30 Toxicological emergencies
Suspected or witnessed poisoning is a common reason for presentation of small animals to emergency clinics, with dogs being much more commonly affected than cats. This chapter presents a general approach to the poisoned patient before going on to describe some of the more commonly implicated poisons in greater detail using a number of case examples.
Approach to the Poisoned Patient
Telephone communication
Initial telephone communication is perhaps no more important than in the intoxicated patient. The important questions to ask are summarized in Box 30.1.
BOX 30.1 Important questions to ask owners ringing with respect to intoxication
Nursing Aspect
Nurses are frequently the first members of staff with whom owners ringing about suspected or witnessed poisoning will communicate. It is therefore imperative that all nurses are well rehearsed in the questions that are important to ask (Box 30.1) and advice should be sought from the veterinary surgeon if there is any concern.
On the basis of the information obtained, a recommendation should be made as to whether the animal needs to be presented to the practice or may be managed conservatively at home. In some cases, the animal will be exhibiting marked clinical signs and questioning should be kept to a minimum with immediate transport to the practice being the only appropriate recommendation. In other cases, it is necessary to obtain further information before a recommendation can be made.
Further information
The purpose of seeking additional information about the poison in question is to ascertain if possible the severity of exposure that has or may have occurred (see Box 30.2).
A number of sources of information are available with respect to veterinary toxicology that includes:
Some information that may be useful when calculating exposure dosages is presented in Box 30.3.
Home management
If the decision is made for the animal to be monitored at home, the owner must be thoroughly briefed both on what signs to observe the animal for and the typical time frame for their onset and progression. In general, if there is any doubt as to the animal’s condition, veterinary examination should be recommended. Owners should be advised on appropriate measures to implement during transportation (e.g. keeping unconscious animals warm or keeping seizuring animals cool and protected from injury).
Inducing emesis at home
In some cases in which poison ingestion has occurred within a suitable period of time, it may be appropriate for the owner to induce emesis at home, for example if financial concerns or practical constraints preclude presentation to the practice. In addition, if a considerable delay is anticipated prior to presentation, and the owner has ready access to an appropriate emetic, inducing emesis prior to departure from home may be advisable to minimize further absorption of the poison in transit. The owner must be questioned carefully to ensure that contraindications to inducing emesis do not exist; these are summarized in Box 30.4.
BOX 30.4 Contraindications to induction of emesis
Agents that may be used to induce emesis at home are shown in Table 30.1.
Table 30.1 Agents for inducing emesis at home
| Agent | Dose | Comments |
|---|---|---|
| Soda crystals (washing soda) | 1 crystal | |
| Syrup of ipecacuanha (7%) | D: 1–2 ml/kg p.o. C: 3.3 ml/kg p.o. |
|
| Hydrogen peroxide (3%) | D, C: 1–3 ml/kg | |
| Table salt (sodium chloride) | Not recommended |
C, cats; D, dogs.
Clinical Tip
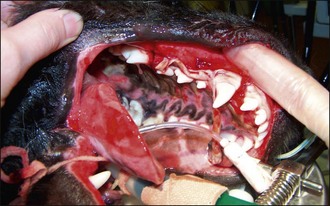
General Clinical Approach
See Box 30.5.
History
Clinical Tip
All the information in Boxes 30.1 and 30.2 should be obtained at the appropriate time. In some emergency patients, clinical signs and progression are compatible with possible intoxication without an immediately suggestive history. In such cases, the owner must be carefully and thoroughly questioned to establish whether a potential source of poison exists that the owner has not considered.
Initial management of ingested poisons
As for all emergency patients, a major body system (cardiovascular, respiratory, neurological) examination (see Ch. 1), including measurement of rectal temperature, should be performed and immediate measures taken to correct potentially life-threatening problems. Intravenous fluid therapy (see Ch. 4) may be required to correct hypovolaemia and/or dehydration. Fluid therapy is also indicated in the management of poisons that are nephrotoxic (e.g. nonsteroidal antiinflammatory agents) and those that are largely dependent on renal excretion. Oxygen supplementation is indicated in patients with respiratory compromise, for example from aspiration following vomiting, and in the context of certain poisons such as carbon monoxide.
Emergency database
An emergency database (see Ch. 3) for the intoxicated patient should consist of the following if possible:
Additional tests that may be indicated at the appropriate time in certain cases include acid–base analysis, urinalysis (e.g. oxalate crystals in ethylene glycol poisoning) and calcium measurement (e.g. hypocalcaemia may occur in ethylene glycol poisoning). Abdominal radiographs taken at the appropriate time may reveal ingestion of items containing heavy metals or certain enteric-coated or sustained-release drug formulations. Routine haematology and biochemistry profiles may become indicated as case management progresses and may also identify pre-existing conditions that may have implications with respect to management of individual patients.
Treatment of seizures and muscle tremors
A variety of poisons include neurotoxicity amongst their mechanisms of action – examples include metaldehyde, pyrethroids, strychnine, caffeine, theobromine, organophosphates and tremorogenic mycotoxins. Seizures and/or muscle tremors are common signs of poisoning requiring symptomatic treatment (see Ch. 24).
In patients intoxicated with tremorogenic poisons (e.g. metaldehyde, permethrin), it can be difficult to differentiate severe muscle tremors from seizure activity. However, if the poison in question is known to be one associated with severe tremors or sufficient clinical suspicion exists, the use of methocarbamol may be indicated and may avoid the need for anaesthesia. This is a centrally acting muscle relaxant related to guaiphenesin whose precise mechanism of action remains unclear. The manufacturer’s recommended dose in dogs and cats is 44–220 mg/kg i.v. with a typical upper limit of 330 mg/kg in a 24 hour period. However, to the author’s knowledge, an injectable preparation of this agent is not currently available in the United Kingdom. In the absence of methocarbamol, the management of patients with tremors is the same as for those with seizures.
Hyperthermia is a potentially serious development in patients suffering from seizures or muscle tremors. Hypothermia may also occur following bathing or sedation. Close monitoring of rectal temperature for either development and appropriate intervention (see Chs 16 and 17) is therefore required.
Gastrointestinal decontamination (GID)
Toxin exposure is usually via ingestion and gastrointestinal decontamination (GID) is frequently indicated. This consists of emptying of the stomach followed by administration of an adsorbent to minimize absorption of any poison remaining in the gastrointestinal tract.
Gastric emptying
In the absence of contraindications (see Box 30.4), induction of emesis is the most expeditious means to empty the stomach; feeding a small meal first may increase the effectiveness of this approach. The author has performed gastric emptying up to 4–6 hours following ingestion with some good results. Drugs used to induce emesis are summarized in Table 30.2.
Table 30.2 Drugs used to induce emesis
| Drug | Dose | Comments |
|---|---|---|
| Apomorphine | D: 0.04–0.1 mg/kg s.c., i.m., ocular | |
| C: 0.01–0.02 mg/kg s.c., i.m. | ||
| Medetomidine | C: 20 µg/kg i.m., i.v. | |
| Xylazine | C: 0.4–0.5 mg/kg i.m., s.c. |
C, cats; CNS, central nervous system; D, dogs; i.m., intramuscular; i.v., intravenous; s.c., subcutaneous.
Activated charcoal
Activated charcoal acts as an adsorbent binding to toxins and allowing their passage through the gastrointestinal tract while preventing or minimizing further systemic absorption. It is typically administered once gastric emptying has been performed and should be given as soon as possible. If an emetic has been employed for gastric emptying, enough time must be allowed for the emetic effects to subside before activated charcoal is administered. In some cases activated charcoal is administered initially via stomach tube at the end of gastric lavage. The recommended dose in dogs and cats is 1–5 g/kg to be repeated as necessary (typically every 4–6 hours) until black faeces are detected. Doses up to 8 g/kg have been used. Activated charcoal is often successfully administered in food to dogs. However, compliance is likely to be much poorer in cats. Various proprietary preparations are available with accompanying dosing guidelines, including powdered formulations (e.g. BCK Granules®, Fort Dodge) that can be added to food or made into a slurry and administered by mouth, and suspensions (e.g. Charcodote®, Pliva Pharma Ltd).
Different substances are bound to different degrees by activated charcoal. However, unless contraindicated, the use of activated charcoal in almost all cases of oral poisoning is probably reasonable and may also help following topical poisoning (see below). Activated charcoal should not be used following ingestion of caustic or corrosive substances, in patients that are vomiting or seizuring, or where there is any possibility of gastrointestinal perforation. Vomiting and constipation following administration of multiple doses are the main complications reported.
Gastric and colorectal lavage
In patients in which induction of emesis is contraindicated or unsuccessful, gastric lavage may be appropriate for gastric emptying (see p. 296). However, it is contraindicated following ingestion of caustic or corrosive substances and where the risks of general anaesthesia are considered unacceptable.
In some cases (e.g. metaldehyde poisoning), following gastric lavage, the author will perform additional lavage via a stomach tube inserted as proximally as possible per rectum. Colorectal lavage is continued until clear fluid is returned and activated charcoal suspension is then instilled. The anus may be plugged with a swab for example for a short period of time to minimize leakage of the activated charcoal.
Topical poisoning
Washing the patient is recommended to minimize irritation of and absorption via the skin; this should also minimize absorption through ingestion following grooming. Washing is usually done using mild soap or detergent, followed by copious rinsing with water and then drying the animal as thoroughly as possible. Powdered toxins may be vacuumed off before washing. All individuals involved in handling the animal should take care to wear gloves and preferably an apron so as to avoid self-contamination. In some cases it may be appropriate for the owner to wash the animal at home. However, in compromised or noncompliant animals, veterinary care is recommended.
Clipping the coat of long-haired patients may help to maximize decontamination. Chemical restraint may be preferable during washing to allow protection of the eyes and in some cases general anaesthesia with endotracheal intubation is safest to minimize the risk of aspiration. Vital parameters including rectal temperature should be monitored closely throughout.
Oily substances may be more successfully removed using commercial hand-cleaning degreaser formulations (e.g. Swarfega Hand Cleaner® products) but it is important to ensure that such preparations are thoroughly washed off the animal subsequently.
The use of activated charcoal is generally recommended following topical poisoning. This is to minimize gastrointestinal absorption that may occur following ingestion from grooming. In addition, some poisons undergo enterohepatic circulation following absorption from the skin and thereby become available in the gastrointestinal tract.
In cases in which the skin has come into contact with an acidic or caustic substance, the affected area should be lavaged very thoroughly (’the solution to pollution is dilution’) using normal saline or indeed warm water. The same is true in cases of ocular contamination and in both cases the animal should be given appropriate analgesia and chemically restrained to allow comprehensive lavage to be performed. Damaged skin is highly susceptible to mechanical injury and gentle lavage is therefore mandatory.
Diuresis
Diuresis is most indicated in the treatment of poisoning by agents for which renal excretion of either the primary intoxicant or its metabolites is a significant feature (e.g. phenobarbital, salicylates). Standard intravenous isotonic crystalloid therapy is employed to promote renal excretion with or without additional diuretic administration. Isotonic crystalloid therapy is administered at a rate of 1–4 ml/kg/hr above calculated fluid requirements and the patient must be monitored closely to ensure that adequate urine production occurs. In cases of aggressive diuresis, close monitoring of hydration status and electrolyte concentrations is indicated.
Antidotes
In a significant proportion of canine and feline patients a diagnosis of poisoning is made presumptively with the poison in question remaining unknown. Furthermore, specific antidotes or antagonists do not exist for the majority of potentially poisonous substances to which dogs and cats may be exposed. That said, the majority of clinical cases reported are due to a relatively small number of poisons for which, in some cases, specific treatments are available. These will be alluded to in the discussion of specific poisons that follows.
Specific Poisons
Pyrethrins and pyrethroids
Theory refresher
Pyrethrins are naturally occurring insecticidal esters of chrysanthemic acid and pyrethric acid extracted from the Chrysanthemum cinerariaefolium plant; pyrethroids (e.g. permethrin) are synthetic pyrethrins. Many topical and household insecticidal preparations containing these compounds are marketed for the control of flea and lice infestations amongst others in dogs and cats. These preparations are widely available from a variety of outlets.
Clinical Tip
Toxic dose
The toxic dose for permethrin in cats is unknown. Dermal application of 100 mg/kg permethrin may prove life-threatening if untreated.
Mechanism of toxicity
Pyrethrins and pyrethroids alter the kinetics of neuronal sodium channels causing repeated nerve firing. In addition, some of these compounds may also inhibit gamma aminobutyric acid (GABA) receptors, resulting in hyperexcitability of nervous tissue. Glucuronidation is one pathway involved in the metabolism of some of these compounds and glucuronyl transferase deficiency may be part of the explanation for the apparent sensitivity of cats to pyrethroids.
Case example 1
Presenting Signs and Case History
A 1-year-old female neutered domestic short hair cat (2.5 kg) presented 3 hours after topical application of a spot-on preparation containing 744 mg permethrin per millilitre. The preparation used was registered for use in dogs only but the owner had thought it would be safe to administer half of the pipette to the cat. The cat had developed clinical signs within 1 hour of administration and at the time of presentation was showing severe muscle tremors.
Clinical Tip
Case management
Diazepam (5 mg) was administered per rectum to facilitate intravenous catheter placement and blood was obtained via the catheter for an emergency database to be performed; this was found to be unremarkable. Major body system examination was unremarkable except for the muscle tremors that were less severe but still marked.
Intravenous fluid therapy was commenced and anaesthesia was induced using propofol and maintained using isoflurane. The cat was washed thoroughly in a mild detergent and dried before being recovered with an Elizabethan collar in place. Muscle tremors were apparent in recovery but of lesser severity and a single intravenous bolus of midazolam (0.2 mg/kg) was administered.
Tremors resolved fully over the next 24 hours, during which time vital parameters, in particular temperature, were monitored closely and appropriate supportive and nursing measures instituted.
Metaldehyde
Theory refresher
Metaldehyde is a cyclic tetramer of acetaldehyde that is commonly used as a pesticide against slugs and snails (molluscicide). Commercial pellet preparations usually contain 1.5–8% metaldehyde w/w (see Box 30.3) in a cereal base. The pellets are often blue or green in colour and the cereals and other additives make slug/snail baits palatable to dogs. Cats as usual are more discerning and metaldehyde poisoning has only been reported in a few cases. Liquid preparations containing higher concentrations of metaldehyde are also available, as are granular and powdered preparations. Metaldehyde baits sometimes contain additional herbicides and pesticides, most commonly carbamate insecticides.
Toxic dose
A fatal oral dosage of metaldehyde in dogs of 60 mg/kg body weight has been reported although some authors quote much higher values. Clinical signs may be expected to occur at a range of typically much lower dosages. A lethal dose of 207 mg/kg has been reported in cats.
Toxicokinetics
Metaldehyde may be absorbed intact from the gastrointestinal tract but its subsequent distribution, metabolism and excretion remain to be fully clarified. Acid hydrolysis occurs in the stomach producing acetaldehyde which is then absorbed rapidly and converted to carbon dioxide (probably eliminated via the lungs) or excreted in urine. The half-life of metaldehyde in people is estimated at approximately 27 hours but is unknown in dogs.
Mechanism of toxicity
The precise mechanism of toxicity of metaldehyde remains to be elucidated but it is known to cross the blood–brain barrier readily and impairment of GABA activity is currently thought to be most implicated. As GABA is an inhibitory neurotransmitter, a decrease in its activity may be responsible for the increased neurological activity seen in metaldehyde poisoning. A decrease in the concentration of other neurotransmitters in the central nervous system (CNS), such as serotonin (5-HT) and noradrenaline (norepinephrine), may also be involved.
Case example 2
Presenting Signs and Case History
A previously healthy 2-year-old male neutered black Labrador retriever (30 kg) presented recumbent with marked twitching, muscle tremors, mydriasis and hyperthermia. The dog had been out unattended in the garden earlier that afternoon and would have had access to the shed in which the owner kept slug pellets. One hour after returning inside the dog was found to be twitching and salivating and the owner had rung the hospital for advice.
Given the dog’s signalment and history, there was a high index of suspicion for poisoning and the owner was advised to come straight down to the hospital (a 15-minute car journey). The owner was advised not to travel alone and to ensure that the dog was kept cool and protected from injury during the journey. Meanwhile another member of the family went to investigate the shed where indeed it was found that the slug bait container had been disrupted. Details from the container were communicated directly to the practice while the dog was en route. Preparations had been made in the interim to manage the dog for a suspected diagnosis of metaldehyde poisoning.
Clinical Tip
Case management
After checking the dog’s temperature, diazepam (1 mg/kg) was administered per rectum and an intravenous catheter subsequently placed. A further intravenous bolus of diazepam (0.5 mg/kg) was administered and a major body system examination performed. Anaesthesia was then induced using propofol and maintained using isoflurane following endotracheal intubation. Thorough gastric lavage was performed and revealed blue-green coloured material. Activated charcoal was then administered into the stomach. Colorectal lavage was also done and an activated charcoal enema administered.
Clinical Tip
The dog was maintained on a propofol infusion for the next 12 hours, during which phenobarbital loading was performed (3 mg/kg i.v. q 1 hr for six doses); thereafter the dog was gradually weaned off propofol (over 4 hours). External stimulation was kept to a minimum (e.g. cotton wool balls/swabs in the ears, dim lighting, minimal noise and passage of personnel). The dog remained markedly depressed for the following 24 hours but gradually became ambulatory and although ataxic initially went on to make a full recovery over the following 3 days. Fluid therapy and appropriate nursing measures were instituted throughout. Phenobarbital (3 mg/kg i.v. then p.o. q 12 hr) therapy was discontinued at discharge.
The slug bait container contained 1 kg of blue pellets with a metaldehyde composition of 4% w/w. Assuming the dog had ingested all 1 kg of bait, this was equal to 40 mg (i.e. 4% × 1000 mg) of metaldehyde and a maximum dosage of only 1.3 mg/kg.
Ethylene glycol
Although ethylene glycol (EG) is a common cause of poisoning in companion animals in North America, its incidence is significantly lower in the United Kingdom. EG is a sweet-tasting liquid used primarily as an antifreeze, screen wash and windshield de-icing agent. Antifreeze ingestion is the most common source of EG poisoning, with many common preparations containing as much as 95% EG. Companion animal exposure is generally the result of environmental contamination from improper disposal or insecure storage, and poisoning is most likely to occur in late autumn and early spring when antifreeze usage increases.
Toxic dose
The minimum lethal dose of undiluted EG is reported to be 4.4–6.6 ml/kg in dogs and 1.5 ml/kg in cats.
Toxicokinetics
EG is rapidly absorbed from the gastrointestinal tract and distributed systemically. Its plasma half-life is approximately 3 hours and a variable proportion is excreted unchanged in urine. The remainder is metabolized predominantly in the liver. The first step in this metabolism is oxidation of EG to glycoaldehyde by alcohol dehydrogenase (ADH), a conversion that can be saturated. Glycoaldehdye is then converted to glycolic acid, glyoxylic acid and finally to oxalic acid. EG metabolic pathways may vary between species.
Calcium binds to oxalic acid, resulting in the formation of calcium oxalate crystals that are deposited widely but especially in the renal tubules, and calcium oxalate crystalluria is a common finding.
Mechanism of toxicity
Clinical Tip
Metabolism of EG generates free oxygen radicals that are potentially cytotoxic to a variety of tissues. In addition, the organic acid metabolites produced interfere with normal cellular processes. Central nervous system (CNS) dysfunction is thought to be predominantly due to the effects of glycoaldehyde along with calcium oxalate deposition in nervous tissue. Both hypocalcaemia secondary to binding of calcium to oxalic acid and metabolic acidosis may contribute further to CNS signs. Metabolic acidosis may be severe and is due to the accumulation of acid metabolites, most notably glycolic acid.
Clinical signs
Clinical signs of EG poisoning are dose-dependent and in companion animals occur in two phases. The first is predominantly associated with EG itself prior to metabolism. Signs develop within an hour of ingestion and may persist for 12 hours. They include CNS depression, somnolence, ataxia, impairment of conscious proprioception, nausea, vomiting, and osmotic diuresis with consequent polyuria/polydipsia. In severe cases seizures, coma and death may occur. In dogs these signs may seemingly resolve with apparent recovery although they may occur earlier and be more persistent in cats.
The second phase of clinical signs is associated with the highly toxic metabolites of EG and is predominantly related to acute renal failure. Signs usually develop within 24–72 hours of ingestion in dogs, often earlier in cats, and may include reduced mentation from depression through to coma, seizures, anorexia, vomiting, and oliguria with low urine specific gravity or isosthenuria. Anuria may develop 3–4 days post-ingestion.
An intermediate phase between the two phases already described consists of cardiopulmonary manifestations (tachycardia, tachypnoea, pulmonary oedema) but this is recognized much less commonly in dogs and cats than in people.
Laboratory tests
Biochemistry, acid–base analysis
Early changes are mainly due to accumulation of acid EG metabolites with normochloraemic metabolic acidosis, reduced plasma bicarbonate (HCO3−) and increased anion gap. If plasma HCO3− measurement is not available, reduced concentration may be reflected as a decrease in total carbon dioxide (total CO2). These changes may be detectable 1–3 hours following ingestion and a high anion gap may persist for 12–48 hours. Total hypocalcaemia may occur due to chelation by oxalic acid, hyperglycaemia is also reported, and prerenal azotaemia may be detected with appropriately severe dehydration.
Later biochemical abnormalities reflect renal injury and reduced glomerular filtration rate with renal azotaemia and hyperphosphataemia. These changes are usually detectable 24–72 hours post-ingestion in dogs and earlier in cats.
Urinalysis
Osmotic diuresis and polydipsia (due to increased serum osmolality) result in reduced urine specific gravity, typically within 3 hours of EG ingestion. Dogs are usually isosthenuric (urine specific gravity 1.007–1.015) but urine specific gravity may be higher than this and often is in cats. Urine specific gravity remains low as renal insufficiency and impaired ability to concentrate urine develop in the later stages. Glucosuria with concurrent normoglycaemia may be detected in dogs as a result of proximal renal tubular damage.
Calcium oxalate crystals may be identified in urine 3–6 hours after EG ingestion. Calcium oxalate monohydrate crystals (clear six-sided prisms or dumbbell-shaped (Figure 30.2) crystals) are more common than the dihydrate form (envelopes or Maltese cross-shaped). Aciduria, haematuria, renal epithelial cells and casts are other common urinalysis findings.
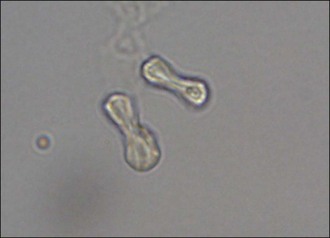
Serum ethylene glycol concentration
Serum EG concentrations peak 1–6 hours post-ingestion and EG is usually no longer detectable in serum (or urine) by 48–72 hours post-ingestion. A commercial test kit (Ethylene Glycol Test Kit®, PRN Pharmacal Inc., Florida; http://www.prnpharmacal.com/egtkit/index.php) is available for estimating serum EG concentrations based on an enzymatic assay; it is both accurate enough for clinical use and relatively inexpensive. Results are available within 30 minutes. These kits have a minimum detectable level of 50 mg/dl of EG in the blood and it is noteworthy that cats may develop clinically significant poisoning despite serum EG levels lower than this threshold.
Clinical Tip
Serum osmolality
EG is an osmotically active substance and ingestion results in an increase in serum osmolality and osmole gap (equivalent to the measured serum osmolality minus the calculated osmolality). Measurement of serum osmolality is useful for identifying early EG poisoning. However, access to measured serum osmolality is likely to be very limited in general emergency practice.
Wood’s lamp
A number of modern antifreeze preparations contain sodium fluorescein to aid in detection of radiator leaks. This dye is excreted in urine following ingestion; excretion usually only occurs for up to 6 hours post-ingestion (but it can be longer). Urine as well as the mouth, paws and vomitus can therefore be examined with a Wood’s lamp for fluorescence due to the presence of this dye. However, it is noteworthy that a negative result does not exclude the possibility of EG ingestion as not all antifreeze preparations contain sodium fluorescein.
Treatment
Treatment of suspected EG poisoning should be instituted as early as possible because it is metabolized rapidly into highly toxic metabolites. Treatment consists of the following components:
Gastrointestinal decontamination
EG is absorbed very rapidly from the gastrointestinal tract and GID may therefore only be of benefit very early post-ingestion (1–2 hr). Activated charcoal decreases ethanol absorption and should not therefore be administered if oral ethanol is being used as an antidote.
Preventing ethylene glycol metabolism
The prevention of EG oxidation by ADH is the most important component of therapy and may be achieved in one of two ways: inhibition of the enzyme or provision of a competitive substrate. Treatment should be administered in all animals presenting within 36 hours of EG ingestion.
Fomepizole
ADH may be inhibited by the administration of fomepizole (4-methylpyrazole, 4-MP). This agent is a synthetic competitive ADH inhibitor and has become the treatment of choice in EG poisoning (Box 30.6). Minimal side effects have been reported following the use of this agent in companion animals and in particular it lacks the CNS depressive effects of ethanol (see below).
Ethanol
Ethanol may be used in the treatment of EG poisoning as it has a higher affinity for ADH than EG and therefore acts as a preferential competitive substrate (Box 30.7). Injectable ethanol should be diluted in saline prior to administration. If a pure preparation for injection is not available, ethanol may be administered orally using an alcoholic beverage.
Perhaps the biggest disadvantage of the therapeutic use of ethanol is the associated CNS depression that may necessitate intensive and supportive care, especially with intermittent bolus administration and in cats. If oral ethanol is being used, direct administration via orogastric intubation may prove necessary in animals that are too sedated to swallow reliably.
Other treatment considerations
Appropriate supportive therapy consists of intravenous fluid therapy to correct hypovolaemia and dehydration and to promote diuresis. Potassium and calcium supplementation should be provided as deemed necessary on the basis of regular monitoring. Patients presenting with oliguric renal failure should be treated with intravenous fluid therapy and diuretic agents to establish diuresis if possible (see Ch. 36). If adequate diuresis cannot be established, then referral for haemodialysis or peritoneal dialysis should be considered if available and affordable.
Renal tubular damage caused by EG may be reversible but can take weeks to months and urine concentrating ability may never return in some cases. Most dogs and cats surviving the acute renal failure phase of EG poisoning will eventually regain normal renal function.
Nursing Aspect
Patients suffering from EG poisoning may be severely depressed and recumbent, both from the poisoning and from treatment if ethanol is used. Standard nursing measures for recumbent patients should be implemented, including provision of clean, dry, well-padded bedding, regular turning and bladder management. Regular eye lubrication and oral care may be needed and patients must be monitored closely for hypothermia.
Prognosis
Prognosis depends on the dosage of EG ingested, rate of absorption and, most importantly, the time to institution of specific therapy. The prognosis for dogs treated with fomepizole within 5 hours of ingestion is good, and most will recover if treatment is administered within 8 hours of ingestion. The prognosis is reasonable for cats receiving therapy within 3 hours of ingestion.
However, a grave prognosis for survival is heralded by the onset of oliguric renal failure in both species and unfortunately most animals present at this late stage.
Grapes/raisins
Theory refresher
Grape/raisin poisoning in dogs has been recognized since the late 1990s. No confirmed cases have been reported in cats at the time of writing but susceptibility is suspected. A similar syndrome has not been reported in people.
Toxic dose, toxicokinetics, mechanism of toxicity
Grape/raisin ingestion may be associated with renal toxicity but the toxin or toxins involved have yet to be identified and the mechanism of toxicity is unknown. There does not appear to be a correlation between the quantity of grapes/raisins consumed and the subsequent renal pathology and clinical progression; an idiosyncratic reaction is suspected.
Case example 3
Presenting Signs and Case History
A previously healthy 3-year-old male entire Labrador retriever (30 kg) presented with a several hour history of acute onset severe vomiting. After extensive questioning the owners revealed that the dog had eaten 350 g of raisins the day before.
Clinical Tip
Major body system examination
Physical examination revealed the dog to be very depressed and moderately dehydrated but was otherwise unremarkable.
Emergency database
An intravenous catheter was placed in a cephalic vein and blood obtained via the catheter for an emergency database. This revealed severe azotaemia (blood urea nitrogen 45 mmol/l, reference range 3–9.10 mmol/l; creatinine 1250 µmol/l, reference range 98–163 µmol/l) and moderate hyperkalaemia (6.0 mmol/l, reference range 4.1–5.3 mmol/l). Dehydration was confirmed (manual packed cell volume 55%, reference range 37–55%; plasma total solids 75 g/l, reference range 49–71 g/l). The severity of azotaemia was consistent with a primary renal or postrenal cause but some prerenal contribution due to dehydration was also likely.
Case management
A soft indwelling Foley urethral catheter was placed with ease, thereby excluding urethral obstruction, and an emergency abdominal ultrasound revealed no free peritoneal fluid, thus excluding uroperitoneum; both kidneys had grossly normal architecture. Urinalysis on a sample collected via the urethral catheter revealed a specific gravity of 1.020 and mild proteinuria, glucosuria and haematuria. The azotaemia was diagnosed as renal in origin (with the caveat that the ureters had not been imaged to exclude completely a postrenal cause or component). A presumptive diagnosis of acute renal failure due to raisin poisoning was therefore made.
The dog was not felt to be hypovolaemic but moderate dehydration (7%) was suspected, and he was started on 0.9% sodium chloride (normal, physiological saline) at 10 ml/kg/hr to provide rehydration and maintenance requirements over approximately 8 hours. The bladder was emptied via the urethral catheter at the outset. After 10 hours of fluid therapy, urine output was calculated and the dog was found to have produced only 0.5 ml/kg/hr of urine. A diagnosis of oliguric acute renal failure was therefore made. The decision was made at this point to refer the dog.
Despite aggressive diuretic therapy at the referral centre, using furosemide and mannitol, oliguria persisted and the dog became overhydrated, showing chemosis, diffuse subcutaneous oedema and mild peritoneal and pleural effusion. Peritoneal dialysis was considered appropriate in this case (young previously healthy dog with potentially curable disease) and was performed successfully. Four months following discharge from the referral hospital mild persistent azotaemia was present (blood urea nitrogen 13 mmol/l, creatinine 190 µmol/l) but the dog was clinically well. Appropriate dietary management and on-going monitoring were recommended.
Clinical Tip
Lilies
Domestic cats are the only animals thus far reported to be susceptible to lily nephrotoxicity. Earliest reports involved Easter lily (Lilium longiflorum) but it is now suspected that all species of the Lilium genera, including Tiger lily, as well as day lilies (Hemerocallis genera) may be potentially nephrotoxic to cats. All parts of the plants, including the flowers, are associated with poisoning and exposure is usually via access to household plants.
Toxic dose, toxicokinetics, mechanism of toxicity
Even very small amounts of plant ingest may be poisonous to cats and rapid absorption from the gastrointestinal tract is suspected as some cats still develop renal insufficiency despite early gastrointestinal decontamination. The toxin or toxins involved have yet to be identified and the metabolism is unknown. The precise mechanism of toxicity is unknown but renal tubular epithelial necrosis and subsequent acute renal failure are known to occur.
Pancreatitis and pancreatic degeneration have also been reported in cats with lily poisoning, as have seizures of unconfirmed pathogenesis.
Clinical signs
Clinical signs may develop in as little as 5–10 minutes following ingestion. Within 1–3 hours vomiting, salivation, depression, lethargy and anorexia may be apparent. Polyuria and consequent dehydration occur 12–30 hours following ingestion and this is followed by anuria.
Anuria typically occurs 24–48 hr following ingestion and death may occur within 7 days of exposure. Renomegaly and abdominal pain may be detected in some cats.
Laboratory tests
Depending on the time elapsed since ingestion, clinicopathological findings are likely to reflect acute renal failure with azotaemia, hyperphosphataemia and possible hyperkalaemia. These abnormalities are usually evident within 24–72 hours of ingestion. Urinalysis may reveal isosthenuria, tubular casts (Figure 30.3), proteinuria and glucosuria.
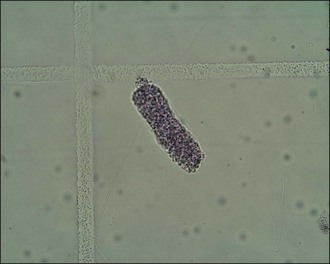
Treatment
Depending on the time elapsed since ingestion, routine GID may be indicated to minimize absorption. Fluid diuresis should be implemented for 24–72 hours, with regular monitoring of serum biochemistry and urinalysis. Diuretic therapy (furosemide, mannitol) may be beneficial in cats with oliguric renal failure but is unlikely to induce urine production in anuric cats for which referral for dialysis may be the only treatment option if available and affordable. Mannitol should not be used in anuric animals.
Prognosis
Prognosis depends in large part on the time between ingestion and presentation for treatment. Early presentation with GID and fluid diuresis carries a good prognosis for clinical recovery although some chronic renal dysfunction may persist. The prognosis is grave with standard medical therapy following the onset of anuria. The prognosis for anuric animals receiving dialysis may be favourable but remains to be elucidated.
Nonsteroidal antiinflammatory agents
Nonsteroidal antiinflammatory agents (NSAIAs) are widely used in human and veterinary medicine as both prescription-only and over-the-counter (OTC) preparations. A large heterogeneous variety of agents exists but they share certain therapeutic antiinflammatory, antipyretic and analgesic effects as well as undesirable adverse effects. The following discussion presents general information about NSAIA intoxication.
Toxic dose
The pharmacokinetics of NSAIAs show differences between species and doses used in people should not be extrapolated to dogs and cats.
Toxicokinetics, mechanism of toxicity
NSAIAs are direct inhibitors of cyclooxygenase (COX) and cause reduced production of prostaglandins and thromboxane. Some NSAIAs are reported to be more COX-2 selective; COX-2 is induced during tissue injury and inflammation. However, it is important to remember that at excessive dosages both COX-1 (constitutive/housekeeping form responsible for physiological functions) and COX-2 inhibition is likely.
NSAIAs are generally readily absorbed following oral exposure. Metabolism is usually hepatic. In dogs, a number of NSAIAs undergo enterohepatic circulation. Adverse effects of NSAIAs have been reported to involve predominantly the gastrointestinal tract, the kidneys, coagulation, and the liver.
Gastrointestinal tract
Gastrointestinal injury is the most common adverse effect associated with excessive NSAIA exposure in dogs and cats. These agents have ulcerogenic effects in the stomach and duodenum as a result of both impaired synthesis of prostaglandins that are important in mucosal cytoprotection and local topical mucosal irritation.
Kidneys
Under normal conditions prostaglandins are of little importance in maintaining renal blood flow. However, they exert a compensatory vasodilatory effect to maintain renal perfusion and function in conditions that cause renal vasoconstriction. Thus nephrotoxicity and consequent renal insufficiency may occur in animals that are exposed to NSAIAs while in a state of volume depletion or hypotension, as well as following exposure to massive overdose.
Coagulation
NSAIAs can induce coagulopathy by inhibiting COX within platelets and thereby impairing the production of thromboxane. Among other functions thromboxane facilitates platelet aggregation.
Adverse effects associated with NSAIAs are more likely to occur in animals exposed to excessive dosages as well as in those with volume depletion, hypotension or pre-existing gastrointestinal or renal disease. Bleeding tendency may be more common in animals with other concurrent coagulation abnormalities (e.g. von Willebrand’s disease). Hepatic injury is not a prominent feature of acute NSAIA intoxication.
Clinical signs
Early clinical signs of NSAIA intoxication in dogs are likely to reflect gastrointestinal complications and include vomiting, with or without haematemesis, and diarrhoea, with or without melaena (or haematochezia); inappetence, lethargy, depression, weakness and possible abdominal pain may occur. Dehydration and potentially hypovolaemia may be present. Cats may show gastrointestinal signs less frequently but tachypnoea may be more common than in dogs. In severe cases gastrointestinal perforation may occur with subsequent peritonitis that may be accompanied by abdominal pain and severe cardiovascular compromise.
Animals may also present with oliguric or polyuric acute renal failure.
Laboratory and other tests
Depending on the time elapsed since ingestion, clinicopathological findings may reflect gastrointestinal blood loss, with anaemia and possible panhypoproteinaemia. Anaemia may be preregenerative or regenerative (see Ch. 3). Prerenal azotaemia may be identified and urea may be disproportionately elevated compared to creatinine due to gastrointestinal haemorrhage. Renal insufficiency will likely manifest as azotaemia, hyperphosphataemia, possible hypercalcaemia and isosthenuria. Proteinuria may also be detected.
If gastrointestinal perforation and peritonitis are suspected, additional testing is mandatory. Abdominal radiography may show loss of serosal detail due to free peritoneal fluid, and free peritoneal gas. Abdominal ultrasonography may reveal free peritoneal fluid and may be used to guide abdominocentesis (see p. 293). Alternatively, blind abdominocentesis may be performed. Any fluid obtained should be examined cytologically and will typically be consistent with a purulent exudate if perforation has occurred.
Treatment
Depending on the time elapsed since ingestion, routine GID including the use of activated charcoal may be indicated to minimize further drug absorption. Intravenous fluid therapy using an isotonic crystalloid solution is indicated to correct hypovolaemia or dehydration and recurrence must be prevented to minimize the risk of nephrotoxicity.
Medical therapy designed to minimize further gastrointestinal compromise and to promote ulcer healing should be provided (Table 30.3). Routine use of anti-emetics may be required.
Table 30.3 Medical therapy for nonsteroidal antiinflammatory agent (NSAIA) intoxication
| Drug | Dose | Comments |
|---|---|---|
| Misoprostol | D: 2–5 µg/kg p.o. q 8–12 hr | |
| Omeprazole | D, C: 0.5–1 mg/kg slow i.v., p.o. q 24 hr | |
| Histamine (H2) receptor antagonists | Doses as per Appendix 1 |
|
| Sucralfate |
C, cats; D, dogs; i.v., intravenous; p.o., per os.
Surgical intervention is required in animals with gastrointestinal perforation and standard management of acute renal failure (see Ch. 36) is indicated in appropriate cases. NSAIA-mediated acute renal insufficiency is often reversible with adequate supportive care.
Aspirin
Toxic dose
In dogs, aspirin administration at 50 mg/kg q 12 hr has been reported to cause vomiting, and haematemesis and gastric ulcer perforation have been reported at 100–300 mg/kg/day for 1–4 weeks. Due to deficiencies in aspirin metabolism, cats are more susceptible and the toxic dose is lower than for dogs.
Toxicokinetics, mechanism of toxicity, clinical signs
Aspirin (acetylsalicylic acid) is a synthetic NSAIA. Following ingestion, aspirin is readily absorbed and is then metabolized in the liver to salicylic acid. Some local hydrolysis to salicylic acid also occurs in the gastrointestinal tract. Salicylic acid is the active form and the form that is absorbed into the circulation. Elimination from the circulation depends on conjugation with glucuronic acid. At high dosages this conjugation becomes overwhelmed, resulting in delayed clearance and accumulation of the active drug. Cats have a defective glucuronic acid conjugation system that results in prolonged drug elimination compared to dogs and an increased susceptibility to aspirin poisoning.
Gastrointestinal irritation and injury are the most common adverse effects of aspirin ingestion and nephrotoxicity is also reported. Metabolic acidosis is responsible for the majority of clinical signs. The metabolic acidosis in salicylate poisoning is a high anion gap acidosis. Severe and fatal acidaemia may occur within a few hours of aspirin ingestion.
Salicylates also have toxic effects in the CNS although the exact mechanism of toxicity is not known. Seizures have been reported in both dogs and cats secondary to salicylate toxicity. Noncardiogenic pulmonary oedema is the most common cause of major morbidity in people and might be related to a salicylate-induced increase in the permeability of the pulmonary vasculature. Salicylates inhibit vitamin K-dependent synthesis of coagulation factors (II, VII, IX and X), leading to a prolonged prothrombin time, and also inhibit prostaglandin-dependent platelet aggregation. Aspirin-induced hepatitis has been reported in cats following excessive exposure.
Paracetamol (acetaminophen)
Theory refresher
Paracetamol (acetaminophen in the USA) is used extensively by people and is widely available over the counter. It is contained in a variety of preparations, either solely or in conjunction with other drugs, including aspirin and opioids. It is an antipyretic and analgesic agent. Paracetamol has been used therapeutically in dogs but should not be administered to cats.
Toxic dose
A dosage of 100–200 mg/kg has been reported to cause clinical signs in dogs although higher dosages may be required and occasionally signs are seen with lower dosages. Cats are much more sensitive and signs of poisoning are generally seen at 50–100 mg/kg but may occur with dosages as low as 10 mg/kg.
Toxicokinetics
Paracetamol is rapidly and almost completely absorbed from the gastrointestinal tract and undergoes hepatic metabolism. In dogs, low doses of paracetamol are predominantly metabolized via capacity-limited glucuronidation and sulphation. This produces nontoxic metabolites that are excreted in bile and urine. However, any metabolism (oxidation) via the cytochrome P450 pathway produces N-acetyl-para-benzoquinoneimine (NAPQI) that is highly reactive and toxic.
NAPQI is usually conjugated with cellular glutathione to produce nontoxic metabolites that are excreted in urine. As the dosage of paracetamol increases, a greater proportion undergoes metabolism via the cytochrome P450 pathway (due to saturation of the glucuronidation and sulphation pathways), resulting in greater production of NAPQI. Glutathione stores, especially in the liver and red blood cells, are subsequently exhausted, resulting in higher concentrations of toxic unconjugated NAPQI. At higher doses paracetamol also inhibits glutathione synthesis, further compromising this metabolic pathway.
Cats are less able to metabolize paracetamol via glucuronidation (and sulphation) and the end result is greater metabolism via the cytochrome P450 pathway and an increased susceptibility to poisoning compared to dogs.
Mechanism of toxicity
Glutathione is important in protecting cells from oxidative injury and depletion of glutathione by conjugation with NAPQI makes cells susceptible to oxidative damage. In dogs, hepatocellular oxidative injury and necrosis are most common, with consequent liver failure possible.
In cats, red blood cells are most susceptible, with Heinz body formation, methaemoglobinaemia and haemolytic anaemia possible. Feline haemoglobin is more prone to oxidation than canine haemoglobin. As methaemoglobin is unable to transport oxygen, methaemoglobinaemia compromises oxygen-carrying capacity, causing tissue hypoxia and characteristic muddy brown mucous membranes. Glutathione, deficient in these cases, is also required to reduce methaemoglobin to haemoglobin.
Clinical signs
In dogs, clinical signs usually relate to hepatotoxicity and include vomiting, anorexia, abdominal pain and icterus. Methaemoglobinaemia may be seen with higher exposures, resulting in muddy/chocolate brown or cyanotic mucous membranes. It is unusual for methaemoglobinaemia to occur in dogs without subsequent signs of hepatotoxicity but this has been reported. Mucous membranes may also be pale due to anaemia secondary to intravascular haemolysis. Oedema of the face and/or paws may also be identified. Neurological signs may be present with severe liver dysfunction and hepatic encephalopathy and are also potentially associated with severe methaemoglobinaemia.
In cats, the most common clinical signs are muddy brown, cyanotic or pale mucous membranes, oedema of the face (especially mandibular region) and/or paws, and respiratory compromise; vomiting, depression, hypothermia and pruritus may also occur. Neurological signs may be present with severe methaemoglobinaemia and coma is associated with poor prognosis. Icterus may occur and at lower exposures is predominantly the result of red blood cell lysis. Clinically significant hepatotoxicity may be seen at higher exposures.
Antidote therapy
Paracetamol is one type of poisoning for which specific antidote therapy is available and treatment is recommended even if there is a significant delay in institution as a successful clinical outcome may still be obtained. N-acetylcysteine is rapidly hydrolysed to cysteine in vivo that is required for intracellular glutathione synthesis. N-acetylcysteine administration thus attempts to address cellular glutathione deficiency. Glutathione itself cannot be used therapeutically as it is not readily taken up by cells. N-acetylcysteine also acts directly on NAPQI, facilitating its excretion, and is oxidized to sulphur in the liver, increasing the capacity of the sulphation pathway.
If N-acetylcysteine is not available, or in severe cases of poisoning, additional sources of sulphur donors may be used. S-adenosylmethionine (SAMe) is one such product that may also have other additional beneficial effects (see Appendix 1 for protocol). The use of cimetidine has been recommended in paracetamol poisoning. This agent can inhibit cytochrome P450-mediated oxidation and may therefore reduce the formation of NAPQI. Given this mechanism of action, cimetidine would need to be given very early on to be effective and is considered an adjunctive therapy only.
Methaemoglobin reduction
In addition to the above therapy, treatment designed to reduce methaemoglobin to haemoglobin may be administered. Methylene blue has been employed here but is typically not readily available. However, ascorbic acid (vitamin C) may be used and oral preparations can usually be easily obtained. Poor compliance may preclude administration in cats. The author has administered ascorbic acid in drinking water to dogs that were not vomiting. Preparations of ascorbic acid for intravenous use are available and the treatment regimen is the same as for oral preparations (see Appendix 1).
Case example 4
Presenting Signs and Case History
A 3-year-old male neutered Beagle was presented as an emergency for suspected paracetamol poisoning. The owners had been out that evening and had returned to find the dog ataxic with evidence of probable ingestion of a large number of paracetamol tablets.
Major body system examination
On presentation the dog was ambulatory but depressed. Tachycardia (heart rate 130 beats per minute) and brown mucous membranes were identified but the rest of the examination was unremarkable.
Emergency database
An intravenous catheter was placed in a cephalic vein and blood obtained via the catheter for an emergency database that was found to be unremarkable.
Case management
Additional venepuncture was performed and the dog’s blood was confirmed to be dark brown in colour, suggestive of methaemoglobinaemia (Figure 30.4). An in-house coagulation analyser was available and prothrombin time (PT) and activated partial thromboplastin time (APTT) were found to be within normal limits.
Clinical Tip
Emesis was induced using apomorphine (0.02 mg/kg s.c.) and the vomitus contained a minimum of 30 paracetamol tablets (500 mg per tablet). The dog’s body weight was 20 kg and an exposure of 750 mg/kg was therefore possible, although it was not clear exactly how many more tablets, if any, the dog had ingested. In addition, the degree of absorption prior to emesis could not be known.
Initial therapy consisted of intravenous N-acetylcysteine (initial loading dose of 140 mg/kg i.v.; further five doses of 70 mg/kg i.v. q 6 hr) and oral ascorbic acid (vitamin C) (30 mg/kg p.o. in drinking water q 6 hr for five doses). Isotonic crystalloid intravenous fluid therapy was provided using compound sodium lactate at 4 ml/kg/hr and oxygen supplemented via an oxygen cage.
Twenty-four hours following presentation clotting tests were prolonged and moderate elevations in serum alanine aminotransferase (ALT) and bilirubin were detected. Mild anaemia was also identified (manual packed cell volume 30%, reference range 37–55%). A type-specific fresh whole blood transfusion (15 ml/kg over 4 hr) (see Ch. 40) was administered for the coagulopathy and anaemia, and additional therapy with S-adenosylmethionine (40 mg/kg p.o.) was commenced.
The dog continued to deteriorate, however, with evidence of haemolysis in the form of severe haemoglobinaemia and haemoglobinuria, with worsening anaemia despite blood transfusion, and with progressive severe dyspnoea. A haemoglobin-based oxygen-carrying solution (Oxyglobin®, Biopure Corporation; see Ch. 4) was administered (6 ml/kg i.v. over 1 hr) with no obvious clinical effect and the dog was euthanased approximately 36 hours after ingestion of the paracetamol tablets. A presumptive diagnosis of fulminant acute hepatic failure and severe haemolysis due to paracetamol poisoning was made.
Anticoagulant rodenticides
Anticoagulant rodenticides usually contain derivatives of either 4-hydroxycoumarin (e.g. brodifacoum, bromadiolone, difenacoum) or indane-1,3-dione (e.g. diphacinone, chlorophacinone). These preparations have a variable potency and duration of action that may be related to the generation type of the constituent compound. Second generation compounds are typically longer acting and have largely replaced older first generation ones. A variety of different commercial preparations are available.
Toxic dose
Given the large number of anticoagulant rodenticide substances in use it is beyond the scope of this chapter to detail toxic doses for each individual substance and the reader is recommended to consult other texts or a veterinary poisons database.
Toxicokinetics
Anticoagulant rodenticides are generally absorbed slowly but substantially from the gastrointestinal tract. A long plasma half-life potentially of a number of days is typical, and the duration of action can be very prolonged – even up to several weeks in some cases. These compounds undergo slow metabolism by hepatic microsomal mixed-function oxidases to form inactive metabolites that are excreted in urine or bile.
Mechanism of toxicity
Vitamin K1 hydroquinone is required for the conversion of inactive precursor coagulation factors to their active forms. During this conversion vitamin K1 hydroquinone is oxidized to vitamin K1 epoxide. Following absorption, anticoagulant rodenticides inhibit hepatic vitamin K1 epoxide-reductase which is responsible for the conversion of vitamin K1 epoxide back to vitamin K1 hydroquinone. Anticoagulant rodenticides therefore impair vitamin K1 ‘recycling’ by the liver, leading to its depletion as existing stores are exhausted; they thereby prevent conversion of several inactive coagulation factors to their active forms. The vitamin K1-dependent coagulation factors are factors II (prothrombin), VII, IX and X.
A delay in the onset of clinical signs following anticoagulant rodenticide ingestion is usually seen. This is due to the presence of circulating active vitamin K-dependent coagulation factors that must be exhausted for clinical signs to become apparent. Of the vitamin K-dependent factors, factor VII has the shortest plasma half-life. This factor is traditionally classified as part of the extrinsic coagulation pathway that is evaluated using the prothrombin time (PT). This explains the early clinical usefulness of measuring PT in cases of suspected anticoagulant rodenticide poisoning. Activated partial thromboplastin time (APTT) and activated clotting time (ACT) are used to evaluate the intrinsic (includes factors II, IX and X) coagulation pathway and are expected to become prolonged subsequently also.
Clinical signs
Clinical signs usually develop from 3–5 days after exposure and may persist for more than 2 weeks without intervention. Clinical signs reflect bleeding tendency as described above and may be accompanied by a variety of nonspecific signs such as lethargy, depression and reduced appetite. Anticoagulant rodenticide poisoning may manifest with signs of respiratory distress, most commonly due to haemothorax but also secondary to pulmonary haemorrhage, and coughing (including haemoptysis) is reported. Bleeding into the peritoneal cavity and mediastinum is also reported. There may be evidence of external bleeding (e.g. nasal or gingival) and gastrointestinal haemorrhage may manifest as melaena, haematemesis and abdominal pain. Petechiae, ecchymoses and excessive bleeding at venepuncture sites may be identified. Bleeding in other sites will manifest with expected clinical signs (e.g. lameness secondary to bleeding into joints or neurological signs secondary to CNS haemorrhage).
In animals that have become anaemic secondary to blood loss, anticipated physical examination findings such as pale mucous membranes, tachycardia and hyperdynamic pulse quality will be present. If blood loss is considerable and rapid, evidence of hypoperfusion secondary to hypovolaemic shock may be identified.
Laboratory and other tests
In any animal presenting following suspected anticoagulant rodenticide ingestion a baseline minimum database should be established. This should include manual packed cell volume, plasma total solids, peripheral blood smear, and coagulation profile (in particular PT) taken before initiation of therapy (see below). PT is prolonged first in poisoned patients as factor VII becomes depleted earliest but prolongation of APTT and ACT usually also occurs before the onset of clinical signs.
A peripheral blood smear should be evaluated for platelet count and, where anaemia is identified, for evidence of regeneration. Mild to moderate thrombocytopenia is a common finding. Lack of a regenerative red blood cell response may represent preregenerative (as opposed to nonregenerative) anaemia (see Ch. 3). Blood typing may also be appropriate. Low serum total solids are expected in anaemia secondary to blood loss.
Thoracic and abdominal diagnostic imaging may identify major sites of haemorrhage, with the thoracic cavity being the most common site of bleeding. Thoracocentesis and abdominocentesis will likely reveal a nonclotting sanguineous effusion with packed cell volume similar to that of the patient’s peripheral blood.
Treatment
Treatment of anticoagulant rodenticide poisoning is dependent largely on whether the patient is showing evidence of bleeding at the time of presentation. Routine GID, including the use of activated charcoal, is indicated in asymptomatic patients presenting within an appropriate timeframe. PT should be measured and no additional therapy is required if PT is within normal limits. Repeat testing of PT should be performed within 2–3 days. If a significant delay in obtaining the results of this test is unavoidable, it may be appropriate in individual cases to commence antidotal therapy with synthetic vitamin K1 (phytomenadione) once blood sampling has been performed and to discontinue therapy if normal results are subsequently obtained. Guidelines for vitamin K1 therapy in anticoagulant rodenticide intoxication are summarized in Box 30.8.
BOX 30.8 Guidelines for vitamin K1 therapy in anticoagulant rodenticide intoxication
If symptomatic at presentation or prothrombin time (PT) prolonged (or significant delay anticipated in obtaining PT), administer vitamin K1 2–5 mg/kg s.c. at multiple sites using the smallest possible needle. Preferably continue parenteral administration until PT normalizes, then change to oral administration at same daily dose, i.e. 2–5 mg/kg daily divided into two or three administrations.
Vitamin K1 therapy should typically be continued for 2–6 weeks depending on the type of anticoagulant ingested (second generation compounds typically require a longer course of vitamin K1 therapy). If the type of anticoagulant is unknown, a 2-week course of treatment is reasonable. After this time PT is rechecked and treatment discontinued as long as PT is normal. Prothrombin time is rechecked 2–3 days after stopping treatment:
If asymptomatic at presentation and PT within normal limits, withhold treatment; recheck PT within 3 days.
If vitamin K1 therapy is commenced a significant period of time prior to sampling for testing of PT, subsequent results may be affected and a definitive diagnosis of coagulopathy secondary to rodenticide poisoning cannot be established. Subsequent vitamin K1 therapy in these cases should then be managed as recommended for animals presenting with clinical signs of haemorrhage or with prolongation of PT.
Clinical Tip
Replacement of clotting factors may be performed in symptomatic patients by administering appropriate blood products. This is typically achieved through the use of fresh frozen plasma but fresh whole blood may be used to treat severe anaemia as well as coagulopathy in appropriate cases. Alternatively, a combination of fresh frozen plasma and packed red blood cells may be employed in these cases (see Ch. 40).
Prognosis
Prognosis is generally good with adequate and timely treatment but is partly dependent on site and severity of haemorrhage at the time of presentation.
Rodenticides containing vitamin D
Some rodenticide preparations contain calciferol (vitamin D2) or cholecalciferol (vitamin D3), either alone or together with anticoagulant agents. Calciferol/cholecalciferol is metabolized to calcitriol (1,25-dihydroxycholecalciferol) which induces hypercalcaemia via increased intestinal absorption, increased renal reabsorption and enhanced bone resorption. Hyperphosphataemia is also consistently present.
Clinical signs of hypercalcaemia are most commonly associated with the neurological, cardiovascular and gastrointestinal systems and with the kidneys. Depending on the preparation consumed, signs may be seen 8–48 hours post-ingestion. Treatment of hypercalcaemia involves: promoting calciuresis using intravenous 0.9% sodium chloride (normal saline) and furosemide; corticosteroid therapy to suppress bone resorption, reduce intestinal calcium absorption and promote calciuresis; possible additional use of salmon calcitonin; and, more recently, treatment with a bisphosphonate drug. Animals poisoned with anticoagulant rodenticides containing vitamin D may have severe morbidity.
Chocolate (theobromine)
Theobromine is a methylxanthine-derived alkaloid occurring naturally in cacao beans and found in chocolate, cocoa powder and other products produced from these beans. In addition, chocolate contains a lesser amount of caffeine, also a methylxanthine.
Clinical Tip
Toxic dose
Fatal doses of theobromine are reported to be in the range of 90–250 mg/kg in dogs and 80–150 mg/kg in cats.
Toxicokinetics
Absorption of theobromine from the gastrointestinal tract is relatively slower in dogs compared to people, with complete absorption potentially taking up to 10 hours. Metabolism is primarily hepatic and enterohepatic circulation occurs. Excretion is considerably slower than in people.
Mechanism of toxicity
Methylxanthines inhibit cyclic nucleotide phosphodiesterases and also act as adenosine receptor antagonists. As with other methylxanthines, theobromine (and caffeine) causes CNS stimulation with consequent cardiac and respiratory effects. It directly stimulates the myocardium and skeletal muscle causing increased contractility and competitively inhibits cerebral benzodiazepine receptors. Theobromine also causes smooth muscle relaxation, especially of the bronchi, and renal diuresis.
Clinical signs
Clinical signs usually develop within 24 hours of ingestion and typically much sooner. Signs may persist for 48–72 hours in some cases. Commonly reported clinical signs include vomiting, abdominal discomfort, restlessness, excitability and hyperactivity, ataxia, tachycardia, and tachypnoea or panting. In more severe cases muscle rigidity, muscle tremors, hyperthermia, seizures and dysrhythmias have been reported. Urinary incontinence, polyuria and polydipsia may also occur. Severe seizures and/or cardiovascular compromise are typically reported in fatal cases.
Treatment
Routine GID is indicated in appropriate cases. As theobromine is absorbed slowly in dogs, induction of emesis may be appropriate even after a significant delay; however, it may be best avoided in animals that are very hyperactive. Theobromine undergoes enterohepatic circulation so repeated use of charcoal may enhance elimination. There is no specific antidote for theobromine poisoning and therapy is otherwise symptomatic. This may include intravenous fluid therapy, antiemetic administration, sedation if excitability is excessive, and routine treatment of seizures. Antidysrhythmic therapy may also be indicated in some cases.
Xylitol
Xylitol is a naturally occurring sugar alcohol found in low concentrations in a variety of fruits and vegetables. It is extracted commercially and used as a sweetener, being popular for use for example by diabetics and in low-carbohydrate diets. The use of xylitol has also been increasing due to its effects in reducing the formation of dental caries (anticariogenic effect) that has led to its inclusion in a number of products including chewing gum, sweets, toothpaste and other oral care products. Xylitol is also found in proprietary baked goods and is commercially available as a powder for baking. Manufacturers are not obliged to specify the xylitol content of products in all cases and sometimes only the total sugar alcohol content (including e.g. sorbitol, isomalt) is listed.
Clinical Tip
Toxic dose
Hypoglycaemia has been reported to occur in dogs at dosages greater than 0.15 g/kg and a dosage of 0.5 g/kg has been associated with hepatic failure. However, it remains unclear whether the hepatotoxic effect of xylitol is dose-related or idiosyncratic, and the possibility of hepatic injury at doses less than 0.5 g/kg cannot therefore be excluded.
Toxicokinetics
Xylitol is a normal intermediate product in the glucuronic acid cycle but excessive exposure occurs through ingestion. Subsequent absorption varies between species but is rapid and almost complete in dogs. Slow release from ingested foodstuffs may delay absorption and explain the potentially sustained hypoglycaemia seen in dogs. Metabolism is predominantly hepatic and occurs rapidly. Virtually no urinary excretion occurs.
Mechanism of toxicity
In dogs xylitol acts as a potent and dose-dependent stimulator of pancreatic insulin release (this effect is negligible or only mild in people). It also causes hepatic injury and probable acute hepatic necrosis by a currently unknown mechanism. Hypoglycaemia may occur as a result of the insulin release and may be both severe and sustained. Hypokalaemia may also result from insulin release. Clinically significant coagulopathy is one potential consequence of hepatic injury and consequent dysfunction. Hepatic failure may also contribute to hypoglycaemia and is thought to be responsible for this finding in dogs that do not show earlier evidence of hypoglycaemia attributable to insulin release.
Clinical signs
Clinical signs associated with hypoglycaemia often develop within an hour of exposure but may be delayed. Signs include lethargy, weakness, vomiting, ataxia, altered mentation from depression through to coma, and seizures. Signs associated with liver failure are more delayed in onset (up to 72 hours after exposure) and may occur with or without earlier signs of hypoglycaemia. Coagulopathy may manifest as petechiae/ecchymoses, haemorrhagic faeces, and excessive bleeding from venepuncture sites.
Laboratory tests
Blood glucose should be evaluated as part of the emergency database and may be normal or mildly to severely reduced. Occasionally dogs will present with hyperglycaemia that then progresses to hypoglycaemia. Regular monitoring is recommended even in dogs that are normoglycaemic at presentation. Electrolyte screening may reveal hypokalaemia that is usually mild or perhaps moderate.
If hepatic injury has occurred, a serum biochemistry profile may reveal a marked increase in serum alanine transaminase (ALT) activity, a mild to moderate increase in serum alkaline phosphatase (ALP) activity, and hyperbilirubinaemia. Prolongation of PT and APTT may be detected in coagulopathic animals, and mild to moderate thrombocytopenia is commonly reported. Regular monitoring of these parameters is recommended for 3–4 days following exposure to a toxic dosage, including in dogs that are normoglycaemic at presentation.
Treatment
Routine GID is indicated in appropriate cases. Emesis should not be induced in dogs with marked neurological compromise secondary to hypoglycaemia. Activated charcoal should be administered empirically but may have limited benefit due to low binding of xylitol (demonstrated in an in vitro study).
Hypoglycaemia is treated using standard parenteral and possibly oral glucose supplementation therapy that may need to be both aggressive and prolonged. Regular small meals and possible oral sugar supplementation may be sensible in asymptomatic dogs. Coagulopathy secondary to hepatic dysfunction may necessitate fresh frozen plasma (FFP) administration (see Ch. 40) and vitamin K1 should be administered. As with any severe coagulopathy, animals should be handled gently, subjected to minimum stress and undergo exercise restriction. Venepuncture should only be performed using peripheral veins and with the smallest needle possible; adequate prolonged pressure should be applied to the site following sampling.
Empirical use of antioxidant hepatoprotectants such as SAMe, N-acetylcysteine and silymarin is reasonable. In cases of severe hepatic failure, treatment for possible hepatic encephalopathy may need to be instituted. Therapy is otherwise supportive and symptomatic.
Prognosis
The prognosis associated with hypoglycaemia alone from xylitol poisoning is good with timely and appropriate management. The prognosis for animals with evidence of hepatic dysfunction is guarded to poor, and grave for acute hepatic failure. Survival from xylitol poisoning may not be correlated with exposure dosage.