Chapter 21 Smoking and air pollution
 Smoking involves the regular inhalation of a variety of toxic compounds that stimulate airway irritant receptors and activate inflammatory pathways in the lung.
Smoking involves the regular inhalation of a variety of toxic compounds that stimulate airway irritant receptors and activate inflammatory pathways in the lung.The air we breathe is rarely a simple mixture of oxygen, nitrogen and water vapour. For much of the world’s population air also contains a variety of other, more noxious, gases and particles. In addition, a substantial proportion of people choose to further contaminate the air that they, and others, breathe with tobacco smoke.
Tobacco Smoke
In the Americas tobacco was used for medicinal purposes for many centuries before being introduced from the New World into Europe in the 16th century. Through his acquaintance with Queen Elizabeth I, Sir Walter Raleigh made smoking tobacco an essential fashionable activity of every gentleman. Thereafter the practice steadily increased in popularity until the explosive growth of the habit following the First World War (1914–1918).
There have always been those opposed to smoking and King James I (1603–1625) described it as ‘a custom loathsome to the eye, hateful to the nose, harmful to the brain and dangerous to the lungs’. However, firm evidence to support his last conclusion was delayed by some 350 years. Only relatively recently did it become clear that smokers had a higher mortality and that the causes of the excess mortality included many respiratory diseases.1,2 The proportion of the population who smoke has generally declined since evidence of serious health consequences emerged, though in the UK the proportion of the adult population who smoke is falling only slowly, reaching 24% in 2005. The global health costs of tobacco smoking are enormous: a third of people who smoke will die as a result of their habit, and it is estimated that smoking will cause 1000 million premature deaths worldwide in this century.3
Constituents of Tobacco Smoke
More than 2000 potentially noxious constituents have been identified in tobacco smoke, some in the gaseous phase and others in the particulate or tar phase. The particulate phase is defined as the fraction eliminated by passing smoke through a filter of pore size 0.1 μm. This is not to be confused with the ‘filter tip’, which allows passage of considerable quantities of particulate matter.
There is great variation in the yields of the different constituents between different brands and different types of cigarettes. This is achieved by using leaves of different species of plants, by varying the conditions of curing and cultivation, and by using filter tips. Ventilated filters have a ring of small holes in the paper between the filter tip and the tobacco. These holes admit air during a puff and dilute all constituents of the smoke. By these means, it is possible to have wide variations in the different constituents of smoke, which do not bear a fixed relationship to one another.
The gaseous phase. Carbon monoxide is present in cigarette smoke at a concentration issuing from the butt of the cigarette during a puff of around 1–5%, which is far into the toxic range. A better indication of the extent of carbon monoxide exposure is the percentage of carboxyhaemoglobin in blood. For non-smokers, the value is normally less than 1.5% but is influenced by exposure to air pollution and other people’s cigarette smoke (see below). Typical values for smokers range from 2% to 12%. The value is influenced by the number of cigarettes smoked, the type of cigarette and the pattern of inhalation of smoke.
Tobacco smoke also contains very high concentrations (about 400 parts per million) of nitric oxide (page 382) and trace concentrations of nitrogen dioxide, the former being slowly oxidised to the latter in the presence of oxygen. The toxicity of these compounds is well known. Nitrogen dioxide hydrates in alveolar lining fluid to form a mixture of nitrous and nitric acids. In addition, the nitrite ion converts haemoglobin to methaemoglobin.
Other constituents of the gaseous phase include hydrocyanic acid, cyanogen, aldehydes, ketones, nitrosamines and volatile polynuclear aromatic hydrocarbons.
The particulate phase. The material removed by a Cambridge filter is known as the ‘total particulate matter’, with aerosol particle size in the range 0.2–1 μm. The particulate phase comprises water, nicotine and ‘tar’. Nicotine ranges from 0.05 to 2.5 mg per cigarette and ‘tar’ from 0.5 to 35 mg per cigarette.
Individual Smoke Exposure
Individual smoke exposure is a complex function of the quantity of cigarettes that are smoked and the pattern of inhalation.
The quantity of cigarettes smoked. Exposure is usually quantified in ‘pack years’. This equals the product of the number of packs (20 cigarettes) smoked per day, multiplied by the number of years that that pattern was maintained. The totals for each period are then summated for the lifetime of the subject.
The pattern of inhalation. There are very wide variations in patterns of smoking. Air is normally drawn through the cigarette in a series of ‘puffs’ with a volume of about 25–50 ml per puff. The puff may be simply drawn into the mouth and rapidly expelled without appreciable inhalation. However, the habituated smoker will either inhale the puff directly into the lungs or, more commonly, pass the puff from the mouth to the lungs by inhaling air either through the mouth or else through the nose while passing the smoke from the mouth into the pharynx by apposing the tongue against the palate and so obliterating the gas space in the mouth. The inspiration is often especially deep, to flush into the lung any smoke remaining in the dead space.
It will be clear that the quantity of nicotine, tar and carbon monoxide obtainable from a single cigarette is highly variable, and the number and type of cigarettes smoked are not the sole determinants of effective exposure. There is good evidence that the habituated smoker adjusts his smoking pattern to maintain a particular blood level of nicotine.4 For example, after changing to a brand with a lower nicotine yield, it is common practice to modify the pattern of inhalation to maximise nicotine absorption.
Respiratory Effects of Smoking
Cigarette smoking has extensive effects on respiratory function and is clearly implicated in the aetiology of a number of respiratory diseases, particularly chronic obstructive pulmonary disease (COPD) and bronchial carcinoma, which are discussed in Chapters 28 and 30 respectively. Why only around one-fifth of smokers go on to develop COPD remains uncertain, but possibly relates to a genetic susceptibility to the effects of tobacco smoke.5 For example smokers who have developed COPD have different patterns of expression of oxidant/antioxidant pathway genes compared with smokers who have not developed COPD.6
Airway mucosa.7 There are conflicting reports regarding the sensitivity of airway reflexes in smokers, with increased sensitivity demonstrated in response to inhalation of small concentrations of ammonia vapour,8 and decreased sensitivity in response to capsaicin.9 The same workers also found different effects of smoking cessation on the cough reflex.8,10 The reasons for these varied results are unclear, but there are multiple ways in which smoking could alter cough reflexes, for example by desensitisation of stimulant receptors or by covering of the receptors with the excessive mucous present in the airways of smokers.
Ciliary function is inhibited by both particulate and gas phase compounds in vitro, but in-vivo studies have shown contradictory results, with some studies showing increased ciliary activity in response to cigarette smoke.
There is agreement that mucous production is increased in long-term smokers, who have hyperplasia of submucosal glands and increased numbers of goblet cells even when asymptomatic. In spite of the inconsistent findings regarding ciliary activity, mucous clearance is universally found to be impaired in smokers, which, coupled with increased mucous production and airway sensitivity, gives rise to the normally productive smoker’s cough. Three months after smoking cessation, many of these changes are reversed except in those patients who have developed airway damage from long-term airway inflammation.
Airway diameter. Airway diameter is reduced acutely with smoking as a result of reflex bronchoconstriction in response to inhaled particles and the increased mucous production already described. Airway narrowing is greatest in those subjects with known bronchial hypersensitivity such as asthmatics. Long-term small airway inflammation causes chronic airway narrowing that has a multitude of effects on lung function. Airway narrowing promotes premature airway closure during expiration, which results in an increase in closing volume and disturbed ventilation/perfusion relationships. Distribution of inspired gas as indicated by the single-breath nitrogen test (page 123) is therefore often abnormal in smokers. Small airway narrowing over many years gives rise to a progressive reduction in the forced expiratory volume in one second (FEV1) described below. Many of these changes are at an advanced stage before smokers develop respiratory symptoms.
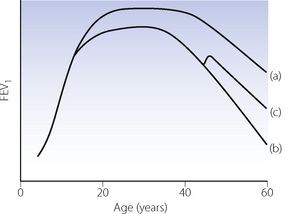
Ventilatory capacity.11 FEV1 normally reaches a peak in early adulthood, remains constant for some years, and then declines steadily as the subject grows older (Figure 21.1). Longitudinal studies of FEV1 in smokers reveal a very different picture, illustrated in Figure 21.1. Most smokers begin smoking in early adulthood, and the rate of increase of FEV1 immediately slows, resulting in a delayed and lower plateau. The plateau in FEV1 is also shorter, before a more rapid decline begins. Smoking cessation is followed by a small improvement in FEV1, followed by a return to the normal rate of decline, but rarely demonstrates a return to non-smoker values. Eventually, this decline in lung function results in lung pathology, with one in every five smokers developing COPD.
Passive Smoking
A non-smoker is exposed to all the constituents of tobacco smoke whilst indoors in the presence of smokers. Exposure varies with many factors, including size and ventilation of the room, number of people smoking and absorption of smoke constituents on soft furnishings and clothing. Carbon monoxide concentrations of 20 ppm have been reported, which is above the recommended environmental concentration (see below). ‘Side-stream’ smoke from a smouldering cigarette stub produces greater quantities of potentially noxious substances than ‘main-stream’ smoke produced when a cigarette burns in a stream of air drawn through it during a puff. On average, ‘side-stream’ smoke is generated during 58 seconds in each minute of cigarette smoking and this is not included in the measured yield of a cigarette.
Evidence for adverse health effects of passive smoking is now convincing: passive smoking by adults has been linked to lung cancer, cardiovascular disease, asthma and COPD.12
Maternal smoking. Infants whose mothers smoke during pregnancy have low birthweight, are more likely to be born prematurely and are at greater risk of sudden infant death syndrome (page 256). Up to two years of age, infants with smoking parents are more prone to lower respiratory tract illnesses and episodes of wheezing, and when older they have reduced lung volumes, higher carboxyhaemoglobin levels and a greater likelihood of developing asthma.11,13 It is uncertain whether this results from passive smoking in utero or from post-natal exposure to tobacco smoke in the home, but evidence for an in utero contribution is mounting, mediated by reduced innate immunity in the child born to a mother who smokes.12,13 Studies have found reduced expiratory flow rates and other markers of impaired lower airway function in neonates even before they leave hospital when their exposure to atmospheric cigarette smoke begins.14 The same finding in neonates born on average 7 weeks before their expected date15 indicates that maternal smoking adversely affects lung development at a crucial stage when terminal airways and alveoli are being formed (page 250). The increased risk of lower respiratory tract illness in passive-smoking infants is believed to result from smaller airway calibre at birth causing a greater propensity to airway closure with the normal infective or allergic challenges of infancy.14 After a few years of normal growth, airway size increases sufficiently to reduce symptoms, and the child ‘grows out’ of their susceptibility to respiratory illness, though their lung function remains worse than children who did not have lower airways disease in early life.16
Smoking and Perioperative Complications17,18,19
The increased sensitivity of the airway to inhaled irritants seen in smokers causes a greater incidence of adverse events such as cough, breath hold or laryngospasm on induction of general anaesthesia, even in passive smokers.20 These complications are not necessarily associated with decreases in oxygen saturation, though episodes of desaturation in the recovery period may be more common in smokers, or even in passively smoking children.21 It is worth noting that commercially available pulse oximeters record carboxyhaemoglobin as if it were oxyhaemoglobin (page 210), and so will consistently overestimate oxygen saturation in recent smokers.
There is ample evidence that smokers have an increased incidence of postoperative pulmonary complications (PPCs), which in comparison to non-smokers are several times more common in smokers,19,22 depending on the definitions used and type of surgery undertaken. This is attributable both to increased secretion and impaired clearance of mucus and to small airway narrowing. Almost all studies of the perioperative effects of smoking have been undertaken in patients having major surgery, usually coronary artery revascularisation or upper abdominal surgery. The high incidence of respiratory complications in this group makes them an ideal study population, but there remains little information regarding the respiratory effects of perioperative smoking and more minor surgery.
Preoperative smoking cessation is vital. Nicotine, which is responsible for many untoward cardiovascular changes, has a half-life of only 30 minutes, whilst carboxyhaemoglobin has a half-life of 4 hours when breathing air. A smoking fast of just a few hours will therefore effectively remove the risks associated with carbon monoxide and nicotine. The duration of smoking abstinence required to reduce the high incidence of PPCs is controversial.18,19 Some studies, mostly in patients having cardiac surgery, found a greater incidence of PPC in patients who stopped smoking for less than 8 weeks compared with those who continued smoking until the day before surgery. Other studies have failed to demonstrate this effect, and there are concerns that the original investigations had insufficient statistical power to conclusively prove a benefit to continuing to smoke before major surgery.18 Current advice is therefore that smokers should always strive to stop smoking preoperatively, and that the longer the period of cessation the greater will be the benefit in terms of avoiding PPCs.
Mechanisms of Smoking Related Lung Damage23
Many of the compounds present in cigarette smoke have direct irritant and toxic effects on the lungs. There are three other mechanisms by which lung damage occurs.
Oxidative Injury24
There is compelling evidence that oxidative injury, including peroxidation of membrane lipids, is an important component of the pulmonary damage caused by cigarette smoke.
Direct oxidative damage. The tar phase contains quinone, the semiquinone free radical and hydroquinone in a polymeric matrix, and the gas phase contains nitric oxide. These compounds can reduce oxygen in the body, to yield the superoxide free radical and thence the highly damaging hydroxyl free radical (Figure 26.3).
Cell-mediated oxidative damage. This results from smoking-induced activation of, or enhancement of, neutrophil and macrophage activity in the respiratory tract. Bronchoalveolar lavage in humans has shown that smokers have larger numbers of intra-alveolar macrophages and also significant numbers of neutrophils that are not normally present in non-smokers.25 It is the particulate component of smoke that is responsible for the recruitment and activation of neutrophils in the alveoli. This suggests that the interaction of particulate matter and alveolar macrophages releases a neutrophil chemattractant and that neutrophils are subsequently activated to release either proteases or reactive oxygen species. This activation may be a direct response to cigarette smoke or may represent excessive reactive oxygen species production in response to minor infective challenge in smokers.
Evidence of in vivo oxidative stress in smokers is based mainly on measures of antioxidant activity in both the lungs and blood. Compared with non-smokers, human smokers have reduced levels of vitamin E in alveolar fluid, reduced plasma concentrations of vitamin C, and greatly increased superoxide dismutase and catalase activity in alveolar macrophages. These abnormalities of oxidant-antioxidant activity are being used to try and find therapeutic agents that may mitigate the damage done by smoking.24
Carcinogenesis
Smoking contributes to the development of cancer in many organs, but the respiratory tract clearly receives the greatest exposure to tobacco smoke carcinogens, and this topic is described in more detail on page 439. There are two groups of compounds with carcinogenic activity, found mostly in the tar of the particulate phase. Some hydrocarbons, in particular polynuclear aromatic hydrocarbons (PAH), are carcinogenic, whilst others such as aromatic phenols (phenol, indole and catechol) are cocarcinogens and tumour promoters, without which the carcinogenic compounds are relatively innocuous. Tobacco-related nitrosamines and nicotine derivatives are also carcinogenic and, because of their ease of absorption into the blood, are responsible for cancer formation not only in the respiratory tract and oesophagus but also in more distant organs such as the pancreas. Knowledge about these carcinogens has led to many attempts to reduce their concentration in smoke by modifying the cigarette, and tar levels in cigarettes have declined almost three-fold since 1955. However, these changes have had little impact on the incidence of lung cancer (page 438) and smoking cessation remains the best way of avoiding all smoking-related cancers.
Immunological Activation26
Smokers have elevated serum IgE levels compared with non-smokers, the cause of which is uncertain but may be two-fold. Direct toxicity and oxidative cell damage result in greater airway mucosal cell permeability, allowing better access for allergens to underlying immunologically active cells. Smoking also increases the activity of some T-lymphocyte subsets that are responsible for producing interleukin-4, a cytokine well known for stimulating IgE production, and is known to produce a long term systemic inflammatory response.
Air Pollution27
Detrimental effects of air pollution were first recognised in the 13th century, though it is only in the last 50 years that effective control of pollution has been achieved. In spite of these controls, increased overall energy requirements and the internal combustion engine have ensured that air pollution remains a current problem. At the same time as levels of pollutants have been reduced in many parts of the world evidence of their harmful effects on health has increased, and air pollution remains a global problem.28 Many of the pollutants described below, like tobacco smoke, create oxidative stress systemically29 and therefore have effects not only on the lungs but are also implicated in causing cardiovascular diseases.30 Recent epidemiological research has found significant effects of air pollution on lung function in children, showing that the closer children live to busy roads the slower is the rate at which their forced expiratory volume in one second (FEV1) increases as they grow,31 and the more likely they are to develop respiratory disease, including asthma.32,33,34 Similar observations have been made relating lung function in adults with proximity to road traffic.35 Fine particulate matter (see below) seems to be the main pollutant responsible for the increased mortality,36 and recent work has found that some individuals are genetically susceptible to these effects on long term health.34,37 These recent developments have led to renewed calls to further reduce ‘acceptable’ levels of all air pollutants.38
Sources of Pollutants
Primary pollutants are substances that are released into the atmosphere directly from the polluting source, and are mostly derived from the combustion of fossil fuels. Petrol engines that ignite the fuel in an oxygen-restricted environment produce varying quantities of carbon monoxide, nitrogen oxides and hydrocarbons such as benzene and polycyclic aromatic compounds. All of these pollutants are reduced by the use of a catalytic converter. In contrast, diesel engines burn fuel with an excess of oxygen and so produce little carbon monoxide but more nitrogen oxides and particulate matter. Burning of coal and oil is now restricted almost entirely to power generation, and the pollutants produced depend on the type of fuel used and the amount of effort expended on ‘cleaning’ the emissions. However, particulates and nitrogen oxides are invariably produced, and this remains the major source of sulphur dioxide.
Secondary pollutants are formed in the atmosphere from chemical changes to primary pollutants. Nitric oxide produced from vehicle engines is quickly converted to nitrogen dioxide, and in doing so may react with ozone, reducing the atmospheric concentration of the latter. Alternatively, when exposed to sunlight in the lower atmosphere both NO and NO2 react with oxygen to produce ozone (O3).
Meteorological conditions influence air pollution. In conditions of strong wind, pollutants are quickly dispersed; in cloudy weather the development of secondary pollutants is unlikely. Ground level pollution in urban areas is exacerbated by clear, calm weather when ‘temperature inversion’ can occur. On a clear night, heat is lost from the ground to the atmosphere by radiation and the ground level air cools dramatically (Figure 21.2A). At dawn the ground is quickly heated by the sun’s radiation and warms the air, which lifts a blanket of cool air to approximately 50–100 m high. Because in still conditions mixing of air masses is slow to occur, the relatively cold air sits on top of the warm air below. In the meantime, the morning rush hour produces large amounts of pollutants that are unable to disperse and become trapped near the ground (Figure 21.2B).
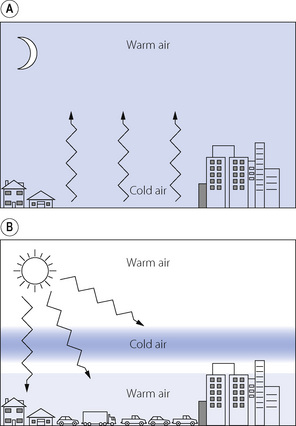
Fig. 21.2 Temperature inversion producing pollution in the morning rush hour. (A) At night, the ground loses heat to the atmosphere by radiation and ground level air cools. (B) In the morning, with strong sun and still conditions, the ground heats up quickly and displaces the blanket of cold air upwards, so preventing effective air mixing and trapping vehicular pollution at ground level.
Respiratory Effects of Pollutants27,39
Recommended maximum levels of common pollutants are shown in Table 21.1. The extent to which these levels are achieved varies greatly between different countries and from year to year.
Table 21.1 World Health Organization air quality guidelines40
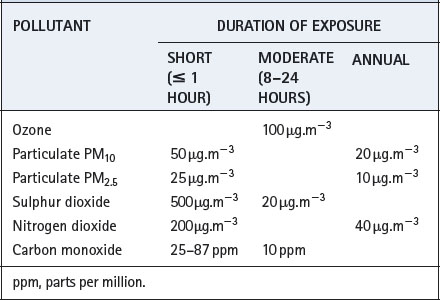
Carbon monoxide (CO) is found in the blood of patients, in trace concentrations, as a result of its production in the body, but mainly as a result of smoking and air pollution. The amount of carboxyhaemoglobin formed when breathing air polluted with CO will depend on the subject’s minute volume. One study reported carboxyhaemoglobin levels of 0.4–9.7% in London taxi drivers but the highest level in a non-smoking driver was 3%.41 Recommended levels shown in Table 21.1 are calculated to result in a carboxyhaemoglobin concentration of less than 2.5% even during moderate exercise. Carbon monoxide levels similar to those seen in smokers are only likely to occur during severe outdoor pollution episodes, though indoor pollution with CO may be more common (see below).
Nitrogen dioxide is mainly a primary pollutant, but a small amount is produced from nitric oxide. In the UK, about half of atmospheric NO2 is derived from vehicles. Indoor levels of NO2 commonly exceed outdoor levels, and the respiratory effects of NO2 are therefore described in the next section.
Ozone is a secondary pollutant formed by the action of sunlight on nitrogen oxides, and therefore highest levels tend to occur in rural areas downwind from cities and roads. In all areas, the dependence on sunlight means that ozone levels slowly increase throughout the day reaching peak levels shortly after the evening rush hour. Ozone is toxic to the respiratory tract, with effects being dependent on both concentration and duration of exposure. Exposure to concentrations of 200 μg.m−3 for just a few hours commonly causes throat irritation, chest discomfort and cough, resulting from both direct stimulation of irritant receptors in the airway and activation of inflammatory pathways. Bronchoconstriction may occur accompanied by a decrease in FEV1, and exercise capacity is limited. There is a large variability between individuals in their spirometric response to ozone, with approximately 10% of subjects having a severe response. This variability in response is partly a result of differing genetic susceptibility.27 It is interesting that laboratory studies have failed to demonstrate that asthmatic subjects are more susceptible to ozone-induced pulmonary symptoms. Even so, there is good evidence that high ozone concentrations are associated with increased hospital attendance, respiratory problems (particularly in children42) and with an increased risk of death from respiratory causes.43
Sulphur dioxide. Declining use of coal has substantially reduced the production of sulphur dioxide in recent years, and two-thirds of production in the UK now originates from oil-burning power stations. Normal atmospheric levels have no short-term effect on healthy subjects, but asthmatic patients may develop bronchoconstriction at between 100 and 250 ppb.
Particulate matter consists of a mixture of soot, liquid droplets, recondensed metallic vapours and organic debris. The disparate nature of particulate pollution reflects its very varied origins, but in the urban environment, diesel engines are a major source. Only particles of less than 10 μm diameter are considered to be ‘inhalable’ into the lung (page 220), so particulate pollution is measured as the concentration of particles less than this diameter, known as PM10. Particulate matter is further sub-divided into:
 Coarse particles, between 2.5–10 μm diameter, make up a smaller proportion of PM10 than the other particles and are less well studied, but are still believed to contribute significantly to the adverse health effects of particulate pollution.44 Fine particles, or PM2.5, are less than 2.5 μm in diameter, the most numerous particle present in air pollution, and responsible for most of the respiratory effects of particulate pollution. Ultrafine particles are carbon particles less than 0.1 μm in size.45 Their small size means they should be breathed in and out without being trapped by the airway lining fluid (page 220). However, some particles are retained in the lung, where they remain in the long term, probably contained within macrophages, and without any evidence of systemic absorption. Their contribution to the health effects of particulate pollution is uncertain.
Coarse particles, between 2.5–10 μm diameter, make up a smaller proportion of PM10 than the other particles and are less well studied, but are still believed to contribute significantly to the adverse health effects of particulate pollution.44 Fine particles, or PM2.5, are less than 2.5 μm in diameter, the most numerous particle present in air pollution, and responsible for most of the respiratory effects of particulate pollution. Ultrafine particles are carbon particles less than 0.1 μm in size.45 Their small size means they should be breathed in and out without being trapped by the airway lining fluid (page 220). However, some particles are retained in the lung, where they remain in the long term, probably contained within macrophages, and without any evidence of systemic absorption. Their contribution to the health effects of particulate pollution is uncertain.Acute effects of particles on lung function again include airway irritation and small reductions in lung volumes such as FEV1 and FVC.46 It is, however, associations between PM10 levels and overall mortality that have been the focus of much research. Even when smoking habits are taken into account, particulate pollution is associated with an increased risk of death from lung cancer or other cardiopulmonary diseases.27 Particulate pollution has widespread pro-inflammatory effects on lung epithelial cells and alveolar macrophages, causing inflammatory responses both locally, in the lung, and in distant sites where activation of clotting pathways may explain PM10-induced increases in death from cardiovascular disease.
Indoor Air Pollution47
Worldwide, the most common form of indoor air pollution is smoke produced by fires used for cooking. Burning biomass fuels produces large amounts of particulate matter, containing smaller particle sizes than diesel engines. As for the outdoor pollution described above these pollutants are associated with the development of COPD,48 and also respiratory infections in children.49
In the developed world energy-efficient homes have become the norm in recent years, with effective heating systems and extensive insulation. This has led to dramatic changes in indoor air quality, including warmer temperatures, higher humidity levels and reduced ventilation. It is estimated that most people spend in excess of 80% of their time indoors, so indoor air pollution may have a considerable impact on public health. The respiratory effects of passive smoking are described above (page 319), and the impact of environmental radon exposure on lung cancer are discussed on page 439.
Indoor air quality generally reflects that of the outdoor air except that ozone levels are invariably low indoors due to the rapid reaction of ozone with the synthetic materials that make up much of the indoor environment. In addition to pollutants from outside, there are three specific indoor pollutants.
Allergens. Warm moist air, poor ventilation and extensive floor coverings provide ideal conditions for house dust mite infestation and the retention of numerous other allergens. This is believed to contribute to the recent upsurge in the prevalence of atopic diseases such as asthma, and is discussed in Chapter 28.
Carbon monoxide.50 Malfunctions of heating equipment in the home may release CO into the indoor environment. Acute CO poisoning from this cause is common, but the occurrence of prolonged low-level exposure to indoor CO may be underestimated. Headache, malaise and flu-like symptoms are all features of long-term CO poisoning, though these symptoms are believed to be completely reversible once the exposure to CO is stopped. Smokers, who have permanently elevated carboxyhaemoglobin levels, appear to be resistant to these symptoms.
Nitrogen dioxide. Gas-fired cookers, stoves and boilers all produce NO2, the amount being dependent on the arrangements for waste gas exclusion.51 In this respect, gas cookers are the worst culprits as they are rarely associated with chimneys and flues, and normally discharge their waste gases directly into the kitchen atmosphere. During cooking, NO2 levels may reach over 750 μg.m−3, which is well in excess of outdoor pollution targets (Table 21.1). Mild airway irritant effects are seen at levels of around 550 μg.m−3 in asthmatic subjects, or at 1800 μg.m−3 in non-asthmatic subjects,52 so acute effects are probably uncommon. However, long-term exposure does seem to be clinically significant by, for example, causing worsening of asthma symptoms in children.53
References
1. Doll R, Hill AB. Smoking and carcinoma of the lung. BMJ. 1950;2:739-748.
2. Anderson DO, Ferris BG. Role of tobacco smoking in the causation of chronic respiratory disease. N Engl J Med.. 1962;267:787-794.
*3. Frieden TR, Bloomberg MR. How to prevent 100 million deaths from tobacco. Lancet. 2007;369:1758-1761.
4. Ashton H, Stepney R, Thompson PW. Self-titration by cigarette smokers. BMJ. 1979;2:357-360.
5. Anthonisen NR. “Susceptible” smokers? Thorax. 2006;61:924-925.
6. Pierrou S, Broberg P, O’Donnell RA, et al. Expression of genes involved in oxidative stress responses in airway epithelial cells of smokers with chronic obstructive pulmonary disease. Am J Respir Crit Care Med.. 2007;175:577-586.
7. Wanner A, Salathe M, O’Riordan TG. Mucociliary clearance in the airways. Am J Respir Crit Care Med.. 1996;154:1868-1902.
8. Erskine RJ, Murphy PJ, Langton JA. Sensitivity of upper airway reflexes in cigarette smokers: effect of abstinence. Br J Anaesth.. 1994;73:298-302.
9. Dicpinigaitis PV. Cough reflex sensitivity in cigarette smokers. Chest. 2003;123:685-688.
10. Dicpinigaitis PV, Sitkauskiene B, Stravinskaite K, Appel DW, Negassa A, Sakalauskas R. Effect of smoking cessation on cough reflex sensitivity. Eur Respir J. 2006;28:786-790.
11. Samet JM, Lange P. Longitudinal studies of active and passive smoking. Am J Respir Crit Care Med.. 1996;154:S257-S265.
12. Eisner MD, Forastiere F. Passive smoking, lung function, and public health. Am J Respir Crit Care Med.. 2006;173:1184-1185.
13. Le Souëf PN. Adverse effects of maternal smoking during pregnancy on innate immunity in infants. Eur Respir J. 2006;28:675-677.
14. Morgan WJ, Martinez FD. Maternal smoking and infant lung function: Further evidence for an in utero effect. Am J Respir Crit Care Med.. 1998;158:689-690.
15. Hoo AF, Henschen M, Dezateux C, Costeloe K, Stocks J. Respiratory function among preterm infants whose mothers smoked during pregnancy. Am J Respir Crit Care Med.. 1998;158:700-705.
16. Martinez FD, Wright AL, Taussig LM, et al. Asthma and wheezing in the first six years of life. N Engl J Med.. 1995;332:133-138.
17. Nel MR, Morgan M. Smoking and anaesthesia revisited. Anaesthesia. 1996;51:309-311.
*18. Warner DO. Perioperative abstinence from cigarettes. Physiologic and clinical consequences. Anesthesiology. 2006;104:356-367.
19. Tønnesen H, Nielsen PR, Lauritzen JB, Møller AM. Smoking and alcohol intervention before surgery: evidence for best practice. Br J Anaesth.. 2009;102:297-306.
20. Schwilk B, Bothner U, Schraag S, Georgieff M. Perioperative respiratory events in smokers and nonsmokers undergoing general anaesthesia. Acta Anaesthesiol Scand.. 1997;41:348-355.
21. Lyons B, Frizelle H, Kirby F, Casey W. The effect of passive smoking on the incidence of airway complications in children undergoing general anaesthesia. Anaesthesia. 1996;51:324-326.
22. Møller AM, Maaløe R, Pedersen T. Postoperative intensive care admittance: The role of tobacco smoking. Acta Anaesthesiol Scand.. 2001;45:345-348.
23. Yanbaeva DG, Dentener MA, Creutzberg EC, Wesseling G, Wouters EFM. Systemic effects of smoking. Chest. 2007;131:1557-1566.
24. Kinnula VL. Focus on antioxidant enzymes and antioxidant strategies in smoking related airway diseases. Thorax. 2005;60:693-700.
25. Hunninghake GW, Crystal RG. Cigarette smoking and lung destruction. Accumulation of neutrophils in the lungs of cigarette smokers. Am Rev Respir Dis.. 1983;128:833-838.
26. Villar MTA, Holgate ST. IgE, smoking and lung function. Clin Exp Allergy. 1995;25:206-209.
*27. Brunekreef B, Holgate ST. Air pollution and health. Lancet. 2002;360:1233-1242.
28. Thurston G. Air pollution, human health, climate change and you. Thorax. 2007;62:748-749.
29. Romieu I, Castro-Giner F, Kunzli N, Sunyer J. Air pollution, oxidative stress and dietary supplementation: a review. Eur Respir J. 2008;31:179-196.
30. Dockery DW, Stone PH. Cardiovascular risks from fine particulate air pollution. N Engl J Med.. 2007;356:511-513.
31. Gauderman WJ, Vora H, McConnell R, et al. Effect of exposure to traffic on lung development from 10 to 18 years of age: a cohort study. Lancet. 2007;369:571-577.
32. Jerrett M. Does traffic-related air pollution contribute to respiratory disease formation in children? Eur Respir J. 2007;29:825-826.
33. Morgenstern V, Zutavern A, Cyrys J, et al. Atopic diseases, allergic sensitization, and exposure to traffic-related air pollution in children. Am J Respir Crit Care Med.. 2008;177:1331-1337.
34. Sandström T, Kelly FJ. Traffic-related air pollution, genetics and asthma development in children. Thorax. 2009;64:98-99.
35. Holguin F. Traffic related exposures and lung function in adults. Thorax. 2007;62:837-838.
36. Pope CA, Ezzati M, Dockery DW. Fine-particulate air pollution and life expectancy in the United States. N Engl J Med.. 2009;360:376-386.
37. Baccarelli A. Breathe deeply into your genes! Genetic variants and air pollution effects. Am J Respir Crit Care Med.. 2009;179:431-432.
38. Gauderman WJ. Air pollution and children – an unhealthy mix. N Engl J Med.. 2006;355:78-79.
39. Committee on the Medical Effects of Air Pollution. The Quantification of the Effects of Air Pollution on Health in the United Kingdom. London: The Stationery Office; 1998.
40. World Health Organization. WHO Air Quality Guidelines for Particulate Matter, Ozone, Nitrogen Dioxide and Sulfur Dioxide, Global update 2005. Summary of risk assessment. Geneva: WHO. 2006.
41. Jones RD, Commins BT, Cernik AA. Blood lead and carboxyhaemoglobin levels in London taxi drivers. Lancet. 1972;2:302-303.
42. Pinkerton KE, Balmes JR, Fanucchi MV, Rom WN. Ozone a malady for all. Am J Respir Crit Care Med.. 2007;176:107-112.
43. Jerrett M, Burnett RT, Pope CA, et al. Long-term ozone exposure and mortality. N Engl J Med.. 2009;360:1085-1095.
44. Brunekreef B, Forsberg B. Epidemiological evidence of effects of coarse airborne particles on health. Eur Respir J. 2005;26:309-318.
45. Möller W, Felten K, Sommerer K, et al. Deposition, retention, and translocation of ultrafine particles from the central airways and lung periphery. Am J Respir Crit Care Med.. 2008;177:426-432.
46. Lippmann M. Health effects of airborne particulate matter. N Engl J Med.. 2007;357:2395-2397.
*47. Samet JM, Spengler JD. Indoor environments and health: Moving into the 21st century. Am J Public Health. 2003;93:1489-1493.
48. Liu Y. Where there’s smoke there’s lung disease. Thorax. 2007;62:838-839.
49. Emmelin A, Wall S. Indoor air pollution. A poverty-related cause of mortality among the children of the world. Chest. 2007;132:1615-1623.
50. Townsend CL, Maynard RL. Effects on health of prolonged exposure to low concentrations of carbon monoxide. Occup Environ Med.. 2002;59:708-711.
51. Fuhlbrigge A, Weiss S. Domestic gas appliances and lung disease. Thorax. 1997;52(supp 3):S58-S62.
52. Committee on the Medical Effects of Air Pollutants. Handbook on Air Pollution and Health. London: The Stationery Office; 1997.
53. Belanger K, Gent JF, Triche EW, Bracken MB, Leaderer BP. Association of indoor nitrogen dioxide exposure with respiratory symptoms in children with asthma. Am J Respir Crit Care Med.. 2006;173:297-303.