Chapter 11 Hematopoietic and Lymphoid Systems
See Targeted Therapy available online at studentconsult.com
The hematopoietic and lymphoid systems are affected by a wide spectrum of diseases. One way to organize these disorders is based on whether they primarily affect red cells, white cells, or the hemostatic system, which includes platelets and clotting factors. The most common red cell disorders are those that lead to anemia, a state of red cell deficiency. White cell disorders, by contrast, are most often associated with excessive proliferation, as a result of malignant transformation. Hemostatic derangements may result in hemorrhagic diatheses (bleeding disorders). Finally, splenomegaly, a feature of numerous diseases, is discussed at the end of the chapter, as are tumors of the thymus.
Although these divisions are useful, in reality the production, function, and destruction of red cells, white cells, and components of the hemostatic system are closely linked, and pathogenic derangements primarily affecting one cell type or component of the system often lead to alterations in others. For example, in certain conditions B cells make autoantibodies against components of the red cell membrane. The opsonized red cells are recognized and destroyed by phagocytes in the spleen, which becomes enlarged. The increased red cell destruction causes anemia, which in turn drives a compensatory hyperplasia of red cell progenitors in the bone marrow.
Other levels of interplay and complexity stem from the anatomically dispersed nature of the hematolymphoid system, and the capacity of both normal and malignant white cells to “traffic” between various compartments. Hence, a patient who is diagnosed with lymphoma by lymph node biopsy also may be found to have neoplastic lymphocytes in their bone marrow and blood. The malignant lymphoid cells in the marrow may suppress hematopoiesis, giving rise to low blood cell counts (cytopenias), and the further seeding of tumor cells to the liver and spleen may lead to organomegaly. Thus, in both benign and malignant hematolymphoid disorders, a single underlying abnormality can result in diverse systemic manifestations. Keeping these complexities in mind, we will use the time-honored classification of hematolymphoid disorders based on predominant involvement of red cells, white cells, and the hemostatic system.
Red Cell Disorders
Disorders of red cells can result in anemia or, less commonly, polycythemia (an increase in red cells also known as erythrocytosis). Anemia is defined as a reduction in the oxygen-transporting capacity of blood, which usually stems from a decrease in the red cell mass to subnormal levels.
Anemia can result from bleeding, increased red cell destruction, or decreased red cell production. These mechanisms serve as one basis for classifying anemias (Table 11–1). In some entities overlap occurs, for example, in thalassemia where reduced red cell production and early destruction give rise to anemia. With the exception of anemias caused by chronic renal failure or chronic inflammation (described later), the decrease in tissue oxygen tension that accompanies anemia triggers increased production of the growth factor erythropoietin from specialized cells in the kidney. This in turn drives a compensatory hyperplasia of erythroid precursors in the bone marrow and, in severe anemias, the appearance of extramedullary hematopoiesis within the secondary hematopoietic organs (the liver, spleen, and lymph nodes). In well-nourished persons who become anemic because of acute bleeding or increased red cell destruction (hemolysis) the compensatory response can increase the production of red cells five- to eight-fold. The rise in marrow output is signaled by the appearance of increased numbers of newly formed red cells (reticulocytes) in the peripheral blood. By contrast, anemias caused by decreased red cell production (aregenerative anemias) are associated with subnormal reticulocyte counts (reticulocytopenia).
Table 11–1 Classification of Anemia According to Underlying Mechanism
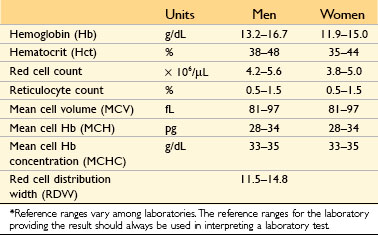
Anemias also can be classified on the basis of red cell morphology, which often points to particular causes. Specific features that provide etiologic clues include the size, color and shape of the red cells. These features are judged subjectively by visual inspection of peripheral smears and also are expressed quantitatively using the following indices:
• Mean cell volume (MCV): the average volume per red cell, expressed in femtoliters (cubic microns)
• Mean cell hemoglobin (MCH): the average mass of hemoglobin per red cell, expressed in picograms
• Mean cell hemoglobin concentration (MCHC): the average concentration of hemoglobin in a given volume of packed red cells, expressed in grams per deciliter
• Red cell distribution width (RDW): the coefficient of variation of red cell volume
Red cell indices are directly measured or automatically calculated by specialized instruments in clinical laboratories. The same instruments also determine the reticulocyte count, a simple measure that distinguishes between hemolytic and aregenerative anemias. Adult reference ranges for these tests are shown in Table 11–2. Depending on the differential diagnosis, a number of other blood tests also may be performed to evaluate anemia, including (1) iron indices (serum iron, serum iron-binding capacity, transferrin saturation, and serum ferritin concentrations), which help distinguish among anemias caused by iron deficiency, chronic disease, and thalassemia; (2) plasma unconjugated bilirubin, haptoglobin, and lactate dehydrogenase levels, which are abnormal in hemolytic anemias; (3) serum and red cell folate and vitamin B12 concentrations, which are low in megaloblastic anemias; (4) hemoglobin electrophoresis, which is used to detect abnormal hemoglobins; and (5) the Coombs test, which is used to detect antibodies or complement on red cells in suspected cases of immunohemolytic anemia. In isolated anemia, tests performed on the peripheral blood usually suffice to establish the cause. By contrast, when anemia occurs along with thrombocytopenia and/or granulocytopenia, it is much more likely to be associated with marrow aplasia or infiltration; in such instances, a marrow examination usually is warranted.
As discussed later, the clinical consequences of anemia are determined by its severity, rapidity of onset, and underlying pathogenic mechanism. If the onset is slow, the deficit in O2-carrying capacity is partially compensated for by adaptations such as increases in plasma volume, cardiac output, respiratory rate, and levels of red cell 2,3-diphosphoglycerate, a glycolytic pathway intermediate that enhances the release of O2 from hemoglobin. These changes mitigate the effects of mild to moderate anemia in otherwise healthy persons but are less effective in those with compromised pulmonary or cardiac function. Pallor, fatigue, and lassitude are common to all forms of anemia. Anemias caused by the premature destruction of red cells (hemolytic anemias) are associated with hyperbilirubinemia, jaundice, and pigment gallstones, all related to increases in the turnover of hemoglobin. Anemias that stem from ineffective hematopoiesis (the premature death of erythroid progenitors in the marrow) are associated with inappropriate increases in iron absorption from the gut, which can lead to iron overload (secondary hemochromatosis) with consequent damage to endocrine organs and the heart. If left untreated, severe congenital anemias such as β-thalassemia major inevitably result in growth retardation, skeletal abnormalities, and cachexia.
 Summary
Summary

Anemia Of Blood Loss: Hemorrhage
With acute blood loss exceeding 20% of blood volume, the immediate threat is hypovolemic shock rather than anemia. If the patient survives, hemodilution begins at once and achieves its full effect within 2 to 3 days; only then is the full extent of the red cell loss revealed. The anemia is normocytic and normochromic. Recovery from blood loss anemia is enhanced by a compensatory rise in the erythropoietin level, which stimulates increased red cell production and reticulocytosis within a period of 5 to 7 days.
With chronic blood loss, iron stores are gradually depleted. Iron is essential for hemoglobin synthesis and erythropoiesis, and its deficiency leads to a chronic anemia of underproduction. Iron deficiency anemia can occur in other clinical settings as well; it is described later along with other anemias caused by decreased red cell production.
Hemolytic Anemias
Normal red cells have a life span of about 120 days. Anemias caused by accelerated red cell destruction are termed hemolytic anemias. Destruction can stem from either intrinsic (intracorpuscular) red cell defects, which are usually inherited, or extrinsic (extracorpuscular) factors, which are usually acquired. Examples of each type of hemolytic anemia are listed in Table 11–1.
Features shared by all uncomplicated hemolytic anemias include (1) a decreased red cell life span, (2) a compensatory increase in erythropoiesis, and (3) the retention of the products of degraded red cells (including iron) by the body. Because the recovered iron is efficiently recycled, red cell regeneration may almost keep pace with the hemolysis. Consequently, hemolytic anemias are associated with erythroid hyperplasia in the marrow and increased numbers of reticulocytes in the peripheral blood. In severe hemolytic anemias, extramedullary hematopoiesis may appear in the liver, spleen, and lymph nodes.
Destruction of red cells can occur within the vascular compartment (intravascular hemolysis) or within tissue macrophages (extravascular hemolysis). Intravascular hemolysis can result from mechanical forces (e.g., turbulence created by a defective heart valve) or biochemical or physical agents that damage the red cell membrane (e.g., fixation of complement, exposure to clostridial toxins, or heat). Regardless of cause, intravascular hemolysis leads to hemoglobinemia, hemoglobinuria, and hemosiderinuria. The conversion of heme to bilirubin can result in unconjugated hyperbilirubinemia and jaundice. Massive intravascular hemolysis sometimes leads to acute tubular necrosis (Chapter 13). Haptoglobin, a circulating protein that binds and clears free hemoglobin, is completely depleted from the plasma, which also usually contains high levels of lactate dehydrogenase (LDH) as a consequence of its release from hemolyzed red cells.
Extravascular hemolysis, the more common mode of red cell destruction, primarily takes place within the spleen and liver. These organs contain large numbers of macrophages, the principal cells responsible for the removal of damaged or immunologically targeted red cells from the circulation. Because extreme alterations of shape are necessary for red cells to navigate the splenic sinusoids, any reduction in red cell deformability makes this passage difficult and leads to splenic sequestration and phagocytosis. As described later in the chapter, diminished deformability is a major cause of red cell destruction in several hemolytic anemias. Extravascular hemolysis is not associated with hemoglobinemia and hemoglobinuria, but often produces jaundice and, if long-standing, leads to the formation of bilirubin-rich gallstones (pigment stones). Haptoglobin is decreased, as some hemoglobin invariably escapes from macrophages into the plasma, and LDH levels also are elevated. In most forms of chronic extravascular hemolysis there is a reactive hyperplasia of mononuclear phagocytes that results in splenomegaly.
We now turn to some of the common hemolytic anemias.
Hereditary Spherocytosis
This disorder stems from inherited (intrinsic) defects in the red cell membrane that lead to the formation of spherocytes, nondeformable cells that are highly vulnerable to sequestration and destruction in the spleen. Hereditary spherocytosis is usually transmitted as an autosomal dominant trait; a more severe, autosomal recessive form of the disease affects a small minority of patients.
 Pathogenesis
Pathogenesis
Hereditary spherocytosis is caused by abnormalities in the membrane skeleton, a network of proteins that underlies lipid bilayer of the red cell (Fig. 11–1). The major membrane skeleton protein is spectrin, a long, flexible heterodimer that self-associates at one end and binds short actin filaments at its other end. These contacts create a two-dimensional meshwork that is linked to the overlying membrane through ankyrin and band 4.2 to the intrinsic membrane protein called band 3, and through band 4.1 to glycophorin.
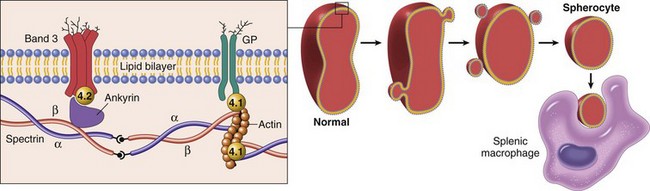
Figure 11–1 Pathogenesis of hereditary spherocytosis. Left panel, Normal organization of the major red cell membrane skeleton proteins. Mutations in α-spectrin, β-spectrin, ankyrin, band 4.2, and band 3 that weaken the association of the membrane skeleton with the overlying plasma membrane cause red cells to shed membrane vesicles and transform into spherocytes (right panel). The nondeformable spherocytes are trapped in the splenic cords and phagocytosed by macrophages. GP, glycophorin.
The mutations in hereditary spherocytosis most frequently involve ankyrin, band 3, and spectrin, but mutations in other components of the skeleton have also been described. A shared feature of the pathogenic mutations is that they weaken the vertical interactions between the membrane skeleton and the intrinsic membrane proteins. This defect somehow destabilizes the lipid bilayer of the red cells, which shed membrane vesicles into the circulation as they age. Little cytoplasm is lost in the process and as a result the surface area to volume ratio decreases progressively over time until the cells become spherical (Fig. 11–1).
The spleen plays a major role in the destruction of spherocytes. Red cells must undergo extreme degrees of deformation to pass through the splenic cords. The floppy discoid shape of normal red cells allows considerable latitude for shape changes. By contrast, spherocytes have limited deformability and are sequestered in the splenic cords, where they are destroyed by the plentiful resident macrophages. The critical role of the spleen is illustrated by the beneficial effect of splenectomy; although the red cell defect and spherocytes persist, the anemia is corrected.
 Morphology
Morphology
On smears, spherocytes are dark red and lack central pallor (Fig. 11–2). The excessive red cell destruction and resultant anemia lead to a compensatory hyperplasia of red cell progenitors in the marrow and an increase in red cell production marked by reticulocytosis. Splenomegaly is more common and prominent in hereditary spherocytosis than in any other form of hemolytic anemia. The splenic weight usually is between 500 and 1000 g. The enlargement results from marked congestion of the splenic cords and increased numbers of tissue macrophages. Phagocytosed red cells are seen within macrophages lining the sinusoids and, in particular, within the cords. In long-standing cases there is prominent systemic hemosiderosis. The other general features of hemolytic anemias also are present, including cholelithiasis, which occurs in 40% to 50% of patients with hereditary spherocytosis.
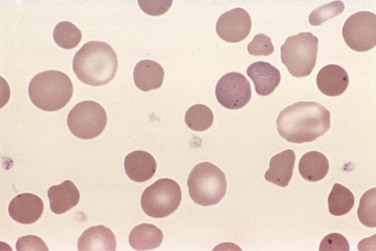
Figure 11–2 Hereditary spherocytosis—peripheral blood smear. Note the anisocytosis and several hyperchromic spherocytes. Howell-Jolly bodies (small nuclear remnants) are also present in the red cells of this asplenic patient.
(Courtesy of Dr. Robert W. McKenna, Department of Pathology, University of Texas Southwestern Medical School, Dallas, Texas.)
Clinical Features
The characteristic clinical features are anemia, splenomegaly, and jaundice. The anemia is highly variable in severity, ranging from subclinical to profound; most commonly it is of moderate degree. Because of their spherical shape, red cells in hereditary spherocytosis have increased osmotic fragility when placed in hypotonic salt solutions, a characteristic that can help establish the diagnosis.
The clinical course often is stable but may be punctuated by aplastic crises. The most severe crises are triggered by parvovirus B19, which infects and destroys erythroblasts in the bone marrow. Because red cells in hereditary spherocytosis have a shortened life span, a lack of red cell production for even a few days results in a rapid worsening of the anemia. Such episodes are self-limited, but some patients need supportive blood transfusions during the period of red cell aplasia.
There is no specific treatment for hereditary spherocytosis. Splenectomy provides relief for symptomatic patients by removing the major site of red cell destruction. The benefits of splenectomy must be weighed against the risk of increased susceptibility to infections, particularly in children. Partial splenectomy is gaining favor, because this approach may produce hematologic improvement while maintaining protection against sepsis.
Sickle Cell Anemia
The hemoglobinopathies are a group of hereditary disorders caused by inherited mutations that lead to structural abnormalities in hemoglobin. Sickle cell anemia, the prototypical (and most prevalent) hemoglobinopathy, stems from a mutation in the β-globin gene that creates sickle hemoglobin (HbS). Other hemoglobinopathies are infrequent and beyond the scope of this discussion.
Normal hemoglobins are tetramers composed of two pairs of similar chains. On average, the normal adult red cell contains 96% HbA (α2β2), 3% HbA2 (α2δ2), and 1% fetal Hb (HbF, α2γ2). HbS is produced by the substitution of valine for glutamic acid at the sixth amino acid residue of β-globin. In homozygotes, all HbA is replaced by HbS, whereas in heterozygotes, only about half is replaced.
Incidence
Sickle cell anemia is the most common familial hemolytic anemia in the world. In parts of Africa where malaria is endemic, the gene frequency approaches 30% as a result of a small but significant protective effect of HbS against Plasmodium falciparum malaria. In the United States, approximately 8% of blacks are heterozygous for HbS, and about 1 in 600 have sickle cell anemia.
Pathogenesis
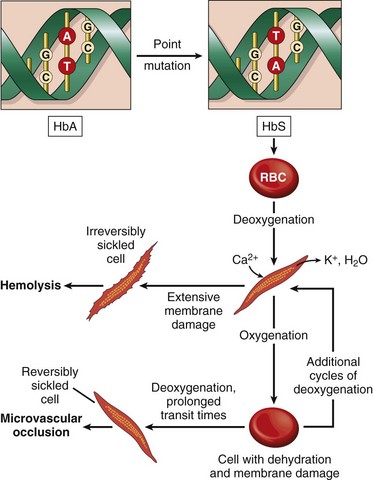
On deoxygenation, HbS molecules form long polymers by means of intermolecular contacts that involve the abnormal valine residue at position 6. These polymers distort the red cell, which assumes an elongated crescentic, or sickle, shape (Fig. 11–3). The sickling of red cells initially is reversible upon reoxygenation. However, the distortion of the membrane that is produced by each sickling episode leads to an influx of calcium, which causes the loss of potassium and water and also damages the membrane skeleton. Over time, this cumulative damage creates irreversibly sickled cells, which are rapidly hemolyzed.
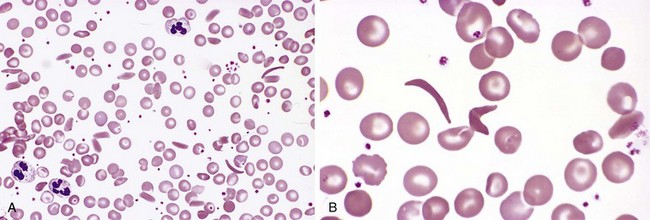
Figure 11–3 Sickle cell anemia—peripheral blood smear. A, Low magnification shows sickle cells, anisocytosis, poikilocytosis, and target cells. B, Higher magnification shows an irreversibly sickled cell in the center.
(Courtesy of Dr. Robert W. McKenna, Department of Pathology, University of Texas Southwestern Medical School, Dallas, Texas.)
Many variables influence the sickling of red cells in vivo. The three most important factors are
• The presence of hemoglobins other than HbS. In heterozygotes approximately 40% of Hb is HbS and the remainder is HbA, which interacts only weakly with deoxygenated HbS. Because the presence of HbA greatly retards the polymerization of HbS, the red cells of heterozygotes have little tendency to sickle in vivo. Such persons are said to have sickle cell trait. HbC, another mutant β-globin, has a lysine residue instead of the normal glutamic acid residue at position 6. About 2.3% of American blacks are heterozygous carriers of HbC; as a result, about 1 in 1250 newborns are compound heterozygotes for HbC and HbS. Because HbC has a greater tendency to aggregate with HbS than does HbA, HbS/HbC compound heterozygotes have a symptomatic sickling disorder called HbSC disease. HbF interacts weakly with HbS, so newborns with sickle cell anemia do not manifest the disease until HbF falls to adult levels, generally around the age of 5 to 6 months.
• The intracellular concentration of HbS. The polymerization of deoxygenated HbS is strongly concentration-dependent. Thus, red cell dehydration, which increases the Hb concentration, facilitates sickling. Conversely, the coexistence of α-thalassemia (described later), which decreases the Hb concentration, reduces sickling. The relatively low concentration of HbS also contributes to the absence of sickling in heterozygotes with sickle cell trait.
• The transit time for red cells through the microvasculature. The normal transit times of red cells through capillaries are too short for significant polymerization of deoxygenated HbS to occur. Hence, sickling in microvascular beds is confined to areas of the body in which blood flow is sluggish. This is the normal situation in the spleen and the bone marrow, two tissues prominently affected by sickle cell disease. Sickling also can be triggered in other microvascular beds by acquired factors that retard the passage of red cells. As described previously, inflammation slows the flow of blood by increasing the adhesion of leukocytes and red cells to endothelium and by inducing the exudation of fluid through leaky vessels. In addition, sickle red cells have a greater tendency than normal red cells to adhere to endothelial cells, apparently because repeated bouts of sickling causes membrane damage that make them sticky. These factors conspire to prolong the transit times of sickle red cells, increasing the probability of clinically significant sickling.
Two major consequences arise from the sickling of red cells (Fig. 11–4). First, the red cell membrane damage and dehydration caused by repeated episodes of sickling produce a chronic hemolytic anemia. The mean life span of red cells in sickle cell anemia is only 20 days (one sixth of normal). Second, red cell sickling produces widespread microvascular obstructions, which result in ischemic tissue damage and pain crises. Vaso-occlusion does not correlate with the number of irreversibly sickled cells and therefore appears to result from factors such as infection, inflammation, dehydration, and acidosis that enhance the sickling of reversibly sickled cells.
Morphology
The anatomic alterations in sickle cell anemia stem from (1) the severe chronic hemolytic anemia, (2) the increased breakdown of heme to bilirubin, and (3) microvascular obstructions, which provoke tissue ischemia and infarction. In peripheral smears, elongated, spindled, or boat-shaped irreversibly sickled red cells are evident (Fig. 11–3). Both the anemia and the vascular stasis lead to hypoxia-induced fatty changes in the heart, liver, and renal tubules. There is a compensatory hyperplasia of erythroid progenitors in the marrow. The cellular proliferation in the marrow often causes bone resorption and secondary new bone formation, resulting in prominent cheekbones and changes in the skull resembling a “crewcut” in radiographs. Extramedullary hematopoiesis may appear in the liver and spleen.
In children there is moderate splenomegaly (splenic weight up to 500 g) due to red pulp congestion caused by entrapment of sickled red cells. However, the chronic splenic erythrostasis produces hypoxic damage and infarcts, which over time reduce the spleen to a useless nubbin of fibrous tissue. This process, referred to as autosplenectomy, is complete by adulthood.
Vascular congestion, thrombosis, and infarction can affect any organ, including the bones, liver, kidney, retina, brain, lung, and skin. The bone marrow is particularly prone to ischemia because of its sluggish blood flow and high rate of metabolism. Priapism, another frequent problem, can lead to penile fibrosis and erectile dysfunction. As with the other hemolytic anemias, hemosiderosis and gallstones are common.
Clinical Course
Homozygous sickle cell disease usually is asymptomatic until 6 months of age when the shift from HbF to HbS is complete. The anemia is moderate to severe; most patients have hematocrits 18% to 30% (normal range, 36% to 48%). The chronic hemolysis is associated with hyperbilirubinemia and compensatory reticulocytosis. From its onset, the disease runs an unremitting course punctuated by sudden crises. The most serious of these are the vaso-occlusive, or pain, crises. The vaso-occlusion in these episodes can involve many sites but occurs most commonly in the bone marrow, where it often progresses to infarction.
A feared complication is the acute chest syndrome, which can be triggered by pulmonary infections or fat emboli from infarcted marrow. The blood flow in the inflamed, ischemic lung becomes sluggish and “spleenlike,” leading to sickling within hypoxemic pulmonary beds. This exacerbates the underlying pulmonary dysfunction, creating a vicious circle of worsening pulmonary and systemic hypoxemia, sickling, and vaso-occlusion. Another major complication is stroke, which sometimes occurs in the setting of the acute chest syndrome. Although virtually any organ can be damaged by ischemic injury, the acute chest syndrome and stroke are the two leading causes of ischemia-related death.
A second acute event, aplastic crisis, is caused by a sudden decrease in red cell production. As in hereditary spherocytosis, this usually is triggered by the infection of erythroblasts by parvovirus B19 and, while severe, is self-limited.
In addition to these crises, patients with sickle cell disease are prone to infections. Both children and adults with sickle cell disease are functionally asplenic, making them susceptible to infections caused by encapsulated bacteria, such as pneumococci. In adults the basis for “hyposplenism” is autoinfarction. In the earlier childhood phase of splenic enlargement, congestion caused by trapped sickled red cells apparently interferes with bacterial sequestration and killing; hence, even children with enlarged spleens are at risk for development of fatal septicemia. Patients with sickle cell disease also are predisposed to Salmonella osteomyelitis, possibly in part because of poorly understood acquired defects in complement function.
In homozygous sickle cell disease, irreversibly sickled red cells are seen in routine peripheral blood smears. In sickle cell trait, sickling can be induced in vitro by exposing cells to marked hypoxia. The diagnosis is confirmed by electrophoretic demonstration of HbS. Prenatal diagnosis of sickle cell anemia can be performed by analyzing fetal DNA obtained by amniocentesis or biopsy of chorionic villi.
The clinical course is highly variable. As a result of improvements in supportive care, an increasing number of patients are surviving into adulthood and producing offspring. Of particular importance is prophylactic treatment with penicillin to prevent pneumococcal infections. Approximately 50% of patients survive beyond the fifth decade. By contrast, sickle cell trait causes symptoms rarely and only under extreme conditions, such as after vigorous exertion at high altitudes.
A mainstay of therapy is hydroxyurea, a “gentle” inhibitor of DNA synthesis. Hydroxyurea reduces pain crises and lessens the anemia through several beneficial intracorpuscular and extracorpuscular effects, including (1) an increase in red cell levels of HbF; (2) an anti-inflammatory effect due to the inhibition of white cell production; (3) an increase in red cell size, which lowers the mean cell hemoglobin concentration; and (4) its metabolism to NO, a potent vasodilator and inhibitor of platelet aggregation. Encouraging results also have been obtained with allogeneic bone marrow transplantation, which has the potential to be curative.
Thalassemia
The thalassemias are inherited disorders caused by mutations that decrease the synthesis of α- or β-globin chains. As a result, there is a deficiency of Hb and additional red cell changes due to the relative excess of the unaffected globin chain. The mutations that cause thalassemia are particularly common among populations in Mediterranean, African, and Asian regions in which malaria is endemic. As with HbS, it is hypothesized that globin mutations associated with thalassemia are protective against falciparum malaria.
Pathogenesis
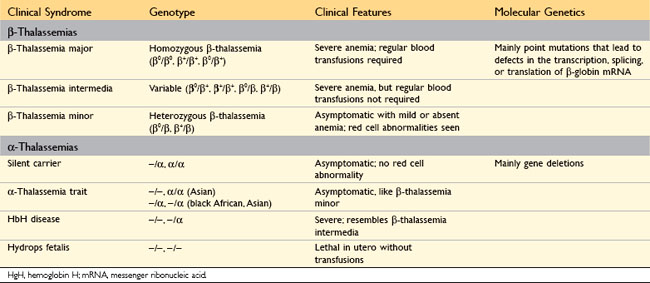
A diverse collection of α-globin and β-globin mutations underlies the thalassemias, which are autosomal codominant conditions. As described previously, adult hemoglobin, or HbA, is a tetramer composed of two α chains and two β chains. The α chains are encoded by two α-globin genes, which lie in tandem on chromosome 11, while the β chains are encoded by a single β-globin gene located on chromosome 16. The clinical features vary widely depending on the specific combination of mutated alleles that are inherited by the patient (Table 11–3), as described next.
β-Thalassemia
The mutations associated with β-thalassemia fall into two categories: (1) β0, in which no β-globin chains are produced; and (2) β+, in which there is reduced (but detectable) β-globin synthesis. Sequencing of β-thalassemia genes has revealed more than 100 different causative mutations, a majority consisting of single-base changes. Persons inheriting one abnormal allele have β-thalassemia minor (also known as β-thalassemia trait), which is asymptomatic or mildly symptomatic. Most people inheriting any two β0 and β+ alleles have β-thalassemia major; occasionally, persons inheriting two β+ alleles have a milder disease termed β-thalassemia intermedia. In contrast with α-thalassemias (described later), gene deletions rarely underlie β-thalassemias (Table 11–3).
The mutations responsible for β-thalassemia disrupt β-globin synthesis in several different ways (Fig. 11–5):
• Mutations leading to aberrant RNA splicing are the most common cause of β-thalassemia. Some of these mutations disrupt the normal RNA splice junctions; as a result, no mature mRNA is made and there is a complete failure of β-globin production, creating β0. Other mutations create new splice junctions in abnormal positions—within an intron, for example. Because the normal splice sites are intact, both normal and abnormal splicing occurs, and some normal β-globin mRNA is made. These alleles are designated β+.
• Some mutations lie within the β-globin promoter and lower the rate of β-globin gene transcription. Because some normal β-globin is synthesized, these are β+ alleles.
• Other mutations involve the coding regions of the β-globin gene, usually with severe consequences. For example, some single-nucleotide changes create termination (“stop”) codons that interrupt the translation of β-globin mRNA and completely prevent the synthesis of β-globin.
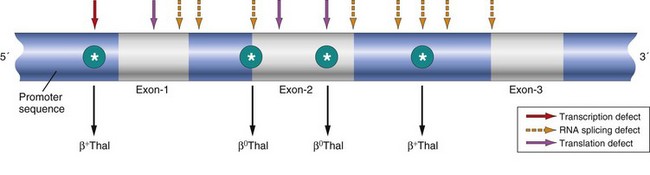
Figure 11–5 Distribution of β-globin gene mutations associated with β-thalassemia. Arrows denote sites at which point mutations giving rise to β+ or β0 thalassemia have been identified.
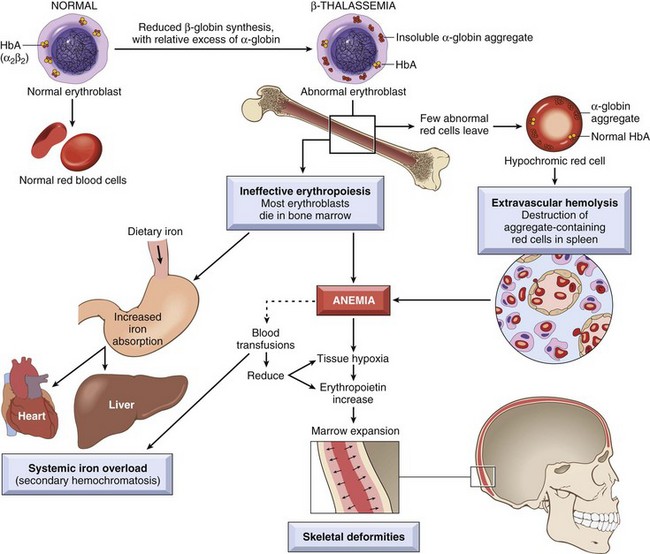
Two mechanisms contribute to the anemia in β-thalassemia. The reduced synthesis of β-globin leads to inadequate HbA formation and results in the production of poorly hemoglobinized red cells that are pale (hypochromic) and small in size (microcytic). Even more important is the imbalance in β-globin and α-globin chain synthesis, as this creates an excess of unpaired α chains that aggregate into insoluble precipitates, which bind and severely damage the membranes of both red cells and erythroid precursors. A high fraction of the damaged erythroid precursors die by apoptosis (Fig. 11–6), a phenomenon termed ineffective erythropoiesis, and the few red cells that are produced have a shortened life span due to extravascular hemolysis. Ineffective hematopoiesis has another untoward effect: It is associated with an inappropriate increase in the absorption of dietary iron, which without medical intervention inevitably leads to iron overload. The increased iron absorption is caused by inappropriately low levels of hepcidin, which is a negative regulator of iron absorption (see later).
α-Thalassemia
Unlike β-thalassemia, α-thalassemia is caused mainly by deletions involving one or more of the α-globin genes. The severity of the disease is proportional to the number of α-globin genes that are missing (Table 11–3). For example, the loss of a single α-globin gene produces a silent-carrier state, whereas the deletion of all four α-globin genes is lethal in utero because the red cells have virtually no oxygen-delivering capacity. With loss of three α-globin genes there is a relative excess of β-globin or (early in life) γ-globin chains. Excess β-globin and γ-globin chains form relatively stable β4 and γ4 tetramers known as HbH and Hb Bart, respectively, which cause less membrane damage than the free α-globin chains that are found in β-thalassemia; as a result, ineffective erythropoiesis is less pronounced in α-thalassemia. Unfortunately, both HbH and Hb Bart have an abnormally high affinity for oxygen, which renders them ineffective at delivering oxygen to the tissues.
Morphology
A range of pathologic features are seen, depending on the specific underlying molecular lesion. On one end of the spectrum is β-thalassemia minor and α-thalassemia trait, in which the abnormalities are confined to the peripheral blood. In smears the red cells are small (microcytic) and pale (hypochromic), but regular in shape. Often seen are target cells, cells with an increased surface area-to-volume ratio that allows the cytoplasm to collect in a central, dark-red “puddle.” On the other end of the spectrum, in β-thalassemia major, peripheral blood smears show marked microcytosis, hypochromia, poikilocytosis (variation in cell size), and anisocytosis (variation in cell shape). Nucleated red cells (normoblasts) are also seen that reflect the underlying erythropoietic drive. β-Thalassemia intermedia and HbH disease are associated with peripheral smear findings that lie between these two extremes.
The anatomic changes in β-thalassemia major are similar in kind to those seen in other hemolytic anemias but profound in degree. The ineffective erythropoiesis and hemolysis result in a striking hyperplasia of erythroid progenitors, with a shift toward early forms. The expanded erythropoietic marrow may completely fill the intramedullary space of the skeleton, invade the bony cortex, impair bone growth, and produce skeletal deformities. Extramedullary hematopoiesis and hyperplasia of mononuclear phagocytes result in prominent splenomegaly, hepatomegaly, and lymphadenopathy. The ineffective erythropoietic precursors consume nutrients and produce growth retardation and a degree of cachexia reminiscent of that seen in cancer patients. Unless steps are taken to prevent iron overload, over the span of years severe hemosiderosis develops (Fig. 11–6). HbH disease and β-thalassemia intermedia are also associated with splenomegaly, erythroid hyperplasia, and growth retardation related to anemia, but these are less severe than in β-thalassemia major.
Clinical Course
β-Thalassemia minor and α-thalassemia trait (caused by deletion of two α-globin genes) are often asymptomatic. There is usually only a mild microcytic hypochromic anemia; generally, these patients have a normal life expectancy. Iron deficiency anemia is associated with a similar red cell appearance and must be excluded by appropriate laboratory tests (described later).
β-Thalassemia major manifests postnatally as HbF synthesis diminishes. Affected children suffer from growth retardation that commences in infancy. They are sustained by repeated blood transfusions, which improve the anemia and reduce the skeletal deformities associated with excessive erythropoiesis. With transfusions alone, survival into the second or third decade is possible, but systemic iron overload gradually develops owing to inappropriate uptake of iron from the gut and the iron load in transfused red cells. Unless patients are treated aggressively with iron chelators, cardiac dysfunction from secondary hemochromatosis inevitably develops and often is fatal in the second or third decade of life. When feasible, bone marrow transplantation at an early age is the treatment of choice. HbH disease (caused by deletion of three α-globin genes) and β-thalassemia intermedia are not as severe as β-thalassemia major, since the imbalance in α- and β-globin chain synthesis is not as great and hematopoiesis is more effective. Anemia is of moderate severity and patients usually do not require transfusions. Thus, the iron overload that is so common in β-thalassemia major is rarely seen.
The diagnosis of β-thalassemia major can be strongly suspected on clinical grounds. Hb electrophoresis shows profound reduction or absence of HbA and increased levels of HbF. The HbA2 level may be normal or increased. Similar but less profound changes are noted in patients affected by β-thalassemia intermedia. Prenatal diagnosis of β-thalassemia is challenging due to the diversity of causative mutations, but can be made in specialized centers by DNA analysis. In fact, thalassemia was the first disease diagnosed by DNA-based tests, opening the way for the field of molecular diagnostics. The diagnosis of β-thalassemia minor is made by Hb electrophoresis, which typically reveals a reduced level of HbA (α2β2) and an increased level of HbA2 (α2δ2). HbH disease can be diagnosed by detection of β4 tetramers by electrophoresis.
Glucose-6-Phosphate Dehydrogenase Deficiency
Red cells are constantly exposed to both endogenous and exogenous oxidants, which are normally inactivated by reduced glutathione (GSH). Abnormalities affecting the enzymes responsible for the synthesis of GSH leave red cells vulnerable to oxidative injury and lead to hemolytic anemias. By far the most common of these anemias is that caused by glucose-6-phosphate dehydrogenase (G6PD) deficiency. The G6PD gene is on the X chromosome. More than 400 G6PD variants have been identified, but only a few are associated with disease. One of the most important variants is G6PD A−, which is carried by approximately 10% of black males in the United States. G6PD A− has a normal enzymatic activity but a decreased half-life. Because red cells do not synthesize proteins, older G6PD A− red cells become progressively deficient in enzyme activity and the reduced form of glutathione. This in turn renders older red cells more sensitive to oxidant stress.
Pathogenesis
G6PD deficiency produces no symptoms until the patient is exposed to an environmental factor (most commonly infectious agents or drugs) that produces oxidants. The drugs incriminated include antimalarials (e.g., primaquine), sulfonamides, nitrofurantoin, phenacetin, aspirin (in large doses), and vitamin K derivatives. More commonly, episodes of hemolysis are triggered by infections, which induce phagocytes to generate oxidants as part of the normal host response. These oxidants, such as hydrogen peroxide, are normally sopped up by GSH, which is converted to oxidized glutathione in the process. Because regeneration of GSH is impaired in G6PD-deficient cells, oxidants are free to “attack” other red cell components including globin chains, which have sulfhydryl groups that are susceptible to oxidation. Oxidized hemoglobin denatures and precipitates, forming intracellular inclusions called Heinz bodies, which can damage the cell membrane sufficiently to cause intravascular hemolysis. Other, less severely damaged cells lose their deformability and suffer further injury when splenic phagocytes attempt to “pluck out” the Heinz bodies, creating so-called bite cells (Fig. 11–7). Such cells become trapped upon recirculation to the spleen and are destroyed by phagocytes (extravascular hemolysis).
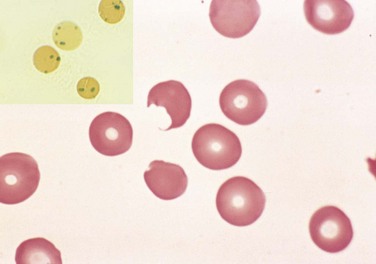
Figure 11–7 Glucose-6-phosphate dehydrogenase deficiency after oxidant drug exposure—peripheral blood smear. Inset, Red cells with precipitates of denatured globin (Heinz bodies) revealed by supravital staining. As the splenic macrophages pluck out these inclusions, “bite cells” like the one in this smear are produced.
(Courtesy of Dr. Robert W. McKenna, Department of Pathology, University of Texas Southwestern Medical School, Dallas, Texas.)
Clinical Features
Drug-induced hemolysis is acute and of variable severity. Typically, patients develop hemolysis after a lag of 2 or 3 days. Since G6PD is X-linked, the red cells of affected males are uniformly deficient and vulnerable to oxidant injury. By contrast, random inactivation of one X chromosome in heterozygous females (Chapter 6) creates two populations of red cells, one normal and the other G6PD-deficient. Most carrier females are unaffected except for those with a large proportion of deficient red cells (a chance situation known as unfavorable lyonization). In the case of the G6PD A− variant, it is mainly older red cells that are susceptible to lysis. Since the marrow compensates for the anemia by producing new resistant red cells, the hemolysis abates even if the drug exposure continues. In other variants such as G6PD Mediterranean, found mainly in the Middle East, the enzyme deficiency and the hemolysis that occur on exposure to oxidants are more severe.
Paroxysmal Nocturnal Hemoglobinuria
Paroxysmal nocturnal hemoglobinuria (PNH) is a rare disorder worthy of mention because it is the only hemolytic anemia that results from an acquired somatic mutation in myeloid stem cells.
Pathogenesis
PNH stems from acquired mutations in gene PIGA, which is required for the synthesis of phosphatidylinositol glycan (PIG), a membrane anchor that is a component of many proteins. Without the “PIG-tail,” these proteins cannot be expressed on the cell surface. The affected proteins include several that limit the activation of complement. As a result, PIGA-deficient precursors give rise to red cells that are inordinately sensitive to complement-mediated lysis. Leukocytes are also deficient in these protective proteins, but nucleated cells are generally less sensitive to complement than are red cells, and as a result the red cells take the brunt of the attack. The paroxysmal nocturnal hemolysis that gives the disorder its name occurs because the fixation of complement is enhanced by the slight decrease in blood pH that accompanies sleep (owing to CO2 retention). However, most patients present less dramatically with anemia due to chronic low-level hemolysis. Another complication that is often serious and sometimes fatal is venous thrombosis. The etiopathogenesis of the prothrombotic state is somehow also related to the activity of the complement membrane attack complex, as inhibitors of this complex (described below) greatly lessen the incidence of thrombosis.
Because PIGA is X-linked, normal cells have only a single active PIGA gene, mutation of which is sufficient to give rise to PIGA deficiency. Because all myeloid lineages are affected in PNH, the responsible mutations must occur in an early myeloid progenitor with self-renewal capacity. Remarkably, many normal individuals harbor small numbers of bone marrow cells bearing PIGA mutations identical to those that cause PNH. It is believed that clinically evident PNH occurs only in rare instances in which the PIGA mutant clone has a survival advantage. One setting in which this may be true is in primary bone marrow failure (aplastic anemia), which most often appears to be caused by immune-mediated destruction or suppression of marrow stem cells. It is hypothesized that PIGA-deficient stem cells somehow escape the immune attack and eventually replace the normal marrow elements. Targeted therapy with an antibody that inhibits the C5b–C9 membrane attack complex is effective at diminishing both the hemolysis and the thrombotic complications, but also places patients at high risk for Neisseria infections, including meningococcal sepsis.
Immunohemolytic Anemias
Some individuals develop antibodies that recognize determinants on red cell membranes and cause hemolytic anemia. These antibodies may arise spontaneously or be induced by exogenous agents such as drugs or chemicals. Immunohemolytic anemias are uncommon and classified on the basis of (1) the nature of the antibody and (2) the presence of predisposing conditions (summarized in Table 11–4).
Table 11–4 Classification of Immunohemolytic Anemias
| Warm Antibody Type |
| Cold Antibody Type |
The diagnosis of immunohemolytic anemias depends on the detection of antibodies and/or complement on red cells. This is done with the direct Coombs antiglobulin test, in which the patient’s red cells are incubated with antibodies against human immunoglobulin or complement. In a positive test result, these antibodies cause the patient’s red cells to clump (agglutinate). The indirect Coombs test, which assesses the ability of the patient’s serum to agglutinate test red cells bearing defined surface determinants, can then be used to characterize the target of the antibody.
Warm Antibody Immunohemolytic Anemias
Warm antibody immunohemolytic anemias are caused by immunoglobulin G (IgG) or, rarely, IgA antibodies that are active at 37°C. More than 60% of cases are idiopathic (primary), while another 25% are secondary to an underlying disease affecting the immune system (e.g., systemic lupus erythematosus) or are induced by drugs. The hemolysis usually results from the opsonization of red cells by the autoantibodies, which leads to erythrophagocytosis in the spleen and elsewhere. In addition, incomplete consumption (“nibbling”) of antibody-coated red cells by macrophages removes membrane. With loss of cell membrane the red cells are transformed into spherocytes, which are rapidly destroyed in the spleen, as described earlier for hereditary spherocytosis. The clinical severity of immunohemolytic anemias is quite variable. Most patients have chronic mild anemia with moderate splenomegaly and require no treatment.
The mechanisms of hemolysis induced by drugs are varied and in some instances poorly understood. Drugs such as α-methyldopa induce autoantibodies against intrinsic red cell constituents, in particular Rh blood group antigens. Presumably, the drug somehow alters the immunogenicity of native epitopes and thereby circumvents T cell tolerance (Chapter 4). Other drugs such as penicillin act as haptens, inducing an antibody response by binding covalently to red cell membrane proteins. Sometimes antibodies recognize a drug in the circulation and form immune complexes that are deposited on red cell membranes. Here they may fix complement or act as opsonins, either of which can lead to hemolysis.
Cold Antibody Immunohemolytic Anemias
Cold antibody immunohemolytic anemias usually are caused by low-affinity IgM antibodies that bind to red cell membranes only at temperatures below 30°C, such as occur in distal parts of the body (e.g., ears, hands, and toes) in cold weather. Although bound IgM fixes complement well, the latter steps of the complement fixation cascade occur inefficiently at temperatures lower than 37°C. As a result, most cells with bound IgM pick up some C3b but are not lysed intravascularly. When these cells travel to warmer areas, the weakly bound IgM antibody is released, but the coating of C3b remains. Because C3b is an opsonin (Chapter 2), the cells are phagocytosed by macrophages, mainly in the spleen and liver; hence, the hemolysis is extravascular. Binding of pentavalent IgM also cross-links red cells and causes them to clump (agglutinate). Sludging of blood in capillaries due to agglutination often produces Raynaud phenomenon in the extremities of affected individuals. Cold agglutinins sometimes also appear transiently during recovery from pneumonia caused by Mycoplasma spp. and infectious mononucleosis, producing a mild anemia of little clinical importance. More important chronic forms of cold agglutinin hemolytic anemia occur in association with certain B cell neoplasms or as an idiopathic condition.
Hemolytic Anemias Resulting from Mechanical Trauma to Red Cells
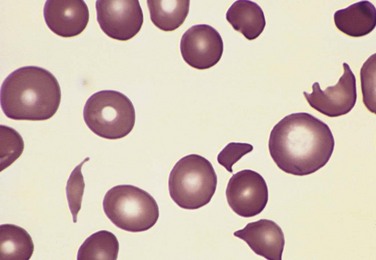
Abnormal mechanical forces result in red cell hemolysis in a variety of circumstances. Traumatic hemolysis can occur incidentally during any activity involving repeated physical blows or their equivalent (e.g., marathon racing, karate chopping, bongo drumming) but is of little clinical importance. More significant mechanical hemolysis is sometimes produced by defective cardiac valve prostheses (the blender effect), which can create sufficiently turbulent blood flow to shear red cells. Microangiopathic hemolytic anemia is observed in pathologic states in which small vessels become partially obstructed or narrowed by lesions that predispose passing red cells to mechanical damage. The most frequent of these conditions is disseminated intravascular coagulation (DIC) (see later), in which vessels are narrowed by the intravascular deposition of fibrin. Other causes of microangiopathic hemolytic anemia include malignant hypertension, systemic lupus erythematosus, thrombotic thrombocytopenic purpura, hemolytic uremic syndrome, and disseminated cancer. The morphologic alterations in the injured red cells (schistocytes) are striking and quite characteristic; “burr cells,” “helmet cells,” and “triangle cells” may be seen (Fig. 11–8). While microangiopathic hemolysis is not usually in and of itself a major clinical problem, it often points to a serious underlying condition.
Malaria
It is estimated that malaria affects 500 million and kills more than 1 million people per year, making it one of the most widespread afflictions of humans. Malaria is endemic in Asia and Africa, but with widespread jet travel cases are now seen all over the world. It is caused by one of four types of protozoa. Of these, the most important is Plasmodium falciparum, which causes tertian malaria (falciparum malaria), a serious disorder with a high fatality rate. The other three species of Plasmodium that infect humans—Plasmodium malariae, Plasmodium vivax, and Plasmodium ovale—cause relatively benign disease. All forms are transmitted by the bite of female Anopheles mosquitoes, and humans are the only natural reservoir.
Pathogenesis
The life cycle of plasmodia is complex. As mosquitoes feed on human blood, sporozoites are introduced from the saliva and within a few minutes infect liver cells. Here the parasites multiply rapidly to form a schizont containing thousands of merozoites. After a period of days to several weeks that varies with the Plasmodium species, the infected hepatocytes release the merozoites, which quickly infect red cells. Intraerythrocytic parasites either continue asexual reproduction to produce more merozoites or give rise to gametocytes capable of infecting the next hungry mosquito. During their asexual reproduction in red cells, each of the four forms of malaria develops into trophozoites with a somewhat distinctive appearance. Thus, the species of malaria that is responsible for an infection can be identified in appropriately stained thick smears of peripheral blood. The asexual phase is completed when the trophozoites give rise to new merozoites, which escape by lysing the red cells.
Clinical Features
The distinctive clinical and anatomic features of malaria are related to the following factors:
• Showers of new merozoites are released from the red cells at intervals of approximately 48 hours for P. vivax, P. ovale, and P. falciparum and 72 hours for P. malariae. The episodic shaking, chills, and fever coincide with this release.
• The parasites destroy large numbers of infected red cells, thereby causing a hemolytic anemia.
• A characteristic brown malarial pigment derived from hemoglobin called hematin is released from the ruptured red cells and produces discoloration of the spleen, liver, lymph nodes, and bone marrow.
• Activation of defense mechanisms in the host leads to a marked hyperplasia of mononuclear phagocytes, producing massive splenomegaly and occasional hepatomegaly.
Fatal falciparum malaria often involves the brain, a complication known as cerebral malaria. Normally, red cells bear negatively charged surfaces that interact poorly with endothelial cells. Infection of red cells with P. falciparum induces the appearance of positively charged surface knobs containing parasite-encoded proteins, which bind to adhesion molecules expressed on activated endothelium. Several endothelial cell adhesion molecules, including intercellular adhesion molecule-1 (ICAM-1), have been proposed to mediate this interaction, which leads to the trapping of red cells in postcapillary venules. In an unfortunate minority of patients, mainly children, this process involves cerebral vessels, which become engorged and occluded. Cerebral malaria is rapidly progressive; convulsions, coma, and death usually occur within days to weeks. Fortunately, falciparum malaria usually pursues a chronic course, which may be punctuated at any time by blackwater fever. The trigger is obscure for this uncommon complication, which is associated with massive intravascular hemolysis, hemoglobinemia, hemoglobinuria, and jaundice.
With appropriate chemotherapy, the prognosis for patients with most forms of malaria is good; however, treatment of falciparum malaria is becoming more difficult with the emergence of drug-resistant strains. Because of the potentially serious consequences of the disease, early diagnosis and treatment are important. The ultimate solution is an effective vaccine, which is long-sought but still elusive.
SummaRy
Hemolytic Anemias
• Autosomal recessive disorder resulting from a mutation in β-globin that causes deoxygenated hemoglobin to self-associate into long polymers that distort (sickle) the red cell
• Blockage of vessels by sickled cells causes pain crises and tissue infarction, particularly of the marrow and spleen
• Red cell membrane damage caused by repeated bouts of sickling results in a moderate to severe hemolytic anemia
Anemias of Diminished Erythropoiesis
The category of anemias involving diminished erythropoiesis includes anemias that are caused by an inadequate dietary supply of nutrients, particularly iron, folic acid, and vitamin B12. Other anemias of this type are those associated with bone marrow failure (aplastic anemia), systemic inflammation (anemia of chronic disease), or bone marrow infiltration by tumor or inflammatory cells (myelophthisic anemia). In this section, some common examples of anemias of these types are discussed individually.
Iron Deficiency Anemia
About 10% of people living in developed countries and 25% to 50% of those in developing countries are anemic. In both settings, the most frequent cause of anemia is iron deficiency. The factors responsible for iron deficiency differ in various populations and are best understood in the context of normal iron metabolism.
The normal total body iron mass is about 2.5 g for women and 3.5 g for men. Approximately 80% of functional body iron is present in hemoglobin, with the remainder being found in myoglobin and iron-containing enzymes (e.g., catalase, cytochromes). The iron storage pool, consisting of hemosiderin and ferritin-bound iron in the liver, spleen, bone marrow, and skeletal muscle, contains on average 15% to 20% of total body iron. Because serum ferritin is largely derived from this storage pool, the serum ferritin level is a good measure of iron stores. Assessment of bone marrow iron is another reliable but more invasive method for estimating iron stores. Iron is transported in the plasma bound to the protein transferrin. In normal persons, transferrin is about 33% saturated with iron, yielding serum iron levels that average 120 µg/dL in men and 100 µg/dL in women. Thus, the normal total iron-binding capacity of serum is 300 to 350 µg/dL.
In keeping with the high prevalence of iron deficiency, evolutionary pressures have yielded metabolic pathways that are strongly biased toward iron retention. There is no regulated pathway for iron excretion, which is limited to the 1 to 2 mg/day that is lost through the shedding of mucosal and skin epithelial cells. Iron balance is maintained largely by regulating the absorption of dietary iron. The normal daily Western diet contains 10 to 20 mg of iron. Most of this is found in heme within meat and poultry, with the remainder present as inorganic iron in vegetables. About 20% of heme iron and 1% to 2% of nonheme iron are absorbable; hence, the average Western diet contains sufficient iron to balance fixed daily losses.
Iron is absorbed in the duodenum (Fig. 11–9). Nonheme iron is carried across the apical and basolateral membranes of enterocytes by distinct transporters. After reduction by ferric reductase, ferrous iron (Fe2+) is transported across the apical membrane by divalent metal transporter-1 (DMT1). A second transporter, ferroportin, then moves iron from the cytoplasm to the plasma across the basolateral membrane. The newly absorbed iron is next oxidized by hephaestin and ceruloplasmin to ferric iron (Fe3+), the form of iron that binds to transferrin. Both DMT1 and ferroportin are widely distributed in the body and are involved in iron transport in other tissues as well. As depicted in Figure 11–9, only a fraction of the iron that enters enterocytes is delivered to transferrin by ferroportin. The remainder is incorporated into cytoplasmic ferritin and lost through the exfoliation of mucosal cells.
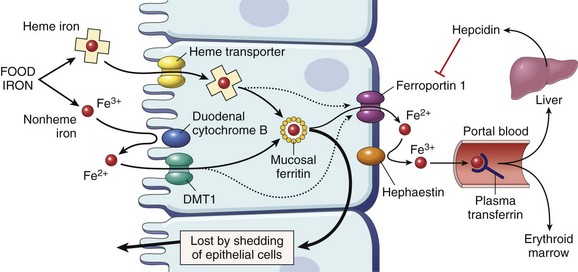
Figure 11–9 Regulation of iron absorption. Duodenal epithelial cell uptake of heme and nonheme iron discussed in the text is depicted. When the storage sites of the body are replete with iron and erythropoietic activity is normal, plasma hepcidin levels are high. This situation leads to downregulation of ferroportin and trapping of most of the absorbed iron, which is lost when duodenal epithelial cells are shed into the gut. Conversely, when body iron stores decrease or erythropoiesis is stimulated, hepcidin levels fall and ferroportin activity increases, allowing a greater fraction of the absorbed iron to be transferred into plasma transferrin. DMT1, divalent metal transporter-1.
When the body is replete with iron, most iron entering duodenal cells is “handed off” to ferritin, whereas transfer to plasma transferrin is enhanced when iron is deficient or erythropoiesis is inefficient. This balance is regulated by hepcidin, a small hepatic peptide that is synthesized and secreted in an iron-dependent fashion. Plasma hepcidin binds ferroportin and induces its internalization and degradation; thus, when hepcidin concentrations are high, ferroportin levels fall and less iron is absorbed. Conversely, when hepcidin levels are low (as occurs in hemochromatosis) (Chapter 15), basolateral transport of iron is increased, eventually leading to systemic iron overload.
Pathogenesis
Iron deficiency arises in a variety of settings:
• Chronic blood loss is the most important cause of iron deficiency anemia in the Western world; the most common sources of bleeding are the gastrointestinal tract (e.g., peptic ulcers, colonic cancer, hemorrhoids) and the female genital tract (e.g., menorrhagia, metrorrhagia, cancers).
• In the developing world, low intake and poor bioavailability due to predominantly vegetarian diets are the most common causes of iron deficiency. In the United States, low dietary intake is an infrequent culprit but is sometimes culpable in infants fed exclusively milk, the impoverished, the elderly, and teenagers subsisting predominantly on junk food.
• Increased demands not met by normal dietary intake occur worldwide during pregnancy and infancy.
• Malabsorption can occur with celiac disease or after gastrectomy (Chapter 14).
Regardless of the cause, iron deficiency develops insidiously. Iron stores are depleted first, marked by a decline in serum ferritin and the absence of stainable iron in the bone marrow. These changes are followed by a decrease in serum iron and a rise in the serum transferrin. Ultimately, the capacity to synthesize hemoglobin, myoglobin, and other iron-containing proteins is diminished, leading to microcytic anemia, impaired work and cognitive performance, and even reduced immunocompetence.
Clinical Features
In most instances, iron deficiency anemia is usually mild and asymptomatic. Nonspecific manifestations, such as weakness, listlessness, and pallor, may be present in severe cases. With long-standing anemia, abnormalities of the fingernails, including thinning, flattening, and “spooning,” may appear. A curious but characteristic neurobehavioral complication is pica, the compunction to consume nonfoodstuffs such as dirt or clay.
In peripheral smears red cells are microcytic and hypochromic (Fig. 11–10). Diagnostic criteria include anemia, hypochromic and microcytic red cell indices, low serum ferritin and iron levels, low transferrin saturation, increased total iron-binding capacity, and, ultimately, response to iron therapy. For unclear reasons, the platelet count often is elevated. Erythropoietin levels are increased, but the marrow response is blunted by the iron deficiency; thus, marrow cellularity usually is only slightly increased.
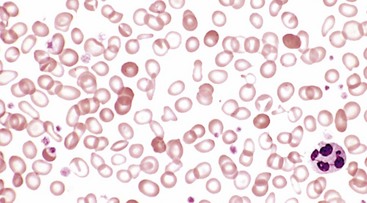
Figure 11–10 Iron deficiency anemia—peripheral blood smear. Note the increased central pallor of most of the red cells. Scattered, fully hemoglobinized cells, from a recent blood transfusion, stand out in contrast.
(Courtesy of Dr. Robert W. McKenna, Department of Pathology, University of Texas Southwestern Medical School, Dallas, Texas.)
Persons often die with iron deficiency anemia, but virtually never of it. An important point is that in well-nourished persons, microcytic hypochromic anemia is not a disease but rather a symptom of some underlying disorder.
Anemia of Chronic Disease
Anemia associated with chronic disease is the most common form of anemia in hospitalized patients. It superficially resembles the anemia of iron deficiency but arises instead from the suppression of erythropoiesis by systemic inflammation. It occurs in a variety of disorders associated with sustained inflammation, including:
• Chronic microbial infections, such as osteomyelitis, bacterial endocarditis, and lung abscess
• Chronic immune disorders, such as rheumatoid arthritis and regional enteritis
• Neoplasms, such as Hodgkin lymphoma and carcinomas of the lung and breast
Pathogenesis
The anemia of chronic disease stems from high levels of plasma hepcidin, which blocks the transfer of iron to erythroid precursors by downregulating ferroportin in macrophages. The elevated hepcidin levels are caused by pro-inflammatory cytokines such as IL-6, which increase hepatic hepcidin synthesis. In addition, chronic inflammation blunts erythropoietin synthesis by the kidney, lowering red cell production by the marrow. The functional advantages of these adaptations in the face of systemic inflammation are unclear; they may serve to inhibit the growth of iron-dependent microorganisms or to augment certain aspects of host immunity.
Clinical Features
As in anemia of iron deficiency, the serum iron levels usually are low in the anemia of chronic disease, and the red cells may even be slightly hypochromic and microcytic. Unlike iron deficiency anemia, however, storage iron in the bone marrow is increased, the serum ferritin concentration is elevated, and the total iron-binding capacity is reduced. Administration of erythropoietin and iron can improve the anemia, but only effective treatment of the underlying condition is curative.
Megaloblastic Anemias
The two principal causes of megaloblastic anemia are folate deficiency and vitamin B12 deficiency. Both vitamins are required for DNA synthesis and the effects of their deficiency on hematopoiesis are essentially identical. However, the causes and consequences of folate and vitamin B12 deficiency differ in important ways.
Pathogenesis
The morphologic hallmark of megaloblastic anemia is the presence of megaloblasts, enlarged erythroid precursors that give rise to abnormally large red cells (macrocytes). Granulocyte precursors are also increased in size. Underlying this cellular gigantism is a defect in DNA synthesis that impairs nuclear maturation and cell division. Because the synthesis of RNA and cytoplasmic elements proceeds at a normal rate and thus outpaces that of the nucleus, the hematopoietic precursors show nuclear-cytoplasmic asynchrony. This maturational derangement contributes to the anemia in several ways. Many megaloblasts are so defective in DNA synthesis that they undergo apoptosis in the marrow (ineffective hematopoiesis). Others mature into red cells but do so after fewer cell divisions, further diminishing the output of red cells. Granulocyte and platelet precursors are also affected (although not as severely) and most patients present with pancytopenia (anemia, thrombocytopenia, and granulocytopenia).
Morphology
Certain morphologic features are common to all forms of megaloblastic anemia. The bone marrow is markedly hypercellular and contains numerous megaloblastic erythroid progenitors. Megaloblasts are larger than normal erythroid progenitors (normoblasts) and have delicate, finely reticulated nuclear chromatin (indicative of nuclear immaturity) (Fig. 11–11). As megaloblasts differentiate and acquire hemoglobin, the nucleus retains its finely distributed chromatin and fails to undergo the chromatin clumping typical of normoblasts. The granulocytic precursors also demonstrate nuclear-cytoplasmic asynchrony, yielding giant metamyelocytes. Megakaryocytes may also be abnormally large and have bizarre multilobed nuclei.
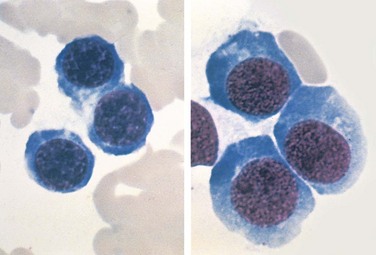
Figure 11–11 Comparison of normoblasts (left) and megaloblasts (right)—bone marrow aspirate. Megaloblasts are larger, have relatively immature nuclei with finely reticulated chromatin, and abundant basophilic cytoplasm.
(Courtesy of Dr. José Hernandez, Department of Pathology, University of Texas Southwestern Medical School, Dallas, Texas.)
In the peripheral blood the earliest change is the appearance of hypersegmented neutrophils, which appear before the onset of anemia. Normal neutrophils have three or four nuclear lobes, but in megaloblastic anemias they often have five or more. The red cells typically include large, egg-shaped macro-ovalocytes; the mean cell volume often is greater than 110 fL (normal, 82 to 92 fL). Although macrocytes appear hyperchromic, in reality the mean cell hemoglobin concentration is normal. Large, misshapen platelets also may be seen. Morphologic changes in other systems, especially the gastrointestinal tract, also occur, giving rise to some of the clinical manifestations.
Folate (Folic Acid) Deficiency Anemia
Megaloblastic anemia secondary to folate deficiency is not common, but marginal folate stores occur with surprising frequency even in apparently healthy persons. The risk of clinically significant folate deficiency is high in those with a poor diet (the economically deprived, the indigent, and the elderly) or increased metabolic needs (pregnant women and patients with chronic hemolytic anemias).
Folate is present in nearly all foods but is destroyed by 10 to 15 minutes of cooking. Thus, the best sources are fresh uncooked vegetables and fruits. Food folates are predominantly in polyglutamate form and must be split into monoglutamates for absorption, a conversion that is hampered by concurrent consumption of acidic foods and substances found in beans and other legumes. Phenytoin (dilantin) and a few other drugs also inhibit folate absorption, while others, such as methotrexate, inhibit folate metabolism. The principal site of intestinal absorption is the upper third of the small intestine; thus, malabsorptive disorders that affect this level of the gut, such as celiac disease and tropical sprue, can impair folate uptake.
Pathogenesis
The metabolism and functions of folate are complex. Here, it is sufficient to note that after absorption folate is transported in the blood mainly as a monoglutamate. Within cells it is further metabolized to several derivatives, but its conversion from dihydrofolate to tetrahydrofolate by dihydrofolate reductase is particularly important. Tetrahydrofolate acts as an acceptor and donor of one-carbon units in several reactions that are required for the synthesis of purines and thymidylate, the building blocks of DNA, and its deficiency accounts for the defect in DNA replication that underlies megaloblastic anemia.
Clinical Features
The onset of the anemia of folate deficiency is insidious, being associated with nonspecific symptoms such as weakness and easy fatigability. The clinical picture may be complicated by the coexistent deficiency of other vitamins, especially in alcoholics. Because the cells lining the gastrointestinal tract, like the hematopoietic system, turn over rapidly, symptoms referable to the alimentary tract, such as sore tongue, are common. Unlike in vitamin B12 deficiency, neurologic abnormalities do not occur.
The diagnosis of a megaloblastic anemia is readily made from examination of smears of peripheral blood and bone marrow. The anemia of folate deficiency is best distinguished from that of vitamin B12 deficiency by measuring serum and red cell folate and vitamin B12 levels.
Vitamin B12 (Cobalamin) Deficiency Anemia (Pernicious Anemia)
Inadequate levels of vitamin B12 (also known as cobalamin) result in a megaloblastic anemia identical to that seen with folate deficiency. However, vitamin B12 deficiency can also cause a demyelinating disorder of the peripheral nerves and the spinal cord. There are many causes of vitamin B12 deficiency. The term pernicious anemia, a relic of days when the cause and therapy of this condition were unknown, applies to vitamin B12 deficiency that results from defects involving intrinsic factor. Intrinsic factor plays a critical role in the absorption of vitamin B12, a multistep process that proceeds as follows:
1 Peptic digestion releases dietary vitamin B12, allowing it to bind a salivary protein called haptocorrin.
2 On entering the duodenum, haptocorrin–B12 complexes are processed by pancreatic proteases; this releases B12, which attaches to intrinsic factor secreted from the parietal cells of the gastric fundic mucosa.
3 The intrinsic factor–B12 complexes pass to the distal ileum and attach to cubulin, a receptor for intrinsic factor, and are taken up into enterocytes.
4 The absorbed vitamin B12 is transferred across the basolateral membranes of enterocytes to plasma transcobalamin, which delivers vitamin B12 to the liver and other cells of the body.
Pathogenesis
Long-standing malabsorption underlies the vast majority of cases of vitamin B12 deficiency. Vitamin B12 is abundant in all food derived from animals, including eggs and dairy products, and is resistant to cooking and boiling. Even bacterial contamination of water and nonanimal foods can provide adequate amounts. As a result, deficiencies due to diet are rare, being confined to strict vegans. Once vitamin B12 is absorbed, the body handles it very efficiently. It is stored in the liver, which normally contains reserves sufficient to support bodily needs for 5 to 20 years.
Pernicious anemia is the most frequent cause of vitamin B12 deficiency. This disease seems to stem from an autoimmune reaction against parietal cells and intrinsic factor itself, which produces gastric mucosal atrophy (Chapter 14). Several associations favor an autoimmune basis:
• Autoantibodies are present in the serum and gastric juice of most patients. Three types of antibodies have been found: parietal canalicular antibodies, which bind to the mucosal parietal cells; blocking antibodies, which disrupt the binding of vitamin B12 to intrinsic factor; and intrinsic factor–B12 complex antibodies, which prevent the complex from binding to cubulin.
• Pernicious anemia frequently occurs concomitantly with other autoimmune diseases, such as Hashimoto thyroiditis, Addison disease, and type 1 diabetes mellitus.
• Serum antibodies to intrinsic factor are often present in patients with other autoimmune diseases.
Chronic vitamin B12 malabsorption is also seen after gastrectomy (owing to loss of intrinsic factor–producing cells) or ileal resection (owing to loss of intrinsic factor–B12 complex–absorbing cells), and in disorders that disrupt the function of the distal ileum (such as Crohn disease, tropical sprue, and Whipple disease). Particularly in older persons, gastric atrophy and achlorhydria may interfere with the production of acid and pepsin, which are needed to release the vitamin B12 from its bound form in food.
The metabolic defects responsible for the anemia are intertwined with folate metabolism. Vitamin B12 is required for recycling of tetrahydrofolate, the form of folate that is needed for DNA synthesis. In keeping with this relationship, the anemia of vitamin B12 deficiency is reversed with administration of folate. By contrast, folate administration does not prevent and may in fact worsen the neurologic symptoms. The main neurologic lesions associated with vitamin B12 deficiency are demyelination of the posterior and lateral columns of the spinal cord, sometimes beginning in the peripheral nerves. In time, axonal degeneration may supervene. The severity of the neurologic manifestations is not related to the degree of anemia. Indeed, the neurologic disease may occur in the absence of overt megaloblastic anemia.
Clinical Features
The manifestations of vitamin B12 deficiency are nonspecific. As with all anemias, findings include pallor, easy fatigability, and, in severe cases, dyspnea and even congestive heart failure. The increased destruction of erythroid progenitors may give rise to mild jaundice. Gastrointestinal signs and symptoms similar to those of folate deficiency are seen. The spinal cord disease begins with symmetric numbness, tingling, and burning in feet or hands, followed by unsteadiness of gait and loss of position sense, particularly in the toes. Although the anemia responds dramatically to parenteral vitamin B12, the neurologic manifestations often fail to resolve. As discussed in Chapter 14, patients with pernicious anemia have an increased risk for the development of gastric carcinoma.
The diagnostic features of pernicious anemia include (1) low serum vitamin B12 levels, (2) normal or elevated serum folate levels, (3) serum antibodies to intrinsic factor, (4) moderate to severe megaloblastic anemia, (5) leukopenia with hypersegmented granulocytes, and (6) a dramatic reticulocytic response (within 2 to 3 days) to parenteral administration of vitamin B12.
Aplastic Anemia
Aplastic anemia is a disorder in which multipotent myeloid stem cells are suppressed, leading to bone marrow failure and pancytopenia. It must be distinguished from pure red cell aplasia, in which only erythroid progenitors are affected and anemia is the only manifestation.
Pathogenesis
In more than half of the cases, aplastic anemia is idiopathic. In the remainder, an exposure to a known myelotoxic agent, such as a drug or a chemical, can be identified. With some agents, the marrow damage is predictable, dose-related, and reversible. Included in this category are antineoplastic drugs (e.g., alkylating agents, antimetabolites), benzene, and chloramphenicol. In other instances, marrow toxicity occurs as an “idiosyncratic” or hypersensitivity reaction to small doses of known myelotoxic drugs (e.g., chloramphenicol) or to drugs such as sulfonamides, which are not myelotoxic in other persons. Aplastic anemia sometimes arises after certain viral infections, most often community-acquired viral hepatitis. The specific virus responsible is not known; hepatitis viruses A, B, and C are not the culprits. Marrow aplasia develops insidiously several months after recovery from the hepatitis and follows a relentless course.
The pathogenic events leading to marrow failure remain vague, but it seems that autoreactive T cells play an important role. This is supported by a variety of experimental data and clinical experience showing that aplastic anemia responds to immunosuppressive therapy aimed at T cells in 70% to 80% of cases. Much less clear are the events that trigger the T cell attack on marrow stem cells; viral antigens, drug-derived haptens, and/or genetic damage may create neoantigens within stem cells that serve as targets for the immune system.
Rare but interesting genetic conditions also are associated with marrow failure. From 5% to 10% of patients with “acquired” aplastic anemia have inherited defects in telomerase, which as noted earlier is needed for the maintenance and stability of chromosomes. It is hypothesized that the defect in telomerase leads to premature senescence of hematopoietic stem cells. Of further interest, the bone marrow cells in up to 50% of sporadic cases have unusually short telomeres, possibly as a consequence of as-yet undiscovered defects in telomerase, or of excessive replication of hematopoietic stem cells, which may lead to premature senescence. Some children with Fanconi anemia, an inherited disorder of DNA repair, also develop marrow aplasia.
Morphology
The bone marrow in aplastic anemia is markedly hypocellular, with greater than 90% of the intertrabecular space being occupied by fat. The limited cellularity often consists only of lymphocytes and plasma cells. Anemia may cause fatty change in the liver. Thrombocytopenia and granulocytopenia may result in hemorrhages and bacterial infections, respectively. The requirement for transfusions may eventually lead to hemosiderosis.
Clinical Course
Aplastic anemia affects persons of all ages and both sexes. The slowly progressive anemia causes the insidious development of weakness, pallor, and dyspnea. Thrombocytopenia often manifests with petechiae and ecchymoses. Granulocytopenia may be manifested by frequent and persistent minor infections or by the sudden onset of chills, fever, and prostration. It is important to separate aplastic anemia from anemias caused by marrow infiltration (myelophthisic anemia), “aleukemic leukemia,” and granulomatous diseases, which may have similar clinical presentations but are easily distinguished on examination of the bone marrow. Aplastic anemia does not cause splenomegaly; if it is present, another diagnosis should be sought. Typically, the red cells are normochromic and normocytic or slightly macrocytic. Reticulocytes are reduced in number (reticulocytopenia).
The prognosis is unpredictable. Withdrawal of drugs sometimes leads to remission, but this is the exception rather than the rule. The idiopathic form carries a poor prognosis if left untreated. Bone marrow transplantation often is curative, particularly in nontransfused patients younger than 40 years of age. Transfusions sensitize patients to alloantigens, producing a high rate of engraftment failure; thus, they must be minimized in persons eligible for bone marrow transplantation. Successful transplantation requires “conditioning” with high doses of immunosuppressive radiation or chemotherapy, reinforcing the notion that autoimmunity has an important role in the disease. As mentioned earlier, patients who are poor transplantation candidates often benefit from immunosuppressive therapy.
Myelophthisic Anemia
Myelophthisic anemia is caused by extensive infiltration of the marrow by tumors or other lesions. It most commonly is associated with metastatic breast, lung, or prostate cancer. Other tumors, advanced tuberculosis, lipid storage disorders, and osteosclerosis can produce a similar clinical picture. The principal manifestations include anemia and thrombocytopenia; in general, the white cell series is less affected. Characteristically misshapen red cells, some resembling teardrops, are seen in the peripheral blood. Immature granulocytic and erythrocytic precursors also may be present (leukoerythroblastosis) along with mild leukocytosis. Treatment is directed at the underlying condition.
Polycythemia
Polycythemia, or erythrocytosis, denotes an increase in red cells per unit volume of peripheral blood, usually in association with an increase in hemoglobin concentration. Polycythemia may be absolute (defined as an increase in total red cell mass) or relative. Relative polycythemia results from dehydration, such as occurs with water deprivation, prolonged vomiting, diarrhea, or the excessive use of diuretics. Absolute polycythemia is described as primary when the increased red cell mass results from an autonomous proliferation of erythroid progenitors, and secondary when the excessive proliferation stems from elevated levels of erythropoietin. Primary polycythemia (polycythemia vera) is a clonal, neoplastic myeloproliferative disorder considered later in this chapter. The increases in erythropoietin that cause secondary forms of absolute polycythemia have a variety of causes (Table 11–5).
Table 11–5 Pathophysiologic Classification of Polycythemia
| Relative |
| Absolute |
| Primary |
| Secondary |