Chapter 17 High altitude and flying
 The low inspired oxygen partial pressure at altitude causes immediate hyperventilation, which increases further with acclimatisation to produce hypocapnia and improve oxygen levels. The rate of ascent and altitude achieved are determinants of altitude-related illnesses, which vary from mild acute mountain sickness to potentially lethal high altitude pulmonary oedema.
The low inspired oxygen partial pressure at altitude causes immediate hyperventilation, which increases further with acclimatisation to produce hypocapnia and improve oxygen levels. The rate of ascent and altitude achieved are determinants of altitude-related illnesses, which vary from mild acute mountain sickness to potentially lethal high altitude pulmonary oedema.With increasing altitude, the barometric pressure falls, but the fractional concentration of oxygen in the air (0.21) and the saturated vapour pressure of water at body temperature (6.3 kPa or 47 mmHg) remain constant. The Po2 of the inspired air is related to the barometric pressure as follows:
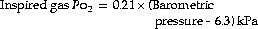
or
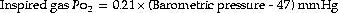
The influence of the saturated vapour pressure of water becomes relatively more important until, at an altitude of approximately 19 000 m or 63 000 feet, the barometric pressure equals the water vapour pressure, and alveolar Po2 and Pco2 become zero.
Table 17.1 is based on the standard table relating altitude and barometric pressure. However, there are important deviations from the predicted barometric pressure under certain circumstances, particularly at low latitudes.1 At the summit of Everest, the actual barometric pressure was found to be 2.4 kPa (18 mmHg) greater than predicted, and this was crucial to reaching the summit without oxygen. The uppermost curve in Figure 17.1 shows the expected Po2 of air as a function of altitude, while the crosses indicate observed values in the Himalayas that have been consistently higher than expected.
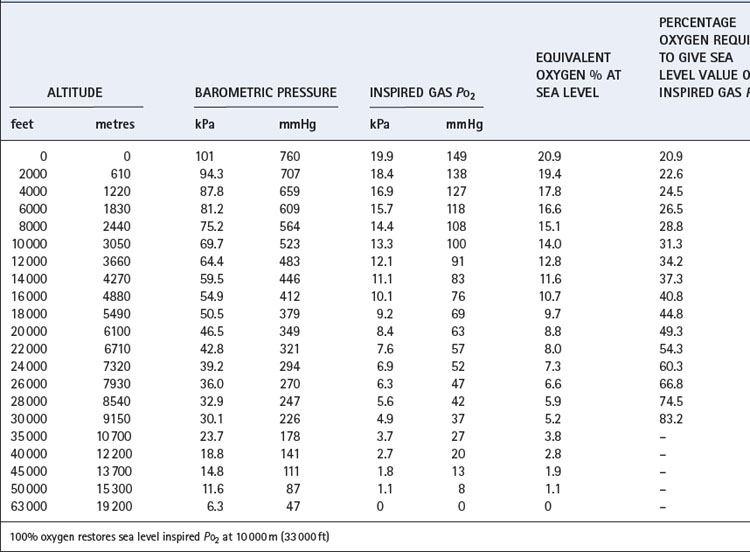
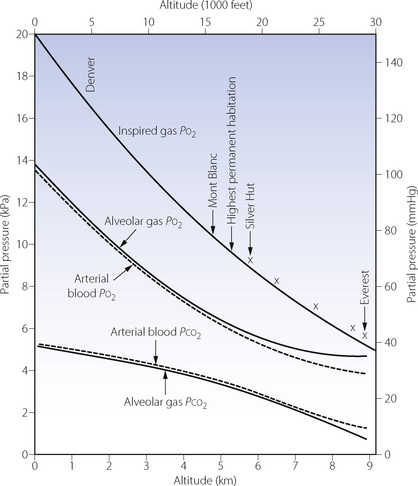
Fig. 17.1 Inspired, alveolar and arterial gas partial pressures at rest, as a function of altitude. The curve for inspired Po2 is taken from standard data in Table 17.1, but the crosses show actual measurements in the Himalayas. The alveolar gas data are from reference 2, and agree remarkably well with the arterial blood data from the simulated ascent of Everest.3
Equivalent Oxygen Concentration
The acute effect of altitude on inspired Po2 may be simulated by reduction of the oxygen concentration of gas inspired at sea level (Table 17.1). This technique is extensively used for studies of hypoxia and for clinical assessment of patients before flying (see below), but there are theoretical reasons why the same inspired Po2 at normal and low barometric pressure may have different physiological effects. These include the density of the gas being breathed and different Pn2 values in the tissues.4
Up to 10 000 m (33 000 ft), it is possible to restore the inspired Po2 to the sea level value by an appropriate increase in the oxygen concentration of the inspired gas (also shown in Table 17.1). Lower inspired Po2 values may be obtained between 10 000 and 19 000 m, above which body fluids boil.
Respiratory System Responses to Altitude1
Ascent to altitude presents three main challenges to the respiratory system, resulting from progressively reduced inspired Po2, low relative humidity, and, in outdoor environments, extreme cold. Hypoxia is by far the most important of these, and requires significant physiological changes to allow continuation of normal activities at altitude. The efficiency of these changes depends on many factors such as the normal altitude at which the subject lives, the rate of ascent, the altitude attained and the health of the subject.
Acute Exposure to Altitude
Transport technology now permits altitude to be attained quickly and without the exertion of climbing. Within a few hours, rail, air, cable car or motor transport may take a passenger from near sea level to as high as 4000 m (13 100 ft).
Ventilatory changes. At high altitude the decrease in inspired gas Po2 reduces alveolar and therefore arterial Po2. The actual decrease in alveolar Po2 is mitigated by hyperventilation caused by the hypoxic drive to ventilation. However, on acute exposure to altitude, the ventilatory response to hypoxia is very short lived due to a combination of the resultant hypocapnia and hypoxic ventilatory decline (page 73 and Figure 5.6). During the first few days at altitude, this disadvantageous negative feedback is reversed by acclimatisation (see below).
Signs and symptoms. Impairment of night vision is the earliest sign of hypoxia, and may be detected as low as 1200 m (4000 ft). However, the most serious aspect of acute exposure to altitude is impairment of mental performance, culminating in loss of consciousness, which usually occurs on acute exposure to altitudes in excess of 6000 m (about 20 000 ft). The time to loss of consciousness varies with altitude and is of great practical importance to pilots in the event of loss of pressurisation (Figure 17.2). The shortest possible time to loss of consciousness (about 15 seconds) applies above about 16 000 m (52 000 ft) and is governed by lung-to-brain circulation time and the capacity of high energy phosphate stores in the brain (page 363). Impaired mental function as a result of hypoxia is due to both a direct effect of hypoxia on brain tissue and from cerebral vasoconstriction from the resulting hypocapnia.5
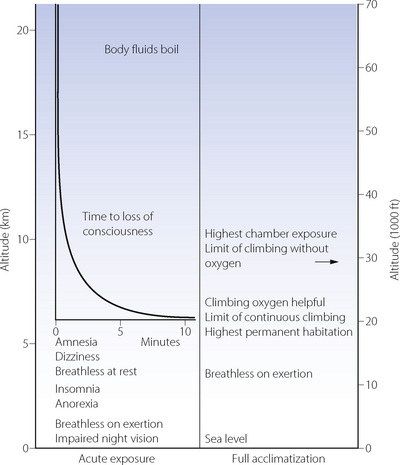
Acclimatisation to Altitude
Acclimatisation refers to the processes by which tolerance and performance are improved over a period of hours to weeks after an individual who normally lives at relatively low altitude ascends to a much higher area. Everest has been climbed without oxygen by well-acclimatised lowlanders, although the barometric pressure on the summit would cause rapid loss of consciousness without acclimatisation (Figure 17.2). Adaptation to altitude (described below) refers to physiological differences in permanent residents at high altitude and is quite different from acclimatisation.
Earlier studies of acclimatisation took place in the attractive, though somewhat hostile, environment of high altitude expeditions in many mountain ranges. Technical limitations in these conditions led to two experiments, named Operation Everest II and III, in which volunteers lived in a decompression chamber in which an ascent to the summit of Everest was simulated.6,7 These conditions permitted extensive physiological research to be undertaken at rest and during exercise.
Ventilatory control. Prolonged hypoxia results in several complex changes in ventilation and arterial blood gases, which are shown in Figure 17.3.8 The initial hypoxic drive to ventilation on acute exposure is short lived, and after about 30 minutes ventilation returns to only slightly above normoxic levels with Pco2 just below control levels (Figure 17.3). This poor ventilatory response causes significant arterial hypoxaemia and results in many of the symptoms seen during the first few hours and days at altitude. Over the next few days, ventilation slowly increases with an accompanying reduction of Pco2 and increase in arterial Po2. This increase in Po2 is of relatively small magnitude and can never correct Po2 to normal (sea level) values, but it does seem to be enough to ameliorate most of the symptoms of exposure to acute altitude.
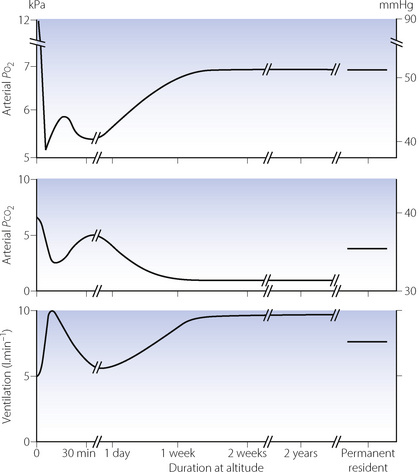
Fig. 17.3 Effects of prolonged hypoxia (equivalent to 4300 m, 14 100 ft) on ventilation and blood gases. The first section of the graph shows the acute hypoxic response and hypoxic ventilatory decline described in Chapter 5. Acclimatisation then takes place, partially restoring Po2 by means of long-term hyperventilation and hypocapnia, a situation that is maintained indefinitely while remaining at altitude. Individuals who reside throughout life at this altitude maintain similar Po2 values with lesser degrees of hyperventilation, but still have a minute ventilation greater than sea level normal.
(After reference 8.)
There are significant differences between species in the rate at which acclimatisation takes place, being just a few hours in most animals, and several days or weeks in humans.4 Both the rate of ascent and the altitude attained influence the speed at which ventilatory acclimatisation occurs, but in humans, most subjects are fully acclimatised within one week.
There are many possible mechanisms to explain the ventilatory changes seen with acclimatisation.8,9 In spite of the low blood Pco2, stimulation of the central chemoreceptors almost certainly plays a part in the hyperventilation that occurs with acclimatisation. It was first suggested, in 1963, that the restoration of cerebrospinal fluid (CSF) pH, by means of bicarbonate transport, might explain this acclimatisation of ventilation to altitude.10 Shortly afterwards Severinghaus and his colleagues measured their own CSF pH during acclimatisation to altitude and showed that it did indeed tend to return towards its initial value of 7.2.10 Subsequent work showed that the time course of changes in CSF pH did not match changes in ventilation,8 with most studies finding a persistent increase in CSF pH during continued exposure to hypoxia.9 Changes in CSF pH therefore seem unlikely to represent an important mechanism of acclimatisation. Other studies, mainly in animals, indicate that acclimatisation represents an increase in the responsiveness of the respiratory centre to hypoxia from both direct effects of prolonged hypoxia on the central nervous system and prolonged maximal afferent input from the peripheral chemoreceptors. This increased responsiveness may be mediated by alterations in the sensitivity to neurotransmitters involved in respiratory control (see Figure 5.4). For example, increased sensitivity to glutamate will directly increase ventilation, or decreasing GABA sensitivity will effectively reduce hypoxic ventilatory decline (page 73).9
In addition to changes affecting the central chemoreceptors, there is evidence that peripheral chemoreceptor sensitivity is increased during prolonged hypoxia, so contributing to the progressive hyperventilation seen with acclimatisation. In humans, the acute hypoxic ventilatory response is increased during the first few days at altitude and for several days after return to sea level. The mechanism of this increased sensitivity to hypoxia is not known, but is independent of changes in Pco2,11 and may reside either with increased sensitivity of the carotid bodies themselves or with the increased responsiveness of the respiratory centre described in the previous paragraph.8,9
Respiratory alkalosis at altitude is counteracted, over the course of a few days, by renal excretion of bicarbonate, resulting in a degree of metabolic acidosis that will tend to increase respiratory drive (see Figure 5.5). This was formerly thought to be the main factor in the ventilatory adaptation to altitude but it now appears to be of minor importance compared to the changes in the central and peripheral chemoreceptors.
Blood gas tensions. Figure 17.3 shows the time course of blood gas changes during acclimatisa-tion and Figure 17.1 shows changes in alveolar gas tensions with altitude in fully acclimatised mountaineers. Alveolar Po2 was found to be unexpectedly well preserved at extreme altitude, and above 8000 m (26 000 ft) tended to remain close to 4.8 kPa (36 mmHg).2 Operations Everest II and III found arterial Po2 values of 3.62 and 4.08 kPa (27 and 31 mmHg) at a pressure equivalent to the summit of Everest (Table 17.2), with an alveolar/arterial Po2 difference of less than 0.3 kPa (2 mmHg) at rest.12 The recent Caudwell Extreme Everest expedition obtained arterial blood samples at 8400 m (27 559 ft) with an average Po2 of 3.28 kPa (24.6 mmHg). There was also a significant alveolar to arterial Po2 difference of 0.72 kPa (5.4 mmHg) which the authors suggested may have resulted from a diffusion barrier to oxygen at such low levels, possibly as a result of sub-clinical pulmonary oedema.13
Table 17.2 Cardiorespiratory data obtained at rest and during exercise at extreme reduction of ambient pressure during the simulated ascent of Everest in a low pressure chamber
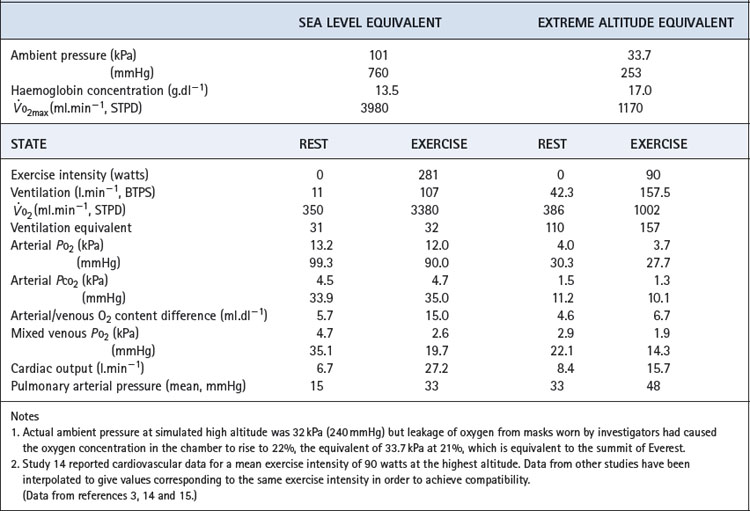
Haemoglobin concentration and oxygen affinity. Anincrease in haemoglobin concentration was the earliest adaptation to altitude to be demonstrated. The recent data from subjects at 8400 m (27 559 ft) reported an increase from 14.8 to 19.3 g.dl−1 which, at the resting value of 54% saturation, maintained an arterial oxygen content of almost 15 ml.dl−1.13 Plasma erythropoietin levels begin to increase within a few hours at altitude, reaching a maximum at 24–48 hours and then declining.16
The haemoglobin dissociation curve at altitude is affected by changes in both pH and 2,3-diphosphoglycerate (DPG) concentration (page 193). 2,3-DPG concentrations increased from 1.7 to 3.8 mmol.l−1 on Operation Everest II.3 It has been estimated that the resultant effect is a leftward shift at extreme altitude, where oxygen loading in the lung takes priority over maintaining Po2 at the point of release.1
Adaptation to Altitude1,17
Adaptation refers to physiological and genetic changes that occur over a period of years to generations by those who have taken up permanent residence at high altitude. There are qualitative as well as quantitative differences between acclimatisation and adaptation but each is remarkably effective. High altitude residents have a remarkable ability to exercise under grossly hypoxic conditions, but their adaptations show many striking differences from those in acclimatised lowlanders. Residents in different high altitude areas of the world have differing adaptations.18
Long-term residence at altitude leads to a reduced ventilatory response to hypoxia, the magnitude of which relates to the product of altitude level and years of residence there.8 This results in a reduction of ventilation compared with an acclimatised lowlander, and a rise in Pco2, though neither of these returns to sea level values (Figure 17.3). High altitude residents maintain similar arterial Po2 values as acclimatised lowlanders in spite of the reduced ventilation and therefore lower alveolar Po2. Pulmonary diffusing capacity must therefore be increased, and depends on anatomical pulmonary adaptations increasing the area available for diffusion by the generation of greater numbers of alveoli and associated capillaries. This adaptation seems not to be inherited, but occurs in children and infants who spend their formative years at altitude. In humans, the development of alveoli by septation of saccules formed in utero occurs mostly after birth (page 250), and it is this process that must be stimulated by hypoxia, though the mechanism of this stimulation remains unknown.19 An adult moving permanently to high altitude will therefore never achieve the same degree of adaptation as a native of the area, so explaining the ability of high altitude residents to exercise to a much greater degree than their non-resident visitors.
Research has found that residents of high altitude areas of the Andes hyperventilate less than residents at equivalent altitude in Tibet.18 This higher ventilation in Tibetans may explain their reduced susceptibility, in comparison with populations in the Andes, to chronic mountain sickness (see below) and some complications of pregnancy that are normally associated with high altitude life.20 Human occupation of Tibet is believed to have begun earlier than other high-altitude areas of the world, and these differences in Tibetan physiology could represent a more advanced genetic adaptation to the physiologically hostile environment.
Polycythaemia is normal and influenced by altitude, population and sex,21 for example male subjects of the Chinese Han population residing at 5200 m (17 060 ft) on the Tibetan plateau have an average haemoglobin concentration of 19.6 g.dl−1. Another major adaptation to altitude by long-term residents appears to be increased vascularity of heart and striated muscles, a change that is also important for the trained athlete. For the high altitude resident, increased perfusion appears to compensate very effectively for the reduced oxygen content of the arterial blood.
Recent data regarding the mortality of climbers attempting to reach the summit of Mount Everest illustrates how effective adaptations to altitude are compared with acclimatised lowlanders. Sherpa residents had a mortality of only 0.4%, compared with 2.7% amongst climbers.22
Chronic mountain sickness (Monge’s disease).1 A small minority of those who dwell permanently at very high altitude develop this dangerous illness. It is characterised by an exceptionally poor ventilatory response to hypoxia resulting in low arterial Po2 and high Pco2. There is cyanosis, high haematocrit, finger clubbing, pulmonary hypertension, right heart failure, dyspnoea and lethargy.
Exercise at High Altitude23,24
The summit of Everest was attained without the use of oxygen in 1978 by Messner and Habeler, and by many other climbers since that date. Studies of exercise have been made at various altitudes up to and including the summit, and on the simulated ascents in Operations Everest II and III. Of necessity, these observations are largely confined to very fit subjects.
Capacity for work performed. There is a progressive decline in the external work that can be performed as altitude increases. On Operation Everest II, 300–360 watts was attained at sea level, 240–270 watts at 440 mmHg pressure (equivalent to 4300 m, 14 000 ft) and 120 watts at 280 mmHg (Everest summit), very close to the results obtained on Everest.25 declined in accord with altitude to 1177 ml.min−1 at 240 mmHg pressure.15 Resting cardiac output is unchanged at moderate altitude and only slightly increased at extreme altitude. During exercise, for a given power expenditure, the increase in cardiac output at altitude is the same as at sea level.3
Ventilation equivalent of oxygen consumption. Figure 15.5 shows that ventilation as a function of is comparatively constant. The length of the line increases with training but the slope of the linear portion remains the same. With increasing altitude, the slope and intercept are both dramatically increased up to four times the sea level value3,15 with maximal ventilation approaching 200 l.min−1 (Figure 17.4). This is because ventilation is reported at body temperature and pressure saturated (BTPS) and oxygen consumption at standard temperature and pressure dry (STPD) – see Appendix C.
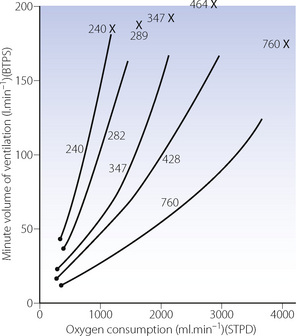
Fig. 17.4 The relationship between minute volume of ventilation and oxygen consumption at rest and during exercise at altitude. The relationship is radically changed at altitude primarily because ventilation is reported at body temperature and pressure (saturated), whereas oxygen consumption is reported at standard temperature and pressure (dry). Numbers in the figure indicate barometric pressure.  , resting points; ×, values at
, resting points; ×, values at 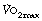 from reference 15.
from reference 15.
(Data from reference 3.)
Fortunately, the density of air is reduced in proportion to the barometric pressure at altitude. Resistance to turbulent flow is decreased and there-fore the work of breathing at a particular minute volume of respiration is less. Even with this mitigation, the extra ventilation needed to deliver the oxygen requirement at altitude means that the energy expenditure upon breathing for a given intensity of exercise is considerably higher than at sea level.
Pco2 and Po2. During exercise at altitude, alveolar Pco2 falls and alveolar Po2 rises.3,26 Arterial Pco2 falls with alveolar Pco2 but the alveolar/arterial Po2 difference increases more than the alveolar Po2 rises26 and there is a consistent decrease in arterial Po2 during exercise at altitude, leading to very low values for Po2. The lower alveolar to pulmonary capillary Po2 gradient, along with a faster pulmonary capillary transit time during exercise, causes diffusion-limitation of oxygen uptake. There is also some evidence that ventilation perfusion inequality occurs during exercise at altitude, and adversely affects oxygenation.23
Altitude Illness27,28,29
Acute Mountain Sickness
Acute mountain sickness (AMS) is characterised by headache, nausea, fatigue, anorexia, dyspnoea, difficulty in sleeping (see below) and impaired climbing performance (Figure 17.2). Symptoms normally begin to occur at above 2000 m (6600 ft), with an abrupt increase in the incidence above 4500 m (14 760 ft), affecting about half of trekkers at this altitude.30 The unacclimatised subject has extreme dyspnoea on exertion at this level and has dyspnoea at rest. Severity varies greatly from a mild inconvenient headache to a severe life threatening illness involving cerebral and pulmonary oedema.
The likelihood of developing AMS relates to altitude (particularly sleeping altitude), the rate of ascent and the degree of exertion. The mountaineer is therefore affected by altitude in a manner that differs from that of the aviator because his physical exertion is much greater and the time course of exposure is different. Rate of ascent seldom exceeds 2000 m (6500 ft) per day from sea level, decreasing to only 300 m (1000 ft) per day at very high altitude.
High Altitude Pulmonary Oedema (HAPE)1,31
A small amount of sub-clinical pulmonary oedema probably occurs in all subjects at high altitude, but in a few percent the oedema becomes progressive and life-threatening.1 As for AMS, the proportion of subjects who develop HAPE depends on the altitude, the speed of ascent, the amount of strenuous exercise performed, and an individual susceptibility to the condition.28,31 It is most commonly seen in the unacclimatised and overambitious climber. Clinical features include cough, dyspnoea and hypoxia with clinical and radiological signs of pulmonary oedema. Untreated, HAPE has a mortality rate of almost 50%, but with appropriate treatment this is normally less than 3%.
The pathophysiology of HAPE is complex.27,31,32 Subjects with HAPE have significant pulmonary hypertension secondary to hypoxia, and low pulmonary capillary wedge pressures indicating normal left ventricular function. Subjects who are susceptible to HAPE seem to have an excessive hypoxic pulmonary vasoconstriction response to hypoxia, and this may in part be due to impaired release of endothelial relaxing factors such as nitric oxide (page 107). Compared with subjects who are not susceptible to HAPE, susceptible subjects exhaled lower concentrations of nitric oxide during a high-altitude trip.33 Pulmonary vasoconstrictors such as endothelin-1 are found in higher concentrations in HAPE susceptible subjects, who also have greater sympathetic responses to hypoxia. Pulmonary shadows on chest X-rays with HAPE are typically patchy, indicating that pulmonary vasoconstriction is non-uniform, such that some areas of lung have little blood flow whilst others have greatly increased blood flow. High capillary flow in some areas is postulated to lead to ‘stress failure’ of capillaries. This mechanism would explain the association between exercise and HAPE, with increased cardiac output causing huge blood flows through less vasoconstricted regions of lung. Magnetic resonance imaging of the lung during hypoxia has shown that hypoxic pulmonary vasoconstriction is patchy in all subjects, but the uneven vasoconstriction is more pronounced in HAPE-susceptible subjects.34 Although inflammation is not believed to be a primary event in the pathogenesis of HAPE, it does occur in severe cases and explains why coincidental lung inflammation from, for example, lower respiratory tract infections, may exacerbate or even cause HAPE.
Other Respiratory Problems at Altitude
Cerebral oedema is also potentially lethal and is manifest in the early stages by ataxia, irritability and irrational behaviour and may progress to hallucinations, drowsiness and coma. Pulmonary and cerebral forms of severe acute mountain sickness may both be present in the same patient, but a common aetiology has not been found. Mild, or localised, brain swelling is thought to occur in all people ascending to high altitudes, but it is unclear whether this always represents cerebral oedema.27
Following return to low altitude, cerebral disturbance may persist. Investigations up to 30 days after expeditions to very high altitudes have shown a variety of impairments, including visual long-term memory.35
Cough.36 Almost half of trekkers in Nepal complain of a cough, which may be severe. Coughing normally develops after a few days at altitude and airway sensitivity to irritants is increased as a result of hyperventilation of low humidity cold air. Development of a cough may however be the first manifestation of impending HAPE.
Sleep disturbance.1 Periodic breathing (page 76) occurs in most individuals during the first few nights above about 4000 m (13 000 ft). There are cyclical changes in tidal volume, often associated with central (rather than obstructive) apnoeas, with or without arousal from sleep (see Figure 16.2). Apnoeas may result in considerable additional hypoxaemia at high altitude. A study at 4500 m (15 000 ft) found nocturnal reductions in saturation of 8% which reduced median nocturnal saturation to just 50%.37 The primary problem is an abnormality of respiratory control, with arousal occurring at the end of a period of apnoea, presumably secondary to hypoxia. The severity of periodic breathing is related to the strength of the subject’s hypoxic ventilatory drive, and it is seldom seen in high altitude residents, who have a much attenuated hypoxic drive. The onset of sleep disturbance and severe nocturnal hypoxia may also contribute to the symptoms of AMS, and subjects developing HAPE have lower oxygen saturations during sleep.37
Therapy for Altitude-Induced Illness28,38
For any severe form of AMS, administration of oxygen and descent to a lower altitude are the first essentials. Without these simple interventions, patients with cerebral oedema or HAPE will have a high mortality. Nifedipine is now an established treatment for HAPE, and when used prophylactically prevents HAPE developing in susceptible individuals.27,28 It is an effective drug for treating pulmonary hypertension, and the convenience of administration by oral or sublingual route makes it a popular choice for mountaineers.
People with milder degrees of AMS do not need to be removed from high altitude. With acclimatisation, most symptoms of AMS will resolve but, if time is limited or symptoms interfere with planned activities, acetazolamide may be useful.39 This carbonic anhydrase inhibitor (page 161) interferes with the transport of carbon dioxide out of cells, causing an intracellular acidosis that includes the cells of the medullary chemoreceptors and so drives respiration, in effect accelerating acclimatisation. Acetazolamide also improves sleep-induced periodic breathing, possibly by an effect on the carotid body responses to hypoxia, and via its renal effects also induces a metabolic acidosis that may further drive respiration via the central chemoreceptors.39
Sildenafil is a pulmonary vasodilator that acts via inhibition of phosphodiesterase (page 112) and may be taken orally. Sildenafil has been shown to be effective at reducing the hypoxia induced rise in pulmonary arterial pressure at altitude, and therefore has potential as a useful treatment for HAPE.40
Flying
Only a very small number of people will ever visit places of high enough altitude to induce any of the respiratory changes described in this chapter so far. However, worldwide, almost 2 billion people per year fly in commercial aircraft, so it is useful to consider the respiratory effects of aviation.41
Altitude Exposure42
For reasons of fuel economy and avoidance of weather systems, commercial aircraft operate at between 9000–12 000 m (30–40 000 ft). The passenger cabin must therefore be pressurised, and a typical design aims for a cabin pressure equivalent to less than 2400 m (8000 ft), often referred to as the ‘cabin altitude’.43 Cabin pressure is maintained by indrawing and compression of external air whilst limiting cabin air outflow to maintain the desired pressure. In practice, a differential pressure is established, which represents the absolute pressure difference between the outside and inside of the aircraft. Differential pressure is increased as the aircraft climbs, and vice versa. Thus cabin pressure changes in parallel with altitude, but to a much lesser degree than the external pressure.
Supersonic flight requires much higher operating altitude to reduce air resistance, for example Concorde’s cruise altitude was 18 300 m (60 000 ft). The differential pressure must therefore be greater to sustain a normal cabin environment at this altitude, which was facilitated in Concorde by the significantly more powerful engines from which compressed air was drawn. Military aircraft fly prolonged reconnaissance missions at an altitude of 22 400 m (73 500 ft), with the cockpit pressurised to an equivalent altitude of 9000 m (30 000 ft). Pilots must therefore breathe 100% oxygen by mask to maintain an inspired Po2 close to sea level to facilitate the required mental performance. At this altitude, military pilots are also at risk of altitude decompression sickness, which is discussed on page 300.
In theory, cabin altitudes of below 2400 m (8000 ft) should represent a minimal physiological challenge to healthy individuals resulting in a drop of only a few per cent in oxygen saturation. In practice, a study of healthy cabin crew during normal flight patterns showed that over half had saturation drops to less than 90%.43 The effects of this degree of hypoxia on performance are controversial, though impaired night vision or colour recognition may occur at this altitude (page 280).37 For passengers, average oxygen saturation during a flight is approximately 92%, though this may be worse during exercise and sleep (Figure 17.5).44,45
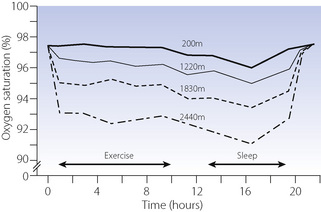
Fig. 17.5 Oxygen saturation in healthy subjects breathing air in a hypobaric chamber simulating four different altitudes: 200 m (650 ft), 1220 m (4000 ft), 1830 m (6000 ft) and 2440 m (8000 ft), the last of which is the highest ‘cabin altitude’ used in commercial flying. During the exercise period subjects walked on a treadmill for 10 minutes in each hour; during the sleep period subjects slept in coach class aircraft seats.
After reference 45 with permission. Copyright © 2007 Massachusetts Medical Society. All rights reserved.
Depressurisation. Loss of cabin pressure at altitude either through equipment failure or accident is extremely rare. In the case of slow loss of cabin pressure, oxygen is provided for passengers as an interim measure until the aircraft can descend: 100% oxygen provides adequate protection from loss of consciousness up to an altitude of about 12 000 m (40 000 ft), where the atmospheric pressure is roughly equal to the sea level atmospheric Po2.
There are sporadic reports of stowaway passengers undertaking long haul flights in the wheel well of modern aircraft.46 This environment affords little protection against the cold and severe hypoxia of altitude levels well above that of Everest. That half of these stowaways die is not surprising, but it is remarkable that half of them survive. Severe hypothermia is believed to protect them against the effects of hypoxia.
Air Travel in Patients with Respiratory Disease47,48
To patients with respiratory disease flying may present a significant problem, particularly if arterial hypoxaemia already exists at sea level, and careful preflight assessment is required. A variety of preflight clinical evaluations and investigations have been recommended to determine whether an individual patient would require supplemental oxygen during flight. British Thoracic Society guidelines47 have provided recommendations for assessing patients with respiratory disease, a summary of which is shown in Table 17.3. For patients with an oxygen saturation whilst breathing air of less than 92%, or in those with 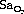 of 92–95% with other risk factors (Table 17.3), then a hypoxic challenge test is recommended. This test involves measurement of arterial Po2 whilst simulating flying conditions by using a hypoxic gas mixture, usually 15% oxygen. This inspired Po2 equates to a cabin altitude of 2400 m (8000 ft) and represents the lowest oxygen tension that should be experienced during a commercial flight (Table 17.1).
of 92–95% with other risk factors (Table 17.3), then a hypoxic challenge test is recommended. This test involves measurement of arterial Po2 whilst simulating flying conditions by using a hypoxic gas mixture, usually 15% oxygen. This inspired Po2 equates to a cabin altitude of 2400 m (8000 ft) and represents the lowest oxygen tension that should be experienced during a commercial flight (Table 17.1).
Table 17.3 British Thoracic Society recommendations on assessing the need for in-flight supplemental oxygen in patients with respiratory disease47
| ASSESSMENT RESULT | ACTION |
|---|---|
| Screening: | |
| Sao2 > 95% | Oxygen not required |
| Sao2 92–95% and no risk factor† | Oxygen not required |
| Sao2 92–95% and additional risk factor† | Hypoxic challenge test |
| Sao2 < 92% | In-flight oxygen |
| Hypoxic challenge test: | |
| Pao2 > 7.4 kPa (>55 mmHg) | Oxygen not required |
| Pao2 6.6–7.4 kPa (50–55 mmHg) | Borderline – walk test may be helpful |
| Pao2 < 6.6 kPa (<50 mmHg) | In-flight oxygen |
Screening test is oxygen saturation whilst breathing air at sea level. Hypoxic challenge test is arterial oxygen tension after breathing 15% oxygen for 20 minutes. †, Additional risk factors include hypercapnia; FEV1 <50% of predicted; lung cancer; restrictive lung disease involving the parenchyma, chest wall (kyphoscoliosis) or respiratory muscles; ventilator support, cerebrovascular or cardiac disease; within 6 weeks of discharge for an exacerbation of chronic lung or cardiac disease.
Cabin Air Quality42,49
Aircraft ventilation systems deliver 4–8 l.sec−1 of air per passenger during flight. However, compression and temperature regulation of fresh air from outside is expensive in energy terms, and more recent designs of aircraft incorporate cabin air recirculation systems.50 Total air delivered remains the same, but up to 50% may be recirculated rather than fresh. This recirculation of cabin air has caused concerns about the potential transmission of airborne pathogens between passengers. These fears seem to be unfounded: recirculated air passes through a high efficiency particulate air filter before re-entering the cabin,49 and studies comparing passengers travelling on aircraft with recirculated compared with 100% fresh air ventilation systems found no difference in the likelihood of developing a common cold after the flight.50
Carbon dioxide concentration in aircraft often exceeds the generally accepted ‘comfort’ level of 1000 ppm, and would be expected to be higher in aircraft with greater amounts of recirculation air-conditioning. Concentrations observed in aircraft vary between around 700 and 1700 ppm,51 and are highest when the aircraft is occupied, but on the ground, and lowest whilst flying at cruise alti-tude. Carbon dioxide itself does not cause respiratory problems at these levels, but is used more as a marker of the adequacy of ventilation.
Humidity is invariably low in aircraft, with most studies finding relative humidity to average 14–19% during flight compared with in excess of 50% in most sea level environments.52 Like carbon dioxide, cabin humidity is maximal when on the ground and minimal at cruise altitude.51 The low humidity occurring in aircraft is responsible for many minor symptoms such as irritation of the eyes and upper airway, though these symptoms are unusual with less than 3–4 hours of exposure.52
With the exception of low humidity, there is therefore little evidence that the cabin air of aircraft poses any threat to healthy passengers. The numerous symptoms reported following air travel almost certainly have their origins in other activities associated with air travel, in particular the consumption of alcohol and differing time zones.
The Respiratory System of Birds53-55
Many species of birds fly at high altitude, including bar-headed geese that fly over the Himalayas twice each year on their migration, though the highest recorded bird is a Rüppell’s griffon vulture that unfortunately collided with a commercial airliner at 11 285 m (37 900 ft). Compared with mammals, birds have a higher body temperature (40°C) and are generally more active, and so expend more energy per unit body mass. The activity of flying is strenuous, with oxygen consumption in a flying bird reaching 13–30 times resting values depending on conditions. To supply this high oxygen consumption whilst at altitude requires the architecture of the avian respiratory system to be fundamentally different to that of mammals, and evolution of birds led to the development of a lung-air-sac system.
Much of a bird’s body volume consists of air sacs, which can be inflated or deflated by the muscles of the body cavities. The air sacs are fundamental to a bird’s breathing, but also have numerous other functions such as reducing body weight and voice production by the passage of air through the syrinx. Avian lungs make up a much smaller proportion of a bird’s body than in mammals, and are almost rigid, being firmly fixed to the ribs. Rather than the tidal breathing used by mammals, in birds the various air sacs are used to pass gas through the lungs, in the same direction, during both inspiration and expiration so the lungs are, in effect, simply acting as a passive gas exchanger. This is achieved with two main groups of air sacs, which vary widely between bird species, but may be approximately divided into caudal and cephalad groups (Figure 17.6). During inspiration, inspired air passes through the larynx and syrinx into each primary intrapulmonary bronchus, from which some air enters the dorsal secondary bronchus and the remainder inflates the caudal air sacs. From the secondary bronchus air passes through the numerous parabronchi, where gas exchange occurs, and on through the ventral secondary bronchus into the cephalad air sacs. On expiration, the caudal air sacs empty into the dorsal secondary bronchus, the air passing through the lung and being expired through the primary bronchus whilst at the same time the cephalad air sacs empty, the gas also being expired. As a result of this pattern of breathing there is an almost continuous flow of inspired air through the lung in a caudad to cephalad direction, whilst pulmonary blood flow is in a cephalad to caudad direction, providing an efficient counter-current gas exchange system.
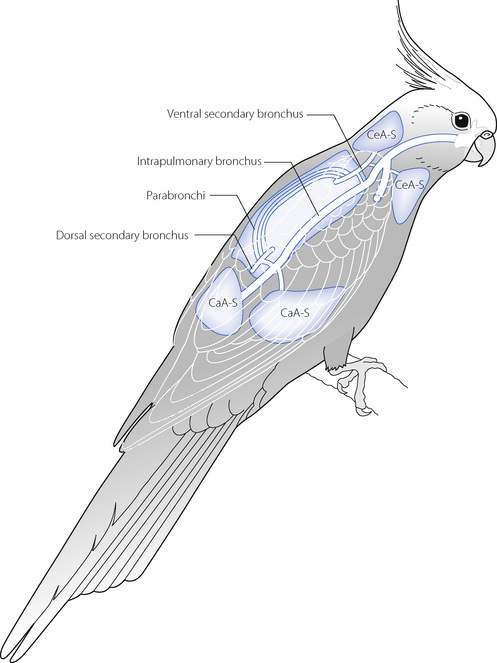
Fig. 17.6 Schematic diagram of the respiratory system of a bird. See text for details. CeA-S, cephalad air sacs; CaA-S, caudal air sacs.
Other features of the avian respiratory system further increase its gas exchange efficiency. At a microscopic level within the avian lung, the blind ending air capillaries (equivalent to a mammalian alveolus, but more tubular in shape) that arise from the parabronchi are structurally arranged with the blood capillaries to provide a further counter current system. Finally, the blood–gas barrier in avian lungs is much thinner than in mammals because the lack of repetitive movement required by tidal breathing requires less structural strength.55 Two separate counter-current gas exchange systems and the reduced blood–gas barrier mean that in birds the Po2 of inspired gas and pulmonary venous blood are almost equal.
References
*1. West JB, Schoene RB, Milledge JS. High Altitude Medicine and Physiology. London: Hodder Arnold; 2007.
2. West JB, Hackett PH, Maret KH, et al. Pulmonary gas exchange on the summit of Mount Everest. J Appl Physiol. 1983;55:678-687.
3. Sutton JT, Reeves JT, Wagner PD, et al. Operation Everest II: oxygen transport during exercise at extreme simulated altitude. J Appl Physiol. 1988;64:1309-1321.
4. Conkin J, Wessell JH. Critique of the equivalent air altitude model. Aviat Space Environ Med. 2008;79:975-982.
5. van Dorpe E, Los M, Dirven P, et al. Inspired carbon dioxide during hypoxia: effects on task performance and cerebral oxygen saturation. Aviat Space Environ Med. 2007;78:666-672.
6. Houston CS, Sutton JR, Cymerman A, Reeves JT. Operation Everest II: man at extreme altitude. J Appl Physiol. 1987;63:877-882.
7. Richalet JP, Robach P, Jarrot S, et al. Operation Everest III (COMEX ‘97): effects of prolonged and progressive hypoxia on humans during a simulated ascent to 8848 m in a hypobaric chamber. Adv Exp Med Biol. 1999;474:297-317.
8. Bisgard GE, Forster HV. Ventilatory responses to acute and chronic hypoxia. In: Fregly MJ, Blatteis CM, editors. Handbook of Physiology, Section 4: Environmental Physiology. New York & Oxford: Oxford University Press; 1996:1207-1239.
*9 Powell FL, Huey KA, Dwinell MR. Central nervous system mechanisms of ventilatory acclimatisation to hypoxia. Respir Physiol. 2000;121:223-236.
10. Severinghaus JW, Mitchell RA, Richardson BW, Singer MM. Respiratory control at high altitude suggesting active transport regulation of CSF pH. J Appl Physiol. 1963;18:1155-1166.
11. Tansley JG, Fatemian M, Howard LSGE, Poulin MJ, Robbins PA. Changes in respiratory control during and after 48 h of isocapnic and poikilocapnic hypoxia in humans. J Appl Physiol. 1998;85:2125-2134.
12. Wagner PD, Sutton JT, Reeves JT, Cymerman A, Groves BM, Malconian MK. Operation Everest II: pulmonary gas exchange during a simulated ascent of Mt. Everest. J Appl Physiol. 1987;63:2348-2359.
13. Grocott MPW, Martin DS, Levett DZH, et al. Arterial blood gases and oxygen content in climbers on mount Everest. N Engl J Med. 2009;360:140-149.
14. Groves BM, Reeves JT, Sutton JT, et al. Operation Everest II: elevated high-altitude pulmonary resistance unresponsive to oxygen. J Appl Physiol. 1987;63:521-530.
15. Cymerman A, Reeves JT, Sutton JT, et al. Operation Everest II: maximal oxygen uptake at extreme altitude. J Appl Physiol. 1989;66:2446-2453.
16. Ward MP, Milledge JS, West JB. High Altitude Medicine and Physiology. London: Chapman and Hall; 1995.
17. Ramirez G, Bittle PA, Rosen R, Rabb H, Pineda D. High altitude living: genetic and environmental adaptation. Aviat Space Environ Med. 1999;70:73-81.
18. Moore LG. Comparative human ventilatory adaptation to high altitude. Respir Physiol. 2000;121:257-276.
19. Massaro D, Massaro GD. Pulmonary alveoli: formation, the ‘call for oxygen’, and other regulators. Am J Physiol Lung Cell Mol Physiol. 2002;282:L345-L348.
*20. Moore LG, Armaza F, Villena M, Vargas E. Comparative aspects of high-altitude adaptation in human populations. Adv Exp Med Biol. 2000;475:45-62.
21. Wu T, Wang X, Wei C, et al. Hemoglobin levels in Qinghai-Tibet: different effects of gender for Tibetans vs. Han. J Appl Physiol. 2005;98:598-604.
22. Firth PG, Zheng H, Windsor JS, et al. Mortality on Mount Everest, 1921–2006: descriptive study. BMJ. 2008;337:a2654.
23. Schoene RB. Limits of human lung function at high altitude. J Exp Biol. 2001;204:3121-3127.
24. Schoene RB. Limits of respiration at high altitude. Clin Chest Med. 2005;26:405-414.
25. West JB, Boyer SJ, Graber DJ, et al. Maximal exercise at extreme altitudes on Mount Everest. J Appl Physiol. 1983;55:688-698.
26. Pugh LGCE, Gill MB, Lahiri S, Milledge JS, Ward MP, West JB. Muscular exercise at great altitudes. J Appl Physiol. 1964;19:431-440.
27. Basnyat B, Murdoch DR. High-altitude illness. Lancet. 2003;361:1967-1974.
28. Schoene RB. Illnesses at high altitude. Chest. 2008;134:402-416.
29. Maloney JP, Broeckel U. Epidemiology, risk factors, and genetics of high-altitude-related pulmonary disease. Clin Chest Med. 2005;26:395-404.
30. Vardy J, Vardy J, Judge K. Acute mountain sickness and ascent rates in trekkers above 2500 m in the Nepali Himalaya. Aviat Space Environ Med. 2006;77:742-744.
31. Bärtsch P, Mairbäurl H, Maggiorini M, Swenson ER. Physiological aspects of high-altitude pulmonary edema. J Appl Physiol. 2005;98:1101-1110.
32. Jerome EH, Severinghaus JW. High-altitude pulmonary edema. N Engl J Med. 1996;334:662-663.
33. Duplain H, Sartori C, Lepori M, et al. Exhaled nitric oxide in high-altitude pulmonary edema. Role in the regulation of pulmonary vascular tone and evidence for a role against inflammation. Am J Respir Crit Care Med. 2000;162:221-224.
34. Dehnert C, Risse F, Ley S, et al. Magnetic resonance imaging of uneven pulmonary perfusion in hypoxia in humans. Am J Respir Crit Care Med. 2006;174:1132-1138.
35. Hornbein TH, Townes BD, Schoene RB, Sutton JR, Houston CS. The cost to the central nervous system of climbing to extremely high altitude. N Engl J Med. 1989;321:1714-1719.
36. Barry PW, Mason NP, Riordan M, O’Callaghan C. Cough frequency and cough-receptor sensitivity are increased in man at altitude. Clin Sci. 1997;93:181-186.
37. Eichenberger U, Weiss E, Riemann D, Oelz O, Bärtsch P. Nocturnal periodic breathing and the development of acute high altitude illness. Am J Respir Crit Care Med. 1996;154:1748-1754.
38. Luks AM, Swenson ER. Medication and dosage considerations in the prophylaxis and treatment of high-altitude illness. Chest. 2008;133:744-755.
39. Leaf DE, Goldfarb DS. Mechanisms of action of acetazolamide in the prophylaxis and treatment of acute mountain sickness. J Appl Physiol. 2007;102:1313-1322.
40. Richalet J-P, Gratadour P, Robach P, et al. Sildenafil inhibits altitude-induced hypoxemia and pulmonary hypertension. Am J Respir Crit Care Med. 2005;171:275-281.
41. Silverman D, Gendreau M. Medical issues associated with commercial flights. Lancet. 2008;373:2067-2077.
42. Harding RM, Mills FJ. Aviation Medicine. London: BMJ Publishing Group; 1993.
43. Cottrell JJ, Lebovitz BL, Fennell RG, Kohn GM. Inflight arterial saturation: continuous monitoring by pulse oximetry. Aviat Space Environ Med. 1995;66:126-130.
44. Kelly PT, Swanney MP, Frampton C, Seccombe LM, Peters MJ, Beckert LE. Normobaric hypoxia inhalation test vs. response to airline flight in healthy passengers. Aviat Space Environ Med. 2006;77:1143-1147.
45. Muhm JM, Rock PB, McMullin DL, et al. Effect of aircraft-cabin altitude on passenger discomfort. N Engl J Med. 2007;357:18-27.
46. Veronneau SJH, Mohler SR, Pennybaker AL, Wilcox BC, Sahiar F. Survival at high altitudes: Wheel-well passengers. Aviat Space Environ Med. 1996;67:784-786.
47. British Thoracic Society Standards of Care Committee. Managing passengers with respiratory disease planning air travel: British Thoracic Society recommendations. Thorax. 2002;57:289-304.
48. Dine CJ, Kreider ME. Hypoxia altitude simulation test. Chest. 2008;133:1002-1005.
*49. Rayman RB. Cabin air quality: an overview. Aviat Space Environ Med. 2002;73:211-215.
50. Zitter JN, Mazonson PD, Miller DP, Hulley SB, Balmes JR. Aircraft cabin air recirculation and symptoms of the common cold. JAMA. 2002;288:483-486.
51. Lindgren T, Norbäck D. Cabin air quality: indoor pollutants and climate during intercontinental flights with and without tobacco smoking. Indoor Air. 2002;12:263-272.
52. Nagda NL, Hodgson M. Low relative humidity and aircraft cabin air quality. Indoor Air. 2001;11:200-214.
53. Maina JN. Development, structure, and function of a novel respiratory organ, the lung-air sac system of birds: to go where no other vertebrate has gone. Biol Rev. 2006;81:545-579.
54. Brown RE, Brain JD, Wang N. The avian respiratory system: a unique model for studies of respiratory toxicosis and for monitoring air quality. Environ Health Perspect. 1997;105:188-200.
55. West JB, Watson RR, Fu Z. The human lung: did evolution get it wrong? Eur Respir J. 2007;29:11-17.