acidifies the bile. The H+ secreted by the gallbladder protonates the intraluminal contents. This action greatly increases the solubility of calcium salts in bile and reduces the likelihood of calcium salt precipitation and gallstone formation. Common pigment gallstones contain one or more of several calcium salts, including carbonate, bilirubinate, phosphate, and fatty acids. The solubility of each of these compounds is significantly increased by the acidification of bile.
Mucus secretion by gallbladder epithelial cells results in the formation of a polymeric gel that protects the apical surface of the gallbladder epithelium from the potentially toxic effects of bile salts. However, excessive mucin synthesis can be deleterious. For example, in animal models of cholesterol cholelithiasis (i.e., formation of gallstones made of cholesterol), a marked increase in mucin release precedes crystal and stone formation.
Bile exiting the liver and flowing down the common hepatic duct reaches a bifurcation that permits flow either into the cystic duct and then into the gallbladder or into the common bile duct, through the sphincter of Oddi, and into the duodenum (Fig. 46-4). The extent to which bile takes either path depends on the relative resistance of the two pathways.
The sphincter of Oddi—which also controls the flow of pancreatic secretions into the duodenum—corresponds functionally to a short (4- to 6-mm) zone within the wall of the duodenum. The basal pressure within the lumen of the duct at the level of the sphincter is 5 to 10 mm Hg. The pressure in the lumen of the resting common bile duct is also 5 to 10 mm Hg, compared with a pressure of ~0 mm Hg inside the duodenum.
The basal contraction of the sphincter prevents reflux of the duodenal contents into the common bile duct. In its basal state, the sphincter exhibits high-pressure, phasic contractions several times per minute. These contractions are primarily peristaltic and directed in antegrade fashion to provide a motive force toward the duodenum. Thus, the sphincter of Oddi acts principally as an adjustable occluding mechanism and a regulator of bile flow.
Both hormonal and cholinergic mechanisms appear to be involved in gallbladder emptying. Dietary lipid stimulates the release of cholecystokinin (CCK) from duodenal I cells (see Chapter 44). This CCK not only stimulates pancreatic secretion but also causes smooth muscle contraction and evacuation of the gallbladder. The coordinated response to CCK also includes relaxation of the sphincter of Oddi, thus enhancing bile flow into the duodenum.
Bile acids are important for promoting the absorption of dietary lipids in the intestine. The quantity of bile acid that the liver normally secretes in a day varies with the number of meals and the fat content of these meals, but it typically ranges between 12 and 36 g. The liver’s basal rate of synthesis of bile acids from cholesterol (Fig. 46-9) is only ~600 mg/day in healthy humans, sufficient to replace the equivalent losses of bile acid in the feces. Obviously, the gastrointestinal tract must have an extremely efficient mechanism for recycling the bile acids secreted by the liver (Fig. 46-13). This recycling, known as the enterohepatic circulation, occurs as the terminal ileum and colon reabsorb bile acids and return them to the liver in the portal blood. The total pool of bile acids in the gastrointestinal tract is ~3 g. This pool must recirculate ~ 4 to 12 times per day, or as many as 5 or more times for a single fat-rich meal. If reabsorption of bile acids is defective, as can happen after resection of the ileum, de novo synthesis of bile acids by the liver can be as high as 4 to 6 g/day.
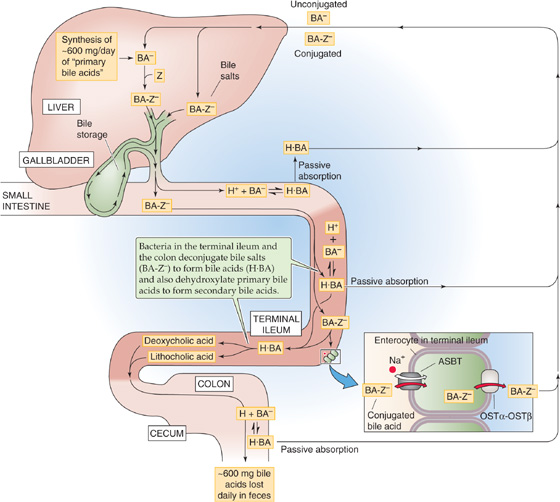
Figure 46-13 Enterohepatic circulation of bile acids. The bile acids that the liver delivers to the duodenum in the bile are primarily conjugated to taurine or glycine (BA-Z−). Most bile acids are reabsorbed as conjugated bile salts (BA-Z−) in the terminal ileum through an Na+-coupled cotransporter (ASBT). Also in the terminal ileum and colon, bacteria deconjugate a small amount of these bile salts to form unconjugated bile acids (H · BA ↔ H+ + BA−), thereby allowing H · BA to be passively absorbed by nonionic diffusion. In addition, bacteria in the terminal ileum and colon dehydroxylate primary bile acids to form secondary bile acids (see Fig. 46-9). Some of these are passively absorbed, and the rest are excreted in the feces. The absorbed bile acids return to the liver through the portal blood and are then taken up into the hepatocyte for secretion again.
Cholestasis
The term cholestasis refers to the suppression of bile secretion. Biliary constituents may therefore be retained within the hepatocyte and regurgitated into the systemic circulation. Cholestasis causes three major groups of negative effects: first, regurgitation of bile components (bile acids, bilirubin) into the systemic circulation gives rise to the symptoms of jaundice and pruritus (itching). Second, cholestasis damages hepatocytes, as evidenced by the release of liver enzymes (e.g., alkaline phosphatase) into the plasma. Third, because the bile acids do not arrive in the duodenum, lipid digestion and absorption may be impaired.
Many acute and chronic liver diseases produce cholestasis by mechanically obstructing the extrahepatic bile ducts or by impairing bile flow at the level of the hepatocytes or intrahepatic bile ducts. The mechanisms underlying the obstructive and functional forms of cholestasis are complex and have not been completely defined. Experimental models of cholestasis have produced multiple abnormalities: (1) altered plasma membrane composition and fluidity; (2) inhibition of membrane proteins, including the Na-K pump; (3) reduced expression of genes encoding transporters for bile acids and other organic anions; (4) increased permeability of the paracellular pathway, with backdiffusion of biliary solutes into the plasma; (5) altered function of microfilaments, with decreased contractions of bile canaliculi; and (6) loss of the polarized distribution of some plasma membrane proteins. Cholestatic conditions, such as bile duct obstruction, markedly increase the basolateral expression of MRP3—which normally is expressed only minimally. This induction of MRP3 allows the efflux of bile acids and other cholephilic anions from the hepatocyte into sinusoidal blood.
Most of the bile secreted into the duodenum is in the conjugated form. Very little of these bile salts are reabsorbed into the intestinal tract until they reach the terminal ileum, an arrangement that allows the bile salts to remain at high levels throughout most of the small intestine, where they can participate in lipid digestion and absorption (see Chapter 44). However, the enterohepatic circulation must eventually reclaim 95% or more of these secreted bile salts. Some of the absorption of bile acids by the intestines is passive and occurs along the entire small intestine and colon. Nevertheless, the major component of bile acid absorption is active and occurs only in the terminal ileum (Fig. 46-13).
Passive absorption of bile acids occurs along the entire small intestine and colon (Fig. 46-13), but it is less intensive than active absorption. The mechanism of bile acid uptake across the apical membrane may consist of either ionic or nonionic diffusion. Nonionic diffusion—or passive diffusion of the protonated or neutral form of the bile acid—is 10-fold greater than ionic diffusion. The extent of nonionic diffusion for a given bile acid depends on the concentration of its neutral, protonated form, which is maximized when the luminal pH is low and the pK of the bile acid is high. At the normal intestinal pH of 5.5 to 6.5, few of the taurine-conjugated bile salts are protonated, a small amount of the glycine-conjugated bile salts are protonated, and ~50% of unconjugated bile acids are protonated. Thus, the unconjugated bile acids are in the best position to be reabsorbed by nonionic diffusion, followed by the glycine-conjugated bile acids and then finally by the taurine-conjugated bile acids. Among these unconjugated bile acids, more lipophilic bile acids, such as chenodeoxycholate and deoxycholate, diffuse more readily through the apical membrane than do hydrophilic bile acids such as cholic acid. Nonionic diffusion also depends on the total concentration of the bile acid (i.e., neutral plus charged form), which, in turn, depends on the maximum solubilizing capacity of bile salt micelles for that bile acid.
Active absorption of bile acids in the intestine is restricted to the terminal ileum. This active process preferentially absorbs the negatively charged conjugated bile salts—the form not well absorbed by the passive mechanisms. Active uptake of bile salts involves saturation kinetics, competitive inhibition, and a requirement for Na+ (Fig. 46-13). The Na+- dependent transporter responsible for the apical step of active absorption is known as the apical Na+/bile salt transporter ASBT (SLC10A2), a close relative of the hepatocyte transporter NTCP (Fig. 46-5C). Once bile salts have entered ileal enterocytes across the apical membrane, they exit across the basolateral membrane via a heteromeric organic solute transporter (Ostα/Ostβ).
Because the most polar bile salts are poorly absorbed by nonionic diffusion, it is not surprising that the ASBT in the apical membrane of the enterocytes of the terminal ileum has the highest affinity and maximal transport rates for these salts. For example, the ASBT is primarily responsible for absorbing the ionized, taurine-conjugated bile salts in the ileum. Conversely, the ASBT in the ileum is relatively poor at absorbing the more lipophilic bile acids, which tend to be absorbed passively in the upper intestine.
On their entry into portal blood, the bile acids are predominantly bound to albumin and, to a lesser extent, lipoproteins. The liver removes or clears these bile acids from portal blood by the transport mechanisms outlined earlier in Figure 46-5C. Hepatic clearance of bile acids is often expressed as the percentage of bile acids removed during a single pass through the liver. The hepatic extraction of bile acids is related to bile acid structure and the degree of albumin binding. It is greatest for hydrophilic bile acids and is least for protein-bound, hydrophobic bile acids.
The small fraction of bile acids that escapes active or passive absorption in the small intestine is subject to bacterial modification in the colon. This bacterial modification takes two forms. First, the bacteria deconjugate the bile. Second, the bacteria perform a 7α-dehydroxylation reaction with the formation of secondary bile acids. These secondary bile acids include deoxycholate and lithocholate (Fig. 46-9). The deconjugated secondary bile acids may then be either absorbed passively in the colon or excreted in the feces; their fate depends on their physicochemical properties and their binding to luminal contents. Up to one third of the deoxycholate formed in the colon may be reabsorbed by nonionic diffusion. Lithocholate, which is relatively insoluble, is absorbed to a much lesser extent. The secondary bile acids formed by colonic bacteria and recycled back to the liver may undergo biotransformation through conjugation to glycine and taurine.
Gallstones
Most gallstones (~80%) consist mainly of cholesterol. Thus, cholelithiasis may be regarded as a disturbance of bile secretion and cholesterol elimination. When cholesterol and phospholipids are secreted together into the bile, they form unilamellar bilayered vesicles. These vesicles become incorporated into mixed micelles that form because of the amphiphilic properties of bile acids. Micellation allows cholesterol to remain in solution in its passage through the biliary tree. However, if the concentration of bile acids is insufficient to maintain all the cholesterol in the form of mixed micelles, the excess cholesterol is left behind as vesicles in the aqueous phase. These cholesterol-enriched vesicles are relatively unstable and are prone to aggregate and form large multilamellar vesicles, from which cholesterol crystals nucleate. Growth of crystals may result in the formation of gallstones. An excess of biliary cholesterol in relation to the amount of phospholipids and bile acids can result from hypersecretion of cholesterol, inadequate secretion of bile acids, or both. Cholelithiasis may be further promoted by other factors, such as gallbladder mucin and other nonmucous glycoproteins, as well as by stasis of bile in the gallbladder.
Thus, the enterohepatic circulation of bile acids is driven by two mechanical pumps: (1) the motor activity of the gallbladder and (2) peristalsis of the intestines to propel the bile acids to the terminal ileum and colon. It is also driven by two chemical pumps: (1) energy-dependent transporters located in the terminal ileum and (2) energy-dependent transporters in the hepatocyte.
The bile acid receptor FXR, a member of the nuclear receptor family, controls multiple components of the enterohepatic circulation of bile acids. Primary bile acids are potent agonists of FXR, which transcriptionally regulates several genes involved in bile acid homeostasis. Four examples of negative feedback by activated FXR are as follows: (1) FXR inhibits the expression of cholesterol 7α-hydroxylase (Fig. 46-9), the rate-limiting enzyme for bile acid synthesis; (2) FXR induces the expression of an inhibitory transcription factor—the small heterodimer partner (SHP)—which controls the activity of another nuclear receptor, the liver receptor homologue-1 (LRH-1), which is required for CYP7a1 expression; (3) FXR upregulates BSEP (increasing bile acid secretion; Fig. 46-5C) and downregulates NTCP (decreasing bile acid uptake; Fig. 46-5C) by SHP-dependent mechanisms; and (4) FXR, through SHP, downregulates ASBT and thereby reduces ileal bile acid uptake. Thus, FXR coordinates bile acid synthesis and transport by the liver and intestine.
The liver is a metabolically active and highly aerobic organ. It receives ~28% of the total blood flow and extracts ~20% of the oxygen used by the body. The liver is responsible for the synthesis and degradation of carbohydrates, proteins, and lipids. The small molecules that are products of digestion are efficiently sorted in the liver for metabolism, storage, or distribution to extrahepatic tissues for energy. The liver provides energy to other tissues mainly by exporting two substrates that are critical for oxidization in the peripheral tissues, glucose and ketone bodies (e.g., acetoacetate).
The liver is one of the key organs that maintain blood glucose concentrations within a narrow range, in a dynamic process involving endogenous glucose production and glucose utilization. The fasting blood [glucose] is normally 4 to 5 mM. Between meals, when levels of insulin are relatively low and levels of glucagon are high (see Chapter 50), the liver serves as a source of plasma glucose, both by synthesizing glucose and by generating it from the breakdown of glycogen. The de novo synthesis of glucose, or gluconeogenesis (see Fig. 51-12), is one of the liver’s most important functions; it is essential for maintaining a normal plasma concentration of glucose, which is the primary energy source for most tissues (see Chapter 57). Glucose is synthesized in the lumen of the ER, principally from amino acids and lactate. Dietary fructose and galactose are also largely converted to glucose. Glucose exits the ER by facilitated diffusion (mediated by GLUT7) and then passes into the blood through another facilitated diffusion mechanism (GLUT2), which has a low affinity and high capacity and is located in the hepatocyte’s basolateral membrane.
The second way in which the liver delivers glucose to blood plasma is by glycogenolysis. Stored glycogen may account for as much as 7% to 10% of the total weight of the liver. Glycogenolysis in the liver yields glucose as its major product, whereas glycogen breakdown in muscle produces lactic acid (see Chapter 59).
After a meal, when levels of insulin are relatively high, the liver does just the opposite: it acts as a sink for glucose by taking it up from the portal blood and either breaking it down to pyruvate or using it to synthesize glycogen (see Fig. 50-8). Glucose oxidation has two phases. In the anaerobic phase, glucose is broken down to pyruvic acid (glycolysis). In the aerobic phase, pyruvic acid is completely oxidized to H2O and CO2 through the citric acid cycle.
The liver also consumes glucose by using it for glycogen synthesis. Carbohydrate that is not stored as glycogen or oxidized is metabolized to fat.
All the aforementioned processes are regulated by hormones such as insulin and glucagon (see Chapter 50), which enable rapid responses to changes in the metabolic requirements of the body.
Protein Synthesis One of the major functions of the liver is to produce a wide array of proteins for export to the blood plasma (Table 46-3). These products include major plasma proteins that are important for maintaining the colloid osmotic pressure of plasma (see Chapter 19). Other products include factors involved in hemostasis (blood clotting) and fibrinolysis (breakdown of blood clots), carriage proteins that bind and transport hormones and other substances in the blood, prohormones, and lipoproteins (Table 46-4). The liver synthesizes plasma proteins at a maximum rate of 15 to 50 g/day.
Table 46-3 Proteins Made by the Liver for Export
Albumin |
α1-Fetoprotein |
Plasma fibronectin (an α2-glycoprotein) |
C-reactive protein |
α2-Microglobulin |
Coagulation: fibrinogen and all others except factor VIII |
Inhibitors of coagulation: α1-antitrypsin and antithrombin III, α2-macroglobulin, protein S, protein C |
Fibrinolysis: plasminogen |
Inhibitors of fibrinolysis: α2-antiplasmin |
Ceruloplasmin |
Corticosteroid-binding globulin (CBG; also called transcortin) |
GH-binding protein (low-affinity form) |
Haptoglobin |
Hemopexin |
IGF-binding proteins |
Retinol-binding protein |
Sex hormone–binding globulin (SHBG) |
Thyroid-binding globulin (TBG) |
Transferrin |
Transthyretin |
Apo A-I |
Apo A-II |
Apo A-IV |
Apo B-100 |
Apo C-II |
Apo D |
GH, growth hormone; IGF, insulin-like growth factor.
Table 46-4 Major Classes of Lipoproteins
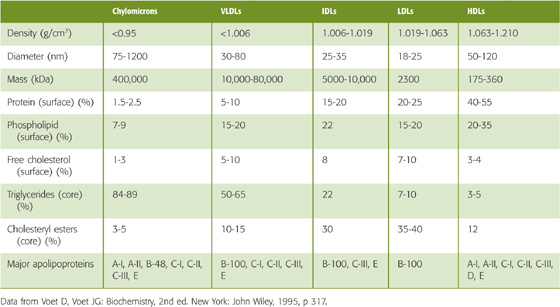
Amino Acid Uptake A major role of the liver is to take up and metabolize dietary amino acids that are absorbed by the gastrointestinal tract (see Chapter 44) and are transported to the liver in portal blood. These amino acids are taken up by both Na+- dependent and Na+- in dependent transporters that are identical to some of the amino acid transporters in the kidney, small intestine, and other tissues (see Table 35-1). An unusual feature of the liver is that, with few exceptions, the same amino acid transporter may be located on both the basolateral and apical membranes. For example, Na+-dependent glutamate uptake by the excitatory amino acid transporters SLC1A1 (EAAT3) and SLC1A2 (EAAT2) occurs primarily at the apical membrane, but dexamethasone (corticosteroid) treatment can induce their expression at the basolateral membrane. In general, hepatic amino acid transporters are highly regulated at the transcriptional and post-translational levels. (See Note: Glutamate Transporters)
Amino Acid Metabolism Under physiological conditions, total and individual plasma concentrations of amino acids are tightly regulated. The liver controls the availability of amino acids in the systemic blood, activating ureagenesis after a high protein meal and repressing it during fasting or low protein intake. Unlike glucose, which can be stored, amino acids must either be used immediately (e.g., for the synthesis of proteins) or broken down. The breakdown of α-amino acids occurs by deamination to α-keto acids and NH+4 (Fig. 46-14). The α-keto acids (“carbon skeleton”), depending on the structure of the parent amino acid, are metabolized to pyruvate, various intermediates of the citric acid cycle (see Fig. 57-9), acetyl coenzyme A (acetyl CoA), or acetoacetyl CoA. The liver detoxifies ~95% of the NH+4 through a series of reactions known as the urea cycle (Fig. 46-14); the liver can also use NH+4—together with glutamate—to generate glutamine. Individual deficiencies in each of the enzymes involved in the urea cycle have been described and result in life-threatening hyperammonemia. The urea generated by the urea cycle exits the hepatocyte through a urea channel, which is, in fact, AQP9. The urea then enters the blood and is ultimately excreted by the kidneys (see Chapter 37). The glutamine synthesized by the liver also enters the blood. Some of this glutamine is metabolized by the kidney to yield glutamate and NH+4, which is exported in the urine (see Chapter 37). (See Note: Hepatic Detoxification of Ammonium by Formation of Glutamine)
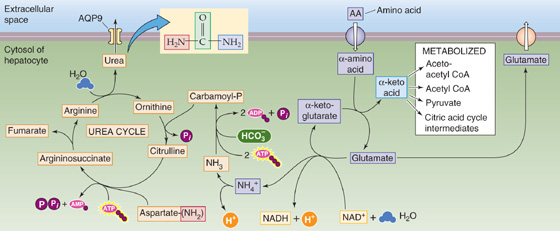
Figure 46-14 Amino acid metabolism and urea formation in hepatocytes. When a hepatocyte takes up an α-amino acid, it either must use it immediately in protein synthesis or deaminate it. The deamination reaction transfers the amino group of the α-amino acid to α-ketoglutarate, thus yielding glutamate and the corresponding α-keto acid. Depending on the backbone of the α-keto acid, it may be metabolized into acetoacetyl CoA, acetyl CoA, pyruvate, or a variety of citric acid cycle intermediates. The NH+4 results from the regeneration of the α-ketoglutarate consumed in the urea cycle. The other amino group of the urea is derived from the amino group of aspartate. The C = O moiety of urea is derived from CO2. The liver then exports the urea, which exits the hepatocyte through AQP9.
The liver is also the main site for the synthesis and secretion of glutathione. GSH is critical for detoxification (in conjugation reactions in the liver) and for protection against oxidative stress in multiple organs. Thus, erythrocytes that have low levels of GSH are more prone to hemolysis. Because more than 90% of the GSH in the circulation is synthesized in the liver, GSH efflux across the basolateral membrane from the hepatocyte to the sinusoid is important. Bidirectional transport of glutathione across the basolateral membrane may occur in part by one of the OATPs as well as MRP4 by cotransport with bile acids. In addition, MRP2 exports some conjugated GSH across the canalicular membrane into bile, as stated earlier, and an unidentified transporter can similarly export smaller amounts of unconjugated GSH.
As discussed in Chapter 44, enterocytes in the small intestine process fatty acids consumed as dietary triglycerides and secrete them into the lymph primarily in the form of extremely large proteolipid aggregates called chylomicrons (Fig. 46-15). These chylomicrons—made up of triglycerides (80% to 90%), phospholipids, cholesterol, and several apoproteins (Chapter 44)—pass from the lymph to the blood through the thoracic duct. As discussed later in Chapter 58, lipoprotein lipase (LPL) on the walls of the capillary endothelium in adipose tissue and muscle then partially digests the triglycerides in these chylomicrons. The results of this digestion are glycerol, fatty acids, and smaller or “remnant” chylomicrons, which are triglyceride depleted and thus enriched in cholesterol. The glycerol and fatty acids generated by LPL enter adipocytes and muscle cells. The cholesterol-rich remnant chylomicrons, conversely, remain in the blood and make their way to the liver, where they enter hepatocytes by a process that is saturable, specific, and of high affinity. Although low-density lipoprotein (LDL) receptors can recognize chylomicron remnants, another member of the LDL receptor gene family, the LDL-related receptor, is of greater importance. The chylomicron remnant binds to this receptor on the basolateral membrane, enters the hepatocyte through receptor-mediated endocytosis (see Chapter 2), and is degraded in lysosomes. Thus, chylomicrons transport dietary triglycerides to adipose tissue and muscle, whereas their remnants transport dietary triglycerides and cholesterol to hepatocytes.
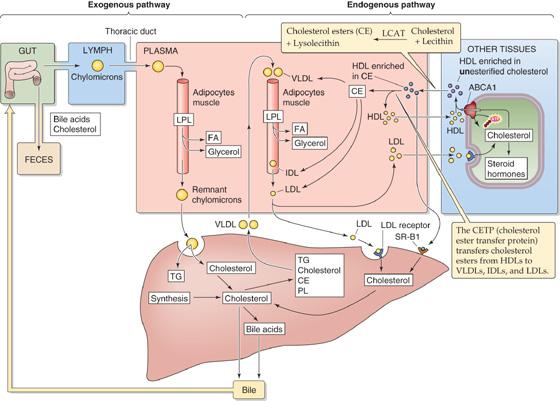
Figure 46-15 Cholesterol metabolism. CE, cholesteryl ester; FA, fatty acid.
The hepatocyte can also take up across its basolateral membrane the long-chain fatty acids liberated by LPL but not used by other tissues. Fatty acid uptake occurs by at least two different mechanisms: facilitated uptake and a nonspecific “flip-flop” across the lipid bilayer.
To extract energy from the neutral fats derived from the remnant chylomicrons, hepatocytes must first split the triglycerides into glycerol and fatty acids. The fatty acids thus derived from remnant chylomicrons, as well as those that enter the hepatocyte directly, are metabolized by β oxidation into two-carbon acetyl intermediates that then form acetyl CoA. Acetyl CoA can enter the citric acid cycle, where it is oxidized to produce large amounts of energy. The acetyl CoA that is not used by the liver is converted by the condensation of two molecules of acetyl CoA to yield acetoacetic acid. The liver is the only organ that produces acetoacetate for metabolism by muscle, brain, and kidney, but it does not use this substrate for its own energy needs. In fasting or in poorly controlled diabetes, in which the supply of acetyl CoA is in excess, the acetyl CoA is diverted to produce acetoacetate, which, in turn, can yield β-hydroxybutyrate and acetone. Together, acetoacetate, β-hydroxybutyrate, and acetone are referred to as ketone bodies. Another fate of fatty acids in the liver is that they can be re-esterified to glycerol, with the formation of triglycerides that can either be stored or exported as very-low-density lipoproteins (VLDLs) and released into the circulation for use by peripheral tissues.
The body’s major pools of cholesterol include the cholesterol and cholesterol derivatives in bile, cholesterol in membranes, cholesterol carried as lipoproteins in blood (Table 46-4), and cholesterol-rich tissues. Cholesterol is present in membranes and bile mainly as free cholesterol. In plasma and in some tissues, cholesterol is esterified with long-chain fatty acids. The major sources of cholesterol are dietary uptake of cholesterol and de novo synthesis of cholesterol by various cells (Table 46-5). The major fates of cholesterol are secretion into bile, excretion in the feces when intestinal cells are sloughed, sloughing of skin, and synthesis of steroid hormones. However, in mammals, the most important route for the elimination of cholesterol is the hepatic conversion of cholesterol into bile acids. In the steady state, the liver must excrete an amount of sterol (as cholesterol and bile acids) that equals the amount of cholesterol that is synthesized in the various organs and absorbed from the diet.
Table 46-5 Sources and Fates of Cholesterol in Humans
Process |
Flow (g/day for a 70-kg Human) |
Intestinal absorption |
0.1-0.5 |
Synthesis |
1.05 |
Biliary secretion |
0.9 |
Cholesterol consumed in synthesis of bile acids |
0.5 |
Cholesterol secreted in VLDLs by the liver |
0.25-1.75 |
Data from Cooper AD, Ellsworth JL: In Zakim D, Boyer TD (eds): Hepatology. Philadelphia: WB Saunders, 1996.
The liver is the major organ for controlling cholesterol metabolism (Fig. 46-15). The liver obtains cholesterol from three major sources. First, the intestine packages dietary cholesterol as chylomicrons, which travel through the lymph to blood vessels in adipocytes and muscle, where LPL hydrolyzes triglycerides to fatty acids and glycerol. The resultant cholesterol-enriched remnant chylomicrons are then delivered as cholesterol to the liver. Second, the liver synthesizes cholesterol de novo. Third, the liver takes up cholesterol in the guise of LDLs. However, the liver exports cholesterol in two major ways: (1) the liver uses cholesterol to synthesize bile acids and also includes cholesterol and cholesteryl esters in the bile, and (2) the liver also exports cholesterol to the blood in the form of VLDLs.
Synthesis of Cholesterol The de novo synthesis of cholesterol occurs in many extrahepatic tissues, as well as in the intestine and liver. The synthesis of cholesterol proceeds from acetyl CoA in a multistep process that takes place in the SER and cytosol (Fig. 46-16). The hepatic synthesis of cholesterol is inhibited by dietary cholesterol and by fasting and is increased with bile drainage (fistula) and bile duct obstruction. The rate-limiting step in cholesterol synthesis is the conversion of 3-hydroxy-3-methylglutaryl-CoA (HMG-CoA) to mevalonate by HMG-CoA reductase, the level of which is decreased—in typical negative feedback fashion—by cholesterol levels in the cell. The most potent cholesterol-lowering agents that are clinically available today—the “statins”—are inhibitors of HMG-CoA reductase. (See Note: Treatment of Hyperlipidemia)
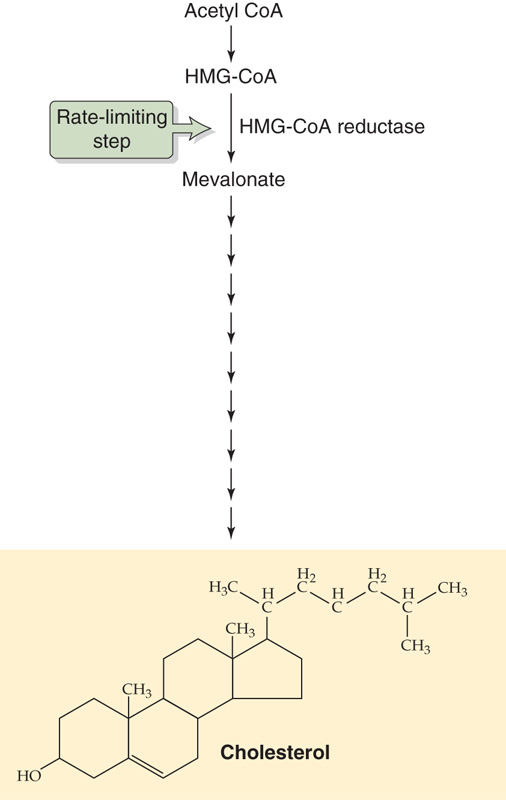
Figure 46-16 Cholesterol synthesis. The liver synthesizes cholesterol de novo from acetyl CoA in a multistep process that occurs in the SER and cytosol. The rate-limiting step is the conversion of HMG-CoA to mevalonate by HMG-CoA reductase.
The liver is the hub of cholesterol metabolism. It is the central feature of an exogenous loop in which the liver takes up dietary cholesterol as remnant chylomicrons and exports cholesterol and cholesterol metabolites into bile. The liver is also the central feature of an endogenous loop in which the liver exports cholesterol and other lipids as VLDLs and takes them up from the blood as LDLs. Table 46-4 summarizes the properties of these lipoproteins, as well as two others: the intermediate-density lipoproteins (IDLs) and the high-density lipoproteins (HDLs). As we move from left to right in Table 46-4, the size of the particles decreases, the density increases (because the fractional mass of protein increases), the fractional amount of triglycerides decreases, and the fractional amount of phospholipids increases.
Regardless of the source of the cholesterol, the liver can package cholesterol along with other lipids and apoproteins as VLDLs. The VLDLs are large—and therefore less dense—when the availability of triglycerides is high (e.g., obesity, diabetes), but they are small when triglyceride availability is low. VLDLs enter the bloodstream (Fig. 46-15) and eventually make their way to the blood vessels of adipose tissue and muscle, where the same LPL that degrades chylomicrons degrades the VLDLs on the luminal surface of blood vessel endothelial cells. In the process, fatty acids are released to the tissues. Thus, VLDLs act as lipid shuttles that transport endogenous triglycerides to adipose tissue for storage as fat or to muscle for immediate use. As a result of the LPL activity, the large, buoyant VLDLs rapidly shrink to become the smaller IDLs and the even smaller LDLs. The half-life of VLDLs is less than an hour. In plasma, only minute amounts of IDLs are present.
Both the liver and extrahepatic tissue can take up LDLs—and to a lesser extent, IDLs—by the process of receptor-mediated endocytosis (see Chapter 2). LDLs are the major carriers of cholesterol in plasma. The half-life of LDLs is 2 to 3 days. Of course, uptake of LDL by the liver is a major pathway of cholesterol input to the liver. The liver degrades ~40% to 60% of LDLs, and no other tissue takes up more than ~10%. LDL uptake by other tissues provides a mechanism for the delivery of cholesterol that can be used for the synthesis of cell membranes and steroid hormones or for storage as cholesteryl ester droplets.
The other major player in cholesterol metabolism is HDL, which is composed of cholesterol, phospholipids, triglycerides, and apoproteins (Table 46-4). The two major apoproteins of HDLs, A-I and A-II, are made by the intestines as part of chylomicrons, as well as by the liver. As LPL digests VLDLs on endothelial cells, some excess surface material (i.e., cholesterol and phospholipids) of these rapidly shrinking particles is transferred to the HDLs. In peripheral cells such as macrophages, the cholesterol transporter ABCA1 facilitates the efflux of cholesterol, which then combines with lipid-poor HDLs. An enzyme that is associated with HDL, lecithin cholesterol acyltransferase (LCAT)—synthesized in the liver, then takes an acyl group from lecithin and esterifies it to cholesterol to produce a cholesterol ester (CE).
When the CE-enriched HDL (HDL-CE) reaches the liver, it binds to the scavenger receptor class B type 1 (SR-B1), which mediates selective uptake of HDL-CE. The cholesterol moiety is targeted for biliary excretion. This pathway may also process LDL-CE and free cholesterol. In contrast to the LDL receptor, the selective uptake of CE by SR-B1 does not involve endocytosis. Instead, a poorly defined mechanism first moves CE to a reversible plasma membrane pool and then to an irreversible pool within the cell. (See Note: Scavenger Receptor Class B Type 1 (SR-B1))
In addition to traveling to the liver for handling through SR-B1, HDL-CE can transfer its CE to apolipoprotein B–containing lipoproteins (i.e., VLDL, IDLs, LDLs) through the action of CE transfer protein (CETP). These less-dense lipoproteins can now move to the liver for uptake by the LDL receptor. The HDL-mediated removal of cholesterol from peripheral tissues—through both SR-B1 and CETP—for transport to the liver and excretion in bile is known as reverse cholesterol transport. This process is thought to protect against atherosclerosis.
We discuss the intestinal uptake of the fat-soluble vitamins in Chapter 44.
Vitamin A Vitamin A (retinol and its derivatives)—like dietary vitamin D, as well as vitamins E and K—is absorbed from the intestine and is then transported in newly synthesized chylomicrons or VLDLs. After some peripheral hydrolysis of its triglyceride, the remnant chylomicrons are taken up by the liver. In the hepatocyte, retinyl esters may be hydrolyzed to release free retinol, which can then be transported into the sinusoids bound to retinol-binding protein (RBP) and prealbumin. Alternatively, retinyl esters may be stored in the hepatocyte or transported as RBP-bound retinol to stellate (Ito) cells, the storage site of more than 80% of hepatic vitamin A under normal conditions. Retinol may also undergo oxidation to retinal and conversion to retinoic acid, which plays a key role in phototransduction (see Chapter 13). Retinoic acid is conjugated to glucuronide and is secreted into bile, where it undergoes enterohepatic circulation and excretion. Liver disease resulting in cholestasis may lead to a secondary vitamin A deficiency by interfering with absorption in the intestine (lack of the bile needed for digestion/absorption of vitamin A) or by impairing delivery to target tissues because of reduced hepatic synthesis of RBP.
Control of Cholesterol Synthesis
HMG-CoA reductase, the rate-limiting enzyme in cholesterol biosynthesis, is a 97-kDa enzyme intrinsic to the ER. The protein is anchored in the ER by a 339–amino acid domain that spans the membrane eight times and is required for activity of the protein. In the short term, decreased intracellular levels of ATP lead to phosphorylation of HMG-CoA reductase, which reduces the activity of the enzyme, presumably preserving energy stores in the cell.
Far more important than the short-term regulation of HMA-CoA reductase is the long-term control of the amount of the enzyme, which can increase as much as 200-fold. The amount of HMG-CoA reductase protein increases after cells are depleted of mevalonate or when the demand for mevalonate-derived metabolites is increased. HMG-CoA reductase activity is upregulated by a combination of enhanced gene transcription, increased mRNA translation, and increased stability of the enzyme. These changes are reversed by the addition of cholesterol or mevalonate to the cells. The transmembrane domain of HMG-CoA reductase may serve as a receptor for the regulatory effects of sterols.
Sterol repression of HMG-CoA reductase gene expression—as well as the expression of LDL and HMG-CoA synthase genes—is mediated by specific transcription factors and specific sequence elements within the 5′ upstream region to which trans-acting proteins bind. Several related sterol regulatory elements whose activities mediate sterol repression of gene expression, such as SRE-1, provide a specific sequence required for the recognition of DNA-binding proteins. These SRE-binding proteins (SREBPs; see Chapter 5) are membrane-bound basic helix-loop-helix transcriptional activators (see Chapter 5) that control genes involved in the synthesis and receptor-mediated uptake of cholesterol and fatty acids.
Vitamin D Skin cells—under the influence of ultraviolet light—synthesize vitamin D3 (see Chapter 51). Dietary vitamin D can come from either animal sources (D3) or plant sources (D2). In either case, the first step in activation of vitamin D is the 25-hydroxylation of vitamin D, catalyzed by a hepatic cytochrome P-450 enzyme. This hydroxylation is followed by 1-hydroxylation in the kidney to yield a product (1, 25-dihydroxyvitamin D) with full biological activity. Termination of the activity of 1, 25-dihydroxyvitamin D also occurs in the liver by hydroxylation at carbon 24, mediated by another cytochrome P-450 enzyme.
Vitamin E This fat-soluble vitamin is absorbed from the intestine primarily in the form of α-and γ-tocopherol. It is incorporated into chylomicrons and VLDLs with other products of dietary lipid digestion. As noted earlier, these particles reach the systemic circulation through the lymphatics and undergo some triglyceride hydrolysis. In the process, some vitamin E is transferred to other tissues. The α-and γ-tocopherol remaining in the remnant chylomicrons is transported into the liver, which is the major site of discrimination between the two forms. The α-tocopherol is secreted again as a component of hepatically derived VLDL and perhaps HDL. The γ-tocopherol appears to be metabolized or excreted by the liver. A hepatic tocopherol-binding protein may play a role in this discriminatory process.
Vitamin K Vitamin K is a fat-soluble vitamin produced by intestinal bacteria. This vitamin is essential for the γ-carboxylation—by the ER enzyme γ-glutamyl carboxylase—of certain glutamate residues in coagulation factors II, VII, IX, and X as well as anticoagulants protein C and protein S and certain other proteins. Intestinal absorption and handling of vitamin K—which is present in two forms, K1 and K2—are similar to those of the other fat-soluble vitamins: A, D, and E. Common causes of vitamin K deficiency, which can lead to a serious bleeding disorder, include extrahepatic or intrahepatic cholestasis, fat malabsorption, biliary fistulas, and dietary deficiency, particularly in association with antibiotic therapy.
Copper The trace element copper is essential for the function of cuproenzymes such as cytochrome c oxidase and superoxide dismutase (see Chapter 57). Approximately half the copper in the diet (recommended dietary allowance, 1.5 to 3 mg/day) is absorbed in the jejunum and reaches the liver in the portal blood, mostly bound to albumin. A small fraction is also bound to amino acids, especially histidine. More than 80% of the copper absorbed by the jejunum each day is excreted in bile, for a total of 1.2 to 2.4 mg/day.
The copper-transport protein Ctr1 imports copper across the hepatocyte basolateral membrane. Copper then binds to a family of intracellular metallochaperones that direct the metal to the appropriate pathway for incorporation into cuproenzymes or for biliary excretion. The copper chaperone Atox1 moves copper to the secretory pathway and directly interacts with ATP7B, a member of the P1B subfamily of P-type ATPases (see Chapter 3). ATP7B, which is mutated in Wilson disease, is located predominantly in the trans-Golgi network and delivers copper either for incorporation into ceruloplasmin (which is released into the blood; see later) or for excretion into bile. Another chaperone, Murr1, appears to be involved in transport of copper to the canalicular membrane for excretion into the bile, complexed to large proteins, such as metallothioneins. The biliary copper-protein complexes cannot be reabsorbed by the small intestine, and hence represent a pathway for copper excretion. Processes that impair the biliary excretion of copper result in the accumulation of copper, initially in the lysosomal fraction of hepatocytes, with subsequent elevation of plasma copper levels.
Wilson Disease
Wilson disease is inherited as an autosomal recessive illness caused by a mutation in ATP7B, the pump responsible for copper accumulation in the trans-Golgi network. The impaired biliary excretion of copper causes a buildup of copper in cells, which produces toxic effects in the liver, brain, kidney, cornea, and other tissues. The disease is rare, but it must be considered in the differential diagnosis of anyone younger than 30 years with evidence of significant liver disease. Patients most often have neuropsychiatric complications, including ataxia, tremors, increased salivation, and behavioral changes. Slit-lamp examination of the cornea reveals the diagnostic Kayser-Fleischer rings at the limbus of the cornea.
Because of the lack of functional ATP7B, the apoceruloplasmin in the trans-Golgi network cannot bind copper to form ceruloplasmin. As a result, the hepatocytes secrete apoceruloplasmin, which lacks the ferroxidase activity of ceruloplasmin. Moreover, the serum concentrations of ceruloplasmin are low. Indeed, the best way to confirm the diagnosis of Wilson disease is the detection of a low serum ceruloplasmin level and elevated urinary copper excretion. A few affected patients have normal ceruloplasmin levels, and the diagnosis must then be sought with liver biopsy. The disease can be treated by chelating the excess copper with penicillamine.
Ceruloplasmin, an α2-globulin synthesized by the liver, binds 95% of copper present in the systemic circulation. Ceruloplasmin has ferroxidase activity but has no critical role in the membrane transport or metabolism of copper.
Iron Dietary iron is absorbed by the duodenal mucosa and is then transported through the blood bound to transferrin (see Chapter 44), a protein synthesized in the liver. The liver also takes up, secretes, and stores iron. Entry of iron into hepatocytes is mediated through specific cell surface transferrin receptors (see Chapter 5). Within the cell, a small pool of soluble iron is maintained for intracellular enzymatic reactions, primarily for those involved in electron transport. However, iron is also toxic to the cell. Hence, most intracellular iron is complexed to ferritin. The toxicity of iron is clearly evident when normal storage mechanisms become overwhelmed, as occurs in hemochromatosis (see the box on hereditary hemochromatosis in Chapter 44), an autosomal recessive disease in which regulation of iron absorption is uncoupled from total body storage levels.
Books and Reviews
Boyer JL: Mechanisms of bile secretion and hepatic transport. In Andreoli TE, Hoffman JF, Fanestil DD, Schultz SG, (eds): Physiology of Membrane Disorders. New York: Plenum, 1986.
Cooper AD, Ellsworth JL: Lipoprotein metabolism. In Zakim D, Boyer TD (eds): Hepatology. Philadelphia: WB Saunders, 1996.
Kipp H, Arias IM: Trafficking of canalicular ABC transporters in hepatocytes. Annu Rev Physiol 2002; 64:595-608.
Kullak-Ublick GA, Stieger B, Meier PJ: Enterohepatic bile salt transporters in normal physiology and liver disease. Gastroenterology 2004; 126:322-342.
Rhainds D, Brissette L: The role of scavenger receptor class B type I (SR-BI) in lipid trafficking: Defining the rules for lipid traders. Int J Biochem Cell Biol 2004; 36:39-77.
Small DM: Role of ABC transporters in secretion of cholesterol from liver into bile. Proc Natl Acad Sci U S A 2003; 100:4-6.
Tao TY, Gitlin JD: Hepatic copper metabolism: Insights from genetic disease. Hepatology 2003; 37:1241-1247.
Journal Articles
Bull LN, van Eijk MJT, Pawlikowska L: A gene encoding a P-type ATPase mutated in two forms of hereditary cholestasis. Nat Genet 1998; 18:219-224.
Hagenbuch B, Meier PJ: Molecular cloning, chromosomal localization and functional characterization of a human liver Na+/bile acid cotransporter. J Clin Invest 1994; 93:1326-1331.
Kullak-Ublick GA, Hagenbuch B, Stieger B: Molecular and functional characterization of an organic anion transporting polypeptide cloned from human liver. Gastroenterology 1995; 109:1274-1282.
Oude Elferink RPJ, Meijer DKF, Kuipers F, et al: Hepatobiliary secretion of organic compounds: Molecular mechanisms of membrane transport. Biochim Biophys Acta 1995; 1241:215-268.