White Cell Disorders
Disorders of white cells include deficiencies (leukopenias) and proliferations, which may be reactive or neoplastic. Reactive proliferation in response to a primary, often microbial, disease is common. Neoplastic disorders, though less common, are more ominous: They cause approximately 9% of all cancer deaths in adults and a staggering 40% in children younger than 15 years of age.
Presented next are brief descriptions of some non-neoplastic conditions, followed by more detailed considerations of the malignant proliferations of white cells.
Non-Neoplastic Disorders of White Cells
Leukopenia
Leukopenia results most commonly from a decrease in granulocytes, the most numerous circulating white cells. Lymphopenia is much less common; it is associated with rare congenital immunodeficiency diseases, advanced human immunodeficiency virus (HIV) infection, and treatment with high doses of corticosteroids. Only the more common leukopenias of granulocytes are discussed here.
Neutropenia/Agranulocytosis
A reduction in the number of granulocytes in blood is known as neutropenia or, when severe, agranulocytosis. Neutropenic persons are susceptible to bacterial and fungal infections, in whom they can be fatal. The risk of infection rises sharply as the neutrophil count falls below 500 cells/µL.
 Pathogenesis
Pathogenesis
The mechanisms underlying neutropenia can be divided into two broad categories:
• Decreased granulocyte production. Clinically important reductions in granulopoiesis are most often caused by marrow failure (as occurs in aplastic anemia), extensive replacement of the marrow by tumor (such as in leukemias), or cancer chemotherapy. Alternatively, some neutropenias are isolated, with only the differentiation of committed granulocytic precursors being affected. The forms of neutropenia are most often caused by certain drugs or, less commonly, by neoplastic proliferations of cytotoxic T cells and natural killer (NK) cells.
• Increased granulocyte destruction. This can be encountered with immune-mediated injury (triggered in some cases by drugs) or in overwhelming bacterial, fungal, or rickettsial infections due to increased peripheral utilization. Splenomegaly also can lead to the sequestration and accelerated removal of neutrophils.
 Morphology
Morphology
The alterations in the bone marrow depend on the underlying cause of the neutropenia. Marrow hypercellularity is seen when there is excessive neutrophil destruction or ineffective granulopoiesis, such as occurs in megaloblastic anemia. By contrast, agents such as drugs that cause neutropenia do so by suppressing granulocytopoiesis, thus decreasing the numbers of granulocytic precursors. Erythropoiesis and megakaryopoiesis can be normal if the responsible agent specifically affects granulocytes, but most myelotoxic drugs reduce marrow elements from all lineages.
Clinical Features
The initial symptoms often are malaise, chills, and fever, with subsequent marked weakness and fatigability. Infections constitute the major problem. They commonly take the form of ulcerating, necrotizing lesions of the gingiva, floor of the mouth, buccal mucosa, pharynx, or other sites within the oral cavity (agranulocytic angina). Owing to the lack of leukocytes, such lesions often contain large masses or sheets of microorganisms. In addition to removal of the offending drug and control of infection, treatment efforts may also include granulocyte colony-stimulating factor, which stimulates neutrophil production by the bone marrow.
Reactive Leukocytosis
An increase in the number of white cells in the blood is common in a variety of inflammatory states caused by microbial and nonmicrobial stimuli. Leukocytoses are relatively nonspecific and are classified according to the particular white cell series that is affected (Table 11–6). As discussed later on, in some cases reactive leukocytosis may mimic leukemia. Such “leukemoid” reactions must be distinguished from true white cell malignancies. Infectious mononucleosis merits separate consideration because it gives rise to a distinctive syndrome associated with lymphocytosis.
Table 11–6 Causes of Leukocytosis
Infectious Mononucleosis
Infectious mononucleosis is an acute, self-limited disease of adolescents and young adults that is caused by Epstein-Barr virus (EBV), a member of the herpesvirus family. The infection is characterized by (1) fever, sore throat, and generalized lymphadenitis and (2) a lymphocytosis of activated, CD8+ T cells. Of note, cytomegalovirus infection induces a similar syndrome that can be differentiated only by serologic methods.
EBV is ubiquitous in all human populations. In the developing world, EBV infection in early childhood is nearly universal. At this age, symptomatic disease is uncommon, and even though infected hosts mount an immune response (described later), more than half continue to shed virus. By contrast, in developed countries with better standards of hygiene, infection usually is delayed until adolescence or young adulthood. For unclear reasons, only about 20% of healthy seropositive persons in developed countries shed the virus, and only about 50% of those who are exposed to the virus acquire the infection.
Pathogenesis
Transmission to a seronegative “kissing cousin” usually involves direct oral contact. It is hypothesized (but has not been proved) that the virus initially infects oropharyngeal epithelial cells and then spreads to underlying lymphoid tissue (tonsils and adenoids), where mature B cells are infected. The infection of B cells takes one of two forms. In a minority of cells, the infection is lytic, leading to viral replication and eventual cell lysis accompanied by the release of virions. In most cells, however, the infection is nonproductive, and the virus persists in latent form as an extrachromosomal episome. B cells that are latently infected with EBV undergo polyclonal activation and proliferation, as a result of the action of several EBV proteins (Chapter 5). These cells disseminate in the circulation and secrete antibodies with several specificities, including the well-known heterophil anti-sheep red cell antibodies that are detected in diagnostic tests for mononucleosis. During acute infections, EBV is shed in the saliva; it is not known if the source of these virions is oropharyngeal epithelial cells or B cells.
A normal immune response is extremely important in controlling the proliferation of EBV-infected B cells and the spread of the virus. Early in the course of the infection, IgM antibodies are formed against viral capsid antigens. Later the serologic response shifts to IgG antibodies, which persist for life. More important in the control of the EBV-positive B cell proliferation are cytotoxic CD8+ T cells and NK cells. Virus-specific CD8+ T cells appear in the circulation as atypical lymphocytes, a finding that is characteristic of mononucleosis. In otherwise healthy persons, the fully developed humoral and cellular responses to EBV act as brakes on viral shedding. In most cases, however, a small number of latently infected EBV-positive B cells escape the immune response and persist for the life of the patient. As described later, impaired T cell immunity in the host can have disastrous consequences.
Morphology
The major alterations involve the blood, lymph nodes, spleen, liver, central nervous system, and occasionally other organs. There is peripheral blood leukocytosis; the white cell count is usually between 12,000 and 18,000 cells/µL. Typically more than half of these cells are large atypical lymphocytes, 12 to 16 µm in diameter, with an oval, indented, or folded nucleus and abundant cytoplasm with a few azurophilic granules (Fig. 11–12). These atypical lymphocytes, which are sufficiently distinctive to suggest the diagnosis, are mainly CD8+ T cells.
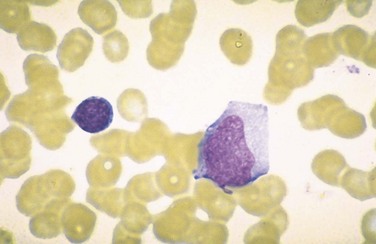
Figure 11–12 Atypical lymphocytes in infectious mononucleosis—peripheral blood smear. The cell on the left is a normal small resting lymphocyte with a compact nucleus and scant cytoplasm. By contrast, the atypical lymphocyte on the right has abundant cytoplasm and a large nucleus with dispersed chromatin.
Lymphadenopathy is common and is most prominent in the posterior cervical, axillary, and groin regions. On histologic examination, the enlarged nodes are flooded by atypical lymphocytes, which occupy the paracortical (T cell) areas. A few cells resembling Reed-Sternberg cells, the hallmark of Hodgkin lymphoma, often are seen. Because of these atypical features, special tests are sometimes needed to distinguish the reactive changes of mononucleosis from lymphoma.
The spleen is enlarged in most cases, weighing between 300 and 500 g, and exhibits a heavy infiltration of atypical lymphocytes. As a result of the rapid increase in splenic size and the infiltration of the trabeculae and capsule by the lymphocytes, such spleens are fragile and prone to rupture after even minor trauma.
Atypical lymphocytes usually also infiltrate the portal areas and sinusoids of the liver. Scattered apoptotic cells or foci of parenchymal necrosis associated with a lymphocytic infiltrate also may be present—a picture that can be difficult to distinguish from that in other forms of viral hepatitis.
Clinical Features
Although mononucleosis classically manifests with fever, sore throat, lymphadenitis, and the other features mentioned earlier, atypical presentations are not unusual. Sometimes there is little or no fever and only fatigue and lymphadenopathy, raising the specter of lymphoma; fever of unknown origin, unassociated with lymphadenopathy or other localized findings; hepatitis that is difficult to differentiate from one of the hepatotropic viral syndromes (Chapter 15); or a febrile rash resembling rubella. Ultimately, the diagnosis depends on the following findings, in increasing order of specificity: (1) lymphocytosis with the characteristic atypical lymphocytes in the peripheral blood, (2) a positive heterophil reaction (Monospot test), and (3) a rising titer of antibodies specific for EBV antigens (viral capsid antigens, early antigens, or Epstein-Barr nuclear antigen). In most patients, mononucleosis resolves within 4 to 6 weeks, but sometimes the fatigue lasts longer. Occasionally, one or more complications supervene. Perhaps the most common of these is hepatic dysfunction, associated with jaundice, elevated hepatic enzyme levels, disturbed appetite, and, rarely, even liver failure. Other complications involve the nervous system, kidneys, bone marrow, lungs, eyes, heart, and spleen (including fatal splenic rupture).
EBV is a potent transforming virus that plays a role in the pathogenesis of a number of human malignancies, including several types of B cell lymphoma (Chapter 5). A serious complication in those lacking T cell immunity (such as organ and bone marrow transplant recipients and HIV-infected individuals) is unimpeded EBV-driven B cell proliferation. This process can be initiated by an acute infection or the reactivation of a latent B cell infection and generally begins as a polyclonal proliferation that transforms to overt monoclonal B cell lymphoma over time. Reconstitution of immunity (e.g., by cessation of immunosuppressive drugs) is sometimes sufficient to cause complete regression of the B cell proliferation, which is uniformly fatal if left untreated.
The importance of T cells and NK cells in the control of EBV infection is driven home by X-linked lymphoproliferative syndrome, a rare inherited immunodeficiency characterized by an ineffective immune response to EBV. Most affected boys have mutations in the SH2D1A gene, which encodes a signaling protein that participates in the activation of T cells and NK cells and in antibody production. In more than 50% of cases, EBV causes an acute overwhelming infection that is usually fatal. Others succumb to lymphoma or infections related to hypogammaglobulinemia, the basis of which is not understood.
Reactive Lymphadenitis
Infections and nonmicrobial inflammatory stimuli often activate immune cells residing in lymph nodes, which act as defensive barriers. Any immune response against foreign antigens can lead to lymph node enlargement (lymphadenopathy). The infections causing lymphadenitis are varied and numerous, and may be acute or chronic. In most instances the histologic appearance of the lymph node reaction is nonspecific. A somewhat distinctive form of lymphadenitis that occurs with cat-scratch disease is described separately later.
Acute Nonspecific Lymphadenitis
This form of lymphadenitis may be isolated to a group of nodes draining a local infection, or be generalized, as in systemic infectious and inflammatory conditions.
Morphology
Inflamed nodes in acute nonspecific lymphadenitis are swollen, gray-red, and engorged. Histologically, there are large germinal centers containing numerous mitotic figures. When the cause is a pyogenic organism, a neutrophilic infiltrate is seen around the follicles and within the lymphoid sinuses. With severe infections, the centers of follicles can undergo necrosis, leading to the formation of an abscess.
Affected nodes are tender and may become fluctuant if abscess formation is extensive. The overlying skin is frequently red and may develop draining sinuses. With control of the infection the lymph nodes may revert to a normal “resting” appearance or if damaged undergo scarring.
Chronic Nonspecific Lymphadenitis
Depending on the causative agent, chronic nonspecific lymphadenitis can assume one of three patterns: follicular hyperplasia, paracortical hyperplasia, or sinus histiocytosis.
Morphology
Follicular Hyperplasia
This pattern occurs with infections or inflammatory processes that activate B cells, which migrate into B cell follicles and create the follicular (or germinal center) reaction. The reactive follicles contain numerous activated B cells, scattered T cells, and phagocytic macrophages containing nuclear debris (tingible body macrophages), and a meshwork of antigen-presenting follicular dendritic cells. Causes of follicular hyperplasia include rheumatoid arthritis, toxoplasmosis, and early HIV infection. This form of lymphadenitis can be confused morphologically with follicular lymphoma (discussed later). Findings that favor follicular hyperplasia are (1) the preservation of the lymph node architecture; (2) variation in the shape and size of the germinal centers; (3) the presence of a mixture of germinal center lymphocytes of varying shape and size; and (4) prominent phagocytic and mitotic activity in germinal centers.
Paracortical Hyperplasia
This pattern is caused by immune reactions involving the T cell regions of the lymph node. When activated, parafollicular T cells transform into large proliferating immunoblasts that can efface the B cell follicles. Paracortical hyperplasia is encountered in viral infections (such as EBV), after certain vaccinations (e.g., smallpox), and in immune reactions induced by drugs (especially phenytoin).
Sinus Histiocytosis
This reactive pattern is characterized by distention and prominence of the lymphatic sinusoids, owing to a marked hypertrophy of lining endothelial cells and an infiltrate of macrophages (histiocytes). It often is encountered in lymph nodes draining cancers and may represent an immune response to the tumor or its products.
Cat-Scratch Disease
Cat-scratch disease is a self-limited lymphadenitis caused by the bacterium Bartonella henselae. It is primarily a disease of childhood; 90% of the patients are younger than 18 years of age. It manifests with regional lymphadenopathy, most frequently in the axilla and the neck. The nodal enlargement appears approximately 2 weeks after a feline scratch or, less commonly, after a splinter or thorn injury. An inflammatory nodule, vesicle, or eschar is sometimes visible at the site of the skin injury. In most patients the lymph node enlargement regresses over a period of 2 to 4 months. Encephalitis, osteomyelitis, or thrombocytopenia may develop in rare patients.
Morphology
The nodal changes in cat-scratch disease are quite characteristic. Initially sarcoid-like granulomas form, but these then undergo central necrosis associated with an infiltrate of neutrophils. These irregular stellate necrotizing granulomas are similar in appearance to those seen in a limited number of other infections, such as lymphogranuloma venereum. The microbe is extracellular and can be visualized with silver stains. The diagnosis is based on a history of exposure to cats, the characteristic clinical findings, a positive result on serologic testing for antibodies to Bartonella, and the distinctive morphologic changes in the lymph nodes.
Neoplastic Proliferations of White Cells
Tumors are the most important disorders of white cells. They can be divided into three broad categories based on the origin of the tumor cells:
• Lymphoid neoplasms, which include non-Hodgkin lymphomas (NHLs), Hodgkin lymphomas, lymphocytic leukemias, and plasma cell neoplasms and related disorders. In many instances tumors are composed of cells resembling some normal stage of lymphocyte differentiation, a feature that serves as one of the bases for their classification.
• Myeloid neoplasms arise from progenitor cells that give rise to the formed elements of the blood: granulocytes, red cells, and platelets. The myeloid neoplasms fall into three fairly distinct subcategories: acute myeloid leukemias, in which immature progenitor cells accumulate in the bone marrow; myeloproliferative disorders, in which an inappropriate increase in the production of formed blood elements leads to elevated blood cell counts; and myelodysplastic syndromes, which are characteristically associated with ineffective hematopoiesis and cytopenias.
• Histiocytic neoplasms include proliferative lesions of macrophages and dendritic cells. Of special interest is a spectrum of proliferations of Langerhans cells (Langerhans cell histiocytoses).
Lymphoid Neoplasms
The numerous lymphoid neoplasms vary widely in their clinical presentation and behavior, and thus present challenges to students and clinicians alike. Some characteristically manifest as leukemias, with involvement of the bone marrow and the peripheral blood. Others tend to manifest as lymphomas, tumors that produce masses in lymph nodes or other tissues. Plasma cell tumors usually arise within the bones and manifest as discrete masses, causing systemic symptoms related to the production of a complete or partial monoclonal immunoglobulin. While these tendencies are reflected in the names given to these entities, in reality all lymphoid neoplasms have the potential to spread to lymph nodes and various tissues throughout the body, especially the liver, spleen, bone marrow, and peripheral blood. Because of their overlapping clinical behavior, the various lymphoid neoplasms can be distinguished with certainty only by the morphologic and molecular characteristics of the tumor cells. Stated another way, for purposes of diagnosis and prognostication, it is most important to focus on what the tumor cell is, not where it resides in the patient.
Two groups of lymphomas are recognized: Hodgkin lymphomas and non-Hodgkin lymphomas. Although both arise most commonly in lymphoid tissues, Hodgkin lymphoma is set apart by the presence of distinctive neoplastic Reed-Sternberg giant cells (see later), which usually are greatly outnumbered by non-neoplastic inflammatory cells. The biologic behavior and clinical treatment of Hodgkin lymphoma also are different from those of NHLs, making the distinction of practical importance.
Historically, few areas of pathology evoked as much controversy and confusion as the classification of lymphoid neoplasms, which is perhaps inevitable in view of the intrinsic complexity of the immune system, from which they arise. Great progress has been made over the past several decades, however, and an international working group of pathologists, molecular biologists, and clinicians working on behalf of the World Health Organization (WHO) has formulated a widely accepted classification scheme that relies on a combination of morphologic, phenotypic, genotypic, and clinical features. As background for the subsequent discussion of this classification, certain important principles warrant consideration:
• B and T cell tumors often are composed of cells that are arrested at or derived from a specific stage of their normal differentiation (Fig. 11–13). The diagnosis and classification of these tumors rely heavily on tests (either immunohistochemistry or flow cytometry) that detect lineage-specific antigens (e.g., B cell, T cell, and NK cell markers) and markers of maturity. By convention, many such markers are identified by their cluster of differentiation (CD) number.
• The most common lymphomas are derived from germinal center or post–germinal center B cells. This conclusion is drawn from molecular analyses showing that most B cell lymphomas have undergone somatic hypermutation, an event confined to germinal center B cells. Normal germinal center B cells also undergo immunoglobulin class switching, an event that allows B cells to express immunoglobulins other than IgM. Class switching and somatic hypermutation are mistake-prone forms of regulated genomic instability, which places germinal center B cells at high risk for potentially transforming mutations. In fact, many of the recurrent chromosomal translocations found in mature B cell malignancies involve the immunoglobulin loci and appear to stem from “accidents” during attempted diversification of the immunoglobulin genes. In this regard, it is interesting that mature T cells, which are genomically stable, give rise to lymphomas infrequently and only very rarely have chromosomal translocations involving the T cell receptor loci.
• All lymphoid neoplasms are derived from a single transformed cell and are therefore clonal. As described in Chapter 4, differentiating precursor B and T cells rearrange their antigen receptor genes, thereby ensuring that each lymphocyte makes a single, unique antigen receptor. Because antigen receptor gene rearrangement virtually always precedes transformation, the daughter cells derived from a given malignant progenitor share the same antigen receptor gene configuration and synthesize identical antigen receptor proteins (either immunoglobulins or T cell receptors). Thus, analyses of antigen receptor genes and their protein products can be used to differentiate clonal neoplasms from polyclonal, reactive processes.
• Lymphoid neoplasms often disrupt normal immune function. Both immunodeficiency (as evident by increased susceptibility to infection) and autoimmunity may be seen, sometimes in the same patient. Ironically, patients with inherited or acquired immunodeficiency are themselves at high risk for the development of certain lymphoid neoplasms, particularly those associated with EBV infection.
• Although NHLs often manifest at a particular tissue site, sensitive molecular assays usually show the tumor to be widely disseminated at diagnosis. As a result, with few exceptions, only systemic therapies are curative. By contrast, Hodgkin lymphoma often arises at a single site and spreads in a predictable fashion to contiguous lymph node groups. For this reason, early in its course, it is sometimes treated with local therapy alone.
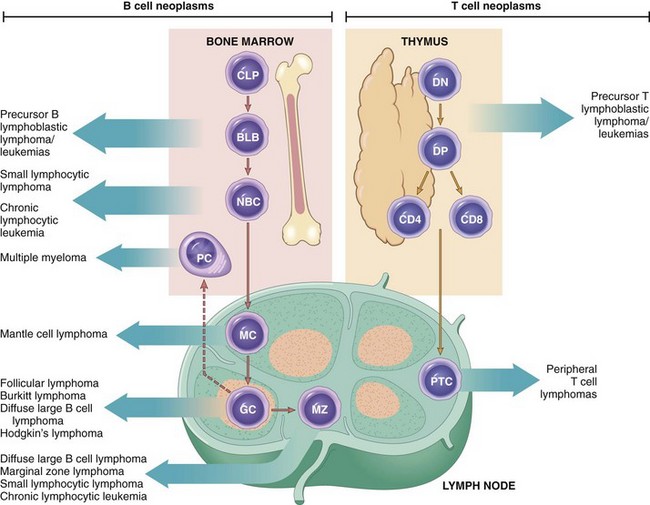
Figure 11–13 Origin of lymphoid neoplasms. Stages of B and T cell differentiation from which specific lymphoid and tumors emerge are shown. BLB, pre-B lymphoblast; CLP, common lymphoid progenitor; DN, CD4−/CD8− (double-negative) pro-T cell; DP, CD4+/CD8+ (double-positive) pre-T cell; GC, germinal center B cell; MC, mantle zone B cell; MZ, marginal zone B cell; NBC, naive B cell; PC, plasma cell; PTC, peripheral T cell.
The WHO classification of lymphoid neoplasms considers the morphology, cell of origin (determined by immunophenotyping), clinical features, and genotype (e.g., karyotype, presence of viral genomes) of each entity. It encompasses all lymphoid neoplasms, including leukemias and multiple myeloma, and separates them on the basis of origin into three major categories: (1) tumors of B cells, (2) tumors of T cells and NK cells, and (3) Hodgkin lymphoma.
An updated version of the WHO classification of lymphoid neoplasms is presented in Table 11–7. As is evident, the diagnostic entities are numerous. The focus here is on the following subsets of neoplasms:
• Precursor B and T cell lymphoblastic lymphoma/leukemia—commonly called acute lymphoblastic leukemia (ALL)
• Chronic lymphocytic leukemia/small lymphocytic lymphoma
• Diffuse large B cell lymphomas
Table 11–7 WHO Classification of Lymphoid Neoplasms*
NK, natural killer; WHO, World Health Organization.
* Entries in italics are among the most common lymphoid tumors.
Together these neoplasms constitute more than 90% of the lymphoid tumors seen in the United States.
The salient features of the more common lymphoid leukemias, non-Hodgkin lymphomas, and plasma cell tumors are summarized in Table 11–8. Hodgkin lymphomas will be discussed later. Also included in the following discussion are a few of the uncommon entities with distinctive clinicopathologic features.
Table 11–8 Characteristics of the More Common Lymphoid Leukemias, Non-Hodgkin Lymphomas, and Plasma Cell Tumors
Acute Lymphoblastic Leukemia/Lymphoblastic Lymphoma
Acute lymphoblastic leukemia (ALL) and lymphoblastic lymphoma are aggressive tumors, composed of immature lymphocytes (lymphoblasts), that occur predominantly in children and young adults. The various lymphoblastic tumors are morphologically indistinguishable, often cause similar signs and symptoms, and are treated similarly. These tumors are therefore considered together here.
Just as B cell precursors normally develop within the bone marrow, pre-B cell tumors usually manifest in the bone marrow and peripheral blood as leukemias. Similarly, pre-T cell tumors commonly manifest as masses involving the thymus, the normal site of early T cell differentiation. However, pre-T cell “lymphomas” often progress rapidly to a leukemic phase, and other pre-T cell tumors seem to involve only the marrow at presentation. Hence, both pre-B and pre-T cell tumors usually take on the clinical appearance of ALL at some time during their course. As a group, ALLs constitute 80% of childhood leukemia, peaking in incidence at age 4, with most cases being of pre-B cell origin. The pre-T cell tumors are most common in male patients between 15 and 20 years of age.
The pathogenesis, laboratory findings, and clinical features of ALL closely resemble those of acute myeloid leukemia (AML), the other major type of acute leukemia. Because of these similarities, the features common to the acute leukemias are reviewed first, followed by a discussion of those specific to ALL.
Pathogenesis
The principal pathogenic defect in acute leukemia and lymphoblastic lymphoma is a block in differentiation. This “maturation arrest” stems from acquired mutations in specific transcription factors that regulate the differentiation of immature lymphoid or myeloid progenitors. Normal B cell, T cell, and myeloid differentiation are regulated by different lineage-specific transcription factors; accordingly, the mutated transcription factor genes found in acute leukemias derived from each of these lineages also are distinct. The most commonly mutated transcription factor genes are TEL1, AML1, E2A, PAX5, and EBF in ALLs of B cell origin (B-ALLs) and TAL1 and NOTCH1 in T cell ALLs (T-ALLs).
Acute leukemias also are associated with complementary acquired mutations that allow the tumor cells to proliferate in a growth factor–independent fashion. In B-ALL, one of the most important mutations of this type is a BCR-ABL fusion gene created by a (9;22) translocation (the so-called Philadelphia chromosome, for the city of its discovery). As discussed later on, the same translocation also is found in chronic myelogenous leukemia (CML). The BCR-ABL fusion gene encodes a BCR-ABL tyrosine kinase that constitutively activates the same pathways that are normally stimulated by growth factors. Some T-ALLs are associated with a different ABL fusion gene, NUP214-ABL, which has functional consequences similar to those of BCR-ABL.
In tumors manifesting as “leukemias,” blasts accumulating in the marrow suppress the growth of normal hematopoietic cells by physical displacement and by other, poorly understood mechanisms. Eventually this suppression produces bone marrow failure, which accounts for the major clinical manifestations. The therapeutic goal, therefore, is to reduce the leukemic clone sufficiently to allow normal hematopoiesis to resume.
Clinical Features of Acute Leukemias
Acute leukemias have the following characteristics:
• Abrupt, stormy onset. Most patients present for medical attention within 3 months of the onset of symptoms.
• Clinical signs and symptoms related to suppressed marrow function, including fatigue (due to anemia), fever (reflecting infections resulting from neutropenia), and bleeding (petechiae, ecchymoses, epistaxis, gum bleeding) secondary to thrombocytopenia
• Bone pain and tenderness, resulting from marrow expansion and infiltration of the subperiosteum
• Generalized lymphadenopathy, splenomegaly, and hepatomegaly due to dissemination of the leukemic cells. These are more pronounced in ALL than in AML.
• Central nervous system manifestations, including headache, vomiting, and nerve palsies resulting from meningeal spread. These are more common in children than in adults and in ALL than in AML.
Laboratory Findings in Acute Leukemias
The diagnosis of acute leukemia rests on the identification of blasts. The peripheral blood sometimes contains no blasts (aleukemic leukemia); in such cases the diagnosis can be established only by marrow examination.
The white cell count is variable; it may be greater than 100,000 cells/µL but in about half of the patients is less than 10,000 cells/µL. Anemia is almost always present, and the platelet count usually is below 100,000/µL. Neutropenia is another common finding.
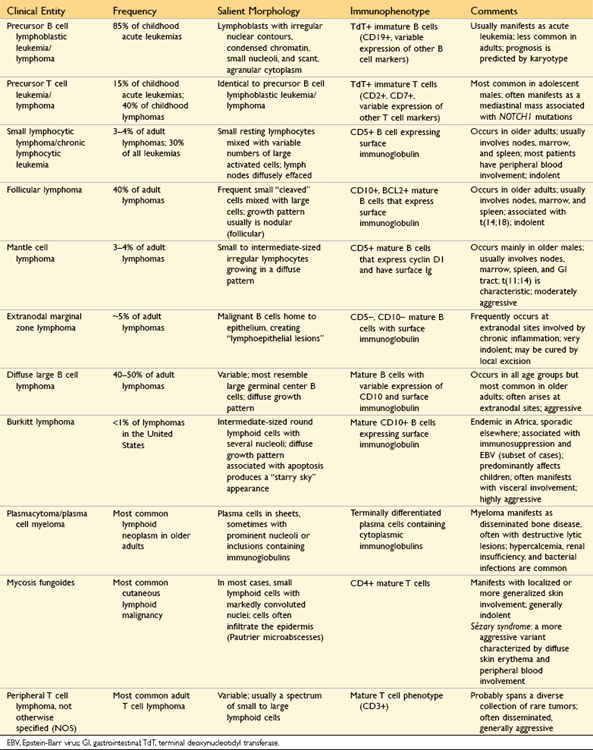
Morphology
Because of differing responses to therapy, it is of great practical importance to distinguish ALL from AML. By definition, in ALL blasts compose more than 25% of the marrow cellularity. In Wright-Giemsa–stained preparations, lymphoblasts have coarse, clumped chromatin, one or two nucleoli, and scant agranular cytoplasm (Fig. 11–14, A), whereas myeloblasts have nuclei with finer chromatin and more cytoplasm, which often contains granules (Fig. 11–14, B). Lymphoblasts also often contain cytoplasmic glycogen granules that are periodic acid–Schiff–positive, whereas myeloblasts are often peroxidase-positive.
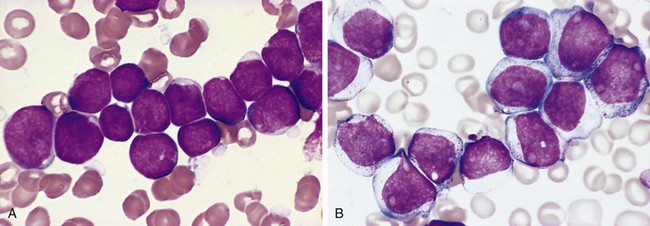
Figure 11–14 Morphologic comparison of lymphoblasts and myeloblasts. A, Lymphoblastic leukemia/lymphoma. Lymphoblasts have condensed nuclear chromatin, small nucleoli, and scant agranular cytoplasm. B, Acute myeloid leukemia. Myeloblasts have delicate nuclear chromatin, prominent nucleoli, and fine azurophilic cytoplasmic granules.
(Courtesy of Dr. Robert W. McKenna, Department of Pathology, University of Texas Southwestern Medical School, Dallas, Texas.)
With completion of the foregoing “short course” in acute leukemia, our focus now returns to the ALLs; the AMLs are discussed later.
Genetic Features
Approximately 90% of ALLs have nonrandom karyotypic abnormalities. Most common in childhood pre-B cell tumors are hyperdiploidy (more than 50 chromosomes/cell) and the presence of a cryptic (12;21) translocation involving the TEL1 and AML1 genes, while about 25% of adult pre-B cell tumors harbor the (9;22) translocation involving the ABL and BCR genes. Pre-T cell tumors are associated with diverse chromosomal aberrations, including frequent translocations involving the T cell receptor loci and transcription factor genes such as TAL1.
Immunophenotypic Features
Immunophenotyping is very useful in subtyping lymphoblastic tumors and distinguishing them from AML. Terminal deoxynucleotidyl transferase (TdT), an enzyme specifically expressed in pre-B and pre-T cells, is present in more than 95% of cases. Further subtyping of ALL into pre-B and pre-T cell types relies on stains for lineage-specific markers, such as CD19 (B cell) and CD3 (T cell).
Prognosis
Treatment of childhood ALL is one of the great success stories in oncology. Children 2 to 10 years of age have the best prognosis; with intensive chemotherapy up to 80% are cured. Other groups of patients do less well. Variables correlated with worse outcomes include male gender; age younger than 2 or older than 10 years; a high leukocyte count at diagnosis; and molecular evidence of persistent disease on day 28 of treatment. Age-dependent differences in the frequencies of various karyotypic abnormalities largely explain the relationship of age to outcome. Tumors with “good prognosis” chromosomal aberrations (such as the t[12;21] and hyperdiploidy) are common in the 2- to 10-year age group. By contrast, rearrangements of the gene MLL or the presence of a BCR-ABL fusion gene, both associated with poor outcomes in B cell tumors, are most common in children younger than 2 years of age and adults, respectively. None of the chromosomal rearrangements found in pre-T cell tumors is predictive of outcome.
Chronic Lymphocytic Leukemia/Small Lymphocytic Lymphoma
Chronic lymphocytic leukemia (CLL) and small lymphocytic lymphoma (SLL) are essentially identical, differing only in the extent of peripheral blood involvement. Somewhat arbitrarily, if the peripheral blood lymphocyte count exceeds 4000 cells/µL, the patient is diagnosed with CLL; if it does not, a diagnosis of SLL is made. Most patients with lymphoid neoplasms fit the diagnostic criteria for CLL, which is the most common leukemia of adults in the Western world. By contrast, SLL constitutes only 4% of NHLs. For unclear reasons, CLL/SLL is much less common in Asia.
Pathogenesis
CLL/SLL is an indolent, slowly growing tumor, suggesting that increased tumor cell survival is more important than tumor cell proliferation in this disease. In line with this idea, the tumor cells contain high levels of BCL2, a protein that inhibits apoptosis (Chapters 1 and 5). Unlike in follicular lymphoma (discussed later), the BCL2 gene is not rearranged. Some evidence suggests that BCL2 is upregulated in the tumor cells as a consequence of the loss of several regulatory micro-RNAs that are encoded on chromosome 13.
Another important pathogenic aspect of CLL/SLL is immune dysregulation. Through unclear mechanisms, the accumulation of CLL/SLL cells suppresses normal B cell function, often resulting in hypogammaglobulinemia. Paradoxically, approximately 15% of patients have autoantibodies against their own red cells or platelets. When present, the autoantibodies are made by nonmalignant bystander B cells, indicating that the tumor cells somehow impair immune tolerance. As time passes the tumor cells tend to displace the normal marrow elements, leading to anemia, neutropenia, and eventual thrombocytopenia.
Morphology
In SLL/CLL, sheets of small lymphocytes and scattered ill-defined foci of larger, actively dividing cells diffusely efface involved lymph nodes (Fig. 11–15, A). The predominant cells are small, resting lymphocytes with dark, round nuclei, and scanty cytoplasm (Fig. 11–15, B). The foci of mitotically active cells are called proliferation centers, which are pathognomonic for CLL/SLL. In addition to the lymph nodes, the bone marrow, spleen, and liver are involved in almost all cases. In most patients there is an absolute lymphocytosis featuring small, mature-looking lymphocytes. The circulating tumor cells are fragile and during the preparation of smears frequently are disrupted, producing characteristic smudge cells. Variable numbers of larger activated lymphocytes are also usually present in the blood smear.
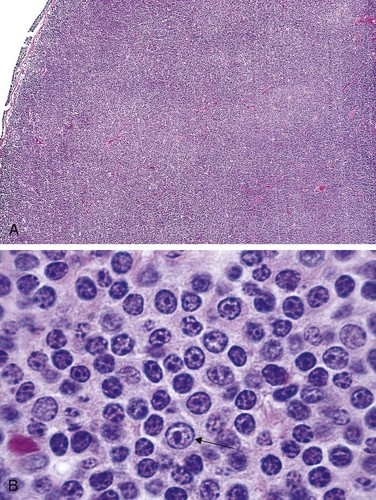
Figure 11–15 Small lymphocytic lymphoma/chronic lymphocytic leukemia—lymph node. A, Low-power view shows diffuse effacement of nodal architecture. B, At high power, a majority of the tumor cells have the appearance of small, round lymphocytes. A “prolymphocyte,” a larger cell with a centrally placed nucleolus, also is present in this field (arrow).
(A, Courtesy of Dr. José Hernandez, Department of Pathology, University of Texas Southwestern Medical School, Dallas, Texas.)
Immunophenotypic and Genetic Features
CLL/SLL is a neoplasm of mature B cells expressing the pan-B cell markers CD19, CD20, and CD23 and surface immunoglobulin heavy and light chains. The tumor cells also express CD5. This is a helpful diagnostic clue, since among B cell lymphomas only CLL/SLL and mantle cell lymphoma (discussed later) commonly express CD5. Approximately 50% of tumors have karyotypic abnormalities, the most common of which are trisomy 12 and deletions of chromosomes 11, 13, and 17. “Deep sequencing” of CLL/SLL cell genomes has identified activating mutations in the Notch1 receptor in a subset of cases that predict a worse outcome.Unlike in other B cell neoplasms, chromosomal translocations are rare.
Clinical Features
When first detected, CLL/SLL is often asymptomatic. The most common clinical signs and symptoms are nonspecific and include easy fatigability, weight loss, and anorexia. Generalized lymphadenopathy and hepatosplenomegaly are present in 50% to 60% of patients. The leukocyte count may be increased only slightly (in SLL) or may exceed 200,000 cells/µL. Hypogammaglobulinemia develops in more than 50% of the patients, usually late in the course, and leads to an increased susceptibility to bacterial infections. Less commonly autoimmune hemolytic anemia and thrombocytopenia are seen. The course and prognosis are extremely variable. Many patients live more than 10 years after diagnosis and die of unrelated causes. The median survival is 4 to 6 years, however, and as time passes, CLL/SLL tends to transform to more aggressive tumors that resemble either prolymphocytic leukemia or diffuse large B cell lymphoma. Once transformation occurs, the median survival is less than 1 year.
Follicular Lymphoma
This relatively common tumor constitutes 40% of the adult NHLs in the United States. Like CLL/SLL, it occurs much less frequently in Asian populations.
Pathogenesis
As in CLL/SLL, the neoplastic cells characteristically express BCL2, a protein that is absent from normal germinal center B cells. Greater than 85% of tumors have a characteristic (14;18) translocation that fuses the BCL2 gene on chromosome 18 to the IgH locus on chromosome 14. This chromosomal rearrangement explains the inappropriate “overexpression” of BCL2 protein in the tumor cells and contributes to tumor cell survival. Whole genome sequencing of follicular lymphomas has identified loss-of-function mutations in several genes encoding histone acetyl transferases in about a third of cases, suggesting that epigenetic changes also contribute to the genesis of these tumors.
Morphology
Lymph nodes usually are effaced by a distinctly nodular proliferation (Fig 11–16, A). The tumor cells resemble normal germinal center B cells. Most commonly the predominant neoplastic cells are slightly larger than resting lymphocytes that have angular “cleaved” nuclei with prominent indentations and linear infoldings (Fig. 11–16, B). The nuclear chromatin is coarse and condensed, and nucleoli are indistinct. These small, cleaved cells are mixed with variable numbers of larger cells with vesicular chromatin, several nucleoli, and modest amounts of cytoplasm. In most tumors, large cells are a minor component of the overall cellularity, mitoses are infrequent, and single necrotic cells (cells undergoing apoptosis) are not seen. These features help to distinguish follicular lymphoma from follicular hyperplasia, in which mitoses and apoptosis are prominent. Uncommonly, large cells predominate, a histologic pattern that correlates with a more aggressive clinical behavior.
Figure 11–16 Follicular lymphoma—lymph node. A, Nodular aggregates of lymphoma cells are present throughout. B, At high magnification, small lymphoid cells with condensed chromatin and irregular or cleaved nuclear outlines (centrocytes) are mixed with a population of larger cells with nucleoli (centroblasts).
(A, Courtesy of Dr. Robert W. McKenna, Department of Pathology, University of Texas Southwestern Medical School, Dallas, Texas.)
Immunophenotypic Features
These tumors express pan-B cell markers (CD19 and CD20), CD10, and BCL6, a transcription factor required for the generation of germinal center B cells.
Clinical Features
Follicular lymphoma mainly occurs in adults older than 50 years of age and affects males and females equally. It usually manifests as painless, generalized lymphadenopathy. The bone marrow is almost always involved at diagnosis, while visceral disease is uncommon. While the natural history is prolonged (median survival, 7 to 9 years), follicular lymphoma is not curable, an unfortunate feature shared with most other relatively indolent lymphoid malignancies. As a result, therapy with cytotoxic drugs and rituximab (anti-CD20 antibody) is reserved for those with bulky, symptomatic disease. In about 40% of patients, follicular lymphoma progresses to diffuse large B cell lymphoma. This transformation is an ominous event, as tumors arising from such conversions are much less curable than de novo diffuse large B cell lymphomas, described later.
Mantle Cell Lymphoma
Mantle cell lymphoma is composed of cells resembling the naive B cells found in the mantle zones of normal lymphoid follicles. It constitutes approximately 4% of all NHLs and occurs mainly in men older than 50 years of age.
Morphology
Mantle cell lymphoma may involve lymph nodes in a diffuse or vaguely nodular pattern. The tumor cells usually are slightly larger than normal lymphocytes and have an irregular nucleus, inconspicuous nucleoli, and scant cytoplasm. Less commonly, the cells are larger and morphologically resemble lymphoblasts. The bone marrow is involved in most cases and the peripheral blood in about 20% of cases. The tumor sometimes arises in the gastrointestinal tract, often manifesting as multifocal submucosal nodules that grossly resemble polyps (lymphomatoid polyposis).
Immunophenotypic and Genetic Features
Almost all tumors have an (11;14) translocation that fuses the cyclin D1 gene to the IgH locus. This translocation dysregulates the expression of cyclin D1, a cell cycle regulator (Chapter 5), and is believed to be an important mediator of uncontrolled tumor cell growth. The tumor cells usually coexpress surface IgM and IgD, pan-B cell antigens (CD19 and CD20), and CD5. Mantle cell lymphoma is most readily distinguished from CLL/SLL by the absence of proliferation centers and the presence of cyclin D1 protein.
Diffuse Large B Cell Lymphoma
Diffuse large B cell lymphoma is the most common type of lymphoma in adults, accounting for approximately 50% of adult NHLs. It includes several subtypes that share an aggressive natural history.
Pathogenesis
About one third of tumors have rearrangements of the BCL6 gene, located on 3q27, and an even higher fraction of tumors have activating point mutations in the BCL6 promoter. Both aberrations result in increased levels of BCL6 protein, an important transcriptional regulator of gene expression in germinal center B cells. Another 30% of tumors have a (14;18) translocation involving the BCL2 gene that results in overexpression of BCL2 protein. Some of these tumors may represent “transformed” follicular lymphomas. Indeed, like follicular lymphoma, about a third of diffuse large B cell lymphomas have loss-of-function mutations in genes encoding histone acetyl transferases, pointing to a potential role for epigenetic alterations in this tumor.
Morphology
The neoplastic B cells are large (at least three to four times the size of resting lymphocytes) and vary in appearance from tumor to tumor. In many tumors, cells with round or oval nuclear contours, dispersed chromatin, several distinct nucleoli, and modest amounts of pale cytoplasm predominate (Fig. 11–17). In other tumors, the cells have a round or multilobate vesicular nucleus, one or two prominent centrally placed nucleoli, and abundant pale or basophilic cytoplasm. Occasionally, the tumor cells are highly anaplastic and include tumor giant cells resembling Reed-Sternberg cells, the malignant cells of Hodgkin lymphoma.
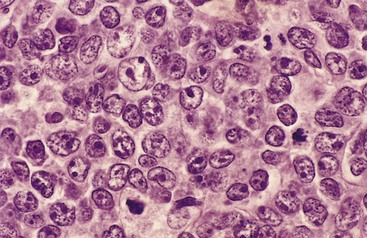
Immunophenotypic Features
These mature B cell tumors express pan-B cell antigens, such as CD19 and CD20. Many also express surface IgM and/or IgG. Other antigens (e.g., CD10, BCL2) are variably expressed.
Subtypes of Diffuse Large B Cell Lymphoma
Several distinctive clinicopathologic subtypes are included in the category of diffuse large B cell lymphoma. EBV-associated diffuse large B cell lymphomas arise in the setting of the acquired immunodeficiency syndrome (AIDS), iatrogenic immunosuppression (e.g., in transplant recipients), and the elderly. In the post-transplantation setting, these tumors often begin as EBV-driven polyclonal B cell proliferations that may regress if immune function is restored. Otherwise, transformation to clonal large B cell lymphoma is observed over weeks to months. Kaposi sarcoma herpesvirus (KSHV), also called human herpesvirus type 8 (HHV-8), is associated with rare primary effusion lymphomas, which may arise within the pleural cavity, pericardium, or peritoneum. These lymphomas are latently infected with KSHV, which encodes proteins homologous to several known oncoproteins, including cyclin D1, and are confined to immunosuppressed hosts. Of note, KSHV also is associated with Kaposi sarcoma in patients with AIDS (Chapters 4 and 9). Mediastinal large B cell lymphoma occurs most often in young women and shows a predilection for spread to abdominal viscera and the central nervous system.
Clinical Features
Although the median age at presentation is about 60 years, diffuse large B cell lymphomas can occur at any age; they constitute about 15% of childhood lymphomas. Patients typically present with a rapidly enlarging, often symptomatic mass at one or several sites. Extranodal presentations are common. Although the gastrointestinal tract and the brain are among the more frequent extranodal sites, tumors can appear in virtually any organ or tissue. Unlike the more indolent lymphomas (e.g., follicular lymphoma), involvement of the liver, spleen, and bone marrow is not common at diagnosis.
Without treatment, diffuse large cell B cell lymphomas are aggressive and rapidly fatal. With intensive combination chemotherapy and anti-CD20 immunotherapy, complete remissions are achieved in 60% to 80% of the patients; of these, approximately 50% remain free of disease and appear to be cured. For those not so fortunate, other aggressive treatments (e.g., high-dose chemotherapy and hematopoietic stem cell transplantation) offer some hope. Microarray-based molecular profiling of these tumors can predict response to current therapies and is being used to identify new, targeted therapy approaches.
Burkitt Lymphoma
Burkitt lymphoma is endemic in parts of Africa and occurs sporadically in other geographic areas, including the United States. Histologically, the African and nonendemic diseases are identical, although there are clinical and virologic differences.
Pathogenesis
Burkitt lymphoma is highly associated with translocations involving the MYC gene on chromosome 8. Most translocations fuse MYC with the IgH gene on chromosome 14, but variant translocations involving the κ and λ light chain loci on chromosomes 2 and 22, respectively, are also observed. The net result of each is the same—the dysregulation and overexpression of MYC protein. The role of MYC in transformation is discussed in Chapter 5. In most endemic cases and about 20% of sporadic cases, the tumor cells are latently infected with EBV, but the role of EBV in the genesis of this tumor remains uncertain.
Morphology
The tumor cells are intermediate in size and typically have round or oval nuclei and two to five distinct nucleoli (Fig. 11–18). There is a moderate amount of basophilic or amphophilic cytoplasm that often contains small, lipid-filled vacuoles (a feature appreciated only on smears). Very high rates of proliferation and apoptosis are characteristic, the latter accounting for the presence of numerous tissue macrophages containing ingested nuclear debris. These benign macrophages often are surrounded by a clear space, creating a “starry sky” pattern.
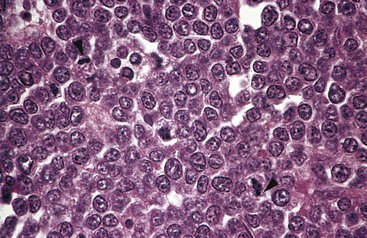
Figure 11–18 Burkitt lymphoma—lymph node. The tumor cells and their nuclei are fairly uniform, giving a monotonous appearance. Note the high level of mitotic activity (arrowheads) and prominent nucleoli. The “starry sky” pattern produced by interspersed, lightly staining, normal macrophages is better appreciated at a lower magnification.
(Courtesy of Dr. Robert W. McKenna, Department of Pathology, University of Texas Southwestern Medical School, Dallas, Texas.)
Immunophenotypic Features
These B cell tumors express surface IgM, the pan-B cell markers CD19 and CD20, and the germinal center B cell markers CD10 and BCL6.
Clinical Features
Both the endemic and nonendemic sporadic forms affect mainly children and young adults. Burkitt lymphoma accounts for approximately 30% of childhood NHLs in the United States. In both settings, the disease usually arises at extranodal sites. Endemic tumors often manifest as maxillary or mandibular masses, whereas abdominal tumors involving the bowel, retroperitoneum, and ovaries are more common in North America. Leukemic presentations are uncommon but do occur and must be distinguished from ALL, which is treated with different drug regimens. Burkitt lymphoma is among the fastest-growing human neoplasms; however, with very aggressive chemotherapy regimens, a majority of patients can be cured.
Multiple Myeloma and Related Plasma Cell Tumors
In virtually all cases, multiple myelomas and related plasma cell tumors secrete a single complete or partial immunoglobulin. Because these immunoglobulins can be detected in the serum, these disorders are also referred to as monoclonal gammopathies, and the associated immunoglobulin is often referred to as an M protein. Although M proteins may be indicative of overt malignancy, they also are found surprisingly often in otherwise normal elderly persons—a condition called monoclonal gammopathy of undetermined significance (MGUS), described later. Collectively, these disorders account for approximately 15% of the deaths that are caused by tumors of white blood cells. They are most common in middle-aged and elderly persons.
The plasma cell neoplasms can be divided into six major variants: (1) multiple myeloma, (2) solitary plasmacytoma, (3) lymphoplasmacytic lymphoma, (4) heavy-chain disease, (5) primary amyloidosis, and (6) MGUS. The focus here is on the most important of these disorders, multiple myeloma and lymphoplasmacytic lymphoma, with a brief discussion of the other disorders.
Multiple Myeloma
Multiple myeloma is one of the most common lymphoid malignancies; approximately 20,000 new cases are diagnosed in the United States each year. The median age at diagnosis is 70 years of age, and it is more common in males and in people of African origin. It principally involves the bone marrow and usually is associated with lytic lesions throughout the skeletal system.
The most frequent M protein produced by myeloma cells is IgG (60%), followed by IgA (20% to 25%); only rarely are IgM, IgD, or IgE M proteins observed. In the remaining 15% to 20% of cases, the plasma cells produce only κ or λ light chains. Because of their low molecular weight, free light chains are excreted in the urine, where they are termed Bence Jones proteins. Even more commonly, malignant plasma cells secrete both complete immunoglobulins and free light chains and thus produce both M proteins and Bence Jones proteins. As described later on, the excess light chains have important pathogenic effects.
Solitary Plasmacytoma
Sometimes plasma tumors manifest as solitary plasmacytomas involving the skeleton or the soft tissues. Solitary skeletal plasmacytomas tend to occur in the same locations as does multiple myeloma and usually progress to full-blown multiple myeloma over a period of 5 to 10 years; these tumors probably are best thought of as an early stage of multiple myeloma. Modestly elevated M proteins are present in some cases at diagnosis. By contrast, plasmacytomas that occur in soft tissues (most often the upper respiratory tract) spread infrequently and often are cured by local resection.
Pathogenesis Of Myeloma
As with most other B cell malignancies, myelomas often have chromosomal translocations involving the IgH locus on chromosome 14 and various other genes, including cyclin D1, fibroblast growth factor receptor 3, and cyclin D3 genes. Late in the course, translocations involving MYC are sometimes also observed. As might be surmised from this list of genes, dysregulation of D cyclins is common in multiple myeloma.
The proliferation of myeloma cells is supported by the cytokine interleukin 6 (IL-6), which is produced by fibroblasts and macrophages in the bone marrow stroma. The characteristic bone resorption results from the secretion of certain cytokines (e.g., IL-1β, tumor necrosis factor, IL-6) by myeloma cells. These cytokines stimulate production of another cytokine called RANK-ligand, which stimulates the differentiation and absorptive activity of osteoclasts (Chapter 20).
Patients with myeloma are immunosuppressed. Through uncertain mechanisms, myeloma cells interfere with the function of normal plasma cells, leading to defects in antibody production. Thus, although the plasma usually contains increased immunoglobulin owing to the presence of an M protein, the levels of functional antibodies often are profoundly depressed, leaving patients at high risk for bacterial infections.
Renal dysfunction is a common, serious problem in myeloma. It stems from several pathologic effects that may occur alone or in combination. Most important are obstructive proteinaceous casts, which often form in the distal convoluted tubules and the collecting ducts. The casts consist mostly of Bence Jones proteins along with variable amounts of complete immunoglobulins, Tamm-Horsfall protein, and albumin. Light chain deposition in the glomeruli or the interstitium, either as amyloid or linear deposits, also may contribute to renal dysfunction. Completing the assault on the kidney is hypercalcemia, which may lead to dehydration and renal stones, and frequent bouts of bacterial pyelonephritis, which stem in part from the hypogammaglobulinemia.
Monoclonal Gammopathy of Undetermined Significance
Monoclonal gammopathy of undetermined significance (MGUS) is the term applied to an asymptomatic monoclonal gammopathy. M proteins are found in the serum of 1% to 3% of otherwise healthy persons older than age 50 years, making this the most common plasma cell proliferation. Despite its name, it is increasingly apparent that MGUS is a precursor lesion with a tendency to evolve to multiple myeloma. Among patients with MGUS, a symptomatic plasma cell tumor, most commonly multiple myeloma, develops at a rate of 1% per year. Moreover, the clonal plasma cells in MGUS contain the same chromosomal translocations found in full-blown multiple myeloma. A diagnosis of MGUS should be made only after careful exclusion of other monoclonal gammopathies, particularly multiple myeloma. In general, patients with MGUS have less than 3 g/dL of monoclonal protein in the serum and no Bence Jones proteinuria.
Lymphoplasmacytic Lymphoma
Lymphoplasmacytic lymphoma is included in the plasma cell neoplasms because the tumor cells secrete an M protein, most commonly IgM, but is otherwise distinct. It is composed of a mixture of B cells ranging from small lymphocytes to plasmacytic lymphocytes to plasma cells. It behaves like an indolent B cell lymphoma and commonly involves the lymph nodes, bone marrow, and spleen at presentation. Often the high levels of IgM cause the blood to become viscous, producing a syndrome called Waldenström macroglobulinemia, described later on. Other symptoms are related to the infiltration of various tissues, particularly the bone marrow, by tumor cells. The synthesis of immunoglobulin heavy and light chains usually is balanced, so free light chains and Bence Jones proteinuria are not seen. Unlike myeloma, this tumor does not produce lytic bone lesions and is only rarely associated with amyloidosis.
Heavy-Chain Disease
Heavy-chain disease is not a specific entity but represents a group of proliferations in which only heavy chains are produced, most commonly IgA. IgA heavy-chain disease shows a predilection for lymphoid tissues in which IgA normally is produced, such as the small intestine and respiratory tract, and may represent a variant of extranodal marginal zone lymphoma (discussed later). The less common IgG heavy-chain disease often manifests with diffuse lymphadenopathy and hepatosplenomegaly and histologically resembles lymphoplasmacytic lymphoma.
Primary Amyloidosis
As noted earlier (Chapter 4), a monoclonal proliferation of plasma cells that secrete free light chains underlies primary amyloidosis. The amyloid deposits (of AL type) consist of partially degraded light chains.
Morphology
Multiple myeloma usually manifests with multifocal destructive skeletal lesions, which most commonly involve the vertebral column, ribs, skull, pelvis, femur, clavicle, and scapula. The lesions generally arise in the medullary cavity, erode cancellous bone, and progressively destroy the cortical bone. This destructive process in turn often leads to pathologic fractures, most frequently in the vertebral column or femur. The bone lesions usually appear as punched-out defects 1 to 4 cm in diameter (Fig. 11–19, A), but in some cases diffuse skeletal demineralization is evident. Microscopic examination of the marrow reveals increased numbers of plasma cells, which usually constitute greater than 30% of the cellularity. Myeloma cells may resemble normal plasma cells but more often show abnormal features such as prominent nucleoli or abnormal cytoplasmic inclusions containing immunoglobulin (Fig. 11–19, B). With disease progression, plasma cells may infiltrate the spleen, liver, kidneys, lungs, lymph nodes, and other soft tissue sites. In terminal stages, a leukemic picture may emerge.
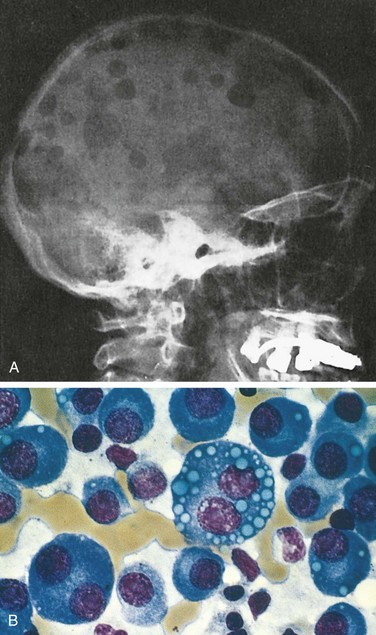
Figure 11–19 Multiple myeloma.
A, Radiograph of the skull, lateral view. The sharply punched-out bone defects are most obvious in the calvaria. B, Bone marrow aspirate. Normal marrow cells are largely replaced by plasma cells, including atypical forms with multiple nuclei, prominent nucleoli, and cytoplasmic droplets containing immunoglobulin.
Renal involvement (myeloma nephrosis) is associated with proteinaceous casts in the distal convoluted tubules and the collecting ducts, consisting mostly of Bence Jones proteins along with variable amounts of complete immunoglobulins, Tamm-Horsfall protein, and albumin. Multinucleate giant cells derived from macrophages usually surround the casts. Very often the epithelial cells adjacent to the casts become necrotic or atrophic owing to the toxic effects of Bence Jones proteins. Other common pathologic processes involving the kidney include metastatic calcification, stemming from bone resorption and hypercalcemia; light chain (AL) amyloidosis, involving the renal glomeruli and vessel walls; and bacterial pyelonephritis, secondary to the increased susceptibility to bacterial infections. Rarely, interstitial infiltrates of neoplastic plasma cells are seen.
In contrast with multiple myeloma, lymphoplasmacytic lymphoma is not associated with lytic skeletal lesions. Instead the neoplastic cells diffusely infiltrate the bone marrow, lymph nodes, spleen, and sometimes the liver. Infiltrations of other organs also occur, particularly with disease progression. The cellular infiltrate consists of lymphocytes, plasma cells, and plasmacytic lymphocytes of intermediate differentiation. The remaining forms of plasma cell neoplasms have either already been described (e.g., primary amyloidosis) (Chapter 4) or are too rare to merit further description.
Clinical Features
The clinical manifestations of plasma cell tumors are varied. They result from the destructive or otherwise damaging effects of tumor cells in various tissues and complications related to the complete or partial immunoglobulins secreted by the tumor cells.
The common clinicopathologic features of multiple myeloma can be summarized as follows:
• Bone pain, due to pathologic fractures. Pathologic fractures of vertebrae may lead to spinal cord impingement, an oncologic emergency.
• Hypercalcemia stemming from bone resorption leads to neurologic manifestations such as confusion and lethargy and contributes to renal dysfunction.
• Anemia, due to marrow replacement by tumor cells as well as suppression of hematopoiesis through uncertain mechanisms
• Recurrent infections with bacteria such as S. aureus, S. pneumoniae, and E. coli, resulting from the marked suppression of normal humoral immunity
• Renal insufficiency (in up to 50% of patients), resulting from the deleterious effect of Bence Jones proteins on renal tubular cells, as well as bacterial infections, hypercalcemia, and amyloidosis
• AL-type amyloidosis (5% to 10% of patients)
• Symptoms related to hyperviscosity may occur owing to excessive production and aggregation of M proteins but this clinical presentation is much more characteristic of lymphoplasmacytic lymphoma.
Multiple myeloma should be suspected when the characteristic focal, punched-out skeletal defects are present—especially when located in the vertebrae or calvaria. Electrophoresis of the serum and urine is an important diagnostic tool. In 99% of cases, either a monoclonal complete immunoglobulin or a monoclonal free immunoglobulin light chain is present in the serum, the urine, or both. In the few remaining cases, monoclonal free immunoglobulins can usually be detected within the plasma cells; in such cases the lesion is sometimes called nonsecretory myeloma. Examination of the bone marrow is used to confirm the presence of a plasma cell proliferation.
Lymphoplasmacytic lymphoma affects older persons; the peak incidence is between the sixth and seventh decades. Most clinical signs and symptoms are caused by secretion of IgM (macroglobulin) from the tumor cells. Because of their size, macroglobulins cause the blood to become viscous, giving rise to a syndrome, known as Waldenström macroglobulinemia, which is associated with the following features:
• Visual impairment, related to striking tortuosity and distention of retinal veins; retinal hemorrhages and exudates can also contribute to the visual problems
• Neurologic problems such as headaches, dizziness, tinnitus, deafness, and stupor, stemming from sluggish blood flow and sludging
• Bleeding, related to the formation of complexes between macroglobulins and clotting factors as well as interference with platelet function
• Cryoglobulinemia, related to precipitation of macroglobulins at low temperatures and producing symptoms such as Raynaud phenomenon and cold urticaria
Multiple myeloma is a progressive disease, with median survival of around 4 to 6 years. The picture for patients has brightened somewhat with the development of several new therapies, including proteasome inhibitors, which induce plasma cell apoptosis, and thalidomide analogues, which somehow alter the marrow microenvironment in a manner that inhibits myeloma cell growth and survival (recall that thalidomide was removed from the market because of its teratogenic effects in pregnant females). Lymphoplasmacytic lymphoma responds well for a time to relatively gentle chemotherapy regimens and plasmapheresis, which removes the macroglobulins; the median survival time is 4 to 5 years. At present, neither myeloma or lymphoplasmacytic lymphoma is curable.
Hodgkin Lymphoma
Hodgkin lymphoma encompasses a distinctive group of neoplasms that are characterized by the presence of a tumor giant cell, the Reed-Sternberg cell. Unlike most NHLs, Hodgkin lymphomas arise in a single lymph node or chain of lymph nodes and typically spread in a stepwise fashion to anatomically contiguous nodes. Although the Hodgkin lymphomas are now understood to be unusual tumors of B cell origin, they are distinguished from the NHLs by their unusual pathologic and clinical features.
Classification
Five subtypes of Hodgkin lymphoma are recognized: (1) nodular sclerosis, (2) mixed cellularity, (3) lymphocyte rich, (4) lymphocyte depletion, and (5) lymphocyte predominance. In the first four subtypes the Reed-Sternberg cells share certain morphologic and immunophenotypic features (described later), leading some researchers to lump together these entities under the rubric “classical Hodgkin lymphoma.” The lymphocyte predominance type is set apart by the expression of germinal center B markers by the Reed-Sternberg cells. This subtype and the two most common forms of classical Hodgkin lymphoma, the nodular sclerosis and mixed-cellularity types, are discussed next.
Morphology
The sine qua non of Hodgkin lymphoma is the Reed-Sternberg (RS) cell (Fig. 11–20), a very large cell (15 to 45 µm in diameter) with an enormous multilobate nucleus, exceptionally prominent nucleoli and abundant, usually slightly eosinophilic cytoplasm. Particularly characteristic are cells with two mirror-image nuclei or nuclear lobes, each containing a large (inclusion-like) acidophilic nucleolus surrounded by a clear zone, features that impart an owl-eye appearance. The nuclear membrane is distinct. Typical RS cells and variants have a characteristic immunophenotype, as they express CD15 and CD30 and fail to express CD45 (common leukocyte antigen), B cell antigens, and T cell antigens. As we shall see, “classic” RS cells are common in the mixed-cellularity subtype, uncommon in the nodular sclerosis subtype, and rare in the lymphocyte-predominance subtype; in these latter two subtypes, other characteristic RS cell variants predominate.
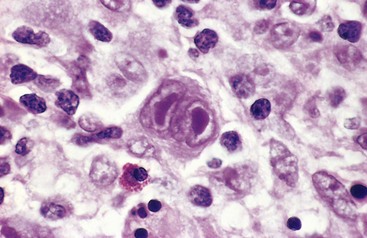
Figure 11–20 Hodgkin lymphoma—lymph node.
A binucleate Reed-Sternberg cell with large, inclusion-like nucleoli and abundant cytoplasm is surrounded by lymphocytes, macrophages, and an eosinophil.
(Courtesy of Dr. Robert W. McKenna, Department of Pathology, University of Texas Southwestern Medical School, Dallas, Texas.)
Nodular sclerosis Hodgkin lymphoma is the most common form. It is equally frequent in men and in women and has a striking propensity to involve the lower cervical, supraclavicular, and mediastinal lymph nodes. Most patients are adolescents or young adults, and the overall prognosis is excellent. It is characterized morphologically by
• The presence of a particular variant of the RS cell, the lacunar cell (Fig. 11–21). This large cell has a single multilobate nucleus, multiple small nucleoli and abundant, pale-staining cytoplasm. In sections of formalin-fixed tissue, the cytoplasm often is torn away, leaving the nucleus lying in an empty space (a lacune). The immunophenotype of lacunar variants is identical to that of other RS cells found in classical subtypes.
• The presence of collagen bands that divide involved lymphoid tissue into circumscribed nodules (Fig. 11–22). The fibrosis may be scant or abundant and the cellular infiltrate contains varying proportions of lymphocytes, eosinophils, histiocytes, and lacunar cells.
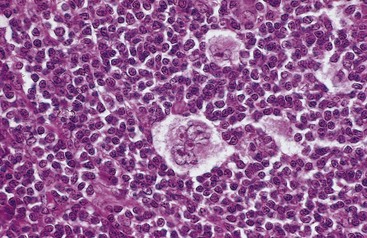
Figure 11–21 Hodgkin lymphoma, nodular sclerosis type—lymph node.
A distinctive “lacunar cell” with a multilobed nucleus containing many small nucleoli is seen lying within a clear space created by retraction of its cytoplasm. It is surrounded by lymphocytes.
(Courtesy of Dr. Robert W. McKenna, Department of Pathology, University of Texas Southwestern Medical School, Dallas, Texas.)
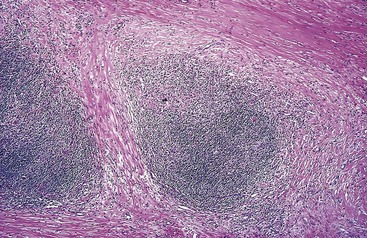
Figure 11–22 Hodgkin lymphoma, nodular sclerosis type—lymph node.
A low-power view shows well-defined bands of pink, acellular collagen that have subdivided the tumor cells into nodules.
(Courtesy of Dr. Robert W. McKenna, Department of Pathology, University of Texas Southwestern Medical School, Dallas, Texas.)
Mixed-cellularity Hodgkin lymphoma is the most common form of Hodgkin lymphoma in patients older than 50 years of age and comprises about 25% of cases overall. There is a male predominance. Classic RS cells are plentiful within a heterogeneous inflammatory infiltrate containing small lymphocytes, eosinophils, plasma cells, and macrophages (Fig. 11–23). This subtype is more likely to be disseminated and to be associated with systemic manifestations than the nodular sclerosis subtype.
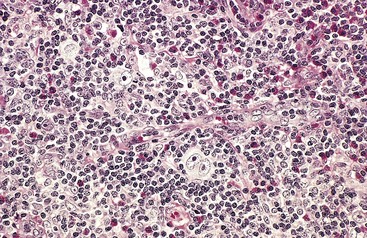
Figure 11–23 Hodgkin lymphoma, mixed-cellularity type—lymph node.
A diagnostic, binucleate Reed-Sternberg cell is surrounded by eosinophils, lymphocytes, and histiocytes.
(Courtesy of Dr. Robert W. McKenna, Department of Pathology, University of Texas Southwestern Medical School, Dallas, Texas.)
Lymphocyte-Predominance Hodgkin lymphoma. This subtype, accounting for about 5% of Hodgkin lymphoma, is characterized by the presence of lymphohistiocytic (L&H) variant RS cells that have a delicate multilobed, puffy nucleus resembling popped corn (“popcorn cell”). L&H variants usually are found within large nodules containing mainly small resting B cells admixed with a variable number of macrophages (Fig. 11–24). Other types of reactive cells, such as eosinophils, neutrophils, and plasma cells, are scanty or absent, and typical RS cells are rare. Unlike the Reed-Sternberg variants in “classical” forms of Hodgkin lymphoma, L&H variants express B cell markers (e.g., CD20) and usually fail to express CD15 and CD30. Most patients with this subtype present with isolated cervical or axillary lymphadenopathy, and the prognosis typically is excellent.
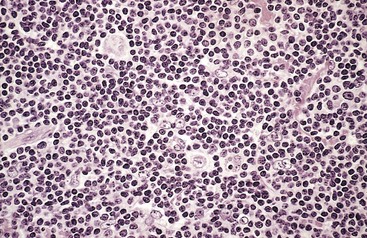
Figure 11–24 Hodgkin lymphoma, lymphocyte-predominance type—lymph node.
Numerous mature-looking lymphocytes surround scattered, large, pale-staining lymphocytic and histiocytic variants (“popcorn” cells).
(Courtesy of Dr. Robert W. McKenna, Department of Pathology, University of Texas Southwestern Medical School, Dallas, Texas.)
Other Considerations in Histologic Diagnosis. It is apparent that Hodgkin lymphoma spans a wide range of histologic patterns and that certain forms, with their characteristic fibrosis, eosinophils, neutrophils, and plasma cells, come deceptively close to simulating an inflammatory reactive process. The histologic diagnosis of Hodgkin lymphoma rests on the definitive identification of RS cells or variants in the appropriate background of reactive cells. Immunophenotyping plays an important adjunct role in distinguishing Hodgkin lymphoma from reactive conditions and other forms of lymphoma. In all subtypes, involvement of the spleen, liver, bone marrow, and other organs may appear in due course and takes the form of irregular nodules composed of a mixture of RS cells and reactive cells, similar to what is observed in lymph nodes.
Pathogenesis
The origin of RS cells remained mysterious through the 19th and most of the 20th centuries but was finally solved by elegant molecular studies performed on single microdissected RS cells. These showed that every RS cell from any given case possessed the same immunoglobulin gene rearrangements. In addition these studies revealed that the rearranged immunoglobulin genes had undergone somatic hypermutation. As a result, it is now agreed that Hodgkin lymphoma is a neoplasm arising from germinal center B cells.
The events that transform these cells and alter their appearance and gene expression programs are still unclear. One clue stems from the involvement of EBV. EBV is present in the RS cells in as many as 70% of cases of the mixed-cellularity subtype and a smaller fraction of other “classical” forms of Hodgkin lymphoma. More important, the integration of the EBV genome is identical in all RS cells in a given case, indicating that infection precedes (and therefore may be related to) transformation and clonal expansion. Thus, as in Burkitt lymphoma and B cell lymphomas in immunodeficient patients, EBV infection probably is one of several steps contributing to tumor development, particularly of the mixed-cellularity subtype.
The characteristic non-neoplastic, inflammatory cell infiltrate is generated by a number of cytokines, some secreted by RS cells, including IL-5 (which attracts and activates eosinophils), transforming growth factor-β (a fibrogenic factor), and IL-13 (which may stimulate RS cells through an autocrine mechanism). Conversely, the responding inflammatory cells, rather than being innocent bystanders, produce additional factors such as CD30 ligand that aid the growth and survival of RS cells and contribute further to the tissue reaction.
Staging and Clinical Features
Hodgkin lymphomas, like NHLs, usually manifest as painless lymphadenopathy. Although a definitive distinction from NHL can be made only by examination of a lymph node biopsy, several clinical features favor the diagnosis of Hodgkin lymphoma (Table 11–9). Once the diagnosis is established, staging is used to guide therapy and determine prognosis (Table 11–10). Younger patients with the more favorable subtypes tend to present with stage I or II disease and usually are free of systemic manifestations. Patients with advanced disease (stages III and IV) are more likely to have systemic complaints such as fever, weight loss, pruritus, and anemia. Due to the long-term complications of radiotherapy, even patients with stage I disease are now treated with systemic chemotherapy. More advanced disease generally is also treated with chemotherapy, sometimes with involved field radiotherapy at sites of bulky disease. The outlook for patients with Hodgkin lymphoma, even those with advanced disease, is very good. The 5-year survival rate for patients with stage I-A or II-A disease is close to 100%. Even with advanced disease (stage IV-A or IV-B), the overall 5-year disease-free survival rate is around 50%. Among long-term survivors treated with radiotherapy, a much higher risk of certain malignancies, including lung cancer, melanoma, and breast cancer, has been reported. These sobering results have spurred the development of new treatment regimens that minimize the use of radiotherapy and employ less toxic chemotherapeutic agents. Anti-CD30 antibodies have produced excellent responses in patients with disease that has failed conventional therapies and represent a promising “targeted” therapy.
Table 11–9 Clinical Differences Between Hodgkin and Non-Hodgkin Lymphomas
| Hodgkin Lymphoma | Non-Hodgkin Lymphoma |
|---|---|
| More often localized to a single axial group of nodes (cervical, mediastinal, para-aortic) | More frequent involvement of multiple peripheral nodes |
| Orderly spread by contiguity | Noncontiguous spread |
| Mesenteric nodes and Waldeyer ring rarely involved | Mesenteric nodes and Waldeyer ring commonly involved |
| Extranodal involvement uncommon | Extranodal involvement common |
Table 11–10 Clinical Staging of Hodgkin and Non-Hodgkin Lymphomas (Ann Arbor Classification)*
| Stage | Distribution of Disease |
|---|---|
| I | Involvement of a single lymph node region (I) or involvement of a single extralymphatic organ or tissue (IE) |
| II | Involvement of two or more lymph node regions on the same side of the diaphragm alone (II) or with involvement of limited contiguous extralymphatic organs or tissue (IIE) |
| III | Involvement of lymph node regions on both sides of the diaphragm (III), which may include the spleen (IIIS), limited contiguous extralymphatic organ or site (IIIE), or both (IIIES) |
| IV | Multiple or disseminated foci of involvement of one or more extralymphatic organs or tissues with or without lymphatic involvement |
* All stages are further divided on the basis of the absence (A) or presence (B) of the following systemic symptoms and signs: significant fever, night sweats, unexplained loss of more than 10% of normal body weight.
From Carbone PT, et al: Symposium (Ann Arbor): staging in Hodgkin disease. Cancer Res 31:1707, 1971.
Miscellaneous Lymphoid Neoplasms
Among the many other forms of lymphoid neoplasia in the WHO classification, several with distinctive or clinically important features merit brief discussion.
Extranodal Marginal Zone Lymphoma
This indolent B cell tumor arises most commonly in epithelial tissues such as the stomach, salivary glands, small and large bowel, lungs, orbit, and breast. Extranodal marginal zone lymphomas tend to develop in the setting of autoimmune disorders (such as Sjögren syndrome and Hashimoto thyroiditis) or chronic infection (such as H. pylori gastritis), suggesting that sustained antigenic stimulation contributes to its development. In the case of H. pylori–associated gastric marginal zone lymphoma, eradication of the organism with antibiotic therapy often leads to regression of the tumor cells, which depend on cytokines secreted by H. pylori–specific T cells for their growth and survival (Chapter 5). When arising at other sites, these lymphomas are often cured by local excision or radiotherapy. Several recurrent cytogenetic abnormalities are recognized, the most common of which is a (11;18) translocation involving the MALT1 and IAP2 genes. Of clinical importance, the presence of the t(11;18) is highly predictive of the failure of gastric tumors to respond to antibiotic treatment.
Hairy Cell Leukemia
Hairy cell leukemia is an uncommon, indolent B cell neoplasm characterized by the presence of leukemic cells with fine, hairlike cytoplasmic projections. The tumor cells express pan-B cell markers (CD19 and CD20), surface immunoglobulin, and CD11c and CD103; the latter two antigens are not present on most other B cell tumors, making them diagnostically useful. Virtually all cases are associated with activating mutations in the serine/threonine kinase BRAF, which is also mutated in diverse other cancers (Chapter 5).
Hairy cell leukemia occurs mainly in older males and its manifestations result from infiltration of bone marrow and spleen. Splenomegaly, often massive, is the most common and sometimes only physical finding. Pancytopenia, resulting from marrow infiltration and splenic sequestration, is seen in more than half of cases. Lymph node involvement is seen only rarely. Leukocytosis is uncommon, being present in 15% to 20% of patients, but scattered “hairy cells” can be identified in the peripheral blood smear in most cases. The disease is indolent but progressive if untreated; pancytopenia and infections cause the major clinical problems. Unlike most other indolent lymphoid neoplasms, this tumor is extremely sensitive to chemotherapeutic agents, particularly purine nucleosides. Complete durable responses are the rule and the overall prognosis is excellent.
Mycosis Fungoides and Sézary Syndrome
These tumors of neoplastic CD4+ T cells home to the skin; as a result, they often are referred to as cutaneous T cell lymphoma. Mycosis fungoides usually manifests as a nonspecific erythrodermic rash that progresses over time to a plaque phase and then to a tumor phase. Histologically, neoplastic T cells, often with a cerebriform appearance produced by marked infolding of the nuclear membranes, infiltrate the epidermis and upper dermis. With disease progression, both nodal and visceral dissemination appear. Sézary syndrome is a clinical variant characterized by (1) a generalized exfoliative erythroderma and (2) tumor cells (Sézary cells) in the peripheral blood. Circulating tumor cells also are present in as many as 25% of cases of plaque- or tumor-phase mycosis fungoides. Patients diagnosed with early-phase mycosis fungoides often survive for many years, whereas patients with tumor-phase disease, visceral disease, or Sézary syndrome survive on average for 1 to 3 years.
Adult T Cell Leukemia/Lymphoma
This is a neoplasm of CD4+ T cells that is caused by a retrovirus, human T cell leukemia virus type 1 (HTLV-1). HTLV-1 infection is endemic in southern Japan, the Caribbean basin, and West Africa, and occurs sporadically elsewhere, including in the southeastern United States. The pathogenesis of this tumor is discussed in Chapter 5. In addition to lymphoid malignancies, HTLV-1 infection also can cause transverse myelitis, a progressive demyelinating disease affecting the central nervous system and the spinal cord.
Adult T cell leukemia/lymphoma commonly is associated with skin lesions, lymphadenopathy, hepatosplenomegaly, hypercalcemia, and variable numbers of malignant lymphocytes in the peripheral blood. In addition to CD4, the leukemic cells express high levels of CD25, the IL-2 receptor α chain. In most cases the tumor is very aggressive and responds poorly to treatment. The median survival time is about 8 months. In 15% to 20% of patients the disease follows a chronic course resembling that of mycosis fungoides.
Peripheral T Cell Lymphomas
This heterogeneous group of tumors makes up 10% to 15% of adult NHLs. Although several rare distinctive subtypes fall under this heading, most tumors in this group are unclassifiable. In general, these are aggressive tumors that respond poorly to therapy. Moreover, because these are tumors of functional T cells, patients often suffer from symptoms related to tumor-derived inflammatory products, even when the tumor burden is relatively low.
 Summary
Summary
Lymphoid Neoplasms
• Classification is based on cell of origin and stage of differentiation.
• Most common types in children are acute lymphoblastic leukemias/lymphoblastic lymphomas derived from precursor B and T cells.
 These highly aggressive tumors manifest with signs and symptoms of bone marrow failure, or as rapidly growing masses. Tumor cells contain genetic lesions that block differentiation, leading to the accumulation of immature, nonfunctional blasts.
These highly aggressive tumors manifest with signs and symptoms of bone marrow failure, or as rapidly growing masses. Tumor cells contain genetic lesions that block differentiation, leading to the accumulation of immature, nonfunctional blasts.• Most common types in adults are non-Hodgkin lymphomas derived from germinal center B cells.
• This heterogeneous group of mature B cell tumors shares a large cell morphology and aggressive clinical behavior and represents the most common type of lymphoma.
• Rearrangements or mutations of BCL6 gene are recognized associations; one third arise from follicular lymphomas and carry a (14;18) translocation involving BCL2.
• This unusual tumor consists mostly of reactive lymphocytes, macrophages, and stromal cells.
• Malignant Reed-Sternberg cells make up a minor part of the tumor mass.
Table 11–8 lists features of specific entities.
Myeloid Neoplasms
Myeloid neoplasms arise from hematopoietic progenitors and typically give rise to clonal proliferations that replace normal bone marrow cells. There are three broad categories of myeloid neoplasia. In the acute myeloid leukemias (AMLs), the neoplastic cells are blocked at an early stage of myeloid cell development. Immature myeloid cells (blasts) accumulate in the marrow and replace normal elements, and frequently circulate in the peripheral blood. In the myeloproliferative disorders, the neoplastic clone continues to undergo terminal differentiation but exhibits increased or dysregulated growth. Commonly, these are associated with an increase in one or more of the formed elements (red cells, platelets, and/or granulocytes) in the peripheral blood. In the myelodysplastic syndromes, terminal differentiation occurs but in a disordered and ineffective fashion, leading to the appearance of dysplastic marrow precursors and peripheral blood cytopenias.
Although these three categories provide a useful starting point, the divisions between the myeloid neoplasms sometimes blur. Both myelodysplastic syndromes and myeloproliferative disorders often transform to AML, and some neoplasms have features of both myelodysplasia and myeloproliferative disorders. Because all myeloid neoplasms arise from early multipotent progenitors, the close relationship among these disorders is not surprising.
Acute Myeloid Leukemia
AML primarily affects older adults; the median age is 50 years. It is very heterogeneous, as discussed later on. The clinical signs and symptoms closely resemble those produced by ALL and usually are related to the replacement of normal marrow elements by leukemic blasts. Fatigue, pallor, abnormal bleeding, and infections are common in newly diagnosed patients, who typically present within a few weeks of the onset of symptoms. Splenomegaly and lymphadenopathy generally are less prominent than in ALL, but on rare occasions AML mimics a lymphoma by manifesting as a discrete tissue mass (a so-called granulocytic sarcoma). The diagnosis and classification of AML are based on morphologic, histochemical, immunophenotypic, and karyotypic findings. Of these, the karyotype is most predictive of outcome.
Pathogenesis
Most AMLs harbor mutations in genes encoding transcription factors that are required for normal myeloid cell differentiation. These mutations interfere with the differentiation of early myeloid cells, leading to the accumulation of myeloid precursors (blasts) in the marrow. Of particular interest is the (15;17) translocation in acute promyelocytic leukemia, which results in the fusion of the retinoic acid receptor α (RARA) gene on chromosome 17 and the PML gene on chromosome 15. The chimeric gene produces a PML/RARα fusion protein that blocks myeloid differentiation at the promyelocytic stage, probably in part by inhibiting the function of normal retinoic acid receptors. Remarkably, pharmacologic doses of all-trans retinoic acid (ATRA), an analogue of vitamin A (Chapter 7), overcome this block and induce the neoplastic promyelocytes to rapidly differentiate into neutrophils. Because neutrophils die after an average lifespan of 6 hours, ATRA treatment rapidly clears the tumor. The effect is very specific; AMLs without translocations involving RARA do not respond to ATRA. More recently, it has been noted that the combination of ATRA and arsenic trioxide, a salt that induces the degradation of the PML/RARA fusion protein, is even more effective than ATRA alone, producing cures in more than 80% of patients. This is an important example of a highly effective therapy targeted at a tumor-specific molecular defect.
Other work using transgenic or “knock-in” mice has shown that the mutations in transcription factors found in AML are not sufficient to cause the disease. Some of the other mutations implicated in AML have no effect on differentiation but instead enhance cellular proliferation and survival. One example involves FLT3, a receptor tyrosine kinase that is activated by mutations in a number of AML subtypes, including acute promyelocytic leukemia. Putative collaborating mutations in several other tyrosine kinase genes and in RAS, an oncogene that is mutated in diverse forms of cancer, also have been detected.
Morphology
By definition, in AML myeloid blasts or promyelocytes make up more than 20% of the bone marrow cellular component. Myeloblasts (precursors of granulocytes) have delicate nuclear chromatin, three to five nucleoli, and fine azurophilic cytoplasmic granules (Fig. 11–14, B). Auer rods, distinctive red-staining rodlike structures, may be present in myeloblasts or more differentiated cells; they are particularly numerous in acute promyelocytic leukemia (Fig. 11–25). Auer rods are specific for neoplastic myeloblasts and thus a helpful diagnostic clue when present. In other subtypes of AML, monoblasts, erythroblasts, or megakaryoblasts predominate.
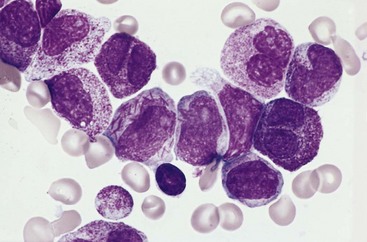
Figure 11–25 Acute promyelocytic leukemia—bone marrow aspirate. The neoplastic promyelocytes have abnormally coarse and numerous azurophilic granules. Other characteristic findings include the presence of several cells with bilobed nuclei and a cell in the center of the field that contains multiple needle-like Auer rods.
(Courtesy of Dr. Robert W. McKenna, Department of Pathology, University of Texas Southwestern Medical School, Dallas, Texas.)
Classification
AMLs are diverse in terms of genetics, cellular lineage, and degree of maturation. The WHO classification relies on all of these features to divide AML into four categories (Table 11–11): (1) AMLs associated with specific genetic aberrations, which are important because they predict outcome and guide therapy; (2) AMLs with dysplasia, many of which arise from myelodysplastic syndromes; (3) AMLs occurring after genotoxic chemotherapy; and (4) AMLs lacking any of the foregoing features. AMLs in the last category are subclassified on the basis of the predominant line of differentiation that the tumor exhibits.
Table 11–11 WHO Classification of Acute Myeloid Leukemia (AML)
| Class | Prognosis |
|---|---|
| I. AML with Recurrent Chromosomal Translocations | |
| AML with t(8;21)(q22;q22); CBFA/ETO fusion gene | Favorable |
| AML with inv(16)(p13;q22); CBFB/MYH11 fusion gene | Favorable |
| AML with t(15;17)(q22;q21.1); PML/RARA fusion gene | Favorable |
| AML with t(11q23;variant); MLL fusion genes | Poor |
| AML with mutated NPM1 | Variable |
| II. AML with Multilineage Dysplasia | |
| With previous myelodysplastic syndrome | Very poor |
| Without previous myelodysplastic syndrome | Poor |
| III. AML, Therapy-Related | |
| Alkylating agent–related | Very poor |
| Epipodophyllotoxin-related | Very poor |
| IV. AML, Not Otherwise Classified | |
| Subclasses defined by extent and type of differentiation (e.g., myelocytic, monocytic) | Intermediate |
NPM1, nucleophosmin 1; WHO, World Health Organization.
Immunophenotype
The expression of immunologic markers is heterogeneous in AML. Most tumors express some combination of myeloid-associated antigens, such as CD13, CD14, CD15, CD64, or CD117 (cKIT). CD33 is expressed on pluripotent stem cells but is retained on myeloid progenitor cells. Such markers are helpful in distinguishing AML from ALL (as shown in Fig. 11–14) and in identifying AMLs with only minimal differentiation.
Prognosis
AML is a devastating disease. Tumors with “good-risk” karyotypic abnormalities (t[8;21], inv[16]) are associated with a 50% chance of long-term disease-free survival, but the overall survival in all patients is only 15% to 30% with conventional chemotherapy. A bright spot is the improvement in outcomes in acute promyelocytic leukemia brought about by targeted treatment with ATRA and arsenic salts. An increasing number of patients with AML are being treated with more aggressive approaches, such as allogeneic hematopoietic stem cell transplantation.
Myelodysplastic Syndromes
In myelodysplastic syndromes (MDSs), the bone marrow is partly or wholly replaced by the clonal progeny of a transformed multipotent stem cell that retains the capacity to differentiate into red cells, granulocytes, and platelets, but in a manner that is both ineffective and disordered. As a result, the marrow usually is hypercellular or normocellular, but the peripheral blood shows one or more cytopenias. The abnormal stem cell clone in the bone marrow is genetically unstable and prone to the acquisition of additional mutations and the eventual transformation to AML. Most cases are idiopathic, but some develop after chemotherapy with alkylating agents or exposure to ionizing radiation therapy.
Pathogenesis
The pathogenesis of MDS is poorly understood. Cytogenetic studies reveal clonal abnormalities in up to 70% of cases. Translocations generally are lacking, whereas losses or gains of whole chromosomes or parts thereof are frequent. Some common karyotypic abnormalities include monosomy 5 or 7; deletions of 5q, 7q, and 20q; and trisomy 8. Recent work suggests that the critical region deleted on 5q contains genes encoding a ribosomal protein and several microRNAs. Loss of all of these genes appears to contribute to a subtype of MDS called the 5q− syndrome. This syndrome occurs more often in women, is associated with severe anemia and preserved or elevated platelet counts, and often responds to treatment with analogs of thalidomide, which are believed to influence the interaction of hematopoietic progenitors and marrow stromal cells.
Morphology
In MDSs, the marrow is populated by abnormal-appearing hematopoietic precursors. Some of the more common abnormalities include megaloblastoid erythroid precursors resembling those seen in the megaloblastic anemias, erythroid forms with iron deposits within their mitochondria (ringed sideroblasts), granulocyte precursors with abnormal granules or nuclear maturation, and small megakaryocytes with single small nuclei or multiple separate nuclei.
Although these syndromes often are described as rare, it is now appreciated that MDS is about as common as AML, affecting up to 15,000 patients per year in the United States. Most persons with MDS are between 50 and 70 years of age. As a result of cytopenias, many suffer from infections, symptoms related to anemia, and hemorrhages. The response to conventional chemotherapy usually is poor, perhaps because MDS arises in a background of stem cell damage. Transformation to AML occurs in 10% to 40%. The prognosis is variable; the median survival time ranges from 9 to 29 months and is worse in patients with increased marrow blasts or cytogenetic abnormalities at the time of diagnosis.
Chronic Myeloproliferative Disorders
Chronic myeloproliferative disorders are marked by the hyperproliferation of neoplastic myeloid progenitors that retain the capacity for terminal differentiation; as a result, there is an increase in one or more formed elements of the peripheral blood. The neoplastic progenitors tend to seed secondary hematopoietic organs (the spleen, liver, and lymph nodes), resulting in hepatosplenomegaly (caused by neoplastic extramedullary hematopoiesis). A common theme is the association of these disorders with activating mutations in tyrosine kinases, which generate constitutive signals mimicking those that are normally produced in response to hematopoietic growth factors. This insight provides a satisfying explanation for the observed overproduction of myeloid cells and is important therapeutically because of the availability of tyrosine kinase inhibitors.
Four major diagnostic entities are recognized: chronic myelogenous leukemia (CML), polycythemia vera, primary myelofibrosis, and essential thrombocythemia. CML is separated from the others by its association with a characteristic abnormality, the BCR-ABL fusion gene, which produces a constitutively active BCR-ABL tyrosine kinase. The remaining BCR-ABL–negative myeloproliferative disorders show considerable clinical and genetic overlap. The most common genetic abnormalities in the “BCR-ABL–negative” myeloproliferative disorders are activating mutations in the tyrosine kinase JAK2, which occur in virtually all cases of polycythemia vera and about 50% of cases of primary myelofibrosis and essential thrombocythemia. Some rare myeloproliferative disorders are associated with activating mutations in other tyrosine kinases, such as platelet-derived growth factor receptor-α and platelet-derived growth factor receptor-β. In addition, all myeloproliferative disorders have variable propensities to transform to a “spent phase” resembling primary myelofibrosis or to a “blast crisis” identical to acute leukemia, both presumably triggered by the acquisition of other somatic mutations. Only CML, polycythemia vera, and primary myelofibrosis are presented here, as essential thrombocythemia and other myeloproliferative disorders are too infrequent to merit discussion.
Chronic Myelogenous Leukemia
CML principally affects adults between 25 and 60 years of age. The peak incidence is in the fourth and fifth decades of life. About 4500 new cases are diagnosed per year in the United States.
Pathogenesis
CML is always associated with the presence of a BCR-ABL fusion gene. In about 95% of cases, the BCR-ABL gene is the product of a balanced (9;22) translocation that moves ABL from chromosome 9 to a position on chromosome 22 adjacent to BCR. In the remaining 5% of cases, a BCR-ABL fusion gene is created by cytogenetically cryptic or complex rearrangements involving more than two chromosomes. The BCR-ABL fusion gene is present in granulocytic, erythroid, megakaryocytic, and B cell precursors, and in some cases T cell precursors as well, indicating that the tumor arises from a transformed hematopoietic stem cell. Although the Ph chromosome is highly characteristic of CML, it also is present in 25% of adult B cell–ALLs and a small subset of AMLs.
As described in Chapter 5, the BCR-ABL gene encodes a fusion protein consisting of portions of BCR and the tyrosine kinase domain of ABL. Normal myeloid progenitors depend on signals generated by growth factors and their receptors for growth and survival. The growth factor dependence of CML progenitors is greatly decreased by constitutive signals generated by BCR-ABL that mimic the effects of growth factor receptor activation. Importantly, because BCR-ABL does not inhibit differentiation, the early disease course is marked by excessive hematopoiesis. Although the BCR-ABL fusion gene is present in multiple lineages, for unclear reasons the pro-growth effects of BCR-ABL are confined mainly to the granulocyte and megakaryocyte lineages.
Morphology
The peripheral blood findings are highly characteristic. The leukocyte count is elevated, often exceeding 100,000 cells/µL. The circulating cells are predominantly neutrophils, metamyelocytes, and myelocytes (Fig. 11–26), but basophils and eosinophils are also prominent and platelets are usually increased. A small proportion of myeloblasts, usually less than 5%, are often seen in the peripheral blood. The bone marrow is hypercellular owing to increased numbers of granulocytic and megakaryocytic precursors. Myeloblasts usually are only slightly increased. The red pulp of the enlarged spleen resembles bone marrow because of the presence of extensive extramedullary hematopoiesis. This burgeoning proliferation often compromises the local blood supply, leading to splenic infarcts.
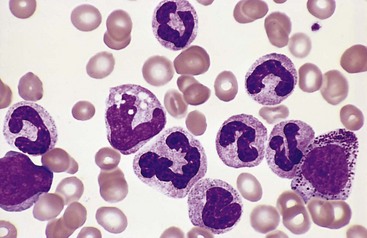
Clinical Features
The onset of CML often is insidious, as the initial symptoms usually are nonspecific (e.g., easy fatigability, weakness, weight loss). Sometimes the first symptom is a dragging sensation in the abdomen caused by splenomegaly. On occasion it may be necessary to distinguish CML from a “leukemoid reaction,” a dramatic elevation of the granulocyte count in response to infection, stress, chronic inflammation, and certain neoplasms. This distinction can be achieved definitively by testing for the presence of the BCR-ABL fusion gene, which can be done by karyotyping, fluorescence in situ hybridization, or PCR assay.
The natural history of CML initially is one of slow progression. Even without treatment, the median survival is 3 years. After a variable (and unpredictable) period, approximately half of CML cases enter an accelerated phase marked by increasing anemia and new thrombocytopenia, the appearance of additional cytogenetic abnormalities, and finally transformation into a picture resembling acute leukemia (blast crisis). In the remaining 50% of cases, blast crisis occurs abruptly, without an accelerated phase. Of note, in 30% of cases the blast crisis resembles precursor-B cell ALL, further attesting to the origin of CML from hematopoietic stem cells. In the remaining 70% of cases, the blast crisis resembles AML. Less commonly, CML progresses to a phase of extensive bone marrow fibrosis resembling primary myelofibrosis.
Fortunately for those affected, the natural history of CML has been altered dramatically by the emergence of targeted therapy. Inhibitors of the BCR-ABL tyrosine kinase, such as imatinib and nilotinib, induce complete remission in a high fraction of patients with little associated toxicity. Treatment with tyrosine kinase inhibitors, particularly in patients with early disease, induces sustained remissions and may prevent the appearance of blast crisis by suppressing the proliferative drive that leads to the acquisition of additional mutations. When patients on tyrosine kinase inhibitors relapse, their tumors frequently are found to have acquired mutations in the kinase domain of BCR-ABL that prevent the drugs from binding. In retrospective studies, these mutations have been shown to be present in small numbers of cells at the time of diagnosis. The selective outgrowth of these cells is explained by the powerful antitumor effects of BCR-ABL inhibitors, and indicates that many resistant tumors are still “addicted” to the pro-growth signals created by BCR-ABL. For those with resistant disease, hematopoietic stem cell transplantation is curative in 70% of patients, but carries with it substantial risks, particularly in the aged.
Polycythemia Vera
Polycythemia vera is characterized by an excessive proliferation of erythroid, granulocytic, and megakaryocytic elements (panmyelosis), but most clinical signs and symptoms are related to an absolute increase in red cell mass. Polycythemia vera must be distinguished from relative polycythemia, which results from hemoconcentration. Unlike reactive forms of absolute polycythemia, polycythemia vera is associated with low levels of serum erythropoietin, which is a reflection of the growth factor–independent growth of the neoplastic clone. This behavior stems from the presence of activating mutations in JAK2, a tyrosine kinase that acts in the signaling pathways downstream of the erythropoietin receptor and other growth factor receptors. The most common JAK2 mutation, a valine-to-phenylalanine substitution at residue 617, sharply lowers the dependence of hematopoietic cells on growth factors for growth and survival, suggesting that it is an important part of the pathogenesis of the disorder.
Morphology
The major anatomic changes in polycythemia vera stem from increases in blood volume and viscosity brought about by the polycythemia. Plethoric congestion of many tissues is characteristic. The liver is enlarged and often contains small foci of extramedullary hematopoiesis. The spleen usually is slightly enlarged (250 to 300 g) due to vascular congestion. As a result of the increased viscosity and vascular stasis, thromboses and infarctions are common, particularly in the heart, spleen, and kidneys. Hemorrhages also occur in about a third of the patients, probably as a result of excessive distention of blood vessels and abnormal platelet function. These most often affect the gastrointestinal tract, oropharynx, or brain. Hemorrhage may occur spontaneously but more often follows some minor trauma or surgical procedure. Platelets produced from the neoplastic clone often are dysfunctional, a derangement that contributes to the elevated risk of thrombosis and bleeding. As in CML, the peripheral blood often shows basophilia.
The bone marrow is hypercellular owing to increased numbers of erythroid, myeloid, and megakaryocytic forms. In addition, some degree of marrow fibrosis is present in 10% of patients at the time of diagnosis. In a subset of patients, this progresses to a spent phase where the marrow is largely replaced by fibroblasts and collagen.
Clinical Course
Polycythemia vera appears insidiously, usually in late middle age. Patients are plethoric and often somewhat cyanotic. Histamine released from the neoplastic basophils may contribute to pruritus and also may account for the increased incidence of peptic ulceration. Other complaints are referable to the thrombotic and hemorrhagic tendencies and to hypertension. Headache, dizziness, gastrointestinal symptoms, hematemesis, and melena are common. Because of the high rate of cell turnover, symptomatic gout is seen in 5% to 10% of cases, and many more patients have asymptomatic hyperuricemia.
The diagnosis usually is made in the laboratory. Red cell counts range from 6 to 10 million/µl and the hematocrit is often 60% or greater. The granulocyte count can be as high as 50,000 cells/µL, and the platelet count is often over 400,000/µL. Basophilia is common. The platelets are functionally abnormal in most cases, and giant platelets and megakaryocyte fragments are often seen in the blood. About 30% of patients develop thrombotic complications, usually affecting the brain or heart. Hepatic vein thrombosis giving rise to the Budd-Chiari syndrome (Chapter 15) is an uncommon but grave complication. Minor hemorrhages (e.g., epistaxis and bleeding from gums) are common and life-threatening hemorrhages occur in 5% to 10% of patients. In those receiving no treatment, death occurs from vascular complications within months; however, the median survival is increased to about 10 years by lowering the red cell count to near normal through repeated phlebotomy.
Unfortunately, prolonged survival has revealed a propensity for polycythemia vera to evolve to a “spent phase” resembling primary myelofibrosis. After an average interval of 10 years, 15% to 20% of cases undergo such a transformation. Owing to the extensive marrow fibrosis, hematopoiesis shifts to the spleen, which enlarges markedly. Transformation to a “blast crisis” identical to AML also occurs but much less frequently than in CML. Inhibitors that target JAK2 are in clinical trials at present.
Primary Myelofibrosis
In the myeloproliferative disorder known as primary myelofibrosis, a “spent phase” of marrow fibrosis supervenes early in the disease course, often after a brief period of granulocytosis and thrombocytosis. As hematopoiesis shifts from the fibrotic marrow to the spleen, liver, and lymph nodes, extreme splenomegaly and hepatomegaly develop. The extramedullary hematopoiesis at these sites is disordered and inefficient, resulting in anemia and thrombocytopenia. Neutropenia also may occur but tends to be mild.
Pathogenesis
The fibroblasts that lay down the collagen in the marrow are not neoplastic. Instead, the marrow fibrosis is secondary to derangements in the hematopoietic cells. It is believed that the fibroblast proliferation is stimulated by platelet-derived growth factor and transforming growth factor β released from neoplastic megakaryocytes. By the time patients come to clinical attention, marrow fibrosis and marked extramedullary hematopoiesis usually are evident. Less commonly, marrow fibrosis is mild at diagnosis and the clinical picture resembles that of other myeloproliferative disorders.
Of pathogenic and possible therapeutic importance, the same JAK2 mutation that is found in polycythemia vera (a valine-to-phenylalanine mutation at amino acid residue 617) is present in around half of cases of primary myelofibrosis (as well as a similar proportion of essential thrombocythemia cases)—a commonality that emphasizes the overlap between these entities. It is not yet known why tumors with the same mutation have such varied clinical pictures.
Morphology
The peripheral blood smear is markedly abnormal (Fig. 11–27). Red cells often exhibit bizarre shapes (poikilocytes, teardrop cells), and nucleated erythroid precursors are commonly present along with immature white cells (myelocytes and metamyelocytes), a combination of findings referred to as “leukoerythroblastosis.” Abnormally large platelets often are present as well. The spleen usually is markedly enlarged, sometimes weighing as much as 4000 g, from the extensive extramedullary hematopoiesis. Subcapsular splenic infarcts, often multiple, are commonly present. Within areas of extramedullary hematopoiesis, megakaryocytes usually are numerous and often display bizarre morphologic features. Moderate hepatomegaly due to extramedullary hematopoiesis is commonplace. The lymph nodes also are involved by extramedullary hematopoiesis, but not to a degree sufficient to cause appreciable enlargement. The bone marrow in advanced cases is hypocellular and diffusely fibrotic, while early in the course it may be hypercellular and have only focal areas of fibrosis. Throughout the course, marrow megakaryocytes usually are increased in number and dysplastic.
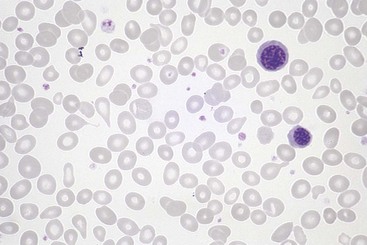
Figure 11–27 Primary myelofibrosis—peripheral blood smear. Two nucleated erythroid precursors and several teardrop-shaped red cells (dacryocytes) are evident. Immature myeloid cells were present in other fields. An identical histologic picture can be seen in other diseases producing marrow distortion and fibrosis.
Clinical Course
By the time most patients come to clinical attention, the disease has progressed to the phase of marrow fibrosis. Early stage disease may have features suggestive of CML, but the Ph chromosome is absent. Most patients have moderate to severe anemia at the time of diagnosis. The white blood cell count may be normal, reduced, or markedly elevated. Early in the course the platelet count is normal or elevated, but eventually patients develop thrombocytopenia. Because of a high rate of cell turnover, hyperuricemia and gout may complicate the picture.
The outcome of this disease is variable, but the median survival time is 4 to 5 years. There is a constant threat of thrombotic and hemorrhagic episodes stemming from platelet abnormalities. Splenic infarctions are common. From 5% to 15% of affected persons develop a blast crisis resembling AML.
Summary
Myeloid Neoplasms
Myeloid tumors occur mainly in adults and fall into three major groups:
• Acute myeloid leukemias (AMLs)
Aggressive tumors comprised of immature myeloid lineage blasts, which replace the marrow and suppress normal hematopoiesis Associated with diverse acquired mutations that lead to expression of abnormal transcription factors, which interfere with myeloid differentiation• Myeloproliferative disorders
Myeloid tumors in which production of formed myeloid elements is initially increased, leading to high blood counts and extramedullary hematopoiesis Commonly associated with acquired mutations that lead to constitutive activation of tyrosine kinases, which mimic signals from normal growth factors. The most common pathogenic kinases are BCR-ABL (associated with CML) and mutated JAK2 (associated with polycythemia vera and primary myelofibrosis).Histiocytic Neoplasms
Langerhans Cell Histiocytoses
The term histiocytosis is an “umbrella” designation for a variety of proliferative disorders of dendritic cells or macrophages. Some, such as very rare histiocytic lymphomas, are highly malignant neoplasms. Others, such as most histiocytic proliferations in lymph nodes, are completely benign and reactive. Between these two extremes lie a group of relatively rare tumors comprised of Langerhans cells, the Langerhans cell histiocytoses. As described in Chapter 4, Langerhans cells are immature dendritic cells found in the epidermis; similar cells are found in many other organs, and function to capture antigens and display them to T cells.
Langerhans cell proliferations take on different clinical forms, but all are believed to be variations of the same basic disorder. The proliferating Langerhans cells express MHC class II antigens, CD1a, and langerin. Langerin is a transmembrane protein found in Birbeck granules, cytoplasmic pentalaminar rodlike tubular structures that in electron micrographs have a characteristic periodicity and sometimes a dilated terminal end (“tennis racket” appearance). Under the light microscope, the proliferating Langerhans cells do not resemble their normal dendritic counterparts. Instead, they have abundant, often vacuolated cytoplasm and vesicular nuclei, an appearance more akin to that of tissue macrophages (called histiocytes by morphologists)—hence the term Langerhans cell histiocytosis.
Langerhans cell histiocytoses manifest as three relatively distinctive clinicopathologic entities. Multisystem Langerhans cell histiocytosis (Letterer-Siwe disease) usually occurs in children younger than 2 years of age. It typically manifests with multifocal cutaneous lesions that grossly resemble seborrheic skin eruptions and are composed of Langerhans cells. Most affected patients have hepatosplenomegaly, lymphadenopathy, pulmonary lesions, and (later in the course) destructive osteolytic bone lesions. Extensive infiltration of the marrow often leads to pancytopenia and predisposes the patient to recurrent infections such as otitis media and mastoiditis. The disease is rapidly fatal if untreated. With intensive chemotherapy, 50% of patients survive 5 years.
Unisystem Langerhans cell histiocytosis (eosinophilic granuloma) may be unifocal or multifocal. It is characterized by expanding, erosive accumulations of Langerhans cells, usually within the medullary cavities of bones or less commonly in the skin, lungs, or stomach. The Langerhans cells are admixed with variable numbers of lymphocytes, plasma cells, neutrophils, and eosinophils, which are usually, but not always, prominent. Virtually any bone in the skeletal system may be involved; the calvaria, ribs, and femur are most commonly affected. Unifocal disease most often involves the skeletal system. It may be asymptomatic or cause pain, tenderness and pathologic fractures. It is an indolent disorder that may heal spontaneously or be cured by local excision or irradiation. Multifocal unisystem disease usually affects children and typically manifests with multiple erosive bony masses that sometimes extend into soft tissues. In about 50% of cases, involvement of the posterior pituitary stalk of the hypothalamus leads to diabetes insipidus. The combination of calvarial bone defects, diabetes insipidus, and exophthalmos is referred to as the Hand-Schüller-Christian triad. Many patients experience spontaneous regressions; others are treated effectively with chemotherapy.
A clue to the pathogenesis of Langerhans cell tumors lies in the discovery that the different clinical forms are frequently associated with an acquired mutation in the serine/threonine kinase BRAF, a valine to glutamate substitution in residue 600 that leads to hyperactivity of the kinase. This same mutation is found in a variety of other tumors, including hairy cell leukemia, benign nevi, malignant melanoma, papillary thyroid carcinoma, and some colon cancers (Chapter 5). BRAF is a component of the Ras signaling pathway that drives cellular proliferation and survival, effects that likely contribute to the growth of the neoplastic Langerhans cells.