Chapter 28 Airways disease
 Whatever the cause, airway narrowing leads to expiratory flow limitation, gas trapping, and hyperinflation of the lung, which manifests itself as breathlessness. Asthma involves intermittent, reversible, airway obstruction caused by airway inflammation and bronchial smooth muscle contraction, both as a result of mediators released from mast cells and eosinophils.
Whatever the cause, airway narrowing leads to expiratory flow limitation, gas trapping, and hyperinflation of the lung, which manifests itself as breathlessness. Asthma involves intermittent, reversible, airway obstruction caused by airway inflammation and bronchial smooth muscle contraction, both as a result of mediators released from mast cells and eosinophils.This chapter considers the physiological changes seen in the three most common diseases of the pulmonary airways: asthma, chronic obstructive pulmonary disease (COPD) and cystic fibrosis. The first two of these have many clinical and physiological features in common, and together constitute the vast majority of respiratory disease seen in clinical practice.
Asthma
Lung diseases resulting from air pollution and infection have decreased dramatically in recent decades, but have been almost entirely replaced by asthma. It is estimated that 300 million people have asthma worldwide, with a prevalence that has increased by approximately 50% per decade. In developed countries the increasing prevalence is believed to have now levelled off at 10–15% of the population, but asthma may be continuing to become more common in the developing world (see below).1-3 In contrast to many respiratory diseases, the onset of asthma is usually in early childhood or in young adulthood. The prevalence of asthma amongst children in the developed world increased 2–3-fold in the last 50 years, though this has also now stopped increasing.4 Although the prevalence is no longer increasing, hospital admissions for asthma worldwide continue to rise, though fortunately deaths attributable to asthma have been falling consistently since the 1980s.1,5
Clinical Features
Asthma causes recurrent episodes of chest ‘tightness’, wheezing, breathlessness and coughing as a result of airway narrowing from a combination of inflammation of the small airways and contraction of bronchial smooth muscle in the lower airway. The term ‘asthma’ includes a wide spectrum of illnesses, varying from a wheezy 6-month-old baby with a viral infection to a young adult with multiple allergies manifested as wheeze or an older patient with chronic lung disease. In the last case, clinical features of asthma merge with those of COPD, and differentiation between the two is difficult. Whatever the clinical presentation, there are three closely related phases of an episode of asthma, as follows.
Bronchoconstriction occurs early in an asthma ‘attack’. This is particularly prominent in allergic asthma when, within minutes of exposure to an allergen, wheezing develops. Narrowing of small airways occurs due to contraction of airway smooth muscle in response to the cellular mechanisms described below. Bronchoconstriction in different regions of lung is not uniform, and positron emission tomography (PET) studies have shown that ventilation becomes patchy with clusters of poorly ventilated lung regions.6,7 This might be expected to cause maldistribution of ventilation and perfusion, but another study using PET showed that blood flow to the poorly ventilated areas is also reduced,8 presumably illustrating the efficiency of hypoxic pulmonary vasoconstriction (page 108). Heterogeneity of bronchoconstriction has significant implications for inhaled therapy, as these studies infer that most of an inhaled drug will be deposited in the better ventilated regions rather than where it is most needed.6
With more severe bronchoconstriction airway closure begins to occur during expiration, gas trapping occurs, and the lungs become hyperinflated.9 Eventually, the patient is attempting to breath in when the lungs are almost at total lung capacity, and a sensation of inspiratory dyspnoea results, even though the defect is with expiration. Physiological effects of hyperinflation are described on page 411.
Bronchoconstriction may quickly subside, either spontaneously or with treatment, but more commonly progresses to a late phase reaction.
Late-phase reactions are characterised by inflammation of the airway and develop a few hours after the acute bronchoconstriction. Airway obstruction continues, and cough with sputum production develops. Asthma precipitated by respiratory tract infection may ‘bypass’ the acute bronchoconstriction phase and the onset of symptoms is then more gradual.
Airway hyper-responsiveness (AHR) describes the observation that asthmatic subjects become wheezy in response to a whole range of stimuli that have little effect on normal individuals. Stimuli include such things as cold air, exercise, pollution (page 322) or inhaled drugs and occur via the neural pathways present in normal lungs (page 50). Methacholine or histamine can be used to measure AHR accurately by determining the inhaled concentration that gives rise to a 20% reduction in forced expiratory volume in one second (FEV1).10 Inhaled adenosine also causes airway narrowing, but unlike histamine and methacholine it does not act directly on bronchial smooth muscle.11 Bronchoconstriction in response to adenosine involves release of mediators from inflammatory cells, so the response is sensitive to the inflammatory state of the airway. For this reason, it is hoped that adenosine provocation may prove useful for monitoring the effectiveness of anti-inflammatory treatment of asthma patients, or even for differentiating between asthma and COPD.11
The degree of AHR seen in patients with asthma is highly variable. Severe asthma is associated with continuous AHR, whilst in mild asthma, the patient’s response will be normal between wheezy episodes.
Cellular Mechanisms of Asthma12-14
Many cell types are involved in the pathophysiology of asthma. A summary of the interactions between these cells is shown in Figure 28.1, which also shows the principal cytokines that facilitate communication between the cells.
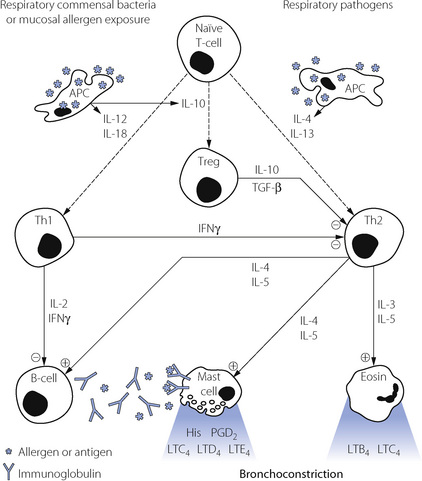
Fig. 28.1 Inflammatory cells involved in the pathogenesis of asthma, and the cytokines by which they communicate with each other. For details see text. The immunological pathways shown are based on a combination of animal and human studies. Eosin, eosinophil; Th2 and Th1, sub-types of T-lymphocyte ‘helper’ cells; Treg, regulatory lymphocyte; B-cell, B-lymphoctye; APC, antigen presenting cell; IL, interleukin; IFN, interferon; TGF, transforming growth factor.
Mast cells are plentiful in the walls of airways and alveoli and also lie free in the lumen of the airways where they may be recovered by bronchial lavage. Mast cell activation is the main cause of the immediate bronchospasm seen in allergen-provoked asthma. The surface of the mast cell contains a large number of binding sites for the immunoglobulin IgE. Activation of the cell results from antigen bridging of only a small number of these receptors, and may also be initiated by complement fractions C3a, C4a and C5a, substance P, physical stimulation and many drugs and other organic molecules.
The triggering mechanism of the mast cell is thus extremely sensitive, and is mediated by an increase in inositol triphosphate and intracellular calcium ions. Within 30 seconds of activation, there is degranulation with discharge of a range of preformed mediators listed in Table 28.1. Histamine acts directly on H1 receptors in the bronchial smooth muscle fibres to cause contraction, on other H1 receptors to increase vascular permeability, and on H2 receptors to increase mucus secretion. The granules also contain proteases, mainly tryptase, which can detach epithelial cells from the basement membrane resulting in desquamation and possibly activating neuronal reflexes causing further bronchospasm.
Table 28.1 Mediators released from mast cells when activated by IgE
| PREFORMED MEDIATORS | NEWLY GENERATED MEDIATORS | CYTOKINES |
|---|---|---|
| Histamine Heparin Serotonin Lysosomal enzymes: Tryptase Chymase β-Galactosidase β-Glucuronidase Hexosaminidase |
Prostaglandin D2 Thromboxane A2 Leukotrienes C4, D4 and E4 |
Interleukins 3, 4, 5, 6 and 13 Granulocyte/macrophage-colony stimulating factor Tumour necrosis factor Platelet activating factor |
The second major event after mast cell activation is the initiation of synthesis of arachidonic acid derivatives (see Figure 12.3). The most important derivative of the cyclo-oxygenase pathway is prostaglandin PGD2, which is a bronchoconstrictor, although its clinical significance is still not clear. The lipoxygenase pathway results in the formation of leukotriene (LT) C4, from which two further peptide leukotrienes, LTD4 and LTE4, are formed (see Figure 4.9).
Finally, mast cells also release a variety of cytokines, some of which are contained within the granules whilst others are generated de novo on activation of the cell. Interleukin-5 (IL-5) and granulocyte/macrophage colony stimulating factor (GM-CSF) are chemotactic for eosinophils whilst IL-4 stimulates IgE production by B-lymphocytes and so amplifies the activation of mast cells.
Eosinophils are freely distributed alongside mast cells in the submucosa, and are believed to be the principal cell involved in the late-phase reaction of asthma. In particular, they release LTB4 and LTC4, which are potent bronchoconstrictors with a prolonged action. They are attracted to the area by GM-CSF, which is released by many inflammatory cells, before being activated by IL-5 and IL-3 originating from mast cells and lymphocytes.
Lymphocytes have an important role in the control of mast cell and eosinophil activation.14 Activated B-lymphocytes are responsible for production of the antigen specific IgE needed to cause mast cell degranulation. B-cells are in turn controlled by two subsets of T ‘Helper’ lymphocytes, known as Th1 and Th2 cells.
Th2 cells are important pro-inflammatory cells in asthma, promoting both bronchospasm and inflammation by stimulation of mast cells, eosinophils and B-lymphocytes with IL-3, IL-4 and IL-5. The Th2 cell is non-specific in its response, and relies on stimulation by IL-4 and IL-13 from antigen presenting cells (APC) both for its generation from naïve T-cells and its subsequent activation to produce its own pro-inflammatory cytokines. It is not clear from where APCs originate, but they are probably located in the airway mucosa. Once activated by their specific antigen, the APC migrates to lymphoid tissue in the lungs to control the division of naïve lymphocytes into their various sub-types. In the case of Th2 stimulation, the APC is responding to a range of lung pathogens and this is the immunological pathway involved in normal pulmonary defences against infection.
Th1 cells are also generated from naïve T-cells in lymphoid tissue in response to cytokines released by activated APCs, but for Th1 generation the cytokines concerned are IL-12 and IL-18. Th1 cells normally act as anti-inflammatory cells by producing interferon and IL-2, which inhibit the activity of Th2 and B-cells.
The relative activity of the opposing effects of Th1 and Th2 lymphocytes was, until recently, believed to play an important role in the development and severity of asthma. However, this convenient explanation, based mainly on studies in animals, is now thought to be an over-simplification of the situation in humans, particularly with respect to the generation of Th1 cells.15
A third sub-type of T-lymphocyte is now thought to be involved in immune regulation of the lung.14,15,16 Regulatory T-cells (Treg) are again generated from naïve T-cells, this time in response to IL-10 released by activated APCs. Activation of the APCs to produce the anti-inflammatory cytokines IL-10, IL-12 and IL-18 is believed to occur in response to antigens from respiratory tract commensal bacteria or from exposure to high levels of allergens.14 Treg cells exert an anti-inflammatory effect by secretion of IL-10 and transforming growth factor β (TGF-β), which modify the activities of both Th1 and Th2 cells.
Nitric oxide (NO) is detectable in small concentrations in the expired air of normal subjects.17 It is produced from the mucosa of the whole respiratory tract, including the nose and nasal sinuses. Nitric oxide acts as the neurotransmitter for the non-cholinergic parasympathetic bronchodilator pathway in normal lungs (page 51), is involved in control of vascular tone in all tissues and is present in blood. In asthmatic patients with active disease, NO concentration in expired air is two to ten times greater than non-asthmatics (page 414).18 In this situation, the extra NO is derived from inducible NO-synthase (iNOS, page 107) in the airway mucosa. Cytokines produced by the inflammatory cells already described are believed to result in increased production of iNOS.19
Causes of Airway Obstruction in Asthma20
Airway smooth muscle (ASM). Stimulation of bronchial smooth muscle by the substances shown in Figure 28.1 and Table 28.1 explains some of the airway narrowing seen in asthma, particularly during the acute and early stages. During deliberately induced bronchoconstriction ASM cells in asthmatic subjects also respond differently to stretching (by taking a deep inspiration) compared with in non-asthmatic subjects. In the normal lung deep inspiration causes ASM relaxation which ameliorates the bronchoconstriction, whilst in asthmatic subjects the ASM fails to respond or even contracts, exacerbating the bronchoconstriction.21 This is a poorly understood aspect of ASM physiology.22
Inflammation. Airway narrowing during the late phase response, or in severe asthma, results from inflammation of the airway. Many cytokines released during asthma have effects on blood vessel permeability and therefore cause oedema of the epithelium and basement membrane.23 Protease enzymes break down normal epithelial architecture generating defects in the epithelial barrier, leading to further inflammation and eventually detachment of the epithelium from the basement membrane. Finally, hypersecretion of mucous and impaired mucociliary clearance are both recognised features of asthma, and this correlates with the flow limitation seen in individual patients. These changes in the thickness of the airway lining translate into a significant reduction in airway cross-sectional area, and thus a large increase in resistance (page 44). Mucous, inflammatory cells and epithelial debris cause obstruction of small airways, compounded by flow limitation preventing an effective cough.
Airway remodelling.13,24,25 Repeated activation of inflammatory pathways inevitably leads to attempts by the body to repair the tissue concerned. In the lung, this results in morphological changes to both the airway smooth muscle and the respiratory epithelium. Hyperplasia of smooth muscle cells causes thickening of the airway wall even when the muscle is relaxed, and exacerbates the airway narrowing that occurs with muscle contraction because a lesser degree of muscle shortening now causes a greater reduction in the airway lumen. Goblet cell hyperplasia occurs, worsening the hypersecretion of mucous seen with airway inflammation. Finally, in asthmatic patients, there is thickening of the lamina reticularis of the epithelial basement membrane and changes to the extracellular matrix, thought to be mediated by Th2 secreted cytokines, and ultimately resulting in collagen deposition and long term loss of lung function. The clinical significance of airway remodelling in asthma is unknown, but remodelling is believed to be responsible for the long-term decline in lung function seen in some asthma patients. Airway remodelling may begin before asthma becomes severe, or is even diagnosed at all,26 and though reducing airway inflammation with steroids may delay remodelling, drugs to reverse the structural changes are as yet undiscovered.27
Aetiology of Asthma2
Genetics.28 Asthma, along with other allergic diseases, has a substantial genetic component with several genomic regions known to be linked with developing the disease.29 Environmental factors invariably contribute to the development of clinical disease, but genetic susceptibility to asthma is strong. Two reasons explain this observation. First, the genes for most of the cytokines involved in asthma are found close together on chromosome 5, and asthmatic patients may have increased expression of these, so encouraging formation of an allergic phenotype.12 Secondly, human lymphocyte antigens (HLA), which are involved in sensitisation of APCs to specific antigens, are part of the major histocompatibility complex allowing immunological ‘self recognition’, and so are inherited. It is possible that some HLA types are particularly active in the processing of common allergens and thus the stimulation of Th2 cells or the suppression of Treg cells.
A genome-wide scan of patients with asthma identified a specific gene that is strongly associated with bronchial hyperresponsiveness.29,30 The gene codes for a protein named ADAM33, part of a large family of proteins with diverse functions, including the control of cell–cell and cell–matrix interactions. In lung tissue ADAM33 protein is found in smooth muscle and fibroblasts, but not epithelial cells, indicating its possible role in airway remodelling in asthma.
Maternal allergic disease is more likely to be passed to offspring than paternal disease, though this may relate to modification of the fetal immune system in utero rather than a true genetic influence. During pregnancy, lymphocyte subsets Th1 and Th2 are closely involved in the prevention of maternal rejection, and abnormalities at this stage may influence the activity of Th1 and Th2 cells in the offspring’s immune system, leading to allergic diseases, including asthma, in later life.31
Allergy. Changes in living conditions are believed to have contributed to the increase in asthma prevalence. In the developing world, population shifts from rural to urban environments have reduced exposure to parasitic infections and increased exposure to other allergens, and it seems likely that the extensive IgE and mast cell systems that formally inactivated parasites now respond to urban allergens. In the developed world, changes in living conditions have resulted in a dramatic increase in allergen exposure, in particular house dust mite (HDM, Dermatophagoides pteronyssinus), domestic animals and fungi. Asthma is more common in affluent families, and correlates with exposure to HDM, which thrives in warm, humid houses with extensive carpeting and bedding. These conditions are ideal for the HDM and its food supply of shed skin flakes. Simply inhaling allergens is only part of the explanation of how allergen exposure causes asthma, and once again pregnancy plays a role. Allergen taken in by the mother is believed to cross the placenta and influence immunological development before birth. Neonatal T-lymphocytes taken from children who subsequently develop asthma already show a reduced production of interferon-γ in response to allergen, indicating an existing immunological susceptibility to asthma.32
Infection.33 Viral respiratory tract infections cause wheezing in many asthmatics and account for over half of acute exacerbations of asthma. In infants, respiratory syncytial or parainfluenza viruses are common, whilst in adults a ‘common cold’ rhinovirus is the most usual pathogen. Viral infection gives rise to an immune response involving many cells and cytokines, but T-lymphocytes are particularly important and undergo both virus-specific and generalised activation. Inevitably, Th2 activity is increased giving rise to wheeze and airway inflammation by the mechanisms described above (Figure 28.1). In addition, stimulation of allergic mechanisms in susceptible individuals continues for some time after the viral symptoms have subsided. Thus, for example, after a simple rhinovirus infection allergen-induced histamine production and eosinophil-induced late-phase reactions remain increased for 4–6 weeks.34
Hygiene hypothesis.35 This hypothesis to explain the rising incidence of asthma claims that in the clean, hygienic, developed world children are exposed to fewer infections or other environmental antigens than only a few decades ago. It is known that some infections may have a protective role in preventing the initiation of asthma in early childhood.33 Children who are exposed to more infections in early life, such as those with older siblings or children living on farms, are less likely to develop allergic disease. This led to a suggestion that lower infection rates in the population at large and effective immunisation programmes may have contributed to the rising incidence of asthma. Measles virus, Mycobacterium tuberculosis, respiratory and gastro-intestinal commensal bacteria, some respiratory viral infections and hepatitis A virus all have the potential to reduce asthma development by modification of the lymphocyte sub-types shown in Figure 28.1. Other micro-organisms to which the modern human is now less commonly exposed, termed ‘old friends’ by the authors,36 include lactobacilli from untreated dairy products, saprophytic mycobacteria found in mud, and helminths (worms). All three are known to promote activity of Treg cells and so potentially protect against the development of asthma (Figure 28.1). For many of these micro-organisms exposure to the entire microbe is not required and beneficial immune responses may be gained from exposure to antigens found in the dust and dirt of the environment.
Pollution. Trends in air pollution have not generally followed trends in asthma prevalence over recent decades, the levels of many pollutants declining whilst asthma becomes more common. Laboratory evidence described on page 322 describes how, in comparison with normal subjects, asthmatics develop wheeze when exposed to lower inhaled concentrations of nitrogen dioxide and sulphur dioxide. The levels required to cause wheezing are still higher than commonly encountered in the atmosphere, and though there is some evidence linking air pollution episodes to respiratory problems the effect is believed to be small.
A role for air pollution in the initiation of asthma has also remained elusive, though there is now some evidence that exposure to traffic pollution may increase asthma incidence in children (page 321). Animal experiments indicate that common air pollutants can sensitise the airway to allergens, probably by disturbance of mucociliary clearance.
Gastric reflux.37 Gastro-oesophageal reflux symptoms are common in asthmatics, and are believed to be involved in the production of cough or wheeze in many patients. Acid in the distal oesophagus may, via a vagally mediated reflex, provoke either bronchoconstriction itself or airway hypersensitivity to allergen. In more severe cases, oesophageal reflux leading to aspiration of small amounts of acid into the airway can provoke severe bronchospasm. In patients with asthma who are resistant to treatment or have mainly nocturnal symptoms, reflux should be considered as a cause, though treatment of the reflux has an inconsistent effect on the asthma symptoms.38
Paracetamol. Depletion of glutathione in the lung (page 385) and oxidative stress are potential mechanisms to explain a link between asthma and paracetamol.39 In a cohort study of over 121 000 adults, frequent use of paracetamol was associated with the development of asthma.40 A recent study of over 200 000 children aged 6–7 years has also found an association between use of paracetamol in the first year of life and developing childhood asthma.41 Other explanations may explain this association apart from the paracetamol, for example, children who have frequent infections may be given paracetamol more often. However, given that the increasing use of paracetamol in children has followed a similar timescale to the rising incidence in childhood asthma, further research may soon reveal a relatively simple explanation for the current childhood asthma epidemic.
Aspirin-Induced Asthma (AIA)42,43
The involvement of arachidonic acid derivatives in the normal control of bronchial smooth muscle (see Table 4.2 and page 53) predicts that drugs blocking these pathways may influence the airways of asthma patients. This is indeed the case, with aspirin, and the closely related non-steroidal anti-inflammatory drugs, sometimes causing bronchospasm in asthma patients. Based on patient history alone only 2.7% of asthma patients report wheezing in response to aspirin, but when provocation with oral aspirin is carried out 21% of patients develop a reduction in FEV1.44 Many asthmatic patients who are sensitive to aspirin have a characteristic clinical presentation. Typically, AIA develops in patients at around 30 years of age, preceded for a few years by rhinitis and nasal polyps, and occurs in more female than male patients.
Mechanism of aspirin sensitivity. Inhibitors of the cyclo-oxygenase (COX) pathway in the airway will reduce synthesis of the bronchodilator prostaglandin PGE2. Reduced synthesis of PGE2 cannot alone account for AIA; patients with AIA also have increased production of LTE4, a potent bronchoconstrictor.42 This effect on the lipo-oxygenase pathway is not mediated by aspirin itself, and possibly results from loss of inhibition of lipo-oxygenase by PGE2. Genetic polymorphisms for the enzymes involved in leukotriene production may explain why some patients are aspirin sensitive.45 Multiple isoforms of COX exist (page 224) and COX-1 seems to be responsible for most cases of AIA. Coxibs, a group of drugs that specifically inhibit COX-2, seem to be safe for use in AIA patients.42 The analgesic effects of paracetamol (acetaminophen) may be mediated by inhibition of COX-3,46 and a small subset of patients with AIA develop bronchospasm in response to paracetamol.44 This sensitivity to paracetamol usually involves only a mild reaction in response to high doses of the drug, and occurs in less than 2% of asthmatic patients.
Principles of Therapy47
Detailed guidelines on the treatment of asthma are published for both the UK48 and the USA,49 and are beyond the scope of this book. Except in mild asthma treatment has now moved away from the traditional bronchodilator inhaler ‘when needed’ approach of the past. The emphasis is now on continuous treatment with drugs and other strategies aimed at preventing exacerbations and suppressing airway inflammation. Therapeutic approaches include the following.
Bronchodilators remain a common treatment for relief of acute bronchospasm. The β2-adrenoceptor agonists (page 51) are widely used, and recent developments include wider use of longer acting drugs,50 though concerns remain about the mortality for patients using these drugs in the long-term.51 Other bronchodilator drugs include inhibitors of leukotriene receptors on bronchial smooth muscle (page 53), blocking the effects of LTC4, LTD4 and LTE4. They are effective in treating asthma, including the bronchospasm seen in the late-phase reaction, and may be particularly useful in patients with exercise-induced or aspirin-induced asthma.52
Steroids,53,54 either inhaled or oral, are an invaluable method of prophylaxis and treatment in asthma. The anti-inflammatory effect of steroids is complex and incompletely elucidated. Steroids act on a glucocorticoid receptor found in the cytoplasm of cells, following which, the receptor–drug complex can enter the nucleus and regulate the transcription of numerous genes.53 By a combination of direct and indirect effects on transcription, steroids inhibit the synthesis of a wide range of inflammatory proteins including cytokines, adhesion molecules and inflammatory receptors.
Allergen-avoidance is an attractive strategy for the prevention of asthma in patients with known allergies. Low humidity is very effective in reducing HDM, and therefore at high altitude (above 1500 m or 5000 ft) HDM allergen is non-existent. Several studies have used this to compare asthma severity in normal and HDM-free high altitude environments, and have found improvements in both clinical and cellular measures of asthma severity.55 However, the rather drastic intervention of moving to high altitude is clearly not practical, and reduction of allergen load in the home is considerably less effective. Measures include removing carpets, reducing temperature and humidity, application of acaricides to kill HDM and encasing mattresses in allergen-impermeable membranes. Some studies have reported clinical benefits, but a meta-analysis did not support this approach.56
Chronic Obstructive Pulmonary Disease
Clinical features of COPD are similar to those of asthma with wheeze, cough and dyspnoea, but the airflow limitation is poorly reversible with bronchodilators. Much older patients are affected by COPD than asthma, and the progressive nature of the process leads to more serious interruption of normal activities and eventually respiratory failure (page 393). COPD accounts for between 3.8% and 4.9% of deaths worldwide, rates which are similar in high, middle and low-income countries and believed to still be increasing.57,58,59
Unlike asthma, where airway obstruction is usually intermittent, COPD is characterised by progressive chronic airflow limitation along with intermittent exacerbations, particularly in winter.60 These exacerbations vary from a slight worsening of symptoms to a life-threatening deterioration, and are usually caused by either viral or bacterial infections,61 possibly exacerbated by air pollution.
Aetiology of COPD62
Smoking is the major aetiological factor in COPD. The accelerated decline in FEV1 seen with smoking is shown in Figure 21.1, and the 15–20% of smokers who develop COPD probably represent an extreme response to this effect of tobacco smoke. Attempts to identify the genes responsible for this susceptibility to COPD in smokers are at an early stage (page 318).63,64
Both asthma and COPD are characterised pathologically by airway narrowing and inflammation, but the causes and clinical course of the two diseases are quite different. Improved understanding of the pathology of COPD and asthma has uncovered a variety of major differences between the two, and these are shown in Figure 28.2. It is believed that around 10% of patients have a mixture of the two disease processes.65
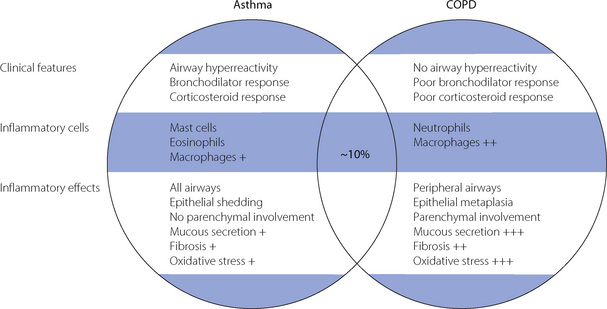
Fig. 28.2 Clinical and pathological differences between chronic obstructive pulmonary disease and asthma. Approximately 10% of patients have features characteristic of both diseases and may be described as having ‘wheezy bronchitis’.
(After reference 65 by permission of the author and publishers of Chest.)
The cellular mechanisms underlying airway inflammation in COPD relate to the disease’s strong association with smoking, with activation of neutrophils and macrophages (page 320) rather than the eosinophils and mast cells seen in asthma. Neutrophil activation causes the release of several protease enzymes, including neutrophil elastase, which degrades pulmonary elastin leading to the loss of lung tissue elasticity that is a characteristic feature of COPD. Smoking also induces oxidative stress in the airways, again potentially leading to irreversible tissue damage (page 320).
Three pathophysiological changes give rise to COPD – emphysema, mucous hypersecretion of larger airways, and small airway obstruction.
Emphysema may be defined as permanent enlargement of airspaces distal to the terminal bronchiole accompanied by destruction of alveolar walls.66 The process begins by enlargement of normal interalveolar holes, followed by destruction of the entire alveolar septum. Both ventilation and perfusion of the emphysematous area are therefore reduced, and, though some mismatch of ventilation and perfusion may occur in widespread emphysema, localised areas, as usually seen in COPD, have little effect. The loss of elastic tissue contained within the alveolar septa is, however, important, and reduces the elastic recoil of the pulmonary tissue so contributing to closure of small airways, particularly during expiration.
Current views on the cellular defect responsible for emphysema involve the relationship between protease and antiprotease activity in the lung.66-68 These enzymes are normally released following activation of neutrophils (e.g. neutrophil elastase) or macrophages in response to tobacco smoke or infection. A deficiency of the most well known antiprotease, α1-antitrypsin, is a significant risk factor for early development of emphysema (page 221). Disturbances of less well understood protease–antiprotease systems, such as the matrix metalloproteases group of enzymes,68 are now also believed to be involved in the generation of emphysema, as these proteases are normally involved in remodelling of the extracellular lung matrix.66 Proteases with activity against elastin are likely to be responsible for generating emphysema. Elastin deposition in the lung occurs early in life, and is minimal beyond late adolescence. Later, any pulmonary elastin lost through disease is likely to be replaced with collagen so reducing lung elasticity, and probably explaining the general decline in lung recoil throughout life.
Small airway obstruction plays a major role in COPD, but its aetiology is controversial.62,69 Part of the expiratory airflow limitation results from emphysema as described above. It is also likely that changes in the airway wall itself contribute. Inflammatory changes in small airways are ubiquitous in COPD, and may lead to mucosal thickening, hypertrophy of bronchial smooth muscle and ultimately to deposition of collagen in the outer airway wall.62
Large airway disease consists of goblet cell hyperplasia, mucosal oedema and production of excessive amounts of mucous. Recurrent respiratory tract infections and smoking undoubtedly contribute, and a chronic productive cough is the result. This feature of COPD is not always present, and its contribution to overall airway obstruction is variable. In some patients, extensive and long-standing inflammation of the large airways gives rise to permanent thickening of the airway wall within the cartilaginous airways, and so causes clinically important degrees of obstruction.69
Hyperinflation. Airflow limitation in small airways results from a combination of airway narrowing and loss of elastic recoil of lung tissue. The latter is of major importance in maintaining the patency of airways less than 1 mm in diameter (page 18), which lack supporting cartilage in their walls. Expiratory flow limitation leads to prolonged expiratory time constants in affected lung units, and incomplete expiration (gas trapping).70 Lung volume is therefore forced to increase and the patient becomes dyspnoeic, particularly during any situation that requires a greater minute volume such as exercise. Hyperinflation of the lung will, in theory, tend to oppose expiratory airway closure (see Figure 4.5), but it also causes a significant reduction in the efficiency of the respiratory muscles. In particular, the diaphragm becomes displaced caudally and, in severe disease, flattened, reducing the zone of apposition (see Figure 6.1) and causing much of the muscle activity to either oppose the opposite side of the diaphragm or pull the lower ribcage inwards rather outwards (see Figure 6.2). In time, lung hyperinflation becomes permanent with expansion of the chest wall (barrel chest) and irreversible flattening of the diaphragm.
When a patient with COPD develops an acute exacerbation airway obstruction worsens acutely and dynamic hyperinflation occurs. This is the same concept as seen during artificial ventilation (page 476), when the expiratory time becomes so long that expiration is incomplete, and end-expiratory lung volume increases acutely. Apart from the inevitable severe dyspnoea, the respiratory muscles also become even less efficient, ventilation/perfusion mismatching worsens, and gas exchange deteriorates.71
Respiratory muscles in COPD. The diaphragm, and to a lesser extent the intercostal muscles, are abnormal in patients with COPD. The fibre type shifts towards fatigue-resistant Type 1 fibres (page 91), and the contractile mechanisms of the fibres becomes less efficient.61,72 Whether these changes result from the chronic stretching of the diaphragm caused by hyperinflation or from systemic inflammation as a result of the COPD is unknown, but the effect is to further impair the ventilatory capacity.
Principles of Therapy73,74
As for asthma, detailed guidelines for the treatment of COPD have been published.75 Surgical procedures used to treat COPD are described on page 496.
Smoking cessation is central to all forms of treatment for COPD. The rate of the progressive decline in lung function returns to that of a non-smoker (see Figure 21.1) and symptoms improve. Patients with COPD have often been heavy smokers for a considerable number of years, and smoking cessation may therefore need great determination. Patients usually only become permanent non-smokers after multiple attempts at quitting, though nicotine replacement and other drug therapies may improve this poor success rate.
Medical treatment.76 Inhaled bronchodilators may be used. Their efficacy depends on the reversibility of the airways disease in each patient. Both β2-agonists and anticholinergic drugs (page 51) are used, and long-acting drugs becoming more widely used though concerns continue about the mortality of COPD patients using long-acting β2-agonists.77 Corticosteroids are not as effective for treating COPD as they are for asthma.78 The inflammatory cells involved are different (Figure 28.2), and may be less susceptible to steroid suppression. Oxidative stress present in COPD airways from neutrophil activation and smoking may inhibit one of the transcription enzymes normally stimulated by steroid drugs.79
Medical treatment also involves active management of exacerbations. Management of the underlying disease with antibiotics and oxygen is required. Artificial ventilation is commonly required, and non-invasive ventilation (page 464) is now accepted as the best initial option for these patients.80
Supplemental oxygen81 at low inspired concentrations for 15–24 hours a day improves survival in patients with severe COPD associated with hypoxia. There may also be some benefit of oxygen therapy for COPD patients who desaturate on exercise, both in terms of preventing the desaturation and improving their exercise capacity.
Oxygen Therapy in COPD82
The previous classification of patients with advanced COPD into ‘pink puffers’ and ‘blue bloaters’ has now been replaced with Type 1 and Type 2 respiratory failure respectively (page 393). Which pattern occurs in an individual patient depends on the relative contributions of airway disease, emphysema and loss of lung elasticity, along with their central chemoreceptor sensitivity to carbon dioxide. Whatever the type of their respiratory failure, administration of oxygen to patients with severe COPD can lead to hypercapnia. Two main mechanisms are believed to be responsible.
Ventilatory depression by oxygen.82 Patients with type 2 respiratory failure may be relying on their hypoxic drive to maintain ventilation. If this is abolished, as, for example, by the achievement of a high arterial Po2, hypoventilation or even apnoea may result. However, studies investigating oxygen-induced hypercapnia in COPD have failed to find consistent changes in minute ventilation during either periods of stable respiratory symptoms83 or acute exacerbations.84 Reduction in minute ventilation in response to oxygen was either too small to explain adequately the changes in Pco2, or only transient, returning towards baseline ventilation after a few minutes. Nevertheless, in one of these reports,84 of 22 subjects studied, two developed severe respiratory depression leading to dangerous hypercapnia after just 15 minutes of breathing 100% oxygen. A small proportion of patients with COPD therefore seem to be susceptible to oxygen induced respiratory depression.
Altered ventilation perfusion relationships with oxygen have been proposed to explain hypercapnia seen in COPD patients in whom minute ventilation remains essentially unchanged.83,84,85 Alveolar Po2 is known to contribute to hypoxic pulmonary vasoconstriction (page 108) and so help to minimise 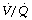 mismatch. Administration of oxygen may therefore abolish hypoxic pulmonary vasoconstriction in poorly ventilated areas, increasing blood flow to these areas, and so reducing blood flow to other lung regions with normal or high ventilation to perfusion ratios.83 These areas will then contribute further to alveolar dead space and so cause an increase in arterial Pco2 (page 130).
mismatch. Administration of oxygen may therefore abolish hypoxic pulmonary vasoconstriction in poorly ventilated areas, increasing blood flow to these areas, and so reducing blood flow to other lung regions with normal or high ventilation to perfusion ratios.83 These areas will then contribute further to alveolar dead space and so cause an increase in arterial Pco2 (page 130).
Which of these mechanisms predominates in an individual patient is currently difficult to predict. Administration of oxygen to patients with COPD must therefore be undertaken with great care, and accompanied by suitable monitoring of both oxygenation and arterial Pco2.
Cystic Fibrosis86
Cystic fibrosis (CF) is an autosomal recessive genetic disorder affecting Caucasian individuals of whom 1 in 25 carry the gene. The disease affects approximately 1 in 2500 births and abnormal CF genes can be identified prenatally. Prediction of phenotype from genetic screening is complex because there is a wide spectrum of clinical disease,87 the severity of which is determined by environmental factors (e.g. smoking) and genetic modifiers of the abnormal CF gene.88 Mortality from CF remains high, but has been improving dramatically for some years, and the anticipated life expectancy for a person born with CF in the year 2000 is now 50 years.89 Thus, although the number of CF births is constant, improved survival means that the prevalence of CF is increasing steadily.
Cystic fibrosis affects epithelial cell function in many body systems, but gastrointestinal and respiratory functions are the most important; this chapter discusses only the latter. Abnormalities of pulmonary airway defence mechanisms lead to lifelong colonisation of the CF lung with bacteria. Recurrent airway infection produces hypersecretion of mucous, cough and over many years, destruction of normal lung architecture including bronchiectasis.
Aetiology of CF
Biochemical abnormality
The molecular mechanisms of CF have been the focus of extensive research for many years, which has led to CF being one of the most completely understood of inherited diseases. As long ago as 1989 the gene responsible for CF was identified.90 It is located on chromosome 7, and codes for a protein named cystic fibrosis transmembrane regulator (CFTR) found in epithelial cells. The CFTR protein functions as a membrane-bound active chloride channel, and plays a major role in controlling salt concentration in epithelial secretions. Sweat production is influenced by CFTR function, allowing measurement of the sodium concentration in sweat to remain a relatively simple investigation for diagnosis, being over twice normal in CF patients.
The CFTR comprises three types of protein sub-unit.91 A ring of membrane spanning domains form a channel through the lipid bilayer of the cell wall (Figure 28.3). Attached to the intracellular aspect of these are two nucleotide-binding domains (NBDs) that use ATP when the channel is activated. Finally, a single regulatory domain (R) protein is loosely attached to the NBDs and can move away from the NBDs to ‘open’ the channel and allow chloride to pass into or out of the cell (Figure 28.3). Intracellular protein kinase A activates the channel by binding to the regulatory domain of CFTR, whilst ATP provides the energy and is dephosphorylated by the NBDs. Over 1000 different mutations of the CF gene have been identified, and can result in no CFTR being formed, failure of the different protein domains to align correctly or failure of the CFTR to become incorporated into the cell membrane. In normal subjects the CFTR protein also acts by inhibiting nearby epithelial sodium channels, a link which is believed to be defective in CF further impairing the regulation of airway lining fluid (page 219).92
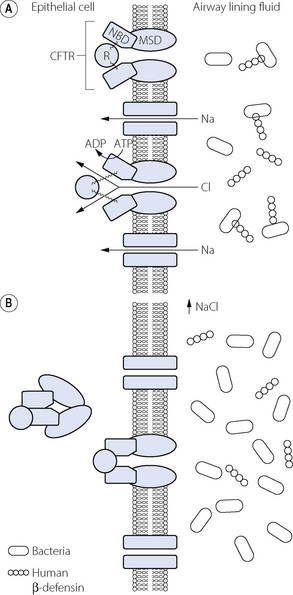
Fig. 28.3 Sodium and chloride transport across the pulmonary epithelial cell wall in cystic fibrosis. (A) Normal lung. Cystic fibrosis transmembrane regulator (CFTR) chloride channel in the closed (upper) and open (lower) positions showing movement of the regulator domain (R). Sodium transport follows chloride via passive Na channels due to altered transmembrane potentials. Bacteria in the airway lining fluid may be inactivated by human β-defensin. (B) Cystic fibrosis. The CFTR proteins are defective so do not locate in the membrane, or are non-functional when they do. Sodium and chloride concentration is therefore abnormally high in the airway, which may inactivate human β-defensin or alter airway lining fluid function and so allow bacterial proliferation. MSD, membrane spanning domain; NBD, nucleotide binding domain.
Causes of lung disease
The sequence of events by which abnormal CFTR function leads to pulmonary pathology remains controversial. Abnormalities of the airway lining fluid and mucous result in poor defences against inhaled pathogens. Bacterial colonisation occurs early in the disease process, and CF patients have an exaggerated inflammatory response to a variety of airway pathogens. A cycle becomes established in which bacterial infection leads to airway inflammation, mucous production and more infection, associated with progressive lung tissue damage. Abnormal CFTR function may adversely affect the ability of the airway to remove inhaled pathogens by a variety of mechanisms, as follows.
Salt-defensin hypothesis. The human lung produces a variety of endogenous antibiotics, of which the most-studied is human β-defensin (HBD), which may play an important role in preventing pulmonary infection. Consisting of a 64 amino acid peptide, HBD has been shown to be inactivated by increased sodium chloride concentrations, so allowing proliferation of bacteria in CF lungs (Figure 28.3).93
Inflammation first hypothesis. This proposes that airway inflammation is the primary event in CF lungs, possibly caused by abnormal cytokine production. Inflammatory changes in the airways then lead to excessive and abnormal mucous production and colonisation with pathogens.
Cell-receptor hypothesis. In normal lung, the CFTR found on epithelial cells, along with a range of cell surface glycoproteins, binds many bacterial pathogens as part of the normal process for killing inhaled microorganisms. Abnormal pH around epithelial cells from CF lung inhibits the binding of lung pathogens found in CF.
Depleted airway surface liquid hypothesis. Despite the altered sodium and chloride transport in the CF lung epithelial cells, the ‘sol’, or periciliary layer of the airway surface liquid (page 219) is believed to be isotonic.87 However, the volume of periciliary fluid is reduced, and this disturbs the physical linkage between the cilia and the periciliary and mucous layers of ASL, effectively preventing the normal clearance of the ASL. The mucous layer becomes abnormally deep and viscous, which inhibits the function of endogenous antimicrobial systems such as HBD, lactoferrin and lysozyme, and also creates a layer of hypoxic mucous in which anaerobic bacteria can thrive.94
Principles of Therapy
Conventional treatment95,96 involves assisting the clearance of airway secretions by physiotherapy, postural drainage and exercise. The viscous mucous layer of ASL results in part from degradation of the numerous inflammatory cells found in infected airways, and it is DNA from these cells that can aggregate and further increase viscosity. Treatment with inhaled recombinant human DNAase reduces the viscosity of sputum, and is a useful adjunct to physical methods of mucous clearance. Antibiotic therapy, for both infective exacerbations and maintenance therapy, is now used for all patients and is believed to be the main reason for improved survival in CF.
Lung transplantation is now a recognised treatment for CF, and is described in Chapter 33.
Gene therapy has held great potential for therapy ever since the CF gene was identified, but unfortunately this potential has not been realised.97,98 A normal CFTR gene can be produced, but the problem arises in incorporating the gene into the airway cells and stimulating its expression into functioning CFTR in vivo. Gene delivery either in liposomes or viral vectors has been attempted, but the functional effect is poor with only transient or small changes in CFTR expression. A more promising approach is to incorporate the normal gene into the fetus, which bypasses immunological reactions and should provide a permanent correction of the defective gene. This has been achieved in mice,99 but studies of this type in humans are currently prohibited by international ethical convention.
Assessment of Airway Disease by Exhaled Breath Analysis
Exhaled nitric oxide. Nitric oxide is detectable in small concentrations in the expired air of normal subjects.17 It is produced from the mucosa of the whole respiratory tract, including the nose and nasal sinuses. Nitric oxide acts as the neurotransmitter for the non-cholinergic parasympathetic bronchodilator pathway in normal lungs (page 51), is involved in control of vascular tone in all tissues and is present in blood. By varying the size of breath and expiratory flow rates it may be possible to measure NO released from different areas of the respiratory system.100 When inflammation is present in the airways, extra NO is produced by inducible NO-synthase (iNOS, page 107) in the airway mucosa, and the exhaled NO level increases. For example in asthmatic patients with active disease, NO concentration in expired air is two to ten times greater than non-asthmatics.18 Exhaled NO levels may be used as a non-specific marker of airway inflammation in asthma, including in children,101 and this technique is now beginning to be advocated for use in clinical practice though its exact role remains unclear.102,103
Exhaled breath condensate (EBC).104 This technique is at an earlier stage of development than exhaled NO, but has enormous potential. Liquid condensate from the lungs is analysed for a range of substances that reflect lung pathology. The concentrations of the biomarkers are very low, often close to the detection limits of the analysers, giving rise to a significant variability in results. Nevertheless, EBC has been used to assess the oxidative stress of the lungs by measuring hydrogen peroxide or lung inflammation using condensate pH or prostaglandin levels.104,105 In future, by analysing DNA obtained from EBC, a non-invasive test for lung cancer is a possibility, which would be a major advance considering that a majority of lung cancers have spread beyond the primary tumour when diagnosed.106
References
1. Braman SS. The global burden of asthma. Chest. 2006;130:4S-12S.
*2. Eder W, Ege MJ, von Mutius E. The asthma epidemic. N Engl J Med.. 2006;355:2226-2235.
3. Holgate ST. The epidemic of asthma and allergy. J R Soc Med.. 2004;97:103-110.
4. Anderson HR, Gupta R, Strachan DP, Limb ES. 50 years of asthma: UK trends from 1955 to 2004. Thorax. 2007;62:85-90.
5. Wijesinghe M, Weatherall M, Perrin K, Crane J, Beasley R. International trends in asthma mortality rates in the 5- to 34-year age group. A call for closer surveillance. Chest. 2009;135:1045-1049.
*6. Venegas JG, Winkler T, Musch G, et al. Self-organised patchiness in asthma as a prelude to catastrophic shifts. Nature. 2005;434:777-782.
7. Venegas J. Linking ventilation heterogeneity and airway hyper-responsiveness in asthma. Thorax. 2007;62:653-654.
8. Harris RS, Winkler T, Tgavalekos N, et al. Regional pulmonary perfusion, inflation, and ventilation defects in bronchoconstricted patients with asthma. Am J Respir Crit Care Med.. 2006;174:245-253.
9. Cormier Y, Lecours R, Legris C. Mechanisms of hyperinflation in asthma. Eur Respir J. 1990;3:619-624.
10. Lötvall J, Inman M, O’Byrne P. Measurement of airway hyper-responsiveness: new considerations. Thorax. 1998;53:419-424.
11. Polosa R, Rorke S, Holgate ST. Evolving concepts on the value of adenosine hyper-responsiveness in asthma and chronic obstructive pulmonary disease. Thorax. 2002;57:649-654.
12. Lee TH. Cytokine networks in the pathogenesis of bronchial asthma: implications for therapy. J R Coll Phys Lond.. 1998;32:56-64.
13. Bousquet J, Jeffery PK, Busse WW, Johnson M, Vignola AM. Asthma: from bronchoconstriction to airways inflammation and remodeling. Am J Respir Crit Care Med.. 2000;161:1720-1745.
*14. Umetsu DT, McIntyre J, Akbari O, Macaubas C, DeKruyff RH. Asthma: an epidemic of dysregulated immunity. Nat Immunol.. 2002;3:715-720.
15. van Oosterhout AJM, Motta AC. Th1/Th2 paradigm: not seeing the forest for the trees? Eur Respir J. 2005;25:591-593.
16. Larché M. Regulatory T cells in allergy and asthma. Chest. 2007;132:1007-1014.
17. DuBois AB, Kelley PM, Douglas JS, Mohsenin V. Nitric oxide production and absorption in trachea, bronchi, bronchioles, and respiratory bronchioles of humans. J Appl Physiol.. 1999;86:159-167.
18. Frank TL, Adisesh A, Pickering AC, et al. Relationship between exhaled nitric oxide and childhood asthma. Am J Respir Crit Care Med.. 1998;158:1032-1036.
19. Hamid Q, Springall DR, Riveros-Moreno V, et al. Induction of nitric oxide synthase in asthma. Lancet. 1993;342:1510-1513.
20. Nadel JA, Busse WW. Asthma. Am J Respir Crit Care Med.. 1998;157:S130-S138.
21. Berend N, Salome C. Can a deep breath blow away the fog surrounding airway hyper-responsiveness? Am J Respir Crit Care Med.. 2007;176:109-111.
22. Bates JHT. How should airway smooth muscle be punished for causing asthma? J Appl Physiol.. 2008;104:575-576.
23. Jeffery PK, Godfrey RW, Ädelroth E, Nelson F, Rogers A, Johansson S-A. Effects of treatment on airway inflammation and thickening of basement membrane reticular collagen in asthma. Am Rev Respir Dis.. 1992;145:890-899.
24. McParland BE, Macklem PT, Paré PD. Airway wall remodelling: friend or foe. J Appl Physiol.. 2003;95:426-434.
25. Fixman ED, Stewart A, Martin JG. Basic mechanisms of development of airway structural changes in asthma. Eur Respir J. 2007;29:379-389.
26. Pohunek P, Roche WR, Tarzikova J, Kurdmann J, Warner JO. Eosinophilic inflammation in the bronchial mucosa in children with bronchial asthma. Eur Respir J. 2000;11(Supp 25):160S.
27. Boulet L-P, Sterk PJ. Airway remodelling: the future. Eur Respir J. 2007;30:831-834.
*28. Martinez FD. Genes, environments, development and asthma: a reappraisal. Eur Respir J. 2007;29:179-184.
29. Van Eerdewegh P, Little RD, Dupuis J, et al. Association of the ADAM33 gene with asthma and bronchial hyper-responsiveness. Nature. 2002;418:426-430.
30. Holgate ST, Holloway JW. Is big beautiful? The continuing story of ADAM33 and asthma. Thorax. 2005;60:263-264.
31. Warner JA, Jones AC, Miles EA, Colwell BM, Warner JO. Maternofetal interaction and allergy. Allergy. 1996;51:447-451.
32. Tang MLK, Kemp AS, Thorburn J, Hill DJ. Reduced interferon-γ secretion in neonates and subsequent atopy. Lancet. 1994;344:983-985.
33. Folkerts G, Busse WW, Nijkamp FP, Sorkness R, Gern JE. Virus-induced airway hyper-responsiveness and asthma. Am J Respir Crit Care Med.. 1998;157:1708-1720.
34. Calhoun WJ, Dick EC, Schwartz LB, Busse WW. A common cold virus, rhinovirus 16, potentiates airway inflammation after segmental antigen bronchoprovocation in allergic subjects. J Clin Invest.. 1994;94:2200-2208.
35. Cullinan P. Childhood allergies, birth order and family size. Thorax. 2006;61:3-5.
36. Rook GAW, Adams V, Hunt J, Palmer R, Martinelli R, Brunet LR. Mycobacteria and other environmental organisms as immunomodulators for immunoregulatory disorders. Springer Semin Immunopathol.. 2004;25:237-255.
37. Harding SM, Richter JE. The role of gastroesophageal reflux in chronic cough and asthma. Chest. 1997;111:1389-1402.
38. Jain S. Proton-pump inhibitor therapy for gastroesophageal reflux disease. Does it treat the asthma? Chest. 2005;127:1097-1098.
39. Eneli I, Sadri K, Camargo C, Barr RG. Acetaminophen and the risk of asthma. The epidemiologic and pathophysiologic evidence. Chest. 2005;127:604-612.
40. Barr RG, Wentowski CC, Curhan GC, et al. Prospective study of acetaminophen use and newly diagnosed asthma among women. Am J Respir Crit Care Med.. 2004;169:836-841.
41. Beasley R, Clayton T, Crane J, et al. Association between paracetamol use in infancy and childhood, and risk of asthma, rhinoconjunctivitis, and eczema in children aged 6–7 years: analysis from phase three of the ISAAC programme. Lancet. 2008;372:1039-1048.
42. Farooque S, Lee TH. Aspirin sensitivity and eicosanoids. Thorax. 2008;63:2-4.
43. Farooque SP, Lee TH. Aspirin-sensitive respiratory disease. Annu Rev Physiol.. 2009;71:465-487.
44. Jenkins C, Costello J, Hodge L. Systematic review of prevalence of aspirin induced asthma and its implications for clinical practice. BMJ. 2004;328:434-437.
45. Szczeklik A, Sanak M. Genetic mechanisms in aspirin-induced asthma. Am J Respir Crit Care Med.. 2000;161:S142-S146.
46. Schwab JM, Schluesener HJ, Laufer S. COX-3: just another COX or the solitary elusive target of paracetamol? Lancet. 2003;361:981-982.
47. Fanta CH. Asthma. N Engl J Med.. 2009;360:1002-1014.
48. British Thoracic Society, Scottish Intercollegiate Guidelines Network. British guideline on the management of asthma. Thorax. 2008;63:iv1-iv121.
49. National Heart, Lung, and Blood Institute. National Asthma Education and Prevention Program, Expert Panel. Bethesda, MD: National Institutes of Health; 2007. Report 3: guidelines for the diagnosis and management of asthma. Publication No. 07-4051
50. Tattersfield AE, Harrison TW. β-adrenoceptor polymorphisms: focus moves to long-acting β-agonists. Am J Respir Crit Care Med.. 2006;173:473-474.
51. Beasley R, Martinez FD, Hackshaw A, Rabe KF, Sterke PJ, Djukanovic R. Safety of long-acting β-agonists: urgent need to clear the air remains. Eur Respir J. 2009;33:3-5.
52. Busse W, Kraft M. Cysteinyl leukotrienes in allergic inflammation: strategic target for therapy. Chest. 2005;127:1312-1326.
53. Barnes PJ. Molecular mechanisms of corticosteroids in allergic disease. Allergy. 2001;56:928-936.
54. Adcock IM, Barnes PJ. Molecular mechanisms of corticosteroid resistance. Chest. 2008;134:394-401.
55. Custovic A, Simpson A, Chapman MD, Woodcock A. Allergen avoidance in the treatment of asthma and atopic disorders. Thorax. 1998;53:63-72.
56. Gotzsche PC, Hammarquist C, Burr M. House dust mite control measures in the management of asthma: meta-analysis. BMJ. 1998;317:1105-1110.
57. Calverley PMA, Walker P. Chronic obstructive pulmonary disease. Lancet. 2003;362:1053-1061.
*58. Viegi G, Pistelli F, Sherrill DL, Maio S, Baldacci S, Carrozzi L. Definition, epidemiology and natural history of COPD. Eur Respir J. 2007;30:993-1013.
59. Mannino DM, Buist AS. Global burden of COPD: risk factors, prevalence, and future trends. Lancet. 2007;370:765-773.
60. Aaron SD. COPD exacerbations. Predicting the future from the recent past. Am J Respir Crit Care Med.. 2009;179:335-336.
61. Ottenheijm CAC, Heunks LMA, Sieck GC, et al. Diaphragm dysfunction in chronic obstructive pulmonary disease. Am J Respir Crit Care Med.. 2005;172:200-205.
*62. Barnes PJ, Shapiro SD, Pauwels RA. Chronic obstructive pulmonary disease: molecular and cellular mechanisms. Eur Respir J. 2003;22:672-688.
63. Sandford AJ, Silverman EK. Chronic obstructive pulmonary disease 1: Susceptibility factors for COPD – the genotype–environment interaction. Thorax. 2002;57:736-741.
64. Anthonisen NR. ‘Susceptible’ smokers? Thorax. 2006;61:924-925.
65. Barnes PJ. Mechanisms in COPD. Differences from asthma. Chest. 2000;117:10S-14S.
66. Hogg JC, Senior RM. Chronic obstructive pulmonary disease 2: Pathology and biochemistry of emphysema. Thorax. 2002;57:830-834.
67. Turino GM. Emphysema in COPD: consequences and causes. Thorax. 2006;61:1031-1032.
68. Turino GM. Proteases in COPD. A critical pathway to injury. Chest. 2007;132:1724-1725.
69. Tiddens HAWM, Paré PD, Hogg JC, Hop WCJ, Lambert R, De Jongste JC. Cartilaginous airway dimensions and airflow obstruction in human lungs. Am J Respir Crit Care Med.. 1995;152:260-266.
70. Man SFP, McAlister FA, Anthonisen NR, Sin DD. Contemporary management of chronic obstructive pulmonary disease. Clinical applications. JAMA. 2003;290:2313-2316.
71. O’Donnell DE, Parker CM. COPD exacerbations. 3: Pathophysiology. Thorax. 2006;61:354-361.
72. McKenzie D. To breathe or not to breathe: the respiratory muscles and COPD. J Appl Physiol.. 2006;101:1279-1280.
73. Celli BR. Update on the management of COPD. Chest. 2008;133:1451-1462.
*74. Sutherland ER, Cherniak RM. Management of chronic obstructive pulmonary disease. N Engl J Med.. 2004;350:2689-2697.
75. Pauwels RA, Buist AS, Calverley PMA, Jenkins CR, Hurd SS. Global strategy for the diagnosis, management, and prevention of chronic obstructive pulmonary disease. NHLBI/WHO Global Initiative for Chronic Obstructive Lung Disease (GOLD) Workshop summary. Am J Respir Crit Care Med.. 2001;163:1256-1276.
76. Barnes PJ. Emerging pharmacotherapies for COPD. Chest. 2008;134:1278-1286.
77. Sears MR. Long-acting bronchodilators in COPD. Chest. 2008;133:1057-1058.
78. Niewoehner DE, Wilt TJ. Inhaled corticosteroids for chronic obstructive pulmonary disease. Am J Respir Crit Care Med.. 2007;175:103-104.
79. Barnes PJ, Ito K, Adcock IM. Corticosteroid resistance in chronic obstructive pulmonary disease: inactivation of histone deacetylase. Lancet. 2004;363:731-733.
80. Plant PK, Elliott MW. Chronic obstructive pulmonary disease 9: Management of ventilatory failure in COPD. Thorax. 2003;58:537-542.
81. Drummond MB, Wise RA. Oxygen therapy in COPD: what do we know? Am J Respir Crit Care Med.. 2007;176:321-326.
*82. O’Driscoll BR, Howard LS, Davison AG. BTS guideline for emergency oxygen use in adult patients, on behalf of the British Thoracic Society. Thorax, 63. 2008: vi1-vi68.
83. Sassoon CSH, Hassell KT, Mahutte CK. Hyperoxic-induced hypercapnia in stable chronic obstructive pulmonary disease. Am Rev Respir Dis.. 1987;135:907-911.
84. Aubier M, Murciano D, Milic-Emili J, et al. Effects of the administration of O2 on ventilation and blood gases in patients with chronic obstructive pulmonary disease during acute respiratory failure. Am Rev Respir Dis.. 1980;122:747-754.
85. Crossley DJ, McGuire GP, Barrow PM, Houston PL. Influence of inspired oxygen concentration on deadspace, respiratory drive, and Paco2 in intubated patients with chronic obstructive pulmonary disease. Crit Care Med.. 1997;25:1522-1526.
86. O'Sullivan BP, Freedman SD. Cystic fibrosis. Lancet. 2009;373:1891-1904.
*87. Boucher RC. New concepts of the pathogenesis of cystic fibrosis lung disease. Eur Respir J. 2004;23:146-158.
88. Vanscoy LL, Blackman SM, Collaco JM, et al. Heritability of lung disease severity in cystic fibrosis. Am J Respir Crit Care Med.. 2007;175:1036-1043.
89. Dodge JA, Lewis PA, Stanton M, Wilsher J. Cystic fibrosis mortality and survival in the UK: 1947–2003. Eur Respir J. 2007;29:522-526.
90. Kerem B, Rommens JM, Buchanan JA, et al. Identification of the cystic fibrosis gene: genetic analysis. Science. 1989;245:1073-1080.
91. Stern M, Geddes D. Cystic fibrosis: basic chemical and cellular mechanisms. Br J Hosp Med.. 1996;55:237-240.
92. Donaldson SH, Boucher RC. Sodium channels and cystic fibrosis. Chest. 2007;132:1631-1636.
93. Goldman MJ, Anderson GM, Stolzenberg ED, Kari UP, Zasloff M, Wilson J. Human β-defensin-1 is a salt-sensitive antibiotic in lung that is inactivated in cystic fibrosis. Cell. 1997;88:553-560.
94. Tunney MM, Field TR, Moriarty TF, et al. Detection of anaerobic bacteria in high numbers in sputum from patients with cystic fibrosis. Am J Respir Crit Care Med.. 2008;177:995-1001.
95. Yankaskas JR, Marshall BC, Sufian B, Simon RH, Rodman D. Cystic fibrosis adult care: consensus conference report. Chest. 2004;125(suppl 1):1S-39S.
96. Flume PA, O’Sullivan BP, Robinson KA, et al. Cystic fibrosis pulmonary guidelines. Chronic medications for maintenance of lung health. Am J Respir Crit Care Med.. 2007;176:957-969.
97. Kolb M, Martin G, Medina M, Ask K, Gauldie J. Gene therapy for pulmonary diseases. Chest. 2006;130:879-884.
98. Driskell RA, Engelhardt JF. Current status of gene therapy for inherited lung diseases. Annu Rev Physiol.. 2003;65:585-612.
99. Larson JE, Morrow SL, Happel L, Sharp JF, Cohen JC. Reversal of cystic fibrosis phenotype in mice by gene therapy in utero. Lancet. 1997;349:619-620.
100. George SC. How accurately should we estimate the anatomical source of exhaled nitric oxide? J Appl Physiol.. 2008;104:909-911.
101. Brussee JE, Smit HA, Kerkhof M, et al. Exhaled nitric oxide in 4-year-old children: relationship with asthma and atopy. Eur Respir J. 2005;25:455-461.
102. Grob NM, Dweik RA. Exhaled nitric oxide in asthma from diagnosis, to monitoring, to screening: are we there yet? Chest. 2008;133:837-839.
103. Pedersen S, O’Byrne PM. Exhaled nitric oxide in guideline-based asthma management. Lancet. 2008;372:1015-1017.
*104. Barnes PJ, Chowdhury B, Kharitonov SA, et al. Pulmonary biomarkers in chronic obstructive pulmonary disease. Am J Respir Crit Care Med.. 2006;174:6-14.
105. Holz O. Catching breath: monitoring airway inflammation using exhaled breath condensate. Eur Respir J. 2005;26:371-372.
106. Powell CA. Waiting to exhale. Am J Respir Crit Care Med.. 2008;177:246-247.