Chapter 20 Bones, Joints, and Soft Tissue Tumors
See Targeted Therapy available online at studentconsult.com
The musculoskeletal system and the integrated neural connections enable locomotion by the human body. Aside from providing the fulcrums and levers against which muscles contract to allow movement, the skeleton is critical for mineral (particularly calcium) homeostasis and also protects viscera and supplies an environment conducive to both hematopoietic and mesenchymal stem cell development. The term diseases of the bones and joints embraces a large number of conditions ranging from localized, benign tumors of bone and soft tissue such as the osteochondroma and lipoma, respectively, to generalized disorders such as osteoporosis and osteogenesis imperfecta. In this chapter we will first consider some of the more common conditions affecting the bones and joints, then discuss tumors arising in the various soft tissues of the body. Diseases of the muscles and peripheral nerves are discussed in Chapter 21.
Bones
The skeletal system is composed of 206 bones that vary in size and shape and are interconnected by a variety of joints that allow for a wide range of movement and promote structural stability. Bones are composed of a unique type of mineralized connective tissue that undergoes mineralization with a distinctive admixture of organic matrix (35%) and inorganic elements (65%). The inorganic mineral component consists mainly of calcium hydroxyapatite [Ca10(PO4)6(OH)2]. This mineral gives bone strength and hardness and serves as the storehouse for 99% of the body’s calcium, 85% of the body’s phosphorus, and 65% of the body’s sodium and magnesium. The organic component includes the cells of bone and the proteinaceous osteoid. The bone-forming cells include osteoblasts and osteocytes, while cells of the bone-digesting lineage include osteoclast precursor cells and mature functional osteoclasts (Fig. 20–1).
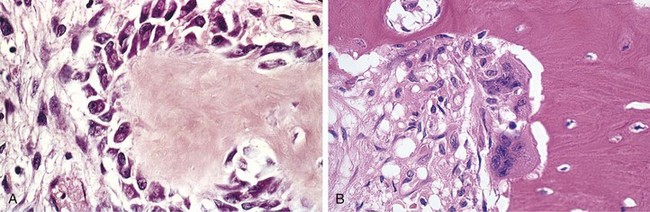
Figure 20–1 Cells of bone. A, Active osteoblasts synthesizing bone matrix proteins. The surrounding spindle cells are osteoprogenitor cells. B, Two osteoclasts resorbing bone. The smaller blue nuclei surrounded by a halo of clearing in the dense pink lamellar bone are osteocytes in their individual lacunae.
To the uninitiated, bone appears to be an inert, stable tissue, but in fact it is very dynamic and subject to constant breakdown and renewal, a process referred to as remodeling. The net effects of remodeling may be bone maintenance, bone loss, or bone deposition, with the balance being determined by the relative activities of osteoblasts, which deposit bone, and osteoclasts, which resorb bone (Fig. 20–1, A and B). As might be imagined, osteoblast and osteoclast activity is highly regulated and tightly integrated under normal circumstances, both by local crosstalk between these two cell types and by circulating factors that impact their activity, such as vitamin D and parathyroid hormone.
Among the local factors that regulate bone remodeling, the most important are RANK (receptor activator for nuclear factor-κB), RANK ligand (RANKL), and osteoprotegerin (OPG) (Fig 20–2). RANK, a member of the tumor necrosis factor (TNF) receptor family, is expressed on the cell membranes of preosteoclasts and mature osteoclasts. Its ligand, RANKL, is expressed by osteoblasts and marrow stromal cells. RANK stimulation by RANKL leads to activation of the transcription factor NF-κB, which drives the expression of genes that stimulate osteoclast formation, fusion, differentiation, function, and survival. RANKL production is upregulated by factors that stimulate osteoclastic activity. The actions of RANKL can be blocked by another member of the TNF receptor family, OPG, which is a “decoy” receptor produced by a number of tissues including bone, hematopoietic marrow, and immune cells. OPG competitively binds to RANKL, preventing RANK from interacting with RANKL. OPG production is regulated by signals similar to those that stimulate RANKL. Therefore, these molecules enable osteoblasts and stromal cells to control osteoclast development and activity and provide a mechanism for a wide variety of biologic mediators (hormones, cytokines, growth factors) to influence the homeostasis of bone tissue and bone mass.
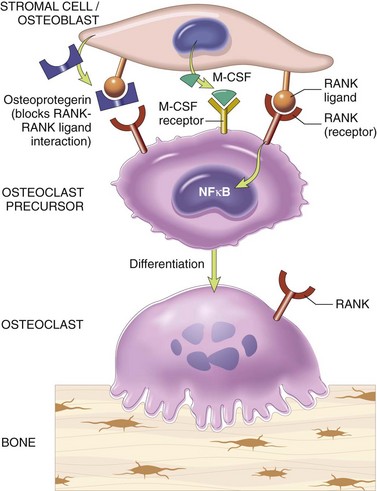
Figure 20–2 Paracrine mechanisms regulating osteoclast formation and function. Osteoclasts are derived from the same stem cells that produce macrophages. RANK (receptor activator for nuclear factor-κB) receptors on osteoclast precursors bind RANK ligand (RANKL) expressed by osteoblasts and marrow stromal cells. Along with macrophage colony-stimulating factor (M-CSF), the RANK-RANKL interaction drives the differentiation of functional osteoclasts. Stromal cells also secrete osteoprotegerin (OPG), which acts as a decoy receptor for RANKL, preventing it from binding the RANK receptor on osteoclast precursors. Consequently, OPG prevents bone resorption by inhibiting osteoclast differentiation.
Primary and secondary diseases of bone are varied and numerous and are classified in this chapter according to their perceived biologic defect or pathologic process.
Congenital Disorders of Bone and Cartilage
Congenital disorders of the skeleton are various and, depending on the resulting defect, become manifest at different ages. The most severe produce developmental abnormalities that are evident from the earliest stages of skeletogenesis.
• Developmental anomalies resulting from localized problems in the migration of mesenchymal cells and the formation of condensations are called dysostoses and may affect individual or a group of bones and can result from mutations in specific homeobox genes. The more common lesions include aplasia (e.g., congenital absence of a digit or rib), the formation of extra bones (e.g., supernumerary digits or ribs), and abnormal fusion of bones (e.g., premature closure of the cranial sutures or congenital fusion of the ribs). Such malformations may occur as isolated, sporadic lesions or as components of a more complex syndrome.
• Mutations that interfere with bone or cartilage formation, growth, and/or maintenance of normal matrix components have more diffuse effects; such disorders are called dysplasias—more specifically, osteodysplasias and chondrodysplasias. Dysplasia in this context refers to abnormal growth and does not imply precancerous lesions, as it does in other tissues (Chapter 5). They number well over 350, and only select examples are discussed here.
• Other genetic metabolic disorders not usually thought of as primary skeletal diseases (e.g., mucopolysaccharidoses such as Hurler syndrome) also affect the bone matrix; such conditions are discussed briefly with other genetic disorders in Chapter 6.
Osteogenesis Imperfecta
Osteogenesis imperfecta (OI), also known as “brittle bone disease,” is actually a group of genetic disorders caused by defective synthesis of type I collagen. Because type I collagen is a major component of extracellular matrix in other parts of the body, there are also numerous extraskeletal manifestations (affecting skin, joints, teeth, and eyes, for example). The mutations underlying OI characteristically involve the coding sequences for α1 or α2 chains of type I collagen. Because collagen synthesis and extracellular export require formation of a complete and intact triple helix, any primary defect in a collagen chain tends to disrupt the entire structure and results in its premature degradation (an example of a dominant negative mutation) (Chapter 6). As a consequence, most defects manifest as autosomal dominant disorders and may be associated with severe malformations. There is, however, a broad spectrum of severity, and mutations that result in qualitatively normal collagen but at only reduced levels generally have milder manifestations.
The fundamental abnormality in all forms of OI is too little bone, resulting in extreme skeletal fragility. Four major subtypes are recognized. The type II variant is uniformly fatal in utero or immediately postpartum as a consequence of multiple fractures that occur before birth. By contrast, patients with type I OI have a normal lifespan, with only a modestly increased proclivity for fractures during childhood (decreasing in frequency after puberty). The classic finding of blue sclerae in type I OI is attributable to decreased scleral collagen content; this deficit causes a relative transparency that allows the underlying choroid to be seen. Hearing loss can be related to conduction defects in the middle and inner ear bones, and small misshapen teeth are a result of dentin deficiency.
Achondroplasia and Thanatophoric Dwarfism
Achondroplasia is the most common form of dwarfism. It is caused by activating point mutations in fibroblast growth factor receptor 3 (FGFR3), a receptor with tyrosine kinase activity that transmits intracellular signals. Signals transmitted by FGFR3 inhibit the proliferation and function of growth plate chondrocytes; consequently, the growth of normal epiphyseal plates is suppressed, and the length of long bones is severely stunted. The disorder can be inherited in autosomal dominant fashion, but many cases arise from new spontaneous mutations.
Achondroplasia affects all bones that develop by enchondral ossification. The most conspicuous changes include short stature, disproportionate shortening of the proximal extremities, bowing of the legs, and frontal bossing with midface hypoplasia. The cartilage of the growth plates is disorganized and hypoplastic.
Thanatophoric dwarfism is a lethal variant of dwarfism, affecting 1 in every 20,000 live births (thanatophoric means “death-loving”). This disease is caused by missense or point mutations most commonly located in the extracellular domains of FGFR3. Affected heterozygotes exhibit extreme shortening of the limbs, frontal bossing of the skull, and an extremely small thorax, which is the cause of fatal respiratory failure in the perinatal period.
Osteopetrosis
Osteopetrosis is a group of rare genetic disorders characterized by defective osteoclast-mediated bone resorption. Osteopetrosis (literally, “bone-that-is-like-stone disorder”) is an appropriate name, since the bones are dense, solid, and stone-like. Paradoxically, because turnover is decreased, the persisting bone tissue becomes weak over time and predisposed to fractures like a piece of chalk. Several variants are known, the two most common being an autosomal dominant adult form with mild clinical manifestations, and autosomal recessive infantile, with a severe/lethal phenotype.
The defects that cause osteopetrosis are categorized into those that disturb osteoclast function and those that interfere with osteoclast formation and differentiation. The precise nature of the osteoclast dysfunction is unknown in many cases. Nevertheless, in some cases the abnormalities have been identified. These include carbonic anhydrase II deficiency, proton pump deficiency and chloride channel defect, all of which interfere with the ability of osteoclasts to resorb bone. A mouse model of osteopetrosis is caused by mutations in the monocyte-colony stimulating factor (M-CSF), which is required for osteoclast differentiation. No comparable defect has been identified in humans.
Besides fractures, patients with osteopetrosis frequently have cranial nerve palsies (due to compression of nerves within shrunken cranial foramina), recurrent infections because of reduced marrow size and activity, and hepatosplenomegaly caused by extramedullary hematopoiesis resulting from reduced marrow space. Morphologically, the primary spongiosa, which normally is removed during growth, persists, filling the medullary cavity, and bone is deposited in increased amounts woven into the architecture. Because osteoclasts are derived from marrow monocyte precursors, hematopoietic stem cell transplantation holds the promise of repopulating recipients with progenitor cells capable of differentiating into fully functional osteoclasts. Indeed, many of the skeletal abnormalities appear to be reversible once normal precursor cells are provided.
 Summary
Summary
Congenital Disorders of Bone and Cartilage
• Abnormalities in a single or group of bones are called dysostoses and can result in the absence of bones, supernumerary bones, or inappropriately fused bones; some of these result from mutations in homeobox genes affecting localized migration and condensation of primitive mesenchymal cells.
• Abnormalities in bone or cartilage organogenesis are called dysplasias; these can be caused by mutations that affect signal transduction pathways or components of the extracellular matrix:
 Achondroplasia and thanatophoric dwarfism occur as a consequence of constitutive FGFR3 activation, resulting in defective cartilage synthesis at growth plates.
Achondroplasia and thanatophoric dwarfism occur as a consequence of constitutive FGFR3 activation, resulting in defective cartilage synthesis at growth plates.Acquired Diseases of Bone
Many nutritional, endocrine, and systemic disorders affect the development of the skeletal system. Nutritional deficiencies causing bone disease include deficiencies of vitamin C (involved in collagen cross-linking; deficiency causes scurvy) and vitamin D (involved in calcium uptake; deficiency causes rickets and osteomalacia). Both of these are discussed in greater detail with other nutritional diseases in Chapter 7. Primary and secondary forms of hyperparathyroidism (discussed in Chapter 19) also cause significant skeletal changes, which are briefly reviewed in this section. Many of these disorders are characterized by inadequate osteoid, also called osteopenia; the most important clinically significant osteopenia is osteoporosis.
Osteoporosis
Osteoporosis is an acquired condition characterized by reduced bone mass, leading to bone fragility and susceptibility to fractures. The bone loss may be confined to certain bones or regions, as in disuse osteoporosis of a limb, or be generalized, involving the entire skeleton. Generalized osteoporosis may be primary or occur secondary to a large variety of insults, including metabolic diseases, vitamin deficiencies, and drug exposures (Table 20–1).
Table 20–1 Categories of Generalized Osteoporosis
| Primary |
| Postmenopausal |
| Senile |
| Secondary |
| Endocrine Disorders |
| Hyperparathyroidism |
| Hypo or hyperthyroidism |
| Hypogonadism |
| Pituitary tumors |
| Diabetes, type 1 |
| Addison disease |
| Neoplasia |
| Multiple myeloma |
| Carcinomatosis |
| Gastrointestinal Disorders |
| Malnutrition |
| Malabsorption |
| Hepatic insufficiency |
| Vitamin C, D deficiencies |
| Idiopathic disease |
| Drugs |
| Anticoagulants |
| Chemotherapy |
| Corticosteroids |
| Anticonvulsants |
| Alcohol |
| Miscellaneous |
| Osteogenesis imperfecta |
| Immobilization |
| Pulmonary disease |
| Homocystinuria |
| Anemia |
Primary forms of osteoporosis are most common and may be associated with aging (senile osteoporosis) or the postmenopausal state in women. The drop in estrogen following menopause tends to exacerbate the loss of bone that occurs with aging, placing older women at high risk of osteoporosis relative to men. The risk of osteoporosis with aging is related to the peak bone mass earlier in life, which is influenced by genetic, nutritional, and environmental factors. Bone mass peaks during young adulthood; the greater the peak bone mass, the greater the delay in onset of osteoporosis. In both men and women, beginning in the third or fourth decade of life, bone resorption begins to outpace bone formation. The bone loss, averaging 0.5% per year, is a seemingly inevitable consequence of aging and is most prominent in areas containing abundant trabecular bone—namely the spine and femoral neck. The amount of bone loss with each cycle of remodeling is accelerated after menopause; hence, the vulnerability of women to osteoporosis and its complications. Regardless of the underlying cause, the progressive loss of bone mass is clinically significant because of the resultant increase in the risk of fractures. Roughly 1.5 million Americans each year experience an osteoporosis-related fracture, with those of greatest clinical significance involving the vertebrae and the hips. All told, the annual health care costs associated with osteoporosis-related fractures in the United States exceeds $18 billion.
 Morphology
Morphology
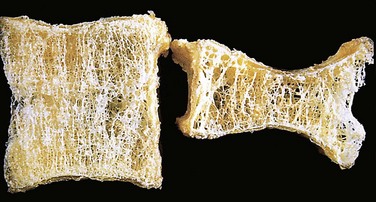
The hallmark of osteoporosis is a loss of bone. The cortices are thinned, with dilated haversian canals, and the trabeculae are reduced in thickness and lose their interconnections. Osteoclastic activity is present but is not dramatically increased, and the mineral content of the bone tissue is normal. Once enough bone is lost, susceptibility to fractures increases (Fig. 20–3). In postmenopausal osteoporosis, trabecular bone loss often is severe, resulting in compression fractures and collapse of vertebral bodies. In senile osteoporosis, cortical bone loss is prominent, predisposing to fractures in other weight-bearing bones, such as the femoral neck.
 Pathogenesis
Pathogenesis
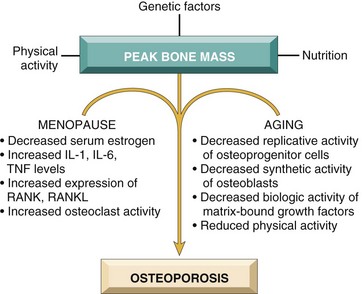
Osteoporosis occurs when the dynamic balance between bone formation by osteoblasts and bone resorption by osteoclasts (Fig. 20–2) tilts in favor of resorption. Several factors may tip the scales (Fig. 20–4):
• Age-related changes. With increasing age, the replicative and matrix production activities of osteoblasts progressively diminish. The various growth factors deposited in the extracellular matrix also diminish with time. Unfortunately, while new bone synthesis wanes with advancing age, osteoclasts retain their youthful vigor.
• Hormonal influences. The decline in estrogen levels associated with menopause correlates with an acceleration of cortical bone and trabecular (cancellous) bone loss. Over 30 to 40 years, this can result in the loss of up to 35% of cortical bone and 50% of trabecular bone! It is therefore not surprising that roughly half of postmenopausal women will suffer an osteoporotic fracture (compared with 2% to 3% of men of comparable age). It appears that the postmenopausal drop in estrogen leads to increased cytokine production (especially IL-1, IL-6, and TNF), presumably from cells in the bone. These stimulate RANK–RANK ligand activity and suppress OPG production (Fig. 20–2). There is some compensatory osteoblastic activity, but it is inadequate to keep pace with osteoclastic bone resorption. While estrogen replacement can ameliorate some of the bone loss, such therapy is increasingly associated with cardiovascular risks (Chapter 10).
• Physical activity. Because mechanical forces stimulate bone remodeling, reduced physical activity increases bone loss. This effect is obvious in an immobilized limb and also occurs throughout the skeleton in astronauts working in a gravity-free environment. Decreased physical activity in older persons also contributes to senile osteoporosis. Because the magnitude of skeletal loading influences bone density more than does the number of load cycles, the type of physical activity is important. Thus, resistance exercises such as weight training increase bone mass more effectively than endurance activities such as jogging.
• Genetic factors. Vitamin D receptor polymorphisms appear to influence the peak bone mass early in life. Additional genetic variables can influence either calcium uptake or PTH synthesis and responses.
• Calcium nutritional state. A majority of adolescent girls (but not boys) have insufficient dietary calcium. Unfortunately, this calcium deficiency occurs during a period of rapid bone growth. As a result, girls typically do not achieve the peak bone mass that could be otherwise expected and are accordingly more likely to develop clinically significant osteoporosis at an earlier age than their male counterparts.
• Secondary causes of osteoporosis. These include prolonged glucocorticoid therapy, which increases bone resorption and reduces bone synthesis. Cigarette smoking and excess alcohol also can result in reduced bone mass.
Clinical Course
The clinical outcome with osteoporosis depends on which bones are involved. Thoracic and lumbar vertebral fractures are extremely common, leading to loss of height and various deformities, including kyphoscoliosis, which can compromise respiratory function. Pulmonary embolism and pneumonia are common complications of fractures of the femoral neck, pelvis, or spine and result in as many as 50,000 deaths annually.
Osteoporosis is difficult to diagnose because it is asymptomatic until skeletal fragility is announced with a fracture. Moreover, it cannot be reliably detected in plain radiographs until 30% to 40% of bone mass has already disappeared; serum levels of calcium, phosphorus, and alkaline phosphatase are notoriously insensitive. Current state-of-the-art methods for bone loss estimation consist of specialized radiographic techniques to assess bone mineral density, such as dual-energy absorptiometry and quantitative computed tomography.
Osteoporosis prevention and treatment begin with adequate dietary calcium intake, vitamin D supplementation, and a regular exercise regimen—starting before the age of 30—to maximize the peak bone mass. Calcium and vitamin D supplements later in life can also modestly reduce bone loss. Pharmacologic treatments include use of antiresorptive and osteoanabolic agents. The antiresorptive agents, such as bisphosphonates, calcitonin, estrogen, and denosumab, decrease bone resorption by osteoclasts. The main anabolic agent is parathyroid hormone or an analogue, given in amounts that stimulate osteoblastic activity.
Paget Disease (Osteitis Deformans)
This unique skeletal disease is characterized by repetitive episodes of frenzied, regional osteoclastic activity and bone resorption (osteolytic stage), followed by exuberant bone formation (mixed osteoclastic-osteoblastic stage), and finally by an apparent exhaustion of cellular activity (osteosclerotic stage). The net effect of this process is a gain in bone mass; however, the newly formed bone is disordered and weak, so bones may become enlarged and misshapen.
Paget disease usually presents in mid- to late adulthood. Marked variation in prevalence has been reported in different populations: The disorder is rare in Scandinavia, China, Japan, and Africa and relatively common in much of Europe, Australia, New Zealand, and the United States, affecting up to 2.5% of the adult populations. Of interest, it appears that the incidence of Paget disease is decreasing.
Morphology
Paget disease may manifest as a solitary lesion (monostotic) or may occur at multiple sites (polyostotic) usually asynchronously. In the initial lytic phase, osteoclasts (and their associated Howship lacunae) are numerous, abnormally large, and have increased numbers of nuclei. Osteoclasts persist in the mixed phase, but the bone surfaces become lined by prominent osteoblasts. The marrow is replaced by loose connective tissue containing osteoprogenitor cells, as well as numerous blood vessels needed to meet the increased metabolic demands of the tissue. The newly formed bone may be woven or lamellar, but eventually all of it is remodeled into abnormal lamellar bone with a pathognomonic mosaic pattern (likened to a jigsaw puzzle) due to prominent haphazardly arranged cement lines (Fig. 20–5). As the osteoblastic activity ceases, the periosseous fibrovascular tissue recedes and is replaced by normal marrow. Although thickened, the resulting cortex is softer than normal and prone to deformation and fracture under stress.
Pathogenesis
When he first described the disease, Sir James Paget attributed the skeletal changes to an inflammatory process, and assigned the moniker osteitis deformans. After many years and multiple alternative theories, Paget’s original idea may prove to be correct. It has long been postulated that a paramyxovirus infection (a slow virus) underlies Paget disease. Paramyxovirus antigens and particles resembling paramyxovirus can be demonstrated in osteoclasts. The causal connection is that paramyxoviruses can induce IL-1 and IL-6 secretion from infected cells, and these cytokines—as well as macrophage colony-stimulating factor (M-CSF)—are produced in large amounts in pagetic bone. As noted earlier, these potently activate osteoclasts. Nevertheless, as intriguing as these observations are, no infectious virus has been isolated from affected tissue. About 10% of affected patients have germline mutations in the gene SQSTM1, which encodes a protein that appears to increase osteoclastogenesis; these mutations are associated with earlier onset disease, a greater number of affected bones, and an increased incidence of fractures.
Clinical Course
The clinical findings depend on the extent and site of the disease. Paget disease is monostotic (tibia, ilium, femur, skull, vertebrae, and humerus) in about 15% of cases and polyostotic in the remainder; the axial skeleton or the proximal femur is involved in as many as 80% of cases. Involvement of the ribs, fibulae, and small bones of the hands and feet is unusual. Although Paget disease can produce a plethora of skeletal, neuromuscular, and cardiovascular complications, most cases are clinically mild, and the bone changes are discovered only incidentally in radiographs. Elevations in serum alkaline phosphatase and increased urinary excretion of hydroxyproline reflect exuberant bone turnover.
In some patients, the early hypervascular bone lesions cause warmth of the overlying skin and subcutaneous tissue. With extensive polyostotic disease, hypervascularity can result in high-output congestive heart failure. In the proliferative phase of the disease involving the skull, common symptoms attributable to nerve impingement include headache and visual and auditory disturbances. Vertebral lesions cause back pain and may be associated with disabling fractures and nerve root compression. Affected long bones in the legs often are deformed, as a consequence of the inability of pagetoid bone to remodel appropriately in response to the stress of weight bearing. Brittle long bones in particular are subject to chalkstick fractures.
The development of sarcoma is a dreaded but fortunately rare complication of Paget disease, occurring in only an estimated 1% of patients. The sarcomas usually are osteogenic, although other histologic variants can occur. The distribution of osteosarcoma generally parallels that of the Paget disease lesions, with the exception of vertebral bodies, which rarely harbor malignancy. The prognosis for patients who develop secondary sarcomas is exceedingly poor, but otherwise Paget disease usually follows a relatively benign course. Most patients have mild symptoms that are readily controlled with bisphosphonates, drugs that interfere with bone resorption.
Rickets and Osteomalacia
Both rickets and osteomalacia are manifestations of vitamin D deficiency or its abnormal metabolism (and are detailed in Chapter 7). The fundamental defect is an impairment of mineralization and a resultant accumulation of unmineralized matrix. This contrasts with osteoporosis, in which the mineral content of the bone is normal and the total bone mass is decreased. Rickets refers to the disorder in children, in which it interferes with the deposition of bone in the growth plates. Osteomalacia is the adult counterpart, in which bone formed during remodeling is undermineralized, resulting in predisposition to fractures.
Hyperparathyroidism
As discussed in Chapter 19, parathyroid hormone (PTH) plays a central role in calcium homeostasis through the following effects:
• Osteoclast activation, increasing bone resorption and calcium mobilization. PTH mediates the effect indirectly by increased RANKL expression on osteoblasts.
• Increased resorption of calcium by the renal tubules
• Increased urinary excretion of phosphates
• Increased synthesis of active vitamin D, 1,25(OH)2-D, by the kidneys, which in turn enhances calcium absorption from the gut and mobilizes bone calcium by inducing RANKL on osteoblasts
The net result of the actions of PTH is an elevation in serum calcium, which, under normal circumstances, inhibits further PTH production. However, excessive or inappropriate levels of PTH can result from autonomous parathyroid secretion (primary hyperparathyroidism) or can occur in the setting of underlying renal disease (secondary hyperparathyroidism) (see also Chapter 19).
In either setting, hyperparathyroidism leads to significant skeletal changes related to unabated osteoclast activity. The entire skeleton is affected, although some sites can be more severely affected than others. PTH is directly responsible for the bone changes seen in primary hyperparathyroidism, but additional influences contribute to the development of bone disease in secondary hyperparathyroidism. In chronic renal insufficiency there is inadequate 1,25-(OH)2-D synthesis, which ultimately affects gastrointestinal calcium absorption. The hyperphosphatemia of renal failure also suppresses renal α1-hydroxylase, further impairing vitamin D synthesis; additional influences include metabolic acidosis and aluminum deposition in bone. As bone mass decreases, affected patients are increasingly susceptible to fractures, bone deformation, and joint problems. Fortunately, a reduction in PTH levels to normal can completely reverse the bone changes.
Morphology
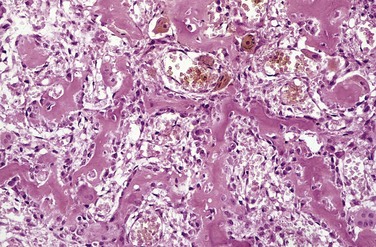
The hallmark of PTH excess is increased osteoclastic activity, with bone resorption. Cortical and trabecular bone are diminished and replaced by loose connective tissue. Bone resorption is especially pronounced in the subperiosteal regions and produces characteristic radiographic changes, best seen along the radial aspect of the middle phalanges of the second and third fingers. Microscopically, there are increased numbers of osteoclasts boring into the centers of bony trabeculae (dissecting osteitis) and expanding haversian canals (cortical cutting cones) (Fig. 20–6, A). The marrow space contains increased amounts of loose fibrovascular tissue. Hemosiderin deposits are present, reflecting episodes of hemorrhage resulting from microfractures of the weakened bone. In some instances, collections of osteoclasts, reactive giant cells, and hemorrhagic debris form a distinct mass termed a brown tumor of hyperparathyroidism (Fig. 20–6, B). Cystic change is common in such lesions (hence the name osteitis fibrosa cystica), which can be confused with primary bone neoplasms.
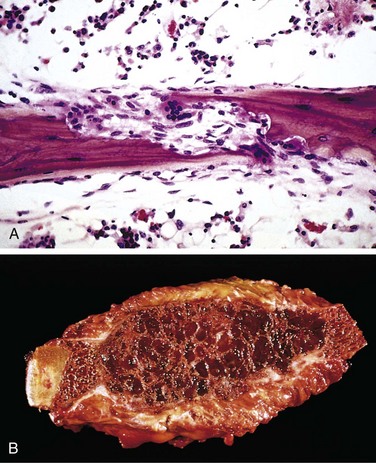
Summary
Acquired Diseases of Bone Development and Mass
• Nutritional deficiencies can affect bone integrity by altering the quality of the organic matrix (e.g., vitamin C is involved in collagen cross-linking) or by influencing bone mineralization (e.g., vitamin D is involved in calcium uptake).
• Osteoporosis results from decreased bone mass and is clinically significant because it predisposes bone to fracture. Although osteoporosis is multifactorial, the two most common forms are senile osteoporosis due to aging-related losses of osteoblast function, and postmenopausal osteoporosis due to increased osteoclastic activity caused by the relative absence of estrogen.
• Paget disease may result from a paramyxovirus infection in genetically susceptible persons and is caused by aberrant and excessive osteoclast activity, followed by exuberant—but structurally unsound—osteoblast deposition of bone.
• Primary or secondary (due to renal failure) overproduction of PTH (hyperparathyroidism) results in increased osteoclast activity and bone resorption, leading to fractures and deformities.
Fractures
Fractures rank among the most common pathologic conditions of bone. They are classified as follows:
• Closed, in which the overlying tissue is intact, or compound, in which the fracture extends into the overlying skin
If the break occurs at the site of previous disease (e.g., a bone cyst, a malignant tumor, or a brown tumor associated with elevated PTH), it is termed a pathologic fracture. A stress fracture develops slowly over time as a collection of microfractures associated with increased physical activity, especially with new repetitive mechanical loads on bone (as sustained in military bootcamp activities).
In all cases, the repair of a fracture is a highly regulated process that involves overlapping stages:
• The trauma of the bone fracture ruptures associated blood vessels; the resulting blood clot creates a fibrin mesh scaffold to recruit inflammatory cells, fibroblasts, and endothelium. Degranulated platelets and marauding inflammatory cells subsequently release a host of cytokines (e.g., platelet-derived growth factor, fibroblast growth factor) that activate bone progenitor cells, and within a week, the involved tissue is primed for new matrix synthesis. This soft tissue callus can hold the ends of the fractured bone in apposition but is noncalcified and cannot support weight bearing.
• Bone progenitors in the periosteum and medullary cavity deposit new foci of woven bone, and activated mesenchymal cells at the fracture site differentiate into cartilage-synthesizing chondroblasts. In uncomplicated fractures, this early repair process peaks within 2 to 3 weeks. The newly formed cartilage acts as a nidus for endochondral ossification, recapitulating the process of bone formation in epiphyseal growth plates. This connects the cortices and trabeculae in the juxtaposed bones. With ossification, the fractured ends are bridged by a bony callus.
• Although excess fibrous tissue, cartilage, and bone are produced in the early callus, subsequent weight bearing leads to remodeling of the callus from nonstressed sites; at the same time there is fortification of regions that support greater loads. This process restores the original size, shape, and integrity of the bone.
The healing of a fracture can be disrupted by many factors:
• Displaced and comminuted fractures frequently result in some deformity; devitalized fragments of splintered bone require resorption, which delays healing, enlarges the callus, and requires inordinately long periods of remodeling and may never completely normalize.
• Inadequate immobilization permits constant movement at the fracture site, so that the normal constituents of callus do not form. In such instances, the healing site is composed mainly of fibrous tissue and cartilage, perpetuating the instability and resulting in delayed union and nonunion. Too much motion along the fracture gap (as in nonunion) causes the central portion of the callus to undergo cystic degeneration; the luminal surface can actually become lined by synovial-type cells, creating a false joint, or pseudoarthrosis. In the setting of a nonunion or pseudoarthrosis, normal healing can be achieved only if the interposed soft tissues are removed and the fracture site is stabilized.
• Infection (a risk in comminuted and open fractures) is a serious obstacle to fracture healing. The infection must be eradicated before successful bone reunion and remodeling can occur.
• Bone repair obviously will be impaired in the setting of inadequate levels of calcium or phosphorus, vitamin deficiencies, systemic infection, diabetes, or vascular insufficiency.
With uncomplicated fractures in children and young adults, practically perfect reconstitution is the norm. When fractures occur in older age groups or in abnormal bones (e.g., osteoporotic bone), repair frequently is less than optimal without orthopedic intervention.
Osteonecrosis (Avascular Necrosis)
Ischemic necrosis with resultant bone infarction occurs relatively frequently. Mechanisms contributing to bone ischemia include
• Vascular compression or disruption (e.g., after a fracture)
• Thromboembolic disease (e.g., nitrogen bubbles in caisson disease—see Chapter 3)
• Primary vessel disease (e.g., vasculitis)
• Sickle cell crisis (Chapter 11)
Most cases of bone necrosis are due to fracture or occur after corticosteroid use, but in many instances the etiology is unknown.
Morphology
The pathologic features of bone necrosis are the same regardless of cause. Dead bone with empty lacunae is interspersed with areas of fat necrosis and insoluble calcium soaps. The cortex usually is not affected, because of collateral blood supply; in subchondral infarcts, the overlying articular cartilage also remains viable because the synovial fluid can provide nutritive support. With time, osteoclasts can resorb some of the necrotic bony trabeculae; any dead bone fragments that remain act as scaffolding for new bone formation, a process called creeping substitution.
Clinical Course
Symptoms depend on the size and location of injury. Subchondral infarcts initially present with pain during physical activity that becomes more persistent with time. Medullary infarcts usually are silent unless large in size (as may occur with Gaucher disease, caisson disease, or sickle cell disease). Medullary infarcts usually are stable, but subchondral infarcts often collapse and may lead to severe osteoarthritis. Roughly 50,000 joint replacements are performed each year in the United States to treat the consequences of osteonecrosis.
Osteomyelitis
Osteomyelitis is defined as inflammation of bone and marrow, but in common use it is virtually synonymous with infection. Osteomyelitis can be secondary to systemic infection but more frequently occurs as a primary isolated focus of disease; it can be an acute process or a chronic, debilitating illness. Although any microorganism can cause osteomyelitis, the most common etiologic agents are pyogenic bacteria and Mycobacterium tuberculosis.
Pyogenic Osteomyelitis
Most cases of acute osteomyelitis are caused by bacteria. The offending organisms reach the bone by one of three routes: (1) hematogenous dissemination (most common); (2) extension from an infection in adjacent joint or soft tissue; or (3) traumatic implantation after compound fractures or orthopedic procedures. Overall, Staphylococcus aureus is the most frequent causative organism; its propensity to infect bone may be related to the expression of surface proteins that allow adhesion to bone matrix. Escherichia coli and group B streptococci are important causes of acute osteomyelitis in neonates, and Salmonella is an especially common pathogen in persons with sickle cell disease. Mixed bacterial infections, including anaerobes, typically are responsible for osteomyelitis secondary to bone trauma. In as many as 50% of cases, no organisms can be isolated.
Morphology
The morphologic changes in osteomyelitis depend on the chronicity and location of the infection. Causal bacteria proliferate, inducing an acute inflammatory reaction, with consequent cell death. Entrapped bone rapidly becomes necrotic; this non-viable bone is called a sequestrum. Bacteria and inflammation can percolate throughout the haversian systems to reach the periosteum. In children, the periosteum is loosely attached to the cortex; therefore, sizable subperiosteal abscesses can form and extend for long distances along the bone surface. Lifting of the periosteum further impairs the blood supply to the affected region, and both suppurative and ischemic injury can cause segmental bone necrosis. Rupture of the periosteum can lead to abscess formation in the surrounding soft tissue that may lead to a draining sinus. Sometimes the sequestrum crumbles, releasing fragments that pass through the sinus tract.
In infants (and uncommonly in adults), epiphyseal infection can spread into the adjoining joint to produce suppurative arthritis, sometimes with extensive destruction of the articular cartilage and permanent disability. An analogous process can involve vertebrae, with an infection destroying intervertebral discs and spreading into adjacent vertebrae.
After the first week of infection, chronic inflammatory cells become more numerous. Leukocyte cytokine release stimulates osteoclastic bone resorption, fibrous tissue ingrowth, and bone formation in the periphery. Reactive woven or lamellar bone can be deposited; when it forms a shell of living tissue around a sequestrum, it is called an involucrum (Fig. 20–7). Viable organisms can persist in the sequestrum for years after the original infection.
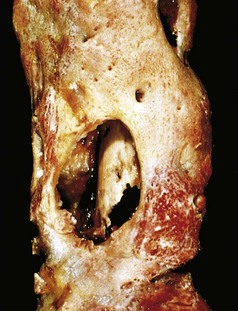
Clinical Features
Osteomyelitis classically manifests as an acute systemic illness, with malaise, fever, leukocytosis, and throbbing pain over the affected region. Symptoms also can be subtle, with only unexplained fever, particularly in infants, or only localized pain in the adult. The diagnosis is suggested by characteristic radiologic findings: a destructive lytic focus surrounded by edema and a sclerotic rim. In many untreated cases, blood cultures are positive, but biopsy and bone cultures are usually required to identify the pathogen. A combination of antibiotics and surgical drainage usually is curative, but up to a quarter of cases do not resolve and persist as chronic infections. Chronicity may develop with delay in diagnosis, extensive bone necrosis, abbreviated antibiotic therapy, inadequate surgical debridement, and/or weakened host defenses. Besides occasional acute flareups, chronic osteomyelitis also may be complicated by pathologic fracture, secondary amyloidosis, endocarditis, sepsis, development of squamous cell carcinoma if the infection creates a sinus tract, and rarely osteosarcoma.
Tuberculous Osteomyelitis
Mycobacterial infection of bone has long been a problem in developing countries; with the resurgence of tuberculosis (due to immigration patterns and increasing numbers of immunocompromised persons) it is becoming an important disease in other countries as well.
Bone infection complicates an estimated 1% to 3% of cases of pulmonary tuberculosis. The organisms usually reach the bone through the bloodstream, although direct spread from a contiguous focus of infection (e.g., from mediastinal nodes to the vertebrae) also can occur. With hematogenous spread, long bones and vertebrae are favored sites. The lesions often are solitary but can be multifocal, particularly in patients with an underlying immunodeficiency. Because the tubercle bacillus is microaerophilic, the synovium, with its higher oxygen pressures, is a common site of initial infection. The infection then spreads to the adjacent epiphysis, where it elicits typical granulomatous inflammation with caseous necrosis and extensive bone destruction. Tuberculosis of the vertebral bodies is a clinically serious form of osteomyelitis. Infection at this site causes vertebral deformity, collapse, and posterior displacement (Pott disease), leading to neurologic deficits. Spinal deformities due to Pott disease afflicted several men of letters (including Alexander Pope and William Henley) and likely served as the inspiration for Victor Hugo’s Hunchback of Notre Dame. Extension of the infection to the adjacent soft tissues with the development of psoas muscle abscesses is fairly common.
Bone Tumors
Primary bone tumors are considerably less common than bone metastases from other primary sites; metastatic disease is discussed at the end of this section.
Primary bone tumors exhibit great morphologic diversity and clinical behaviors—from benign to aggressively malignant. Most are classified according to the normal cell counterpart and line of differentiation; Table 20–2 lists the salient features of the most common primary bone neoplasms, excluding multiple myeloma and other hematopoietic tumors. Overall, matrix-producing and fibrous tumors are the most common, and among the benign tumors, osteochondroma and fibrous cortical defect occur most frequently. Osteosarcoma is the most common primary bone cancer, followed by chondrosarcoma and Ewing sarcoma. Benign tumors greatly outnumber their malignant counterparts, particularly before the age of 40 years; bone tumors in elderly persons are much more likely to be malignant.
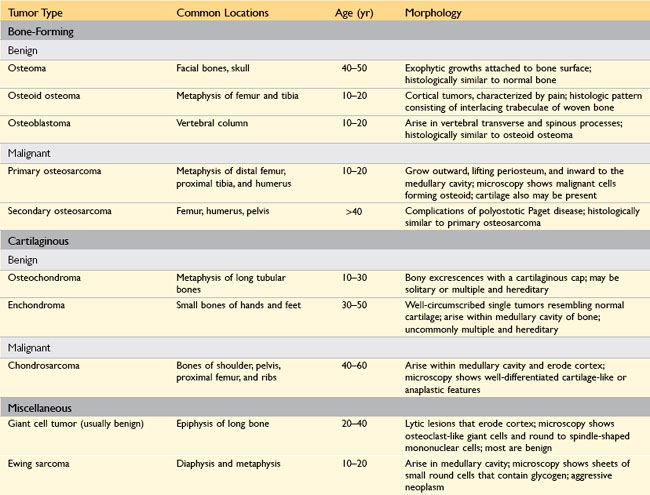
Most bone tumors develop during the first several decades of life and have a propensity to originate in the long bones of the extremities. Nevertheless, specific tumor types target certain age groups and anatomic sites; these associations are often helpful in arriving at the correct diagnosis. For instance, most osteosarcomas occur during adolescence, with half arising around the knee, either in the distal femur or proximal tibia. By contrast, chondrosarcomas tend to develop during mid- to late adulthood and involve the trunk, limb girdles, and proximal long bones.
Most bone tumors arise without any previous known cause. Nevertheless, genetic syndromes (e.g., Li-Fraumeni and retinoblastoma syndromes) (Chapter 5) are associated with osteosarcomas, as are (rarely) bone infarcts, chronic osteomyelitis, Paget disease, irradiation, and use of metal orthopedic devices.
In terms of clinical presentation, benign lesions frequently are asymptomatic and are detected as incidental findings. Others produce pain or a slowly growing mass. Occasionally, a pathologic fracture is the first manifestation. Radiologic imaging is critical in the evaluation of bone tumors; however, biopsy and histologic study and, in some cases, molecular tests are necessary for diagnosis.
Bone-Forming Tumors
The tumor cells in the following neoplasms all produce bone that usually is woven and variably mineralized.
Osteoma
Osteomas are benign lesions most commonly encountered in the head and neck, including the paranasal sinuses, but which can occur elsewhere as well. They typically present in middle age as solitary, slowly growing, hard, exophytic masses on a bone surface. Multiple lesions are a feature of Gardner syndrome, a hereditary condition discussed later. On histologic examination, osteomas recapitulate cortical-type bone and are composed of a mixture of woven and lamellar bone. Although they may cause local mechanical problems (e.g., obstruction of a sinus cavity) and cosmetic deformities, they are not locally aggressive and do not undergo malignant transformation.
Osteoid Osteoma and Osteoblastoma
Osteoid osteomas and osteoblastomas are benign neoplasms with very similar histologic features. Both lesions typically appear during the teenage years and 20s, with a male predilection (2 : 1 for osteoid osteomas). They are distinguished from each other primarily by their size and clinical presentation. Osteoid osteomas arise most often beneath the periosteum or within the cortex in the proximal femur and tibia or posterior spinal elements and are by definition less than 2 cm in diameter, whereas osteoblastomas are larger. Localized pain, most severe at night, is an almost universal complaint with osteoid osteomas, and usually is relieved by aspirin. Osteoblastomas arise most often in the vertebral column; they also cause pain, although it often is more difficult to localize and is not responsive to aspirin. Local excision is the treatment of choice; incompletely resected lesions can recur. Malignant transformation is rare unless the lesion is treated with irradiation.
Morphology
On gross inspection, both lesions are round-to-oval masses of hemorrhagic, gritty-appearing tan tissue. A rim of sclerotic bone is present at the edge of both types of tumors; however, it is much more conspicuous in osteoid osteomas. On microscopic examination, both neoplasms are composed of interlacing trabeculae of woven bone surrounded by osteoblasts (Fig. 20–8). The intervening stroma is loose, vascular connective tissue containing variable numbers of giant cells.
Osteosarcoma
Osteosarcoma is a bone-producing malignant mesenchymal tumor. After myeloma and lymphoma, osteosarcoma is the most common primary malignant tumor of bone, accounting for approximately 20% of primary bone cancers; a little over 2000 cases are diagnosed annually in the United States. Osteosarcomas occur in all age groups, but about 75% of patients are younger than 20 years of age, with a second peak occurring in elderly persons, usually in association with other conditions, including Paget disease, bone infarcts, and previous irradiation. Men are more commonly affected than women (1.6 : 1). Although any bone can be involved, most tumors arise in the metaphyseal region of the long bones of the extremities, with almost 60% occurring about the knee, 15% around the hip, 10% at the shoulder, and 8% in the jaw. Several subtypes of osteosarcoma are distinguished on the basis of the site of involvement within the bone (e.g., medullary versus cortical), degree of differentiation, number of involved sites, presence of underlying disease, and histologic features; the most common type of osteosarcoma is primary, solitary, intramedullary, and poorly differentiated, producing a predominantly bony matrix.
Morphology
On gross evaluation, osteosarcomas are gritty-appearing, gray-white tumors, often exhibiting hemorrhage and cystic degeneration. Tumors frequently destroy the surrounding cortices, producing soft tissue masses (Fig. 20–9, A). They spread extensively in the medullary canal, infiltrating and replacing the marrow but only infrequently penetrating the epiphyseal plate or entering the joint space. Tumor cells vary in size and shape and frequently have large hyperchromatic nuclei; bizarre tumor giant cells are common, as are mitotic figures. The production of mineralized or unmineralized bone (osteoid) by malignant cells is essential for diagnosis of osteosarcoma (Fig. 20–9, B). The neoplastic bone typically is coarse and lacelike but also can be deposited in broad sheets. Cartilage and fibroblastic differentiation can also be present in varying amounts. When malignant cartilage is abundant, the tumor is called a chondroblastic osteosarcoma. Vascular invasion is common, as is spontaneous tumor necrosis.
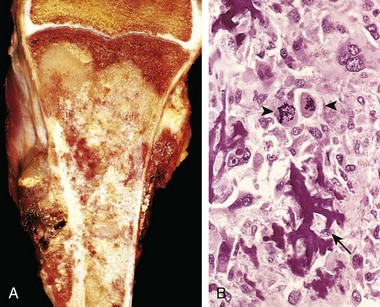
Figure 20–9 Osteosarcoma. A, Mass involving the upper end of the tibia. The tan-white tumor fills most of the medullary cavity of the metaphysis and proximal diaphysis. It has infiltrated through the cortex, lifted the periosteum, and formed soft tissue masses on both sides of the bone. B, Histologic appearance, with coarse, lacelike pattern of neoplastic bone (arrow) produced by anaplastic tumor cells. Note the wildly aberrant mitotic figures (arrowheads).
Pathogenesis
Several mutations are closely associated with the development of osteosarcoma. In particular, RB gene mutations occur in 60% to 70% of sporadic tumors, and persons with hereditary retinoblastomas (due to germline mutations in the RB gene) have a thousand-fold greater risk for development of osteosarcoma. Like many other cancers, spontaneous osteosarcomas also frequently exhibit mutations in TP53 and in genes that regulate the cell cycle, including cyclins, cyclin-dependent kinases, and kinase inhibitors. Many osteosarcomas develop at sites of greatest bone growth, perhaps because rapidly dividing cells provide a fertile soil for mutations.
Clinical Features
Osteosarcomas typically manifest as painful enlarging masses, although a pathologic fracture can be the first sign. Radiographic imaging usually shows a large, destructive, mixed lytic and blastic mass with indistinct infiltrating margins. The tumor frequently breaks through the cortex and lifts the periosteum, resulting in reactive periosteal bone formation. A triangular shadow on the x-ray film between the cortex and raised periosteum (Codman triangle) is characteristic of osteosarcomas. Osteosarcomas typically spread hematogenously; at the time of diagnosis, approximately 10% to 20% of patients have demonstrable pulmonary metastases, and a larger number have microscopic metastases.
Despite aggressive behavior, standard treatment with chemotherapy and limb salvage therapy currently yields long-term survivals of 60% to 70%.
Secondary osteosarcomas occur in older adults most commonly in the setting of Paget disease or previous radiation exposure. Like primary osteosarcomas, secondary osteosarcomas are highly aggressive tumors, but they do not respond well to therapy and are usually fatal.
Cartilage-Forming Tumors
Cartilage-forming tumors produce hyaline or myxoid cartilage; fibrocartilage and elastic cartilage are rare components. Like the bone-forming tumors, cartilaginous tumors constitute a spectrum from benign, self-limited growths to highly aggressive malignancies; again, benign cartilage tumors are much more common than malignant ones. Only the more common types are discussed here.
Osteochondroma
Osteochondromas are relatively common benign, cartilage-capped tumors attached by a bony stalk to the underlying skeleton. Solitary osteochondromas typically are first diagnosed in late adolescence and early adulthood (male-to-female ratio of 3 : 1); multiple osteochondromas become apparent during childhood, occurring as multiple hereditary osteochondromas, an autosomal dominant disorder. Inactivation of both copies of the EXT1 or EXT2 genes through mutation and loss of heterozygosity in chondrocytes of the growth plate is implicated in both sporadic and hereditary osteochondromas. These tumor suppressor genes encode glycosyltransferases essential for polymerization of heparin sulfate, an important component of cartilage. This finding and other molecular genetic studies support the concept that osteochondromas are true neoplasms and not developmental malformations.
Osteochondromas develop only in bones of endochondral origin arising at the metaphysis near the growth plate of long tubular bones, especially about the knee; they tend to stop growing once the normal growth of the skeleton is completed (Fig. 20–10). Occasionally they develop from bones of the pelvis, scapula, and ribs and in these sites frequently are sessile. Rarely, osteochondromas arise in the short tubular bones of hands and feet.
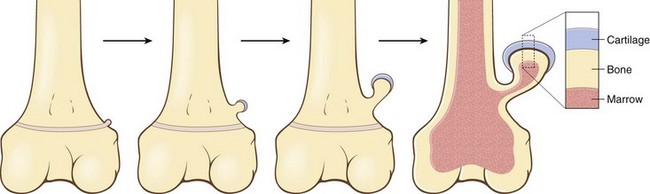
Figure 20–10 The development of an osteochondroma, beginning with an outgrowth from the epiphyseal cartilage.
Morphology
Osteochondromas range from 1 to 20 cm in size and have a cartilaginous cap that is usually less than 2 cm in thickness. The hyaline cartilage resembles a disorganized growth plate undergoing endochondral ossification. Newly formed bone forms the inner portion of the head and stalk, with the stalk cortex and central region merging with the cortex and medullary cavity, respectively, of the host bone.
Clinical Features
Osteochondromas are slow-growing masses that can be painful if they impinge on a nerve or if the stalk is fractured. In many cases, they are incidental findings. In multiple hereditary osteochondromas, deformity of the underlying bone suggests an associated disturbance in epiphyseal growth. Solitary osteochondromas rarely progress to chondrosarcoma or other sarcomas, but malignant transformation occurs more frequently in those with multiple hereditary osteochondromas.
Chondroma
Chondromas are benign neoplasms of hyaline cartilage. When they arise within the medulla, they are termed enchondromas; when on the bone surface, they are called juxtacortical chondromas. Enchondromas usually are diagnosed in persons between the ages of 20 and 50 years; they typically are solitary and located in the metaphyseal region of tubular bones, the favored sites being the short tubular bones of the hands and feet. Ollier disease is characterized by multiple chondromas preferentially involving one side of the body, and Maffucci syndrome is characterized by multiple chondromas associated with soft tissue spindle cell hemangiomas.
Pathogenesis
Enchondromas occurring in Ollier disease and Maffucci syndrome frequently contain point mutations in either isocitrate dehydrogenase I (IDH1) or IDH2 that create a new enzyme activity. The same IDH mutations occur as somatic mutations in acute myeloid leukemias and gliomas, but in Ollier and Maffucci disease the mutations are also found at low frequency in normal tissues, suggesting the mutations occurred early during embryonic development, an example of genetic mosaicism.
Morphology
Enchondromas are gray-blue, translucent nodules usually smaller than 5 cm in greatest dimension. On microscopic examination, they are well circumscribed and composed of hyaline cartilage containing cytologically benign chondrocytes. At the periphery, there is endochondral ossification, while the center frequently calcifies and dies. In the hereditary multiple chondromatoses, the islands of cartilage exhibit greater cellularity and atypia, making them more difficult to distinguish from chondrosarcoma.
Clinical Features
Most enchondromas are detected as incidental findings; occasionally they are painful or cause pathologic fractures. On x-ray imaging, the unmineralized nodules of cartilage produce well-circumscribed oval lucencies surrounded by thin rims of radiodense bone (O-ring sign). Calcified matrix manifests as irregular opacities. The growth potential of chondromas is limited, and most remain stable, although they can recur if incompletely excised. Solitary chondromas rarely undergo malignant transformation, but those associated with enchondromatoses are at increased risk for such change. Maffucci syndrome is associated with an increased risk for development of other types of malignancies, including ovarian carcinomas and brain gliomas.
Chondrosarcoma
Chondrosarcoma is a malignant connective tissue tumor (sarcoma) whose cells manufacture and secrete neoplastic cartilage matrix. It is subclassified according to site (e.g., intramedullary versus juxtacortical) and histologic variants (discussed next). Chondrosarcomas occur roughly half as frequently as osteosarcomas; most patients are age 40 or older, with men affected twice as frequently as women.
Morphology
Conventional chondrosarcoma, the most common variant, arises within the medullary cavity of the bone to form an expansile glistening mass that often erodes the cortex (Fig. 20–11, A). It is composed of malignant hyaline and myxoid cartilage. Myxoid chondrosarcomas are viscous and gelatinous in consistency, and the matrix oozes from the cut surface. The adjacent cortex is thickened or eroded, and the tumor grows with broad pushing fronts into marrow spaces and the surrounding soft tissue. Tumor grade is determined by cellularity, degree of cytologic atypia, and mitotic activity (Fig. 20–11, B). Low-grade tumors may be difficult to distinguish from enchondroma. Higher-grade lesions contain pleomorphic chondrocytes with frequent mitotic figures.
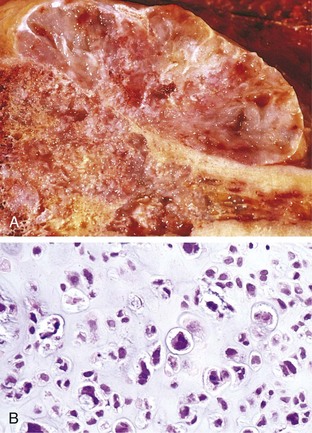
Figure 20–11 Chondrosarcoma. A, Islands of hyaline and myxoid cartilage expand the medullary cavity and grow through the cortex to form a sessile paracortical mass. B, Anaplastic chondrocytes within a chondroid matrix.
Approximately 10% of patients with conventional low-grade chondrosarcomas have a second high-grade poorly differentiated component (dedifferentiated chondrosarcomas) that includes foci of fibro- or osteosarcomas. Other histologic variants include clear cell and mesenchymal chondrosarcomas.
Clinical Features
Chondrosarcomas commonly arise in the pelvis, shoulder, and ribs; in contrast with enchondromas, chondrosarcomas rarely involve the distal extremities. They typically manifest as painful, progressively enlarging masses. A slowly growing low-grade tumor causes reactive thickening of the cortex, whereas a more aggressive high-grade neoplasm destroys the cortex and forms a soft tissue mass; consequently, the more radiolucent the tumor the greater the likelihood that it is high grade. There is also a direct correlation between grade and biologic behavior of the tumor. Fortunately, most conventional chondrosarcomas are indolent and low-grade, with a 5-year survival rate of 80% to 90% (versus 43% for grade 3 tumors); grade 1 tumors rarely metastasize, whereas 70% of the grade 3 tumors disseminate. Size is another prognostic feature, with tumors larger than 10 cm being significantly more aggressive than smaller tumors. Chondrosarcomas metastasize hematogenously, preferentially to the lungs and skeleton. Conventional chondrosarcomas are treated with wide surgical excision; chemotherapy is added for the mesenchymal and dedifferentiated variants because of their aggressive clinical course.
Fibrous and Fibroosseous Tumors
Fibrous tumors of the skeleton are extremely common and exhibit a wide diversity of morphologic variants.
Fibrous Cortical Defect and Nonossifying Fibroma
Fibrous cortical defects are probably developmental abnormalities rather than true neoplasms. The vast majority are smaller than 0.5 cm in diameter and arise eccentrically in the metaphysis of the distal femur or proximal tibia; almost 50% are bilateral or multiple. Larger lesions (5 to 6 cm) develop into nonossifying fibromas.
Morphology
Fibrous cortical defects and nonossifying fibromas both manifest as sharply demarcated radiolucencies surrounded by a thin zone of sclerosis. On gross inspection, they are gray to yellow-brown; microscopic examination shows cellular lesions composed of cytologically benign fibroblasts and activated macrophages, including multinucleate forms. The fibroblasts classically exhibit a storiform (pinwheel) pattern (Fig. 20–12). Hemorrhage and hemosiderin deposits are a common finding.
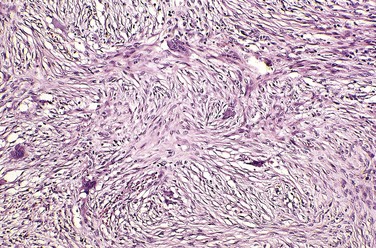
Clinical Features
Fibrous cortical defects are asymptomatic and typically are detected only as incidental radiographic lesions. The usual clinical course is characterized by spontaneous differentiation into normal cortical bone within a few years, so as a rule, biopsy is not required. The few that enlarge into nonossifying fibromas can manifest with pathologic fracture; in such cases, biopsy is necessary to rule out other tumors.
Fibrous Dysplasia
Fibrous dysplasia is a benign tumor in which all components of normal bone are present, but they fail to differentiate into mature structures. Fibrous dysplasia manifests with one of three clinical patterns: (1) involvement of a single bone (monostotic); (2) involvement of multiple bones (polyostotic); and (3) polyostotic disease, associated with café au lait skin pigmentations and endocrine abnormalities, especially precocious puberty (McCune-Albright syndrome). Mutations of the GNAS gene, resulting in a constitutively active Gs protein (Chapter 2), are responsible for all forms of fibrous dysplasia. The mutation occurs during embryogenesis (somatic mutations) resulting in mosaicism in the fetus and adult. The extent of manifestation (mono-ostotic, polyostotic, or McCune-Albright syndrome) depends on (1) the stage of embryogenesis when the mutation is acquired and (2) the fate of the cell harboring the initial mutation.
Monostotic fibrous dysplasia accounts for 70% of cases. The tumor usually arises during the second and third decades of life; there is no gender predilection. In descending order of frequency, ribs, femur, tibia, jawbones, calvariae, and humerus are most commonly affected. Lesions often are asymptomatic and frequently are discovered incidentally. However, fibrous dysplasia can cause marked enlargement and distortion of bone, so that if the face or skull is involved, disfigurement can occur, or it can cause pain and pathologic fracture.
Polyostotic fibrous dysplasia without endocrine dysfunction accounts for a majority of the remaining cases. It manifests at a slightly earlier age than that for the monostotic type. In descending order of frequency, femur, skull, tibia, and humerus are most commonly involved. Craniofacial involvement is present in 50% of patients with moderate skeletal involvement and in 100% of patients with extensive skeletal disease. Polyostotic disease tends to involve the shoulder and pelvic girdles, resulting in severe deformities and spontaneous fractures.
McCune-Albright syndrome accounts for 3% of all cases. The associated endocrinopathies include sexual precocity (girls more often than boys), hyperthyroidism, growth hormone–secreting pituitary adenomas, and primary adrenal hyperplasia. The severity of manifestations depends on the number and cell types that harbor the G protein mutation. The bone lesions may be unilateral, and the skin pigmentation usually is limited to the same side of the body. The cutaneous macules classically are large, dark to light brown (café au lait), and irregular in configuration.
Morphology
On gross inspection, fibrous dysplasia is characterized by well-circumscribed, intramedullary lesions of varying sizes; large masses expand and distort the bone. Lesional tissue is tan-white and gritty-appearing; on microscopic examination, it exhibits curved trabeculae of woven bone (mimicking Chinese characters), without osteoblastic rimming, surrounded by a moderately cellular fibroblastic proliferation (Fig. 20–13).
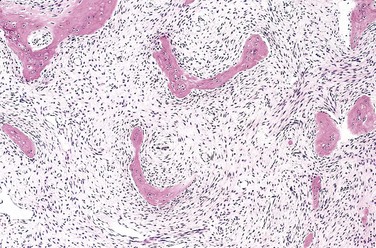
Clinical Course
The natural history depends on the extent of skeletal involvement; patients with monostotic disease usually have minimal symptoms. On x-ray imaging, lesions exhibit a characteristic ground glass appearance with well-defined margins. Symptomatic lesions are readily cured by conservative surgery. Polyostotic involvement frequently is associated with progressive disease and more severe skeletal complications (e.g., fractures, long bone deformities, craniofacial distortion). Rarely, polyostotic disease can transform into osteosarcoma, especially after radiotherapy.
Miscellaneous Bone Tumors
Ewing Sarcoma and Primitive Neuroectodermal Tumor
Ewing sarcoma and primitive neuroectodermal tumors (PNETs) are primary malignant small round cell tumors of bone and soft tissue. They share certain molecular features (described below) and are best viewed as variants of the same tumor, differing only in degree of neuroectodermal differentiation and clinical features. PNETs demonstrate clear neural differentiation, whereas Ewing sarcomas are undifferentiated.
Ewing sarcoma accounts for 6% to 10% of primary malignant bone tumors. After osteosarcoma, it is the second most common pediatric bone sarcoma. Most patients are 10 to 15 years of age, and 80% are younger than 20 years. Boys are affected slightly more frequently than girls, and there is a striking racial predilection for whites; blacks and Asians are rarely afflicted. The common chromosomal abnormality is a translocation that causes fusion of the EWS gene on 22q12 with a member of the ETS family of transcription factors. The most common fusion partners are the FL1 gene on 11q24 and the ERG gene on 21q22. The resulting chimeric protein functions as a transcription factor, but precisely how it contributes to oncogenesis remains uncertain; effects on differentiation, proliferation, and survival have all been proposed. At a practical level, these translocations are of diagnostic importance, as approximately 95% of tumors have t(11;22)(q24;q12) or t(21;22)(q22;q12).
Morphology
Ewing sarcoma/PNET arises in the medullary cavity and invades the cortex and periosteum to produce a soft tan-white tumor mass, frequently with hemorrhage and necrosis. It is composed of sheets of uniform small, round cells that are slightly larger than lymphocytes; typically, there are few mitotic figures and little intervening stroma (Fig. 20–14). The cells have scant glycogen-rich cytoplasm. The presence of Homer-Wright rosettes (tumor cells circled about a central fibrillary space) indicates neural differentiation.
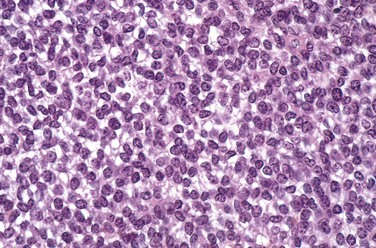
Clinical Features
Ewing sarcoma/PNET typically manifests as a painful enlarging mass in the diaphyses of long tubular bones (especially the femur) and the pelvic flat bones. Some patients have systemic signs and symptoms suggestive of infection. Imaging studies show a destructive lytic tumor with infiltrative margins and extension into surrounding soft tissues. There is a characteristic periosteal reaction with deposition of bone in an onion-skin pattern.
Treatment includes chemotherapy and surgical excision with or without irradiation. The 5-year survival rate is currently 75% for patients presenting with localized tumors.
Giant Cell Tumor of Bone
Giant cell tumors (GCTs) contain prominent by multinucleate osteoclast-type giant cells—hence the synonym osteoclastoma. GCT is a relatively common benign but locally aggressive bone tumor, usually arising in persons in their 20s to 40s. Despite the name, molecular analyses have shown that it is the mononuclear cells in the tumor that are neoplastic. These cells may be related to osteoblast precursor cells, as they express RANK ligand, which may stimulate the development of surrounding non-neoplastic osteoclast-like cells.
Morphology
GCTs are large and red-brown, and often show cystic degeneration. They are composed of uniform oval mononuclear cells and scattered osteoclast-type giant cells containing 100 or more nuclei (Fig. 20–15). Mitotic figures are typically frequent. Necrosis, hemorrhage, and reactive bone formation also are commonly present.
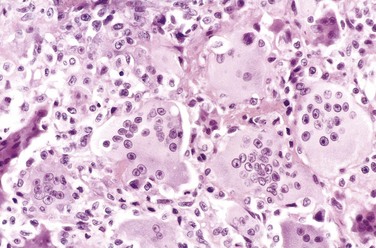
Clinical Course
Although almost any bone may be involved, a majority of GCTs arise in the epiphysis and involve the metaphysis of long bones around the knee (distal femur and proximal tibia), frequently causing pain. Occasionally, GCTs manifest with pathologic fractures. Most are solitary tumors. Radiographically, GCTs are large, purely lytic, and eccentric; the overlying cortex frequently is destroyed, producing a bulging soft tissue mass with a thin shell of reactive bone. Although GCTs are considered benign, roughly half recur after simple curettage, and as many as 2% spread to the lungs as localized lesions that are cured by local excision.
Metastatic Disease
Metastatic tumors are the most common malignant tumors involving bone. Pathways of spread include (1) direct extension, (2) lymphatic or hematogenous dissemination, and (3) intraspinal seeding. Any cancer can spread to bone, but certain tumors exhibit a distinct skeletal predilection. In adults more than 75% of skeletal metastases originate from cancers of the prostate, breast, kidney, and lung. In children, neuroblastoma, Wilms tumor, osteosarcoma, Ewing sarcoma, and rhabdomyosarcoma are the common sources of bony metastases.
Most metastases involve the axial skeleton (vertebral column, pelvis, ribs, skull, sternum), proximal femur, and humerus, in descending order. The red marrow in these areas, with its rich capillary network, slow blood flow, and nutrient environment rich in growth factors, facilitates tumor cell implantation and growth.
The radiologic appearance of metastases can be purely lytic, purely blastic, or both. In lytic lesions (e.g., with kidney and lung tumors and melanoma), the metastatic cells secrete substances such as prostaglandins, interleukins, and PTH-related protein (PTHrP) that stimulate osteoclastic bone resorption; the tumor cells themselves do not directly resorb bone. Similarly, metastatic tumors that elicit an osteoblastic response (e.g., prostate adenocarcinoma) do so by stimulating osteoblastic bone formation. Most metastases induce a mixed lytic and blastic reaction.
Summary
Bone Tumors
• Most bone tumors are categorized according to their normal tissue counterpart; chondroid and bony matrices are roughly equally represented. Benign lesions far outnumber malignant tumors. Metastatic tumors are the most common form of skeletal malignancy.
Joints
The joints are subject to a wide variety of disorders, including degeneration, infections, immune-mediated injury, metabolic derangements, and neoplasms. Discussed in this section are the most common forms of arthritis—namely, osteoarthritis or degenerative joint disease, select autoimmune arthritides, gout, and infectious arthritis—along with the two most common benign joint tumors.
Arthritis
Osteoarthritis
Osteoarthritis, or degenerative joint disease, is the most common joint disorder. It is a frequent, if not inevitable, part of aging and is an important cause of physical disability in persons older than 65 years of age. The fundamental feature of osteoarthritis is degeneration of the articular cartilage; structural changes in the underlying bone are likely secondary. Although the term osteoarthritis implies an inflammatory disease, osteoarthritis is primarily a degenerative disorder of articular cartilage in which the chondrocytes respond to biomechanical and biologic stresses in a way that results in breakdown of the matrix.
In most cases, osteoarthritis appears insidiously with age and without apparent initiating cause (primary osteoarthritis). In such cases the disease usually is oligoarticular (i.e., affecting only a few joints), with the joints of the hands, knees, hips, and spine most commonly affected. In the unusual circumstance (less than 5% of cases) when osteoarthritis strikes in youth, there is typically some predisposing condition, such as previous trauma, developmental deformity, or underlying systemic disease such as ochronosis, hemochromatosis, or marked obesity. In these settings the disease is called secondary osteoarthritis and often involves one or several predisposed joints. Gender has some influence; knees and hands are more commonly affected in women, whereas hips are more commonly affected in men. It is estimated that the economic toll of osteoarthritis in the United States is more than $33 billion annually.
Morphology
The early changes in osteoarthritis include alterations in the composition and structure of the matrix. The chondrocytes have limited capacity to proliferate, and some divide to form small clones of cells that secrete newly synthesized matrix. Subsequently, vertical and horizontal fibrillation and cracking of the matrix occur as the superficial layers of the cartilage are degraded (Fig. 20–16, A). Gross examination at this stage reveals a soft granular-appearing articular cartilage surface, a condition known as chondromalacia. Eventually, full-thickness portions of the cartilage are lost, and the subchondral bone plate is exposed and is smoothened and burnished by friction, giving it the appearance of polished ivory (bone eburnation) (Fig. 20–16, B). The underlying cancellous bone becomes rebuttressed by osteoblastic activity. Small fractures can dislodge pieces of cartilage and subchondral bone into the joint, forming loose bodies (joint mice). The fracture gaps allow synovial fluid to be forced into the subchondral regions to form fibrous walled cysts. Mushroom-shaped osteophytes (bony outgrowths) develop at the margins of the articular surface. In severe disease, a fibrous synovial pannus covers the peripheral portions of the articular surface.
Pathogenesis
Articular cartilage bears the brunt of the degenerative changes in osteoarthritis. Normal articular cartilage performs two functions: (1) Along with the synovial fluid, it provides virtually friction-free movement within the joint; and (2) in weight-bearing joints, it spreads the load across the joint surface in a manner that allows the underlying bones to absorb shock and weight. These functions require the cartilage to be elastic (i.e., to regain normal architecture after compression) and to have high tensile strength. These attributes are provided by proteoglycans and type II collagen, respectively, both produced by chondrocytes. As with adult bone, articular cartilage constantly undergoes matrix degradation and replacement. Normal chondrocyte function is critical to maintain cartilage synthesis and degradation; any imbalance can lead to osteoarthritis.
Chondrocyte function is affected by a variety of influences. Although osteoarthritis is not exclusively a wear-and-tear phenomenon, mechanical stresses and aging nevertheless figure prominently. Genetic factors, including polymorphisms and mutations in genes encoding components of the matrix and signaling molecules, contribute to osteoarthritis susceptibility. The risk of osteoarthritis also is increased with increasing bone density, as well as sustained high estrogen levels.
Regardless of the inciting stimulus, there is an imbalance in the expression, activity, and signaling of cytokines and growth factors that results in degradation and loss of matrix. Early osteoarthritis is marked by degenerating cartilage containing more water and less proteoglycan (the proteoglycan component conveys turgor and elasticity). The type II collagen network also is diminished, presumably as a result of decreased local synthesis and increased breakdown; chondrocyte apoptosis is increased. Overall, cartilage tensile strength and resilience are compromised. In response to these degenerative changes, chondrocytes proliferate and attempt to “repair” the damage by synthesizing new collagen and proteoglycans. Although these reparative changes initially are able to keep pace, matrix changes and chondrocyte loss eventually predominate.
Clinical Course
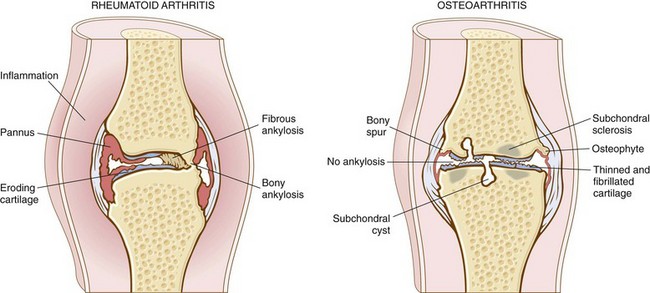
Osteoarthritis is an insidious disease, predominantly affecting patients beginning in their 50s and 60s. Characteristic symptoms and signs include deep, aching pain exacerbated by use, morning stiffness, crepitus (grating or popping sensation in the joint), and limitation in range of movement. Osteophyte impingement on spinal foramina can cause nerve root compression with radicular pain, muscle spasms, muscle atrophy, and neurologic deficits. Hips, knees, lower lumbar and cervical vertebrae, proximal and distal interphalangeal joints of the fingers, first carpometacarpal joints, and first tarsometatarsal joints of the feet are commonly involved. Heberden nodes in the fingers, representing prominent osteophytes at the distal interphalangeal joints, are characteristic in women. Aside from complete inactivity, there is no predicted way to prevent or halt the progression of primary osteoarthritis; it can stabilize for years but generally is slowly progressive. With time, significant joint deformity can occur, but unlike in rheumatoid arthritis (discussed next), fusion does not take place. Treatment usually is based on symptoms, with joint replacement in severe cases. A comparison of the important morphologic features of these two disorders is presented in Figure 20–17.
Rheumatoid Arthritis
Rheumatoid arthritis (RA) is a systemic, chronic inflammatory autoimmune disease affecting many tissues but principally attacking the joints. It causes a nonsuppurative proliferative synovitis that frequently progresses to destroy articular cartilage and underlying bone with resulting disabling arthritis. When extraarticular involvement develops—for example, of the skin, heart, blood vessels, muscles, and lungs—RA may resemble lupus or scleroderma.
RA is a relatively common condition, with a prevalence of approximately 1%; it is three to five times more common in women than in men. The peak incidence is in the second to fourth decades of life, but no age is immune.
Pathogenesis
RA is an autoimmune disease involving complex, and still poorly understood, interactions of genetic risk factors, environment, and the immune system. The pathologic changes are caused mainly by cytokine-mediated inflammation, with CD4+ T cells being the principal source of the cytokines (Fig. 20–18). Many patients also produce antibodies against cyclic citrullinated peptides (CCPs), which may contribute to the joint lesions. CCPs are derived from proteins in which arginine residues are converted to citrulline residues posttranslationally. In RA, antibodies to citrullinated fibrinogen, type II collagen, α-enolase, and vimentin are the most important and may form immune complexes that deposit in the joints. These antibodies are a diagnostic marker for the disease and may be involved in tissue injury.
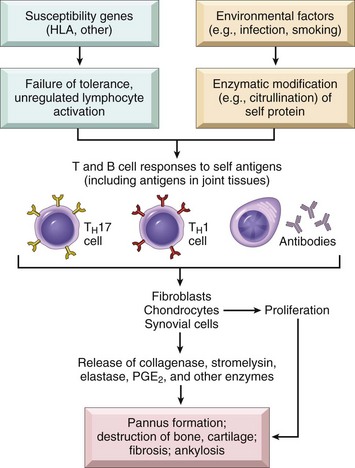
Like other autoimmune diseases, RA is a disorder in which genetic and environmental factors contribute to the breakdown of tolerance to self-antigens.
• Genetic factors: It is estimated that 50% of the risk of developing RA is related to genetic factors. Susceptibility to rheumatoid arthritis is linked to the HLA-DRB1 locus. Recent linkage and genome-wide association studies have revealed a large number of non-HLA genes in which polymorphisms are associated with RA. There is a strong association with a polymorphism in the PTPN22 gene, which encodes a tyrosine phosphatase that is postulated to inhibit T cell activation.
• Environmental factors: Many candidate infectious agents whose antigens may activate T or B cells have been considered, but none has been conclusively implicated. As mentioned above, in at least 70% of patients the blood contains anti-CCP antibody, which may be produced during inflammation. Inflammatory and environmental insults such as smoking and infections may induce the citrullination of some self proteins, creating new epitopes that trigger autoimmune reactions.
It is proposed that the disease is initiated in a genetically predisposed person by activation of CD4+ helper T cells responding to some arthritogenic agent, possibly microbial, or to a self-antigen such as CCP (Fig. 20–18). CD4+ TH1 and TH17 cells, activated B lymphocytes, plasma cells, and macrophages, as well as other inflammatory cells, are found in the inflamed synovium, and in severe cases, well-formed lymphoid follicles with germinal centers may be present. Numerous cytokines, including IL-1, IL-8, TNF, IL-6, IL-17, and interferon-γ, have been detected in the synovial fluid. Cytokines produced by the activated T cells recruit leukocytes such as macrophages, whose products cause tissue injury, and also activate resident synovial cells to produce proteolytic enzymes, such as collagenase, that mediate destruction of the cartilage, ligaments, and tendons of the joints. Increased osteoclast activity in the joints contributes to the bone destruction in rheumatoid arthritis; this may be caused by the production of the TNF family cytokine RANK ligand by activated T cells. Despite the plethora of cytokines produced in the joint in RA, TNF appears to play a pivotal role. This is demonstrated by the remarkable effectiveness of TNF antagonists in patients with the disease, even those who are resistant to other therapies.
It is suspected from a variety of experimental and clinical observations that antibodies also play a role in the disease. The contribution of anti-CCP was previously mentioned. About 80% of patients have serum immunoglobulin M (IgM) (and, less frequently, IgA) autoantibodies that bind to the Fc portions of their own (self) IgG. These autoantibodies are called rheumatoid factor. They may form immune complexes with self-IgG that deposit in joints and other tissues, leading to inflammation and tissue damage. However, the role of rheumatoid factor in the pathogenesis of the joint or extraarticular lesions has not been established. Of interest, there seem to be two variants of RA, one characterized by presence of anti-CCP and rheumatoid factor and another in which these autoantibodies are lacking.
Morphology
A broad spectrum of morphologic alterations are seen in RA; the most severe occur in the joints. RA typically manifests as symmetric arthritis, principally affecting the small joints of the hands and feet, ankles, knees, wrists, elbows, and shoulders. Most often, the proximal interphalangeal and metacarpophalangeal joints are affected, but distal interphalangeal joints are spared. Axial involvement, when it occurs, is limited to the upper cervical spine; similarly, hip joint involvement is extremely uncommon. On histologic examination, the affected joints show chronic papillary synovitis, characterized by (1) synovial cell hyperplasia and proliferation; (2) dense perivascular inflammatory cell infiltrates (frequently forming lymphoid follicles) in the synovium composed of CD4+ T cells, plasma cells, and macrophages; (3) increased vascularity due to angiogenesis; (4) neutrophils and aggregates of organizing fibrin on the synovial surface and in the joint space; and (5) increased osteoclast activity in the underlying bone, leading to synovial penetration and periarticular bone erosion. The classic appearance is that of a pannus, formed by proliferating synovial lining cells admixed with inflammatory cells, granulation tissue, and fibrous connective tissue; the overgrowth of this tissue is so exuberant that the usually thin, smooth synovial membrane is transformed into lush, edematous, frondlike (villous) projections (Fig. 20–19, A–C). With full-blown inflammatory joint involvement, periarticular soft tissue edema usually develops, classically manifested first by fusiform swelling of the proximal interphalangeal joints. With progression of the disease, the articular cartilage subjacent to the pannus is eroded and, in time, virtually destroyed. The subarticular bone also may be attacked and eroded. Eventually the pannus fills the joint space, and subsequent fibrosis and ossification may cause permanent ankylosis. The radiographic hallmarks are joint effusions and juxtaarticular osteopenia with erosions and narrowing of the joint space and loss of articular cartilage. Destruction of tendons, ligaments, and joint capsules produces the characteristic deformities, including radial deviation of the wrist, ulnar deviation of the fingers, and flexion-hyperextension abnormalities of the fingers (swan-neck deformity, boutonnière deformity).
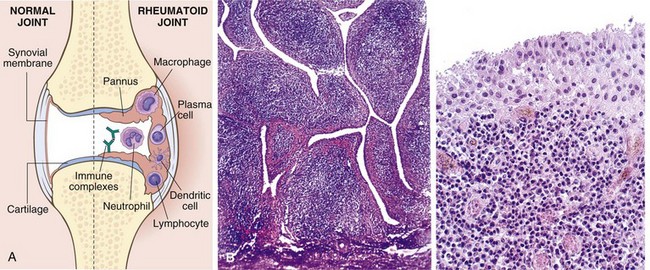
Figure 20–19 Rheumatoid arthritis. A, A joint lesion. B, Synovium demonstrating papillary hyperplasia caused by dense inflammatory infiltrate. C, Hypertrophied synoviocytes with numerous underlying lymphocytes and plasma cells.
(A, Modified with permission from Feldmann M: Development of anti-TNF therapy for rheumatoid arthritis. Nat Rev Immunol 2:364, 2002.)
Rheumatoid subcutaneous nodules develop in about one fourth of patients, occurring along the extensor surface of the forearm or other areas subjected to mechanical pressure; rarely, they can form in the lungs, spleen, heart, aorta, and other viscera. Rheumatoid nodules are firm, nontender, oval or rounded masses as large as 2 cm in diameter. They are characterized microscopically by a central focus of fibrinoid necrosis surrounded by a palisade of macrophages, which in turn is rimmed by granulation tissue and lymphocytes (Fig. 20–20).
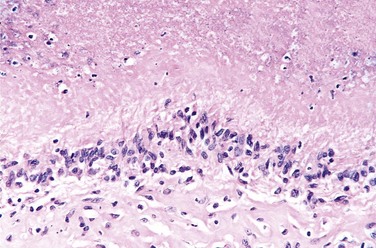
Figure 20–20 Rheumatoid nodule. Serpiginous area of necrobiotic collagen surrounded by palisading histiocytes.
Patients with severe erosive disease, rheumatoid nodules, and high titers of rheumatoid factor are at risk of developing vasculitic syndromes; acute necrotizing vasculitis may involve small or large arteries. Serosal involvement may manifest as fibrinous pleuritis or pericarditis or both. Lung parenchyma may be damaged by progressive interstitial fibrosis. Ocular changes such as uveitis and keratoconjunctivitis (similar to those seen in Sjögren syndrome; see Chapter 4) may be prominent in some cases.
Clinical Features
Although RA is basically a symmetric polyarticular arthritis, there also may be constitutional symptoms such as weakness, malaise, and low-grade fever. Many of the systemic manifestations result from the same mediators that cause joint inflammation (e.g., IL-1, TNF). The arthritis first appears insidiously, with aching and stiffness of the joints, particularly in the morning. As the disease advances, the joints become enlarged, motion is limited, and in time complete ankylosis may appear. Vasculitic involvement of the extremities may give rise to Raynaud phenomenon and chronic leg ulcers. Such multisystem involvement must be distinguished from lupus, scleroderma, polymyositis, dermatomyositis, and Lyme disease, as well as other forms of arthritis. Helpful in making the correct diagnosis are (1) characteristic radiographic findings; (2) sterile, turbid synovial fluid with decreased viscosity, poor mucin clot formation, and inclusion-bearing neutrophils; and (3) anti-CCP and rheumatoid factor (80% of patients).
The clinical course of RA is highly variable. In a minority of patients, the disease may stabilize or even regress; in most patients, however, it pursues a chronic, remitting-relapsing course. Historically, the natural history of the disease has been one of progressive joint destruction leading to disability after 10 to 15 years. The outcome has been dramatically improved by recent advances in therapy, including aggressive treatment of early RA and the introduction of highly effective biologic agents that antagonize TNF. RA is an important cause of reactive amyloidosis (Chapter 4), which develops in 5% to 10% of these patients, particularly those with long-standing severe disease.
Juvenile Rheumatoid Arthritis
Juvenile rheumatoid arthritis (JRA) is not a single disease but a group of multifactorial disorders with environmental and genetic components. These disorders are of unknown etiology and are classified according to their presentation into oligoarthritis, polyarthritis, and systemic (Still’s disease) variants. Large joints often are affected, and symptoms and signs such as joint swelling, warmth, pain, and loss of function begin before the age of 16 years and persist for more than 6 weeks. Extraarticular inflammatory manifestations such as uveitis also may be present. Common risk factors include genetic susceptibility (such as particular HLA and PTPN22 gene variants) and perhaps infection. As in adult RA, the pathogenesis is likely to involve activation of TH1 and TH17 cells, which in turn activate B cells, macrophages, and fibroblasts to produce antibodies and a variety of cytokines including TNF, IL-1, and IL-6, which eventually result in damage to articular structures.
Seronegative Spondyloarthropathies
Clinical, morphologic, and genetic features distinguish these disorders from rheumatoid arthritis and other arthritides. The spondyloarthropathies are characterized by the following:
• Pathologic changes that begin in the ligamentous attachments to bone rather than in the synovium
• Involvement of the sacroiliac joints, with or without arthritis in other peripheral joints
• Absence of rheumatoid factor (hence the designation seronegative)
This group of disorders includes several clinical entities, of which ankylosing spondylitis is the prototype. Others include Reiter syndrome, psoriatic arthritis, spondylitis associated with inflammatory bowel diseases, and reactive arthropathies after infections (e.g., with Yersinia, Shigella, Salmonella, Helicobacter, or Campylobacter). Sacroiliitis is a common manifestation in all of these disorders; they are distinguished from one another by the particular peripheral joints involved, as well as on the basis of associated extraskeletal manifestations (for example, urethritis, conjunctivitis, and uveitis are characteristic of Reiter syndrome). Although triggering infections and immune mechanisms are thought to underlie most of the seronegative spondyloarthropathies, their pathogenesis remains obscure.
Gout
Gout affects about 1% of the population, and shows a predeliction for males. It is caused by excessive amounts of uric acid, an end product of purine metabolism, within tissues and body fluids. Monosodium urate crystals precipitate from supersaturated body fluids and induce an acute inflammatory reaction. Gout is marked by recurrent episodes of acute arthritis, sometimes accompanied by the formation of large crystalline aggregates called tophi, and eventual permanent joint deformity. Although an elevated level of uric acid is an essential component of gout, not all such persons develop gout, and genetic and environmental factors also contribute to its pathogenesis. Gout traditionally is divided into primary and secondary forms, accounting for about 90% and 10% of cases, respectively (Table 20–3). Primary gout designates cases in which the basic cause is unknown or (less commonly) in which the disorder is due to an inborn metabolic defect that causes hyperuricemia. In secondary gout, the cause of the hyperuricemia is known, but gout is not necessarily the main or even dominant clinical disorder.
Table 20–3 Classification of Gout
| Clinical Category | Metabolic Defect |
|---|---|
| Primary Gout (90% of cases) | |
| Enzyme defects—unknown (85% to 90% of cases) | Overproduction of uric acid |
| Normal excretion (majority) | |
| Increased excretion (minority) | |
| Underexcretion of uric acid with normal production | |
| Known enzyme defects—e.g., partial HGPRT deficiency (rare) | Overproduction of uric acid |
| Secondary Gout (10% of cases) | |
| Associated with increased nucleic acid turnover—e.g., leukemias | Overproduction of uric acid with increased urinary excretion |
| Chronic renal disease | Reduced excretion of uric acid with normal production |
| Inborn errors of metabolism | Overproduction of uric acid with increased urinary excretion, e.g., complete HGPRT deficiency (Lesch-Nyhan syndrome) |
HGPRT, hypoxanthine guanine phosphoribosyl transferase.
Morphology
The major morphologic manifestations of gout are acute arthritis, chronic tophaceous arthritis, tophi in various sites, and gouty nephropathy.
Acute arthritis is characterized by a dense neutrophilic infiltrate permeating the synovium and synovial fluid. Long, slender, needle-shaped monosodium urate crystals frequently are found in the cytoplasm of the neutrophils as well as in small clusters in the synovium. The synovium is edematous and congested and contains scattered mononuclear inflammatory cells. When the episode of crystallization abates and the crystals resolubilize, the attack remits.
Chronic tophaceous arthritis evolves from repetitive precipitation of urate crystals during acute attacks. The urates can heavily encrust the articular surfaces and form visible deposits in the synovium (Fig. 20–21, A). The synovium becomes hyperplastic, fibrotic, and thickened by inflammatory cells, forming a pannus that destroys the underlying cartilage, leading to juxtaarticular bone erosions. In severe cases, fibrous or bony ankylosis ensues, resulting in loss of joint function.
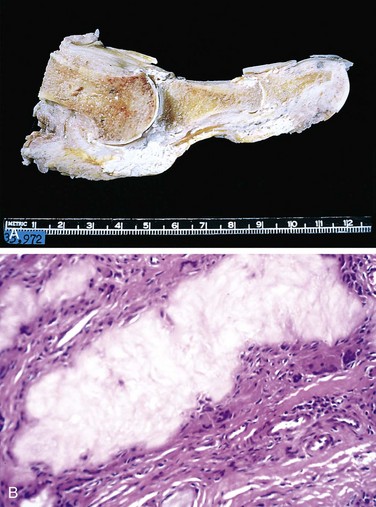
Figure 20–21 Gout. A, Amputated great toe with white tophi involving the joint and soft tissues. B, Photomicrograph of a gouty tophus. An aggregate of dissolved urate crystals is surrounded by reactive fibroblasts, mononuclear inflammatory cells, and giant cells.
Tophi are pathognomonic for gout. They are formed by large aggregations of urate crystals surrounded by an intense inflammatory reaction of lymphocytes, macrophages, and foreign-body giant cells, attempting to engulf the masses of crystals (Fig. 20–21, B). Tophi can appear in the articular cartilage of joints and in the periarticular ligaments, tendons, and soft tissues, including the ear lobes, nasal cartilages, and skin of the fingertips. Superficial tophi can lead to large ulcerations of the overlying skin.
Gouty nephropathy refers to renal complications associated with urate deposition, variously forming medullary tophi, intratubular precipitations, or free uric acid crystals and renal calculi. Secondary complications such as pyelonephritis can occur, especially when there is urinary obstruction.
Pathogenesis
Elevated uric acid levels can result from overproduction or reduced excretion of uric acid, or both (Table 20–3). Most cases of gout are characterized by a primary overproduction of uric acid. Less commonly, uric acid is produced at normal rates, and hyperuricemia occurs because of decreased renal excretion of urate. For an understanding of these influences, a brief review of normal uric acid synthesis and excretion is warranted.
• Uric acid synthesis. Uric acid is the end product of purine catabolism; consequently increased urate synthesis typically reflects some abnormality in purine nucleotide production. The synthesis of purine nucleotides involves two different but interlinked pathways: the de novo and salvage pathways.
• The de novo pathway is involved in the synthesis of purine nucleotides from nonpurine precursors.
• The salvage pathway is involved in the synthesis of purine nucleotides from free purine bases, derived from dietary intake and by catabolizing nucleic acids and purine nucleotides.
• Uric acid excretion. Circulating uric acid is freely filtered by the glomerulus and virtually completely resorbed in the proximal tubules of the kidney. A small fraction of the resorbed uric acid is subsequently secreted by the distal nephron and excreted in the urine.
Although the cause of excessive uric acid biosynthesis in primary gout is unknown in most cases, rare patients have identifiable enzymatic defects. For example, complete lack of HGPRT, an enzyme essential in the salvage pathway, gives rise to the Lesch-Nyhan syndrome. In secondary gout, hyperuricemia can be caused by increased urate production (e.g., rapid cell lysis during chemotherapy for lymphoma or leukemia) or decreased excretion (chronic renal insufficiency), or both. Reduced renal excretion may also be caused by drugs such as thiazide diuretics, presumably because of effects on uric acid tubular transport.
Whatever the cause, increased levels of uric acid in the blood and other body fluids (e.g., synovium) lead to the precipitation of monosodium urate crystals. This, in turn, triggers a chain of events that culminate in joint injury (Fig. 20–22). Urate crystals are thought to directly activate the complement system, leading to production of chemotactic and pro-inflammatory mediators. The crystals are phagocytosed by macrophages and recognized by the intracellular sensor called the inflammasome (Chapter 2), which is activated and stimulates the production of the cytokine IL-1. IL-1 is a mediator of inflammation, and causes local accumulation of neutrophils and macrophages in the joints and synovial membranes. These cells become activated, leading to the release of a host of additional mediators including chemokines, other cytokines, toxic free radicals, and leukotrienes—particularly leukotriene B4. The activated neutrophils also liberate destructive lysosomal enzymes. The cytokines can also directly activate synovial cells and cartilage cells to release proteases (e.g., collagenase) that exacerbate tissue injury. The resulting acute arthritis typically remits in days to weeks, even if untreated. Repeated bouts, however, can lead to the permanent damage seen in chronic tophaceous arthritis.
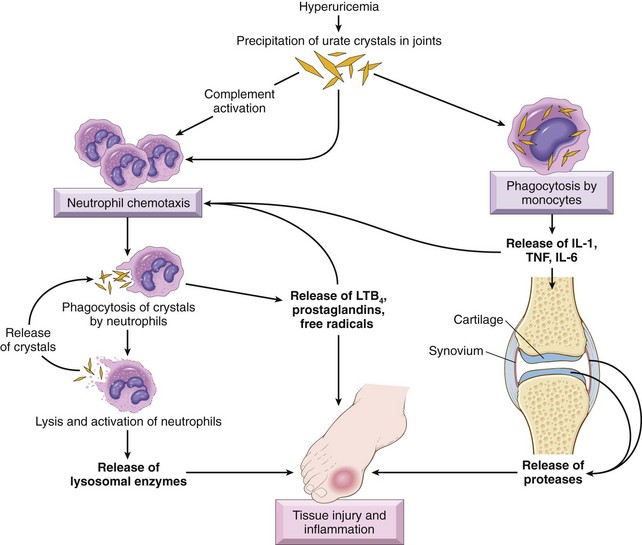
Clinical Features
Gout is more common in men than in women; it does not usually cause symptoms before the age of 30. Risk factors for the disease include obesity, excess alcohol intake, consumption of purine-r ich foods, diabetes, the metabolic syndrome, and renal failure. Polymorphisms in genes involved in the transport and homeostasis of urate such as URAT1 and GLUT9 also are associated with hyperuricemia and gout.
Four stages are classically recognized: (1) asymptomatic hyperuricemia, (2) acute gouty arthritis, (3) “intercritical” gout, and (4) chronic tophaceous gout. Asymptomatic hyperuricemia appears around puberty in males and after menopause in women. After a long interval of years, acute arthritis appears in the form of sudden onset, excruciating joint pain associated with localized erythema, and warmth; constitutional symptoms are uncommon, except possibly mild fever. The vast majority of first attacks are monoarticular; 50% occur in the first metatarsophalangeal joint (great toe), and 90% in the instep, ankle, heel, or wrist. Untreated, acute gouty arthritis may last for hours to weeks, but it gradually completely resolves, and the patient enters an asymptomatic intercritical period. Although some fortunate persons never have another attack, most experience a second episode within months to a few years. In the absence of appropriate therapy, the attacks recur at shorter intervals and frequently become polyarticular. Eventually, after a decade or so, symptoms fail to resolve completely after each attack, and the disease progresses to chronic tophaceous gout. At this stage, radiographs show characteristic juxtaarticular bone erosion caused by the crystal deposits and loss of the joint space. Progression leads to severe crippling disease.
Renal manifestations of gout can appear as renal colic associated with the passage of gravel and stones, and can evolve into chronic gouty nephropathy. About 20% of persons with chronic gout die of renal failure.
Numerous drugs are available to abort or prevent acute attacks of arthritis and mobilize tophaceous deposits. Their use is important, because many aspects of gout are related to the duration and severity of hyperuricemia. Generally, gout does not materially shorten the life span, but it can certainly impair quality of life.
Pseudogout
Pseudogout also is known as chondrocalcinosis or, more formally, calcium pyrophosphate crystal deposition disease. The crystal deposits first appear in structures composed of cartilage such as menisci, intervertebral discs, and articular surfaces. When the deposits enlarge enough, they may rupture, inducing an inflammatory reaction. Pseudogout typically first occurs in persons 50 years of age or older, becoming more common with increasing age, and eventually reaching a prevalence of 30% to 60% in those age 85 or older. There is no gender or race predilection.
Although pathways leading to crystal formation are not understood, they are likely to involve the overproduction or decreased breakdown of pyrophosphate, resulting in its accumulation and eventual crystallization with calcium in the matrix surrounding chondrocytes. Mutations in a transmembrane pyrophosphate transporter are associated with a rare familial form of the disease, in which crystals develop relatively early in life and there is severe osteoarthritis.
Much of the subsequent joint pathology in pseudogout involves the recruitment and activation of inflammatory cells and is similar to that in gout (see earlier). Duration of clinical signs can be from several days to weeks, and joint involvement may be monoarticular or polyarticular; the knees, followed by the wrists, elbows, shoulders, and ankles, are most commonly affected. Ultimately, approximately 50% of patients experience significant joint damage. Therapy is supportive; no known treatment prevents or retards crystal formation.
Infectious Arthritis
Microorganisms of any type can lodge in joints during hematogenous dissemination. Articular structures can also become infected by direct inoculation or by contiguous spread from osteomyelitis or a soft tissue abscess. Infectious arthritis is serious because it can cause rapid joint destruction and permanent deformities.
Suppurative Arthritis
Bacteria can seed joints during episodes of bacteremia; joint infection with such microorganisms almost uniformly results in a suppurative arthritis. Although virtually any bacteria can be causal, Haemophilus influenzae predominates in children younger than 2 years of age, S. aureus is the main causative agent in older children and adults, and the gonococcus is prevalent in older adolescents and young adults. Patients with sickle cell disease are prone to Salmonella infection at any age. Both genders are affected equally, except for gonococcal arthritis, which occurs mainly in sexually active women. In this group, those with deficiency of certain complement proteins (C5, C6, and C7) are particularly susceptible to disseminated gonococcal infections and hence arthritis.
The classic presentation is one of sudden onset of pain, redness, and swelling of the affected joint(s), with restricted range of motion. Fever, leukocytosis, and elevated erythrocyte sedimentation rate are common. In gonococcal infections, the course tends to be more subacute. In 90% of cases of nongonococcal suppurative arthritis, the infection involves only a single joint—usually the knee—followed in order by hip, shoulder, elbow, wrist, and sternoclavicular joints. Joint aspiration typically yields a purulent fluid in which the causal agent can be identified.
Lyme Arthritis
Lyme disease is caused by infection with the spirochete Borrelia burgdorferi, transmitted by deer ticks of the Ixodes ricinus complex; it is named for the Connecticut town where the disease was first recognized in the 1970s. With more than 20,000 cases reported annually, it is the leading arthropod-borne disease in the United States. As with another major spirochetal disease, syphilis, Lyme disease involves multiple organ systems and in its classic form progresses through three successive stages. In stage 1, Borrelia spirochetes multiply at the site of the tick bite and cause an expanding area of redness, often with an indurated or pale center. This skin lesion, called erythema chronicum migrans, may be accompanied by fever and lymphadenopathy but usually disappears in a few weeks’ time. In stage 2, the early disseminated stage, spirochetes spread hematogenously and cause secondary annular skin lesions, lymphadenopathy, migratory joint and muscle pain, cardiac arrhythmias, and meningitis, often with cranial nerve involvement. Diagnostically useful antibodies (usually both IgM and IgG) against Borrelia antigens appear in the serum at this stage of the disease. Some spirochetes, however, escape host antibody and T cell responses by sequestering themselves in the central nervous system or within endothelial cells. In stage 3, the late disseminated stage, which occurs 2 or 3 years after the initial bite, Lyme Borrelia organisms cause a chronic arthritis, sometimes with severe damage to large joints, and an encephalitis that ranges in severity from mild to debilitating.
Lyme arthritis develops in roughly 60% to 80% of untreated patients and is the dominant feature of late disease. The arthritis may be caused by immune responses against Borrelia antigens that cross-react with proteins in the joints, but the exact mechanisms are not yet understood. The disease tends to be migratory, with remissions and relapses. It involves mainly large joints, especially the knees, shoulders, elbows, and ankles, in descending order of frequency. Histologic examination reveals a chronic papillary synovitis with synoviocyte hyperplasia, fibrin deposition, mononuclear cell infiltrates, and onion-skin thickening of arterial walls; in severe cases, the morphology closely resembles that of rheumatoid arthritis. In only 25% of cases do silver stains reveal a sprinkling of organisms, and formal diagnosis of Lyme arthritis may depend on the clinical picture, including history, and/or appropriate serologic studies. Chronic arthritis with pannus formation and permanent deformities develops in roughly 1 in 10 patients.
Summary
Arthritis
• Osteoarthritis (degenerative joint disease) is by far the most common joint disease; it is primarily a degenerative disorder of articular cartilage in which matrix breakdown exceeds synthesis. Inflammation is secondary. The vast majority of cases occur without apparent precipitating cause except increasing age. Local production of pro-inflammatory cytokines and other mediators (IL-1, TNF, nitric oxide) may contribute to the progression of the joint degeneration.
• Rheumatoid arthritis (RA) is a chronic autoimmune inflammatory disease that affects mainly the joints, especially small joints, but can affect multiple tissues. RA is caused by an autoimmune response against self-antigen(s) such as citrullinated proteins, which leads to T cell reactions in the joint with production of cytokines that activate phagocytes that damage tissues and stimulate proliferation of synovial cells (synovitis). The cytokine TNF plays a central role, and antagonists against TNF are of great clinical benefit. Antibodies may also contribute to the disease.
• Gout and pseudogout. Increased circulating levels of uric acid (gout) or calcium pyrophosphate (pseudogout) can lead to crystal deposition in the joint space. Resulting inflammatory cell recruitment and activation lead to cartilage degradation, fibrosis, and arthritis.
• Either direct infection of a joint space (suppurative arthritis) or cross-reactive immune responses to systemic infections (e.g., in some cases of Lyme arthritis) can lead to joint inflammation and injury.
Joint Tumors and Tumor-Like Lesions
Primary neoplasms of joints are uncommon and usually benign; in general, they reflect the cells and tissue types (synovial membrane, vessels, fibrous tissue, and cartilage) native to the joints. Benign tumors are much more frequent than their malignant counterparts. The rare malignant neoplasms of these structures are discussed below with the soft tissue tumors. In comparison, reactive tumor-like lesions such as ganglions and synovial cysts are much more common than neoplasms; these typically result from trauma or degenerative processes. Here we discuss the more common or clinically significant tumor-like lesions and neoplasms of joints and associated soft tissues.
Ganglion and Synovial Cysts
A ganglion is a small cyst (less than 1.5 cm in diameter) located near a joint capsule or tendon sheath; the wrist is an especially common site. Lesions manifest as firm to fluctuant pea-sized nodules that are translucent to light. Microscopically, they consist of fluid-filled spaces that lack a true cell lining, apparently because they stem from cystic degeneration of connective tissue. Coalescence of adjacent cysts can produce multilocular lesions. The cyst contents resemble synovial fluid, although often there is no communication with the joint space. Ganglions typically are completely asymptomatic. Classically, these can be treated by “Bible therapy”: Whacking the affected area with a large tome usually is sufficient to rupture the cyst, but reaccumulation may recur. Despite their name, they have no relationship to ganglia of the nervous system.
Herniation of synovium through a joint capsule or massive enlargement of a bursa can produce a synovial cyst. A good example is the Baker cyst that occurs in the popliteal fossa.
Tenosynovial Giant Cell Tumor
Tenosynovial giant cell tumor (TGCT) is a catchall term for several closely related benign neoplasms of synovium. Although these lesions previously were considered reactive proliferations (hence the earlier designation synovitis), they are consistently associated with an acquired (1;2) translocation that fuses the promoter of the collagen 6A3 gene to the coding sequence of the growth factor M-CSF. Classic examples are diffuse tenosynovial giant cell tumor, previously known as pigmented villonodular synovitis (PVNS), involving joint synovium, and localized tenosynovial giant cell tumor, also known as giant cell tumor of tendon sheath. Both types typically arise in people in their 20s to 40s, without gender predilection.
Morphology
Grossly, TGCTs are red-brown to orange-yellow. In the diffuse variant the joint synovium becomes a contorted mass of red-brown folds, finger-like projections, and nodules (Fig. 20–23, A). By contrast, the localized type is well circumscribed and contained. Tumor cells in both lesions resemble synoviocytes, and numerous hemosiderin-laden macrophages, osteoclast-like giant cells and hyalinized stromal collagen also are present (Fig. 20–23, B). The tumor cells spread along the surface and infiltrate the subsynovial compartment. In localized TGCT, the cells grow in a solid nodular aggregate. Other typical findings include hemosiderin deposits, foamy macrophages, multinucleate giant cells, and zones of scarring.
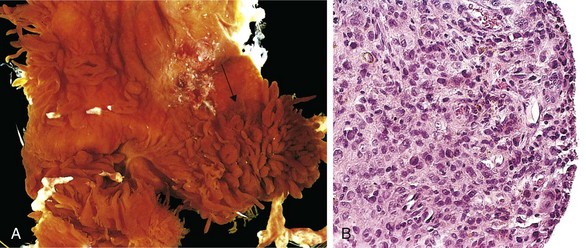
Clinical Features
Diffuse TGCT usually mimics a monoarticular arthritis; it affects the knee in 80% of cases, followed by the hip and ankle. Patients typically complain of pain, locking, and recurrent swelling. Tumor progression limits the range of movement of the joint. Aggressive lesions erode into adjacent bones and soft tissues, causing confusion with other tumors. In contrast, localized TGCT presents as a solitary, slowly growing, painless mass frequently involving wrist and finger tendon sheaths; it is the most common soft tissue tumor of the hand. Cortical erosion of adjacent bone occurs in approximately 15% of cases. Both lesions are amenable to surgical resection, but also prone to local recurrence. Recognition of the association of TGCT and M-CSF gene rearrangement and overexpression has inspired trials of antagonists of M-CSF or its cognate receptor (M-CSFR, a tyrosine kinase); some excellent responses have been reported.
Soft Tissue
By convention, the term soft tissue describes any nonepithelial tissue other than bone, cartilage, central nervous system, hematopoietic, and lymphoid tissues. The focus of this section is on soft tissue tumors, which are classified according to the tissue type they recapitulate, including fat, fibrous tissue, and neurovascular tissue (Table 20–4). In some soft tissue neoplasms, however, no corresponding normal counterpart is known. Although soft tissue tumors are classified based on recognizable lines of differentiation, current evidence indicates that these tumors arise from pluripotent mesenchymal stem cells and are not the result of malignant transformation of mature mesenchymal cells. With the exception of skeletal muscle neoplasms (see further on), benign soft tissue tumors outnumber their malignant counterparts by at least a hundred-fold. In the United States, approximately 12,000 soft tissue sarcomas are diagnosed annually, representing less than 1% of all invasive malignancies. However, they cause 2% of all cancer deaths, reflecting their lethal nature.
| Tumors of Adipose Tissue |
| Lipomas |
| Liposarcoma |
| Tumors and Tumor-Like Lesions of Fibrous Tissue |
| Nodular fasciitis |
| Fibromatoses |
| Superficial fibromatoses |
| Deep fibromatoses |
| Fibrosarcoma |
| Fibrohistiocytic Tumors |
| Fibrous histiocytoma |
| Dermatofibrosarcoma protuberans |
| Pleomorphic fibroblastic sarcoma/pleomorphic undifferentiated sarcoma (malignant fibrous histiocytoma) |
| Tumors of Skeletal Muscle |
| Rhabdomyoma |
| Rhabdomyosarcoma |
| Tumors of Smooth Muscle |
| Leiomyoma |
| Smooth muscle tumors of uncertain malignant potential |
| Leiomyosarcoma |
| Vascular Tumors |
| Hemangioma |
| Lymphangioma |
| Hemangioendothelioma |
| Angiosarcoma |
| Peripheral Nerve Tumors |
| Neurofibroma |
| Schwannoma |
| Granular cell tumor |
| Malignant peripheral nerve sheath tumors |
| Tumors of Uncertain Histogenesis |
| Synovial sarcoma |
| Alveolar soft part sarcoma |
| Epithelioid sarcoma |
Most soft tissue tumors arise without antecedent causes; rarely, radiation exposure, burn injury, or toxin exposure is implicated. Kaposi sarcoma (Chapter 9) is associated with the human herpesvirus 8, but viruses probably are not important in the pathogenesis of most sarcomas in humans. A small minority of soft tissue tumors are associated with genetic syndromes, most notably neurofibromatosis type 1 (neurofibroma, malignant schwannoma), Gardner syndrome (fibromatosis), Li-Fraumeni syndrome (soft tissue sarcoma), and Osler-Weber-Rendu syndrome (telangiectasia). Specific chromosomal abnormalities and genetic derangements in these syndromes provide important clues to the genesis of the neoplasms. Like their mesenchymal brethren, the hematopoietic neoplasms, many soft tissue tumors are associated with highly characteristic chromosomal rearrangements, most commonly translocations that provide insight into pathogenesis and are diagnostically useful. Indeed, some tumors, such as synovial sarcoma, are virtually defined by their associated translocations.
Soft tissue tumors can arise in any location, but approximately 40% of sarcomas occur in the lower extremities, especially the thigh. While the overall incidence of sarcomas increases with age, 15% arise in children. Certain sarcomas tend to appear in certain age groups—for example, rhabdomyosarcoma in childhood, synovial sarcoma in young adulthood, and liposarcoma and pleomorphic fibroblastic or undifferentiated sarcomas in later adult life. Soft tissue sarcomas usually are treated with wide surgical excision (frequently limb-sparing), with irradiation and systemic therapy reserved for large high-grade tumors.
Several features of soft tissue sarcomas influence prognosis:
• Diagnostic classification. This is based not only on histology, but also on immunohistochemistry, electron microscopy, cytogenetics, and molecular genetics, which are indispensable in assigning the correct diagnosis in some cases.
• Grading. Grading, usually on a scale of I to III, is based on the degree of differentiation, the average number of mitoses per high-power field, cellularity, pleomorphism, and an estimate of the extent of necrosis (presumably a reflection of rate of growth). Mitotic counts and necrosis are the most important predictors.
• Staging. With tumors larger than 20 cm, metastases develop in 80% of cases; by contrast, for tumors 5 cm or smaller, metastases occur in only 30% of cases.
• Location. In general, tumors arising in superficial locations (e.g., skin) have a better prognosis than deep-seated lesions; overall, the 10-year survival rate for sarcomas is approximately 40%.
Presented next is an overview of the individual tumors and tumor-like lesions; only the more common forms are covered here. Others are covered elsewhere in the book.
Tumors of Adipose Tissue
Lipoma
Lipomas are benign tumors of fat and are the most common soft tissue tumors in adults. Most lipomas are solitary lesions; multiple lipomas usually suggest the presence of rare hereditary syndromes. Lipomas can be subclassified on the basis of their histologic features and/or characteristic chromosomal rearrangements. Most lipomas are mobile, slowly enlarging, painless masses (although angiolipomas can manifest with local pain); complete excision usually is curative.
Conventional lipomas (the most common subtype) are soft, yellow, well-encapsulated masses of mature adipocytes; they can vary considerably in size. On histologic examination, they consist of mature white fat cells with no pleomorphism.
Liposarcoma
Liposarcomas are malignant neoplasms with adipocyte differentiation. They occur most commonly in the fifth and sixth decades of life. Most liposarcomas arise in the deep soft tissues or in the retroperitoneum. The prognosis of liposarcomas is greatly influenced by the histologic subtype; well-differentiated tumors grow slowly and are associated with a more favorable outlook than the aggressive myxoid/round cell and pleomorphic variants, which tend to recur after excision and metastasize to lungs. Amplification of a region of 12q is common in well-differentiated liposarcomas; this region contains the MDM2 gene, whose product binds and degrades the p53 protein. A t(12;16) chromosomal translocation is associated with myxoid/round cell liposarcomas; this rearrangement creates a fusion gene encoding an abnormal transcription factor that may interfere with adipocyte differentiation.
Morphology
Liposarcomas usually manifest as relatively well-circumscribed lesions. Several different histologic subtypes are recognized, including the low-grade variant, well-differentiated liposarcoma, and the myxoid/round cell liposarcoma, characterized by abundant, mucoid extracellular matrix. Some well-differentiated lesions can be difficult to distinguish from lipomas, whereas very poorly differentiated tumors can resemble various other high-grade malignancies. In most cases, cells indicative of fatty differentiation known as lipoblasts are present; they have cytoplasmic lipid vacuoles that scallop the nucleus (Fig. 20–24), and appearance recapitulating that of fetal fat cells.
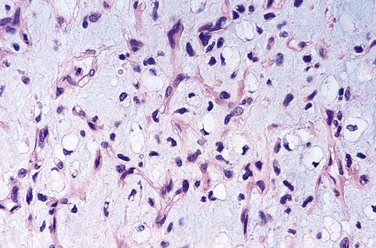
Fibrous Tumors and Tumor-Like Lesions
Fibrous tissue proliferations are a heterogeneous group of lesions. At one end of the spectrum, nodular fasciitis is not a true tumor but rather a reactive, self-limited proliferation. At the other end, fibrosarcomas are highly malignant neoplasms that tend to recur locally and can metastasize. Fibromatoses fall somewhere in the middle; these are benign lesions that infiltrate locally and often can defy attempts at surgical excision. Distinguishing among the various lesions requires considerable skill and diagnostic experience.
Reactive Proliferations
Nodular Fasciitis
Nodular fasciitis is a self-limited fibroblastic proliferation (Fig. 20–25) that typically occurs in adults on the volar aspect of the forearm, the chest, or the back. Patients characteristically present with a several-week history of a solitary, rapidly growing, and occasionally painful mass. Preceding trauma is noted in 10% to 15% of cases. Nodular fasciitis rarely recurs after excision.
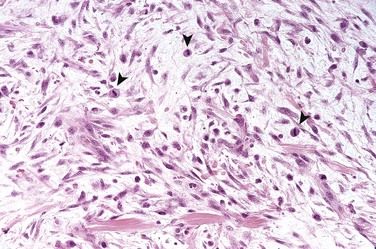
Myositis Ossificans
Myositis ossificans is distinguished from other fibroblastic proliferations by the presence of metaplastic bone. It usually develops in the proximal muscles of the extremities in athletic adolescents and young adults after trauma. The affected area initially is swollen and painful, and subsequently evolves into a painless, hard, well-demarcated mass. It is critical to distinguish this lesion from extraskeletal osteosarcoma. Simple excision usually is curative.
Fibromatoses
The fibromatoses are a group of fibroblastic proliferations distinguished by their tendency to grow in an infiltrative fashion and, in many cases, to recur after surgical removal. Although some lesions are locally aggressive, they do not metastasize. The fibromatoses are divided into two major clinicopathologic groups: superficial and deep.
• The superficial fibromatoses arise in the superficial fascia and include such entities as palmar fibromatosis (Dupuytren contracture) and penile fibromatosis (Peyronie disease). Superficial lesions are genetically distinct from their deep-seated cousins and are generally more innocuous (they can be associated with trisomy 3 and 8); they also come to clinical attention earlier, because they cause deformity of the involved structure.
• The deep fibromatoses include the so-called desmoid tumors that arise in the abdominal wall and muscles of the trunk and extremities, and within the abdomen (mesentery and pelvic walls). They can be isolated lesions, or multiple, as a component of Gardner syndrome, an autosomal dominant disorder including colonic adenomatous polyps and osteomas. Mutations in the APC or β-catenin genes are present in a majority of these tumors. Deep fibromatoses tend to grow in a locally aggressive manner and often recur after excision.
Morphology
Fibromatoses are gray-white, firm to rubbery, poorly demarcated, infiltrative masses 1 to 15 cm in greatest dimension. On histologic examination, they are composed of plump spindle cells arranged in broad sweeping fascicles that penetrate the adjacent tissue; mitoses are few in number. Immunohistochemical and ultrastructural studies show that the tumor cells are fibroblasts and myofibroblasts. Some lesions may be quite cellular, particularly early in their evolution, whereas others contain abundant dense collagen.
In addition to being disfiguring or disabling, fibromatoses occasionally are painful. Although curable by adequate excision, they often recur when incompletely removed due to their infiltrative nature. For those tumors that cannot be resected, therapeutic options include watchful waiting, radiation therapy, and chemotherapy.
Fibrosarcoma
Fibrosarcomas are malignant neoplasms composed of fibroblasts. Most occur in adults, typically in the deep tissues of the thigh, knee, and retroperitoneal area. They tend to grow slowly, and have usually been present for several years at the time of diagnosis. As with other sarcomas, fibrosarcomas often recur locally after excision (in more than 50% of cases) and can metastasize hematogenously (in greater than 25% of cases), usually to the lungs.
Morphology
Fibrosarcomas are soft unencapsulated, infiltrative masses that frequently contain areas of hemorrhage and necrosis. Better-differentiated lesions can appear deceptively circumscribed. Histologic examination discloses all degrees of differentiation, from tumors that closely resemble fibromatoses, to densely packed lesions with spindle cells growing in a herringbone fashion (Fig. 20–26), while others have a myxoid stroma (myxofibrosarcoma), and some are highly cellular neoplasms exhibiting architectural disarray, pleomorphism, frequent mitoses, and necrosis.
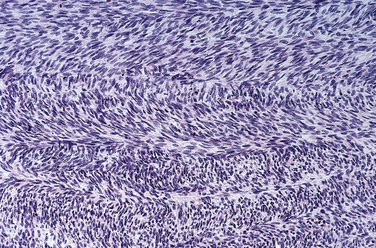
Fibrohistiocytic Tumors
Fibrohistiocytic tumors are composed of a mixture of fibroblasts and phagocytic, lipid-laden cells resembling activated tissue macrophages (also called histiocytes by morphologists). The neoplastic cells in many cases are fibroblasts and myofibroblasts. Consequently, the term fibrohistiocytic is descriptive and does not connote a specific line of differentiation. These tumors span a broad range of histologic patterns and biologic behavior, from self-limited benign lesions to aggressive high-grade sarcomas.
Benign Fibrous Histiocytoma (Dermatofibroma)
Dermatofibromas are relatively common benign lesions in adults manifesting as circumscribed, small (less than 1 cm) mobile nodules in the dermis or subcutaneous tissue. On histologic evaluation, these typically consist of bland, interlacing spindle cells admixed with foamy, lipid-rich histiocyte-like cells. The borders of the lesions tend to be infiltrative, but extensive local invasion does not occur. They are cured by simple excision. The pathogenesis of these lesions is uncertain.
Pleomorphic Fibroblastic Sarcoma/Pleomorphic Undifferentiated Sarcoma
Tumors of this type originally fell under the diagnostic rubric “malignant fibrous histiocytoma,” but with use of objective immunohistochemical markers it became evident that this was a wastebasket diagnostic category that included a number of poorly differentiated sarcomas, such as leiomyosarcomas and liposarcomas. The common histologic features that define this group of poorly differentiated sarcomas are cytologic pleomorphism, the presence of bizarre multinucleate cells, and storiform architecture. Currently, tumors with this histologic appearance that exhibit fibroblastic differentiation are designated undifferentiated pleomorphic spindle cell sarcoma or pleomorphic fibroblastic sarcoma (Fig. 20–27). They usually are large (5 cm to 20 cm), gray-white unencapsulated masses that often appear deceptively circumscribed. They usually arise in the musculature of the proximal extremities or in the retroperitoneum. Most of these tumors are extremely aggressive, recur unless widely excised, and have a metastatic rate of 30% to 50%.
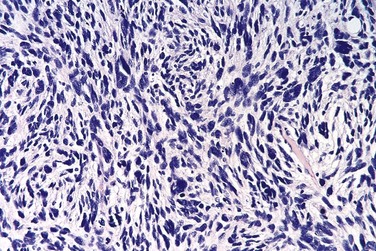
Skeletal Muscle Tumors
Tumors of skeletal muscle differentiation are almost all malignant. Rhabdomyoma, a benign type of skeletal muscle tumor, is rare and is most often found in the heart (Chapter 10).
Rhabdomyosarcoma
Rhabdomyosarcoma is the most common soft tissue sarcoma of childhood and adolescence, usually appearing before age 20. Of interest, it occurs most commonly in the head and neck or genitourinary tract, usually at sites where there is little, if any, normal skeletal muscle.
This tumor occurs in three different histologic types, as described below. Chromosomal translocations are found in most cases of the alveolar variant; the more common t(2;13) translocation fuses the PAX3 gene on chromosome 2 with the FKHR gene on chromosome 13. PAX3 functions upstream of genes that control skeletal muscle differentiation, and tumor development probably involves dysregulation of muscle differentiation by the chimeric PAX3-FKHR protein.
Morphology
Rhabdomyosarcoma is histologically subclassified into the embryonal, alveolar, and pleomorphic variants. The gross appearance of these tumors is variable. Some, particularly the embryonal variant when arising near the mucosal surfaces of the bladder or vagina, can manifest as soft, gelatinous, grapelike masses, designated sarcoma botryoides. In other cases they are poorly defined, infiltrating tan-white masses. The rhabdomyoblast is the diagnostic cell in all types; it has granular eosinophilic cytoplasm rich in thick and thin filaments. The rhabdomyoblasts may be round or elongated; the latter are known as tadpole or strap cells (Fig. 20–28) and may contain cross-striations visible by light microscopy. The diagnosis of rhabdomyosarcoma is based on the demonstration of skeletal muscle differentiation, either in the form of sarcomeres under the electron microscope or by immunohistochemical demonstration of skeletal muscle–specific transcription factors such as myogenin and MYOD-1, and the muscle-associated intermediate filament desmin.
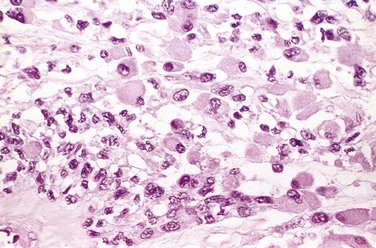
Rhabdomyosarcomas are aggressive neoplasms treated with a combination of surgery, chemotherapy, and radiation. Location, histologic appearance, and tumor genetics all impact the likelihood of cure, with progressively worsening rates for embryonal, pleomorphic, and alveolar variants, in that order. The malignancy is curable in almost two thirds of children; the prognosis is much less favorable in adults with the pleomorphic type.
Smooth Muscle Tumors
Leiomyoma
Benign smooth muscle tumors, or leiomyomas, are common, well-circumscribed neoplasms that can arise from smooth muscle cells anywhere in the body but are encountered most commonly in the uterus (Chapter 18) and the skin.
Leiomyosarcoma
Leiomyosarcomas account for 10% to 20% of soft tissue sarcomas. They occur in adults, more commonly females. Skin and deep soft tissues of the extremities and retroperitoneum (inferior vena cava) are common sites. These neoplasms manifest as firm, painless masses; retroperitoneal tumors can be large and bulky and cause abdominal symptoms. Histologic examination shows spindle cells with cigar-shaped nuclei arranged in interwoven fascicles. Treatment depends on the size, location, and grade of the tumor. Superficial or cutaneous leiomyosarcomas usually are small and carry a good prognosis, whereas retroperitoneal tumors are large and difficult to excise and cause death by both local extension and metastatic spread.
Synovial Sarcoma
Synovial sarcoma was originally believed to recapitulate synovium; however, the phenotype of the neoplastic cells bears no resemblance to a synoviocyte, and despite the name, less than 10% of tumors are intra-articular. Synovial sarcomas account for approximately 10% of all soft tissue sarcomas, typically occurring in persons in their 20s to 40s. They usually develop in deep soft tissues around the large joints of the extremities, with 60% to 70% occurring around the knee; many have been present for several years at the time of presentation. Most synovial sarcomas show a characteristic (X;18) translocation that produces a fusion gene encoding a chimeric transcription factor.
Morphology
On histologic examination, synovial sarcomas may be biphasic or monophasic. Classic biphasic synovial sarcoma exhibits differentiation of tumor cells into both epithelial-type cells and spindle cells. The epithelial cells are cuboidal to columnar and form glands or grow in solid cords or aggregates. The spindle cells are arranged in densely cellular fascicles that surround the epithelial cells (Fig. 20–29). Many synovial sarcomas are monophasic—that is, composed of spindle cells only. Lesions composed solely of spindle cells are easily mistaken for fibrosarcomas or malignant peripheral nerve sheath tumors. Immunohistochemistry is helpful, because the tumor cells are positive for keratin and epithelial membrane antigen, differentiating them from most other sarcomas.
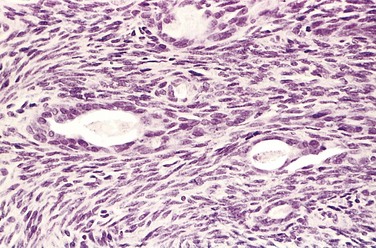
Synovial sarcomas are treated aggressively with limb-sparing surgery and chemotherapy. Common metastatic sites are lung, bone, and regional lymph nodes. Reported 5-year survival rates range from 25% to 62%, and only 10% to 30% of the patients live longer than 10 years.
Bovée JV. EXTra hit for mouse osteochondroma. Proc Natl Acad Sci U S A. 2010;107:1813. [Good explanation of the current understanding of the molecular and cellular genesis of osteochondroma and osteochondromatosis.]
Bovée JV, Hogendoorn PC, Wunder JS, Alman BA. Cartilage tumours and bone development: molecular pathology and possible therapeutic targets. Nat Rev Cancer. 2010;10:481. [A good review of the known genetic abnormalities in these tumors.]
Cao L, Yu Y, Bilke S, et al. Genome-wide identification of PAX-3FKHR binding sites in rhabdomyosarcoma reveals candidate target genes important for development and cancer. Cancer Res. 2010;70:6497. [Scholarly discussion of the PAX3-FKHR genetic translocation and its implications regarding target genes in alveolar rhabdomyosarcomas.]
Flanagan AM, Delaney D, O’Donnell P. Benefits of molecular pathology in the diagnosis of musculoskeletal disease: part II of a two-part review: bone tumors and metabolic disorders. Skeletal Radiol. 2010;39:213. [A good overview of molecular aberrations in bone tumors and select metabolic conditions.]
Goldring M, Goldring S. Osteoarthritis. J Cell Physiol. 2007;213:626. [Excellent review of biologic and biomechanical factors underlying the disorder.]
Goldring M, Goldring S. Articular cartilage and subchondral bone in the pathogenesis of osteoarthritis. Ann N Y Acad Sci. 2010;1111:230. [A succinct and thoughtful presentation of the review of the role of articular structures in the development of osteoarthritis.]
Gorlick R, Khanna C. Osteosarcoma. J Bone Miner Res. 2010;25:6831. [Current overview of the underlying genetic and pathologic basis for osteosarcoma.]
Iliopoulou BP, Huber BT. Infectious arthritis and immune dysregulation: lessons learned from Lyme disease. Curr Opin Rheumatol. 2010;22:451. [Current overview of the immune mechanisms underlying Lyme arthritis.]
Jain S, Xu R, Prieto VG, Lee P. Molecular classification of soft tissue sarcomas and clinical implications. Int J Clin Exp Pathol. 2010;23:416. [Succinct summary of the molecular alterations in a variety of soft tissue sarcomas and their clinical utility.]
Kumar R, Thompson JR. The regulation of parathyroid hormone secretion and synthesis. J Am Soc Nephrol. 2011;22:216. [Good review of mechanisms controlling parathyroid synthesis in health and renal disease.]
Mazzaferro S, Pasquali M, Pirrò G, et al. The bone and the kidney. Arch Biochem Biophys. 2010;503:95. [A well-written discussion about the interplay of kidney and bone in metabolic bone disease.]
Pinto A, Dickman P, Parham D. Pathobiologic markers of the Ewing sarcoma family of tumors: state of the art and prediction of behaviour. Sarcoma. 2011;2011:856190. [Excellent summary of the clinical findings in and molecular basis of Ewing sarcoma.]
Prince F, Otten M, van Suijlekom-Smit LW. Diagnosis and management of juvenile idiopathic arthritis. BMJ. 2011;342:95. [Concise discussion of the definition, etiology, and treatment of the disease.]
Riminucci M, Robey PG, Saggio I, Bianco P. Skeletal progenitors and the GNAS gene: fibrous dysplasia of bone read through stem cells. J Mol Endocrinol. 2010;45:355. [Excellent discussion of how a mutation can affect skeletal progenitor cells and cause the clinical expression of fibrous dysplasia.]
Scott D, Wolfe FM, Huizinga T. Rheumatoid arthritis. Lancet. 2011;376:1094. [Good review and update of pathogenesis and treatment of the disease.]
Singer F. The etiology of Paget disease of bone: viral and genetic interactions. Cell Metab. 2011;13:1. [Very good review about the viral and genetic pathways in Paget disease.]
Sipos W, Pietschmann P, Rauner M, et al. Pathophysiology of osteoporosis. Wien Med Wochenschr. 2009;159:230. [Good review and update of pathogenesis of osteoporosis.]
Smith JL, Riedel RF. Emerging therapeutic targets for soft tissue sarcoma. Curr Oncol Rep. 2011. [Interesting review of how elucidation of the genetics and cell pathways active in soft tissue sarcoma has provided new targets for molecular therapy.] April 27 Epub ahead of print
Takahashi N, Maeda K, Ishihara A, et al. Regulatory mechanism of osteoclastogenesis by RANKL and Wnt signals. Front Biosci. 2011;16:21. [Good review of the mechanisms of activating osteoclasts, and of the role of these pathways in disease.]
Van Dijk FS, Pals G, Van Rijn RR, et al. Classification of osteogenesis imperfecta revisited. Eur J Med Genet. 2010;53:1. [An overview of the current classification of osteogenesis based on clinical and molecular findings.]
Vanitallie TB. Gout: epitome of painful arthritis. Metabolism. 2010;59(Suppl 1):S32. [Excellent summary of the recent developments in the molecular and cellular biology underlying gout.]