Chapter 24 Hypoxia
 Intracellular acidosis from anaerobic metabolism occurs soon after the onset of cellular hypoxia, and is worse when there is a plentiful supply of glucose to the cell. Lack of high energy substrates such as ATP and direct effects of hypoxia both inhibit the activity of ion channels, decreasing the transmembrane potential of the cell, leading to increased intracellular calcium levels.
Intracellular acidosis from anaerobic metabolism occurs soon after the onset of cellular hypoxia, and is worse when there is a plentiful supply of glucose to the cell. Lack of high energy substrates such as ATP and direct effects of hypoxia both inhibit the activity of ion channels, decreasing the transmembrane potential of the cell, leading to increased intracellular calcium levels.Chapter 1 explained how all but the simplest forms of life have evolved to exploit the immense advantages of oxidative metabolism. The price they have paid is to become dependent on oxygen for their survival. The essential feature of hypoxia is the cessation of oxidative phosphorylation (page 199) when the mitochondrial Po2 falls below a critical level. Anaerobic pathways, in particular the glycolytic pathway (see Figure 11.13), then come into play. These trigger a complex series of cellular changes leading first to reduced cellular function and ultimately to cell death.
Biochemical Changes in Hypoxia
Depletion of High-Energy Compounds
Anaerobic metabolism produces only one-nineteenth of the yield of the high-energy phosphate compound adenosine triphosphate (ATP) per mole of glucose, when compared with aerobic metabolism (page 200). In organs with a high metabolic rate such as the brain, it is impossible to increase glucose transport sufficiently to maintain the normal level of ATP production. Therefore, during hypoxia, the ATP/ADP ratio falls and there is a rapid decline in the level of all high energy compounds (Figure 24.1). Very similar changes occur in response to arterial hypotension. These changes will rapidly block cerebral function, but organs with a lower energy requirement will continue to function for a longer time and are thus more resistant to hypoxia (see below).
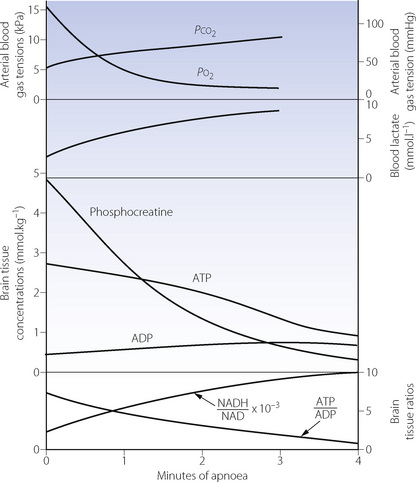
Fig. 24.1 Biochemical changes during 4 minutes of respiratory arrest in rats previously breathing 30% oxygen. Recovery of all values, except blood lactate, was complete within 5 minutes of restarting pulmonary ventilation.
(Data from reference 1.)
Under hypoxic conditions, there are two ways in which reductions in ATP levels may be minimised, both of which are effective for only a short time. First, the high energy phosphate bond in phosphocreatine may be used to create ATP,2 and initially this slows the rate of reduction of ATP (Figure 24.1). Secondly, two molecules of ADP may combine to form one of ATP and one of AMP (the adenylate kinase reaction). This reaction is driven forward by the removal of AMP (adenosine monophosphate), which is converted to adenosine (a potent vaso-dilator) and thence to inosine, hypoxanthine, xanthine and uric acid, with irreversible loss of adenine nucleotides. The implications for production of reactive oxygen species are discussed on page 383.
End-Products Of Metabolism
The end-products of aerobic metabolism are carbon dioxide and water, both of which are easily diffusible and lost from the body. The main anaerobic pathway produces hydrogen and lactate ions which, from most of the body, escape into the circulation, where they may be conveniently quantified in terms of the base deficit, excess lactate or lactate/pyruvate ratio. However, the blood–brain barrier is relatively impermeable to charged ions, and therefore hydrogen and lactate ions are retained within the neurones of the hypoxic brain. Lactacidosis can only occur when circulation is maintained to provide the large quantities of glucose required for conversion to lactic acid.
In severe cerebral hypoxia, a major part of the dysfunction and damage is due to intracellular acidosis rather than simply depletion of high energy compounds (see below). Gross hypoperfusion is more damaging than total ischaemia, because the latter limits glucose supply and therefore the formation of lactic acid. Similarly, patients who have an episode of cerebral ischaemia whilst hyperglycaemic (e.g. a stroke) have been found to have more severe brain injury than those with normal or low blood glucose levels at the time of the hypoxic event.3
Initiation of Glycolysis4
The enzyme 6-phosphofructokinase (PFK) is the rate-limiting step of the glycolytic pathway (see Figure 11.13). Activity of PFK is enhanced by the presence of ADP, AMP and phosphate, which will rapidly accumulate during hypoxia, thus accelerating glycolysis. PFK is, however, inhibited by acidosis, which will therefore quickly limit the formation of ATP from glucose. The intracellular production of phosphate from ATP breakdown also promotes the activity of glycogen phosphorylase, which cleaves glycogen molecules to produce fructose-1,6-diphosphate. This enters the glycolytic pathway below the rate-limiting PFK reaction, and also avoids the expenditure of two molecules of ATP in its derivation from glucose. Therefore four molecules of ATP are produced from one of fructose-1,6-diphosphate in comparison with two from one molecule of glucose. There is no subsequent stage in the glycolytic pathway that is significantly rate-limited by acidosis. Provided glycogen is available within the cell, this second pathway therefore provides a valuable reserve for the production of ATP.
Mechanisms of Hypoxic Cell Damage
Many mechanisms contribute to cell damage or death from hypoxia. The precise role of each is unclear, but there is general agreement that different tissues respond to hypoxia in quite varied ways. Also, the nature of the hypoxic insult has a large effect with differing speed of onset, degree of hypoxia, blood flow, blood glucose concentration and tissue metabolic activity all influencing the resulting tissue dysfunction.
Immediate Cellular Responses to Hypoxia5
Because of the dramatic clinical consequences of nervous system damage, neuronal cells are the most widely studied and therefore form the basis for the mechanisms described in this section.2 Changes in the transmembrane potential of a hypoxic neurone are shown in Figure 24.2, along with the major physiological changes that occur. At the onset of anoxia, CNS cells immediately become either slightly hyperpolarised (as shown in Figure 24.2) or depolarised, depending on the cell type. This is followed by a gradual reduction in membrane potential until a ‘threshold’ value is reached, when a spontaneous rapid depolarisation occurs. At this stage there are gross abnormalities in ion channel function and the normal intracellular and extracellular ionic gradients are abolished, leading to cell death.
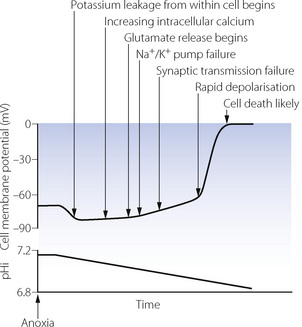
Fig. 24.2 Changes in transmembrane potential and intracellular pH (pHi) in a neuronal cell following the sudden onset of anoxia. Significant physiological events in the course of the hypoxic insult are shown. Once membrane potential reaches zero, cell death is almost inevitable (see text for details). The time between anoxia and rapid depolarisation is highly variable, between about 4 minutes with complete ischaemia to almost an hour with hypoxia and preserved blood flow.
(After reference 2.)
Potassium and sodium flux. Hypoxia has a direct effect on potassium channels (page 71), increasing transmembrane K+ conductance and causing the immediate hyperpolarisation. Potassium begins to leak out from the cell, increasing the extracellular K+ concentration thus tending to depolarise the cell membrane. Potassium leakage, along with sodium influx, are accelerated when falling ATP levels cause failure of the Na/K ATPase pump. Following rapid depolarisation, sodium and potassium channels probably simply remain open, allowing free passage of ions across the cell membrane leading to cellular destruction.
Calcium. Intracellular calcium concentration increases shortly after the onset of hypoxia. Voltage gated calcium channels open in response to the falling transmembrane potential and the increasing intracellular sodium concentration causes the membrane-bound Na/Ca exchanger to reverse its activity. An altered transmembrane potential is detected within the cell by ryonidine receptors on intracellular organelles leading to release of calcium from the endoplasmic reticulum and mitochondria.6 This increase in intracellular calcium is generally harmful, causing the activation of ATPase enzymes just when ATP may be critically low, the activation of proteases to damage sarcolemma and the cytoskeleton, and the uncontrolled release of neurotransmitters (see below). At this stage, the cell has probably not been irretrievably damaged by spontaneous depolarisation, but derangement of calcium channel function effectively prevents normal synaptic transmission and therefore cellular function. Extracellular adenosine, formed from the degradation of AMP, is also believed to play a role in blocking calcium channels during anoxia.2
Excitatory amino acid release.7 The excitatory amino acids glutamate and aspartate are released from many neurones at concentrations of 2–5× normal early in the course of a hypoxic insult, followed by further dramatic increases following rapid depolarisation. Glutamate reuptake mechanisms also fail, and extracellular concentrations quickly reach neurotoxic levels,7,8 acting via the N-methyl-d-aspartate (NMDA) receptor. Cells with depleted energy stores are particularly susceptible, but the mechanism by which glutamate and aspartate bring about cell damage is unknown.
Delayed Cellular Responses to Hypoxia
Following brain injury in humans, cerebral oedema often continues to develop for some hours after the initial insult. There are several possible explanations for this delayed neuronal damage with activation of many different cellular systems being implicated. However, it is a quite different clinical problem that has recently focussed attention on cellular adaptations to hypoxia. The core of many solid malignant tumours has a poor blood supply, caused by the failure of angiogenesis to keep up with the rapid tumour growth. Tumour hypoxia is associated with highly malignant, aggressive tumours, which often respond poorly to treatment. For this reason, much recent research has focussed on understanding the cellular effects of hypoxia, with a view to developing new therapeutic approaches.
Table 24.1 shows the numerous genes that may be induced by hypoxia. Most of the systems activated by hypoxia assist the cell in overcoming the hypoxic conditions, for example erythropoietin to increase haemoglobin concentration, or glycolytic enzymes to increase anaerobic ATP formation. Some activated genes may accelerate cell proliferation and therefore increase tumour malignancy, while other genes are activated that encourage apoptosis and impair tumour growth.9
Table 24.1 Genes induced by hypoxia and their effects
| FUNCTION | GENE | BIOLOGICAL ACTION |
|---|---|---|
| Oxygen transport | Erythropoietin | Stimulation of red cell production |
| Transferrin | Iron transport | |
| Increased blood flow | VEGF | Angiogenesis |
| NO synthase | Vasodilatation | |
| ATP production | Glucose transporter-1 | Transfer of glucose into cell |
 |
||
| pH correction | Carbonic anhydrase | Buffering of metabolic acidosis |
| Inflammation | Interleukin-6, -8 | Activation of inflammatory cells |
VEGF, Vascular endothelial growth factor; NO, nitric oxide.
Hypoxia inducible factor 1 (HIF-1).10-12 Many of these cellular adaptations to hypoxia are mediated by a transcription regulating protein called HIF-1. Under normal conditions cytoplasmic HIF-1 is ubiquitous, but a prolyl-hydroxylase protein (PHD-1) rapidly hydroxylates HIF-1 rendering it inactive. Oxygen is required as a cosubstrate for this reaction such that when cellular hypoxia occurs hydroxylation by PHD-1 fails and HIF-1 remains stable for long enough to initiate transcription of some of the hypoxia induced genes shown in Table 24.1. The HIF-1 system is now seen as a major potential target for therapeutic agents to treat malignancies prone to tumour hypoxia.
Ischaemic Preconditioning13,14
Prior exposure of a tissue to a series of short periods of hypoxia, interspersed with normal oxygen levels, has been found to influence the tissue’s subsequent response to a prolonged ischaemic insult, a phenomenon known as ischaemic preconditioning. Though mostly studied in heart muscle, ischaemic preconditioning has been demonstrated in other tissues.
Early protection. Reduction in the damage occurring from an ischaemic period begins immediately after the preconditioning has occurred, and lasts for 2–3 hours. Activation of sarcolemmal and mitochondrial ATP-dependent K channels (KATP) is believed to be the main mechanism by which protection from ischaemia occurs. After preconditioning, the enhanced activity of KATP channels helps to maintain the transmembrane potential nearer to normal values, and so slows the rate of progression of the immediate cellular responses to hypoxia described above. During prolonged hypoxia, fluid and electrolyte imbalances also occur across the mitochondrial membrane impairing the ability of the cell to make the best use of any oxygen remaining in the cell. Activated mitochondrial KATP channels will again reduce the rate at which these changes occur. Extracellular triggers that bring about preconditioning include adenosine, purines, bradykinin or catecholamines, all acting via G-proteins and protein kinase C to cause activation of the KATP channels.
Late protection. This describes the protection from ischaemia seen about 12 hours after the preconditioning and is less effective than early protection. It is again mediated by activation of KATP channels, this time brought about by gene transcription of proteins such as inducible nitric oxide synthase, super-oxide dismutase (page 385) or cyclo-oxygenase (page 224).
Agents used for preconditioning. Several drugs, but particularly inhalational anaesthetics, can precondition cardiac muscle in a manner similar to brief ischaemic episodes.13,15 The mechanism is also similar, with most of the effective drugs somehow enhancing KATP channel activity. Similar responses occur to the noble gases helium and xenon, possibly mediated by modulation of nitric oxide.16 Unfortunately, impressive laboratory studies of ischaemic preconditioning have so far failed to translate into clinically useful benefits, possibly because of an inability of diseased cardiac muscle to show the same response to preconditioning as that of normal myocardium.17
Po2 Levels at which Hypoxia Occurs
Cellular Po2
‘Critical Po2’ refers to the oxygen tension below which oxidative cellular metabolism fails. For isolated mitochondria, this is known to be below 0.13 kPa (1 mmHg), and possibly as low as 0.01 kPa (0.1 mmHg) in muscle cells4 despite their large oxygen consumption. Venous Po2 approximates to end-capillary Po2, and though highly variable this is usually in excess of 3 kPa (≈20 mmHg) even in maximally working skeletal muscle. Thus with the minimal Po2 in the nearby capillary being approximately 200 times greater than that required by the mitochondria, it is difficult to envisage how cellular hypoxia can occur in all but the most extreme situations. There are reasons why this is not the case in vivo.
Measurement of intracellular Po2 is difficult. The most widely used technique is applicable only to muscle cells and involves measurement of myoglobin saturation, from which Po2 may be determined. These studies have indicated that intracellular Po2 is in the range 0.5–2 kPa (3–15 mmHg) depending on cell activity.4 Many studies have also indicated a minimal difference between the Po2 in extracellular fluid and within cells.17 This would indicate a possibly substantial barrier to oxygen diffusion between the capillary and extracellular fluid. Finally, diffusion of oxygen within cells is believed to be slow because of the proteinaceous nature of the cytoplasm, and therefore large variations in intracellular Po2 are likely to exist. Thus in intact cells, as opposed to isolated mitochondria, critical Po2 is more likely to be of the order of 0.5–1.3 kPa (3–10 mmHg), much closer to the end-capillary value.18
Critical Arterial Po2 for Cerebral Function
The minimal safe level of arterial Po2 is that which will maintain a safe tissue Po2. This will depend on many factors besides arterial Po2, including haemoglobin concentration, tissue perfusion and tissue oxygen consumption. These factors accord with Barcroft’s classification of ‘anoxia’ into anoxic, anaemic and stagnant (page 203), which has previously been shown as a Venn diagram (see Figure 11.16).
This argument may be extended to consider in which circumstances the venous Po2 (and by implication tissue Po2) may fall below its critical level corresponding, in normal blood, to 32% saturation and oxygen content of 6.4 ml.dl−1. If the brain has a mean oxygen consumption of 46 ml.min−1 and a blood flow of 620 ml.min−1, the arterial/venous oxygen content difference will be 7.4 ml.dl−1. Therefore, with normal cerebral perfusion, haemoglobin concentration, pH, etc., this would correspond to a critical arterial oxygen content of 13.8 ml.dl−1, saturation 68% and Po2 4.8 kPa (36 mmHg). This calculation and others under various different conditions are set out in Table 24.2.
Table 24.2 Lowest arterial oxygen levels compatible with a cerebral venous Po2 of 2.7 kPa (20 mmHg) under various conditions
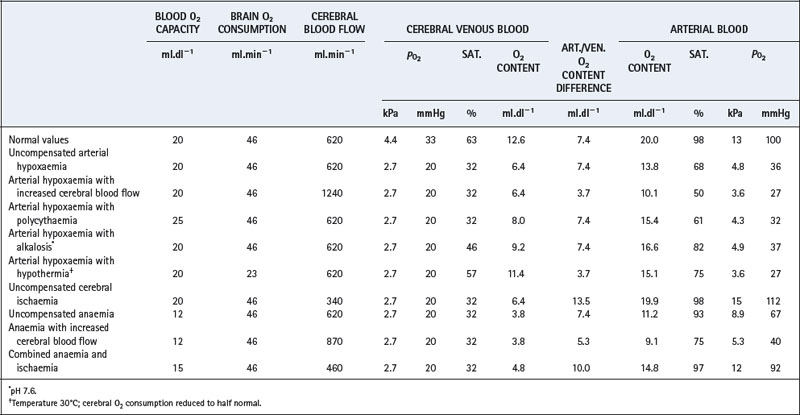
However, the other factors in italics (above) will probably not be normal. They may be unfavourable as a result of multiple pathologies in the patient (e.g. anaemia or a decreased cerebral blood flow). Alternatively, there may be favourable factors, such as polycythaemia in chronic arterial hypoxaemia, or reduced cerebral oxygen requirements during hypothermia or anaesthesia. The possible combinations of circumstances are so great that it is not feasible to consider every possible situation. Instead, certain important examples have been selected which illustrate the fundamentals of the problem, and these are shown in Table 24.2.
Uncompensated ischaemia is dangerous and, with a 45% reduction in cerebral blood flow, any reduction of arterial Po2 exposes the brain to risk of hypoxia. Uncompensated anaemia is almost equally dangerous, although an increase in cerebral blood flow restores a satisfactory safety margin. In the example in Table 24.2, a 40% reduction of blood oxygen carrying capacity and a 40% increase of cerebral blood flow permits the arterial Po2 to fall to 5.3 kPa (40 mmHg) without the cerebral venous Po2 falling below 2.7 kPa (20 mmHg). The last line in Table 24.2 shows the very dangerous combination of anaemia (haemoglobin concentration 11 g.dl−1) and cerebral blood flow three-quarters of normal. Neither abnormality is very serious considered separately, but in combination the arterial Po2 cannot be reduced below its normal value without the risk of cerebral hypoxia.
Table 24.2 is not to be taken too literally, because there are many minor factors that have not been considered. However, it is a general rule that maximal cerebral vasodilatation may be expected to occur in any condition (other than cerebral ischaemia) that threatens cerebral oxygenation. Also, there are circumstances in which the critical organ is not the brain but the heart, liver or kidney.
The most important message of this discussion is that there is no simple answer to the question ‘What is the safe lower limit of arterial Po2?’. Acclimatised mountaineers have remained conscious at high altitude with arterial Po2 values as low as 3.28 kPa (25 mmHg) (Chapter 17). Patients presenting with severe respiratory disease tend to remain conscious down to similar levels of arterial Po2. However, both acclimatised mountaineers and patients with chronic respiratory disease have compensatory polycythaemia and maximal cerebral vasodilatation. Uncompensated subjects who are acutely exposed to hypoxia are unlikely to remain conscious at such low values for arterial Po2, but considerable individual variation must be expected.
Effects of Hypoxia
Hypoxia presents a serious threat to the body, and compensatory mechanisms usually take priority over other changes. Thus, for example, in hypoxia with concomitant hypocapnia, hyperventilation and an increase in cerebral blood flow occur in spite of the decreased Pco2. Certain compensatory mechanisms will come into play whatever the reason for the hypoxia, although their effectiveness will depend to a large extent on the cause. For example, hyperventilation will be largely ineffective in stagnant or anaemic hypoxia because hyperventilation while breathing air can do little to increase the oxygen content of arterial blood, and usually nothing to increase perfusion.
Hyperventilation results from a decreased arterial Po2 but the response is non-linear (see Figure 5.7). There is little effect until arterial Po2 is reduced to about 7 kPa (52.5 mmHg): maximal response is at 4 kPa (30 mmHg). The interrelationship between hypoxia and other factors in the control of breathing is discussed in Chapter 5.
Pulmonary distribution of blood flow is improved by hypoxia as a result of hypoxic pulmonary vasoconstriction (page 108).
The sympathetic system is concerned in many of the responses to hypoxia, particularly the increase in organ perfusion. The immediate response is reflex and is initiated by chemoreceptor stimulation: it occurs before there is any measurable increase in circulating catecholamines, although this does occur in due course. Reduction of cerebral and probably myocardial vascular resistance is not dependent on the autonomic system but depends on local responses in the vicinity of the vessels themselves. With the exception of pulmonary vessels, hypoxia causes vasodilatation of blood vessels almost everywhere in the body. This results mainly from a direct effect of adenosine and other metabolites generated by hypoxia.
Cardiac output is increased by hypoxia, together with the regional blood flow to almost every major organ, particularly the brain.
Haemoglobin concentration is not increased in acute hypoxia in humans but it is increased in chronic hypoxia due to residence at altitude or respiratory disease.
The oxyhaemoglobin dissociation curve is displaced to the right by an increase in 2,3-DPG and by acidosis which may also be present. This tends to increase tissue Po2 (see Figure 11.10).
Anaerobic metabolism is increased in severe hypoxia in an attempt to maintain the level of ATP (see above).
References
1. Kaasik AE, Nilsson L, Siesjö BK. The effect of asphyxia upon the lactate, pyruvate and bicarbonate concentrations of brain tissue and cisternal CSF, and upon the tissue concentrations of phosphocreatine and adenine nucleotides in anesthetized rats. Acta Physiol Scand.. 1970;78:433-437.
2. Martin RL, Lloyd HGE, Cowan AI. The early events of oxygen and glucose deprivation: setting the scene for neuronal death? Trends Neurosci.. 1994;17:251-256.
3. Candelise L, Landi G, Orazio EN, Boccardi E. Prognostic significance of hyperglycaemia in acute stroke. Arch Neurol.. 1985;42:661-663.
4. Connett RJ, Honig CR, Gayeski TEJ, Brooks GA. Defining hypoxia: a systems view of 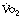 , glycolysis, energetics, and intracellular Po2. J Appl Physiol.. 1990;68:833-842.
, glycolysis, energetics, and intracellular Po2. J Appl Physiol.. 1990;68:833-842.
*5. Ransom BR, Brown AM. Intracellular Ca2+ release and ischemic axon injury: the Trojan horse is back. Neuron. 2003;40:2-4.
6. Katchman AN, Hershkowitz N. Early anoxia-induced vesicular glutamate release results from mobilization of calcium from intracellular stores. J Neurophysiol.. 1993;70:1-7.
7. Choi DW. Cerebral hypoxia: some new approaches and unanswered questions. J Neurosci.. 1990;10:2493-2501.
8. Ohmori T, Hirashima Y, Kurimoto M, Endo S, Takaku A. In vitro hypoxia of cortical and hippocampal CA1 neurons: glutamate, nitric oxide, and platelet activating factor participate in the mechanism of selective neural death in CA1 neurons. Brain Res.. 1996;743:109-115.
9. Harris AL. Hypoxia – A key regulatory factor in tumour growth. Nat Rev Cancer. 2002;2:38-46.
10. Höpfl G, Ogunshola O, Gassman M. HIFs and tumors – causes and consequences. Am J Physiol Regul Integr Comp Physiol.. 2004;286:R608-R623.
*11. Semenza GL. HIF-1: mediator of physiological and pathophysiological responses to hypoxia. J Appl Physiol.. 2000;88:1474-1480.
*12. Berchner-Pfannschmidt U, Frede S, Wotzlaw C, Fandrey J. Imaging of the hypoxia-inducible factor pathway: insights into oxygen sensing. Eur Respir J. 2008;32:210-217.
13. Zaugg M, Lucchinetti E, Uecker M, Pasch T, Schaub MC. Anaesthetics and cardiac preconditioning. Part I. Signalling and cytoprotective mechanisms. Br J Anaesth.. 2003;91:551-565.
14. Zaugg M, Lucchinetti E, Garcia C, Pasch T, Spahn DR, Schaub MC. Anaesthetics and cardiac preconditioning. Part II. Clinical implications. Br J Anaesth.. 2003;91:566-576.
15. Tanaka K, Ludwig LM, Kersten JR, Pagel PS, Warltier DC. Mechanisms of cardioprotection by volatile anaesthetics. Anesthesiology. 2004;100:707-721.
16. Pagel PS, Krolikowski JG, Pratt PF, et al. The mechanism of helium-induced preconditioning: a direct role for nitric oxide in rabbits. Anesth Analg.. 2008;107:762-768.
17. Rumsey WL, Wilson DF. Tissue capacity for mitochondrial oxidative phosphorylation and its adaptation to stress. In: Fregly MJ, Blatteis CM, editors. Handbook of Physiology, Section 4: Environmental Physiology. New York & Oxford: Oxford University Press; 1996:1095-1114.
18. Epstein FH, Agmon Y, Brezis M. Physiology of renal hypoxia. Ann N Y Acad Sci.. 1995;718:72-81.