Chapter 19 Endocrine System
See Targeted Therapy available online at studentconsult.com
The endocrine system is a highly integrated and widely distributed group of organs that orchestrate a state of metabolic equilibrium, or homeostasis, among the various tissues of the body. Signaling by extracellular secreted molecules can be classified as one of three types—autocrine, paracrine, or endocrine—according to the distance over which the signal acts (Chapter 2). In endocrine signaling, the secreted molecules, which frequently are called hormones, act on target cells distant from their site of synthesis. An endocrine hormone typically is carried by the blood from its site of release to its target. Increased activity of the target tissue often downregulates the activity of the gland that secretes the stimulating hormone, a process known as feedback inhibition.
Hormones can be classified into several broad categories, based on the nature of their receptors:
• Hormones that trigger biochemical signals upon interacting with cell surface receptors: This large class of compounds is composed of two groups: (1) peptide hormones, such as growth hormone and insulin, and (2) small molecules, such as epinephrine. Binding of these hormones to cell surface receptors leads to an increase in intracellular molecules, termed second messengers, such as cyclic adenosine monophosphate (cAMP); production of mediators from membrane phospholipids (e.g., inositol 1,4,5-trisphosphate); and shifts in intracellular levels of ionized calcium. Elevated levels of one or more of these compounds can change proliferation, differentiation, survival, and functional activity of cells, mainly by regulating the expression of specific genes.
• Hormones that diffuse across the plasma membrane and interact with intracellular receptors: Many lipid-soluble hormones pass through the plasma membrane by diffusion to interact with receptors in the cytosol or the nucleus. The resulting hormone-receptor complexes bind specifically to promoter and enhancer elements in DNA, thereby affecting the expression of specific target genes. Hormones of this type include the steroids (e.g., estrogen, progesterone, glucocorticoids), the retinoids (vitamin A), and thyroxine.
Several processes may disturb the normal activity of the endocrine system, including impaired synthesis or release of hormones, abnormal interactions between hormones and their target tissues, and abnormal responses of target organs to their hormones. Endocrine diseases can be generally classified as (1) diseases of underproduction or overproduction of hormones, with associated biochemical and clinical consequences, or (2) diseases associated with the development of mass lesions, which may be nonfunctional or may be associated with overproduction or underproduction of hormones.
With the exception of mass lesions, study of endocrine diseases relies heavily on biochemical measurements of the levels of hormones, their regulators, and other metabolites.
Pituitary
The pituitary gland is a small, bean-shaped structure that lies at the base of the brain within the confines of the sella turcica. It is intimately related to the hypothalamus, with which it is connected by both a stalk, composed of axons extending from the hypothalamus, and a rich venous plexus constituting a portal circulation. Along with the hypothalamus, the pituitary has a central role in the regulation of most of the other endocrine glands. The pituitary is composed of two morphologically and functionally distinct components: the anterior lobe (adenohypophysis) and the posterior lobe (neurohypophysis). Diseases of the pituitary, accordingly, can be divided into those that primarily affect the anterior lobe and those that primarily affect the posterior lobe.
The anterior pituitary, or adenohypophysis, is composed of epithelial cells derived embryologically from the developing oral cavity. In routine histologic sections, a colorful array of cells containing basophilic cytoplasm, eosinophilic cytoplasm, or poorly staining (chromophobic) cytoplasm is present (Fig. 19–1). Detailed studies using electron microscopy and immunocytochemical techniques have demonstrated that the staining properties of these cells are related to the presence of various trophic polypeptide hormones within their cytoplasm. The release of trophic hormones is in turn under the control of factors produced in the hypothalamus; while most hypothalamic factors are stimulatory and promote pituitary hormone release, others (e.g., somatostatin and dopamine) are inhibitory in their effects (Fig. 19–2). Rarely, signs and symptoms of pituitary disease may be caused by excess or lack of the hypothalamic factors, rather than by a primary pituitary abnormality.
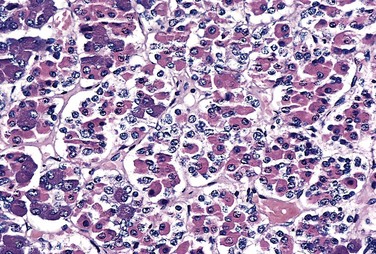
Figure 19–1 Normal architecture of the anterior pituitary. The gland is populated by several distinct cell types containing a variety of stimulating (trophic) hormones. Each of the hormones has different staining characteristics, resulting in a mixture of cell types in routine histologic preparations. Note also the presence of a fine reticulin network.
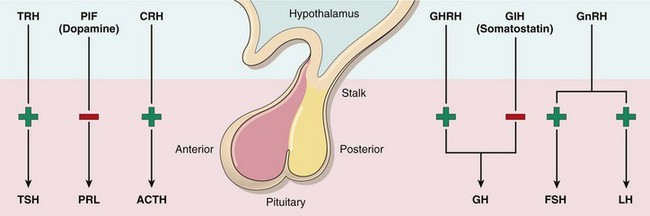
Figure 19–2 The adenohypophysis (anterior pituitary) releases six hormones: adrenocorticotropic hormone (ACTH), or corticotropin; follicle-stimulating hormone (FSH); growth hormone (GH), or somatotropin; luteinizing hormone (LH); prolactin (PRL); and thyroid-stimulating hormone (TSH), or thyrotropin. These hormones are in turn under the control of various stimulatory and inhibitory hypothalamic releasing factors. The stimulatory releasing factors are corticotropin-releasing hormone (CRH), growth hormone–releasing hormone (GHRH), gonadotropin-releasing hormone (GnRH), and thyrotropin-releasing hormone (TRH). The inhibitory hypothalamic factors are growth hormone inhibitory hormone (GIH), or somatostatin, and prolactin inhibitory factor (PIF), which is the same as dopamine.
Symptoms and signs of pituitary disease can be grouped as follows:
• Hyperpituitarism-related effects: Hyperpituitarism arises from excessive secretion of trophic hormones. It most often results from an anterior pituitary adenoma but also may be caused by other pituitary and extrapituitary lesions, as described subsequently. The symptoms and signs of hyperpituitarism are discussed in the context of individual tumors, later in the chapter.
• Hypopituitarism-related effects: Hypopituitarism is caused by deficiency of trophic hormones and results from a variety of destructive processes, including ischemic injury, surgery or radiation, and inflammatory reactions. In addition, nonfunctional pituitary adenomas may encroach upon and destroy adjacent normal anterior pituitary parenchyma, causing hypopituitarism.
• Local mass effects: Among the earliest changes referable to mass effect are radiographic abnormalities of the sella turcica, including sellar expansion, bony erosion, and disruption of the diaphragma sellae. Because of the close proximity of the optic nerves and chiasm to the sella, expanding pituitary lesions often compress decussating fibers in the optic chiasm. This altered neuroanatomy gives rise to visual field abnormalities, classically in the form of defects in the lateral (temporal) visual fields—a so-called bitemporal hemianopsia. As in the case of any expanding intracranial mass, pituitary adenomas may produce signs and symptoms of elevated intracranial pressure, including headache, nausea, and vomiting. Pituitary adenomas that extend beyond the sella turcica into the base of the brain (invasive pituitary adenoma) produce seizures or obstructive hydrocephalus; involvement of cranial nerves can result in cranial nerve palsy. On occasion, acute hemorrhage into an adenoma is associated with clinical evidence of rapid enlargement of the lesion and depression of consciousness, a situation appropriately termed pituitary apoplexy. Acute pituitary apoplexy constitutes a neurosurgical emergency, because it may be rapidly fatal.
Hyperpituitarism and Pituitary Adenomas
The most common cause of hyperpituitarism is an adenoma arising in the anterior lobe. Other, less common, causes include hyperplasia and carcinomas of the anterior pituitary, secretion of hormones by some extrapituitary tumors, and certain hypothalamic disorders. Some salient features of pituitary adenomas are as follows:
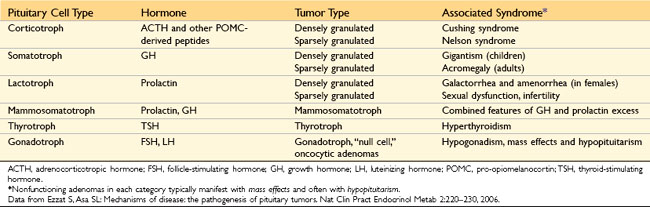
• Pituitary adenomas are classified on the basis of hormone(s) produced by the neoplastic cells, which are detected by immunohistochemical stains performed on tissue sections (Table 19–1).
• Pituitary adenomas can be functional (i.e., associated with hormone excess and clinical manifestations thereof) or nonfunctioning (i.e., demonstration of hormone production at the tissue level only, without clinical manifestations of hormone excess). Both functional and nonfunctioning pituitary adenomas usually are composed of a single cell type and produce a single predominant hormone, but there are some exceptions. Some pituitary adenomas can secrete two different hormones (growth hormone and prolactin being the most common combination); rarely, pituitary adenomas are plurihormonal. At the other end of the spectrum, pituitary adenomas also may be truly “hormone negative,” as indicated by absence of immunohistochemical reactivity or ultrastructural evidence of hormone production.
• Most pituitary adenomas occur as sporadic (i.e., non-familial) lesions. In about 5% of cases, however, adenomas occur as a result of an inherited predisposition (see later).
• Pituitary adenomas are designated, somewhat arbitrarily, as microadenomas if they are less than 1 cm in diameter and macroadenomas if they exceed 1 cm in diameter.
• Nonfunctioning and hormone-negative adenomas are likely to come to clinical attention at a later stage and are, therefore, more likely to be macroadenomas than are lesions associated with endocrine abnormalities. In addition, nonfunctioning adenomas may cause hypopituitarism as they encroach on and destroy adjacent anterior pituitary parenchyma.
 Pathogenesis
Pathogenesis
With recent advances in molecular techniques, substantial insight has been gained into the genetic abnormalities associated with pituitary adenomas:
• Guanine nucleotide–binding protein (G protein) mutations are the best-characterized molecular abnormalities in these neoplasms. G proteins have a critical role in signal transduction, transmitting signals from cell surface receptors (e.g., growth hormone–releasing hormone receptor) to intracellular effectors (e.g., adenyl cyclase), which then generate second messengers (e.g., cAMP). Gs is a stimulatory G protein that has a pivotal role in signal transduction in several endocrine organs, including the pituitary. Gs exists as an inactive protein, with guanosine diphosphate (GDP) bound to the guanine nucleotide–binding site of the alpha subunit of Gs, encoded by the GNAS1 gene. On triggering of the hormone receptor, GDP dissociates, and guanosine triphosphate (GTP) binds to Gsα, activating the G protein. GTP-bound Gsα directly interacts with and activates its effectors (such as adenyl cyclase), with a resultant increase in intracellular cAMP. The cAMP acts as a potent mitogenic stimulus for a variety of endocrine cell types, promoting cellular proliferation and hormone synthesis and secretion. The activation of Gsα and the resultant generation of cAMP are transient because of an intrinsic GTPase activity in the α-subunit, which hydrolyzes GTP into GDP. A mutation in the α-subunit that interferes with its intrinsic GTPase activity therefore results in constitutive activation of Gsα, persistent generation of cAMP, and unchecked cellular proliferation. Approximately 40% of growth hormone–secreting somatotroph cell adenomas and a minority of adrenocorticotropic hormone (ACTH)–secreting corticotroph cell adenomas bear GNAS1 mutations.
• As stated previously, approximately 5% of pituitary adenomas arise as a consequence of an inherited predisposition. Four genes have been identified thus far as a cause of familial pituitary adenomas: MEN1, CDKN1B, PRKAR1A, and AIP. Germline inactivating mutations of the MEN1 gene are responsible for multiple endocrine neoplasia syndrome type 1 (MEN-1) (discussed in detail later on). The product of the CDKN1B gene is the cell cycle checkpoint regulator p27 or KIP1; germline mutations of CDKN1B are responsible for a subset of patients with a “MEN-1 like” syndrome who lack MEN1 abnormalities. The gene encoding the aryl hydrocarbon receptor–interacting protein (AIP) is a recently described pituitary adenoma predisposition gene, and patients with AIP germline mutations often develop GH-secreting adenomas at a younger age (before 35 years) than that typical for sporadic GH adenoma patients.
• Mutations of TP53 in pituitary adenomas are associated with a propensity for aggressive behavior, such as invasion and recurrence.
 Morphology
Morphology
The usual pituitary adenoma is a well-circumscribed, soft lesion that may, in the case of smaller tumors, be confined by the sella turcica. Larger lesions may compress the optic chiasm and adjacent structures (Fig. 19–3), erode the sella turcica and anterior clinoid processes, and extend locally into the cavernous and sphenoidal sinuses. In as many as 30% of cases, the adenomas are nonencapsulated and infiltrate adjacent bone, dura, and (uncommonly) brain. Foci of hemorrhage and/or necrosis are common in larger adenomas.
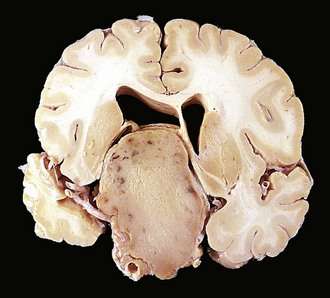
Figure 19–3 Pituitary adenoma. This massive, nonfunctioning adenoma has grown far beyond the confines of the sella turcica and has distorted the overlying brain. Nonfunctioning adenomas tend to be larger at the time of diagnosis than those that secrete a hormone.
Pituitary adenomas are composed of relatively uniform, polygonal cells arrayed in sheets, cords, or papillae. Supporting connective tissue, or reticulin, is sparse, accounting for the soft, gelatinous consistency of many of these tumors. The nuclei of the neoplastic cells may be uniform or pleomorphic. Mitotic activity usually is scanty. The cytoplasm of the constituent cells may be acidophilic, basophilic, or chromophobic, depending on the type and amount of secretory product within the cell, but it is fairly uniform throughout the neoplasm. This cellular monomorphism and the absence of a significant reticulin network distinguish pituitary adenomas from non-neoplastic anterior pituitary parenchyma (Fig. 19–4). The functional status of the adenoma cannot be reliably predicted from its histologic appearance. Adenomas that harbor TP53 mutations often demonstrate brisk mitotic activity and higher proliferation rates and are designated atypical adenomas to reinforce their potential for aggressive behavior.
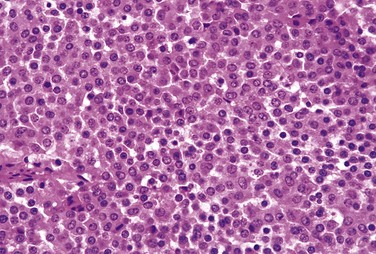
Figure 19–4 Pituitary adenoma. The monomorphism of these cells contrasts markedly with the admixture of cells seen in the normal anterior pituitary in Figure 19–1. Note also the absence of reticulin network.
 Summary
Summary
Hyperpituitarism
• The most common cause of hyperpituitarism is an anterior lobe pituitary adenoma.
• Pituitary adenomas can be macroadenomas (greater than 1 cm in diameter) or microadenomas (less than 1 cm across), and on clinical evaluation, they can be functional or nonfunctioning.
• Macroadenomas may potentially lead to mass effects, including visual disturbances.
• Functioning adenomas are associated with distinct endocrine signs and symptoms.
• Mutation of the GNAS1 gene, which results in constitutive activation of a stimulatory G protein, is one of the more common genetic alterations.
• The two distinctive morphologic features of most adenomas are their cellular monomorphism and absence of a reticulin network.
Prolactinomas
Prolactinomas are the most common type of hyperfunctioning pituitary adenoma. They range in size from small microadenomas to large, expansile tumors associated with considerable mass effect. Prolactin is demonstrable within the cytoplasm of the neoplastic cells by immunohistochemical techniques.
Hyperprolactinemia causes amenorrhea, galactorrhea, loss of libido, and infertility. Because many of the manifestations of hyperprolactinemia (e.g., amenorrhea) are more obvious in premenopausal women than in men or postmenopausal women, prolactinomas usually are diagnosed at an earlier stage in women of reproductive age than in other persons so affected. By contrast, hormonal manifestations may be quite subtle in men and older women, in whom the tumor may reach considerable size before coming to clinical attention. Hyperprolactinemia may be caused by conditions or factors other than prolactin-secreting pituitary adenomas, including pregnancy, high-dose estrogen therapy, renal failure, hypothyroidism, hypothalamic lesions, and dopamine-inhibiting drugs (e.g., reserpine). In addition, any mass in the suprasellar compartment may disturb the normal inhibitory influence of hypothalamus on prolactin secretion, resulting in hyperprolactinemia—a mechanism known as the stalk effect. Thus, mild elevations of serum prolactin (less than 200 µg/L) in a patient with a pituitary adenoma do not necessarily indicate a prolactin-secreting neoplasm.
Growth Hormone–Producing (Somatotroph Cell) Adenomas
Growth hormone–producing neoplasms (somatotroph cell adenomas), including those that produce a mixture of growth hormone and other hormones (e.g., prolactin), constitute the second most common type of functional pituitary adenoma. Because the clinical manifestations of excessive growth hormone may be subtle, somatotroph cell adenomas may be quite large by the time they come to clinical attention. On microscopic examination, growth hormone–producing adenomas are composed of densely or sparsely granulated cells, and immunohistochemical staining demonstrates growth hormone within the cytoplasm of the neoplastic cells. Small amounts of immunoreactive prolactin often are present as well.
Persistent hypersecretion of growth hormone stimulates the hepatic secretion of insulin-like growth factor I (somatomedin C), which causes many of the clinical manifestations. If a growth hormone–secreting adenoma occurs before the epiphyses close, as is the case in prepubertal children, excessive levels of growth hormone result in gigantism. This condition is characterized by a generalized increase in body size, with disproportionately long arms and legs. If elevated levels of growth hormone persist, or develop after closure of the epiphyses, affected persons develop acromegaly, in which growth is most conspicuous in soft tissues, skin, and viscera and in the bones of the face, hands, and feet. Enlargement of the jaw results in its protrusion (prognathism), with broadening of the lower face and separation of the teeth. The hands and feet are enlarged, with broad, sausage-like fingers. In clinical practice, the gigantism typically is accompanied by evidence of acromegaly.
Growth hormone excess also is associated with a number of other disturbances, including abnormal glucose tolerance and diabetes mellitus, generalized muscle weakness, hypertension, arthritis, osteoporosis, and congestive heart failure. Prolactin is demonstrable in a number of growth hormone–producing adenomas and in some cases may be released in sufficient quantities to produce signs and symptoms of hyperprolactinemia.
Adrenocorticotropic Hormone–Producing (Corticotroph Cell) Adenomas
Most corticotroph cell adenomas are small (microadenomas) at the time of diagnosis. These adenomas stain positively with periodic acid–Schiff (PAS) stains, as a result of the accumulation of glycosylated ACTH protein. As in the case of other pituitary hormones, the secretory granules can be detected by immunohistochemical methods. By electron microscopy they appear as membrane-bound, electron-dense granules averaging 300 nm in diameter.
Corticotroph cell adenomas may be clinically silent or may cause hypercortisolism, manifested clinically as Cushing syndrome, because of the stimulatory effect of ACTH on the adrenal cortex. Cushing syndrome, discussed in more detail later with diseases of the adrenal gland, may be caused by a wide variety of conditions in addition to ACTH-producing pituitary neoplasms. When the hypercortisolism is caused by excessive production of ACTH by the pituitary, the process is designated Cushing disease, because it is the pattern of hypercortisolism originally described by Dr. Harvey Cushing. Large, clinically aggressive corticotroph cell adenomas may develop after surgical removal of the adrenal glands for treatment of Cushing syndrome. In most instances, this condition, known as Nelson syndrome, results from loss of the inhibitory effect of adrenal corticosteroids on a preexisting corticotroph microadenoma. Because the adrenals are absent in persons with Nelson syndrome, hypercortisolism does not develop. Instead, patients present with the mass effects of the pituitary tumor. In addition, because ACTH is synthesized as part of a larger prohormone substance that includes melanocyte-stimulating hormone (MSH), hyperpigmentation also may be a feature.
Other Anterior Pituitary Neoplasms
• Gonadotroph (luteinizing hormone [LH]–producing and follicle-stimulating hormone [FSH]–producing) adenomas can be difficult to recognize, because they secrete hormones inefficiently and variably, and the secretory products usually do not cause a recognizable clinical syndrome. They are typically detected when the tumors have become large enough to cause neurologic signs and symptoms, such as impaired vision, headaches, diplopia, or pituitary apoplexy. The neoplastic cells usually demonstrate immunoreactivity for the common gonadotropin α-subunit and the specific β-FSH and β-LH subunits; FSH usually is the predominant secreted hormone.
• Thyrotroph (thyroid-stimulating hormone [TSH]–producing) adenomas account for about 1% of all pituitary adenomas and constitute a rare cause of hyperthyroidism.
• Nonfunctioning pituitary adenomas comprise both clinically silent counterparts of the functioning adenomas just described (for example, a silent gonadotroph adenoma) and true hormone-negative (null cell) adenomas; the latter are quite infrequent, and as noted, many have been reclassified using improved diagnostic techniques. Nonfunctioning adenomas constitute approximately 25% of all pituitary tumors. Not surprisingly, the typical presentation with nonfunctioning adenomas is one characterized by mass effects. These lesions also may compromise the residual anterior pituitary sufficiently to produce hypopituitarism.
• Pituitary carcinomas are exceedingly rare. In addition to local extension beyond the sella turcica, these tumors virtually always demonstrate distant metastases.
Summary
Clinical Manifestations of Pituitary Adenomas
• Prolactinomas: amenorrhea, galactorrhea, loss of libido, and infertility
• Growth hormone (somatotroph cell) adenomas: gigantism (children), acromegaly (adults), and impaired glucose tolerance and diabetes mellitus
• Corticotroph cell adenomas: Cushing syndrome, hyperpigmentation
• All pituitary adenomas, particularly nonfunctioning adenomas, may be associated with mass effects and hypopituitarism.
Hypopituitarism
Hypofunction of the anterior pituitary may occur with loss or absence of 75% or more of the anterior pituitary parenchyma. This may be congenital (exceedingly rare) or may result from a wide range of acquired abnormalities that are intrinsic to the pituitary. Less frequently, disorders that interfere with the delivery of pituitary hormone–releasing factors from the hypothalamus, such as hypothalamic tumors, also may cause hypofunction of the anterior pituitary. Hypopituitarism accompanied by evidence of posterior pituitary dysfunction in the form of diabetes insipidus (see later) is almost always of hypothalamic origin. Most cases of anterior pituitary hypofunction are caused by
• Nonfunctioning pituitary adenomas (discussed earlier)
• Ischemic necrosis of the anterior pituitary, an important cause of pituitary insufficiency. The anterior pituitary has substantial reserve capacity; as a result, destruction of large amounts of the anterior pituitary (75% or greater) must occur before signs and symptoms of hypopituitarism develop. Sheehan syndrome, or postpartum necrosis of the anterior pituitary, is the most common form of clinically significant ischemic necrosis of the anterior pituitary. During pregnancy, the anterior pituitary enlarges considerably, largely because of an increase in the size and number of prolactin-secreting cells. This physiologic enlargement of the gland, however, is not accompanied by an increase in blood supply from the low-pressure portal venous system. The enlarged gland is thus vulnerable to ischemic injury, especially in women who experience significant hemorrhage and hypotension during the peripartal period. The posterior pituitary, because it receives its blood directly from arterial branches, is much less susceptible to ischemic injury and therefore usually is not affected. Clinically significant pituitary necrosis also may be encountered in other conditions, including disseminated intravascular coagulation, sickle cell anemia, elevated intracranial pressure, traumatic injury, and shock of any origin. The residual gland is shrunken and scarred.
• Ablation of the pituitary by surgery or irradiation
• Other, less common causes of anterior pituitary hypofunction, including inflammatory lesions such as sarcoidosis or tuberculosis, trauma, and metastatic neoplasms involving the pituitary.
The clinical manifestations of anterior pituitary hypofunction depend on the specific hormones that are lacking. In children, growth failure (pituitary dwarfism) may occur as a result of growth hormone deficiency. Gonadotropin or gonadotropin-releasing hormone (GnRH) deficiency leads to amenorrhea and infertility in women and to decreased libido, impotence, and loss of pubic and axillary hair in men. TSH and ACTH deficiencies result in symptoms of hypothyroidism and hypoadrenalism, respectively, and are discussed later in the chapter. Prolactin deficiency results in failure of postpartum lactation. The anterior pituitary also is a rich source of MSH, synthesized from the same precursor molecule that produces ACTH; therefore, one of the manifestations of hypopituitarism is pallor from loss of stimulatory effects of MSH on melanocytes.
Posterior Pituitary Syndromes
The posterior pituitary, or neurohypophysis, is composed of modified glial cells (termed pituicytes) and axonal processes extending from nerve cell bodies in the supraoptic and paraventricular nuclei of the hypothalamus. The hypothalamic neurons produce two peptides: antidiuretic hormone (ADH) and oxytocin. They are stored in axon terminals in the neurohypophysis and released into the circulation in response to appropriate stimuli. Oxytocin stimulates the contraction of smooth muscle in the pregnant uterus and of muscle surrounding the lactiferous ducts of the mammary glands. Impairment of oxytocin synthesis and release has not been associated with significant clinical abnormalities. The clinically important posterior pituitary syndromes involve ADH production. They include diabetes insipidus and secretion of inappropriately high levels of ADH.
ADH is a nonapeptide hormone synthesized predominantly in the supraoptic nucleus. In response to several different stimuli, including increased plasma oncotic pressure, left atrial distention, exercise, and certain emotional states, ADH is released from axon terminals in the neurohypophysis into the general circulation. The hormone acts on the collecting tubules of the kidney to promote the resorption of free water. ADH deficiency causes diabetes insipidus, a condition characterized by excessive urination (polyuria) caused by an inability of the kidney to properly resorb water from the urine. Diabetes insipidus can result from several causes, including head trauma, neoplasms, and inflammatory disorders of the hypothalamus and pituitary, and from surgical procedures involving the hypothalamus or pituitary. The condition sometimes arises spontaneously (idiopathic) in the absence of an underlying disorder. Diabetes insipidus from ADH deficiency is designated as central, to differentiate it from nephrogenic diabetes insipidus as a result of renal tubular unresponsiveness to circulating ADH. The clinical manifestations of both diseases are similar and include the excretion of large volumes of dilute urine with an inappropriately low specific gravity. Serum sodium and osmolality are increased as a result of excessive renal loss of free water, resulting in thirst and polydipsia. Patients who can drink water generally can compensate for urinary losses; patients who are obtunded, bedridden, or otherwise limited in their ability to obtain water may develop life-threatening dehydration.
In the syndrome of inappropriate ADH (SIADH) secretion, ADH excess is caused by several extracranial and intracranial disorders. This condition leads to resorption of excessive amounts of free water, with resultant hyponatremia. The most common causes of SIADH include the secretion of ectopic ADH by malignant neoplasms (particularly small cell carcinomas of the lung), non-neoplastic diseases of the lung, and local injury to the hypothalamus or neurohypophysis. The clinical manifestations of SIADH are dominated by hyponatremia, cerebral edema, and resultant neurologic dysfunction. Although total body water is increased, blood volume remains normal, and peripheral edema does not develop.
Thyroid
The thyroid gland consists of two bulky lateral lobes connected by a relatively thin isthmus, usually located below and anterior to the larynx. The thyroid gland develops embryologically from an evagination of the developing pharyngeal epithelium that descends from the foramen cecum at the base of the tongue to its normal position in the anterior neck. This pattern of descent explains the occasional presence of ectopic thyroid tissue, most commonly located at the base of the tongue (lingual thyroid) or at other sites abnormally high in the neck.
The thyroid is divided into lobules, each composed of about 20 to 40 evenly dispersed follicles. The follicles range from uniform to variable in size and are lined by cuboidal to low columnar epithelium, which is filled with thyroglobulin, the iodinated precursor protein of active thyroid hormone. In response to trophic factors from the hypothalamus, TSH (also called thyrotropin) is released by thyrotrophs in the anterior pituitary into the circulation. The binding of TSH to its receptor on the thyroid follicular epithelium results in activation and conformational change in the receptor, allowing it to associate with a stimulatory G protein (Fig. 19–5). Activation of the G protein eventually results in an increase in intracellular cAMP levels, which stimulates thyroid hormone synthesis and release mediated by cAMP-dependent protein kinases. Thyroid follicular epithelial cells convert thyroglobulin into thyroxine (T4) and lesser amounts of triiodothyronine (T3). T4 and T3 are released into the systemic circulation, where most of these peptides are reversibly bound to circulating plasma proteins, such as T4-binding globulin, for transport to peripheral tissues. The binding proteins serve to maintain the serum unbound (free) T3 and T4 concentrations within narrow limits while ensuring that the hormones are readily available to the tissues. In the periphery the majority of free T4 is deiodinated to T3; the latter binds to thyroid hormone nuclear receptors in target cells with 10-fold greater affinity than that observed for T4 and has proportionately greater activity. The interaction of thyroid hormone with its nuclear thyroid hormone receptor (TR) results in the formation of a hormone-receptor complex that binds to thyroid hormone response elements (TREs) in target genes, regulating their transcription. Thyroid hormone has diverse cellular effects, including upregulation of carbohydrate and lipid catabolism and stimulation of protein synthesis in a wide range of cells. The net result of these processes is an increase in the basal metabolic rate.
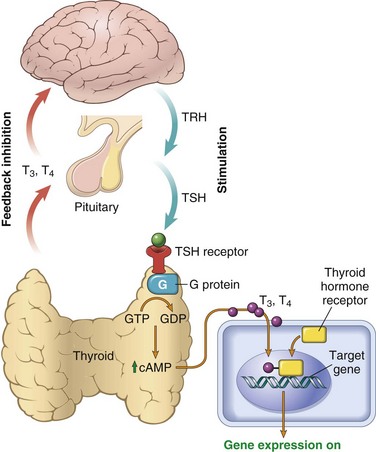
Figure 19–5 Homeostasis in the hypothalamus-pituitary-thyroid axis and mechanism of action of thyroid hormones. Secretion of thyroid hormones (T3 and T4) is controlled by trophic factors secreted by both the hypothalamus and the anterior pituitary. Decreased levels of T3 and T4 stimulate the release of thyrotropin-releasing hormone (TRH) from the hypothalamus and thyroid-stimulating hormone (TSH) from the anterior pituitary, causing T3 and T4 levels to rise. Elevated T3 and T4 levels, in turn, suppress the secretion of both TRH and TSH. This relationship is termed a negative-feedback loop. TSH binds to the TSH receptor on the thyroid follicular epithelium, which causes activation of G proteins, release of cyclic AMP (cAMP), and cAMP-mediated synthesis and release of thyroid hormones (i.e., T3 and T4). In the periphery, T3 and T4 interact with the thyroid hormone receptor (TR) and form a complex that translocates to the nucleus and binds to so-called thyroid response elements (TREs) on target genes, thereby initiating transcription.
Clinical recognition of diseases of the thyroid is important, because most are amenable to medical or surgical management. Such diseases include conditions associated with excessive release of thyroid hormones (hyperthyroidism), those associated with thyroid hormone deficiency (hypothyroidism), and mass lesions of the thyroid. Considered next are the clinical consequences of disturbed thyroid function, followed by an overview of the disorders that generate these problems.
Hyperthyroidism
Thyrotoxicosis is a hypermetabolic state due to elevated circulating levels of free T3 and T4. Because it is caused most commonly by hyperfunction of the thyroid gland, thyrotoxicosis often is referred to as hyperthyroidism. In certain conditions, however, the oversupply either is related to excessive release of pre-formed thyroid hormone (e.g., in thyroiditis) or comes from an extrathyroidal source, rather than a hyperfunctioning gland (Table 19–2). Thus, strictly speaking, hyperthyroidism is only one (albeit the most common) category of thyrotoxicosis. Despite this clear distinction, the following discussion adheres to the common practice of using the terms thyrotoxicosis and hyperthyroidism interchangeably.
Table 19–2 Causes of Thyrotoxicosis
| Associated with Hyperthyroidism |
| Primary |
| Diffuse toxic hyperplasia (Graves disease) |
| Hyperfunctioning (“toxic”) multinodular goiter |
| Hyperfunctioning (“toxic”) adenoma |
| Iodine-induced hyperthyroidism |
| Secondary |
| TSH-secreting pituitary adenoma (rare)* |
| Not Associated with Hyperthyroidism |
| Granulomatous (de Quervain) thyroiditis (painful) |
| Subacute lymphocytic thyroiditis (painless) |
| Struma ovarii (ovarian teratoma with thyroid) |
| Factitious thyrotoxicosis (exogenous thyroxine intake) |
TSH, thyroid-stimulating hormone.
* Associated with increased TSH; all other causes of thyrotoxicosis associated with decreased TSH.
The clinical manifestations of thyrotoxicosis are truly protean and include changes referable to the hypermetabolic state induced by excessive amounts of thyroid hormone as well as those related to overactivity of the sympathetic nervous system:
• Constitutional symptoms: The skin of thyrotoxic persons tends to be soft, warm, and flushed; heat intolerance and excessive sweating are common. Increased sympathetic activity and hypermetabolism result in weight loss despite increased appetite.
• Gastrointestinal: Stimulation of the gut results in hypermotility, malabsorption, and diarrhea.
• Cardiac: Palpitations and tachycardia are common; elderly patients may develop congestive heart failure as a consequence of aggravation of preexisting heart disease.
• Neuromuscular: Patients frequently experience nervousness, tremor, and irritability. Nearly 50% develop proximal muscle weakness (thyroid myopathy).
• Ocular manifestations: a wide, staring gaze and lid lag are present because of sympathetic overstimulation of the levator palpebrae superioris (Fig. 19–6). However, true thyroid ophthalmopathy associated with proptosis is a feature seen only in Graves disease (discussed later).
• Thyroid storm is used to designate the abrupt onset of severe hyperthyroidism. This condition occurs most commonly in patients with underlying Graves disease, probably resulting from an acute elevation in catecholamine levels, as might be encountered during stress. Thyroid storm constitutes a medical emergency: A significant number of untreated patients die of cardiac arrhythmias.
• Apathetic hyperthyroidism refers to thyrotoxicosis occurring in elderly persons, in whom the typical features of thyroid hormone excess seen in younger patients are blunted. In these patients the diagnosis is often made during laboratory workup for unexplained weight loss or worsening cardiovascular disease.
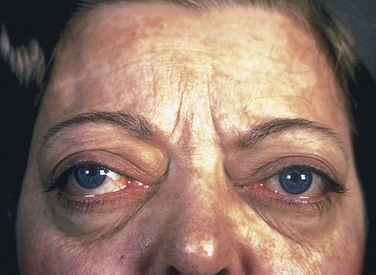
Figure 19–6 Patient with hyperthyroidism. A wide-eyed, staring gaze, caused by overactivity of the sympathetic nervous system, is one of the classic features of this disorder. In Graves disease, one of the most important causes of hyperthyroidism, accumulation of loose connective tissue behind the orbits also adds to the protuberant appearance of the eyes.
The diagnosis of hyperthyroidism is based on clinical features and laboratory data. The measurement of serum TSH is the most useful single screening test for hyperthyroidism, because TSH levels are decreased even at the earliest stages, when the disease may still be subclinical. In rare cases of pituitary- or hypothalamus-associated (secondary) hyperthyroidism, TSH levels are either normal or raised. A low TSH value usually is associated with increased levels of free T4. In the occasional patient, hyperthyroidism results predominantly from increased circulating levels of T3 (T3 toxicosis). In such cases, free T4 levels may be decreased, and direct measurement of serum T3 may be useful. Once the diagnosis of thyrotoxicosis has been confirmed by a combination of TSH and free thyroid hormone assays, measurement of radioactive iodine uptake by the thyroid gland often is valuable in determining the etiology. For example, such scans may show diffusely increased (whole-gland) uptake in Graves disease, increased uptake in a solitary nodule in toxic adenoma, or decreased uptake in thyroiditis.
Hypothyroidism
Hypothyroidism is caused by any structural or functional derangement that interferes with the production of adequate levels of thyroid hormone. As in the case of hyperthyroidism, this disorder is sometimes divided into primary and secondary categories, depending on whether the hypothyroidism arises from an intrinsic abnormality in the thyroid or from hypothalamic or pituitary disease (Table 19–3). Worldwide, the most common cause of hypothyroidism is dietary deficiency of iodine (see further on), while in most developed nations, autoimmune causes predominate. Genetic defects that perturb thyroid development itself (thyroid dysgenesis) or the synthesis of thyroid hormone (dyshormonogenetic goiter) are relatively rare.
Table 19–3 Causes of Hypothyroidism
| Primary |
| Rare developmental abnormalities (thyroid dysgenesis): mutations in PAX8, FOXE1 |
| Congenital biosynthetic defect (dyshormonogenetic goiter)* |
| Postablative |
| Surgery, radioiodine therapy, or external irradiation |
| Autoimmune hypothyroidism |
| Hashimoto thyroiditis* |
| Iodine deficiency* |
| Drugs (lithium, iodides, p-aminosalicylic acid)* |
| Secondary (Central) |
| Pituitary failure |
| Hypothalamic failure (rare) |
FOXE1, forkhead box E1 gene; PAX8, paired box 8 gene.
* Associated with enlargement of thyroid (“goitrous hypothyroidism”). Hashimoto thyroiditis and postablative hypothyroidism account for a majority of cases of hypothyroidism in developed countries.
The clinical manifestations of hypothyroidism include cretinism and myxedema.
• Cretinism refers to hypothyroidism developing in infancy or early childhood. This disorder formerly was fairly common in areas of the world where dietary iodine deficiency is endemic, including mountainous areas such as the Himalayas and the Andes (endemic cretinism). It is now much less frequent because of the widespread supplementation of foods with iodine. By contrast, enzyme defects that interfere with thyroid hormone synthesis are a cause of sporadic cretinism. Clinical features of cretinism include impaired development of the skeletal system and central nervous system, with severe mental retardation, short stature, coarse facial features, a protruding tongue, and umbilical hernia. The severity of the mental impairment in cretinism seems to be directly influenced by the timing of onset of the deficient state in utero. Normally, maternal hormones that are critical to fetal brain development, including T3 and T4, cross the placenta. If maternal thyroid deficiency is present before the development of the fetal thyroid gland, mental retardation is severe. By contrast, reduction in maternal thyroid hormones later in pregnancy, after the fetal thyroid has developed, allows normal brain development.
• Hypothyroidism developing in older children and adults results in a condition known as myxedema. Myxedema, or Gull disease, was first linked with thyroid dysfunction in 1873 by Sir William Gull in a paper addressing the development of a cretinoid state in adults. Manifestations of myxedema include generalized apathy and mental sluggishness that in the early stages of disease may mimic depression. Patients with myxedema are listless, cold intolerant, and often obese. Mucopolysaccharide-rich edematous fluid accumulates in skin, subcutaneous tissue, and a number of visceral sites, with resultant broadening and coarsening of facial features, enlargement of the tongue, and deepening of the voice. Bowel motility is decreased, resulting in constipation. Pericardial effusions are common; in later stages, the heart is enlarged, and heart failure may supervene.
Laboratory evaluation has a vital role in the diagnosis of suspected hypothyroidism. As in the case of hyperthyroidism, measurement of serum TSH is the most sensitive screening test for this disorder. The serum TSH is increased in primary hypothyroidism because of a loss of feedback inhibition of thyrotropin-releasing hormone (TRH) and TSH production by the hypothalamus and pituitary, respectively. The TSH concentration is not increased in persons with hypothyroidism caused by primary hypothalamic or pituitary disease. Serum T4 is decreased in patients with hypothyroidism of any origin.
Thyroiditis
Thyroiditis, or inflammation of the thyroid gland, encompasses a diverse group of disorders characterized by some form of thyroid inflammation. These diseases include conditions that result in acute illness with severe thyroid pain (e.g., infectious thyroiditis, granulomatous [de Quervain] thyroiditis) and disorders in which relatively little inflammation occurs and the illness is manifested primarily by thyroid dysfunction (subacute lymphocytic [“painless”] thyroiditis and fibrous [Reidel] thyroiditis). This section focuses on the more common and clinically significant types of thyroiditis: (1) Hashimoto thyroiditis (or chronic lymphocytic thyroiditis); (2) granulomatous (de Quervain) thyroiditis; and (3) subacute lymphocytic thyroiditis.
Chronic Lymphocytic (Hashimoto) Thyroiditis
Hashimoto thyroiditis is the most common cause of hypothyroidism in areas of the world where iodine levels are sufficient. It is characterized by gradual thyroid failure secondary to autoimmune destruction of the thyroid gland. It is most prevalent between the ages of 45 and 65 years and is more common in women than in men, with female predominance in a ratio of 10:1 to 20:1. Although it is primarily a disease of older women, it can occur in children and is a major cause of nonendemic goiter in children.
Pathogenesis
Hashimoto thyroiditis is caused by a breakdown in self-tolerance (Chapter 4) to thyroid autoantigens. Thus, circulating autoantibodies against thyroid antigens are present in the vast majority of patients, who demonstrate progressive depletion of thyroid epithelial cells (thyrocytes) and their replacement by mononuclear cell infiltration and fibrosis. The inciting events leading to breakdown in self-tolerance have not been fully elucidated, but multiple immunologic mechanisms that may contribute to thyrocyte damage have been identified (Fig. 19–7), including
• CD8+ cytotoxic T cell–mediated cell death: CD8+ cytotoxic T cells may cause thyrocyte destruction.
• Cytokine-mediated cell death: Excessive T cell activation leads to the production of inflammatory cytokines such as interferon-γ in the thyroid gland, with resultant recruitment and activation of macrophages and damage to follicles.
• Binding of antithyroid antibodies (antithyroglobulin, and antithyroid peroxidase antibodies), followed by antibody-dependent cell–mediated cytotoxicity (Chapter 4).
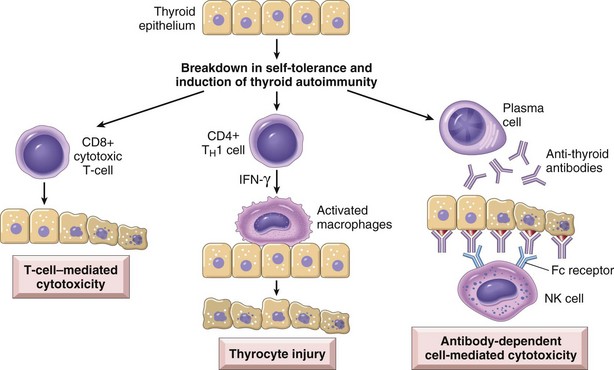
Figure 19–7 Pathogenesis of Hashimoto thyroiditis. Breakdown of immune tolerance to thyroid autoantigens results in progressive autoimmune destruction of thyrocytes by infiltrating cytotoxic T cells, locally released cytokines, or antibody-dependent cytotoxicity.
A significant genetic component to the disease pathogenesis is supported by the concordance of disease in as many as 40% of monozygotic twins, as well as the presence of circulating antithyroid antibodies in approximately 50% of asymptomatic siblings of affected patients. Increased susceptibility to Hashimoto thyroiditis is associated with polymorphisms in multiple immune regulation–associated genes, the most significant of which is the linkage to cytotoxic T lymphocyte–associated antigen-4 gene (CTLA4), which codes for a negative regulator of T cell function (Chapter 4).
Morphology
The thyroid usually is diffusely and symmetrically enlarged, although more localized enlargement may be seen in some cases. The cut surface is pale and gray-tan in appearance, and the tissue is firm and somewhat friable. Microscopic examination reveals widespread infiltration of the parenchyma by a mononuclear inflammatory infiltrate containing small lymphocytes, plasma cells, and well-developed germinal centers (Fig. 19–8). The thyroid follicles are atrophic and are lined in many areas by epithelial cells distinguished by the presence of abundant eosinophilic, granular cytoplasm, termed Hürthle, or oxyphil, cells. This is a metaplastic response of the normally low cuboidal follicular epithelium to ongoing injury; on ultrastructural examination, the Hürthle cells are characterized by numerous prominent mitochondria. Interstitial connective tissue is increased and may be abundant. Less commonly, the thyroid is small and atrophic as a result of more extensive fibrosis (fibrosing variant). Unlike in Reidel thyroiditis, the fibrosis does not extend beyond the capsule of the gland.
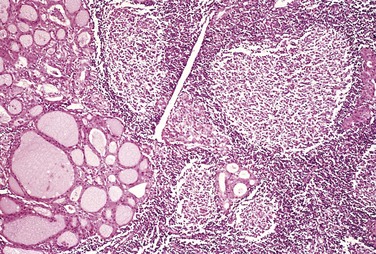
Clinical Features
Hashimoto thyroiditis comes to clinical attention as painless enlargement of the thyroid, usually associated with some degree of hypothyroidism, in a middle-aged woman. The enlargement of the gland usually is symmetric and diffuse, but in some cases it may be sufficiently localized to raise suspicion for neoplasm. In the usual clinical course, hypothyroidism develops gradually. In some cases, however, it may be preceded by transient thyrotoxicosis caused by disruption of thyroid follicles, with secondary release of thyroid hormones (hashitoxicosis). During this phase, free T4 and T3 concentrations are elevated, TSH is diminished, and radioactive iodine uptake is decreased. As hypothyroidism supervenes, T4 and T3 levels progressively fall, accompanied by a compensatory increase in TSH. Patients with Hashimoto thyroiditis often have other autoimmune diseases and are at increased risk for the development of B cell non-Hodgkin lymphomas (Chapter 11), which typically arise within the thyroid gland. The relationship between Hashimoto disease and thyroid epithelial cancers remains controversial, with some morphologic and molecular studies suggesting a predisposition to papillary carcinomas.
Subacute Granulomatous (de Quervain) Thyroiditis
Subacute granulomatous thyroiditis, also known as de Quervain thyroiditis, is much less common than Hashimoto disease. De Quervain thyroiditis is most common between the ages of 30 and 50 and, like other forms of thyroiditis, occurs more frequently in women than in men. Subacute thyroiditis is believed to be caused by a viral infection or an inflammatory process triggered by viral infections. A majority of patients have a history of an upper respiratory infection just before the onset of thyroiditis. By contrast with autoimmune thyroid disease, the immune response is not self-perpetuating, so the process is limited.
Morphology
The gland is firm, with an intact capsule, and may be unilaterally or bilaterally enlarged. Histologic examination reveals disruption of thyroid follicles, with extravasation of colloid leading to a polymorphonuclear infiltrate, which is replaced over time by lymphocytes, plasma cells, and macrophages. The extravasated colloid provokes an exuberant granulomatous reaction with giant cells, some containing fragments of colloid. Healing occurs by resolution of inflammation and fibrosis.
Clinical Features
The onset of this form of thyroiditis often is acute, characterized by pain in the neck (particularly with swallowing), fever, malaise, and variable enlargement of the thyroid. Transient hyperthyroidism may occur, as in other cases of thyroiditis, as a result of disruption of thyroid follicles and release of excessive thyroid hormone. The leukocyte count and erythrocyte sedimentation rates are increased. With progression of disease and gland destruction, a transient hypothyroid phase may ensue. The condition typically is self-limited, with most patients returning to a euthyroid state within 6 to 8 weeks.
Subacute Lymphocytic Thyroiditis
Subacute lymphocytic thyroiditis also is known as silent or painless thyroiditis; in a subset of patients the onset of disease follows pregnancy (postpartum thyroiditis). This disease is most likely to be autoimmune in etiology, because circulating antithyroid antibodies are found in a majority of patients. It mostly affects middle-aged women, who present with a painless neck mass or features of thyroid hormone excess. The initial phase of thyrotoxicosis (which is likely to be secondary to thyroid tissue damage) is followed by return to a euthyroid state within a few months. In a minority of affected persons the condition eventually progresses to hypothyroidism. Except for possible mild symmetric enlargement, the thyroid appears normal on gross inspection. The histologic features consist of lymphocytic infiltration and hyperplastic germinal centers within the thyroid parenchyma.
Other Forms of Thyroiditis
Riedel thyroiditis, a rare disorder of unknown etiology, is characterized by extensive fibrosis involving the thyroid and contiguous neck structures. Clinical evaluation demonstrates a hard and fixed thyroid mass, simulating a thyroid neoplasm. It may be associated with idiopathic fibrosis in other sites in the body, such as the retroperitoneum. The presence of circulating antithyroid antibodies in most patients suggests an autoimmune etiology.
Summary
Thyroiditis
• Chronic lymphocytic (Hashimoto) thyroiditis is the most common cause of hypothyroidism in regions where dietary iodine levels are sufficient.
• Hashimoto thyroiditis is an autoimmune disease characterized by progressive destruction of thyroid parenchyma, Hürthle cell change, and mononuclear (lymphoplasmacytic) infiltrates, with or without extensive fibrosis.
• Multiple autoimmune mechanisms account for Hashimoto disease, including cytotoxicity mediated by CD8+ T cells, cytokines (IFN-γ), and antithyroid antibodies.
• Subacute granulomatous (de Quervain) thyroiditis is a self-limited disease, probably secondary to a viral infection, and is characterized by pain and the presence of a granulomatous inflammation in the thyroid.
• Subacute lymphocytic thyroiditis is a self-limited disease that often occurs after a pregnancy (postpartum thyroiditis), typically is painless, and is characterized by lymphocytic inflammation in the thyroid.
Graves Disease
In 1835 Robert Graves reported on his observations of a disease characterized by “violent and long continued palpitations in females” associated with enlargement of the thyroid gland. Graves disease is the most common cause of endogenous hyperthyroidism. It is characterized by a triad of manifestations:
• Thyrotoxicosis, caused by a diffusely enlarged, hyperfunctional thyroid, is present in all cases.
• An infiltrative ophthalmopathy with resultant exophthalmos is noted in as many as 40% of patients.
• A localized, infiltrative dermopathy (sometimes designated pretibial myxedema) is seen in a minority of cases.
Graves disease has a peak incidence between the ages of 20 and 40, with women being affected up to seven times more commonly than men. This very common disorder is estimated to affect 1.5% to 2.0% of women in the United States. Genetic factors are important in the causation of Graves disease; the incidence is increased in relatives of affected patients, and the concordance rate in monozygotic twins is as high as 60%. As with other autoimmune disorders, a genetic susceptibility to Graves disease is associated with the presence of certain human leukocyte antigen (HLA) haplotypes, specifically HLA-DR3, and polymorphisms in genes encoding the inhibitory T cell receptor CTLA-4 and the tyrosine phosphatase PTPN22.
Pathogenesis
Graves disease is characterized by a breakdown in self-tolerance to thyroid autoantigens, of which the most important is the TSH receptor. The result is the production of multiple autoantibodies, including:
• Thyroid-stimulating immunoglobulin: An IgG antibody that binds to the TSH receptor and mimics the action of TSH, stimulating adenyl cyclase, with resultant increased release of thyroid hormones. Almost all persons with Graves disease have detectable amounts of this autoantibody, which is relatively specific for Graves disease.
• Thyroid growth-stimulating immunoglobulins: Also directed against the TSH receptor, these antibodies have been implicated in the proliferation of thyroid follicular epithelium.
• TSH-binding inhibitor immunoglobulins: These anti-TSH receptor antibodies prevent TSH from binding to its receptor on thyroid epithelial cells and in so doing may actually inhibit thyroid cell function. The coexistence of stimulating and inhibiting immunoglobulins in the serum of the same patient is not unusual—a finding that may explain why some patients with Graves disease spontaneously develop episodes of hypothyroidism.
A T cell–mediated autoimmune phenomenon also is involved in the development of the infiltrative ophthalmopathy characteristic of Graves disease. In Graves ophthalmopathy, the volume of the retroorbital connective tissues and extraocular muscles is increased as a result of several causes, including (1) marked infiltration of the retroorbital space by mononuclear cells, predominantly T cells; (2) inflammatory edema and swelling of extraocular muscles; (3) accumulation of extracellular matrix components, specifically hydrophilic glycosaminoglycans such as hyaluronic acid and chondroitin sulfate; and (4) increased numbers of adipocytes (fatty infiltration). These changes displace the eyeball forward, potentially interfering with the function of the extraocular muscles.
Autoimmune disorders of the thyroid thus span a continuum on which Graves disease, characterized by hyperfunction of the thyroid, lies at one extreme and Hashimoto disease, manifesting as hypothyroidism, occupies the other end. Sometimes hyperthyroidism may supervene on preexisting Hashimoto thyroiditis (hashitoxicosis), while at other times persons with Graves disease may spontaneously develop thyroid hypofunction; occasionally, Hashimoto thyroiditis and Graves disease may coexist within an affected kindred. Not surprisingly, there is also an element of histologic overlap between the autoimmune thyroid disorders (most characteristically, prominent intrathyroidal lymphoid cell infiltrates with germinal center formation). In both disorders, the frequency of other autoimmune diseases, such as systemic lupus erythematosus, pernicious anemia, type 1 diabetes, and Addison disease, is increased.
Morphology
In the typical case of Graves disease, the thyroid gland is enlarged (usually symmetrically) due to diffuse hypertrophy and hyperplasia of thyroid follicular epithelial cells. The gland is usually smooth and soft, and its capsule is intact. On microscopic examination, the follicular epithelial cells in untreated cases are tall, columnar, and more crowded than usual. This crowding often results in the formation of small papillae, which project into the follicular lumen (Fig. 19–9). Such papillae lack fibrovascular cores, in contrast with those of papillary carcinoma. The colloid within the follicular lumen is pale, with scalloped margins. Lymphoid infiltrates, consisting predominantly of T cells, with fewer B cells and mature plasma cells, are present throughout the interstitium; germinal centers are common.
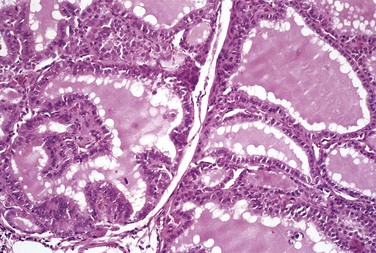
Figure 19–9 Graves disease. The thyroid is diffusely hyperplastic. The follicles are lined by tall columnar epithelial cells that project into the lumina. These cells actively resorb the colloid in the centers of the follicles, resulting in the “scalloped” appearance of the edges of the colloid.
Changes in extrathyroidal tissues include generalized lymphoid hyperplasia. In persons with ophthalmopathy, the tissues of the orbit are edematous because of the presence of hydrophilic glycosaminoglycans. In addition, there is infiltration by lymphocytes, mostly T cells. Orbital muscles initially are edematous but may undergo fibrosis late in the course of the disease. The dermopathy, if present, is characterized by thickening of the dermis, as a result of deposition of glycosaminoglycans and lymphocyte infiltration.
Clinical Features
The clinical manifestations of Graves disease include those common to all forms of thyrotoxicosis (discussed earlier), as well as those associated uniquely with Graves disease: diffuse hyperplasia of the thyroid, ophthalmopathy, and dermopathy. The degree of thyrotoxicosis varies from case to case, and the related changes may sometimes be less conspicuous than other manifestations of the disease. Increased flow of blood through the hyperactive gland often produces an audible bruit. Sympathetic overactivity produces a characteristic wide, staring gaze and lid lag. The ophthalmopathy of Graves disease results in abnormal protrusion of the eyeball (exophthalmos). The extraocular muscles often are weak. The exophthalmos may persist or progress despite successful treatment of the thyrotoxicosis, sometimes resulting in corneal injury. The infiltrative dermopathy, or pretibial myxedema, most commonly involves the skin overlying the shins, where it manifests as scaly thickening and induration of the skin. The skin lesions may be slightly pigmented papules or nodules and often have an orange peel texture. Laboratory findings in Graves disease include elevated serum free T4 and T3 and depressed serum TSH. Because of ongoing stimulation of the thyroid follicles by TSIs, radioactive iodine uptake is increased, and radioiodine scans show a diffuse uptake of iodine.
Summary
Graves Disease
• Graves disease, the most common cause of endogenous hyperthyroidism, is characterized by the triad of thyrotoxicosis, ophthalmopathy, and dermopathy.
• Graves disease is an autoimmune disorder caused by autoantibodies to the TSH receptor that mimic TSH action and activate TSH receptors on thyroid epithelial cells.
• The thyroid in Graves disease is characterized by diffuse hypertrophy and hyperplasia of follicles and lymphoid infiltrates; glycosaminoglycan deposition and lymphoid infiltrates are responsible for the ophthalmopathy and dermopathy.
• Laboratory features include elevations in serum free T3 and T4 and decreased serum TSH.
Diffuse and Multinodular Goiter
Enlargement of the thyroid, or goiter, is the most common manifestation of thyroid disease. Diffuse and multinodular goiters reflect impaired synthesis of thyroid hormone, most often caused by dietary iodine deficiency. Impairment of thyroid hormone synthesis leads to a compensatory rise in the serum TSH, which in turn causes hypertrophy and hyperplasia of thyroid follicular cells and, ultimately, gross enlargement of the thyroid gland. The compensatory increase in functional mass of the gland is enough to overcome the hormone deficiency, ensuring a euthyroid metabolic state in the vast majority of affected persons. If the underlying disorder is sufficiently severe (e.g., a congenital biosynthetic defect), the compensatory responses may be inadequate to overcome the impairment in hormone synthesis, resulting in goitrous hypothyroidism. The degree of thyroid enlargement is proportional to the level and duration of thyroid hormone deficiency.
Goiters can be endemic or sporadic.
• Endemic goiter occurs in geographic areas where the soil, water, and food supply contain little iodine. The designation endemic is used when goiters are present in more than 10% of the population in a given region. Such conditions are particularly common in mountainous areas of the world, including the Himalayas and the Andes. With increasing availability of dietary iodine supplementation, the frequency and severity of endemic goiter have declined significantly.
• Sporadic goiter occurs less commonly than endemic goiter. The condition is more common in females than in males, with a peak incidence in puberty or young adulthood, when there is an increased physiologic demand for T4. Sporadic goiter may be caused by several conditions, including the ingestion of substances that interfere with thyroid hormone synthesis at some level, such as excessive calcium and vegetables belonging to the Brassicaceae (also called Cruciferae) family (e.g., cabbage, cauliflower, Brussels sprouts, turnips). In other instances, goiter may result from hereditary enzymatic defects that interfere with thyroid hormone synthesis (dyshormonogenetic goiter). In most cases, however, the cause of sporadic goiter is not apparent.
Morphology
In most cases, TSH-induced hypertrophy and hyperplasia of thyroid follicular cells result initially in diffuse, symmetric enlargement of the gland (diffuse goiter). The follicles are lined by crowded columnar cells, which may pile up and form projections similar to those seen in Graves disease. If dietary iodine subsequently increases, or if the demands for thyroid hormone decrease, the stimulated follicular epithelium involutes to form an enlarged, colloid-rich gland (colloid goiter). The cut surface of the thyroid in such cases usually is brown, somewhat glassy-appearing, and translucent. On microscopic examination, the follicular epithelium may be hyperplastic in the early stages of disease or flattened and cuboidal during periods of involution. Colloid is abundant during the latter periods. With time, recurrent episodes of hyperplasia and involution combine to produce a more irregular enlargement of the thyroid, termed multinodular goiter. Virtually all long-standing diffuse goiters convert into multinodular goiters. Multinodular goiters typically are hormonally silent, although a minority (approximately 10% over 10 years) can manifest with thyrotoxicosis secondary to the development of autonomous nodules that produce thyroid hormone independent of TSH stimulation. This condition, known as toxic multinodular goiter or Plummer syndrome, is not accompanied by the infiltrative ophthalmopathy and dermopathy of Graves disease–associated thyrotoxicosis.
Multinodular goiters are multilobulate, asymmetrically enlarged glands, which may attain massive size. On cut surface, irregular nodules containing variable amounts of brown, gelatinous colloid are evident (Fig. 19–10, A). Older lesions often show areas of fibrosis, hemorrhage, calcification, and cystic change. The microscopic appearance includes colloid-rich follicles lined by flattened, inactive epithelium and areas of follicular epithelial hypertrophy and hyperplasia, accompanied by the regressive changes just noted (Fig. 19–10, B).
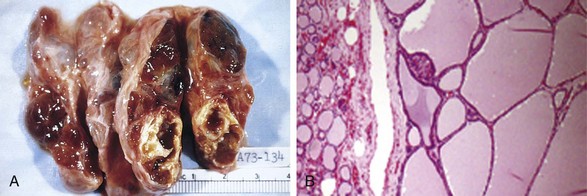
Figure 19–10 Multinodular goiter. A, Gross morphologic appearance. The coarsely nodular gland contains areas of fibrosis and cystic change. B, Photomicrograph of specimen from a hyperplastic nodule, with compressed residual thyroid parenchyma on the periphery. The hyperplastic follicles contain abundant pink “colloid” within their lumina. Note the absence of a prominent capsule, a feature distinguishing such lesions from neoplasms of the thyroid.
(B, Courtesy of Dr. William Westra, Department of Pathology, Johns Hopkins University, Baltimore, Maryland.)
Clinical Features
The dominant clinical features of goiter are those caused by the mass effects of the enlarged gland. In addition to the obvious cosmetic problem of a large neck mass, goiters also may cause airway obstruction, dysphagia, and compression of large vessels in the neck and upper thorax (so-called superior vena cava syndrome). As stated, a hyperfunctioning (toxic) nodule may develop within a long-standing goiter, resulting in hyperthyroidism. The incidence of malignancy in long-standing multinodular goiters is low (less than 5%) but not zero, and concern for malignancy arises with goiters that demonstrate sudden changes in size or associated symptoms (e.g., hoarseness).
Neoplasms of the Thyroid
The thyroid gland gives rise to a variety of neoplasms, ranging from circumscribed, benign adenomas to highly aggressive, anaplastic carcinomas. From a clinical standpoint, the possibility of a tumor is of major concern in patients who present with thyroid nodules. Fortunately, the overwhelming majority of solitary nodules of the thyroid prove to be either follicular adenomas or localized, non-neoplastic conditions (e.g., a dominant nodule in multinodular goiter, simple cysts, or foci of thyroiditis). Carcinomas of the thyroid, by contrast, are uncommon, accounting for much less than 1% of solitary thyroid nodules. Several clinical criteria provide a clue to the nature of a given thyroid nodule:
• Solitary nodules, in general, are more likely to be neoplastic than are multiple nodules.
• Nodules in younger patients are more likely to be neoplastic than are those in older patients.
• Nodules in males are more likely to be neoplastic than are those in females.
• A history of radiation treatment to the head and neck region is associated with an increased incidence of thyroid malignancy.
• Nodules that take up radioactive iodine in imaging studies (hot nodules) are more likely to be benign than malignant, reflecting well-differentiated cells.
Such statistics and general trends, however, are of little significance in the evaluation of a given patient, in whom the timely recognition of a malignancy, however uncommon, can be lifesaving. Ultimately, it is the morphologic evaluation of a given thyroid nodule by fine needle aspiration, combined with histologic study of surgically resected thyroid parenchyma, that provides the most definitive information about its nature. This section presents an overview of the major thyroid neoplasms, including adenomas and carcinomas of various types.
Adenomas
Adenomas of the thyroid are benign neoplasms derived from follicular epithelium. As in the case of all thyroid neoplasms, follicular adenomas usually are solitary. On clinical and morphologic grounds, they may be difficult to distinguish from a dominant nodule in multinodular goiter, for example, or from the less common follicular carcinomas. Although the vast majority of adenomas are nonfunctional, a small proportion produce thyroid hormones (toxic adenomas), causing clinically apparent thyrotoxicosis. In general, follicular adenomas are not forerunners to carcinomas; nevertheless, shared genetic alterations support the possibility that at least a subset of follicular carcinomas arise in preexisting adenomas (see further on).
Pathogenesis
The TSH receptor signaling pathway plays an important role in the pathogenesis of toxic adenomas. Activating (gain-of-function) somatic mutations in one of two components of this signaling system—most often the gene encoding the TSH receptor itself (TSHR) and, less commonly, the α-subunit of Gs (GNAS)—allow follicular cells to secrete thyroid hormone independent of TSH stimulation (thyroid autonomy). The result of this overabundance is symptomatic hyperthyroidism, with a “hot” thyroid nodule seen on imaging studies. Overall, somatic mutations in the TSH receptor signaling pathway seem to be present in slightly over half of toxic adenomas. Not surprisingly, such mutations also are observed in a subset of autonomous nodules that give rise to toxic multinodular goiters, as described earlier. A minority of nonfunctioning follicular adenomas (less than 20%) exhibit mutations of RAS or phosphatidylinositol-3-kinase (PIK3CA), or bear a PAX8/PPARG fusion gene, all of which are genetic alterations shared with follicular carcinomas. These are discussed in further detail under “Carcinomas” (see further on).
Morphology
The typical thyroid adenoma is a solitary, spherical lesion that compresses the adjacent non-neoplastic thyroid. The neoplastic cells are demarcated from the adjacent parenchyma by a well-defined, intact capsule (Fig. 19–11, A). These features are important in making the distinction from multinodular goiters, which contain multiple nodules on their cut surface (even though the patient may present clinically with a solitary dominant nodule), do not demonstrate compression of the adjacent thyroid parenchyma, and lack a well-formed capsule. On microscopic examination, the constituent cells are arranged in uniform follicles that contain colloid (Fig. 19–11, B). Papillary growth patterns, if present, should raise suspicion for an encapsulated papillary carcinoma (discussed later). Occasionally, the neoplastic cells acquire brightly eosinophilic granular cytoplasm (oxyphil or Hürthle cell change) (Fig. 19–12); the clinical presentation and behavior of a Hürthle cell adenoma are no different from those of a conventional adenoma. Similar to endocrine tumors at other anatomic sites, even benign follicular adenomas may, on occasion, exhibit focal nuclear pleomorphism, atypia, and prominent nucleoli (endocrine atypia); by themselves, these features do not constitute evidence of malignancy. The hallmark of all follicular adenomas is the presence of an intact well-formed capsule encircling the tumor. Careful evaluation of the integrity of the capsule is therefore critical in distinguishing follicular adenomas from follicular carcinomas, which demonstrate capsular and/or vascular invasion (see later).
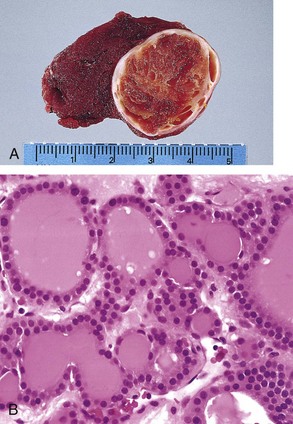
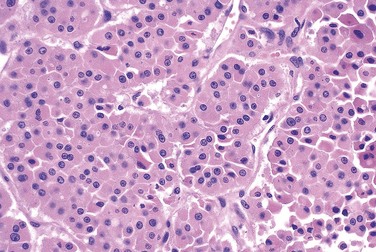
Clinical Features
Most adenomas of the thyroid manifest as painless nodules, often discovered during a routine physical examination. Larger masses may produce local symptoms such as difficulty in swallowing. As previously stated, persons with toxic adenomas can present with features of thyrotoxicosis. After injection of radioactive iodine, most adenomas take up iodine less avidly than normal thyroid parenchyma. On radionuclide scanning, therefore, adenomas appear as cold nodules relative to the adjacent normal thyroid gland. Toxic adenomas, however, will appear as warm or hot nodules in the scan. As many as 10% of cold nodules eventually prove to be malignant. By contrast, malignancy is rare in hot nodules. Essential techniques used in the preoperative evaluation of suspected adenomas are ultrasonography and fine needle aspiration biopsy. Because of the need for evaluating capsular integrity, the definitive diagnosis of thyroid adenoma can be made only after careful histologic examination of the resected specimen. Suspected adenomas of the thyroid are therefore removed surgically to exclude malignancy. Thyroid adenomas carry an excellent prognosis and do not recur or metastasize.
Carcinomas
Carcinomas of the thyroid are relatively uncommon in the United States, accounting for about 1.5% of all cancers. A female predominance has been noted among patients who develop thyroid carcinoma in the early and middle adult years. By contrast, cases manifesting in childhood and late adult life are distributed equally between males and females. Most thyroid carcinomas (except medullary carcinomas) are derived from the thyroid follicular epithelium, and of these, the vast majority are well-differentiated lesions. The major subtypes of thyroid carcinoma and their relative frequencies are
• Papillary carcinoma (accounting for more than 85% of cases)
• Follicular carcinoma (5% to 15% of cases)
• Anaplastic (undifferentiated) carcinoma (less than 5% of cases)
Because of the unique clinical and biologic features associated with each variant of thyroid carcinoma, these subtypes are described separately. Presented next is an overview of the molecular pathogenesis of all thyroid cancers.
Pathogenesis
Both genetic and environmental factors are implicated in the pathogenesis of thyroid cancers.
Genetic Factors
Distinct molecular events are involved in the pathogenesis of the four major variants of thyroid cancer. As stated, medullary carcinomas do not arise from the follicular epithelium. Genetic alterations in the three follicular cell–derived malignancies are clustered along two oncogenic pathways—the mitogen-activated protein (MAP) kinase pathway and the phosphatidylinositol-3-kinase (PI-3K)/AKT pathway (Fig. 19–13). In normal cells, these pathways are transiently activated by binding of soluble growth factor ligands to the extracellular domain of receptor tyrosine kinases, which results in autophosphorylation of the cytoplasmic domain of the receptor, permitting intracellular signal transduction. In thyroid carcinomas, as with many solid cancers (Chapter 5), gain-of-function mutations along components of these pathways lead to constitutive activation even in the absence of ligand, thus promoting carcinogenesis.
• Papillary thyroid carcinomas: Activation of the MAP kinase pathway is a feature of most papillary carcinomas and can occur by one of two major mechanisms. The first mechanism involves rearrangements of RET or NTRK1 (neurotrophic tyrosine kinase receptor 1), both of which encode transmembrane receptor tyrosine kinases, and the second mechanism involves activating point mutations in BRAF, whose product is an intermediate signaling component in the MAP kinase pathway (Fig. 19–13). The RET gene is not normally expressed in thyroid follicular cells. In papillary cancers, chromosomal rearrangements place the tyrosine kinase domain of RET under the transcriptional control of genes that are constitutively expressed in the thyroid epithelium. The novel fusion proteins that are so formed are known as RET/PTC (papillary thyroid carcinoma) and are present in approximately 20% to 40% of papillary thyroid cancers. The frequency of RET/PTC rearrangements is significantly higher in papillary cancers arising in the backdrop of radiation exposure. Similarly, rearrangements of NTRK1 are present in 5% to 10% of papillary thyroid cancers, and the resultant fusion proteins are constitutively expressed in thyroid cells, leading to activation of MAP kinase pathways. One third to one half of papillary thyroid carcinomas harbor a gain-of-function mutation in the BRAF gene, which most commonly is a valine-to-glutamate change on codon 600 (BRAFV600E). Since chromosomal rearrangements of the RET or NTRK1 genes and mutations of BRAF have redundant effects on the thyroid epithelium (both mechanisms result in activation of the MAP kinase signaling pathway), papillary thyroid carcinomas demonstrate either one or the other molecular abnormality, but not both. RET/PTC rearrangements and BRAF point mutations are not observed in follicular adenomas or carcinomas.
• Follicular thyroid carcinomas: Approximately one third to one half of follicular thyroid carcinomas harbor mutations in the PI-3K/AKT signaling pathway, resulting in constitutive activation of this oncogenic pathway. This subset of tumors includes those with gain-of-function point mutations of RAS and PIK3CA, those with amplification of PIK3CA, and those with loss-of-function mutations of PTEN, a tumor suppressor gene and negative regulator of this pathway. The progressive increase in the prevalence of RAS and PIK3CA mutations from benign follicular adenomas to follicular carcinomas to anaplastic carcinomas (see next) suggests a shared histogenesis and molecular evolution among these follicular cell–derived tumors. A unique (2;3)(q13;p25) translocation has been described in one third to one half of follicular carcinomas. This translocation creates a fusion gene composed of portions of PAX8, a paired homeobox gene that is important in thyroid development, and the peroxisome proliferator–activated receptor gene (PPARG), whose gene product is a nuclear hormone receptor implicated in terminal differentiation of cells. Less than 10% of follicular adenomas harbor PAX8/PPARG fusion genes, and thus far these have not been documented in other thyroid neoplasms.
• Anaplastic carcinomas: These highly aggressive and lethal tumors can arise de novo or, more commonly, by dedifferentiation of a well-differentiated papillary or follicular carcinoma. Molecular alterations present in anaplastic carcinomas include those also seen in well-differentiated carcinomas (e.g., RAS or PIK3CA mutations), albeit at a significantly higher rate, suggesting that the presence of these mutations might predispose existing thyroid neoplasms to transform. Other genetic hits, such as inactivation of TP53, are essentially restricted to anaplastic carcinomas and may also relate to their aggressive behavior.
• Medullary thyroid carcinomas: In contrast with the subtypes described earlier, these neoplasms arise from the parafollicular C cells, rather than the follicular epithelium. Familial medullary thyroid carcinomas occur in multiple endocrine neoplasia type 2 (MEN-2) (see later) and are associated with germline RET proto-oncogene mutations that lead to constitutive activation of the receptor. RET mutations are also seen in approximately one half of nonfamilial (sporadic) medullary thyroid cancers. Chromosomal rearrangements involving RET, such as the RET/PTC translocations reported in papillary cancers, are not seen in medullary carcinomas.
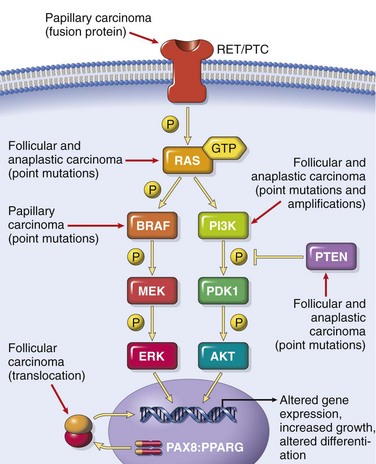
Environmental Factors
The major risk factor predisposing to thyroid cancer is exposure to ionizing radiation, particularly during the first 2 decades of life. In keeping with this finding, there was a marked increase in the incidence of papillary carcinomas among children exposed to ionizing radiation after the Chernobyl nuclear disaster in 1986. Deficiency of dietary iodine (and by extension, an association with goiter) is linked with a higher frequency of follicular carcinomas.
Papillary Carcinoma
As mentioned earlier, papillary carcinomas represent the most common form of thyroid cancer. These tumors may occur at any age, and they account for the vast majority of thyroid carcinomas associated with previous exposure to ionizing radiation.
Morphology
Papillary carcinomas may manifest as solitary or multifocal lesions within the thyroid. In some cases, they may be well circumscribed and even encapsulated; in other instances, they infiltrate the adjacent parenchyma with ill-defined margins. The lesions may contain areas of fibrosis and calcification and often are cystic. On cut surface, they may appear granular and sometimes contain grossly discernible papillary foci (Fig. 19–14, A). The definitive diagnosis of papillary carcinoma can be made only after microscopic examination. In current practice, the diagnosis of papillary carcinoma is based on nuclear features even in the absence of a papillary architecture. The nuclei of papillary carcinoma cells contain very finely dispersed chromatin, which imparts an optically clear appearance, giving rise to the designation ground glass or “Orphan Annie eye” nuclei (Fig. 19–14, C and D). In addition, invaginations of the cytoplasm may give the appearance of intranuclear inclusions (hence the designation pseudoinclusions) in cross-sections. A papillary architecture is common (Fig. 19–14, B); unlike hyperplastic papillary lesions seen in Graves disease, the neoplastic papillae have dense fibrovascular cores. Concentrically calcified structures termed psammoma bodies often are present within the papillae. Foci of lymphatic permeation by tumor cells are often present, but invasion of blood vessels is relatively uncommon, particularly in smaller lesions. Metastases to adjacent cervical lymph nodes are estimated to occur in about half of cases. There are over a dozen variants of papillary thyroid carcinoma, but the most common is one composed predominantly or exclusively of follicles (follicular variant of papillary thyroid carcinoma). The follicular variant more frequently is encapsulated and is associated with a lower incidence of lymph node metastases and extrathyroidal extension than that typical for conventional papillary carcinomas.
Figure 19–14 Papillary carcinoma of the thyroid. A–C, A papillary carcinoma with grossly discernible papillary structures. In this particular example, well-formed papillae (B) are lined by cells with characteristic empty-appearing nuclei, sometimes termed “Orphan Annie eye” nuclei (C). D, Cells obtained by fine-needle aspiration of a papillary carcinoma. Characteristic intranuclear inclusions are visible in some of the aspirated cells (arrows).
(Courtesy of Dr. S. Gokasalan, Department of Pathology, University of Texas Southwestern Medical School, Dallas, Texas.)
Clinical Features
Papillary carcinomas are nonfunctional tumors, so they manifest most often as a painless mass in the neck, either within the thyroid or as metastasis in a cervical lymph node. A preoperative diagnosis usually can be established by fine-needle aspiration on the basis of the characteristic nuclear features described earlier. Papillary carcinomas are indolent lesions, with 10-year survival rates in excess of 95%. Of interest, the presence of isolated cervical nodal metastases does not seem to have a significant influence on the generally good prognosis of these lesions. In a minority of patients, hematogenous metastases are present at the time of diagnosis, most commonly to the lung. The long-term survival of patients with papillary thyroid cancer is dependent on several factors, including age (in general, the prognosis is less favorable among patients older than 40 years), the presence of extrathyroidal extension, and presence of distant metastases (stage).
Follicular Carcinoma
Follicular carcinomas account for 5% to 15% of primary thyroid cancers. They are more common in women (occurring in a ratio of 3:1) and manifest at an older age than that typical for papillary carcinomas, with a peak incidence between the ages of 40 and 60 years. Follicular carcinoma is more frequent in areas with dietary iodine deficiency (accounting for 25% to 40% of thyroid cancers), while its incidence has either decreased or remained stable in iodine-sufficient areas of the world.
Morphology
On microscopic examination, most follicular carcinomas are composed of fairly uniform cells forming small follicles, reminiscent of normal thyroid (Fig. 19–15); in other cases, follicular differentiation may be less apparent. As with follicular adenomas, Hürthle cell variants of follicular carcinomas may be seen. Follicular carcinomas may be widely invasive, infiltrating the thyroid parenchyma and extrathyroidal soft tissues, or minimally invasive. The latter type are sharply demarcated lesions that may be impossible to distinguish from follicular adenomas on gross examination. This distinction requires extensive histologic sampling of the tumor capsule–thyroid interface, to exclude capsular and/or vascular invasion (Fig. 19–16). As mentioned earlier, follicular lesions in which the nuclear features are typical of papillary carcinomas should be regarded as follicular variants of papillary cancers.
Figure 19–15 Follicular carcinoma of the thyroid. A few of the glandular lumina contain recognizable colloid.
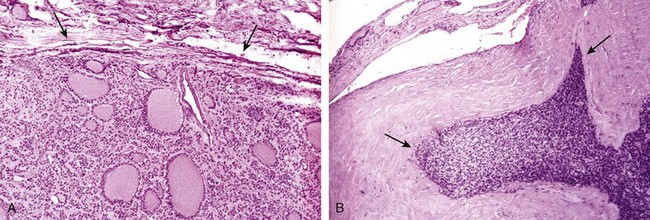
Figure 19–16 Capsular invasion in follicular carcinoma. Evaluating the integrity of the capsule is critical in distinguishing follicular adenomas from follicular carcinomas. A, In adenomas, a fibrous capsule, usually thin but occasionally more prominent, surrounds the neoplastic follicles and no capsular invasion is seen (arrows); compressed normal thyroid parenchyma usually is present external to the capsule (top). B, By contrast, follicular carcinomas demonstrate capsular invasion (arrows) that may be minimal, as in this case, or widespread, with extension into local structures of the neck.
Clinical Features
Follicular carcinomas manifest most frequently as solitary cold thyroid nodules. In rare cases, they may be hyperfunctional. These neoplasms tend to metastasize through the bloodstream (hematogenous dissemination) to the lungs, bone, and liver. In contrast with papillary carcinomas, regional nodal metastases are uncommon. As many as half of patients with widely invasive carcinomas succumb to their disease within 10 years, while less than 10% of patients with minimally invasive follicular carcinomas die within the same time span. Follicular carcinomas are treated with surgical excision. Well-differentiated metastases may take up radioactive iodine, which can be used to identify and also ablate such lesions. Because better-differentiated lesions may be stimulated by TSH, patients usually are placed on a thyroid hormone regimen after surgery to suppress endogenous TSH.
Anaplastic Carcinoma
Anaplastic carcinomas are undifferentiated tumors of the thyroid follicular epithelium, accounting for less than 5% of thyroid tumors. They are aggressive, with a mortality rate approaching 100%. Patients with anaplastic carcinoma are older than those with other types of thyroid cancer, with a mean age of 65 years. Approximately a quarter of patients with anaplastic thyroid carcinomas have a past history of a well-differentiated thyroid carcinoma, and another quarter harbor a concurrent well-differentiated tumor in the resected specimen.
Morphology
Anaplastic carcinomas manifest as bulky masses that typically grow rapidly beyond the thyroid capsule into adjacent neck structures. On microscopic examination, these neoplasms are composed of highly anaplastic cells, which may take on any of several histologic patterns, including those populated by
Foci of papillary or follicular differentiation may be present in some tumors, suggesting origin from a better-differentiated carcinoma.
Medullary Carcinoma
Medullary carcinomas of the thyroid are neuroendocrine neoplasms derived from the parafollicular cells, or C cells, of the thyroid. Like normal C cells, medullary carcinomas secrete calcitonin, the measurement of which plays an important role in the diagnosis and postoperative follow-up evaluation of patients. In some cases, the tumor cells elaborate other polypeptide hormones such as somatostatin, serotonin, and vasoactive intestinal peptide (VIP). Medullary carcinomas arise sporadically in about 70% of cases. The remaining 30% are familial cases occurring in the setting of MEN syndrome 2A or 2B, or familial medullary thyroid carcinoma without an associated MEN syndrome, as discussed later. Of note, both familial and sporadic medullary forms demonstrate activating RET mutations. Sporadic medullary carcinomas, as well as familial cases without an associated MEN syndrome, occur in adults, with a peak incidence in the fifth and sixth decades. Cases associated with MEN-2A or MEN-2B, by contrast, have been reported in younger patients, including children.
Morphology
Medullary carcinomas may arise as a solitary nodule or may manifest as multiple lesions involving both lobes of the thyroid. Multicentricity is particularly common in familial cases. Larger lesions often contain areas of necrosis and hemorrhage and may extend through the capsule of the thyroid. On microscopic examination, medullary carcinomas are composed of polygonal to spindle-shaped cells, which may form nests, trabeculae, and even follicles. Amyloid deposits, derived from altered calcitonin molecules, are present in the adjacent stroma in many cases (Fig. 19–17) and are a distinctive feature. Calcitonin is readily demonstrable both within the cytoplasm of the tumor cells and in the stromal amyloid by immunohistochemical methods. Electron microscopy reveals variable numbers of intracytoplasmic membrane-bound, electron-dense granules (Fig. 19–18). One of the peculiar features of familial medullary carcinomas is the presence of multicentric C cell hyperplasia in the surrounding thyroid parenchyma, a feature usually absent in sporadic lesions. Foci of C cell hyperplasia are believed to represent the precursor lesions from which medullary carcinomas arise.
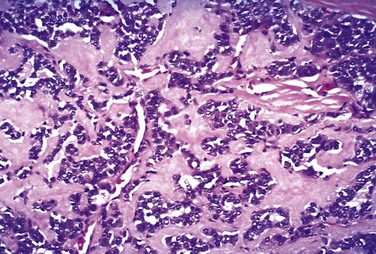
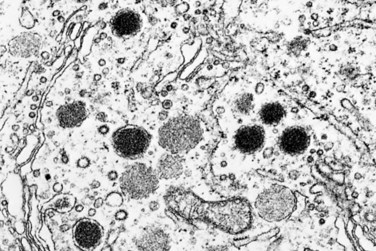
Clinical Features
In the sporadic cases, medullary carcinoma manifests most often as a mass in the neck, sometimes associated with compression effects such as dysphagia or hoarseness. In some instances, the initial manifestations are caused by the secretion of a peptide hormone (e.g., diarrhea caused by the secretion of VIP). Screening of the patient’s relatives for elevated calcitonin levels or RET mutations permits early detection of tumors in familial cases. As discussed at the end of this chapter, all members of MEN-2 kindreds carrying RET mutations are offered prophylactic thyroidectomies to preempt the development of medullary carcinomas; often, the only histologic finding in the resected thyroid of these asymptomatic carriers is the presence of C cell hyperplasia or small (less than 1 cm) micromedullary carcinomas. Recent studies have shown that specific RET mutations correlate with an aggressive behavior in medullary carcinomas.
Summary
Thyroid Neoplasms
• Most thyroid neoplasms manifest as solitary thyroid nodules, but only 1% of all thyroid nodules are neoplastic.
• Follicular adenomas are the most common benign neoplasms, while papillary carcinoma is the most common malignancy.
• Multiple genetic pathways are involved in thyroid carcinogenesis. Some of the genetic abnormalities that are fairly unique to thyroid cancers include PAX8/PPARG fusion (in follicular carcinoma), chromosomal rearrangements involving the RET oncogene (in papillary cancers), and mutations of RET (in medullary carcinomas).
• Follicular adenomas and carcinomas both are composed of well-differentiated follicular epithelial cells; the latter are distinguished by evidence of capsular and/or vascular invasion.
• Papillary carcinomas are recognized based on nuclear features (ground glass nuclei, pseudoinclusions) even in the absence of papillae. These neoplasms typically metastasize by way of lymphatics, but the prognosis is excellent.
• Anaplastic carcinomas are thought to arise by dedifferentiation of more differentiated neoplasms. They are highly aggressive, uniformly lethal cancers.
• Medullary cancers are nonepithelial neoplasms arising from the parafollicular C cells and can occur in either sporadic (70%) or familial (30%) settings. Multicentricity and C cell hyperplasia are features of familial cases. Amyloid deposits are a characteristic histologic finding.
Parathyroid Glands
The parathyroid glands are derived from the developing pharyngeal pouches that also give rise to the thymus. They normally lie in close proximity to the upper and lower poles of each thyroid lobe but may be found anywhere along the pathway of descent of the pharyngeal pouches, including the carotid sheath and the thymus and elsewhere in the anterior mediastinum. Most of the gland is composed of chief cells. On hematoxylin-eosin (H&E) staining, the chief cells range from light to dark pink, depending on their glycogen content. They contain secretory granules of parathyroid hormone (PTH). Oxyphil cells are found throughout the normal parathyroid either singly or in small clusters. They are slightly larger than the chief cells, have acidophilic cytoplasm, and are tightly packed with mitochondria. The activity of the parathyroid glands is controlled by the level of free (ionized) calcium in the bloodstream, rather than by trophic hormones secreted by the hypothalamus and pituitary. Normally, decreased levels of free calcium stimulate the synthesis and secretion of PTH, with the following effects:
• Increase in renal tubular reabsorption of calcium
• Increase in urinary phosphate excretion, thereby lowering serum phosphate levels (since phosphate binds to ionized calcium)
• Increase in the conversion of vitamin D to its active dihydroxy form in the kidneys, which in turn augments gastrointestinal calcium absorption
• Enhancement of osteoclastic activity (i.e., bone resorption, thus releasing ionized calcium), mediated indirectly by promoting the differentiation of osteoclast progenitor cells into mature osteoclasts
The net result of these activities is an increase in the level of free calcium, which inhibits further PTH secretion. Abnormalities of the parathyroids include both hyperfunction and hypofunction. Tumors of the parathyroid glands, unlike thyroid tumors, usually come to attention because of excessive secretion of PTH, rather than mass effects.
Hyperparathyroidism
Hyperparathyroidism occurs in two major forms, primary and secondary, and, less commonly, as tertiary hyperparathyroidism. The first condition represents an autonomous, spontaneous overproduction of PTH, while the latter two conditions typically occur as secondary phenomena in patients with chronic renal insufficiency.
Primary Hyperparathyroidism
Primary hyperparathyroidism is a common endocrine disorder, and an important cause of hypercalcemia. There has been a dramatic increase in the detection of cases in the latter half of the last century, mainly as a result of the routine inclusion of serum calcium assays in testing for a variety of clinical conditions that bring a patient to the hospital. The frequency of occurrence of the various parathyroid lesions underlying the hyperfunction is as follows:
In more than 95% of cases, primary hyperparathyroidism is caused by a sporadic parathyroid adenoma or sporadic hyperplasia. The genetic defects identified in familial primary hyperparathyroidism include multiple endocrine neoplasia syndromes, specifically MEN-1 and MEN-2A (see further on). Familial hypocalciuric hypercalcemia is a rare cause of hyperparathyroidism, caused by inactivating mutations in the calcium-sensing receptor gene on parathyroid cells, leading to constitutive PTH secretion.
PathogEnesis
Although the details of genetic alterations in sporadic parathyroid tumors are beyond the scope of this discussion, abnormalities in two specific genes are commonly associated with these tumors:
• Cyclin D1 gene inversions: Cyclin D1 is a positive regulator of the cell cycle. A chromosomal inversion on chromosome 11 results in relocation of the cyclin D1 gene (normally on 11q), so that it is now positioned adjacent to the 5′-flanking region of the PTH gene (on 11p), leading to abnormal expression of cyclin D1 protein and increased proliferation. Between 10% and 20% of adenomas have this clonal genetic defect. In addition, cyclin D1 is overexpressed in approximately 40% of parathyroid adenomas, suggesting that mechanisms other than cyclin D1 gene inversion can lead to its overexpression.
• MEN1 mutations: Approximately 20% to 30% of parathyroid tumors not associated with the MEN-1 syndrome have mutations in both copies of the MEN1 gene (see later). The spectrum of MEN1 mutations in the sporadic tumors is virtually identical to that in familial parathyroid adenomas.
Morphology
The morphologic changes seen in primary hyperparathyroidism include those in the parathyroid glands as well as those in other organs affected by elevated levels of calcium. In 75% to 80% of cases, one of the parathyroids harbors a solitary adenoma, which, like the normal parathyroids, may lie in close proximity to the thyroid gland or in an ectopic site (e.g., the mediastinum). The typical parathyroid adenoma is a well-circumscribed, soft, tan nodule, invested by a delicate capsule. By definition, parathyroid adenomas are almost invariably confined to single glands (Fig. 19–19), and the remaining glands are normal in size or somewhat shrunken, as a result of feedback inhibition by elevated serum calcium. Most parathyroid adenomas weigh between 0.5 and 5 g. On microscopic examination, parathyroid adenomas are composed predominantly of chief cells (Fig. 19–20). In most cases, at least a few nests of larger oxyphil cells also are present. A rim of compressed, non-neoplastic parathyroid tissue, generally separated by a fibrous capsule, often is visible at the edge of the adenoma. This finding constitutes a helpful internal control, since the chief cells of the adenoma are larger and show greater nuclear size variability than that typical for the normal chief cells. Cells with bizarre and pleomorphic nuclei are often seen within adenomas (so-called endocrine atypia) and must not be taken as a sign of malignancy. Mitotic figures are rare. In contrast with the normal parathyroid parenchyma, adipose tissue is inconspicuous within adenomas.
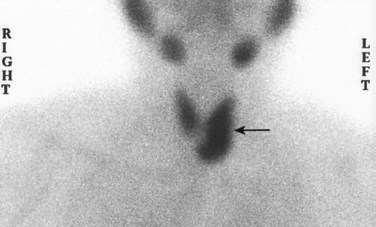
Figure 19–19 Technetium-99 radionuclide scan demonstrates an area of increased uptake corresponding to the left inferior parathyroid gland (arrow). This proved to be a parathyroid adenoma. Preoperative scintigraphy is useful in localizing and distinguishing adenomas from parathyroid hyperplasia, in which more than one gland will demonstrate increased uptake.
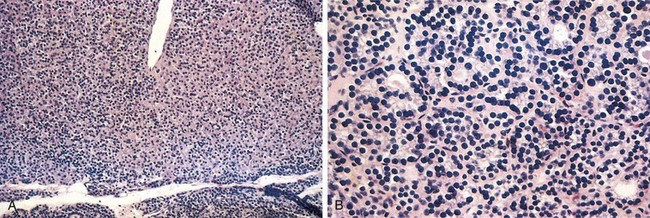
Figure 19–20 Chief cell parathyroid adenoma. A, On this low-power view, a solitary adenoma is clearly delineated from the residual gland below. B, High-power detail shows slight variation in nuclear size and tendency to follicular formation but no anaplasia.
Parathyroid hyperplasia is typically a multiglandular process. In some cases, however, enlargement may be grossly apparent in only one or two glands, complicating the distinction between hyperplasia and adenoma. The combined weight of all glands rarely exceeds 1.0 g and often is less. Microscopically, the most common pattern seen is that of chief cell hyperplasia, which may involve the glands in a diffuse or multinodular pattern. Less commonly, the constituent cells contain abundant clear cytoplasm as a consequence of accumulation of glycogen—a condition designated “water-clear cell hyperplasia.” As in the case of adenomas, stromal fat is inconspicuous within foci of hyperplasia.
Parathyroid carcinomas may be circumscribed lesions that are difficult to distinguish from adenomas, or they may be clearly invasive neoplasms. These tumors enlarge one parathyroid gland and consist of gray-white, irregular masses that sometimes exceed 10 g in weight. The cells usually are uniform and resemble normal parathyroid cells. They are arrayed in nodular or trabecular patterns with a dense, fibrous capsule enclosing the mass. There is general agreement that a diagnosis of carcinoma based on cytologic detail is unreliable, and invasion of surrounding tissues and metastasis are the only definitive criteria. Local recurrence occurs in one third of cases, and more distant dissemination occurs in another third.
Morphologic changes in other organs deserving special mention are found in the skeleton and kidneys. Skeletal changes include increased osteoclastic activity, which results in erosion of bone matrix and mobilization of calcium salts, particularly in the metaphyses of long tubular bones. Bone resorption is accompanied by increased osteoblastic activity and the formation of new bone trabeculae. In more severe cases the cortex is grossly thinned and the marrow contains increased amounts of fibrous tissue accompanied by foci of hemorrhage and cysts (osteitis fibrosa cystica) (Chapter 20). Aggregates of osteoclasts, reactive giant cells, and hemorrhagic debris occasionally form masses that may be mistaken for neoplasms (brown tumors of hyperparathyroidism). PTH-induced hypercalcemia favors the formation of urinary tract stones (nephrolithiasis) as well as calcification of the renal interstitium and tubules (nephrocalcinosis). Metastatic calcification secondary to hypercalcemia also may be seen in other sites, including the stomach, lungs, myocardium, and blood vessels.
Clinical Features
Primary hyperparathyroidism usually is a disease of adults and is much more common in women than in men (gender ratio of nearly 4:1). The most common manifestation of primary hyperparathyroidism is an increase in serum ionized calcium. In fact, primary hyperparathyroidism is the most common cause of clinically silent hypercalcemia. Of note, other conditions also may produce hypercalcemia (Table 19–4). The most common cause of clinically apparent hypercalcemia in adults is paraneoplastic syndromes associated with malignancy and bone metastases (Chapter 5). The prognosis for patients with malignancy-associated hypercalcemia is poor, because it often occurs in those with advanced cancers. In persons with hypercalcemia caused by parathyroid hyperfunction, serum PTH is inappropriately elevated, whereas serum PTH is low to undetectable in those with hypercalcemia caused by nonparathyroid diseases, including malignancy. Other laboratory alterations referable to PTH excess include hypophosphatemia and increased urinary excretion of both calcium and phosphate.
Table 19–4 Causes of Hypercalcemia
| Raised PTH | Decreased PTH |
|---|---|
PTH, parathyroid hormone; PTH-rP, PTH-related protein.
* Primary hyperparathyroidism is the most common cause of hypercalcemia overall. Malignancy is the most common cause of symptomatic hypercalcemia. Primary hyperparathyroidism and malignancy together account for nearly 90% of cases of hypercalcemia.
† Secondary and tertiary hyperparathyroidism are most commonly associated with progressive renal failure.
Primary hyperparathyroidism traditionally has been associated with a constellation of symptoms that included “painful bones, renal stones, abdominal groans, and psychic moans.” Pain, secondary to fractures of bones weakened by osteoporosis or osteitis fibrosa cystica and resulting from renal stones, with obstructive uropathy, was at one time a prominent manifestation of primary hyperparathyroidism. Because serum calcium is now routinely assessed in the workup of most patients who need blood tests for unrelated conditions, clinically silent hyperparathyroidism is detected early. Hence, many of the classic clinical manifestations, particularly those referable to bone and renal disease, are seen much less frequently. Additional signs and symptoms that may be encountered in some cases include
• Gastrointestinal disturbances, including constipation, nausea, peptic ulcers, pancreatitis, and gallstones
• Central nervous system alterations, including depression, lethargy, and seizures
• Neuromuscular abnormalities, including weakness and hypotonia
Although some of these alterations, for example, polyuria and muscle weakness, are clearly related to hypercalcemia, the pathogenesis of many of the other manifestations of the disorder remains poorly understood.
Secondary Hyperparathyroidism
Secondary hyperparathyroidism is caused by any condition associated with a chronic depression in the serum calcium level, because low serum calcium leads to compensatory overactivity of the parathyroids. Renal failure is by far the most common cause of secondary hyperparathyroidism. The mechanisms by which chronic renal failure induces secondary hyperparathyroidism are complex and not fully understood. Chronic renal insufficiency is associated with decreased phosphate excretion, which in turn results in hyperphosphatemia. The elevated serum phosphate levels directly depress serum calcium levels and thereby stimulate parathyroid gland activity. In addition, loss of renal substances reduces the availability of α1-hydroxylase enzyme necessary for the synthesis of the active form of vitamin D, which in turn reduces intestinal absorption of calcium (Chapter 7).
Morphology
The parathyroid glands in secondary hyperparathyroidism are hyperplastic. As in the case of primary hyperplasia, the degree of glandular enlargement is not necessarily symmetric. On microscopic examination, the hyperplastic glands contain an increased number of chief cells, or cells with more abundant, clear cytoplasm (water-clear cells), in a diffuse or multinodular distribution. Fat cells are decreased in number. Bone changes similar to those seen in primary hyperparathyroidism also may be present. Metastatic calcification may be seen in many tissues.
Clinical Features
The clinical manifestations of secondary hyperparathyroidism usually are dominated by those related to chronic renal failure. Bone abnormalities (renal osteodystrophy) and other changes associated with PTH excess are, in general, less severe than those seen in primary hyperparathyroidism. Serum calcium remains near normal because the compensatory increase in PTH levels sustains serum calcium. The metastatic calcification of blood vessels (secondary to hyperphosphatemia) occasionally may result in significant ischemic damage to skin and other organs—a process sometimes referred to as calciphylaxis. In a minority of patients, parathyroid activity may become autonomous and excessive, with resultant hypercalcemia—a process sometimes termed tertiary hyperparathyroidism. Parathyroidectomy may be necessary to control the hyperparathyroidism in such patients.
Summary
Hyperparathyroidism
• Primary hyperparathyroidism is the most common cause of asymptomatic hypercalcemia.
• In a majority of cases, primary hyperparathyroidism is caused by a sporadic parathyroid adenoma and, less commonly, by parathyroid hyperplasia.
• Parathyroid adenomas are solitary, while hyperplasia typically is a multiglandular process.
• Skeletal manifestations of hyperparathyroidism include bone resorption, osteitis fibrosa cystica, and brown tumors. Renal changes include nephrolithiasis (stones) and nephrocalcinosis.
• The clinical manifestations of hyperparathyroidism can be summarized as “painful bones, renal stones, abdominal groans, and psychic moans.”
• Secondary hyperparathyroidism most often is caused by renal failure, and the parathyroid glands are hyperplastic.
• Malignancies are the most important cause of symptomatic hypercalcemia, which results from osteolytic metastases or release of PTH-related protein from nonparathyroid tumors.
Hypoparathyroidism
Hypoparathyroidism is far less common than hyperparathyroidism. The major causes of hypoparathyroidism include the following:
• Surgically induced hypoparathyroidism: The most common cause is inadvertent removal of parathyroids during thyroidectomy or other surgical neck dissections.
• Congenital absence: This occurs in conjunction with thymic aplasia (Di George syndrome) and cardiac defects, secondary to deletions on chromosome 22q11.2 (Chapter 6)
• Autoimmune hypoparathyroidism: This is a hereditary polyglandular deficiency syndrome arising from autoantibodies to multiple endocrine organs (parathyroid, thyroid, adrenals, and pancreas). Chronic fungal infections involving the skin and mucous membranes (mucocutaneous candidiasis) are sometimes encountered in affected persons. This condition is caused by mutations in the autoimmune regulator gene (AIRE) and is discussed more extensively later on, in the context of autoimmune adrenalitis. As one consequence of the failure of self-tolerance, some of these patients make autoantibodies against their own IL-17, accounting for the increased susceptibility to Candida infections (in which the TH17 response plays an important protective role).
The major clinical manifestations of hypoparathyroidism are secondary to hypocalcemia and include increased neuromuscular irritability (tingling, muscle spasms, facial grimacing, and sustained carpopedal spasm or tetany), cardiac arrhythmias, and, on occasion, increased intracranial pressures and seizures. Morphologic changes generally are inconspicuous but may include cataracts, calcification of the cerebral basal ganglia, and dental abnormalities.