Chapter 26 Integrated Cardiovascular Responses
1. Heart failure is compensated for by both Starling’s mechanism and the arterial baroreflex.
2. Serious complications secondary to heart failure include exercise intolerance, edema, salt and water retention, uremia, kidney failure, septic shock, and decompensation.
3. The immediate cardiovascular effects of hemorrhage are minimized by compensations initiated by the atrial volume receptor reflex and the arterial baroreceptor reflex.
4. The blood volume lost in hemorrhage is restored through a combination of capillary fluid shifts and hormonal and behavioral responses.
5. The initiation of exercise involves an interplay of local and neural changes that increases cardiac output and delivers increased flow to exercising muscle.
Chapters 18 to 25 describe the various elements of cardiovascular function and control. An understanding of these individual elements is not sufficient, however, to provide a basis for the diagnosis and treatment of cardiovascular dysfunction. The veterinary clinician must understand the interaction of these elements in both normal and abnormal situations. Therefore this chapter discusses three fundamentally important, integrated cardiovascular responses: (1) the response to heart failure, (2) the response to hemorrhage, and (3) the response to exercise. In addition to elucidating important, integrated responses, this discussion provides a review and summary of key concepts of cardiovascular physiology.
Heart Failure Is Compensated for by Both Starling’s Mechanism and the Arterial Baroreflex
There are many types and causes of heart failure. Some clinicians use the term very broadly to refer to any condition in which a problem in the heart limits its ability to deliver a normal cardiac output to the body tissues. Such conditions would include various valve defects, arrhythmias, and even heartworm infestation. A more restrictive definition, and one favored by physiologists, is that heart failure is any condition in which a depressed cardiac contractility limits the ability of the heart to deliver a normal cardiac output. The broad definition of heart failure encompasses virtually any problem with the heart as a pump; a common synonym is pump failure. The more restrictive definition, as used in this chapter, equates heart failure with myocardial failure, a depressed contractility of the heart muscle itself.
A depressed cardiac contractility can result from coronary artery disease, cardiac hypoxia, myocarditis, toxins, drug effects, or electrolyte imbalances. If the decrease in contractility affects both sides of the heart, the condition is called bilateral heart failure. In other circumstances, failure may be restricted primarily to either the left ventricle or the right ventricle and thus is called left-sided heart failure or right-sided heart failure.
Ventricular function curves provide a helpful way to envision the consequences of heart failure and the compensations for heart failure. In Figure 26-1 the curve labeled Normal indicates the relationship between stroke volume and preload for a normal ventricle (for a review, see Figure 21-3, C). The curve labeled Initially severe failure shows that a ventricle in failure has a depressed contractility (i.e., a smaller stroke volume for any given preload). If a normal heart suddenly goes into severe failure, stroke volume changes from its normal value (shown by point 1) to the low value (shown by point 2). For purposes of illustration, imagine that these curves define the function of the left ventricle and that the left ventricle is the one that fails. A decrease in left ventricular stroke volume causes a decrease in left ventricular output, which results in a decrease in mean arterial blood pressure. If there is inadequate compensation for this fall in blood pressure, severe exercise intolerance is certain, inadequate perfusion of the critical organs is likely, and death is probable. However, several mechanisms react rapidly, within seconds to minutes, to compensate for heart failure and to minimize its adverse effects.
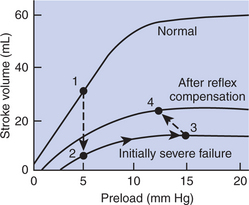
FIGURE 26-1 Ventricular function curves depicting the consequences and compensations for heart failure in terms of changes in preload (end-diastolic ventricular pressure) and stroke volume. See text for details.
One compensation for heart failure is Starling’s mechanism. If the left ventricle suddenly decreases its stroke volume, blood dams up (accumulates) in the left atrium and pulmonary veins because the right ventricle, at least for a few heartbeats, maintains a higher stroke volume than the failing left ventricle. Some of the excess blood pumped by the right ventricle accumulates in the left atrium, so left atrial pressure increases. The increase in left atrial pressure creates an increase in left ventricular preload, which leads to an increase in left ventricular end-diastolic volume and (by Starling’s mechanism) an increase in stroke volume. In other words, instead of remaining at the point on the graph labeled 2 (Figure 26-1), the function of the left ventricle moves to the right along the curve of initially severe failure, to point 3, with a higher preload and a somewhat higher stroke volume. This sequence of events, whereby an increase in preload helps offset the fall in stroke volume, is also diagrammed in Figure 26-2 (top left loop). Note that this compensation does not return stroke volume to its normal value, but does bring it to a level somewhat higher than before the compensation.
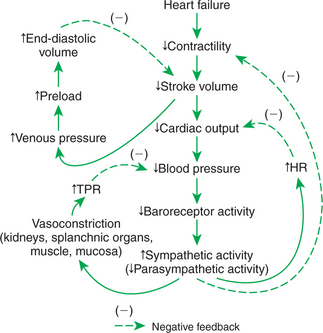
FIGURE 26-2 Consequences and compensations for heart failure. The changes described here include those presented graphically in Figure 26-1. See text for details. HR, Heart rate; TPR, total peripheral resistance.
The arterial baroreflex is another mechanism that reacts rapidly to compensate for heart failure. Because stroke volume remains below normal, even after compensation by Starling’s mechanism, left ventricular output also remains below normal, as does arterial blood pressure. Therefore, baroreceptor activity is below normal. The central nervous system (CNS) responds reflexively by increasing sympathetic efferent activity to the heart and blood vessels and by decreasing parasympathetic activity to the heart.
The sympathetic effect on the heart increases ventricular contractility. Contractility is not restored to normal but is brought to a higher level than existed in the absence of reflex compensation. Graphically, the effect of the baroreflex is to move the failing ventricle to a function curve that is intermediate between the “normal” curve and the curve of “initially severe failure” (see point 4 in Figure 26-1). Note that the increase in contractility also brings stroke volume back toward (but not reaching) its normal level.
Sympathetic actions on the heart increase heart rate above normal and decrease the systolic duration; these changes also help to restore cardiac output back toward normal despite the persistently depressed stroke volume. Finally, sympathetic activation causes vasoconstriction, particularly in the “noncritical” organs, which increases total peripheral resistance (TPR) above normal. This also helps to return blood pressure toward its normal level.
The net effect of the compensations by way of Starling’s mechanism and the baroreflex is that blood pressure can be maintained near its normal level, at least when the animal is at rest, despite a severe ventricular failure. Figure 26-2 summarizes these reflex effects. Note that after compensation by Starling’s mechanism and the baroreflex, contractility, stroke volume, cardiac output, and blood pressure remain at least somewhat below normal. By contrast, preload, sympathetic activity, heart rate, and TPR are above normal.
Serious Complications Secondary to Heart Failure Include Exercise Intolerance, Edema, Salt and Water Retention, Uremia, Kidney Failure, Septic Shock, and Decompensation
Even though Starling’s mechanism and the baroreflex can compensate to a remarkable degree for severe heart failure, important secondary complications often develop. These complications make heart failure a serious clinical problem, even in cases where compensatory mechanisms can maintain cardiac output and arterial pressure at near-normal levels when the animal is at rest.
The first complication secondary to heart failure is exercise intolerance. In a normal animal the ability of the heart to increase cardiac output during exercise depends on sympathetically mediated increases in stroke volume and heart rate. In a patient with heart failure, however, the ability of sympathetic activation to increase cardiac output is almost exhausted simply to restore cardiac output toward normal in the resting state. Therefore the patient’s attempt to exercise is not accompanied by an effective, further increase in sympathetic activity or cardiac output. The failing heart cannot provide the increased cardiac output required to meet the blood flow requirements of exercising skeletal muscle. In the absence of an adequate increase in cardiac output, metabolic vasodilation in the exercising muscle results in a large decrease in arterial pressure and inadequate blood flow to all organs, including the exercising muscle. The patient exhibits lethargy and weakness; even mild exercise leads quickly to exhaustion.
Edema is another serious complication secondary to heart failure. As noted, blood dams up in the atrium and veins behind a failing ventricle. In the case of left ventricular failure, left atrial pressure increases, as does pressure in the pulmonary veins and pulmonary capillaries. The increase in pulmonary capillary hydrostatic pressure leads to an increase in the filtration of capillary fluid into the interstitial spaces of the lungs. Pulmonary edema develops. These events are summarized in Figure 26-3 (top left). Excess interstitial fluid in the lungs slows the transfer of oxygen from the lung alveoli into the lung capillaries and can result in systemic hypoxia. In extreme cases, interstitial fluid leaks into the intrapleural space (pleural effusion) or in the alveolar air spaces, which causes a further reduction in lung function. The resulting systemic hypoxia can be fatal. In a patient with right ventricular failure the increase in venous pressure occurs in the systemic circulation. Therefore the resulting edema occurs in the systemic organs, particularly in dependent extremities and in the abdomen.
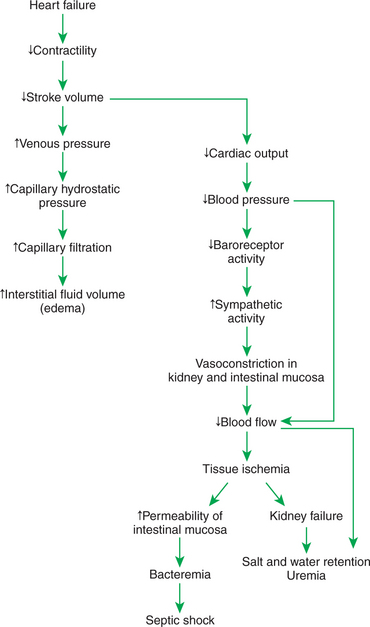
FIGURE 26-3 Heart failure leads to exercise intolerance. Additional, life-threatening complications secondary to heart failure are diagrammed here, including edema, salt and water retention, uremia, kidney failure, and septic shock. See text for details.
Whether the edema is in the lungs or in the systemic circulation, its degree is limited by the three safety factors previously discussed (see Figure 23-5). These safety factors would probably keep the edema of heart failure well controlled were it not for an additional factor that exaggerates the elevation of venous pressure in heart failure. As long as arterial pressure remains subnormal in a patient with heart failure, the baroreceptor reflex and some mechanisms involving the kidneys work to raise blood volume above normal. These volume-increasing mechanisms include increasing thirst (which raises fluid intake), increasing the release of antidiuretic hormone (ADH) from the pituitary (which decreases fluid loss in the urine), and activating the renin-angiotensin-aldosterone system (which decreases sodium loss in the urine). These effects of the baroreflex were mentioned briefly in Chapter 25, and the mechanisms involving the kidneys are described in more detail in Chapters 41 and 43.
The point for now is that the patient with severe heart failure experiences a substantial and persistent increase in blood volume. The excess blood accumulates particularly in the veins and capillaries upstream from the failing ventricle and often leads to an increase in capillary filtration that overwhelms the normal safety factors against edema. In fact, one of the main goals in the clinical treatment of heart failure is to counteract the buildup of excessive blood and interstitial fluid volume. Diuretic drugs are the main therapies used for this purpose (see Chapter 43).
Severe, persistent heart failure leads to several additional adverse effects. The baroreceptor reflex responds to an abnormally low arterial pressure in heart failure by initiating arteriolar vasoconstriction, primarily in the kidneys, splanchnic organs, and resting skeletal muscle (the “noncritical” organs). In severe failure the skin and mucous membranes are also vasoconstricted. Vasoconstriction in these organs helps compensate for heart failure by permitting the available cardiac output to be routed to the “critical” organs (brain, heart, and working skeletal muscle). However, persistent vasoconstriction leads to the additional complications of uremia, kidney failure, and septic shock.
Vasoconstricted kidneys cannot form urine in a normal manner and therefore do not rid the body of the excess volume of blood and interstitial fluid that accumulates in heart failure. The compromised kidney function also allows nitrogenous and acidic waste products to accumulate in the body. The condition is called uremia, which literally means “urine in the blood.” To make matters worse, after a prolonged period of intense vasoconstriction, the kidney tissue becomes irreversibly damaged. At this stage, uremia, acidosis, and salt and water retention may persist even if clinical treatment is temporarily successful in returning cardiac output and blood pressure close to normal. In fact, renal failure often is the terminal event in chronic heart failure.
Intense and prolonged vasoconstriction in the splanchnic circulation can also have lethal consequences. The mucosa of the gastrointestinal tract is particularly susceptible to ischemic damage. Normally, the intestinal mucosa creates a barrier between the intestinal lumen and the bloodstream. Ischemic damage to the intestinal mucosa allows bacteria and bacterial toxins to pass into the bloodstream or the peritoneum. The resulting bacteremia or peritonitis can cause septic shock and death. The causes and consequences of renal and splanchnic ischemia are summarized in Figure 26-3 (bottom right).
Cardiac decompensation is an additional (and frequently terminal) complication secondary to heart failure. The basic concept of decompensation is that when heart failure reaches a certain degree of severity, the compensations by the body for heart failure end up making the heart failure worse. Vicious “decompensatory” cycles develop and can lead to death within a few hours unless there is vigorous medical intervention.
The specific mechanisms of the decompensatory cycles are very complex, but two examples illustrate the concept. As explained, in the case of left ventricular failure, the damming up of blood in the left atrium is compensatory because it increases left ventricular preload, which helps boost stroke volume back toward normal. However, the increased left ventricular preload leads to the secondary complication of pulmonary edema. If severe, pulmonary edema interferes with the oxygenation of blood. One tissue that depends critically on an adequate supply of oxygen is cardiac muscle; hypoxia depresses the contractility of cardiac muscle. Thus a vicious cycle can develop: severely depressed ventricular contractility → severe pulmonary edema → inadequate oxygenation of blood → hypoxia of the left ventricular muscle, which further depresses ventricular contractility.
For a second example of a vicious decompensatory cycle, consider again the effects of the baroreflex on the kidneys. Renal vasoconstriction is compensatory for heart failure in that it helps increase TPR, which helps raise arterial pressure back toward normal, which helps keep perfusion pressure high enough to deliver adequate blood flow to the critical organs. However, intense and prolonged renal vasoconstriction leads to the accumulation of acidic and nitrogenous waste products in the blood (uremia) and in various body tissues, including the heart. The accumulation of acidic and nitrogenous waste products in cardiac muscle tissue depresses cardiac contractility. Thus, another vicious cycle can develop: severe ventricular failure → intense and prolonged renal vasoconstriction → uremia → accumulation of metabolic waste products in cardiac muscle, which further depresses ventricular contractility.
Other decompensatory cycles develop in cases of severe, prolonged heart failure, but these examples illustrate the basic concept and show why decompensation is such a serious development.
In summary, in addition to exercise intolerance, the life-threatening complications secondary to heart failure include edema, salt and water retention, uremia, kidney failure, septic shock, and decompensation. Careful clinical diagnosis and prompt treatment of heart failure are imperative, even in cases in which compensatory mechanisms have maintained blood pressure near its normal level.
In evaluating the severity of heart failure and the extent of compensation, it is clinically useful to group the signs of heart failure into two categories. The first category is referred to as backward heart failure. The signs of backward heart failure include the changes in the circulation upstream from the failing ventricle: increased atrial pressure, increased venous pressure, excessive capillary filtration, edema, and the functional changes secondary to edema (e.g., respiratory failure). The category forward heart failure refers to the consequences of heart failure downstream from the failing ventricle: decreased cardiac output, decreased arterial blood pressure, and the consequences of excessive vasoconstriction in the systemic organs, especially the kidneys and intestines.
The Immediate Cardiovascular Effects of Hemorrhage Are Minimized by Compensations Initiated by the Atrial Volume Receptor Reflex and the Arterial Baroreceptor Reflex
Figures 26-4 and 26-5 summarize the cardiovascular responses to hemorrhage. The curve labeled Normal in Figure 26-4 shows that the maintenance of a normal stroke volume depends on the maintenance of a normal level of ventricular preload. When hemorrhage occurs, blood is lost from the whole cardiovascular system, particularly from the veins, which are the blood reservoirs of the body. Hemorrhage therefore decreases central venous volume, central venous pressure, and atrial pressure. The decrease in atrial pressure decreases ventricular preload and end-diastolic ventricular volume. In the absence of any compensation, the stroke volume decreases from point 1 in Figure 26-4 to point 2. Note that the normal ventricular function curve is rather steep to the left of the normal operating point (point 1). Therefore a 40% hemorrhage results in approximately 40% reductions in central venous pressure, left atrial pressure, ventricular preload, and stroke volume. In the absence of compensations, cardiac output and mean arterial pressure (MAP) would also decrease by 40%. MAP would then be inadequate to sustain normal function in the critical organs, and the animal would die. With intact compensatory mechanisms, however, a normal animal can withstand a 40% hemorrhage without death and have only about a 10% decrease in MAP.
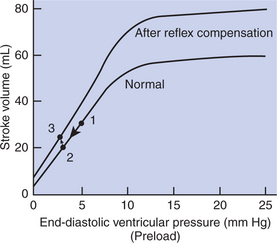
FIGURE 26-4 Direct effect of hemorrhage is to decrease ventricular preload, which decreases stroke volume (transition from point 1 to point 2). Stroke volume is restored toward normal by a reflex increase in sympathetic activity, which increases ventricular contractility above normal (transition from point 2 to point 3).
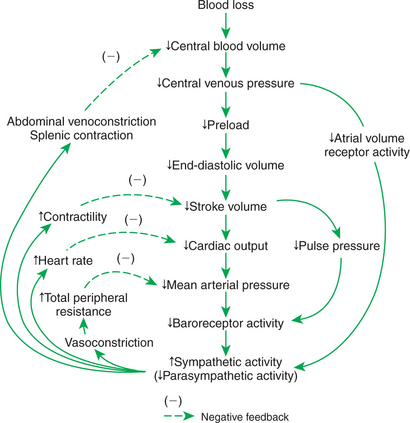
FIGURE 26-5 Summary of the consequences of hemorrhage and the rapid compensations from the arterial baroreflex and atrial volume receptor reflex. The changes described here include those presented in Figure 26-4.
The immediate compensations for hemorrhage are initiated by the arterial baroreflex and atrial volume receptor reflex. Hemorrhage decreases MAP, which decreases the activity of arterial baroreceptors. The baroreflex response is to increase sympathetic activity and to decrease parasympathetic activity. The increased sympathetic activity acts on the heart to increase cardiac contractility. This helps restore stroke volume back toward normal, despite a persistent, subnormal preload and end-diastolic volume. The effect of this sympathetic compensation is diagrammed in Figure 26-4 as point 3. Although stroke volume is returned toward normal, it remains low; after compensation for a 40% hemorrhage, the stroke volume may remain 25% below normal.
Additional compensations help restore blood pressure closer to normal despite the persistence of low stroke volume. First, heart rate increases above normal, which brings cardiac output back to within about 20% of its normal level. In addition, sympathetic vasoconstriction in the noncritical organs raises TPR above normal, resulting in a MAP that remains within approximately 10% of its normal level, despite a persistent 20% drop in cardiac output. The reader can review the compensations described thus far by locating them on Figure 26-5.
You may wonder why baroreflex compensatory actions continue if MAP is returned most of the way toward normal. Compensatory baroreflex responses are sustained because baroreceptors are responsive to changes in pulse pressure as well as to changes in MAP, and pulse pressure remains low. There are two reasons for the subnormal pulse pressure: (1) the persistent decrease in stroke volume and (2) the increase in heart rate above normal. Thus, even if MAP is returned substantially toward normal after compensation for a hemorrhage, baroreceptor activity (action potential frequency) remains below normal.
The atrial volume receptor reflex also contributes to the sustained increase in sympathetic activity after hemorrhage. As shown in Figure 26-5, hemorrhage leads to a persistent decrease in central venous pressure and atrial pressure. Therefore the activity of the atrial volume receptors is decreased below normal. The CNS responds to this decreased afferent activity from atrial volume receptors by elevating sympathetic efferent activity and by decreasing cardiac parasympathetic efferent activity. Thus the atrial volume receptor reflex and the arterial baroreflex work synergistically to compensate for hemorrhage.
In severe hemorrhage the reflex increases in sympathetic activity affect not only the heart and resistance vessels but also the veins. The abdominal veins in particular are constricted when sympathetic activation is intense. Sympathetic venoconstriction displaces blood from the abdominal veins and moves it toward the central circulation, which helps to increase central venous pressure, atrial pressure, and preload back toward normal (Figure 26-5). Sympathetic activation also constricts the blood vessels within the spleen and the muscular capsule around the spleen. The blood that is sequestered in the spleen is expelled into the abdominal veins, and then it moves toward the heart. In species that have large spleens (e.g., dog and horse), splenic contraction can mobilize a volume of blood equal to 10% of the total blood volume. An additional, adaptive feature of the blood sequestered in the spleen is that it has a higher-than-normal hematocrit. The mobilization of these sequestered red blood cells helps to offset the fall in hematocrit that is a normal consequence of interstitial fluid reabsorption after hemorrhage (as described next).
The arterial baroreceptor reflex and the atrial volume receptor reflex act within a few seconds to restore blood pressure toward its normal level after a hemorrhage. Other compensations come into play in the minutes and hours after hemorrhage to restore the lost fluid volume.
The Blood Volume Lost in Hemorrhage Is Restored Through a Combination of Capillary Fluid Shifts and Hormonal and Behavioral Responses
Hemorrhage causes both venous and arterial pressures to fall below normal, so capillary hydrostatic pressure also falls below normal throughout the body. This alters the balance of hydrostatic and oncotic pressures acting on water in a direction that favors reabsorption of interstitial fluid back into the capillaries (Figure 26-6). The volume of interstitial fluid that can be reabsorbed by this process in 1 hour is approximately 10% of the volume lost in the hemorrhage. However, the rate of reabsorption of interstitial fluid becomes limited after 3 to 4 hours. As interstitial fluid is reabsorbed, there is a decrease in interstitial fluid hydrostatic pressure (it becomes even more negative than normal), and this opposes further reabsorption. Also, as interstitial fluid is reabsorbed, the interstitial fluid protein concentration increases because proteins in the interstitial fluid are not reabsorbed. The resulting increase in interstitial fluid oncotic pressure also opposes further reabsorption. Despite these limits, the reabsorption of interstitial fluid is an important compensation for hemorrhage in the first few hours.
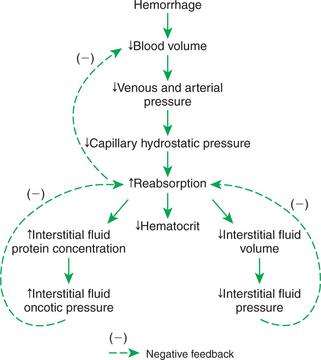
FIGURE 26-6 During the first 3 to 4 hours after a hemorrhage, interstitial fluid is reabsorbed into the bloodstream, which helps compensate for the lost blood volume. A complication is that the hematocrit decreases. Reabsorption is limited by decreases in interstitial fluid hydrostatic pressure and by increases in interstitial fluid oncotic pressure.
A complication arising from the reabsorption of interstitial fluid after hemorrhage is that the reabsorbed fluid contains no plasma proteins or blood cells. Therefore the proteins and cells already in the bloodstream are diluted as interstitial fluid is reabsorbed. The concentration of plasma proteins in blood decreases, as does the hematocrit. This is why a decreasing hematocrit over a few hours in an otherwise-normal patient is presumptive evidence that a hemorrhage has occurred recently or is continuing to occur. In the absence of an obvious hemorrhage, such a patient should be examined for evidence of internal bleeding.
The restoration of blood volume after hemorrhage also involves hormonal and behavioral responses (Figure 26-7). As mentioned, hemorrhage leads to a decrease in the action potential frequency of both the arterial baroreceptors and the atrial volume receptors. The immediate, compensatory reflex responses have already been described (see Figure 26-5). The arterial baroreflex and the atrial volume receptor reflex also trigger important hormonal and behavioral effects. The increase in sympathetic activity (coupled with a decrease in arterial pressure) acts on the kidneys to increase their release of the hormone renin. As mentioned in Chapter 25, renin works through the additional hormones angiotensin and aldosterone to decrease sodium excretion by the kidneys. Decreased activity of the baroreceptors and atrial volume receptors also triggers increased ADH secretion from the pituitary gland. ADH circulates to the kidneys, where it reduces urine formation. Through the combined actions of renal vasoconstriction, the renin-angiotensin-aldosterone system, and ADH, sodium excretion and water excretion are both decreased. Note that these actions conserve the available blood volume after hemorrhage, but they do not restore it to normal. The actual restoration of blood volume after hemorrhage requires increased fluid intake. The baroreceptor reflex and the atrial volume receptor reflex act through the hypothalamus to increase the sensation of thirst. If water is available, fluid intake increases until the lost blood volume is restored to normal. This may take 1 to 2 days.
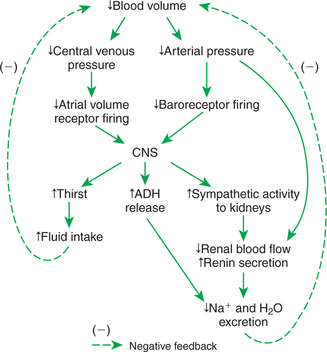
FIGURE 26-7 Behavioral and humoral responses after hemorrhage include increased fluid intake and retention of body fluid and electrolytes. ADH, Antidiuretic hormone; CNS, central nervous system.
The final compensations for hemorrhage involve the restoration of the lost plasma proteins and blood cells. The plasma proteins are synthesized by the liver, and the blood cells are produced by the bone marrow. The time required may be several days for the plasma proteins and a few weeks for the blood cells.
The preceding discussion focused on the effects of severe hemorrhage. All the same compensations occur to a lesser degree after mild hemorrhage. For example, when a human donates blood, about 10% of the blood volume (0.5 L) is removed. All the compensations just described are evident after this 10% hemorrhage.
In humans and in some large animals, the transition from a supine to a standing posture elicits many of the same cardiovascular responses as hemorrhage. You can understand the reason for this if you consider the effect of gravity on the blood contained within the blood vessels of the body. In a standing subject, gravity increases the distending pressure in the dependent vessels (those below heart level), particularly in the leg vessels. The gravitational effect does not cause much accumulation of blood in the arteries and arterioles because these vessels are not easily distensible (i.e., they have low compliance). However, the gravitational effect causes a significant distention of the dependent veins because of their much greater compliance. The extra blood in the dependent veins is blood that would otherwise have returned to the central circulation. Therefore, in an upright subject, there is a decrease in central blood volume and central venous pressure, just as there would be after hemorrhage. Furthermore, upright posture causes decreased cardiac filling, decreased stroke volume, decreased cardiac output, and so on. In a normal human the assumption of an upright posture is equivalent to a 10% hemorrhage. In small animals the gravitational effect of standing is negligible. In large animals, such as horses and cattle, the volume of blood that pools in the leg veins is minimized by the relatively small size of veins in the extremities.
The Initiation of Exercise Involves an Interplay of Local and Neural Changes That Increases Cardiac Output and Delivers Increased Flow to Exercising Muscle
As discussed in Chapter 24, local metabolic control mechanisms dilate skeletal muscle arterioles during exercise. Metabolic products accumulate in exercising muscle, and the local oxygen concentration decreases. The metabolic products and hypoxia both cause dilation of the arterioles within the exercising muscle. This vasodilation is a local response, not dependent on nerves or hormones. The result is an increased blood flow to the exercising muscle (active hyperemia). The increased blood flow delivers more oxygen and removes some of the accumulated metabolic vasodilating products. In this way, muscle blood flow is matched to metabolic rate (Figure 26-8, top).
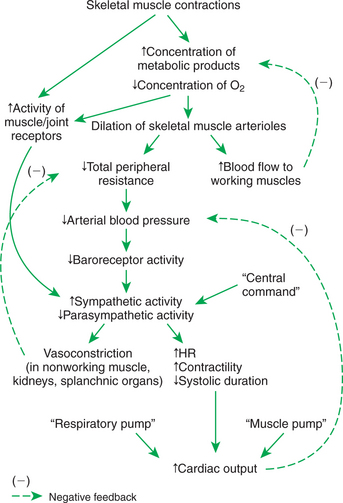
FIGURE 26-8 Cardiovascular responses to exercise involve a complex interplay of local metabolic control mechanisms: central command, reflexes, and the blood-pumping effects of muscle contraction and respiration. The overall result is increased blood flow in exercising muscle, decreased blood flow in the noncritical organs, decreased total peripheral resistance, increased cardiac output, and (normally) maintenance of arterial blood pressure near its normal level. HR, Heart rate.
Metabolic control of blood flow in exercising muscle can succeed only if arterial blood pressure is maintained at a level sufficient to provide the needed additional blood flow. This necessitates a substantial increase in cardiac output and, in extreme exercise, vasoconstriction in the noncritical organs (which makes more blood flow available for the exercising muscle and other critical organs). These adjustments are brought about by three neural mechanisms: central command, the exercise reflex, and the arterial baroreflex.
Central command is a psychogenic effect. In preparation for exercise (and continuing during exercise) the CNS increases sympathetic activity to the heart and blood vessels and decreases parasympathetic activity to the heart. The sympathetic and parasympathetic changes are graded, depending on the intensity of the exercise. In effect, central command represents a “guess” by the brain as to the levels of sympathetic and parasympathetic activity that will be needed during the exercise to match cardiac output to the needs of the systemic organs.
The exercise reflex is the second mechanism that helps set the level of sympathetic and parasympathetic activity during exercise. The exercise reflex is initiated by specialized nerve endings within muscles and joints. An increase in muscular work and in the movement of the body joints activates these muscle and joint receptors. The resulting increased afferent neural activity initiates a reflex increase in sympathetic (and decrease in parasympathetic) efferent drive. Although the mechanism for excitation of the muscle and joint receptors is not completely understood, it is clear that the activation of these receptors is necessary to keep blood pressure from falling during exercise.
The arterial baroreceptor reflex is the third major controller of sympathetic and parasympathetic activity during exercise. The baroreflex serves to fine-tune autonomic drive to the heart and arterioles to keep arterial pressure at its set point. If central command and the exercise reflex do not set sympathetic activity to a sufficiently high level during a particular bout of exercise, arterial pressure falls below normal. The arterial baroreceptors detect this low pressure, and the baroreflex responds by increasing sympathetic activity. Conversely, if central command and the exercise reflex set sympathetic activity too high, arterial pressure rises above normal. The response of the baroreflex is to decrease sympathetic activity.
In effect, central command and the exercise reflex initiate the autonomic adjustments for exercise, and the arterial baroreflex performs the fine-tuning to keep arterial pressure near its set point (Figure 26-8).
Two additional, nonneural mechanisms also help to increase the cardiac output during exercise. The first of these is the muscle pump (Figure 26-9). When skeletal muscles contract, they tend to squeeze down on the blood vessels contained within them. One consequence of this is the tendency for a muscle to restrict its blood flow during a sustained contraction (see Chapter 24). If the contractions are rhythmical, however, each contraction causes blood to be expelled out of the muscle veins and thus toward the central circulation. Minimal backflow of blood occurs from the central circulation into the veins during muscular relaxation because the veins have one-way valves within them. Thus, by massaging the veins, exercising muscles exert a pumping action that displaces venous blood toward the central circulation and increases central venous pressure. The consequence is an increase in ventricular preload above the level that would otherwise exist.
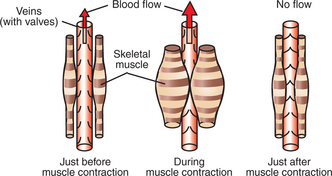
FIGURE 26-9 During dynamic exercise, the rhythmical contractions of the skeletal muscles squeeze venous blood back toward the central circulation. This so-called muscle pump helps increase central venous pressure in an exercising animal.
The second nonneural mechanism that helps to increase cardiac output during exercise is the respiratory pump. Vigorous exercise involves an increase in the rate and the depth of respiration. During each inspiration, a subatmospheric pressure is generated within the intrapleural space. This negative pressure distends the airways of the lungs and expands them. It also increases the distending pressure on the central veins and the heart. Distention of the central veins and heart helps promote the flow of blood from the abdominal veins into the central veins and heart. In addition, the diaphragm muscle moves caudally during inspiration and compresses the abdominal organs. The resulting increase in intraabdominal pressure “squeezes” blood out of the abdominal veins and back toward the central veins. Overall, the respiratory pumping action helps to increase venous return, central venous volume, and ventricular preload during exercise.
Cardiac output can increase four to six times its resting level during vigorous exercise as a result of the combined effects of sympathetic and parasympathetic responses, the muscle pump, and the respiratory pump. Note that the success of the mechanisms that increase cardiac output during exercise depends on the heart’s ability to respond normally both to increased sympathetic drive and to increases in preload. As mentioned earlier, during heart failure the autonomic mechanisms available to increase cardiac contractility and heart rate are invoked simply to maintain a normal cardiac output at rest. Therefore the autonomic nervous system in a patient with heart failure has a limited ability to bring about further increases in cardiac output during the initiation of exercise. For this reason, patients with heart failure typically exhibit exercise intolerance.
Maximal exercise ability in normal humans and animals appears to be limited by cardiac output. That is, the respiratory system can oxygenate as much blood as the heart can deliver to the lungs, and skeletal muscle can take up and metabolize as much oxygen as the heart can deliver to it. When cardiac output has reached a maximal level, however, oxygen transport from the lungs to the skeletal muscle also is maximized. This sets the upper limit to the level of exercise that can be sustained.
CLINICAL CORRELATIONS
Exercise Intolerance Secondary to Congestive Heart Failure
History.
An 8-year-old female Great Dane has been diagnosed previously with idiopathic dilative cardiomyopathy. Severe, generalized cardiac enlargement was evident on thoracic radiographs. The dog has been losing weight and is unable to complete daily walks with her owners.
Clinical Examination.
Femoral pulses are weak but regular at 140 beats/min. The mucous membranes are pale, and the capillary refill time is prolonged. Heart sounds are muffled, and a murmur is heard on the left side over the atrioventricular valve. Respiratory rate is greater than normal (45breaths/min). Auscultation reveals increased bronchovesicular (respiratory) sounds. The abdomen is distended, and the abdominal organs are difficult to palpate. The electrocardiogram shows sinus tachycardia with broad, high-voltage QRS complexes. Thoracic radiography reveals a greatly enlarged heart and moderate pulmonary edema. Echocardiography reveals dilation of all four cardiac chambers. Ejection fraction is below normal, and there is mitral regurgitation.
Additional diagnostic tests are conducted to help assess the degree of complications secondary to the heart failure. The percentage saturation of hemoglobin in arterial blood is 78% (normal, 95%–100%), the difference in oxygen content between arterial and venous blood is 8.5 mL of O2 per deciliter of blood (normal, 4–6mL), the serum creatinine concentration is 3mg/dL (normal, <1mg/dL), urine specific gravity is 1.036 (high normal), and central venous pressure is 14 mm Hg (normal, 0–3 mm Hg).
When persuaded to exercise, the dog appears to become tired after walking less than 1 minute. Her legs begin to tremble, and then she collapses. Her pulse rate is 180 beats/min, and her mucous membranes are dark and cyanotic (blue).
Comment.
Chronic heart failure secondary to cardiomyopathy is common in large dogs older than 4 years. Often the cardiomyopathy is idiopathic (of unknown cause). The case presented here is fairly typical of advanced heart failure. All the clinical findings are either direct consequences of the heart failure or consequences of attempts by the body to compensate for the heart failure (see Figures 26-1, 26-2, and 26-3). In brief, ventricular failure (decreased contractility) leads to decreased stroke volume, cardiac output, and blood pressure.
Compensations for the heart failure involve reflex decreases in parasympathetic activity, increases in sympathetic activity, and increases in the release of ADH and renin. Heart rate is increased, which helps raise cardiac output back toward normal. Pulse pressure, judged by palpation of the femoral pulse, is reduced (because heart rate is high and stroke volume is low). There is decreased blood supply to the mucosa, splanchnic organs, kidneys, and resting skeletal muscle because of vasoconstriction. This helps support arterial pressure and reserves the available cardiac output for the heart and brain. The vasoconstriction is evident in the pale color and slow refilling of the mucous membranes. Renal vasoconstriction reduces the rate of urine formation. Urinary loss of salt and water is further reduced by the actions of ADH and the renin-angiotensin-aldosterone system. The urine that does form has a high solute concentration (high specific gravity). Metabolic products (e.g., creatinine) that are normally eliminated by the kidneys accumulate in the blood. Salt and water retention increases blood volume above normal.
Most of the excess blood volume is in the veins, so venous and atrial pressures are above normal. The elevated atrial pressure (preload) increases ventricular end-diastolic volume above normal, which helps the failing heart to pump a larger stroke volume than it otherwise would. However, the excessive volume and pressure of blood in the veins also cause systemic edema (distended abdomen caused by ascites) and pulmonary edema (visible on the radiograph). Pulmonary edema impairs the ability of the lungs to oxygenate blood. Therefore the hemoglobin saturation and the oxygen content of arterial blood are both below normal in this dog. The tissues of the body respond to the low rate of oxygen delivery by unloading as much oxygen as they can from the blood as it flows through the tissue. This makes the arteriovenous difference in oxygen content greater than normal. The general inadequacy of cardiovascular transport leads to poor gastrointestinal function and metabolic stresses on the tissues of the dog, and weight loss occurs.
Despite many compensatory mechanisms, this dog is unable to deliver a normal amount of well-oxygenated blood to the body tissues, even at rest. When the dog tries to exercise, cardiac output increases very little. Therefore, when exercise-induced vasodilation occurs in the exercising muscles and total peripheral resistance decreases, blood pressure falls dramatically. There is a further decrease in blood flow in the tissues of the systemic circulation that were already vasoconstricted (e.g., mucous membranes), and these tissues become hypoxic and cyanotic. Inadequate blood flow in the exercising skeletal muscles leads to hypoxia and acidosis, and the dog collapses.
Treatment.
The ideal treatment strategy for this dog is to improve the contractile performance of the myocardium. Theoretically, β-adrenergic agonists or cardiac glycosides could be administered to increase cardiac contractility. However, currently available drugs are either ineffective or only mildly effective in dogs with severe, chronic heart failure. Therefore, treatment should emphasize symptomatic therapy, with the goals of controlling pulmonary congestion and improving cardiac output. Diuretics or venodilators reduce venous pressures and are usually effective in controlling signs of congestion. They must be used cautiously, however, because they create the risk of lowering preload and therefore exacerbating the low cardiac output. Arteriolar vasodilators can augment the output of a failing heart by reducing the afterload (arterial pressure) against which the heart must eject blood. An appropriate initial treatment for this dog includes a diuretic (furosemide) and a cardiac glycoside (digitalis). If digitalis fails to improve cardiac contractility in this advanced case of cardiomyopathy, an arteriolar vasodilator (hydralazine) or a mixed vasodilator-venodilator (enalapril) can be added to the furosemide regimen.
Despite therapy, the prognosis for a dog with such severe, chronic heart failure is poor.
Cow with “Hardware Disease”
History.
A 4-year-old, pregnant Holstein cow is presented for lethargy, poor appetite, and edema. She is due to calve in 2 months. The producer noticed that over the last few weeks the cow has seemed progressively more lethargic and reluctant to move. He observed swelling below her jaw and in her brisket. She has lost 75 to 125 pounds.
Clinical Examination.
The cow appears depressed. She is dehydrated. Her mucous membranes are dark (indicating poor perfusion), and capillary refill time is prolonged. She has marked brisket and submandibular edema. Her jugular veins are prominent. She grunts when she moves. Her temperature, pulse, and respiratory rates are all increased. Her heart sounds are muffled (as if heard through fluid), and she has a murmur (“washing machine” murmur). She has increased bronchovesicular (respiratory) sounds dorsally, but the sounds are muffled ventrally. Peripheral pulses are weak. Rumen contractions are decreased (one every 1-3 minutes). Feces are scant. Blood is submitted for a complete blood count and chemistry profile. Results indicate that the white blood cell count is low and serum creatinine concentration is increased. Fibrinogen, globulins, and total protein are all increased. Calcium and potassium levels are low.
An electrocardiogram reveals decreased amplitude of QRS complexes and ST segment elevation. Echocardiography reveals excessive fluid and gas in the pericardial space. Fibrin tags are also present. The right atrium and right ventricle appear to “collapse” during diastole, which is consistent with cardiac tamponade (excessive pericardial fluid pushing in on the heart). The left ventricle also contracts less forcefully and less completely than normal during systole (“decreased left ventricular free wall motion”).
With guidance from the echocardiogram, a sample of pericardial fluid is obtained. The fluid is reddish in color (rather than clear) and has a distinct, bad odor. Laboratory analysis reveals elevated protein concentration and an elevated count of white blood cells (primarily neutrophils) in the pericardial fluid. Culture reveals that both aerobic and anaerobic bacteria are present.
Comment.
This cow has traumatic reticuloperitonitis with pericarditis. Traumatic reticuloperitonitis (TRP), or “hardware disease,” is common in cattle. Cattle are indiscriminate eaters, and they accidentally swallow sharp metal objects that get mixed into their feed. Metal objects settle in the reticulum of the rumen. Contractions of the reticulum may push sharp objects through the wall of the reticulum and into the peritoneum. Bacteria follow and cause peritonitis. Subsequently, the sharp object may penetrate the diaphragm, which is located just cranial to the reticulum, and may then move on to penetrate the pericardium. The consequence is pericarditis (inflammation of the pericardium). Sequelae include formation of scar tissue (seen as fibrin tags), pericardial bacterial infection, and accumulation of inflammatory fluid in the pericardium. The pericardial fluid presses on the cardiac chambers, restricting their filling during diastole, and this causes pump failure.
Evidence of congestive pump failure in this cow includes poor perfusion (weak pulses, dark mucous membranes, and prolonged capillary refill time), cardiac abnormalities (subnormal right atrial and ventricular filling, decreased left ventricular motion), elevated heart rate, distended jugular veins, edema, and lethargy.
Treatment.
Prognosis is poor in this case because of the combination of pericardial infection and congestive pump failure. The producer could try to treat the infection, in hopes of delivering a live calf. Because the cow is already in pump failure with very limited cardiac output, however, it is likely that the calf is not receiving sufficient blood flow and oxygen. The calf could die in utero and could be aborted by the cow.
Boulpaep EL. Integrated control of the cardiovascular system. In: Boron WF, Boulpaep EL. Medical physiology: a cellular and molecular approach. Philadelphia: Saunders, 2005.
Bulmer BJ, Sisson DD. Therapy of heart failure. Ettinger SJ, Feldman EC. Textbook of veterinary internal medicine: diseases of the dog and cat, ed 6, St Louis: Saunders, 2005.
De Morais HA, Schwartz DS. Pathophysiology of heart failure. Ettinger SJ, Feldman EC. Textbook of veterinary internal medicine: diseases of the dog and cat, ed 6, St Louis: Saunders, 2005.
Guard C. Traumatic reticuloperitonitis. Smith BP, ed. Large animal internal medicine, ed 3, St Louis: Mosby, 2002.
Guyton AC, Hall JE. Textbook of medical physiology, ed 11. Philadelphia: Saunders, 2006.
Hamlin RL, Stokhof AA. Pathophysiology of cardiovascular disease. In: Dunlop RH, Malbert CH. Veterinary pathophysiology. Oxford: Blackwell Publishing, 2004.
Katz AM. Physiology of the heart, ed 4. Baltimore: Lippincott Williams & Wilkins, 2005.
King AS. The cardiorespiratory system: integration of normal and pathological structure and function. London: Blackwell Scientific, 1999.
Kittleson MD. Feline myocardial disease. Ettinger SJ, Feldman EC. Textbook of veterinary internal medicine, diseases of the dog and cat, ed 6, St Louis: Saunders, 2005.
Levy MN, Pappano AJ. Cardiovascular physiology, ed 9. Philadelphia: Mosby, 2007.
Lilly LS. Pathophysiology of heart disease: a collaborative project of medical students and faculty, ed 4. Baltimore: Williams & Wilkins, 2006.
Marr C, ed. Cardiology of the horse. London: Saunders, 1999.
Meurs KM. Primary myocardial disease in the dog. Ettinger SJ, Feldman EC. Textbook of veterinary internal medicine: diseases of the dog and cat, ed 6, St Louis: Saunders, 2005.
Mohrman DE, Heller LJ. Cardiovascular physiology, ed 6. New York: Lange Medical Books/McGraw-Hill, 2006.
Patteson MW. Acquired cardiac disease. In: Robinson NE, ed. Current therapy in equine medicine 4. Philadelphia: Saunders, 1997.
Reed SM, Bayly M, Sellon DC. Equine internal medicine. Philadelphia: Saunders, 2004.
Rowell LB. Human cardiovascular control. New York: Oxford University Press, 1993.
PRACTICE QUESTIONS
1. During experimental trials on a new artificial aortic valve, a dog is anesthetized and placed on cardiac bypass for 1hour (i.e., a heart-lung machine is substituted for the dog’s own heart and lungs). After successful installation of the artificial valve, the dog is taken off bypass, and the normal circulation is restored. Ten minutes later, the dog’s central venous pressure is 20 mm Hg, mean arterial pressure is 90 mm Hg, and heart rate is 130beats/min. The cardiac output is not measured, but the surgeon suspects that it is too low and therefore the patient’s tissues are not being adequately supplied with blood. Which of the following measures would be best to improve the patient’s condition?
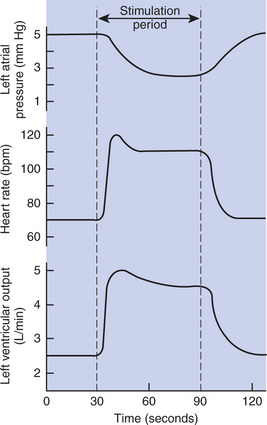
2. One of the nerves leading to a dog’s heart is stimulated for 1 minute while left atrial pressure, heart rate, and left ventricular output are measured (Figure 26-10). During this stimulation:
3. One hour after a severe hemorrhage, a dog’s arterial pulse pressure, mean pressure, and hematocrit are all below normal. Which of the following statements is true?
4. When a sheep is held in a vertical, head-up position, arterial pressure decreases because: