Chapter 14 Pregnancy, neonates and children
 Hormonal changes of pregnancy stimulate breathing, causing an increase in tidal volume and hypocapnia. Human lung development is incomplete at birth with new alveoli continuing to form until around 3 years of age.
Hormonal changes of pregnancy stimulate breathing, causing an increase in tidal volume and hypocapnia. Human lung development is incomplete at birth with new alveoli continuing to form until around 3 years of age.Respiratory Function in Pregnancy1
Several physiological changes occur during pregnancy that affect respiratory function. Fluid retention resulting from increasing oestrogen levels causes oedema throughout the airway mucosa, and increases blood volume, substantially increasing oxygen delivery. Progesterone levels rise six-fold through pregnancy and have significant effects on the control of respiration and therefore arterial blood gases. Finally, in the last trimester of pregnancy, the enlarging uterus has a direct impact on respiratory mechanics. A summary of the changes for common respiratory measurements is shown in Table 14.1.
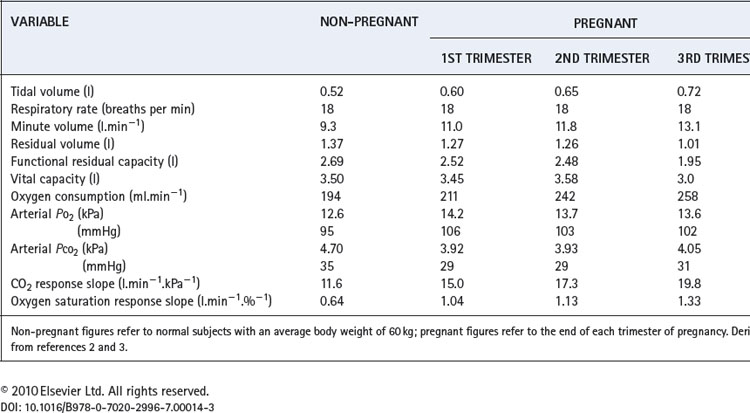
Lung volumes. During the last third of pregnancy the diaphragm becomes displaced cephalad by the expansion of the uterus into the abdomen. This reduces both the residual volume (by about 20%) and expiratory reserve volume, such that functional residual capacity is greatly reduced (Table 14.1). This is particularly true in the supine position, and effectively removes one of the largest stores of oxygen available to the body, making pregnant women very susceptible to hypoxia during anaesthesia or with respiratory disease.
Vital capacity, forced expiratory volume in one second and maximal breathing capacity are normally unchanged during pregnancy.
Oxygen consumption. Oxygen consumption increases throughout pregnancy peaking at between 15% and 30% above normal at full term.2,4 The increase is mainly attributable to the demands of the fetus, uterus and placenta, such that when oxygen consumption is expressed per kg of body weight there is little change.
Ventilation. Respiratory rate remains unchanged whilst tidal volume, and therefore minute volume of ventilation, increase by up to 40% above normal at full term. The increase in ventilation is beyond the requirements of the enhanced oxygen uptake or carbon dioxide production so alveolar and arterial Pco2 are reduced to about 4 kPa (30 mmHg).3 This must facilitate clearance of carbon dioxide by the fetus. There is also an increase in alveolar and arterial Po2 of about 1 kPa (7.5 mmHg).
The hyperventilation is attributable to progesterone, and the mechanism is assumed to be a sensitisation of the central chemoreceptors. Pregnancy gives rise to a three-fold increase in the slope of a Pco2/ventilation response curve.2 The hypoxic ventilatory response is increased two-fold, most of the change occurring before the mid-point of gestation, at which time oxygen consumption has hardly begun to increase.5
Dyspnoea occurs in more than half of pregnant women, often beginning early in pregnancy, before the mass effect of the uterus becomes apparent. Dyspnoeic pregnant women, compared with non-dyspnoeic controls, show a greater degree of hyperventilation in spite of having similar plasma progesterone levels.2 Dyspnoea early in pregnancy therefore seems to arise from a greater sensitivity of the chemoreceptors to the increase in progesterone levels. In the third trimester, when dyspnoea on mild exercise is almost universal, the extra effort required by the respiratory muscles to increase tidal volume is believed to be responsible for breathlessness rather than an altered perception of respiratory discomfort.6
The Lungs Before Birth
Embryology
The lungs develop in four stages, under the control of a host of transcriptional factors:7-11
The lungs begin to contain surfactant and are first capable of function by approximately 24–26 weeks, this being a major factor in the viability of premature infants.
Lung Liquid
Fetal lungs contain ‘lung liquid’ (LL) which is secreted by the pulmonary epithelial cells and flows out through the developing airway into the amniotic fluid or gastrointestinal tract, flushing debris from the airways as it does so. A more important function of LL seems to be to prevent the developing lung tissues from collapsing. It is thought that LL maintains the lung at a slight positive pressure relative to the amniotic fluid, and that this expansion is responsible for stimulating cell division and lung growth, particularly with respect to airway branching.10 The respiratory tract in late pregnancy contains some 40 ml of LL, but its turnover is rapid, believed to be of the order of 500 ml per day. Its volume corresponds approximately with the functional residual capacity (FRC) after breathing is established.9
Fetal breathing movements also contribute to lung development. In humans they begin in the middle trimester of pregnancy, and are present for over 20 minutes per hour in the last trimester,13 normally during periods of general fetal activity. During episodes of breathing, the frequency is about 45 breaths per minute and the diaphragm seems to be the main muscle concerned, producing an estimated fluid shift of about 2 ml at each ‘breath’.
Maintenance of a positive pressure in the developing lung requires the upper airway to offer some resistance to the outflow of LL.9 During apnoea, elastic recoil of the lung tissue and continuous production of LL are both opposed by intrinsic laryngeal resistance and a collapsed pharynx. Fetal inspiratory activity, as in the adult, includes dilation of the upper airway. With quiet breathing this would allow increased efflux of LL from the airway, but simultaneous diaphragmatic contraction opposes this. During vigorous breathing movements with the mouth open, pharyngeal fluid may be ‘sucked’ into the airway thus contributing to the expansion of the lungs. Thus fetal breathing movements are believed to contribute to maintaining lung expansion, and their abolition is known to impair lung development.9
Lung Development and Lung Function Later in Life
Complications of pregnancy such as maternal hypertension, pre-eclampsia or the use of antibiotics have all been shown to increase the incidence of wheezing in childhood.14 Small abnormalities of lung development in utero or in the early post-natal phase can adversely affect lung function beyond childhood and into adult life.10,15 For example, in genetically susceptible individuals maternal smoking causes irreversible abnormalities of airway calibre. Abnormalities of amniotic fluid turnover may impair normal lung development in utero, and premature birth alters the normal process of alveolar formation with long term impairment in lung function (see below).
The Fetal Circulation
The fetal circulation differs radically from the postnatal circulation (Figure 14.1). Blood from the right heart is deflected away from the lungs, partly through the foramen ovale and partly through the ductus arteriosus. Less than 10% of the output of the right ventricle reaches the lungs, the remainder passing to the systemic circulation and the placenta. Right atrial pressure exceeds left atrial pressure and this maintains the patency of the foramen ovale. Furthermore, because the vascular resistance of the pulmonary circulation exceeds that of the systemic circulation before birth, pressure in the right ventricle exceeds that in the left ventricle and these factors control the direction of flow through the ductus arteriosus.
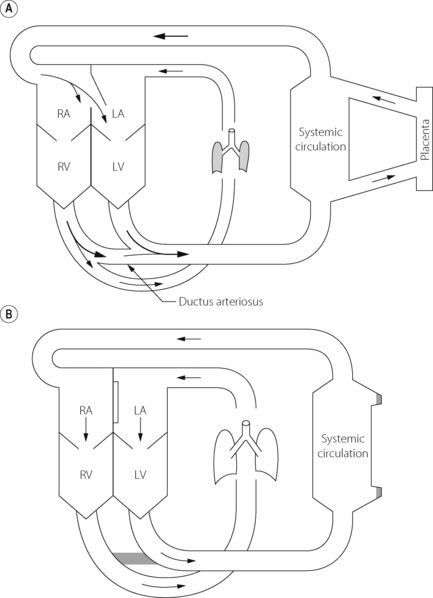
Fig. 14.1 Fetal circulation (A) compared with adult circulation (B). The foramen ovale is between right atrium (RA) and left atrium (LA). RV and LV, right and left ventricles.
The umbilical veins drain via the ductus venosus into the inferior vena cava, which therefore contains better oxygenated blood than the superior vena cava. The anatomy of the atria and the foramen ovale is such that the better oxygenated blood from the inferior vena cava passes preferentially into the left atrium and thence to the left ventricle and so to the brain. (This is not shown in Figure 14.1). Overall gas tensions in the fetus are of the order of 6.4 kPa (48 mmHg) for Pco2 and 4 kPa (30 mmHg) for Po2. The fact that the fetus remains apnoeic for much of the time in utero with these blood-gas levels is probably in part attributable to central hypoxic ventilatory depression (page 74).
Events at Birth
Oxygen stores in the fetus are small and it is therefore essential that air breathing and oxygen uptake be established within a few minutes of birth. This requires radical changes in the function of both lungs and circulation.
Factors in the Initiation of Breathing
Most infants take their first breath within 20 s of delivery, and rhythmic respiration is usually established within 90 s. Thoracic compression during vaginal delivery followed by recoil of the rib cage causes air to be drawn passively into the lungs. However, the major stimuli to breathing are probably the cooling of the skin and mechanical stimulation, both acting via the respiratory centre. Without this, babies born via Caesarean section would suffer greatly from immediate respiratory difficulties, which is not the case in practice. Hypoxaemia, resulting from apnoea or clamping of the cord, is unlikely to be a reliable respiratory stimulus at this time because of central hypoxic ventilatory depression (see above).
Fate of the fetal lung liquid. The volume of LL decreases just before and during labour. Some of the residual fluid may be expressed during a vaginal delivery but this is not thought to be a major factor. During in-utero life, the pulmonary epithelium actively secretes lung liquid but at birth this process reverses and the epithelial cells switch to absorption of fluid from the airway.16 Absorption of fluid from airways and alveoli is an active process facilitated by a sodium channel (page 421). The sodium channels are primed during the third trimester by thyroid and steroid hormones, and at birth, fetal adrenaline and oxygen trigger the channels to become active. Aquaporin, a trans-membrane protein that facilitates water transport across membranes, is present in lungs at birth but its role remains uncertain.16,17
Changes in the Circulation
The geometry of the circulation changes radically and quickly at birth. The establishment of spontaneous breathing causes a massive decrease in the vascular resistance of the pulmonary circulation, due partly to mechanical factors and partly to changes in blood gases. Simultaneously there is an increase in the resistance of the systemic circulation, due partly to vasoconstriction and partly to cessation of the placental circulation. As a result, the right atrial pressure falls below the left atrial pressure, to give the relationship that is then maintained throughout life. This normally results in closure of the foramen ovale (Figure 14.1), which is followed by closure of the ductus arteriosus as a result of active vasoconstriction of its smooth muscle layer in response to increased Po2. The circulation is thus converted from the foetal mode, in which the lungs and the systemic circulation are essentially in parallel, to the adult mode in which they are in series.
Mechanism of reduced pulmonary vascular resistance at birth. Pulmonary vascular resistance declines due to a combination of ventilation of the lung and changes in blood gases, particularly increasing Po2. Clearly this is a difficult area to study in humans, and most work has been performed in other mammals,9 but there is no reason to expect humans to differ significantly. Removal of LL from the lung establishes an air–liquid interface that is responsible for a rapid increase in lung recoil pressure, which, possibly along with changes in chest wall compliance, results in a negative intrapleural pressure as in adult lungs. This creates the transmural pressure gradient between the alveoli and pleura, which physically dilates the pulmonary capillaries (page 104). These mechanical forces leading to a reduction in pulmonary vascular resistance are believed to account for over half of the observed changes at birth.
Further reductions in pulmonary vascular resistance as a result of increased Po2 and decreased Pco2 are believed to be endothelium dependent.18 Though many mediators may be involved, prostaglandins, endothelin and nitric oxide are the most widely studied. The first two groups have conflicting effects on the neonatal pulmonary circulation, with in vitro studies showing both vasodilating and vasoconstricting effects of different individual mediators. Prostacyclin seems to be involved in the in vivo vasodilation of pulmonary blood vessels at birth, but the effect is minor. In animals, inhibition of nitric oxide synthase prior to birth attenuates the reduction in pulmonary vascular resistance on delivery.
Persistent pulmonary hypertension of the newborn (PPHN)19 occurs in about 1 in 1000 births. Pulmonary vascular resistance remains elevated, so right heart pressures remain high and a significant right-to-left shunt continues with resulting hypoxaemia. Although PPHN may occur with other parenchymal lung problems such as meconium aspiration or respiratory distress syndrome, it also occurs in isolation. Mechanical changes at birth leading to pulmonary vasodilation, as described above, still occur and are probably responsible for bringing about sufficient pulmonary blood flow for immediate survival. However, structural abnormalities of pulmonary vessels are common in PPHN, and may limit the vasodilation obtained by mechanical factors. There is undoubtedly an element of abnormal pulmonary vasoconstriction, or at least a failure of oxygen-stimulated vasodilation, in babies with PPHN and abnormalities of both endothelin and nitric oxide activity are implicated.
The Apgar Score
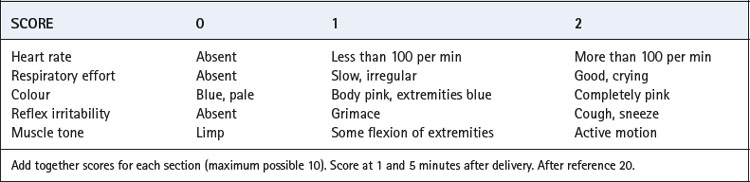
The scoring system devised many years ago by Virginia Apgar is still widely accepted as an assessment of the overall condition of the neonate. This is based on scoring of a scale of 0–2 for five attributes, two of which are related to respiration (Table 14.2).20 The total score is the sum of each of the five constituent scores and is best undertaken 1 and 5 minutes after delivery. Scores of 8–10 are regarded as normal.
Neonatal Lung Function
Mechanics of breathing. Functional residual capacity is about 30 ml.kg−1 and total respiratory compliance 50 ml.kPa−1 (5 ml.cmH2O−1). Most of the impedance to expansion is due to the lung and depends primarily on the presence of surfactant in the alveoli. The chest wall of the neonate is highly compliant. This contrasts with the adult, in whom compliance of lung and chest wall are approximately equal. Total respiratory resistance is of the order of 7 kPa.l−1.s (70 cmH2O.l−1.s), most of which is in the bronchial tree. Compliance is about one-twentieth that of an adult and resistance about 15 times greater. At the first breath the infant is capable of generating a subatmospheric intra-thoracic pressure of the order of 7 kPa (70 cmH2O).
Ventilation and gas exchange. For a 3 kg neonate, the minute volume is about 0.6 l.min−1, with a high respiratory frequency of 25–40 breaths per minute. Dead space is close to a half of tidal volume, giving a mean alveolar ventilation of about 0.3 l.min−1 for a neonate of average size. There is a shunt of about 10% immediately after birth. However, distribution of gas is better than in the adult and there is, of course, a negligible hydrostatic pressure gradient in the vertical axis of the tiny lungs of an infant.
Oxygen consumption is of the order of 20–30 ml.min−1, depending on weight in the range 2–4 kg. Arterial Pco2 is close to 4.5 kPa (34 mmHg) and Po2 9 kPa (68 mmHg). Because of the shunt of 10%, there is an alveolar/arterial Po2 gradient of about 3.3 kPa (25 mmHg) compared with less than half of this in a young adult. Arterial pH is within the normal adult range.
Control of breathing. Animal studies have shown that, in the fetus, carotid chemoreceptors are active but at a much lower Po2 than in adults; the ventilatory response curve is displaced far to the left compared with adults. Prolonged periods of apnoea seen in utero in spite of this carotid sinus activity occur because of brain stem inhibition of the respiratory centre. In contrast to this, cardiovascular responses to hypoxia are well developed in the fetus, bradycardia and vasoconstriction being well recognised responses to hypoxia in neonates as shown by the Apgar score. After birth, there is a very rapid transition towards the adult pattern of respiratory control. Brainstem hypoxic ventilatory depression ceases, and the carotid chemoreceptors ‘reset’ to adult values within a few weeks.21 Ventilatory response to carbon dioxide appears to be similar to that in the adult if allowance is made for body size, although the response is depressed in REM sleep22 and the apnoeic threshold (page 70) may be closer to the normal Pco2 in neonates resulting in a susceptibility to apnoea.23
At birth, changes in respiratory pattern must, by necessity, be substantial as the long periods of apnoea seen in utero are incompatible with life in the outside world. Although most changes occur shortly after birth, complete transition to ‘adult’ respiration may take some weeks to complete, particularly in premature and small babies, and those with other respiratory problems that cause repeated periods of hypoxia.24 In the meantime, newborn infants have a variety of breathing patterns. For example, ‘periodic breathing’ consists of slowly oscillating changes in respiratory rate and tidal volume size; ‘periodic apnoea’ consists of a series of respiratory pauses of over four seconds’ duration with a few normal breaths in between. In normal babies aged under two months of age, there may be in excess of 200 apnoeic episodes and 50 minutes of periodic breathing per day,25 and these may be associated with short lived reductions in saturation.26 The proportion of time spent with regular breathing increases with age, such that, beyond three months old, periodic breathing and apnoeas are significantly less.25 Moderate reductions in inspired oxygen (15%), similar to that seen during flying or at altitude (Chapter 17), cause a dramatic increase in the amount of time 3-month-old infants spend with periodic apnoea, indicating that adult hypoxic ventilatory responses are not fully developed.27
Haemoglobin. Children are normally born polycythaemic with a mean haemoglobin of about 18 g.dl−1 and a haematocrit (packed cell volume) of 53%. Some 70% of the haemoglobin is HbF and the resultant P50 is well below the normal adult value (see Figure 11.9). Arterial oxygen content is close to the normal adult value in spite of the low arterial Po2. The haemoglobin concentration decreases rapidly to become less than the normal adult value by 3 weeks of life. HbF gradually disappears from the circulation to reach negligible values by 6 months, by which time the P50 has already attained the normal adult value.
Premature Birth and the Lungs
Respiratory Distress Syndrome (RDS)28
The syndrome comprises respiratory distress within a few hours of birth and occurs in 2% of all live births, but with a greatly increased incidence in premature infants. The essential lesion is a deficiency of surfactant, which is first detectable in the fetal lung at 20–24 weeks of gestation, but the concentration increases rapidly after the 30th week. Therefore, prematurity is a major factor in the aetiology of RDS, though male babies, Caesarean delivery, perinatal stress or birth asphyxia, and maternal diabetes are all additional risk factors for its development. There is believed to be a genetic susceptibility to developing RDS, possibly resulting from inherited variations in surfactant proteins A and B (page 29).
The disease presents with difficulty in inspiration against the decreased compliance due to the high surface tension of the surfactant-deficient alveolar lining fluid. This progresses to ventilatory failure, alveolar collapse, hyaline membrane deposit, pulmonary oedema leading to denaturing of surfactant, and ultimately interference with gas exchange, resulting in severe hypoxaemia. Increased pulmonary vascular resistance may raise right atrial pressure and reopen the foramen ovale, so increasing the shunt.
Principles of therapy. The physiological basis of therapy is to supplement surfactant activity and employ artificial ventilation as a temporary expedient to spare the infant the excessive work of breathing against stiff lungs.
Surfactant replacement therapy is difficult because endogenous surfactant is complex, consisting of phospholipids and protein components (page 29).29,30 Exogenous surfactants may be either synthetic, consisting mostly of phospholipids, or natural surfactant preparations, obtained from mammalian lungs, which contain both phospholipid and some of the surfactant proteins, though not necessarily of the same type and proportion as in humans. Surfactant proteins are important to facilitate spreading of the surfactant around the lung following administration by intra-tracheal instillation, and natural surfactants may therefore be more effective as therapeutic agents.29 Surfactant replacement therapy has now been conclusively shown to improve survival and reduce complication rates in RDS, with greater efficacy when given earlier in the course of the disease.31
Artificial ventilation is considered in Chapter 32. A high respiratory frequency is required such that inspiratory and expiratory durations may be as little as 0.3 second, but inflation pressures are similar to those used in adults and do not usually exceed 3 kPa (30 cmH2O). Both the compressible volume of the ventilator circuit and the apparatus dead space tend to be large in relation to the size of very small children so pressure generators are preferable to volume generators.
Extracorporeal membrane oxygenation (ECMO) is described in Chapter 32. In contrast to its use in adults, ECMO is of proven benefit in infants (page 487) reducing mortality and long term disability in severe neonatal respiratory failure from a variety of causes,32 including RDS. Unfortunately, most cases of RDS cannot be treated with ECMO as a result of technical problems in babies of less than 2 kg weight or 35 weeks gestation.
Partial liquid ventilation with perflubron, a synthetic oxygen carrier (page 197), has been successfully used in neonates with severe RDS.33 A volume of Perflubron approximately equal to the infant’s FRC is instilled into the lungs, and positive pressure ventilation by conventional methods continued. The liquid improves lung function by replacing the alveolar air–liquid interface, by physically preventing alveolar collapse, and by increasing lung compliance allowing more effective ventilation. Chest radiographs show the extent to which partial liquid ventilation replaces normal gas-filled lung, and also shows that clearance of Perflubron by evaporation from the lung takes some time (Figure 14.2).
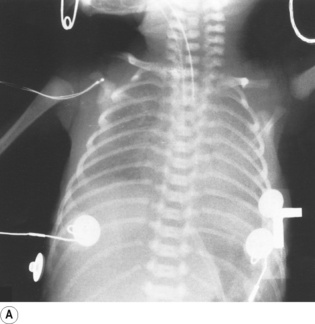
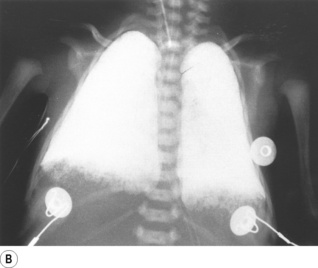
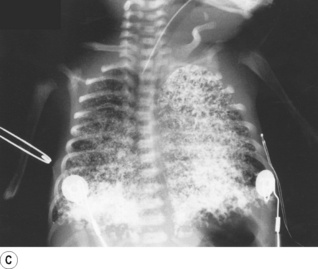
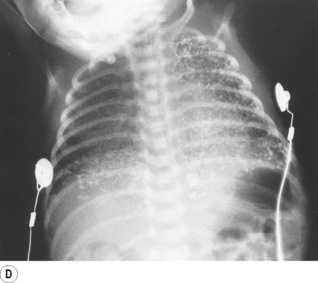
Fig. 14.2 Chest radiographs of an infant receiving partial liquid ventilation for severe respiratory distress syndrome. (A) Conventional ventilation for respiratory distress syndrome. (B) partial liquid ventilation with Perflubron. (C) 48 hours after termination of liquid ventilation, and (D) three weeks later.
(After reference 33 with permission of the authors and the publishers of New England Journal of Medicine. Copyright © 1996 Massachusetts Medical Society. All rights reserved.)
Bronchopulmonary Dysplasia30
Bronchopulmonary dysplasia describes a condition in which a neonate requires oxygen therapy for more than 28 days because of poor lung function. It is a common complication of RDS, and may simply be a form of pulmonary barotrauma (page 482) in the ventilated neonate or may represent abnormal lung development as a result of prematurity. Airway damage, including smooth muscle hypertrophy, inflammation and fibrosis all occur, and the alveolar stage of lung development (page 250) may be abnormal. Long-term impairment of lung function results, at least into childhood and possibly throughout the patient’s whole life.30,34
Sudden Infant Death Syndrome (SIDS)35,36
This is defined simply as the sudden death of an infant younger than 1 year of age that remains unexplained after review of the clinical history, examination of the circumstances of the death and a post-mortem examination. The peak incidence is at 2–3 months of age and there remains a multitude of theories regarding the aetiology, though the respiratory system is implicated in most. A triple risk hypothesis is proposed, which suggests that an infant must be vulnerable to SIDS, must be at a critical stage of development of homeostatic control and must receive a stressor such as an infection, parental smoking or sleeping in the prone position.36
Abnormal homeostatic control may include respiratory disturbances. The apnoea hypothesis remains popular, mainly because of the frequent periods of apnoea and desaturation observed in almost all babies under 3 months old (see above). The peak incidence of SIDS corresponds to the period of development when the fetal and adult systems for ventilatory control are swapping over, and it is believed that this may make the infant susceptible to respiratory disturbances. Normal patterns of arousal from sleep are altered in babies who subsequently become SIDS victims.37 Post-mortem studies have found decreased binding of serotonin in several areas of the brain including an area which, in adults, is believed to be crucial in controlling arousal from sleep.35 Despite the popularity of the apnoea hypothesis evidence that these episodes of periodic breathing or apnoeas contribute directly to SIDS is lacking.
Sleeping position. There is a substantial body of agreement that the prone sleeping position is commoner in infants dying of SIDS, though the mechanism remains uncertain. In the late 1980s and early 1990s many countries introduced national health educational policies to encourage the avoidance of the prone sleeping position, and the incidence of SIDS was reduced by 50–90%.
Development of Lung Function During Childhood
The lungs continue to develop during childhood. Chest wall compliance, which is very high at birth, decreases rapidly for the first two years of life, when it becomes approximately equal to lung compliance as in the adult.38 Below the age of 8 years, measurement of lung volumes is difficult, but beyond this age many studies of normal lung function are available. Because of large variations in the rate at which children grow, reference values are usually related to height rather than age or weight. Equations relating lung volumes to height are available,39 and some are shown in graphical form in Figure 14.3.
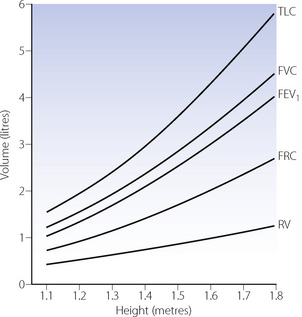
Fig. 14.3 Changes in lung volumes as a function of stature. When considering reference values for children, height in metres is used in preference to age to allow for large differences in growth rate. Each graph represents the mean for both boys and girls, though boys generally have greater values at equivalent heights.
Various indices of respiratory function are independent of age and body size so that adult values can be used. These include forced expiratory volume (1 second) as a fraction of vital capacity, functional residual capacity and peak expiratory flow rate as a fraction of total lung capacity, specific airway conductance and specific compliance (page 34) and probably dead space/tidal volume ratio.39
Blood gases and the control of breathing. Arterial Pco2 and alveolar Po2 do not change appreciably during childhood but arterial Po2 increases from the neonatal value to reach a maximum of about 13 kPa (98 mmHg) at young adulthood. Much of this increase occurs during the first year of life. There are obvious difficulties in determining the normal arterial Po2 in children. Ventilatory responses to both hypercapnia and hypoxia are at their highest in early childhood and decrease progressively into adulthood.40 The changes are small for hypoxic responses, but quite marked for hypercapnia, and are believed to relate to the higher metabolic rate in children.
References
1. Elkus R, Popovich J. Respiratory physiology in pregnancy. Clin Chest Med.. 1992;13:555-565.
2. Garcia-Rio F, Pino JM, Gomez L, Alvarez-Sala R, Villasnate C, Villamor J. Regulation of breathing and perception of dyspnoea in healthy pregnant women. Chest. 1996;110:446-453.
3. Templeton A, Kelman GR. Maternal blood-gases, (Pao2-Pao2), physiological shunt and Vd/Vt in normal pregnancy. Br J Anaesth.. 1976;48:1001-1004.
4. Pernol ML, Metcalfe J, Schlenker TL, Welch JE, Matsumoto JA. Oxygen consumption at rest and during exercise in pregnancy. Respir Physiol.. 1975;25:285-292.
5. Moore LG, McCullough RE, Weil JV. Increased HVR in pregnancy: relationship to hormonal and metabolic changes. J Appl Physiol.. 1987;62:158-163.
6. Jensen D, Webb KA, Davies GAL, O’Donnell DE. Mechanical ventilatory constraints during incremental cycle exercise in human pregnancy: implications for respiratory sensation. J Physiol.. 2008;586:4735-4750.
7. Merkus PJFM, ten Have-Opbroek AAW, Quanjer PH. Human lung growth: A review. Ped Pulmonol.. 1996;21:383-397.
*8. Jeffery PK. The development of large and small airways. Am J Respir Crit Care Med.. 1998;157:S174-S180.
9. Harding R, Hooper SB. Regulation of lung expansion and lung growth before birth. J Appl Physiol.. 1996;81:209-224.
*10. Shi W, Bellusci S, Warburton D. Lung development and adult lung diseases. Chest. 2007;132:651-656.
11. Maeda Y, Davé V, Whitsett JA. Transcriptional control of lung morphogenesis. Physiol Rev.. 2007;87:219-244.
12. Massaro GD, Massaro D. Formation of pulmonary alveoli and gas-exchange surface area: Quantitation and regulation. Annu Rev Physiol.. 1996;58:73-92.
13. Patrick J, Campbell K, Carmichael L, Natale R, Richardson B. Patterns of human fetal breathing during the last 10 weeks of pregnancy. Obstet Gynecol.. 1980;56:24-30.
14. Rusconi F, Galassi C, Forastiere F, et al. Maternal complications and procedures in pregnancy and at birth and wheezing phenotypes in children. Am J Respir Crit Care Med.. 2007;175:16-21.
15. Bush A, Annesi-Maesano I. Beam Me Up, Scotty!. Am J Respir Crit Care Med.. 2007;175:1-2.
16. Barker PM, Olver RE. Clearance of lung liquid during the perinatal period. J Appl Physiol.. 2002;93:1542-1548.
17. Verkman AS, Matthay MA, Song Y. Aquaporin water channels and lung physiology. Am J Physiol Lung Cell Mol Physiol.. 2002;278:L867-L879.
18. Ziegler JW, Ivy DD, Kinsella JP, Abman SH. The role of nitric oxide, endothelin, and prostaglandins in the transition of the pulmonary circulation. Clin Perinatol.. 1995;22:387-403.
19. Steinhorn RH, Millard SL, Morin FC. Persistent pulmonary hypertension of the newborn. Clin Perinatol.. 1995;22:405-428.
20. McIntosh N. The newborn. In: Campbell AGM, McIntosh N, editors. Forfar and Arneil’s Textbook of Pediatrics. Edinburgh: Churchill Livingstone, 1998.
21. Donnelly DF. Assisting Mother Nature in postnatal chemoreceptor maturation. J Appl Physiol.. 2008;104:1260-1261.
22. Cohen G, Xu C, Henderson-Smart D. Ventilatory response of the sleeping newborn to CO2 during normoxic rebreathing. J Appl Physiol.. 1991;71:168-174.
23. Khan A, Qurashi M, Kwiatkowski K, Cates D, Rigatto H. Measurement of the CO2 apneic threshold in newborn infants: possible relevance for periodic breathing and apnea. J Appl Physiol.. 2005;98:1171-1176.
24. Greer J. Development of respiratory rhythm generation. J Appl Physiol.. 2008;104:1211-1212.
25. Richards JM, Alexander JR, Shinebourne EA, de Swiet M, Wilson AJ, Southall DP. Sequential 22-hour profiles of breathing patterns and heart rate in 110 full-term infants during their first 6 months of life. Pediatrics. 1984;74:763-777.
26. Stebbens VA, Poets CF, Alexander JR, Arrowsmith WA, Southall DP. Oxygen saturation and breathing patterns in infancy. 1: Full term infants in the second month of life. Arch Dis Child. 1991;66:569-573.
27. Parkins KJ, Poets CF, O’Brien LM, Stebbens VA, Southall DP. Effect of exposure to 15% oxygen on breathing patterns and oxygen saturation in infants: interventional study. BMJ. 1998;316:887-894.
28. Copland IB, Post M. Understanding the mechanisms of infant respiratory distress and chronic lung disease. Am J Respir Cell Mol Biol.. 2002;26:261-265.
29. Merrill JD, Ballard RA. Pulmonary surfactant for neonatal respiratory disorders. Curr Opin Pediatrics. 2003;15:149-154.
30. Baraldi E, Filippone M. Chronic lung disease after premature birth. N Engl J Med.. 2007;357:1946-1955.
*31. Stevens TP, Sinkin RA. Surfactant replacement therapy. Chest. 2007;131:1577-1582.
32. UK Collabarative ECMO Trial Group. UK collaborative trial of neonatal extracorporeal membrane oxygenation. Lancet. 1996;348:75-82.
33. Leach CL, Greenspan JS, Rubenstein SD, et al. Partial liquid ventilation with perflubron in premature infants with severe respiratory distress syndrome. The LiquiVent study group. N Engl J Med.. 1996;335:761-767.
34. Vrijlandt EJLE, Gerritsen J, Boezen HM, Grevink RG, Duiverman EJ. Lung function and exercise capacity in young adults born prematurely. Am J Respir Crit Care Med.. 2006;173:890-896.
35. Hunt CE. Sudden infant death syndrome and other causes of infant mortality. Diagnosis, mechanisms, and risk for recurrence in siblings. Am J Respir Crit Care Med.. 2001;164:346-357.
*36. Moon RY, Horne RSC, Hauck FR. Sudden infant death syndrome. Lancet. 2007;370:1578-1587.
37. Harper RM. Impaired arousals and sudden infant death syndrome. Pre-existing neuronal injury? Am J Respir Crit Care Med.. 2003;168:1262-1263.
38. Papastamelos C, Panitch HB, England SE, Allen JL. Developmental changes in chest wall compliance in infancy and early childhood. J Appl Physiol.. 1995;78:179-184.
39. Cotes JE, Chinn DJ, Miller MR. Lung Function. Physiology, Measurement and Application in Medicine. Oxford: Blackwell Publishing; 2006.
40. Marcus CL, Glomb WB, Basinski DJ, Davidson SL, Keens TG. Developmental pattern of hypercapnic and hypoxic ventilatory responses from childhood to adulthood. J Appl Physiol.. 1994;76:314-320.