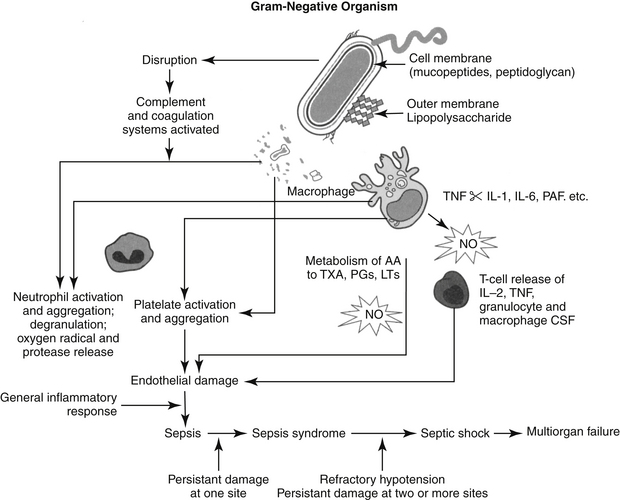
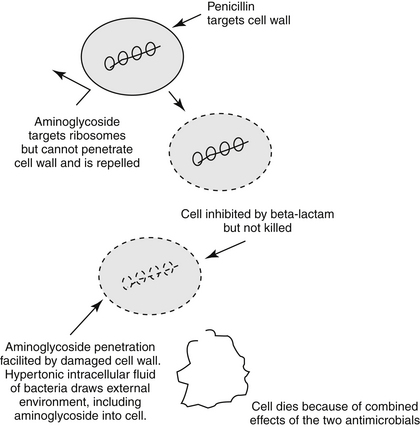
Microbial Factors that Affect Antimicrobial Efficacy
Among the most obvious ways that microbes can affect antimicrobial efficacy is the advent of resistance. However, microbes can negatively affect antimicrobials through mechanisms that do not influence MIC. These effects are not as obvious to detect as resistance but nonetheless can profoundly affect therapeutic success.
Inoculum Size
The larger the bacterial inoculum at the target site, the greater the concentration (number of molecules) of antimicrobial necessary to kill the organism. Further, more CFUs are more likely to produce greater amounts of enzymes or other materials that can destroy the drug. The “inoculum effect” of ESBL resistance describes the increasing MIC of the organisms toward cephalosporins at a larger (107) compared with smaller (105) inoculum.92 In addition, the larger the inoculum, the greater the risk that spontaneous mutation will contribute to resistance or virulence. Note that resistance and virulence do not necessarily co-exist. In general, emerging resistance appears to be associated with decreased rather than increased virulence, although increasingly studies are identifying exceptions. For example, community-acquired infections may be associated with increased virulence, but less resistance. For example, although hospital-acquired infections tend to be caused by nonvirulent organisms, community-acquired infections reflect virulent organisms that can infect even the overtly healthy patient. Concern regarding MRSA reflects, in part, its apparent acquisition of virulence factors that have facilitated its transition from a hospital to community-acquired infection.
Virulence Factors
The degree of pathogenicity of bacteria (virulence) will affect antimicrobial efficacy indirectly by facilitating infection. The ability of microbes to cause disease reflects the size of the inocululm, the effectiveness of host defense mechanisms, and the intrinsic pathogenicity of the microbes resulting from the presence of virulence factors. Like biochemical mechanisms of resistance, virulence factors generally involve proteins encoded by DNA of chromosomal or shared (e.g., plasmids, transduction) origin. Contributing to the negative impact of virulence factors is host response to their effects. Virulence factors facilitate adhesion to host cell surfaces, colonization (e.g., urease of Helicobacter pylori, which protects it from gastric acidity), invasion (facilitated by disruption of host cell membranes or stimulation of endocytosis), immunosuppression (e.g., antibody-binding proteins), or bacterial toxins that cause local, distant, or both (e.g., endotoxin) host damage. Pathogen attachment to host cells is a crucial early step in mucosal infections and is facilitated in epithelial tissues by bacterial adherence. Adherence is a specific two-phase process involving bacterial virulence factors called adhesins and complementary receptors of the host epithelial cells.93,94 Adhesins are generally found on the surface of microbes, (e.g., bacterial fimbriae) and along with other virulence factors facilitating infection, may be targets for alternative (to antimicrobial)therapy. Species differences exist among the types of receptors in the host epithelial cells. The predominant receptor type in humans is glycolipid in nature, and its presence varies with blood cell types, implying individual variation in susceptibility to bacterial adherence in several body systems. Bacterial adherence is discussed with regard to specific body systems in Chapter 8.
Another virulence factors that facilitate infection are invasins. Invasins are enzymes that damage physical barriers presented by tissue matrices or cell membranes, facilitating rapid bacterial spread. Examples include clostridial hyaluronidase, which is able to destroy connective tissue, and lecithinases and phospholipases of clostridial and gram-positive organisms. Bacteria have developed siderophores, which are specialized virulence factors that mediate the release or scavenging of iron critical for microbial virulence. Bacteria also have developed specialized transport systems that secrete toxic materials into the extracellular matrix. It is not clear whether the efflux proteins that transport toxins are related to those that transport drugs (see the discussion of resistance). Bacteria also facilitate invasion through materials (e.g., proteins, “slime”) that prevent phagocytosis or, if the microbe is phagocytized, preclude intracellular killing. Examples include lytic enzymes of gram-positive cocci or exotoxin A produced by P. aeruginosa. Toxins include both endotoxins (discussed in depth later) and exotoxins. Bacterial exotoxins are among the most potent toxins known, acting on either the cell surface (e.g., E. coli hemolysins, “superantigens” of S. aureus or Streptococcus pyogenes); membrane; or, once the membrane is penetrated, intracellular targets (e.g., A/B toxins).
Biofilm
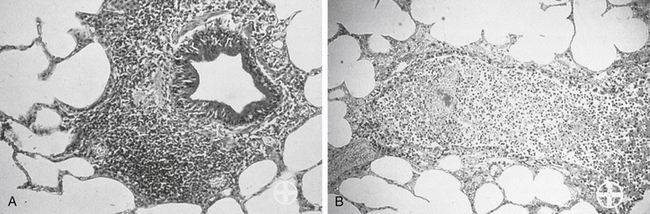
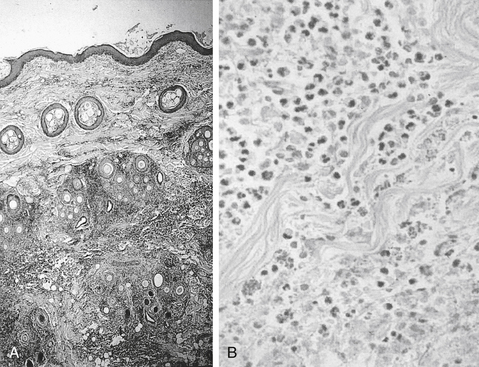
Among the most effective and probably least appreciated protective microbial factors is biofilm. Bacteria exist in either a planktonic (free floating) or sessile (attached) state; while it is the former state that characterizes C&S testing, but it is the latter state that enables persistence of the resident population, as well as the formation of biofilm.95-97 Biofilm is defined as a biopolymer, matrix-enclosed bacterial population in which bacteria adhere either to one another or to a surface.95 The outer layer of the biofilm may lose water such that it is hardened, thus providing better protection from the environment, including exposure to antimicrobials. The inner sactum of the biofilm is largely aqueous, composed of glycocalyx or slime (e.g., Staphylococcus spp.). In addition to passive diffusion, aqueous pores permeate the structure, allowing movement of nutrients and metabolic debris. Biofilm populations containing normal microflora in the skin or mucous membranes (e.g., urinary bladder) are lost with shedding of the skin (or bladder) surface or by the excretion of mucus; new cells and mucus are rapidly colonized by biofilm-forming bacteria. Microbes released from the surface may colonize new surfaces and subsequently produce new biofilms and new (e.g., persistent or recurring) infections. Bacterial communication during biofilm formation is sophisticated, involving quorum-sensing systems that ultimately may be targets of microbial therapy.96 Biofilm may facilitate and protect growth of normal or pathogenic flora on foreign surfaces and can facilitate subsequent translocation of microbes to otherwise sterile tissues. Persistent, chronic bacterial infections may reflect biofilm-producing bacteria; persistent inflammation associated with immune complexes contributes to clinical signs. Dental plaque is a prototypic example of the impact that biofilm might have on preventing antimicrobial penetration. Cystic fibrosis associated with Pseudomonas is a disease in which biofilm contributes to mortality. Pathogens associated with biofilm in veterinary medicine include, but are by no means limited to, Acinetobacter, Actinobacillus, Klebsiella, P. aeruginosa, and Staphylococcus (aureus and pseudintermedius).95 Glycocalyx may contribute to protective mechanisms of other organisms as well (e.g., sulfur granules and Nocardia; Figure 6-14). Not all pathogens associated with biofilm cause infection (e.g., urinary catheters). However, because they ultimately may be the source of infection, clinical resolution may not be possible until the biofilm is destroyed. Yet, its nature is difficult to predict based on the planktonic growth of individuals in cultures compared to the consortium that occurs in vivo.97 Catheters (urinary or intravascular), orthopedic fixation devices, and materials used in wound management are examples of surfaces on which biofilm might develop.
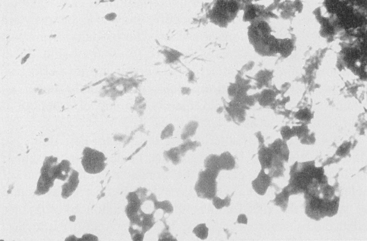
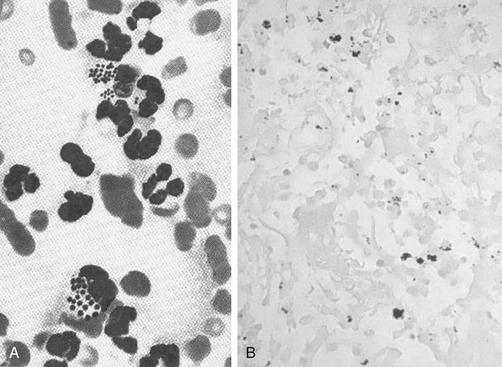
Figure 6-14 An example of combined host and microbial factors that negatively impacts therapy, Nocardia causes a marked inflammatory response by the host. Additionally, the organism causes secretion of calcium that combines with its biofilm, resulting in the formation of “sulfur” granules that protect the organisms from drug penetrations.
Antimicrobial Resistance
The role of resistance in therapeutic failure of antimicrobials is well established.23,98 The use of antimicrobials increasingly is associated with emergence of resistance. For each class of antimicrobial drugs approved for use in human medicine, resistance generally has emerged within 1 to 2 decades of use. Clinically relevant resistance toward sulfonamides, the first class of antimicrobials approved in the United States (1930s) was documented by the 1940s. Penicillins, tetracyclines, streptomycin (aminoglycoside), and erythromycin (macrolides) were all approved within a 10-year span, with resistance documented within 5 years for methicillin versus approximately 10 years for streptomycin. Resistance to nalidixic acid, the progenitor of fluoroquinolones (approved in 1950), took 3 decades to emerge, perhaps convincing manufacturers that resistance to fluoroquinolones would emerge very slowly. However, resistance to norfloxacin, the first fluoroquinolone approved in the United States, took less than 3 years to emerge, despite the fact that the lack of plasmid-mediated resistance was among the attributes of this class. Resistance to extended-spectrum cephalosporins emerged within 4 years of approval and to amoxicillin–clavulanic acid, within 5 years. Resistance to vancomycin, specifically developed to treat MRSA, emerged in its second decade of use.
KEY POINT 6-21
Antimicrobial resistance increasingly will prevent the successful empirical selection of antimicrobial drugs.
Inherent Versus Acquired Resistance
Antimicrobial resistance might be inherent to the microorganisms or acquired, either through chromosomal mutations or transfer of genetic information.99 Generally, spectrums of antimicrobials (listed on package inserts and elsewhere) reflect inherent resistance patterns rather than acquired resistance patterns. Examples include limited efficacy of aminoglycosides toward anaerobic organisms because the drugs must be actively transported into the cell (oxygen dependent) or the resistance of gram-positive organisms, which lack an outer cell membrane, to polymyxin B, which targets the same. Acquired resistance, on the other hand, generally renders a previously susceptible organism resistant. As such, it is not necessarily predictable and can occur during the course of therapy (leading to changes in a C&S pattern). More problematically, it is often shared among microbes.
Shared resistance among bacteria reflects the ability of bacteria to incorporate extrachromosomal DNA carrying the information for resistance from other organisms. Extrachromosomal DNA (including plasmids and bacteriophages) encode for resistance to multiple drugs and can be transmitted vertically (to progeny) or horizontally, across species and genera. Transposons are individual or clusters of resistance genes bound by integrons, which move resistance genes back and forth between chromosomes to plasmids. Consequently, bacterial resistance is extremely mobile and can spread rapidly.101 Among the mechanisms by which genetic resistance information is shared is (sexual) conjugation. Conjugation occurs particularly in gram-negative organisms and may be accompanied by genetic material that confers bacterial pathogenicity as well as altered metabolic functions. However, Enterococcus spp. and selected other gram-positive bacteria also transfer resistance to glycopeptides through conjugative transposons.101 Transduction, which requires a specific receptor, involves transfer of information by a bacterial virus (bacteriophage) and is implemented especially by Staphylococcus spp. Resistance, including methicillin resistance, can be transferred between coagulase-negative and -positive Staphylococcus.1 Transformation involves transfer of naked DNA from one lysed bacterium to another; this mechanism of transfer tends to be limited (in humans) to pneumococcal meningitis.
Although present for eons, acquired antimicrobial resistance increasingly is becoming problematic. The impact of antimicrobial resistance can be extensive. In some human intensive care units, selected isolates are characterized by a resistance prevalence of 86%. The impact of resistance on the patient includes increased morbidity, mortality, and increased hospital costs.107 Patterns of resistance have emerged in veterinary medicine, although differences appear to occur in the ability of organisms to develop resistance to an antimicrobial, varying with species and strain. Many organisms remain predictably susceptible to selected drugs (e.g., Brucella, Chlamydia), whereas others are becoming problematic (e.g., P. multocida). Several organisms traditionally have developed resistance that can rapidly impair efficacy of new antimicrobials (e.g., E. coli, K. pneumoniae, Salmonella, S. aureus, S. pneumoniae). In general, these organisms have developed multidrug resistance (MDR). MDR is now considered the normal response to antimicrobials for gram-positive cocci pneumococci, enterococci, and staphylococci.102 Among these, Staphylococcus spp. is considered most problematic: it is intrinsically virulent, is able to adapt to many different environmental conditions, increasingly is associated with resistance to other classes of antimicrobials, and tends to be associated with life-threatening infections.102,103 In a veterinary teaching hospital the percentage of patients with S. intermedius susceptible to cephalexin and amoxicillin–clavulanic acid decreased from a high of 96% in 2005 to < 60% in 2007, a trend that appears to be emerging in other veterinary hospitals.110
E. coli is among the organisms that have developed multi-drug resistance.104,105 Fluoroquinolone-resistant E. coli emerged as early as 1998, little over a decade after the approval of enrofloxacin for dogs or cats.28 Multidrug-resistant E. coli has emerged as a cause of nosocomial infections in dogs108 and UTIs in canine critical care patients.104,109 The presentation is similar to the that in human critical care patients, with risk factors such as sex (males), hospital stay, and previous antimicrobial therapy being similar for both.
Factors Contributing to the Emergence of Resistance
Development of antimicrobial resistance is facilitated by several factors111; among the most important is exposure to antimicrobials. In the individual patient, single-dose ciprofloxacin prophylaxis increased the prevalence of ciprofloxacin-resistant fecal E. coli from 3% to 12% in humans.112 Ciprofloxacin treatment for prostatitis resulted in posttreatment fecal colonization with quinolone-resistant E. coli that was genetically distinct from the infection-causing strains after treatment in 50% of the patients.113 Our laboratory has demonstrated that standard doses of either amoxicillin or enrofloxacin given orally will cause close to 100% of fecal E. coli to become resistant to the treatment drug within 3 to 9 days of therapy; for enrofloxacin the isolates generally are multidrug resistant. As with MRSA or MRSI (S. intermedius), the advent of resistance by E. coli and other gram-negative organisms has been associated with increased cephalosporin use.1
The gastrointestinal flora offers a natural environment that exemplifies the impact of antimicrobials on selection pressure. The normal flora of the gastrointestinal tract is extremely diverse, with anaerobes predominating. Among the aerobes, E. coli are the major gram-negative and Enterococcus the major gram-positive organisms.101 Environmental microbes maintain an ecologic niche through suppression of the competition by either consumption of nutrients or secretion of antibiotics. Therefore commensal organisms are constantly being exposed to antibiotics, and are “primed” to develop resistance.101 However, the microbes producing the antibiotic, as well as surrounding normal flora, are resistant to the antibiotic. Thus genes for resistance develop along with genes directing antibiotic production.
Rapid microbial turnover in the gastrointestinal tract supports the development of resistance by ensuring active DNA replication and thus mutation potential (see previous discussion). Chromosomal (DNA) mutations (10-14 to 10-10 per cell division) are DNA mistakes that have been missed by bacterial repair mechanisms. These mistakes occur spontaneously and randomly, regardless of whether the antibiotic is present. If the mutation that confers resistance to an antimicrobial occurs in the presence of the antimicrobial when it is administered to the patient, the surviving mutant, reflecting its single-step mutation, confers a low level of resistance (see the discussion of mutant prevention concentration). The MIC of the organism is likely to increase. Further microbial turnover and continued therapy can lead to multistep mutations and rapid emergence of high-level resistance characterized by increasingly higher MIC. Stepwise mutations can lead to specific resistance such as that demonstrated toward fluorinated quinolones (stepwise mutation in the DNA gyrase gene). Nonspecific mechanisms of resistance, including that shared among organisms, are more likely to result in MDR. Microflora of the gastrointestinal tract can serve as a reservoir of resistance genes; a single drug, via integrons, plasmids, and transposons, facilitates the rapid transfer of MDR among organisms. The gastrointestinal environment exemplifies a pattern whereby resistance can emerge as a result of a combination of selection pressure and mutation. Clinically, similar mechanisms of emerging resistance are likely to occur at sites of infection.
Mutant Prevention Concentration
Drlica and coworkers114 have hypothesized the mutant selection window, (see Figure 6-15) comprised of a lower threshold represented by the culture MIC of the infecting organism and an upper threshold or boundary, the MPC. Should a dose be designed such that drug concentrations fall within this window (i.e., between the MIC and MPC) at the site of infection, the mutant isolate is likely to emerge as a resistant colony. The practical application of the hypothesis explains the observed behavior of mycobacterium organisms toward fluoroquinolones (FQs). Increasing concentrations of the FQs inhibits the nonresistant (wild-type) organisms and colony numbers rapidly decrease. But this period of decline is followed by a plateau period of minimal or no growth. During this plateau phase, remaining resistant isolates recover and start to multiply again. The resistance of this emerging, second population presumably reflect a single-step (chromosomal or plasmid-mediated) mutation that resulted in an increase in the MIC to low-level resistance (e.g., MIC is close to the breakpoint). However, when these first-step mutants are exposed to even higher drug concentrations, a second rapid decline in numbers occurs, this time reflecting inhibition of the mutated, resistant organisms. Again, once sufficient bacteria recover, a second plateau occurs as the first-step mutants mutate. This stepwise or multistep mutation confers high level resistance (MIC exceeds the breakpoint several fold) that can be overcome only by very high concentrations of the FQ. The mutant selection window, which is to be avoided with initial therapy, describes drug concentrations on either side of the initial plateau for the single-step mutants. The lower boundary is defined by those drug concentrations sufficiently high to remove the majority of the wild-type competitors (MIC), whereas the higher boundary (the MPC) is defined by the concentrations necessary to inhibit the least susceptible (most resistant) isolates (the single-step mutants).115 Above this concentration, a second mutation step (which is very rare) would be required for a population of resistant organisms to develop; the risk of this happening is reduced by preventing microbial turnover (i.e., killing all isolates).
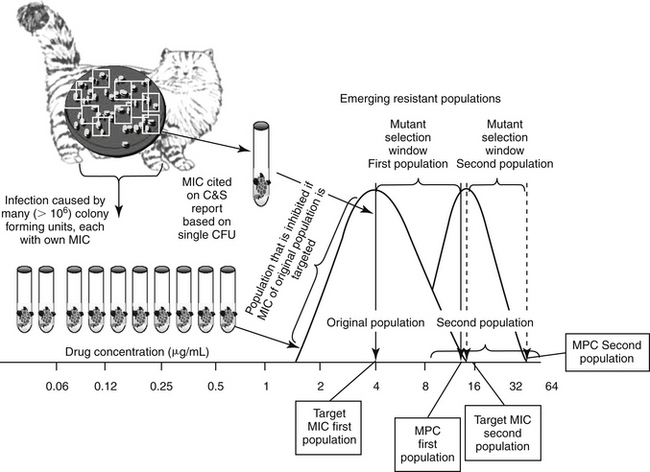
Figure 6-15 Stepwise mutation can emerge as a result of selection pressure induced by antimicrobial therapy that targets the minimum inhibitory concentration (MIC) of the infecting microbe. The mutant prevention concentration (MPC) is the concentration of drug that is necessary to inhibit first-step mutants, or the MIC of the least susceptible isolate in a resident population of pathogens. As the resident population or inoculum of wild (nonresistant) pathogen isolates reaches 108-10 colony-forming units (CFUs), some isolates will spontaneously mutate such that resistance emerges to the drug of interest. However, when cultured, the MIC reported for the population is likely to represent the mode (the most commonly reported MIC), which in a normally distributed population, is also the MIC50 for the population. In contrast, the MIC of the first-step mutant will be the high end of the population MIC range. This is the concentration that should be targeted to inhibit the entire population—that is, the MPC. If the dosing regimen is designed to target the the mutant selection window, that is, the MIC of the wild population rather than the MPC (the MIC of the first-step mutant)—treatment with the drug will inhibit all isolates at or below the MIC. The void in isolates will allow the remaining, more resistant first-step mutants to recover, particularly in patients not sufficiently healthy to suppress recovering microbes. As this new population expands, a second distribution curve emerges. If recultured, the MIC of the second first-step mutant population will be higher than the wild population. If the population reaches a sufficient size (e.g., 108 CFUs), a second, spontaneous mutation is likely to occur, resulting in a new, higher MPC. Targeting the MPC is particularly important when using drugs for which resistance emerges in response to mutations.
KEY POINT 6-23
The mutant prevention concentration (MPC) is the highest MIC of any of the colony-forming units causing infection in the patient. Failure to achieve this concentration may allow resistant microbes to emerge, particularly in the at-risk patient.
On the basis of this observation, Drlica and coworkers contend that MIC-based strategies used to design dosing regimens readily select for resistant mutants.115 Their contention is based on the observation that only one resistant mutation is needed for bacteria to grow in the presence of an antimicrobial and that infections generally contain an adequate number of CFUs for several first-step resistant mutants to be present prior to treatment. They coined the term MPC as an in vitro measure of preferred antimicrobial concentration target. If the MPC (rather than the MIC) is achieved at the site of infection, the risk of resistance is minimized because isolates that exceed the MPC concentration must have undergone a second concurrent resistance mutation step prior to therapy. As such, the MPC, not the MIC, would be the concentration targeted at the site of infection in the patient. Indeed, simply achieving the reported MIC of the infecting microbe at the site of infection is probably the approach that is most likely to yield clinically resistant organisms. Accordingly, consideration should be given to assuring that “dead bugs don’t mutate.” If the least susceptible of the isolates is inhibited with the dosing regimen, then the recovering population should not be resistant.
Drlica115 has demonstrated that MPCs do not correlate to MICs. In vitro, the MPC would be defined in vitro as the (lowest) drug concentration (in the media) that yields no recovered organisms when over 1010 CFUs (mimicking bacterial load in the patient) are plated. Currently, determining the MPC is costly, requiring multiple testing steps and large numbers of cells; for example, standard culture procedures are based on 106 CFUs, whereas determination of the MPC requires at least 108-10 CFUs. However, an MPC-based strategy to dosing clinically makes sense and should be an effective means of blocking the growth of first-step resistant mutants. Such a strategy would force wild-type cells to acquire two resistance mutations for growth, an event that is rare. Experimental in vitro data114 have confirmed that MPC levels of an FQ do indeed inhibit strains that harbor first-step gyrA mutations (the mechanism of microbial resistance to FQ).116 Application of the MPC is most appropriate for drugs and organisms that develop resistance by chromosomally mediated point mutations (e.g., the fluorquinolones).117 However, the spirit of targeting the MPC might be assumed even for other drugs, in order to minimize the impact of selection pressure on emergent resistant populations. The mutant selection window can be narrowed if more than two bacterial sites are targeted, such as might occur with combination antimicrobial therapy, or with drugs that simultaneously target more than one site (e.g., the FQs).114
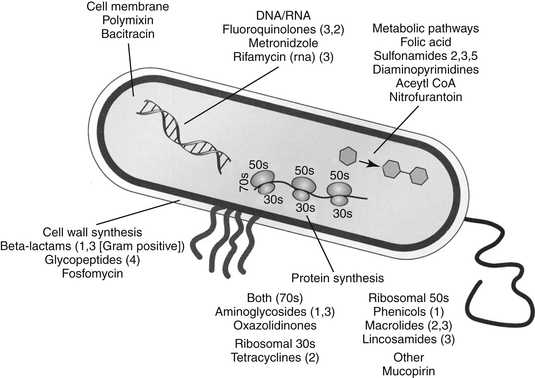
Biochemical Mechanisms of Resistance
Bacteria often respond to the presence of the antimicrobial by altering their physiology such that resistance occurs, often to multiple drugs. Microbes develop antimicrobial resistance by two primary mechanisms: modification of the target site or altered intracellular drug concentration. Methods by which intracellular drug concentration can be decreased include changes in porin sizes for gram-negative organisms (e.g., most drugs; see Figure 6-3). Porins are transmembrane proteins (e.g., OmpF) that form an aqueous channel that allows passive movement of large hydrophilic molecules. Porins are one of the few means by which drugs can gain access to intracellular targets. A change in porin size (i.e., by the addition of side chains that filter out drugs) or number increases antimicrobial resistance, as is demonstrated by the loss of the OprD protein that imparts resistance to imepenem by Pseudomonas spp. Closely associated with the porin proteins are efflux proteins that pump drug out of the organism; the pumps often are associated with porin proteins (e.g., FQs and tetracyclines). Most of these pumps are fueled by energy associated with proton exchange, the most notable in gram negative organisms being of the RND (resistance nodulation division) family. The best characterized in this family is the Acr-AB/TolC system, which is a complex bacterial stress response system that allows bacteria to pump out toxic molecules.101 These and other pump systems are often characterized by a wide range of substrate specificities and, along with porins, are a common mechanism whereby an isolate can express multidrug resistance. In contrast, a number of microbes generate enzymes that destroy antimicrobials (e.g., aminoglycoside acetylases, beta-lactamases that destroy penicillins or cephalosporins, transferases that destroy chloramphenicol); in such instances resistance conferred by these mechanisms is generally limited to a single drug or drug class. Enzymatic inactivation is more likely for natural drugs to which microbes have previously been exposed (and thus presented with a greater opportunity to develop enzymes). In contrast, enzymes are less likely to destroy synthetic drugs.118 However, plasmid-mediated enzymatic destruction of FQs has recently been described, once again highlighting the resourcefulness of bacteria.119 Changes in target structure are another major mechanism of resistance. Example targets that have been modified include, but are not limited to, cell wall proteins (e.g., penicillin-binding proteins [PBs], particularly for MRSA [PB2] or Enterococcus [PB5]), or binding sites (i.e., on ribosomes, as for aminoglycosides, or DNA gyrase for FQs).120,121 Organisms often are characterized by more than one mechanism of resistance. Multiple mechanisms are well documented for some organisms against selected beta-lactams and have been described against FQs (e.g., altered DNA gyrase and increased efflux pumps) and others. Resistance can be induced, as is exemplified by beta-lactamase formation in Staphylococcus spp. which greatly increases in the presence of a beta-lactam antibiotic, or for fluoroquinolone, for which efflux pump activity is markedly upregulated. Discussion of specific mechanisms of resistance will be addressed with the appropriate drugs (see Chapter 7).
Avoiding Antimicrobial Resistance
Among the approaches to reducing resistance are pharmacologic manipulations and changes in antimicrobial use practices. Pharmaceutical manufacturers have been able to manipulate antimicrobial drugs in a variety of ways such that resistance is minimized, and these options can be selected in an attempt to minimize resistance. For example, bacterial resistance has been decreased by synthesizing smaller molecules that can penetrate smaller porins (e.g., the extended-spectrum penicillins ticarcillin and piperacillin); synthesizing larger molecules that force the microbe to develop more than one point mutation (e.g., later-generation FQs), “protecting” the antimicrobial from enzymatic destruction (e.g., with clavulanic acid, which diverts the beta-lactamase from the penicillin); modifying the compound so that it is more difficult to destroy (e.g., amikacin, which is a larger and more difficult to reach molecule than gentamicin and carbapenems, later generation cephalosporins); and developing lipid-soluble compounds that are more able to achieve effective concentrations at the site of infection (e.g., doxycycline compared with other tetracyclines). Increasingly, drug design–based tactics will be implemented to minimize emergent resistance. Increasingly the role of the practitioner is equally important. A three “D”s approach might reduce the risk of emergent resistance: De-escalate antimicrobial use, design a treatment regimen that minimizes resistance (dead bugs don’t mutate), and decontaminate the environment through proper hygiene. These approaches are exemplified by strategies implemented by intensive care units to reduce antimicrobial resistance that often involve a multitiered approach (Box 6-3). Actions include the following:
KEY POINT 6-24
The goal of antimicrobial therapy is twofold: resolving clinical signs associated with infection and avoiding emergent resistance. The two goals are not mutually inclusive.
Risk factors for emerging resistance in the hospital or community setting include but are not limited to increased antimicrobial use, host factors such as severity of illness and length of stay, and lack of adherence to infection control practices.107 Consequently, among the de-escalation efforts implemented in human hospital and community environments is restricted antimicrobial use. In humans the increasing presence of drug-resistant bacterial infections among hospitalized patients is linked to the greater numbers of patients receiving inappropriate antimicrobial treatment.123 A recent on-line report found that in human medicine, antimicrobials were often prescribed despite infection being an infrequent cause of the illness (i.e., pharyngitis). Further, the chosen antimicrobial often was inappropriate for those bacteria potentially causing infection in the treated body system.124 Accordingly, reducing inappropriate antimicrobial use has become a priority in human medicine.
Among the more rational paradigms for antibacterial de-escalation, is an approach to empirical antimicrobial use in the hospital setting for patients with serious bacterial infections.123 Such antimicrobial de-escalation attempts to balance the need to provide appropriate initial antibacterial treatment while limiting the emergence of antimicrobial resistance. The goal of de-escalation in this setting is to prescribe an initial antimicrobial regimen that will cover the most likely bacterial pathogens associated with infection while minimizing the risk of emerging antimicrobial resistance.123 The three-pronged approach includes narrowing the antimicrobial regimen through culture, assessing isolate susceptibility for dose determination, and choosing the shortest course of therapy clinically acceptable. Judicious antimicrobial use combined with restricted use of ceftazidime led to a decreased antimicrobial resistance to beta-lactams, in general, in a human teaching hospital environment.126 Note that this strategy does not exclude the use of “big gun” antimicrobials. The approach of withholding use of high-impact drugs (e.g., meropenem or vancomycin) in patients whose need for effective therapy is critical to avoid emerging resistance that might limit drug use in later patients may not be rational or in the best interest of the patient. A more appropriate approach is to use the drug correctly. However, routine use of less powerful drugs is appropriate but only if these alternatives are just as effective. Regardless of the choice, once the decision is made to use an antimicrobial, attention must be paid to dosing regimens that minimize the advent of resistance by ensuring that infecting microbes are eradicated.
Another strategy to decrease the impact of antimicrobial use on resistance is a decreased duration of therapy (see the discussion of enhancing antimicrobial efficacy). One study in human critical care patients found that reducing the duration of antimicrobial therapy from 14 days to 10 days decreased the emergence of resistance.127 Increasingly, clinical trials will focus on demonstrating efficacy of shorter (i.e., < 5 to 7 days) treatment regimens.