Mechanisms of Microbial Infections
The goal of this chapter is to provide a mechanistic overview of the key steps involved in understanding the pathogenesis of infectious diseases. Coverage is not intended to be encyclopedic nor will every fine distinction of individual microbial strains, genera, or families be discussed. Because the knowledge base for some veterinary diseases is limited, some sections of this chapter are based on (1) extrapolations from known experimental systems, (2) established mechanisms of injury covered in the basic pathology chapters of this book, and (3) assumptions anchored in the characteristics of macroscopic and microscopic lesions that arise with each disease. This chapter assumes that students have prior experiences with infectious diseases through coursework in microbiology, virology, mycology, and parasitology and with innate and adaptive defense mechanisms through coursework in immunology. Subject matter presented in this chapter changes rapidly, often daily, through new discoveries; nevertheless, every attempt has been made to assure that material presented is current. However, new discoveries that occur between the writing of this chapter and the publication of this book may not be included in this chapter. Although every effort has been made to minimize errors, this chapter is an attempt to meld information from many different disciplines, often with conflicting conclusions and/or differing terminologies and interpretations of how cells and tissues respond to injury. Errors in interpretation, although inadvertent, may occur.
This chapter will also discuss and illustrate selected “especially dangerous and contagious microorganisms” because diseases caused by these pathogens can have disastrous impact on livestock health and production and on the economies of affected countries. The location in this textbook of coverage of these diseases considered by the USDA/APHIS and the World Organization for Animal Health (OIE) as “Foreign Animal Diseases” or “OIE Reportable Diseases” is listed in Table 4-1.
TABLE 4-1
Location in this Textbook of the Coverage of Especially Dangerous and Contagious Microorganisms

Synopsis
Infectious microorganisms like bacteria, viruses, fungi, protozoa, and prions cause many of the diseases common to the practice of veterinary medicine. For convenience, this chapter discusses individual diseases using an organ system approach under the headings: Bacterial Diseases, Viral Diseases, Fungal Diseases, Protozoal Diseases, and Prion Diseases. Students are advised to initially read (1) the opening portions of this chapter that discuss and illustrate background information about mechanisms of disease and (2) the opening portions of the sections covering bacterial diseases and viral diseases that discuss and illustrate background information about the chronologic sequences of events in disease processes. These materials provide the context for understanding individual animal diseases.
Infectious microorganisms follow chronologic sequences of events regulated by virulence determinates to infect target cells unique to specific organ systems and cause disease (Fig. 4-1). They most commonly enter the body through ingestion, inhalation, or cutaneous penetration and interact with mucosae or skin. If their target cell is not in mucosae or skin, they may spread to submucosal and subcutaneous lymphoid nodules such as in tonsils or Peyer’s patches, then to regional lymph nodes, and then systemically in the circulatory and/or lymphatic systems to other organ systems. They often infect macrophages, lymphocytes, and/or dendritic cells and use these cells to spread via leukocyte trafficking to target cells in organ systems as they migrate through these systems as part of their normal immunologic surveillance activities. Cell and tissue specificity is based on ligand-receptor interactions in which proteins (ligands) expressed on the surface of infectious organisms bind to receptors on membranes of host target cells, mucus associated with these cells, vascularized extracellular matrix (ECM) tissues beneath these cells, or macrophages, lymphocytes, and dendritic cells supporting these cells. Once bound to receptors, a sequence of events facilitated by virulence determinates is initiated that colonizes the surface of these cells or invades the cells through phagocytosis or endocytosis. The organism then establishes control of normal metabolic systems of these cells and uses them to replicate in and spread to other organ systems. The outcome of this process is usually cell dysfunction and/or death and thus clinical disease.
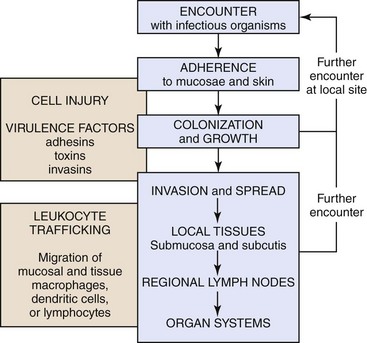
Fig. 4-1 Sequence of events in infection.
The chronologic sequence of events used by infectious microorganisms to colonize and invade mucosae and skin, spread to local tissues and regional and systemic organ systems, and cause disease. (Courtesy Dr. J. F. Zachary, College of Veterinary Medicine, University of Illinois.)
Portals of Entry
Except for contact with carrier animals, the chronologic sequence of events leading to disease caused by infectious organisms are not random events. Contact places susceptible animals in close proximity to infected animals, where infectious organisms can be spread through direct contact, grooming, licking, bite wounds, sneezing, and other normal physiologic body processes in fomites from body fluids, water droplets, snot, sputum, urine, and feces. Infection depends on creating an initial beachhead to establish, sustain, and spread the infection when organisms first encounter tissues of the body. Generally, the initial beachhead is established in one of two tissue types: (1) mucosae of the respiratory system (nasal cavity, nasal pharynx, conductive system [see Chapter 9]), alimentary (small intestine) system (see Chapter 7), lower urinary system (see Chapter 11), reproductive systems (see Chapters 18 and 19), or ear and eye (conjunctiva) (see Chapter 20) or (2) subcutaneous tissues, including muscle and endothelial cells of the skin (Fig. 4-2). In these beachheads, organisms gain access and attach to, enter, and/or replicate in mucosal- and connective tissue-associated macrophages, lymphocytes, and dendritic cells. It is from these beachheads that they then spread locally (submucosae and subcutis), regionally (lymph nodes), and/or systemically (organ systems) to other target cells to sustain and amplify the infection and cause disease (Fig. 4-3).
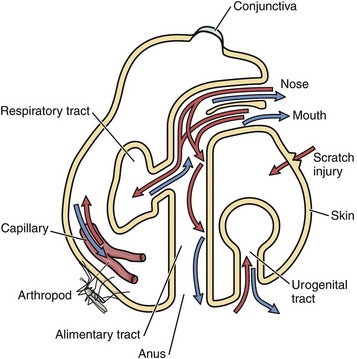
Fig. 4-2 Portals of entry.
Infectious microorganisms commonly enter the body through ingestion (alimentary portal), inhalation (respiratory portal), cutaneous penetration (skin portal), and ascending infection (lower urinary and reproductive portals) and interact with epithelial cells, macrophages, dendritic cells, and lymphocytes of the mucosae or skin. (Modified from Goering R, Dockrell H, Roitt I, et al: Mims’ medical microbiology, ed 4, St. Louis, 2008, Mosby.)
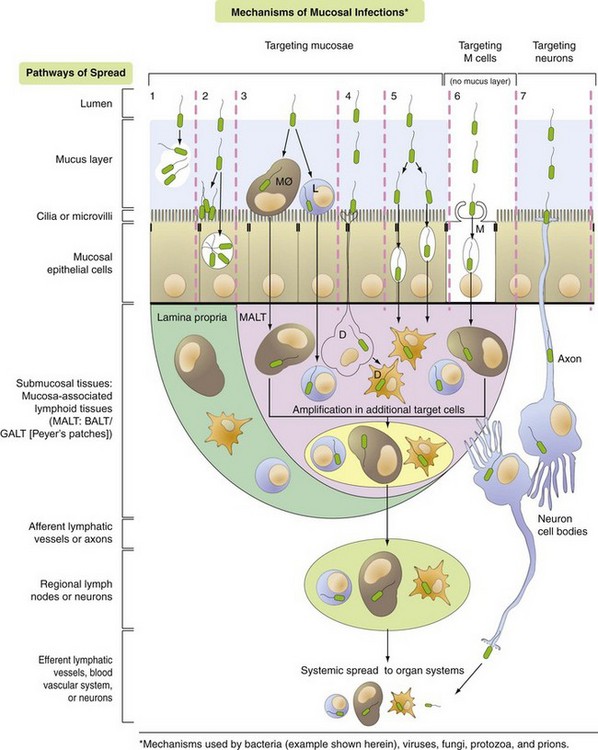
Fig. 4-3 Mechanisms of Microbial Infections and Pathways of Spread.
Pathway 1: Bacteria target the mucus layer.
Pathway 2: Bacteria target cilia or microvilli and/or mucosal epithelial cells.
Pathway 3: Bacteria target MALT via mucosal macrophages (MØ) and/or lymphocytes (L).
Pathway 4: Bacteria target MALT via dendritic cells (D).
Pathway 5: Bacteria target MALT via transcytosis or intercellular (junctional complexes) spread.
Pathway 6: Bacteria target MALT via M cells and transcytosis.
Pathway 7: Bacteria target nerve endings in mucosa and enter the brain via retrograde axonal transport. (Courtesy Dr. J. F. Zachary, College of Veterinary Medicine, University of Illinois.)
A concept central to the pathogenesis of disease is the ability of an infectious organism to reach a site in the body suitable for growth and replication. Ingestion, inhalation, cutaneous penetration, and ascending infection are the most common portals of entry for infectious organisms to gain access to mucosae of the respiratory and alimentary systems; the epidermis, dermis, and subcutis of the integumentary system; and the lower urinary system and reproductive systems (see Fig. 4-2). The mucosae of the alimentary and respiratory systems are covered by a protective mucus gel composed predominantly of mucin glycoproteins synthesized and secreted by goblet cells (Fig. 4-4). The mucus layer forms a barrier system that attempts to do the following:
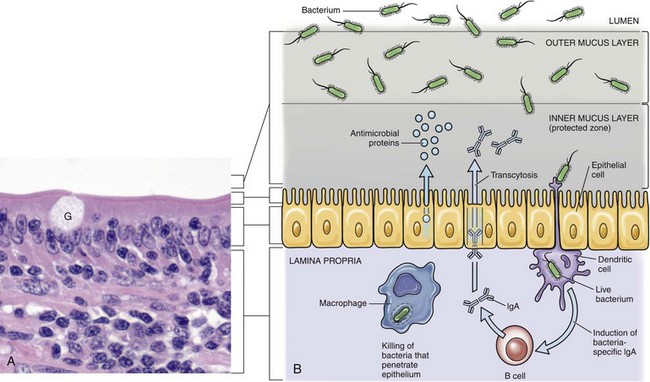
Fig. 4-4 Mucus layer of alimentary and respiratory mucosae.
A, Mucosa of the intestine (shown here) and of the conductive respiratory airways are covered by a mucus layer (not visible in H&E sections) secreted by goblet cells (G). The mucus covers the microvilli or cilia of these systems. H&E stain. B, The mucus layer has an outer layer that traps microorganisms (infectious and noninfectious) and other particles and an inner layer in which the cilia beat and which contains antimicrobial substances that diffuse into the outer layer. Dendritic cells and mucosa-associated macrophages and lymphocytes play central roles in preventing infection of mucosa. (A courtesy Dr. J. F. Zachary, College of Veterinary Medicine, University of Illinois.)
1. Block infectious organisms from reaching target cells.
2. Trap infectious organisms so they can be phagocytosed by mucosal macrophages and neutrophils.
3. Trap infectious organisms so they can be exposed to bacteriostatic and bacteriocidal molecules sequestered in the mucin matrix.
4. Facilitate phagocytosis of infectious organisms via mucosa-associated macrophages, mucosal dendritic cells, and microfold (M) cells.
5. Deliver microbial antigens to local lymphoid tissues like Peyer’s patches or bronchial-associated lymphoid tissues (BALT) and then to regional lymph nodes via afferent lymphatic drainage.
Normal microflora such as bacteria are observed within the outer luminal zone of the mucus layer, indicating the importance of mucus gel in preventing direct adherence of bacteria to epithelial cells. Changes in the function of goblet cells and the chemical composition of mucus can occur through the release of bioactive factors from infectious organisms or by activation of immune cells. Additionally, predisposing management stressors, such as dehydration, shipping, humidity, and ventilation, combined with weather changes can also change the function of goblet cells and the chemical composition of mucus, making mucosae more susceptible to infection.
Infectious organisms use three mechanisms to penetrate the mucus layer and gain access to target cells. More information is known about the interactions of bacteria with the mucus layer, when compared to other infectious organisms, especially viruses. These mechanisms include penetrating motility, digestion of mucus via enzymes and its consumption as an energy source, and evasion of the mucus layer in areas around Peyer’s patches and M cells, areas devoid of mucus. Mucus also provides pathogenic advantages to bacteria as follows: (1) mucin oligosaccharides represent a direct source of carbohydrates, peptides, and exogenous nutrients, including vitamins and minerals; (2) bacteria that colonize mucus avoid rapid expulsion out of the alimentary system by peristalsis; and (3) adhesion to specific molecules within the mucin facilitates colonization of the mucus layer by infectious organisms. Microbial mucolysis, the ability to enzymatically degrade mucus, appears to be a common trait among bacteria and provides access to readily available sources of carbon and energy and enables bacteria to reach the surface of epithelial cells. Mucins are classified as neutral and acidic types, with the latter being further categorized as sulfated (sulfomucins) or nonsulfated (sialomucins). These biochemical differences likely explain some of the segmental target cell specificity (i.e., localization in one area of the organ over another) of some diseases of the alimentary and respiratory systems. Localization and colonization of specific zones of mucus by certain organisms likely occurs through the expression of adherence molecules unique to specific types of mucins.
Ingestion
Organisms enter the alimentary system (see Chapter 7) by ingestion of infectious fomites. Through the processes of mastication, swallowing, and peristalsis, infectious organisms gain access to and are trapped in the mucus layer of the oral pharynx and intestines. Mucosae most commonly involved include tonsillar epithelium, villus epithelium, crypt epithelium, and epithelium containing M cells overlying Peyer’s patches. Infectious organisms then must penetrate the mucus layer to reach targets such as mucosal epithelial cells, dendritic cells, and tissue macrophages. M cells are also targets but lack a mucus covering. Mucus in the alimentary system is produced by goblet cells distributed among mucosal epithelial cells in the villi and crypts where it covers and protects microvilli (see Fig. 4-4). The mucus layer is a (1) physical and (2) biologic barrier protecting the intestine against infectious organisms via (1) its thickness and viscosity, (2) binding to bacterial adhesins, (3) serving as a reservoir for immunoglobulin A (IgA) and lysozyme, and (4) acting as a free radical scavenger. Additionally, the mucus layer is a habitat for beneficial enteric microflora.
Generally, there are more goblet cells in the large intestine than the small intestine, more in crypts than in villi, and more in the ileum than in the jejunum or duodenum. It appears that mucus covers all intestinal epithelial surfaces to varying degrees of thickness and viscosity and is composed of an inner gel layer and an outer soluble layer. The mucus layer is thickest in the colon (≈830 µm) and thinnest in the jejunum (≈123 µm). It is less prominent over absorptive enterocytes with microvilli when compared to crypt enterocytes. M cells are not covered by a mucus layer; therefore infectious organisms can readily interact with their cell membranes. Once entrapped in the mucus layer, infectious organisms must then penetrate it to gain access to target cells for infection. Additionally, infectious organisms also encounter mucosal fluids, such as gastric acid, mucins, secretions such as lysozyme, and humoral mediators such as immunoglobulins, and also compete with normal microflora for resources. Mucosae-associated lymphoid tissues (MALT), such as Peyer’s patches (Fig. 4-5), are submucosal lymphoid nodules located in the distal jejunum and ileum that surround groups of intestinal crypts. Nodules are composed of lymphocytes, macrophages, and dendritic cells. They are covered by modified epithelial cells of intestinal crypts called M cells that transfer antigens in the lumen of the intestine across the mucosa to dendritic cells and immune cells like T lymphocytes in the nodule. M cells are the interface between materials in the lumen of intestinal crypts and the lymphoid nodules (see Fig. 4-5). Peyer’s patches have afferent lymphatic vessels that drain to regional mesenteric lymph nodes.
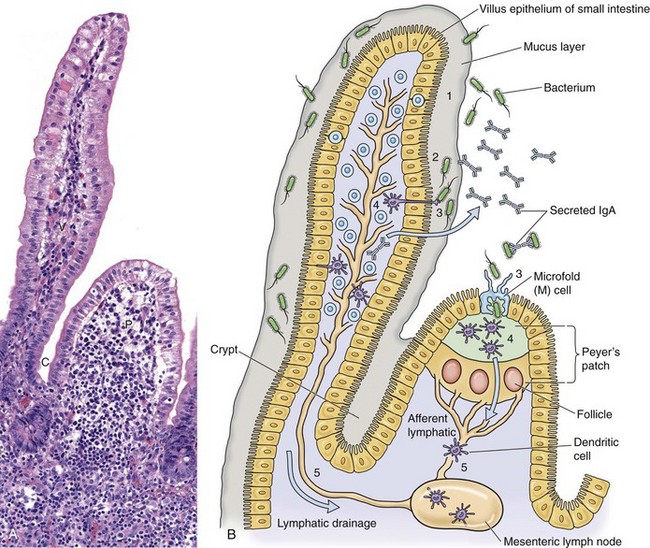
Fig. 4-5 Microbial interactions with a barrier system: intestinal mucosa.
A, Mucosa that cover intestinal villi (V) and Peyer’s patches (P) and line crypts (C) form a barrier system that attempts to prevent the spread of infectious organisms into the underlying lamina propria. H&E stain. B, Schematic diagram of the responses of bacteria (or viruses) trapped in the mucus layer (1). Bacterial proteins (virulence determinates) act to allow them to penetrate the mucus layer and come into contact with the mucosal epithelium (2). IgA secreted by mature plasma cells in the lamina propria passes through mucosal epithelial cells into the lumen and can act as an “opsonizing” defense mechanism thus preventing infection. Bacteria then interact with mucosal epithelial cells, dendritic cells (D), or M cells (3). They then encounter lymphoid cells in the lamina propria or Peyer’s patches (4) and spread in lymphocytes or as free virus in lymph from this location via efferent lymphatic vessels to regional lymph nodes (5). Note the absence of a mucus layer over M cells and follicle-associated epithelium. Also see Fig. 4-7 for an example of barrier system: respiratory mucosae. (A courtesy Dr. J. F. Zachary, College of Veterinary Medicine, University of Illinois.)
Inhalation
In the respiratory system (see Chapter 9), infectious organisms are inhaled through the nostrils and are deposited on mucosae of the nasal turbinates, nasal pharynx, and/or the conductive system based on physical properties of the agent such as size, shape, weight, and electrostatic charge (Fig. 4-6). Groups of infectious organisms ranked from smallest to largest include: viruses (5 to 300 nm [1 × 10−9 m] in diameter), prions (≈16 nm in diameter), bacteria (0.5 to 5 µm [1 × 10−6 m] in diameter), fungi 5 to 60 µm in diameter, and protozoa 1 to 300 µm in diameter. Although it is convenient to compare infectious organisms based on size, rarely are these organisms inhaled as free infectious organisms. Most commonly, they are enclosed in fomites (i.e., inanimate objects or substances capable of carrying infectious organisms) such as dust particles, soil, septum, or body fluids. Thus the physical properties of infectious fomites determine where they are deposited on mucosal surfaces of the respiratory system and cause disease. When inhaled, larger infectious fomites, such as bacteria and fungi, are deposited and trapped in the nasal turbinates, whereas in a gradient of descending size, smaller ones are able to reach the pharynx, larynx, trachea, and bronchi before they are deposited and trapped in the mucosae. The nasal cavity and turbinates trap 70% to 80% of particulate matter 3 to 5 µm or greater in diameter; 60% of particulate matter 2 µm or greater but cannot trap particles with sizes <1 µm in diameter. In a normal functioning respiratory system, only infectious fomites of 1 µm or less in diameter, such as viruses and some bacteria, can be inhaled into bronchioles, alveolar ducts, and alveoli, which are the oxygen-carbon dioxide (O2-CO2) exchange portion of the respiratory system.
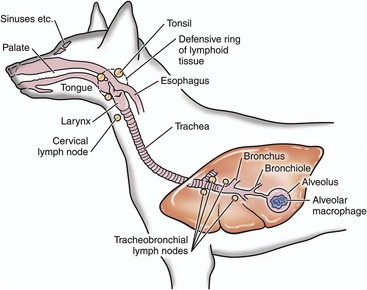
Fig. 4-6 Deposition of infectious microorganisms.
Infectious microorganisms inhaled through the nostrils are deposited on mucosa of the nasal turbinates, nasal pharynx, and/or the conductive system of the respiratory tract. The site of deposition depends on the physical properties of the agent such as size, shape, weight, and electrostatic charge. (Modified from Goering R, Dockrell H, Roitt I, et al: Mims’ medical microbiology, ed 4, St. Louis, 2008, Mosby.)
When infectious fomites are inhaled, they first encounter the nasal turbinates. The movement of air through the turbinates causes centrifugal turbulence that forces them against the mucosae where they are trapped in its overlying mucus layer for removal. If the size of fomites allows them to pass through the turbinates and be carried to the pharynx, larynx, trachea, or bronchi, inertial turbulence forces fomites against airway mucosae where they are trapped in the mucus layer for removal. Inertial turbulence occurs when a laminar stream of airflow is disrupted by a septum within the conductive portion of the respiratory system when airways branch. When the flow is split by a septum, the flow rotates centrifugally on either side of the septum and forces fomites against the mucosae. Depending on the species, airways can branch 23 times en route from the trachea to the alveoli. The O2-CO2 exchange portion of the respiratory system (the bronchioles, alveolar ducts, and alveoli) are not ciliated and have no protective mucus layer because of their gas exchange function. The outcome of these turbulence mechanisms is to trap infectious fomites in the mucus layer overlying ciliated mucosal epithelial cells. When infectious fomites are trapped in the mucus layer, they are (1) acted on by other components of the innate immune system, such as phagocytes like alveolar macrophages and neutrophils, and microbicidal molecules, such as lysozyme and immunoglobulins, and (2) removed by the mucociliary apparatus (see Chapter 9).
The mucociliary apparatus is composed of the mucus layer and ciliated mucosal epithelial cells and is an important defensive mechanism in the respiratory system (Fig. 4-7). The mucus layer, produced by goblet cells and submucosal glands, is biphasic and consists of a luminal viscoelastic or gel layer used to trap infectious fomites and a serous inner layer in which the cilia of ciliated mucosal epithelial cells beat (see Fig. 4-7). The tips of the cilia just slightly enter the gel layer and their beating moves the gel and fomites. In the nasal cavity and sinuses, cilia move mucus and debris downward toward the pharynx for swallowing; in the conductive portion of the respiratory system, cilia move mucus and debris upward toward the pharynx for swallowing. The directionality of mucus flow is determined by the rhythmic unidirectional beating pattern of the cilia. The conductive portion of the respiratory system has an anterior and ventral flow pattern of distribution such that gravity influences the deposition of infectious fomites, if the mucus layer and/or the mucociliary apparatus are dysfunctional. Therefore injury to mucosal epithelial cells by specific infectious organisms, such as influenza and bovine rhinotracheitis viruses, can disrupt the function of the mucociliary apparatus, thereby exacerbating an existing disease or creating an opportunity for a secondary microbial infection of dependent lung usually prevented by this clearance mechanism. Swallowing of infectious organism-infected mucus may be a mechanism to clear certain bacteria; however, it provides other bacteria, such as Rhodococcus equi, the opportunity to gain access to the alimentary system and cause disease. The mucosae of the conductive portion of the respiratory system also contain dendritic cells and alveolar and tissue macrophages that commonly migrate through the mucosae and the mucus layer during their normal patterns of leukocyte trafficking (see Chapters 5 and 13). Because these cells can phagocytose and kill infectious organisms, they serve as a primary defense mechanism against infections. However, certain infectious organisms have virulence determinates that allow them to evade killing by phagocytes and use them like a “Trojan horse” to spread the agent and infect other cells and tissues. These cells are common targets for infectious organisms and along with mucosal epithelia serve as the initial beachhead infection before organisms spread locally often to the tonsil, regionally to lymph nodes, and systemically to other organ systems. bronchial-associated lymphoid tissues (BALTs) are submucosal lymphoid nodules located subjacent to ciliated mucosae, usually in areas in which inertial turbulence deposits foreign material on mucosae (see Fig. 4-5). Nodules are composed of lymphocytes, macrophages, and dendritic cells and function much like Peyer’s patches. BALTs have afferent lymphatic vessels that drain to regional tracheobronchial lymph nodes.
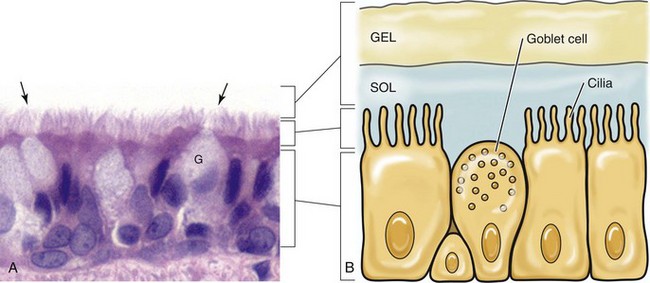
Fig. 4-7 Mucociliary apparatus.
A, Cilia (arrows) of the bronchiole mucosal epithelial cells and the mucus layer (not visible) form the mucociliary apparatus of the conductive component of the respiratory system. The mucus layer is not visible because it has been removed during histologic processing of tissue. H&E stain. B, Diagram of the mucociliary apparatus. The mucus layer is biphasic and consists of a luminal viscoelastic or gel layer used to trap bacteria and a serous inner layer in which the cilia of ciliated mucosal epithelial cells beat unidirectionally to move bacteria upward in the airways to be swallowed or expectorated. G, Goblet cell. (Courtesy Dr. J. F. Zachary, College of Veterinary Medicine, University of Illinois.)
Cutaneous Penetration
Infectious organisms enter the skin, dermis, and subcutaneous tissues (see Chapter 17) via penetration through abrasions, scratches, and bite wounds or through bites of insect vectors such as mosquitoes that spread the agent into subcutaneous tissues such as muscle, blood vessels, and connective tissue (see Fig. 4-2). In these tissues, organisms encounter a limited range of host target cells such as epithelial cells in the skin, dendritic cells (Langerhans’ cells), tissue macrophages, endothelial cells of the vascular system, and connective tissues and muscle of the dermis and subcutis. They may also be deposited directly in the blood vascular system via penetration of a capillary or venule by an insect proboscis. Additionally, organisms encounter body fluids, such as blood and plasma proteins, that serve as resources for survival, infection, and replication.
Ascending Infection
Infectious organisms can enter the lower urinary system and reproductive systems via ascending infection from coitus or the use of contaminated instruments, insemination straws, or semen. Traumatic injury of mucosae resulting in abrasions or penetrating wounds increases the likelihood of colonization by infectious microorganisms. The mechanisms of colonization and infection are similar to those discussed above.
Defense Mechanisms
Mucosal and cutaneous epithelia of the respiratory, alimentary, integumentary, lower urinary, and reproductive systems are the interface between the outside and inside of the body and are held together by occluding junctions such as tight junctions, desmosomes, and adherens junctions and to the basement membrane and ECM by anchoring junctions. Thus these cells form barrier systems that protect the body against entry by infectious organisms. In the respiratory and alimentary systems (and likely in other mucosae), the surface of an epithelial cell located above its junctional complexes and exposed to the lumen is called the apical domain, whereas the surface below junctional complexes on the sides and base make up the basolateral domain (Fig. 4-8). This relationship establishes a polarity to the cell, and it has been shown experimentally that such polarity is often reflected in the expression of different sets of cell membrane receptors that can potentially be used by infectious organisms to attach to and enter the cells. As examples, parvoviruses use receptors expressed only on the basolateral surfaces to infect small intestinal crypt cells via Peyer’s patches, whereas influenza viruses use receptors expressed only on apical surfaces to infect respiratory epithelial cells via the airway. Infectious organisms enter cells through a process called receptor-mediated endocytosis most commonly at the apical surface and exit the cell from the basolateral surface via a mechanism called exocytosis.
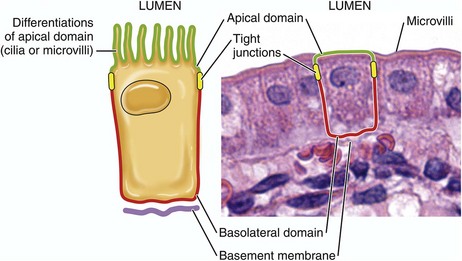
Fig. 4-8 Domains of polarized epithelial cells in mucosal barriers.
Infectious organisms use the apical or basolateral domains of mucosal epithelial cells to enter and exist from these cells. Apical or basolateral cell surface receptors may facilitate entry into the cell. (Courtesy Dr. J. F. Zachary, College of Veterinary Medicine, University of Illinois.)
Innate and Adaptive Immune Responses
The innate and adaptive immune responses are covered in detail in Chapters 3 and 5.
Monocyte-Macrophage System
The monocyte-macrophage system is covered in detail in Chapter 5. In summary, cells of the monocyte-macrophage system form a network of phagocytic and immune cells like alveolar macrophages, mucosal macrophages, and tissue macrophages (Fig. 4-9) that migrate throughout the body in the circulatory and lymphatic systems looking for foreign matter and infectious organisms and return to local, regional, and systemic lymphoid tissues to share antigens with resident immune effector cells. These cells are important in phagocytizing and killing infectious organisms (Fig. 4-10) and commonly encounter infectious organisms, such as bacteria and viruses, in mucosae and the mucus layer. They migrate through the mucus layer, phagocytizing infectious organisms and material they recognize as foreign and carry it via cell migration through the mucosal epithelium to local lymphoid tissue using a mechanism called leukocyte trafficking (Fig. 4-11). Local lymphoid tissues, such as Peyer’s patches and BALT, contain lymphocytes and macrophages that can be infected with organisms present in trafficking macrophages (see Fig. 4-5). From these local sites, infected lymphocytes and macrophages spread via lymphatic vessels to regional lymph nodes where additional cells are infected and then spread via lymphatic vessels to the thoracic duct and the circulatory system. From here, infected cells spread systemically to other organ systems in which specific target cells are infected, including lymphoid organs such as the spleen, lymph nodes, and bone marrow. Under normal conditions, tissue macrophages are derived from two sources: blood monocytes and tissue macrophage progenitor cells that are distributed throughout body tissues during organogenesis of the embryo. Precursor monocytes in bone marrow are capable of providing monocytes that migrate to and differentiate into macrophages in tissues. Tissue macrophages are also replenished locally and in large numbers by proliferation of tissue macrophage progenitor cells. These two populations of cells give rise to tissue macrophages that form the functional basis for innate and adaptive responses to microbes in tissues and organs (see Chapters 5 and 13). Tissues and organs that utilize cells of the monocyte-macrophage system include (1) lung (alveolar macrophages), (2) liver sinusoids (Kupffer cells), (3) lymph nodes (free and fixed macrophages), (4) spleen (free and fixed macrophages), (5) bone marrow (fixed macrophages), (6) connective tissue (histiocytes), (7) serous fluids (pleural and peritoneal macrophages), and (8) skin (histiocytes, Langerhans’ cells) (see Fig. 4-9).
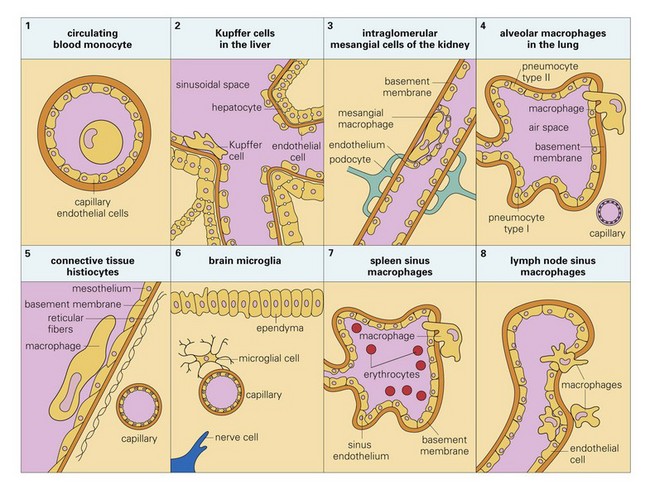
Fig. 4-9 Tissue locations of cells of the monocyte-macrophage system. (From Goering R, Dockrell H, Roitt I, et al: Mims’ medical microbiology, ed 4, St. Louis, 2008, Mosby.)
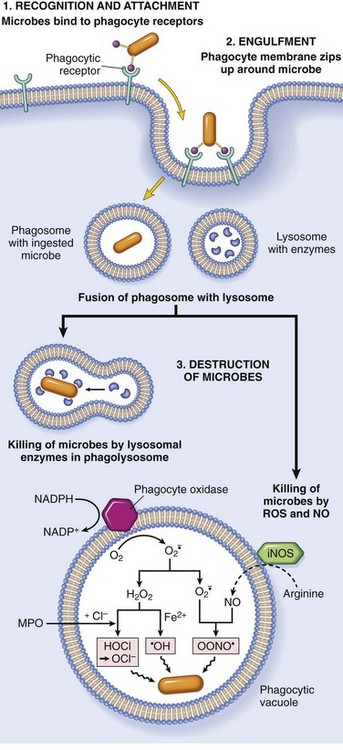
Fig. 4-10 Phagocytosis and intracellular destruction of microbes.
Phagocytosis of a particle (e.g., infectious microorganism) involves binding to receptors on the leukocyte membrane, engulfment, and fusion of lysosomes with phagocytic vacuoles. This process is followed by destruction of ingested particles within the phagolysosomes by lysosomal enzymes and by reactive oxygen and nitrogen species. The microbicidal products generated from superoxide are hypochlorite (HOCl·) and hydroxyl radical (·OH), and from nitric oxide (NO) it is peroxynitrite (OONO·). During phagocytosis, granule contents may be released into extracellular tissues (not shown). MPO, Myeloperoxidase; iNOS, inducible NO synthase. (From Kumar V, Abbas A, Fausto N, et al: Robbins & Cotran pathologic basis of disease, ed 8, Philadelphia, 2009, Saunders.)
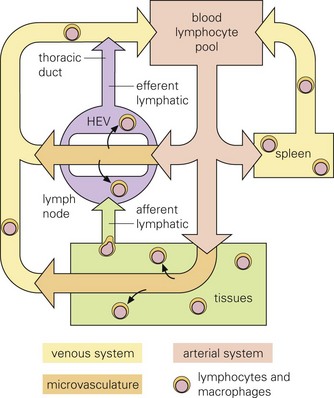
Fig. 4-11 Leukocyte trafficking.
Infectious microorganisms often use macrophages, lymphocytes, and/or dendritic cells to spread themselves to other organ systems as these cells migrate through these systems as part of their normal immunologic surveillance activities. As an example, lymphocytes move through the circulation and enter lymph nodes via specialized endothelial cells of postcapillary venules (HEVs), leave through efferent lymphatic vessels and pass through other nodes, finally entering the thoracic duct, which empties into the circulation. Lymphocytes enter white pulp of the spleen, then pass into sinusoids of the red pulp and leave via the splenic vein. (From Goering R, Dockrell H, Roitt I, et al: Mims’ medical microbiology, ed 4, St. Louis, 2008, Mosby.)
Dendritic Cells
Dendritic cells are covered in detail in Chapters 3 and 5. In summary, dendritic cells are migratory phagocytic antigen-processing and -presenting cells of mucosae (see Fig. 4-5) and skin (e.g., Langerhans’ cells) that are commonly found intermixed with epithelial cells. Infected dendritic cells can migrate from mucosae and skin to local and regional lymphoid tissues via lymphatic vessels. Infectious organisms through their surface proteins are able to bind to receptors expressed on the apical domains of these cells and infect them, then exit via the basolateral domain via exocytosis, gain access to lymphoid nodules (tissues commonly associated with dendritic cells), and establish a local infection in lymphocytes and macrophages. Infected lymphocytes and macrophages spread the agent via leukocyte trafficking from local sites to regional lymph nodes and then systemically to other organ systems as discussed for the monocyte-macrophage system.
Genetic Resistance of Animals to Infectious Diseases
The resistance of animals to disease depends on the effective interplay of many structural and functional (physiologic) components of the body, including cutaneous and mucosal barrier systems and the immune system, respectively. Distinct networks of genes play central roles in structural and functional activities of the body. They control the development, maturation, and maintenance of epithelial cells, mucus, and supporting ECM tissues, such as collagen, that form the barrier systems. Additionally, they control similar structural activities for a variety of cell lineages of the innate and adaptive immune systems like T lymphocytes, macrophages, neutrophils, and dendritic cells and the expression of proteins that form pattern recognition receptors in the membranes of these cells (see Chapter 5). These receptors recognize pathogen-associated molecular patterns (PAMPs) expressed by infectious organisms and are discussed in greater detail in Chapters 3 and 5. Genes also play a central role in functional processes of cells, including adhesion, chemotaxis, phagocytosis, phagosome-lysosome fusion, intracellular killing of microbes, and antigen processing (see Chapters 3 and 5) involved in innate and adaptive responses of the immune system. Thus genetic resistance to infectious diseases is a polygenic trait regulated mainly by the immune system and its interactions with barrier systems and environmental factors such as weather conditions and nutritional status.
In animals, the genetics of disease resistance is most closely associated with the major histocompatibility complex (MHC), a tightly linked group of genes that encode proteins involved in immune responses. This genetic region in cattle has been given the abbreviated name BoLA, and similar regions have been identified in other animal species. Few specific genes or genetic markers related to disease resistance have been identified in domestic animals; however, genes involved in antigen processing appear to be important in resistance to infectious diseases.
Disorders of Barrier Systems
Barrier systems most commonly involved in infectious diseases of animals include mucosae and the skin and were discussed previously. Additionally, mucosae of the conjunctiva and urinary systems also serve as barriers to infectious organisms. These systems and their physical barriers develop embryologically under strict genetic control and when mature are functionally maintained, regulated, and repaired through processes dependent on transcription and translation of genes. Structural and/or functional alterations in these barriers can make animals more susceptible to infectious organisms.
An example of a genetic disorder that predisposes animals to infectious disease that occurs because of an alteration in the development of the basic structure of a barrier is epitheliogenesis imperfecta. It is an autosomal recessive hereditary disease of young horses, cattle, and pigs characterized by loss of epithelium affecting the skin and mucosae of the oral cavity and tongue likely caused by alterations in the subbasal plate and its hemidesmosomes and laminin-5 (see Chapter 17). Loss of the skin or mucosae exposes underlying vascularized ECM tissues to environmental contamination with feces and other matter allowing bacterial pathogens access to matrix tissues and capillary beds.
An example of a genetic disorder that predisposes animals to infectious disease that occurs because of an alteration in the function of a barrier is primary ciliary dyskinesia of the dog. It appears to be an autosomal recessive hereditary disease of young dogs, but an autosomal dominant mutation has not been excluded. This disorder is caused by ciliary dysfunction attributable to immotile or dyskinetic cilia caused by defects of proteins in the outer and/or inner dynein arms of cilia, which give them their motility. This outcome leads to dysfunction of the mucociliary apparatus and the retention of infectious organisms in the respiratory system leading to bacterial bronchitis and pneumonia. Other examples of genetic alterations of barrier systems that predispose animals to increased susceptibility to infectious disease are discussed in the organ system chapters of this book.
Disorders of the Innate Immune Response
The innate immune system provides animals with an immediate defense against infectious organisms and is discussed in detail in Chapters 3 and 5. In summary, this system involves the initial encounter with infectious organisms often at the barriers formed by mucosae and skin and the fluidic and cellular processes that ensue (see Chapter 3). It is essentially acute inflammation and the cellular and chemical mediators associated with the process, such as phagocytic cells like macrophages, neutrophils, and dendritic cells; effector cells, such as T lymphocytes and mast cells; chemical mediators of the complement system; and the vascular system. The purpose of acute inflammation is to dilute and isolate infectious organisms in edema fluid and fibrin, phagocytose and kill them, and process and present their antigens to effector cells of the adaptive immune response. When epithelial cells, endothelial cells, or mucosal or cutaneous macrophages of barrier systems are injured by or infected with infectious organisms, they release large quantities of cytokines into the surrounding tissues. These cytokines recruit, via chemotaxis, inflammatory cells from capillaries in vascularized ECM tissues and cause vasodilation and increased permeability of these blood vessels (i.e., edema fluid and fibrin). Inflammatory cells also release cytokines and other chemical mediators that act to recruit additional inflammatory cells, activate the complement cascade to identify bacteria and kill infectious organisms, promote phagocytosis of dead cells and infectious organisms by phagocytic cells, and activate the adaptive immune system through antigen processing and presentation to immune cells such as T and B lymphocytes. Phagocytic and effector cells of the acute inflammatory response are recruited from capillaries and migrate along a chemoattractant gradient formed by chemical mediators and molecules released from infectious organisms to the inflammatory focus. In inflammatory foci, these cells express Toll-like receptors (TLRs), also known as pattern-recognition receptors (PRRs) that recognize molecules on infectious agents called PAMPs (see Chapters 3 and 5) (Fig. 4-12). These cells also express interleukin-1 (IL-1) receptors that act in concert with PRRs to initiate and sustain the innate immune response through phagocytosis.
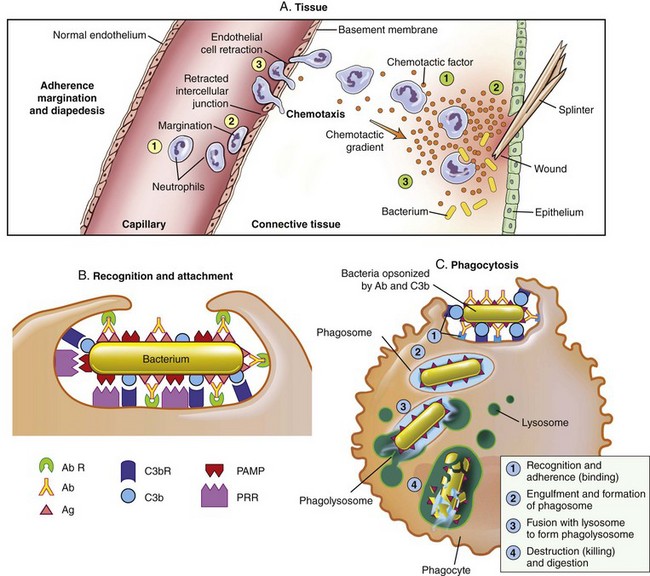
Fig. 4-12 Toll-like receptors in the process of phagocytosis.
The process that results in phagocytosis is characterized by three interrelated steps: adherence and diapedesis, tissue invasion by chemotaxis, and phagocytosis. A, Adherence, margination, diapedesis, and chemotaxis. The primary phagocyte in the blood is the neutrophil, which usually moves freely within the vessel (1). At sites of inflammation, the neutrophil progressively develops increased adherence to the endothelium, leading to accumulation along the vessel wall (margination or pavementing) (2). At sites of endothelial cell retraction the neutrophil exits the blood by means of diapedesis (3). Chemotaxis: In the tissues, the neutrophil detects chemotactic factor gradients through surface receptors (1) and migrates towards higher concentrations of the factors (2). The high concentration of chemotactic factors at the site of inflammation immobilizes the neutrophil (3). B, Specific receptors for recognition and attachment. C, Phagocytosis. Opsonized microorganisms bind to the surface of a phagocyte through specific receptors (1). The microorganism is engulfed (ingested) into a phagocytic vacuole, or phagosome (2). Lysosomes fuse with the phagosome, resulting in the formation of a phagolysosome (3). During this process the microorganism is exposed to products of the lysosomes, including a variety of enzymes and products of the hexose-monophosphate-shunt (e.g., H2O2, O2−). The microorganism is killed and digested (4). Ab, Antibody; AbR, antibody receptor; C3b, complement component C3b; C3bR, complement C3b receptor; PAMP, pathogen-associated molecular pattern; PRR, pattern recognition receptor. (From McCance KL: Pathophysiology: the biologic basis for disease in adults and children, ed 6, St. Louis, 2010, Mosby.)
Genetic disorders can affect all of the steps involved in the innate immune response from initial recognition of infectious organisms to their phagocytosis and killing and are discussed in many chapters of this book. Examples of genetic disorders of the innate immune system that predispose animals to infectious disease, most commonly bacterial diseases, include leukocyte adhesion deficiencies and granulocytopathy syndromes. Leukocyte adhesion deficiency occurs in dogs and cattle and has an autosomal recessive mode of inheritance. It is characterized by alterations in the leukocyte adhesion cascade (see Chapter 3) involving deficiencies or dysfunction of integrins and selectins resulting in the inability of neutrophils to adhere to endothelial cells in the wall of blood vessels and migrate into sites of bacterial infection. Granulocytopathy syndrome occurs in dogs and cattle and has an autosomal recessive mode of inheritance. It is characterized by alterations in the ability of neutrophils to kill bacteria in phagosomes and is linked to reduced nicotinamide adenine dinucleotide phosphate (NADPH) concentrations that may arise from a metabolic anomaly in the hexose monophosphate shunt (see Fig. 4-10). This deficiency may lead to reduced concentrations of hydrogen peroxide in phagosome-lysosome fusion and the bactericidal capability of the neutrophil. The process of phagocytosis is usually normal. Affected animals have shortened lifespan, long-term febrile disease, dermatitis, oral ulcers, lymphadenitis, and poor healing, attributable to irresolvable and repeated bacterial infections.
Genetic disorders of the innate immune system can also be caused by failure of leukocytes to correctly develop and mature in the bone marrow. Cyclic hematopoiesis occurs in dogs and has an autosomal recessive mode of inheritance. It is caused by an abnormality of stem cells in the bone marrow resulting in periodic declines, every 10 to 12 days, in neutrophil concentrations followed by hyperplasia and a return to normal. Abnormal concentrations of purine and pyrimidine metabolites in affected stems cells suggest that a metabolic derangement in purine or pyrimidine metabolism may be the cause of this genetic disorder. This outcome increases susceptibility to bacterial infection, often leading to periodic fever, joint pain, or other signs of ocular, respiratory, or skin infections. Other examples of genetic alterations of the innate immune response that predisposes animals to increased susceptibility to infectious disease are discussed in the organ system chapters of this book.
Disorders of the Adaptive Immune Response
Genetic disorders of the adaptive immune response are disorders in which affected animals are incapable of generating antigen-specific immune responses (see Chapter 5). Such genetic diseases are closely associated with genes that regulate the expression of the MHC, especially those genes involved in antigen processing and presentation. Examples of this type of disorder include agammaglobulinemia (a B lymphocyte immunodeficiency) and severe combined immunodeficiency (a B and T lymphocyte immunodeficiency). T lymphocyte, macrophage, and complement immunodeficiencies also occur but are not discussed here. Agammaglobulinemia has a X-linked recessive mode of inheritance and thus is a disorder of young colts. It is likely caused by dysfunction of cytoplasmic tyrosine kinase resulting in blockage in the differentiation of B lymphocyte lineages and a nearly complete absence of B lymphocytes and plasma cells. Clinically, this type of immunodeficiency results in colts with chronic bacterial diseases leading to pneumonia, enteritis, dermatitis, arthritis, and laminitis. Severe combined immunodeficiency occurs in dogs and Arabian horses and has an autosomal recessive mode of inheritance. In dogs, an X chromosome–linked mode of inheritance has also been indentified. Affected animals produce no antibodies after infection or immunization because of an absence of B lymphocytes and have no or nonfunctional T lymphocytes when present. This genetic disorder occurs when lymphocyte precursors fail to differentiate into mature T or B lymphocytes, which is likely because of the result of mutations within recombinase-activating genes or within genes encoding DNA-dependent protein kinase or when differentiated lymphocytes are incapable of completing signal transduction pathways because of defects in cell surface receptors for interleukins. Other examples of genetic alterations of the adaptive immune response that predisposes animals to increased susceptibility to infectious disease are discussed in the organ system chapters of this book.
Bacterial Diseases
The pathogenicity (i.e., ability to cause disease) of a bacterium is regulated by its virulence determinates. Virulence determinates are molecules, often glycoproteins or glycolipids, derived from bacterial genes. Their expression establishes the processes used by bacteria to successfully colonize mucosae, infect cells, grow and replicate, and cause cell death. Virulence determinates are used to kill phagocytic cells such as neutrophils and macrophages, block phagocytosis, evade fusion with lysosomes and microbial killing, and enhance replication within phagocytes. Important actions (Fig. 4-13) of virulence determinates include the following:
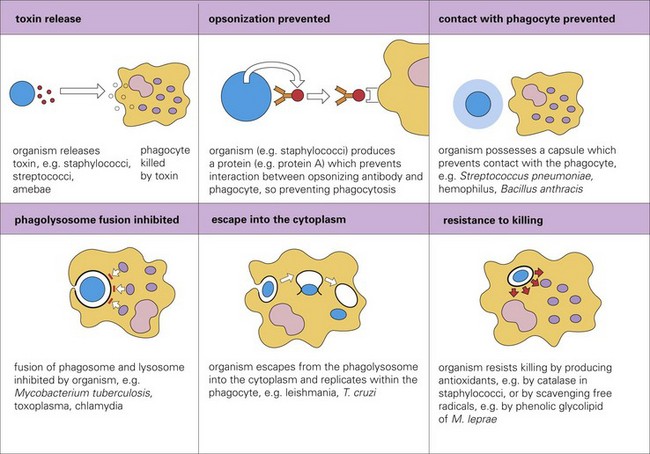
Fig. 4-13 Mechanisms adopted by infectious microorganisms to avoid phagocytosis. (From Kumar V, Abbas A, Fausto N, et al: Robbins & Cotran pathologic basis of disease, ed 8, Philadelphia, 2009, Saunders.)
1. Production of bacterial toxins that kill phagocytes.
2. Synthesis of bacterial proteins that prevent phagocytosis by blocking the interaction of opsonins with phagosomes.
3. Synthesis of a bacterial capsule that can block contact with the microbe and prevent phagocytosis.
4. Inhibition of fusion of the phagosome containing microbes with lysosomes.
5. Facilitate escape of the microbe into the cytoplasm before the microbe is killed in the phagolysosome.
6. Production of bacterial antioxidants (i.e., catalase) that block killing in phagolysosomes.
The interaction between an animal and a bacterial pathogen is back and forth, each acting to influence the activities and functions of the other. The outcome depends on the virulence of the pathogen and the resistance or susceptibility of the animal. Resistance or susceptibility to disease in healthy animals is derived from (1) innate defenses such as cellular and mucus barrier systems, acute inflammation (including neutrophil phagocytosis), the monocyte-macrophage system (phagocytosis), and normal bacterial flora and (2) adaptive defenses provided by the immune system such as passive immunity via colostrum and active immunity via T and B lymphocytes. In general, bacterial pathogenicity is determined by two characteristics: (1) its ability to infect cells and (2) its ability to produce toxins and damage cells and their ECM tissues such as collagen. To infect cells, infectious organisms use mechanisms such as adhesion, multiplication, colonization, tissue invasion, and circumvention of animal defense mechanisms. To damage cells via toxins, infectious organisms use mechanisms such as cytolysis and invasion of vascularized ECM tissues (locally or systemically) incited by bacteria-derived exotoxins (Gram-positive bacteria) and endotoxins (Gram-negative bacteria). Virulence determinates determine the sum of the characteristics that allow bacteria to cause disease and thus provide a pathogenicity profile or fingerprint for each bacterium.
Virulence Determinates
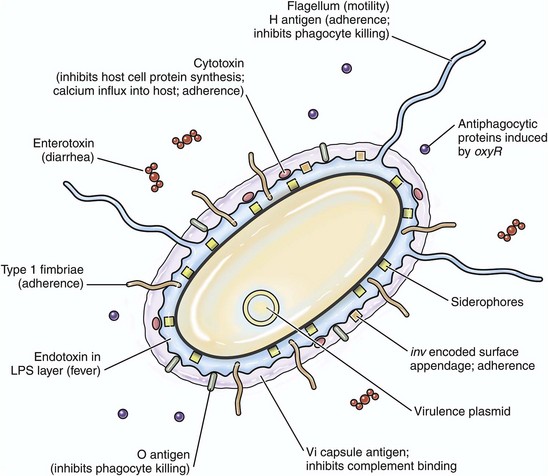
Bacterial virulence determinates are molecules that influence interactions between bacteria and cells, including events such as adherence to cell membranes; mucosal colonization; cell entry via endocytosis or phagocytosis; growth and replication; local, regional, and systemic spread; and cell disruption, inflammation, injury, or death by acting as toxins (Fig. 4-14). In general, they are coded for by more than one microbial gene. Furthermore, they inhibit immune responses and allow the organism to hide from defense mechanisms and proliferate in harsh environments. Bacterial virulence is determined in part by the type and number of factors the bacterium expresses to successfully complete its life cycle in an animal. Other things that can indirectly influence the success of these virulence determinates include physical and environmental issues, such as weather, access to food and water, and management (shipping) or housing stressors (ventilation, humidity, or overcrowding).
Initial Encounters
Before infecting cells, bacteria must often penetrate the mucus layer of mucosal barrier systems to gain access to mucosal epithelial cells, submucosal cells, and vascularized ECM tissues. Bacteria that are trapped in the mucus layer of mucosae may be phagocytosed by macrophages and dendritic cells that migrate in, on, and through the mucus layer and the mucosae. These cells may carry bacteria to mucosal epithelial cells and cells in submucosal lymph nodules, regional lymph nodes, and other organ systems. However, in many diseases, it is unclear how bacteria ultimately invade or penetrate the mucus layer to gain access to mucosal epithelial cells (target cells). Several mechanisms are likely used, including (1) motility, (2) digestion and consumption of the mucus layer, and (3) random discovery of mucosae lacking a mucus layer. As examples in the alimentary system, some bacteria, such as the spirochetes, are motile and can penetrate the mucus layer and reach target cells. Other bacteria, such as Clostridium septicum, digest the mucus layer with bacterial enzymes and then consume oligosaccharides such as N-acetylglucosamine, galactose, and N-acetylgalactosamine in the mucus layer as a carbon source during intense periods of replication. Finally, some bacteria use M cells to gain access to target cells; these cells are not covered by mucus and their surface membranes and receptors are available to passing infectious organisms.
Adhesion, Colonization, and Invasion
Some virulence determinates mediate adhesion, colonization, and invasion; others act indirectly to minimize the animal’s defense mechanisms by providing resistance to antibiotics, antiphagocytic properties, and weakened immune responses by acting as inhibitors. Adhesion, colonization, and invasion factors include bacterial membrane proteins, polysaccharide capsules, secretory proteins, cell wall and outer membrane components, and other miscellaneous molecules. Specific virulence determinates derived from membrane proteins assist the bacterium in adhering to, colonizing, and invading epithelia of barrier system portals of entry as in the alimentary, respiratory, integumentary, urinary, and reproductive systems. Because these epithelial cells are replaced continually (≈48 hour lifespan) and these systems have defensive mechanisms such as peristalsis, unidirectional mucociliary undulations, and urination, pathogenic bacteria must be able to adhere to, colonize (replicate), and/or invade into or between these epithelial cells to avoid being swept away. Attachment occurs when membrane proteins called adhesins (a broad term) bind to receptors on cell membranes. Attachment is a typical ligand-receptor interaction; a protein on the bacterium binds to a receptor on a host target cell. Some bacteria express adhesins, such as microbial surface cell recognition adhesion matrix molecules, that bind the bacterium to the surface of the cell. Other bacteria utilize extensions of their cell membranes called fimbriae or pili to bind to animal cells. Fimbriae and pili have adhesins, such as pilus-associated proteins, fimbrial antigens, or fimbrial adhesins, that bind to receptors on microvilli of the glycocalyx or in the mucus layer (Fig. 4-15). Fimbriae and pili may also act to inhibit phagocytosis. For example, uropathogenic and enterotoxic Escherichia coli, causes of urinary tract infections and diarrhea in animals, express fimbrial (type I, P, and S/F1C) and pilus (K99) adhesins, respectively. In the urinary tract, P-fimbriae is an important attachment adhesin and allows the bacterium to attach to the transitional epithelium and initiate the disease known as acute necrohemorrhagic urocystitis. Other virulence determinates, such as α-hemolysin and cytotoxic necrotizing factor type 1, cause necrosis and hemorrhage later. In the small intestine, K99 pilus adhesin allows E. coli to adhere to enterocytes and reduces their loss in number via peristalsis. When large numbers of E. coli are attached to the small intestine, they produce other virulence determinates, such as enterotoxin, that act directly on enterocytes, leading to diarrhea.
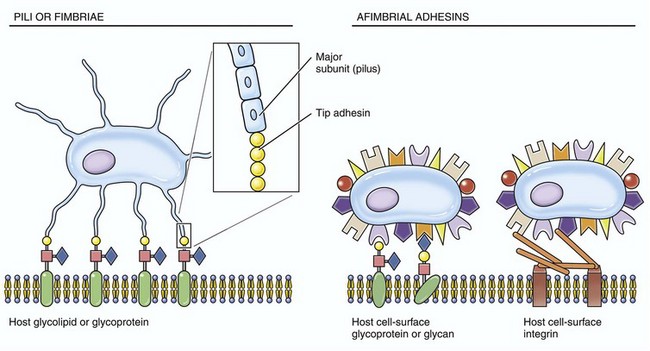
Fig. 4-15 Fimbrial (pilus) and afimbrial adhesins.
These structures are used by infectious microorganisms to attach and bind to protein receptors on membranes of target cells (especially mucosal epithelial cells) or to molecules of the mucus layer or vascularized extracellular matrix (connective) tissues.
Ligand-receptor interactions are likely common to most bacterial diseases; however, in many veterinary diseases, specific bacterial ligands and their host cell receptors have not been identified. The attachment of sufficient numbers of bacteria at the appropriate portal of entry initiates an early stage of bacterial infection termed colonization. After colonization, another group of virulence determinates called invasins, or spreading factors, are produced by infectious organisms. These factors include hyaluronidase, collagenase, kinases, lecithinase, and phospholipase and act to break down barrier systems formed by mucosae (mucus layer) and skin, cell junctional complexes, and ECM molecules like collagen (Table 4-2). This process allows bacteria to spread rapidly into and through intercellular spaces and protect itself in safe areas isolated from unfavorable host environments or host-derived defensive molecules. As examples, Clostridium chauvoei, the bacterium that causes blackleg in cattle, produces sufficient lecithinases and phospholipases to punch holes in cell membranes and cause death of myocytes and endothelial cells. Listeria monocytogenes, the cause of listeriosis in the nervous system of cattle, produces invasins that induce endocytosis of the bacterium for colonization by acting on host cell actin filaments. Other proteins of bacteria, such as surface components and polysaccharide capsules, are virulence determinates that allow bacteria to avoid phagocytosis and evade recognition by cells of the innate and/or adaptive immune systems. They disrupt or block one or more steps used by neutrophils, monocytes, or macrophages in the phagocytic process such as initial contact, engulfment, phagosome formation, phagosome-lysosome fusion, and killing and digestion. As examples, Streptococcus pyogenes, a cause of bovine mastitis, utilizes M-protein and a hyaluronic acid capsule to inhibit phagocytosis and the same hyaluronic acid capsule to evade recognition of the immune system.
TABLE 4-2
Biological Actions of Invasins or Spreading Factors
| Virulence Determinate | Action |
| Hyaluronidase | Depolymerizes hyaluronic acid in the extracellular matrix |
| Collagenase | Breaks down collagen fibers especially in muscle tissue |
| Neuraminidase | Degrades neuraminic acid (sialic acid) that serves to keep epithelial cells attached to basement membrane in the mucosa |
| Kinases | Digest fibrin and prevent clotting of the blood needed to wall-off bacteria |
| Lecithinase | Punch holes through or break down cell membranes |
| Phospholipase | Punch holes through or break down cell membranes |
Toxins
Certain virulence determinates are toxins like exotoxins, lipoteichoic acid, and endotoxins (lipopolysaccharide [LPS]) that damage cells, and their ECMs, such as collagen, are expressed by Gram-negative and Gram-positive bacteria (Fig. 4-16). Functionally, these molecules act to kill cells via cytolysis cells or through activation of the inflammatory cascade, often initiated via the complement pathway. In certain diseases, these toxins are named (and grouped) according to their biologic activity such as leukotoxins and neurotoxins.
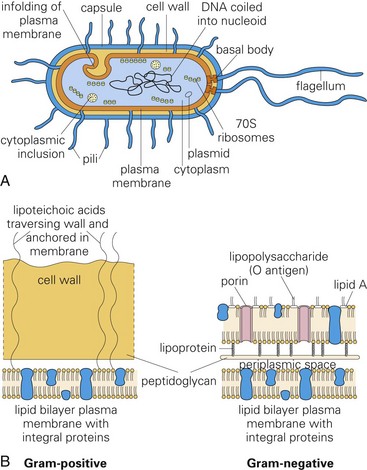
Fig. 4-16 Morphology and molecules of Gram-positive and Gram-negative bacteria.
Molecules like exotoxins, lipoteichoic acid, and endotoxins (lipopolysaccharide [LPS]) that form the structure of bacterial cell walls are often toxic. They act as virulence determinates that damage cells and their extracellular matrices such as collagen. A, Morphology of a typical bacterium. B, Gram-positive bacteria have a thick layer of peptidoglycan (left). Gram-negative bacteria have a thin peptidoglycan layer and an outer membrane of LPS (right). (A and B from Goering R, Dockrell H, Roitt I, et al: Mims’ medical microbiology, ed 4, St. Louis, 2008, Mosby.)
Exotoxins and Lipoteichoic Acid: Exotoxins (usually from Gram-positive bacteria) are secreted from viable bacteria and are potent toxins (Fig. 4-17, A). Some act directly on cells to cause cytolysis; others act via the A-B toxin system and bind to cell membranes with a receptor (B subunit) and deliver a second toxic molecule (A subunit) into the cytoplasm. As examples, A-B toxin systems are used in botulism (Clostridium botulinum), tetanus (Clostridium tetani), and diseases caused by Corynebacterium spp. Another virulence determinate, lipoteichoic acid, is located in the cell wall of Gram-positive bacteria like Staphylococcus aureus. It behaves as a Gram-positive endotoxin because its actions mimic LPS. Lipoteichoic acid binds to endothelial cells, interacts with circulating antibodies, activates the complement cascade, and triggers the release of reactive oxygen and nitrogen species, acid hydrolases, highly cationic proteinases, bactericidal cationic peptides, growth factors, and cytotoxic cytokines from neutrophils and macrophages. Vacuolating toxin of Helicobacter pylori, E. coli hemolysin, and superantigens belonging to Streptococcus pyogenes and Staphylococcus aureus are surface-acting toxins. Surface-acting toxins bind to cell membranes and form pores through which cell death occurs. Staphylococcus aureus also has pore-forming cytotoxins called α-toxin.
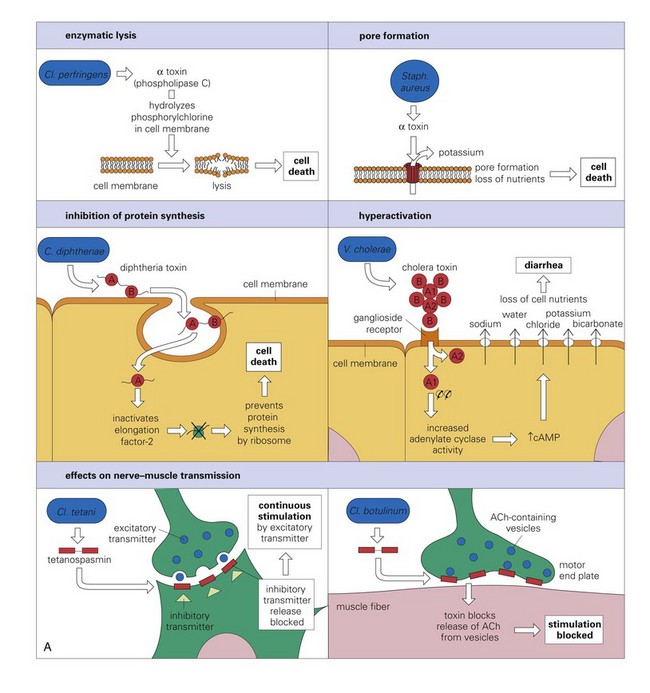
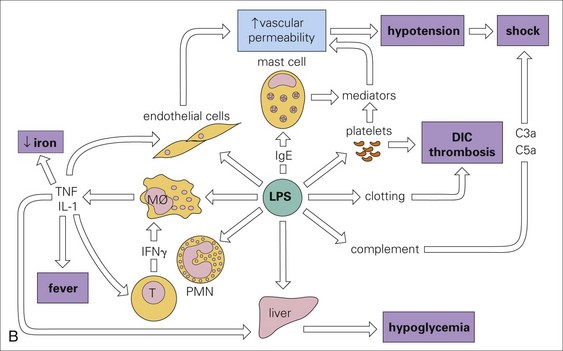
Fig. 4-17 Actions of bacterial toxins (virulence determinates) on the structure and function of target cells.
A, The mode of action of some exotoxins. Bacterial toxins act in a variety of ways. Often the toxin is a two-chain molecule, one chain being concerned with entry into cells while the other has inhibitory activity against some vital function. ACh, Acetylcholine; cAMP, cyclic adenosine monophosphate; C, Corynebacterium; Cl, Clostridium; Staph, Staphylococcus; V, Vibrio. B, The activities of bacterial endotoxin. Lipopolysaccharide (LPS) activates almost every immune mechanism, as well as the clotting pathway, and as a result, LPS is one of the most powerful immune stimuli known. DIC, Disseminated intravascular coagulation; IFN, interferon; IL, interleukin; M, macrophage; PMN, polymorphonuclear leukocyte; TNF, tumor necrosis factor. (A and B from Goering R, Dockrell H, Roitt I, et al: Mims’ medical microbiology, ed 4, St. Louis, 2008, Mosby.)
Endotoxins: Endotoxins (usually from Gram-negative bacteria) are toxic to most animal cells (especially endothelial cells and macrophages), tissues, and organs and can be lethal if large quantities are released into the circulatory system, causing the activation of proinflammatory cytokines and nitric oxide (NO) from macrophages and endothelial cells. This result leads to the activation of the complement and coagulation cascades and endotoxic shock (Fig. 4-17, B). Pathogenic bacteria, such as E. coli, Salmonella spp., Pseudomonas spp., Haemophilus spp., and Bordetella spp., can release endotoxins into tissue when they die.
Other Virulence Determinates
Secretion Systems: Secretion systems, of which six types (type I to VI) have been described, are bacterial organelles that secrete or inject bacterial-derived toxins into the cytoplasm of host target cells. The type III secretion system is best known and occurs in some Gram-negative bacteria such as Salmonella spp. and E. coli. It injects, like a needle, specialized bacterial protein toxins like exotoxins into the cytoplasm of cells. These protein toxins often disrupt cell signal transduction and other cellular processes leading to cell death.
Siderophores: Some pathogenic bacteria require iron for colonization of mucosae. Iron is plentiful in cells but unavailable to bacteria because it is tightly bound in heme, ferritin, transferrin, or lactoferrin molecules. Siderophores are virulence determinates that mediate the release of iron from intracellular iron stores (see Fig. 4-14). An example is enterochelin from E. coli and Salmonella spp.; this molecule scavenges bound iron from animal cells and makes it available for the bacteria.
Biofilms/Intracellular Bacterial Communities: Bacterial colonization and commensalism often occur through virulence determinates that form an exopolysaccharide matrix called a biofilm on mucosal surfaces lining, for example, the oral and nasal cavities and the mammary duct system. Bacteria embedded in biofilms are not susceptible to phagocytosis by macrophages and embedded bacteria can become resistant to antibiotics. For example, a surface protein, biofilm-associated protein (Bap), has been implicated in the formation of a Staphylococcus aureus biofilm in chronic bovine mastitis. Similarly, infections caused, for example, by certain strains of uropathogenic E. coli can result in the formation of intracellular bacterial communities affecting mucosal epithelial cells of the urinary bladder, which behave functionally much like a biofilm.
Role of Bacterial Genes in Susceptibility and/or Resistance to Disease
Virulence determinates are encoded in and translated from genes in chromosomal DNA, bacteriophage DNA, or plasmids of bacteria. They can be readily transferred horizontally between bacteria (e.g., virulence determinates for antibiotic resistance) via pathogenicity islands and/or virulence plasmids. Pathogenicity islands are clusters of genes that code for virulence determinates found in bacterial chromosomes. Virulence plasmids are clusters of self-replicating extrachromosomal genes for virulence determinates located in plasmids within the cytoplasm of the bacteria. Most bacteria have only one chromosome but may contain hundreds of copies of a specific virulence plasmid. Plasmids replicate independently of cell division, and when a bacterium containing plasmids divides, the plasmids distribute randomly between the two resulting bacteria. Chromosomal or plasmid genes express virulence determinates such as bacterial adhesins, colonization factors, protein toxins like hemolysins, other types of toxins, and molecules that affect the innate and adaptive immune responses. Strains of bacteria lacking pathogenicity islands and/or virulence plasmids usually do not cause disease. The number and type of virulence determinates in a given bacterial strain are constantly changing, usually through genomic selection of those factors that favor the survival of the bacterium in the animal host. Each bacterial genera and strains within genera have their own unique virulence determinate profile; therefore the total number of determinates that have been identified in all bacteria genera combined are in the hundreds. As an example, strains of Rhodococcus equi that cause disease have chromosomal virulence determinates for capsular polysaccharide, cholesterol oxidase, phospholipase C, lecithinase, and cell wall mycolic acids and plasmid virulence determinates for virulence-associated protein (VAP).
Antibiotic Resistance
Antibiotic resistance, the ability of bacteria to withstand the static or lytic affects of antibiotics, evolves via natural selection of randomly mutated bacterial genes. These genes code for bacterial molecules (i.e., virulence determinates) that cause resistance through the following four key mechanisms (Fig. 4-18):
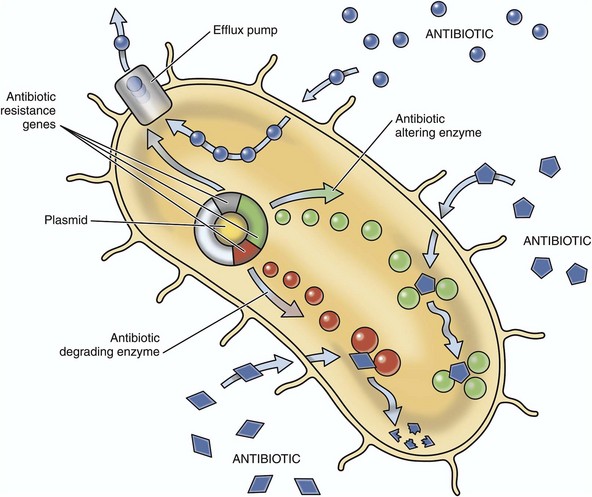
1. Enzymatic deactivation (antibiotic inactivation or modification) as occurs with β-lactamases and extended-spectrum β-lactamases (resistant to cephalosporins and monobactams) produced by bacteria such as Klebsiella pneumoniae, Pseudomonas aeruginosa, E. coli, and Salmonella typhimurium.
2. Alteration of antibiotic binding sites (penicillin-binding proteins [PBPs]) such as occurs in infections with methicillin-resistant Staphylococcus aureus (MRSA) and other penicillin/methicillin/oxacillin-resistant bacteria such as Streptococcus pneumoniae, vancomycin-resistant enterococci (VRE), and penicillin-resistant Streptococcus pneumoniae (PRSP).
3. Alteration of a metabolic pathway, such as occurs with some sulfonamide-resistant bacteria that utilize preformed folic acid in place of para-aminobenzoic acid (PABA), a precursor for the synthesis of folic acid in bacteria inhibited by sulfonamides.
4. Reduced antibiotic accumulation in bacteria through decreased membrane permeability to the antibiotic and/or enhanced efflux via membrane pumps.
Bacterial Transfer of Antibiotic Resistance: If a bacterium contains several antibiotic resistance genes, it is called a multiresistant pathogen. Although a human disease, MRSA infections are beginning to appear in animals. Such pathogens have multiple resistance genes that protect them from most if not all broad-spectrum antibiotics commonly used to treat the disease. These resistance genes are transferred between and among bacteria of related and different genera by vertical and horizontal gene transfer.
Bacterial Gene Transfer
Vertical Gene Transfer: The time required for bacteria to divide or a colony of bacteria to double in number is called the generation time and can be as short as 15 minutes. Although gene mutations for antibiotic resistance in bacteria are very rare events, because of fast generation times and the ability to reach extremely high absolute numbers of bacteria via binary fission in short periods of time, if unchecked it does not take long before antibiotic resistance develops. The spontaneous mutation rate for antibiotic resistance is approximately 1 × 108 to 1 × 109; this means that one out of every 100 million to 1 trillion bacteria in an infection develops resistance through a mutation. The use of antibiotics is a form of environmental pressure on bacteria; those having a favorable genetic mutation (i.e., virulence determinate for antibiotic resistance) survive therapy and continue to reproduce. Through vertical gene transfer, they then pass this virulence determinate for antibiotic resistance during DNA replication to their offspring, resulting in a fully resistant colony. As a result, the overuse of broad-spectrum antibiotics in humans and animals is a serious concern.
Horizontal Gene Transfer: Bacteria can also transfer antibiotic resistance genes between bacteria via horizontal gene transfer (Fig. 4-19) as follows:
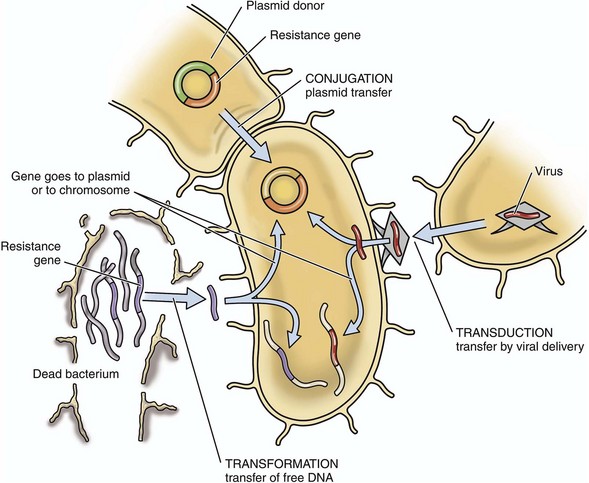
Fig. 4-19 Horizontal gene transfer.
Mechanisms used by bacteria to transfer resistance to an antibiotic to other bacteria.
1. Direct bacteria-bacteria contact (conjugation) via plasmids (the most common form).
2. Chromosomal DNA (transformation) in which pieces of DNA coded for antibiotic resistance and free in extracellular fluid as a result of death of its host bacterium are taken up by a viable bacteria.
3. Bacteria-specific viruses (bacteriophages) that transfer DNA (transduction) between two closely related bacteria.
Bacterial Diseases by Body Systems
Although the same bacterial disease often affects several different organ systems, diseases in this section are placed into a specific organ system based on which organ system demonstrates the primary gross lesion or lesions that are most commonly used to initially recognize and identify the disease. Bacterial diseases are identified by a primary mechanism of injury in Table 4-3.
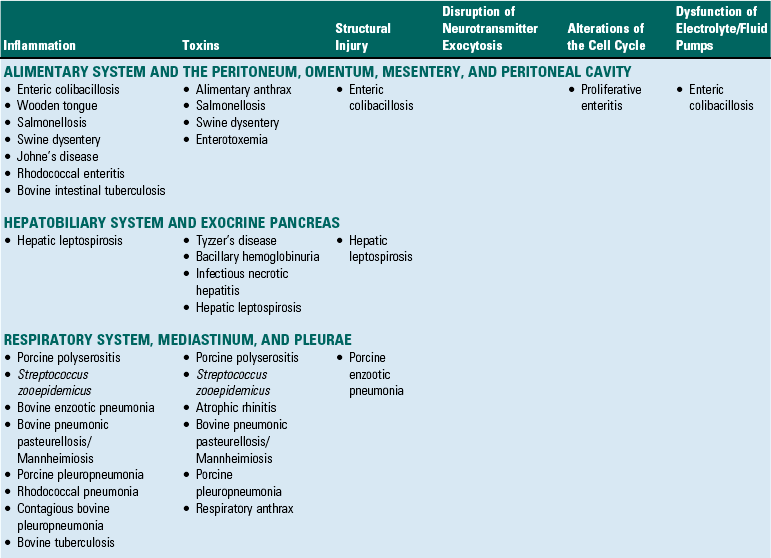
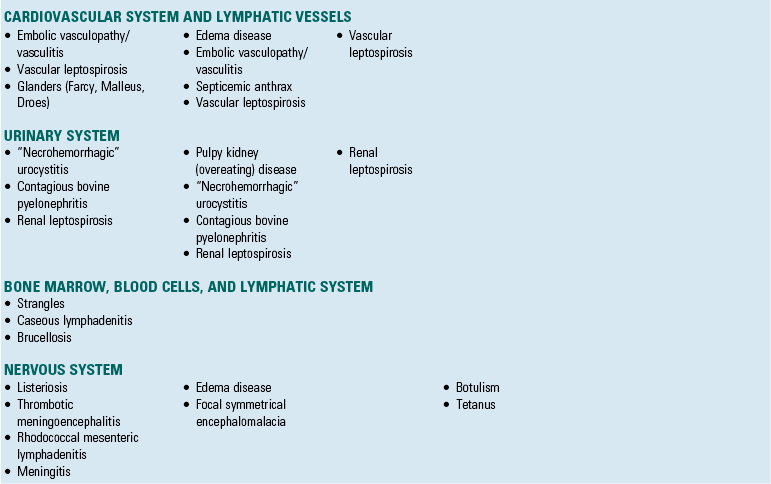

Alimentary System and the Peritoneum, Omentum, Mesentery, and Peritoneal Cavity
Enteric Colibacillosis (Escherichia coli): E. coli strains that cause disease in animals have been named enterotoxigenic, enteropathogenic, and enterohemorrhagic based on the mechanisms and virulence determinates that they use to cause diarrhea. The mechanisms of injury in enteric colibacillosis are (1) nonstructural alterations in the function of cell membrane ion and fluid transport systems (enterotoxigenic bacterial strains) and (2) acute coagulative necrosis of cells caused by bacterial toxins and by acute inflammation and its mediators and degradative enzymes (enteropathogenic and enterohemorrhagic bacterial strains). Enterotoxigenic and enteropathogenic strains do not invade enterocytes; enterohemorrhagic strains do invade enterocytes. Enterotoxigenic strains secrete toxins that functionally, but not structurally, affect enterocytes causing alterations in electrolyte and fluid secretion that result in a secretory diarrhea. Enteropathogenic strains structurally affect the microvillus border of enterocytes causing alterations in electrolyte and fluid secretion that result in an osmotic diarrhea (malabsorption) and a less significant secretory diarrhea. Enterohemorrhagic strains structurally affect the enterocytes of the colon causing cell death (necrosis), inflammation, and hemorrhage that leads to reduced absorption of colonic fluids and a malabsorption diarrhea. Endotoxins (LPS) likely directly or indirectly play a role in diseases caused by these three strains. There are no gross lesions in enterotoxigenic colibacillosis, whereas in enteropathogenic and enterohemorrhagic colibacillosis, the mucosae are rough and granular (enterocyte necrosis, villus atrophy) with areas of hemorrhage, acute inflammation, and fibrin exudation (see Fig. 7-157).
Animals encounter E. coli through ingestion of bacteria in fomites contaminated with fecal material. The bacterium is swallowed and gains access via peristalsis to the mucus layer and mucosae of the intestines. It is likely that flagella expressed by some strains of E. coli facilitate their penetration of the mucus layer to gain access to microvilli of enterocytes. Enterotoxigenic E. coli expresses K99 (F5) or F41 fimbrial adhesins that allow it to bind to receptor molecules in the mucus layer and to ganglioside and glycoprotein receptors on cell membranes of microvilli of enterocytes. When the mucosa is colonized, large numbers of bacteria are produced (Fig. 4-20), and they secrete heat labile (LT) and heat stable (ST) enterotoxins that diffuse in the mucus layer and microvilli, bind to specific receptors on the microvillus border of enterocytes, disrupt the function of cell membrane electrolyte and fluid transport systems, and cause secretory diarrhea. This process results in a functional lesion; no structural changes are observed grossly. LT and ST enterotoxins bind to glycolipid receptors on apical surfaces of enterocytes. After binding, these complexes are endocytosed and interact with a series of second messenger systems (epithelial cell signal transduction systems) that ultimately results in increased concentrations of intracellular cyclic adenosine monophosphate (cAMP, LT enterotoxin) and cyclic guanosine monophosphate (cGMP, ST enterotoxin). These molecules operate to open chloride channels (cystic fibrosis transmembrane regulator) in enterocyte cell membranes, thus acting irreversibly to move intracellular chloride ions extracellularly into the lumen of the intestine. Excessive chloride ion secretion also pulls water with it into the lumen of the intestine, thereby increasing the volume of fluid in the intestine. This volume ultimately exceeds the ability of the intestine to absorb the excessive fluid.
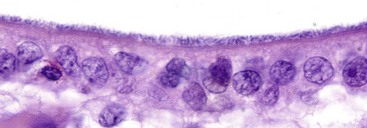
Fig. 4-20 Colonization of mucosa in enteric colibacillosis.
Escherichia coli attach to microvilli, thus forming a uniform layer of blue-staining (hematoxylin) coccobacilli. Note the lack of epithelial cell injury. H&E stain. (Courtesy Dr. J. F. Zachary, College of Veterinary Medicine, University of Illinois.)
Enteropathogenic E. coli (EPEC) colonize the mucosae in a manner similar to that used by enterotoxigenic E. coli. EPEC do not produce LT or ST enterotoxins but do express adhesins such as fimbriae, EPEC adherence factor, and intimin (nonfimbrial outer membrane protein). Integrins may serve as host cell membrane receptors for intimin, and this interaction appears to produce a tight bond between the bacterium and the enterocyte. After colonization and growth, bacterial virulence determinates injure the brush border, leading to the loss of microvilli at the site of colonization. Such virulence determinates appear to involve processes that disrupt cytoskeletal functions of the microvilli through interference with actin filaments, actin polymerization, and other cytoskeletal components and by causing alterations in intracellular calcium concentrations. This type of change has been called attaching and effacing injury and has resulted in naming the bacterium attaching and effacing E. coli. Injury to and loss of microvilli leads to decreased digestive enzyme activity in the glycocalyx (osmotic diarrhea) and disruption of ion transport systems (secretory diarrhea). EPEC also secretes bacterial proteins and likely injects them into the cytoplasm of enterocytes through a type-III secretion system. These proteins, EspA, EspB, and EspD, activate a number of signal transduction pathways in the host cell, which appear to be involved in the pathogenesis of microvillus disruption. Additionally, acute inflammation occurs at the site of binding between the bacterium and the microvillus, likely contributing to the attaching and effacing lesion. Some attaching and effacing strains also secrete a virulence determinate called verotoxin, which kills enterocytes and cells of the lamina propria (vascularized ECM tissues), leading to mucosal erosions and ulcers, intestinal edema and hemorrhage, and increased denuded mucosal surfaces for the absorption of endotoxins.
Enterohemorrhagic E. coli appear to colonize the mucus layer and mucosae in a manner similar to that used by the other two strains of the bacterium; however, enterocytes of the colon are the primary target cells. This specificity could be mediated through ligand-receptor interactions, and bacterial fimbriae have been shown to act as adhesins and attach to enterocyte cell membranes. Chemical gradients, such as the concentration of iron in target cells, could also provide the basis for colonic specificity. Once attached to colonic enterocytes, the bacterium replicates in large numbers and secretes a verotoxin that elicits an intense acute inflammatory response. It is also able to invade enterocytes, and verotoxin kills the cells. Enterohemorrhagic E. coli do not produce LT or ST enterotoxins. Thus mucosal lesions (hemorrhagic colitis) characteristic of this strain of E. coli appear to result from a combination of inflammatory enzymes and mediators and toxins, all of which cause cell death and expose the underlying denuded lamina propria to a variety of harmful luminal molecules such as LPS that are readily available for absorption. Endotoxin, especially when in the blood, can lead to inflammation, capillary damage, vasculitis, thrombosis, intravascular coagulation, tissue degradation and infarction, endotoxic shock, and death. These mechanisms likely underlie the occurrence of acute adrenal cortical hemorrhage and necrosis (see Fig. 12-25) observed in endotoxemia with E. coli and other coliform infections. Septicemic colibacillosis, likely resulting from an enteropathogenic strain of E. coli, may begin as an alimentary manifestation and then progress to enterotoxemic colibacillosis or septicemic colibacillosis. In these forms, the bacterium and its enterotoxins most likely gain access to the circulatory system via invasion into and absorption through capillary beds in the lamina propria of injured mucosae. Enterotoxemic colibacillosis and its toxins cause edema disease of the nervous system (see section on Nervous System; also see Chapter 14) leading to fibrinoid arteriopathy/arteriolopathy of the brain and ischemia and malacia, whereas septicemic colibacillosis and its bacterium and toxins cause disease of the cardiovascular system (see Chapter 10) leading to death via toxic and endotoxic shock and cardiovascular collapse.
Wooden Tongue (Actinobacillus lignieresii): The mechanism of injury in wooden tongue is incessant pyogranulomatous inflammation and fibrotic reparative responses. Gross lesions include a firm enlarged tongue that protrudes from the oral cavity. Cut surfaces have numerous randomly distributed yellow-white granulomas intermixed with broad bands of fibrous connective tissue (see Fig. 7-27). Actinobacillus lignieresii is a normal commensal bacterium of mucosae of the oral cavity of cattle and sheep. During chewing, the bacterium is carried through the mucosae into submucosal connective tissues via penetrating wounds such as those caused by sharp foreign bodies like sticks or wires. The bacterium colonizes submucosal connective tissue, and LPS of the bacterial cell wall, in part, likely plays a role in the pyogranulomatous inflammatory and concurrent fibrotic responses that occur. Little is known about how virulence determinates, ligand-receptor interactions, target cells, toxins, capsule antiphagocytic molecules, or other factors contribute to the pathogenicity of this bacterium. However, it appears likely that the bacterium is able to evade killing by neutrophils and macrophages, thus colonizing itself in abscesses in tissues of the tongue and oral cavity. Repeated cycles of phagocytosis, bacterial growth and replication, death of macrophages, release of large numbers of new bacteria, recruitment of additional naïve inflammatory cells, and reparative responses like fibrosis likely perpetuate and expand the scope of the disease process. Fibrosis and encapsulation occur concurrently and appear to represent a last ditch attempt to isolate and wall of the bacterium and exudate from the vascularized tissue in the tongue and oral cavity. Actinobacillus lignieresii can spread via lymphatic vessels to regional lymph nodes and cause a similar inflammatory response in these nodes.
Alimentary Anthrax (Bacillus anthracis): The mechanism of injury in alimentary anthrax is acute coagulative necrosis of cells caused by bacterial toxins. Gross lesions include segments of the small intestine or the entire small intestine that are dark red to purple-black (hemorrhagic enteritis) and accompanied by mucosal, submucosal, and serosal edema and hemorrhage (see Fig. 7-135). Additionally, mesenteric lymph nodes can be enlarged, edematous, and hemorrhagic. These toxins are produced by bacteria that are lined up in a laminar fashion along the surfaces of the mucosae, and as toxins are produced, they diffuse into the mucosa and lamina propria, causing in addition to necrosis, thrombosis of mucosal and submucosal vessels. This latter lesion leads to ischemic necrosis of tissues supplied by these blood vessels. Inflammation usually does not occur.
Animals, most commonly cattle, encounter Bacillus anthracis through the ingestion of fomites contaminated with endospores and/or vegetative forms of the bacterium. The bacterium exists most commonly in soil and water as an endospore, a dormant, nonreproductive form that is resistant to UV radiation, dehydration, extremely cold and hot temperatures, and chemical disinfectants. These conditions are harmful to the vegetative form, the form of the bacterium that produces toxin and causes disease. Animals can ingest endospores that subsequently germinate into vegetative forms in the alimentary tract. However, animals can also ingest vegetative forms as the result of environmental conditions that allow the vegetative form to persist for a limited period of time. Heavy rain after a drought can cause the germination of endospores in areas contaminated with endospores and the multiplication of vegetative forms. Endospores present in undercooked or poorly processed meat wastes and by-products can germinate into vegetative forms, persist for a limited period of time, and then be ingested when fed to animals.
Endospores and/or vegetative forms are swallowed, evade destruction by gastric acidity, and gain access to the mucosae of the small intestine via peristalsis. The sequence of events from ingestion of endospores or vegetative forms to the occurrence of lesions is largely unknown. It has been suggested that endospores can gain access to the mucosae and lamina propria via ulcers, cuts, and puncture wounds of the alimentary system, geminate into vegetative forms, colonize the tissue, and secrete toxins causing disease. It is also likely that vegetative forms gain access to regional mesenteric lymph nodes via afferent lymphatic vessels as cell-free bacteria or within macrophages that migrate via leukocyte trafficking in lymphatic vessels to these nodes. Vegetative forms probably use mechanisms discussed next to cause disease in the injured mucosae. Alternatively, the need for mucosal injury as an initiating event in the disease is open to discussion. Hypothetically, three possible mechanisms could result in the production of vegetative forms, toxins, and lesions:
1. Endospores* could be trapped in the mucus layer of the mucosa, endospores germinate to vegetative forms, vegetative forms use mucus to grow and replicate, and vegetative forms produce toxins that diffuse into the mucosa and submucosa, resulting in lesions.
2. Endospores* could be trapped in the mucus layer of the mucosa, phagocytosed by macrophages or dendritic cells and carried to Peyer’s patches where they germinate into vegetative forms, and vegetative forms produce toxins that diffuse into the mucosa and submucosa, resulting in lesions.
3. Endospores* are phagocytosed by M cells and carried to Peyer’s patches where they germinate into vegetative forms, and vegetative forms produce toxins that diffuse into the mucosa and submucosa resulting in lesions.
Primary virulence determinates produced by Bacillus anthracis are in plasmid genes and include those that form the capsule and anthrax toxins. The capsule is important in establishing the infection, whereas anthrax toxins cause the lesions, disease, and death. The capsule consists of poly-d-glutamic acid that is nontoxic, protects the bacterium from destructive antibodies and bactericidal components of plasma, and inhibits phagocytosis, killing, and digestion of vegetative forms of the bacterium by macrophages and neutrophils. Anthrax toxins consist of three exotoxins that act together to cause cell death. One exotoxin, called protective antigen, facilitates the entry of itself into cells via endocytosis and then creates a pore in the cell membrane through which the remaining two toxins, edema and lethal factors, can enter the cell. Exotoxins must first bind to receptors on target cells. Protective antigen binds to two different cell surface receptors, tumor endothelial marker 8 (TEM8) and capillary morphogenesis protein 2 (CMG2). These receptors appear to explain the vascular orientation of the disease and the circulatory system collapse that results. In addition to vascular tissues, these receptors are also commonly expressed on cells in many other organ systems, likely accounting for the various forms (inhalation, cutaneous, and gastrointestinal) of anthrax. Once inside cells, protective antigen combines with edema factor to form edema toxin, which disrupts cell membrane water and electrolyte transport systems, resulting in edema and also blocks phagocytosis of vegetative forms by neutrophils and macrophages. Additionally, protective antigen combines with lethal factor to form lethal toxin. This toxin stimulates the production of a variety of cytokines that act to cause cell death, especially affecting phagocytic cells such as macrophages and endothelial cells of capillaries. Because of injury to the mucosae and lamina propria, capillary beds in the ECM can absorb edema and lethal toxins, as well as a variety of intestinal endotoxins. Cytokines, anthrax toxins, and endotoxins have profound systemic effects on the cardiovascular system, all of which contribute to cardiogenic and circulatory shock and death. Chromosomal genes of the vegetative form of the bacterium also express a capsule virulence determinate, which makes it resistant to phagocytosis by mucosae-associated tissue macrophages. Additionally, the bacterium has several chromosomal virulence determinates for hemolysins, phospholipases, and iron acquisition proteins that can contribute to or cause cell death.
Salmonellosis (Salmonella spp.): The mechanism of injury in salmonellosis is acute coagulative necrosis of cells caused by bacterial toxins and by acute inflammation and its mediators and degradative enzymes. Three forms of salmonellosis occur: peracute, acute, and chronic. Gross lesions in the peracute form include petechiation and a blue discoloration (cyanosis) of the skin, fibrinous polyserositis, and disseminated intravascular coagulopathy (see Fig. 7-128). These lesions are rooted in injury of the vascular system with vasculitis and thrombosis caused by bacterial toxins. In the acute form, lesions affect the mucosae of the small intestine, large intestine, and cecum (fibrinonecrotic ileotyphlocolitis) and are characterized by a rough and granular mucosal surface (necrosis) mixed with mucus, fibrin, and occasionally blood (see Fig. 7-128). The content is malodorous (septic tank odor). This pattern of injury is caused by bacterial toxins, acute inflammation, and their effects on enterocytes and blood vessels within the lamina propria. Bacteria can spread via the portal vein to the liver leading to the formation of foci of bacteria-induced necrosis and inflammation (paratyphoid nodules) (see Fig. 8-49). This spread likely occurs via leukocyte trafficking and infection of Kupffer cells, but bacteremia may also occur. Leukocyte trafficking also spreads the bacteria to mesenteric and systemic lymph nodes and the gall bladder (fibrinous cholecystitis). In the chronic form, injury is associated with discrete foci of necrosis and ulceration of the mucosae (button ulcers). These lesions are rooted in injury of the vascular system with vasculitis and thrombosis caused by bacterial toxins diffusing within the submucosa of the intestine.
Animals encounter Salmonella spp. through ingestion of bacteria in fomites contaminated with fecal material. The bacterium is swallowed and gains access via peristalsis to the mucus layer and mucosae of the intestines. Salmonella spp. appear to use two mechanisms to colonize the mucosae and gain access to the lamina propria and its capillary beds. The first mechanism uses M cells in intestinal crypts. Because a mucus layer is absent over M cells, the bacteria have direct contact with apically positioned cell membranes of M cells. The second mechanism uses apically positioned cell membranes of enterocytes to gain access to cells; however, these cells are covered with a mucus layer. Because Salmonella spp. are motile bacteria, they can penetrate through the mucus layer and gain access to these membranes. The bacterium must adhere to the apical surfaces of M cells and enterocytes to begin the process of colonizing the intestine, which is a process mediated through ligand-receptor interactions. Although unproved, it is likely that one or some of the fimbria possessed by Salmonella spp. are involved in the initial adherence between the bacterium and these cells. Which fimbriae are used to bind to host cells may vary depending on whether the target cell is an M cell or an enterocyte. Once bound to host cell membrane, a type III secretion system is used to inject bacterial proteins into the host cell that stimulate phagocytosis through the mobilization of actin filaments in the cytoplasm. It is thought, though unproved, that fimbria may determine susceptibility of the various strains of Salmonella spp. for animal species and for specific target cells within each species. Colonization of the mucosae only occurs if bacterial LPS is present in the bacterial cell, and it likely plays a role in adherence to target cells through its participation in bacterial cell wall stability and resistance to bile salts, cell surface hydrophobicity, and the correct insertion and folding of membrane proteins such as those that occur in fimbria. Salmonella spp. that enter the mucosa via M cells have direct access to macrophages and lymphoid cells in Peyer’s patches, whereas those that enter via enterocytes are likely phagocytized by mucosal macrophages and carried via leukocyte trafficking in afferent lymphatic vessels to Peyer’s patches. Salmonella spp. are able to inhibit phagosome-lysosome fusion, thereby blocking their killing within macrophages. If phagosome-lysosome fusion occurs, bacteria are able to block the effects of lysosomal enzymes, acidity, and free radicals. In fact, the bacterium resides and replicates in a phagosome and/or phagolysosome until released from the macrophage after the death of the macrophage caused by toxins produced by the bacterium. Once in Peyer’s patches, macrophages infected with bacteria die and release bacteria that will infect additional macrophages through ligand-receptor interactions. Salmonella spp. can kill macrophages via apoptosis using a type I secretory system that leads to activation of caspase-1 in macrophages. These macrophages are often recruited as monocytes from the systemic circulation as a component of the acute inflammatory response. Once infected with the bacterium, macrophages migrate in efferent lymphatic vessels via leukocyte trafficking to regional mesenteric lymph nodes using mechanisms similar to those described in Peyer’s patches and then systemically via the thoracic duct and circulatory system. Macrophages also likely gain access to the systemic circulatory system through capillaries and postcapillary venules in lymph nodes. It also appears that dendritic cells in the intestinal mucosa are infected by the bacterium and these cells likely spread the bacterium to Peyer’s patches.
In all of these situations, acute inflammation occurs concurrently in response to bacterial toxins and antigens, resulting in vascular permeability changes and injury, and in the recruitment of neutrophils and their degradative enzymes, which can cause additional tissue injury. As part of this response, the interaction of M cells and enterocytes with bacteria appear to cause the release of chemokines and other chemoattractants for neutrophils into surrounding vascularized ECM. It appears that the form of disease (peracute, acute, or chronic) that occurs depends on which steps used in the chronology (as described previously) are emphasized through the expression of virulence determinates by different strains of Salmonella spp. The peracute form likely favors spread to regional lymph nodes and then systemically with the release of toxins leading to vascular injury, failure of the circulatory system, and death. The acute form likely favors mucosal adherence and colonization leading to mucosal necrosis mediated by bacterial toxins. Through this process and the acute inflammation that ensues, capillary beds in the lamina propria are permeable and likely bacteria, bacterial toxins, and bacteria-infected macrophages enter the venous circulatory system and spread via the portal vein to the liver. The chronic form likely favors invasion of the lamina propria and submucosa (motile bacteria) with direct effects on the vasculature that supplies the intestine with blood. However, it is possible that button ulcers observed in this form are a manifestation of septicemia with attachment of bacteria to vascular endothelium resulting in vasculitis, thrombosis, ischemia, and infarction. The lesions and clinical signs that occur in diseases caused by Salmonella spp. are in part attributable to (1) an enterotoxin (exotoxin) that produces a secretory diarrhea, (2) a cytotoxin that inhibits protein synthesis, and (3) endotoxins and LPSs that cause membrane injury and cell death. Acute inflammation and cell and tissue injury that ensue are also important causes of the lesions.
Enterotoxemia (Clostridium perfringens): The mechanism of injury in enterotoxemia is acute coagulative necrosis of cells and tissues caused by bacterial toxins. Gross lesions include segments of the small intestine or the entire small intestine that are dark red to purple-black (hemorrhagic enteritis) and accompanied by mucosal, submucosal, and serosal edema and hemorrhage (see Figs. 7-131, 7-132, and 7-147). Clostridium perfringens attach in a laminar fashion to mucosal surfaces, and as toxins are released, they diffuse into the mucosa and lamina propria, causing in addition to necrosis, thrombosis of mucosal and submucosal vessels. Inflammation usually does not occur. Clostridium perfringens causes disease syndromes that are categorized based on bacterial type (type A-E), toxin type (α-, β-, ε-, and ι-toxins), species affected, and/or age of the animal affected.
Cattle, sheep, goats, pigs, and horses encounter Clostridium perfringens through ingestion of bacterial spores in the soil or from contact with fomites contaminated with the vegetative form of the bacterium from carrier animals. The vegetative form of Clostridium perfringens can be a normal inhabitant of the alimentary system of domestic animals. It appears that under the proper conditions in the intestine that are usually linked to changes in the diet or the ingestion of an energy source rich in carbohydrates, spores germinate into vegetative forms and proliferate or the ingested vegetative form proliferates. It has been shown experimentally that dietary trypsin can inactivate β-toxin, thus it is thought that diets deficient in trypsin may increase the likelihood of disease. Additionally, sudden dietary change can also alter the composition of normal intestinal microflora providing opportunities for the vegetative form of the bacterium to proliferate and produce toxins. Vegetative forms are nonmotile and gain access to mucosae via random motion from peristalsis. They colonize the mucus layer by using bacterial proteases to expose receptors in mucus and then use bacterial adhesins to bind to these receptors. Within the mucus layer, bacteria are protected from acids and enzymes in intestinal content. The bacterium also consumes mucus as an energy source for bacterial growth and replication, and this process is thought to activate bacterial genes that regulate the production of toxins. Once the mucus layer is colonized, bacteria then interact with microvilli of enterocytes by attachment and retraction of type IV pili (gliding motility) and eventually attach to the apical surfaces of enterocytes. Attachment is likely mediated by ligand-receptor interactions.
Experimental studies suggest that toxins may first injure endothelial cells in capillaries of the lamina propria before the attachment of bacteria to the apical surfaces of enterocytes and that attachment may require changes in the membranes of apical enterocytes that are induced directly by the effect of toxins at the apical surfaces of enterocytes and indirectly by ischemia. At this phase of the disease, injury is primarily limited to enterocytes. Once mucosae are colonized, bacteria replicate in immense numbers and the disease enters a second phase characterized by the production of abundant potent cytotoxins, which spread via diffusion as a wave into the mucosae, lamina propria, submucosa, and muscle layers. Some clinical forms of enterotoxemia remain in the first phase of the disease; others progress into the second phase. Discussion of these various clinical forms is outside of the scope of this chapter. Potent cytotoxins produced by the bacteria include α-, β-, ε-, and ι-toxins, which behave as enterocyte membrane toxins (α, β), such as phospholipases, lecithinases, and pore-forming toxins, as well as ECM toxins, such as collagenase, hyaluronidase, and sialidase. ε-Toxin has the unique ability to increase enterocyte and endothelial cell permeability by acting on the cytoskeleton and probably altering the function of junctional complexes, thus affecting the absorption of toxins by the vascular system resulting in systemic effects. Iota toxin disrupts the cytoskeleton, which leads to cell death. Because of the abundance of toxins produced in the second phase of enterotoxemia, toxins freely move in the intestinal lumen via peristalsis to interact with uncolonized normal enterocytes, thus the lesions are quickly spread to other areas within the intestine. Toxins cause death and sloughing of enterocytes of villi and crypts followed by further colonization by bacteria, additional proliferation and toxin production, and toxin-induced massive necrosis resulting in structural breakdown and hemorrhage of the entire intestinal wall. Because ε-toxin is a permease that alters cell permeability, the vascular beds in affected intestinal tissues readily absorb toxins from the lumen of the intestine into the circulatory system. Toxins are then carried to the brain, kidney, and other tissues in which the increase in vascular permeability leads to the release of blood plasma containing toxins into the interstitium and body cavities, resulting in edema and effusions. In the brain and kidney, these toxins cause focal symmetric encephalomalacia and pulpy kidney disease, respectively. However, it appears that the mechanisms leading to these two diseases occur in the first phase or early in the second phase of enterotoxemia before toxin-induced massive necrosis of the intestine occurs.
Proliferative Enteritis/Hemorrhagic Bowel Syndrome (Lawsonia intracellularis): Although proliferative enteritis/hemorrhagic bowel syndrome is most commonly recognized as a disease of pigs, a syndrome resembling the proliferative form of this disease in pigs also occurs in horses and the pathogenesis is likely similar to that described in pigs. Lawsonia intracellularis appears to cause two distinct disease syndromes in a single disease continuum. The mechanism of injury of the first syndrome is characterized by biologic events that result in cell proliferation (i.e., proliferative enteritis), whereas the mechanism in the second syndrome is characterized by biologic events that result in cell death and hemorrhage (i.e., hemorrhagic bowel syndrome). It seems that the first syndrome can occur and resolve without transitioning into the second syndrome. The reverse scenario does not appear to occur. The proliferative form of this disease is also known as porcine intestinal adenomatosis, proliferative ileitis, regional ileitis, garden hose disease, and other similar names. The hemorrhagic form of this disease is also known as necrotic enteritis or acute proliferative hemorrhagic enteropathy. The mechanism of injury is initially cellular hypertrophy and hyperplasia (proliferation) that can be followed by cell death resulting from ischemia and necrosis. Gross lesions in proliferative enteritis include a circumferentially firm and thickened ileum with a mucosa that fills the lumen and bulges into the lumen on cut surface (see Fig. 7-153). Gross lesions in hemorrhagic bowel syndrome include segmental necrosis and hemorrhage with fibrinous diphtheritic membranes attached to the mucosa, luminal blood clots, and thinned intestinal walls (see Fig. 7-155).
Pigs encounter Lawsonia intracellularis through ingestion of the bacterium in manure-contaminated fomites from the environment. It is swallowed and gains access via peristalsis to the mucosae of the small intestine. It is unclear how the bacterium initially colonizes the mucus layer and mucosae and why it targets epithelial cells in the ileum. Ligand-receptor interactions are likely involved in this ileal specificity; however, specific bacterial adhesins or cell membrane receptors have not been identified. Alternatively, possibly in conjunction with ligand-receptor interactions, ileal specificity may be attributed to unidentified metabolic or growth factors required by the bacterium that are provided only by intestinal crypt cells (see later section for more detail). In addition, it is unclear how the bacterium is able to penetrate the mucus layer and gain direct access to the luminal membrane of epithelial cells. Lawsonia intracellularis is regarded as a nonmotile bacterium; however, there is scant experimental evidence suggesting that the bacterium develops a transient bacterial appendage that behaves as flagellum and could provide motility into and through the mucus layer. Colonization also appears to be enhanced by the presence of other anaerobic bacterial species in the mucus layer. The significance of these findings is unclear, but could be related to these species providing molecules required by Lawsonia intracellularis for colonization and replication. Lawsonia intracellularis infects cells of the crypts located in the proliferative zone (Fig. 4-21). Following colonization, the bacterium interacts with the brush border and then is taken into the cell via endocytosis and resides in a phagosome within the cytoplasm. The bacterium rapidly escapes from the phagosome before phagosome-lysosome fusion occurs and resides free in cell cytoplasm. A phospholipase bacterial virulence determinate, mediated through a type III secretion system, may be involved in the escape mechanism. Once free in the cytoplasm, bacteria remain in close proximity to the apical (luminal) cell membrane where they grow and replicate within the cytoplasm (see Fig. 7-156). In this location, bacteria appear to aggregate near mitochondria; thus it has been suggested that they may need preformed triphosphates for growth.
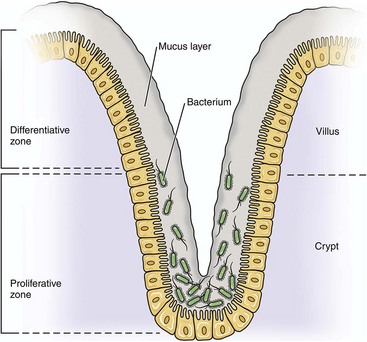
Fig. 4-21 Pathogenesis of proliferative enteritis in pigs.
Lawsonia intracellularis infects cells of the crypts located in the proliferative zone.
A unique feature of the pathogenesis of proliferative enteritis is the finding that proliferation of bacteria intracellularly (growth and replication) occurs concurrently with proliferation of crypt enterocytes (hypertrophy and hyperplasia) (see Fig. 7-154). One process does not occur without the other and vice versa. Under normal conditions, crypt cells within the proliferative zone are dividing cells that differentiate into nondividing cells of the differentiative zone as these cells migrate along basement membrane up the villus to its apex (see Fig. 4-21). It appears that once crypt cells of the proliferative zone are infected with bacteria, the bacterium is able to inhibit the normal maturation of crypt cells, probably through disruption of the cell cycle. Additionally, infection of crypt cells by bacteria dramatically increases the rate of crypt cell division. Therefore, when crypt cells are infected, they do not mature but remain in an undifferentiated proliferative state and divide continuously, thus resulting in massive thickening of the mucosal surface by proliferating crypt cells. Proliferating cells continue to migrate to the apices of villi where they die and are extruded into the lumen of the intestine. This outcome provides a source of bacteria to infect additional uninfected crypt enterocytes and spread through feces into the environment. Normal villus structure is lost and replaced by a branching glandular pattern, where cells lining the hyperplastic glands are crowded into a mucosal layer of up to 10 to 15 epithelial cells in thickness. Mitotic activity is prominent. Inflammation does not occur.
The literature does not provide any discussion of the relationship between proliferative enteritis and hemorrhagic bowel syndrome and how the former transitions into the latter or if it does. Histologically, it has been shown that lesions occurring in necrotic enteritis (hemorrhagic bowel syndrome) include acute coagulative necrosis of the proliferating crypt epithelium. This lesion is consistent with ischemia or the direct effects of toxins (burnlike injury). Necrotic enteritis may simply be a manifestation of proliferating cells outgrowing an available blood supply, becoming ischemic, and then dying via acute coagulative necrosis. No cell in the body can survive if it is farther than 100 µm away from a source of oxygen, either from a capillary or highly oxygenated body fluid. Additionally, Lawsonia intracellularis is a Gram-negative bacterium, and endotoxin or other toxic molecules may directly cause injury and cell death consistent with acute coagulative necrosis. Acute inflammation with hemorrhage and fibrinogenesis commonly occur concurrently with acute coagulative necrosis.
Swine Dysentery (Brachyspira hyodysenteriae): The mechanism of injury in swine dysentery is death of mucosal epithelial cells of the colon and cecum caused by bacterial hemolysins and proteases and from inflammation and its mediators and degradative enzymes. Gross lesions include a mucohemorrhagic necrofibrinous colitis and typhlitis with diphtheritic membranes covering intestinal mucosae formed by abundant mucus, hemorrhage, fibrin, plasma proteins, and cellular debris arising from necrotic mucosal epithelial cells and inflammatory cells (see Fig. 7-151).
Pigs encounter Brachyspira hyodysenteriae through ingestion of the bacterium in manure-contaminated fomites in the environment. The bacterium is swallowed and gains access via peristalsis to the intestine, especially the cecum and colon. Goblet cell mucus is important as a physical matrix and chemical substrate for colonization of the mucus layer by the bacterium, thus goblet cells play a central role in the pathogenesis of lesions affecting mucosal epithelial cells and their junctional complexes. Brachyspira hyodysenteriae, an anaerobic motile spirochete, is able to actively move through the mucus layer to gain access to mucosal epithelial and goblet cells. It is unclear why the bacterium infects the cecum and colon, but it appears that it prefers to initially replicate in mucigen droplets within goblet cells. Mucigen droplets fill the apical cytoplasm of goblet cells, and the nucleus is displaced to the basal region of the cell. Because the relative number of such cells is much greater in the cecum and colon when compared to other segments of the alimentary system, this quantitative difference may account for the location of the lesions. Additionally, mucins (e.g., the fucose and l-serine components) are strong chemoattractants for spirochetes, and because there are significant biochemical and pH differences in mucins, such as those that are synthesized and released from goblet cells, it is plausible that the chemical composition of the mucins may account for the locations of the lesions. Once the bacterium infects mucigen droplets within goblet cells, it appears to be able to activate the goblet cell to increase the production of mucus. Thus there is a large increase in the volume of mucus secreted by these cells so that mucosal surfaces are covered with a thick grayish gelatinous layer. It is unclear how the bacterium activates the goblet cell to produce and release large quantities of mucus. It is plausible that one or more bacterial virulence determinates influence the cellular processes of transcription, translation, assembly, and packaging of mucigen droplets thereby producing abundant quantities of mucus to enhance its opportunity to colonize the mucosa. Concurrently with infecting goblet cells, the bacterium begins the process of colonizing the thickened mucus layer covering the mucosal epithelium. It appears that mucus and their mucins are central to both the colonization and replication processes and result in the accumulation of large numbers of bacteria in close proximity to cell membranes and junctional complexes of mucosal epithelial cells.
Recently, it has been demonstrated experimentally that more virulent strains of the bacterium express increased numbers of genes for carbohydrate and amino acid metabolism and transport that potentially could be linked to energy and carbon sources that are available in the mucus layer. Additionally, highly fermentable feeds favor colonization of the mucus layer by bacteria. Fermentation may provide the bacterium with an energy source or other molecules required for colonization and replication. Colonization is also enhanced by the presence of other anaerobic bacterial species in the mucus layer. The significance of this finding is unclear, but again could be related to these species providing molecules required by Brachyspira hyodysenteriae for colonization and replication. Finally, because the bacterium is an anaerobe, it synthesizes high concentrations of nicotinamide adenine dinucleotide hydrogen (NADH) oxidase (a virulence determinate) that is used to protect itself from oxidative stress and toxic oxygen molecules in the oxygen-rich environment of the mucus layer.
The bacterium does not attach to luminal (apical) membranes of colonic and cecal epithelial cells; however, experimental studies have reported that the bacterium invades the epithelium and the lamina propria because they have been identified in these areas. The largest quantity of bacteria appears to exist in the mucus layer just overlying the epithelium. Therefore it is unclear as to whether this invasion is a direct and targeted process or merely an innocent bystander phenomenon, where the motility of the bacterium carries it into these locations to cause injury. Injury and death of colonic and cecal epithelial cells (enterocytes), as well as penetration through junctional complexes into the superficial lamina propria, are likely caused by one or more proteases and hemolysins and endotoxic effects of lipooligosaccharide (LOS) from the cell wall of the bacterium. Death and loss of the mucosal epithelium results in hemorrhage and the opportunity for infectious organisms, such as other anaerobic bacteria and the protozoan Balantidium coli, to invade the lamina propria. Additionally, denuded mucosa also provides a mechanism for the absorption of endotoxins, cytotoxins from inflammatory cells, and other toxic molecules that could cause endotoxic shock locally and systemically via the blood vascular system.
Johne’s Disease (Mycobacterium avium ssp. paratuberculosis): The mechanisms of injury in Johne’s disease are (1) dysfunction and death of the epithelial cells and ECM proteins forming the junctional barrier systems of mucosae of the small intestinal, (2) dysfunction of the drainage of afferent lymphatic vessels in the lamina propria of villi of the small intestine, and (3) death of cells of the monocyte-macrophage system and of all cell populations in the lamina propria of intestinal villi from inflammation and its mediators and degradative enzymes. Gross lesions include granulomatous enteritis and mesenteric granulomatous lymphadenitis, lymphangitis, and lymphangiectasia (see Fig. 7-146). Granulomatous enteritis is characterized by a thickened intestinal wall, most commonly affecting the ileum and ileal-cecal junction with a yellow-white exudate exemplified by infiltrating granulomatous inflammatory cells. Mesenteric granulomatous lymphadenitis is characterized by enlarged mesenteric lymph nodes that on cut surfaces have discrete and coalescing areas of yellow-white caseous exudate, occasionally mineralized, infiltrating, and compressing contiguous parenchyma.
The young of cattle, sheep, and goats encounter Mycobacterium avium ssp. paratuberculosis through ingestion of the bacterium in manure-contaminated fomites in the environment. The bacterium is swallowed and gains access via peristalsis to the alimentary system. Bacteria appear to bind to receptors on luminal (apical) surfaces of M cells and are likely translocated in endocytotic vesicles or phagosomes to the basal membranes of the cell and are released into Peyer’s patches where they can be phagocytosed by tissue macrophages. Unlike most other regions of the intestine, the luminal surface of M cells lacks a covering of mucus. Therefore bacteria have direct access to M cells. Lesions appear to have a segmental pattern of occurrence most commonly affecting the ileocecal intestine. Although attachment and phagocytosis of the bacterium by M cells likely involves ligand-receptor interactions, this mechanism does not explain why the lesions are most severe in ileocecal intestine.
Mycobacterium avium ssp. paratuberculosis requires iron for growth inside phagosomes of tissue macrophages. For an unknown reason, the concentration and availability of iron is greatest in tissue macrophages of the ileocecal intestine when compared to the concentration in other types of tissue macrophages. Therefore this gradient of iron appears to establish tissue specificity for lesions in Johne’s disease. In macrophages, iron is stored as ferritin, but it is not accessible by the bacterium. Mycobacteria secrete iron-chelating proteins called exochelins, iron-reductases, and potentially siderophores and use these enzymes to acquire iron from ferritin stored in macrophages. Additionally, as the severity of inflammation increases, there is a concurrent increase in the concentration of ferritin available for use by the bacterium in cells and tissues in the areas of inflammation. Mycobacterial siderophores or reductases may also serve to block iron-dependent bactericidal reactions of tissue macrophages such as Fe3+-dependent conversion of H2O2 into highly toxic hydroxyl radicals.
In ileocecal tissues, phagocytosis of the bacterium by tissue macrophages likely involves ligand-receptor interactions. Toll-like receptors may also be involved in attachment and phagocytosis. The cell walls of mycobacteria contain a variety of complex lipoglycans, glycoproteins, and lipoproteins such as lipoarabinomannan (LAM), 19-kDa lipoprotein, and the mycolyl-arabinogalactan-peptidoglycan complex that can serve as ligands. The cell membranes of tissue macrophages express receptors for these specific molecules and they are probably involved in the recognition, attachment, and adherence of the bacterium to the macrophage cell membrane. Additionally, complement receptors and other receptors, including mannose and CD14 receptors, expressed on tissue macrophages are the major receptors involved in phagocytosis of the bacterium, whereas integrin receptors, Toll-like receptors (TLRs), mannose receptors, CD14 receptors, scavenger receptors, and immunoglobulin Fc receptors are involved in early recognition and cell signaling in response to interaction with the bacterium. Generally, these signaling pathways initiate production of a variety of cytokines, chemokines, and antimicrobial metabolites that control mycobacterial infections; however, the bacterium through these signaling pathways is able to attenuate macrophage activation responses induced by interferon-γ (IFN-γ) and the secretion of IFN-γ. These interactions do not involve opsonin-mediated phagocytosis, thus the induction of a respiratory burst to kill the internalized bacteria does not occur and the bacterium persists in the phagosome. Receptors for opsonins expressed by tissue macrophages might also play a role in phagocytosis of the bacterium. Fibronectin may bind to the surface of macrophages and serve as a ligand to facilitate phagocytosis by macrophages. However, when employing opsonization as a means of entry into a phagosome, the bacterium must also use a mechanism to inhibit the respiratory burst to prevent its death.
The time from initial encounter with the bacterium to the expression of clinical disease is usually 12 months or longer. An explanation for this extended delay is unknown; it may simply be a slow-growing bacterium. However, bacterial growth likely involves the (1) interplay of bacterium-infected tissue macrophages and cells of the immune system mediated by proinflammatory and antiinflammatory cytokines, (2) migration of tissue macrophages locally from Peyer’s patches into the lamina propria and submucosal tissues, (3) time it takes the bacterium to replicate in sufficient quantities to activate the adaptive immune response, and (4) progression of severity resulting from the death of bacterium-infected macrophages leading to the recruitment of additional macrophages.
After phagocytosis by tissue macrophages in Peyer’s patches, the bacterium is confined within phagosomes and phagolysosomes. It appears to be able to disrupt phagosome-lysosome fusion and if fusion occurs, block the degradative actions of lysosomal enzymes and molecules via the structure and composition of its cell envelope and through the production of peroxidases. When a phagolysosome forms, the fused lysosome releases an acidic cytosol; proteases; and antibacterial substances, such as defensins and toxic oxygen and nitrogen intermediates, into the phagosome, all of which can injure and kill the bacterium. In general, mycobacterial species can (1) inhibit acidification of the phagosome, phagosome-lysosome fusion, and lysosomal enzyme activities; (2) block injury from toxic oxygen and nitrogen intermediates; and (3) suppress the ability of macrophages to be activated by cytokines such as INF-γ. Although highly probable, it is not known which or if any of these mechanisms are used by Mycobacterium avium ssp. paratuberculosis. Tissue macrophages, once infected with the bacterium, are activated and begin to secrete proinflammatory cytokines that act to recruit and activate additional macrophages. Additionally, because the lifespan of fully differentiated tissue macrophages is approximately 10 to 30 days, death of these cells related to aging and bacterial-induced injury releases bacterium into adjacent tissue where they are phagocytosed by newly recruited macrophages only to endlessly repeat this process. The severity and extent of the inflammatory response, concurrently with tissue injury, grows through the recruitment of additional monocytes and tissue macrophages from the circulatory system and regional lymph nodes. Bacteria-infected tissue macrophages can also spread via leukocyte trafficking in afferent lymphatic vessels within the intestinal mesentery to mesenteric lymph nodes (ileocecal lymph nodes) leading to a pyogranulomatous lymphadenitis and lymphangiectasia through the same progressive mechanism of inflammation.
Rhodococcal Enteritis (Rhodococcus equi): See the section on the Respiratory System, Mediastinum, and Pleurae for discussion of the early pathogenesis of the alimentary form of rhodococcal enteritis. Gross lesions include (1) chronic active pyogranulomatous lymphadenitis (see Fig. 7-137) characterized by enlarged firm lymph nodes that on a cut surface have discrete and coalescing areas of yellow-white exudate infiltrating and compressing contiguous parenchyma and (2) ulcerative enteritis (see Fig. 7-136) characterized by discrete foci of ulceration and hemorrhage centered over Peyer’s patches.
In the alimentary system, foals encounter Rhodococcus equi by swallowing mucus (sputum), exudate, and cellular debris contaminated with bacteria that move into the oral pharynx via the positive pressure of coughing and the upward rhythmic movement of cilia in the mucociliary apparatus. The bacterium then gains access to the alimentary system via intestinal peristalsis. Bacteria probably bind to receptors on luminal surfaces of M cells and are then transported in endocytotic vesicles to the basal membranes of the cell and released into the Peyer’s patches where they can be phagocytosed by tissue macrophages or dendritic cells. Unlike most other regions of the intestine, the luminal surface of M cells lacks a covering of mucus. Therefore bacteria have direct access to M cells. It is likely that ligand-receptor mechanisms (as discussed in the section on the Respiratory System, Mediastinum, and Pleurae) are applicable to M cells and tissue macrophages in Peyer’s patches. Once tissue macrophages are infected, the pathogenesis of the disease appears to progress much like that which occurs in the lung resulting in pyogranulomatous lymphadenitis. Ulcerative enteritis, characteristic of the alimentary form of the disease, occurs over affected Peyer’s patches. Although unknown, it is probable that mediators and degradative enzymes from inflammation diffuse into contiguous tissues, causing direct injury to the mucosae or indirectly via vascular injury and occlusion leading to infarction and ulceration. Bacteria-infected tissue macrophages can also spread via leukocyte trafficking in afferent lymphatic vessels within the intestinal mesentery to mesenteric lymph nodes leading to a pyogranulomatous lymphadenitis and then systemically to lymph nodes and lymphoid tissues such as the spleen.
Bovine Intestinal Tuberculosis (Mycobacterium bovis): Because the pathogenesis and lesions of bovine intestinal tuberculosis are identical to those observed in the pneumonic form, see the section on the Respiratory System, Mediastinum, and Pleurae. Also, the section on Johne’s disease should be reviewed because it mimics intestinal tuberculosis. It appears that intestinal tuberculosis commonly begins as the pneumonic form and is spread to the intestine by (1) coughing up and swallowing sputum containing macrophages infected with bacteria and/or “free” bacteria and (2) hematogenous or lymphatic spread of infected macrophages via leukocyte trafficking to intestinal lymph nodes and Peyer’s patches. Via the alimentary route, intestinal M cells and possibly dendritic cells are used to phagocytose bacteria and then release them via exocytosis from basolateral surfaces into Peyer’s patches where they are phagocytosed by macrophages and granulomatous inflammation and granuloma formation follow. Intestinal tuberculosis is associated with mucosal ulceration overlying Peyer’s patches. Ulceration appears to result from vasculitis, thrombosis, ischemia, and infarction secondary to inflammation in Peyer’s patches, but could also be caused directly by inflammatory mediators released from granulomas diffusing to and acting on blood vessels or mucosae.
Tyzzer’s Disease (Clostridium piliforme [Bacillus piliformis]): See Tyzzer’s disease in the section on the Hepatobiliary System and Exocrine Pancreas.
Hepatobiliary System and Exocrine Pancreas
Tyzzer’s Disease (Clostridium piliforme [Bacillus piliformis]): The mechanism of injury in Tyzzer’s disease is necrosis of hepatocytes, intestinal mucosal epithelial cells, and adjacent vascular and stromal tissues. Gross lesions include hepatomegaly and numerous white-gray-yellow foci (<2-mm diameter) of hepatocyte necrosis distributed at random, usually throughout all lobes of the liver (see Fig. 8-48). In severe cases, the center of these foci may be depressed and red (hemorrhage).
The young of all animal species can contract Tyzzer’s disease; however, foals appear to be the most susceptible and encounter Clostridium piliforme, an obligate intracellular bacterium, through ingestion of spores present in soil or vegetative forms in fecal fomites from infected animals. The disease is less common in dogs, cats, and calves. Although the bacterium uses the intestinal mucosa as an initial beachhead, it ultimately infects, replicates in, and injures the liver. The mechanism of spread from ingestion to the liver is unclear. In other diseases caused by Clostridium spp., such as blackleg, cells of the monocyte-macrophage system and M cells are probably used to spread spores and/or vegetative forms and hide (sequester) spores. After ingestion, it is likely that spores or vegetative forms are carried by normal peristaltic activities through the oral pharynx, esophagus, and stomach to their final destination, the small intestine. It is unknown if and how the bacterium interacts with and gains access to intestinal mucosal epithelial cells and/or mucosal macrophages. Vegetative forms of the bacterium are motile and may be able to penetrate the mucus layer and encounter mucosal epithelial cells of the small intestine. How they enter these cells is unknown, although direct penetration or receptor-mediated endocytosis through ligand-receptor interactions could be involved. Spores could be taken up through endostosis by intestinal mucosal epithelial cells, but how the spores penetrate the mucus layer and gain access to epithelial cells is unknown. They could be phagocytosed by mucosal macrophages in the mucus layer and carried to or through the mucosae by leukocyte trafficking. Additionally, spores could bind with receptors on the surface of M cells, which lack a mucus layer, enter though endocytosis, germinate into vegetative forms, infect and replicate in these cells, and then spread to adjacent mucosal epithelial cells.
In either case, spores or vegetative form are able to infect mucosal epithelial cells of the intestine through their apical surfaces and then replicate in the cells. It is not known what type of ligand-receptor interactions are involved in this process of entry into the cell. The bacterium appears to adhere to the apical cell membrane, be phagocytosed, and then escape the phagosome to reside and replicate in the cytoplasm of the cell.
It has not been shown how the bacterium spreads from mucosal epithelial cells or from M cells systemically to the liver. Because Clostridium piliforme is a motile bacterium, it has been suggested that it leaves epithelial cells of the intestine (possibly via their basal surfaces) and enters the circulatory system, likely by penetrating capillaries in the lamina propria that drain through the portal system to the liver. The bacterium could also potentially be transported to the liver by macrophages (leukocytic trafficking). If the bacterium is able to infect and replicate in M cells, the integration of M cells with Peyer’s patches provides the opportunity for the bacterium to interact with macrophages or gain access to capillaries within submucosal ECM tissues. Macrophages could phagocytose the bacterium using ligand-receptor interactions and carry it via leukocyte trafficking in afferent lymphatic vessels to mesenteric lymph nodes and then systemically via the thoracic duct and venous system into the circulatory system and ultimately the liver via the hepatic artery. However, in a mouse model of Tyzzer’s disease, depletion of macrophages did not change the course of infection. This outcome suggests that leukocyte trafficking may not be involved in the spread of the bacterium from the intestine to the liver.
The bacterium encounters endothelial cells lining hepatic sinusoids through spread in the circulatory system, likely via the portal vein. As a motile bacterium, it is free in the circulatory system and could (1) directly penetrate the endothelium and enter and infect hepatocytes, (2) infect and replicate in endothelial cells and then spread to adjacent hepatocytes, or (3) infect and replicate in Kupffer cells and then spread to adjacent hepatocytes. Hemorrhage occurs in Tyzzer’s disease, and this lesion suggests that vascular injury occurs either from direct penetration of the blood vessels or by lysis of the endothelial cells after replication of the bacterium. Although direct penetration of endothelial cells and hepatocytes is a possible mechanism of entry into these cells, typical ligand-receptor interactions may also be involved. The bacterium enters hepatocytes probably via receptor-mediated endocytosis and then escapes the phagosome to reside and replicate in the cytoplasm. The replication of Clostridium piliforme in hepatocytes eventually results in hepatocellular necrosis. The mechanisms causing necrosis are unknown. Bacterial cytotoxic proteins and several cellular cytokines like interleukin and tumor necrosis factor (TNF) have been implicated as the cause of hepatocellular necrosis, but experimental results are inconclusive. It appears that the bacterium first causes hepatocyte necrosis, which then incites an acute inflammatory response with abundant neutrophils and occasional macrophages in affected tissues. In the same mouse model of Tyzzer’s disease, the number of bacteria in hepatocytes and the severity of lesions were much worse in mice depleted of neutrophils and NK cells. This outcome suggests that the acute inflammation plays an important role as an innate defense mechanism in the disease.
Bacillary Hemoglobinuria (Clostridium haemolyticum): The mechanism of injury in bacillary hemoglobinuria has a local and a systemic component. Local injury is cell death (necrosis) of hepatocytes (necrotizing hepatitis), whereas systemic injury is death of erythrocytes in the blood vascular system. Injury in both components is caused by phospholipase C and other toxins released from Clostridium haemolyticum. Gross lesions include vasculitis, infarction, coagulative necrosis, and hemorrhage in the liver (see Fig. 8-68) and hemoglobinuria in the urinary system.
Cattle and sheep probably encounter Clostridium haemolyticum through ingestion of spores present in the soil. Although the bacterium ultimately resides in and injures the liver, the mechanism of spread from ingestion to the liver is unknown. In other diseases caused by Clostridium spp., such as blackleg, cells of the monocyte-macrophage, M cells, and dendritic systems are likely used to spread and hide spores. It is plausible that after ingestion, spores are carried by normal peristaltic activities through the oral pharynx, esophagus, abomasum, and rumen to their final destination, the small intestine. It is unknown how spores interact with and gain access to epithelial cells and mucosal macrophages. The mucus layer of the small intestine probably presents a significant barrier to spores; therefore spores could bind with receptors on the surface of M cells or dendritic cells and through transcytosis gain access to macrophages and lymphocytes located in Peyer’s patches contiguous with these cells. Macrophages could also phagocytose spores using ligand-receptor interactions and carry the spores via leukocyte trafficking in afferent lymphatic vessels to mesenteric lymph nodes and then systemically via the thoracic duct and venous system into the circulatory system. Although unproved, spores are likely the form of the bacterium that spreads systemically to liver. Trafficking macrophages could enter the sinusoids of the liver and transfer spores to Kupffer cells embedded in the endothelium. Spores then hide in Kupffer cells until they are activated to germinate and produce vegetative bacteria. Tropism for Kupffer cells is probably mediated by ligand-receptor interactions.
The occurrence of bacillary hemoglobinuria follows injury to the liver caused by migration of liver flukes (Fasciola hepatica, Fascioloides magna). Thus bacillary hemoglobinuria only occurs in geographic locations that have these flukes. The flukes migrate through the liver and injure intrahepatic veins, causing thrombosis, ischemia, and infarction of associated hepatocytes. Infarcted areas of liver are anaerobic and have a lowered oxidation-reduction (redox) potential required for germination of spores released from dead Kupffer cells. Spores germinate into vegetative bacteria and they produce large quantities of phospholipase C (also known as lecithinase C, an α-toxin) and hemolysins that destroy cell membranes and cause hepatocyte death. These toxins are also absorbed into the venous system within viable liver, resulting in entry into the systemic circulation leading to erythrocyte membrane injury, death of erythrocytes, release of hemoglobin, and hemoglobinuria.
Infectious Necrotic Hepatitis (Clostridium novyi): The pathogenesis and lesions of infectious necrotic hepatitis are similar to those of bacillary hemoglobinuria; however, the disease lacks hemoglobinuria, which is likely attributable to the absence of toxins that injure erythrocyte membranes.
Hepatic Leptospirosis (Leptospira spp.): The pathogenesis of hepatic leptospirosis begins as vascular leptospirosis caused by Leptospira spp., and this disease of the cardiovascular system and lymphatic vessels should be reviewed. The mechanisms used by Leptospira spp. to infect the liver are likely similar to those used in the kidney and are covered in the urinary system section of this chapter. Gross lesions include discrete and coalescing white-to-gray foci of hepatic necrosis scattered at random throughout hepatic parenchyma that are intermixed with hemorrhage.
Respiratory System, Mediastinum, and Pleurae
Porcine Polyserositis (Haemophilus suis/parasuis, Actinobacillus suis, Streptococcus suis, Escherichia coli): Several bacteria cause porcine polyserositis, but it is most commonly associated with Haemophilus suis/parasuis, the bacterium that causes Glasser’s disease. Gross lesions include vasculitis leading to variable quantities of a gray-white friable material (fibrin) on serosal surfaces (fibrinous polyserositis) of the lungs (fibrinous pleuritis), heart (fibrinous pericarditis), and abdominal cavity (fibrinous peritonitis) (see Fig. 7-157). The cavities formed by these anatomic structures may also contain a fibrinous exudate and edema fluid. Opposing serosal surfaces are often loosely attached to each other by fibrinous exudate, making normal physiologic processes like respiration more difficult. The serosa and cavities of meninges, joints, and testis can also be affected. Information about the mechanisms used by these bacteria to cause porcine polyserositis is limited. Thus portions of this section are speculative and based on (1) what is known mechanistically about other diseases of the respiratory system caused by the Pasteurellaceae family of bacteria and (2) a reasonable probability that inflammation, responses to injury, and lesions that have been described in porcine polyserositis are the result of underlying and known pathobiologic mechanisms.
Pigs likely encounter Haemophilus suis/parasuis, Actinobacillus suis, and Streptococcus suis through inhalation of these bacteria in contaminated fomites or fluid droplets. They appear to be commensal organisms of respiratory mucosae existing in biofilms in the nasopharynx and tonsil of healthy pigs. Environmental stressors, such as overcrowding, poor ventilation and humidity, or abrupt changes in ambient air temperature, alter the mucus layer and the commensal relationship, allowing bacteria to replicate in sufficient numbers to begin colonization of respiratory mucosae and to spread to other animals. Preceding or concurrent viral infections could also damage the mucociliary apparatus, allowing bacteria to colonize the mucus layer or mucosae. In the respiratory system, the bacteria are deposited on mucosae of the conductive component by centrifugal and inertial turbulence and trapped in the mucus layer. The bacterium is nonmotile, and it has not been clearly shown how it penetrates the mucus layer; gains access to cilia of mucosal epithelial cells; expresses virulence determinates such as adhesins, capsular molecules, fimbriae, and outer membrane proteins required for ligand-receptor interactions; and colonizes the mucosae. Once the mucus layer and/or mucosae are colonized and nonciliated and ciliated epithelial cells are injured by LPS, potentially by a neuraminidase, and by a suggested bacterial toxin, focal acute inflammation ensues and is followed to a limited extent by the death of these cells. In fact, receptors necessary for colonization of the mucus layer and/or mucosae or for the invasion of host cells may be exposed by the “potential” neuraminidase.
It has also been suggested that bacteria gain access to the lamina propria by altering the function of junctional complexes, allowing them to move between adjacent mucosal epithelial cells. The outcome of these processes is the loss of normal mucosal barriers, which provide the bacteria with direct access to the vascularized ECM of the lamina propria. It has not been determined how the bacteria actually cross this altered barrier, reach the capillary beds, penetrate the endothelium, and spread into the blood vascular system. Mechanisms, such as a cell-free bacteremia or leukocyte trafficking in alveolar or intravascular macrophages, lymphocytes, or dendritic cells, are hypothetic possibilities. A study of Haemophilus suis has shown that mucosal macrophages contain structures resembling phagolysosomes indicative of phagocytic activity. Lesions suggest these bacteria may have a tropism for vascular endothelial cells of serosae. It is unclear why this occurs, but it is probably linked to the expression of bacterial virulence determinates and ligand-receptor interactions with host endothelial cells. Additionally, it is thought that bacterial endotoxins may contribute to vascular injury and permeability changes leading to the leakage of fibrinogen and its polymerization to fibrin on serosal surfaces and in some cases to microthrombus formation and disseminated intravascular coagulation (DIC) in other organ systems.
Strep Zoo (Streptococcus equi ssp. zooepidemicus): The mechanism of injury in “strep zoo” is injury and death of mucosal epithelial cells and vascular endothelial cells from bacterial toxins and from inflammation and its mediators and degradative enzymes. Gross lesions include vasculitis leading to (1) a lung with a firm texture attributable to the leakage of variable quantities of fibrin into alveoli and alveolar septa (fibrinous pneumonia) from injured blood vessels and (2) the appearance of variable quantities of a gray-white friable material (fibrin) often mixed with hemorrhage on serosal surfaces (fibrinous polyserositis) of the lungs (fibrinous pleuritis), heart (fibrinous pericarditis), and abdominal cavity (fibrinous peritonitis) (see Fig. 10-14). The cavities formed by these anatomic structures may contain a fibrinous exudate and edema fluid mixed with hemorrhage. Opposing serosal surfaces are often loosely attached to each other by fibrinous exudate making normal physiologic processes such as respiration more difficult. The serosa and cavities of meninges, joints, and testis can also be affected. Information about the mechanisms used by this bacterium is limited. Some of this section is speculative and based on (1) what is known about virulence determinates used by other members of the Streptococceae family, especially Streptococcus equi ssp. equi, to cause disease and (2) a reasonable probability that inflammation, responses to injury, and lesions that have been described in strep zoo are the result of underlying and known pathobiological mechanisms.
Streptococcus equi ssp. zooepidemicus is a zoonosis. Horses and dogs likely encounter Streptococcus equi ssp. zooepidemicus through inhalation of the bacterium in fomites or fluid droplets. The bacterium appears to be a commensal organism of mucous membranes of the nasal and oral pharynxes, probably existing in biofilms of healthy animals. Environmental stressors, such as overcrowding, poor ventilation and humidity, or abrupt changes in ambient air temperature, alter the mucus layer and the commensal relationship, allowing the bacteria to replicate in sufficient numbers to colonize the respiratory mucosae and spread the bacterium to other animals. Preceding or concurrent viral infections could also damage the mucociliary apparatus, allowing these bacteria to colonize the mucus layer or mucosae. In the respiratory system, the bacteria are deposited on mucosae of the conductive component by centrifugal and inertial turbulence and trapped in the mucus layer. Although Streptococcus equi ssp. zooepidemicus can express many of the virulence determinates expressed by Streptococcus equi ssp. equi, a causal link of these factors to disease is unclear. The bacterium is nonmotile, and it has not been clearly shown how it penetrates the mucus layer; gains access to mucosal epithelial cells or cilia; expresses virulence determinates such as adhesins, capsular molecules, fimbriae, and outer membrane proteins required for ligand-receptor interactions; and colonizes the mucosae. Once mucosae are colonized and epithelial cells are injured, acute inflammation ensues, leading to death of these cells and loss of the ciliated mucosal barrier. Injury to ciliated epithelial cells likely alters the function of the mucociliary apparatus, allowing the bacteria to gain access to terminal bronchioles and alveoli by dependent settling due to gravity. Herein, bacteria colonize mucosae of the terminal bronchioles and alveoli, spread into vascularized ECM tissues, interact with and injure blood vessels of the air-blood barrier, and leak fibrinogen into the alveoli (fibrinous pneumonia). This process accounts for a fibrinous pneumonia but may not satisfactorily account for the fibrinous polyserositis so characteristic of this disease.
By an undetermined mechanism, bacteria likely gain access to the lamina propria of the respiratory system and have direct access to the vascularized ECM. It has not been determined how bacteria actually cross this altered barrier, reach the capillary beds, penetrate the endothelium, and spread into the blood vascular system. Mechanisms, such as a cell-free bacteremia or leukocyte trafficking in alveolar or intravascular macrophages, lymphocytes, or dendritic cells, are hypothetical possibilities. The characteristic gross lesions of fibrinohemorrhagic polyserositis suggest these bacteria may have a tropism for vascular endothelial cells of serosae. It is unclear why this occurs, but it is probably linked to the expression of bacterial virulence determinates and ligand-receptor interactions with host endothelial cells. Additionally, it is possible that bacterial toxins may contribute to vascular injury and permeability changes leading to the leakage of fibrinogen and its polymerization to fibrin on serosal surfaces and in some cases to microthrombus formation and DIC in other organ systems.
Disease caused by Streptococcus equi ssp. zooepidemicus in dogs appears to follow the same chronologic sequence of events as in horses, but virulence determinates involved in the disease appear to cause more severe lesions to the vascular system and a greater degree of hemorrhage.
Strangles (Streptococcus equi ssp. equi): See the section on Bone Marrow, Blood Cells, and the Lymphatic System for a discussion of strangles.
Porcine Enzootic Pneumonia (Mycoplasma hyopneumoniae): The mechanism of injury in porcine enzootic pneumonia is injury of ciliated epithelial cells of the bronchi and bronchioles, causing dysfunction and death of these cells. This outcome results initially in dysfunction of cilia, loss of cilia, and then cell death followed by a secondary bacterial infection leading to additional cell death of all cell types in the lung from chronic inflammation and its mediators and degradative enzymes. The susceptibility to and severity of porcine enzootic pneumonia can be enhanced by environmental stressors (abrupt or long-term changes in ventilation, temperature, and/or humidity) and earlier or concurrent viral (porcine reproductive and respiratory virus, swine influenza virus) or bacterial (Pasteurella multocida) infections. Gross lesions characteristic of this disease are firm (consolidation) yellow-tan-gray anterior-ventral lung lobes (see Fig. 9-83). Pleural surfaces are usually not involved, indicating that vascular injury and permeability changes and their association with the expression of bacterial virulence determinates are not significant in the pathogenesis of the disease.
Pigs encounter Mycoplasma hyopneumoniae through inhalation of the bacterium in fomites or fluid droplets. The bacterium is deposited on mucosae of the conductive component by centrifugal and inertial turbulence and trapped in the mucus layer. The bacterium is nonmotile, and it has not been clearly shown how it penetrates the mucus layer, gains access to cilia of mucosal epithelial cells, and colonizes the mucosae. Colonization of the respiratory system appears to probably involve adherence and binding of the bacterium to cell membrane receptors, using fimbria and pili by ligand-receptor interactions, because it has been shown that the bacterium attaches to cilia and line up in parallel rows along the surface of the cells. A molecule called cilium adhesin expressed on the surface of the bacterium appears to be involved in the attachment and binding process, where it is thought to interact with glycosaminoglycan and heparin on cell membranes. Additionally, the bacterium probably expresses glycosaminoglycans, such as heparin, heparin sulfate, and chondroitin sulfate B, or coats itself with such molecules that bind to ECM molecules, such as fibronectin, vitronectin, laminin, and collagen, but little is actually known about this process. This interaction ultimately results in dysfunction of cilia (ciliostasis) and death of the epithelial cells and reduced function of the mucociliary apparatus. Virulence determinates used by the bacterium to cause dysfunction and death of these cells have not been determined. Mycoplasma hyopneumoniae does not produce toxins, but some mildly toxic molecules do occur. Because of dysfunction of the mucociliary apparatus, Mycoplasma hyopneumoniae and other bacteria are able to reach distal aspects of terminal bronchioles and alveoli by dependent settling caused by forces of gravity. The bacteria replicate in these sites and cause chronic anterior-ventral bronchopneumonia.
Atrophic Rhinitis (Bordetella bronchiseptica and Pasteurella multocida): Although Bordetella bronchiseptica and Pasteurella multocida can each separately cause clinical forms of atrophic rhinitis in pigs, the classic form of the disease from a pathologist’s perspective appears to be caused by these two bacteria interacting synergistically. Information about the mechanisms used by Bordetella bronchiseptica and Pasteurella multocida to cause atrophic rhinitis in pigs is limited. Thus portions of this section are speculative and based on (1) what is known mechanistically about other diseases of the respiratory system caused by Pasteurella spp. and (2) a reasonable probability that inflammation, responses to injury, and lesions that have been described in atrophic rhinitis are the result of underlying and known pathobiologic mechanisms. The mechanism of injury is (1) death of ciliated epithelial and stromal cells of turbinate mucosae and (2) concurrent activation and suppression of osteoclasts and osteoblasts, respectively resulting in osteolysis of bone of the turbinates leading to turbinate atrophy. Gross lesions include varying degrees of loss (atrophy) and remodeling of turbinate scrolls and the nasal septum, with ventral scrolls usually being most severely affected (see Fig. 9-21).
Pigs encounter Bordetella bronchiseptica and Pasteurella multocida through inhalation of the bacteria in fomites or fluid droplets. These bacteria are likely commensal organisms that reside in the nasopharynx of healthy pigs, but environmental stressors, such as overcrowding, poor ventilation and humidity, or abrupt changes in ambient air temperature, alter the commensal relationship, allowing the bacteria to replicate in sufficient numbers to colonize the mucosae and spread the bacterium to other animals. Colonization appears to be a two-stage process; a Bordetella bronchiseptica phase followed by a Pasteurella multocida phase. When inhaled, Bordetella bronchiseptica is deposited on and trapped in the mucus layer of mucosae. The bacterium is nonmotile and it has not been clearly shown how the bacterium penetrates the mucus layer, gains access to cilia of mucosal epithelial cells, and colonizes the mucosae. Bordetella bronchiseptica does produce a dermonecrotic toxin (virulence determinate) and possibly adenylate cyclase-hemolysin toxin that likely affect the mucus layer and ciliated epithelial cells, making them more susceptible to colonization. The types of adhesins (ligands) and receptors used in colonization have not been determined but may include adhesins such as filamentous hemagglutinin, pertactin, and fimbriae proteins. The bacterium has an outer membrane protein called pertactin that may act as an adhesin, allowing the bacterium to colonize turbinate mucosae that have been injured by dermonecrotic toxin. Receptors for pertactin on mucosal epithelial cells have not been identified, but once in contact with cell membranes, bacteria likely adhere and bind to cell membrane receptors using fimbria and pili. This process results in colonization of mucosae by Bordetella bronchiseptica.
Under normal conditions, Pasteurella multocida has limited and weak virulence determinates for attaching to and colonizing turbinate mucosae. Initial colonization by Bordetella bronchiseptica leads to disruption of the mucus layer and the mucosal barrier system and results in mucosae that are more suitable for colonization by Pasteurella multocida in the second phase of infection. Mucosal lesions begin as focal erosions and ulcerations accompanied by acute inflammation (neutrophils), which subsequently spread into underlying lamina propria, ECM tissues, and bone of the turbinates. These lesions result in altered clearance mechanisms of ciliated epithelial cells and exposure of its lamina propria, where Pasteurella multocida can adhere to and colonize vascularized ECM tissues. Once the mucosae and lamina propria are colonized, the primary virulence determinate expressed by the bacterium is Pasteurella multocida toxin (PMT), a dermonecrotic toxin. The toxin, a typical A-B toxin, causes turbinate atrophy and snout deformation through chronic inflammation leading to bone remodeling and fibrous osteodystrophy of periosteal fibroblast origin. Mechanistically, the toxin initially acts to stimulate osteoblasts, which in turn act to increase the numbers (hyperplasia) and activity of osteoclasts. As toxin concentrations increase, it acts by blocking the function of osteoblasts, and cell degeneration and death may ensue. Overall, Pasteurella multocida toxin causes turbinate atrophy by increasing osteoclast numbers and activities through osteolysis of existing turbinate bone, as well as by inhibiting osteoblastic activities and the formation of new bone, both resulting in turbinate atrophy.
Bovine Enzootic Pneumonia (Pasteurella multocida ssp. multocida serogroup A): Mechanistically, the virulence determinates and mechanisms used by Pasteurella multocida ssp. multocida to cause bovine enzootic pneumonia are very similar functionally to those used by Mannheimia haemolytica to cause bovine pneumonic Mannheimiosis. However, in bovine enzootic pneumonia, bacterial pathogenicity is noticeably reduced and reflected by a slow onset and insidious inflammatory response and a nearly complete absence of cell necrosis, vasculitis, permeability changes, and fibrinogenesis. The mechanism of injury in bovine enzootic pneumonia is injury of all cell populations in the respiratory system attributable to inflammation and its mediators and degradative enzymes. The susceptibility to and severity of bovine enzootic pneumonia can be enhanced by environmental stressors and earlier or concurrent viral infection such as those caused by bovine respiratory syncytial virus, bovine viral diarrhea virus, infectious bovine rhinotracheitis virus, or parainfluenza III virus. Gross lesions include firm (consolidation) yellow-gray anterior-ventral lung lobes (see Fig. 9-56). Pleural surfaces are usually not involved, indicating that vascular injury and permeability changes and their association with the expression of bacterial virulence determinates are not significant in the pathogenesis of the disease.
Bovine Pneumonic Pasteurellosis/Mannheimiosis (Mannheimia [Pasteurella] haemolytica): The mechanism of injury in bovine pneumonic pasteurellosis/Mannheimiosis is injury and death (coagulative necrosis) of all cell populations in the respiratory system. In addition to injury caused by bacterial toxins (leukotoxin), acute inflammation and its mediators and degradative enzymes significantly contribute to the pathogenesis of the disease. Mannheimia haemolytica can cause severe pneumonic disease independent of other contributory factors; however, the susceptibility to and severity of disease can be enhanced by environmental stressors and earlier or concurrent viral infection. Gross lesions include severe fibrinonecrotic (often hemorrhagic) pneumonia and vasculitis attributable to necrosis and apoptosis, especially affecting type I pneumocytes and capillary endothelial cells forming the air-blood barrier of alveolar septa and the vascular system (severe necrotizing vasculitis) (see Figs. 9-58, 9-71, and 9-72).
Cattle (and probably sheep and goats) encounter Mannheimia haemolytica through inhalation of the bacterium in fomites or fluid droplets. The bacterium is a commensal organism that resides in the nasopharynx and tonsils of healthy animals, but environmental stressors, such as weaning, adverse weather conditions, changes in diet, and shipping, can alter the commensal relationship, allowing the bacteria to replicate in sufficient numbers to colonize the respiratory mucosae and spread the bacterium to other animals. Colonization appears to be a two-stage process, first affecting the conductive component (terminal bronchioles and alveoli), then affecting the O2-CO2 (terminal bronchioles and alveoli) exchange component. When inhaled, bacteria are deposited on and trapped in the mucus layer of the mucosae of the conductive component by centrifugal and inertial turbulence. Mannheimia haemolytica is a nonmotile bacterium, and it has not been clearly shown how the bacterium penetrates the mucus layer to gain access to cilia of mucosal epithelial cells. Neuraminidase produced by Mannheimia haemolytica reduces the viscosity of the mucus, making it less dense and a more fluidic layer, thus allowing the bacterium better access to cell membranes via gravity and random Brownian movement. Additionally, neuraminidase cleaves sialic acid from the surface of cell membranes, thus decreasing the net negative surface charge and allowing closer contact of the bacterium with membranes. Once in contact with cell membranes, bacteria adhere and bind to receptors using fimbria and pili by ligand-receptor interactions. This process results in bacterial colonization of the mucosae. The types of adhesins (ligands) and receptors used in colonization have not been determined. Once colonization occurs, the bacteria replicate in large numbers in the conductive component of the respiratory system and produce enzymes, such as neuraminidase, and toxins, such as leukotoxin and LPS that injure and disrupt the function of the mucociliary apparatus.
Additionally, polysaccharides of the bacterial capsule (virulence determinate) inhibit phagocytosis of the bacterium by neutrophils and mucosal macrophages. Because of mucociliary dysfunction, bacteria spread via gravity to dependent portions of the lung, including terminal bronchioles and alveoli of the O2-CO2 exchange component. Once bacteria arrive in this location, the second stage of the process, which is more severe than the first stage, begins. The difference in severity is, in part, based on three factors: (1) extensive replication of bacteria in stage one that settle into the O2-CO2 exchange component and their subsequent amplification through replication, (2) the large surface area of lung tissue affected, and (3) the greater vulnerability of the air-blood barrier and septa in the O2-CO2 exchange component to injury. All of these factors contribute to the severity of the acute inflammatory responses and tissue injury.
The single most important virulence determinate in bovine pneumonic Mannheimiosis is leukotoxin. Leukotoxin, a member of the RTX group of toxins, is a cytotoxin that causes death and apoptosis of alveolar macrophages and neutrophils. RTX toxins attach to cells through passive adsorption and cell surface receptors, the latter being the transmembrane receptor CD18. At high concentrations, it causes necrosis by creating pores in cell membranes leading to cell swelling and death, whereas at lower concentrations, it causes apoptosis. Additionally, at lower concentrations, it activates neutrophils and induces the production of proinflammatory cytokines. When bacteria are phagocytosed by alveolar macrophages, leukotoxin is used to kill the macrophages and release the bacteria back into vascularized ECM tissue and alveolar spaces. Iron is also required for optimal growth of the bacterium and production of leukotoxin. LPS and leukotoxin also activate the complement system and the release of proinflammatory cytokines resulting in vascular injury and severe acute inflammation. Vascular injury leads to permeability changes with edema and the release of fibrinogen that polymerizes to fibrin in alveolar spaces, interalveolar septa, interlobular and interlobar septa, and on pulmonary serosal surfaces (fibrinonecrotic pneumonia). Vascular injury can also lead to pulmonary hemorrhage.
Acute inflammation is characterized by the recruitment of large numbers of neutrophils from the circulation into affected lung tissue followed by activation of these cells via a respiratory burst and the release of degradative enzymes. The bacterium has several mechanisms (see later) to minimize the effects of neutrophils, but innocent bystander lung tissue, such as terminal bronchioles and cells that form the air-blood barrier, are severely injured by the molecules and enzymes released from activated neutrophils. Capsular polysaccharides, outer membrane proteins, and LPS of the bacterium are also important in the pathogenesis of the disease, especially as related to acute inflammation and vascular injury. Polysaccharides are virulence determinates that facilitate adherence, colonization, and likely invasion of respiratory mucosae; inhibit phagocytosis by neutrophils; and disrupt complement-mediated death of bacteria. Outer membrane proteins are chemotactic for neutrophils, but when in contact with neutrophils, they disrupt phagocytosis and intracellular killing of bacteria. LPS binds with cell membrane CD14, β2-integrins, and TLRs on alveolar macrophages inducing the synthesis of proinflammatory cytokines, arachidonic acid metabolites, and NO that injure cells in inflammation. LPS may also injure endothelial cells directly and through molecules released from macrophages such as those listed in the previous sentence.
Pulmonary Histophilosis (Histophilus somni): The pathogenesis of pulmonary histophilosis is likely very similar to the mechanisms as described previously for bovine pneumonic pasteurellosis/Mannheimiosis.
Porcine Pleuropneumonia (Actinobacillus pleuropneumoniae): The mechanism of injury in porcine pleuropneumonia is injury and death (coagulative necrosis) of all cell populations in the respiratory system, especially those of the vascular system (severe necrotizing vasculitis), secondary to the effects of bacterial toxins and to acute inflammation and its mediators and degradative enzymes. Gross lesions, attributable to vascular injury affecting lung and regional lymph nodes, include (1) edema and alterations in vascular permeability; (2) hemorrhage; and (3) fibrinous and hemorrhagic pneumonic, pleural, and pericardial effusions and acute necrotizing inflammation and pneumonia (see Fig. 9-84).
Pigs encounter Actinobacillus pleuropneumoniae through inhalation of the bacterium in fomites or fluid droplets. In the respiratory system, the bacterium is deposited on mucosae of the conductive system and probably on the exchange system by centrifugal and inertial turbulence. The characteristic distribution pattern (dorsal-diaphragmatic) of gross lesions may reflect the fact that droplet size and inertial turbulence results in the initial deposit being near branch points of conductive airways. The bacterium must first colonize the mucosae by adhering and binding to the membranes of epithelial cells using ligand-receptor interactions mediated through fimbriae. Following colonization, Actinobacillus pleuropneumoniae requires iron to grow and replicate and is capable of utilizing porcine transferrin as a source of iron. However, it appears that iron is also obtained from blood and the death of red blood cells caused by bacterial hemolysins and proteases. After the death of red blood cells, LPS and outer membrane proteins of the bacterial cell wall can bind hemoglobin and assist in the transfer of the iron molecule required for growth and replication into the bacterium. In part, this requirement may explain the severe hemorrhage that occurs in this disease.
It has been shown that the bacterium binds poorly to cilia and the epithelium of the trachea and bronchi, whereas it binds strongly to cilia and membranes of terminal bronchioles and membranes of alveolar epithelial cells. This selective pattern of binding and inertial turbulence (see previous discussion) may account for the distribution of lesions observed in porcine pleuropneumonia. LPS and LOS are known to play important roles in the pathogenesis of Gram-negative infections; however, their role in porcine pleuropneumonia is unclear and may involve acting as ligands for adherence to host cell surfaces. Glycosphingolipids present in membranes of epithelial cells may serve as receptors for these ligands. It has not been determined if and how Actinobacillus pleuropneumoniae penetrates the mucus layer to gain access to cilia and cell membranes of epithelial cells. The suppression of mucus production and ciliary activity increase the severity of porcine pleuropneumonia by decreasing clearance of the bacteria through the mucociliary apparatus mechanism. Once Actinobacillus pleuropneumoniae is bound to cilia and cell membranes of terminal bronchioles and alveolar epithelial cells, the bacterium is positioned to be phagocytosed by alveolar, interstitial, and intravascular macrophages. Although all of these types of macrophages are phagocytic, intravascular macrophages also have strong cytolytic activities that may also account, in part, for the hemorrhage characteristic of pulmonary vascular lesions.
Neutrophils are not involved in the initial phagocytic response to the bacterium, but once the process has been started by macrophages and chemokines are released from activated macrophages, neutrophils are recruited from the vasculature into the inflammatory response to phagocytose the bacteria. After phagocytosis, it has been shown that neutrophils can immediately kill Actinobacillus pleuropneumoniae, whereas macrophages cannot. In fact, the bacterium can survive for more than 90 minutes in a phagosome of a macrophage, during which it grows, replicates, and synthesizes and releases Apx toxins, leading to the death of these macrophages and release of the bacterium. Additionally, during this time, infected macrophages can move into alveolar and lobular septa, alveolar lumina, and perivascular and peribronchiolar tissues. Thus, when an infected macrophage is killed, large numbers of bacteria are released into vascularized ECM, leading to more inflammation, recruitment of neutrophils and macrophages, and exacerbation of injury to surrounding tissues.
Actinobacillus pleuropneumoniae has several virulence determinates that allow it to survive in phagosomes and resist the effects of phagosome-lysosome fusion, including a capsule, cell-wall LPS, copper-zinc superoxide dismutase, stress proteins, and ammonia. The capsule, cell-wall molecules, and superoxide dismutase participate in removing oxygen free radicals. The bacterium produces ammonia within phagosomes through the release of a potent urease, which inhibits phagosome-lysosome fusion and disrupts acid hydrolase activity in lysosomes. Finally, at low concentrations probably early in the disease process, RTX toxins (ApxI-III) produced by the bacterium also impair chemotaxis and phagocytosis by macrophages and neutrophils by likely acting to disrupt actin-myosin–directed movements of the cell or its organelles. At higher concentrations, such as those that occur after the bacteria has gone through several replication and kill cycles in macrophages, ApxI and ApxIII are highly toxic and ApxII is moderately toxic for macrophages and neutrophils. Additionally, ApxI-III are highly toxic to surrounding tissues, such as blood and lymphatic vessels, and ECM tissues, resulting in loss of barrier systems, increased vascular permeability with fibrin leakage and polymerization, hemorrhage, and vasculitis. In fact, it should be remembered that all of these infected cells and innocent bystander cells and tissues are within several hundred micrometers of one another and the vascular system. In particular, vascular injury appears to result from the activation and killing of intravascular macrophages and endothelial cells by Apx toxins and LPS. Activation results in the release of oxygen free radicals (superoxide anion, hydrogen peroxide, and hydroxyl radical) as well as proteolytic enzymes and various cytokines, all of which can injure endothelial cells of capillaries and postcapillary venules. Injury leads to activation of the coagulation, fibrinolysis, and kinin systems with concurrent hemorrhage, edema, effusions, platelet activation, and the formation of thrombi, ischemia, and subsequent coagulative necrosis of lung.
Rhodococcus Pneumonia (Rhodococcus equi): The mechanism of injury in rhodococcal pneumonia is death of cells of the monocyte-macrophage system and of all cell populations in the respiratory system secondary to inflammation and its mediators and degradative enzymes. Gross lesions include (1) cranioventral chronic active pyogranulomatous pneumonia characterized by consolidated firm yellow-white lung parenchyma attributable to infiltrating inflammatory cells, abscesses, and granulomas in the affected lung tissue (see Fig. 9-67) and (2) necrotizing pyogranulomatous lymphadenitis of tracheobronchial lymph nodes of the lung exemplified by enlarged firm lymph nodes that on a cut surface have discrete and coalescing areas of yellow-white exudate infiltrating and compressing contiguous parenchyma (see Fig. 7-137). The latter lesion occurs through leukocyte trafficking of bacteria-infected alveolar macrophages as described below.
Foals encounter Rhodococcus equi through inhalation of the bacterium in manure-contaminated fomites or water droplets from the environment. It is a common bacterium of the soil, grows optimally at 30° C in the manure of most animal species, and has a very rapid generation time. When inhaled, the bacterium is deposited on the mucosae of the conductive and exchange systems by centrifugal and inertial turbulence. Here, bacteria encounter cells of the monocyte-macrophage system, including alveolar macrophages and dendritic cells, which phagocytose bacteria in the mucus layer of the mucociliary apparatus. These cells carry bacteria via leukocyte trafficking to local lymphoid tissues, such as BALT, through peribronchiolar and alveolar septal connective tissue, and to regional lymph nodes via afferent lymphatic vessels. Rhodococcus equi replicates intracellularly in alveolar and other tissue macrophages. Alveolar macrophages phagocytize Rhodococcus equi through ligand-receptor interactions. The bacterium must initially adhere to macrophages and requires that the bacterium be opsonized with either antibody or complement fragments that occur through complement fixation and the activation of the alternative complement pathway. In nonimmune foals, complement is the primary opsonin. The bacterium also expresses uncharacterized surface molecules that bind to membrane receptors expressed on alveolar macrophages before phagocytosis can occur. Leukocyte complement receptor, Mac-1, other complement receptors, mannose receptors, and potentially TLRs are expressed on alveolar macrophages.
Opsonized bacteria and the products of complement fixation facilitate the process of adhesion and invasion through phagocytosis into alveolar macrophages. Phagocytosis is mediated by bacterial virulence determinates that appear to restrict tropism to specific types of phagocytic cells. After phagocytosis by alveolar macrophages, the bacterium is confined within phagosomes. Experimental results from studies on fusion of phagosomes with lysosomes to form phagolysosomes are contradictory. Some studies suggest that Rhodococcus equi can block the fusion of lysosomes with phagosomes, which allows for the survival, persistence, and replication of bacteria intracellularly. Other studies suggest that Rhodococcus equi is not able to block phagosome-lysosome fusion; however, the bacterium is able to produce molecules that suppress acidification of phagolysosomes, resulting in their survival and replication in alveolar macrophages. The mechanism used to block fusion is unknown but appears to involve bacterium-directed compartmentalization of the fusion process so that they are selectively isolated from lysosomal effector molecules, such as acid, reactive oxygen, NO compounds, and lysosomal hydrolases within phagosomes. Other proteins and molecules appear to contribute to persistence and replication in alveolar macrophages. For example, strains of Rhodococcus equi that cause disease have chromosomal virulence determinates for capsular polysaccharide, cholesterol oxidase, phospholipase C, lecithinase, and cell-wall mycolic acids and plasmid virulence determinates for virulence-associated protein (VAP). It is also likely that mycolic acids from the bacterial cell wall are involved in the pathogenesis of the pyogranulomatous pneumonia characteristic of the disease.
Because Rhodococcus equi is able to disrupt normal phagosome-lysosome killing in alveolar macrophages and the generation of an oxidative burst that could kill the bacterium, it is able to persist and replicate. Studies suggest that rapid replication of bacterium within phagosomes and molecules such as cholesterol oxidase produced by the bacterium contribute to premature death of alveolar macrophages, leading to the release of large numbers of infectious organisms into adjacent tissue. Additionally, because the lifespan of fully differentiated alveolar macrophages is approximately 10 to 30 days, death of these cells related to aging and bacteria-induced injury releases large numbers of bacteria into adjacent tissue where they are phagocytosed by macrophages only to endlessly repeat the process. The severity and extent of the inflammatory response, concurrently with tissue injury, grows through the recruitment of additional monocytes and tissue macrophages from the circulatory system and regional lymph nodes. Neutrophils are active in the acute inflammatory response against Rhodococcus equi. They are able to phagocytose the bacterium, fuse the phagosome with the lysosome to form a phagolysosome, initiate an oxidative burst, and kill the bacterium. However, this process is an ineffective mechanism in controlling the disease and results in extensive tissue destruction through the release of lysosomal enzymes and reactive oxygen species, thus contributing to the cyclic and progressive destruction of pulmonary parenchyma. This damage allows large numbers of bacteria to gain access to the alveoli and bronchioles and encounter mucus of the mucous membranes and the mucociliary apparatus. In general, the mucociliary apparatus is not directly affected by Rhodococcus equi, thus the bacterium is moved up the conductive system to the nasopharynx where it is swallowed and gains access via peristalsis to the alimentary system.
Respiratory Anthrax (Bacillus anthracis): The mechanism of injury in respiratory anthrax is cell death caused by bacterial toxins that act directly on cell membranes leading to acute coagulative necrosis. Gross lesions include pulmonary and lymph node edema, hemorrhage, and necrosis.
Animals encounter Bacillus anthracis through inhalation of fomites contaminated with endospores from soil. Fomites containing endospores must be less than 5 µm in diameter to reach the O2-CO2 exchange portion of the respiratory system. Infected fomites are deposited on the mucosae, where they are then phagocytosed either by alveolar macrophages migrating through and on the surface of the mucosae or by dendritic cells. Infected macrophages and dendritic cells spread the bacterium to regional lymph nodes (bronchiolar and mediastinal) via afferent lymphatic vessels through leukocyte trafficking. During the migration process, endospores germinate into vegetative bacteria, so on arrival at the lymph nodes, bacteria are already producing anthrax toxins, which kill infected cells and release the bacteria into the ECM of the lymph nodes. In lymph nodes, bacteria continue to replicate and produce anthrax toxins killing additional lymphoid and endothelial cells, leading to edema and hemorrhage. The bacterium and its toxins enter lymphatic vessels and spread via the thoracic duct into the circulatory system as a septicemia, where endothelial cells and cells of other organ systems are injured, leading to edema, hemorrhage, and cell necrosis.
Contagious Bovine Pleuropneumonia (Mycoplasma mycoides var. mycoides Small Colony): Little is known about the mechanisms used by Mycoplasma mycoides var. mycoides small colony (SC) to cause disease in the respiratory system of cattle, thus much of this section is speculative and based on a reasonable probability that lesions that occur are the result of underlying and known pathobiologic mechanisms. The mechanism of injury in contagious bovine pleuropneumonia is cell death likely caused by inflammation and its mediators and degradative enzymes and by vasculitis leading to thrombosis, ischemia, and infarction of lung tissue. Gross lesions include (1) fibrinous pleural effusion and fibrinous pleuritis with hemorrhage and (2) fibrinous pleuropneumonia with prominent interlobular septa filled with fibrinous effusion and fibrin thrombi (Fig. 4-22). Infarcts occur in affected lung tissues probably arising from vascular injury leading to permeability changes, vasculitis with activation of clotting cascades, thrombosis, and infarction. Infarcted lung often appears as sequestra likely arising from reparative mechanisms that isolate the dead infarcted tissue via fibrosis. It is unclear how infarcts occur in lung tissue when it has a dual blood supply, unless infarcts occur in areas that lack a dual supply or vasculitis and thrombosis concurrently affect vessels of each arterial source.
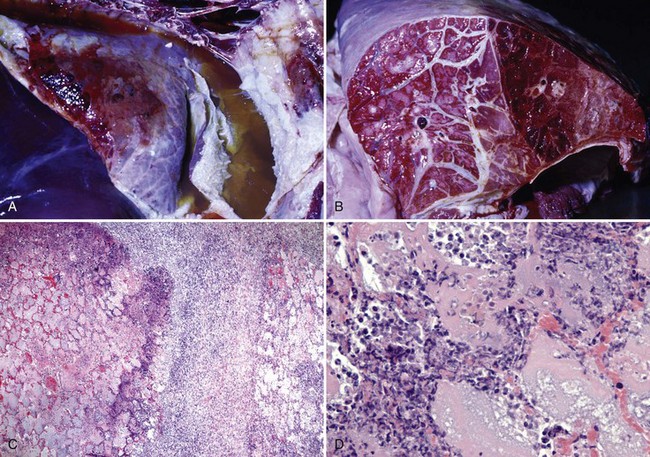
Fig. 4-22 Contagious bovine pleuropneumonia.
A, Thoracic cavity. The thoracic cavity is filled with a fibrinous pleural effusion and the visceral and parietal pleurae are covered by fibrin (fibrinous pleuritis). Also note the areas of hemorrhage affecting the pleurae and the subjacent lung. B, Transverse section of lung. Note the prominent interlobular septa filled with fibrinous effusion and fibrin thrombi and additionally the area of hemorrhage (right half of the section). Infarcts with pulmonary sequestra (not shown here) can occur in affected lung tissues likely arising from vascular injury leading to infarction. C, The interlobular septum (center) is filled with acute inflammatory cells mixed with a fibrinous effusion. Alveoli are filled with highly proteinaceous edema fluid, fibrinous effusion, and acute inflammatory cells. There is extensive necrosis of all tissues at the interface between alveoli and the interlobar septum (dark blue staining band). H&E stain. D, Higher magnification of C. The dark blue color is attributable to necrosis of cells, including neutrophils leading to escape and coagulation of nucleic acids from degenerate nuclei in the inflammatory exudate. Alveoli are filled with edema fluid and acute inflammatory cells. H&E stain. (A and B courtesy Dr. D. Gregg, Plum Island Animal Disease Center and Noah’s Arkive, College of Veterinary Medicine, The University of Georgia. C and D courtesy Dr. J. F. Zachary, College of Veterinary Medicine, University of Illinois.)
Cattle (and probably sheep and goats) encounter Mycoplasma mycoides var. mycoides SC through inhalation of fomites and fluid droplets. These droplets are deposited on mucosae of the conductive component of the respiratory system by centrifugal and inertial turbulence where they are trapped in the mucus layer and are subsequently phagocytosed by alveolar macrophages. Ligand-receptor interactions are probably involved in target cell specificity. Alveolar macrophages in all probability spread the bacterium to local lymphoid tissue such as BALT, in which the bacterium replicates and kills infected macrophages releasing bacterium into bronchiolar and alveolar interstitium, causing severe acute inflammation and the characteristic fibrinous lesions and vasculitis. How the bacterium evades killing by phagosome-lysosome fusion, produces toxic molecules that injure and kill cells, spreads to blood vessels, and causes vasculitis and thrombosis is unknown. Bacterial membrane lipoprotein LppQ, a common antigen of Mycoplasma mycoides ssp. mycoides, could be involved in some of these processes. Highly virulent strains of the bacterium are known to produce and release large quantities of H2O2 that are cytotoxic for all cells. H2O2 release appears to be correlated with adhesion of the bacterium to host cell membranes. It also appears that macrophages may spread the bacterium via leukocyte trafficking systemically to lymph nodes and synovium and joint space of joints such as the carpus where inflammation characteristic of the disease also occurs.
Bovine Tuberculosis (Mycobacterium bovis): The mechanism of injury in bovine tuberculosis is death of cells of the monocyte-macrophage system and of all cell populations in the lung and associated regional lymph nodes secondary to granulomatous inflammation and its mediators and degradative enzymes. Gross lesions include (1) enlarged lymph nodes that contain discrete and coalescing granulomas (tubercles) formed by dry and gritty (mineralized) yellow-white to green-white caseous exudate often encapsulated by fibrous connective tissue (see Fig. 1-19) and (2) lung parenchyma that contains similar granulomas distributed at random in some or all lung lobes (see Figs. 9-65 and 9-68).
Cattle (and probably sheep and goats) encounter Mycobacterium bovis through inhalation of fomites and fluid droplets contaminated with the bacterium. These droplets are deposited on mucosae of the conductive component of the respiratory system by centrifugal and inertial turbulence where they are trapped in the mucus layer and are subsequently phagocytosed by alveolar and tissue macrophages. Macrophages appear to use several routes to spread the bacterium across mucosal barriers and then to regional lymph nodes and the lung. Macrophages in the pharynx cross the mucosal barrier, migrate to the tonsil, and spread the bacterium to infect naïve macrophages in tonsillar tissues. Macrophages in other regions of mucosae encounter and phagocytose bacteria and spread them via lymphatic vessels to local lymphoid tissues and then via lymphatic vessels to regional lymph nodes such as the retropharyngeal and parotid nodes to infect naïve macrophages in these nodes.
Bacteria that are deposited in the mucus layer of mucosae of bronchi and bronchioles are phagocytosed by alveolar macrophages and spread via lymphatic vessels to local lymphoid tissues (BALT) and then via lymphatic vessels to regional lymph nodes, such as the tracheobronchial and mediastinal nodes, to infect naïve macrophages in these nodes. The primary goal of Mycobacterium bovis is to be phagocytosed by macrophages. In the mucus layer, macrophages encounter trapped bacteria via random movement. Once bacteria are in contact with macrophages, bacteria adhere and bind to pathogen PRRs on macrophage cell membranes. The process used by macrophages to phagocytose Mycobacterium bovis involves ligand-receptor interactions. In fact, the bacterium appears to utilize multiple membrane pathogen PRRs such as those for complement (CR1, 3, and 4), mannose, surfactant protein, and CD14 protein to enter macrophages. Some receptors are probably used in the early mucosal phases of infection when inflammation is minimal, whereas other receptors, such as those for complement, are used when vascular changes in lymph nodes and lung induced by acute inflammation result in permeability changes and the release of plasma proteins and complement into inflamed tissues.
Mycobacterium bovis is able to activate the alternative pathway of complement and use C3b and C3bi fragments to opsonize its surface and then bind to complement receptors CR1, 3, and/or 4 on cell membranes of macrophages. This binding results in phagocytosis of the bacterium in phagosomes. Mannose receptors, surfactant proteins and receptors, and lipoarabinomannan (LAM) and CD14 receptors are also involved in phagocytosis. It appears that the use of multiple ligands and pathogen PRRs ensures that the bacterium once inhaled or ingested can be phagocytosed by monocytes, macrophages, and/or even neutrophils that migrate to the site of local infection in response to chemokines secreted by infected macrophages. Subsequently, these cells can then be used to spread the infection to other regional or systemic sites, such as the liver, spleen, lymph nodes, and intestines, via leukocyte trafficking in the blood or lymphatic vascular systems.
Once present in a phagosome, Mycobacterium bovis is able to disrupt phagosome-lysosome fusion and prevent activation of macrophage antimicrobial mechanisms such as production of reactive oxygen or nitrogen intermediates and phagosome acidification. The bacterium is able to grow and replicate in the phagosome, but with cellular aging, infected macrophages die and release bacteria into the vascularized ECM tissues. This outcome results in repetitive cycles of inflammation and the recruitment of additional monocytes, macrophages, and neutrophils into the granuloma (tubercle). The formation of granulomas (tubercles) is covered in detail in Chapter 3, but components of the poorly digestible, waxy cell wall of the bacterium, such as sulpholipids and LAM, appear to contribute to the type of inflammatory response that ensues and the formation of granulomas.