Chapter 30 Diseases of the lung parenchyma and pleura
 Lung collapse occurs either from compression of lung tissue or by absorption of gas from lung units with occluded, or severely narrowed, airways. Many forms of interstitial lung disease exist, varying from purely inflammatory conditions (alveolitis), to those involving progressive fibrosis with minimal lung inflammation. Lung fibrosis arises from an imbalance between the cellular systems responsible for inflammation and tissue repair.
Lung collapse occurs either from compression of lung tissue or by absorption of gas from lung units with occluded, or severely narrowed, airways. Many forms of interstitial lung disease exist, varying from purely inflammatory conditions (alveolitis), to those involving progressive fibrosis with minimal lung inflammation. Lung fibrosis arises from an imbalance between the cellular systems responsible for inflammation and tissue repair.Pulmonary Collapse
Pulmonary collapse may be defined as an acquired state in which the lungs or part of the lungs become airless. Atelectasis is strictly defined as a state in which the lungs of a newborn have never been expanded, but the term is widely used as a synonym for regional pulmonary collapse.
Collapse may be caused by two different mechanisms. The first of these is loss of the forces opposing the elastic recoil of the lung, which then decreases in volume to the point at which airways are closed and gas is trapped behind the closed airways. The second is obstruction of airways at normal lung volume, which may be due to many different causes. This also results in trapping of gas behind the obstructed airway. Whatever the cause of the airway closure, there is rapid absorption of the trapped gas because the total partial pressure of gases in mixed venous blood is always less than atmospheric (see Table 26.2). This generates a sub-atmospheric pressure more than sufficient to overcome any force tending to hold the lung expanded.
Pulmonary collapse during anaesthesia is described in Chapter 22.
Loss of Forces Opposing Retraction of the Lung
The lungs are normally prevented from collapse by the outward elastic recoil of the ribcage and any resting tone of the diaphragm. The pleural cavity normally contains no gas but, if a small bubble of gas is introduced, its pressure is subatmospheric (see Figure 3.5). Pulmonary collapse due to loss of forces opposing lung retraction may be considered under five headings as follows.
Voluntary reduction of lung volume. It seems unlikely that voluntary reduction of lung volume below closing capacity will cause overt collapse of lung in a subject breathing air. However, in older subjects, there is an increase in the alveolar/arterial Po2 gradient, suggesting trapping of alveolar gas (see Figure 22.11).
Excessive external pressure. Ventilatory failure is the more prominent aspect of an external environmental pressure in excess of about 6 kPa (60 cmH2O), which is not communicated to the airways (page 396). However, some degree of pulmonary collapse could also occur and this is a normal consequence of the great depths attained by diving mammals while breath holding. An approximately normal lung volume is maintained during conventional diving operations when respired gas is maintained at the surrounding water pressure, though this does not occur with surface diving or snorkelling (page 297).
Loss of integrity of the rib cage. Multiple rib fractures or the old operation of thoracoplasty may impair the elastic recoil of the ribcage to the point at which partial lung collapse results. This depends entirely on the extent of the injury to the ribcage, but multiple adjacent ribs fractured in two places will usually result in collapse. However, extensive trauma to the rib cage also causes interference with the mechanics of breathing which is generally more serious than collapse (page 396).
Intrusion of abdominal contents into the chest. Extensive atelectasis results from a congenital defect of the diaphragm. Abdominal contents may completely fill one-half of the chest with total atelectasis of that lung. In adults, similar changes may occur with a large hiatus hernia, or ascites may push the diaphragm into the thoracic cavity. Paralysis of one side of the diaphragm causes the diaphragm to lie higher in the chest, with a tendency to basal collapse on that side.
Space occupation of the pleural cavity. Air introduced into the pleural cavity (pneumothorax) reduces the forces opposing retraction of the lung and this is a potent cause of collapse. The same effect occurs when the pleural cavity is occupied by an effusion, empyema or haemothorax. Pleural disease is discussed on page 444 et seq.
Absorption of Trapped Gas
Absorption of alveolar gas trapped beyond obstructed airways may be the consequence of reduction in lung volume by the mechanisms described above. However, it is the primary cause of collapse when there is total or partial airway obstruction at normal lung volume. Obstruction is commonly due to secretions, pus, blood or tumour but may be due to intense local bronchospasm or airway oedema.
Gas trapped beyond the point of airway closure is absorbed by the pulmonary blood flow. The total of the partial pressures of the gases in mixed venous blood is always less than atmospheric (see Table 26.2), although pressure gradients for the individual component gases between alveolar gas and mixed venous blood may be quite different.
The effect of respired gases. If the patient has been breathing 100% oxygen prior to obstruction the alveoli will contain only oxygen, carbon dioxide and water vapour. Because the last two together normally amount to less than 13.3 kPa (100 mmHg), the alveolar Po2 will usually be in excess of 88 kPa (660 mmHg). However, the Po2 of the mixed venous blood is unlikely to exceed about 6.7 kPa (50 mmHg), so the alveolar/mixed venous Po2 gradient will be of the order of 80% of an atmosphere. Absorption collapse will thus be rapid and there will be no nitrogen in the alveolar gas to maintain inflation. This has important implications during anaesthesia, when 100% oxygen is commonly administered (page 336).
The situation is much more favourable in a patient who has been breathing air, as most of the alveolar gas is then nitrogen, which is at a tension only about 0.5 kPa (4 mmHg) below that of mixed venous blood.1 Alveolar nitrogen tension rises above that of mixed venous blood as oxygen is absorbed and eventually the nitrogen will be fully absorbed. Collapse must eventually occur but the process is much slower than in the patient who has been breathing oxygen. Figure 30.1 shows a computer simulation of the time required for collapse with various gas mixtures.2 Nitrous oxide/oxygen mixtures may be expected to be absorbed almost as rapidly as 100% oxygen. This is partly because nitrous oxide is much more soluble in blood than nitrogen, and partly because the mixed venous tension of nitrous oxide is usually much less than the alveolar tension, except after a long period of inhalation.
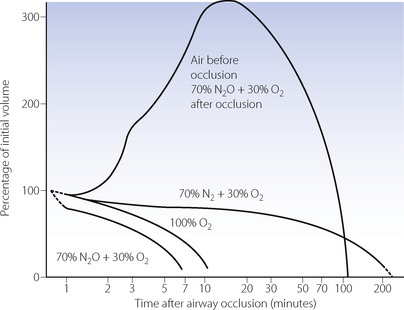
Fig. 30.1 Predicted rates of absorption from alveoli of differing gas mixtures. The lower curves show the rate of absorption of the contents of sections of the lung whose air passages are obstructed, resulting in sequestration of the contents. The upper curve shows the expansion of the sequestered gas when nitrous oxide is breathed by a patient who has recently developed regional airway obstruction whilst breathing air. In all other cases, it is assumed that the inspired gas is not changed after obstruction has occurred. Similar considerations apply to closed gas cavities elsewhere in the body.
(Reproduced from reference 2 by permission of the authors and the publishers of Anaesthesia.)
When the inspired gas composition is changed after obstruction and trapping occur, complex patterns of absorption may ensue. The inhalation of nitrous oxide, after airway occlusion has occurred while breathing air, results in temporary expansion of the trapped volume (Figure 30.1). This is caused by large volumes of the more soluble nitrous oxide passing from blood to alveolus in exchange for smaller volumes of the less soluble nitrogen passing in the reverse direction. This phenomenon also applies to any closed air space in the body, such as closed pneumothorax, gas emboli, bowel, and the middle ear with a blocked pharyngotympanic (Eustachian) tube. It is potentially dangerous and may contraindicate the use of nitrous oxide as an anaesthetic.
Magnitude of the pressure gradients. It needs to be stressed that the forces generated by the absorption of trapped gases are very large. The total partial pressure of gases in mixed venous blood is normally 87.3 kPa (655 mmHg). The corresponding pressure of the alveolar gases is 95.1 kPa (713 mmHg), allowing for water vapour pressure at 37°C. The difference, 7.8 kPa (58 mmHg or 78 cmH2O), is sufficient to overcome any forces opposing recoil of the lung. Absorption collapse after breathing air may therefore result in drawing the diaphragm up into the chest, reducing ribcage volume or displacing the mediastinum. If the patient has been breathing oxygen, the total partial pressure of gases in the mixed venous blood is barely one-tenth of an atmosphere (see Table 26.2) and absorption of trapped alveolar gas generates enormous forces.
Effect of reduced ventilation/perfusion ratio. Absorption collapse may still occur in the absence of total airway obstruction provided that the ventilation/perfusion (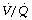 ) ratio is sufficiently reduced. Older subjects, as well as those with a pathological increase in scatter of
) ratio is sufficiently reduced. Older subjects, as well as those with a pathological increase in scatter of 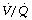 ratios, may have substantial perfusion of areas of lung with
ratios, may have substantial perfusion of areas of lung with 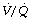 ratios in the range 0.01–0.1. This shows as a characteristic ‘shelf’ in the plot of perfusion against
ratios in the range 0.01–0.1. This shows as a characteristic ‘shelf’ in the plot of perfusion against 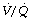 (Figure 30.2). These grossly hypoventilated areas are liable to collapse if the patient breathes oxygen (Figure 30.2B). If the
(Figure 30.2). These grossly hypoventilated areas are liable to collapse if the patient breathes oxygen (Figure 30.2B). If the 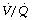 ratio is less than 0.05, ventilation even with 100% oxygen cannot supply the oxygen that is removed (assuming the normal arterial/mixed venous oxygen content difference of 0.05 ml.ml−1). As the
ratio is less than 0.05, ventilation even with 100% oxygen cannot supply the oxygen that is removed (assuming the normal arterial/mixed venous oxygen content difference of 0.05 ml.ml−1). As the 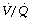 ratio decreases below 0.05, so the critical inspired oxygen concentration necessary for collapse also decreases (Figure 30.2C). The flat part of the curve between
ratio decreases below 0.05, so the critical inspired oxygen concentration necessary for collapse also decreases (Figure 30.2C). The flat part of the curve between 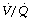 ratios of 0.001 and 0.004 means that small differences in inspired oxygen concentration in the range 20–30% may be very important in determining whether collapse occurs or not. There is no difficulty in demonstrating that pulmonary collapse may be induced in healthy middle-aged subjects breathing oxygen close to residual volume.3,4
ratios of 0.001 and 0.004 means that small differences in inspired oxygen concentration in the range 20–30% may be very important in determining whether collapse occurs or not. There is no difficulty in demonstrating that pulmonary collapse may be induced in healthy middle-aged subjects breathing oxygen close to residual volume.3,4
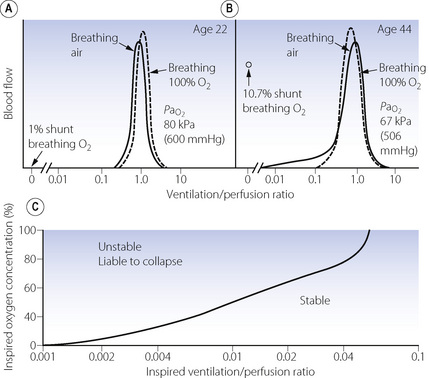
Fig. 30.2 Inspiration of 100% oxygen causes collapse of alveoli with very low 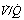 ratios. (A) The minor change in the distribution of blood flow (in relation to
ratios. (A) The minor change in the distribution of blood flow (in relation to 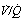 ratio) when a young subject breathes oxygen. Collapse is minimal and a shunt of 1% develops. (B) The changes in an older subject with a ‘shelf’ of blood flow distributed to alveoli with very low
ratio) when a young subject breathes oxygen. Collapse is minimal and a shunt of 1% develops. (B) The changes in an older subject with a ‘shelf’ of blood flow distributed to alveoli with very low 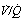 ratios. Breathing oxygen causes collapse of these alveoli and this is manifested by disappearance of the shelf and development of an intrapulmonary shunt of 10.7%. (C) The inspired oxygen concentration relative to the inspired
ratios. Breathing oxygen causes collapse of these alveoli and this is manifested by disappearance of the shelf and development of an intrapulmonary shunt of 10.7%. (C) The inspired oxygen concentration relative to the inspired 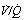 ratio that is critical for absorption collapse.
ratio that is critical for absorption collapse.
(After reference 5 by permission of the authors and the publishers of the Journal of Clinical Investigation, and from reference 6 by permission of the authors and the publishers of Journal of Applied Physiology.)
Diagnosis of Pulmonary Collapse
The diagnosis may be made on physical signs of decreased air entry and chest dullness but reliance is usually placed on chest radiography. Pulmonary opacification is seen, along with indirect signs of thoracic volume loss such as displacement of interlobular fissures, raised diaphragms, and displaced hilar or mediastinal structures.7 In the upright position, collapse is commonest in the basal segments, often concealed behind the cardiac shadow unless the exposure is appropriate. Areas of atelectasis are clearly seen with computerised tomography (see Figure 22.10).
Collapse results in a reduction in pulmonary compliance, but the value of this in diagnosis is limited by the wide scatter of normal values. A sudden reduction in compliance may give an indication of collapse, provided, of course, that control measurements were available before collapse. Collapse also reduces the functional residual capacity and arterial Po2. However, in a patient with impaired oxygenation a reduction in arterial Po2 cannot distinguish between the three very common conditions of pulmonary collapse, consolidation and oedema.
Principles of Therapy
Therapy depends on the physiological abnormality. Factors opposing the elastic recoil of the lung should be removed wherever possible. For example, pneumothorax, pleural effusion and ascites may be corrected. In other cases, particularly impaired integrity of the chest wall, it may be necessary to treat the patient with artificial ventilation. Re-expansion of collapsed lung often requires high pressures to be applied (page 336), but it is usually possible to restore normal lung volume.
When collapse is caused by regional airway obstruction, the most useful methods in both treatment and prevention are by chest physiotherapy, combined when necessary with tracheobronchial toilet, through either a tracheal tube or a bronchoscope. Fibreoptic bronchoscopy alone will often clear an obstructed airway and permit re-expansion, particularly with lobar atelectasis.8
Voluntary maximal inspirations are effective in clearing areas of absorption collapse in subjects who had been breathing oxygen near residual volume.4 This manoeuvre is the basis of the ‘incentive spirometer’, which is used to prevent postoperative lung collapse.
With artificial ventilation a logical approach is hyperinflation of the chest or an artificial ‘sigh’. Some ventilators were designed to provide an intermittent ‘sigh’ but evidence of its efficacy was never found. Current strategies to prevent pulmonary collapse during artificial ventilation are described in Chapter 32.
Pulmonary Consolidation (Pneumonia)
Inflammation of areas of lung parenchyma, usually due to infection, can lead to the accumulation of exudate within the alveoli and small airways, causing consolidation. Areas of consolidation may be patchy, and referred to as bronchopneumonia, or confined to discrete areas of the lung, forming lobar pneumonia. Pulmonary collapse frequently occurs in conjunction with pneumonia as a result of airway narrowing in surrounding lung areas. Clinical features of pyrexia, cough, sputum production and dyspnoea occur with signs of consolidation such as bronchial breathing, chest dullness and inspiratory crackles, though physical signs may be absent in bronchopneumonia. Diagnosis again relies on chest radiography, where consolidation appears as pulmonary shadowing, sometimes accompanied by an ‘air bronchogram’. With resolution of the infection, cough becomes more productive, and the lung returns to normal within a few weeks.
Effects on gas exchange. Patients with pneumonia are commonly hypoxic. Consolidated areas of lung behave in a similar fashion to collapse, forming an intrapulmonary shunt through which mixed venous blood flows. In addition, there is an increase in areas with low 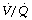 ratios (<0.1), but the contribution of these areas to impaired oxygenation is believed to be small because of hypoxic pulmonary vasoconstriction. Administration of oxygen to patients with pneumonia causes a further widening of the scatter of
ratios (<0.1), but the contribution of these areas to impaired oxygenation is believed to be small because of hypoxic pulmonary vasoconstriction. Administration of oxygen to patients with pneumonia causes a further widening of the scatter of 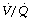 ratios, implying a reduction in hypoxic pulmonary vasoconstriction,9 but nevertheless results in a considerable improvement in arterial Po2. In comparison with collapsed lung, consolidation is commonly associated with a worse pulmonary shunt and therefore more severe hypoxia. Many of the inflammatory mediators released as part of the response to infection act as local pulmonary vasodilators, in effect over-riding hypoxic pulmonary vasoconstriction.
ratios, implying a reduction in hypoxic pulmonary vasoconstriction,9 but nevertheless results in a considerable improvement in arterial Po2. In comparison with collapsed lung, consolidation is commonly associated with a worse pulmonary shunt and therefore more severe hypoxia. Many of the inflammatory mediators released as part of the response to infection act as local pulmonary vasodilators, in effect over-riding hypoxic pulmonary vasoconstriction.
Pathophysiology10
Airway inflammation was described in detail in Chapter 28. Invasion of the lower respiratory tract with viruses and bacteria leads to further inflammatory changes characterised by migration of neutrophils from the circulation into the lung tissue. Depending on the pathogen involved, the stimulus for this migration may originate from the lung epithelial cells or alveolar macrophages. Chemokines released from these cells initiate neutrophil margination, and a range of proinflammatory cytokine pathways begin. Once in the lung tissue and activated, neutrophils are highly effective killers of the invading pathogen (page 455). As part of this process an inflammatory exudate develops that leads to consolidation of the lung tissue. The exudate is a complex mixture or invading organisms, inflammatory cells (dead and alive), immunoglobulins and other immune mediators, fluid transudate from increased capillary permeability, and products resulting from destruction of lung tissue as a result of protease activity.
Margination of neutrophils. Before a neutrophil can contribute to the inflammatory response it must stick to the blood vessel wall (margination), migrate across the endothelium, interstitium and epithelium, and become activated ready to contribute to pathogen removal (see Figure 31.2). These activities are controlled by an extensive series of cytokines in a very similar fashion to airway inflammation (see Figure 28.1). Lymphocytes again play an important role, but in parenchymal inflammation macrophages have an important control function instead of the eosinophils and mast cells involved in airway inflammation.
Neutrophil margination has been extensively studied in the systemic circulation. Selectins expressed on the surface of endothelial cells transiently bind the neutrophil causing it to roll along the blood vessel wall. Eventually, different adhesion molecules on the endothelial cell (e.g. intercellular adhesion molecule-1, ICAM-1) bind to specific receptors on the neutrophil surface (e.g. β2 integrins CD11/CD18) causing a more firm adhesion to the endothelium. Once ‘caught’ by the endothelial cell, cytokines are released and neutrophil activation begins. The way in which neutrophils are marginated in the lung differs from elsewhere in the body.11,12 Adhesion to endothelial cells occurs predominantly in the pulmonary capillary, rather than in venules as in the systemic circulation. Adhesion to the capillary wall can occur by either a CD11/CD18-dependent mechanism, or by another mechanism that seems to be independent of all the adhesion molecules normally required for margination in a systemic capillary.13 Selectin-induced rolling of neutrophils may not occur. Adhesion is facilitated by a slow transit time for neutrophils across pulmonary capillaries. Human neutrophils are of similar size to red blood cells, but are much less deformable, so neutrophils take up to 120 seconds to traverse a pulmonary capillary compared with less than a second for a red blood cell.11 Inflammatory mediators may cause changes to the biomechanical properties of neutrophils, in particular, a stiffening of the cell that will further impede its movement through the pulmonary capillary.14 Once adhered to the pulmonary capillary wall neutrophils may become flattened, leaving some capillary lumen available for blood flow. In this position, emigration into the pulmonary tissue begins and the neutrophil moves through small holes in the capillary basal laminae, being guided by chemokines released from epithelial cells and possibly assisted by fibroblasts in the interstitial space (Figure 30.3).15
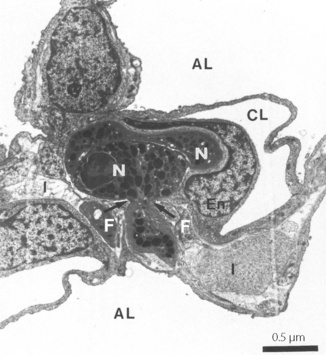
Fig. 30.3 Neutrophil emigration in rabbit lung during streptococcal pneumonia. This electron micrograph shows that the neutrophils (N), which are normally the same diameter as a pulmonary capillary, are elongated so leaving capillary lumen (CL) partly patent. These neutrophils have already emigrated from the capillary lumen across the endothelium (En), and one is now passing into the interstitium (I) through a small hole in the capillary basement membrane (arrows). The pseudopod of the neutrophil is in close contact with fibroblasts (F), which may be guiding the neutrophil through the defect in the basement membrane. AL, alveolar lumen.
(Figure kindly provided by Professor DC Walker. Reproduced from reference 15 by permission of the author and publishers of Microvascular Research.)
Interstitial Lung Disease and Pulmonary Fibrosis
Diffuse pulmonary inflammation occurs in a wide variety of conditions, which are summarised in Table 30.1. Pneumonitis may simply resolve, as in pneumonia, leaving no permanent damage, but with long-term inflammation varying degrees of pulmonary fibrosis develop.
Table 30.1 Causes of interstitial pneumonitis and pulmonary fibrosis
| CAUSES | SUBGROUPS | EXAMPLES |
|---|---|---|
| Drug induced | Anti-cancer Antibiotics Others |
Bleomycin, busulphan, cyclophosphamide, methotrexate Isoniazid, nitrofurantoin, sulphonamides Amiodarone |
| Dust | Inorganic Organic |
Silicosis Asbestosis Farmer’s lung |
| Infections | Viral Other |
Viral pneumonia HIV Mycoplasma Opportunistic infections |
| Systemic disease | Connective tissue disease Others |
Rheumatoid arthritis, scleroderma systemic lupus erythematosus, ankylosing spondylitis Sarcoidosis, histiocytosis, uraemia |
| Miscellaneous | Acute inflammation Inhalation injury Radiation lung damage Cryptogenic fibrosing alveolitis |
Acute lung injury Smoke, cadmium, sulphur dioxide |
Clinical features. vary according to the aetiology. Pneumonitis alone (i.e. without fibrosis) may be asymptomatic at first, progressing to a cough and dyspnoea, and in severe cases gives rise to systemic symptoms such as fever. When accompanied by fibrosis, dyspnoea becomes worse, and basal inspiratory crackles are present on examination. Lung function tests show a typical restrictive pattern with similar reductions in both forced vital capacity and forced expiratory volume in one second (page 96). Diffuse reticular shadows develop on chest radiography, and high-resolution CT scanning of the lungs shows either ‘ground glass’ appearances, which correlate with pneumonitis, or ‘honeycombing’, which represent more advanced fibrosis.
Causes of Pulmonary Fibrosis
These have been summarised in Table 30.1.
Drug induced fibrosis may follow lung injury induced by oxygen toxicity (page 387) precipitated by, for example, bleomycin, but the mechanism of this response is poorly understood.
Inorganic dusts.16 Occupational exposure to asbestos fibres (asbestosis) or silica (silicosis) for many years leads to pulmonary fibrosis. Inhaled dust particles between 1 and 3 μm in diameter reach the alveoli and are ingested by macrophages. Different dust types have variable persistence in the lung, some being rapidly cleared and others persisting within the pulmonary macrophage for many years. In addition, the total (lifetime) fibre burden probably correlates with the degree of resulting fibrosis.
Organic dusts may cause lung inflammation by an immune mechanism, a condition referred to as extrinsic allergic alveolitis. The allergen is normally derived from a fungus to which the patient has occupational exposure, giving rise to a host of disease names such as farmer’s lung, malt worker’s lung etc. Bird fancier’s lung differs in that it is precipitated by exposure to IgA derived from domestic birds. In extrinsic allergic alveolitis, pneumonitis results from activation of T-lymphocytes and IgG mediated inflammation. If caught early enough, and avoidance measures taken, allergic alveolitis resolves completely, but with continued exposure fibrosis develops.
Systemic diseases that lead to fibrosis are numerous and the mechanisms obscure. Many of the diseases associated with lung fibrosis have an immunological basis. For example, sarcoidosis results from T-lymphocyte activation in response to an unknown stimulus, whilst many connective tissue diseases are known to have an autoimmune aetiology. These immune changes are therefore likely to cause activation of the pulmonary inflammatory cells described below.
Radiation lung damage17 is seen following radiotherapy for tumours in or near the chest. Radiation pneumonitis develops over several weeks following radiotherapy, while fibrosis may take up to 2 years to develop. Cellular radiation damage occurs when cell division occurs, so susceptible cells in the lung are those with the greatest rate of turnover. Thus radiation injury begins with damage to type II pneumocytes and capillary endothelial cells, which results in altered surfactant and interstitial pulmonary oedema (page 423) respectively. A cascade of inflammatory cell activation will then follow, often proceeding to fibrosis.18
Idiopathic pulmonary fibrosis (IPF),19 synonymous with cryptogenic fibrosing alveolitis, includes all cases of pulmonary fibrosis in which no cause can be found. It is the most common type of pulmonary fibrosis, occurs more commonly in males and is of uncertain aetiology. Patients with CFA have extensive activation of pulmonary inflammatory cells and cytokines as described below. There is also accumulation of neutrophils, and this indicates a role for pulmonary oxidant injury (page 382) in IPF. Whatever the cause, IPF can be rapidly progressive with a median survival from diagnosis of just a few years.
Cellular Mechanisms of Pulmonary Fibrosis20,21
Lung inflammation has been described earlier in this chapter as well as in Chapters 28 and 31. Progression to pulmonary fibrosis is not inevitable, but predicting which patients, and which underlying diseases, do progress is important clinically. There has been extensive research into the mechanisms of fibrosis, though a useful prognostic test remains a distant prospect.
Inflammation anywhere in the body is naturally succeeded by a cellular healing process that involves the laying down of new collagen. The lung is no exception, and pulmonary fibrosis is a result of excessive deposition of collagen in the lung extracellular matrix.
In pulmonary fibrosis the initial disease process is diverse (Table 30.1) and may cause changes in either type I or type II alveolar epithelial cells, pulmonary macrophages, neutrophils or T-lymphocytes.22 Interactions between these cells produce numerous cytokines, which amplify the inflammatory response and initiate cellular repair mechanisms. Once these repair mechanisms are established, apoptosis occurs in the inflammatory cells and tissue repair proceeds. Transforming growth factor-β (TGF-β) is believed to be the most important cytokine involved in stimulating tissue repair, and probably acts as the final common pathway for most mechanisms leading to fibrosis.23 Caveolin-1, which is a structural protein forming caveoli on the plasma membrane of many cells, is believed to be the endogenous regulator of TGF-β activity.20 Myofibroblasts are the cells responsible for repairing the extracellular matrix in lung tissue, this matrix forming the scaffolding on which new lung tissue is formed. Once myofibroblasts have completed their task, they too undergo apoptosis.
In most causes of pulmonary fibrosis this well-controlled sequence of events is abnormal. The activity of acute inflammatory cells may not subside once the stimulus has been removed, and prolonged stimulation of repair mechanisms will occur. Alternatively, the normal mechanisms that terminate myofibro-blast activity may be defective. The combination of inherited differences in the expression of cytokines or their receptors and the range of environmental stimuli described above is believed to result in pulmonary fibrosis. For example, an inherited defect in the expression of caveolin-1 may allow the actions of TGF-β to go unchecked. The intriguing possibility of abnormal apoptosis in lung cells has been suggested to explain pulmonary fibrosis.24 Type I alveolar epithelial cells may undergo premature apoptosis and so prolong the inflammatory stimulus by continued exposure of underlying tissue. Alternatively, once tissue repair is complete, myofibroblasts may fail to respond to normal apoptotic stimuli and continue to remodel the extracellular matrix.
In a similar fashion to emphysema (page 410), excessive myofibroblast activity leads to a reduction in the amount of elastin present. Synthesis of elastin in normal lung is minimal in adults, and though there is some evidence of increased production in pulmonary fibrosis the elastic fibres formed are abnormal and probably non-functional.25 Loss of elasticity by this mechanism causes collapse of both alveolar and small airway walls leading to a reduction in compliance and the area available for gas exchange.
Principles of Therapy26,27
Where feasible, removal of the stimulant for lung inflammation or fibrosis is vital. Though this may not halt the development of fibrosis, for example following irradiation, it may limit the degree of pulmonary damage that occurs. Few patients with IPF gain any benefit from treatment with steroids, and predicting who will respond is difficult. Second-line treatment is with cytotoxic drugs such as azathioprine and cyclophosphamide, though with similar poor results. Recent elucidation of the cytokines involved in pulmonary fibrosis has led to optimism about future therapeutic approaches,23 with many drugs that affect these processes currently under development.27
Lung Cancer28
At the start of the 20th century lung cancer was a rare disease, but by the end of the century improved longevity and greater exposure to environmental carcinogens had led to lung cancer becoming one of the most common preventable causes of death in the world.29,30 Improvements in the success rates for treatment of lung cancer have been less than for malignancies in other organs, and the overall 5-year survival rate for lung cancer remains poor at only 15.7%.29 It has been estimated that 1.2 million people worldwide died of lung cancer in 2002.30
Epidemiology
Occupational exposure to lung carcinogens such as asbestos was one of the earliest causative factors for lung malignancy to be identified, with several other occupational agents subsequently being linked with lung cancer such as arsenic, cadmium, beryllium and silica. Co-existing lung disease and diet have also been shown to be linked with the development of lung cancer. The role of these factors in causing lung cancer is however now known to be insignificant compared with exposure to environmental radon and the overwhelming role of tobacco smoke.
Tobacco.30 Tobacco smoking (Chapter 21) is responsible for 90% of lung cancers in countries where smoking is common, and on a population scale lung cancer rates mirror smoking rates with an approximate lag time of 20 years. This is an ominous statistic for many currently developing countries where smoking rates are still increasing. Both the number of cigarettes smoked per day and the duration of being a smoker are positively correlated with the risk of developing lung cancer, though the latter is the more strong association. Quitting smoking has the predictable opposite effect with the risk of developing lung cancer decreasing with every year of continued abstinence, though the risk never falls as low as that for a lifetime non-smoker. Changes to cigarettes over the last 60 years such as the introduction of filtered cigarettes, and low tar and nicotine brands, were all aimed at reducing the health risks of smoking. Although some studies have demonstrated a reduced disease burden of smoking, the overall effect of these changes on lung cancer risk is believed to be negligible.
Smoking prevalence amongst men was at a peak approximately 20 years before the peak for women, so at present the lung cancer incidence in men is declining whilst in women the incidence continues to increase, and lung cancer is now the commonest cause of cancer-related death in women. The greater incidence of lung cancer in women is not caused solely by differences in smoking prevalence, but on a dose-for-dose basis women also seem to be more susceptible to the carcinogens found in cigarette smoke with an odds ratio of between 1.2 and 1.7 compared with men for developing lung cancer with equivalent smoking habits.31 Studies from several countries have also shown that lung cancer incidence is higher in lower socio-economic groups, an observation which is again not entirely explained by differing smoking habits.
Most of the carcinogens in tobacco smoke are found amongst the 3500 compounds that make up the particulate phase, or ‘tar’, of cigarette smoke (page 318) and their mechanism of carcinogenesis is described below.
Radon. The second most important cause of lung cancer is environmental exposure to radon gas.32 Radon is part of the natural decay series of uranium (Figure 30.4) and both elements are ubiquitous in the soil and rocks of the world though in widely varying concentrations. Radon gas is approximately eight times heavier than air and therefore tends to accumulate in the cellars and basements of dwellings, making it an important indoor pollutant.33 The highest concentrations are found in mines, in particular uranium mines, therefore miners are the group most exposed to radon and an association between these occupations and lung cancer has been described for centuries. Residential exposure to radon may account for 10% of deaths from lung cancer or make an even greater contribution to the relatively rare cases of lung cancer in non-smokers.32,34
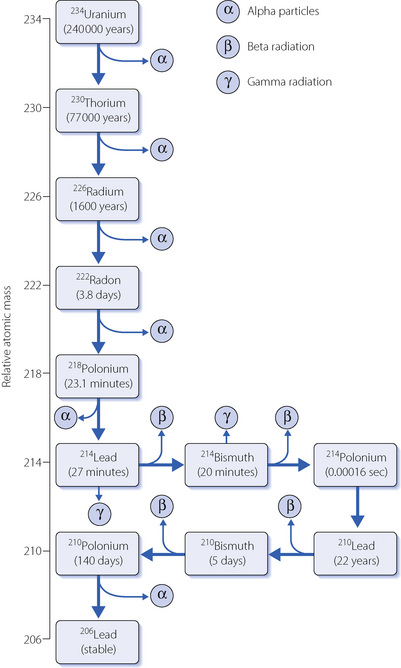
Fig. 30.4 Decay series of 234uranium to stable 206lead. An inhaled 222radon molecule will decay to 210lead within minutes, releasing three alpha and two beta particles in the process.
Radon is an inert gas, so when inhaled into the lung there will be no chemical reaction with other molecules, and with a molecular weight of 222 its diffusion within the alveolus (page 147) and absorption into the blood will both be slow. Most inhaled radon will therefore be exhaled in the same breath, but the most common environmental isotope, 222radon, has a half life of only 3.8 days so whilst in the airway some of the radon will decay. The decay products are mostly solids which may be deposited in the airways, and also have short half-lives (Figure 30.4). As a result, inhaled radon and its progeny are a source of large quantities of alpha irradiation.
In comparison with beta and gamma radiation the alpha particle, made up of two protons and two neutrons, contains an enormous amount of energy and is therefore more harmful to biological molecules. For a sub-atomic particle, an alpha particle has a large mass and when travelling at around 15 000 km. s−1 this equates to a large kinetic energy. The strong positive charge of the alpha particle causes the electron shells of nearby atoms to rapidly slow the particle, dissipating its energy in a much smaller area than other forms of radiation. Alpha particles travel only a few centimetres in air, and probably only 30–50 μm in living tissue. It is unknown whether the alpha particles released in the lung by radon inhalation can penetrate deeply enough into the airway epithelium to damage the rapidly dividing epithelial stem cells, which are far more likely to be a source of a malignancy than superficial, non-dividing, cells. An alternative explanation is that the radioactive progeny of radon are absorbed by other pulmonary cells such as macrophages and carried deeper into lung tissue.
Carcinogenesis of Lung Cancer34,35
Radiation. There are three postulated mechanisms by which an alpha particle may initiate malignancy. Firstly, when individual cells are traversed by a single alpha particle only 20% die, and the molecular damage in the survivors doubles their gene mutation rate.36 Second, the cells surrounding those hit by the alpha particle are damaged by molecular products released from the directly hit cell, an observation known as the bystander effect. Third, much of the positive charge of an alpha particle is neutralised in tissue by removal of electrons from the abundant and nearby water molecules, initiating the production of a range of reactive oxygen species (Chapter 26).
Tobacco smoke carcinogens. Tobacco smoke contains 44 known human carcinogens, but two groups are of significance to the formation of lung cancer, polycyclic aromatic hydrocarbons (PAHs) and nitros-amines. As for radiation, much of the molecular damage is caused by the carcinogens generating reactive oxygen species. The normal defence mechanisms (page 385) are overwhelmed, and DNA, RNA, lipids and protein molecules are damaged by oxidation reactions. Many tobacco carcinogens also directly react with DNA either by causing methylation of bases within the DNA or simply by forming adducts between themselves and the DNA molecule. These various chemical changes to the DNA molecule will either interfere with transcription immediately, or induce mutations when the DNA replicates during subsequent cell divisions, so explaining why rapidly dividing cells are more susceptible to malignant change.
Molecular mechanisms of carcinogenesis. In order to appreciate how the various molecular injuries bring about malignant change it is useful to review the normal biochemical systems that produce functioning proteins within a cell from the genes within the cell nucleus, and the normal phases of the cell cycle. Transcription of genes in eukaryotic cells has several complex stages:
As part of the normal cell cycle, all cells pass through various stages of division:
 G0 – the cell is quiescent, i.e. it is metabolically active, performing its normal functions, and not moving through the cell cycle towards division;
G0 – the cell is quiescent, i.e. it is metabolically active, performing its normal functions, and not moving through the cell cycle towards division;Regulation of progression between these phases, particularly G1 to S and G2 to M, is controlled by a complex group of proteins called cyclin-dependent kinases (CDK). These proteins may, for example, be required to ‘hold’ the cell in the S phase until all the DNA has been replicated, and failure of this system will lead to premature replication of the cell, producing two progeny each with an abnormal DNA complement. Much of the activity of CDK is post-transcriptional, i.e. new CDK molecules are not being produced. Instead, control of the protein is exerted by phosphorylation and dephosphorylation of the various CDK components, with the degree of phosphorylation affecting the structure, and therefore activity, of the CDK. Alteration of a single base pair in the gene for a CDK will change a single amino acid of the CDK molecule and so fundamentally disturb the regulation of cell division.
A further way in which molecular damage within a cell may promote malignant change is via apoptosis, or programmed physiological cell death. Apoptosis is regulated by many of the same genes as those responsible for control of cell division so abnormalities of these systems may also prolong the life of a cell beyond its normal physiological term, so contributing to tumour growth.
Considering the continuous bombardment with toxic chemicals and radiation that airway cells receive and the myriad steps at which the production and function of a protein could be harmed, and cell division disturbed, it seems surprising that not everybody develops a lung malignancy. Many cells will be killed by the radiation or tobacco constituents, but the resulting tissue damage is quickly repaired, and though in the long term this repeated inflammation and repair cycle may itself damage lung tissue (see Figure 21.1), lung cancer does not result. Even more cells will be damaged, but the body’s extensive and incompletely understood cellular repair mechanisms prevent malignancy developing. For a cell to become malignant, the cellular damage must fundamentally alter the cell’s passage through the cell cycle or progress towards apoptosis in such a way that tissue growth becomes uncontrolled, which is the fundamental characteristic of a malignant cell.
The immune system has a role in preventing the development of cancer. Cell-mediated immunity involves T-lymphocytes recognising the body’s own cells via the major histocompatibility (MHC) antigens present on the surface of all cells. In cancer cells, the damage to DNA and its transcription into proteins may produce abnormal or absent MHC proteins or may cause the cell to display other abnormal peptide molecules that the T-lymphocytes recognise as abnormal. An example is the presence on the cell surface of a malignant cell of peptide chains that are similar enough to those displayed by cells infected with a virus to cause the T-lymphocyte to attack the cell. By this mechanism we are protected against the formation of clinically apparent cancers, and differences in immune responsiveness may explain why some individuals are more vulnerable to developing malignancies. Modulation of the immune response to cancer cells is a potentially very useful future strategy to improve cancer treatment and prevention, for example by immunisation.
Target genes for pulmonary carcinogenesis.35,37 Abnormal functioning of two groups of genes contribute to causing lung cancer – oncogenes and tumour suppressor genes. Oncogenes involved include:
ras genes, which code for a G-protein involved in signal transduction of growth factor receptors on the cell surface. Mutations of the ras gene in lung cancer stimulate excessive cell growth even with normal levels of growth factor. myc proteins are transcription factors that are involved in controlling the transition of cells from the G0 to G1 phases of cell division, and though the myc gene is of normal structure in lung cancer it is over-expressed, amplifying its effect. bcl-2 oncogene is normally involved in controlling cell division in the embryo or in adult stem cells, and it also has a role in controlling the timing of apoptosis. This gene is also over-expressed in some lung cancers, so facilitating cell proliferation and delaying apoptosis.The physiological function of tumour suppressor genes is to respond to stress signals within a cell. Thus if a cell undergoes a period of hypoxia, oxidative stress, or incurs damage to its DNA, these genes are activated and will either delay progress through the cell cycle to allow time for damage to be repaired, or hasten apoptosis to prevent further cellular dysfunction. Tumour suppressor genes involved in lung cancer include:
p53. Depending on the circumstances, activation of the p53 gene holds the cell in the G1 phase or induces apoptosis. A wide range of p53 mutations occur in lung cancer including deletions and altered splicing of the pre-mRNA. RB gene, which codes for one of the proteins involved in controlling transition from the G1 to S phase of cell division. Mutation of the RB gene probably produces a protein structure that is only slightly altered, but unable to be phosphorylated as required to hold the cell in the G1 phase so allowing cell division to progress too rapidly.Better understanding of the genetic basis of lung cancer should lead to improved survival from the disease, though realisation of this ambition has been limited so far. Early detection of lung cancer by identifying abnormal gene expression in bronchial epithelial cells has been hindered by the finding of many abnormalities in histologically normal cells.37 Many of the genetic abnormalities described above are associated with poor response to treatment, which is of little help to the patient involved, but hopefully will, in future, allow the best form of treatment to be determined.
Clinical Aspects38
Unfortunately, in a majority of patients their lung cancer has already spread beyond the primary tumour by the time symptoms develop. This remains the main reason for the continued poor outcomes for lung cancer treatment compared with many other malignancies. There is therefore a desperate need for a useable screening test for lung cancer, but none has yet emerged, though low-dose CT and various biomarkers39 from the blood or exhaled gases (page 415) are currently being considered. In the meantime, avoidance of tobacco smoke and radon32 remain the best options for prevention of lung cancer.
Pathology. Lung cancers can be divided into small cell lung cancer (SCLC) and non-small cell lung cancer (NSCLC), which is further divided into squamous cell carcinoma and adenocarcinoma. Squamous cell carcinomas account for around one-third of lung cancers, and mostly arise from central airways, often growing peri-bronchially to cause airway narrowing without necessarily being visible from within the airway lumen. They tend to be slow growing, metastasise late, and in the periphery of the lung may undergo central necrosis and cavitation. Adenocarcinomas also account for around one-third of lung cancers, but predominantly arise in the periphery of the lung, are faster growing than squamous cell carcinomas, and metastasise early via the blood or lymphatics. Finally, the NSCLC tumours include a range of different pathological malignancies, all of which share highly malignant characteristics including early spread via the lymphatic system.
Clinical features. Cough is the most common symptom of lung cancer, occurring in most patients at some stage of their disease, though cough is such a common complaint in smokers that this remains a very non-specific symptom. A cough arises from the lung cancer normally by direct irritation of the airway wall, either from within the lumen or from the peribronchial tissue, and is typically positional as the tumour presses on the airway in specific postures. Haemoptysis is the second commonest symptom, occurring in half of patients, and varying from staining of expectorated sputum to massive haemoptysis if the tumour erodes into a major thoracic vessel. Wheezing as a result of small airway occlusion by a peripheral lung tumour occurs in around 10% of patients and may be misdiagnosed as adult onset asthma. Narrowing of larger airways causes stridor, though this is only believed to occur when the cross-sectional airway is reduced by more than 75%. Dyspnoea, chest pain and chest infections (usually distal to an airway obstructed by tumour) are other pulmonary symptoms resulting from lung tumours. Invasion of nearby thoracic structures by a lung cancer causes a host of other presentations, such as pleural effusion (page 445), Horners or Pancoast’s syndrome from invasion of the sympathetic chain and brachial plexus nerves respectively, or obstruction of the superior vena cava. Finally, any lung cancer, but particularly NSCLC, may present with the symptoms and signs of distant metastases.
Principles of Therapy for Lung Cancer
A detailed description of the complex subject of treating lung cancer is beyond the scope of this book, and guidelines are available.40 There are three main therapeutic options and their use is dependent on many factors, of which the two most important are the type of tumour and the stage at which the disease presents:
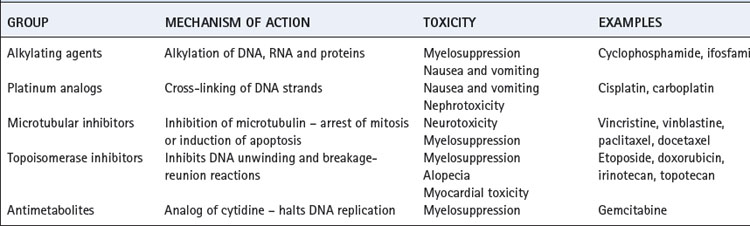
Chemotherapy may be used as an adjuvant treatment for patients having surgical management, or may be used as the mainstay of treatment in more advanced NSCLC. Some form of chemotherapy is invariably used for treatment of SCLC. Most chemotherapy regimes involve administering drugs from two or more different groups to maximise the chances of successfully killing the malignant cells. Chemotherapy is normally given in multiple short courses, mostly to allow the patient to recover from the inevitable toxicity (Table 30.2). Also, this pattern of administration may increase the efficacy of the cytotoxic drugs as repeated hits by the drug encourages the malignant cells to all become aligned in the same phase of the cell cycle. Chemotherapy alone is unlikely to ever be curative. A 1-cm lung cancer is estimated to contain 109 malignant cells.35 If a dose of chemotherapy kills 99.9% of those cells, then 106 still remain after the treatment, and this number will increase during the recovery period between treatments. Thus many treatments are required, with the associated toxicity, and in theory it is not possible to kill every malignant cell. However, given that the immune system is also known to have significant cytotoxic abilities, chemotherapy can reduce the tumour cell burden to such an extent that T-lymphocytes may completely remove the cancer.
A major determinant of the sensitivity of a tumour cell to killing by radiation is the Po2 in the cell when it is irradiated, with most mammalian tumour cells requiring two to three times more radiation to cause cell death when hypoxic. This observation supports the hypothesis that much of the molecular damage induced by radiation is mediated via reactive oxygen species. Animal studies have demonstrated that many solid tumours have hypoxic centres, an observation which is believed to result from an inability of angiogenesis to keep pace with the rapidly growing tumour so leaving some regions with no blood supply. Positron emission tomography may be used to detect hypoxic tissue within tumours, and in a study of patients with NSCLC 48% of the tumour volume was found to be hypoxic, with a majority of tumours containing areas with an estimated Po2 below 0.27 kPa (2 mmHg), a level at which radiation sensitivity would be poor.41
Pleural Disease
Physiology of the Pleural Space
Two pleural layers exist: the first lines the inside of the thoracic cavity (parietal pleura) including the diaphragm, and the second (visceral pleura) covers the lung from the hilum outwards including the major and minor pulmonary fissures. The opposing elastic forces of lung and chest wall (Chapter 3) cause a pressure of 3–5 cmH2O below atmospheric to exist in the pleural space. The pleural space facilitates mechanical coupling between the chest wall and the lungs and to do this efficiently, that is with minimal loss of energy, there should be minimal friction between the two structures. The visceral and parietal pleura must therefore slide easily against each other, and this is achieved by the presence of a small amount of pleural fluid and a layer of surfactant molecules on the surface of the mesothelial cells lining both pleural membranes.42
An average 70 kg human has a total pleural surface area of 4000 cm2 containing approximately 18 ml of pleural fluid,43 which is an ultrafiltrate of plasma containing only small amounts of protein (approximately 1 g.dl−1). Pleural fluid production is determined by the same Starling’s forces that determine movement of fluid across capillary walls (page 420). In the parietal pleura, which is supplied by the systemic circulation, the negative intra-pleural pressure results in an increased hydrostatic pressure gradient, so favouring fluid movement out of the capillary, but the pleural mesothelial cells are less permeable to protein than systemic capillaries so producing an oncotic gradient that opposes fluid movement out of the capillary. The net effect in parietal pleura is a gradient of about 6 cmH2O for fluid to move from the capillary into the pleural space.38 The blood supply of the visceral pleura derives from the bronchial circulation or the pulmonary circulation, both of which drain to the low pressure pulmonary venous system, and the hydrostatic pressure gradient is therefore much smaller than in parietal pleura, and no fluid movement is believed to occur from the visceral pleura into the pleural space. Another source of pleural fluid, particularly under pathological conditions, is direct flow from the interstitial space of the lung.
Fluid leaves the pleural space via the lymphatic system draining directly through openings, called stomata, between the parietal pleural and lymphatic channels. Stoma are up to 6 μm in diameter so permitting fluid, proteins and cells to pass through, and are probably more numerous in the caudal and diaphragmatic regions of the pleura, where pleural fluid accumulates due to gravity. Under physiological conditions pleural fluid turnover is about 0.01 ml.kg−1.h−1 but when excess fluid accumulates in the pleural space drainage can increase by about 28 times.38
Pleural Effusion
Excessive production of pleural fluid will eventually overwhelm the ability of the lymphatic system to drain the pleural space, and fluid accumulates. There are numerous reasons for excessive pleural fluid production, and these are divided into two main groups:38
Investigation of pleural effusions requires care to avoid unnecessary drainage of the effusion with the associated possibility of introducing an infection. Provided the patient’s serum protein levels are normal then the protein level in the effusion fluid will differentiate between exudates and transudates, and cytology of the cells in the effusion fluid will allow relatively easy diagnosis in 60% of malignant effusions.44
Re-expansion of lung following drainage of a pleural effusion can result in pulmonary oedema of the expanded lung, and it is suggested that no more than 1 litre of fluid should be removed at one time. There is some evidence that recently expanded lung tissue has a leaky microvasculature,45 caused either by physical loss of integrity of the tight junctions between endothelial cells or by the generation of negative interstitial hydrostatic pressures favouring movement of fluid out of the capillary (page 420). It remains unclear whether the likelihood of pulmonary oedema occurring relates to the volume of fluid removed or the amount of negative pressure created by its removal. A recent study of 185 patients found that re-expansion pulmonary oedema only occurred in 2.7% of patients and was unrelated to the volume of fluid removed provided that the intrathoracic pressure was not allowed to fall below −20 cmH2O.46
Pneumothorax
This occurs when air enters the pleural cavity either from the outside across a defect in the chest wall and parietal pleura or from the lung or mediastinum through a defect in the visceral pleura. The many causes of pneumothorax are usually divided into spontaneous and acquired aetiology and are outlined in Table 30.3.
Primary spontaneous pneumothorax is the most common cause (Figure 30.5A) and is postulated to result from rupture of small, thin-walled cysts in the immediate subpleural lung tissue, referred to as blebs if less than 2 cm in diameter or bullae if greater than 2 cm (Figure 30.5B).47 Blebs or bullae are seen in most patients presenting with a primary spontaneous pneumothorax,47 but are also present in 6% of the normal healthy population, so their role in causing pneumothorax remains controversial.48 Whatever the cause, varying degrees of collapse of a lung will inevitably impair gas exchange, and arterial hypoxaemia occurs in three-quarters of patients with a pneumothorax on presentation, with the hypoxaemia being worse with larger pneumo-thoraces or underlying lung disease.49
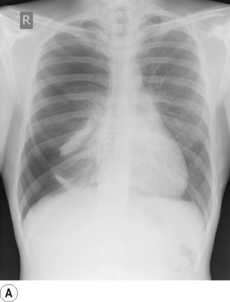
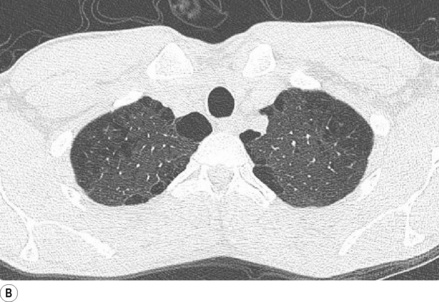
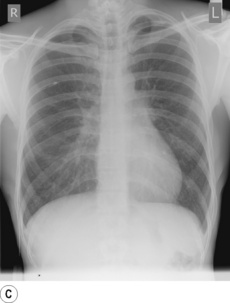
Fig. 30.5 (A) Spontaneous pneumothorax with almost complete collapse of the right lung. (B) Computerised tomography scan of the lung apices in the same patient showing multiple lung blebs. (C) The pneumothorax has been treated by the insertion of a chest drain, with complete re-expansion of the lung.
Tension pneumothorax. Occasionally, the defect in the lung or chest wall through which air gains entry to the pleura forms a valve mechanism and a tension pneumothorax occurs. During inspiration, air is sucked into the pleural space but cannot leave the pleura during expiration. A large pneumothorax develops and increased respiratory effort reduces intrapleural pressure further, until the pressure in the affected hemithorax remains above atmospheric throughout almost the entire respiratory cycle. Ventilation of the lung on the affected side ceases, the lung collapses and severe hypoxaemia occurs. A more dramatic effect is the shift of the mediastinum away from the pneumothorax, which causes a sudden and catastrophic reduction in venous return and therefore cardiac output. Insertion of a cannula into the affected hemithorax relieves the pressure, creates an open pneumothorax, and invariably saves the patient’s life.
Principles of therapy for pneumothorax
Treatment of a pneumothorax depends on its size and the patient’s symptoms.49 If the patient is not breathless and the rim of air between the lung and the inside of chest wall on a chest radiograph is less than 2 cm wide then no intervention is required. For larger or symptomatic pneumothoraces aspiration of air is performed and the chest radiograph reviewed to confirm lung expansion. If this is unsuccessful, then a chest drain is placed, complete lung re-expansion confirmed (Figure 30.5C), and the drain left in place until there is no further air leak and full lung expansion for 24 hours. If the lung fails to re-expand with a chest drain in situ or an air leak persists for days, then surgery is usually required. During surgery any visible blebs or bullae at the lung apex are resected, and a pleurodesis (page 496) performed, following which the lung is re-expanded under direct vision.
Absorption of air from the pleura. For small asymptomatic pneumothoraces, or following treatment as described above, complete resolution of a closed pneumothorax requires air to be reabsorbed from the pleura. How quickly this occurs depends on the partial pressure gradients of the various gases between the pleura and the circulation, in particular the venous blood where partial pressures are lowest (see Table 26.2). Two phases of gas reabsorption will theoretically occur: phase 1, when gases in the pleura come into equilibrium with the venous blood and phase 2, when the gas is absorbed. For phase 1, the cause of the pneumothorax is important in that the gas entering the pleura may be either air from the outside or alveolar gas from the lung. When ambient air, which is likely to be dry, enters the pleural space the first change to occur is a small increase in volume as water vapour humidifies the gas. Oxygen will be absorbed into the blood and carbon dioxide diffuses into the pleural space, but these two volume changes should be approximately equal and cause little change in volume.
For partial pressures, the loss of oxygen from the pleural air is partially offset by the gain of water vapour and carbon dioxide, but overall the nitrogen partial pressure increases slightly, and some nitrogen will slowly be absorbed into the circulation. When alveolar gas, which is already humidified and contains carbon dioxide, enters the pleural space the only change will be the absorption of a small amount of oxygen and a slight reduction in the volume of the pneumothorax, and therefore lung re-expansion. In patients breathing oxygen at the time of a pneumo-thorax (originating from the lung) the situation is, in theory, more favourable as the alveolar gas is now almost entirely oxygen and most of the pneumothorax will be quickly absorbed into the blood stream. A similar phenomenon has been observed if the pneumothorax involves carbon dioxide entering the pleural space during laparoscopic surgery (technically a capnothorax) following which complete resolution occurred within two hours.50 In both these situations nitrogen from the blood will diffuse into the pleural space, but this process is very slow compared with oxygen or carbon dioxide absorption.
In phase 2 of reabsorption of a pneumothorax the partial pressures of each gas in the pleural space are in equilibrium with the venous blood. Fortunately the subatmospheric total gas partial pressure of venous blood (Table 26.2) maintains small gradients that facilitate slow reabsorption of the pneumothorax. A further theoretical benefit of breathing oxygen may be obtained during phase 2 of pneumothorax reabsorption. The greater the inspired oxygen concentration the lower is the blood Pn2, so increasing the rate of diffusion of nitrogen from the pleura into the blood. An opposite problem occurs when using nitrous oxide in patients with a closed pneumothorax, when the nitrous oxide in the blood diffuses down its concentration gradient into the pneumothorax, increasing its volume.
These theoretical considerations are harder to demonstrate in practice. Though not investigated for some years, there is agreement that absorption of gas from a pneumothorax is slow, with the most widely quoted estimate being that 1.8% of the hemithorax volume is absorbed per day.51 This means that a small pneumothorax occupying 15% of hemithorax volume will take approximately 10 days to resolve fully. Animal studies have shown a dose-dependent reduction in the time taken for a pneumothorax to resolve with increasing inspired oxygen fraction, the duration being approximately halved by breathing 50% oxygen compared with air.52 A small study in humans from 1971 found that during periods breathing an unspecified high oxygen concentration resolution of the pneumothorax was approximately 4 times faster than when breathing air.53
Empyema38
Empyema thoracis is a condition resulting from bacterial infection of the normally sterile pleural space. Almost two-thirds of patients with pneumonia develop a ‘simple’, non-infected, pleural effusion by direct movement of interstitial fluid from the infected lung tissue into the pleural space.54 In around 10% of these effusions bacterial spread from the underlying pneumonia follows, and an empyema develops. Other, less common, causes of empyema include its development as a complication of trauma, thoracic surgery, pneumothorax or diagnostic thoracocentesis. The bacterial infection of the pleural fluid follows the normal stages of inflammation, with influx of white cells eventually causing the formation of pus. In empyema, deposition of fibrin begins early and is aggressive, and within a few weeks a thick layer of collagen (referred to as ‘rind’ or ‘peel’) is deposited on both pleural spaces. If left untreated the process continues until pleural fibrosis causes contraction of the chest wall and lung (fibrothorax).
A restrictive pattern of reduced lung function occurs with forced expiratory volume in one second (FEV1) and forced vital capacity (FVC) values of half predicted normal being typical.55 Early intervention with antibiotics, fibrinolysis or chest drainage may limit the progression of the fibrotic process.56 However if these less invasive therapies fail or restrictive lung disease is already apparent, more extensive intervention is required in the form of surgical drainage or decortication, in which the peel formed on the pleura, particularly the parietal layer, is stripped off to allow the underlying lung to re-expand. Decortication is associated with a significant mortality and numerous other serious complications but improves lung function significantly,55 though return to normal lung volumes rarely occurs.38
References
1. Klocke FJ, Rahn H. The arterial-alveolar inert gas (‘N2’) difference in normal and emphysematous subjects, as indicated by the analysis of urine. J Clin Invest.. 1961;40:286-294.
2. Webb SJS, Nunn JF. A comparison between the effect of nitrous oxide and nitrogen on arterial Po2. Anaesthesia. 1967;22:69-81.
3. Nunn JF, Coleman AJ, Sachithanandan T, Bergman NA, Laws JW. Hypoxaemia and atelectasis produced by forced expiration. Br J Anaesth.. 1965;37:3-12.
4. Nunn JF, Williams IP, Jones JG, Hewlett AM, Hulands GH, Minty BD. Detection and reversal of pulmonary absorption collapse. Br J Anaesth.. 1978;50:91-100.
5. Wagner PD, Laravuso RB, Uhl RR, West JB. Continuous distributions of ventilation-perfusion ratios in normal subjects breathing air and 100% O2. J Clin Invest.. 1974;54:54-68.
6. Dantzker DR, Wagner PD, West JB. Instability of lung units with low VA/Q ratios during O2 breathing. J Appl Physiol.. 1975;38:886-895.
7. Ashizawa K, Hayashi K, Aso N, Minami K. Lobar atelectasis: diagnostic pitfalls on chest radiography. Br J Radiol.. 2001;74:89-97.
8. Kreider ME, Lipson DA. Bronchoscopy for atelectasis in the ICU. Chest. 2003;124:344-350.
9. Gea J, Roca J, Torres A, Agusti AGN, Wagner PD, Rodriguez-Roisin R. Mechanisms of abnormal gas exchange in patients with pneumonia. Anesthesiology. 1991;75:782-789.
*10. Mizgerd JP. Acute lower respiratory tract infection. N Engl J Med.. 2008;358:716-727.
11. Hogg JC, Walker BA. Polymorphonuclear leucocyte traffic in lung inflammation. Thorax. 1995;50:819-820.
12. D’Ambrosio D, Mariani M, Panina-Bordignon P, Sinigaglia F. Chemokines and their receptors guiding T lymphocyte recruitment in lung inflammation. Am J Respir Crit Care Med.. 2001;164:1266-1275.
*13. Doerschuk CM, Tasaka S, Wang Q. CD11/CD18-dependent and -independent neutrophil emigration in the lungs. How do neutrophils know which route to take. Am J Respir Cell Mol Biol.. 2000;23:133-136.
14. Doerschuk CM, Mizgerd JP, Kubo H, Qin L, Kumasaka T. Adhesion molecules and cellular biomechanical changes in acute lung injury. Chest. 1999;116:37S-43S.
15. Walker DC, Behzad AR, Chu F. Neutrophil migration through preexisting holes in the basal laminae of alveolar capillaries and epithelium during streptococcal pneumonia. Microvasc. Res.. 1995;50:397-416.
16. Mossman BT, Churg A. Mechanisms in the pathogenesis of asbestosis and silicosis. Am J Respir Crit Care Med.. 1998;157:1666-1680.
17. Movsas B, Raffin TA, Epstein AH, Link CJ. Pulmonary radiation injury. Chest. 1997;111:1061-1076.
18. Rubin P, Johnston CJ, Williams JP, McDonald S, Finkelstein JN. A perpetual cascade of cytokines postirradiation leads to pulmonary fibrosis. Int J Radiat Oncol Biol Phys.. 1995;33:99-109.
19. Noth I, Martinez FJ. Recent advances in idiopathic pulmonary fibrosis. Chest. 2007;132:637-650.
20. Verma S, Slutsky AS. Idiopathic pulmonary fibrosis – new insights. N Engl J Med.. 2007;356:1371-1372.
*21. Thannickal VJ, Toews GB, White ES, Lynch JP, Martinez FJ. Mechanisms of pulmonary fibrosis. Annu Rev Med.. 2004;55:395-417.
22. Kumar RK, Lykke AWJ. Messages and handshakes: cellular interactions in pulmonary fibrosis. Pathology. 1995;27:18-26.
23. Bartram U, Speer CP. The role of transforming growth factor β in lung development and disease. Chest. 2004;125:754-765.
24. Uhal BD. Apoptosis in lung fibrosis and repair. Chest. 2002;122:293S-298S.
25. Pierce RA, Mariani TJ, Senior RM. Elastin in lung development and disease. Ciba Found Symp.. 1995;192:199-214.
26. Gross TJ, Hunninghake GW. Idiopathic pulmonary fibrosis. N Engl J Med.. 2001;345:517-525.
27. Bouros D, Antoniou KM. Current and future therapeutic approaches in idiopathic pulmonary fibrosis. Eur Respir J. 2005;26:693-702.
28. Herbst RS, Heymach JV, Lippman SM. Lung cancer. N Engl J Med.. 2008;359:1367-1380.
29. Alberg AJ, Ford JG, Samet JM. Epidemiology of lung cancer. Chest. 2007;132:29S-55S.
30. Parkin DM, Bray F, Ferlay J, Pisani P. Global cancer statistics, 2002. CA Cancer J Clin.. 2005;55:74-108.
31. Zang EA, Wynder EL. Differences in lung cancer risk between men and women: Examination of the evidence. J Natl Cancer Inst.. 1996;88:183-192.
32. Gray A, Read S, McGale P, Darby S. Lung cancer deaths from indoor radon and the cost effectiveness and potential of policies to reduce them. BMJ. 2009;338:215-218.
33. Samet J. Residential radon and lung cancer: End of the story? J Toxicol Environ Health Part A. 2006;69:527-531.
34. Alavanja MCR. Biologic damage resulting from exposure to tobacco smoke and from radon: implication for preventive interventions. Oncogene. 2002;21:7365-7375.
35. Pass HI, Mitchell JB, Jihnson DH, Turrisi AT, Minna JD, editors. Lung Cancer, Principles and Practice. Philadelphia: Lippincott Williams & Wilkins, 2000.
36. Hei TK, Wu L-J, Liu S-X, Vannais D, Waldren CA, Randers-Pehrson G. Mutagenic effects of a single and an exact number of particles in mammalian cells. Proc Natl Acad Sci.. 1997;94:3765-3770.
37. Sun S, Schiller JH, Spinola M, Minna JD. New molecular targeted therapies for lung cancer. J Clin Invest.. 2007;117:2740-2750.
38. Shields TW, LoCicero J, Ponn RB, Rusch VW, editors. General Thoracic Surgery, 6th ed., Philadelphia: Lippincott Williams & Wilkins, 2005.
39. Hirschowitz EA. Biomarkers for lung cancer screening: interpretation and implications of an early negative advanced validation study. Am J Respir Crit Care Med.. 2009;179:1-3.
40. Alberts WM. Diagnosis and management of lung cancer. Executive summary: ACCP Evidence-based clinical practice guidelines. Chest. 2007;132:1-19.
41. Rasey JS, Koh W, Evans ML, et al. Quantifying regional hypoxia in human tumors with positron emission tomography of [18F] fluoromisonidazole: A pretherapy study of 37 patients. Int J Radiat Oncol Biol Phys.. 1996;136:417-428.
42. Miserocchi G. Physiology and pathophysiology of pleural fluid turnover. Eur Respir J. 1997;10:219-225.
*43. Zocchi L. Physiology and pathophysiology of pleural fluid turnover. Eur Respir J. 2002;20:1545-1558.
44. Maskell NA, Butland RJA. BTS guidelines for the investigation of a unilateral pleural effusion in adults. Thorax. 2003;58:8-17.
45. Wilkinson PD, Keegan J, Davies SW, Bailey J, Rudd RM. Changes in pulmonary microvascular permeability accompanying re-expansion oedema: evidence from dual isotope scintigraphy. Thorax. 1990;45:456-459.
46. Feller-Kopman D, Berkowitz D, Boiselle P, Ernst A. Large-volume thoracentesis and the risk of re-expansion pulmonary edema. Ann Thorac Surg.. 2007;84:1656-1662.
47. Amjadi K, Alvarez GG, Vanderhelst E, Velkeniers B, Lam M, Noppen M. Prevalence of blebs or bullae among young healthy adults. A thoracoscopic investigation. Chest. 2007;132:1140-1150.
48. Baumann MH. To bleb or not to bleb? Chest. 2007;132:1110-1112.
49. Henry M, Arnold T, Harvey J. BTS guidelines for the management of spontaneous pneumothorax. Thorax. 2003;58(suppl 1):39-52.
50. Karayiannakis AJ, Anagnostoulis S, Michailidis K, Vogiatzaki T, Polychronidis A, Simopoulos C. Spontaneous resolution of massive right-sided pneumothorax occurring during laparoscopic cholecystectomy. Surg Laparosc Endosc Percutan Tech.. 2005;15:100-103.
51. Flint K, Al-Hillawi AH, Johnson NMcI. Conservative management of spontaneous pneumothorax. Lancet. 1984;323:687-688.
52. England GJ, Hill RC, Timberlake GA, et al. Resolution of experimental pneumothorax in rabbits by graded oxygen therapy. J Trauma.. 1998;45:333-334.
53. Northfield TC. Oxygen therapy for spontaneous pneumothorax. BMJ. 1971;4:86-88.
54. Anon. Managing empyema in adults. Drug Ther Bull.. 2006;44:17-21.
55. Rzyman W, Skokowski J, Romanowicz G, Lass P, Dziadziuszko R. Decortication in chronic pleural empyema – effect on lung function. Eur J Cardiothorac Surg.. 2002;21:502-507.
56. Davies CWH, Gleeson FV, Davies RJO. BTS guidelines for the management of pleural infection. Thorax. 2003;58(suppl 2):18-28.