Pediatric Diseases
As mentioned earlier and illustrated by several examples, many diseases of infancy and childhood are of genetic origin. Others, although not genetic, either are unique to children or take distinctive forms in this patient population and thus merit the designation pediatric diseases. During each stage of development, infants and children are prey to a somewhat different group of diseases (Table 6–6). Clearly, diseases of infancy (i.e., the first year of life) pose the highest risk of death. During this phase, the neonatal period (the first 4 weeks of life) is unquestionably the most hazardous time.
Table 6–6 Causes of Death by Age
| Cause* | Rate† |
|---|---|
| Under 1 Year | 677.3 |
|
Congenital malformations, deformations, and chromosomal anomalies Disorders related to short gestation and low birth weight Sudden infant death syndrome (SIDS) Newborn affected by maternal complications of pregnancy Newborn affected by complications of placenta, cord, and membranes Respiratory distress syndrome of newborn |
|
| 1–4 Years | 28.2 |
|
Accidents (unintentional injuries) Congenital malformations, deformations, and chromosomal abnormalities Diseases of the heart‡ |
|
| 5–9 Years | 13.6 |
| 10–14 Years | 16.7 |
* Causes are listed in decreasing order of frequency. All causes and rates are final 2007 statistics.
† Rates are expressed per 100,000 population from all causes within each age group.
‡ Excludes congenital heart disease.
Data from Heron MP, Sutton PD, Xu J, et al: Annual Summary of Vital Statistics: 2007. Pediatrics 125:4, 2010.
Once the infant survives the first year of life, the outlook brightens considerably. However, it is sobering to note that between 1 year and 14 years of age, injuries resulting from accidents are the leading cause of death. Not all conditions listed in Table 6–6 are described in this chapter; only a select few that are most common are considered here. Although general principles of neoplastic disease and specific tumors are discussed elsewhere, a few tumors of children are described, to highlight the differences between pediatric and adult neoplasms.
Congenital Anomalies
Congenital anomalies are structural defects that are present at birth, although some, such as cardiac defects and renal anomalies, may not become clinically apparent until years later. As will be evident from the ensuing discussion, the term congenital does not imply or exclude a genetic basis for birth defects. It is estimated that about 120,000 babies are born with a birth defect each year in the United States, an incidence of 1 in 33. As indicated in Table 6–6, congenital anomalies constitute an important cause of infant mortality. Moreover, they continue to be a significant cause of illness, disability, and death throughout the early years of life.
Before consideration of the etiology and pathogenesis of congenital anomalies, it is essential to define some of the terms used to describe errors in morphogenesis.
• Malformations represent primary errors of morphogenesis. In other words, there is an intrinsically abnormal developmental process. Malformations usually are multifactorial, rather than the result of a single gene or chromosomal defect. They may manifest in any of several patterns. In some presentations, such as congenital heart diseases, single body systems may be involved, whereas in others, multiple malformations involving many organs and tissues may coexist (Fig. 6–21).
• Disruptions result from secondary destruction of an organ or body region that was previously normal in development; thus, in contrast with malformations, disruptions arise from an extrinsic disturbance in morphogenesis. Amniotic bands, denoting rupture of amnion with resultant formation of “bands” that encircle, compress, or attach to parts of the developing fetus, constitute the classic example of a disruption (Fig. 6–22). A variety of environmental agents may cause disruptions (see below). Disruptions are not heritable, of course, and thus are not associated with risk of recurrence in subsequent pregnancies.
• Deformations, like disruptions, also represent an extrinsic disturbance of development rather than an intrinsic error of morphogenesis. Deformations are common problems, affecting approximately 2% of newborn infants to various degrees. Fundamental to the pathogenesis of deformations is localized or generalized compression of the growing fetus by abnormal biomechanical forces, leading eventually to a variety of structural abnormalities. The most common cause of such deformations is uterine constraint. Between weeks 35 and 38 of gestation, rapid increase in the size of the fetus outpaces the growth of the uterus, and the relative amount of amniotic fluid (which normally acts as a cushion) also decreases. Thus, even the normal fetus is subjected to some degree of uterine constraint. However, several variables increase the likelihood of excessive compression of the fetus, including maternal conditions such as first pregnancy, small uterus, malformed (bicornuate) uterus, and leiomyomas. Causes relating to the fetus, such as presence of multiple fetuses, oligohydramnios, and abnormal fetal presentation, also may be involved.
• Sequence refers to multiple congenital anomalies that result from secondary effects of a single localized aberration in organogenesis. The initiating event may be a malformation, deformation, or disruption. An excellent example is the oligohydramnios (or Potter) sequence (Fig. 6–23, A). Oligohydramnios, denoting decreased amniotic fluid, may be caused by a variety of unrelated maternal, placental, or fetal abnormalities. Chronic leakage of amniotic fluid due to rupture of the amnion, uteroplacental insufficiency resulting from maternal hypertension or severe toxemia, and renal agenesis in the fetus (because fetal urine is a major constituent of amniotic fluid) all are potential causes of oligohydramnios. The fetal compression associated with significant oligohydramnios in turn results in a classic phenotype in the newborn infant, including flattened facies and positional abnormalities of the hands and feet (Fig. 6–23, B). The hips may be dislocated. Growth of the chest wall and the contained lungs also is compromised, sometimes to such an extent that survival is not possible. If the embryologic connection between these defects and the initiating event is not recognized, a sequence may be mistaken for a malformation syndrome.
• Malformation syndrome refers to the presence of several defects that cannot be explained on the basis of a single localizing initiating error in morphogenesis. Syndromes most often arise from a single causative condition (e.g., viral infection or a specific chromosomal abnormality) that simultaneously affects several tissues.
• In addition to these global definitions, some general terms are applied to organ-specific malformations: Agenesis refers to the complete absence of an organ or its anlage, whereas aplasia and hypoplasia indicate incomplete development and underdevelopment, respectively, of an organ. Atresia describes the absence of an opening, usually of a hollow visceral organ or duct such as intestines and bile ducts.
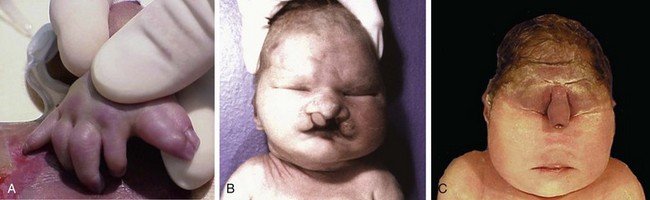
Figure 6–21 Human malformations can range in severity from the incidental to the lethal. A, Polydactyly (one or more extra digits) and syndactyly (fusion of digits), have little functional consequence when they occur in isolation. B, Similarly, cleft lip, with or without associated cleft palate, is compatible with life when it occurs as an isolated anomaly; in this case, however, the child had an underlying malformation syndrome (trisomy 13) and expired because of severe cardiac defects. C, Stillbirth representing a severe and essentially lethal malformation, in which the midface structures are fused or ill-formed; in almost all cases, this degree of external dysmorphogenesis is associated with severe internal anomalies such as maldevelopment of the brain and cardiac defects.
(A and C, Courtesy of Dr. Reade Quinton, Department of Pathology, University of Texas Southwestern Medical Center, Dallas, Texas. B, Courtesy of Dr. Beverly Rogers, Department of Pathology, University of Texas Southwestern Medical Center, Dallas, Texas.)
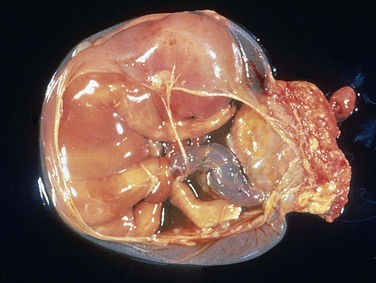
Figure 6–22 Disruptions occur in a developing organ because of an extrinsic abnormality that interferes with normal morphogenesis. Amniotic bands are a frequent cause of disruptions. In the gross specimen shown, the placenta is at the right of the diagram, and the band of amnion extends from the top portion of the amniotic sac to encircle the leg of the fetus.
(Courtesy of Dr. Theonia Boyd, Children’s Hospital of Boston, Boston, Massachusetts.)
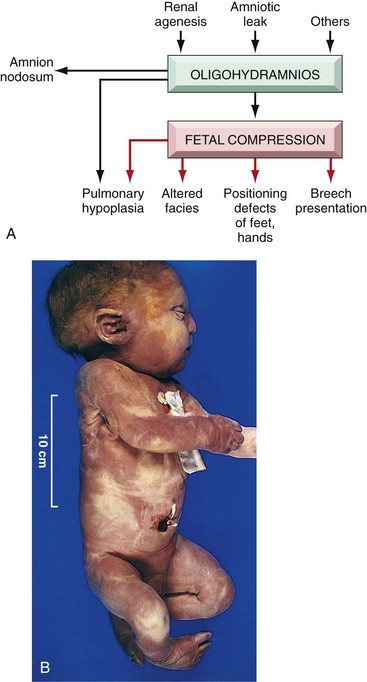
Figure 6–23 A, Pathogenesis of the oligohydramnios (Potter) sequence. B, Infant with oligohydramnios (Potter) sequence. Note flattened facial features and deformed foot (talipes equinovarus).
Etiology
Known causes of errors in human malformations can be grouped into three major categories: genetic, environmental, and multifactorial (Table 6–7). The cause has not been identified for almost half of the reported cases.
Table 6–7 Causes of Congenital Malformations in Humans
| Cause | Frequency of Malformations* (%) |
|---|---|
| Genetic | |
| Chromosomal aberrations | 10–15 |
| Mendelian inheritance | 2–10 |
| Environmental | |
| Maternal/placental infections | 2–3 |
| Rubella Toxoplasmosis Syphilis Cytomegalovirus infection Human immunodeficiency virus infection |
|
| Maternal disease states | 6–8 |
| Diabetes Phenylketonuria Endocrinopathies |
|
| Drugs and chemicals | ∼1 |
| Alcohol Folic acid antagonists Androgens Phenytoin Thalidomide Warfarin 13-Cis-retinoic acid Others |
|
| Irradiation | ~1 |
| Multifactorial | 20–25 |
| Unknown | 40–60 |
Data from Stevenson RE, Hall JG, Goodman RM (eds): Human Malformations and Related Anomalies. New York, Oxford University Press, 1993, p 115.
Genetic causes of malformations include all of the previously discussed mechanisms of genetic disease. Virtually all chromosomal syndromes are associated with congenital malformations. Examples are Down syndrome and other trisomies, Turner syndrome, and Klinefelter syndrome. Most chromosomal disorders arise during gametogenesis and hence are not familial. Single-gene mutations, characterized by mendelian inheritance, may underlie major malformations. For example, holoprosencephaly is the most common developmental defect of the forebrain and midface in humans (see Chapter 22); the Hedgehog signaling pathway plays a critical role in the morphogenesis of these structures, and loss-of-function mutations of individual components within this pathway are reported in families with a history of recurrent holoprosencephaly.
Environmental influences, such as viral infections, drugs, and radiation to which the mother was exposed during pregnancy, may cause fetal malformations (the appellation of “malformation” is used loosely in this context, since technically, these anomalies represent disruptions). Among the viral infections listed in Table 6–7, rubella was a major scourge of the 19th and early 20th centuries. Fortunately, maternal rubella and the resultant rubella embryopathy have been virtually eliminated in developed countries as a result of vaccination. A variety of drugs and chemicals have been suspected to be teratogenic, but perhaps less than 1% of congenital malformations are caused by these agents. The list includes thalidomide, alcohol, anticonvulsants, warfarin (oral anticoagulant), and 13-cis-retinoic acid, which is used in the treatment of severe acne. For example, thalidomide, once used as a tranquilizer in Europe and currently used for treatment of certain cancers, causes an extremely high incidence (50% to 80%) of limb malformations. Alcohol, perhaps the most widely used agent today, is an important environmental teratogen. Affected infants show prenatal and postnatal growth retardation, facial anomalies (microcephaly, short palpebral fissures, maxillary hypoplasia), and psychomotor disturbances. These features in combination are designated the fetal alcohol syndrome. While cigarette smoke–derived nicotine has not been convincingly demonstrated to be a teratogen, there is a high incidence of spontaneous abortions, premature labor, and placental abnormalities among pregnant smokers; babies born to mothers who smoke often have a low birth weight and may be prone to the sudden infant death syndrome (SIDS). In light of these findings, it is best to avoid nicotine exposure altogether during pregnancy. Among maternal conditions listed in Table 6–7, diabetes mellitus is a common entity, and despite advances in antenatal obstetric monitoring and glucose control, the incidence of major malformations in infants of diabetic mothers stands between 6% and 10% in most reported series. Maternal hyperglycemia–induced fetal hyperinsulinemia results in fetal macrosomia (organomegaly and increased body fat and muscle mass); cardiac anomalies, neural tube defects, and other CNS malformations are some of the major anomalies seen in diabetic embryopathy.
Multifactorial inheritance, which implies the interaction of environmental influences with two or more genes of small effect, is the most common genetic cause of congenital malformations. Included in this category are some relatively common malformations such as cleft lip and palate and neural tube defects. The importance of environmental contributions to multifactorial inheritance is underscored by the dramatic reduction in the incidence of neural tube defects by periconceptional intake of folic acid in the diet. The recurrence risks and mode of transmission of multifactorial disorders are described earlier in this chapter.
 Pathogenesis
Pathogenesis
The pathogenesis of congenital anomalies is complex and still poorly understood, but two general principles of developmental pathology are relevant regardless of the etiologic agent:
1 The timing of the prenatal teratogenic insult has an important impact on the occurrence and the type of anomaly produced. The intrauterine development of humans can be divided into two phases: (1) the embryonic period, occupying the first 9 weeks of pregnancy, and (2) the fetal period, terminating at birth.
 In the early embryonic period (first 3 weeks after fertilization), an injurious agent damages either enough cells to cause death and abortion or only a few cells, presumably allowing the embryo to recover without developing defects. Between the third and the ninth weeks, the embryo is extremely susceptible to teratogenesis, and the peak sensitivity during this period occurs between the fourth and the fifth weeks. During this period organs are being crafted out of the germ cell layers. The fetal period that follows organogenesis is marked chiefly by the further growth and maturation of the organs, with greatly reduced susceptibility to teratogenic agents. Instead, the fetus is susceptible to growth retardation or injury to already formed organs. It is therefore possible for a given agent to produce different anomalies if exposure occurs at different times of gestation.
In the early embryonic period (first 3 weeks after fertilization), an injurious agent damages either enough cells to cause death and abortion or only a few cells, presumably allowing the embryo to recover without developing defects. Between the third and the ninth weeks, the embryo is extremely susceptible to teratogenesis, and the peak sensitivity during this period occurs between the fourth and the fifth weeks. During this period organs are being crafted out of the germ cell layers. The fetal period that follows organogenesis is marked chiefly by the further growth and maturation of the organs, with greatly reduced susceptibility to teratogenic agents. Instead, the fetus is susceptible to growth retardation or injury to already formed organs. It is therefore possible for a given agent to produce different anomalies if exposure occurs at different times of gestation.2 The complex interplay between environmental teratogens and intrinsic genetic defects is exemplified by the fact that features of dysmorphogenesis caused by environmental insults often can be recapitulated by genetic defects in the pathways targeted by these teratogens. Some representative examples follow:
Cyclopamine is a plant teratogen. Pregnant sheep who feed on plants containing cyclopamine give birth to lambs that have severe craniofacial abnormalities including holoprosencephaly and cyclopia (single fused eye—hence the origin of the moniker cyclopamine). This compound is a potent inhibitor of Hedgehog signaling in the embryo, and as stated previously, mutations of Hedgehog genes are present in subsets of fetuses with holoprosencephaly. Valproic acid is an antiepileptic and a recognized teratogen. Valproic acid disrupts expression of a family of highly conserved developmentally critical transcription factors known as homeobox (HOX) proteins. In vertebrates, HOX proteins have been implicated in the patterning of limbs, vertebrae, and craniofacial structures. Not surprisingly, mutations in HOX family genes are responsible for congenital anomalies that mimic features observed in valproic acid embryopathy. The vitamin A (retinol) derivative all-trans-retinoic acid is essential for normal development and differentiation, and its absence during embryogenesis results in a constellation of malformations affecting multiple organ systems, including the eyes, genitourinary system, cardiovascular system, diaphragm, and lungs (see Chapter 7 for vitamin A deficiency in the postnatal period). Conversely, excessive exposure to retinoic acid also is teratogenic. Infants born to mothers treated with retinoic acid for severe acne have a predictable phenotype (retinoic acid embryopathy), including CNS, cardiac, and craniofacial defects, such as cleft lip and cleft palate. The last entity may stem from retinoic acid–mediated deregulation of components of the transforming growth factor-β (TGF-β) signaling pathway, which is involved in palatogenesis. Mice with knockout of the Tgfb3 gene uniformly develop cleft palate, once again underscoring the functional relationship between teratogenic exposure and signaling pathways in the causation of congenital anomalies. Summary
Summary
Congenital Anomalies
• Congenital anomalies result from intrinsic abnormalities (malformations) as well as extrinsic disturbances (deformations, disruptions).
• Congenital anomalies can result from genetic (chromosomal abnormalities, gene mutations), environmental (infections, drugs, alcohol), and multifactorial causes.
• The timing of the in utero insult has profound influence on the extent of congenital anomalies, with earlier events usually demonstrating greater impact.
• The interplay between genetic and environmental causes of anomalies is demonstrated by the fact that teratogens often target signaling pathways in which mutations have been reported as a cause for the same anomalies.
Perinatal Infections
Infections of the fetus and neonate may be acquired transcervically (ascending infections) or transplacentally (hematologic infections).
• Transcervical, or ascending, infections involve spread of infection from the cervicovaginal canal and may be acquired in utero or during birth. Most bacterial infections (e.g., α-hemolytic streptococcal infection) and a few viral infections (e.g., herpes simplex) are acquired in this manner. In general, the fetus acquires the infection by “inhaling” infected amniotic fluid into the lungs or by passing through an infected birth canal during delivery. Fetal infection usually is associated with inflammation of the placental membranes (chorioamnionitis) and inflammation of the umbilical cord (funisitis). This mode of spread is typical for pneumonia and, in severe cases, sepsis and meningitis.
• Transplacental infections gain access to the fetal bloodstream by crossing the placenta via the chorionic villi, and may occur at any time during gestation or occasionally, as may be the case with hepatitis B and human immunodeficiency virus, at the time of delivery via maternal-to-fetal transfusion. Most parasitic (e.g., toxoplasma, malaria) and viral infections and a few bacterial infections (i.e., from Listeria and Treponema) demonstrate this mode of hematogenous transmission. The clinical manifestations of these infections are highly variable, depending largely on the gestational timing and the microorganism involved. The most important transplacental infections can be conveniently remembered by the acronym TORCH. The elements of the TORCH complex are Toxoplasma (T), rubella virus (R), cytomegalovirus (C), herpesvirus (H), and any of a number of other (O) microbes such as Treponema pallidum. These agents are grouped together because they may evoke similar clinical and pathologic manifestations. TORCH infections occurring early in gestation may cause chronic sequelae in the child, including growth restriction, mental retardation, cataracts, and congenital cardiac anomalies, whereas infections later in pregnancy result primarily in tissue injury accompanied by inflammation (encephalitis, chorioretinitis, hepatosplenomegaly, pneumonia, and myocarditis).
Prematurity and Fetal Growth Restriction
Prematurity is the second most common cause of neonatal mortality (second only to congenital anomalies), and is defined by a gestational age less than 37 weeks. As might be expected, infants born before completion of gestation also weigh less than normal (below 2500 gm). The major risk factors for prematurity include premature rupture of membranes; intrauterine infection leading to inflammation of the placental membranes (chorioamnionitis); structural abnormalities of the uterus, cervix, and placenta; and multiple gestation (e.g., twin pregnancy). It is well established that children born before completion of the full period of gestation demonstrate higher morbidity and mortality rates than those reported for full-term infants. The immaturity of organ systems in preterm infants makes them especially vulnerable to several important complications:
• Respiratory distress syndrome (RDS), also called hyaline membrane disease
• Necrotizing enterocolitis (NEC)
• Intraventricular and germinal matrix hemorrhage (Chapter 22)
Although birth weight is low in preterm infants, it usually is appropriate once adjusted for gestational age. By contrast, as many as one third of infants who weigh less than 2500 gm are born at term and are therefore undergrown rather than immature. These small-for-gestational-age (SGA) infants suffer from fetal growth restriction. Fetal growth restriction may result from fetal, maternal, or placental abnormalities, although in many cases the specific cause is unknown.
• Fetal factors: This category consists of conditions that intrinsically reduce growth potential of the fetus despite an adequate supply of nutrients from the mother. Prominent among such fetal conditions are chromosomal disorders, congenital anomalies, and congenital infections. Chromosomal abnormalities may be detected in as many as 17% of fetuses evaluated for fetal growth restriction and in as many as 66% of fetuses with documented ultrasonographic malformations. Fetal infection should be considered in all growth-restricted neonates, with the TORCH group of infections (see earlier) being a common cause. When the causation is intrinsic to the fetus, growth retardation is symmetric (i.e., affects all organ systems equally).
• Placental factors: Placental causes include any factor that compromises the uteroplacental supply line. This may result from placenta previa (low implantation of the placenta), placental abruption (separation of placenta from the decidua by a retroplacental clot), or placental infarction. With placental (and maternal) causes of growth restriction, the growth retardation is asymmetric (i.e., the brain is spared relative to visceral organs such as the liver).
• Maternal factors: This category comprises by far the most common causes of the growth deficit in SGA infants. Important examples are vascular diseases such as preeclampsia (“toxemia of pregnancy”) (Chapter 18) and chronic hypertension. The list of other maternal conditions associated with fetal growth restriction is long, but some of the avoidable influences are maternal narcotic abuse, alcohol intake, and heavy cigarette smoking (as noted previously, many of these same causes also are involved in the pathogenesis of congenital anomalies). Drugs causing fetal growth restriction in similar fashion include teratogens, such as the commonly administered anticonvulsant phenytoin (Dilantin), as well as nonteratogenic agents. Maternal malnutrition (in particular, prolonged hypoglycemia) also may affect fetal growth, but the association between growth restriction in infants and the nutritional status of the mother is complex.
Not only is the growth-restricted infant handicapped in the perinatal period, but the deficits also persist into childhood and adult life. Affected persons are thus more likely to have cerebral dysfunction, learning disabilities, and sensory (i.e., visual and hearing) impairment.
Respiratory Distress Syndrome of the Newborn
There are many causes of respiratory distress in the newborn, including excessive sedation of the mother, fetal head injury during delivery, aspiration of blood or amniotic fluid, and intrauterine hypoxia secondary to compression from coiling of the umbilical cord about the neck. The most common cause, however, is respiratory distress syndrome (RDS), also known as hyaline membrane disease because of the formation of “membranes” in the peripheral air spaces observed in infants who succumb to this condition. An estimated 24,000 cases of RDS are reported annually in the United States, and improvements in management of this condition have sharply decreased deaths due to respiratory insufficiency from as many as 5000 per year a decade ago to less than 900 cases yearly.
Pathogenesis
RDS is basically a disease of premature infants. It occurs in about 60% of infants born at less than 28 weeks of gestation, 30% of those born between 28 to 34 weeks’ gestation, and less than 5% of those born after 34 weeks’ gestation. There are also strong though not invariable associations with male gender, maternal diabetes, and delivery by cesarean section.
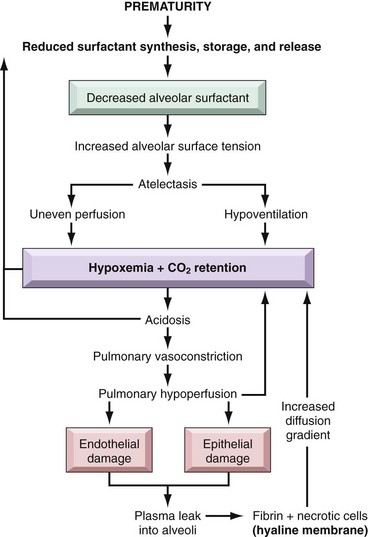
The fundamental defect in RDS is the inability of the immature lung to synthesize sufficient surfactant. Surfactant is a complex of surface-active phospholipids, principally dipalmitoylphosphatidylcholine (lecithin) and at least two groups of surfactant-associated proteins. The importance of surfactant-associated proteins in normal lung function can be gauged by the occurrence of severe respiratory failure in neonates with congenital deficiency of surfactant caused by mutations in the corresponding genes. Surfactant is synthesized by type II pneumocytes and, with the healthy newborn’s first breath, rapidly coats the surface of alveoli, reducing surface tension and thus decreasing the pressure required to keep alveoli open. In a lung deficient in surfactant, alveoli tend to collapse, and a relatively greater inspiratory effort is required with each breath to open the alveoli. The infant rapidly tires from breathing, and generalized atelectasis sets in. The resulting hypoxia sets into motion a sequence of events that lead to epithelial and endothelial damage and eventually to the formation of hyaline membranes (Fig. 6–24). As discussed later, this classical picture of surfactant deficiency is greatly modified by surfactant treatment.
Surfactant synthesis is regulated by hormones. Corticosteroids stimulate the formation of surfactant lipids and associated proteins. Therefore, conditions associated with intrauterine stress and fetal growth restriction that increase corticosteroid release lower the risk of developing RDS. Surfactant synthesis can be suppressed by the compensatory high blood levels of insulin in infants of diabetic mothers, which counteracts the effects of steroids. This may explain, in part, why infants of diabetic mothers are at higher risk for developing RDS. Labor is known to increase surfactant synthesis; accordingly, cesarean section performed before the onset of labor may be associated with increased risk for RDS.
 Morphology
Morphology
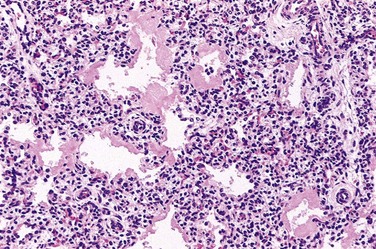
The lungs in infants with RDS are of normal size but are heavy and relatively airless. They have a mottled purple color, and on microscopic examination the tissue appears solid, with poorly developed, generally collapsed (atelectatic) alveoli. If the infant dies within the first several hours of life, only necrotic cellular debris will be present in the terminal bronchioles and alveolar ducts. Later in the course, characteristic eosinophilic hyaline membranes line the respiratory bronchioles, alveolar ducts, and random alveoli (Fig. 6–25). These “membranes” contain necrotic epithelial cells admixed with extravasated plasma proteins. There is a remarkable paucity of neutrophilic inflammatory reaction associated with these membranes. The lesions of hyaline membrane disease are never seen in stillborn infants or in live-born infants who die within a few hours of birth. If the infant dies after several days, evidence of reparative changes, including proliferation of type II pneumocytes and interstitial fibrosis, is seen.
Clinical Features
The classic clinical presentation before the era of treatment with exogenous surfactant was described earlier. Currently, the actual clinical course and prognosis for neonatal RDS vary, depending on the maturity and birth weight of the infant and the promptness of institution of therapy. A major thrust in the control of RDS focuses on prevention, either by delaying labor until the fetal lung reaches maturity or by inducing maturation of the lung in the fetus at risk. Critical to these objectives is the ability to assess fetal lung maturity accurately. Because pulmonary secretions are discharged into the amniotic fluid, analysis of amniotic fluid phospholipids provides a good estimate of the level of surfactant in the alveolar lining. Prophylactic administration of exogenous surfactant at birth to extremely premature infants (born before a gestational age of 28 weeks) has been shown to be very beneficial, such that it is now uncommon for infants to die of acute RDS.
In uncomplicated cases, recovery begins to occur within 3 or 4 days. In affected infants, oxygen is required. Use of high concentrations of ventilator-administered oxygen for prolonged periods, however, is associated with two well-known complications: retrolental fibroplasia (also called retinopathy of prematurity) in the eyes and bronchopulmonary dysplasia (BPD):
• Retinopathy of prematurity has a two-phase pathogenesis. During the hyperoxic phase of RDS therapy (phase I), expression of the pro-angiogenic vascular endothelial growth factor (VEGF) is markedly decreased, causing endothelial cell apoptosis; VEGF levels rebound after return to relatively hypoxic room air ventilation (phase II), inducing retinal vessel proliferation (neovascularization) characteristic of the lesions in the retina.
• The major abnormality in BPD is a striking decrease in alveolar septation (manifested as large, simplified alveolar structures) and a dysmorphic capillary configuration. Thus, the current view is that BPD is caused by a potentially reversible impairment in the development of alveolar septation at the so-called saccular stage. Multiple factors—hyperoxemia, hyperventilation, prematurity, inflammatory cytokines, and vascular maldevelopment—contribute to BPD and probably act additively or synergistically to promote injury. The levels of a variety of pro-inflammatory cytokines (TNF and the interleukins IL-1β, IL-6, and IL-8) are increased in the alveoli of infants in whom BPD subsequently develops, suggesting a role for these cytokines in arresting pulmonary development.
Fortunately, both complications are now significantly less common as a result of gentler ventilation techniques, antenatal glucocorticoid therapy, and prophylactic surfactant treatments.
Infants who recover from RDS also are at increased risk for developing a variety of other complications associated with preterm birth; most important among these are patent ductus arteriosus, intraventricular hemorrhage, and necrotizing enterocolitis (NEC). Thus, although technologic advances help save the lives of many infants with RDS, they also bring to the surface the exquisite fragility of the immature neonate.
Summary
Neonatal Respiratory Distress Syndrome
• Neonatal RDS (hyaline membrane disease) is a disease of prematurity; most cases occur in neonates born before 28 weeks of gestational age.
• The fundamental abnormality in RDS is insufficient pulmonary surfactant, which results in failure of lungs to inflate after birth.
• The characteristic morphologic pattern in RDS is the presence of hyaline membranes (consisting of necrotic epithelial cells and plasma proteins) lining the airways.
• RDS can be ameliorated by prophylactic administration of steroids, surfactant therapy, and by improved ventilation techniques.
• Long-term sequelae associated with RDS therapy include retinopathy of prematurity and BPD; the incidence of both complications has decreased with improvements in management of RDS.
Necrotizing Enterocolitis
Necrotizing enterocolitis (NEC) most commonly occurs in premature infants, with the incidence of the disease being inversely proportional to the gestational age. It occurs in approximately 1 of 10 very-low-birth-weight infants (less than 1500 gm). In addition to prematurity, most cases are associated with enteral feeding, suggesting that some postnatal insult (such as introduction of bacteria) sets in motion the cascade culminating in tissue destruction. While infectious agents are likely to play a role in NEC pathogenesis, no single bacterial pathogen has been linked to the disease. A large number of inflammatory mediators have been associated with NEC. One particular mediator, platelet-activating factor (PAF), has been implicated in increasing mucosal permeability by promoting enterocyte apoptosis and compromising intercellular tight junctions, thereby “adding fuel to the fire.”
NEC typically involves the terminal ileum, cecum, and right colon, although any part of the small or large intestine may be involved. The involved segment typically is distended, friable, and congested (Fig. 6–26), or it can be frankly gangrenous; intestinal perforation with accompanying peritonitis may be seen. On microscopic examination, mucosal or transmural coagulative necrosis, ulceration, bacterial colonization, and submucosal gas bubbles are all features associated with NEC. Evidence of reparative changes, such as granulation tissue and fibrosis, may be seen shortly after resolution of the acute episode.
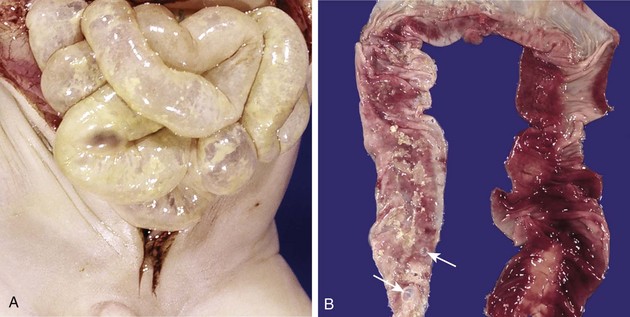
Figure 6–26 Necrotizing enterocolitis. A, At postmortem examination in a severe case, the entire small bowel was markedly distended with a perilously thin wall (usually this appearance implies impending perforation). B, The congested portion of the ileum corresponds to areas of hemorrhagic infarction and transmural necrosis seen on microscopy. Submucosal gas bubbles (pneumatosis intestinalis) can be seen in several areas (arrows).
The clinical course is fairly typical, with the onset of bloody stools, abdominal distention, and development of circulatory collapse. Abdominal radiographs often demonstrate gas within the intestinal wall (pneumatosis intestinalis). When detected early, NEC often can be managed conservatively, but many cases (20% to 60%) require operative intervention including resection of the necrotic segments of bowel. NEC is associated with high perinatal mortality; infants who survive often develop post-NEC strictures from fibrosis caused by the healing process.
Sudden Infant Death Syndrome
Sudden infant death syndrome (SIDS) is a disease of unknown cause. The National Institute of Child Health and Human Development defines SIDS as “the sudden death of an infant under 1 year of age which remains unexplained after a thorough case investigation, including performance of a complete autopsy, examination of the death scene, and review of the clinical history.” An aspect of SIDS that is not stressed in the definition is that the infant usually dies while asleep—hence the lay terms crib death and cot death. SIDS is the leading cause of death between the ages of 1 month and 1 year in U.S. infants, and the third leading cause of death overall in this age group, after congenital anomalies and diseases of prematurity and low birth weight. In 90% of cases, the infant is younger than 6 months; most are between the ages of 2 and 4 months.
Pathogenesis
The circumstances surrounding SIDS have been explored in great detail, and the general consensus is that it is a multifactorial condition, with a variable mixture of contributing causes in a given case. A proposed “triple-risk” model of SIDS postulates the intersection of three overlapping variables: (1) a vulnerable infant, (2) a critical developmental period in homeostatic control, and (3) one or more exogenous stressors. According to this model, several factors make the infant vulnerable to sudden death during the critical developmental period (i.e., 1 month to 1 year). These vulnerability factors may be specific to the parents or the infant, while the exogenous stressor(s) is attributable to the environment (Table 6–8). Although numerous factors have been proposed to account for a vulnerable infant, the most compelling hypothesis is that SIDS reflects a delayed development of arousal and cardiorespiratory control. Regions of the brain stem, particularly the arcuate nucleus located in the ventral medullary surface, play a critical role in the body’s arousal response to noxious stimuli such as hypercarbia, hypoxia, and thermal stress encountered during sleep. In addition, these areas regulate breathing, heart rate, and body temperature. In certain infants, for as-yet inexplicable reasons, there may be a maldevelopment or delay in maturation of this region, compromising the arousal response to noxious stimuli. Certain polymorphic variants in genes related to serotonergic signaling and autonomic innervation have been identified at a higher frequency in SIDS babies than in the general population, suggesting that genetic factors may play a role in predisposing the infant to impaired arousal. Among the potential environmental causes, prone sleeping position, sleeping on soft surfaces, and thermal stress are possibly the most important modifiable risk factors for SIDS. The prone position increases the infant’s vulnerability to one or more recognized noxious stimuli (hypoxia, hypercarbia, and thermal stress) during sleep. In addition, the prone position also is associated with decreased arousal responsiveness compared with the supine position. Results of studies from Europe, Australia, New Zealand, and the United States showed clearly increased risk for SIDS in infants who sleep in a prone position, prompting the American Academy of Pediatrics to recommend placing healthy infants on their back when laying them down to sleep. This “Back to Sleep” campaign has resulted in substantial decreases in SIDS-related deaths since its inception in 1994.
Table 6–8 Factors Associated with Sudden Infant Death Syndrome (SIDS)
| Parental |
| Infant |
| Environment |
| Postmortem Abnormalities Detected in Cases of Sudden Unexpected Infant Death* |
C4, complement component 4; KCNQ1, potassium voltage-gated channel; LCHAD, long-chain 3-hydroxyacyl coenzyme A dehydrogenase; MCAD, medium-chain acyl coenzyme A dehydrogenase; MTCYB, mitochondrial cytochrome b; SCHAD, short-chain 3-hydroxyacyl coenzyme A dehydrogenase; SCN5A, sodium channel, voltage-gated.
* SIDS is not the only cause of sudden unexpected death in infancy; instead, it is a diagnosis of exclusion. Therefore, performance of an autopsy may reveal findings that would explain the cause of sudden unexpected death. These cases should not, strictly speaking, be designated SIDS.
Morphology
Anatomic studies of victims have yielded inconsistent histologic findings. Multiple petechiae are the most common finding in the typical SIDS autopsy (in approximately 80% of cases); these usually are present on the thymus, visceral and parietal pleura, and epicardium. On gross examination, the lungs usually are congested, and vascular engorgement with or without pulmonary edema is demonstrable on microscopic examination in a majority of cases. Sophisticated morphometric studies have revealed quantitative brain stem abnormalities such as hypoplasia of the arcuate nucleus or a subtle decrease in brain stem neuronal populations in several cases; these observations are not uniform, however, and use of such studies is not feasible in most “routine” autopsy procedures.
Of note, SIDS is not the only cause of sudden unexpected death in infancy. In fact, SIDS is a diagnosis of exclusion, requiring careful examination of the death scene and a complete postmortem examination. The latter can reveal an unsuspected cause of sudden death in as many as 20% or more of babies presumed to have died of SIDS (Table 6–8). Infections (e.g., viral myocarditis or bronchopneumonia) are the most common causes of sudden “unexpected” death, followed by an unsuspected congenital anomaly. As a result of advancements in molecular diagnostics, several genetic causes of sudden “unexpected” infant death have emerged. For example, fatty acid oxidation disorders, characterized by defects in mitochondrial fatty acid oxidative enzymes, may be responsible for as many as 5% of sudden deaths in infancy; of these, a deficiency in medium-chain acyl-coenzyme A dehydrogenase is the most common. Retrospective analyses in cases of sudden infant death originally designated SIDS also have revealed mutations of cardiac sodium and potassium channels, which result in a form of cardiac arrhythmia characterized by prolonged QT intervals; these cases account for no more than 1% of SIDS deaths. SIDS in an earlier sibling is associated with a five-fold relative risk of recurrence; traumatic child abuse must be carefully excluded under these circumstances.
Summary
Sudden Infant Death Syndrome
• SIDS is a disorder of unknown cause, defined as the sudden death of an infant younger than 1 year of age that remains unexplained after a thorough case investigation including performance of an autopsy. Most SIDS deaths occur between the ages of 2 and 4 months.
• The most likely basis for SIDS is a delayed development in arousal reflexes and cardiorespiratory control.
• Numerous risk factors have been proposed, of which the prone sleeping position is best recognized—hence the success of the “Back to Sleep” program in reducing the incidence of SIDS.
Fetal Hydrops
Fetal hydrops refers to the accumulation of edema fluid in the fetus during intrauterine growth. The causes of fetal hydrops are manifold; the most important are listed in Table 6–9. In the past, hemolytic anemia caused by Rh blood group incompatibility between mother and fetus (immune hydrops) was the most common cause, but with the successful prophylaxis of this disorder during pregnancy, causes of nonimmune hydrops have emerged as the principal culprits. Notably, the intrauterine fluid accumulation can be quite variable, ranging in degree from progressive, generalized edema of the fetus (hydrops fetalis), a usually lethal condition, to more localized and less marked edematous processes, such as isolated pleural and peritoneal effusions or postnuchal fluid collections (cystic hygroma), that often are compatible with life (Fig. 6–27). The mechanism of immune hydrops is discussed first, followed by other important causes of fetal hydrops.
Table 6–9 Major Causes of Fetal Hydrops*
| Cardiovascular |
| Chromosomal |
| Thoracic |
| Fetal Anemia |
| Twin Gestation |
| Infection (excluding parvovirus) |
| Genitourinary tract malformations |
| Tumors |
| Genetic/metabolic disorder |
* The cause of fetal hydrops may be “idiopathic” in as many as 20% of cases.
Modified from Machin GA: Hydrops, cystic hygroma, hydrothorax, pericardial effusions, and fetal ascites. In Gilbert-Barnes E (ed): Potter’s pathology of fetus and infant. St. Louis, Mosby, 1997.
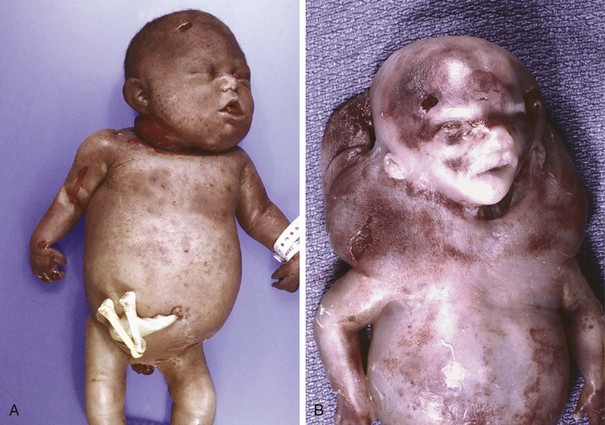
Figure 6–27 Hydrops fetalis. A, Generalized accumulation of fluid in the fetus. B, Fluid accumulation particularly prominent in the soft tissues of the neck. This condition has been termed cystic hygroma. Cystic hygromas are characteristically seen with, but not limited to, constitutional chromosomal anomalies such as 45,X karyotypes.
(Courtesy of Dr. Beverly Rogers, Department of Pathology, University of Texas Southwestern Medical Center, Dallas, Texas.)
Immune Hydrops
Immune hydrops results from an antibody-induced hemolytic disease in the newborn that is caused by blood group incompatibility between mother and fetus. Such an incompatibility occurs only when the fetus inherits red cell antigenic determinants from the father that are foreign to the mother. The most common antigens to result in clinically significant hemolysis are the Rh and ABO blood group antigens. Of the numerous antigens included in the Rh system, only the D antigen is a major cause of Rh incompatibility. Fetal red cells may reach the maternal circulation during the last trimester of pregnancy, when the cytotrophoblast is no longer present as a barrier, or during childbirth itself (fetomaternal bleed). The mother then becomes sensitized to the foreign antigen and produces antibodies that can freely traverse the placenta to the fetus, in which they cause red cell destruction. With initiation of immune hemolysis, progressive anemia in the fetus leads to tissue ischemia, intrauterine cardiac failure, and peripheral pooling of fluid (edema). As discussed later, cardiac failure may be the final pathway by which edema occurs in many cases of nonimmune hydrops as well.
Several factors influence the immune response to Rh-positive fetal red cells that reach the maternal circulation:
• Concurrent ABO incompatibility protects the mother against Rh immunization, because the fetal red cells are promptly coated by isohemagglutinins (pre-formed anti-A or anti-B antibodies) and removed from the maternal circulation.
• The antibody response depends on the dose of immunizing antigen, so hemolytic disease develops only when the mother has experienced a significant transplacental bleed (more than 1 mL of Rh-positive red cells).
• The isotype of the antibody is important, because immunoglobulin G (IgG) (but not IgM) antibodies can cross the placenta. The initial exposure to Rh antigen evokes the formation of IgM antibodies, so Rh disease is very uncommon with the first pregnancy. Subsequent exposure during the second or third pregnancy generally leads to a brisk IgG antibody response.
Appreciation of the role of previous sensitization in the pathogenesis of Rh-hemolytic disease of the newborn has led to its therapeutic control. Currently, Rh-negative mothers are given anti-D globulin soon after the delivery of an Rh-positive baby. The anti-D antibodies mask the antigenic sites on the fetal red cells that may have leaked into the maternal circulation during childbirth, thus preventing long-lasting sensitization to Rh antigens.
As a result of the remarkable success achieved in prevention of Rh hemolysis, fetomaternal ABO incompatibility currently is the most common cause of immune hemolytic disease of the newborn. Although ABO incompatibility occurs in approximately 20% to 25% of pregnancies, hemolysis develops in only a small fraction of infants born subsequently, and in general the disease is much milder than Rh incompatibility. ABO hemolytic disease occurs almost exclusively in infants of blood group A or B who are born to mothers of blood group O. The normal anti-A and anti-B isohemagglutinins in group O mothers usually are of the IgM type and therefore do not cross the placenta. However, for reasons not well understood, certain group O women possess IgG antibodies directed against group A or B antigens (or both) even without previous sensitization. Therefore, the firstborn may be affected. Fortunately, even with transplacentally acquired antibodies, lysis of the infant’s red cells is minimal. There is no effective method of preventing hemolytic disease resulting from ABO incompatibility.
Nonimmune Hydrops
The major causes of nonimmune hydrops include those disorders associated with cardiovascular defects, chromosomal anomalies, and fetal anemia. Both structural cardiovascular defects and functional abnormalities (i.e., arrhythmias) may result in intrauterine cardiac failure and hydrops. Among the chromosomal anomalies, 45,X karyotype (Turner syndrome) and trisomies 21 and 18 are associated with fetal hydrops; the basis for this disorder usually is the presence of underlying structural cardiac anomalies, although in Turner syndrome there may be an abnormality of lymphatic drainage from the neck leading to postnuchal fluid accumulation (resulting in cystic hygromas). Fetal anemias due to causes other than Rh or ABO incompatibility also may result in hydrops. In fact, in some parts of the world (e.g., Southeast Asia), severe fetal anemia caused by homozygous α-thalassemia probably is the most common cause of fetal hydrops. Transplacental infection by parvovirus B19 is increasingly recognized as an important cause of fetal hydrops. The virus gains entry into erythroid precursors (normoblasts), where it replicates. The ensuing cellular injury leads to the death of the normoblasts and aplastic anemia. Parvoviral intranuclear inclusions can be seen within circulating and marrow erythroid precursors (Fig. 6–28). The basis for fetal hydrops in fetal anemia of both immune and nonimmune cause is tissue ischemia with secondary myocardial dysfunction and circulatory failure. Additionally, secondary liver failure may occur, with loss of synthetic function contributing to hypoalbuminemia, reduced plasma osmotic pressure, and edema.
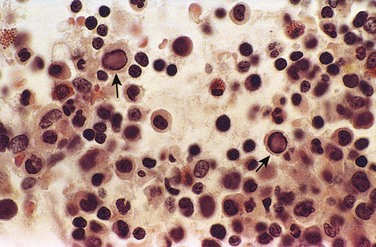
Figure 6–28 Bone marrow from an infant infected with parvovirus B19. The arrows point to two erythroid precursors with large homogeneous intranuclear inclusions and a surrounding peripheral rim of residual chromatin.
Morphology
The anatomic findings in fetuses with intrauterine fluid accumulation vary with both the severity of the disease and the underlying etiologic disorder. As previously noted, hydrops fetalis represents the most severe and generalized manifestation (Fig. 6–27), and lesser degrees of edema such as isolated pleural, peritoneal, or postnuchal fluid collections can occur. Accordingly, infants may be stillborn, die within the first few days, or recover completely. The presence of dysmorphic features suggests underlying constitutional chromosomal abnormalities; postmortem examination may reveal a cardiac anomaly. In hydrops associated with fetal anemia, both fetus and placenta are characteristically pale; in most cases, the liver and spleen are enlarged as a consequence of cardiac failure and congestion. Additionally, the bone marrow shows compensatory hyperplasia of erythroid precursors (parvovirus-associated aplastic anemia being a notable exception), and extramedullary hematopoiesis is present in the liver, the spleen, and possibly other tissues such as the kidneys, the lungs, the lymph nodes, and even the heart. The increased hematopoietic activity accounts for the presence in the peripheral circulation of large numbers of normoblasts, and even more immature erythroblasts (erythroblastosis fetalis) (Fig. 6–29).
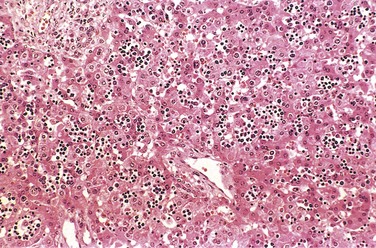
Figure 6–29 Numerous islands of extramedullary hematopoiesis (small blue cells) are scattered among mature hepatocytes in this histologic preparation from an infant with nonimmune hydrops fetalis.
The presence of hemolysis in Rh or ABO incompatibility is associated with the added complication of increased circulating bilirubin from the red cell breakdown. The CNS may be damaged when hyperbilirubinemia is marked (usually above 20 mg/dL in full-term infants, but often less in premature infants). The circulating unconjugated bilirubin is taken up by the brain tissue, on which it apparently exerts a toxic effect. The basal ganglia and brain stem are particularly prone to deposition of bilirubin pigment, which imparts a characteristic yellow hue to the parenchyma (kernicterus) (Fig. 6–30).
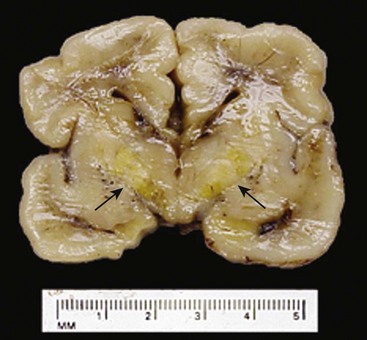
Figure 6–30 Kernicterus. Severe hyperbilirubinemia in the neonatal period—for example, secondary to immune hydrolysis—results in deposition of bilirubin pigment (arrows) in the brain parenchyma. This occurs because the blood–brain barrier is less well developed in the neonatal period than it is in adulthood. Infants who survive develop long-term neurologic sequelae.
Clinical Course
Early recognition of intrauterine fluid accumulation is imperative, since even severe cases can sometimes be salvaged with currently available therapy. Fetal hydrops that results from Rh incompatibility may be more or less accurately predicted, because severity correlates well with rapidly rising Rh antibody titers in the mother during pregnancy. Amniotic fluid obtained by amniocentesis may show high levels of bilirubin. The human anti-globulin test (Coombs test) (Chapter 11) using fetal cord blood yields a positive result if the red cells have been coated by maternal antibody. Antenatal exchange transfusion is an effective form of therapy. Postnatally, phototherapy is helpful, because visible light converts bilirubin to readily excreted dipyrroles. As already discussed, in an overwhelming majority of cases, administration of anti-D globulins to the mother can prevent the occurrence of immune hydrops in subsequent pregnancies. Group ABO hemolytic disease is more difficult to predict but is readily anticipated by awareness of the blood incompatibility between mother and father and by hemoglobin and bilirubin determinations in the vulnerable newborn infant. In fatal cases of fetal hydrops, a thorough postmortem examination is imperative to determine the cause and to exclude a potentially recurring cause such as a chromosomal abnormality.
Summary
Fetal Hydrops
• Fetal hydrops refers to the accumulation of edema fluid in the fetus during intrauterine growth.
• The degree of fluid accumulation is variable, from generalized hydrops fetalis to localized cystic hygromas.
• The most common causes of fetal hydrops are nonimmune (chromosomal abnormalities, cardiovascular defects, and fetal anemia), while immune hydrops has become less frequent as a result of Rh antibody prophylaxis.
• Erythroblastosis fetalis (due to circulating immature erythroid precursors) is a characteristic finding of fetal anemia-associated hydrops.
• Hemolysis-induced hyperbilirubinemia can result in kernicterus in the basal ganglia and brain stem, particularly in premature infants.
Tumors and Tumor-Like Lesions of Infancy and Childhood
Malignant neoplasms constitute the second most common cause of death in children between the ages of 4 and 14 years; only accidents exact a higher toll. Benign tumors are even more common than cancers.
It is difficult to segregate, on morphologic grounds, true tumors from tumor-like lesions in the infant and child. In this context, two special categories of tumor-like lesions should be recognized:
• Heterotopia or choristoma refers to microscopically normal cells or tissues that are present in abnormal locations. Examples are a pancreatic tissue “rest” found in the wall of the stomach or small intestine and a small mass of adrenal cells found in the kidney, lungs, ovaries, or elsewhere. Heterotopic rests usually are of little clinical significance, but on the basis of their appearance they can be confused with neoplasms.
• Hamartoma refers to an excessive but focal overgrowth of cells and tissues native to the organ in which it occurs. Although the cellular elements are mature and identical to those found in the remainder of the organ, they do not reproduce the normal architecture of the surrounding tissue. Hamartomas can be thought of as the linkage between malformations and neoplasms. The line of demarcation between a hamartoma and a benign neoplasm frequently is tenuous and is variously interpreted. Hemangiomas, lymphangiomas, rhabdomyomas of the heart, and adenomas of the liver are considered by some researchers to be hamartomas and by others to be true neoplasms.
Benign Tumors
Virtually any tumor may be encountered in the pediatric age group, but three—hemangiomas, lymphangiomas, and sacrococcygeal teratomas—deserve special mention here because they occur commonly in childhood.
Hemangiomas are the most common tumors of infancy. Both cavernous and capillary hemangiomas may be encountered (Chapter 9), although the latter often are more cellular than in adults and thus may be deceptively worrisome-appearing. In children, most hemangiomas are located in the skin, particularly on the face and scalp, where they produce flat to elevated, irregular, red-blue masses; the flat, larger lesions are referred to as port wine stains. Hemangiomas may enlarge as the child gets older, but in many instances they spontaneously regress (Fig. 6–31). The vast majority of superficial hemangiomas have no more than a cosmetic significance; rarely, they may be the manifestation of a hereditary disorder associated with disease within internal organs, such as the von Hippel-Lindau and Sturge-Weber syndromes (Chapter 9). A subset of CNS cavernous hemangiomas can occur in the familial setting; affected families harbor mutations in one of three cerebral cavernous malformation (CCM) genes.
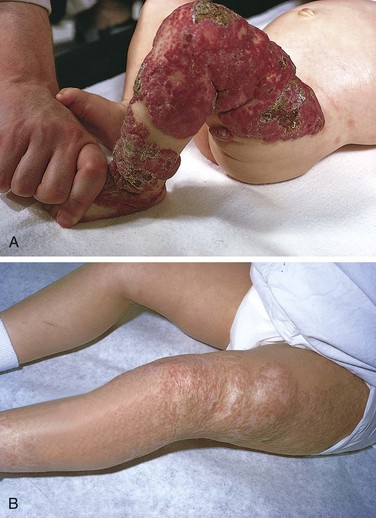
Figure 6–31 Congenital capillary hemangioma at birth (A) and at 2 years of age (B) after the lesion had undergone spontaneous regression.
(Courtesy of Dr. Eduardo Yunis, Children’s Hospital of Pittsburgh, Pittsburgh, Pennsylvania.)
Lymphangiomas represent the lymphatic counterpart of hemangiomas. Microscopic examination shows cystic and cavernous spaces lined by endothelial cells and surrounded by lymphoid aggregates; the spaces usually contain pale fluid. They may occur on the skin but, more important, also are encountered in the deeper regions of the neck, axilla, mediastinum, and retroperitoneum. Although histologically benign, they tend to increase in size after birth and may encroach on mediastinal structures or nerve trunks in axilla.
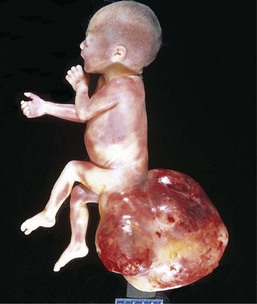
Sacrococcygeal teratomas are the most common germ cell tumors of childhood, accounting for 40% or more of cases (Fig. 6–32). In view of the overlap in the mechanisms underlying teratogenesis and oncogenesis, it is interesting that approximately 10% of sacrococcygeal teratomas are associated with congenital anomalies, primarily defects of the hindgut and cloacal region and other midline defects (e.g., meningocele, spina bifida) not believed to result from local effects of the tumor. Approximately 75% of these tumors are histologically mature with a benign course, and about 12% are unmistakably malignant and lethal (Chapter 17). The remainder are designated immature teratomas, and their malignant potential correlates with the amount of immature tissue elements present. Most of the benign teratomas are encountered in younger infants (4 months of age or younger), whereas children with malignant lesions tend to be somewhat older.
Malignant Tumors
The organ systems involved most commonly by malignant neoplasms in infancy and childhood are the hematopoietic system, neural tissue, and soft tissues (Table 6–10). This distribution is in sharp contrast with that in adults, in whom tumors of the lung, heart, prostate, and colon are the most common forms. Malignant tumors of infancy and childhood differ biologically and histologically from those in adults. The main differences are as follows:
• Relatively frequent demonstration of a close relationship between abnormal development (teratogenesis) and tumor induction (oncogenesis)
• Prevalence of constitutional genetic abnormalities or syndromes that predispose to cancer
• Tendency of fetal and neonatal malignancies to spontaneously regress or undergo “differentiation” into mature elements
• Improved survival or cure of many childhood tumors, so that more attention is now being paid to minimizing the adverse delayed effects of chemotherapy and radiotherapy in survivors, including the development of second malignancies
Table 6–10 Common Malignant Neoplasms of Infancy and Childhood
| 0–4 Years of Age | 5–9 Years of Age | 10–14 Years of Age |
|---|---|---|
CNS, central nervous system.
Many malignant pediatric neoplasms are histologically unique. In general, they tend to have a primitive (embryonal) rather than pleomorphic-anaplastic microscopic appearance, and frequently they exhibit features of organogenesis specific to the site of tumor origin. Because of their primitive histologic appearance, many childhood tumors have been collectively referred to as small, round, blue cell tumors. These are characterized by sheets of cells with small, round nuclei. The tumors in this category include neuroblastoma, lymphoma, rhabdomyosarcoma, Ewing sarcoma (peripheral neuroectodermal tumor), and some cases of Wilms tumor. Sufficient distinctive features usually are present to permit definitive diagnosis on the basis of histologic examination alone, but when necessary, clinical and radiographic findings, combined with ancillary studies (e.g., chromosome analysis, immunoperoxidase stains, electron microscopy) are used. Three common tumors—neuroblastoma, retinoblastoma, and Wilms tumor—are described here to highlight the differences between pediatric tumors and those in adults.
Neuroblastoma
The term neuroblastic includes tumors of the sympathetic ganglia and adrenal medulla that are derived from primordial neural crest cells populating these sites; neuroblastoma is the most important member of this family. It is the second most common solid malignancy of childhood after brain tumors, accounting for 7% to 10% of all pediatric neoplasms, and as many as 50% of malignancies diagnosed in infancy. Neuroblastomas demonstrate several unique features in their natural history, including spontaneous regression and spontaneous or therapy-induced maturation. Most occur sporadically, but 1% to 2% are familial, with autosomal dominant transmission, and in such cases the neoplasms may involve both of the adrenals or multiple primary autonomic sites. Germ line mutations in the anaplastic lymphoma kinase gene (ALK) recently have been identified as a major cause of familial predisposition to neuroblastoma. Somatic gain-of-function ALK mutations also are observed in a subset of sporadic neuroblastomas. It is envisioned that tumors harboring ALK mutations in either the germline or somatic setting will be amenable to treatment using drugs that target the activity of this kinase.
Morphology
In childhood, about 40% of neuroblastomas arise in the adrenal medulla. The remainder occur anywhere along the sympathetic chain, with the most common locations being the paravertebral region of the abdomen (25%) and posterior mediastinum (15%). Macroscopically, neuroblastomas range in size from minute nodules (the in situ lesions) to large masses weighing more than 1 kg. In situ neuroblastomas are reported to be 40 times more frequent than overt tumors. The great preponderance of these silent lesions spontaneously regress, leaving only a focus of fibrosis or calcification in the adult. Some neuroblastomas are sharply demarcated with a fibrous pseudocapsule, but others are far more infiltrative and invade surrounding structures, including the kidneys, renal vein, and vena cava, and envelop the aorta. On transection, they are composed of soft, gray-tan, brainlike tissue. Larger tumors have areas of necrosis, cystic softening, and hemorrhage.
Histologically, classic neuroblastomas are composed of small, primitive-appearing cells with dark nuclei, scant cytoplasm, and poorly defined cell borders growing in solid sheets (Fig. 6–33, A). Mitotic activity, nuclear breakdown (“karyorrhexis”), and pleomorphism may be prominent. The background often demonstrates a faintly eosinophilic fibrillary material (neuropil) that corresponds to neuritic processes of the primitive neuroblasts. Typically, so-called Homer-Wright pseudo-rosettes can be found in which the tumor cells are concentrically arranged about a central space filled with neuropil (the absence of an actual central lumen garners the designation “pseudo-”). Other helpful features include immunochemical detection of neuron-specific enolase and demonstration on ultrastructural studies of small, membrane-bound, cytoplasmic catecholamine-containing secretory granules.
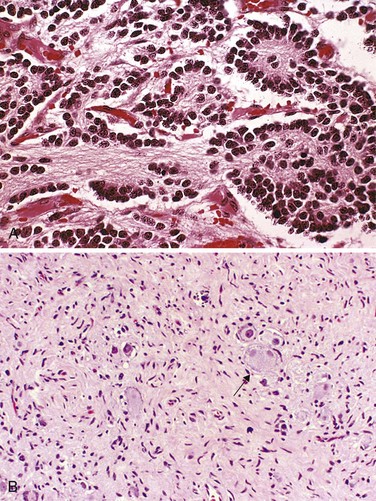
Figure 6–33 A, Neuroblastoma. This tumor is composed of small cells embedded in a finely fibrillar matrix (neuropil). A Homer-Wright pseudo-rosette (tumor cells arranged concentrically around a central core of neuropil) is seen in the upper right corner. B, Ganglioneuromas, arising from spontaneous or therapy-induced maturation of neuroblastomas, are characterized by clusters of large cells with vesicular nuclei and abundant eosinophilic cytoplasm (arrow), representing neoplastic ganglion cells. Spindle-shaped Schwann cells are present in the background stroma.
Some neoplasms show signs of maturation, either spontaneous or therapy-induced. Larger cells having more abundant cytoplasm with large vesicular nuclei and a prominent nucleolus, representing ganglion cells in various stages of maturation, may be found in tumors admixed with primitive neuroblasts (ganglioneuroblastoma). Even better-differentiated lesions contain many more large cells resembling mature ganglion cells in the absence of residual neuroblasts; such neoplasms merit the designation ganglioneuroma (Fig. 6–33, B). Maturation of neuroblasts into ganglion cells usually is accompanied by the appearance of Schwann cells. In fact, the presence of a “schwannian stroma” composed of organized fascicles of neuritic processes, mature Schwann cells, and fibroblasts is a histologic prerequisite for the designation of ganglioneuroblastoma and ganglioneuroma; ganglion cells in and of themselves do not fulfill the criteria for maturation.
Clinical Course and Prognosis
Many factors influence prognosis, but the most important are the stage of the tumor and the age of the patient.
• Staging of neuroblastomas (Table 6–11) assumes great importance in establishing a prognosis. Special note should be taken of stage 4S (S means special), because the outlook for these patients is excellent, despite the spread of disease. As noted in Table 6–11, the primary tumor would be classified as stage 1 or 2 but for the presence of metastases, which are limited to liver, skin, and bone marrow, without bone involvement. Infants with 4S tumors have an excellent prognosis with minimal therapy, and it is not uncommon for the primary or metastatic tumors to undergo spontaneous regression. The biologic basis for this welcome behavior is not clear.
• Age is the other important determinant of outcome, and the outlook for children younger than 18 months is much more favorable than for older children at a comparable stage of disease. Most neoplasms diagnosed in children during the first 18 months of life are stage 1 or 2, or stage 4S (“low” risk category in Table 6–11), while neoplasms in older children fall into the “intermediate” or “high” risk category.
• Morphology is an independent prognostic variable in neuroblastic tumors; evidence of schwannian stroma and gangliocytic differentiation is indicative of a favorable histologic pattern.
• Amplification of the NMYC oncogene in neuroblastomas is a molecular event that has profound impact on prognosis. NMYC amplification is present in about 25% to 30% of primary tumors, most in advanced-stage disease; the greater the number of copies, the worse the prognosis. NMYC amplification is currently the most important genetic abnormality used in risk stratification of neuroblastic tumors and automatically renders a tumor as “high” risk, irrespective of stage or age.
• Deletion of the distal short arm of chromosome 1, gain of the distal long arm of chromosome 17, and overexpression of telomerase all are adverse prognostic factors, while expression of TrkA, a high-affinity receptor for nerve growth factor that is indicative of differentiation toward sympathetic ganglia lineage, is associated with favorable prognosis.
Table 6–11 Staging of Neuroblastomas
| Stage 1 | Localized tumor completely excised, with or without microscopic residual disease; representative ipsilateral nonadherent lymph nodes negative for tumor (nodes adherent to the primary tumor may be positive for tumor) |
| Stage 2A | Localized tumor resected incompletely grossly; representative ipsilateral nonadherent lymph nodes negative for tumor microscopically |
| Stage 2B | Localized tumor with or without complete gross excision, ipsilateral nonadherent lymph nodes positive for tumor; enlarged contralateral lymph nodes, which are negative for tumor microscopically |
| Stage 3 | Unresectable unilateral tumor infiltrating across the midline with or without regional lymph node involvement; or localized unilateral tumor with contralateral regional lymph node involvement |
| Stage 4 | Any primary tumor with dissemination to distant lymph nodes, bone, bone marrow, liver, skin, and/or other organs (except as defined for stage 4S) |
| Stage 4S* | Localized primary tumor (as defined for stage 1, 2A, or 2B) with dissemination limited to skin, liver, and/or bone marrow (<10% of nucleated cells are constituted by neoplastic cells; >10% involvement of bone marrow is considered as stage 4); stage 4S limited to infants younger than 1 year of age |
Adapted from Brodeur GM, Pritchard J, Berthold F, et al: Revisions of the international neuroblastoma diagnosis, staging, and response to treatment. J Clin Oncol 11:1466, 1993.
Children younger than 2 years with neuroblastomas generally present with protuberant abdomen resulting from an abdominal mass, fever, and weight loss. In older children the neuroblastomas may remain unnoticed until metastases cause hepatomegaly, ascites, and bone pain. Neuroblastomas may metastasize widely through the hematogenous and lymphatic systems, particularly to liver, lungs, bones, and the bone marrow. In neonates, disseminated neuroblastomas may manifest with multiple cutaneous metastases associated with deep blue discoloration to the skin (earning the rather unfortunate moniker of “blueberry muffin baby”). As already noted, many variables can influence the prognosis of neuroblastomas, but as a rule of thumb, stage and age are the paramount determinants. Tumors of all stages diagnosed in the first 18 months of life, as well as low-stage tumors in older children, have a favorable prognosis, while high-stage tumors diagnosed in children younger than 18 months of age have the poorest outcome. About 90% of neuroblastomas, regardless of location, produce catecholamines (similar to the catecholamines associated with pheochromocytomas), which constitutes an important diagnostic feature (i.e., elevated blood levels of catecholamines and elevated urine levels of catecholamine metabolites such as vanillylmandelic acid [VMA] and homovanillic acid [HVA]). Despite the elaboration of catecholamines, hypertension is much less frequent with these neoplasms than with pheochromocytomas (Chapter 19).
Summary
Neuroblastoma
• Neuroblastomas and related tumors arise from neural crest–derived cells in the sympathetic ganglia and adrenal medulla.
• Neuroblastomas are undifferentiated, whereas ganglioneuroblastomas and ganglioneuromas demonstrate evidence of differentiation (schwannian stroma and ganglion cells). Homer-Wright pseudo-rosettes are characteristic of neuroblastomas.
• Age, stage, and NMYC amplification are the most important prognostic features; children younger than 18 months usually have a better prognosis than older children, while children with higher-stage tumors or NMYC amplification fare worse.
• Neuroblastomas secrete catecholamines, whose metabolites (VMA/HVA) can be used for screening patients.
Retinoblastoma
Retinoblastoma is the most common primary intraocular malignancy of children. The molecular genetics of retinoblastoma has been discussed previously (Chapter 5). Approximately 40% of the tumors are associated with a germline mutation in the RB1 gene and are therefore heritable. The remaining 60% of the tumors develop sporadically, and these have somatic RB1 gene mutations. Although the name retinoblastoma might suggest origin from a primitive retinal cell that is capable of differentiation into both glial and neuronal cells, it is now clear that the cell of origin of retinoblastoma is neuronal. As noted earlier, in approximately 40% of cases, retinoblastoma occurs in persons who inherit a germ line mutation of one RB allele. Familial cases typically are associated with development of multiple tumors that are bilateral, although they may be unifocal and unilateral. All of the sporadic, nonheritable tumors are unilateral and unifocal. Patients with familial retinoblastoma also are at increased risk for the development of osteosarcoma and other soft tissue tumors.
Morphology
Retinoblastomas tend to be nodular masses, usually in the posterior retina, often with satellite seedings (Fig. 6–34, A). On light microscopic examination, undifferentiated areas of these tumors are found to be composed of small, round cells with large hyperchromatic nuclei and scant cytoplasm, resembling undifferentiated retinoblasts.
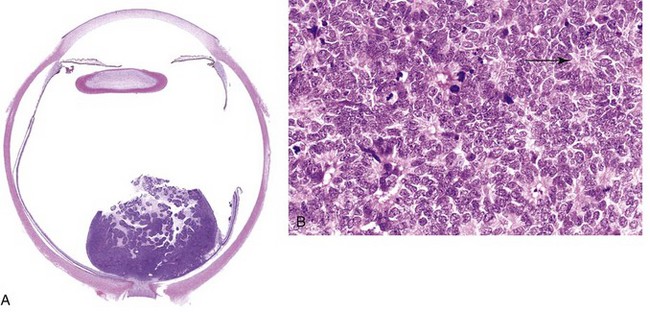
A, Poorly cohesive tumor in retina is seen abutting the optic nerve. B, Higher-power view showing Flexner-Wintersteiner rosettes (arrow) and numerous mitotic figures.
Differentiated structures are found within many retinoblastomas, the most characteristic of these being Flexner-Wintersteiner rosettes (Fig. 6–34, B). These structures consist of clusters of cuboidal or short columnar cells arranged around a central lumen (in contrast with the pseudo-rosettes of neuroblastoma, which lack a central lumen). The nuclei are displaced away from the lumen, which by light microscopy appears to have a limiting membrane resembling the external limiting membrane of the retina.
Tumor cells may disseminate beyond the eye through the optic nerve or subarachnoid space. The most common sites of distant metastases are the CNS, skull, distal bones, and lymph nodes.
Clinical Features
The median age at presentation is 2 years, although the tumor may be present at birth. The presenting findings include poor vision, strabismus, a whitish hue to the pupil (“cat’s eye reflex”), and pain and tenderness in the eye. Untreated, the tumors usually are fatal, but after early treatment with enucleation, chemotherapy, and radiotherapy, survival is the rule. As noted earlier, some tumors spontaneously regress, and patients with familial retinoblastoma are at increased risk for the development of osteosarcoma and other soft tissue tumors.
Wilms Tumor
Wilms tumor, or nephroblastoma, is the most common primary tumor of the kidney in children. Most cases occur in children between 2 and 5 years of age. This tumor illustrates several important concepts of childhood tumors: the relationship between congenital malformation and increased risk of tumors, the histologic similarity between tumor and developing organ, and finally, the remarkable success in the treatment of childhood tumors. Each of these concepts is evident in the following discussion.
Three groups of congenital malformations are associated with an increased risk for development of Wilms tumor. Of patients with the WAGR syndrome (i.e., Wilms tumor, aniridia, genital abnormalities, and mental retardation), approximately one in three will go on to develop this tumor. Another group of patients, those with the so-called Denys-Drash syndrome (DDS), also have an extremely high risk (approximately 90%) for the development of Wilms tumor. This syndrome is characterized by gonadal dysgenesis and renal abnormalities. Both of these conditions are associated with abnormalities of the Wilms tumor 1 gene (WT1), located on 11p13. The nature of genetic aberration differs, however: Patients with WAGR syndrome demonstrate loss of genetic material (i.e., deletions) of WT1, and persons with DDS harbor a dominant negative inactivating mutation in a critical region of the gene. (A dominant negative mutation interferes with the function of the remaining wild-type allele.) The WT1 gene is critical to normal renal and gonadal development; it is not surprising, therefore, that constitutional inactivation of one copy of this gene results in genitourinary abnormalities in humans. A third group of patients, those with the Beckwith-Wiedemann syndrome (BWS), also are at increased risk for the development of Wilms tumor. These patients exhibit enlargement of individual body organs (e.g., tongue, kidneys, or liver) or entire body segments (hemihypertrophy); enlargement of adrenal cortical cells (adrenal cytomegaly) is a characteristic microscopic feature. BWS is an example of a disorder of genomic imprinting (see earlier). The genetic locus that is involved in these patients is in band p15.5 of chromosome 11 distal to the WT1 locus. Although this locus is called “WT2” for the second Wilms tumor locus, the gene involved has not been identified. This region contains at least 10 genes that normally are expressed from only one of the two parental alleles, with transcriptional silencing of the other parental homologue by methylation of the promoter region, located upstream of the transcription start site. Of all candidate “WT2” genes, imprinting abnormalities of insulin-like growth factor-2 (IGF2) have the strongest relationship to tumor predisposition in persons with BWS. IGF2 normally is expressed solely from the paternal allele, while the maternal allele is imprinted (i.e., silenced) by methylation. In some Wilms tumors, loss of imprinting (i.e., reexpression of IGF2 by the maternal allele) can be demonstrated, leading to overexpression of the IGF2 protein, which is postulated to result in both organ enlargement and tumorigenesis. Thus, these associations suggest that in some cases, congenital malformations and tumors represent related manifestations of genetic lesions affecting a single gene or closely linked genes. In addition to Wilms tumors, patients with BWS also are at increased risk for the development of hepatoblastoma, adrenocortical tumors, rhabdomyosarcomas, and pancreatic tumors. Recently, gain-of-function mutations of the gene encoding β-catenin (Chapter 5) have been demonstrated in approximately 10% of sporadic Wilms tumors.
Morphology
As seen on gross examination, Wilms tumor typically is a large, solitary, well-circumscribed mass, although 10% are either bilateral or multicentric at the time of diagnosis. On cut section, the tumor is soft, homogeneous, and tan to gray, with occasional foci of hemorrhage, cystic degeneration, and necrosis (Fig. 6–35).
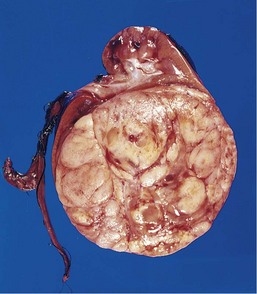
Figure 6–35 Wilms tumor in the lower pole of the kidney with the characteristic tan to gray color and well-circumscribed margins.
On microscopic examination, Wilms tumors are characterized by recognizable attempts to recapitulate different stages of nephrogenesis. The classic triphasic combination of blastemal, stromal, and epithelial cell types is observed in most lesions, although the percentage of each component is variable (Fig. 6–36, A). Sheets of small blue cells, with few distinctive features, characterize the blastemal component. Epithelial “differentiation” usually takes the form of abortive tubules or glomeruli. Stromal cells are usually fibrocytic or myxoid in nature, although skeletal muscle “differentiation” is not uncommon. Rarely, other heterologous elements are identified, including squamous or mucinous epithelium, smooth muscle, adipose tissue, cartilage, and osteoid and neurogenic tissue. Approximately 5% of tumors contain foci of anaplasia (cells with large, hyperchromatic, pleomorphic nuclei and abnormal mitoses) (Fig. 6–36, B). The presence of anaplasia correlates with the presence of acquired TP53 mutations, and the emergence of resistance to chemotherapy. The pattern of distribution of anaplastic cells within the primary tumor (focal versus diffuse) has important implications for prognosis (see further on).
Figure 6–36 A, Wilms tumor with tightly packed blue cells consistent with the blastemal component and interspersed primitive tubules, representing the epithelial component. Although multiple mitotic figures are seen, none are atypical in this field. B, Focal anaplasia was present in other areas within this Wilms tumor, characterized by cells with hyperchromatic, pleomorphic nuclei and abnormal mitoses.
Nephrogenic rests are putative precursor lesions of Wilms tumors and are sometimes present in the renal parenchyma adjacent to the tumor. Nephrogenic rests have a spectrum of histologic appearances, from expansile masses that resemble Wilms tumors (hyperplastic rests) to sclerotic rests consisting predominantly of fibrous tissue with occasional admixed immature tubules or glomeruli. It is important to document the presence of nephrogenic rests in the resected specimen, since these patients are at an increased risk for the development of Wilms tumors in the contralateral kidney.
Clinical Course
Patients’ complaints usually are referable to the tumor’s enormous size. Commonly, there is a readily palpable abdominal mass, which may extend across the midline and down into the pelvis. Less often, the patient presents with fever and abdominal pain, with hematuria or, occasionally, with intestinal obstruction as a result of pressure from the tumor. The prognosis for Wilms tumor generally is very good, and excellent results are obtained with a combination of nephrectomy and chemotherapy. Anaplasia is a harbinger of adverse prognosis, but careful analyses by the National Wilms’ Tumor Study group in the United States have shown that as long as the anaplasia is focal and confined within the resected nephrectomy specimen, the outcome is no different from that for tumors without evidence of anaplasia. By contrast, Wilms tumors with diffuse anaplasia, especially those exhibiting extrarenal spread, have the least favorable outcome, underscoring the need for correctly identifying this histologic pattern.
Summary
Wilms Tumor
• Wilms tumor is the most common renal neoplasm of childhood.
• Patients with three syndromes are at increased risk for Wilms tumors: Denys-Drash, Beckwith-Wiedemann, and Wilms tumor, aniridia, genital abnormalities, and mental retardation syndrome.
• Wilms tumor, aniridia, genital abnormalities, and mental retardation syndrome and Denys-Drash syndrome are associated with WT1 inactivation, while Beckwith-Wiedemann arises through imprinting abnormalities at the WT2 locus, principally involving the IGF2 gene.
• The morphologic components of Wilms tumor include blastema (small, round blue cells) and epithelial and stromal elements.
Molecular Diagnosis of Mendelian and Complex Disorders
In the past decade, few disciplines within pathology have seen a surge in both “supply” and “demand” comparable with that observed for molecular diagnostics. In the era predating the ready availability of molecular diagnostic assays, rendering the diagnosis of a genetic disorder depended on the identification of abnormal gene products (e.g., mutant hemoglobin or abnormal metabolites) or their clinical effects, such as mental retardation (e.g., in PKU). The nascent field of molecular diagnostics emerged in the latter half of the 20th century, with the application of low-throughput approaches such as conventional karyotyping for recognition of cytogenetic disorders (e.g., Down syndrome) and DNA-based assays such as Southern blotting for the diagnosis of Huntington disease. Several factors have since enabled the rapid expansion of molecular diagnostics from the esoteric realm to an almost ubiquitous presence in both academic and commercial pathology laboratories (with current estimates for the “worldwide market” running into tens of billions of dollars). These factors include (1) the sequencing of the human genome and the deposition of these data in publicly available databases; (2) the availability of numerous “off the shelf” polymerase chain reaction (PCR) kits tailor-made for the identification of specific genetic disorders; (3) the availability of high-resolution microarrays (“gene chips”) that can interrogate both DNA and RNA on a genome-wide scale using a single platform; and finally, (4) the emergence of automated and extremely high-throughput, next-generation (“NextGen”) sequencing technologies. The last two advances have been especially useful in the context of new research to elucidate the genetic basis for both mendelian and complex disorders. While a detailed discussion of molecular diagnostics is beyond the scope of this book, some of the better-known approaches are highlighted in the ensuing paragraphs. One important caveat at this juncture is that irrespective of the technique used, the genetic aberration being queried can be either in the germ line (i.e., present in each and every cell of the affected person, as with the CFTR mutation in a patient with CF) or somatic (i.e., restricted to specific tissue types or lesions, as with the NMYC amplification in neuroblastoma cells). This consideration will determine the nature of the sample (e.g., peripheral blood lymphocytes [PBLs], saliva, tumor tissue) used for the assay.
Molecular Diagnosis of Copy Number Abnormalities
As already discussed in this chapter, various diseases may occur as a result of copy number abnormalities, at the level of either the entire chromosome (trisomy 21), chromosomal segments (22q11 deletion syndrome), or submicroscopic intragenic deletions (WAGR syndrome). Karyotype analysis of chromosomes by G banding remains the classic approach for identifying changes at the chromosomal level; however, as might be expected, the resolution with this technique is fairly low. In order to identify subchromosomal alterations, both focused analysis of chromosomal regions by FISH and global genomic approaches such as comparative genomic hybridization (CGH) have become popular.
Fluorescence in Situ Hybridization (FISH)
FISH utilizes DNA probes that recognize sequences specific to chromosomal regions of greater than 100 kilobases in size, which defines the limit of resolution with this technique for identifying chromosomal changes. Such probes are labeled with fluorescent dyes and applied to metaphase spreads or interphase nuclei. The probe hybridizes to its complementary sequence on the chromosome and thus labels the specific chromosomal region that can then be visualized under a fluorescence microscope. The ability of FISH to circumvent the need for dividing cells is invaluable when a rapid diagnosis is warranted (e.g., in a critically ill infant suspected of having an underlying genetic disorder). Such analysis can be performed on prenatal samples (e.g., cells obtained by amniocentesis, chorionic villus biopsy, or umbilical cord blood), peripheral blood lymphocytes, and even archival tissue sections. FISH has been used for detection of numeric abnormalities of chromosomes (aneuploidy) (Fig. 6–37, A); for the demonstration of subtle microdeletions (Fig. 6–37, B) or complex translocations not detectable by routine karyotyping; for analysis of gene amplification (e.g., NMYC amplification in neuroblastomas); and for mapping newly isolated genes of interest to their chromosomal loci.
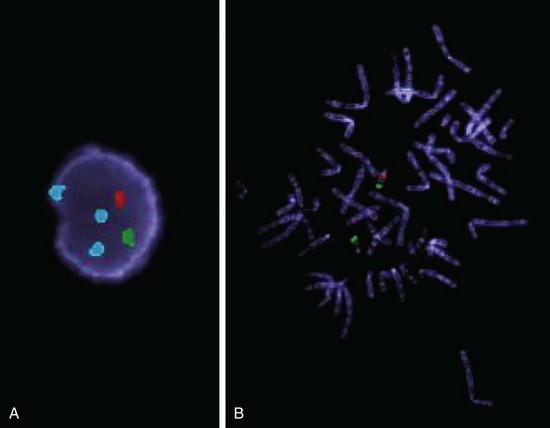
Figure 6–37 Fluorescence in situ hybridization (FISH). A, Interphase nucleus from a male patient with suspected trisomy 18. Three different fluorescent probes have been used in a “FISH cocktail”; the green probe hybridizes to the X chromosome centromere (one copy), the red probe to the Y chromosome centromere (one copy), and the aqua probe to the chromosome 18 centromere (three copies). B, A metaphase spread in which two fluorescent probes have been used, one hybridizing to chromosome region 22q13 (green) and the other hybridizing to chromosome region 22q11.2 (red). There are two 22q13 signals. One of the two chromosomes does not stain with the probe for 22q11.2, indicating a microdeletion in this region. This abnormality gives rise to the 22q11.2 deletion syndrome (DiGeorge syndrome).
(Courtesy of Dr. Nancy R. Schneider and Jeff Doolittle, Cytogenetics Laboratory, University of Texas Southwestern Medical Center, Dallas, Texas.)
Array-Based Genomic Hybridization
It is obvious from the preceding discussion that FISH requires previous knowledge of the one or few specific chromosomal regions suspected of being altered in the test sample. However, chromosomal abnormalities may also be detected without previous knowledge of what these aberrations may be, using a global strategy known as array-based CGH. Here the test DNA and a reference (normal) DNA are labeled with two different fluorescent dyes (most commonly, Cy5 and Cy3, which fluoresce red and green, respectively). The differentially labeled samples are then hybridized to an array of segments of genomic DNA “spotted” on a solid matrix, usually a glass slide (Fig. 6–38, A). These segments of DNA are representations of the human genome at regularly spaced intervals, and cover all 22 autosomes and the sex chromosome (Fig. 6–38, A). Amplifications and deletions in the test sample produce an increase or decrease in signal relative to the normal DNA that can be detected down to a 10-kilobase (kb) resolution (Fig. 6–38, B). Newer generations of microarrays using single-nucleotide polymorphisms (SNPs) (see further on) provide even higher resolution (with more than 1 million SNPs from the human genome on a single microarray!) and are currently being used to uncover copy number abnormalities in a variety of diseases, from cancer to autism.
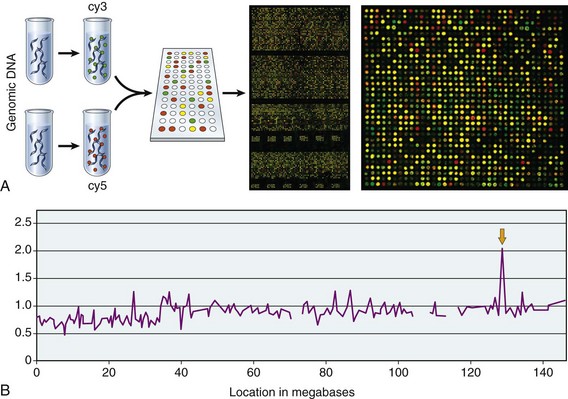
Figure 6–38 Array comparative genomic hybridization (CGH) is performed by hybridization of fluorescently labeled test DNA and control DNA on a slide that contains thousands of probes corresponding to defined chromosomal regions across the human genome. The resolution with most currently available array CGH technology is on the order of approximately 10 kb. A, Higher-power view of the array demonstrates copy number aberrations in the test Cy5-labeled sample (red), including regions of amplification (spots with excess of red signal) and deletion (spots with excess of green signal); yellow spots correspond to regions of normal (diploid) copy number. B, The hybridization signals are digitized, resulting in a virtual karyotype of the genome of the “test” sample. In the illustrated example, array CGH of a cancer cell line identifies an amplification on the distal long arm of chromosome 8, which corresponds to increased copy number of the oncogene MYC.
(A, From Snijders AM, Nowak N, Segraves R, et al: Assembly of microarrays for genome-wide measurement of DNA copy number. Nat Genet 29:263, 2001; Web Figure A. Copyright 2001. Reprinted by permission from Macmillan Publishers Ltd.)
Direct Detection of DNA Mutations by Polymerase Chain Reaction (PCR) Analysis
PCR analysis, which involves exponential amplification of DNA, is now widely used in molecular diagnosis. If RNA is used as the substrate, it is first reverse-transcribed to obtain cDNA and then amplified by PCR. This method involving reverse transcription (RT) often is abbreviated as RT-PCR. One prerequisite for direct detection is that the sequence of the normal gene must be known. To detect the mutant gene, two primers that bind to the 3′ and 5′ ends of the normal sequence are designed. By utilizing appropriate DNA polymerases and thermal cycling, the target DNA is greatly amplified, producing millions of copies of the DNA sequence between the two primer sites. The subsequent identification of an abnormal sequence can then be performed in several ways:
• The DNA can be sequenced to obtain a readout of the order of nucleotides, and by comparison with a normal (wild-type) sequence, mutations can be identified. Most DNA sequencing machines are automated and use a fluorescent dye–based version of a sequencing technology originally named after its inventor, Frederick Sanger. More recently, gene chips (microarrays) also have become available that can be used for sequencing genes or portions of genes. Short sequences of DNA (oligonucleotides) that are complementary to the wild-type sequence and to known mutations are “tiled” adjacent to each other on the gene chip, and the DNA sample to be tested is hybridized to the array (Fig. 6–39). Before hybridization, the sample is labeled with fluorescent dyes. The hybridization (and, consequently, the fluorescent signal emitted) will be strongest at the oligonucleotide that is complementary to the wild-type sequence if no mutations are present, while the presence of a mutation will cause hybridization to occur at the complementary mutant oligonucleotide. Computerized algorithms can then rapidly “decode” the DNA sequence for hundreds of thousands of base pairs of sequence from the fluorescent hybridization pattern on the chip, to identify potential mutations.
• Another approach for identifying mutations at a specific nucleotide position (say, a codon 12 mutation in the KRAS oncogene that converts glycine [GGT] to aspartic acid [GAT]) would be to add fluorescently labeled nucleotides C and T to the PCR mixture, which are complementary to either the wild-type (G) or mutant (A) sequence, respectively. Since these two nucleotides are labeled with different fluorophores, the fluorescence emitted by the resulting PCR product can be of one or another color, depending on whether a C or a T becomes incorporated in the process of primer extension (Fig. 6–40). The advantage of this allele-specific extension strategy is that it can detect the presence of mutant DNA even in heterogeneous mixtures of normal and abnormal cells (for example, in clinical specimens obtained from patients suspected of harboring a malignancy). Many variations on this theme have been developed and are being currently used for mutation detection in the laboratory and clinical settings.
• In this context, one would be remiss not to mention next-generation (“NextGen”) sequencing technologies, so named because the Sanger sequencing mentioned earlier is now considered “first generation.” The availability of NextGen sequencing technology has the potential to alter molecular diagnostics radically, by the sheer volume of sequencing data (more than 1 giga–base pairs or 1,000,000,000 base pairs of DNA per day!) at relatively cheap costs. The entire human genome has a little over 3 gigabases, so true “whole genome sequencing” can be performed several times over in a matter of days. In contrast with Sanger sequencing, NextGen sequencing technologies utilize platforms where sequencing of multiple fragments of the human genome (DNA or cDNA) can occur in parallel (“massively parallel sequencing”), significantly enhancing its speed (Fig. 6–41). Fluorescently labeled nucleotides are incorporated complementary to the template DNA strands, which are immobilized on a solid phase, with one nucleotide added per template per cycle. The cycles are repeated until a sufficient length of “read” is generated that can then be mapped back to the human genome using sophisticated bioinformatics. Deep sequencing is now being utilized to sequence somatic mutations in some of the most common tumor types, while germline sequencing has started identifying the hitherto unknown genetic basis of rare mendelian disorders.
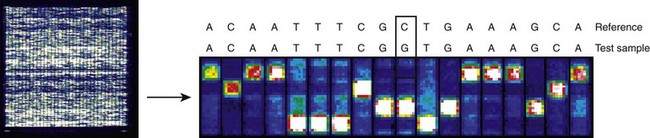
Figure 6–39 Microarray-based DNA sequencing. Left panel, A low-power digitized scan of a “gene chip” that is no larger than a nickel in size but is capable of sequencing thousands of base pairs of DNA. High-throughput microarrays have been used for sequencing whole organisms (such as viruses), organelles (such as the mitochondria), and entire human chromosomes. Right panel, A high-resolution view of the gene chip illustrates hybridization patterns corresponding to a stretch of DNA sequence. Typically, a computerized algorithm is available that can convert the individual hybridization patterns across the entire chip into actual sequence data within a matter of minutes (“conventional” sequencing technologies would require days to weeks for such analysis). Here, the sequence on top is the reference (wild-type) sequence, while the lower one corresponds to the test sample sequence. As shown, the computerized algorithm has identified a C→G mutation in the test sample.
(Adapted from Maitra A, Cohen Y, Gillespie SE, et al: The Human MitoChip: a high-throughput sequencing microarray for mitochondrial mutation detection. Genome Res 14:812, 2004.)
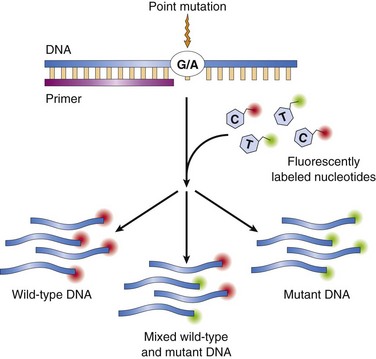
Figure 6–40 Allele-specific polymerase chain reaction (PCR) analysis for mutation detection in a heterogeneous sample containing an admixture of normal and mutant DNA. Nucleotides complementary to the mutant and wild-type nucleotides at the queried base position are labeled with different fluorophores such that incorporation into the resulting PCR product yields fluorescent signals of variable intensity in accordance with the ratio of mutant to wild-type DNA present.
Figure 6–41 Principle of next-generation sequencing. Several alternative approaches currently are available for “NextGen” sequencing, and one of the more commonly used platforms is illustrated. A, Short fragments of genomic DNA (“template”) between 100 and 500 base pairs in length are immobilized on a solid phase platform such as a glass slide, using universal capture primers that are complementary to adapters that have previously been added to ends of the template fragments. The addition of fluorescently labeled complementary nucleotides, one per template DNA per cycle, occurs in a “massively parallel” fashion, at millions of templates immobilized on the solid phase at the same time. A four-color imaging camera captures the fluorescence emanating from each template location (corresponding to the specific incorporated nucleotide), following which the fluorescent dye is cleaved and washed away, and the entire cycle is repeated. B, Powerful computational programs can decipher the images to generate sequences complementary to the template DNA at the end of one “run,” and these sequences are then mapped back to the reference genomic sequence, in order to identify alterations.
(Reproduced with permission from Metzker M: Sequencing technologies—the next generation. Nat Rev Genet 11:31–46, 2010, © Nature Publishing Group.)
Linkage Analysis and Genome-Wide Association Studies
Direct diagnosis of mutations is possible only if the gene responsible for a genetic disorder is known and its sequence has been identified. In several diseases that have a genetic basis, including some common disorders, direct genetic diagnosis is not possible, either because the causal gene has not been identified or because the disease is multifactorial (polygenic) and no single gene is involved. In such cases, two types of analyses can be performed for unbiased identification of disease-associated gene(s): linkage analysis and genome-wide association studies (GWASs). In both instances, surrogate markers in the genome, also known as marker loci, must be used to localize the chromosomal regions of interest, based on their linkage to one or more putative disease-causing genes. The marker loci utilized are naturally occurring variations in DNA sequences known as polymorphisms. The most common DNA polymorphisms—SNPs—occur at a frequency of approximately one nucleotide in every 1000–base pair stretch and are found throughout the genome (e.g., in exons and introns and in regulatory sequences). SNPs serve both as a physical landmark within the genome and as a genetic marker whose transmission can be followed from parent to child.
Two technologic breakthroughs have enabled the application of SNPs to high-throughput “gene hunting”: First is the completion of the so-called HapMap project, which has provided linkage disequilibrium patterns in three major ethnoracial groups, based on genome-wide SNP mapping. The entire human genome can now be divided into blocks known as “haplotypes,” which contain varying numbers of contiguous SNPs on the same chromosome that are in linkage disequilibrium and hence inherited together as a cluster. As a result, rather than querying every single SNP in the human genome, comparable information about shared DNA can be obtained simply by looking for shared haplotypes, using single or a small number of SNPs that “tag” or identify a specific haplotype. Second, it is now possible to simultaneously genotype hundreds of thousands to a million SNPs at one time, in a cost-effective way, using high-density SNP chip technology.
• Linkage analysis deals with assessing shared marker loci (i.e., SNPs) in family members exhibiting the disease or trait of interest, with the assumption that SNPs in linkage disequilibrium with the disease allele are transmitted through pedigrees. With time it becomes possible to define a “disease haplotype” based on a panel of SNPs, all of which cosegregate with the putative disease allele. Eventually, linkage analysis facilitates localization and cloning of the disease allele. Linkage analysis is most useful in mendelian disorders that are related to one gene with profound effects and high penetrance.
• It is now established that some of the most common human diseases, such as hypertension, diabetes, mental disorders, and asthma, have a polygenic basis, with multiple genetic loci contributing small independent effects, resulting in a disease phenotype. Conventional linkage analyses lack the statistical power to detect such genetic variants. In GWASs, large cohorts of patients with and without a disease (rather than families) are examined across the entire genome for variant SNPs that are overrepresented in persons with the disease. This identifies regions of the genome that contain a variant gene or genes that confer disease susceptibility. The causal variant within the region is then provisionally identified using a “candidate gene” approach, in which genes are selected on the basis of how tightly they are associated with the disease and whether their biologic function seems likely to be involved in the disease under study. In addition to polygenic diseases, GWASs also have led to the identification of genetic loci that modulate common quantitative traits in humans, such as height, body mass, hair and eye color, and bone density.
Indications for Genetic Analysis
The preceding discussion described some of the many techniques available today for the diagnosis of genetic diseases. For judicious application of these methods, it is important to recognize which persons require genetic testing. In general, genetic testing can be divided into prenatal and postnatal analysis. It may involve conventional cytogenetics, FISH, molecular diagnostics, or a combination of these techniques.
Prenatal genetic analysis should be offered to all patients who are at risk of having cytogenetically abnormal progeny. It can be performed on cells obtained by amniocentesis, on chorionic villus biopsy material, or on umbilical cord blood. Some important indications are the following:
• Advanced maternal age (beyond 34 years), which is associated with greater risk of trisomies
• Confirmed carrier status for a balanced reciprocal translocation, Robertsonian translocation, or inversion (in such cases, the gametes may be unbalanced, so the progeny would be at risk for chromosomal disorders)
• A chromosomal abnormality affecting a previous child
• Determination of fetal sex when the patient or partner is a confirmed carrier of an X-linked genetic disorder
Postnatal genetic analysis usually is performed on peripheral blood lymphocytes. Indications are as follows:
• Multiple congenital anomalies
• Unexplained mental retardation and/or developmental delay
• Suspected aneuploidy (e.g., features of Down syndrome)
• Suspected unbalanced autosome (e.g., Prader-Willi syndrome)
• Suspected sex chromosome abnormality (e.g., Turner syndrome)
• Suspected fragile X syndrome
• Infertility (to rule out sex chromosome abnormality)
• Multiple spontaneous abortions (to rule out the parents as carriers of balanced translocation; both partners should be evaluated)
In the context of these and other clinical applications, an important point is that we are currently living in a breath-taking era of so-called genomic medicine. Future years will foretell how the advances in elucidation of the genetic basis of human disease will affect its diagnosis, prevention, and therapy.