Chapter 22 Central Nervous System
See Targeted Therapy available online at studentconsult.com
Degenerative, inflammatory, infectious, and neoplastic disorders of the central nervous system (CNS) are some of the most serious diseases of mankind. The pathology of these diseases has many features that reflect the unique properties of the CNS. In fact, the diagnosis and analysis of CNS disorders requires specialized expertise, a realization that has led to the creation of the field of neuropathology.
Patterns of Injury in The Nervous System
The cells of the nervous system respond to various forms of injury with distinct morphologic changes.
 Morphology
Morphology
Features of Neuronal Injury
In response to injury, a number of changes occur in neurons and their processes (axons and dendrites). Within 12 hours of an irreversible hypoxic-ischemic insult, acute neuronal injury becomes evident on routine hematoxylin and eosin (H&E) staining (Fig. 22–1, A). There is shrinkage of the cell body, pyknosis of the nucleus, disappearance of the nucleolus, and loss of Nissl substance, with intense eosinophilia of the cytoplasm (“red neurons”). Often, the nucleus assumes the angulated shape of the shrunken cell body. Injured axons undergo swelling and show disruption of axonal transport. The swellings (spheroids) can be recognized on H&E stains (Fig. 22–1, B) and can be highlighted by silver staining or immunohistochemistry. Axonal injury also leads to cell body enlargement and rounding, peripheral displacement of the nucleus, enlargement of the nucleolus, and peripheral dispersion of Nissl substance (central chromatolysis) (Fig. 22–1, C). In addition, acute injuries typically result in breakdown of the blood-brain barrier and variable degrees of cerebral edema (described later).
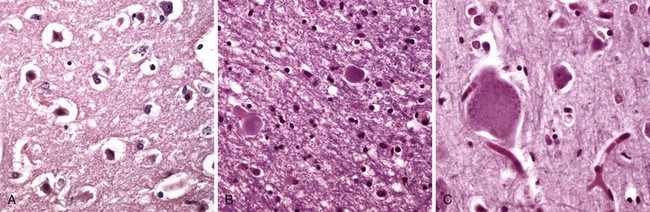
Figure 22–1 Patterns of neuronal injury. A, Acute hypoxic-ischemic injury in cerebral cortex, where the individual cell bodies are shrunken, along with the nuclei. They also are prominently stained by eosin (“red neurons”). B, Axonal spheroids are visible as bulbous swellings at points of disruption, or altered axonal transport. C, With axonal injury there can be swelling of the cell body and peripheral dispersal of the Nissl substance, termed chromatolysis.
Many neurodegenerative diseases are associated with specific intracellular inclusions (e.g., Lewy bodies in Parkinson disease and tangles in Alzheimer disease), also described later. Pathogenic viruses can also form inclusions in neurons, just as they do in other cells of the body. In some neurodegenerative diseases, neuronal processes also become thickened and tortuous; these are termed dystrophic neurites. With age, neurons also accumulate complex lipids (lipofuscin) in their cytoplasm and lysosomes.
Astrocytes in Injury and Repair
Astrocytes are the principal cells responsible for repair and scar formation in the brain, a process termed gliosis. In response to injury, astrocytes undergo both hypertrophy and hyperplasia. The nucleus enlarges and becomes vesicular, and the nucleolus becomes prominent. The previously scant cytoplasm expands and takes on a bright pink hue, and the cell extends multiple stout, ramifying processes (gemistocytic astrocyte). Unlike elsewhere in the body, fibroblasts participate in healing after brain injury to a limited extent except in specific settings (penetrating brain trauma or around abscesses). In long-standing gliosis, the cytoplasm of reactive astrocytes shrinks in size and the cellular processes become more tightly interwoven (fibrillary astrocytes). Rosenthal fibers are thick, elongated, brightly eosinophilic protein aggregates found in astrocytic processes in chronic gliosis and in some low-grade gliomas.
Changes in Other Cell Types
Oligodendrocytes, which produce myelin, exhibit a limited spectrum of specific morphologic changes in response to various injuries. In progressive multifocal leukoencephalopathy, viral inclusions can be seen in oligodendrocytes, with a smudgy, homogeneous-appearing enlarged nucleus.
Microglial cells are bone-marrow–derived cells that function as the resident phagocytes of the CNS. When activated by tissue injury, infection, or trauma, they proliferate and become more prominent histologically. Microglial cells take on the appearance of activated macrophages in areas of demyelination, organizing infarct, or hemorrhage; in other settings such as neurosyphilis or other infections, they develop elongated nuclei (rod cells). Aggregates of elongated microglial cells at sites of tissue injury are termed microglial nodules. Similar collections can be found congregating around and phagocytosing injured neurons (neuronophagia).
Ependymal cells line the ventricular system and the central canal of the spinal cord. Certain pathogens, particularly cytomegalovirus (CMV), can produce extensive ependymal injury, with typical viral inclusions. Choroid plexus is in continuity with the ependyma, and its specialized epithelial covering is responsible for the secretion of cerebrospinal fluid (CSF).
Edema, Herniation, and Hydrocephalus
The brain and spinal cord exist within the protective and rigid skull and spinal canal, with nerves and blood vessels passing through specific foramina. The advantage of housing the delicate CNS within such a protective environment is obvious, but this arrangement provides little room for brain parenchymal expansion in disease states. Disorders that may cause dangerous increases in brain volume within the fixed space of the skull include generalized cerebral edema, hydrocephalus, and mass lesions such as tumors.
Cerebral Edema
Cerebral edema is the accumulation of excess fluid within the brain parenchyma. There are two types, which often occur together particularly after generalized injury.
• Vasogenic edema occurs when the integrity of the normal blood-brain barrier is disrupted, allowing fluid to shift from the vascular compartment into the extracellular spaces of the brain. Vasogenic edema can be either localized (e.g., increased vascular permeability due to inflammation or in tumors) or generalized.
• Cytotoxic edema is an increase in intracellular fluid secondary to neuronal and glial cell membrane injury, as might follow generalized hypoxic-ischemic insult or after exposure to some toxins.
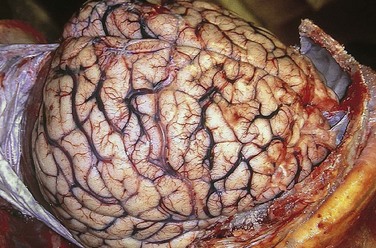
The edematous brain is softer than normal and often appears to “over fill” the cranial vault. In generalized edema the gyri are flattened, the intervening sulci are narrowed, and the ventricular cavities are compressed (Fig. 22–2).
Hydrocephalus
After being produced by the choroid plexus within the ventricles, CSF circulates through the ventricular system and flows through the foramina of Luschka and Magendie into the subarachnoid space, where it is absorbed by arachnoid granulations. The balance between rates of generation and resorption regulates CSF volume.
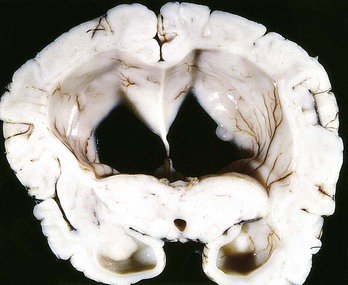
Hydrocephalus refers to the accumulation of excessive CSF within the ventricular system. This disorder most often is a consequence of impaired flow or resorption; overproduction of CSF, typically seen with tumors of the choroid plexus, only rarely causes hydrocephalus. If there is a localized obstacle to CSF flow within the ventricular system, then a portion of the ventricles enlarges while the remainder does not. This pattern is referred to as noncommunicating hydrocephalus and most commonly is caused by masses obstructing the foramen of Monro or compressing the cerebral aqueduct. In communicating hydrocephalus, the entire ventricular system is enlarged; it is usually caused by reduced CSF resorption.
If hydrocephalus develops in infancy before closure of the cranial sutures, the head enlarges. Once the sutures fuse, hydrocephalus causes ventricular expansion and increased intracranial pressure, but no change in head circumference (Fig. 22–3). In contrast with these states, in which increased CSF volume is the primary process, a compensatory increase in CSF volume can also follow the loss of brain parenchyma (hydrocephalus ex vacuo), as after infarcts or with degenerative diseases.
Herniation
When the volume of tissue and fluid inside the skull increases beyond the limit permitted by compression of veins and displacement of CSF, intracranial pressure rises. The cranial vault is subdivided by rigid dural folds (falx and tentorium), and a focal expansion of the brain displaces it in relation to these partitions. If the expansion is sufficiently large, herniation occurs. Herniation often leads to “pinching” and vascular compromise of the compressed tissue, producing infarction, additional swelling, and further herniation. There are three main types of herniation (Fig. 22–4).
• Subfalcine (cingulate) herniation occurs when unilateral or asymmetric expansion of a cerebral hemisphere displaces the cingulate gyrus under the edge of falx. This may be associated with compression of the anterior cerebral artery.
• Transtentorial (uncinate) herniation occurs when the medial aspect of the temporal lobe is compressed against the free margin of the tentorium. As the temporal lobe is displaced, the third cranial nerve is compromised, resulting in pupillary dilation and impaired ocular movements on the side of the lesion (“blown pupil”). The posterior cerebral artery may also be compressed, resulting in ischemic injury to tissue supplied by that vessel, including the primary visual cortex. If the amount of displaced temporal lobe is large enough, the pressure on the midbrain can compress the contralateral cerebral peduncle against the tentorium, resulting in hemiparesis ipsilateral to the side of the herniation (a so-called false localizing sign). The compression of the peduncle creates a deformation known as Kernohan’s notch. Progression of transtentorial herniation is often accompanied by linear or flame-shaped hemorrhages in the midbrain and pons, termed Duret hemorrhages (Fig. 22–5). These lesions usually occur in the midline and paramedian regions and are believed to be the result of tearing of penetrating veins and arteries supplying the upper brain stem.
• Tonsillar herniation refers to displacement of the cerebellar tonsils through the foramen magnum. This type of herniation is life-threatening, because it causes brain stem compression and compromises vital respiratory and cardiac centers in the medulla.
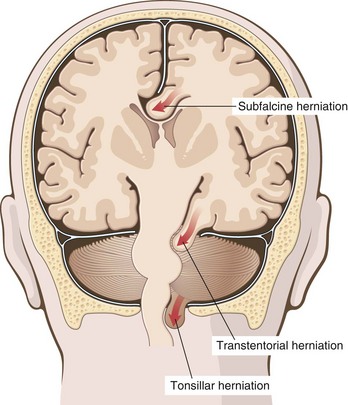
Figure 22–4 Herniation syndromes. Displacement of brain parenchyma across fixed barriers can be subfalcine, transtentorial, or tonsillar (into the foramen magnum).
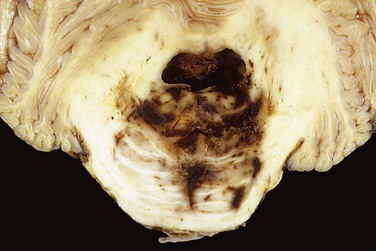
Figure 22–5 Duret hemorrhage. As mass effect displaces the brain downward, there is disruption of the vessels that enter the pons along the midline, leading to hemorrhage.
 Summary
Summary
Edema, Herniation, and Hydrocephalus
• Cerebral edema is the accumulation of excess fluid within the brain parenchyma. Hydrocephalus is defined as an increase in CSF volume within all or part of the ventricular system.
• Increases in brain volume (as a result of increased CSF volume, edema, hemorrhage, or tumor) raise the pressure inside the fixed capacity of the skull.
• Increases in pressure can damage the brain either by decreasing perfusion or by displacing tissue across dural partitions inside the skull or through openings in the skull (herniations).
Cerebrovascular Diseases
Cerebrovascular diseases—the broad category of brain disorders caused by pathologic processes involving blood vessels—constitute a major cause of death in the developed world and are the most prevalent cause of neurologic morbidity. The three main pathogenic mechanisms are (1) thrombotic occlusion, (2) embolic occlusion, and (3) vascular rupture. Stroke is the clinical designation applied to all of these conditions when symptoms begin acutely. Thrombosis and embolism have similar consequences for the brain: loss of oxygen and metabolic substrates, resulting in infarction or ischemic injury of regions supplied by the affected vessel. Similar injury occurs globally when there is complete loss of perfusion, severe hypoxemia (e.g., hypovolemic shock), or profound hypoglycemia. Hemorrhage accompanies rupture of vessels and leads to direct tissue damage as well as secondary ischemic injury. Traumatic vascular injury is discussed separately in the context of trauma.
Hypoxia, Ischemia, and Infarction
The brain is a highly oxygen-dependent tissue that requires a continual supply of glucose and oxygen from the blood. Although it constitutes no more than 2% of body weight, the brain receives 15% of the resting cardiac output and is responsible for 20% of total body oxygen consumption. Cerebral blood flow normally remains stable over a wide range of blood pressure and intracranial pressure because of autoregulation of vascular resistance. The brain may be deprived of oxygen by two general mechanisms:
• Functional hypoxia, caused by a low partial pressure of oxygen (e.g., high altitude), impaired oxygen-carrying capacity (e.g., severe anemia, carbon monoxide poisoning), or inhibition of oxygen use by tissue (e.g., cyanide poisoning)
• Ischemia, either transient or permanent, due to tissue hypoperfusion, which can be caused by hypotension, vascular obstruction, or both
Global Cerebral Ischemia
Widespread ischemic-hypoxic injury can occur in the setting of severe systemic hypotension, usually when systolic pressures fall below 50 mm Hg, as in cardiac arrest, shock, and severe hypotension. The clinical outcome varies with the severity and duration of the insult. When the insult is mild, there may be only a transient postischemic confusional state, with eventual complete recovery. Neurons are more susceptible to hypoxic injury than are glial cells, and the most susceptible neurons are the pyramidal cells of the hippocampus and neocortex and Purkinje cells of the cerebellum. In some individuals, even mild or transient global ischemic insults may cause damage to these vulnerable areas. In severe global cerebral ischemia, widespread neuronal death occurs irrespective of regional vulnerability. Patients who survive often remain severely impaired neurologically and in a persistent vegetative state. Other patients meet the clinical criteria for so-called brain death, including evidence of diffuse cortical injury (isoelectric, or “flat,” electroencephalogram) and brain stem damage, including absence of reflexes and respiratory drive. When patients with this form of irreversible injury are maintained on mechanical ventilation, the brain gradually undergoes autolysis, resulting in the so-called “respirator brain.”
Morphology
In the setting of global ischemia, the brain is swollen, with wide gyri and narrowed sulci. The cut surface shows poor demarcation between gray and white matter. The histopathologic changes that accompany irreversible ischemic injury (infarction) are grouped into three categories. Early changes, occurring 12 to 24 hours after the insult, include acute neuronal cell change (red neurons) (Fig. 22–1, A) characterized initially by microvacuolization, followed by cytoplasmic eosinophilia, and later nuclear pyknosis and karyorrhexis. Similar changes occur somewhat later in astrocytes and oligodendroglia. After this, the reaction to tissue damage begins with infiltration by neutrophils (Fig. 22–6, A). Subacute changes, occurring at 24 hours to 2 weeks, include necrosis of tissue, influx of macrophages, vascular proliferation, and reactive gliosis (Fig. 22–6, B). Repair, seen after 2 weeks, is characterized by removal of all necrotic tissue, loss of organized CNS structure, and gliosis (Fig. 22–6, C). The distribution of neuronal loss and gliosis in the neocortex typically is uneven with preservation of some layers and devastation of others—a pattern termed pseudolaminar necrosis.
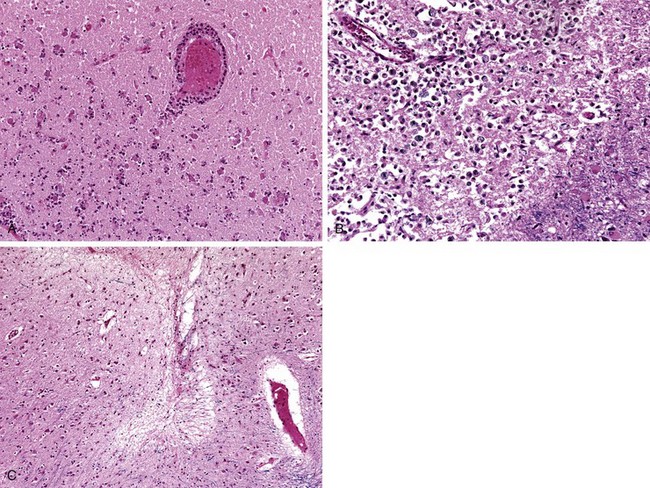
Figure 22–6 Cerebral infarction. A, Infiltration of a cerebral infarction by neutrophils begins at the edges of the lesion where the vascular supply is intact. B, By day 10, an area of infarction shows the presence of macrophages and surrounding reactive gliosis. C, Old intracortical infarcts are seen as areas of tissue loss with a modest amount of residual gliosis.
Border zone (“watershed”) infarcts are wedge-shaped areas of infarction that occur in regions of the brain and spinal cord that lie at the most distal portions of arterial territories. They are usually seen after hypotensive episodes. In the cerebral hemispheres, the border zone between the anterior and the middle cerebral artery distributions is at greatest risk. Damage to this region produces a band of necrosis over the cerebral convexity a few centimeters lateral to the interhemispheric fissure.
Focal Cerebral Ischemia
Cerebral arterial occlusion leads first to focal ischemia and then to infarction in the distribution of the compromised vessel. The size, location, and shape of the infarct and the extent of tissue damage that results may be modified by collateral blood flow. Specifically, collateral flow through the circle of Willis or cortical-leptomeningeal anastomoses can limit damage in some regions. By contrast, there is little if any collateral flow to structures such as the thalamus, basal ganglia, and deep white matter, which are supplied by deep penetrating vessels.
Embolic infarctions are more common than infarctions due to thrombosis. Cardiac mural thrombi are a frequent source of emboli; myocardial dysfunction, valvular disease, and atrial fibrillation are important predisposing factors. Thromboemboli also arise in arteries, most often from atheromatous plaques within the carotid arteries or aortic arch. Other emboli of venous origin cross over to the arterial circulation through cardiac defects and lodge in the brain (paradoxical embolism; see Chapter 3); these include thromboemboli from deep leg veins and fat emboli, usually following bone trauma. The territory of the middle cerebral artery, a direct extension of the internal carotid artery, is most frequently affected by embolic infarction. Emboli tend to lodge where vessels branch or in areas of stenosis, usually caused by atherosclerosis.
Thrombotic occlusions causing cerebral infarctions usually are superimposed on atherosclerotic plaques; common sites are the carotid bifurcation, the origin of the middle cerebral artery, and at either end of the basilar artery. These occlusions may be accompanied by anterograde extension, as well as thrombus fragmentation and distal embolization.
Infarcts can be divided into two broad groups based on their macroscopic and corresponding radiologic appearance (Fig. 22–7). Nonhemorrhagic infarcts result from acute vascular occlusions and can be treated with thrombolytic therapies, especially if identified shortly after presentation. This approach is contraindicated in hemorrhagic infarcts, which result from reperfusion of ischemic tissue, either through collaterals or after dissolution of emboli, and often produce multiple, sometimes confluent petechial hemorrhages (Fig. 22–7, A and B).
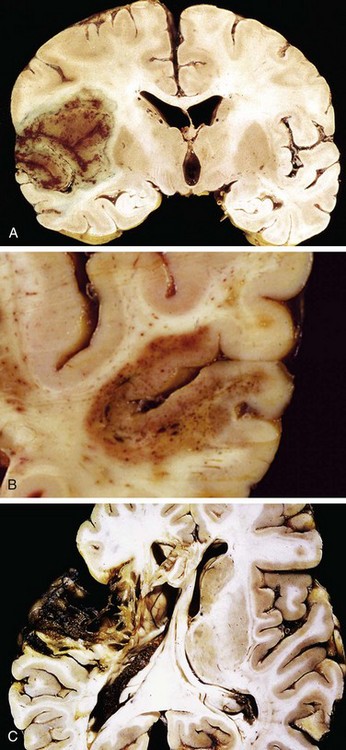
Figure 22–7 Cerebral infarction.
A, Section of the brain showing a large, discolored, focally hemorrhagic region in the left middle cerebral artery distribution (hemorrhagic, or red, infarction). B, An infarct with punctate hemorrhages, consistent with ischemia-reperfusion injury, is present in the temporal lobe. C, Old cystic infarct shows destruction of cortex and surrounding gliosis.
Morphology
The macroscopic appearance of a nonhemorrhagic infarct evolves over time. During the first 6 hours the tissue is unchanged in appearance, but by 48 hours, the tissue becomes pale, soft, and swollen. From days 2 to 10, the brain turns gelatinous and friable, and the boundary between normal and abnormal tissue becomes more distinct as edema resolves in the adjacent viable tissue. From day 10 to week 3, the tissue liquefies, eventually leaving a fluid-filled cavity lined by dark gray tissue, which gradually expands as dead tissue is resorbed (Fig. 22–7, C).
Microscopically, the tissue reaction follows a characteristic sequence. After the first 12 hours, ischemic neuronal change (red neurons) (Fig. 22–1, A) and cytotoxic and vasogenic edema predominate. Endothelial and glial cells, mainly astrocytes, swell, and myelinated fibers begin to disintegrate. Up to 48 hours, there is some neutrophilic emigration, which is followed by mononuclear phagocytic cells during the ensuing 2 to 3 weeks. Macrophages containing myelin or red cell breakdown products may persist in the lesion for months to years. As the process of phagocytosis and liquefaction proceeds, astrocytes at the edges of the lesion progressively enlarge, divide, and develop a prominent network of cytoplasmic extensions.
After several months, the striking astrocytic nuclear and cytoplasmic enlargement regresses. In the wall of the cavity, astrocyte processes form a dense feltwork of glial fibers admixed with new capillaries and a few perivascular connective tissue fibers. In the cerebral cortex, the cavity is delimited from the meninges and subarachnoid space by a gliotic layer of tissue, derived from the molecular layer of the cortex. The pia and arachnoid are not affected and do not contribute to the healing process.
The microscopic picture and evolution of hemorrhagic infarction parallel those of ischemic infarction, with the addition of blood extravasation and resorption. In persons with coagulopathies, hemorrhagic infarcts may be associated with extensive intracerebral hematomas.
Intracranial Hemorrhage
Hemorrhages within the brain are associated with (1) hypertension and other diseases leading to vascular wall injury, (2) structural lesions such as arteriovenous and cavernous malformations, and (3) tumors. Subarachnoid hemorrhages most commonly are caused by ruptured aneurysms but also occur with other vascular malformations. Subdural or epidural hemorrhages usually are associated with trauma.
Primary Brain Parenchymal Hemorrhage
Spontaneous (nontraumatic) intraparenchymal hemorrhages are most common in mid- to late adult life, with a peak incidence at about 60 years of age. Most are due to the rupture of a small intraparenchymal vessel. Hypertension is the leading underlying cause, and brain hemorrhage accounts for roughly 15% of deaths among persons with chronic hypertension. Intracerebral hemorrhage can be clinically devastating when it affects large portions of the brain or extends into the ventricular system; alternatively, it can affect small regions and be clinically silent. Hypertensive intraparenchymal hemorrhages typically occur in the basal ganglia, thalamus, pons, and cerebellum (Fig. 22–8), with the location and the size of the bleed determining its clinical manifestations. If the person survives the acute event, gradual resolution of the hematoma ensues, sometimes with considerable clinical improvement.
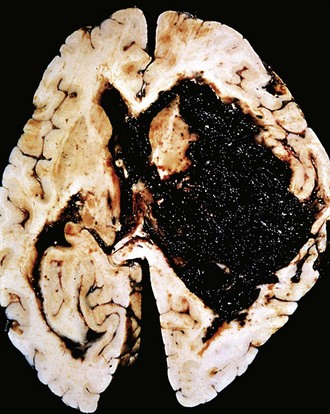
Figure 22–8 Cerebral hemorrhage. Massive hypertensive hemorrhage rupturing into a lateral ventricle.
Morphology
Acute hemorrhages are characterized by extravasated blood, which compresses the adjacent parenchyma. With time, hemorrhages are converted to a cavity with a brown, discolored rim. On microscopic examination, early lesions consist of clotted blood surrounded by brain tissue showing anoxic neuronal and glial changes as well as edema. Eventually the edema resolves, pigment- and lipid-laden macrophages appear, and proliferation of reactive astrocytes becomes visible at the periphery of the lesion. The cellular events then follow the same time course observed after cerebral infarction.
Cerebral Amyloid Angiopathy
Cerebral amyloid angiopathy (CAA) is a disease in which amyloidogenic peptides, typically the same ones found in Alzheimer disease (discussed later), deposit in the walls of medium- and small-caliber meningeal and cortical vessels. The amyloid confers a rigid, pipelike appearance and stains with Congo red. Amyloid deposition weakens vessel walls and increases the risk of hemorrhages, which differ in distribution from those associated with hypertension. Specifically, CAA-associated hemorrhages often occur in the lobes of the cerebral cortex (lobar hemorrhages).
Subarachnoid Hemorrhage and Saccular Aneurysms
The most frequent cause of clinically significant non-traumatic subarachnoid hemorrhage is rupture of a saccular (berry) aneurysm. Hemorrhage into the subarachnoid space also may result from vascular malformation, trauma (usually associated with other signs of the injury), rupture of an intracerebral hemorrhage into the ventricular system, hematologic disturbances, and tumors.
Rupture can occur at any time, but in about one third of cases it is associated with acute increases in intracranial pressure, such as with straining at stool or sexual orgasm. Blood under arterial pressure is forced into the subarachnoid space, and the patient is stricken with sudden, excruciating headache (classically described as “the worst headache I’ve ever had”) and rapidly loses consciousness. Between 25% and 50% of affected persons die from the first bleed, and recurrent bleeds are common in survivors. Not surprisingly, the prognosis worsens with each bleeding episode.
About 90% of saccular aneurysms occur in the anterior circulation near major arterial branch points (Fig. 22–9); multiple aneurysms exist in 20% to 30% of cases. Although they are sometimes referred to as congenital, they are not present at birth but develop over time because of underlying defects in the vessel media. There is an increased risk of aneurysms in patients with autosomal dominant polycystic kidney disease (Chapter 13), as well as those with genetic disorders of extracellular matrix proteins. Overall, roughly 1.3% of aneurysms bleed per year, with the probability of rupture increasing nonlinearly with size. For example, aneurysms larger than 1 cm in diameter have a roughly 50% risk of bleeding per year. In the early period after a subarachnoid hemorrhage, there is an additional risk of ischemic injury from vasospasm of other vessels. Healing and the attendant meningeal fibrosis and scarring sometimes obstruct CSF flow or disrupt CSF resorption, leading to hydrocephalus.
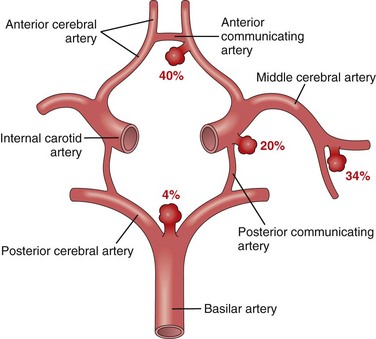
Morphology
An unruptured saccular aneurysm is a thin-walled outpouching of an artery (Fig. 22–10). Beyond the neck of the aneurysm, the muscular wall and intimal elastic lamina are absent, such that the aneurysm sac is lined only by thickened hyalinized intima. The adventitia covering the sac is continuous with that of the parent artery. Rupture usually occurs at the apex of the sac, releasing blood into the subarachnoid space or the substance of the brain, or both.
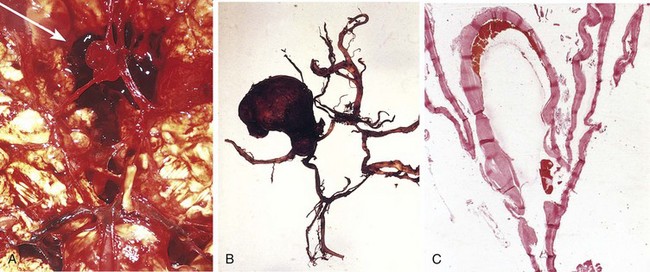
Figure 22–10 Saccular aneurysms. A, View of the base of the brain, dissected to show the circle of Willis with an aneurysm of the anterior cerebral artery (arrow). B, Circle of Willis dissected to show large aneurysm. C, Section through a saccular aneurysm showing the hyalinized fibrous vessel wall. Hematoxylin-eosin stain.
In addition to saccular aneurysms, atherosclerotic, mycotic, traumatic, and dissecting aneurysms also occur intracranially. The last three types (like saccular aneurysms) most often are found in the anterior circulation, whereas atherosclerotic aneurysms frequently are fusiform and most commonly involve the basilar artery. Nonsaccular aneurysms usually manifest with cerebral infarction due to vascular occlusion instead of subarachnoid hemorrhage.
Vascular Malformations
Vascular malformations of the brain are classified into four principal types based on the nature of the abnormal vessels: arteriovenous malformations (AVMs), cavernous malformations, capillary telangiectasias, and venous angiomas. AVMs, the most common of these, affect males twice as frequently as females and most commonly manifest between the ages of 10 and 30 years with seizures, an intracerebral hemorrhage, or a subarachnoid hemorrhage. Large AVMs occurring in the newborn period can lead to high-output congestive heart failure because of blood shunting from arteries to veins. The risk of bleeding makes AVM the most dangerous type of vascular malformation. Multiple AVMs can be seen in the setting of hereditary hemorrhagic telangiectasia, an autosomal dominant condition often associated with mutations affecting the TGFβ pathway.
Morphology
AVMs may involve subarachnoid vessels extending into brain parenchyma or occur exclusively within the brain. On gross inspection, they resemble a tangled network of wormlike vascular channels (Fig. 22–11). Microscopic examination shows enlarged blood vessels separated by gliotic tissue, often with evidence of previous hemorrhage. Some vessels can be recognized as arteries with duplicated and fragmented internal elastic lamina, while others show marked thickening or partial replacement of the media by hyalinized connective tissue.
Cavernous malformations consist of distended, loosely organized vascular channels with thin collagenized walls without intervening nervous tissue. They occur most often in the cerebellum, pons, and subcortical regions, and have a low blood flow without significant arteriovenous shunting. Foci of old hemorrhage, infarction, and calcification frequently surround the abnormal vessels.
Capillary telangiectasias are microscopic foci of dilated thin-walled vascular channels separated by relatively normal brain parenchyma that occur most frequently in the pons. Venous angiomas (varices) consist of aggregates of ectatic venous channels. These latter two types of vascular malformation are unlikely to bleed or to cause symptoms, and most are incidental findings.
Other Vascular Diseases
Hypertensive Cerebrovascular Disease
Hypertension causes hyaline arteriolar sclerosis of the deep penetrating arteries and arterioles that supply the basal ganglia, the hemispheric white matter, and the brain stem. Affected arteriolar walls are weakened and are more vulnerable to rupture. In some instances, minute aneurysms (Charcot-Bouchard microaneurysms) form in vessels less than 300 µm in diameter. In addition to massive intracerebral hemorrhage (discussed earlier), several other pathologic brain processes are related to hypertension.
• Lacunes or lacunar infarcts are small cavitary infarcts, just a few millimeters in size, found most commonly in the deep gray matter (basal ganglia and thalamus), the internal capsule, the deep white matter, and the pons. They are caused by occlusion of a single penetrating branch of a large cerebral artery. Depending on their location, lacunes can be silent clinically or cause significant neurologic impairment.
• Rupture of the small-caliber penetrating vessels may occur, leading to the development of small hemorrhages. In time, these hemorrhages resorb, leaving behind a slitlike cavity (slit hemorrhage) surrounded by brownish discoloration.
• Acute hypertensive encephalopathy most often is associated with sudden sustained rises in diastolic blood pressure to greater than 130 mm Hg. It is characterized by increased intracranial pressure and global cerebral dysfunction, manifesting as headaches, confusion, vomiting, convulsions, and sometimes coma. Rapid therapeutic intervention to reduce the intracranial pressure is essential. Postmortem examination may show brain edema, with or without transtentorial or tonsillar herniation. Petechiae and fibrinoid necrosis of arterioles in the gray and white matter may be seen microscopically.
Vasculitis
A variety of inflammatory processes involving blood vessels may compromise blood flow and cause cerebral infarction. Infectious arteritis of small and large vessels was previously seen mainly in association with syphilis and tuberculosis, but is now more often caused by opportunistic infections (such as aspergillosis, herpes zoster, or CMV) arising in the setting of immunosuppression. Some systemic forms of vasculitis, such as polyarteritis nodosa, may involve cerebral vessels and cause single or multiple infarcts throughout the brain. Primary angiitis of the CNS is a form of vasculitis involving multiple small to medium-sized parenchymal and subarachnoid vessels that is characterized by chronic inflammation, multinucleate giant cells (with or without granuloma formation), and destruction of vessel walls. Affected persons present with a diffuse encephalopathy, often with cognitive dysfunction. Treatment consists of an appropriate regimen of immunosuppressive agents.
Summary
Cerebrovascular Diseases
• Stroke is the clinical term for acute-onset neurologic deficits resulting from hemorrhagic or obstructive vascular lesions.
• Cerebral infarction follows loss of blood supply and can be widespread or focal, or affect regions with the least robust vascular supply (“watershed” infarcts).
• Focal cerebral infarcts are most commonly embolic; with subsequent dissolution of an embolism and reperfusion, a nonhemorrhagic infarct can become hemorrhagic.
• Primary intraparenchymal hemorrhages typically are due to either hypertension (most commonly in white matter, deep gray matter, or posterior fossa contents) or cerebral amyloid angiopathy.
• Spontaneous subarachnoid hemorrhage usually is caused by a structural vascular abnormality, such as an aneurysm or arteriovenous malformation.
Central Nervous System Trauma
Trauma to the brain and spinal cord is a significant cause of death and disability. The severity and site of injury affect the outcome: injury of several cubic centimeters of brain parenchyma may be clinically silent (if in the frontal lobe), severely disabling (spinal cord), or fatal (involving the brain stem).
A blow to the head may be penetrating or blunt; it may cause an open or a closed injury. The magnitude and distribution of resulting traumatic brain lesions depend on the shape of the object causing the trauma, the force of impact, and whether the head is in motion at the time of injury. Severe brain damage can occur in the absence of external signs of head injury, and conversely, severe lacerations and even skull fractures do not necessarily indicate damage to the underlying brain. When the brain is damaged, the injuries may involve the parenchyma, the vasculature, or both.
Recent evidence suggests that repetitive episodes of trauma (such as occurs in athletes participating in contact sports) can lead to later development of neurodegenerative processes. In addition to a long-recognized association of trauma with the risk of Alzheimer disease, a distinct form of trauma-associated degeneration has been described, chronic traumatic encephalopathy, which is characterized by a unique pattern of intraneuronal tau protein inclusions (described later).
Traumatic Parenchymal Injuries
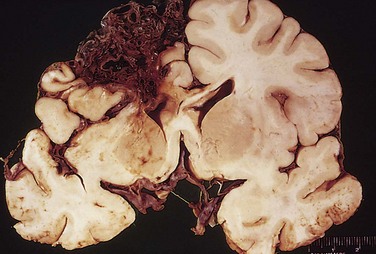
When an object impacts the head, brain injury may occur at the site of impact—a coup injury—or opposite the site of impact on the other side of the brain—a contrecoup injury. Both coup and contrecoup lesions are contusions, with comparable gross and microscopic appearances. A contusion is caused by rapid tissue displacement, disruption of vascular channels, and subsequent hemorrhage, tissue injury, and edema. Since they are closest to the skull, the crests of the gyri are the part of the brain that is most susceptible to traumatic injury. Contusions are common in regions of the brain overlying rough and irregular inner skull surfaces, such as the orbitofrontal regions and the temporal lobe tips. Penetration of the brain by a projectile such as a bullet or a skull fragment from a fracture causes a laceration, with tissue tearing, vascular disruption, and hemorrhage.
Morphology
On cross-section, contusions are wedge-shaped, with the widest aspect closest to the point of impact (Fig. 22–12, A). Within a few hours of injury, blood extravasates throughout the involved tissue, across the width of the cerebral cortex, and into the white matter and subarachnoid spaces. Although functional effects are seen earlier, morphologic evidence of injury in the neuronal cell body (nuclear pyknosis, cytoplasmic eosinophilia, cellular disintegration) takes about 24 hours to appear. The inflammatory response to the injured tissue follows its usual course, with neutrophils preceding the appearance of macrophages. In contrast with ischemic lesions, in which the superficial layer of cortex may be preserved, trauma affects the superficial layers most severely.
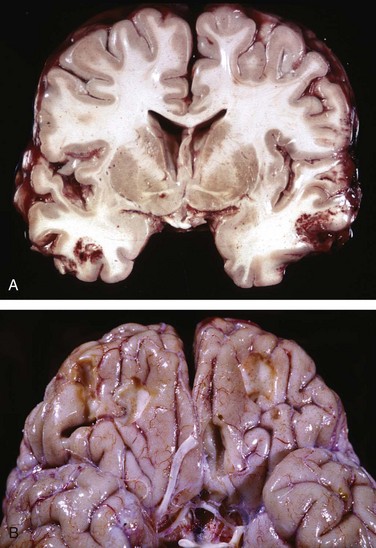
Figure 22–12 Cerebral trauma. A, Acute contusions are present in both temporal lobes, with areas of hemorrhage and tissue disruption. B, Remote contusions, seen as discolored yellow areas, are present on the inferior frontal surface of this brain.
Old traumatic lesions have a characteristic macroscopic appearance: They are depressed, retracted, yellowish brown patches involving the crests of gyri (Fig. 22–12, B). More extensive hemorrhagic regions of brain trauma give rise to larger cavitary lesions, which can resemble remote infarcts. In sites of old contusions, gliosis and residual hemosiderin-laden macrophages predominate.
Although contusions are more easily seen, trauma can also cause more subtle but widespread injury to axons within the brain (called diffuse axonal injury), sometimes with devastating consequences. The movement of one region of brain relative to another is thought to disrupt axonal integrity and function. Angular acceleration, even in the absence of impact, may cause axonal injury as well as hemorrhage. As many as 50% of patients who develop coma shortly after trauma are believed to have white matter damage and diffuse axonal injury. Although these injuries may be widespread, the lesions usually are asymmetric and are most commonly found near the angles of the lateral ventricles and in the brain stem. They take the form of axonal swellings that appear within hours of the injury. These are best demonstrated with silver stains or by immunohistochemical stains for axonal proteins.
Concussion describes reversible altered consciousness from head injury in the absence of contusion. The characteristic transient neurologic dysfunction includes loss of consciousness, temporary respiratory arrest, and loss of reflexes. Although neurologic recovery is complete, amnesia for the event persists. The pathogenesis of the sudden disruption of nervous activity is unknown.
Traumatic Vascular Injury
CNS trauma often directly disrupts vessel walls, leading to hemorrhage. Depending on the affected vessel, the hemorrhage may be epidural, subdural, subarachnoid, or intraparenchymal (Fig. 22–13, A), occurring alone or in combination. Subarachnoid and intraparenchymal hemorrhages most often occur at sites of contusions and lacerations.
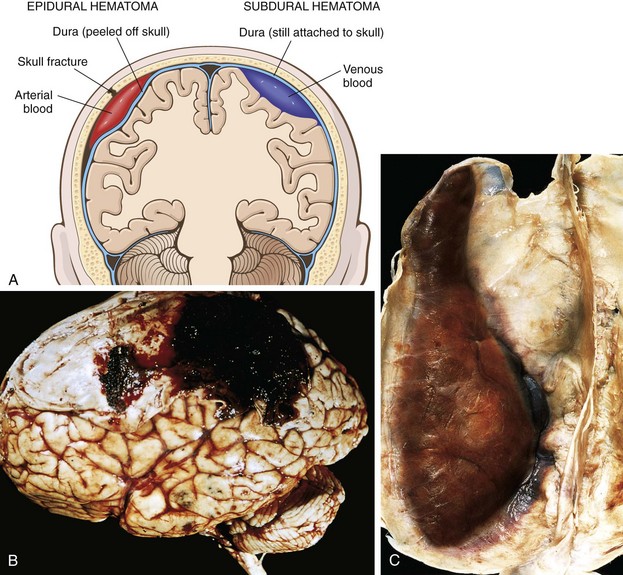
Figure 22–13 Traumatic intracranial hemorrhages. A, Epidural hematoma (left) in which rupture of a meningeal artery, usually associated with a skull fracture, has led to accumulation of arterial blood between the dura and the skull. In a subdural hematoma (right), damage to bridging veins between the brain and the superior sagittal sinus has led to the accumulation of blood between the dura and the arachnoid. B, Epidural hematoma covering a portion of the dura. C, Large organizing subdural hematoma attached to the dura.
(B, Courtesy of the late Dr. Raymond D. Adams, Massachusetts General Hospital, Boston, Massachusetts.)
Epidural Hematoma
Dural vessels—especially the middle meningeal artery—are vulnerable to traumatic injury. In infants, traumatic displacement of the easily deformable skull may tear a vessel, even in the absence of a skull fracture. In children and adults, by contrast, tears involving dural vessels almost always stem from skull fractures. Once a vessel is torn, blood accumulating under arterial pressure can dissect the tightly applied dura away from the inner skull surface (Fig. 22–13, B), producing a hematoma that compresses the brain surface. Clinically, patients can be lucid for several hours between the moment of trauma and the development of neurologic signs. An epidural hematoma may expand rapidly and constitutes a neurosurgical emergency necessitating prompt drainage and repair to prevent death.
Subdural Hematoma
Rapid movement of the brain during trauma can tear the bridging veins that extend from the cerebral hemispheres through the subarachnoid and subdural space to the dural sinuses. Their disruption produces bleeding into the subdural space. In patients with brain atrophy, the bridging veins are stretched out, and the brain has additional space within which to move, accounting for the higher rate of subdural hematomas in elderly persons. Infants also are susceptible to subdural hematomas because their bridging veins are thin-walled.
Subdural hematomas typically become manifest within the first 48 hours after injury. They are most common over the lateral aspects of the cerebral hemispheres and may be bilateral. Neurologic signs are attributable to the pressure exerted on the adjacent brain. Symptoms may be localizing but more often are nonlocalizing, taking the form of headache, confusion, and slowly progressive neurologic deterioration.
Morphology
An acute subdural hematoma appears as a collection of freshly clotted blood apposed to the contour of the brain surface, without extension into the depths of sulci (Fig. 22–13, C). The underlying brain is flattened, and the subarachnoid space is often clear. Typically, venous bleeding is self-limited; breakdown and organization of the hematoma take place over time. Subdural hematomas organize by lysis of the clot (about 1 week), growth of granulation tissue from the dural surface into the hematoma (2 weeks), and fibrosis (1 to 3 months). Organized hematomas are attached to the dura, but not to the underlying arachnoid. Fibrosing lesions may eventually retract, leaving only a thin layer of connective tissue (“subdural membranes”). Subdural hematomas commonly rebleed (resulting in chronic subdural hematomas), presumably from the thin-walled vessels of the granulation tissue, leading to microscopic findings consistent with hemorrhages of varying age. Symptomatic subdural hematomas are treated by surgical removal of the blood and associated reactive tissue.
Summary
Central Nervous System Trauma
• Physical injury to the brain can occur when the inside of the skull comes into forceful contact with the brain.
• In blunt trauma, if the head is mobile there may be brain injury both at the original point of contact (coup injury) and on the opposite side of the brain (contrecoup injury) owing to impacts with the skull.
• Rapid displacement of the head and brain can tear axons (diffuse axonal injury), often causing immediate severe, irreversible neurologic deficits.
• Traumatic tearing of blood vessels leads to epidural hematoma, subdural hematoma, or subarachnoid hemorrhage.
Congenital Malformations and Perinatal Brain Injury
The incidence of CNS malformations, giving rise to mental retardation, cerebral palsy, or neural tube defects, is estimated at 1% to 2%. Malformations of the brain are more common in the setting of multiple birth defects. Prenatal or perinatal insults may either interfere with normal CNS development or cause tissue damage. Since different parts of the brain develop at different times during gestation, the timing of an injury will be reflected in the pattern of malformation; earlier events typically lead to more severe phenotypes. Mutations affecting genes that regulate the differentiation, maturation, or intercellular communication of neurons or glial cells can cause CNS malformation or dysfunction. Additionally, various chemicals and infectious agents have teratogenic effects.
Not all developmental disorders are characterized by specific, recognizable gross or microscopic findings, yet such disorders may nevertheless be associated with profound neuronal dysfunction. Genetic underpinnings for various forms of autism have emerged recently; many of the implicated genes contribute to the development or maintenance of synaptic connections. Similarly, Rett syndrome is an X-linked dominant disorder associated with mutations in the gene encoding methyl-CpG–binding protein-2 (MeCP2), a regulator of epigenetic modifications of chromatin. Development in affected girls initially is normal, but neurologic deficits affecting cognition and movement appear by the age of 1 to 2 years, highlighting the importance of epigenetic processes in neuronal development and synaptic plasticity.
Malformations
Neural Tube Defects
On of the earliest steps in brain development is the formation of the neural tube, which gives rise to the ventricular system, brain and spinal cord. Partial failure or reversal of neural tube closure may lead to one of several malformations, each characterized by abnormalities involving some combination of neural tissue, meninges, and overlying bone or soft tissues. Collectively, neural tube defects constitute the most frequent type of CNS malformation. The overall recurrence risk in subsequent pregnancies is 4% to 5%, suggesting a genetic component. Folate deficiency during the initial weeks of gestation also increases risk through uncertain mechanisms; of clinical importance, prenatal vitamins containing folate can reduce the risk of neural tube defects by up to 70%. The combination of imaging studies and maternal screening for elevated α-fetoprotein has increased the early detection of neural tube defects.
The most common defects involve the posterior end of the neural tube, from which the spinal cord forms. These can range from asymptomatic bony defects (spina bifida occulta) to severe malformation consisting of a flat, disorganized segment of spinal cord associated with an overlying meningeal outpouching. Myelomeningocele is an extension of CNS tissue through a defect in the vertebral column that occurs most commonly in the lumbosacral region (Fig. 22–14). Patients have motor and sensory deficits in the lower extremities and problems with bowel and bladder control. The clinical problems derive from the abnormal spinal cord segment and often are compounded by infections extending from the thin or ulcerated overlying skin.
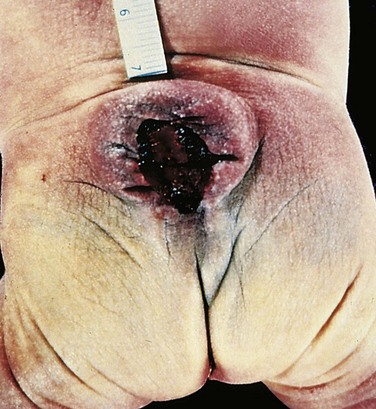
Figure 22–14 Myelomeningocele. Both meninges and spinal cord parenchyma are included in the cystlike structure visible just above the buttocks.
At the other end of the developing CNS, anencephaly is a malformation of the anterior end of the neural tube that leads to the absence of the brain and the top of skull. An encephalocele is a diverticulum of malformed CNS tissue extending through a defect in the cranium. It most often involves the occipital region or the posterior fossa. When it occurs anteriorly, brain tissue can extend into the sinuses.
Forebrain Malformations
In certain malformations, the volume of the brain is abnormally large (megalencephaly) or small (microencephaly). Microencephaly, by far the more common of the two, usually is associated with a small head as well (microcephaly). It has a wide range of associations, including chromosome abnormalities, fetal alcohol syndrome, and human immunodeficiency virus type 1 (HIV-1) infection acquired in utero. The unifying feature is decreased generation of neurons destined for the cerebral cortex. During the early stages of brain development, as progenitor cells proliferate in the subependymal zone, the balance between cells leaving the progenitor population to begin migration to the cortex and those remaining in the proliferating pool affects the overall number of neurons and glial cells generated. If too many cells leave the progenitor pool prematurely, there is inadequate generation of mature neurons, leading to a small brain.
Disruption of neuronal migration and differentiation during development can lead to abnormalities of gyration and the six-layered neocortical architecture, often taking the form of neurons ending up in the wrong anatomic location. Various mutations in genes that control migration result in these malformations, which include the following:
• Lissencephaly (agyria) or, with more patchy involvement, pachygyria, is characterized by absent gyration leading to a smooth-surfaced brain. The cortex is abnormally thickened and usually has only four layers. Many forms of lissencephaly are associated with defects in genes that control neuronal migration.
• Polymicrogyria is characterized by an increased number of irregularly formed gyri that result in a bumpy or cobblestone-like surface. These changes can be focal or widespread. The normal cortical architecture can be altered in various ways, and adjacent gyri often show fusion of the superficial molecular layer.
• Holoprosencephaly is characterized by a disruption of the normal midline patterning. Mild forms may just show absence of the olfactory bulbs and related structures (arrhinencephaly). In severe forms the brain is not divided into hemispheres or lobes, and this anomaly may be associated with facial midline defects such as cyclopia. Holoprosencephaly as well as polymicrogyria can be the result of acquired or genetically determined disruption of normal development. Several single-gene defects including mutations in sonic hedgehog have been linked to holoprosencephaly.
• Other examples are focally disordered cortex (confusingly called dysplastic cortex) and neurons stranded beneath the cortex, sometimes as nodules and other times as bands.
Posterior Fossa Anomalies
The most common malformations in this region of the brain result in misplacement or absence of portions of the cerebellum. The Arnold-Chiari malformation (Chiari type II malformation) combines a small posterior fossa with a misshapen midline cerebellum and downward extension of the vermis through the foramen magnum; hydrocephalus and a lumbar myelomeningocele typically are also present. The far milder Chiari type I malformation has low-lying cerebellar tonsils that extend through the foramen magnum. Excess tissue in the foramen magnum results in partial obstruction of CSF flow and compression of the medulla, with symptoms of headache or cranial nerve deficits often manifesting only in adult life. Surgical intervention can alleviate the symptoms.
Syndromes characterized by “missing” cerebellar tissue include Dandy-Walker malformation, characterized by an enlarged posterior fossa, absence of the cerebellar vermis, and a large midline cyst, and Joubert syndrome, in which there is absence of the vermis and brain stem abnormalities resulting in eye movement problems and disrupted respiratory patterns. A range of recessive genetic lesions have been found to cause Joubert syndrome, with many involving alterations of the primary cilium.
Spinal Cord Abnormalities
In addition to neural tube defects, structural alterations of the spinal cord can occur that are not associated with abnormalities of the bony spine or overlying skin. These include expansions of the ependyma-lined central canal of the cord (hydromyelia) or development of fluid-filled cleftlike cavities in the inner portion of the cord (syringomyelia, syrinx). These lesions are surrounded by dense reactive gliosis, often with Rosenthal fibers. A syrinx also may develop after trauma or with intramedullary spinal tumors.
Perinatal Brain Injury
A variety of exogenous factors can injure the developing brain. Injuries that occur early in gestation may destroy brain tissue without evoking reactive changes, sometimes making them difficult to distinguish from malformations. Brain injury occurring in the perinatal period is an important cause of childhood neurologic disability. Cerebral palsy is a term for nonprogressive neurologic motor deficits characterized by spasticity, dystonia, ataxia or athetosis, and paresis attributable to injury occurring during the prenatal and perinatal periods. Signs and symptoms may not be apparent at birth and only declare themselves later, well after the causal event.
The two major types of injury that occur in the perinatal period are hemorrhages and infarcts. These differ from the otherwise similar lesions in adults in terms of their locations and the tissue reactions they engender. In premature infants, there is an increased risk of intraparenchymal hemorrhage within the germinal matrix, most often adjacent to the anterior horn of the lateral ventricle. Hemorrhages may extend into the ventricular system and from there to the subarachnoid space, sometimes causing hydrocephalus. Infarcts may occur in the supratentorial periventricular white matter (periventricular leukomalacia), especially in premature babies. The residua of these infarcts are chalky yellow plaques consisting of discrete regions of white matter necrosis and mineralization (Fig. 22–15). When severe enough to involve the gray and white matter, large cystic lesions can develop throughout the hemispheres, a condition termed multicystic encephalopathy.

Figure 22–15 Perinatal brain injury. This specimen from a patient with periventricular leukomalacia contains a central focus of white matter necrosis with a peripheral rim of mineralized axonal processes.
Summary
Congenital Malformations and Perinatal Brain Injury
• Malformations of the brain can occur because of genetic factors or external insults.
• The developmental timing and position of the injury determine its pattern and characteristics.
• Various malformations stem from failure of neural tube closure, improper formation of neural structures, and altered neuronal migration.
• Perinatal brain injury mostly takes one of two forms: (1) hemorrhage, often in the region of the germinal matrix with the risk of extension into the ventricular system; and (2) ischemic infarcts, leading to periventricular leukomalacia.
Infections of the Nervous System
The brain and its coverings, as with all other parts of the body, can be sites of infections. Some infectious agents have a relative or absolute predilection for the nervous system (e.g., rabies), while others can affect many other organs as well as the brain (e.g., Staphylococcus aureus). Damage to nervous tissue may be the consequence of direct injury of neurons or glial cells by the infectious agent or microbial toxins, or may be a consequence of the host innate or adaptive immune response.
Infectious agents may reach the nervous system through several routes of entry:
• Hematogenous spread by way of the arterial blood supply is the most common means of entry. There can also be retrograde venous spread, through the anastomoses between veins of the face and the venous sinuses of the skull.
• Direct implantation of microorganisms is almost invariably due to traumatic introduction of foreign material. In rare cases it can be iatrogenic, as when microbes are introduced with a lumbar puncture needle.
• Local extension can occur with infections of the skull or spine. Sources include air sinuses, most often the mastoid or frontal; infected teeth; cranial or spinal osteomyelitis; and congenital malformations, such as meningomyelocele.
• Peripheral nerves also may serve as paths of entry for a few pathogens—in particular, viruses such as the rabies and herpes zoster viruses.
Epidural and Subdural Infections
The epidural and subdural spaces can be involved by bacterial or fungal infections, usually as a consequence of direct local spread. Epidural abscesses arise from an adjacent focus of infection, such as sinusitis or osteomyelitis. When abscesses occur in the spinal epidural space, they may cause spinal cord compression and constitute a neurosurgical emergency. Infections of the skull or air sinuses may also spread to the subdural space, producing subdural empyema. The underlying arachnoid and subarachnoid spaces usually are unaffected, but a large subdural empyema may produce a mass effect. In addition, thrombophlebitis may develop in the bridging veins that cross the subdural space, resulting in venous occlusion and infarction of the brain. Most patients are febrile, with headache and neck stiffness, and if untreated may develop focal neurologic signs referable to the site of the infection, lethargy, and coma. With treatment, including surgical drainage, resolution of the empyema occurs from the dural side; if resolution is complete, a thickened dura may be the only residual finding. With prompt treatment, complete recovery is usual.
Meningitis
Meningitis is an inflammatory process involving the leptomeninges within the subarachnoid space; if the infection spreads into the underlying brain it is termed meningoencephalitis. Meningitis usually is caused by an infection, but chemical meningitis also may occur in response to a nonbacterial irritant introduced into the subarachnoid space. Infectious meningitis can be broadly divided into acute pyogenic (usually bacterial), aseptic (usually viral), and chronic (usually tuberculous, spirochetal, or cryptococcal) subtypes. Examination of the CSF is often useful in distinguishing between various causes of meningitis.
Acute Pyogenic Meningitis (Bacterial Meningitis)
Many bacteria can cause acute pyogenic meningitis, but the most likely organisms vary with patient age. In neonates, common organisms are Escherichia coli and the group B streptococci; in adolescents and in young adults, Neisseria meningitidis is the most common pathogen; and in older individuals, Streptococcus pneumoniae and Listeria monocytogenes are more common. In all age groups, patients typically show systemic signs of infection along with meningeal irritation and neurologic impairment, including headache, photophobia, irritability, clouding of consciousness, and neck stiffness. Lumbar puncture reveals an increased pressure; examination of the CSF shows abundant neutrophils, elevated protein, and reduced glucose. Bacteria may be seen on a smear or can be cultured, sometimes a few hours before the neutrophils appear. Untreated pyogenic meningitis is often fatal, but with prompt diagnosis and administration of appropriate antibiotics, many patients can be saved.
Morphology
In acute meningitis, an exudate is evident within the leptomeninges over the surface of the brain (Fig. 22–16, A). The meningeal vessels are engorged and prominent. From the areas of greatest accumulation, tracts of pus can be followed along blood vessels on the brain surface. When the meningitis is fulminant, the inflammatory cells infiltrate the walls of the leptomeningeal veins and may spread into the substance of the brain (focal cerebritis), or the inflammation may extend to the ventricles, producing ventriculitis. On microscopic examination, neutrophils fill the entire subarachnoid space in severely affected areas or may be found predominantly around the leptomeningeal blood vessels in less severe cases. In untreated meningitis, Gram stain reveals varying numbers of the causative organism. Bacterial meningitis may be associated with abscesses in the brain (Fig. 22–16, B), discussed later. Phlebitis also may lead to venous occlusion and hemorrhagic infarction of the underlying brain. If it is treated early, there may be little or no morphologic residuum.
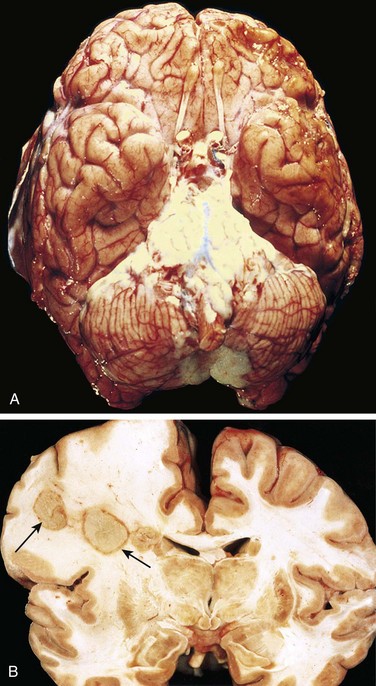
Figure 22–16 Bacterial infections. A, Pyogenic meningitis. A thick layer of suppurative exudate covers the brain stem and cerebellum and thickens the leptomeninges. B, Cerebral abscesses in the frontal lobe white matter (arrows).
(A, From Golden JA, Louis DN: Images in clinical medicine: acute bacterial meningitis. N Engl J Med 333:364, 1994. Copyright © 1994 Massachusetts Medical Society. All rights reserved.)
Aseptic Meningitis (Viral Meningitis)
Aseptic meningitis is a clinical term for an illness comprising meningeal irritation, fever, and alterations in consciousness of relatively acute onset. The clinical course is less fulminant than in pyogenic meningitis. In contrast to pyogenic meningitis, examination of the CSF often shows lymphocytosis, moderate protein elevation, and a normal glucose level. The disease typically is self-limiting. It is believed to be of viral origin in most cases, but it is often difficult to identify the responsible virus. There are no distinctive macroscopic characteristics except for brain swelling, seen in only some instances. On microscopic examination, there is either no recognizable abnormality or a mild to moderate leptomeningeal lymphocytic infiltrate.
Chronic Meningitis
Several pathogens, including mycobacteria and some spirochetes, are associated with chronic meningitis; infections with these organisms also may involve the brain parenchyma.
Tuberculous Meningitis
Tuberculous meningitis usually manifests with generalized signs and symptoms of headache, malaise, mental confusion, and vomiting. There is only a moderate increase in CSF cellularity, with mononuclear cells or a mixture of polymorphonuclear and mononuclear cells; the protein level is elevated, often strikingly so, and the glucose content typically is moderately reduced or normal. Infection with Mycobacterium tuberculosis also may result in a well-circumscribed intraparenchymal mass (tuberculoma), which may be associated with meningitis. Chronic tuberculous meningitis is a cause of arachnoid fibrosis, which may produce hydrocephalus.
Morphology
The subarachnoid space contains a gelatinous or fibrinous exudate, most often at the base of the brain, obliterating the cisterns and encasing cranial nerves. There may be discrete white granules scattered over the leptomeninges. Arteries running through the subarachnoid space may show obliterative endarteritis with inflammatory infiltrates and marked intimal thickening. On microscopic examination there are mixtures of lymphocytes, plasma cells, and macrophages. Florid cases show well-formed granulomas, often with caseous necrosis and giant cells, similar to the lesions of tuberculosis elsewhere.
Spirochetal Infections
Neurosyphilis, a tertiary stage of syphilis, occurs in about 10% of persons with untreated Treponema pallidum infection. Patients with HIV infection are at increased risk for neurosyphilis, which often is more aggressive and severe in this setting. The infection can produce chronic meningitis (meningovascular neurosyphilis), usually involving the base of the brain, often with an obliterative endarteritis rich in plasma cells and lymphocytes. There can also be parenchymal involvement by spirochetes (paretic neurosyphilis), leading to neuronal loss and marked proliferation of rod-shaped microglial cells. Clinically, this form of the disease causes an insidious progressive loss of mental and physical functions, mood alterations (including delusions of grandeur), and eventually severe dementia. Tabes dorsalis is another form of neurosyphilis, resulting from damage to the sensory nerves in the dorsal roots that produces impaired joint position sense and ataxia (locomotor ataxia); loss of pain sensation, leading to skin and joint damage (Charcot joints); other sensory disturbances, particularly characteristic “lightning pains”; and the absence of deep tendon reflexes.
Neuroborreliosis represents involvement of the nervous system by the spirochete Borrelia burgdorferi, the pathogen of Lyme disease. Neurologic signs and symptoms are highly variable and include aseptic meningitis, facial nerve palsies, mild encephalopathy, and polyneuropathies.
Parenchymal Infections
The entire gamut of infectious pathogens (viruses to parasites) can potentially infect the brain, often in characteristic patterns. In general, viral infections are diffuse, bacterial infections (when not associated with meningitis) are localized, while other organisms produce mixed patterns. In immunosuppressed hosts, more widespread involvement with any agent is typical.
Brain Abscesses
Brain abscesses are nearly always caused by bacterial infections. These can arise by direct implantation of organisms, local extension from adjacent foci (mastoiditis, paranasal sinusitis), or hematogenous spread (usually from a primary site in the heart, lungs, or distal bones, or after tooth extraction). Predisposing conditions include acute bacterial endocarditis, from which septic emboli are released that may produce multiple abscesses; cyanotic congenital heart disease, associated with a right-to-left shunt and loss of pulmonary filtration of organisms; and chronic pulmonary infections, as in bronchiectasis, which provide a source of microbes that spread hematogenously.
Abscesses are destructive lesions, and patients almost invariably present with progressive focal deficits as well as general signs related to increased intracranial pressure. The CSF white cell count and protein levels are usually high, while the glucose content tends to be normal. A systemic or local source of infection may be apparent or may have ceased to be symptomatic. The increased intracranial pressure and progressive herniation can be fatal, and abscess rupture can lead to ventriculitis, meningitis, and venous sinus thrombosis. Surgery and antibiotics reduce the otherwise high mortality rate, with earlier intervention leading to better outcomes.
Morphology
Abscesses are discrete lesions with central liquefactive necrosis and a surrounding fibrous capsule (Fig. 22–16, B). On microscopic examination, the necrotic center is surrounded by edema and granulation tissue, often with exuberant vascularization. Outside the fibrous capsule is a zone of reactive gliosis.
Viral Encephalitis
Viral encephalitis is a parenchymal infection of the brain that is almost invariably associated with meningeal inflammation (and therefore is better termed meningoencephalitis). While different viruses may show varying patterns of injury, the most characteristic histologic features are perivascular and parenchymal mononuclear cell infiltrates, microglial nodules, and neuronophagia (Fig. 22–17, A and B). Certain viruses also form characteristic inclusion bodies.
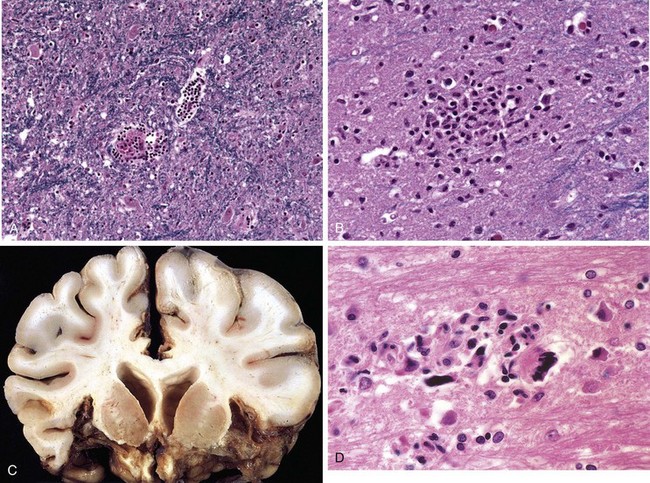
Figure 22–17 Viral infections. A and B, Characteristic findings in many forms of viral meningitis include perivascular cuffing of lymphocytes (A) and microglial nodules (B). C, Herpes encephalitis showing extensive destruction of inferior frontal and anterior temporal lobes. D, Human immunodeficiency virus (HIV) encephalitis. Note the accumulation of microglia forming a microglial nodule and multinucleate giant cell.
(C, Courtesy of Dr. T.W. Smith, University of Massachusetts Medical School, Worcester, Massachusetts.)
The nervous system is particularly susceptible to certain viruses such as rabies virus and poliovirus. Some viruses infect specific CNS cell types, while others preferentially involve particular brain regions (such as the medial temporal lobes, or the limbic system) that lie along the viral route of entry. Intrauterine viral infection may cause congenital malformations, as occurs with rubella. In addition to direct infection of the nervous system, the CNS also can be injured by immune mechanisms after systemic viral infections.
Arboviruses
Arboviruses (arthropod-borne viruses) are an important cause of epidemic encephalitis, especially in tropical regions of the world, and are capable of causing serious morbidity and high mortality. Among the more commonly encountered types are Eastern and Western equine encephalitis and West Nile virus infection. Patients develop generalized neurologic symptoms, such as seizures, confusion, delirium, and stupor or coma, as well as focal signs, such as reflex asymmetry and ocular palsies. The CSF usually is colorless but with a slightly elevated pressure and an early neutrophilic pleocytosis that rapidly converts to a lymphocytosis; the protein level is elevated, but the glucose is normal.
Morphology
Arbovirus encephalitides produce a similar histopathologic picture. Characteristically, there is a perivascular lymphocytic meningoencephalitis (sometimes with neutrophils) (Fig. 22–17, A). Multifocal gray and white matter necrosis is seen, often associated with neuronophagia, the phagocytosis of neuronal debris, as well as localized collections of microglia termed microglial nodules (Fig. 22–17, B). In severe cases there may be a necrotizing vasculitis with associated focal hemorrhages.
Herpesviruses
HSV-1 encephalitis may occur in any age group but is most common in children and young adults. It typically manifests with alterations in mood, memory, and behavior, reflecting involvement of the frontal and temporal lobes. Recurrent HSV-1 encephalitis is sometimes associated with inherited mutations that interfere with Toll-like receptor signaling (specifically that of TLR-3), which has an important role in antiviral defense.
Morphology
Herpes encephalitis starts in, and most severely involves, the inferior and medial regions of the temporal lobes and the orbital gyri of the frontal lobes (Fig. 22–17, C). The infection is necrotizing and often hemorrhagic in the most severely affected regions. Perivascular inflammatory infiltrates usually are present, and large eosinophilic intranuclear viral inclusions (Cowdry type A bodies) can be found in both neurons and glial cells.
HSV-2 also affects the nervous system, usually in the form of meningitis in adults. Disseminated severe encephalitis occurs in many neonates born by vaginal delivery to women with active primary HSV genital infections.
Varicella-zoster virus (VZV) causes chickenpox during primary infection, usually without any evidence of neurologic involvement. The virus establishes latent infection in neurons of dorsal root ganglia. Reactivation in adults manifests as a painful, vesicular skin eruption in the distribution of one or a few dermatomes (shingles). This usually is a self-limited process, but there may be a persistent pain syndrome in the affected region (postherpetic neuralgia). VZV also may cause a granulomatous arteritis that can lead to tissue infarcts. In immunosuppressed patients, acute herpes zoster encephalitis can occur. Inclusion bodies can be found in glial cells and neurons.
Cytomegalovirus
CMV infects the nervous system in fetuses and immunosuppressed persons. All cells within the CNS (neurons, glial cells, ependyma, and endothelium) are susceptible to infection. Intrauterine infection causes periventricular necrosis, followed later by microcephaly with periventricular calcification. When adults are infected, CMV produces a subacute encephalitis, again often most severe in the periventricular region. Lesions can be hemorrhagic and contain typical viral inclusion–bearing cells.
Poliovirus
Poliovirus is an enterovirus that most often causes a subclinical or mild gastroenteritis; in a small fraction of cases, it secondarily invades the nervous system and damages motor neurons in the spinal cord and brain stem (paralytic poliomyelitis). With loss of motor neurons, it produces a flaccid paralysis with muscle wasting and hyporeflexia in the corresponding region of the body. In the acute disease, death can occur from paralysis of respiratory muscles. Long after the infection has resolved, typically 25 to 35 years after the initial illness, a postpolio syndrome of progressive weakness associated with decreased muscle bulk and pain can appear. The cause of this syndrome is unclear. One hypothesis is that motor neurons that survive the initial insult sprout new nerve terminals to compensate for the death of their neighbors, and that over time the additional demands placed on these neurons leads to injury that diminishes function or causes cell death.
Rabies Virus
Rabies is a severe encephalitic infection transmitted to humans from rabid animals, usually by a bite. Various mammals are natural reservoirs. Exposure to some bat species, even without evidence of a bite, is also a risk factor. Virus enters the CNS by ascending along the peripheral nerves from the wound site, so the incubation period depends on the distance between the wound and the brain, usually taking a few months. The disease manifests initially with nonspecific symptoms of malaise, headache, and fever. As the infection advances, the patient shows extraordinary CNS excitability; the slightest touch is painful, with violent motor responses progressing to convulsions. Contracture of the pharyngeal musculature may create an aversion to swallowing even water (hydrophobia). Periods of mania and stupor progress to coma and eventually death, typically from respiratory failure.
Human Immunodeficiency Virus
In the first 15 years or so after recognition of AIDS, neuropathologic changes were demonstrated at postmortem examination in as many as 80% to 90% of cases, owing to direct effects of virus on the nervous system, opportunistic infections, and primary CNS lymphoma. Introduction of highly active antiretroviral therapy (HAART) has decreased the frequency of these secondary effects of HIV infection. However, cognitive dysfunction ranging from mild to full-blown dementia that is lumped under the umbrella term HIV-associated neurocognitive disorder (HAND) continues to be a source of morbidity. The cognitive symptoms are believed to stem from HIV infection of microglial cells in the brain. This leads to activation of innate immune responses, both in infected microglial cells and unaffected bystanders. The ensuing neuronal injury likely stems from a combination of cytokine-induced inflammation and toxic effects of HIV-derived proteins.
Aseptic meningitis occurs within 1 to 2 weeks of onset of primary infection by HIV in about 10% of patients; antibodies to HIV can be demonstrated, and the virus can be isolated from the CSF. The few neuropathologic studies of the early and acute phases of symptomatic or asymptomatic HIV invasion of the nervous system have shown mild lymphocytic meningitis, perivascular inflammation, and some myelin loss in the hemispheres. After the acute phase, an HIV encephalitis (HIVE) commonly can be found if affected persons come to autopsy.
Morphology
HIV encephalitis is best characterized microscopically as a chronic inflammatory reaction with widely distributed infiltrates of microglial nodules, sometimes with associated foci of tissue necrosis and reactive gliosis (Fig. 22–17, D). The microglial nodules also are found in the vicinity of small blood vessels, which show abnormally prominent endothelial cells and perivascular foamy or pigment-laden macrophages. These changes occur especially in the subcortical white matter, diencephalon, and brain stem. An important component of the microglial nodule is the macrophage-derived multinucleate giant cell. In some cases, there is also a disorder of white matter characterized by multifocal or diffuse areas of myelin pallor with associated axonal swellings and gliosis. HIV is present in CD4+ mononuclear and multinucleate macrophages and microglia.
Polyomavirus and Progressive Multifocal Leukoencephalopathy
Progressive multifocal leukoencephalopathy (PML) is caused by JC virus, a polyomavirus, which preferentially infects oligodendrocytes, resulting in demyelination as these cells are injured and then die. Most people show serologic evidence of exposure to JC virus during childhood, and it is believed that PML results from virus reactivation, as the disease is restricted to immunosuppressed persons. Patients develop focal and relentlessly progressive neurologic symptoms and signs, and imaging studies show extensive, often multifocal, ring-enhancing lesions in the hemispheric or cerebellar white matter.
Morphology
The lesions are patchy, irregular, ill-defined areas of white matter destruction that enlarge as the disease progresses (Fig. 22–18). Each lesion is an area of demyelination, in the center of which are scattered lipid-laden macrophages and a reduced number of axons. At the edges of the lesion are greatly enlarged oligodendrocyte nuclei whose chromatin is replaced by glassy-appearing amphophilic viral inclusions. The virus also infects astrocytes, leading to bizarre giant forms with irregular, hyperchromatic, sometimes multiple nuclei that can be mistaken for tumor.
Fungal Encephalitis
Fungal infections usually produce parenchymal granulomas or abscesses, often associated with meningitis. The most common fungal infections have distinctive patterns:
• Candida albicans usually produces multiple microabscesses, with or without granuloma formation.
• Mucormycosis is the term used to describe rhinocerebral infections caused by several fungi belonging to the order Mucorales. It typically presents as an infection of the nasal cavity or sinuses of a diabetic patient with ketoacidosis. It may spread to the brain through vascular invasion or by direct extension through the cribriform plate. The proclivity of Mucor to invade the brain directly sets it apart from other fungi, which tend to reach the brain by hematogenous dissemination from distant sites.
• Aspergillus fumigatus tends to cause a distinctive pattern of widespread septic hemorrhagic infarctions because of its marked predilection for blood vessel wall invasion and subsequent thrombosis.
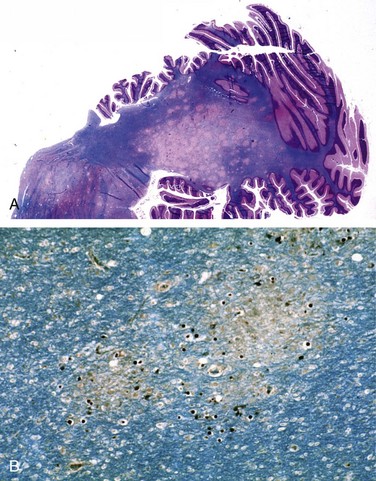
• Cryptococcus neoformans can cause both meningitis and meningoencephalitis, often in the setting of immunosuppression. It can be fulminant and fatal in as little as 2 weeks or may exhibit indolent behavior, evolving over months or years. The CSF may have few cells but elevated protein, and the mucoid encapsulated yeasts can be visualized on India ink preparations. Extension into the brain follows vessels in the Virchow-Robin spaces. As organisms proliferate, these spaces expand, giving rise to a “soap bubble”–like appearance (Fig. 22–19). The diagnosis is usually established by a positive test for cryptococcal antigens in the CSF or the blood.
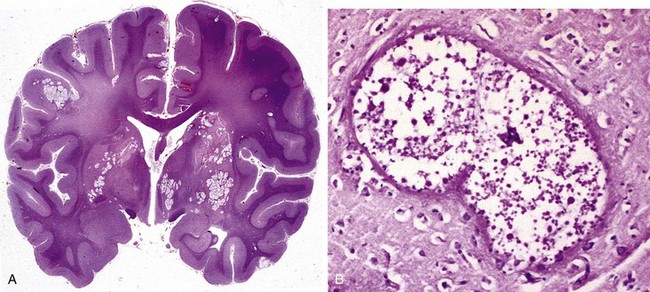
Figure 22–19 Cryptococcal infection. A, Whole-brain section showing the numerous areas of tissue destruction associated with the spread of organisms in the perivascular spaces. B, At higher magnification, it is possible to see the cryptococci in the lesions.
In endemic areas, Histoplasma capsulatum, Coccidioides immitis, and Blastomyces dermatitidis also can infect the CNS, especially in the setting of immunosuppression.
Other Meningoencephalitides
While a wide range of other organisms can infect the nervous system and its covering, only three specific entities are considered here.
Cerebral Toxoplasmosis
Cerebral infection with the protozoan Toxoplasma gondii can occur in immunosuppressed adults or in newborns who acquire the organism transplacentally from a mother with an active infection. In adults, the clinical symptoms are subacute, evolving during a 1- or 2-week period, and may be both focal and diffuse. Due to inflammation and breakdown of the blood-brain barrier at sites of infection, computed tomography and magnetic resonance imaging studies often show edema around lesions (so-called ring enhancing lesions). In newborns who are infected in utero, the infection classically produces the triad of chorioretinitis, hydrocephalus, and intracranial calcifications. Understandably, the CNS abnormalities are most severe when the infection occurs early in gestation during critical stages of brain development. Necrosis of periventricular lesions gives rise to secondary calcifications as well as inflammation and gliosis, which can lead to obstruction of the aqueduct of Sylvius and hydrocephalus.
Morphology
When the infection is acquired in immunosuppressed adults, the brain shows abscesses, frequently multiple, most often involving the cerebral cortex (near the gray-white junction) and deep gray nuclei. Acute lesions consist of central foci of necrosis with variable petechiae surrounded by acute and chronic inflammation, macrophage infiltration, and vascular proliferation. Both free tachyzoites and encysted bradyzoites may be found at the periphery of the necrotic foci (Fig. 22–20).
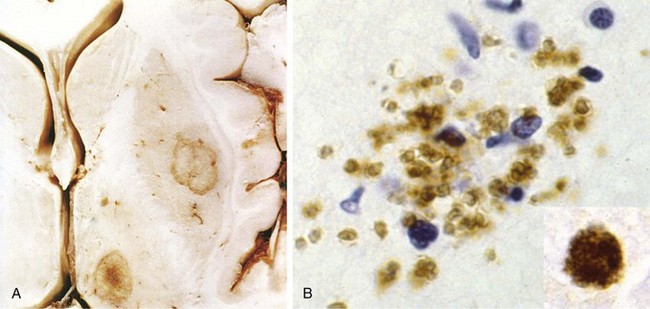
Cysticercosis
Cysticercosis is the consequence of an end-stage infection by the tapeworm Tenia solium. If ingested larval organisms leave the lumen of the gastrointestinal tract, where they would otherwise develop into mature tapeworms, they encyst. Cysts can be found throughout the body but are common within the brain and subarachnoid space. Cysticercosis typically manifests as a mass lesion and can cause seizures. Symptoms can intensify when the encysted organism dies, as happens after therapy.
The organism is found within a cyst with a smooth lining. The body wall and hooklets from mouth parts are most commonly recognized. If the encysted organism has died, there can be an intense inflammatory infiltrate in the surrounding brain, often including eosinophils, which may be associated with marked gliosis.
Amebiasis
Amebic meningoencephalitis manifests with different clinical syndromes, depending on the responsible pathogen. Naegleria spp., associated with swimming in nonflowing warm fresh water, cause a rapidly fatal necrotizing encephalitis. By contrast, Acanthamoeba causes a chronic granulomatous meningoencephalitis.
Prion Diseases
Prion diseases are a group of rare but fascinating disorders that include sporadic, familial, iatrogenic, and variant forms of Creutzfeldt-Jakob disease (CJD), as well as animal diseases such as scrapie in sheep and bovine spongiform encephalopathy in cattle (“mad cow disease”). Unlike in other infectious diseases, the agent in prion diseases is an abnormal form of a cellular protein. This protein, termed prion protein (PrP), may undergo a conformational change from its normal shape (PrPc) to an abnormal conformation called PrPsc (sc for scrapie). PrP normally is rich in α-helices, but PrPsc has a high content of β-sheets, a characteristic that makes it resistant to proteolysis (hence an alternative term for the pathogenic form, PrPres—i.e., protease-resistant). More important, when PrPsc physically interacts with PrP molecules it induces them to also adopt the PrPsc conformation (Fig. 22–21), a property that accounts for the “infectious nature” of PrPsc. Over time, this self-amplifying process leads to the accumulation of a high burden of pathogenic PrPsc molecules in the brain. PrPc also may change its conformation spontaneously (but at an extremely low rate), accounting for sporadic cases of prion disease (sCJD). Certain mutations in the gene encoding PrPc (PRNP) accelerate the rate of spontaneous conformational change; these variants are associated with early-onset familial forms of prion disease (fCJD). Accumulation of PrPsc in neural tissue seems to be the cause of cell injury, but the mechanisms underlying the cytopathic changes and eventual neuronal death are still unknown.
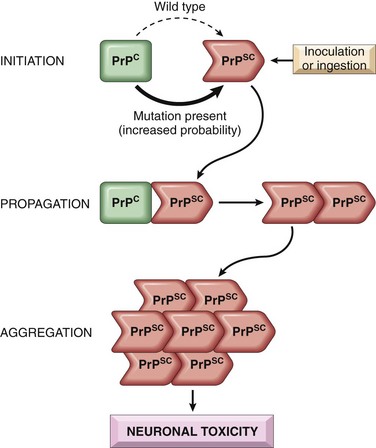
Figure 22–21 Pathogenesis of prion disease. α-Helical PrPc may spontaneously shift to the β-sheet PrPsc conformation, an event that occurs at a much higher rate in familial disease associated with germ line PrP mutations. PrPsc may also be from exogenous sources, such as contaminated food, medical instrumentation, or medicines. Once present, PrPsc converts additional molecules of PrPc into PrPsc through physical interaction, eventually leading to the formation of pathogenic PrPsc aggregates.
Creutzfeldt-Jakob Disease
CJD is a rapidly progressive dementing illness, with a typical duration from first onset of subtle changes in memory and behavior to death in only 7 months. It is sporadic in approximately 85% of cases and has a worldwide annual incidence of about 1 per million. While commonly affecting persons older than 70 years of age, familial forms caused by mutations in PRNP may present in younger people. In keeping with the infectious nature of PrPsc, there are well-established cases of iatrogenic transmission by contaminated deep implantation electrodes and human growth hormone preparations.
Morphology
The progression to death in CJD usually is so rapid that there is little, if any, macroscopic evidence of brain atrophy. On microscopic examination, the pathognomonic finding is a spongiform transformation of the cerebral cortex and deep gray matter structures (caudate, putamen); this multifocal process results in the uneven formation of small, apparently empty, microscopic vacuoles of varying sizes within the neuropil and sometimes in the perikaryon of neurons (Fig. 22–22, A). In advanced cases, there is severe neuronal loss, reactive gliosis, and sometimes expansion of the vacuolated areas into cystlike spaces (“status spongiosus”). No inflammatory infiltrate is present. Immunohistochemical staining demonstrates the presence of proteinase K–resistant PrPsc in tissue, while western blotting of tissue extracts after partial protease digestion allows detection of diagnostic PrPsc.
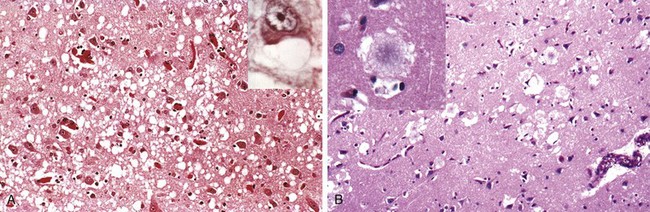
Figure 22–22 Prion disease. A, Histologic features of Creutzfeldt-Jakob disease (CJD) include spongiform change in the cerebral cortex. Inset, High magnification of neuron with vacuoles. B, Variant CJD (vCJD) is characterized by amyloid plaques (see inset) that sit in the regions of greatest spongiform change.
Variant Creutzfeldt-Jakob Disease
Starting in 1995, cases of a CJD-like illness appeared in the United Kingdom. The neuropathologic findings and molecular features of these new cases were similar to those of CJD, suggesting a close relationship between the two illnesses, yet this new disorder differed from typical CJD in several important respects: The disease affected young adults, behavioral disorders figured prominently in early disease stages, and the neurologic syndrome progressed more slowly than in other forms of CJD. Multiple lines of evidence indicate that this new disease, termed variant Creutzfeldt-Jakob disease (vCJD) is a consequence of exposure to the prion disease of cattle, called bovine spongiform encephalopathy. There has now also been documentation of transmission by blood transfusion. This variant form has a similar pathologic appearance to that in other types of CJD, with spongiform change and absence of inflammation. In vCJD, however, there are abundant cortical amyloid plaques, surrounded by the spongiform change (Fig. 22–22, B).
Summary
Infections of the Nervous System
• Pathogens from viruses through parasites can infect the brain; in addition, prion disease is a protein-induced transmissible disease unique to the nervous system.
• Different pathogens use distinct routes to reach the brain, and cause different patterns of disease.
• Bacterial infections may cause meningitis, cerebral abscesses, or a chronic meningoencephalitis.
• Viral infections can cause meningitis or meningoencephalitis.
• HIV can directly cause meningoencephalitis, or indirectly affect the brain by increasing the risk of opportunistic infections (toxoplasmosis, CMV) or CNS lymphoma.
• Prion diseases are transmitted by an altered form of a normal cellular protein. They can be sporadic, transmitted, or inherited.
Primary Diseases of Myelin
Within the CNS, axons are tightly ensheathed by myelin, an electrical insulator that allows rapid propagation of neural impulses. Myelin consists of multiple layers of highly specialized, closely apposed plasma membranes that are assembled by oligodendrocytes. Although myelinated axons are present in all areas of the brain, they are the dominant component in the white matter; therefore, most diseases of myelin are primarily white matter disorders. The myelin in peripheral nerves is similar to the myelin in the CNS but has several important differences: (1) peripheral myelin is made by Schwann cells, not oligodendrocytes; (2) each Schwann cell in a peripheral nerve provides myelin for only one internode, while in the CNS, many internodes are created by processes coming from a single oligodendrocyte; and (3) the specialized proteins and lipids are also different. Therefore, most diseases of CNS myelin do not involve the peripheral nerves to any significant extent, and vice versa.
In general, diseases involving myelin are separated into two broad groups.
• Demyelinating diseases of the CNS are acquired conditions characterized by preferential damage to previously normal myelin. The most common diseases in this group result from immune-mediated injury, such as multiple sclerosis (MS) and related disorders. Other processes that can cause this type of disease include viral infection of oligodendrocytes, as in progressive multifocal leukoencephalopathy (see earlier), and injury caused by drugs and other toxic agents.
• By contrast, in dysmyelinating diseases, myelin is not formed properly or has abnormal turnover kinetics. As would be expected, dysmyelinating diseases are associated with mutations that disrupt the function of proteins that are required for the formation of normal myelin sheaths. The other general term for these diseases is leukodystrophy.
Multiple Sclerosis
MS is an autoimmune demyelinating disorder characterized by distinct episodes of neurologic deficits, separated in time, attributable to white matter lesions that are separated in space. It is the most common of the demyelinating disorders, having a prevalence of approximately 1 per 1000 persons in most of the United States and Europe. The disease may become clinically apparent at any age, although onset in childhood or after age 50 is relatively rare. Women are affected twice as often as men. In most patients with MS, the illness shows relapsing and remitting episodes of neurologic impairment. The frequency of relapses tends to decrease during the course of the illness, but a steady neurologic deterioration is characteristic in a subset of patients.
 Pathogenesis
Pathogenesis
It is believed that MS, like other autoimmune diseases, is caused by a combination of environmental and genetic factors that result in a loss of tolerance to self proteins (in this case, myelin antigens). The nature of the initiating agent, often suggested to be an infectious agent, remains uncertain. Many lines of evidence indicate a significant contribution of genetic factors to the risk of developing MS. The disease risk is 15-fold higher when the disease is present in a first-degree relative, and the concordance rate for monozygotic twins is approximately 25%, with a much lower rate for dizygotic twins. A significant fraction of the genetic risk for MS is attributable to HLA-DR variants, the DR2 allele being the one that most significantly increases the risk for developing MS. Many other genetic polymorphisms have been linked to the disease by genome-wide association studies. Two that have received considerable recent interest are polymorphisms in the genes encoding receptors for the cytokines IL-2 and IL-7, which are known to control the activation and regulation of T cell–mediated immune responses.
In view of the prominence of chronic inflammatory cells within and around MS plaques as well as the genetic evidence, immune-mediated myelin destruction is thought to have a central role in MS. Evidence from human studies as well as from experimental allergic encephalomyelitis—an animal model of MS in which demyelination and inflammation occur after immunization with myelin, myelin proteins, or certain peptides from myelin proteins—has suggested that a range of immune cells contribute to lesion development in MS. A central role for CD4+ T cells has been suggested, with an increase in TH17 and TH1 CD4+ cells thought to be a critical component of the injury to myelin. There is also evidence for important contributions from CD8+ T cells and B cells. While MS is characterized by the presence of demyelination out of proportion to axonal loss, some injury to axons does occur. Toxic effects of lymphocytes, macrophages, and their secreted molecules have been implicated in initiating the process of axonal injury, sometimes even leading to neuronal death.
Morphology
MS is primarily a white matter disease with affected areas showing multiple, well-circumscribed, slightly depressed, glassy-appearing, gray-tan, irregularly shaped lesions termed plaques (Fig. 22–23, A). These commonly arise near the ventricles. They also are frequent in the optic nerves and chiasm, brain stem, ascending and descending fiber tracts, cerebellum, and spinal cord. The lesions have sharply defined borders at the microscopic level (Fig. 22–23, B). In an active plaque there is evidence of ongoing myelin breakdown with abundant macrophages containing myelin debris. Lymphocytes and macrophages are present, mostly as perivascular cuffs. Small active lesions often are centered on small veins. Axons are relatively preserved but may be reduced in number.
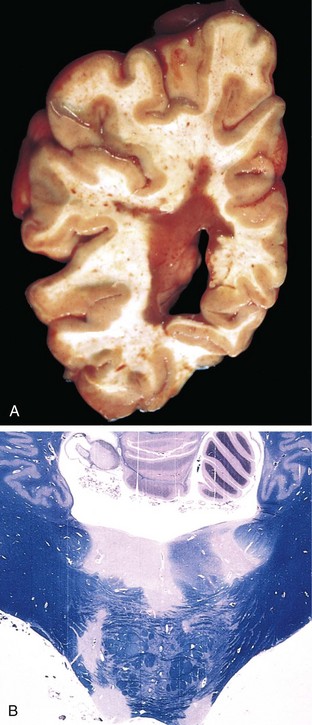
Figure 22–23 Multiple sclerosis (MS). A, Section of fresh brain showing a plaque around occipital horn of the lateral ventricle. B, Unstained regions of demyelination (MS plaques) around the fourth ventricle. Luxol fast blue–periodic acid–Schiff stain for myelin.
Active plaques fall into four classes, only one of which typically is seen in a particular affected patient. The recognized microscopic patterns are type I, which has macrophage infiltrates with sharp margins; type II, which is similar to type I but also shows complement deposition (suggesting an antibody-mediated component); type III, with less well-defined borders and oligodendrocyte apoptosis; and type IV, with nonapoptotic oligodendrocyte loss. When plaques become quiescent (inactive plaques), the inflammation mostly disappears, leaving behind little to no myelin. Instead, astrocytic proliferation and gliosis are prominent.
Clinical Features
The course of MS is variable, but commonly there are multiple relapses followed by episodes of remission; typically, recovery during remissions is not complete. As a consequence, over time there is usually a gradual, often stepwise, accumulation of neurologic deficits. Imaging studies have demonstrated that there are often more lesions in the brains of patients with MS than might be expected from the clinical examination, and that lesions can come and go much more often than was previously suspected. Changes in cognitive function can be present, but are often much milder than the other deficits. In any individual patient, it is hard to predict when the next relapse will occur; most current treatments, which are intended to control the immune response, aim at decreasing the rate and severity of relapses rather than recovering lost function.
The CSF in patients with MS shows a mildly elevated protein level with an increased proportion of immunoglobulin; in one third of cases, there is moderate pleocytosis. When the immunoglobulin is examined further, oligoclonal bands usually are identified. These antibodies are directed against a variety of antigenic targets and can be used as markers of disease activity. Although B cells are clearly involved in the pathogenesis of MS, the contribution of these characteristic antibodies to the disease process is unknown.
Other Acquired Demyelinating Diseases
Immune-mediated demyelination can occur after a number of systemic infectious illnesses, including relatively mild viral diseases. These are not thought to be related to direct spread of the infectious agents to the nervous system. Rather, it is believed that immune cells responding to pathogen-associated antigens are cross reactive against myelin antigens, resulting in myelin damage.
There are two general patterns of postinfectious autoimmune reactions to myelin; unlike in MS, they are associated with acute-onset monophasic illnesses. In acute disseminated encephalomyelitis, symptoms typically develop a week or two after an antecedent infection and are nonlocalizing (headache, lethargy, and coma), in contrast with the focal findings of MS. Symptoms progress rapidly, and the illness is fatal in as many as 20% of cases; in the remaining patients, there is complete recovery. Acute necrotizing hemorrhagic encephalomyelitis is a more devastating related disorder, which typically affects young adults and children.
Neuromyelitis optica (NMO) is an inflammatory demyelinating disease centered on the optic nerves and spinal cord. Previously thought to be a form of MS with stereotypic anatomic regions of susceptibility, this is now recognized to be an antibody-mediated autoimmune disorder. Antibodies to the water channel aquaporin-4 are both diagnostic and pathogenic.
Central pontine myelinolysis is a nonimmune process characterized by loss of myelin involving the center of the pons, most often after rapid correction of hyponatremia. The mechanism of oligodendroglial cell injury is uncertain, but it may be related to edema induced by sudden changes in osmotic pressure. It occurs in a variety of clinical settings including alcoholism and severe electrolyte or osmolar imbalance. Although the most characteristic lesion occurs in the pons, similar lesions can be found elsewhere in the brain. Because of the involvement of fibers in the pons carrying signals to motor neurons in the spinal cord, patients often present with rapidly evolving quadriplegia.
As discussed earlier, progressive multifocal leukoencephalopathy (PML) is a demyelinating disease that occurs after reactivation of JC virus in immunosuppressed patients.
Leukodystrophies
Leukodystrophies exemplify inherited dysmyelinating diseases in which the clinical symptoms derive from abnormal myelin synthesis or turnover. Some of these disorders involve lysosomal enzymes, while others involve peroxisomal enzymes; a few are associated with mutations in myelin proteins. Most are of autosomal recessive inheritance, although X-linked diseases also occur (Table 22–1).
Table 22–1 Selected Leukodystrophies
| Metabolic Disorder | Inheritance Mode | Abnormality |
|---|---|---|
| Metachromatic leukodystrophy | AR | Arylsulfatase A deficiency |
| Krabbe disease | AR | Galactocerebroside β-galactosidase deficiency |
| Adrenoleukodystrophy | AR, X | Peroxisomal defects; elevated very-long-chain fatty acids |
| Canavan disease | AR | Aspartoacylase deficiency |
| Pelizaeus-Merzbacher disease | X | Mutations in proteolipid protein |
| Vanishing white matter disease | AR | Translation initiation factor; link to myelin unclear |
| Alexander disease | AR | Mutations in glial fibrillary acidic protein |
AR, autosomal recessive; X, X-linked.
Morphology
Much of the pathologic change of leukodystrophy is found in the white matter, which is diffusely abnormal in color (gray and translucent) and volume (decreased). Early in their course, some diseases may show patchy involvement, while others have a predilection for occipital lobe involvement. In the end, though, nearly all of the white matter usually is affected. With the loss of white matter, the brain becomes atrophic, the ventricles enlarge, and secondary changes can be found in the gray matter. Myelin loss leads to infiltration of macrophages, which often become stuffed with lipid. Some of these diseases also show specific inclusions created by the accumulation of particular lipids.
Clinical Features
Each of the various leukodystrophies has a characteristic clinical presentation, and most can be diagnosed by genetic or biochemical methods. Despite differences in underlying mechanisms, the leukodystrophies share many features because of the common myelin target. Affected children are normal at birth but begin to miss developmental milestones during infancy and childhood. Diffuse involvement of white matter leads to deterioration in motor skills, spasticity, hypotonia, or ataxia. In general, the earlier the age at onset, the more severe the deficiency and clinical course.
Summary
Primary Diseases of Myelin
• Because of the critical role of myelin in nerve conduction, diseases of myelin can lead to widespread and severe neurologic deficits.
• Diseases of myelin can be grouped into demyelinating diseases (in which normal myelin is broken down for inappropriate reasons—often by inflammatory processes), and dysmyelinating diseases (metabolic disorders that include the leukodystrophies in which myelin structure or its turnover is abnormal).
• Multiple sclerosis, an autoimmune demyelinating disease, is the most common disorder of myelin, affecting young adults. It often pursues a relapsing-remitting course, with eventual progressive accumulation of neurologic deficits.
• Other, less common forms of immune-mediated demyelination often follow infections and are more acute illnesses.
Acquired Metabolic and Toxic Disturbances
Toxic and acquired metabolic diseases are relatively common causes of neurologic illnesses. Because of its high metabolic demands, the brain is particularly vulnerable to nutritional diseases and alterations in metabolic state. Surprisingly, even though metabolic alterations might be expected to affect the entire brain uniformly, there can be very distinct clinical presentations because of unique features or requirements of different anatomic regions. A few of the more common types of injury, particularly those with distinct patterns of damage, are discussed here.
Nutritional Diseases
Thiamine Deficiency
In addition to the systemic effects of thiamine deficiency (beriberi), there also may be abrupt onset of confusion, abnormalities in eye movement, and ataxia—a syndrome termed Wernicke encephalopathy. Treatment with thiamine can reverse these deficits. If the acute stages go untreated, they are followed by largely irreversible profound memory disturbances (Korsakoff syndrome). Because the two syndromes are closely linked, the term Wernicke-Korsakoff syndrome is often applied.
The syndrome is particularly common in the setting of chronic alcoholism but also may be encountered in patients with thiamine deficiency resulting from gastric disorders, including carcinoma and chronic gastritis, or from persistent vomiting.
Morphology
Wernicke encephalopathy is characterized by foci of hemorrhage and necrosis, particularly in the mammillary bodies but also adjacent to the ventricles, especially the third and fourth ventricles. Despite the presence of necrosis, there is relative preservation of many of the neurons in these structures. Early lesions show dilated capillaries with prominent endothelial cells and progress to hemorrhage. As the lesions resolve, a cystic space appears along with hemosiderin-laden macrophages. Lesions in the medial dorsal nucleus of the thalamus seem to best correlate with the memory disturbance in Korsakoff syndrome.
Vitamin B12 Deficiency
In addition to pernicious anemia, deficiency of vitamin B12 may lead to neurologic deficits associated with changes in the spinal cord, collectively termed subacute combined degeneration of the spinal cord. As the name implies, both ascending and descending tracts of the spinal cord are affected. Symptoms develop over weeks. Early clinical signs often include slight ataxia and lower extremity numbness and tingling, which can progress to spastic weakness of the lower extremities; sometimes even complete paraplegia ensues. Prompt vitamin replacement therapy produces clinical improvement; however, if paraplegia has developed, recovery is poor.
Metabolic Disorders
Several systemic derangements may produce CNS dysfunction; only those associated with glucose levels and liver dysfunction are considered here.
Hypoglycemia
Since the brain requires glucose as a substrate for energy production, the cellular effects of diminished glucose generally resemble those of global hypoxia. Hippocampal neurons are particularly susceptible to hypoglycemic injury, while cerebellar Purkinje cells are relatively spared. As with anoxia, if the level and duration of hypoglycemia are sufficiently severe, there may be widespread injury to many areas of the brain.
Hyperglycemia
Hyperglycemia is most common in the setting of inadequately controlled diabetes mellitus and can be associated with either ketoacidosis or hyperosmolar coma. Patients develop confusion, stupor, and eventually coma associated with intracellular dehydration caused by the hyperosmolar state. The hyperglycemia must be corrected gradually, because rapid correction can produce severe cerebral edema.
Hepatic Encephalopathy
Decreased hepatic function may be associated with depressed levels of consciousness and sometimes coma. In the early stages, patients exhibit a characteristic “flapping” tremor (asterixis) when extending the arms with palms facing the observer. Elevated levels of ammonia, which the liver normally clears through the urea cycle, in combination with inflammation and hyponatremia, cause the changes in brain function. Because it is only one contributing factor, ammonia levels in symptomatic patients vary widely. Within the CNS, ammonia metabolism occurs only in astrocytes through the action of glutamine synthetase, and in the setting of hyperammonemia, astrocytes in the cortex and basal ganglia develop swollen, pale nuclei (called Alzheimer type II cells).
Toxic Disorders
The list of toxins with effects on the brain is extremely long. Among the major categories of neurotoxic substances are metals, including lead (often causing a diffuse encephalopathy), as well as arsenic and mercury; industrial chemicals, including organophosphates (in pesticides) and methanol (causing blindness from retinal damage); and environmental pollutants such as carbon monoxide (combining hypoxia with selective injury to the globus pallidus).
Ethanol has a variety of effects on the brain. While acute intoxication is reversible, excessive intake can result in profound metabolic disturbances, including brain swelling and death. Chronic alcohol exposure leads to cerebellar dysfunction in about 1% cases, with truncal ataxia, unsteady gait, and nystagmus, associated with atrophy in the anterior vermis of the cerebellum.
Ionizing radiation, commonly used to treat intracranial tumors, can cause rapidly evolving signs and symptoms including headaches, nausea, vomiting, and papilledema, even months to years after irradiation. Affected brain regions show large areas of coagulative necrosis, adjacent edema, and blood vessels with thickened walls containing intramural fibrin-like material.
Neurodegenerative Diseases
Degenerative diseases of the CNS are disorders characterized by the cellular degeneration of subsets of neurons that typically are related by function, rather than by physical location in the brain. Many of these disorders are associated with the accumulation of abnormal proteins, which serve as histologic hallmarks of specific disorders (Table 22–2). An important but unanswered question is why these abnormal proteins tend to accumulate in and preferentially affect particular kinds of neurons, since the involved proteins typically are widely expressed throughout the nervous system.
Table 22–2 Protein Inclusions in Degenerative Diseases
| Disease | Protein | Location |
|---|---|---|
| Alzheimer disease | Aβ Tau |
Extracellular Neurons |
| Frontotemporal lobar degeneration | Tau | Neurons |
| Progressive supranuclear palsy | Tau | Neurons and glia |
| Corticobasal degeneration | Tau | Neurons and glia |
| Parkinson disease | α-Synuclein | Neurons |
| Multiple system atrophy | α-Synuclein | Glia and some neurons |
| Frontotemporal lobar degenerations | TDP-43 | Neurons |
| Amyotrophic lateral sclerosis | TDP-43 | Neurons |
| SOD-1 (familial disease) | Neurons | |
| Huntington disease | Huntingtin | Neurons |
| Spinocerebellar ataxias | Ataxins (various) | Neurons |
SOD-1, superoxide dismutase-1; TDP-43, TAR DNA-binding protein 43.
Subtle differences among subtypes of neurons are presumed to explain why particular neurons are affected in specific disorders. Understandably, the clinical manifestations of degenerative diseases are dictated by the pattern of neuronal dysfunction: those that affect the cerebral cortical neurons result in loss of memory, language, insight, and planning, all components of dementia; those that affect the neurons of the basal ganglia result in movement disorders; those that affect the cerebellum result in ataxia; and those that affect motor neurons result in weakness. Although many degenerative diseases have primary targets, other brain regions are often affected later in the course of the illness; thus, while Huntington disease often has movement disorders as an early symptom, later cortical involvement typically results in the development of cognitive changes as well. Dementia is defined as the development of memory impairment and other cognitive deficits severe enough to decrease the affected person’s capacity to function at the previous level despite a normal level of consciousness. It arises during the course of many neurodegenerative diseases; it also can accompany numerous other diseases that injure the cerebral cortex (Table 22–3). Dementia is an increasing public health concern as the population ages.
Table 22–3 Some Causes of Dementia or Cognitive Impairment
| Primary Neurodegenerative Disorders |
| Infections |
| Vascular and Traumatic Diseases |
| Metabolic and Nutritional Diseases |
| Miscellaneous |
Alzheimer Disease
Alzheimer disease (AD) is the most common cause of dementia in the elderly population. The disease usually manifests with the insidious onset of impaired higher intellectual function and altered mood and behavior. Later, this progresses to disorientation, memory loss, and aphasia, findings indicative of severe cortical dysfunction, and over another 5 to 10 years, the patient becomes profoundly disabled, mute, and immobile. Death usually occurs from intercurrent pneumonia or other infections. Age is an important risk factor for AD; the incidence is about 3% in persons 65 to 74 years old, 19% in those 75 to 84 years old, and 47% in those older than 84 years. Most cases of AD are sporadic, but at least 5% to 10% are familial. Sporadic cases rarely present before 50 years of age, but early onset is seen with some heritable forms.
Pathogenesis
Study of the familial forms of AD supports a model in which a peptide called beta amyloid, or Aβ, accumulates in the brain over time, initiating a chain of events that result in AD. Aβ is created when the transmembrane protein amyloid precursor protein (APP) is sequentially cleaved by the enzymes β-amyloid converting enzyme (BACE) and γ-secretase (Fig. 22–24). APP also can be cleaved by α-secretase and γ-secretase, which liberates a different peptide that is nonpathogenic. Mutations in APP or in components of γ-secretase (presenilin-1 or presenilin-2) lead to familial AD by increasing the rate at which Aβ is generated. The APP gene is located on chromosome 21, and the risk of AD also is higher in those with an extra copy of the APP gene, such as patients with trisomy 21 (Down syndrome) and persons with small interstitial duplications of APP, presumably because this too leads to greater Aβ generation. The other major genetic risk factor is a variant of apolipoprotein E called ε4 (ApoE4). Each ApoE4 allele that is present increases the risk of AD by approximately 4 fold and also appears to lower the age of onset. How ApoE4 influences Aβ accumulation is unknown; it may increase Aβ aggregation or deposition, or decrease Aβ clearance.
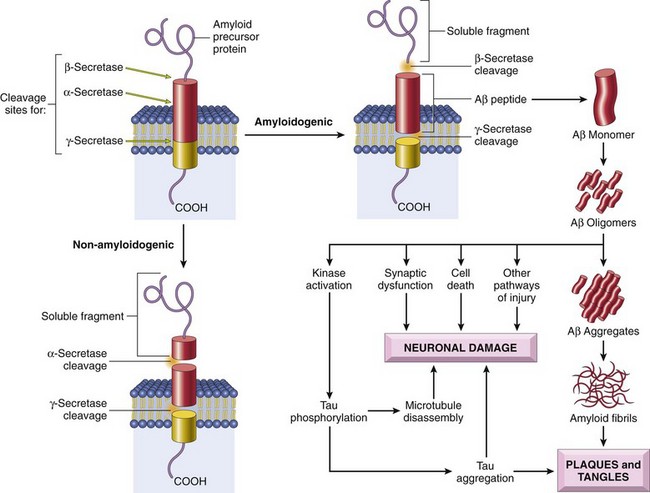
Figure 22–24 Aβ peptide genesis and consequences in Alzheimer disease. Amyloid precursor protein cleavage by α-secretase and γ-secretase produces a harmless soluble peptide, whereas amyloid precursor protein cleavage by β-amyloid–converting enzyme (BACE) and γ-secretase releases Aβ peptides, which form pathogenic aggregates and contribute to the characteristic plaques and tangles of Alzheimer disease.
While large deposits of Aβ are a feature of end-stage AD, small aggregates of Aβ may also be pathogenic, as they alter neurotransmission and are toxic to neurons and synaptic endings. Large deposits, in the form of plaques, also lead to neuronal death, elicit a local inflammatory response that can result in further cell injury, and may cause altered region-to-region communication through mechanical effects on axons and dendrites.
The presence of Aβ also leads to hyperphosphorylation of the neuronal microtubule binding protein tau. This increased level of phosphorylation causes tau to redistribute from axons into dendrites and cell bodies, where it aggregates into tangles, which also contribute to neuronal dysfunction and cell death.
Morphology
Macroscopic examination of the brain shows a variable degree of cortical atrophy, resulting in a widening of the cerebral sulci that is most pronounced in the frontal, temporal, and parietal lobes. With significant atrophy, there is compensatory ventricular enlargement (hydrocephalus ex vacuo). At the microscopic level, AD is diagnosed by the presence of plaques (an extracellular lesion); and neurofibrillary tangles (an intracellular lesion) (Fig. 22–25). Because these may also be present to a lesser extent in the brains of elderly nondemented persons, the current criteria for a diagnosis of AD are based on a combination of clinical and pathologic features. There is a fairly constant progressive involvement of different parts of the brain: pathologic changes (specifically plaques, tangles, and the associated neuronal loss and glial reaction) are evident first in the entorhinal cortex, then in the hippocampal formation and isocortex, and finally in the neocortex. Silver staining or immunohistochemistry methods are extremely helpful in assessing the true lesional burden.
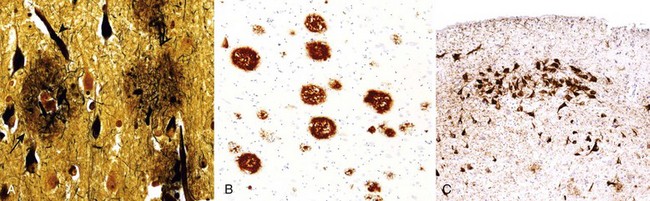
Figure 22–25 Alzheimer disease. A, Plaques (arrow) contain a central core of amyloid and a surrounding region of dystrophic neurites (Bielschowsky stain). B, Immunohistochemical stain for Aβ. Peptide is present in the core of the plaques as well as in the surrounding region. C, Neurons containing tangles stained with an antibody specific for tau.
Neuritic plaques are focal, spherical collections of dilated, tortuous, silver-staining neuritic processes (dystrophic neurites), often around a central amyloid core (Fig. 22–25, A). Neuritic plaques range in size from 20 to 200 µm in diameter; microglial cells and reactive astrocytes are present at their periphery. Plaques can be found in the hippocampus and amygdala as well as in the neocortex, although there usually is relative sparing of primary motor and sensory cortices until late in the disease course. The amyloid core contains Aβ (Fig. 22–25, B). Aβ deposits also can be found that lack the surrounding neuritic reaction, termed diffuse plaques; these typically are found in the superficial cerebral cortex, the basal ganglia, and the cerebellar cortex and may represent an early stage of plaque development.
Neurofibrillary tangles are bundles of paired helical filaments visible as basophilic fibrillary structures in the cytoplasm of the neurons that displace or encircle the nucleus; tangles can persist after neurons die, becoming a form of extracellular pathology. They are commonly found in cortical neurons, especially in the entorhinal cortex, as well as in the pyramidal cells of the hippocampus, the amygdala, the basal forebrain, and the raphe nuclei. A major component of paired helical filaments is abnormally hyperphosphorylated tau (Fig. 22–25, C). Tangles are not specific to AD, being found in other degenerative diseases as well.
Frontotemporal Lobar Degeneration
Another major category of disease that results in dementia is called frontotemporal lobar degeneration (FTLD). These disorders share clinical features (progressive deterioration of language and changes in personality) stemming from the degeneration and atrophy of temporal and frontal lobes; the clinical syndromes commonly are referred to as frontotemporal dementias. When the frontal lobe bears the greatest burden of disease, behavioral changes often dominate, whereas when the disease begins in the temporal lobe, language problems often are the presenting complaint. These symptoms precede memory disturbances, which can assist in their separation from AD on clinical grounds.
On gross inspection, there is atrophy of the brain that predominantly affects the frontal and temporal lobes. Different subgroups are characterized by neuronal inclusions involving the affected regions. In some cases the defining inclusions contain tau (FTLD-tau), but the configuration of the tau inclusions differs from the tau-containing tangles of AD. FTLD-tau sometimes is caused by mutations in the gene encoding tau. One well-recognized subtype of FTLD-tau is Pick disease, which is associated with smooth, round inclusions known as Pick bodies. The other major form of FTLD is characterized by aggregates containing the DNA/RNA-binding protein TDP-43 (FTLD-TDP43). This form of FTLD is associated with predominantly frontal lobe cognitive impairment. It is sometimes caused by mutations in the gene encoding TDP-43, which is also mutated in a subset of cases of amyotrophic lateral sclerosis (described later).
Parkinson Disease
Parkinsonism is a clinical syndrome characterized by tremor, rigidity, bradykinesia and instability. These types of motor disturbances may be seen in a range of diseases that damage dopaminergic neurons, which project from the substantia nigra to the striatum. Parkinsonism can be induced by drugs such as dopamine antagonists or toxins that selectively injure dopaminergic neurons. Among the neurodegenerative diseases, most cases of parkinsonism are caused by Parkinson disease (PD), which is associated with characteristic neuronal inclusions containing α-synuclein. Other diseases in which parkinsonism may be present include multiple system atrophy (MSA), in which α-synuclein aggregates are found in oligodendrocytes; progressive supranuclear palsy (PSP) and corticobasal degeneration (CBD), which are both associated with tau-containing inclusions in neurons and glial cells; and postencephalitic parkinsonism, which was associated with the 1918 influenza pandemic.
Pathogenesis
While PD in most cases is sporadic, both autosomal dominant and recessive forms of the disease also exist. Point mutations and duplications of the gene encoding α-synuclein, a protein involved in synaptic transmission, cause autosomal dominant PD. Even in sporadic PD, the diagnostic feature of the disease—the Lewy body—is an inclusion containing α-synuclein. The linkage between α-synuclein and disease pathogenesis is unclear, but other genetic forms of PD provide some clues. Two other causative genetic loci encode the proteins parkin, an E3 ubiquitin ligase, and UCHL-1, an enzyme involved in recycling of ubiquitin from proteins targeted to the proteasome, suggesting that defects in protein degradation may have a pathogenic role. Another tantalizing clue comes from the association of PD with mutations in a protein kinase called LRRK2; histopathologic examination of cases associated with LRRK2 mutations may show either Lewy bodies containing α-synuclein or tangles containing tau. Finally, some forms of familial PD are associated with mutations in the PARK7 or PINK1 genes, both of which appear to be important for normal mitochondrial function.
Morphology
A typical gross finding at autopsy is pallor of the substantia nigra (Fig. 22–26, A and B) and locus ceruleus. Microscopic features include loss of the pigmented, catecholaminergic neurons in these regions associated with gliosis. Lewy bodies (Fig. 22–26, C) may be found in those neurons that remain. These are single or multiple, intracytoplasmic, eosinophilic, round to elongated inclusions that often have a dense core surrounded by a pale halo. On ultrastructural examination, Lewy bodies consist of fine filaments, densely packed in the core but loose at the rim, composed of α-synuclein and other proteins, including neurofilaments and ubiquitin. The other major histologic finding is Lewy neurites, dystrophic neurites that also contain abnormally aggregated α-synuclein.
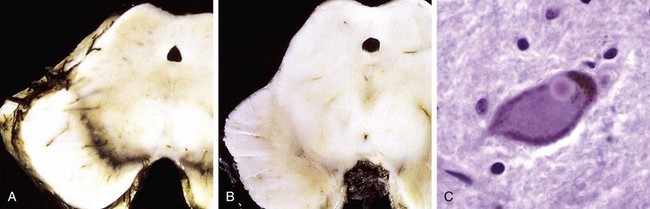
Figure 22–26 Parkinson disease. A, Normal substantia nigra. B, Depigmented substantia nigra in idiopathic Parkinson disease. C, Lewy body in a neuron from the substantia nigra stains pink.
As implied by the occurrence of a broad array of neurologic deficits in PD, immunohistochemical staining for α-synuclein highlights more subtle Lewy bodies and Lewy neurites in many brain regions outside of the substantia nigra and in nondopaminergic neurons. These lesions appear first in the medulla and then in the pons, before involvement of the substantia nigra. As implied by the dementia, Lewy bodies and Lewy neurites eventually appear in the cerebral cortex and subcortical areas, including the cholinergic cells of the basal nucleus of Meynert and the amygdala.
Clinical Features
PD commonly manifests as a movement disorder in the absence of a toxic exposure or other known underlying etiology. The disease usually progresses over 10 to 15 years, eventually producing severe motor slowing to the point of near immobility. Death usually is the result of intercurrent infection or trauma from frequent falls caused by postural instability.
Movement symptoms of PD initially respond to l-dihydroxyphenylalanine (l-DOPA), but this treatment does not slow disease progression. Over time, l-DOPA becomes less effective and begins to cause potentially problematic fluctuations in motor function.
While the movement disorder associated with loss of the nigrostriatal dopaminergic pathway is an important feature of PD, it is clear that the disease has more extensive clinical and pathologic manifestations. Lesions can be found lower in the brain stem (in the dorsal motor nucleus of the vagus and in the reticular formation) in advance of nigral involvement, in line with clinical studies showing that autonomic dysfunction and behavioral disorders often are present in advance of the motor problems. Dementia, typically with a mildly fluctuating course and hallucinations, emerges in many persons with PD and is attributable to involvement of the cerebral cortex. When dementia arises within 1 year of the onset of motor symptoms, it is referred to Lewy body dementia (LBD).
Huntington Disease
Huntington disease (HD) is an autosomal dominant movement disorder associated with degeneration of the striatum (caudate and putamen). The movement disorder is choreiform (dancelike), with increased and involuntary jerky movements of all parts of the body; writhing movements of the extremities are typical. The disease is relentlessly progressive, resulting in death after an average course of about 15 years. Early cognitive symptoms include forgetfulness and thought and affective disorders, and there may be progression to a severe dementia. As a part of these early behavioral changes, HD carries an increased risk of suicide.
Pathogenesis
HD is caused by CAG trinucleotide repeat expansions in a gene located on 4p16.3 that encodes the protein huntingtin. Normal alleles contain 11 to 34 copies of the repeat; in disease-causing alleles the number of repeats is increased, sometimes into the hundreds. There is strong genotype-phenotype correlation, with larger numbers of repeats resulting in earlier-onset disease. Once the symptoms appear, however, the course of the illness is not affected by repeat length. Further expansions of the pathologic CAG repeats can occur during spermatogenesis, so paternal transmission may be associated with earlier onset in the next generation, a phenomenon referred to as anticipation (Chapter 6).
HD appears to be caused by a toxic gain-of-function mutation somehow related to the expanded polyglutamine tract in huntingtin. The mutant protein is subject to ubiquitination and proteolysis, yielding fragments that can form large intranuclear aggregates. As in other degenerative diseases, smaller aggregates of the abnormal protein fragments are suspected to be the critical toxic agent. These aggregates may sequester transcription factors, disrupt protein degradation pathways, perturb mitochondrial function, or alter brain-derived neurotrophic factor (BDNF) signaling. It is likely that some combination of these aberrations contributes to HD pathogenesis.
Morphology
On gross examination, the brain is small and shows striking atrophy of the caudate nucleus and, sometimes less dramatically, the putamen (Fig. 22–27). Pathologic changes develop over the course of the illness in a medial to lateral direction in the caudate and from dorsal to ventral in the putamen. The globus pallidus may be atrophied secondarily, and the lateral and third ventricles are dilated. Atrophy frequently is also seen in the frontal lobe, less often in the parietal lobe, and occasionally in the entire cortex.
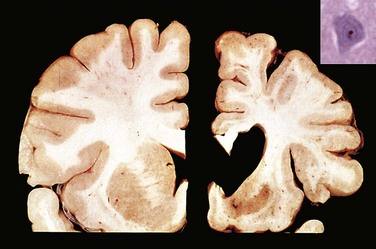
Figure 22–27 Huntington disease. Normal hemisphere on the left compared with the hemisphere with Huntington disease on the right showing atrophy of the striatum and ventricular dilation. Inset, An intranuclear inclusion in a cortical neuron is strongly immunoreactive for ubiquitin.
(Courtesy of Dr. J.P. Vonsattel, Columbia University, New York, New York.)
Microscopic examination reveals severe loss of neurons from affected regions of the striatum. The medium-sized, spiny neurons that release the neurotransmitters γ-aminobutyric acid (GABA), enkephalin, dynorphin, and substance P are especially sensitive, disappearing early in the disease. Also seen is fibrillary gliosis, which is more extensive than in the usual reaction to neuronal loss. There is a strong correlation between the degree of degeneration in the striatum and the severity of motor symptoms; there is also an association between cortical neuronal loss and dementia. In remaining striatal neurons and in the cortex, there are intranuclear inclusions that contain aggregates of ubiquitinated huntingtin protein (Fig. 22–27, inset).
Spinocerebellar Ataxias
Spinocerebellar ataxias (SCAs) are a clinically heterogeneous group of diseases that are frequently caused by trinucleotide repeat expansion mutations. They are distinguished from one another by differences in causative mutations, patterns of inheritance, age at onset, and signs and symptoms. This group of diseases affects, to a variable extent, the cerebellar cortex, spinal cord, other brain regions, and peripheral nerves. As a result, clinical findings may include a combination of cerebellar and sensory ataxia, spasticity, and sensorimotor peripheral neuropathy. Degeneration of neurons, often without distinctive histopathologic changes, occurs in the affected areas and is associated with mild gliosis. The additional clinical symptoms that accompany the ataxia can help distinguish between well-characterized subtypes. Although more than two dozen distinct genetic types of SCA have been identified, there remain many cases which do not fall into one of the already characterized forms.
As with Huntington disease, several forms of SCA (SCA types 1, 2, 3, 6, 7, and 17 and dentatorubropallidoluysian atrophy) are caused by CAG repeat expansions encoding polyglutamine tracts in various genes. In these forms of SCA, neuronal intranuclear inclusions are present containing the abnormal protein and there is an inverse correlation between the degree of repeat expansion and age of onset. Other SCAs are caused by trinucleotide repeat expansions in untranslated regions or by other types of mutations.
Friedreich ataxia is an autosomal recessive disorder that generally manifests in the first decade of life with gait ataxia, followed by hand clumsiness and dysarthria. Most patients develop pes cavus and kyphoscoliosis, and there is a high incidence of cardiac disease and diabetes. The disease usually is caused by a GAA trinucleotide repeat expansion in the gene encoding frataxin, a protein that regulates cellular iron levels, particularly in the mitochondria. The repeat expansion results in decreased protein levels through transcriptional silencing; rare cases in which point mutations produce a nonfunctional frataxin protein also have been described. Decreased frataxin leads to mitochondrial dysfunction as well as increased oxidative damage.
Amyotrophic Lateral Sclerosis
Amyotrophic lateral sclerosis (ALS) results from the death of lower motor neurons in the spinal cord and brain stem, and of upper motor neurons (Betz cells) in the motor cortex. The loss of lower motor neurons results in denervation of muscles, muscular atrophy (the “amyotrophy” of the condition), weakness, and fasciculations, while the loss of upper motor neurons results in paresis, hyperreflexia, and spasticity, along with a Babinski sign. An additional consequence of upper motor neuron loss is degeneration of the corticospinal tracts in the lateral portion of the spinal cord (“lateral sclerosis”). Sensation usually is unaffected, but cognitive impairment does occur, sometimes as a frontotemporal dementia.
The disease affects men slightly more frequently than women and becomes clinically manifest in the fifth decade or later, usually beginning with subtle asymmetric distal extremity weakness. As the disease progresses to involve more of the motor system, muscle strength and bulk diminish and involuntary contractions of individual motor units, termed fasciculations, occur. The disease eventually involves the respiratory muscles, leading to recurrent bouts of pulmonary infection, which is the usual cause of death. The balance between upper and lower motor neuron involvement can vary, although most patients exhibit involvement of both. In some patients, degeneration of the lower brain stem cranial motor nuclei occurs early and progresses rapidly, a pattern of disease referred to as bulbar amyotrophic lateral sclerosis. With this disease pattern, abnormalities of swallowing and speaking dominate.
Pathogenesis
While most cases are sporadic, 5% to 10% are familial, mostly with autosomal dominant inheritance. Familial disease begins earlier in life than sporadic disease, but once symptoms appear, the clinical course is similar in both forms. More than a dozen genes have been implicated, but the most frequent genetic cause (20% of cases) is mutations in the superoxide dismutase gene, SOD-1, on chromosome 21. These mutations are thought to generate abnormal misfolded forms of the SOD-1 protein, which may trigger the unfolded protein response and cause apoptotic death of neurons. The next two most common causative genes both encode DNA/RNA binding proteins, TDP-43 and FUS; how these mutations cause disease is unknown. As already mentioned, mutations in TDP-43 also can cause frontotemporal lobar degeneration (FTLD) or a disease with overlapping features of both ALS and FTLD.
Morphology
The most striking gross changes are found in anterior roots of the spinal cord, which are thin and gray (rather than white). In especially severe cases, the precentral gyrus (motor cortex) may be mildly atrophic. Microscopic examination demonstrates a reduction in the number of anterior horn cell neurons throughout the length of the spinal cord associated with reactive gliosis and loss of anterior root myelinated fibers. Similar findings are found with involvement of motor cranial nerve nuclei except those supplying the extraocular muscles, which are spared except in very longstanding survivors. Remaining lower motor neurons often harbor cytoplasmic inclusions that contain TDP-43, except in those cases in which the underlying cause is a mutation in SOD-1.
Death of upper motor neurons—a finding that may be hard to demonstrate microscopically—results in degeneration of the descending corticospinal tracts. This is usually easily seen in the spinal cord. With the loss of innervation from the death of anterior horn cells, skeletal muscles show neurogenic atrophy.
Summary
Neurodegenerative Diseases
• Neurodegenerative diseases cause symptoms that depend on the pattern of brain involvement. Cortical disease usually manifests as cognitive change, alterations in personality, and memory disturbances; basal ganglia disorders usually manifest as movement disorders.
• Many neurodegenerative diseases preferentially affect a primary set of brain regions, but other regions can be involved later in the disease course. This evolving process can change the phenotype of the disease over time—as with the appearance of cognitive impairments in people initially affected by the movement disorder of Parkinson disease.
• Many of the neurodegenerative diseases are associated with various protein aggregates, which serve as pathologic hallmarks. It is unclear whether these striking inclusions and deposits are critical mediators of cellular degeneration. Familial forms of these diseases are associated with mutations in the genes encoding these proteins or controlling their metabolism.
Tumors
The annual incidence of CNS tumors ranges from 10 to 17 per 100,000 persons for intracranial tumors and 1 to 2 per 100,000 persons for intraspinal tumors; about half to three quarters are primary tumors, and the rest are metastatic. Tumors of the CNS make up a larger proportion of childhood cancers, accounting for as many of 20% of all pediatric tumors. Childhood CNS tumors differ from those in adults in both histologic subtype and location. In childhood, tumors are likely to arise in the posterior fossa; in adults, they are mostly supratentorial.
Tumors of the nervous system have unique characteristics that set them apart from neoplastic processes elsewhere in the body.
• These tumors do not have detectable premalignant or in situ stages comparable to those of carcinomas.
• Even low-grade lesions may infiltrate large regions of the brain, leading to serious clinical deficits, nonresectability, and poor prognosis.
• The anatomic site of the neoplasm can influence outcome independent of histologic classification due to local effects (e.g., a benign meningioma may cause cardiorespiratory arrest from compression of the medulla) or nonresectability (e.g., brain stem gliomas).
• Even the most highly malignant gliomas rarely spread outside of the CNS; in addition to local infiltration, the subarachnoid space allows for spread to distant sites along the neuroaxis.
Gliomas
Gliomas are tumors of the brain parenchyma that are classified histologically on the basis of their resemblance to different types of glial cells. The major types of glial tumors are astrocytomas, oligodendrogliomas, and ependymomas. The most common types are highly infiltrative or “diffuse gliomas,” including astrocytic, oligodendroglial, and mixed forms. In contrast, ependymomas tend to form solid masses.
Astrocytoma
Several different categories of astrocytic tumors are recognized, the most common being diffuse and pilocytic astrocytomas. Different types of astrocytomas have characteristic histologic features, anatomic distributions, and clinical features.
Diffuse Astrocytoma
Diffuse astrocytomas account for about 80% of adult gliomas. They are most frequent in the fourth through the sixth decades of life. They usually are found in the cerebral hemispheres. The most common presenting signs and symptoms are seizures, headaches, and focal neurologic deficits related to the anatomic site of involvement. They show a spectrum of histologic differentiation that correlates well with clinical course and outcome. On the basis of histologic features, they are stratified into three groups: well-differentiated astrocytoma (grade II/IV), anaplastic astrocytoma (grade III/IV), and glioblastoma (grade IV/IV), with increasingly grim prognosis as the grade increases.
Well-differentiated astrocytomas can be static for several years, but at some point they progress; the mean survival is more than 5 years. Eventually, patients suffer rapid clinical deterioration that is correlated with the appearance of anaplastic features and more rapid tumor growth. Other patients present with glioblastoma from the start. Once the histologic features of glioblastoma appear, the prognosis is very poor; with treatment (resection, radiotherapy, and chemotherapy), the median survival is only 15 months.
Astrocytomas are associated with a variety of acquired mutations, which cluster in several important pathways. In glioblastoma, loss-of-function mutations in the p53 and Rb tumor suppressor pathways and gain-of-function mutations in the oncogenic PI3K pathways have central roles in tumorigenesis. Surprisingly, mutations that alter the enzymatic activity of two isoforms of the metabolic enzyme isocitrate dehydrogenase (IDH1 and IDH2) are common in lower-grade astrocytomas. As a result, immunostaining for the mutated form of IDH1 has become an important diagnostic tool in evaluating biopsy specimens for the presence of low-grade astrocytoma.
Morphology
Well-differentiated astrocytomas are poorly defined, gray, infiltrative tumors that expand and distort the invaded brain without forming a discrete mass (Fig. 22–28, A). Infiltration beyond the grossly evident margins is always present. The cut surface of the tumor is either firm or soft and gelatinous; cystic degeneration may be seen. In glioblastoma, variation in the gross appearance of the tumor from region to region is characteristic (Fig. 22–28, B). Some areas are firm and white, others are soft and yellow (the result of tissue necrosis), and still others show regions of cystic degeneration and hemorrhage.
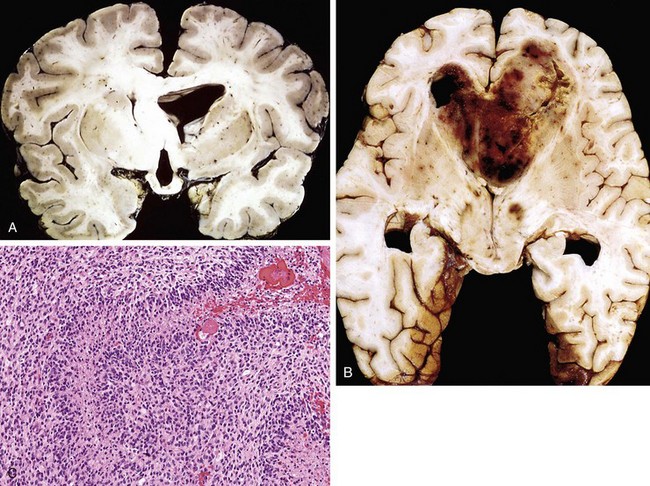
Figure 22–28 Astrocytomas. A, Low-grade astrocytoma is seen as expanded white matter of the left cerebral hemisphere and thickened corpus callosum and fornices. B, Glioblastoma appearing as a necrotic, hemorrhagic, infiltrating mass. C, Glioblastoma is a densely cellular tumor with necrosis and pseudopalisading of tumor cell nuclei.
Well-differentiated astrocytomas are characterized by a mild to moderate increase in the number of glial cell nuclei, somewhat variable nuclear pleomorphism, and an intervening feltwork of fine, glial fibrillary acidic protein (GFAP)-positive astrocytic cell processes that give the background a fibrillary appearance. The transition between neoplastic and normal tissue is indistinct, and tumor cells can be seen infiltrating normal tissue many centimeters from the main lesion. Anaplastic astrocytomas show regions that are more densely cellular and have greater nuclear pleomorphism; mitotic figures are present. Glioblastoma has a histologic appearance similar to that of anaplastic astrocytoma, as well as either necrosis (often with pseudopalisading nuclei) or vascular proliferation (Fig. 22–28, C).
Pilocytic Astrocytoma
Pilocytic astrocytomas are relatively benign tumors, typically affecting children and young adults. Most commonly located in the cerebellum, they also may involve the third ventricle, the optic pathways, spinal cord, and occasionally the cerebral hemispheres. There is often a cyst associated with the tumor, and symptomatic recurrence from incompletely resected lesions is often associated with cyst enlargement, rather than growth of the solid component. Tumors that involve the hypothalamus are especially problematic because they cannot be resected completely.
A high proportion of pilocytic astrocytomas have activating mutations in the serine-threonine kinase BRAF—either a specific point mutation (V600E) that is also found in many other cancers (Chapter 5), or more commonly a partial tandem duplication event. Mutations in IDH1 and IDH2 (common in low-grade diffuse astrocytomas) are not found in pilocytic tumors. These genetic distinctions support the division of these astrocytomas into two diagnostic categories.
Morphology
A pilocytic astrocytoma often is cystic, with a mural nodule in the wall of the cyst; if solid, it is usually well circumscribed. The tumor is composed of bipolar cells with long, thin “hairlike” processes that are GFAP-positive. Rosenthal fibers, eosinophilic granular bodies, and microcysts are often present; necrosis and mitoses are rare.
Oligodendroglioma
Oligodendrogliomas account for 5% to 15% of gliomas and most commonly are detected in the fourth and fifth decades of life. Patients may have had several years of antecedent neurologic complaints, often including seizures. The lesions are found mostly in the cerebral hemispheres, mainly in the frontal or temporal lobes.
Patients with oligodendrogliomas enjoy a better prognosis than that for patients with astrocytomas of similar grade. Treatment with surgery, chemotherapy, and radiotherapy yields an average survival of 10 to 20 years for well-differentiated (WHO grade II) or 5 to 10 years for anaplastic (WHO grade III) oligodendrogliomas. The most common genetic findings are deletions of chromosomes 1p and 19q, alterations that typically occur together. Tumors with deletions of 1p and 19q are usually highly responsive to chemotherapy and radiotherapy.
Morphology
Well-differentiated oligodendrogliomas (WHO grade II/IV) are infiltrative tumors that form gelatinous, gray masses and may show cysts, focal hemorrhage, and calcification. On microscopic examination, the tumor is composed of sheets of regular cells with spherical nuclei containing finely granular-appearing chromatin (similar to that in normal oligodendrocytes) surrounded by a clear halo of cytoplasm (Fig. 22–29, A). The tumor typically contains a delicate network of anastomosing capillaries. Calcification, present in as many as 90% of these tumors, ranges in extent from microscopic foci to massive depositions. Mitotic activity usually is difficult to detect. Anaplastic oligodendroglioma (WHO grade III/IV) is a more aggressive subtype with higher cell density, nuclear anaplasia and mitotic activity.
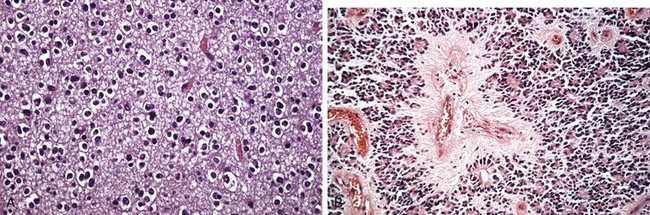
Ependymoma
Ependymomas most often arise next to the ependyma-lined ventricular system, including the central canal of the spinal cord. In the first 2 decades of life, they typically occur near the fourth ventricle and constitute 5% to 10% of the primary brain tumors in this age group. In adults, the spinal cord is their most common location; tumors in this site are particularly frequent in the setting of neurofibromatosis type 2 (Chapter 21). The clinical outcome for completely resected supratentorial and spinal ependymomas is better than for those in the posterior fossa.
Morphology
In the fourth ventricle, ependymomas typically are solid or papillary masses extending from the ventricular floor. The tumors are composed of cells with regular, round to oval nuclei and abundant granular chromatin. Between the nuclei is a variably dense fibrillary background. Tumor cells may form round or elongated structures (rosettes, canals) that resemble the embryologic ependymal canal, with long, delicate processes extending into a lumen (Fig. 23–29, B); more frequently present are perivascular pseudorosettes in which tumor cells are arranged around vessels with an intervening zone containing thin ependymal processes. Anaplastic ependymomas show increased cell density, high mitotic rates, necrosis, and less evident ependymal differentiation.
Neuronal Tumors
Central neurocytoma is a low-grade neoplasm found within and adjacent to the ventricular system (most commonly the lateral or third ventricles), characterized by evenly spaced, round, uniform nuclei and often islands of neuropil.
Gangliogliomas are tumors with a mixture of glial elements, usually a low-grade astrocytoma, and mature-appearing neurons. Most of these tumors are slow-growing, but the glial component occasionally becomes frankly anaplastic, and the disease then progresses rapidly. These lesions often manifest with seizures.
Dysembryoplastic neuroepithelial tumor is a distinctive, low-grade childhood tumor that grows slowly and carries a relatively good prognosis after resection; it often manifests as a seizure disorder. It typically is located in the superficial temporal lobe and consists of small round neuronal cells arranged in columns and around central cores of processes. These typically form multiple discrete intracortical nodules that have a myxoid background. Also present are well-differentiated “floating” neurons within pools of mucopolysaccharide-rich myxoid fluid.
Embryonal (Primitive) Neoplasms
Some tumors of neuroectodermal origin have a primitive “small round cell” appearance that is reminiscent of normal progenitor cells encountered in the developing CNS. Differentiation is often limited, but may progress along multiple lineages. The most common is the medulloblastoma, accounting for 20% of pediatric brain tumors.
Medulloblastoma
Medulloblastoma occurs predominantly in children and exclusively in the cerebellum. Neuronal and glial markers are nearly always expressed, at least to a limited extent. It is highly malignant, and the prognosis for untreated patients is dismal; however, medulloblastoma is exquisitely radiosensitive. With total excision, chemotherapy, and irradiation, the 5-year survival rate may be as high as 75%. Tumors of similar histologic type and a poor degree of differentiation can be found elsewhere in the nervous system, where they are called primitive neuroectodermal tumors (PNETs).
Morphology
In children, medulloblastomas are located in the midline of the cerebellum; lateral tumors occur more often in adults. The tumor often is well circumscribed, gray, and friable and may be seen extending to the surface of the cerebellar folia and involving the leptomeninges (Fig. 22–30, A). Medulloblastomas are extremely cellular, with sheets of anaplastic (“small blue”) cells (Fig. 22–30, B). Individual tumor cells are small, with little cytoplasm and hyperchromatic nuclei; mitoses are abundant. Often, focal neuronal differentiation is seen in the form of the Homer Wright or neuroblastic rosette, which closely resembles the rosettes encountered in neuroblastomas; they are characterized by primitive tumor cells surrounding central neuropil (delicate pink material formed by neuronal processes).
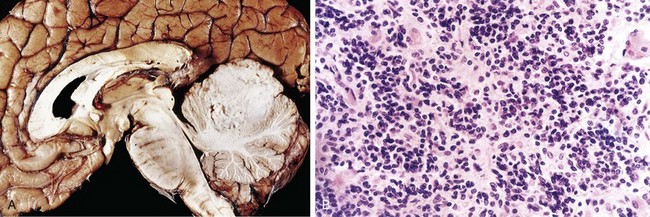
Genetic analysis of medulloblastoma has revealed that morphologically similar tumors commonly exhibit distinct alterations, and that there is a relationship between the underlying mutations and outcome. In general, tumors with MYC amplifications are associated with poor outcomes, while those linked with mutations in genes of the WNT signaling pathway have a more favorable course. Many tumors also have mutations that activate the sonic hedgehog (shh) pathway, which has a critical role in tumorigenesis but an uncertain relationship to outcome. These genetic distinctions are beginning to be used to stratify patients into different risk groups and guide therapy. Ideally, it would be best to avoid CNS radiotherapy in young patients, and it is hoped that new therapies targeting mutated gene products will achieve this goal.
Other Parenchymal Tumors
Primary Central Nervous System Lymphoma
Primary CNS lymphoma, occurring mostly as diffuse large B cell lymphomas, accounts for 2% of extranodal lymphomas and 1% of intracranial tumors. It is the most common CNS neoplasm in immunosuppressed persons, in whom the tumors are nearly always positive for the oncogenic Epstein-Barr virus. In nonimmunosuppressed populations, the age spectrum is relatively wide, with the incidence increasing after 60 years of age. Regardless of the clinical context, primary brain lymphoma is an aggressive disease with relatively poor response to chemotherapy as compared with peripheral lymphomas.
Patients with primary brain lymphoma often are found to have multiple tumor nodules within the brain parenchyma, yet involvement outside of the CNS is an uncommon late complication. Lymphoma originating outside the CNS rarely spreads to the brain parenchyma; when it happens, tumor usually is also within the CSF or involvement of the meninges.
Morphology
Lesions often involve deep gray structures, as well as the white matter and the cortex. Periventricular spread is common. The tumors are relatively well defined as compared with glial neoplasms but are not as discrete as metastases. EBV-associated tumors often show extensive areas of necrosis. The tumors are nearly always aggressive large B-cell lymphomas, although other histologic types can be observed rarely (Chapter 11). Microscopically, malignant cells accumulate around blood vessels and infiltrate the surrounding brain parenchyma.
Germ Cell Tumors
Primary brain germ cell tumors occur along the midline, most commonly in the pineal and the suprasellar regions. They account for 0.2% to 1% of brain tumors in people of European descent but as many as 10% of brain tumors in persons of Japanese ethnicity. They are a tumor of the young, with 90% occurring during the first 2 decades of life. Germ cell tumors in the pineal region show a strong male predominance. The most common primary CNS germ cell tumor is germinoma, a tumor that closely resembles testicular seminoma (Chapter 17). Secondary CNS involvement by metastatic gonadal germ cell tumors also occurs.
Meningiomas
Meningiomas are predominantly benign tumors that arise from arachnoid meningothelial cells. They usually occur in adults and are often attached to the dura. Meningiomas may be found along any of the external surfaces of the brain as well as within the ventricular system, where they arise from the stromal arachnoid cells of the choroid plexus. They usually come to attention because of vague nonlocalizing symptoms, or with focal findings referable to compression of adjacent brain. Although most meningiomas are easily separable from underlying brain, some tumors infiltrate the brain, a feature that is associated with an increased risk of recurrence. The overall prognosis is determined by the lesion size and location, surgical accessibility, and histologic grade.
When a person has multiple meningiomas, especially in association with eighth-nerve schwannomas or glial tumors, the diagnosis of neurofibromatosis type 2 (NF2) should be considered (Chapter 21). About half of meningiomas not associated with NF2 have acquired loss-of-function mutations in the NF2 tumor suppressor gene on the long arm of chromosome 22 (22q). These mutations are found in all grades of meningioma, suggesting that they are involved with tumor initiation. Mutations in NF2 are more common in tumors with certain growth patterns (fibroblastic, transitional, and psammomatous).
Morphology
Meningiomas (WHO grade I/IV) grow as well-defined dura-based masses that may compress the brain but do not invade it (Fig. 22–31, A). Extension into the overlying bone may be present. Among the varied histologic patterns are syncytial, named for whorled clusters of cells without visible cell membranes that sit in tight groups; fibroblastic, with elongated cells and abundant collagen deposition between them; transitional, which shares features of the syncytial and fibroblastic types; psammomatous, with numerous psammoma bodies (Fig. 22–31, B); and secretory, with gland-like PAS-positive eosinophilic secretions known as pseudopsammoma bodies.
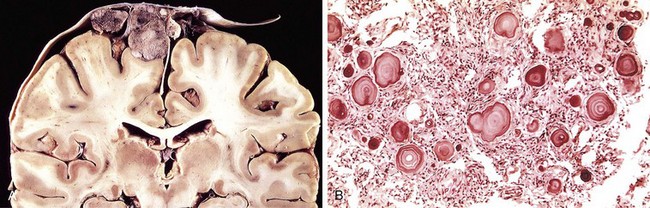
Figure 22–31 Meningioma. A, Parasagittal multilobular meningioma attached to the dura with compression of underlying brain. B, Meningioma with a whorled pattern of cell growth and psammoma bodies.
Atypical meningiomas (WHO grade II/IV) are recognized by the presence of certain histologic features (prominent nucleoli, increased cellularity, pattern-less growth), and often have a higher mitotic rate. These tumors demonstrate more aggressive local growth and a higher rate of recurrence; they may require therapy in addition to surgery.
Anaplastic (malignant) meningiomas (WHO grade III/IV) are highly aggressive tumors that may resemble a high-grade sarcoma or carcinoma, although there usually is some histologic evidence of a meningothelial cell origin.
Metastatic Tumors
Metastatic lesions, mostly carcinomas, account for approximately one fourth to one half of intracranial tumors. The most common primary sites are lung, breast, skin (melanoma), kidney, and gastrointestinal tract—together these account for about 80% of cases. Metastases form sharply demarcated masses, often at the gray-white junction, and elicit edema (Fig. 22–32). The boundary between tumor and brain parenchyma is sharp at the microscopic level as well, with surrounding reactive gliosis.
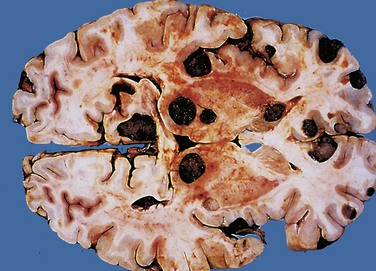
Figure 22–32 Metastatic melanoma. Metastatic lesions are distinguished grossly from most primary central nervous system tumors by their multicentricity and well-demarcated margins. The dark color of the tumor nodules in this specimen is due to the presence of melanin.
In addition to the direct and localized effects produced by metastases, paraneoplastic syndromes may involve the peripheral and central nervous systems, sometimes even preceding the clinical recognition of the malignant neoplasm. Many but not all patients with paraneoplastic syndromes have antibodies against tumor antigens. Some of the more common patterns include
• Subacute cerebellar degeneration resulting in ataxia, with destruction of Purkinje cells, gliosis, and a mild inflammatory infiltrate
• Limbic encephalitis causing a subacute dementia, with perivascular inflammatory cells, microglial nodules, some neuronal loss, and gliosis, all centered in the medial temporal lobe
• Subacute sensory neuropathy leading to altered pain sensation, with loss of sensory neurons from dorsal root ganglia, in association with inflammation
• Syndrome of rapid-onset psychosis, catatonia, epilepsy, and coma associated with ovarian teratoma and antibodies against the N-methyl-d-aspartate (NMDA) receptor
Familial Tumor Syndromes
Several inherited syndromes caused by mutations in various tumor suppressor genes are associated with an increased risk of particular types of cancers. Those with particular involvement of the CNS are discussed here; familial syndromes associated with tumors of the peripheral nervous system are covered in Chapter 21.
Tuberous Sclerosis
Tuberous sclerosis (TSC) is an autosomal dominant syndrome characterized by the development of hamartomas and benign neoplasms involving the brain and other tissues. CNS hamartomas variously consist of cortical tubers and subependymal hamartomas, including a larger tumefactive form known as subependymal giant cell astrocytoma. Because of their proximity to the foramen of Monro, they often present acutely with obstructive hydrocephalus, which requires surgical intervention and/or therapy with an mTOR inhibitor (see below). Seizures are associated with cortical tubers and can be difficult to control with antiepileptic drugs. Extracerebral lesions include renal angiomyolipomas, retinal glial hamartomas, pulmonary lymphangiomyomatosis, and cardiac rhabdomyomas. Cysts may be found at various sites, including the liver, kidneys, and pancreas. Cutaneous lesions include angiofibromas, leathery thickenings in localized patches (shagreen patches), hypopigmented areas (ash leaf patches), and subungual fibromas. TSC results from disruption of either TSC1, which encodes hamartin, or TSC2, which encodes tuberin. The two TSC proteins form a dimeric complex that negatively regulates mTOR, a kinase that “senses” the cell’s nutrient status and regulates cellular metabolism. Loss of either protein leads to increased mTOR activity, which disrupts nutritional signaling and increases cell growth.
Morphology
Cortical hamartomas are firmer than normal cortex and have been likened in appearance to potatoes—hence the appellation “tubers.” They are composed of haphazardly arranged large neurons that lack the normal cortical laminar architecture. These cells may exhibit a mixture of glial and neuronal features, having large vesicular nuclei with nucleoli (like neurons) and abundant eosinophilic cytoplasm (like gemistocytic astrocytes). Similar abnormal cells are present in the subependymal nodules, in which large astrocyte-like cells cluster beneath the ventricular surface.
von Hippel–Lindau Disease
In this autosomal dominant disorder, affected persons develop hemangioblastomas within the cerebellar hemispheres, retina, and, less commonly, the brain stem, spinal cord, and nerve roots. Patients also may have cysts involving the pancreas, liver, and kidneys and have an increased propensity to develop renal cell carcinoma. The disease frequency is 1 in 30,000 to 40,000. Therapy is directed at the symptomatic neoplasms, including surgical resection of cerebellar tumors and laser ablation of retinal tumors. The affected gene, the tumor suppressor VHL, encodes a protein that is part of a ubiquitin-ligase complex that targets the transcription factor hypoxia-inducible factor (HIF) for degradation. Tumors arising in patients with von Hippel–Lindau disease generally have lost all VHL protein function. As a result, these tumors express high levels of HIF, which drives the expression of VEGF, various growth factors, and sometimes erythropoietin, leading to a form of paraneoplastic polycythemia.
Morphology
The cerebellar capillary hemangioblastoma, the principal neurologic manifestation of the disease, is a highly vascular neoplasm that occurs as a mural nodule associated with a large, fluid-filled cyst. On microscopic examination, the lesion consists of numerous capillary-sized or somewhat larger thin-walled vessels separated by intervening stromal cells with vacuolated, lightly PAS-positive, lipid-rich cytoplasm.
Summary
Tumors of the Central Nervous System
• Tumors of the CNS may arise from the cells of the coverings (meningiomas), the brain (gliomas, neuronal tumors, choroid plexus tumors), or other CNS cell populations (primary CNS lymphoma, germ cell tumors), or they may originate elsewhere in the body (metastases).
• Even low-grade or benign tumors can have poor clinical outcomes, depending on where they occur in the brain.
• Distinct types of tumors affect specific brain regions (e.g., cerebellum for medulloblastoma, an intraventricular location for central neurocytoma) and specific age populations (medulloblastoma and pilocytic astrocytomas in pediatric age groups, and glioblastoma and lymphoma in older patients).
• Glial tumors are broadly classified into astrocytomas, oligodendrogliomas, and ependymomas. Increasing tumor malignancy is associated with more cytologic anaplasia, increased cell density, necrosis, and mitotic activity.
• Metastatic spread of brain tumors to other regions of the body is rare, but the brain is not comparably protected against spread of distant tumors. Carcinomas are the dominant type of systemic tumors that metastasize to the nervous system.
In general, many areas of neuropathology and neurologic diseases are well covered in the following standard texts:
Burger, PC, Scheithauer, BW. Tumors of the Central Nervous System. AFIP Atlas of Tumor Pathology: Series 4. Washington, DC: American Registry of Pathology, 2007.
Louis, DN, Frosch, MP, Mena, H, et al. Non-Neoplastic Diseases of the Central Nervous System. AFIP Atlas of Nontumor Pathology: Series 1. Washington, DC: American Registry of Pathology, 2009.
Louis, DN, Ohgaki, H, Wiestler, OD, Cavenee, WK. WHO Classification of Tumours of the Central Nervous System (IARC), 4th ed, Geneva: World Health Organization, 2007.
Love, S, Louis, DN, Ellison, DW. Greenfield’s Neuropathology, 8th ed, Oxford: Oxford University Press, 2008.
Perry A, Brat DJ. Neuropathology patterns and introduction. In: Perry, A, Brat, DJ. Practical Surgical Neuropathology. Philadelphia: Elsevier/Churchill Livingstone, 2010.
Ropper, AH, Samuels, MA. Adams and Victor’s Principles of Neurology, 9th ed, New York: McGraw-Hill Professional, 2009.
For some topics covered in this chapter, there have been recent changes in classification, advances in understanding of pathogenesis, therapeutic interventions, and better understanding of clinicopathologic correlations. For these selected topics, additional reading recommendations are provided.
McKee AC, Cantu RC, Nowinski CJ, et al. Chronic traumatic encephalopathy in athletes: progressive tauopathy after repetitive head injury. J Neuropathol Exp Neurol. 2009;68:709.
Congenital Malformations and Perinatal Brain Injury
Copp AJ, Greene ND. Genetics and development of neural tube defects. J Pathol. 2010;220:217.
Diaz AL, Gleeson JG. The molecular and genetic mechanisms of neocortex development. Clin Perinatol. 2009;36:503.
Kriegstein A, Alvarez-Buylla A. The glial nature of embryonic and adult neural stem cells. Annu Rev Neurosci. 2009;32:149.
Lee JE, Gleeson JG. Cilia in the nervous system: linking cilia function and neurodevelopmental disorders. Curr Opin Neurol. 2011;24:98.
Lim Y, Golden JA. Patterning the developing diencephalon. Brain Res Rev. 2007;53:17.
Na ES, Monteggia LM. The role of MeCP2 in CNS development and function. Horm Behav. 2011;59:364.
Ten Donkelaar HJ, Lammens M. Development of the human cerebellum and its disorders. Clin Perinatol. 2009;36:513.
Thompson BL, Levitt P. The clinical-basic interface in defining pathogenesis in disorders of neurodevelopmental origin. Neuron. 2010;67:702.
Walsh CA, Morrow EM, Rubenstein JL. Autism and brain development. Cell. 2008;135:396.
Infections of the Nervous System
Gambetti P, Cali I, Notari S, et al. Molecular biology and pathology of prion strains in sporadic human prion diseases. Acta Neuropathol. 2011;121:79.
Ironside JW. Variant Creutzfeldt-Jakob disease. Haemophilia. 2010;16(Suppl 5):175.
Johnson T, Nath A. Neurological complications of immune reconstitution in HIV-infected populations. Ann N Y Acad Sci. 2010;1184:106.
Martin-Blondel G, Delobel P, Blancher A, et al. Pathogenesis of the immune reconstitution inflammatory syndrome affecting the central nervous system in patients infected with HIV. Brain. 2011;134(Pt 4):928.
Parchi P, Strammiello R, Giese A, Kretzschmar H. Phenotypic variability of sporadic human prion disease and its molecular basis: past, present, and future. Acta Neuropathol. 2011;121:91.
Singer EJ, Valdes-Sueiras M, Commins D, Levine A. Neurologic presentations of AIDS. Neurol Clin. 2010;28:253.
Wright EJ. Neurological disease: the effects of HIV and antiretroviral therapy and the implications for early antiretroviral therapy initiation. Curr Opin HIV AIDS. 2009;4:447.
Comabella M, Khoury SJ. Immunopathogenesis of multiple sclerosis. Clin Immunol. 2011;10:399.
Hu W, Lucchinetti CF. The pathological spectrum of CNS inflammatory demyelinating diseases. Semin Immunopathol. 2009;31:439.
Jarius S, Wildemann B. AQP4 antibodies in neuromyelitis optica: diagnostic and pathogenetic relevance. Nat Rev Neurol. 2010;6:383.
Oksenberg JR, Baranzini SE, Sawcer S, Hauser SL. The genetics of multiple sclerosis: SNPs to pathways to pathogenesis. Nat Rev Genet. 2008;9:516.
DeKosky ST, Carrillo MC, Phelps C, et al. Revision of the criteria for Alzheimer’s disease: A symposium. Alzheimers Dement. 2011;7:e1.
Durr A. Autosomal dominant cerebellar ataxias: polyglutamine expansions and beyond. Lancet Neurol. 2010;9:885.
Pandolfo M. Friedreich ataxia. Arch Neurol. 2008;65:1296.
Kiernan MC, Vucic S, Cheah BC, et al. Amyotrophic lateral sclerosis. Lancet. 2011;377:942.
Ross CA, Tabrizi SJ. Huntington’s disease: from molecular pathogenesis to clinical treatment. Lancet Neurol. 2011;10:83.
Selkoe DJ. Biochemistry and molecular biology of amyloid beta-protein and the mechanism of Alzheimer’s disease. Handb Clin Neurol. 2008;89:245.
Storch A, Hofer A, Krüger R, et al. New developments in diagnosis and treatment of Parkinson’s disease—from basic science to clinical applications. J Neurol. 2004;251(Suppl 6):VI33.
Thinakaran G, Koo EH. Amyloid precursor protein trafficking, processing, and function. J Biol Chem. 2008;283:29615.
Vidailhet M. Movement disorders in 2010: Parkinson disease-symptoms and treatments. Nat Rev Neurol. 2011;7:70.
Cancer Genome Atlas Research Network. Comprehensive genomic characterization defines human glioblastoma genes and core pathways. Nature. 2008;455:1061.
Cho YJ, Tsherniak A, Tamayo P, et al. Integrative genomic analysis of medulloblastoma identifies a molecular subgroup that drives poor clinical outcome. J Clin Oncol. 2011;29:1424.
Dubuc AM, Northcott PA, Mack S, et al. The genetics of pediatric brain tumors. Curr Neurol Neurosci Rep. 2010;10:215.
Maher ER, Neumann HP, Richard S. von Hippel-Lindau disease: A clinical and scientific review. Eur J Hum Genet. 2011;19:617.
Mawrin C, Perry A. Pathological classification and molecular genetics of meningiomas. J Neurooncol. 2010;99:379.
Orlova KA, Crino PB. The tuberous sclerosis complex. Ann N Y Acad Sci. 2010;1184:87.
Riemenschneider MJ, Jeuken JW, Wesseling P, Reifenberger G. Molecular diagnostics of gliomas: state of the art. Acta Neuropathol. 2010;120:567.