Chapter 12 Non-respiratory functions of the lung
 The entire cardiac output passes through the pulmonary circulation, so the lungs act as a filter, preventing emboli from passing to the left side of the circulation.
The entire cardiac output passes through the pulmonary circulation, so the lungs act as a filter, preventing emboli from passing to the left side of the circulation.The lungs are primarily adapted for the purpose of gas exchange, and to achieve this with such efficiency almost the entire blood volume passes through the lungs during a single circulation. This characteristic makes the lungs ideally suited to undertake many other important functions. The location of the lungs within the circulatory system is ideal for its role as a filter to protect the systemic circulation, not only from particulate matter but also from a wide range of chemical substances that undergo removal or biotransformation in the pulmonary circulation. The pulmonary arterial tree is well adapted for the reception of emboli without resultant infarction, and the very large area of endothelium gives the lung a metabolic role out of proportion to its total mass. This large interface between the external atmosphere and the circulation is not without its own hazards, and the lung must protect the circulation from many potentially harmful inhaled substances.
Filtration
Sitting astride the whole output of the right ventricle, the lung is ideally situated to filter out particulate matter from the systemic venous return. Without such a filter, there would be a constant risk of particulate matter entering the arterial system, where the coronary and cerebral circulations are particularly vulnerable to damaging emboli.
Pulmonary capillaries have a diameter of about 7 μm, but this does not appear to be the effective pore size of the pulmonary circulation when considered as a filter. There is no clear agreement on the maximal diameter of particles that can traverse the pulmonary circulation. Animal studies have demonstrated the passage through perfused lungs of glass beads up to 500 μm.1 It is well known that small quantities of gas and fat emboli may gain access to the systemic circulation in patients without intracardiac shunting. Emboli may bypass the alveoli via some of the pre-capillary anastomoses that are known to exist in the pulmonary circulation (page 99), though the functional role of these anastomoses remains uncertain. More extensive invasion of the systemic arteries may occur in the presence of an overt right-to-left intracardiac shunt, which is now known to be quite common. Post mortem studies show that over 25% of the population have a ‘probe-patent’ foramen ovale, usually in the form of a slit-like defect that acts as a valve, and which is therefore normally kept closed by the left atrial pressure being slightly greater than the right.2 In 10% of normal subjects, a simple Valsalva manoeuvre or cough results in easily demonstrable blood flow between the right and left atria.3 Paradoxical embolism may therefore result from a relative increase in right atrial pressure caused by physiological events or pulmonary embolus (Chapter 29).
So far as the survival of the lung is concerned, the geometry of the pulmonary microcirculation is particularly well adapted to maintaining alveolar perfusion in the face of quite large degrees of embolisation. However, a significant degree of embolisation inevitably blocks the circulation to parts of the lung, disturbing the balance between ventilation and perfusion. This situation is considered in Chapter 29. Pulmonary microembolism with small clumps of fibrin and/or platelets will not have a direct effect on gas exchange until it is very extensive. Plugging of pulmonary capillaries by microemboli does, however, initiate neutrophil activation in the area, leading to an increase in endothelial permeability and alveolar oedema, and has been implicated in the aetiology of acute lung injury (Chapter 31).
Thrombi are cleared more rapidly from the lungs than from other organs. The lung possesses well-developed proteolytic systems not confined to the removal of fibrin. Pulmonary endothelium is known to be rich in plasmin activator, which converts plasminogen into plasmin, which in turn converts fibrin into fibrin degradation products. However, the lung is also rich in thromboplastin, which converts prothrombin to thrombin. To complicate the position further, the lung is a particularly rich source of heparin, and bovine lung is used in its commercial preparation. The lung can thus produce high concentrations of substances necessary to promote or delay blood clotting and also for fibrinolysis. Apart from the lung’s ability to clear itself of thrombo-emboli, it may play a role in controlling the overall coagulability of the blood.
Defence against Inhaled Substances
The skin, gastrointestinal tract and lungs form the major interfaces between the outside world and the carefully controlled internal body systems. Efficient gas exchange in the lung requires a physically very thin interface between air and blood, which leaves the lung vulnerable to invasion by many airborne hazards, both chemical and biological. These are almost entirely prevented from reaching the distal airways by the airway lining fluid found throughout the tracheobronchial tree.
Airway Lining Fluid
Within the airway lining fluid there are two distinct layers,4,5 a periciliary or ‘sol’ layer which is of low viscosity containing water and solutes and in which the cilia are embedded, and a mucous or ‘gel’ layer above.
Mucous layer. Large airways are completely lined by a mucous layer, whilst in smaller, more distal, airways the mucous is found in ‘islands’, and a mucous layer is absent in small bronchioles and beyond. Mucous is composed mostly of glycoproteins called mucins,6,7 which determine the visco-elastic and other properties of the mucous. Mucin is released by rapid (<150 ms) exocytosis from the mucous secreting goblet cells in response to a range of stimuli including direct chemical irritation, inflammatory cytokines8 and neuronal stimulation, predominantly by cholinergic nerves.9,10 Mucins have a core composed of glycoprotein subunits joined by disulphide bonds and their length may extend up to 6 μm. The core is 80% glycosylated with side chains attached via O-glycosidic bonds. Almost all terminate in sialic acid and possess micro-organism binding sites. Mucous plays a vital role in pathogen entrapment and removal, and also has a variety of antimicrobial actions (see below).
Ciliary function.11,12 The mucous layer is propelled cephalad by the ciliated epithelial cells (Figure 12.1) at an average rate of 4 mm.min−1, to be removed by expectoration or swallowing on reaching the larynx and pharynx. The cilia beat mostly within the low-viscosity periciliary layer of airway lining fluid, with the cilia tips intermittently gripping the underside of the mucous layer, so propelling the mucous layer along the airway wall.

Fig. 12.1 Scanning electron micrograph of ciliated epithelial cells beneath the mucous (Mu) lining the larger airways.
(Kindly reproduced by permission of Dr P. K. Jeffery, Imperial College School of Science, Technology and Medicine, London and the publishers of Respiratory Medicine.13)
Cilial beat frequency is 12–14 beats per second and can be affected by pollutants, smoke, anaesthetic agents and infection. Two phases occur for each beat (Figure 12.2). First is the recovery stroke which occupies 75% of the time of each cycle and involves a slow bowing movement away from the resting position by a sideways action of the cilium. There then follows the effective stroke in which the cilium extends to its full height, gripping the mucous layer above with claws on its tip, before moving forward in a plane perpendicular to the cell below and returning to its resting position. Adjacent cilia somehow coordinate their strokes to produce waves of activity that move the mucous layer along, probably by a physical effect of cilia stimulating adjacent cilia during the sideways sweep of the recovery stroke.
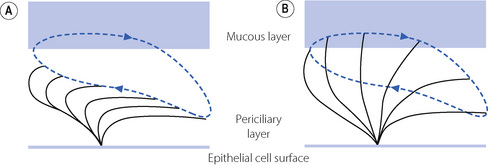
Fig. 12.2 Mechanism of action of a single cilium on the respiratory epithelium. (A) Recovery stroke in which the cilium bows backwards and sideways within the low-viscosity periciliary layer. (B) Effective stroke in which the cilium extends perpendicular to the epithelial cell into the mucous layer and propels it forwards. The blue line shows the trajectory of the cilium tip.
Periciliary layer. For this propulsion system to work effectively, it is crucial that the depth of the periciliary fluid layer be closely controlled,4,5 particularly considering the increasing amount of mucus that will occur each time two smaller airways converge into one larger airway. The depth of both layers of the airway lining fluid is controlled by changes in the volume of secretions and the speed of their re-absorption. If the periciliary layer reduces in depth, the gel layer will compensate for this by donating liquid to the periciliary layer to maintain the correct depth of fluid, an effect probably mediated by simple osmotic gradients between the two layers. The mucous layer may donate fluid to the periciliary layer until its volume is diminished by 70%. The reverse happens as the mucus converges on the larger airways, with the mucous layer absorbing excess periciliary water. The volume of periciliary fluid is therefore effectively determined by its salt concentration, which is in turn controlled by active ion transport on the surface of the epithelial cells. The ion channels responsible for active control are amiloride sensitive Na+ and Cl− channels, the latter better known as the cystic fibrosis transmembrane regulator (CFTR) protein. CFTR is likely to be partially active at rest but is stimulated when Na+ channels are inhibited. The factors responsible for this critical system are unknown, though adenosine diphosphate release in response to cyclical airway movements may be involved.14
In patients with cystic fibrosis an inherited defect of CFTR leads to dysfunction in the regulation of airway lining fluid homeostasis, with severe consequences (Chapter 28).
Humidification. The airway lining fluid acts as a heat and moisture exchanger to humidify and warm inspired gas. During inspiration, relatively cool, dry air causes evaporation of surface water and cooling of the airway lining, then on expiration moisture condenses on the surface of the mucous and warming occurs. Thus only about one half of the heat and moisture needed to condition (fully warm and saturate) each breath is lost to the atmosphere. With quiet nasal breathing, air is conditioned before reaching the trachea, but as ventilation increases smaller airways are recruited until at minute volumes of over 50 l.min−1 airways of 1 mm diameter are involved in humidification.
Inhaled Particles
Where in the respiratory tract inhaled particles are deposited depends on both their size and the breathing pattern during inhalation. Three mechanisms cause deposition:
Aiding all these mechanisms is the high humidity within the respiratory tract. Absorption of water by the particle during its journey along the airways will increase the particle’s weight, and so encourage both inertial impaction and sedimentation to occur. Naturally this affects hygroscopic particles to a greater degree.
Defence against Inhaled Pathogens
As an interface with the outside environment the lung is exposed to a great many organisms carried by the ∼20 000 litres of air breathed each day. Pulmonary defence mechanisms have evolved to protect the respiratory tract from invasion by micro-organisms. They can be subdivided into direct removal of the pathogen, chemical inactivation of the invading organism and, if these fail, immune defences.
Direct removal of pathogens. With normal nasal breathing, a majority of inhaled pathogens impact on the nasal mucosa, which is swept backwards by the ciliated nasal epithelium and swallowed. At higher inspiratory flow rates, for example when dyspnoeic, pathogens will penetrate deeper into the airways and be trapped by the sticky mucous layer of the airway lining fluid, before being removed.
Chemical inactivation of pathogens.15 Airway lining fluid is more than a simple transport mechanism for impacted micro-organisms. Some smaller particles will penetrate far into the bronchial tree and take some time to be transported out of the respiratory tract. To prevent these organisms from causing damage during this time, the airway lining fluid contains multiple systems for directly killing pathogens. Surfactant, in addition to its role in reducing lung compliance (page 30), also acts as part of the innate defences in the lung.16 Surfactant protein A is the most active surfactant protein in pulmonary defence, its actions including stimulating macrophage migration, the production of reactive oxygen species, and the synthesis of immunoglobulins and cytokines. Both surfactant protein A and D are directly bactericidal,17 with activity against many common pulmonary pathogens. Lysozyme is also present in airway lining fluid. This enzyme, secreted by neutrophils, is capable of destroying microbial cell walls causing bacterial lysis, particularly for Gram-positive bacteria.
Finally, airway lining fluid contains a range of natural antimicrobial peptides referred to as defensins. These are small molecular weight (3–5 kD) peptides with a broad antimicrobial range, acting either directly on the bacterial cell wall or indirectly by stimulating respiratory epithelial cells to release chemokines to recruit inflammatory cells. α-defensins are present in the α-granules of neutrophils and have activity against a range of bacteria and the Herpes simplex virus. β-defensins originate from the epithelial cells and at least four human β-defensins (HBD) have been identified. HBD-1 is found in lung secretions in normal individuals while HBD-2 is found in secretions of cystic fibrosis patients as well as those with inflammatory lung disease. These small peptides contribute to inflammation and repair. Defective functioning of HBDs in the airway lining fluid is believed to be a major contributor to chronic airway infection in cystic fibrosis (Chapter 28).18
Protease/antiprotease system. Protease enzymes such as neutrophil elastase and metalloproteinases are normally released in the lung following activation of neutrophils or macrophages in response to pathogens or tobacco smoke. These enzymes are powerful anti-microbial molecules in the airway lining fluid, but if left unchecked, they will damage lung tissue. There are at least two mechanisms that protect the lung from damage by its own protease enzymes. First, the proteases are mostly confined to the mucous layer of the airway surface liquid, so avoiding close contact with underlying epithelial cells whilst being in close proximity to inhaled micro-organisms. Secondly, they are inactivated by conjugation with anti-protease enzymes present in the lung.19 Anti-protease enzymes active in the lung include α1-antitrypsin, α2-macroglobulin and α1-chymotrypsin. α1-antitrypsin is manufactured in the liver and transported to the lung. It constitutes a major proportion of antiprotease activity in the alveoli and is the most active inhibitor of neutrophil elastase.
Inactivation of such powerful protease enzymes presents a significant biochemical challenge, and the way that α1-antitrypsin achieves this has recently been elucidated.19,20 The α1-antitrypsin molecule exists in a semi-stable state, held together by a loop of amino acids that projects from the molecule with a pair of methionine-serine residues at its tip, which acts as a ‘bait’ for protease enzymes. When a protease binds the peptide loop, the α1-antitrypsin structure becomes unstable and rapidly flips the bound protease onto the other side of the molecule, an action that has been likened to a mousetrap. Once flipped to the other side of the molecule the protease becomes bound so tightly within a β-sheet of the α1-antitrypsin that it is effectively crushed, preventing the conformational changes required for its function.
In 1963 a group of patients were described whose plasma proteins were deficient in α1-antitrypsin and who had developed emphysema.21 The enzyme deficiency is inherited as an autosomal recessive gene, with 7.7% of people of European descent being carriers for one of the two common mutations of the α1-antitrypsin gene.19,22 Lower plasma levels of α1-antitrypsin in homozygous patients result not from failed production of α1-antitrypsin, but from failure to secrete the protein from hepatocytes. The retained α1-antitrypsin protein polymerises within the cell and leads to hepatic damage.20 About 1:3000 of the population are believed to be homozygous for the more severe Z mutation of the α1-antitrypsin gene, though many of these are believed to succumb to pulmonary and liver disease before the α1-antitrypsin deficiency is ever found.19,23 Homozygotes do form a higher proportion of patients with emphysema and estimates range from 3% to 26%. These patients tend to have basal emphysema, onset at a younger age and a severe form of the disease. It thus appears that α1-antitrypsin deficiency is an aetiological factor in a small proportion of patients with emphysema (page 411). Smoking, which increases neutrophil protease production (page 321), is associated with more severe lung disease in patients with a deficiency of α1-antitrypsin.23 Disturbances of the less well understood protease-antiprotease systems, such as the matrix metalloproteases group of enzymes, are now also believed to be involved in pathogenesis of a variety of inflammatory lung diseases.24
Immune systems. Humoral immunity is provided in the lung by immunoglobulins found in the airway lining fluid. IgA is the major type present in the nasopharyngeal area and large bronchi. Its role seems to be to prevent the binding of bacteria to the nasal mucosa, and specific IgA has the ability to act as an opsonin and induce complement. Deeper in the respiratory tract IgG is present in larger amounts, becoming the most prevalent immuno-globulin in the alveoli.
Cellular immunity involves the immunologically active epithelial cells and macrophages that are present in normal airways. In response to a variety of stimuli bronchial epithelial cells initiate an inflammatory response, and are probably also responsible for terminating the response and initiating tissue repair.25 This is done by secretion of numerous molecules:
 adhesion molecules (e.g. ICAM-1) to induce margination of inflammatory cells in nearby pulmonary capillaries cytokines (e.g. IL-1, IL-6, tumour necrosis factor) to amplify the inflammatory response by further stimulation of inflammatory cells growth factors (e.g. TGF-β, EGF) to stimulate the cells responsible for tissue repair such as fibroblasts
adhesion molecules (e.g. ICAM-1) to induce margination of inflammatory cells in nearby pulmonary capillaries cytokines (e.g. IL-1, IL-6, tumour necrosis factor) to amplify the inflammatory response by further stimulation of inflammatory cells growth factors (e.g. TGF-β, EGF) to stimulate the cells responsible for tissue repair such as fibroblastsOnce initiated, this response causes large numbers of phagocytic cells to enter the lung tissue. The presence of immunoglobulins, complement and other opsonins enhances the phagocytic cells’ recognition process. In severe infections, the reactive oxygen species used in the killing of micro-organisms by phagocytic cells may spill out of the lysosome and into the lung tissue, exacerbating the tissue injury.
In patients with asthma, the inflammatory cells responsible for airway inflammation are eosinophils and mast cells, while in those with chronic obstructive pulmonary disease and other forms of lung inflammation, neutrophils predominate.
Chemical Hazards
Many factors will influence the fate of inhaled chemicals:26
Particle size, as with biological particles described above, will affect where in the lung deposition occurs.
Water solubility. Once incorporated into the lung tissue, water solubility affects the rate at which chemicals are cleared from the lung, with water soluble substances taking longer than lipid soluble ones to be absorbed into the blood for disposal elsewhere.
Concentration of inhaled chemicals is important as metabolic activity within the lung is easily saturated.
Metabolism of inhaled chemicals is poorly understood in the human lung, and, though it has been extensively investigated in animals, there are known to be large species differences.26 Metabolic activity is found in all cell types of the respiratory mucosa, but in animals is particularly well developed in Clara cells and type II alveolar cells (page 23).26,27 As in the liver, metabolism of toxic chemicals involves two stages:
Metabolic changes to inhaled chemicals may not be beneficial, especially with many synthetic organic compounds and several chemicals in cigarette smoke (page 317). Bioactivation by phase I metabolism converts some quite innocuous compounds into potent carcinogens, while slightly different metabolic conversions may do the reverse.26,28 The balance between activating and inactivating pathways varies between species. What little data is available on human lungs indicates that we are fortunate in having a very favourable ratio, the inactivation of potential carcinogens being 100-fold greater than in rodents.26 Presumably, without this evolutionary advantage, the history of cigarette smoking would have been considerably different.
Processing of Endogenous Compounds by the Pulmonary Vasculature29
Hormones may pass through the lung unchanged, others may be almost entirely removed from the blood during a single pass, and some may be activated during transit (Table 12.1).
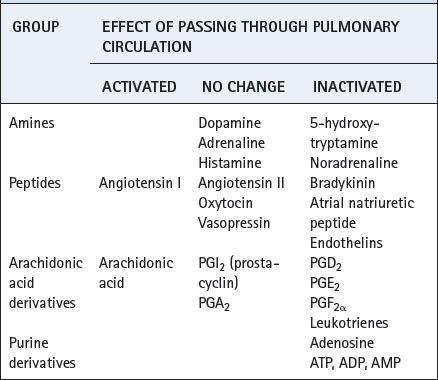
Of the many types of cell in the lungs, it is the endothelium that is most active metabolically. The most important location is the pulmonary capillary but it must be stressed that endothelium from a range of vessels throughout the body have been shown to possess a similar repertoire of metabolic processes. The extensive metabolic actions of the pulmonary endothelium take place in spite of the paucity of organelles that are normally associated with metabolic activity, in particular mitochondria and smooth endoplasmic reticulum or microsomes. Nevertheless, the caveolae result in a major increase in the already extensive surface area of these cells (about 126 m2),30 which is particularly advantageous for membrane bound enzymes.
Catecholamines and Acetylcholine
Noradrenaline (norepinephrine). There is a striking difference in the handling of noradrenaline and adrenaline. Although each catecholamine has a half-life of about 20 seconds in blood, some 30% of noradrenaline is removed in a single pass through the lungs,31 while adrenaline (and isoprenaline and dopamine) are unaffected. Monoamine oxidase and catechol-O-methyl transferase within the endothelial cells will metabolise all amine derivatives with equal efficiency. The specificity of pulmonary endothelium for noradrenaline therefore lies with the cell membrane, which selectively takes up only noradrenaline and 5-hydroxytrypt-amine.32 Extraneuronal uptake of noradrenaline is not confined to the endothelium of the lungs, but uptake by the pulmonary circulation (uptake 1) differs from extraneuronal uptake (uptake 2) in other tissues, which is less specific for noradrenaline.
5-Hydroxytryptamine (5-HT, serotonin) is removed very effectively by the lungs, up to 98% being removed in a single pass. There are considerable similarities to the processing of noradrenaline. 5-HT is taken up by the endothelium, mainly in the capillaries, and is then rapidly metabolised by monoamine oxidase. The half-life of 5-HT in blood is about 1–2 minutes and pulmonary clearance plays the major role in the prevention of its recirculation.
Histamine, dopamine and adrenaline (epinephrine) are not removed from blood on passing through the pulmonary circulation, in spite of the high concentrations of monoamine oxidase in lung tissue. Their removal from the circulation is limited by the lack of a transport mechanism across the blood/endothelium barrier.
Acetylcholine is rapidly hydrolysed in blood where it has a half-life of less than 2 seconds. This tends to overshadow any changes attributable to the lung, which nevertheless does contain acetylcholinesterases and pseudocholinesterases.
Peptides
Angiotensin. It has long been known that angiotensin I, a decapeptide formed by the action of renin on a plasma α2-globulin (angiotensinogen), was converted into the vasoactive octapeptide angiotensin II by incubation with plasma. Angiotensin-converting enzyme (ACE) is found free in the plasma, but is also bound to the surface of endothelium. This appears to be a general property of endothelium but ACE is present in abundance on the vascular surface of pulmonary endothelial cells, also lining the inside of the caveolae and extending onto the projections into the lumen. Some 80% of angiotensin I passing through the lungs is converted to angiotensin II in a single pass. Angiotensin converting enzyme is a zinc containing carboxypeptidase with two active sites, each located within a deep groove in the side of the protein.33 Binding sites in the groove attach the substrate firmly to the protein and the zinc moiety then cleaves either a phenylalanine-histidine bond (angiotensin I) or a phenylalanine-arginine bond (bradykinin). Drugs that inhibit ACE (see below) do so by becoming buried deep within the protein groove, simply covering the active site.33
Bradykinin, a vasoactive nonapeptide, is also very effectively removed during passage through the lung and other vascular beds. The half-life in blood is about 17 seconds but less than 4 seconds in various vascular beds. Like angiotensin I, ACE is the enzyme responsible for metabolism of bradykinin.
By its effects on bradykinin and angiotensin, ACE plays a crucial role in controlling arterial blood pressure. Bradykinin, which promotes blood vessel dilation and a lowering of blood pressure, is inactivated. Conversely, angiotensin II production results in a host of events that increase blood pressure such as renal sodium retention, vasoconstriction and release of noradrenaline. Drugs that inhibit ACE are now widely used in the treatment of cardiovascular disease. However, this also decreases the degradation of bradykinin by ACE, although other enzymes are capable of metabolising bradykinin, so allowing ACE inhibitors to exert their hypotensive effects.
Angiotensin II itself passes through the lung unchanged, as do vasopressin and oxytocin.
Atrial natriuretic peptide (ANP), is largely removed by the lung in many animal species. Methodological problems caused by the secretion of ANP from both left and right atria in humans led to uncertainty about the ability of human lungs to metabolise ANP. Studies using radiolabelled ANP have now shown that in humans, ANP is not metabolised by the lung to any significant extent.34
Endothelins, a group of 21 amino acid peptides with diverse biological activity (page 110) have a plasma half-life of just a few minutes, being cleared by the kidney, liver and lungs. The pulmonary enzymes responsible are not clearly defined, but there are believed to be several different types in humans.35
Arachidonic Acid Derivatives
The lung is a major site of synthesis, metabolism, uptake and release of arachidonic acid metabolites. The group as a whole are 20-carbon carboxylic acids, generically known as eicosanoids. The initial stages of eicosanoid synthesis involve the conversion, by phospholipase A2, of membrane phospholipids into arachidonic acid. Metabolism of arachidonic acid involves its oxygenation by two main pathways for which the enzymes are cyclo-oxygenase (COX) and lipoxygenase (Figures 12.3 and 4.9 respectively). Oxygenation and cyclisation of arachidonic acid by COX produces the prostaglandin PGG2 (the subscript 2 indicates two double bonds in the carbon chain). A non-specific peroxidase then converts PGG2 to PGH2, which is the parent compound for synthesis of the many important derivatives shown in Figure 12.3.
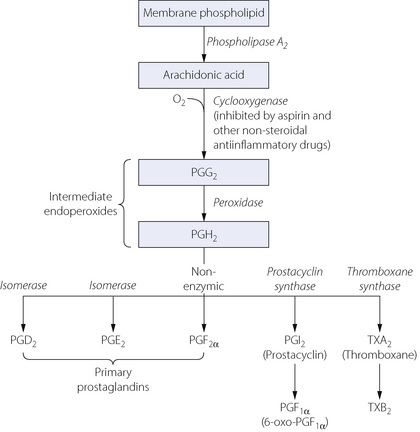
Fig. 12.3 The cyclo-oxygenase pathway for the production of arachidonic acid, and its subsequent conversion to form the prostaglandins and thromboxanes. See text for metabolism taking place in the lungs.
Eicosanoids are not stored preformed, but are synthesised as required by many cell types in the lung, including endothelium, airway smooth muscle, mast cells, epithelial cells and vascular muscle. Activation of phospholipase initiates the pathway, and results from a variety of stimuli such as inflammatory cytokines, complement activation, hormones, allergens or mechanical stimuli. The enzyme for the next step of the pathway, COX, exists in multiple isoforms, including COX-1, which is a constitutive enzyme present at low concentrations, and COX-2, which is induced by inflammatory cytokines. In the normal lung, the physiological role of these COX isoforms is uncertain, but in some patients with asthma, inhibition of COX-1 by aspirin induces bronchospasm, whilst inhibition of COX-2 does not (page 408).
PGF2α, PGD2, PGG2, PGH2 and thromboxane are bronchial and tracheal constrictors, PGF2α and PGD2 being much more potent in asthmatic patients compared with normal subjects. PGE1 and PGE2 are bronchodilators, particularly when administered by aerosol. Prostacyclin (PGI2) has different effects in different species. In humans, it has no effect on airway calibre in doses that have profound cardiovascular effects. PGI2 and PGE1 are pulmonary vasodilators. PGH2 and PGF2α are pulmonary vasoconstrictors.
Various specific enzymes in the lung are responsible for extensive metabolism of PGE2, PGE1 and PGF2α, but PGA2 and PGI2 pass through the lung unchanged. As for catecholamine metabolism, specificity for pulmonary prostaglandin metabolism is in the uptake pathways rather than with the intracellular enzymes.29
Leukotrienes36 are also eicosanoids derived from arachidonic acid but by the lipoxygenase pathway (see Figure 4.9). The leukotrienes LTC4 and LTD4 are mainly responsible for the bronchoconstrictor effects of what was formerly known as slow-reacting substance A or SRS-A. SRS-A also contains LTB4, which is a less powerful bronchoconstrictor but increases vascular permeability. These compounds, which are synthesised by the mast cell, have an important role in asthma and the mechanism of their release is discussed in Chapter 28, whilst drugs that inhibit leukotrienes are described on page 53.
Purine Derivatives
Specific enzymes exist on the surface of pulmonary endothelial cells for the degradation of AMP, ADP and ATP to adenosine. Adenosine itself has potent effects on the circulation, but is also inactivated in the lungs by a rapid uptake mechanism into the endothelial cells, where it is either phosphorylated into AMP or deaminated to produce inosine and ultimately uric acid for excretion.
Pharmacokinetics and the Lung
Drug Delivery37,38
Inhalation of drugs to treat lung disease may be considered as topical administration of the drug to the respiratory tract, though systemic absorption of the drug is likely to be greater than other topical routes. Pulmonary administration of a drug that is intended to work systemically offers many advantages over other routes, such as very rapid delivery into the circulation and avoidance of first pass metabolism in the liver.
For delivery to the alveoli, particles around 3 μm are the optimal size, as larger particles tend to deposit in the airways, and smaller particles tend to be inhaled and exhaled without being deposited in lung tissue (page 220). Targeted delivery of drugs to specific regions of the respiratory tract should be possible, by, for example, modifying the particle size, the timing of its addition to the breath or the breathing pattern during inhalation.38,39 In future even more specific targeting of inhaled drugs may be possible, as demonstrated by a study in which magnetic iron oxide nanoparticles were added to aerosol solutions and a magnetic field used to modify where in the lung the aerosol was deposited.40 In clinical practice, most delivery devices in clinical use produce aerosols containing a wide range of particle sizes, the most commonly used metered-dose inhaler used to treat asthma generating particles between 1–35 μm. Use of a spacer device before inhalation allows the largest particles to fall out of the aerosol before inhalation, and so reduces their impaction in the pharynx where they are responsible for some of the side effects of inhaled drugs.
Drug Elimination
The wide range of mechanisms present in the lung for the processing of endogenous and inhaled substances makes an effect on drug disposition almost inevitable.
Inhaled drugs will be subjected to the same metabolic activity in the airway and alveolar cells as other toxic chemicals described above. Mixed function oxidase and cytochrome P-450 systems are active in the lung and so are presumed to metabolise drugs in the same way as in hepatocytes. Steroids are known to be metabolised in lung airway tissue, as is isoprenaline.29
Pulmonary circulation.29,41,42 Many drugs are removed from the circulation on passing through the lungs. However, in the majority of cases this occurs by retention of the drug in lung tissue rather than actual metabolism. This low activity of metabolic enzymes found in the lung occurs for two reasons. First, access to the metabolic enzymes in endothelial cells is closely controlled by highly specific uptake mechanisms that are vital to allow the highly selective metabolism of endogenous compounds. Secondly, it is possible that the oxidative systems responsible for drug metabolism elsewhere in the body are located mostly in the airways thus preventing blood borne drugs gaining access to them. Drugs that are basic (pKa >8) and lipophilic tend to be taken up in the pulmonary circulation while acidic drugs preferentially bind to plasma proteins.29,42 Drug binding in the pulmonary circulation may act as a first pass filter for any drug administered intravenously.41 This drug reservoir within the lung may then be released slowly, or even give rise to rapid changes in plasma drug levels when the binding sites either become saturated or when one drug is displaced by a different drug with greater affinity for the binding site.
Pulmonary toxicity of drugs. Accumulation of some drugs and other toxic substances in the lung may cause dangerous local toxicity.43 Paraquat is an outstanding example: it is slowly taken up into alveolar epithelial cells where it promotes the production of reactive oxygen species (page 384), with resulting lung damage. Some drugs cause pulmonary toxicity by a similar mechanism, including nitrofurantoin and bleomycin, toxicity from the latter being strongly associated with exposure to high oxygen concentrations. Amiodarone, a highly effective and commonly used antiarrhythmic agent, is also associated with pulmonary toxicity which occurs in 6% of patients given the drug.44 When toxicity occurs it may be severe and is fatal in up to 10% of cases. The cause is unknown, but formation of reactive oxygen species, immunological activation and direct cellular toxicity are all believed to contribute.44
The Endocrine Lung
To qualify as a true endocrine organ, the lung must secrete a substance into the blood, which brings about a useful physiological response in a distant tissue. In spite of its wide-ranging metabolic activities already described, the endocrine functions of the lung remain ill-defined. Contenders include the following.
Inflammatory mediators. Histamine, endothelin and eicosanoids are released from the lung following immunological activation by inhaled allergens (Chapter 28). These mediators are undoubtedly responsible for cardiovascular and other physiological changes in the rest of the body, such as a rash, peripheral vasodilation and a reduction in blood pressure. However, it is doubtful if this can really be regarded as a desirable physiological effect.
Hypoxic endocrine responses.45 Animal studies have demonstrated the presence of clusters of peptide and amine secreting cells in lung tissue. These cells degranulate in the presence of acute hypoxia, but the substances secreted and their effects are not known. The cells belong to the ‘diffuse endocrine system’ and are present in humans, but their role is extremely unclear.
Nitric oxide (NO) plays an important role in the regulation of airway smooth muscle (page 51) and pulmonary vascular resistance (page 107), and is well known for its effects on platelet function and the systemic vasculature elsewhere in the body. There is no evidence that pulmonary endothelium secretes NO into the blood in order to exert an effect elsewhere, mainly because of the rapid uptake of NO by haemoglobin (page 194). However, this does not rule out an indirect effect of pulmonary NO production in influencing peripheral blood flow, which may be controlled by the balance between different forms of NO–haemoglobin complexes (page 195).
References
1. Niden AH, Aviado DM. Effects of pulmonary embolism on the pulmonary circulation with special reference to arteriovenous shunts in the lung. Circ Res. 1956;4:67-73.
2. Kerut EK, Norfleet WT, Plotnick GD, Giles TD. Patent foramen ovale: A review of associated conditions and the impact of physiological size. J Am Coll Cardiol. 2001;38:613-623.
3. Fisher DC, Fisher EA, Budd JH, Rosen SE, Goldman ME. The incidence of patent foramen ovale in 1000 consecutive patients. Chest. 1995;107:1504-1509.
4. Widdicombe JH. Regulation of the depth and composition of airway surface liquid. J Anat. 2002;201:313-318.
5. Boucher RC. Regulation of airway surface liquid volume by human airway epithelia. Pflugers Arch. 2003;445:495-498.
6. Rose MC, Voynow JA. Respiratory tract mucin genes and mucin glycoproteins in health and disease. Physiol Rev. 2006;86:245-278.
7. Hattrup CL, Gendler SJ. Structure and function of the cell surface (tethered) mucins. Annu Rev Physiol. 2008;70:431-457.
8. Burgel P-R, Nadel JA. Epidermal growth factor receptor-mediated innate immune responses and their roles in airway diseases. Eur Respir J. 2008;32:1068-1081.
9. Rogers DF. Airway goblet cells: responsive and adaptable frontline defenders. Eur Respir Journal. 1994;7:1690-1706.
10. Rogers DF. Motor control of airway goblet cells and glands. Respir Physiol. 2000;125:129-144.
11. Houtmeyers E, Gosselink R, Gayan-Ramirez G, Decramer M. Regulation of mucociliary clearance in health and disease. Eur Respir J. 1999;13:1177-1188.
12. Salathe M. Regulation of mammalian ciliary beating. Annu Rev Physiol. 2007;69:401-422.
13. Jeffery PK. Microscopic structure of normal lung. In: Brewis RAL, Corrin B, Geddes DM, Gibson GJ, editors. Respiratory Medicine. London: WB Saunders Company Ltd; 1995:54-72.
14. Button B, Picher M, Boucher RC. Differential effects of cyclic and constant stress on ATP release and mucociliary transport by human airway epithelia. J Physiol. 2007;580:577-592.
15. Ganz T. Antimicrobial polypeptides in host defense of the respiratory tract. J Clin Invest. 2003;109:693-697.
16. Wright JR. Pulmonary surfactant: a front line of lung host defense. J Clin Invest. 2003;111:1453-1455.
17. Wu H, Kuzmenko A, Wan S, et al. Surfactant proteins A and D inhibit the growth of Gram-negative bacteria by increasing membrane permeability. J Clin Invest. 2003;111:1589-1602.
*18. Hiemstra PS. Antimicrobial peptides in the real world: implications for cystic fibrosis. Eur Respir J. 2007;29:617-618.
*19. Carrell RW, Lomas DA. Alpha1-antitrypsin deficiency – a model for conformational diseases. N Engl J Med. 2002;346:45-53.
20. Lomas DA, Mahadeva R. α1-antitrypsin polymerisation and the serpinopathies: pathobiology and prospects for therapy. J Clin Invest. 2002;110:1585-1590.
21. Laurell CB, Eriksson S. The electrophoretic α1-globulin pattern of serum in α1-antitrypsin deficiency. Scand J Clin Lab Invest. 1963;15:132-136.
22. Lomas DA. The selective advantage of α1-antitrypsin deficiency. Am J Respir Crit Care Med. 2006;173:1072-1077.
23. Norman MR, Mowat AP, Hutchinson DCS. Molecular basis, clinical consequences and diagnosis of alpha-1 antitrypsin deficiency. Ann Clin Biochem. 1997;34:230-246.
*24. Greenlee KJ, Werb Z, Kheradmand F. Matrix metalloproteinases in lung: multiple, multifarious, and multifaceted. Physiol Rev. 2007;87:69-98.
25. Smyth RL. The airway epithelium in health and disease: “calm on the surface, paddling furiously underneath.”. Thorax. 2009;64:277-278.
26. Bond JA. Metabolism and elimination of inhaled drugs and airborne chemicals from the lungs. Pharmacol Toxicol. 1993;72(3):36-47.
27. Dahl AR, Gerde P. Uptake and metabolism of toxicants in the respiratory tract. Environ Health Perspect. 1994;102(supp 11):67-70.
28. Kikkawa Y. Diverse role of pulmonary cytochrome P-450 monooxygenase. Lab Invest. 1992;67:535-539.
29. Bahkle YS. Pharmacokinetic and metabolic properties of the lung. Br J Anaesth. 1990;65:79-93.
30. Weibel ER. How does lung structure affect gas exchange. Chest. 1983;83:657-665.
31. Sole MJ, Dobrac M, Schwartz L, Hussain MN, Vaughan-Neil EF. The extraction of circulating catecholamines by the lungs in normal man and in patients with pulmonary hypertension. Circulation. 1979;60:160-163.
32. Gillis CN, Pitt BR. The fate of circulating amines within the pulmonary circulation. Annu Rev Physiol. 1982;44:269-281.
33. Brew K. Structure of human ACE gives new insights into inhibitor binding and design. Trends Pharmacol Sci. 2003;24:391-394.
34. Iervasi G, Clerico A, Pilo A, et al. Atrial natriuretic peptide is not degraded by the lungs in humans. J Clin Endocrinol Metab. 1998;83:2898-2906.
35. Michael JR, Markewitz BA. Endothelins and the lung. Am J Respir Crit Care Med. 1996;154:555-581.
36. Peters-Golden M, Henderson WR. Leukotrienes. N Engl J Med. 2007;357:1841-1854.
*37. Groneberg DA, Witt C, Wagner U, Chung KF, Fischer A. Fundamentals of pulmonary drug delivery. Respir Med. 2003;97:382-387.
38. Bennett WD, Brown JS, Zeman KL, Hu S-C, Scheuch G, Sommerer K. Targeting delivery of aerosols to different lung regions. J Aerosol Med. 2002;15:179-188.
39. Usmani OS, Biddiscombe MF, Barnes PJ. Regional lung deposition and bronchodilator response as a function of β2-agonist particle size. Am J Respir Crit Care Med. 2005;172:1497-1504.
*40. Coates AL. Guiding aerosol deposition in the lung. N Engl J Med. 2008;358:304-305.
41. Upton RN, Doolette DJ. Kinetic aspects of drug deposition in the lungs. Clin Exp Pharmacol Physiol. 1999;26:381-391.
*42. Boer F. Drug handling by the lungs. Br J Anaesth. 2003;91:50-60.
43. Foth H. Role of the lung in accumulation and metabolism of xenobiotic compounds – implications for chemically induced toxicity. Crit Rev Toxicol. 1995;25:165-205.
44. Reasor MJ, Kacew S. An evaluation of possible mechanisms underlying amiodarone-induced pulmonary toxicity. Proc Soc Exp Biol Med. 1996;212:297-305.
45. Gosney JR. The endocrine lung and its response to hypoxia. Thorax. 1994;49:S25-S26.