
Edward G. Moczydlowski and Michael Apkon
The primary function of muscle is to generate force or movement in response to a physiological stimulus. The human body contains three fundamentally different types of muscle adapted to specialized functions. Skeletal muscle is responsible for the voluntary movement of bones that underlie locomotion and work production. Skeletal muscle also controls the breathing cycle of the lungs through contraction of the diaphragm and functions as a pump assisting return of the venous blood supply to the heart. Cardiac muscle is specific to the heart as the biomechanical pump driving the delivery of blood to the lungs and tissues. Smooth muscle provides mechanical control of organ systems such as the digestive, urinary, and reproductive tracts as well as the blood vessels of the circulatory system and the airway passages of the respiratory system.
Contraction of muscles is initiated either by a chemical neurotransmitter or paracrine factor or by direct electrical excitation. All muscles transduce chemical energy released by hydrolysis of ATP into mechanical work. The unique physiological role of each of the three basic muscle types dictates inherent differences in the rate and duration of contraction, metabolism, fatigability, and ability to regulate contractile strength. For example, both skeletal and cardiac muscle must be capable of rapid force development and shortening. However, skeletal muscle must be able to maintain contractile force for relatively long periods. Cardiac muscle contracts only briefly with each heartbeat but must sustain this rhythmic activity for a lifetime. Smooth muscle, like skeletal muscle, must be able to regulate contraction over a wide range of force development and elastic changes in the size of organs such as the urinary bladder and uterus. In some tissues (e.g., sphincters), smooth muscle sustains contraction without fatigue for very long periods. Despite these differences, the trigger for muscle contraction is the same for all three types of muscle: a rise in the free cytosolic Ca2+ concentration ([Ca2+]i).
This chapter describes the fundamental physiology of muscle excitation, the coupling of excitation to contraction, the molecular mechanism of contraction, the regulation of contraction, and the related issues of muscle diversity. We describe general molecular mechanisms shared by all muscle cells and contrast the unique features of skeletal, cardiac, and smooth muscle. Because molecular mechanisms specific to cardiac myocytes are best understood in the unique context of the heart as a pump, we discuss details of cardiac muscle physiology at greater depth in Chapters 22 and 23.
The smallest contractile unit of skeletal muscle is a multinucleated, elongated cell called a muscle fiber or myofiber (Fig. 9-1). A bundle of linearly aligned muscle fibers forms a fascicle. In turn, bundles of fascicles form a muscle, such as the biceps. The whole muscle is contained within an external sheath extending from the tendons called the epimysium. Fascicles within the muscle are enveloped by a sheath called the perimysium. Single muscle fibers within individual fascicles are surrounded by a sheath called the endomysium. The highly organized architecture of skeletal muscle fibers and connective tissue allows skeletal muscle to generate considerable mechanical force in a vectorial manner. Beneath the endomysium surrounding each muscle fiber is the plasma membrane of the muscle cell called the sarcolemma. An individual skeletal muscle cell contains a densely arranged parallel array of cylindrical elements called myofibrils. Each myofibril is essentially an end-to-end chain of regular repeating units—or sarcomeres—that consist of smaller interdigitating filaments called myofilaments, which contain both thin filaments and thick filaments.
Figure 9-1 Structure of skeletal muscle.
As discussed in Chapter 8, a motor nerve axon contacts each muscle fiber to form a synapse near the middle of the fiber called the neuromuscular junction. The specialized region of sarcolemma in closest contact with the presynaptic nerve terminal is called the motor end plate. Although skeletal muscle fibers can be artificially excited by direct electrical stimulation, physiological excitation of skeletal muscle always involves chemical activation by release of acetylcholine (ACh) from the motor nerve terminal. Binding of ACh to the nicotinic receptor gives rise to a graded depolarizing end-plate potential. An end-plate potential of sufficient magnitude triggers a propagating action potential in the region of sarcolemma adjacent to the end plate by raising the membrane potential to the firing threshold.
All skeletal muscle is under voluntary or reflex control by motor neurons of the somatic motor system. Somatic motor neurons are efferent neurons with cell bodies located in the central nervous system (CNS). A single muscle cell responds to only a single motor neuron whose cell body—except for cranial nerves—resides in the ventral horn of the spinal cord. However, the axon of a motor neuron typically branches near its termination to innervate a few or many individual muscle cells. The group of muscle fibers innervated by all of the collateral branches of a single motor neuron is referred to as a motor unit. A whole muscle can produce a wide range of forces and a graded range of shortening by varying the number of motor units excited within the muscle. The innervation ratio of a whole skeletal muscle is defined as the number of muscle fibers innervated by a single motor neuron. Muscles with a small innervation ratio control fine movements involving small forces. For example, fine, high-precision movements of the extraocular muscles that control positioning movements of the eye are achieved through an innervation ratio of as little as ~3 muscle fibers per neuron. Conversely, muscles with a large innervation ratio control coarse movement requiring development of large forces. Postural control by the soleus muscle uses an innervation ratio of ~200. The gastrocnemius muscle, which is capable of developing large forces required in athletic activities such as jumping, has innervation ratios that vary from ~100 to ~1000.
Although the ultimate intracellular signal that triggers and sustains contraction of skeletal, cardiac, or smooth muscle cells is a rise in [Ca2+]i, the three types of muscle cells differ substantially in the detailed mechanism by which a depolarization of the sarcolemmal membrane results in a rise in [Ca2+]i. Ca2+ can enter the cytoplasm from the extracellular space through voltage-gated ion channels, or alternatively, Ca2+ can be released into the cytoplasm from the intracellular Ca2+ storage reservoir of the sarcoplasmic reticulum. Thus, both extracellular and intracellular sources may contribute to the increase in [Ca2+]i. However, the relative importance of these two sources of Ca2+ varies among the different muscle types. The process by which electrical “excitation” of the surface membrane triggers an increase of [Ca2+]i in muscle is known as excitation-contraction coupling or EC coupling.
Action potentials originating at the surface membrane of skeletal and cardiac muscle fibers propagate into the interior of the cell through specialized membrane invaginations. In skeletal and cardiac muscle, these invaginations take the form of radially projecting tubes of membrane called transverse tubules or T tubules (Fig. 9-2A). T tubules penetrate the muscle fiber and surround the myofibrils at two points in each sarcomere: at the junctions of the A and I bands. A cross section through the A-I junction shows a complex, branching array of T tubules penetrating to the center of the muscle cell and surrounding the individual myofibrils. Along its length, the tubule associates with two cisternae, which are specialized regions of the sarcoplasmic reticulum (SR). The SR of muscle cells is a specialized version of the endoplasmic reticulum of noncontractile cells and serves as a storage organelle for intracellular Ca2+. The combination of the T-tubule membrane and its two neighboring cisternae is called a triad; this structure plays a crucial role in the coupling of excitation to contraction in skeletal and cardiac muscle. Smooth muscle, in contrast, has more rudimentary and shallow invaginations called caveolae (Fig. 9-2B).
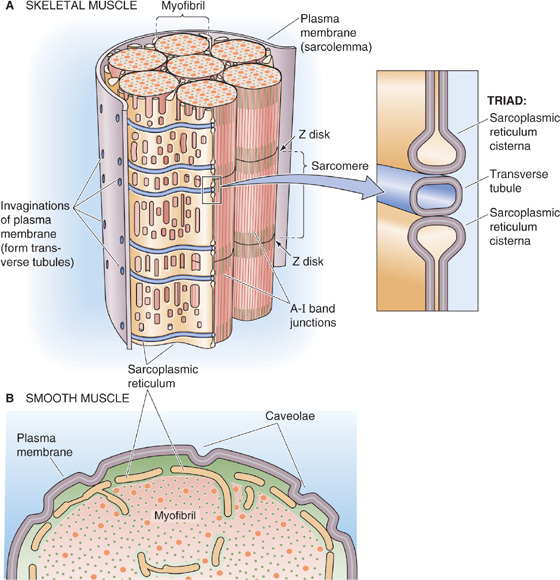
Figure 9-2 Plasma membrane invaginations. A, The transverse tubules (T tubules) are extensions of the plasma membrane, penetrating the muscle cell at two points in each sarcomere: the junctions of the A and I bands. B, Smooth muscle cells have rudimentary invaginations of the plasma membrane, called caveolae, contacting the sarcoplasmic reticulum.
The propagation of the action potential into the T tubules depolarizes the triad region of the T tubules, activating L-type Ca2+ channels (see Chapter 7). These voltage-gated channels cluster in groups of four called tetrads (Fig. 9-3) and have a pivotal role as the voltage sensor in EC coupling. Electron microscopy reveals a checkerboard pattern of projections arising from the T-tubule membrane and extending toward the cisternae of the SR; these projections probably represent the cytoplasmic face of these L-type Ca2+ channels. Functional complexes of L-type Ca2+ channels contain the α1-subunit of the voltage-gated Ca2+ channel as well as the accessory α2-δ, β, and γ subunits (see Fig. 7-12B). The L-type Ca2+ channel is also often referred to as the DHP receptor because it is inhibited by a class of antihypertensive and antiarrhythmic drugs known as dihydropyridines. Depolarization of the T-tubule membrane produces conformational changes in each of the four voltage-activated L-type Ca2+ channels of the tetrad, resulting in two major effects. First, the conformational changes allow Ca2+ to enter through the four channel pores. Second, and more important in skeletal muscle, the conformational changes in the four L-type Ca2+ channels induce a conformational change in each of the four subunits of another channel—the Ca2+-release channel—that is located in the SR membrane.
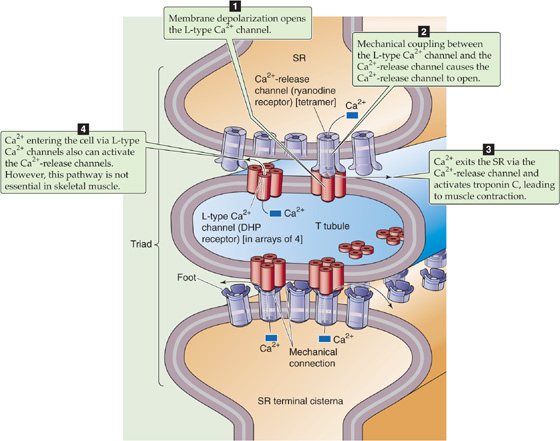
Figure 9-3 EC coupling in skeletal muscle. A tetrad of four L-type Ca2+ channels on the T tubules faces a single Ca2+-release channel of the SR, so that each L-type Ca2+ channel interacts with the foot of one of the four subunits of the Ca2+-release channel. Note that half of the Ca2+-release channels lack associations with L-type Ca2+ channels. DHP, dihydropyridine.
The Ca2+-release channel (see Fig. 6-21S) has a homotetrameric structure quite different from that of the L-type Ca2+ channel that constitutes the voltage sensor for EC coupling. The Ca2+-release channel in the SR is also known as the ryanodine receptor because it is inhibited by a class of drugs that include the plant alkaloids ryanodine and caffeine. Ca2+-release channels cluster in the portion of the SR membrane that faces the T tubules. Each of the four subunits of these channels has a large extension—also known as a foot. These feet project as a regular array into the cytosol. The foot of each of the four Ca2+-release channel subunits is complementary to the cytoplasmic projection of one of the four L-type Ca2+ channels in a tetrad on the T tubule (Fig. 9-3). The physical proximity of these two proteins as well as the ability of both DHP and ryanodine to block muscle contraction suggests that physical and mechanical interaction between these two different Ca2+ channels underlies EC coupling. The precise mechanism of interaction between these proteins is not entirely understood, although it is not solely electrical in nature inasmuch as ionic conductance of the Ca2+-release channel is not strongly voltage dependent. A large cytoplasmic projection on the α1 subunit of the L-type Ca2+ channel appears to be necessary for interaction between the two Ca2+ channels on opposing T-tubule and SR membranes. Thus, it is likely that direct mechanical coupling exists between this projection and the Ca2+-release channel. On this basis, the mechanism of EC coupling in skeletal muscle is termed an electromechanical coupling mechanism.
After depolarization of the L-type Ca2+ channel on the T-tubule membrane and mechanical activation of the Ca2+-release channel in the SR, Ca2+ stored in the SR rapidly leaves through the Ca2+-release channel. The resultant rapid increase in [Ca2+]i activates troponin C, initiating formation of cross-bridges between myofilaments, as described later. EC coupling in skeletal muscle thus includes the entire process we have just described, beginning with the depolarization of the T-tubule membrane to the initiation of the cross-bridge cycle of contraction.
Although we have stressed that EC coupling in skeletal muscle primarily involves direct mechanical coupling between the L-type Ca2+ channel in the T-tubule membrane and the Ca2+-release channel of the SR, a second mechanism for activating the Ca2+-release channel may also contribute. Intracellular Ca2+ can directly activate the Ca2+-release channel in the SR—a process known as Ca2+-induced Ca2+ release (CICR). The source of the elevated [Ca2+]i for CICR is the Ca2+ released from the SR, as well as the Ca2+ that enters the cell via L-type Ca2+ channels during action potentials. However, this influx of external Ca2+ from the lumen of T tubules is not required for contraction of mammalian skeletal muscle. Indeed, skeletal muscle contraction persists even when Ca2+ is absent from the extracellular fluid of the muscle fiber. As described below, Ca2+ influx through L-type Ca2+ channels nevertheless plays an essential role in EC coupling in cardiac muscle.
There are two types of myofilaments: thick filaments composed primarily of a protein called myosin and thin filaments largely composed of a protein called actin (see Chapter 2). The sarcomere is defined as the repeating unit between adjacent Z disks or Z lines (Fig. 9-4A and B). A myofibril is thus a linear array of sarcomeres stacked end to end. The highly organized sarcomeres within skeletal and cardiac muscle are responsible for the striped or striated appearance of muscle fibers of these tissues as visualized by various microscopic imaging techniques. Thus, both skeletal muscle and cardiac muscle are referred to as striated muscle. In contrast, smooth muscle lacks striations because actin and myosin have a less regular pattern of organization in these myocytes.
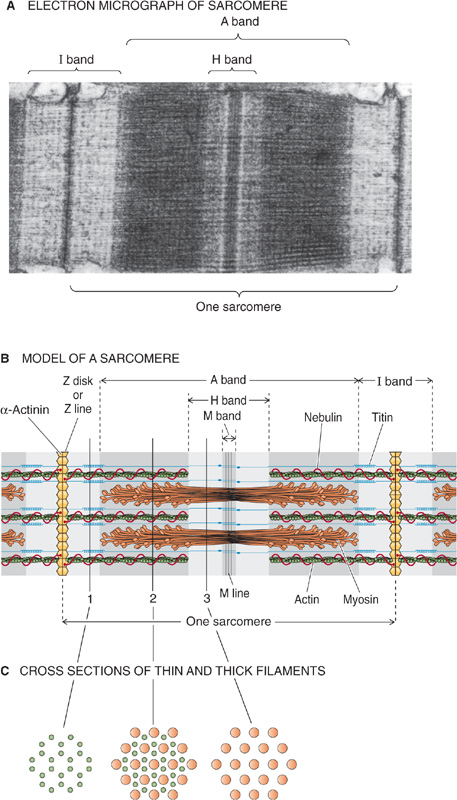
Figure 9-4 Structure of the sarcomere.
In striated muscle, thin filaments are 5 to 8 nm in diameter and 1 μm in length. Thin filaments are tethered together at one end, where they project from a dense disk known as the Z disk (Fig. 9-4B). The Z disk is oriented perpendicular to the axis of the myofibril and has the diameter of the myofibril. Thin filaments project from both faces of the Z disk. Not only do Z disks tether the thin filaments of a single myofibril together, but connections between the Z disks also tether each myofibril to its neighbors and align the sarcomeres.
The thick filaments are 10 nm in diameter and, in striated muscle, 1.6 μm in length (Fig. 9-4B). They lie between and partially interdigitate with the thin filaments. This partial interdigitation results in alternating light and dark bands along the axis of the myofibril. The light bands, which represent regions of the thin filament that do not overlap with thick filaments, are known as I bands because they are isotropic to polarized light as demonstrated by polarization microscopy. The Z disk is visible as a dark perpendicular line at the center of the I band. The dark bands, which represent the myosin filaments, are known as A bands because they are anisotropic to polarized light. During contraction, the A bands are unchanged in length, whereas the I bands shorten.
When the A band is viewed in cross section where the thick and thin filaments overlap, six thin filaments (actin) surround each thick filament (myosin) in a tightly packed hexagonal array (Fig. 9-4C). Within the A bands, the pivoting heads of the thick myosin filaments, the molecular motors, establish cross-bridges to the thin actin filaments. As discussed later, the ATP-dependent cycle of making and breaking cross-bridges causes the actin filament to be drawn over the myosin filament and thereby results in muscle contraction.
Thin Filaments The backbone of the thin filament is a double-stranded α-helical polymer of actin molecules (Fig. 9-5A). Each helical turn of a single strand of filamentous actin or F-actin consists of 13 individual actin monomers and is ~70 nm long. F-actin is associated with two important regulatory, actin-binding proteins: tropomyosin and troponin.
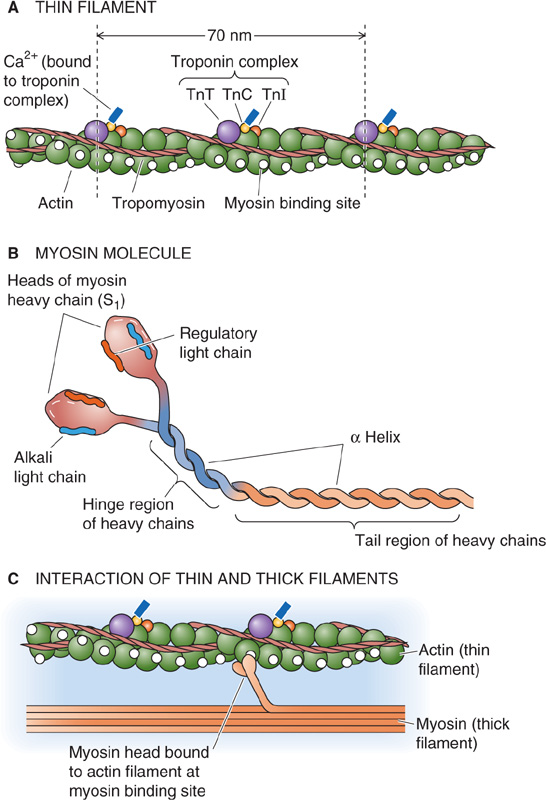
Figure 9-5 Structure of thin and thick filaments.
Individual tropomyosin molecules consist of two identical α helices that coil around each other and sit near the two grooves that are formed by the two helical actin strands. Head-to-tail contact between neighboring tropomyosin molecules results in two nearly continuous helical filaments that shadow the actin double helix. The length of a single tropomyosin molecule corresponds to about seven actin monomers (i.e., a half turn of the actin helix). As we describe later, the role of tropomyosin is to regulate the binding of myosin head groups to actin.
Troponin is a heterotrimer consisting of (1) troponin T (TnT or TNNT), which binds to a single molecule of tropomyosin; (2) troponin C (TnC or TNNC), which binds Ca2+; and (3) troponin I (TnI or TNNI), which binds to actin and inhibits contraction. Troponin C is closely related to another Ca2+-binding protein, calmodulin (CaM; see Chapter 3). Thus, each troponin heterotrimer interacts with a single tropomyosin molecule, which in turn interacts with seven actin monomers. The troponin complex also interacts directly with the actin filaments. The coordinated interaction among troponin, tropomyosin, and actin allows actin-myosin interactions to be regulated by changes in [Ca2+]i.
Thick Filaments Like actin thin filaments, thick filaments are also an intertwined complex of proteins (Fig. 9-5B). A thick filament is a bipolar assembly of multiple myosin II molecules. Each myosin II molecule is a hexamer (actually two trimers) composed of two intertwined heavy chains, two alkali (or essential) light chains, and two regulatory light chains. The two heavy chains have three regions: a rod, a hinge, and a head region. The rod portions are α helices that wrap around each other. At the hinge regions, the molecule flares open to form two globular heads, which are the cross-bridges between the thick and thin filaments of the sarcomere. The heads of the heavy chains—also called S1 fragments—each possess a site for binding actin as well as a site for binding and hydrolyzing ATP. The head portion of each myosin forms a complex with two light chains, one regulatory and one alkali. The alkali light chain plays an essential role in stabilizing the myosin head region. The regulatory light chain, as its name implies, regulates the ATPase activity of myosin. The activity of the myosin regulatory light chain is in turn regulated through phosphorylation by Ca2+-dependent and Ca2+-independent kinases. Figure 9-5C summarizes the interaction between a thin filament and a single pair of myosin head groups from a thick filament.
Running alongside the thick filaments of skeletal muscle is a large protein called titin. Titin is the largest known protein, with a linear sequence of ~25,000 amino acids (~3000 kDa). Titin is tethered from the M line in the middle of the A band to each of the neighboring Z disks. The M line also serves as the attachment site for myosin molecules within the thick filament. Titin appears to be involved in the elastic behavior of muscle by maintaining the resting length of muscle during relaxation. Nebulin is another large protein (600 to 900 kDa) of muscle that runs from the Z disk along the actin thin filaments.
Underlying muscle contraction is a cycle in which myosin II heads bind to actin, these cross-bridges become distorted, and finally the myosin heads detach from actin. Energy for this cycling comes from the hydrolysis of ATP. However, if unregulated, the cycling would continue until the myocyte is depleted of ATP. It is not surprising, then, that skeletal, cardiac, and smooth muscle have particular mechanisms for regulating cross-bridge cycling. In all three cell types, an increase in [Ca2+]i initiates and allows cross-bridge cycling to continue. During this excitatory increase, [Ca2+]i may rise from its resting level of less than 10−7 M to greater than 10−5 M. The subsequent decrease in [Ca2+]i is the signal to cease cross-bridge cycling and to relax. The tightly regulated decrease in [Ca2+]i is achieved by transport processes that remove Ca2+ from the sarcoplasm, the term used for the cytoplasm of a muscle fiber.
Regardless of the muscle type, Ca2+ exerts its effect by binding to regulatory proteins rather than directly interacting with contractile proteins. In the absence of Ca2+, these regulatory proteins act in concert to inhibit actin-myosin interactions, thus inhibiting the contractile process. When Ca2+ binds to one or more of these proteins, a conformational change takes place in the regulatory complex that releases the inhibition of contraction.
In skeletal muscle, the troponin C (TNNC2 subtype) has two pairs of Ca2+-binding sites. Two high-affinity sites—located on the C-lobe of TNNC2—are aways occupied by Ca2+ or Mg2+ under physiological conditions. On the other hand, two low-affinity sites—located on the N-lobe of TNNC2—bind and release Ca2+ as [Ca2+]i rises and falls in the sarcoplasm, thereby regulating the binding of actin to myosin (Fig. 9-6). Binding of Ca2+ to these low-affinity sites induces a conformational change in the troponin complex that has two effects. The first effect is that the C terminus of the inhibitory troponin I moves away from the actin/tropomyosin filament, thereby permitting the tropomyosin molecule to move. According to one hypothesis, the other effect, transmitted through troponin T, is to push tropomyosin away from the myosin binding site on the actin and into the actin groove. With the steric hindrance removed, the myosin head is able to interact with actin and to engage in cross-bridge cycling. (See Note: Tropomyosin–Troponin Interactions—The “Functional Group” of the Thin Filament)
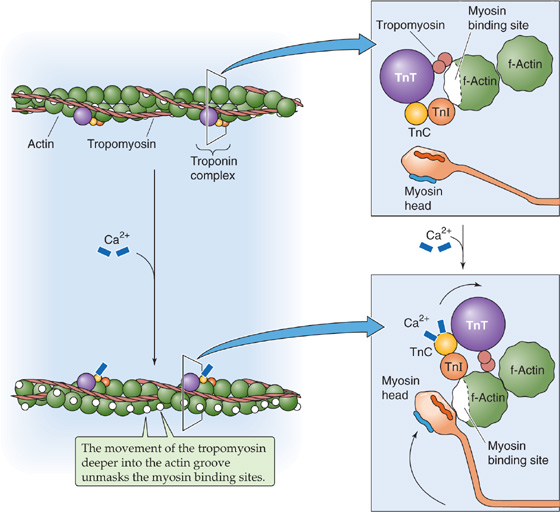
Figure 9-6 The role of Ca2+ in triggering the contraction of skeletal and cardiac muscle.
The cross-bridge cycle that we introduced in the preceding section occurs in five steps (Fig. 9-7). Initially, the myosin head is attached to an actin filament after the “power stroke” from the previous cycle and after the actomyosin complex has released ADP. In the absence of ATP, the system could remain locked in the rigid conformation of the ADP-bound actomyosin complex for an indefinitely long period limited only by protein decomposition. Such a state of extreme muscle rigidity called rigor mortis develops in a corpse soon after death from lack of ATP after cessation of metabolism. In this rigid state, the myosin head is fixed at a 45-degree angle with respect to the actin and myosin filaments.
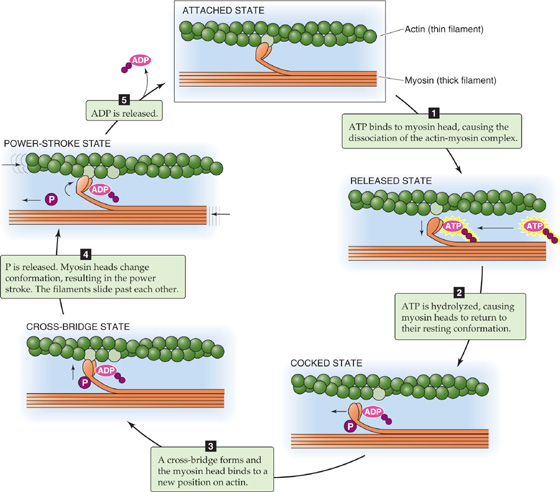
Figure 9-7 The cross-bridge cycle in skeletal and cardiac muscle. Each cycle advances the myosin head by two actin monomers, or ~11 nm.
Step 1: ATP binding. ATP binding to the head of the myosin heavy chain (MHC) reduces the affinity of myosin for actin, causing the myosin head to release from the actin filament. If all cross-bridges in a muscle were in this state, the muscle would be fully relaxed.
Step 2: ATP hydrolysis. The breakdown of ATP to ADP and inorganic phosphate (Pi) occurs on the myosin head; the products of hydrolysis are retained on the myosin. As a result of hydrolysis, the myosin head pivots around the hinge into a “cocked” position (perpendicular or at a 90-degree angle to the thick and thin filaments). This rotation causes the tip of the myosin to move ~11 nm along the actin filament so that it now lines up with a new actin monomer two monomers farther along the actin filament (see the box titled Measuring the Force of a Single Cross-Bridge Cycle). Once again, if all cross-bridges in a muscle were in this state, the muscle would be fully relaxed.
Step 3: Cross-bridge formation. The cocked myosin head now binds to its new position on the actin filament. This binding reflects the increased affinity of the myosin-ADPPi complex for actin.
Step 4: Release of Pi from the myosin. Dissociation of Pi from the myosin head triggers the power stroke, a conformational change in which the myosin head bends ~45 degrees about the hinge and pulls the actin filament ~11 nm toward the tail of the myosin molecule. This conformational change causes the actin filament to be drawn along the myosin filament, thereby generating force and motion.
Step 5: ADP release. Dissociation of ADP from myosin completes the cycle, and the actomyosin complex is left in a rigid state. The myosin head remains in the same position and at a 45-degree angle with respect to the thick and thin filaments. The ADP–free myosin complex remains bound to actin until another ATP molecule binds and initiates another cycle.
The ADP–free myosin complex (“attached state” in Fig. 9-7) would quickly bind ATP at the concentrations of ATP normally found within cells. If unrestrained, this cross-bridge cycling would continue until the cytoplasm is depleted of ATP. At that time, the muscle would remain in the stiff attached state because release of the cross-bridges from actin requires binding of ATP to myosin.
These steps show that [ATP]i does not regulate the cross-bridge cycle of actin-myosin interaction. Skeletal and cardiac muscle control the cycle of contraction at the third step by preventing cross-bridge formation until the tropomyosin moves out of the way in response to an increase in [Ca2+]i.
Each round of the cross-bridge cycle consumes one molecule of ATP. In skeletal muscle, the entire cellular store of ATP is sufficient to allow only a few seconds of continuous maximal contraction. Therefore, the muscle cell must resynthesize ATP from ADP at a rate comparable to the rate of ATP consumption. Skeletal muscle has specialized energy stores that permit rapid regeneration of ATP. The most readily available pool of this energy is the high-energy phosphate bond of phosphocreatine. The enzyme creatine kinase transfers the high-energy phosphate of phosphocreatine to ADP, thereby rephosphorylating ADP to ATP. The phosphocreatine content of skeletal muscle is adequate to replenish the ATP pool several times, but it is still inadequate to sustain the energy needs of contracting muscle for more than 10 seconds.
In comparison with the energy stored as phosphocreatine, glycogen is a far more abundant energy source within skeletal muscle. Glycogen that has been previously stored by muscle can be enzymatically degraded to pyruvic acid. Degradation of glycogen to pyruvate is rapid and liberates energy that the cell invests in phosphorylating ADP to yield ATP. Pyruvate is further metabolized along with other foodstuffs by oxidative metabolism, which during the long term is the primary mechanism for the regeneration of ATP (see Chapter 58). The rate of ATP generation by oxidative metabolism is limited by the rate of oxygen delivery to the muscle. However, glycolytic formation of pyruvate occurs independently of oxygen, as does the conversion of pyruvate to lactate. The pathway of anaerobic metabolism of muscle glycogen ensures that energy stores are sufficient to sustain muscle activity for nearly a minute even when oxygen is unavailable. In Chapter 60, we will discuss the aerobic and anaerobic metabolism of exercising muscle in more depth.
After the action potential in the skeletal muscle has subsided, Ca2+ must be removed from the cytoplasm for contraction to actually cease and for relaxation to occur. Removal of Ca2+ from the sarcoplasm occurs by two mechanisms. Ca2+ may be extruded across the cell plasma membrane or sequestered within intracellular compartments (Fig. 9-8).
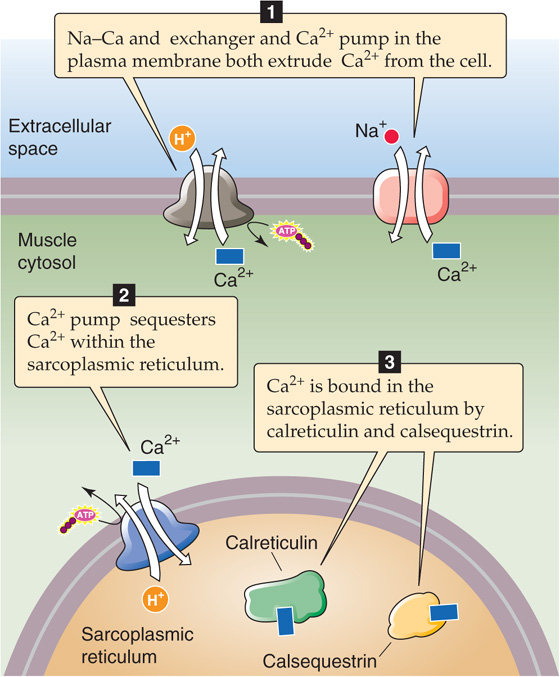
Figure 9-8 Mechanisms of Ca2+ removal from the cytoplasm.
The cell may extrude Ca2+ by use of either the Na-Ca exchanger (NCX or SLC8) or the Ca2+ pump at the plasma membrane (PMCA). Extrusion across the cell membrane, however, would eventually totally deplete the cell of Ca2+ and is therefore a minor mechanism for Ca2+ removal from the cytoplasm. Instead, Ca2+ re-uptake into the SR is the most important mechanism by which the cell returns [Ca2+]i to resting levels. Ca2+ re-uptake by the SR is mediated by a sarcoplasmic and endoplasmic reticulum Ca2+-ATPase (SERCA)–type Ca2+ pump (see Chapter 5). (See Note: SERCA Isoforms)
High [Ca2+] within the SR lumen inhibits the activity of SERCA. This inhibition of SR Ca2+ pump activity is delayed by Ca2+-binding proteins within the SR lumen. These Ca2+-binding proteins buffer the [Ca2+] increase in the SR during Ca2+ re-uptake and thus markedly increase the Ca2+ capacity of the SR. The principal Ca2+-binding protein in skeletal muscle, calsequestrin, is also present in cardiac and some smooth muscle. Calreticulin is a ubiquitous Ca2+-binding protein that is found in particularly high concentrations within the SR of smooth muscle. These proteins have a tremendous capacity to bind Ca2+, with up to 50 binding sites per protein molecule.
Ca2+-binding proteins are not located diffusely within the SR. Rather, calsequestrin is highly localized to the region of the SR immediately beneath the triad junction. Calsequestrin appears to bind directly at the triad, where it forms a complex with the Ca2+-release channel and with two other triad proteins, junctin and triadin. Here, calsequestrin is poised not only to aid muscle relaxation by buffering Ca2+ within the SR lumen but also to unload its Ca2+ in the vicinity of the Ca2+-release channel and thus facilitate EC coupling. It has been hypothesized that EC coupling promotes Ca2+ release from calsequestrin, making Ca2+ available for exit from the SR.
The total force generated by a muscle is the sum of the forces generated by many independently cycling actin-myosin cross-bridges. The number of simultaneously cycling cross-bridges depends substantially on the initial length of the muscle fiber and on the pattern or frequency of muscle cell stimulation. When muscle is stimulated to contract, it exerts a force tending to pull the attachment points at either end toward each other. This force is referred to as the tension developed by the muscle.
Two mechanical—and artificial—arrangements can be used to study muscle contraction. In one, the attachment points are immobile, thereby fixing the muscle length. Here, stimulation causes an increase in tension, but no shortening. Because these contractions occur at constant length, they are referred to as isometric contractions (Fig. 9-9A). In the second arrangement, one of the two attachment points is mobile, and a force—or load—tends to pull this mobile point away from the fixed one. Here, stimulation causes shortening, provided the tension developed by the muscle is greater than the opposing load. Because these shortenings occur at constant load, they are referred to as isotonic contractions (see Fig. 9-9B). Both isometric and isotonic contractions can be examined at different initial muscle lengths. Moreover, they can be measured during individual muscle twitches that are evoked by single muscle action potentials as well as during other patterns of stimulation.
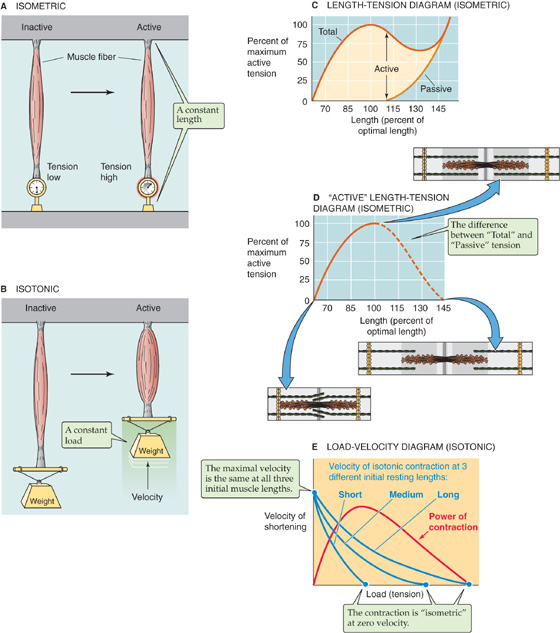
Figure 9-9 Isometric and isotonic contraction. A, Experimental preparation for study of muscle contraction under isometric conditions. B, Experimental preparation for study of muscle contraction under isotonic conditions. C, The passive curve represents the tension that is measured at various muscle lengths before muscle contraction. The total curve represents the tension that is measured at various muscle lengths during muscle contraction. Muscle length is expressed as the percent of “optimal” length, that is, the length at which active isometric tension is maximal. D, The active tension is the difference between the total and the passive tensions in C. E, Each of the three blue curves shows that the velocity of muscle shortening is faster if the muscle lifts a lighter weight—it is easier to lift a feather (left side of each curve/low load) than to lift a barbell (right side of each curve/high load). The three blue curves also show that for any given velocity of shortening, a longer muscle can develop a greater tension than can a shorter muscle.
Malignant Hyperthermia
Malignant hyperthermia (MH) is a genetic disorder affecting between 1 in 10,000 and 1 in 50,000 individuals. Affected individuals are at risk for a potentially life-threatening syndrome on exposure to any of the various inhalation anesthetic agents, particularly halothane. Administration of succinylcholine can also trigger or exaggerate MH. This drug is a short-acting inotropic (nicotinic) ACh receptor antagonist that acts by first opening the ACh receptor channel and then blocking it, thereby resulting in a burst of muscle activity, followed by paralysis. Onset of the syndrome in the setting of the operating room is typified by the development of tachypnea (rapid breathing), low plasma [O2], high plasma [CO2], tachycardia (rapid heart rate), and hyperthermia (rising body temperature) as well as by rigidity, sweating, and dramatic swings in blood pressure. The patient’s temperature may rise as rapidly as 1°C every 5 minutes. The onset of MH is usually during anesthesia, but it can occur up to several hours later. If untreated, the patient will develop respiratory and lactic acidosis, muscle rigidity, and a breakdown of muscle tissue that leads to the release of K+ and thus profound hyperkalemia. These episodes reflect a progressively severe hypermetabolic state in the muscle tissues. Fortunately, our evolving understanding of the physiological process of MH has led to the development of a therapeutic regimen that has greatly improved the once-dismal prognosis.
The major features of the syndrome—hyperthermia, muscle rigidity, and an increased metabolic rate—led early investigators to suggest that MH is a disease of abnormal regulation of muscle contraction. According to this hypothesis, uncontrolled muscle contraction—somehow triggered by the administration of halothane and succinylcholine—causes excessive ATP hydrolysis to provide energy for contraction. The increased rate of ATP hydrolysis leads to an increased metabolic rate as muscle tries to replenish and to sustain its ATP stores. Hyperthermia develops because of the heat liberated by the hydrolysis of ATP.
Further support for this hypothesis came from the observation that more tension developed in muscle fibers obtained by biopsy from susceptible individuals than in fibers from normal individuals when the fibers were exposed to halothane. In muscle fibers from both humans and a strain of swine susceptible to MH, Ca2+-induced Ca2+ release from the SR is enhanced compared with fibers from unaffected subjects. Furthermore, caffeine, which causes the Ca2+-release channels to open, induced greater contractions in fibers from susceptible subjects. Taken together, these observations suggested the possibility that MH results from an abnormality in the Ca2+-release channel in the SR membrane.
In both humans and animals, inheritance of MH follows a mendelian autosomal dominant pattern. Cloning of the gene (RYR1) encoding the Ca2+-release channel (ryanodine receptor) allowed genetic linkage analysis to demonstrate that human MH is closely linked in some families to the RYR1 gene on chromosome 19. In swine, MH results from a single amino acid substitution in RYR1 (Cys for Arg at position 614). An analogous substitution is present in some human kindreds as well. This substitution increases the probability that the Ca2+-release channel will be open. In other families, MH has been associated with other genetic abnormalities in the RYR1 gene. In still others, MH does not appear to be genetically linked to the RYR1 gene. It is possible that defects in other steps along the excitation-contraction cascade can result in abnormal regulation of muscle contraction and the MH phenotype. For example, when they are under anesthesia, patients with some forms of muscular dystrophy may have metabolic crises that resemble MH.
MH also occurs in domestic livestock. The incidence of MH is particularly high in swine, and episodes are triggered by a variety of physical and environmental stresses (porcine stress syndrome). MH in animals has significant economic importance in view of the potential loss from fatal episodes and the devaluation of meat as a result of muscle destruction during nonfatal episodes.
In humans, a condition similar to MH may occur in patients treated with neuroleptic agents such as the phenothiazines or haloperidol. It is called the neuroleptic malignant syndrome and appears to result from abnormally high neuronal input to the muscle cells.
Therapy for MH now involves administration of the drug dantrolene, cessation of anesthesia, and aggressive efforts aimed at cooling the body. Dantrolene is an effective therapeutic agent because it blocks EC coupling between T tubules and the SR, thus interrupting the otherwise uncontrolled progression of muscle contractions. The drug can be given acutely in an effort to abort an ongoing attack, or in a person known to be at risk, it can be given before the initiation of anesthesia to prevent onset of the syndrome. Therapy also includes intravenous hydration and the judicious use of diuretics to keep the urine flowing; this lessens damage to the kidneys from the release of breakdown products, such as myoglobin from the damaged muscles. Sodium bicarbonate is given to counter the lactic acidosis, and patients may be mechanically hyperventilated to blow off the excess CO2.
Despite the intensive protocol just outlined, MH is still associated with high mortality. The relatives of a patient with a documented history of one episode of MH should be carefully screened to see whether they, too, carry the inherited trait; many of the affected relatives may demonstrate baseline elevations in muscle enzyme levels in their blood (e.g., an increase in creatine kinase levels).
The isometric force of contractions depends on the initial length of the muscle fiber. Unstimulated muscle may be elongated somewhat by applying tension and stretching it. The tension measured before muscle contraction is referred to as passive tension (Fig. 9-9C). Because muscle gets stiffer as it is distended, it takes increasing amounts of passive tension to progressively elongate the muscle cell. If at any fixed length (i.e., isometric conditions) the muscle is stimulated to contract, an additional active tension develops because of cross-bridge cycling. The total measured tension is thus the sum of the passive and active tensions. This incremental or active tension—the difference between total tension and passive tension—is quite small when the muscle is less than ~70% of its normal resting length (Fig. 9-9D). As muscle length increases toward its normal length, active tension increases. Active tension is maximal at a length—usually called L0—that is near the normal muscle length. Active tension decreases with further lengthening; thus, active tension is again small when the muscle is stretched beyond 150% of its normal resting length. Although the relationship between muscle length and tension has been best characterized for skeletal muscle, the tension of cardiac and smooth muscle also appears to depend on length in a similar manner.
Measuring the Force of a Single Cross-Bridge Cycle
The force of a single cross-bridge cycle has been measured directly. Finer, Simmons, and Spudich used optical tweezers to manipulate a single actin filament and to place it in proximity to a myosin molecule immobilized on a bead (Fig. 9-10). With the use of video-enhanced microscopy, these investigators were able to detect movements of the actin filament as small as 1 nm. The optical tweezers could also exert an adjustable force opposing movement of the actin filament. When the tweezers applied only a small opposing force and the experiment was conducted in the presence of ATP, they observed that the actin moved over the myosin bead in step-like displacements of 11 nm. This observation, made under “microscopically isotonic” conditions, suggests that the quantal displacement of a single cross-bridge cycle is ~11 nm. When the tweezers applied a force sufficiently large to immobilize the actin filament, the investigators observed step-like impulses of force that averaged ~5 pN. This observation, made under “microscopically isometric” conditions, suggests that the quantal force developed during a single cross-bridge cycle is ~5 pN. Interestingly, these isometric force impulses lasted longer when the ATP concentration was lower. This last finding is consistent with the notion that ATP binding to myosin must occur to allow detachment of the cross-bridges (step 1 in the cycle in Fig. 9-7).
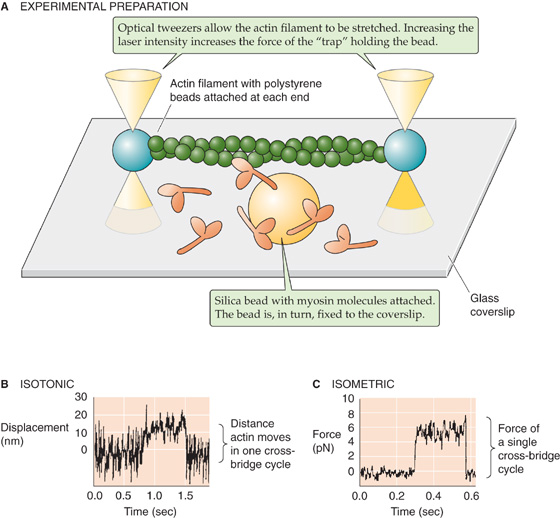
Figure 9-10 Microscopic measurements of cross-bridge force and displacement. A, An actin filament is attached at each end to a polystyrene bead. The optical tweezers, a finely focused beam of laser light, can trap the bead at its focal point and physically move it. By adjusting the laser intensity, the experimenter can alter the strength of the trap (i.e., the force with which the bead is held). In this experiment, two optical tweezers were used to suspend the actin filament above a coverglass. Attached to this coverglass is a silica bead, and myosin molecules are bound to the bead. B, In an isotonic experiment, the force between the actin filament and the fixed myosin/silica bead is kept constant by use of a stable laser intensity. The experimenter measures, as a function of time, the displacement of the polystyrene bead away from the center of the trap. Thus, in one cross-bridge cycle, the myosin-actin interaction pulls the polystyrene bead ~11 nm away from the center of the trap. C, In an isometric experiment, the experimenter measures, as a function of time, the extra force that needs to be applied (i.e., increase in laser intensity) to keep the polystyrene bead at a fixed position near the center of the trap. Thus, in one cross-bridge cycle, the myosin-actin interaction exerts a force of ~5 pN. (Data from Finer JT, Mehta AD, Spudich JA: Characterization of single actinmyosin interactions. Biophys J 1995; 68:291s-296s.)
This length-tension relationship is a direct result of the anatomy of the thick and thin filaments within individual sarcomeres (Fig. 9-9D). As muscle length increases, the ends of the actin filaments arising from neighboring Z disks are pulled away from each other. When length is increased beyond 150% of its resting sarcomere length, the ends of the actin filaments are pulled beyond the ends of the myosin filaments. Under this condition, no interaction occurs between actin and myosin filaments and hence no development of active tension. As muscle length shortens from this point, actin and myosin filaments begin to overlap and tension can develop; the amount of tension developed corresponds to the degree of overlap between the actin and the myosin filaments. As the muscle shortens further, opposing actin filaments slide over one another and the ends of the myosin filaments and—with extreme degrees of shortening—eventually butt up against the opposing Z disks. Under these conditions, the spatial relationship between actin and myosin is distorted and active tension falls. The maximal degree of overlap between actin and myosin filaments, and hence maximal active tension, corresponds to a sarcomere length that is near its normal resting length.
Under isotonic conditions, the velocity of shortening decreases as the applied load opposing contraction of the muscle fiber increases. This point is obvious; anyone can lift a single French fry much faster than a sack of potatoes. As shown for any of the three blue curves in Figure 9-9E—each of which represents a different initial length of muscle—the relationship between velocity and load is hyperbolic. Thus, the smaller the load applied to the muscle, the greater its velocity of shortening. Conversely, the greater the load, the lower the velocity of shortening.
The load (or tension)–velocity relationship is perhaps best understood by considering the situation at maximum load for a resting muscle length (i.e., isometric conditions). This situation is represented by the upper blue curve in Figure 9-9E. At any time, all the available cross-bridges are engaged in resisting the opposing force. None are left over to make the muscle shorten. If the number of engaged cross-bridges were decreased, the muscle would lengthen. At a slightly smaller load but at the same isotonic muscle length, fewer cross-bridges need to be engaged to resist the opposing load. Thus, extra cross-bridges are available to ratchet the thick myosin filaments over the thin actin filaments, but at a very low velocity. At a still lower load, even more cross-bridges are available for ratcheting the myosin over the actin, and the velocity increases further. At very low loads, it is reasonable to expect that as the myosin filament slides along the actin filament, only a tiny fraction of the actin monomers need to interact with myosin heads to overcome the load. Under these conditions of vanishingly small loads, the speed with which the thick and thin filaments slide over each other is limited only by the time that it takes for the ATP-consuming cross-bridge cycle to occur. With increasing velocity, the probability of actin-myosin interactions decreases. Thus, fewer cross-bridges are simultaneously active at higher shortening velocities, and less tension develops.
Note that the upper blue curve in Figure 9-9E applies to a particular initial length of the muscle, that is, the resting length. We already saw in Figure 9-9C that the total isometric tension (i.e., the maximal load that the muscle can sustain at zero velocity) increases with initial muscle length. This principle is confirmed in Figure 9-9E: the longer the initial length, the larger the maximal load under zero-velocity conditions (i.e., the three different intercepts with the abscissa). In contrast to this maximal load, which depends very much on length, maximal velocity is independent of length, as shown by the common intercept of the family of curves with the ordinate. The explanation for this effect, as we have already noted, is that maximal velocity (at no load) depends on the maximal rate of cross-bridge turnover, not on the initial overlap of the thin and thick filaments.
The velocity-tension curve reveals an interesting relationship between muscle power and applied load. Muscle does measurable mechanical work only when it displaces a load. This mechanical work (W) is the product of load (F) and displacement (Δx). Power (P) is the rate at which work is performed, or work per unit time (Δt):

Because velocity (v) is Δx/Δt, it follows that

For a given load (F), we can calculate the power by reading the velocity (v) from the uppermost of the three blue load-velocity relationships in Figure 9-9E. Power is maximal at intermediate loads (where both F and v are moderate) and falls to zero at maximum load (where v = 0) and at zero load (where F = 0).
At sufficiently low stimulation frequencies, the tension developed falls to the resting level between individual twitches (Fig. 9-11A). Single skeletal muscle twitches last between 25 and 200 ms, depending on the type of muscle. Although each twitch is elicited by a single muscle action potential, the duration of contraction is long compared with the duration of the exciting action potential, which lasts only several milliseconds. Because the muscle twitch far exceeds the duration of the action potential, it is possible to initiate a second action potential before a first contraction has fully subsided. When this situation occurs, the second action potential stimulates a twitch that is superimposed on the residual tension of the first twitch, thereby achieving greater isometric tension than the first (compare Fig. 9-11A and B). This effect is known as summation.
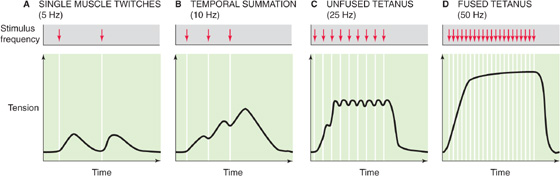
Figure 9-11 Frequency summation of skeletal muscle twitches.
If multiple action potentials occur close enough in time, the multiple twitches can summate and thus greatly increase the tension developed. Summation is more effective at increasing tension when the action potentials are grouped more closely in time, as in Figure 9-11C. In other words, tension is higher when action potentials are evoked at higher frequency. Because this type of tension enhancement depends on the frequency of muscle stimulation, it is referred to as frequency summation.
When the stimulation frequency is increased sufficiently, the individual twitches occur so close together in time that they fuse (Fig. 9-11D) and cause the muscle tension to remain at a steady plateau. The state in which the individual twitches are no longer distinguishable from each other is referred to as tetanus. Tetanus arises when the time between successive action potentials is insufficient to return enough Ca2+ to the SR to lower [Ca2+]i below a level that initiates relaxation. In fact, a sustained increase in [Ca2+]i persists until the tetanic stimulus ceases. At stimulation frequencies above the fusion frequency that causes tetanus, muscle fiber tension increases very little.
In addition to determining the frequency with which it stimulates a single muscle fiber, the CNS can control muscle force by determining the number of individual muscle fibers that it stimulates at a given time. As each additional motor neuron cell body within the spinal cord is excited, those muscle fibers that are part of the motor unit of that motor neuron are added to the contracting pool of fibers (Fig. 9-12). This effect is known as multiple-fiber summation. In general, smaller motor neurons serve motor units consisting of fewer individual muscle fibers. Because a given excitatory stimulus will generate a larger excitatory postsynaptic potential (see Chapter 8) in motor neurons with smaller cell bodies, the small motor units are recruited even with minimal neuronal stimulation. As neuronal stimulation intensifies, larger motor neurons innervating larger motor units are also recruited. The progressive recruitment of first small and then larger and larger motor units is referred to as the size principle. The group of all motor neurons innervating a single muscle is called a motor neuron pool.
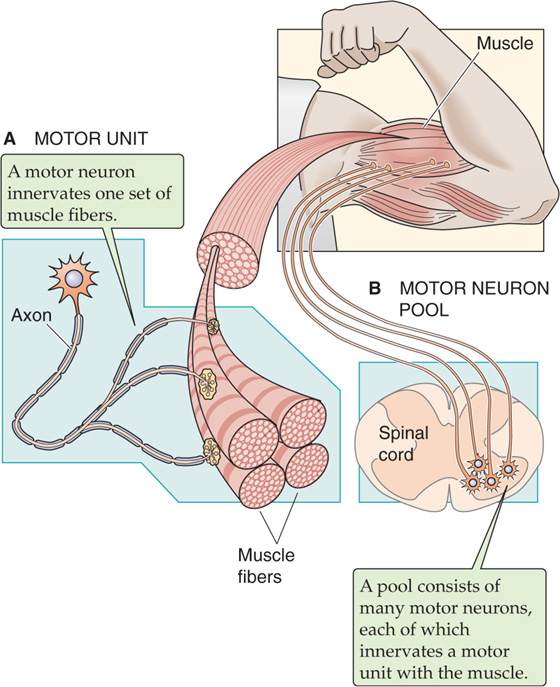
Figure 9-12 The motor unit and the motor neuron pool.
Multiple-fiber summation, sometimes referred to as spatial summation, is an important mechanism that allows the force developed by a whole muscle to be relatively constant in time. It is true that the CNS could direct the force to be relatively constant over time merely by driving a fixed number of motor units within the muscle to tetanus, where the force fluctuations are very small (Fig. 9-11D). However, adding tetanic motor units would increase total muscle force by rather large individual increments. Instead, the CNS can activate individual motor units asynchronously so that some units are developing tension while others are relaxing. Thus, whole-muscle force can be relatively constant with time, even when individual fibers are not stimulated to tetanus. Smooth, nontetanic contraction is essential for fine motor control.
Cardiac muscle and its individual myocytes have a morphological character different from that of skeletal muscle and its cells (Fig. 9-13). Cardiac myocytes are shorter, branched, and interconnected from end to end by structures called intercalated disks, which can be observed as dark lines at the level of a light microscope. The intercalated disks connecting the ends of adjoining cardiac myocytes contain desmosomes that link adjacent cells mechanically and gap junctions (see Chapter 6) that link cells electrically. Cardiac muscle thus acts as mechanical and electrical syncytium of coupled cells, unlike skeletal muscle fibers, which are separate cells bundled together by connective tissue. Like skeletal muscle, cardiac muscle is striated and its sarcomeres contain similar arrays of thin and thick filaments.
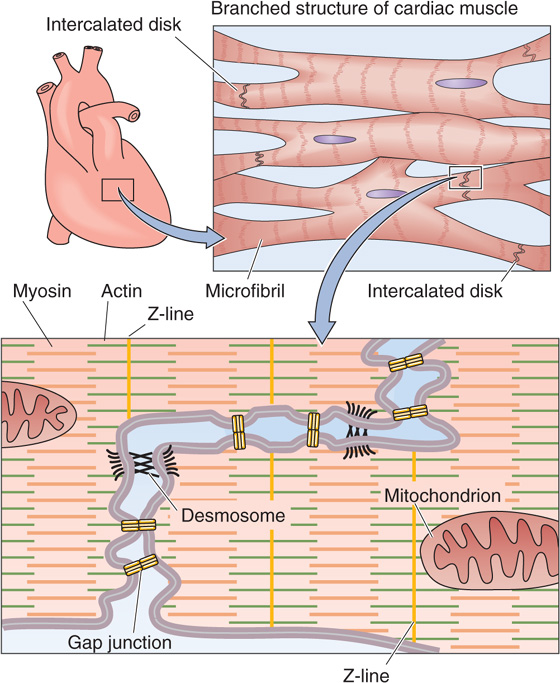
Figure 9-13 Electrical coupling of cardiac myocytes.
Contraction of cardiac muscle cells is not initiated by neurons as in skeletal muscle but by electrical excitation originating from the heart’s own pacemaker, the sinoatrial node (see Chapter 21), which generates spontaneous and periodic action potentials. When an action potential is initiated in one cell, current flows through the gap junctions and depolarizes neighboring cells. If depolarization causes the membrane potential (Vm) to be more positive than threshold, self-propagating action potentials occur in the neighboring cells as well. Thus, the generation of an action potential is just as critical for initiating contraction in cardiac muscle as it is in skeletal muscle but is triggered by the sinoatrial node and the specialized conduction system of the heart as described in Chapter 21.
Cardiac myocytes receive synaptic input from autonomic neurons, but the sympathetic and parasympathetic divisions of the autonomic nervous system (see Chapter 14) use these synapses to modulate rather than to initiate cardiac muscle function.
Whereas EC coupling in skeletal muscle does not require Ca2+ influx through L-type Ca2+ channels, cardiac contraction has an absolute requirement for Ca2+ influx through these channels during the action potential. Because the T-tubule lumen is an extension of the extracellular space, it facilitates the diffusion of Ca2+ from bulk extracellular fluid to the site of the L-type Ca2+ channels on the T-tubule membrane. Thus, the Ca2+ can simultaneously reach superficial and deep regions of the muscle. The increase in [Ca2+]i resulting from Ca2+ influx alone is not, however, sufficient to initiate contraction. Rather, the increase in [Ca2+]i that is produced by the L-type Ca2+ channels is greatly amplified by Ca2+-induced Ca2+ release from the SR through the Ca2+-release channels. Indeed, because the Ca2+-release channels remain open for a longer period than do L-type Ca2+ channels, the contribution of CICR to the rise in [Ca2+]i is greater than the flux contributed by the L-type Ca2+ channels of the T tubules.
It appears that each L-type Ca2+ channel controls only one SR Ca2+-release channel. The physical proximity of L-type Ca2+ channels of the T-tubule membrane and the Ca2+-release channel in the SR at the triad junctions allows this tight local control. Although Ca2+ diffuses in the cytosol away from its SR release site, Ca2+ release at one site does not appear to be able to induce Ca2+ release from a neighboring SR Ca2+-release channel. Thus, Ca2+ release events are not propagated along the myocyte. In fact, the SR Ca2+-release channel does not appear to respond to generalized increases in cytoplasmic [Ca2+]i. Generalized cardiac muscle contractions occur as a result of the spatial and temporal summation of individual CICR events.
Cardiac muscle is generally similar to skeletal muscle in the interaction of the actin and myosin during cross-bridge cycling, the resynthesis of ATP, and the termination of contraction. However, there are a few important differences. For example, the regulatory protein troponin C (Fig. 9-6) of cardiac muscle, which is of the TNNC1 subtype, has just a single, active low-affinity Ca2+ binding site, rather than the two high-affinity and two low-affinity sites of TNNC2 in skeletal muscle. In addition, the termination of cardiac contraction has an additional feature compared with skeletal muscle. In cardiac muscle, SR Ca2+ pump activity is inhibited by the regulatory protein phospholamban. When phospholamban is phosphorylated by cAMP-dependent protein kinase (PKA), its ability to inhibit the SR Ca2+ pump is lost. Thus, activators of PKA, such as the neurotransmitter epinephrine, may enhance the rate of cardiac myocyte relaxation (see Chapter 22).
Whereas frequency summation and multiple-fiber summation are important mechanisms for regulating the strength of skeletal muscle contractions, these mechanisms would not be consistent with the physiological demands of cardiac muscle. Because cardiac muscle must contract only once with each heartbeat and must fully relax between each contraction, frequency summation is precluded. Furthermore, the extensive electrical coupling between cardiac myocytes, as well as the requirement that cardiac muscle contract homogeneously, eliminates the potential for multiple-fiber summation. Therefore, the strength of cardiac muscle contraction must be regulated by modulating the contractile force generated during each individual muscle twitch. This type of regulation is an important part of the adaptive response to exercise and is mediated by norepinephrine, a neurotransmitter released by the sympathetic nervous system.
Because an increase in [Ca2+]i activates contraction by removing the inhibitory influence of the regulatory proteins, it is reasonable to consider that contractile function may be regulated either by modulating the magnitude of the rise in [Ca2+]i or by altering the Ca2+ sensitivity of the regulatory proteins. In fact, both these mechanisms are important in controlling the force of cardiac muscle contraction.
In cardiac muscle, a significant proportion of the activator Ca2+ enters the cell through voltage-gated Ca2+ channels that open during the cardiac action potential. Most of this Ca2+ influx occurs through L-type Ca2+ channels. How does norepinephrine increase the contractile force of the heart? This hormone acts through the β-type adrenergic receptor to increase the generation of cAMP, to activate PKA (see Chapter 3), and in turn to phosphorylate the L-type Ca2+ channels, thereby increasing the passive influx of Ca2+. An increased [Ca2+]i leads to an increase in contractile force. The cAMP pathway also appears to increase the Ca2+ sensitivity of the contractile apparatus by phosphorylating one or more of the regulatory proteins. Thus, cAMP causes an increase in the force generated for any given [Ca2+]i.
Reciprocal control over Ca2+ entry is provided by cGMP-dependent phosphorylation of the L-type Ca2+ channels. ACh, acting through muscarinic ACh receptors, raises intracellular cGMP concentrations. In turn, the cGMP-dependent phosphorylation of L-type Ca2+ channels, at sites distinct from those phosphorylated by the cAMP-dependent kinase, causes a decrease in Ca2+ influx during the cardiac action potential and thus a decrease in the force of contraction.
Ca2+ entry may also be regulated indirectly by modulating other ion channels so that they either change their Ca2+ permeability or alter the duration of the action potential. Norepinephrine, for example, may increase the Ca2+ permeability of voltage-gated Na+ channels. Receptor transduction mechanisms that inhibit voltage-gated K+ currents may prolong the cardiac action potential and thereby increase net Ca2+ influx through L-type Ca2+ channels without modulating the Ca2+ channels themselves.
Like skeletal muscle, smooth muscle receives synaptic input from the nervous system. However, the synaptic input to smooth muscle differs from that of skeletal muscle in two ways. First, the neurons are part of the autonomic nervous system rather than the somatic nervous system (see Chapter 14). Second, the neuron makes multiple contacts with a smooth muscle cell. At each contact point, the axon diameter expands to form a series of swellings called varicosities that contain the presynaptic components for vesicular release of transmitter. Each varicosity is close to the postsynaptic membrane of the smooth muscle cell, but there is relatively little specialization of the postsynaptic membrane. Rather than being closely clustered at the neuromuscular junction, as in skeletal muscle, the neurotransmitter receptors in smooth muscle are spread more widely across the postsynaptic membrane.
The mechanisms of intercellular communication among smooth muscle cells are more diverse than those of skeletal or cardiac muscle. In some organs, smooth muscle is innervated in a manner similar to skeletal muscle in that each smooth muscle cell receives synaptic input. However, a difference is that a smooth muscle cell may receive input from more than one neuron. Moreover, there is little electrical coupling among these smooth muscle cells (i.e., few gap junctions). As a result, each smooth muscle cell may contract independently of its neighbor. Because this type of smooth muscle behaves like multiple, independent cells or groups of cells, it is called multiunit smooth muscle (Fig. 9-14A). Note that the “multi” in multiunit refers to the muscle fibers acting independently of one another as multiple units. Multiunit smooth muscles are capable of finer control. Indeed, multiunit smooth muscle is found in the iris and ciliary body of the eye, the piloerector muscles of the skin, and some blood vessels.
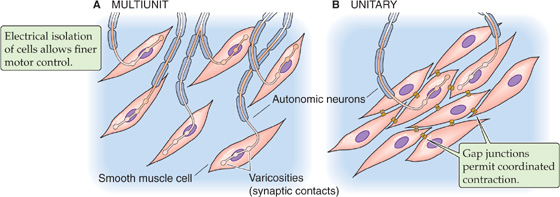
Figure 9-14 Smooth muscle organization. A, Each smooth muscle cell receives its own synaptic input. B, Only a few of the smooth muscle cells receive direct synaptic input.
In contrast to multiunit smooth muscle, the smooth muscle cells of most organs have extensive intercellular communication in the manner of cardiac muscle cells. In this type of smooth muscle, gap junctions permit electrical communication between neighboring cells. This communication allows coordinated contraction of many cells. Because these cells contract as a single unit, this type of smooth muscle is called unitary smooth muscle (Fig. 9-14B). Unitary smooth muscle is the predominant smooth muscle type within the walls of visceral organs such as the gastrointestinal tract, the uterus, and many blood vessels. For this reason, unitary smooth muscle is often referred to as visceral smooth muscle. Among unitary smooth muscles, variation in the strength of intercellular coupling from organ to organ leads to variation in the spatial extent of a single unit. For example, in the bladder, extensive coupling among cells defines large functional units, which allows the muscular wall of the bladder to contract in synchrony. On the other hand, the smooth muscle cells of blood vessels couple to form smaller, independently functioning units that are more akin to multiunit smooth muscle. In fact, electrical coupling of smooth muscle units exhibits a tissue-specific continuum from multiunit to unitary coupling.
Whereas both skeletal muscle and cardiac muscle produce action potentials that initiate contraction, smooth muscle cells produce a wide range of Vm variations that can either initiate or modulate contraction. Action potentials that are similar to those seen in skeletal muscle are observed in unitary smooth muscle and in some multiunit muscle. Like cardiac muscle cells, some smooth muscle cells exhibit prolonged action potentials that are characterized by a prominent plateau. Still other smooth muscle cells cannot generate action potentials at all. In these cells, Vm changes in a graded fashion (see Chapter 7) rather than in the all-or-none manner of action potentials. The stimuli that produce a graded response of Vm include many circulating and local humoral factors as well as mechanical stimuli, such as stretching of the cell. These graded Vm changes may be either hyperpolarizing or depolarizing; they sum temporally as well as spatially. If the summation of graded depolarizations brings Vm above threshold—in a smooth muscle cell capable of producing an action potential—an action potential will then ensue.
Action potentials are usually seen in unitary (visceral) smooth muscle. These action potentials typically have a slower upstroke and longer duration (up to ~100 ms) than do skeletal muscle action potentials (~2 ms). The action potential in a smooth muscle cell can be a simple spike, a spike followed by a plateau, or a series of spikes on top of slow waves of Vm (Fig. 9-15A). In any case, the upstroke or depolarizing phase of the action potential reflects opening of voltage-gated Ca2+ channels. The inward Ca2+ current further depolarizes the cell and thereby causes still more voltage-gated Ca2+ channels to open. Thus, some smooth muscle cells can undergo the same type of regenerative depolarization that is seen in skeletal muscle. However, the rate of rise of the action potential in smooth muscle is lower because Ca2+ channels open more slowly than do Na+ channels in skeletal and cardiac muscle (see Chapter 7). Repolarization of the smooth muscle cell is also relatively slow. Two explanations may be offered for this slower repolarization. First, voltage-gated Ca2+ channels, which are responsible for the depolarization phase of the action potential, inactivate slowly. Second, the repolarization phase of the action potential reflects the delayed activation of voltage-gated K+ channels and, in some cases, Ca2+-activated K+ channels.
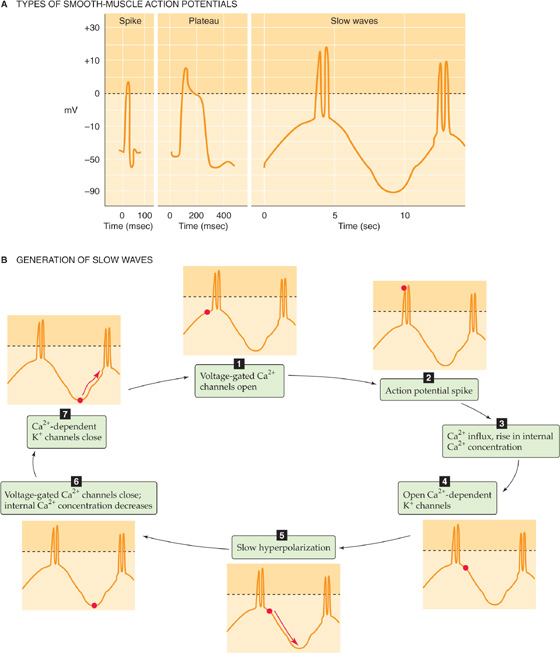
Figure 9-15 Action potentials and slow waves in smooth muscle.
Some smooth muscle cells have fast, voltage-gated Na+ channels. However, even when these channels are present, they do not appear to be necessary for generating an action potential. Their main role may be to allow more rapid activation of voltage-gated Ca2+ channels and thus contribute to a faster rate of depolarization.
In some unitary smooth muscle, repolarization is so delayed that the action potential contour displays a prominent plateau. These plateau potentials may be several hundred milliseconds in duration, as in cardiac muscle. Plateau action potentials occur in smooth muscle of the genitourinary tract, including the ureters, bladder, and uterus. The long Vm plateau allows the entry of Ca2+ to continue for a longer period and thus allows [Ca2+]i to remain high for a longer period, thereby prolonging the contraction.
Although smooth muscle cells undergo changes in Vm in response to neural, hormonal, or mechanical stimulation, many smooth muscle cells are capable of initiating spontaneous electrical activity. In some cells, this spontaneous activity results from pacemaker currents. These currents result from time-and voltage-dependent properties of ion currents that produce either a spontaneous increase in inward, or depolarizing, currents (e.g., voltage-gated Ca2+ currents) or a spontaneous decrease in outward, or hyperpolarizing, currents (e.g., voltage-gated K+ currents). The pacemaker currents cause the cell to depolarize until Vm reaches threshold, triggering an action potential.
In other smooth muscle cells, this spontaneous electrical activity results in regular, repetitive oscillations in Vm. These Vm oscillations occur at a frequency of several oscillations per minute and are referred to as slow waves (Fig. 9-15B). One hypothesis for the origin of slow-wave potentials suggests that voltage-gated Ca2+ channels—active at the resting Vm—depolarize the cell enough to activate more voltage-gated Ca2+ channels. This activation results in progressive depolarization and Ca2+ influx. The increase in [Ca2+]i activates Ca2+-dependent K+ channels, which leads to progressive hyperpolarization and termination of the depolarization phase of the wave. These periodic depolarizations and [Ca2+]i increases cause periodic, tonic contractions of the smooth muscle. When the amplitude of the slow Vm waves is sufficient to depolarize the cell to threshold, the ensuing action potentials lead to further Ca2+ influx and phasic contractions.
Other hypotheses to explain spontaneous electrical and mechanical activity in smooth muscle cells are based on oscillatory changes in other intracellular ions or molecules. For example, increased [Ca2+]i during an action potential might stimulate Na-Ca exchange and lead to a cyclic increase in [Na+]i and thus an increase in the rate of Na+ extrusion by the electrogenic Na-K pump. Alternatively, the inositol 1, 4, 5-trisphosphate (IP3) receptor channel (see Chapter 3) might spontaneously open and release Ca2+. The effect on [Ca2+]i would be self-reinforcing because of Ca2+-activated Ca2+ release through the IP3 receptor. At high [Ca2+]i, this channel is inhibited and the Ca2+ release event is terminated, followed by re-uptake of Ca2+ into the SR. The [Ca2+]i increases may themselves lead to periodic electrical activity by stimulating Ca2+-activated inward and outward currents.
Whereas action potential generation is essential for initiating contraction of skeletal and cardiac muscle, many smooth muscle cells contract despite being unable to generate an action potential. As discussed previously, Vm oscillations can lead to tonic contractions in the absence of action potentials. Action potentials usually do not occur in multiunit smooth muscle. For example, in the smooth muscle that regulates the iris, excitatory neurotransmitters such as norepinephrine and ACh cause a local depolarization, the junctional potential, which is similar to the end-plate potential in skeletal muscle. Junctional potentials spread electrotonically (i.e., in a graded fashion) throughout the muscle fiber, thereby altering Vm and triggering the entry of Ca2+ through voltage-gated slow (L-type) Ca2+ channels. Changes in Vm—by an unknown mechanism—may also modulate the activity of the enzyme phospholipase C, which cleaves phosphoinositides to release the intracellular second messengers diacylglycerol (DAG) and IP3 (see Chapter 3). Both these second messengers are modulators of contractile force. In the absence of action potentials, some unitary smooth muscle, including some vascular smooth muscle, also contracts as a result of graded Vm changes.
Some smooth muscle cells contract without any change in Vm. For example, a neurotransmitter can bind to a receptor, activate a G protein, and lead to the generation of IP3, which in turn leads to the release of Ca2+ from the SR.
Smooth muscle cells use three major pathways—which are not mutually exclusive—for producing the rise in [Ca2+]i that triggers contraction (Fig. 9-16): (1) Ca2+ entry through voltage-gated channels in response to cell depolarization, (2) Ca2+ release from the SR, and (3) Ca2+ entry through voltage-independent channels.
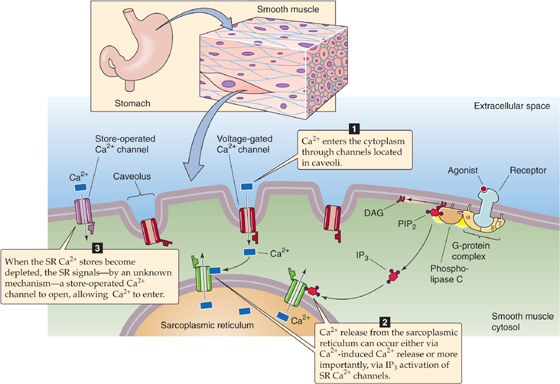
Figure 9-16 EC coupling in smooth muscle.
Ca2+ Entry Through Voltage-Gated Channels Smooth muscle cells respond to stimulation with graded depolarizations or action potentials. In either case, depolarization may produce an influx of Ca2+ through voltage-gated L-type Ca2+ channels.
Ca2+ Release from the SR Sarcoplasmic Ca2+ release may occur by either of two mechanisms: Ca2+-induced Ca2+ release or IP3-mediated Ca2+ release. As we have already seen, CICR plays a key role in EC coupling in cardiac muscle, in which the L-type Ca2+ channels are highly ordered and close to the Ca2+-release channels in the SR. Thus, Ca2+ influx through L-type Ca2+ channels can trigger CICR. In smooth muscle, the relationship between the plasma membrane and the SR is not as regularly organized as it is in striated muscle. Nevertheless, electron-dense couplings have been observed bridging the 8-to 10-nm gap between the cell membranes and elements of the SR in smooth muscle. Although CICR occurs in smooth muscle cells under some conditions, it requires [Ca2+]i levels that are higher than those that typically occur under physiological conditions, and its role remains unclear.
A more important mechanism for Ca2+ release from the SR of smooth muscle is the IP3 pathway. The existence of this pathway is supported by the observation that some extracellular agonists can elicit smooth muscle contraction with minimal depolarization and negligible Ca2+ influx. Furthermore, even for agonists such as serotonin and norepinephrine, which activate a Ca2+-influx pathway, the observed increase in [Ca2+]i is out of proportion to that expected from Ca2+ influx alone. Thus, another pathway must exist for increasing [Ca2+]i. Some agonists cause smooth muscle contraction by triggering the production of IP3, which binds to a specific receptor on the membrane of the smooth muscle SR (see Chapter 3). The IP3 receptor is a ligand-gated Ca2+ channel. Thus, a receptor in the plasma membrane can—via IP3—indirectly induce Ca2+ release from the SR and hence contraction.
Ca2+ Entry Through Voltage-Independent Channels We have just noted that extracellular ligands binding to G-protein–coupled receptors can lead to the release of Ca2+ from the SR. The eventual depletion of Ca2+ stores in the SR somehow leads to the activation of store-operated Ca2+ channels (SOCs), which mediate Ca2+ influx across the cell membrane. The Ca2+ entering through these channels allows [Ca2+]i to remain elevated even after SR depletion and also appears to replenish SR Ca2+ stores. SOCs play an important role in a variety of cell types. In lymphocytes—although perhaps not in smooth muscle—a fall in [Ca2+] inside the endoplasmic reticulum (ER) activates a protein called STIM1 in the ER membrane (see Fig. 6-21T). STIM1, by direct mechanical linkage, then activates a plasma-membrane Ca2+ channel called Orai, which in turn mediates an uptake of Ca2+. Electrophysiologists refer to the Ca2+-release–activated Ca2+ current as ICRAC. Missense mutations in the human Orai1 gene eliminate ICRAC in lymphocytes—where ICRAC plays a vital role in lymphocyte activation—and result in a severe combined immunodeficiency syndrome (SCID). (See Note: Store-Operated Ca2+ Channels (SOCs))
Both the Ca2+ release from the SR and the entry of Ca2+ via SOCs are voltage independent. These two mechanisms provide the Ca2+ that underlies one form of pharmacomechanical coupling. Thus, drugs, excitatory neurotransmitters, and hormones can induce smooth muscle contraction that is independent of action-potential generation, as discussed in the previous section.
As we noted earlier, because smooth muscle actin and myosin are not as highly organized as in skeletal and cardiac muscle, smooth muscle does not exhibit striations characteristic of striated muscle. The actin filaments of smooth muscle are oriented mainly parallel or oblique to the long axis of the cell. Multiple actin filaments appear to join at specialized locations in the cell called dense bodies. Dense bodies are found immediately beneath the cell membrane as well as within the interior of the myocyte. Thick filaments are interspersed among the thin filaments in smooth muscle and are far less abundant than in skeletal or cardiac muscle.
In comparison to skeletal and cardiac muscle, an entirely different mechanism controls cross-bridge turnover in smooth muscle. Here, an increase in [Ca2+]i initiates a slow chain of events that ultimately increases the ATPase activity of the myosin (Fig. 9-17). The first step is the binding of four Ca2+ ions to calmodulin, which is closely related to troponin C of striated muscle. Next, the Ca2+-CaM complex activates an enzyme known as myosin light chain kinase (MLCK), which in turn phosphorylates the regulatory light chain that is associated with the myosin II molecule. Phosphorylation of the light chain alters the conformation of the myosin head, which greatly increases its ATPase activity and allows it to interact with actin and to act as a molecular motor. Thus, in smooth muscle, CaM rather than troponin C is the Ca2+-binding protein responsible for transducing the contraction-triggering increases in [Ca2+]i. Note that in smooth muscle, contraction cannot begin until MLCK increases the ATPase activity of myosin, which is a time-consuming process. In skeletal and cardiac muscle, on the other hand, the ATPase activity of the myosin head is constitutively high, and cross-bridge cycling can begin as soon as the tropomyosin is moved out of the way.
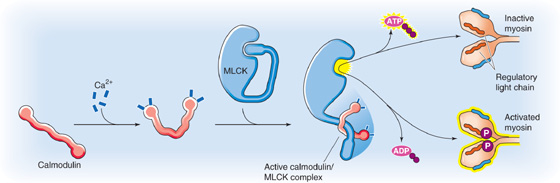
Figure 9-17 The role of Ca2+ in triggering the contraction of smooth muscle.
The mechanism just outlined activates the thick filaments in smooth muscle. Other mechanisms act on the thin filaments of smooth muscle to remove the tonic inhibition to actin-myosin interactions that are caused by steric hindrance. Two proteins, caldesmon and calponin, tonically inhibit the interaction between actin and myosin. Both are Ca2+-CaM–binding proteins, and both bind to actin and tropomyosin. Calponin, which is found in a fixed stoichiometry with tropomyosin and actin (one calponin–one tropomyosin–seven actin monomers), tonically inhibits the ATPase activity of myosin. As we saw earlier, the increase in [Ca2+]i that triggers smooth muscle contraction activates Ca2+-CaM. Besides activating MLCK, this Ca2+-CaM complex has two effects on calponin. First, Ca2+-CaM binds to calponin. Second, Ca2+-CaM activates Ca2+-CaM–dependent protein kinase, which phosphorylates calponin. Both effects reduce calponin’s inhibition of myosin’s ATPase activity. Caldesmon is another regulatory protein of smooth muscle that appears to act like calponin by tonically inhibiting the actin-activated ATPase activity of myosin in smooth muscle. Caldesmon contains binding domains for actin, myosin, tropomyosin, and Ca2+-CaM. It appears to block the interaction of actin with myosin; however, the exact mechanism is controversial.
Although the steps of cross-bridge cycling in smooth muscle are similar to those in skeletal and cardiac muscle (Fig. 9-7), smooth muscle controls the initiation of the cross-bridge cycle differently—at step 2 in Figure 9-7, where Ca2+ confers ATPase activity to the myosin head, as discussed before. Recall that the ATPase activity of striated muscle is always high and Ca2+ regulates the access of the myosin head to the actin. Another difference between smooth and striated muscle is that the frequency of cross-bridge cycling in smooth muscle is less than one tenth that in skeletal muscle. This variation reflects differences in the properties of myosin isoforms that are expressed in various cell types. Even though cross-bridge cycling occurs less frequently in smooth muscle, force generation may be as great or greater, perhaps because the cross-bridges remain intact for a longer period with each cycle. It is likely that this longer period during which the cross-bridges are intact reflects a lower rate of ADP release from the smooth muscle isoform of myosin.
Because Ca2+ triggers smooth muscle contraction by inducing phosphorylation of the myosin regulatory light chain, merely restoring [Ca2+]i to its low resting value may not allow muscle relaxation. Rather, relaxation of smooth muscle requires myosin light chain (MLC) dephosphorylation, which is accomplished by myosin light chain phosphatase. This phosphatase is a heterotrimer consisting of subunits with molecular masses of 130, 20, and 37 kDa. The 130-kDa subunit confers specificity by binding to myosin; the 37-kDa protein is the catalytic subunit responsible for the dephosphorylating activity.
Whereas many excitatory stimuli rely on increases in [Ca2+]i to evoke contraction, some stimuli appear to cause contraction without a measurable increase in [Ca2+]i. One mechanism by which excitatory stimuli might induce Ca2+-independent contractions is by modulating the activity of contractile or regulatory proteins directly. Thus, the amount of force developed at any given [Ca2+]i may vary. This force/[Ca2+]i ratio may be increased or decreased and is generally higher during pharmacomechanical activation than during depolarization-activated contractions. Because phosphorylation of the MLC is a major determinant of contractile force in smooth muscle, Ca2+-independent contractions may result either from an increase in the rate of MLC phosphorylation by MLCK or from a decrease in the rate of MLC dephosphorylation by MLC phosphatase. One second-messenger system that can decrease the activity of phosphatases is protein kinase C (PKC; see Chapter 3). Some excitatory stimuli are therefore capable of initiating smooth muscle contraction by inducing IP3-mediated release of Ca2+ from intracellular stores as well as by producing PKC-mediated decreases in MLC phosphatase activity. These pathways are further examples of pharmacomechanical coupling.
Unlike skeletal muscle, in which force development results from the summation of individual muscle twitches, individual smooth muscle cells can maintain a sustained contraction that can be graded in strength over a wide range. Contractile force in smooth muscle largely depends on the relative balance between the phosphorylation and dephosphorylation of MLCs. The rate of MLC phosphorylation is regulated by the Ca2+-CaM complex, which in turn depends on levels of intracellular Ca2+. Smooth muscle cells can regulate [Ca2+]i over a wider range than in skeletal and cardiac muscle for several reasons. First, some smooth muscle cells do not generate action potentials. Rather, their membrane potential varies slowly in response to neurotransmitters or hormones. This graded response of Vm allows finer regulation of Ca2+ influx through voltage-gated channels. Second, release of Ca2+ from intracellular stores may be modulated through neurotransmitter-induced generation of intracellular second messengers such as IP3. This modulation allows finer control of Ca2+ release than occurs in the SR Ca2+-release channel by L-type Ca2+ channels in skeletal and cardiac muscle.
A second level of control over contractile force occurs by regulating the Ca2+ sensitivity of proteins that regulate contraction. For example, inhibiting myosin light chain phosphatase alters the balance between phosphorylation and dephosphorylation, in effect allowing a greater contraction at a lower [Ca2+]i. Some neurotransmitters act by inhibiting the phosphatase, which appears to occur through activation of G-protein–coupled receptors. Another mechanism for governing the Ca2+ sensitivity of proteins that regulate contraction is alteration of the Ca2+ sensitivity of the myosin light chain kinase. For example, MLCK itself is phosphorylated at specific sites by several protein kinases, including PKA, PKC, and Ca2+-CaM–dependent kinases. Phosphorylation by any of these kinases decreases the sensitivity of MLCK to activation by the Ca2+-CaM complex.
Smooth muscle is often called on to maintain high force for long periods. If smooth muscle consumed ATP at rates similar to striated muscle, metabolic demands would be considerable and the muscle would be prone to fatigue. Unlike striated muscle, however, smooth muscle is able to maintain high force at a low rate of ATP hydrolysis. This low-energy consumption/high-tension state is referred to as the latch state. The latch state in smooth muscle is unique because high tension can be maintained despite a decrease in the degree of muscle activation by excitatory stimuli. As a result, force is maintained at a lower level of MLCK phosphorylation.
The mechanism underlying the latch state is not entirely known, although it appears to be due in large part to changes in the kinetics of actin-myosin cross-bridge formation and detachment. These changes may be a direct result of a decrease in the rate at which dephosphorylated cross-bridges detach. Tension is directly related to the number of attached cross-bridges. Furthermore, the proportion of myosin heads cross-bridged to actin is related to the ratio of attachment rates to detachment rates. Therefore, it is reasonable to expect that a decrease in the detachment rate would allow a greater number of cross-bridges to be maintained and would result in a lower rate of cross-bridge cycling and ATP hydrolysis. Thus, smooth muscle appears to be able to slow down cross-bridge cycling just before detachment, a feat that can be accomplished in skeletal muscle (Fig. 9-7) only at low ATP levels (as in rigor mortis).
As we have seen, each muscle type (skeletal, cardiac, and smooth) is distinguishable on the basis of its unique histology, EC coupling mechanisms, and regulation of contractile function. However, even within each of the three categories, muscle in different locations must serve markedly different purposes, with different demands for strength, speed, and fatigability. This diversity is possible because of differences in the expression of specific isoforms for various contractile and regulatory proteins (Table 9-1).
Table 9-1 Isoform Expression of Contractile and Regulatory Proteins
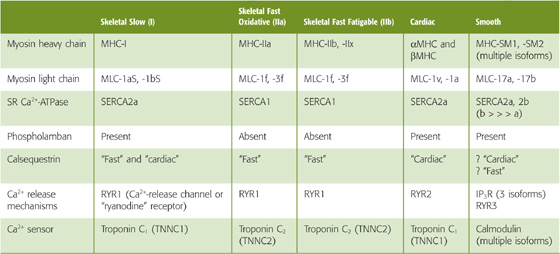
Some skeletal muscles must be resistant to fatigue and be able to maintain tension for relatively long periods, although they need not contract rapidly. Examples are muscles that maintain body posture, such as the soleus muscle of the lower part of the leg. In contrast, some muscles need to contract rapidly, yet infrequently. Examples are the extraocular muscles, which must contract rapidly to redirect the eye as an object of visual interest moves about.
Individual muscle fibers are classified as slow twitch (type I) or fast twitch (type II), depending on their rate of force development. These fiber types are also distinguished by their histologic appearance and their ability to resist fatigue.
Slow-twitch fibers (Table 9-2) are generally thinner and have a denser capillary network surrounding them. These type I fibers also appear red because of a large amount of the oxygen-binding protein myoglobin (see Chapter 29) within the cytoplasm. This rich capillary network together with myoglobin facilitates oxygen transport to the slow-twitch fibers, which mostly rely on oxidative metabolism for energy. The metabolic machinery of the slow-twitch fiber also favors oxidative metabolism because it has low glycogen content and glycolytic enzyme activity but a rich mitochondrial and oxidative enzyme content. Oxidative metabolism is slow but efficient, making these fibers resistant to fatigue.
Table 9-2 Properties of Fast- and Slow-Twitch Muscle Fibers
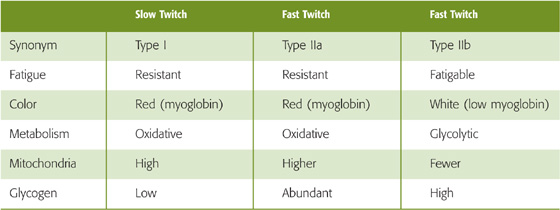
Fast-twitch fibers differ among themselves with respect to fatigability. Some fast-twitch fibers are fatigue resistant; they rely on oxidative metabolism (type IIa) and are quite similar to slow-twitch fibers with respect to myoglobin content (indeed, they are red) and metabolic machinery. One important difference is that fast-twitch oxidative fibers contain abundant glycogen and have a greater number of mitochondria than slow-twitch fibers do. These features ensure adequate ATP generation to compensate for the increased rate of ATP hydrolysis in fast-twitch fibers.
Other fast-twitch fibers are not capable of sufficient oxidative metabolism to sustain contraction. Because these fibers must rely on the energy that is stored within glycogen (and phosphocreatine), they are more easily fatigable. Fatigable fast-twitch fibers (type IIb) have fewer mitochondria and lower concentrations of myoglobin and oxidative enzymes. Because of their low myoglobin content, type IIb muscle fibers are white. They are, however, richer in glycolytic enzyme activity than other fiber types are.
In reality, slow- and fast-twitch fibers represent the extremes of a continuum of muscle fiber characteristics. Moreover, each whole muscle is composed of fibers of each twitch type, although one of the fiber types predominates in any given muscle. The differences between fiber types derive in large part from differences in isoform expression of the various contractile and regulatory proteins (Table 9-1).
Differences in the rate of contraction, for example, may be directly correlated with the maximal rate of myosin ATPase activity. The human genome database lists at least 15 MHC genes, with their respective splice variants. Individual isoform expression varies among muscle types and is developmentally regulated. At least four isoforms of the MHC protein are expressed in skeletal muscle (MHC-I, MHC-IIa, MHC-IIb, MHC-IIx/d). For the most part, a muscle fiber type expresses a single MHC isoform, the ATPase activity of which appears to correspond to the rate of contraction in that fiber type. Whereas most fibers express one of these isoforms, some fibers express a combination of two different isoforms. These hybrid cells have rates of contraction that are intermediate between the two pure fiber types.
Differences in the rates and strength of contraction may also result from differences in myosin light chain isoform expression or from isoform differences among other components of the EC coupling process. Three skeletal muscle isoforms have been identified. MLC-1as and MLC-1bs are expressed in slow-twitch fibers, whereas MLC-1f and MLC-3f are expressed in fast-twitch fibers.
Isoform differences also exist for the SR Ca2+ pump (i.e., the SERCA), calsequestrin, the Ca2+-release channel, and troponin C. Furthermore, some proteins, such as phospholamban, are expressed in one fiber type (slow twitch) and not the other.
One particularly interesting feature of muscle differentiation is that fiber-type determination is not static. Through exercise training or changes in patterns of neuronal stimulation, alterations in contractile and regulatory protein isoform expression may occur. For example, it is possible for a greater proportion of fast-twitch fibers to develop in a specific muscle with repetitive training. It is even possible to induce cardiac-specific isoforms in skeletal muscle, given appropriate stimulation patterns.
Just as skeletal muscle consists of multiple fiber types, so too does heart muscle. The electrophysiological and mechanical properties of cardiac muscle vary with their location (i.e., atria versus conducting system versus ventricle). Moreover, even among cells within one anatomical location, functional differences may exist between muscle cells near the surface of the heart (epicardial cells) and those lining the interior of the same chambers (endocardial cells). As in skeletal muscle, many of these differences reflect differences in isoform expression of the various contractile and regulatory proteins. Although some of the protein isoforms expressed in cardiac tissue are identical to those expressed in skeletal muscle, many of the proteins have cardiac-specific isoforms (Table 9-1). The MHC in heart, for example, exists in two isoforms, α and β, which may be expressed alone or in combination.
When one considers that smooth muscle has a broad range of functions, including regulating the diameter of blood vessels, propelling food through the gastrointestinal tract, regulating the diameter of airways, and delivering a newborn infant from the uterus, it is not surprising that smooth muscle is a particularly diverse type of muscle. In addition to being distinguished as unitary or multiunit muscle, smooth muscle in different organs diverges with respect to nerve and hormonal control, electrical activity, and characteristics of contraction.
Even among smooth muscle cells within the same sort of tissue, important functional differences may exist. For example, vascular smooth muscle cells within the walls of two arterioles that perfuse different organs may vary in their contractile response to various stimuli. Differences may even exist between vascular smooth muscle cells at two different points along one arterial pathway.
The phenotype of smooth muscle within a given organ may change with shifting demands. The uterus, for example, is composed of smooth muscle—the myometrium—that undergoes remarkable transformation during gestation as it prepares for parturition (see Chapter 56). In addition to hypertrophy, greater coupling develops between smooth muscle cells through the increased formation of gap junctions. The cells also undergo changes in their expression of contractile protein isoforms. Changes in the expression of ion channels and hormone receptors facilitate rhythmic electrical activity. This activity is coordinated across the myometrium by propagation of action potentials and increases in [Ca2+]i through the gap junctions. These rhythmic, coordinated contractions develop spontaneously, but they are strongly influenced by the hormone oxytocin, levels of which increase just before and during labor and just after parturition.
These differences in smooth muscle function among various tissues or even over the lifetime of a single cell probably reflect differences in protein composition. Indeed, in comparison to striated muscle, smooth muscle cells express a wider variety of isoforms of contractile and regulatory proteins (Table 9-1). This variety is a result of both multiple genes and alternative splicing (see Chapter 4). This richness in diversity is likely to have important consequences for smooth muscle cell function, although the precise relationship between the structure and function of these protein isoforms is not yet clear.
Perhaps one of the most impressive sources of diversity among smooth muscle cells relates to differences in response to neurotransmitters, environmental factors, and circulating hormones. Smooth muscle cells differ widely with respect to the types of cell surface receptors that mediate the effects of these various mediators. In general, smooth muscle cells each express a variety of such receptors, and receptor stimulation may lead to either contraction or relaxation. Many substances act through different receptor subtypes in different cells, and these receptor subtypes may act through different mechanisms. For example, whereas some neurotransmitter/hormone receptors may be ligand-gated ion channels, others act through heterotrimeric G proteins that either act directly on targets or act through intracellular second messengers such as cAMP, cGMP, or IP3 and DAG.
The list of neurotransmitters, hormones, and environmental factors regulating the function of vascular smooth muscle cells alone is vast (see Chapter 23). A few of these vasoactive substances include epinephrine, norepinephrine, serotonin, angiotensin, vasopressin, neuropeptide Y, nitric oxide, endothelin, and oxygen. Identical stimuli, however, may result in remarkably different physiological responses by smooth muscle in different locations. For example, systemic arterial smooth muscle cells relax when the oxygen concentration around them decreases, whereas pulmonary arterial smooth muscle contracts when local oxygen decreases (see Chapter 31). (See Note: Nitric Oxide (NO))
A summary comparison between muscle types is presented in Table 9-3.
Table 9-3 Summary of Comparisons Between Muscle Types
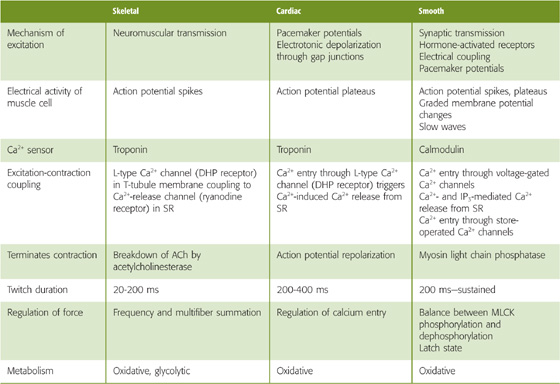
Books and Reviews
Farah CS, Reinach FC: The troponin complex and regulation of muscle contraction. FASEB J 1995; 9:755-767.
Franzini-Armstrong C, Protasi F: Ryanodine receptors of striated muscles: A complex channel capable of multiple interactions. Physiol Rev 1997; 77:699-729.
Holda J, Klishin A, Sedova M, et al: Capacitative calcium entry. News Physiol Sci 1998; 13:157-163.
Horowitz A, Menice CB, Laporte R, Morgan KG: Mechanisms of smooth muscle contraction. Physiol Rev 1996; 76:967-1003.
Parekh AB, Penner R: Store depletion and calcium influx. Physiol Rev 1997; 77:901-930.
Striggow F, Ehrlich BE: Ligand-gated calcium channels inside and out. Curr Opin Cell Biol 1996; 8:490-495.
Journal Articles
Cannell MB, Cheng H, Lederer WJ: The control of calcium release in heart muscle. Science 1995; 268:1045-1049.
Finer JT, Simmons RM, Spudich JA: Single myosin molecule mechanics: Piconewton forces and nanometre steps. Nature 1994; 368:113-119.
Gordon AM, Huxley AF, Julian FJ: The variation in isometric tension with sarcomere length in vertebrate muscle. J Physiol 1966; 184:170-192.
Mickelson JR, Louis CF: Malignant hyperthermia: Excitation-contraction coupling, Ca2+-release channel, and cell Ca2+ regulation defects. Physiol Rev 1996; 76:537-592.