HYDROCEPHALUS AND HYDRANENCEPHALY OF RUMINANTS
Definition and Etiology
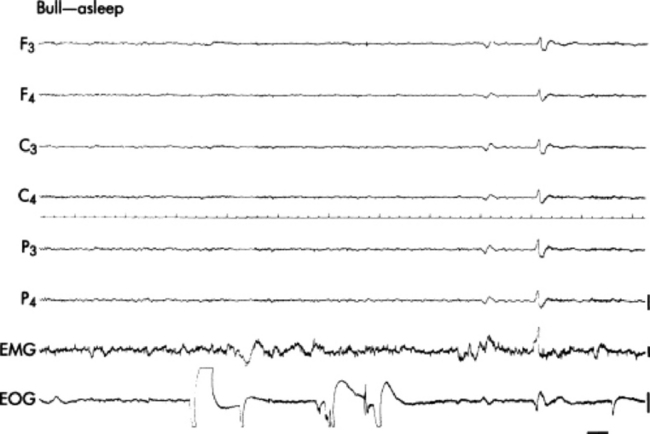
Hydrocephalus/hydranencephaly is a common occurrence in large ruminants. It is underdiagnosed because many of the affected animals die of complications, and the primary condition is overlooked during clinical and pathologic examinations. One study reported that 97 of 155 calves with CNS lesions had hydrocephalus.1209 Hydrocephalus may be classified as hypertensive or normotensive.1210
NORMOTENSIVE HYDROCEPHALUS (HYDRANENCEPHALY)
Normotensive hydrocephalus that develops as a result of a failure of cell growth or cellular necrosis is called hydranencephaly.1210 Most cases of hydranencephaly in domestic livestock are caused by in utero infection of the fetus by the bluetongue, bovine viral diarrhea (BVD), akabane, Cache Valley, aino, or border disease virus.1209,1211-1217 The pathogenesis and epizootiology of the multisystemic virus infections bluetongue and BVD are discussed in detail in Chapter 32. The neurologic effects of border disease virus infection are discussed earlier in this chapter.
The loss of neurons results in flexural contractions of the limbs (arthrogryposis) and inability to nurse. The calves appear blind and are unaware of their surroundings. They usually are unwilling to stand and display a weak suckle. They may exhibit a dysphonia, which resembles a bark. Neonates that are unable to nurse are deprived of colostrum and die of septicemia by 4 days after birth.
Akabane Virus Infection
The akabane virus is a member of the Sindbis serologic subgroup of the Bunyaviridae family of the Arboviridae.1211 It has been isolated from cattle in Africa, Japan, Israel, Korea, and Australia.1212-1217 The host range of akabane virus includes sheep, cattle, and goats. Infection of pregnant, nonimmune dams results in hydranencephaly or arthrogryposis of the fetus.1218 The disease is thought to be transmitted to the cow by various Culicoides species. Experimentally infected calves develop porencephaly and encephalitis when exposed to the akabane virus between gestational days 62 and 96.1219 Studies in naturally infected cattle showed that infection of calves between days 76 and 104 of gestation resulted in hydranencephaly or porencephaly, whereas infection between days 103 and 174 of gestation resulted in arthrogryposis.1220 Lambs are susceptible when exposed to the virus on gestational days 30 to 36.1217 Fetuses that survive the in utero infection are born with arthrogryposis. The CNS lesions apparently are the result of a direct necrotizing effect of the virus on the developing neurons. The pathologic changes of the CNS in experimentally infected calves and lambs are similar to those of naturally acquired infections.1214,1217 Adults occasionally abort when infected by the virus but do not develop clinical disease.
A syndrome of arthrogryposis, facial deformities, kyphoscoliosis, hydranencephaly, and hypoplasia of multiple regions of the brain and spinal cord has been described in Corriedale sheep in Australia. Although resembling the disorder caused by congenital akabane virus infection, breeding trials supported an autosomal recessive inheritance for the disease.1221
Aino Virus Infection
The aino virus causes stillbirths, premature calving, and congenital malformations, including arthrogryposis, cerebellar hypoplasia, and hydranencephaly, in calves of Japan and Australia.1222-1224 Aino virus is antigenically and biologically distinct from akabane virus, but the clinical syndromes of fetal infection by the two viruses are indistinguishable.
Chuzan Virus Infection
Hydrocephalus, hydranencephaly, and cerebellar hypoplasia have been attributed to infection of pregnant cattle with the Chuzan virus.1225-1227 This virus is a relative of the akabane and aino viruses and is classified as a new member of the Palyam subgroup of the genus Orbivirus. The virus has been isolated from Culicoides oxystoma, which may serve as the major vector. The clinical signs are characteristic of hydrocephalus.
Cache Valley Virus Infection
A flock outbreak of arthrogryposis, myositis, hydranencephaly, and a variety of other brain malformations (micrencephaly, cerebellar hypoplasia, porencephaly) in newborn lambs in the southwestern United States was attributed to in utero infection with the Cache Valley virus (family Arboviridae).1228,1229 Cache Valley virus was first isolated from mosquitoes from Utah and has since been isolated from caribou, horses, sheep, and cattle elsewhere. Antibodies have been found in white-tailed deer in the southwestern United States, but the role of this mammal in the survival of the virus and the transmission of the disease to livestock is unknown.1230 In one survey of sheep in the western United States, the seroprevalence for the Cache Valley virus was 19.1%.1228 Vectors for the virus include Anopheles, Aedes, Culex, and Coquellettidia mosquitoes.1231 Infection before 30 days of gestation may cause embryonic death, whereas infection between days 30 and 52 causes fetal malformations.1231
Bluetongue Virus Infection
The bovine fetus is most susceptible to the development of hydranencephaly from bluetongue virus when the dam is infected at approximately 125 days of gestation.1232,1233 Abortions occur when nonimmune dams are infected at other times of gestation. Serotype 11 or serotype 17 of the virus is most frequently isolated from calf and lamb neonates in field epizootics.1233 Calves infected in utero may develop one or more associated birth defects, including hydranencephaly, arthrogryposis, brachygnathia, prognathia, and excessive gingival tissue. In vitro studies have not supported the role of infected calves as reservoirs for the virus.1234 Similar abnormalities and fetal deaths have been reported following vaccination of pregnant ewes with live attenuated virus.1235 (See Chapter 32 for additional information.)
Bovine Viral Diarrhea Virus Infection
Hydranencephaly, hydrocephalus, and cerebellar hypoplasia have been associated with fetal infection of cattle with the BVD virus.1236-1239 Precolostral serum antibody titers for the virus in affected calves vary; some titers range from 1:32 to 1:256, but other calves may have persistent viremias yet no demonstrable antibody. The BVD antibody titer in CSF may range from 1:4 to 1:32. The virus can be isolated from approximately 12% of affected calves.
Other Infectious Agents
A single case of hydrocephalus in a calf aborted at 7 months of gestation was associated with necrotizing encephalitis caused by Neospora caninum.1240
HYPERTENSIVE HYDROCEPHALUS
An increase in CSF volume that results from compressive or obstructive lesions in the ventricular system or from decreased CSF absorption is called hypertensive hydrocephalus.1210 Obstructive lesions of the ventricular system trap the CSF in the ventricles, causing an increase in CSF volume and pressure. Ischemia and CNS degeneration result from the high CSF pressure. The sites of obstruction most often include the lateral apertures, mesencephalic aqueduct, lateral ventricles, interventricular foramina, and fourth ventricle. The obstructions may be either congenital or acquired. Causes of acquired obstructive hydrocephalus include cerebral abscess, cholesteatoma (equines), equine infectious anemia, Coenurus cerebralis infestation, pachymeningitis, and lymphosarcoma. Hypertensive hydrocephalus also may be caused by acute inflammatory disease such as meningitis and vitamin A deficiency. In these diseases the increased pressure is the result of impaired CSF resorption.
CONGENITAL HYPERTENSIVE HYDROCEPHALUS
Congenital hypertensive hydrocephalus is a hereditary condition seen in Hereford, Charolais, Ayrshire, Dexter, Holstein, and Jersey calves.1241-1243 The condition also has been recognized in Arabian foals.1244
At least six forms of congenital hypertensive hydrocephalus (types I through VI) have been identified in cattle.1245 Type I is a communicating hydrocephalus that is unrelated to dwarfism and apparently has a hereditary basis.1241 The mode of inheritance is thought to be a single autosomal recessive character. In highly inbred herds the prevalence of heterozygotes may exceed 20%. Pathologic lesions of this form of hydrocephalus included cranial doming and enlargement of the cerebral cortex and the choroid plexus. All affected calves die by 5 weeks of age.
Type II occurs in Herefords and is characterized by dorsal kinking of the mesencephalon and stenosis of the sylvian aqueduct, without cranial doming. Ventricular dilation is less than that described for type I hydrocephalus.
Type III also occurs in horned Hereford cattle. This type is similar to type II, except that cerebellar hypoplasia, microphthalmia, and muscular degeneration are observed. These are not characteristics of the type II hydrocephalus.
Type IV occurs in white shorthorn calves. The disease is considered to be heritable through either an autosomal recessive gene or a dominant gene with incomplete penetrance. Affected animals develop an obstructive hydrocephalus, microphthalmia, and scoliosis of the thoracolumbar spinal column. Ocular lesions associated with the disease include persistent pupillary membranes, retinal detachment, retinal dysplasia, vitreous hemorrhage, and hypoplasia of the optic tracts. A misshapen sylvian aqueduct apparently causes the fluid accumulation.
Type V is a form of hydrocephalus with congenital achondroplasia that has been reported in Dexter and Jersey calves. The disease is considered to result from a recessive genetic trait. The calves are either aborted or stillborn. Animals that survive to term have arrested development of the nasal bones and maxillae. Anasarca, achondroplasia, kyphosis, and cleft palate also are seen.
Type VI is a form of internal hydrocephalus of Holstein-Friesian calves. The animals are born dead or die shortly after birth. The pathologic abnormalities include fluid enlargement of the lateral ventricles with normal cranial development.1244 The condition is thought to be hereditary.
Clinical Signs
Hydrocephalic animals often are born dead or are weak and die shortly after birth. The most obvious signs in animals that survive include failure to bond to the dam, depression, diminished learning ability, partial failure of suckling, droopy head and ears, muscular fasciculations, head tremor, conscious proprioceptive deficits, blindness, ventrolateral strabismus, nystagmus, dysphonia, tongue flaccidity or paralysis, retention of food material in the cheeks and lips, limb spasticity, hyperreflexia, psychomotor seizures, recumbency, and coma. Occasionally, doming of the calvarium or protrusion of fluid-filled cystic structures through an open fontanelle is seen.1246 Affected neonates often do not ingest sufficient amounts of colostrum and frequently die of septicemia.
In virally induced cases of hydranencephaly, associated skeletal deformities may be observed, including abnormally curved ribs, kyphoscoliosis, flexural deformities of the limbs, domed skulls, and brachygnathia. Patients with hydrocephalus caused by compressive lesions around the ventricular system may show unilateral or bilateral signs of increased intracranial pressure. The clinical signs of unilateral lesions include head tilt (toward the lesion side), ipsilateral mydriasis, and contralateral menace deficit. Signs of hydrocephalus in foals are similar to those in calves. The cause of the condition in horses is unknown.
Antemortem diagnosis of brain malformations has been facilitated by CT and MRI. However, these techniques are rarely warranted or available for use in large animal species. A more practical technique for using ultrasonographic imaging, transorbital echoencephalography, has recently been described and has proved effective for the diagnosis of hydranencephaly.1247
Clinical Pathology
The diagnosis of hydrocephalus in calves and lambs is typically based on the presence of characteristic clinical signs and a domed skull. Whenever hydranencephaly is suspected, blood should be collected for virus isolation, serologic testing, and quantitative immunoglobulin determination. Presuckle serum samples from bovine fetuses that have been infected by the akabane or bluetongue virus in the latter part of gestation may be seropositive. Immunologically competent calves that are infected with the bluetongue virus have serum neutralization indices ranging from 2.5 to 4.1230
Necropsy Findings
The pathologic lesions of hydranencephaly are similar regardless of the etiologic agent. They include microcephaly, cerebellar hypoplasia, hydrocephalus, hydranencephaly, and porencephaly of the cerebral and the cerebellar cortex. Microscopic lesions of hydranencephaly include segmental loss of dorsolateral ventricular ependyma, thinning of the periventricular white matter, porencephalic cysts, and nonsuppurative meningoencephalitis. Lesions in other parts of the CNS may include loss of ventral horn cells in the spinal cord and demyelination in the spinal cord. Nonsuppurative inflammatory changes may be seen in cases caused by viral infections. Polymyositis has been described in affected calves; however, it is unclear if these lesions are caused by viral infection or occur secondary to the denervation. The skeletal deformities associated with virally induced hydranencephalies include rigid extension or contraction of one or more limbs (arthrogryposis), abnormally curved ribs, domed skull, thickening of the calvarium, kyphoscoliosis, and brachygnathia.
Treatment
Except for one report of successful surgical intervention in a calf with a meningocele, no satisfactory therapy is available for the treatment of hydrocephalus or hydranencephaly in large animals.
AMMONIATED FORAGE TOXICOSIS (COW BONKERS)
Exposure of poor-quality forage to anhydrous ammonia improves the nutritional density of the material and reduces certain toxic fungal metabolites, specifically the prolactin-like toxins of the endophytic fungus Acremonium coenophialum.1248 Ammoniation increases dry matter intake, enhances digestibility, and increases the relative value of the protein content of the feed. However, overammoniation of the forage, at a rate exceeding 3% of the forage on a dry matter basis, may result in toxicosis. Studies now suggest that several dialkylimidazoles may be responsible for the neurotoxic effects of ammoniated feedstuffs, superseding previous theories that 4-methylimidazole is the primary neurotoxin.1249,1250 Ammoniated foodstuffs containing high levels of molasses are more toxic than similarly treated grass hay. The toxin may be concentrated in milk; consequently, calves suckling from normal-appearing dams may show clinical signs of intoxication.
Affected animals are hyperesthetic and ataxic. At rest the animals assume a sawhorse stance, but when excited, they become hyperactive, appear to be blind, and circle propulsively. Other clinical signs include vocalization, dysphonia, and walking or running into objects. The periods of frenzy may result in recumbency and convulsions. The spasmodic episodes last for 15 to 20 minutes. Afterward the animals rest quietly, with occasional muscle tremors. Repeated occurrences of the mania may be precipitated by loud noises or other frightening experiences. The concentrations of ammonia in the cerebrospinal fluid (CSF) and blood may be increased. In one report, blood and CSF concentrations of ammonia were 8.16 and 1.05 μg/mL, respectively. Levels of interleukin-6 (IL-6) are elevated in the CSF of affected calves, but not in the systemic circulation.1251 IL-6 is hypothesized to play a key role in ammoniated forage toxicosis. Although specific treatments have not been identified, one report indicated that affected calves benefited from acepromazine (0.045 mg/kg IV) and thiamine (1.14 mg/kg IM).1252
LEAD POISONING
Definition and Etiology
Lead poisoning in ruminants is characterized by an acute encephalopathy. In contrast, lead poisoning in horses is characterized by chronic polyneuritis. Blindness, ataxia, and depressed sensorium are significant clinical signs in cattle, sheep, and goats, whereas in horses the poisoning is associated with weight loss, dysphagia, and secondary aspiration pneumonia. Cattle most often are poisoned because of their tendency to lick or chew on foreign objects, their access to lead-containing materials, and their propensity to drink contaminated petroleum distillates.1253
Clinical Signs
The signs of lead poisoning in ruminants are characteristic of central nervous system (CNS) derangement. During the first stages of lead poisoning, affected cattle stand alone and are depressed.1254 They may show hyperesthesia, muscular fasciculations, and rapid, spastic twitching of the eyelids or other facial muscles. Progression of the disease is associated with ataxia, conscious proprioceptive deficits, blindness, head pressing, odontoprisis, coma, and convulsions.1255,1256 Despite the blindness, the pupillary reflexes usually are normal. Some animals may display episodic running, hyperesthesia, and bellowing. Others may die suddenly without premonitory signs. The more acute and severe the toxicity, the more acute, severe, and excitatory are the clinical signs.1257 Animals with subacute or chronic lead poisoning have signs that are less excitatory and more indicative of CNS depression and have a longer clinical course. Affected cattle may accumulate frothy saliva at the commissures of the lips. Gastrointestinal (GI) signs of bloat, diarrhea, rumen atony, and colic occur in about 60% of lead-poisoned cattle, and the presence of such signs increases the index of suspicion for lead toxicity versus other causes of cerebral dysfunction.1258 Other substances ingested with the lead may contribute to GI disturbances. The clinical signs of lead poisoning in horses include weight loss, lack of coordination, laryngeal or pharyngeal paralysis, dysphonia, roaring, conscious proprioceptive deficits, loss of anal tone, facial paralysis, and difficulty with mastication.1259 Aspiration of pharyngeal debris caused by dysphagia may result in pneumonia. Fine muscular tremors occur intermittently. The poisoned animals die in psychomotor seizures. Horses with lead poisoning are emaciated at death.1260
In cattle, lead produces microscopic changes of the myocardium that result in arterial hypertension (120 to 150 mm Hg) and electrocardiographic abnormalities. These electrical changes, which occur by 30 days after exposure, include increased duration and amplitude of the P wave (0.16 second and 0.06 mV, respectively), prolongation of the PR interval (0.14 to 0.16 second), decreased QT interval (0.32 second), and inverted T wave in lead II.1261
Clinical Pathology
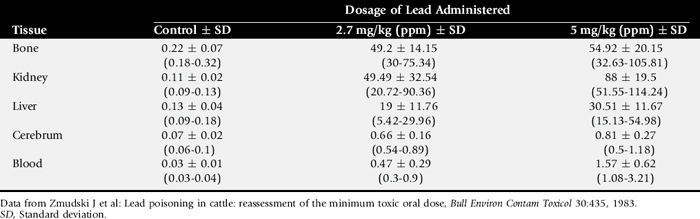
Diagnosis of lead poisoning is based on measurement of increased blood and tissue concentrations of lead. Tissue levels of lead in naturally poisoned cattle can reach 20 to 100 ppm in the liver, 30 ppm in the kidneys, and 5000 ppm in bone. Reported reference blood lead concentrations vary considerably, ranging from 0.05 to 2.5 ppm.1255,1256,1262 Suggested toxic ranges also vary considerably among laboratories. Reference values from earlier colorimetric studies are consistently higher than those from later tests using spectrophotometry.1263 Modern techniques usually report 0.3 ppm as the maximum normal blood lead concentration. When interpreting the results of a lead measurement, consideration of the reference ranges obtained with similar methodology is essential. Table 35-12 compares the lead concentrations of various tissues of experimentally poisoned and control calves. Heparin is the anticoagulant of choice when collecting blood for lead measurement because it does not chelate the lead. The lead concentration of ruminal fluid from acutely poisoned cattle ranges from 0 to 11,875 ppm.1255
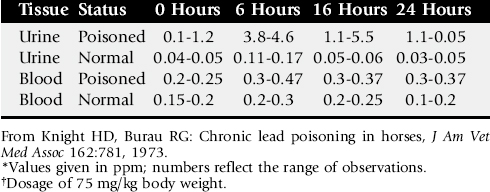
Livestock that are chronically poisoned with low concentrations of lead may have a normal blood lead concentration but a high concentration in the bone. In these cases the poisoning can be diagnosed by administration of calcium disodium ethylenediamine tetraacetic acid (EDTA), which solubilizes the bone lead stores and increases the concentration of lead in the plasma. The soluble lead-EDTA complexes are excreted in the urine. The urinary lead concentration may rise by 40-fold over pretreatment levels within a few hours. Table 35-13 shows the lead concentrations in the urine and blood of naturally exposed horses and the temporal changes that occur after treatment with calcium disodium EDTA (75 mg/kg). In cases of chronic lead poisoning, radiographs of the abdomens of smaller patients may reveal lead-containing radiodense foreign material in the GI tract.1264 “Lead lines” also may be present in the long bones of young animals chronically exposed to lead.
When blood lead concentrations are normal in chronically poisoned animals, measurement of free erythrocyte porphyrins and erythrocyte concentrations of α-aminolevulinic acid (ALA) are the preferred methods of diagnoses. The concentration of porphyrins is increased in the blood, urine, and feces of animals with lead poisoning. The reference range of blood porphyrin concentrations in normal calves is 21.6 ± 11.6 to 45.6 ± 10.3 μg/dL for whole blood and 113 and 142.8 ± 32.4 μg/dL for erythrocytes.1256,1265,1266 In chronically exposed, asymptomatic cattle, the free erythrocyte porphyrin concentrations are frequently greater than 2000 μg/dL. A field test has been developed for determining blood porphyrins.1266
Reference ranges for ALA dehydrase are 45.8 ± 20.6 U, whereas activities ranging from 28 to 33 U have been reported in naturally exposed calves.1260 The urinary concentration of δ-ALA is increased and range above 500 μg/mL.1261,1267,1268 Measurement of ALA in the erythrocytes is more reliable than measurement in urine.1269
Environmental sources of lead can be detected by direct measurement of the lead concentration of the soil or pasture forage. Forage from toxic pastures contains more than 30 ppm of lead, and in some cases the level may exceed 300 ppm.1268,1270
The hematologic abnormalities of lead poisoning are subtle. Most poisoned livestock have a normal hemogram. If present, lead-related changes are characteristic of a hemolytic anemia with an inappropriately large-bone marrow response. The morphologic abnormalities of erythrocytes include anisocytosis, poikilocytosis, polychromasia, hypochromia, Howell-Jolly bodies, metarubricytes, and basophilic stippling.1255,1271 The shape changes begin within hours after ingestion of the lead and peak by 100 days.1255,1271 Blood changes do not occur in all cases of the disease and are not necessarily specific indicators of lead poisoning in cattle, in which elevated blood lead may be present without hematological abnormalities.1272 However, hematologic abnormalities tend to be more consistent in chronic lead toxicity and in lead-poisoned horses.
Lead toxicity has a variety of effects on endocrine function in cattle. Elevated levels of serum T3, T4, estradiol, and cortisol have been demonstrated in cattle with elevated blood lead levels.1273 Parameters of liver function also are affected; serum alanine and aspartate transaminase (ALT and AST) levels are increased, whereas serum lipids, total protein, and albumin are decreased.
In poisoned animals the concentrations of protein and WBCs in CSF are increased, ranging from 50 to 100 mg/μL of protein and 5 to 50 mononuclear cells/mL, respectively. Such changes are relatively nonspecific and found with other causes of polioencephalomalacia.
Pathophysiology
Lead enters the body through the GI tract or less often through the respiratory tract. Metallic lead and the sulfide form are less well absorbed than the acetate, phosphate, carbonate oxide, and hydroxide salts. Metallic lead is poorly absorbed and causes toxicity only when a lead foreign body becomes entrapped in the stomach for prolonged periods. Interaction between lead and other minerals may occur. For example, high levels of dietary calcium reduce the GI absorption of lead. Concomitant exposure to lead and cadmium results in a worsening of the clinical signs of lead poisoning.1274
Acute toxic single doses of lead range from 200 to 600 mg/kg for calves and 600 to 800 mg/kg for adults.1275,1276 Although intestinal absorption of lead is relatively inefficient, significant amounts can cross into the blood if sufficient quantities are ingested. Approximately 1% to 2% of the total oral dose of lead is absorbed by 24 hours.1255 Increases in the blood lead concentration are observed as early as 3 hours after dosing. Most of the lead absorbed from the digestive tract (90%) is bound irreversibly to erythrocyte proteins, resulting in a low lead concentration in the plasma but higher concentrations in whole-blood specimens.1277 At the end of the erythrocytes’ lifespan, the cell-bound lead is metabolized from the erythrocyte proteins and deposited in the bone as the triphosphate salt. A smaller amount of dissolved lead is deposited into the soft tissues as the diphosphate. A portion of the soft tissue lead is excreted through the GI tract via the secretions (pancreatic juices, bile) and direct diffusion. The half-life of blood lead in adult cattle is extremely variable and unpredictable, ranging from 48 to 2507 days in one study.1278 Phenotypic or genotypic factors may affect metabolism and storage of lead; for example, beef cattle store more lead in the liver than the kidneys compared to dairy cattle.1279 The variable time for clearance of lead has an obvious implication for public health, because all carcasses of animals suspected or known to be exposed to lead must be tested before being cleared for human consumption.1280
Lead also crosses the placental barrier and accumulates in fetal bone, liver, and kidneys, but does not substantially accumulate in milk. Infertility, abortions, and fetal malformations may result from exposure to lead.1257 The concentration of lead in milk from lactating cattle fed a daily dose of 13 mg of lead acetate remains less than 5.9 parts per billion (ppb).1281 In one study a logarithmic relationship between blood and milk lead concentrations was found. At blood concentrations below 3.6 μg/dL, milk lead concentrations were 0.8 μg/mL. However, cattle with higher blood lead levels (4.8 μg/dL) had exponentially greater concentrations in milk (2.2 μg/kg).1282 Lead cannot be detected in milk by 7 months after exposure.1283
The toxic effects of lead include inhibition of free sulfhydryl groups found in many enzymes, interference with zinc-containing metalloproteins, and steric inhibition of enzyme activity.1271 Enzymes of heme synthesis are particularly susceptible to injury. These include δ-ALA dehydratase and ferrochelatase. Interference with ferrochelatase inhibits the formation of heme from protoporphyrin, resulting in a buildup of unmetabolized porphyrins, including protoporphyrin I, uroporphyrins, and coproporphyrins. The last two molecules are excreted in the urine and feces, respectively.1254 Protoporphyrin I is retained in the erythrocyte.
Interference with the activity of ALA dehydrase may be partly responsible for the brain damage associated with lead poisoning. The enzyme δ-ALA dehydrase combines two molecules of δ-ALA into a single porphobilinogen molecule. This enzyme is exquisitely sensitive to lead. Inhibition of the enzyme leads to accumulation of ALA, which is excreted into the urine. Concentrations of the synthetic product porphobilinogen in the erythrocytes are reduced.1266,1267,1269,1284
Because of the interference with heme metabolism and the altered function of other erythrocyte proteins, the erythrocyte half-life is shortened, which may result in a normochromic, normocytic anemia in a small proportion of chronically poisoned animals.1284 Iron is not adequately used and is stored in sideroblasts in the bone marrow.1266 Lead also interferes with the activity of pyrimidine-specific 5′-nucleotidase.1285 Loss of activity of this enzyme results in basophilic stippling.
After absorption, lead rapidly enters the brain at a dose-dependent rate. The lead deposition in the CNS results in acute cerebellar hemorrhage and edema from capillary dysfunction.1277 Abnormalities of brain cerebroside content and catecholamine metabolism have also been described in animals with lead poisoning; however, the role of these changes in the pathogenesis of the clinical signs is unknown. The pathogenesis of lead encephalopathy is multifactorial. Encephalitic signs probably originate from a combination of decreased microvasculature, cellular necrosis, brain swelling, neurotransmitter dysfunction, and decreased glucose uptake by the brain.1286
The molecular effects of lead on the myocardium are unknown. Hypertension caused by chronic poisoning is thought to result from an inhibition of sodium-potassium adenosine triphosphatase or an alteration of the juxtaglomerular apparatus.1261
Ingestion of lead also results in aberrations of other minerals. For example, long-term exposure to lead results in competitive inhibition of selenium uptake, thereby diminishing the absorption of selenium by as much as 26%.1276 If selenium intake is marginal, lead toxicosis could manifest as an outbreak of white muscle disease. Lead-induced selenium deficiency may contribute to the pathogenesis of myocardial disease and immune system dysfunction.
Epidemiology
Sources of lead are legion, including lead arsenate defoliants, batteries, used motor oil, linoleum, roofing felt, paint, machinery grease, caulking compounds, improperly compounded mineral supplements, and foliage near lead smelters and battery-recycling plants.1254,1260,1268,1287-1293 Blood lead levels of animals residing in highly contaminated urban environments may be significantly greater than those of their rural-dwelling counterparts, and high lead levels have been reported in grasses growing near busy roadways, but the clinical significance of these findings is unclear.1290,1294,1295 Contamination of preserved feeds, such as silage, can occur before or during processing or during storage. Factors that can increase the likelihood of ingestion of lead-contaminated foodstuffs include lack of alternative feed, hunger, and phosphorus deficiency.1257 The single lethal dose of lead for cattle is estimated to range from 220 to 600 mg/kg for calves, 600 to 800 mg/kg for adult cattle, and 400 mg/kg for goats.1257,1275 Poisonings from cumulative intake are associated with substantially lower daily doses. Although cattle can detect fairly low levels of lead on pasture and have an aversion toward contaminated herbage, continued exposure may lessen this aversion, making animals more prone to ingest contaminated material.1296 Lead poisoning has been induced in cattle by feeding 5 to 6 mg lead/kg body weight/day for 3 years or 6 mg/kg of lead (lead acetate) for 7 days.1268,1297 Lead poisoning has been reported in cattle exposed naturally to 6 to 7 mg/kg/day of lead on foliage and in calves given oral lead acetate at 2.7 to 20 mg/kg/day.1297 The interval for development of clinical signs ranges from 5 to 20 days and is related to the dose and the ionic form of lead administered.1297 Ensiling of contaminated forage results in percolation and concentration of lead at the bottom of the silo.1298
The toxicity of lead is apparently influenced by dietary factors. Calves on a milk diet are more susceptible to lead poisoning than calves fed hay and grain.1262 There appears to be a direct correlation between high levels of vitamin D and enhanced lead absorption, which may explain the greater occurrence of the poisoning during the summer. Elevated copper concentrations in forage, such as may be found in pastures fertilized using pig slurry, may potentiate accumulation of lead in animals consuming it.1299
The estimated cumulative toxic dose of lead for horses is 2.9 mg/kg/day.1300 Poisonings have been reported in horses grazing pastures contaminated with 320 to 440 ppm of lead from a metal smelter; this amounted to a daily intake of 2 g (∼6.4 mg/kg). Metallic lead and the “galena” (insoluble sulfide salt) are less toxic than the acetate and carbonate lead salts.1301
Necropsy Findings
The macroscopic brain lesions of lead poisoning are mild and include brain edema, congestion of vessels of the cerebral cortex, and yellowish discoloration and flattening of the cortical gyri. Lesions tend to be most severe in the occipital lobes. Microscopic changes in the brain include capillary prominence, endothelial cell swelling, edema of the Purkinje cell layer of the cerebral cortex, laminar cortical neuronal necrosis, and edema of the white matter.1302 The lesions are predominantly located on the tips of the gyri. Whether these lesions are caused by a direct effect of lead on the neurons or from vascular damage is unclear.1277 Intranuclear acid-fast inclusion bodies in the renal tubular epithelial cells have been described in experimentally poisoned cattle. Chronic lead exposure also may interfere with normal functioning of the immune system, resulting in an increased susceptibility to infections.1303
Treatment
Therapy for lead poisoning should include removal of the lead from the digestive tract, chelation therapy with calcium disodium EDTA, and fluid and nutritional support of the patient. Treatment with calcium disodium EDTA (calcium versenate) has been shown to be superior to treatment with penicillamine or dimercaprol (BAL). The EDTA chelates osseous but not soft tissue—bound lead.1274 After chelation the unsaturated bone stores reequilibrate with the lead remaining in the soft tissues. In cases of acute lead poisoning, several days are required before reequilibration results in a decreased blood lead concentration. The dose of calcium disodium EDTA is 66 mg/kg/day, divided into several doses daily for 3 to 5 days.1304 After five daily treatments, a 2-day nontreatment period is recommended to reequilibrate the soft tissue and bone lead. After the 2 days’ rest, daily treatments are given for another 5 days. The decision to continue therapy with EDTA should be based on the results of posttreatment blood lead analyses and renal function tests. Another recommendation is for administration of two IV injections of calcium disodium EDTA (110 mg/kg per dose) given 12 hours apart for 2 days.1305 Therapy then is withheld for 2 days, after which the EDTA treatments are reinstituted for 2 more days. The comparative efficacy of this regimen is unknown.
The EDTA also chelates other divalent cations. Consequently, prolonged administration of the drug results in trace-mineral deficiencies, especially of zinc. For this reason, after prolonged EDTA therapy, oral supplementation with zinc should be considered to prevent the development of parakeratosis.
Meso-2,3,-dimercaptosuccinic acid may be a more effective agent for lead chelation, particularly when it comes to removing lead from soft tissue.1306 However, experience with this drug is still limited. There appears to be no advantage to using this drug in conjunction with calcium disodium EDTA.
Reports have indicated that thiamine therapy is an effective adjunctive treatment with EDTA in cases of acute lead poisoning of cattle.1275,1307,1308 Administration of 2 mg/kg thiamine daily was more effective than treatment with disodium EDTA (62 mg/kg twice daily for 4 days) or thiamine plus disodium EDTA in inducing remission of clinical signs of experimentally induced lead poisoning.1309 For clinical treatment of lead poisoning, thiamine dosages of 500 mg for small ruminants and 1 g for cattle weighing 300 kg or 5 mg/kg have been recommended.1307 Administration of daily doses of thiamine (100 mg/calf/day or 5 mg/kg) has protected experimentally exposed calves from clinical signs of lead poisoning and reduced lead deposition in the soft tissues.1308,1310,1311 The nature of the protective effects of thiamine is unclear. Apparently, either lead interferes with thiamine synthesis, or the tissue distribution and deposition of lead are reduced by the formation of rapidly excreted lead-thiamine complexes.
In ruminants, ingested lead is best removed from the digestive tract by means of a rumenotomy.1275 Magnesium sulfate laxatives are administered concomitantly to form insoluble lead sulfides. Because of the possibility of additional lead absorption from the GI tract, oral administration of chelators is contraindicated.
Patients that respond slowly to chelation and thiamine therapy should be given supportive care. These measures should include provision of 40 to 80 mL free water/kg/day for maintenance, oral hyperalimentation, and administration of diazepam or phenobarbital for convulsions (see Table 35-9).
Prevention and Control
Toxic pastures can be made safe by removing contaminated forage. This is best done by cutting, baling, and burying native grasses; burning the stubble; and applying agricultural lime at the rate of 1 ton per acre where the lead concentration of topsoil exceeds 175 ppm.1266 In the case of negligent poisonings, vigorous attempts at laboratory confirmation of the clinical diagnosis should be made. The source of the lead should be established, and the affected animals should be carefully documented. In the United States, insurance liability responsibilities may be covered under homeowner or farm insurance.
TOXICITY FROM GASOLINE, PETROLEUM DISTILLATES, AND RELATED PRODUCTS
Ingestion of natural gas condensate or petroleum distillates can cause neurologic disease in livestock. Affected animals appear to be anesthetized and fail to respond to auditory or visual stimuli. The clinical signs of petroleum distillate poisoning include depression, ataxia, diarrhea, recumbency, coma, semicoma, absent menace response, decreased palpebral reflex, and muscular hypotonia. Constituents of petroleum and related products cause pathology in many organs, including the lungs, kidney, liver, and digestive tract. Thus a variety of clinical signs, such as dyspnea, coughing, and bloat, may be present in poisoned animals, in addition to neurologic abnormalities.1312,1313 The feces and digestive tract contents have a strong odor of petroleum or gasoline. Some animals may die suddenly without premonitory signs. Animals in poor condition or suffering from chronic illness are at greatest risk of toxicity.1314
Necropsy findings in poisoned animals include diffuse serosal hyperemia of the bowel and forestomachs and diffuse serosal ecchymotic hemorrhages. The lungs are firm and mottled, especially in the middle and cranial lobes. These pulmonary changes may be associated with moderate amounts of serofibrinous pleural exudates. Microscopic changes in poisoned animals include myocardial degeneration and necrosis, enteritis, mild renal tubular degeneration, and granular eosinophilic casts. Affected livers develop periacinar fatty degeneration and periportal infiltrations of lymphocytes and plasma cells.
Gas chromatography of the intestinal contents usually reveals peaks of aromatic hydrocarbons. For identification of the source of hydrocarbons, gas chromatographic profiles of the environmental specimens can be compared to those of the rumen liquor. Several oxidative biochemical activities of circulating neutrophils are reversibly depressed in animals exposed experimentally to crude oil or diesel fuel. This effect is dose dependent and may provide a method for determining exposure to oil and petroleum products and for tracking recovery.1315
In early cases of petroleum distillate poisoning, removal of the hydrocarbons by means of a rumenotomy should be considered. Treatment usually is futile when the animal becomes recumbent and unresponsive.1316
ETHYLENE GLYCOL TOXICOSIS (ANTIFREEZE POISONING)
Antifreeze poisoning occurs primarily in ruminants.1317,1318 When ingested, ethylene glycol is enzymatically converted to a number of acidic intermediate compounds, especially glycolic acid, which is further metabolized to oxalic acid. This acid combines with calcium in the kidneys to precipitate as calcium oxalate. Ruminants are thought to be more resistant to the toxic effects of ethylene glycol than monogastric animals because of their ability to metabolize large quantities of oxalate in the rumen. The acute toxic dose of ethylene glycol for adult ruminants ranges from 5 to 10 mL/kg, whereas that for preruminant calves is 2 mL/kg.
Animals that have ingested sufficient amounts of ethylene glycol become ill by 3 to 4 days after ingestion. Clinical signs of ethylene glycol toxicity include blindness, progressive hindlimb ataxia, salivation, depressed sensorium, nystagmus, tonic-clonic seizures, and status epilepticus. Pupillary reflexes usually are intact. Hemolytic anemia and hemoglobinuria occasionally may be seen.1317 The clinicopathologic changes of ethylene glycol toxicosis include azotemia (448 mg/dL), increased serum creatinine, hypophosphatemia, hypocalcemia, acidosis, hyperosmolality, and increased γ-glutamyl transaminase.
The pathologic lesions include slight swelling of the kidneys and pulmonary edema. Oxalate crystals can be demonstrated by microscopic examination of the kidney tissues using polarized light. Ethylene glycol can be detected in the rumen for at least 4 days after ingestion. Mass spectrometry of body fluids may show increased urinary and ocular fluid concentrations of glycolic acid (4.3 μg/mL and 2.3 μg/mL, respectively).
Treatment with 20% ethanol at a rate of 50 mL/hr has been recommended but is unsuccessful in advanced stages of the disease. Some have suggested that ruminants also be given an oral dose of activated charcoal, but the effect of this treatment on long-term survival is unknown.1318
NARDOO FERN POISONING
Sheep that graze extensively on the Nardoo fern (Marsilea drummondii) develop a condition that is indistinguishable from polioencephalomalacia (PEM). Death losses of 2200 of 57,000 sheep have been reported.1319 The clinical signs are indistinguishable from those of PEM. Neuronal necrosis, malacia, perivascular cuffing, vacuolation of the neuropil, vascular dilation and endothelial hypertrophy, and gliosis occur in the central nervous system. The condition responds to a single subcutaneous injection of thiamine (200 mg). The fern is thought to contain a form of thiaminase I.
HELICHRYSUM ARGYROSPHAERUM POISONING
Both naturally occurring and experimental poisoning of sheep and cattle in South Africa by plants of the genus Helichrysum results in blindness and a variety of CNS signs.1320,1321 The clinical signs of intoxication include progressive tetraparesis, depression, nystagmus, mydriasis, blindness, intentional head tremor, and star-gazing attitude. Older sheep may develop lens cataracts 2 to 3 months after eating the plants. The case-attack rate ranges from 1% to 29%. Plants are toxic only in the flowering stage. Helichrysum species have been shown to contain substances that bind at the γ-aminobutyric acid (GABA)—benzodiazepine receptor, suggesting a mechanism for toxic effects on the nervous system.1322
Pathologic findings include widespread status spongiosus of brain white matter, particularly in subependymal areas and in the cerebellar peduncles and brainstem.1321 Myelin edema is present in some cases. Edematous swelling of the optic nerve causes compression of the nerve in the optic canal, with secondary damage to nerve axons and myelin. The toxic principle in Helichrysum plants also causes a primary retinopathy in some animals.
Other members of the Helichrysum genus are being studied for a variety of medicinal properties, including antiviral, antioxidant, antiinflammatory, and free-radical scavenging activities.1323-1325
FLATPEA (LATHYRUS SYLVESTRIS, LATHYRUS COLLIS) POISONING
Ingestion of flatpea (Lathyrus sylvestris, Lathyrus collis) results in a CNS disorder. The condition may be seen by 5 days after consumption of a diet composed of 50% flatpea vines. Toxicosis has been induced in sheep ingesting forage of 35% flatpea vines.1326 Livestock can develop a tolerance for the plant through rumen microbial detoxification. Nevertheless, acclimatized animals can be rendered susceptible by treatment with monensin or by a change in rumen microflora.1327
The toxic constituent of the plant, 2,4-diaminobutyric acid, is known to inhibit ornithine transcarbamylase, an enzyme responsible for urea detoxification. Consequently, the blood ammonia concentration in clinically affected animals ranges from 189 to 263 mmol/mL (normal, 108 to 185 mmol/mL). Diaminobutyric acid also interferes with the uptake of GABA and inhibits GABA transaminase activity.
The clinical signs of flatpea intoxication are depression, muscular tremors, and spasmodic torticollis. Affected animals become recumbent and are reluctant to rise. When stimulated to move, they display circling, head pressing, and odontoprisis. The urine may appear dark brown. The clinical disorder often culminates fatally in a seizure. During the interictal periods the animals may rest, rise, and resume normal behavior and gait. Treatment is empiric and supportive and could include 1 to 2 L of vinegar orally, IV diazepam, and removal from the offending forage.
Lathyrus sylvestris is a leguminous plant with a high protein content that might be an adequate substitute for alfalfa in areas where the latter grows poorly.1328 L. sylvestris harvested in the vegetative state has been fed to lambs in combination with alfalfa and as a sole diet without ill effect.1329 Similarly, when fed as part of a mixed silage in which the concentration of diaminobutyric acid was approximately 1%, L. sylvestris produced an acceptable weight gain in cattle without signs of toxicity.1330
LEUKOENCEPHALOMALACIA (MOLDY CORN DISEASE; EQUINE ENCEPHALOMALACIA; PESTA DE CEGARE; PEN YAN DISEASE; MOLDY CORNSTALK DISEASE; BLIND STAGGERS)
Definition and Etiology
Leukoencephalomalacia (LEM) is an intoxication of horses caused by ingestion of corn contaminated with the fungus Fusarium moniliforme.1331-1335 Fumonisin toxins (B1, B2, and B3) produced by F. moniliforme interfere with sphingolipid metabolism, disrupting endothelial cell walls and basement membranes.1336 Although all three substances are toxic, fumonisin B1 is believed to be mainly responsible for LEM.1337 Outbreaks of multifocal neurologic signs and hepatic disease occur in groups of horses exposed to tainted feedstuffs.
Clinical Signs
The clinical signs of LEM occur suddenly. Occasionally, animals die acutely, without other overt signs,1338 but most horses show a variety of neurologic signs before death. These include somnolence, flaccidity of the facial and pharyngeal muscles, muscle fasciculations over the neck and withers, ataxia, conscious proprioceptive deficits, head pressing, mania, facial desensitization, pharyngeal paralysis, blindness, seizures, and a tendency to circle or lean to one side.1338,1339 Most animals die while convulsing.1331 The few horses that recover usually have permanent neurologic dysfunction. Hepatic involvement occurs in many cases, as evidenced by elevated serum liver enzymes, although hepatic failure is uncommon. Signs of liver disease include icterus, petechiation on mucous membranes, and swelling of the muzzle or lips. Gastrointestinal disease caused by fumonisin toxins has been reported and may manifest as signs of colic.
Unique constellations of clinical signs may predominate within any one outbreak of the disease. Fumonisin toxins cause a variety of clinical syndromes in other species, but horses appear to be particularly susceptible and can show signs when exposed to toxin concentrations as low as 5 to 10 ppm, almost 10 times less than the concentration needed to cause mild signs of inappetence and decreased weight gain in cattle.
Clinical Pathology and Diagnosis
Fumonisin toxicosis has no unique clinicopathologic findings, therefore antemortem diagnosis relies on recognition of the clinical signs with a history of exposure to moldy corn. Specific changes in the cerebrospinal fluid (CSF) of affected horses have not been reported. Serum liver enzymes (aspartate transaminase [AST], γ-glutamyltransferase [GGT], sorbitol dehydrogenase [SDH]) and bilirubin may be elevated. Nonspecific changes in serum chemistry associated with dehydration (increased hematocrit, prerenal azotemia) and recumbency (elevated serum creatine kinase [CK]) also may be present. Anemia, leukocytosis, and leukopenia all have been reported, but none is a consistent finding.
Differential diagnoses include craniocerebral trauma, the arboviral encephalitides, hepatic encephalopathy, equine protozoal myeloencephalitis, Theiler’s disease, and botulism.
Epidemiology
Leukoencephalomalacia occurs worldwide.1333,1340 Corn becomes contaminated during growth rather than in storage, and climatic factors that stress the plants, such as drought, excess moisture, or heat, contribute to the likelihood of mold development. Most cases of equine disease occur during the winter and early spring.1332,1338 In experimental studies the toxic dose of infected corn ranged from 5 to 15 kg (10 to 30 lb), but the amount of corn required to cause the disease is likely to vary considerably depending on the amount of toxin in the grain.1334 A direct link between the onset and severity of clinical signs and the dose of toxin has not been established in naturally occurring cases, but experimental data suggest a dose-related effect.1341 Repeated exposure to the toxin, rather than a single large dose, seems to be associated with the development of clinical signs.1342 Experimental studies with infected corn demonstrated an onset of clinical signs on the ninth day after the beginning of the feeding period. Older animals develop clinical signs of LEM more rapidly than younger animals and thus appear to be most susceptible to the effects of the mycotoxin.1334
The rates of disease in exposed horses vary widely, from 14% to 100% in some reports.1343-1346 Ruminants apparently are more resistant than horses to the effects of the neurotoxin, but this is not a complete resistance because camels and water buffalo have died after ingesting toxic corn. Diplodiosis, a similar neuromycotoxicosis of cattle caused by ingestion of Diplodia maydis, occurs in Africa; however, the toxicologic relationship between these conditions is unknown.
Pathology
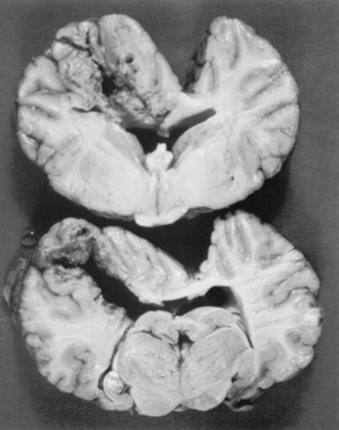
The major pathologic features in the central nervous system (CNS) result from the vascular damage caused by fumonisin toxins, including liquefactive necrosis and degeneration or malacia of the white matter of one or both cerebral hemispheres.1347,1348 The size of the lesions may vary from 0.5 cm in diameter to complete necrosis of the entire cerebral cortex.1331 Flattening of the cortical gyri, enlargement of the cerebral cortex, vascular congestion, cortical softening, yellowish discoloration of the white matter, hemorrhage, and cavitation of the cerebral cortex may be present1331,1338,1340 (Fig. 35-14). A gelatinous fluid can be seen in many of the cavitary lesions.1338 Hemorrhage in the CNS also has been reported.1343 Lesions in the visceral organs, including hepatic congestion, centrilobular hepatic necrosis, hemorrhagic enteritis, and cystitis are found in some horses. The relationship between these lesions in the CNS and those in the liver, urinary bladder, and GI tract is unknown.
Treatment
There is no known specific treatment for LEM, but successful treatment of horses was reported using antiinflammatory medications such as dimethyl sulfoxide (DMSO) (1 g/kg given as 10% solution by slow IV infusion once daily for 3 days) or flunixin meglumine (0.25 to 1 mg/kg), as well as antibiotics and supportive care (thiamine, 5 g IV every 12 hours).1349 In other cases, survivors usually have permanent neurologic dysfunction.
BLUE-GREEN ALGAE TOXICOSIS
Definition and Etiology
Ingestion of stagnant pond water containing certain species of blue-green algae may result in a peracute intoxication of livestock. Blue-green algae poisoning is characterized by convulsions, ataxia, bloody diarrhea, and sudden death.1350-1353 Although often a fatal toxicity, some affected animals can make a full recovery.1351,1354 The algal toxins have been responsible for high losses of livestock and illness in humans and for deaths of domestic dogs.1355,1356 The algal toxins may also be responsible for occasional die-offs of fish and aquatic birds. Toxic algal species include Microcystis aeruginosa, Anabaena flos-aquae, Aphanizomenon flos-aquae, Anacystis cyanea, Gloeotrichia echinulata, Nodularia sphaerocarpa, and Oscillatoria agardhii. Of these, the first three are most toxic.1357 Blue-green algae poisoning most often results in sudden death. Affected animals rarely move far from the source of the toxin. Some of the algae produce hepatotoxins, and animals develop liver failure, diarrhea, and photosensitivity. The development of toxic stands of blue-green algae requires specific environmental conditions, including a water pH above 6, organic pollution, and a water temperature ranging from 15° C to 30°C (59° F to 86°F).
Clinical Signs
Clinical syndromes of blue-green algae poisoning in livestock may be separated into acute and chronic forms. Acutely affected animals may show signs resembling those of milk fever,1354 including muscle tremors, reluctance to rise or move, ataxia, cold extremities, weak rapid pulse, mydriasis, muscle tremors, salivation, colic, rumen atony, mild bloat, pallor, increased capillary refill time, vomiting, ataxia, conscious proprioceptive deficits, and bloody diarrhea. Some of the toxins are absorbed through the oral mucosa. Consequently, the full range of clinical signs, culminating in death from respiratory arrest, can occur within minutes of ingestion of the toxic water. If clinical signs are seen before death, affected animals tend to be afebrile but have significantly increased pulse and respiratory rates. Many animals die suddenly without premonitory symptoms.1358,1359 The deaths often occur in the vicinity of the pond, and dead animals may be covered by the green scum.
In the chronic form of blue-green algae intoxication, affected animals show ataxia, depression, anorexia, hemorrhagic diarrhea, icterus, and photosensitization, which occur secondary to hepatic necrosis.1359 Death from respiratory arrest and circulatory shock may occur within 2 to 72 hours after the toxin is ingested.
Clinical Pathology
The diagnosis of blue-green algae poisoning depends on recognition of a relationship between livestock deaths and ingestion of pond water, identification of toxic algae in the pond water, recognition of hepatic disease in chronically affected animals, and elimination of the possibility of similar clinical conditions, such as cyanide or acute poisoning. Diseases that kill animals suddenly should be considered as differential diagnoses (see Chapter 14). Blue-green algae poisoning should be considered whenever a group of cattle simultaneously develop marked massive hepatic necrosis.
The vegetative cells of the algae can be identified by microscopic examination of rumen contents. The intestinal contents should be split. Half the contents should be placed in 10% neutral buffered formalin for microscopic analysis, and the other half should be refrigerated (not frozen) for mouse bioassay tests or chromatographic identification of the toxin using high-performance liquid chromatography (HPLC).1360 The blood of animals poisoned by microcystin, the toxic principle of Microcystis aeruginosa, shows changes characteristic of hepatic necrosis, including increased concentrations of bilirubin, AST, GGT, alkaline phosphatase (ALP), and arginase. The animals may be secondarily hypocalcemic, which complicates the clinical picture.1354
Pathophysiology
Blue-green algae grow more slowly than other algae in cold water; therefore, highly flushed systems cannot achieve a toxic bloom. The blue-green algae can fix atmospheric nitrogen dissolved in the water, and they have intracellular gas vesicles that accumulate the nitrogen when photosynthesis decreases. If mixing occurs because of the wind, the amount of light reaching the algae decreases because of the turbulence. The buoyancy of the cells increases because of decreased photosynthesis. At night the winds become calmer, and the algae lose their ability to regulate density. The cells float to the surface of the water and form a scum, which is concentrated on the leeward side. For these reasons, poisonings tend to occur in the period of stable weather just after a frontal system has passed.
Direct ingestion of the toxicant is necessary to cause clinical signs. No significant level of toxin was detected in milk of cows fed the toxin experimentally, so calves are not exposed through their dams’ milk.1361
All species of blue-green algae probably produce toxins, which can be classified into the following three groups:
 Aphanizomenon, Oscillatoria,
Aphanizomenon, Oscillatoria, and
Anabaena species.
Aphanizomenon produces two alkaloid toxins that have a structure resembling that of saxitoxin, the agent of paralytic shellfish poisoning. Toxins from
Anabaena species are named anatoxin-a and anatoxin-a(s) and are structural analogs of cocaine. Toxins from
Oscillatoria species resemble those of
Anabaena; they can be absorbed unchanged through the mucous membranes and kill by depolarizing blockade of the neuromuscular junction.
1358,1359,1362-1364 Peptide hepatotoxins. These substances are produced by strains of
Microcystis, Oscillatoria, and
Anabaena algae. Microcystin-LR is the most frequently isolated hepatotoxin.
1365-1369 On a weight basis, this toxin is 20 times more active than cyanide or strychnine. A single intraperitoneal injection of 1 to 2 μg in a mouse is lethal. At least nine structural variants of microcystin have been identified. These toxins act exert their toxic effects on mitochondria.
1370 The toxins can cross the placenta and cause lesions in the fetus.
Lipopolysaccharides. These substances may be produced by most species of blue-green algae.
Necropsy Findings
The pathologic lesions of blue-green algae poisoning are either severe centrilobular hepatic necrosis in animals that die of the chronic poisoning or generalized petechiation and body cavity effusions in animals that die peracutely.1358
Epidemiology
Blue-green algae intoxication occurs worldwide and affects mammals, birds, and fish. The bloom is most abundant during the late summer and early autumn when warm, sunny conditions favor algal growth. Growth is most abundant in ponds with an alkaline pH and in high concentrations of nitrogen, phosphates, carbonates, or organic matter. Release of the toxin is associated with death of the algae and production of a “rotting fish” odor. Most poisonings occur on the leeward side of the pond, where the algae are concentrated by the action of the prevailing wind. Ingestion of approximately 1080 to 1500 mL of heavily contaminated water can be fatal for cattle.1350 Toxicity varies daily and in different parts of the pond or lake.
Treatment
Therapy for blue-green algae poisoning is symptomatic and usually unsuccessful. Experimentally poisoned calves have not recovered, even after 30 hours of artificial respiration, although recovery of cows naturally intoxicated has been reported.1351,1363
Prevention
Methods for control of the disease include restriction of access to infested ponds and treatment of the pond with copper sulfate or algacides.1352 Prevention of blue-green algae poisoning depends on the proper construction of farm ponds and the prophylactic treatment of the water with bluestone (copper sulfate) to achieve a final concentration ranging from 0.5 to 1.0 ppm in acid water and 1.5 to 2.0 ppm in alkaline water. The bluestone is either dissolved in water and sprayed over the pond or dragged through the pond in a burlap sack in lanes that are 5 to 10 feet apart.1352 This amounts to 1.22 kg/acre foot in alkaline water. Cattle should be fenced from the pond for several days after the copper sulfate treatment. The treatment should be repeated whenever the toxic bloom recurs. To prevent algal bloom without application of copper sulfate, farm ponds should be constructed so that they are 80 × 20 feet in length and width and 10 feet in depth. Surrounding drainage areas should be fenced from the livestock. Water should be pumped from the pond to the cattle in polyethylene pipes and delivered into raised water troughs. The water for the troughs should be pumped from the center and bottom of the pond.
NITROFURAZONE TOXICOSIS
Nitrofurazone is an antimicrobial that has been fed to cattle for the treatment and control of respiratory or gastrointestinal diseases. Treatment of food-producing animals with the nitrofurans currently is prohibited by the U.S. FDA. Nervous system signs of nitrofurazone toxicosis occur after 1 to 3 weeks of continuous feeding at dosages exceeding 15 to 30 mg/kg.1371,1372 Lower dosages (7.1 mg/kg) reduce feed intake but do not result in neurologic signs. The nitrofurans inhibit enzymes of the oxidative glycolytic pathways and are thought to interfere with brain metabolism of carbohydrates.
Clinical signs of nitrofurazone toxicosis include hyperirritability, propulsive running, muscular tremors, blindness, convulsions, and death. At lower doses the convulsions may appear intermittently, but as the condition progresses, the signs become continuous.
INTRACAROTID DRUG INJECTION
Definition and Etiology
Intracarotid drug injection is common in horses because the jugular vein and the common carotid artery are closely apposed in the caudal third of the neck. The condition is rarely seen in cattle because the omohyoideus muscle lies between the carotid artery and the jugular vein in the posterior part of the neck. Hypertonic or caustic drugs, including phenothiazine tranquilizers, chloramphenicol, chloral hydrate, barbiturate anesthetics, phenylbutazone, calcium gluconate, sodium iodide, and chloramphenicol, cause cortical necrosis when injected into the carotid artery.1373,1374
Clinical Signs
The onset is peracute. When the drug is injected into the carotid artery, the animal recoils backward and falls over. Some horses strike or rear violently or run wildly without regard to obstructions. Other animals fall down and become comatose without showing severe motor activity. Severely affected animals may die after a variable period, but others regain their footing and recover completely. Residual neurologic deficits may occur in surviving animals. These deficits include contralateral blindness, facial hypalgesia, head tilt (toward the side of the lesion), and a largely contralateral, conscious proprioceptive deficit. If the injection has damaged the ascending vagosympathetic pathways, the animal may display Horner’s syndrome, with signs that include ptosis, miosis, and enophthalmos. Horses with Horner’s syndrome also sweat profusely over the head and neck of the ipsilateral side, whereas cattle with the syndrome fail to sweat on the planum nasale on the ipsilateral side of the lesion.
Pathophysiology
The CNS lesions are caused by vascular endothelial damage. Intracarotid drug injection results in intense vasospasm and profound alterations of the blood-brain barrier (BBB). The vascular damage causes endothelial cell swelling, increased vascular permeability, mural necrosis, hemorrhage, intercellular edema, and thrombosis.1373
Necropsy Findings
Pathologic lesions include diffuse cerebral edema and brain swelling. Microscopic lesions include arteriolar hyalinization, hemorrhage, edema, necrobiosis, and status spongiosus. Vacuolation of the neuropil, perivascular hemorrhage, fibrin, and edema are also seen.
Treatment
No effective treatment exists for an accidental intracarotid drug injection. Violent horses should be placed in a padded stall, sedated with diazepam, and treated with dexamethasone (1 to 2 mg/kg). Administration of mannitol or other osmotic diuretics should be avoided in the first 24 hours because of active bleeding in the CNS and loss of the BBB. Administration of a hypertonic dehydrating agent at that time may result in distribution of the osmotically active drugs into the CNS parenchyma, resulting in a large increase in intracranial pressure. Although most animals eventually recover from the effects of an intracarotid injection, fatalities have been reported.1373-1375
Prevention
Intracarotid injection of drugs is best prevented by the use of large-bore needles or catheters for intravenous injections. This allows better visualization of pulsating oxygenated blood when the carotid artery has been accidentally punctured. In the horse, venipunctures should be performed in the anterior one third of the jugular furrow because the artery and vein are separated by the omohyoideus muscle in this area. Needles should be inserted into the vein while they are separated from the syringe.
COENUROSIS (SHEEP GID; COENURUS CEREBRALIS INFESTATION; TAENIA MULTICEPS INFESTATION)
Definition and Etiology
Coenurosis is caused by invasion of the CNS by Coenurus cerebralis, the intermediate stage of the tapeworm Taenia multiceps. The adult worms live in the intestine of domestic dogs and some wild carnivores, where they shed eggs into the feces. Ruminants eat the eggs from contaminated pastures. The eggs hatch in the small intestine of the ruminant, and the larval stages travel through the blood to the CNS, where they mature into C. cerebralis. The life cycle is completed when the ruminant dies and the brain is eaten by a scavenging carnivore. Coenurus cysts then develop into sexually mature adults in the bowel of the carnivore host.
Many animals, including sheep, goats, cattle, horses, wild ruminants, and humans, are susceptible to C. cerebralis infestation.1376-1378 Outbreaks of coenurosis may occur in previously uninfected sheep that are suddenly exposed to contaminated fecal matter from carnivores. Cases initially occur as early as 2 weeks after the sheep are exposed and continue for as long as 4 months.
Clinical Signs
Signs can occur acutely, during the migratory phase of the larval stage in the intermediate host. Lambs 6 to 8 weeks old are most often affected by this form and develop fever, dullness, and mild neurologic deficits.1379 Occasionally, acute encephalitis occurs, leading to sudden onset of severe neurologic signs and death within a few days. More frequently, the clinical presentation of coenurosis is that of a space-occupying brain lesion; signs include depression, anorexia, ataxia, unilateral or asymmetric loss of vision, facial hemiplegia, head tilt, circling, high-stepping forelimb gait, and hyperesthesia. When the spinal cord is the site of cyst development, hindlimb ataxia and paresis to paralysis is the main clinical sign.1380 As the disease progresses, the sheep assume lateral recumbency and become comatose.1381,1382 In advanced cases the calvarium directly over the parasite enlarges and softens.1378
Pathophysiology
Lesions of the CNS may result from three separate pathogenic mechanisms. These include encephalitis from invasion of the CNS by large numbers of larvae, hypertensive hydrocephalus resulting from interference with CSF drainage, and development of large cerebral cysts that increase intracranial pressure. Full development of the Coenurus cyst requires 6 to 7 months. Mature cysts may reach 5 cm in diameter and displace the bones of the calvarium.
Necropsy Findings
In the acute form of coenurosis, the main finding is coagulation necrosis and inflammation associated with the pathway of the larval form as it migrates through the CNS.1383 This may be visible grossly as yellow to red tracks through the brain parenchyma. Coagulation necrosis and surrounding inflammatory cells, such as degenerate granulocytes, macrophages, and histiocytes, are found microscopically. The mature cysts, up to 7 cm in diameter, are thin walled and contain clear fluid or, occasionally, purulent fluid. Protoscolices, up to many hundreds, can be visualized microscopically within the cysts, which are surrounded by severe and mainly nonsuppurative inflammation. The cysts deform and compress the underlying brain tissue.
Diagnosis
The combination of a characteristic clinical syndrome and location in an endemic area supports a presumptive diagnosis. Radiographs in the lateral and posteroanterior planes may detect radiolucent areas in the calvarium. The optimum diagnostic views in the posteroanterior projection occur whenever the base of the nose is level with the upper margin of the orbit. Computed tomography (CT) effectively demonstrates the presence of cysts but rarely is practicable in large animal species.1384
Treatment
Praziquantel,* 50 to 100 mg/kg orally daily for 3 to 5 days, is effective for the treatment of coenurosis in sheep that do not yet have neurologic signs.1380,1385 Concomitant administration of a nonsteroidal antiinflammatory drug (NSAID) or dexamethasone may enhance the posttreatment survival rate.
The cyst can also be removed surgically,1386 with success rates as high as 90% reported.1387 A craniotomy is performed over the site of the cyst. Approximately 70% of the cysts are located extradurally and can be removed easily with minimal dissection. The other cysts are located on the surface of the pia arachnoid, in which case the dura mater is incised. The cyst usually bulges from under the incised dura and can be removed. When the cyst is located in the cerebral cortex, ultrasound probes placed on the surface of the brain may be used to locate the pocket of fluid.1388-1390
Prevention
In endemic areas the carcasses of affected animals should not be fed to dogs, and dogs in endemic areas should be treated repeatedly with a vermifuge to minimize the possibility of pasture contamination. Appropriate management practices have virtually eliminated this disease from North American sheep flocks. Lyophilized antigens from in vitro—cultured larvae have protected sheep; however, this preparation is not commercially available.
CEROID LIPOFUSCINOSIS
Definition and Etiology
Ceroid lipofuscinosis is a lysosomal storage disease that has been reported in South Hampshire, Swedish Landrace, and Rambouillet sheep, Nubian goats, Devon cattle, and horses.1391-1396 The disease is known to be inherited as an autosomal recessive trait in many cases1397 and is believed to be so in others.1396 It is characterized by the intracellular accumulation of abnormal autofluorescent lipopigments in lysosomes of neurons and other cells throughout the body. The storage material has been shown to consist predominantly of the subunit c of mitochondrial c synthase.1398,1399 The mechanism of neuronal dysfunction is hypothesized to be mediated by N-methyl-D-aspartate (NMDA) receptor excitotoxicity.1400 Affected animals display progressive ataxia and postural abnormalities, blindness due to retinal involvement in many cases, sensory depression, and terminally, coma. Lesions seen on CT scans include enlargement of the lateral ventricles and reduced thickness of the cerebral cortex.1401
Gross pathologic lesions in the CNS may include moderate enlargement of the lateral ventricles, flattening of cerebral gyri, and a yellow to brown discoloration of the brain parenchyma. Accumulation of protein storage material in neuronal lysosomes is evident on microscopic examination and is accompanied by neuronal necrosis and astrocytosis, which may be severe. The lesions sometimes have a lamellar appearance.1400 The disease is ultimately fatal, and no practical method of treatment is currently available.
CITRULLINEMIA
Citrullinemia is a rare genetic defect of Holstein calves that has been reported in Australasia, Europe, and India.1402-1404 The genetic defect has been found in one carrier bull in the United States.1405 The mutation responsible has been traced to offspring of a North American sire named Greyview Crisscross and his son Linmack Kriss King.1406 Approximately 8% of all bulls used for artificial insemination in Australia are heterozygous for the gene, but the gene prevalence appears to be much lower in the United States.1405,1407
Citrullinemia is caused by a defect of argininosuccinate synthetase, an enzyme that processes citrulline in the pathway for the formation of urea. The condition is fatal. Affected calves are normal at birth but become clinically depressed by 24 hours after birth. By 2 to 3 days after birth, affected calves show head pressing, drooling of saliva, bellowing, muzzle twitching, tongue protrusion, and odontoprisis. Convulsions are first seen at 1 to 4 days of age, and death rapidly follows.
The diagnosis may be made by observing an increased concentration of citrulline in the plasma. The concentration of citrulline in normal calves is 0.16 mM and in affected calves is greater than 1.5 mM by the third day after birth. The plasma arginine concentration is decreased to less than 0.02 mM at death.1408 There is a marked hyperammonemia because of the inactivity of the hepatic ornithine-citrulline cycle. The brain concentrations of the transmitter amino acids glutamate, aspartate, and GABA are decreased. Affected calves also have a reduced affinity of postsynaptic glutamate NMDA receptors in the brain.1409 The genetic deficit has been traced to the insertion of a chain termination codon for arginine in the argininosuccinate synthetase genome, which causes a complete loss of enzymatic activity. A PCR test for detection of heterozygotes has been developed.1407 Microscopic brain alterations include astroglial edema and mild to severe spongiform changes in the deep laminae of the cerebral cortex.1410
BRAIN TUMORS
Nervous system tumors of ruminants include medulloblastoma, ependymoblastoma, neurofibrosarcoma, angioblastoma, meningioma, meningeal hemangioma, neurofibroma, schwannoma, choroid plexus papilloma, pituitary adenocarcinoma, primitive neurectodermal tumor, and reticulosis.1411-1415 Central nervous system (CNS) tumors of horses include pituitary adenomas, microgliomas, medulloepithelioma, choroid plexus papilloma, ependymoma, neurofibroma, meningioma, meningeal carcinoma, and reticulosis.1411,1416-1420 Secondary tumors that invade the CNS include melanoma, lymphosarcoma, adenocarcinoma, squamous cell carcinoma, hemangiosarcoma, and osteoma.1411,1420-1424 Of these, lymphosarcoma is most often encountered.1425-1427 Metastatic invasion to the CNS occurs either by vascular routes or by extension along the peripheral nerve rootlets.1428 Local extension from adjacent tissue, such as the paranasal sinuses, also can occur.1421
Clinical signs of BRAIN TUMORS vary with the location and include abnormalities of gait (ataxia, paresis, hypometria/hypermetria), seizures, altered mentation (especially dullness), facial paresis or paralysis, facial anesthesia or analgesia, dysphagia, head tilt, strabismus, nystagmus, and loss of the menace reflex.1421,1428-1430 Migration of facial tumors (squamous cell carcinomas) into the cranial vault through the cranial nerve foramina may also result in facial swelling, exophthalmos, Horner’s syndrome, or asymmetric airflow through the nares.1427 Pituitary adenomas of aged horses (see Chapter 41) rarely cause neurologic disease, but they secrete melanocyte-stimulating hormone, which stimulates the adrenal cortex and causes Cushing’s disease. Some tumors are discovered as incidental findings at necropsy.
Antemortem diagnostic tests include radiographs of the skull (for tumors that spread locally and some metastatic tumors), and electroencephalography (EEG) to elucidate brain dysfunction.1421 Where available, CT or MRI can greatly facilitate diagnosis, but limited availability and considerations of cost restrict their use in most cases.1428,1430-1432
Treatment of BRAIN TUMORS in horses and livestock is generally not feasible because of limitations of cost, nursing care challenges after craniotomy, lack of access for large animals to radiation therapy, and considerations of safety for personnel handling animals with significant neurologic deficits. Palliative treatment, such as corticosteroids, may reduce clinical signs temporarily in some animals. Euthanasia is the choice for most large animals with BRAIN TUMORS.
CHOLESTEROL GRANULOMAS
Cholesteatomas are common lesions in the brains of older horses and frequently are incidental findings at necropsy. Cholesteatomas usually are found in the lateral ventricles.1433 They may form secondary to chronic hemorrhage into the choroid plexuses, but their exact pathogenesis is unknown. Clinical signs of cerebral dysfunction, such as seizures, result only when the masses grow large enough either to obstruct CSF flow from the lateral ventricles or to attenuate the surrounding neuropil directly. Antemortem diagnosis of cholesteatomas in horses can be done by CT scanning of the brain.1434 Cholesteatomas appear grossly as brownish nodular thickenings in the choroid plexuses or less often as large masses filling the ventricle. Light microscopy reveals abundant cholesterol crystals interspersed with empty clefts, hemosiderin, and an inflammatory reaction consisting of both macrophages and giant cells. There is no specific treatment for cholesteatomas, and relief of clinical signs should be symptomatic, including anticonvulsants as appropriate.
EPILEPSY
GEORGE M. STRAIN
MARY O. SMITH
LISLE W. GEORGE
Epilepsy is a condition of recurrent seizures not attributable to other neurologic or metabolic disorders.1435 A seizure (ictus) may be generalized, involving the entire cortex and accompanied by loss of consciousness, or partial (focal), involving a limited cortical region with no loss of conscious. Partial seizures may in turn become generalized. Seizures may result from trauma, infection, tumors, electrolyte disturbances, or cerebral swelling. Some seizures are idiopathic. Seizures may be preceded by a prodromal aura, usually consisting of a stereotypic sensory disturbance and followed by a postictal depression of variable duration. Seizures in very young or old animals frequently are not of epileptic origin.
Seizure activity results from the synchronization of large aggregates of neurons that are driven, at least in the case of partial seizures, by abnormal epileptic neurons in a seizure focus that recruit increasing numbers of connected neurons.1436 Generalized epileptic seizure activity may result from subcortical pacing neurons acting through the excitatory amino acid system.1437 These neurons probably depend heavily on aspartate and glutamate for facilitation.1438
Microscopic abnormalities of the brain may be detectable in focal epilepsies but usually are not in generalized epilepsies. Hippocampal degeneration is a well-recognized pathologic change in humans with some forms of epilepsy and has been described in a single cow with seizures. It was not clear in this report, however, whether the hippocampal changes were the cause or the consequence of the seizures.1439
Status epilepticus, a condition of repetitive seizures with little or no intervening recovery that requires immediate emergency treatment, may be the first detected clinical manifestation of epilepsy. Status epilepticus may result if antiepileptic medication is abruptly discontinued.
Clinical Signs
Generalized seizures may consist of muscle contractions that are tonic, clonic, or alternating between tonic and clonic. Affected animals may fall and display various autonomic signs, such as salivation, urination, and defecation. During the seizure there may be dorsiflexion of the head and neck and rotation of the eyes. Postictal depression may last minutes to days. Blindness lasting minutes to weeks may be present postictally.1440 Partial seizures may consist of tonic or clonic contractions of isolated muscle groups without loss of consciousness. Seizure disorders have been described in Romagnola,1441 Swedish Red,1442 Brown Swiss1443 Hereford,1444 Angus,1445 Brahman,1446 and crossbred1447 cattle. A recent study in Angus cattle suggests that the previously described epilepsy may, in fact, be a cerebellar disease, with episodes of severe cerebellar dysfunction, rather than a true seizure disorder of cerebral origin (see also Bovine Familial Convulsions and Ataxia).1448 Attacks in a 6-month-old Brown Swiss bull were prompted by undue excitement and declined in number with age.1449 Young progeny exhibited similar attacks, suggesting that the trait was transmitted as an autosomal dominant genetic character.
Epilepsy has been poorly documented in horses but occurs in Arabians and in some ponies.1450 There is anecdotal evidence of the existence of the condition in Paso Fino horses of the western United States. A condition known as “benign epilepsy” is seen in foals of many breeds, especially Arabians, but unlike true epilepsy, is usually outgrown. The condition in Arabians is termed “juvenile idiopathic epilepsy.” It occurs in animals of Egyptian lineage, is responsive to antiepileptic drugs (AEDs), and usually resolves by 1 to 2 years of age.1440 Idiopathic epilepsy in mares has been associated with elevated levels of estrogen.1451
An epileptic condition in an 8-year-old Hereford cow has been described.1444 The seizures began at 6½ years of age. Tonic-clonic convulsions could be elicited by administration of pentylenetetrazol at doses of 4 mg/kg, which was well below the convulsant dose for normal animals. EEG showed high-frequency spike bursts, spike and wave, and polyspike and sinusoidal wave abnormalities (Fig. 35-15). No microscopic abnormality could be found in the CNS of the affected animal. Partial epilepsy has been described in a 5-year-old Nubian goat with a 3½-year history of episodic convulsions. Between episodes the goat appeared to be healthy. The seizures appeared more often at times of peak endogenous plasma estrogen concentrations and could be induced by administration of ketamine. The clinical signs included repetitive tremors of the forelimbs and hindlimbs, head tremors, and mastication without loss of consciousness. The postictal depression lasted for several hours.1452
The EEG evaluation of seizure disorders may be performed using evocative drug challenges with pentylenetetrazol, ketamine, or other seizure-inducing agents and established techniques, but care must be taken to prevent injury to the animal. Although EEG may be of some value in investigation of epilepsy and other brain disorders,1453 the need for sedation or anesthesia in large animals, combined with the paucity of data available for comparison, limits its usefulness.
Because of the rarity of epilepsy in large animals, specific drug therapies have not been established; however, Table 35-9 presents several drugs that could be administered. These drugs usually are highly protein bound in plasma and can be displaced or functionally altered by other drugs, including tetracycline and chloramphenicol. Because of these potential interactions, these agents should not be used concomitantly with the anticonvulsants.
All anticonvulsant treatments are begun at a low dose, which is increased daily or every second or third day until the seizures have been controlled. If seizures cannot be controlled without causing depression or ataxia, a second anticonvulsant is added. The dose of the second drug is gradually increased until the seizures stop. This combination treatment is continued for 2 to 4 weeks. Thereafter, the first anticonvulsant is tapered until it is discontinued. If seizures reappear, the dose of this drug is increased until they disappear again. After 1 month the trough blood concentration of all anticonvulsants is monitored. Therapeutic trough concentration of phenobarbital is 15 to 40 μg/mL of plasma and for diphenylhydantoin 5 to 20 μg/mL. Any attempt to withdraw anticonvulsant therapy should be made gradually, because rebound seizures may occur with too rapid withdrawal. Dosage recommendations have been established for potassium bromide in horses.1454 A loading dose of 120 mg/kg daily for 5 days, followed by 90 mg/kg once daily, results in serum concentrations in the range found to be effective for seizure control in other species. However, the efficacy of potassium bromide for seizure control in large animals has yet to be established.
In many animals, epilepsy may be incurable because of its genetic basis. In these cases, treatment is rarely indicated. Horses with epilepsy should not be ridden or used for sporting purposes. Specific methods of control, other than breeding selection against affected animals, are unavailable.
Mares with estral-related seizures may be treated with an ovariectomy. All ruminants experiencing seizures should also immediately be treated with thiamine (10 to 20 mg/kg IM or SC three times daily, or diluted in 5% dextrose or isotonic fluid and given slowly IV) in case the seizural problem is caused by polioencephalomalacia (see previous discussion). The plasma sodium, magnesium, potassium, and calcium levels should be measured in all animals experiencing seizures of unknown origin. Infrequent seizures generally do not justify anticonvulsant treatment, and economic considerations often limit the amount of drug therapy possible. Status epilepticus can be treated with IV diazepam in 5-mg doses until the seizures are controlled or by titrated doses of phenobarbital or pentobarbital.
NARCOLEPSY AND CATAPLEXY
GEORGE M. STRAIN
MARY SMITH
LISLE W. GEORGE
Definition and Etiology
Narcolepsy is a CNS disorder characterized by excessive daytime sleepiness, episodes of muscular weakness (cataplexy), and rapid eye movement (REM)–onset sleep.1455-1457 Narcoleptic attacks differ from convulsions in that they do not involve tonic-clonic muscular activity.1455 Cataplexy, a sudden episode of paralysis of the voluntary muscles, frequently is induced by stimulation of the patient and can range from atonic, areflexic paralysis of all nonrespiratory muscles to weakness of facial neck and forelimb muscles. Episodes of cataplexy last seconds to minutes. Environmental factors that can stimulate cataplectic attacks include active restraint, feeding, or changing the stall environment, grooming, or saddling horses in preparation for work.1455,1458-1460 Excessive daytime sleepiness and REM-onset sleep are difficult to document in large animals, so cataplexy is the most frequently recognized manifestation of sleep disorder in these species.1461 The disorder is considered an intrusion of aspects of REM sleep into the waking stage, especially the active descending paralysis of skeletal muscle. This intrusion results in indistinct boundaries between wakefulness and REM and non-REM sleep. Cataplectic attacks result from sequential activation of pontine α1-adrenergic and muscarinic cholinergic systems. The numbers of muscarinic and dopaminergic receptors also are increased.1456,1462
Attacks of narcolepsy and cataplexy are considered a paradoxic form of sleep because the EEG is characteristic of an alert, awake animal, but the REM is characteristic of deep sleep.1455 Both neonatal-onset and adult-onset syndromes have been described in horses.1463 Narcolepsy has been reported in quarter horses, a Shetland pony, thoroughbreds, Morgans, Paint horses, Arabians, Appaloosas, standardbreds, Welsh ponies, Suffolk sheep, Spanish fighting bulls, a Guernsey bull, and a Brahman bull.1459,1464-1468 A familial occurrence has been reported in American miniature horses.1469
Clinical Signs
The clinical signs of narcolepsy include staggering, drooped head, kneeling posture, flaccidity of the lips, closure of the eyes, loss of the menace reflex, stertorous breathing, and proprioceptive deficits. During severe narcoleptic attacks the animal assumes lateral recumbency and appears comatose. The sensorium returns to normal after a time. In the periods between attacks, the patient appears normal.
The EEG changes of narcolepsy and cataplexy have been reported in cattle.1464 The waveforms vary between low-voltage high-frequency (LVHF) and high-voltage low-frequency (HVLF) patterns, with LVHF corresponding to the actual period of narcoleptic attack (Fig. 35-16). The normal EEG sleep patterns of the horse are usually unavailable, so the technique has limited application for diagnosis of the condition in equine species.
Cataplexy can be induced by IV administration of centrally acting cholinomimetics such as physostigmine salicylate (0.05 to 0.1 mg/kg) or α1-adrenergic blockers such as prazocin (0.02 to 0.06 mg/kg), but attacks cannot be evoked in all animals. Signs can be reversed for several hours by antimuscarinic drugs such as atropine sulfate (0.02 to 0.1 mg/kg IV once for acute signs). Diagnostic drugs must be used with caution because of possibly inducing colic, especially in horses. Pathologic lesions have not been reported in narcolepsy of large animals. In humans, narcolepsy can be caused by lesions in the rostral brainstem.1455
Treatment and Prevention
The tricyclic antidepressant imipramine can be used to treat narcolepsy or cataplexy. Lifelong treatment is required, which must be considered when deciding whether to treat affected large animals. Side effects of imipramine in horses can be serious, including tachycardia, muscle fasciculations, sensitivity to noise, and hemolysis.1470 Such side effects can occur even when the blood level of imipramine is below the therapeutic range. This drug must be used with caution, and oral dosage in horses should not exceed 2 mg/kg; lower doses should be used when possible.1457 Results of oral imipramine therapy are inconsistent1457; duration of effect is about 5 hours.
HEAD SHAKING IN HORSES
JOHN E. MADIGAN
Head shaking in the horse is a well-recognized problem that shows no predilection for age, breed, or gender.1471-1482 Some horses head-shake at rest, whereas most manifest head shaking shortly after the onset of exercise. Head shaking may be vertical, horizontal, or both and often is accompanied by agitation. Suggested causes have included middle ear disorders, ear mites, Trombicula autumnalis (harvest mite) larval infestation, cranial nerve abnormalities, ocular disease, guttural pouch mycosis, dental abnormalities, maxillary sinus mass, and allergic vasomotor rhinitis. Finding a specific cause has been difficult, with most cases termed “idiopathic” head shaking. The disorder can occur at any time, but a spring and early-summer pattern is typically reported, although some cases start in the fall and winter.1480 The research group at University of California, Davis, suggests that neuropharmacologic alterations associated with photoperiod mechanisms, leading to optic trigeminal summation (similar to photic sneezing in humans), may explain the spring onset of head shaking. In many horses, sneezing, snorting, and nose rubbing accompanies head shaking. Most horses continue head shaking for many years, experience periods of remission, and often resume head shaking at the same time each year.
Examination of horses with head shaking includes a complete physical examination; ophthalmic, otoscopic, neurologic, and dental examinations; endoscopic examination of nasal passages, pharynx, and guttural pouches; radiography of the skull, and a complete blood count and chemistry panel. No abnormalities are usually noted in these examinations.1483
Intraorbital neurectomy eliminated head shaking in three to seven horses in one study.1484 These results would seem to support that some cases of head shaking are caused by a tingling (or other uncomfortable sensation) in the muzzle area. It is postulated that light may play a role in stimulating the response, and that exercise may lower the threshold for onset in some horses.1483 It is important to note that infraorbital neurectomy has several complications and is not indicated for the treatment of head shaking. The infraorbital branch of the trigeminal nerve is just one branch from that nerve, and it is believed the problem may reside higher up, at the trigeminal ganglia area.
The etiology of head shaking usually is not known. Most horses show no physical abnormalities on comprehensive physical examination, and no lesions have been seen at necropsy. The symptoms in these horses may best be explained by the presence of neuropathic pain involving the trigeminal nerve. Neuropathic pain is a burning, tingling, itching, or electric-like pain that can be intermittent or continuous. Symptoms include sharp, quick movements of the head; occasional striking at the face with a front hoof; excessive snorting and rubbing of the head on objects; carrying the head very low during exercise; and dragging the nose in the dirt. These behaviors could be manifestations of neuropathic pain. The cause of the assumed dysfunction of the trigeminal nerve is not known. The average age of onset of head shaking is 9 years, and most affected horses are geldings, although about 10% to 20% are mares. Often a period of inactivity precedes the onset of head shaking, which can occur quite abruptly.1485
Cyproheptadine, 0.3 mg/kg orally twice daily, has been used with some success for idiopathic head shaking.1483 Cyproheptadine, a type I histamine and serotonergic (5-hydroxytryptamine) blocking agent, was chosen for treating head shaking in horses because of serotonin’s role in pain sensations in humans,1484 and because of photoperiod-induced increases in serotonin in the CNS in the spring. Side effects are few, but mild colic and lethargy have been reported. The drug cannot be used in show or performance horses because it is not an approved medication. Cyproheptadine appears to be effective only when the horse has an adequate blood level of the drug; therefore, it often is not possible to treat a horse and then withdraw the medication before a show and have the head shaking controlled during the event.
Other medications (e.g., corticosteroids, antihistamines, NSAIDs), chiropractic therapy, and acupuncture have not been successful. A few horses respond to a heavy hair net or a dangling device that makes contact with the nose area or upper forehead (Fig. 35-17). This type of contact may prevent the nerve from “firing,” similar to blocking a sneeze by placing a finger under the nose and applying pressure. In a review of therapies for head shaking in horses, a nose net device was reported to be successful in approximately 30% of cases.1486Box 35-2 lists other treatments used by the author.
Box 35-2 Treatments and Medications for Horses with Head Shaking*
PHYSICAL METHODS
If light is a trigger, consider having the horse wear an ultraviolet (UV) light–blocking sun shade–type fly mask from Guardian Mask, San Diego (
http://www.horsemask.com/Main.html).
If the horse is only bothered during exercise, use nose net or other device that attaches to or dangles from over the nostril and muzzle area (e.g., German dangling device); in one stuidy, 30% of head shakers respond.
1486The ear covering and forehead “dangling” material.
DRUGS TO CONTROL NEUROPATHIC PAIN
Cyproheptidine, 0.3 mg/kg twice daily orally.
Carbamazepine, 1 to 4 g either twice daily or four times daily alone or in combination with cyproheptidine.
Hydroxyzine (Atarax), 400 mg twice daily orally.
Fluphenazine (Reposital), 2 mL IM per horse. Repeat as needed every month or sometimes every 4 months. Use only unopened; if opened, can use refrigerated drug with no discoloration.
Clonidine, 0.0025 mg/kg (0.3-mg tablets); give 4 to 6 tablets orally twice daily to a 500-kg horse.
Phenobarbital, 97.2 mg/tablet; give 16 tablets orally twice daily to 500-kg horse.
OTHER TREATMENTS
Lower protein in diet to about 8%. Allow mild weight loss and continue exercise.
Alteration in photoperiod for seasonal head shakers that begin in spring and stop in late fall or winter. Give 12 to 15 mg melatonin orally starting November 1 every day at 5
PM. Can try starting in midseason, but not as effective. This resets seasonal clock. Give this dose year-round. Horse may not shed and may need body clipping.
Combination
Cyproheptidine, standard dose.
Add magnesium supplement: Quiessence (double dose daily).
Magnesium
Magnesium is an electrolyte that calms nerves by increasing the stimuli needed for depolarization. Supplementing the diet with magnesium may help decrease the stimulation of the trigeminal nerve. One commercial formulation of magnesium, Quiessence, contains 5 g of magnesium per ounce. Feed 2 oz once daily and increase to 4 oz once daily if there is no improvement in the head shaking. Quiessence is available online (
http://www.foxdenequine.com/quies.htm).
Have the veterinarian measure the horse’s serum magnesium level in about 2 weeks and check it once monthly to ensure it is not high.
Melatonin
Give 15 mg (average horse) or 18 mg (larger horse) of melatonin orally (five or six 3-mg tablets) every day at 5
PM. Vitaminshoppe is one source for melatonin (
http://www.vitaminshoppe.com). Twin Labs is a health food or supermarket brand of melatonin that has good bioavailability. This switches the horse’s photoperiod back to winter and sometimes stops the head shaking that starts in the spring.
DISEASES PRESENTING PRINCIPALLY WITH BRAINSTEM AND CRANIAL NERVE DYSFUNCTION
LISLE W. GEORGE
LISTERIOSIS (CIRCLING DISEASE; SILAGE DISEASE; LISTERIA MONOCYTOGENES INFECTION)
Definition and Etiology
Listeriosis is an acute meningoencephalitis caused by the gram-positive bacterium Listeria monocytogenes. The disease has a worldwide distribution but occurs most often in temperate climates. Listeriosis typically affects ruminants, fowl, and humans but is rare in horses. The prevalence of listeric meningoencephalitis in infected herds does not usually exceed 1% of the adult animals at risk.1487
Clinical forms of listeriosis include septicemia of neonates, abortion, neonatal death, ophthalmitis, septicemia and diarrhea of ewes, and neurologic disease.1488 Usually, only one clinical form is recognized during an outbreak, and only one serovar can be isolated from the clinically affected animals. Neurologic listeriosis may manifest as a multifocal brainstem disorder, as a diffuse meningoencephalitis, or as a spinal cord myelitis. The condition usually affects individual animals but occasionally can affect several members of a herd.1489 Asymptomatic intramammary infections apparently occur and may be responsible for outbreaks of listeriosis in humans. Repeated intramammary inoculation of 106 to 107 colony-forming units (CFUs) of L. monocytogenes into cattle resulted in 500 to 50,000 CFUs of Listeria in the milk for as long as 12 months.1490
Clinical Signs
The neurologic signs of listeriosis in adults reflect dysfunction of the caudal brainstem, cerebellar peduncles, or spinal cord.1491 Signs common to most Listeria infections of the central nervous system (CNS) include fever, anorexia, depression, conscious proprioceptive deficits, head pressing, and centrally located cranial nerve deficiencies. Depressed consciousness is the result of lesions of the reticular activating system. Conscious proprioceptive deficits are caused by interference with the descending motor pathways and the ascending proprioceptive fibers in the brainstem and may precede or accompany cranial nerve dysfunction. The fever occurs early in the disease course and often disappears after 3 to 5 days. Head pressing and propulsive walking or compulsive circling are caused by lesions of the basal ganglia.
Cranial nerves (CNs) V through XII usually are dysfunctional in listeric animals. Patients with loss of the trigeminal nerve (CN V) show dropped jaw or asymmetric jaw closure and facial analgesia or anesthesia. Facial analgesia is best detected by stimulation of the nasal septum with a pencil or piece of straw. Animals with lesions of CN VI exhibit a medial strabismus on the ipsilateral side of the lesion. Animals with lesions of CN VII have ptosis, loss of menace response, absent palpebral reflex, drooped ear, loss of levator nasolabialis muscle function, and decreased lip tone (Fig. 35-18). Small ruminants with CN VII loss have a deviated philtrum. The paralysis of the orbicularis oculi muscle results in exposure keratitis and, in chronic cases, panophthalmitis.1492 The loss of levator nasolabialis function is best detected by observation of the muscular contraction on the dorsum of the nose during inspiration. Loss of lip and cheek muscle tone is best detected by observation of drooling of saliva from the ipsilateral side of the mouth and by palpation of the lips and nostrils.
Animals with CN VIII lesions display a nystagmus that changes as the position of the head is altered. Other signs include a head tilt toward the side of the lesion and a tendency to circle or fall to the lesion side. The nystagmus may be horizontal, vertical, or rotatory and usually is inconstant. Goats may lie on their backs with the head curved toward the trunk and tilted with the lesion side toward the ground. If the spinal reflexes can be tested, the affected animals show a mild to moderate hypertonia and hyperreflexia in the limbs opposite the side of the lesion. Lesions of the cerebellar peduncles (juxtarestiform body) may cause paradoxic vestibular signs in which the head tilt and circling are directed away from the side of the lesion and proprioceptive deficits are on the same side as the lesion.1493 This should be suspected whenever the head tilt is directed toward the side opposite that of the other dysfunctional cranial nerves. Animals with acute loss of CNs IX, X, and XII develop stertorous breathing and dysphagia. Animals with dysfunctional CN XII have paresis or paralysis of the tongue. With unilateral lesions the tongue protrudes from the side of the mouth ipsilateral to the lesion. Progression of listeriosis is associated with decreased consciousness, coma, and convulsions.
Lambs may selectively develop spinal myelitis without brainstem disease. This condition results in flaccid paraparesis or hemiparesis without attendant signs of brainstem dysfunction.1494 The clinical signs of myelitis include tetraparesis, tetraplegia, paraparesis, paraplegia, conscious proprioceptive deficits, and recumbency. The sensorium and appetite are normal in some affected animals and greatly depressed in others.1495
Clinical Pathology
The clinical signs of multifocal brainstem disease with fever in a ruminant are suggestive of listeriosis, Haemophilus somnus infection (cattle), or aberrant parasite migration. Examination of the cerebrospinal fluid (CSF) should be helpful for confirming a diagnosis of listeriosis, but the cell and protein concentrations of the specimens do not correlate with the severity of the clinical signs or the prognosis. The protein concentration in the CSF may be more than 40 μg/dL, and the CSF white blood cell (WBC) counts may be more than 12 mononuclear cells/μL.1487,1496,1497 Many cattle with advanced signs of listeriosis develop metabolic acidosis as a result of salivary bicarbonate loss.
Pathology
Pathologic confirmation of listeriosis is based on identification of multifocal microabscesses in the brainstem and isolation of L. monocytogenes from infected brain tissue. The agent is only rarely isolated from CSF and is best recovered from refrigerated nervous tissues. Enrichment of the Listeria organisms may be accomplished by refrigerating slices of brain at 4° C (39.1° F) for 3 months while culturing the tissues weekly. In contrast to septicemic listeriosis of monogastric animals, peripheral monocytosis is not observed in infected ruminants.
Pathophysiology
Listeria organisms produce a hemolysin, listeriolysin-O, a thiol-activated toxin (molecular weight, 58 kD) that is correlated with pathogenicity. The molecular role of the toxin in dissemination of the infection and in cell death is not known.
It is unclear whether infection of the brain by L. monocytogenes occurs hematogenously or by ascent from the cranial nerve rootlets.1498 Morphologic studies of naturally occurring cases of encephalitic listeriosis have demonstrated the bacterium in the axons of the trigeminal nerve rootlets, indicating a possible centripetal migration.1498,1499 Similar findings have been reported in animals infected experimentally.1500 Younger animals may be susceptible because eruption of the permanent teeth may expose trigeminal nerve rootlets. A model for experimental induction of listeriosis by inoculation of the bacterium into the pulp cavity of sheep has been described.1501 Infection of the CNS without bacteremia has been detected, indicating that centripetal migration of the bacterium is possible.1502 Some investigators consider axonal migration as an unlikely mode of pathogenesis because of a lack of nutritional dependency of L. monocytogenes for nervous tissue and the ready ability to produce multifocal brain microabscesses by intravenous (IV) inoculation of the bacteria into susceptible hosts.1498,1503
Epidemiology
L. monocytogenes has 16 major serologic types based on comparison of somatic and flagellar antigens. Most clinical infections are caused by serovars 1/2a, 1/2b, 4a, and 4b. Listeria ivanovii, usually associated with abortions in sheep, also has been classified as L. monocytogenes serovar 5. The serovar 1/2 (a and b subtypes) is most prevalent in livestock. The 1/2b subtype appears to be exclusively related to encephalitic infections, whereas other subtypes, including 1/2a, can be associated with any of the clinical forms of listeriosis.1504 The pathogenicity of serovar 1/2a is hemolysin dependent. Serogroup 1/2 (a or b type) is most often isolated from feedstuffs. Serovar 4b is responsible for most infections in humans.1505
The case-attack rate in ruminants may reach 9% but rarely is greater than 2%.1506 Listeriosis occurs sporadically in weaned lambs confined to a drylot and appears in only a small proportion of the lambs at risk. The encephalitis usually occurs from 4 to 32 days after weaning and at 6 to 12 weeks of age. Between 0.7% and 1.6% of all lambs at risk may develop the infection.1507 In untreated cases the fatality rate is almost 100%.1491 The survival rate in treated animals is considerably higher than in untreated patients. The disease in sheep and goats tends to be more acute and results in a higher case-fatality rate than in cattle. Occasional outbreaks of listeriosis may occur in sheep without access to silage.1491,1508 In these cases the source may be the feces of carrier animals or rotting vegetation on the pastures or feed bunks. During an outbreak of listeriosis, the bacterium can be isolated from the feces of a large percentage of normal animals.1509 It is unclear whether this high rate of asymptomatic infections represents a true carrier state or is simply the result of a high environmental contamination by nonpathogenic isolates.1509 The agent infects the udder but rarely causes clinical mastitis. In sheep, excretion of the bacterium in the milk is greatest during the immediate postlambing period.1510,1511
Listeria monocytogenes can survive for long periods in the environment and in asymptomatic carriers. The bacterium can multiply at low environmental temperatures and is resistant to environmental influences. L. monocytogenes is shed in the feces of asymptomatic carriers, especially at the end of pregnancy and at lambing. Once in the environment, the bacterium can survive for 2 years in dry soil. It is resistant to freezing and thawing in the soil but does not survive for more than 1 to 2 weeks in properly preserved silage. The bacterium proliferates in rotting vegetation in which aerobic conditions exist and the pH is above 5.4.1512-1514 Common sources of contaminated forage include spoiled silage at the ends of trench silos, decaying forage at the bottom of feed bunks, or rotting hay at the periphery of hay stacks.1512,1515 The incidence of listeriosis may be increasing because of the greater use of trench silos and bulk handling methods that result in a greater amount of spoilage than in conventional upright silos. Although L. monocytogenes can be isolated from silage with a pH below 4, fewer bacteria are found in well-preserved forage.1516
A selective enrichment medium has been developed for identifying and enumerating the Listeria organisms in silage. This medium permits semiquantitative enumeration of Listeria bacteria in 10 g of silage. Hemolysin production is measured by overlaying the colonies with bovine blood agar and reincubating the plates. Using this method, outbreaks of listeriosis have been correlated with silage containing 1 million Listeria organisms and more than 1 million enterobacteria per gram of silage. The pH of such silage characteristically is above 7.8.1517
There is a significant public health concern about Listeria contamination of milk products. The 4b serotype of L. monocytogenes is most often responsible for infections in humans.1518 Outbreaks have been traced to ingestion of pasteurized milk, cole slaw, and soft, ripened cheese. The occurrence in cheese has led to concerns that the bacterium may survive the pasteurization process1519; however, heat resistance by Listeria organisms does not appear to be a significant factor in milk-related human exposures. One study indicated that intracellular Listeria bacteria survived after exposure to temperatures as high as 73.9° C (165° F) for 16.4 seconds. Complete killing of the Listeria organisms required temperatures as high 76.4° C (169.5° F) for 15.4 seconds. These temperatures exceeded the minimum temperatures required by the U.S. Food and Drug Administration (71.7° C [161° F] for 15 seconds).1520 The public health implications of these findings are unclear.
Pathologic Lesions
The lesions of listeric meningoencephalitis are most common in the pons and the trapezoid bodies, but they can be located anywhere in the brainstem. Neurologic structures most often affected include the reticular formation and CNs V and VII to X. Macroscopic lesions are limited to mild meningeal congestion and clouding of the CSF. Microscopic lesions include perivascular cuffing with mononuclear cells, multifocal asymmetric brainstem microabscesses, and mononuclear cell meningoencephalitis.1521 The microabscesses are composed predominantly of neutrophils. Other microscopic changes include degeneration of the neuropil and neuronophagia. To enhance the speed and accuracy of pathologic diagnosis, a peroxidase-antiperoxidase method has been developed for use with formalin-fixed nervous tissue. The test detects degraded bacterial proteins, as well as intact bacteria in the suspect tissue. Bacterial antigen is exclusively located in areas of malacia or in the microabscesses.1522
Treatment
The recovery rate is best if treatment is administered early in the disease course. Animals that are recumbent, comatose, or convulsive rarely survive despite intensive antibiotic and supportive therapy. In most cases, treatment must be administered for a prolonged period because recovery may take as long as 1 month. L. monocytogenes is susceptible to most of the common antimicrobial drugs. Recommended treatment is either oxytetracycline, 10 mg/kg intravenously (IV) twice daily, or penicillin G.1492 Specific recommendations for penicillin therapy include an initial dosage of 40,000 IU/kg (IV potassium penicillin G) three or four times daily for 7 days and then 22,000 IU/kg (procaine penicillin) intramuscularly (IM) once daily for 14 to 21 additional days.1492
The plasma concentrations of bicarbonate and potassium should be measured and specific corrective fluid therapy administered. Maintenance fluids also may be administered by gavage. Good footing and nursing care are helpful in the short term but may not influence the overall recovery rate.1523
Prevention
Serologic responses to flagellin and listeriolysin-O develop after oral administration of virulent L. monocytogenes and correlate with protection against listerial bacteremia.1502 Cell-mediated immune responses also are important in protection against virulent challenge. Vaccines of attenuated or killed bacteria have been used successfully to protect sheep and goats. Although these vaccines reduce the incidence of listeriosis in vaccinated flocks, they are not commercially available in the United States.
Although the case-attack rate of listeriosis is low, occasional epizootics may occur in cattle, sheep, or goat herds, invariably associated with high rates of environmental contamination. In such cases the hay and silage should be examined culturally for L. monocytogenes. Rotten vegetation should be discarded, and cattle should be fenced from contaminated areas.
THROMBOEMBOLIC MENINGOENCEPHALITIS (HISTOPHILUS SOMNI [HAEMOPHILUS SOMNUS] INFECTION; SLEEPER CALVES)
Definition and Etiology
Thromboembolic meningoencephalitis (TEME) is a fulminant neurologic disease of cattle that arises from septicemia caused by the pleomorphic, nonencapsulated, gram-negative bacterium Histophilus somni (formerly Haemophilus somnus).1524 The disease was first reported in Colorado in 1956,1525 but characterization of the etiologic agent, H. somni, was not completed until 1960.1526 The disease is economically significant for livestock owners. One study in feedlots of western Canada indicated that the average economic loss from TEME resulted in 15 sick animals and five deaths annually, amounting to $3190 in lost revenue.1527 In addition to TEME, disease syndromes that have been associated with H. somni infection include pneumonia, infertility, metritis, vulvitis, orchitis, conjunctivitis, otitis, and mastitis.1528-1530 The bacterium has been isolated from unthrifty calves, but its causative relationship to that syndrome is unclear.
Cross-agglutination, complement fixation tests and countercurrent immunoelectrophoresis have shown the existence of common surface antigens among isolates of H. somni.1531 Nevertheless, differences in the susceptibility of heterologous isolates of H. somni to antibody and complement have been identified.1532 Isolates from septicemic animals are serum resistant, whereas those from preputial or vaginal mucosa of healthy animals tend to be serum susceptible.
Clinical Signs
Only the neurologic syndrome (TEME) is discussed here. Descriptions of other clinical syndromes of H. somni infection are discussed in Chapters 31, 39 and 43. The neurologic signs of TEME occur peracutely and may be preceded for 1 to 2 weeks by a dry, harsh cough and dyspnea.1533 Death may occur within 36 hours after the onset of neurologic signs. The initial signs of TEME are fever (40° C to 41.6° C; 104° F to 107° F), anorexia, depression, and ataxia.1534-1536 In addition to depression, affected animals show a number of conscious proprioceptive deficits, including knuckling, circumduction, crossing over, and interference.1536 Affected animals may fall while attempting to walk. Signs specifically associated with lesions of the cerebellum and caudal brainstem include head tilt, nystagmus, strabismus, blindness, muscular tremors, opisthotonos, coma, and convulsions.1537 Auscultatory abnormalities in the chest include harsh bronchovesicular sounds and pleural friction rubs. Localization of the bacterium in the joints or lungs may result in lameness, joint swelling, and fluctuant swellings over the joint surface. Other signs observed in some animals include retinal hemorrhages, hyphema, and hypopyon.1535,1538
After recovery from the pneumonia, some affected animals may develop pleuritis, necrotic laryngitis, and weight loss.1539 Some symptomatic animals and as many as 10% of inapparently infected cattle may develop suppurative arthritis of the hock and the stifle joints.
Clinical Pathology
Examination of CSF may be helpful in substantiating a clinical diagnosis of TEME. Specific CSF changes characteristic of hemorrhage include high erythrocyte counts, xanthochromia, and increased concentrations of protein (>100 mg/dL) and neutrophils (>500 WBCs/μL). In untreated cases of TEME, the bacterium may be isolated from pleural fluid, lung sections, aspirated tracheal exudate, urine, blood, and preputial washings. The bacterium can be isolated from 25% to 34% of all fatally infected cattle.1537 Histophilus organisms die rapidly on swabs or transport media, so specimens should be inoculated directly onto a growth medium as soon as they are collected from the patient.1530 The inoculated medium should be incubated in an atmosphere containing 5% carbon dioxide. Isolation of H. somnus from joint fluid and CSF usually is unsuccessful.1540 The kidneys and brain should be collected at postmortem examination because these tissues contain the highest concentrations of H. somni.1540
Initial changes in the peripheral WBC count include neutropenia, left shift, and toxic changes in the neutrophils. A test for serum agglutinins has been developed. Cattle that develop acute TEME invariably have antibody titers greater than 1:400 and show a fourfold increase by 2 to 4 days after infection.1541 Serum agglutination titers greater than 1:1024 are seen in convalescent cattle; lower titers may be seen with inapparent infection or vaccination.1542
Pathophysiology
Infection of cattle by H. somni probably occurs through the respiratory tract. Bacterial proliferation in the lungs and other soft tissues by serum-resistant isolates results in bacteremia. Circulating Histophilus organisms are phagocytosed by neutrophils but are not killed. Blood-borne bacteria also adhere to and may be phagocytosed by the cells of the vascular endothelium. The infected endothelial cells then degenerate and desquamate, exposing the subendothelial collagen and initiating the blood-clotting cascade and thrombosis.1543 Death of neutrophils in the tissues is thought to enhance the tissue damage.1544,1545 The sites of the body most affected by the thrombosis are the brainstem, spinal cord, synovial membranes, pleura, and lungs. Although the name “thromboembolic meningoencephalitis” implies the presence of disseminated coagulopathy, only local thrombus formation likely occurs at the specific vascular lesion. Immunologic mechanisms may play a role in the vascular lesion. Thrombosis occurs most often in animals with high levels of specific agglutinating antibodies and is not seen in colostrum-deprived calves with H. somni septicemia, indicating the importance of antigen-antibody complexes for the development of vasculitis.1546
Cattle probably develop immunity to H. somni infection; however, the presence of serum antibodies does not always confer substantial protection against challenge exposure by virulent bacteria.
Epidemiology
Most cattle develop Histophilus infection by inhalation of contaminated respiratory secretions from carrier animals. Although TEME occurs most frequently in feedlot cattle,1536 outbreaks in the western United States and Canada have been reported in both pasture and dry-lot environments.1534,1547 The disease occasionally may occur in adult cattle.1527 Outbreaks of the neurologic form of TEME tend to occur in the winter months, after shipment or overcrowding, or after additions to the herd in the previous 7 months. Outbreaks usually are preceded by a poorly defined respiratory infection.1527 In feedlot outbreaks the disease frequently is restricted to herdmates in a single pen or pasture.
Transmission of H. somni from asymptomatic carriers to uninfected calves may be enhanced by concomitant infection with infectious bovine rhinotracheitis virus.1535 The anatomic site of bacterial infection in the carrier animals is unknown. Histophilus organisms can be readily isolated from the vaginal and urethral epithelium and from urine, the preputial cavity, and accessory sex glands, but the relationship between these isolates and those found in TEME is unknown.1548 Although random bacteriologic surveys of cattle indicated that 71% of bulls may have H. somni in the preputial orifice, the concentration in the nasal secretions and upper respiratory tract epithelium in cattle with TEME is low.1548 Consequently, some investigators have suggested that the urogenital tract may constitute the primary colonization site in chronically infected cattle.1548 Differences exist in serum susceptibility of isolates from the CNS and from the urogenital tract.1532
The seroprevalence rate may be as high as 100% in some endemic herds and may range from 25% to 56% in herds in which the CNS disease is uncommon. In comparison, the case-attack rate of TEME ranges from 2% to 7.4%.1549 Repeated annual outbreaks can occur in some herds. Estimates of the proportion of carrier animals in feedlots range from 3.2% to 8.8%.
Necropsy Findings
Cattle with the neurologic lesions do not tend to develop fibrinous pneumonia. Macroscopic pathologic lesions of the CNS include disseminated multifocal hemorrhages and 0.1-cm to 0.3-cm infarctions in the spinal cord, brainstem, and cerebral cortex. Bacterial colonies frequently are observed in thrombosed blood vessels and the surrounding infarcted tissues. Ocular lesions are characterized by conjunctivitis, multifocal retinal hemorrhages, and areas of retinal edema. The CSF is cloudy and xanthochromic. A focal fibrinous meningitis is seen, and suppurative otitis may be seen in some cases.
The earliest microscopic lesion of TEME is a vasculitis that progresses to septic infarction and abscessation. The lesions usually are found in the CNS but in severe cases may be disseminated throughout the body.
Nonneurologic lesions of H. somni infection include suppurative arthritis, synovitis, suppurative pleuritis, and bronchopneumonia. Changes associated with the bronchopneumonia include infarction, cranioventral pulmonary consolidation, and hemorrhagic interstitial pneumonia. Simultaneous pulmonary infections with H. somni and Pasteurella multocida result in particularly severe pathologic changes.1550 These lesions include ecchymotic to petechial hemorrhages over the serous surfaces, purulent exudate in the joints, and ulceration of the laryngeal and tracheal mucosa with pseudodiphtheritic membrane formation.1551
Treatment and Prognosis
During an outbreak, cattle must be examined frequently and should be treated when the neurologic signs first appear. H. somni is susceptible to many antibiotics and antimicrobial drugs. Drugs that have been reported to be effective for the treatment of TEME include tetracyclines, penicillin, aminoglycosides, and ampicillin. Parenteral oxytetracycline is regarded as the most cost-effective treatment for infected commercial cattle; dosage is 10 mg/kg of a conventional formulation given IV twice daily for 3 days or 20 mg/kg of a long-acting formulation given IM every other day for three treatments. After oxytetracycline therapy, daily treatment with procaine penicillin (10,000 to 20,000 IU/kg IM) should be continued until complete recovery is observed. Some advocate the addition of chlortetracycline (2.2 mg/kg) to the feed for 10 successive days.1552 This method of mass therapy for a TEME epizootic is considered more efficacious than vaccination when the mortality is less than 2%.1552
Prevention and Control
Antibody responses protective against respiratory challenge have been generated by vaccinating cattle with anionic, heat-stable proteins derived from the bacterial cell wall.1553 In comparison, cattle vaccinated with cationic cell wall proteins developed precipitating antibodies but were not protected against challenge exposure.1553 Bactericidal antibodies may constitute important mechanisms for host resistance against H. somni.1545,1550 Vaccination of cattle with commercial products affords substantial protection from experimentally induced H. somni septicemia and neurologic disease1533,1554 for as long as 95 days after vaccination.1533 (See Chapter 48 for more information about vaccines.)
Mass prophylactic treatment of affected cattle with a parenterally administered, long-acting oxytetracycline formulation may be efficacious for preventing TEME in cattle that have been stressed and exposed to carrier animals.1555 Oxytetracycline feed additives also may be useful for preventing H. somnus infection.1551 One field study indicated that administration of modified live virus vaccines for infectious bovine rhinotracheitis and bovine viral diarrhea on arrival at a feedlot significantly increased the incidence of TEME.1550 Such data indicate that modified live virus vaccines should be administered cautiously to cattle exposed to H. somni.
BACTERIAL OTITIS MEDIA-INTERNA OF RUMINANTS
Otitis media-interna is a common disease of cattle and sheep. The condition usually occurs as a sequel to severe respiratory infections caused by Pasteurella haemolytica, Pasteurella multocida, Corynebacterium pseudotuberculosis, Pseudomonas aeruginosa, Histophilus somni, or Mycoplasma species.1556-1561 Bacterial ear infections are common in feedlot-reared lambs. The incidence may range from 2.9% to 12% of all animals raised under those conditions, and in these animals the condition usually is subclinical.1556-1565
Suppurative bacterial otitis is characterized by thickened mucosae of the vestibular membranes and accumulation of thick fluid in the labyrinths. The infection enters the ear through the eustachian tube. The tympanum may be intact in sheep.1559 In calves, however, it usually is ruptured, and a clear, yellow, proteinaceous fluid is discharged through the external ear canal; the fluid then accumulates at the base of the ear.1558,1566 In chronic cases of bacterial otitis, invasion of the local bone of the skull may result in bone remodeling and hyperostosis.1561
Vestibular disease results from infections that extend into the inner ear. Patients with vestibular signs display heat tilt (toward the side of the lesion), continuous horizontal nystagmus (fast phase away from the side of the lesion), and a tendency to stumble or fall toward the side the lesion. Head shaking with development of an aural hematoma may precede the clinical vestibular signs. Animals may become recumbent1561 and lie with the lesion side toward the ground; if turned, they return to the same position. If the lesion involves the cerebellar peduncles, the animal develops paradoxic signs. In this case the head tilt is away from the lesion side, the fast phase of the nystagmus is toward the lesion side, and the patient circles away from the lesion side. Many animals with otitis media also develop facial nerve dysfunction, which results in ptosis, drooped ear, and flaccid lips and nostrils. In small ruminants, facial nerve paralysis results in deviation of the philtrum toward the normal side. Deviation of the philtrum does not occur in cattle because of the large amount of connective tissue surrounding the planum nasale. Sheep with Pseudomonas otitis may develop necrotizing dermatitis of the ear canal. Some affected sheep also develop signs of cortical or brainstem disease, including unilateral blindness and contralateral mydriasis. These signs occur when the infection extends from the middle ear into the meninges.
Although the clinical signs of peripheral vestibular disease are characteristic, it is important to differentiate this condition from that of central vestibular disturbance. Animals with peripheral vestibular disease usually are appetent, alert, and aware of their surroundings and do not have a significant deficit of postural placement. The nystagmus of these animals is constantly horizontal. In comparison, animals with central vestibular disturbances are systemically depressed, have a nystagmus that varies in direction, and show marked conscious proprioceptive abnormalities.
Treatment of bacterial otitis with oxytetracycline (6 mg/kg IM or IV daily or 20 mg/kg of long-acting formulation IM every other day) or procaine penicillin (40,000 IU/kg IM daily) may be effective. Drug therapy should be continued for several weeks to prevent relapses. Treatment is very effective in obtaining a complete cure in animals with acute otitis but is less so in the chronic form. Otic instillation of aminoglycoside antibiotics is contraindicated. Lincomycin (6.5 mg/kg) and spectinomycin (10 mg/kg) have been used successfully when oxytetracycline or trimethoprim-sulfonamide therapy has failed.1558 Other drugs that have been beneficial for the treatment of otitis include ampicillin, gentamicin, and enrofloxacin. Animals that do not respond should be examined for an abscess or squamous cell carcinoma invading the calvarium or osteomyelitis of the petrous temporal bone.
EAR MITE INFESTATIONS OF RUMINANTS
The ear mite of cattle is Raillietia auris, and that of small ruminants is Psoroptes cuniculi. Cattle that become infected with the ear mite develop a hearing impairment. Severe infestations perforate the tympanum and result in vestibular disease, facial paralysis, and ataxia.1567-1570 Infected sheep and goats shake their heads vigorously and develop aural hematomas.1571 Infestation of cattle can be recognized by observation of ulceration and purulent debris in the auditory canal next to the tympanum. In cattle, mites may be entrapped between the plug and the tympanum and may not be visible during an otoscopic examination. All infested cattle have pus and ulceration of the ear canal.1572 A foul-smelling discharge on the side of the face under the ear canal may be seen in some affected animals. Chronically affected cattle develop a hearing loss for high-frequency sounds.
The psoroptic ear mite of small ruminants does not spread over the remainder of the body. The accumulation of purulent debris and swelling of auricular tissues block the transmission of sound to the tympanum.
Ear mite infestations in cattle are common and in some herds may affect most of the adults. One U.S. study reported a prevalence rate of 66% (29 of 44 cattle).1567 The biologic importance of the infestation may be related to changes in the herding or mothering behavior of range cattle. The economic significance of the infestation is unknown. The parasite can complete a full life cycle by 8 days.
Ear mites have been successfully treated in goats using ear drops containing rotenone* once daily for 5 to 10 days. Good clinical responses can be obtained in cattle with the same treatment. Fenthion (Spotton,† 0.2 mL) drops also have been used successfully. In cases of parasitic otitis in which the discharge has been inspissated, lateral resection of the ear canal should be considered to establish adequate ventral drainage.1573 The addition of nicotine to final concentrations of 2 ppm in dip tanks containing 0.25% toxaphene has been 95% effective in some outbreaks.1574 Plunge-dipping in diazonon, propetamphos, or flumethrin, pour-on preparations of synthetic pyrethroids, and oral dosing with ivermectin all are ineffective.1574 A single subcutaneous (SC) injection of ivermectin at 0.2 mg/kg has been shown to be an effective treatment for P. cuniculi otitis in both sheep and goats.1574,1575
The life cycle of the mite involves two free-living stages, the proto and the deuto nymphs. These forms molt in the vegetation and reinfest cattle as they graze or bed during the evening.1576 Therefore, tilling the soil of the infested pastures or re-treating cattle every 14 to 21 days with insecticide should be considered as part of an eradication scheme.
A recent report of neurologic dysfunction in a cow with Raillietia infestation highlights the importance of thorough evaluation before diagnosis.1577 This cow also had listeriosis, the more likely cause of the neurologic signs.
SPACE-OCCUPYING LESIONS OF CRANIAL NERVES IN CALVES
LISLE W. GEORGE
An epizootic disease of Groningse Blaarkop calves characterized by facial paralysis and vestibulocochlear disease has been identified.1578 The calves showed drooped ear, loss of vision with normal pupillary reflexes, head tilt, dorsolateral strabismus, circling, and depression. One calf had a mild fever, dysphagia, and mandibular paralysis. The CSF contained a high WBC count and an increased total protein level. The disease was caused by multifocal space-occupying lesions that surrounded the cranial nerves at the entrance of the calvarium. The microscopic lesions consisted of granulomatous inflammation of the nerves at the internal acoustic meatus and the facial canal. The inflammatory cells in the lesions consisted of histiocytes, lymphocytes, multinucleated giant cells, and plasma cells. The specific etiologic agent was not identified. The granulomas did not contain acid-fast bacilli, fungal elements, or Listeria monocytogenes. The microscopic appearance of the lesions and their location in the nervous system were similar to cauda equina neuritis in horses. The disease in the calves was nonprogressive, and some of the calves recovered, which differentiates the calf disease from the progressively fatal cauda equina neuritis in horses.
PERIPHERAL VESTIBULAR DISEASE OF HORSES
Definition and Etiology
Vestibular disease of horses is an acute, asymmetric condition with one of several causes: extension of pyogenic bacterial infections from the guttural pouch, polyneuritis equi, viral labyrinthitis, and traumatic skull fractures.1579-1581 Idiopathic labyrinthitis probably represents an acute viral inflammation of the vestibular system that is severe,1580 but spontaneous recovery is common. Lightning strike also has been suspected as the cause of vestibular signs in horses.1582
Vestibular disease may also be caused by pyogenic inflammation of the petrous temporal bone or membranous labyrinths. Staphylococci, streptococci, and Aspergillus species have been isolated from cases of suppurative otitis in horses.1579,1583 Two forms of suppurative otitis media-interna have been identified.1579,1583 In the least severe form, the pyogenic inflammation localizes in the petrous temporal bone but does not spread into the calvarium or rupture the tympanum. This infection results in vestibular signs. This mild form of otitis media-interna also causes dysfunction of CNs VII and VIII.
Clinical Signs
The clinical course of peripheral vestibular disease may be chronic with acute exacerbations. Recovery is common with appropriate therapy. The more severe form occurs when the pyogenic inflammation extends outward into the temporohyoid joint and stylohyoid bone.1583 The inflammatory process fuses the tympanohyoid joint, which fractures during strong contractions of the muscles of the pharynx and neck. The fracture line extends into the calvarium, resulting in hematoma formation in the CNS. Pyogenic agents from the original septic site may then gain access to the CNS and cause meningitis. The clinical signs are peracute, and mortality is high. Extension of the fracture lines along the cranial vault and osteomyelitis result in dysfunction of CNs VII and VIII and the cerebrum. Affected horses show a rapid deterioration in mental status immediately after the initial onset of clinical signs. Involvement of the temporohyoid bone can be recognized on radiographic examination of the head and pharynx. The radiographic changes include thickening and pathologic fracture of the stylohyoid bone and tympanosclerosis.
Early clinical signs may appear to be unrelated to the CNS. The horse may appear to be uncomfortable and may shake the head or rub the affected ear for 2 to 3 weeks before the onset of vestibular signs. Otorrhea is not usually observed. The neurologic signs appear suddenly. Horses with mild disease develop ataxia, head tilt, facial paralysis, and nystagmus. The nystagmus is not changed by movement of the head (rapid phase away from the side of the lesion). There also is a ventrolateral strabismus on the side of the lesion. The affected animals circle or more often lean against the stall walls for support. Frequently, a mild conscious proprioceptive deficit is worse on the affected side. Horses with severe calvarium fractures fall and become recumbent. They lie with the side of the lesion facing the floor. Because of the proximity of the facial nerve to the vestibular apparatus in the petrous temporal bone, most affected horses also show signs of facial palsy, including drooped ear and lips, drooling of saliva, ptosis, exposure keratitis, and deviation of the philtrum toward the opposite side of the lesion (Fig. 35-19). If extensive bleeding into the calvarium has occurred, the animal becomes blind in the eye contralateral to the side of the hematoma. Pressure in the cerebral cortex may result in mydriatic pupils on the ipsilateral side. Lesions located central to the geniculate ganglion denervate the lacrimal glands and result in keratitis sicca.
Horses with lesions of the peripheral vestibular system remain appetent and alert. In comparison, animals with vestibular disease accompanied by petrous temporal bone fractures and meningitis tend to be depressed, febrile, and inappetent. Animals that develop septic meningitis secondary to a temporal bone fracture show rapid deterioration of mental status, rigidity, or flailing of the limbs with mild stimulation, stiffness of the neck, hyperesthesia, fever, otorrhea, and dysphagia.1584
Diagnosis and Treatment
Ancillary diagnostic measures for vestibular disease in horses include skull radiographs and endoscopic examination of the guttural pouch to exclude the possibility of tympanosclerosis, fractured hyoid bone, or fungal otitis. Otic examination in the horse in difficult because of the external ear anatomy in this species and the resistance of horses to this type of examination. Chemical restraint and the use of video otoscopy can facilitate both otoscopic examination and the sampling of material from the external ear for microscopic examination and culture.1585 The rate of tear secretion may be tested with a Schirmer tear test strip. The normal rate of tear secretion is approximately 21 mm/min, whereas deficient tear production is less than 17 mm/min. Brainstem auditory-evoked response testing may help to establish the localization of vestibular signs as peripheral in origin rather than central.1586
Antibiotic treatment of peripheral vestibular disease should include high doses of penicillin (20,000 to 40,000 IU/kg IV four times daily) or, as an alternative, a third-generation cephalosporin or trimethoprim-sulfonamide combination. The alternative drugs should be considered when infection by penicillin-resistant bacteria is suspected. One study reported clinical improvement in patients treated with chloramphenicol (10 mg/kg orally four times daily).1583
Patients with early cases of vestibular disease may benefit from treatment with nonsteroidal antiinflammatory drugs (NSAIDs). Administration of corticosteroids in the acute stages of the disease may ameliorate the clinical signs, but the beneficial antiinflammatory effects of these drugs should be weighed against the potential for nonspecific immune suppression and ultimate extension of pyogenic foci into the CNS along cracks in the calvarium.
Affected horses should be kept in a quiet, heavily bedded stall with good footing. Exposure keratitis may be treated by performing a tarsorrhaphy or by repeatedly administering petrolatum ophthalmic-lubricating ointments. Horses with keratoconjunctivitis sicca may be treated with 0.25% pilocarpine eyedrops four times daily.1587
Horses that recover after long-term antibiotic therapy should be used cautiously because subtle neurologic deficits that interfere with coordinated motor activities could precipitate catastrophic accidents. Relapses may occur in some seemingly recovered patients.
EXOPHTHALMOS AND STRABISMUS OF CATTLE
LISLE W. GEORGE
A heritable exophthalmos and strabismus of Jersey, Holstein, Brown Swiss, and shorthorn cattle has been described.1588-1591 The defect is characterized by protrusion of the eyeballs and anteromedial rotation of the eye around the axis (cross-eyed). The defect does not become evident until the animals are over 6 months of age. Affected animals have defective vision and show difficulty walking in unfamiliar environments. Both genders are affected. The condition in Holsteins is thought to be related to a decreased number of nerve cells in the abducens motor nucleus.
NIGROPALLIDAL ENCEPHALOMALACIA (YELLOW STAR THISTLE POISONING; RUSSIAN KNAPWEED POISONING)
Definition and Etiology
Nigropallidal encephalomalacia is a disease of adult horses characterized by facial dystonia, variable ataxia, mild depression, and food retention in the mouth. The disease is caused by ingestion of large quantities of the plants Centaurea solstitialis (yellow star thistle) or Centaurea repens (Russian knapweed).1592-1595
Clinical Signs
The signs appear suddenly but always after long-term ingestion of large quantities of the plants. Characteristic signs of nigropallidal encephalomalacia common to all cases include weight loss, mild to moderate depression, conscious proprioceptive deficits, yawning, lowered head, protruding tongue, tremor of the tongue and lips, and facial hypertonicity when feed is offered. The facial hypertonicity causes a retraction of the lips, resulting in a fixed grimace with the mouth and lips held half-open (Fig. 35-20). The patient may display constant chewing movements, and prehension, mastication, and deglutition of food are uncoordinated and inefficient. Affected horses can grasp food in their incisors but are unable to chew adequately and propel the food to the back of the mouth. Food retained in the mouth and cheek pouches may protrude from the commissures of the lips. Affected animals may attempt to drink by immersing their muzzles deeply into the bucket to force the water into the back of the pharynx. Once the food or water is in the posterior part of the pharynx, the animal is able to swallow. Affected animals die of starvation or dehydration. Horses that appear to be depressed usually can be aroused by mild stimulation. Motor and sensory deficits include hypertonicity, ataxia, conscious proprioceptive deficits, and occasionally hypermetria. There also may be a transient tendency to walk propulsively or to circle. Occasionally the animals are hyperexcitable. After several days the signs stabilize, and the disease does not progress. Affected animals usually do not recover.
Diagnosis
The CSF of affected horses may show increases in WBC count (75/μL).1594 There are no characteristic changes in the complete blood count (CBC) or the serum chemistries. Magnetic resonance imaging (MRI) was used to make an antemortem diagnosis in one horse with nigropallidal encephalomalacia.1596
Pathophysiology
The toxic principle in the plants has been isolated and chemically characterized. The toxic molecule is called repin, a sesquiterpene lactone with high affinity for neural tissue.1597-1600 Long-term feeding of alcoholic extracts containing repin to monkeys has resulted in collapse, convulsions, and death.1601 Studies in rats suggest that repin exerts its neurotoxic effects by inhibiting dopamine release.1600 Additional neurotoxic compounds, including aspartic and glutamic acids, also have been isolated from Centaurea plants.1602
Necropsy Findings
At necropsy, sharply demarcated areas of yellowish malacia are visible grossly in the substantia nigra and globus pallidus (extrapyramidal system). Lesions are bilaterally symmetric in more than 50% of cases but are asymmetric in a substantial proportion of affected animals. Lesions in other brainstem nuclei are found in a small number of animals.1593,1603 Microscopic lesions include neuronal necrosis, vacuolation with gliosis, liquefactive necrosis, and cavitation in well-developed lesions.1597
Epidemiology
Nigropallidal encephalomalacia has been reported in horses of the United States, Australia, and South America. Yellow star thistle is a common plant in unirrigated pastures in the arid regions of the western United States. The plant is resistant to the effects of saline or alkaline soil conditions and has a minimum moisture requirement. Russian knapweed belongs to the sunflower family and grows predominantly on flood plains, where it can extract deep subterranean moisture. In the United States the plants tend to remain green during the dry months, so most poisonings occur during the summer or late autumn. Most horses are reluctant to eat Centaurea plants unless other vegetation is unavailable, but some develop a craving and selectively seek it out. Horses that develop nigropallidal encephalomalacia usually are being fed a poor-quality, high-roughage diet. Affected horses range from 4 months to 10 years of age (median, 2 years).1597 The case-attack rates of yellow star thistle poisoning range from 3% to 31% of horses on infested pastures.1597 Feeding studies have reported that as much as 59% to 200% of the body weight of yellow star thistle and 59% to 63% of Russian knapweed must be eaten over 3 to 11 weeks to cause clinical disease.1593,1597 Continuous protracted exposure to the weeds seems to be important for expression of clinical disease. The dried plants retain their toxicity.
Treatment and Prevention
There is no known treatment for the poisoning. Prevention is best aimed at correcting the nutritional problem by daily supplementation with 10 to 15 lb of alfalfa hay and by not pasturing horses in areas where the thistle grows.
RUPTURED RECTUS CAPITIS VENTRALIS MUSCLES (TRAUMA TO CRANIAL NERVES IX, X, AND XI)
LISLE W. GEORGE
Traumatic avulsion of the rectus capitis ventralis muscle is seen exclusively in equids. The condition causes dysphagia. The muscle is ruptured when horses fall over backward and hyperextend the neck and head. Tearing of the tendinous insertion of the muscle damages CNs IX, X, and XI. The clinical signs include mild transitory epistaxis, laryngeal hemiplegia, dysphagia, and pharyngeal paralysis. Endoscopic abnormalities of the pharynx and larynx include mucoid discharge from the guttural pouch, pharyngeal and laryngeal paralysis, atonic proximal esophagus, and food particles in the trachea and bronchi.
Radiographic examination of the head and neck may be helpful for substantiating a clinical diagnosis. The radiologic lesions include irregular radiopaque lesions in the guttural pouch and fracture of sclerotic occipital and petrous temporal bones. These cases usually are associated with a preexisting mycotic lesion that results in bone weakness and pathologic fractures. The neurologic lesion usually is reversible, but affected horses may die of aspiration pneumonia before neurologic resolution.1604
HORNER’S SYNDROME
Definition and Etiology
Horner’s syndrome results from interruption of ocular sympathetic pathways. Sympathetic fibers originate from neuronal cell bodies located in the mesencephalic tectum. Axons descend to the first to third thoracic (T1 to T3) segments of the spinal cord, where they enter the gray matter, synapse, and exit through the ventral spinal nerves. From there the nerves pass through the cervicothoracic and middle cervical ganglia (stellate ganglia) and ascend in the cranial vagosympathetic trunk.1605 The nerves enter the cranial ganglion in the petrous temporal bone, where they synapse. The postganglionic fibers are distributed to the sweat glands of the head, ciliary muscles, periorbital smooth muscles, and periarteriolar musculature. Fibers of the vagosympathetic trunk or the cranial cervical ganglion can be injured as they pass through the neck or over the caudodorsal aspect of the guttural pouch.
Specific causes of Horner’s syndrome include mycotic guttural pouch infections; traumatic lesions of the basisphenoid area; cervical trauma; abscesses, tumors, or space-occupying lesions in the anterior aspect of the thorax1606,1607; periorbital abscesses or tumors; parotid duct obstruction and inflammation1608; esophageal rupture; and complications associated with surgical ligation of the carotid artery. Intracranial lesions involving the central components of the sympathetic pathway in the ipsilateral brainstem are rare but have been reported as a consequence of metastatic neoplasia.1609 Horner’s syndrome also has occurred after IV injection of certain drugs, including xylazine, vitamin E or selenium, and phenylbutazone.1610-1612 Horner’s syndrome has also been seen in horses with polyneuritis equi (cauda equina neuritis) syndrome and equine protozoal myeloencephalitis of the cervical spinal cord.1613,1614 Tumors that have resulted in Horner’s syndrome include sclerosing respiratory epithelial carcinoma, squamous cell carcinoma, and melanoma.1606,1607,1610,1615,1616
Clinical Signs
The clinical signs of Horner’s syndrome in horses vary but include miosis, enophthalmos, ptosis, regional hyperthermia, excessive sweating on the ipsilateral side of the face, congested mucous membranes, inspiratory stridor, and dermatitis caused by chronic sweating. Ptosis may be detected by palpation of decreased eyelid tone. The palpebral reflex and the menace response are normal. Facial sweating often disappears 6 to 14 days after sympathectomy. If concomitant damage to the cervical sympathetic nerves is present, sweating of the skin of the neck also may be seen. This is not observed in animals with lesions solely in the tectotegmentospinal pathway. Regional hyperthermia is caused by vasodilation, which results from deficient vasomotor tonus. Sweating is thought to be caused by vasodilation and increased cutaneous blood flow.1617,1618 Increased sweating can be induced by β2-agonists, including IV clenbuterol (200 μg) or isoprenaline (2 mg) or local application of 10% phenylephrine.1619 Dysfunction of adjacent neurologic structures may result in simultaneous facial nerve paralysis and laryngeal hemiplegia. Bilateral Horner’s syndrome has been reported in a horse with metastatic neoplasia affecting the sympathetic innervation of the head bilaterally.1616 This horse had a mixed pattern of both preganglionic and postganglionic denervation caused by widespread metastases at multiple sites along the nerves.
In contrast to the disease in horses, cattle do not sweat on the planum nasale of the affected side. This can be explained by the mediation of normal sweat gland secretion by α-adrenergic receptors in the bovine.1618 The other signs seen in cattle are similar to those in horses. The clinical signs of experimentally induced Horner’s syndrome in sheep and goats are limited to a mild ptosis.1618 Retrobulbar tumors may cause Horner’s syndrome, but in these cases the eyeball proptosis results from the excessive retrobulbar pressure.1615
Diagnosis
The specific site of the denervation of the ocular sympathetic system usually can be located by pharmacologic testing.1605 Hydroxyamphetamine (1% solution) instilled into the eye will result in release of norepinephrine from intact postganglionic sympathetic neurons, causing pupillary dilation, but no response when the postganglionic neurons are damaged. A positive response to this test indicates a preganglionic sympathetic lesion, and a lack of response indicates a postganglionic lesion. In animals with a postganglionic lesion, topical administration of 0.1 mL of 1:1000 epinephrine solution directly activates the iris musculature and produces mydriasis by 20 minutes, whereas the onset of dilation occurs at about 40 minutes in animals with preganglionic lesions. Similarly, 2.5% to 10% phenylephrine solution will produce pupillary dilation in an eye with a postganglionic sympathetic lesion, but not in a normal eye. The increased sensitivity to these direct-acting sympathomimetics in animals with postganglionic sympathetic lesions results from the phenomenon of “denervation supersensitivity,” with numbers and sensitivity of norepinephrine receptors in the iris muscle increasing over days to weeks after postganglionic nerve injury. A positive response is therefore expected only after the nerve lesion has been present for at least several days. Parenteral administration of 1 mL of 1:1000 epinephrine solution causes affected horses to sweat profusely over the affected side of the face.1610 However, this test does not differentiate between preganglionic and postganglionic lesions.
The guttural pouches of horses and the pharynx of all patients should be examined endoscopically to exclude the possibility of pharyngeal or laryngeal paralysis or guttural pouch disease. The jugular furrows should be palpated for swellings. Insertion of a nasogastric tube during palpation may be helpful for detecting subtle lesions on the left side of the neck. Radiographs of the cervical vertebrae should be obtained to exclude spinal cord disease. The thorax should be examined using auscultation and percussion and, if indicated, radiographs taken. The gait and proprioceptive responses should be examined to evaluate the function of the spinal cord. The skin temperature may be measured using thermography1607,1620; on the affected side it is 1° C to 2.5° C (33.4° F to 37.5° F) higher than normal.
Treatment
The treatment for Horner’s syndrome depends on the underlying cause of the denervation. Except for Horner’s syndrome related to IV injection of xylazine, the neurologic signs often are irreversible, even if the primary cause of the condition has been eliminated. When xylazine is administered IV, the condition disappears spontaneously. The situation differs from inadvertent perivascular drug injections, in which permanent neurologic sequelae may occur. The necrotizing effects of perivascular drug injections can be minimized if treatment is administered immediately. These treatments should include aseptic infusion of large volumes of saline at the perivascular injection site and systemic administration of NSAIDs or dexamethasone, or both. Abscesses should be drained, and fungal infections of the guttural pouch should be treated as described in Chapter 31.
GUTTURAL POUCH MYCOSIS, NEUROLOGIC SIGNS (DAMAGE TO CRANIAL NERVES IX THROUGH XII)
Definition and Etiology
In one retrospective survey, mycotic infections of the guttural pouch were the third most common disease of the upper respiratory tract of horses.1621 The clinical signs occur because fungal infections in the medial part of the pouch extend dorsally and damage CNs IX through XII and the internal carotid artery. Mycotic guttural pouch infection usually occurs in older animals; however, horses as young as 3 months have been affected.1622
Clinical Signs
Initially the horse may display head shaking and unilateral nasal discharge. Additional clinical signs are nasal catarrh, dysphagia, head shyness, head shaking, roaring, dysphonia, protrusion of the tongue from the mouth, and epistaxis1622-1624 (Fig. 35-21). Other clinical signs include parotid pain, abnormal head posture, facial sweating, shivering, Horner’s syndrome, colic, hemiparesis to hemiplegia of the tongue on the affected side, and facial paralysis.1622-1628 Epistaxis may be fulminant and life threatening. The abnormal head posture is characterized by a tendency to hold the head in extension or lower to the ground than normal. Atrophy of the brachiocephalicus and trapezius muscles occurs secondary to denervation of the accessory spinal nerve. Horner’s syndrome occurs secondary to damage of the cranial cervical ganglion and sympathetic trunk. The sensorium is intact unless aspiration pneumonia develops or the fungus embolizes into the brain.1629,1630 Occasionally, affected horses die peracutely as a result of exsanguination from a ruptured internal carotid artery. Endoscopic examination of the larynx of affected horses may reveal dorsal displacement of the soft palate, inability to swallow, and unilateral or bilateral laryngeal hemiplegia. Mycotic guttural pouch infection can be definitively diagnosed by endoscopically identifying the characteristic fungal mass in the dorsomedial compartment (Fig. 35-22). Radiographic examination of the pouches shows a poorly defined border of the pouch in the abnormal area. Extension of a mycotic infection from the guttural pouch to the brain through the internal carotid artery has been reported.1630 The infection caused disseminated necrosis and hemorrhage that was most severe in the thalamus, cerebral cortex, and hippocampus. The clinical signs were fever (38.3° C; 100.9° F), epistaxis, dysphagia, laryngeal hemiplegia, pharyngeal paralysis, circling, unilateral blindness, mydriasis, facial paralysis, and apprehension.
Pathophysiology
The guttural pouch is separated into two compartments by the stylohyoid bone and the occipitohyoideus muscle. CNs IX through XII and the internal carotid artery are located in the dorsomedial aspect of the medial compartment and are susceptible to damage from mycotic infections. Extension to the cranial nerves occurs because of inflammation and direct destruction of these structures by the fungal elements. Pathologic studies have shown swelling of the myelin sheaths and Schwann cells. Some sections demonstrate necrosis of the nerves and invasion by fungal elements.1631
In chronic infections the fungal lesion may extend into the tympanic bulla and cause vestibular disease. Extensive growth of the lesion also results in temporomandibular osteoarthropathy and fusion of the temporomandibular joint or osteoarthritis of the atlantooccipital joint. Excessive muscular force on the fused joint can result in avulsion fractures of the petrous temporal bone and calvarium. Hemorrhage or spread of infection into the vestibular apparatus and calvarium results in acute vestibular disease, cerebral hemorrhage, and septic meningitis (see Peripheral Vestibular Disease of Horses). In rare cases, fungal elements may reach into the lateral compartment, invade the wall of the internal maxillary artery, and affect the facial nerve. Facial nerve paralysis also has been observed and results from abscessation of the parotid lymph nodes secondary to a fungal guttural pouch infection.1623 In other rare cases, Horner’s syndrome may be caused by mycotic lesions in the cranial cervical ganglion. The syndrome also may occur iatrogenically during surgical ligation of the external carotid artery.1632 Occasionally, the mycotic infection may extend into the brain and cause encephalitic signs.1629
Treatment
Before the onset of neurologic disturbances, the internal carotid artery may be occluded using a ligature or by insertion of a balloon-tipped catheter.1633 During the exploration the fungal mass is debrided surgically. The surgical procedure is effective for preventing fatal epistaxis but may not reduce the potential for progression of the infection, resulting in nerve deficits or extension of the infection to the CNS. Some studies have shown excellent resolution of the mycotic lesion in horses that underwent carotid artery occlusion with or without additional antifungal therapy,1634-1636 but in other animals the lesion may progress despite complete arterial occlusion.1628 Moreover, optic neuropathy and blindness of the ipsilateral eye are common postsurgical sequelae.1637 Neurologic signs may be permanent, although improvement or recovery over many months has been reported in some horses.1636 Affected horses often are euthanized for humane reasons. (See Chapter 31 for treatment of guttural pouch mycosis.)
DISEASES PRODUCING TREMORS AND ATAXIA; CEREBELLAR DISEASES
CEREBELLAR HYPOPLASIA CAUSED BY CONGENITAL BOVINE VIRAL DIARRHEA VIRUS INFECTION
A complete discussion of bovine viral diarrhea–mucosal disease (BVD-MD) can be found in Chapter 32. BVD virus infection of susceptible pregnant cattle from 90 to 170 days’ gestation results in abortion or stillbirth, hydranencephaly, or cerebellar hypoplasia in the fetus.1638,1639 These signs are also seen in calves born to susceptible dams vaccinated during gestation with a modified live BVD vaccine.1640 The BVD virus infects the developing germinal cells of the cerebellum and kills Purkinje’s cells in the granular layer, resulting in necrosis and inflammation.1641 Such cerebellar lesions tend to be most severe by 21 days after inoculation of the susceptible pregnant dams.1638 The microscopic lesions include necrosis of the external germinal cells, focal parenchymal hemorrhages, and folial edema. After infection the acute inflammatory responses subside by 42 days, when the microscopic changes include cavities ranging from 1 to 7 mm in diameter, thinning of the neuropil, atrophy of the cerebellar folia, axonal torpedoes, and mild reactive astrocytosis. Calves infected with vaccine virus between 90 and 118 days’ gestation may develop hydrocephalus and hydranencephaly.1642
The signs of cerebellar dysfunction usually are present at birth and include truncal ataxia, falling backward, opisthotonos, base-wide stance, coarse intentional head tremors, hypermetria, hyperreflexia, and nystagmus or strabismus (Fig. 35-23).1643,1644 If severely affected, the animal may be unable to stand or lie in sternal recumbency. Excitatory stimuli in these animals precipitate wild oscillations and side-to-side movements of the head, which can be mistaken for convulsions. The affected calves may have a deficient menace response and appear to be blind, especially with concomitant hydranencephaly or microphthalmia. The neurologic condition rarely improves after birth. Other fetal changes that may be induced by the BVD virus include thymic atrophy, retinal degeneration, corneal opacity, failure to grow, and abortion.1638,1643 The diagnosis of cerebellar hypoplasia is based on identification of the specific clinical signs and the recognition of BVD antibodies in precolostral blood specimens. The virus may be cultured from the blood of some affected calves. Viral antigen has been detected in the spleen, kidneys, and lymph nodes of aborted fetuses.1644 The virus cannot usually be isolated from immunocompetent calves after antiviral antibody responses develop but may be recovered repeatedly from immunoincompetent, seronegative calves. Bluetongue virus may also occasionally cause cerebellar lesions in calves and lambs.
CEREBELLAR ABIOTROPHY OF CATTLE
Abiotrophy is defined as a degeneration of formed elements of the nervous system.1645 Cerebellar abiotrophy has been described in Holstein, Angus, and Limousin calves. The condition has been reported in the United States, Canada,1646-1648 Australia,1649,1650 and the United Kingdom.1651 In Holstein calves a recessive mode of inheritance is suspected, which can be traced to a single sire. The etiology of the condition in other breeds of cattle is uncertain. Some calves are affected from birth,1651 whereas others are normal at birth but develop signs at 3 to 9 months, or show signs intermittently, especially when stressed by factors such as inclement weather.1649,1650 Severely affected animals may be in lateral recumbency and unable to rise. Nystagmus and opisthotonos are often seen in these calves. During the physical examination, signs of cerebellar dysfunction may be observed (Fig. 35-24), including intentional head tremors, base-wide stance, hypermetria, hyperesthesia, hyperreflexia, and lack of menace with preservation of eyesight. The clinical condition of affected animals may remain static or progress slowly until the animals become recumbent and unable to rise. Two calves showed gradual resolution of clinical signs when moved to a new location1650; thus environmental factors may play some role in the clinical development of the disease. The pathophysiology of the disease is unknown. There are no macroscopic abnormalities of the cerebellum of affected calves. The microscopic pathologic changes of abiotrophy include noninflammatory focal loss of Purkinje’s cells and cerebellar nuclear neurons, with gliosis in the Purkinje and molecular cell layers. Swellings within Purkinje cell axons also have been described in some cases.1650 The variable pathology described in affected animals suggests that bovine cerebellar atrophy may be several similar but slightly differing disorders rather than a single entity.
HEREDITARY HYPERMETRIA IN SHORTHORN CATTLE
A neurologic disease characterized by symmetric cerebellar signs was reported in 15 shorthorn cattle of Brazil. The animals were affected at birth, and the clinical disorder was not progressive. The affected animals had no pathologic changes in the central nervous system that differentiated the condition from cerebellar hypoplasia and cerebellar abiotrophy. Examination of the relatives of the affected animals indicated a familial distribution. Pedigree analysis suggested an autosomal recessive mode of inheritance.1652
CEREBELLAR MALFORMATIONS OF AYRSHIRE AND JERSEY CALVES
Cerebellar malformations have been reported in two Ayrshire calves from Great Britain.1653 The calves appeared to be normal at birth but displayed characteristic signs of cerebellar dysfunction by 24 hours of age. A similar condition has been reported in Jersey calves.1654,1655 The clinical signs included opisthotonos, base-wide stance, truncal ataxia, hypermetria and hypertonia of all four limbs, and head tremors. Lesions in the calves were present in the cerebellum and the pons; some animals also developed hydrocephalus. The cause of these abnormalities was not definitely determined but was presumed to be a hereditary condition. The relationship of this disease to BVD virus infection is unclear.
BOVINE FAMILIAL CONVULSIONS AND ATAXIA
Bovine familial convulsions and ataxia is a disease of Angus cattle characterized by multiple tetanic tonic-clonic convulsions and a spastic ataxia that persists for several months.1656-1658 A similar condition has been reported in a 9-month-old Charolais calf in the United States, in polled Hereford calves in Australia, and in Angus crossbred calves in Canada.1659-1661 The disorder is believed to result from a defective autosomal dominant gene with incomplete penetrance, although some affected animals did not have a close relationship to blood lines believed to carry the trait.1658
Clinical Signs
The onset of clinical signs ranges from 2 to 3 hours after birth to 3 months of age. Calves may be born dead at or near term or may be aborted. Some aborted calves have a dorsiflexion of the spine. Most affected calves are born alive and rapidly develop intermittent signs of cerebellar dysfunction with multiple “tetaniform seizures” that last 3 to 12 hours. Two forms of seizures have been described. One form is characterized by a generalized stiffness and inability to protract the legs, elevation of the tailhead, and head-neck extension with mild head tremor. The animals are hyperesthetic. A more severe form is characterized by lateral recumbency, loss of consciousness, opisthotonos, hypertonicity, tonic-clonic seizures, and trismus. Animals may improve greatly if supported during the tetaniform activity. The frequency of the seizures declines over several months, but animals are left with a permanent ataxia characterized by cerebellar signs. The attacks may be precipitated by sudden, violent auditory, olfactory, visual, or tactile stimuli. These could include driving by dogs, shipment, flashing lights, loud sounds, or painful tactile stimulation. These features of the attacks, combined with a lack of electroencephalographic (EEG) evidence of seizure activity in the cerebrums of affected calves, suggest that these episodes may be exacerbations of cerebellar signs rather than true seizures.1662 There is no response to treatment with electrolytes, mineral supplements, or B-complex vitamin injections. Seizure-like activity can be controlled by administration of barbiturates or inhalational anesthetics.
Most animals improve when turned out to grass pastures, but episodic relapses associated with excitement may occur for as long as 2 years. Affected animals gradually recover and by 2 years of age either show only mild cerebellar signs or are completely normal. There are no specific diagnostic tests for the disease.
Pathology
The gross appearance of the brain is normal. Microscopic lesions are restricted to the cerebellar cortex and include swelling and vacuolation of Purkinje’s cells, chromatolysis, loss of neurofibrils, and formation of axonal torpedoes. The axonal structures have been defined as “argyrophilic axonal swellings.” They are located in the granular layer of the lingula, uvula, and adjacent parts of the vermis. An early report stated that these lesions were not seen in affected animals under 6 weeks of age, but a more recent study found lesions in affected animals of all ages.1661,1663
Diagnosis
Diagnosis is based on the presence of typical clinical signs, particularly when several related animals are affected. Differential diagnoses include cerebellar hypoplasia caused by congenital BVD virus infection, hypomyelinogenesis, congenital brain malformations affecting structures in the caudal fossa, congenital storage diseases, and the various cerebellar abiotrophies described in cattle.1662 Imaging studies (e.g., CT, MRI) are expected to be within normal limits.1662 Definitive diagnosis can be made only on histologic examination postmortem.
Treatment and Control
There is no known treatment for bovine familial convulsions and ataxia. Affected animals can be fattened and slaughtered but should not be used as breeding stock because the disease is genetically transmitted. Elimination of carrier animals from the breeding population will effectively control the propagation of this disease.
CEREBELLAR ABIOTROPHY (HYPOPLASIA) IN ARABIAN HORSES
Cerebellar abiotrophy occurs in purebred Arabian or Arabian crossbred horses.1664-1669 Clinical signs may be present at birth or may develop after several weeks to months of postnatal life. Signs most frequently develop between 2 and 4 months of age and almost always occur before 6 months of age. The disease was initially reported as a cerebellar hypoplasia, but most descriptions suggest that the degeneration begins postnatally, prompting classification of the condition as an abiotrophy.1670 Some investigators have suggested, however, that the clinical and pathologic course of the disease is not always consistent with a progressive postnatal degeneration of the nervous system.1666
The signs of cerebellar abiotrophy appear suddenly and range from subtle ataxia to complete diffuse cerebellar dysfunction. Head tremor is usually present and may occur in either a vertical or horizontal direction. Some horses show no progression of signs, whereas others progress slowly, followed by a plateau. The initial clinical sign in most foals is mild conscious proprioceptive deficits. As the deficits worsen, affected animals show hypertonia and stiff, hypermetric gaits accentuated by stimulation. In the most severe cases the animal may rear and fall over backward when suddenly startled. More severely affected foals have marked intentional head tremor, truncal ataxia, and hypermetria of all four limbs, often more pronounced in the forelimbs. These deficits are exaggerated by turning the animal sharply, by having the foal step up and down a curb, or by walking the foal on an incline. Most affected animals have a decreased to absent menace response despite normal visual acuity and facial nerve function; however, two affected foals with normal menace responses have been described.1665,1667 Rotary nystagmus occurs in rare cases.
Diagnosis generally is made on the basis of typical clinical signs in an Arabian horse under 6 months of age. Differential diagnoses include head trauma (particularly associated with basisphenoid bone fracture) and atlantooccipital malformation. With head trauma, other evidence of trauma and signs of vestibular dysfunction often are present. Foals with atlantooccipital malformation are ataxic and somewhat weak but do not have intentional head tremors. Muscular strength is preserved in cerebellar abiotrophy. Cerebrospinal fluid (CSF) analysis usually is normal, although CSF creatine kinase occasionally is elevated.
The major histologic finding is a degeneration and loss of Purkinje’s cells in the cerebellum accompanied by gliosis and thinning of the molecular and granular layers of the cerebellum. Evidence indicates that apoptosis may be the primary mechanism underlying Purkinje cell death in Arabian horses with cerebellar abiotrophy.1671 Mineral deposits in the thalamus also are found in horses with cerebellar abiotrophy, but their significance and relationship to the cerebellar changes are unknown.1669
The cause of cerebellar hypoplasia in Arabians is unknown, but there is a familial pattern of occurrence. One survey has reported an 8% prevalence rate in one family of 36 foals and a 6% rate in another family of 67 foals.1664 Two of four full-sibling colts from a mare were affected.1664 Pedigree analysis of 19 affected animals showed a high degree of relationship between the patients.
No effective treatment exists for cerebellar abiotrophy of Arabian foals. Occasional animals are reported to have shown gradual mild improvement, with considerable resolution of the head tremor. They remain unsafe for riding, however, and are not suitable as breeding stock because the disease probably is inherited. Owners of affected foals should be counseled about the probable heritability of the disease and should be encouraged to discontinue use of the parent lineage as breeding stock.
FAMILIAL ATAXIA IN HEREFORD CALVES
A central nervous system (CNS) disorder characterized by lateral recumbency, ataxia, incoordination, pupillary dilation, and abnormal head posture has been reported in Hereford calves.1672 The disease occurred at 24 hours of age. The pathologic lesions could be differentiated from those of hereditary neuraxial edema and familial convulsions and ataxia of Angus cattle. The lesions in the Hereford calves included hypomyelination of the cerebellum, cerebral cortex, medulla, and midbrain and vacuolation of the white matter. No necrosis of the CNS or inflammatory lesions was seen, and the neurons of the cerebellum appeared to be normal. The cause of the condition was not identified, but the dams were highly related, and a genetic etiology was postulated.
MICROGNATHIA AND CEREBELLAR HYPOPLASIA IN ANGUS CALVES
Micrognathia was observed in Angus calves of a small herd in western Missouri. These animals were born dead. The calves had severe brachygnathia and cerebellar hypoplasia. Other somatic changes included hepatomegaly and patent foramen ovale. A pedigree analysis indicated that an autosomal recessive genetic trait derived from a common ancestor may have been responsible for the condition.1673
STORAGE DISEASES AND INBORN ERRORS OF METABOLISM
Storage diseases and inborn errors of metabolism are characterized by intraneuronal accumulation of indigestible metabolic products. The material accumulates in the cells because of a deficient activity of one of several lysosomal catabolic enzymes. Neurons have a long lifespan and are rich in gangliosides and glycolipids, which are continuously degraded and resynthesized. In normal animals the metabolic products are internalized by the intraneuronal lysosomes and are degraded into constituent amino acids, monosaccharides, fatty acids, alcohols, and simple lipids by acidic catabolic enzymes. Disturbances of ganglioside metabolism result in accumulation of the degraded by-products in the neurons and other cells.1674 Overloading of the lysosomes by the undigested material produces profound neurologic dysfunction.
Storage diseases can be classified as either genetic or acquired. Acquired storage diseases are caused by ingestion of plants that contain specific inhibitors of one or more lysosomal catabolic enzymes. Genetic storage diseases are caused by the production of an inactive lysosomal enzyme. One such storage disease is ceroid lipofuscinosis, described earlier.
The genetic storage diseases are named according to the metabolic by-product that accumulates in the lysosomes. When tissues of affected animals are sectioned, processed, and examined microscopically, the metabolic storage product is dissolved from the tissue sections by the normal dehydrating and fixative agents. The spaces that contain the product appear as intraneuronal vacuoles when examined by light microscopy. Special fixation and staining procedures may be used to preserve and identify the metabolic product.1674
α-MANNOSIDOSIS (PSEUDOLIPIDOSIS)
α-Mannosidosis is a genetic defect of the enzyme α-mannosidase that is inherited as an autosomal recessive trait in Angus, Murray Gray, Simmental, Galloway, and Holstein cattle.1675 At least two different mutations in the gene coding for α-mannosidase occur in different breeds of cattle and have been characterized in Angus and Galloway breeds.1676 In animals deficient in α-mannosidase, the final cleavage between N-acetylglucosamine and mannose cannot occur, and the oligosaccharide accumulates in the lysosomes of the macrophages, neurons, and reticuloendothelial cells.1674 Other abnormalities of glycoprotein metabolism occurring in affected animals may be the result of the multiple functions that have been ascribed to α-mannosidases.1677
The clinical signs first appear by 1 week to 15 months of age. Affected calves tend to be less well developed than age-matched herdmates.1678,1679 The first symptom usually is a mild ataxia of the pelvic limbs that develops after exercise. Other signs include mild intentional head tremor, hypermetria, base-wide stance, and unwarranted aggressiveness.1678 When galloping, the rear limbs are overflexed, and the animal’s hindquarters appear to be sunken. The nervous system signs become much worse when the animals are excited. Most patients develop diarrhea, become recumbent after 3 to 4 months, and die shortly thereafter. A few affected animals survive for as long as 4 years. The neurologic signs of these animals remain constant, but they usually fail to grow normally. Other clinical manifestations in calves include premature delivery, abortion, stillbirth, and superior brachygnathia. Mannosidosis in Galloway calves is associated with somatic abnormalities such as arthrogryposis, hydrocephalus, and hepatic and renal enlargement.1675,1680 Phenotypic variations affect the severity and onset of the neurologic signs.
The concentration of α-mannosidase in the plasma can be measured with an enzymatic assay. Heterozygotes have less activity than genetically normal animals; however, occasional overlapping between the phenotypes of homozygotes and heterozygotes can confound attempts at classification. Three isoenzymes of α-mannosidase exist, but only one form is inactive in diseased animals. A delay in separation of the plasma from the cells or the use of serum for enzymatic assays results in a leakage of other isoenzymes from tissue compartments and uncertainty in interpretation of the results. In most cases, two populations of animals usually are evident, the homozygous normal and the heterozygote. The mean plasma concentration of α-mannosidase in heterozygotes is 6.6 nmol/min/mL, whereas the mean plasma enzyme activity in homozygotes is 29.1 nmol/min/mL. The test is most accurate at detecting heterozygotes over 18 months of age.
The brain enzyme activity of heterozygotes ranges from 0.03 to 0.05 IU/g of tissue; the reference range of enzyme activity is 1.8 to 3.1 IU/g. Heterozygotes also may be detected by measuring the relative concentration of α-mannosidase and hexosaminidase in purified peripheral blood neutrophils. This test has been recommended for confirmation of a carrier animal whenever the plasma mannosidase assay is questionable.
Pathology
The diagnosis of a clinical case of mannosidosis can be substantiated pathologically by observation of cytoplasmic vacuolation in the neurons of the cerebrum, cerebellum, brainstem, and spinal cord.1675 There is also a mild to marked internal hydrocephalus. The microscopic appearance of the brain of affected calves is characterized by vacuolation of the neurons and astrocytes and by reactive astrocytosis.1675 The vacuolation is not restricted to the nervous system, however, and can be seen in Kupffer’s cells, pancreatic exocrine cells, fibrocytes, and macrophages of the spleen and lymph nodes.1681 The vacuoles are lined by a single membrane and are thought to be part of the Golgi apparatus.1681 Associated neuronal changes include axonal swelling and spheroids.
Because of the hereditary nature of the disease, the ancestry of affected animals should be traced, and the biochemical phenotypes of related individuals should be tested. Both biochemical and molecular testing facilitate early detection of carrier animals, enabling their elimination from breeding programs.1682 Although bone marrow transplantation has been shown to correct the defect in the feline and murine models of α-mannosidosis,1683-1685 there is currently no effective and practical treatment for α-mannosidosis of cattle.