ALTERATIONS OF LEUKOCYTE, LYMPHOID, AND HEMOSTATIC FUNCTION



The many disorders involving leukocytes range from deficiencies in the quality and quantity of leukocytes (leukopenia) to increased numbers of leukocytes (leukocytosis) in response to infections to proliferative disorders, such as leukemia. Many hematologic disorders are malignancies, and many nonhematologic malignancies metastasize to bone marrow, affecting leukocyte production. Thus a large portion of this chapter is devoted to malignant disease.
The primary role of clotting (hemostasis) is to stop bleeding through an interaction among vascular endothelium, platelets, and the clotting system. Many disease states are associated with clinically significant aberrations in any of these three necessary components of clotting. This chapter discusses various components of clotting and their control systems.
ALTERATIONS OF LEUKOCYTE FUNCTION
Leukocyte function is affected if too many or too few white cells are present in the blood or if the cells that are present are structurally or functionally defective. Quantitative leukocyte disorders result from decreased production in the bone marrow or accelerated destruction of cells in the circulation. Other quantitative alterations, however, occur in response to infections.
Qualitative leukocyte disorders consist of disruptions of leukocyte function. Phagocytic cells (granulocytes, monocytes, macrophages) may lose their capacity to function as effective phagocytes. Lymphocytes may lose their capacity to respond to antigens. (Qualitative disruptions of inflammatory and immune processes caused by leukocyte disorders are described in Chapter 8.) Other leukocyte alterations include infectious mononucleosis and cancers of the blood—leukemia and multiple myeloma.
Quantitative Alterations of Leukocytes
Leukocytosis is a leukocyte count that is higher than normal; conversely, leukopenia is a count that is lower than normal. Leukocytosis or leukopenia may affect all cell types or only a specific type of leukocyte and may result from a variety of physiologic conditions and alterations.
Leukocytosis occurs as a normal protective response to physiologic stressors, such as infection, strenuous exercise, emotional changes, temperature changes, anesthesia, surgery, pregnancy, and some drugs, hormones, and toxins. It is also caused by pathologic conditions, such as malignancies and hematologic disorders. Unlike leukocytosis, leukopenia is never normal. When the leukocyte count decreases to less than 1000/mm3, the individual is at increased risk for infection. With counts less than 500/mm3, the possibility for life-threatening infections is high. Leukopenia can be caused by radiation, anaphylactic shock, autoimmune disease (e.g., systemic lupus erythematosus), immune deficiencies (see Chapter 8), and exposure to certain chemotherapeutic agents.
Granulocytes and Monocytes
Increased numbers of circulating granulocytes (neutrophils, eosinophils, basophils) and monocytes are primarily a response to infection. Increased numbers also occur as a result of myeloproliferative disorders (i.e., polycythemia vera, chronic myelogenous leukemia, chronic neutrophilic leukemia, chronic eosinophilic leukemia) that increase stem cell proliferation in bone marrow.
Decreased numbers occur when infectious processes exhaust the supply of circulating granulocytes and monocytes by drawing them out of the circulation and into infected tissues faster than they can be replaced. Decreases also can be caused by disorders that suppress marrow function.
Granulocytosis—an increase in granulocytes (neutrophils, eosinophils, basophils)—begins with the release of stored leukocytes from the venous sinuses of the marrow. Neutrophilia is another term that may be used to describe granulocytosis because neutrophils are the most numerous of the granulocytes (Table 27-1). Neutrophilia occurs in the early stages of infection or inflammation and is established when the absolute neutrophil count exceeds 7500/μL. Stored neutrophils are approximately 20 to 40 times greater in number than circulating neutrophils. When the neutrophil count increases greatly—more than 100,000/μL (usually seen only in those with myelocytic leukemia)—the blood viscosity may increase greatly so that thrombosis or occlusion of blood vessels occurs. Release and depletion of stored neutrophils from the venous sinuses stimulate granulopoiesis to replenish neutrophil reserves. Specific conditions associated with neutrophilia are identified in Table 27-1.
Table 27-1
Other Conditions Associated with Neutrophils, Eosinophils, Basophils, Monocytes, and Lymphocytes

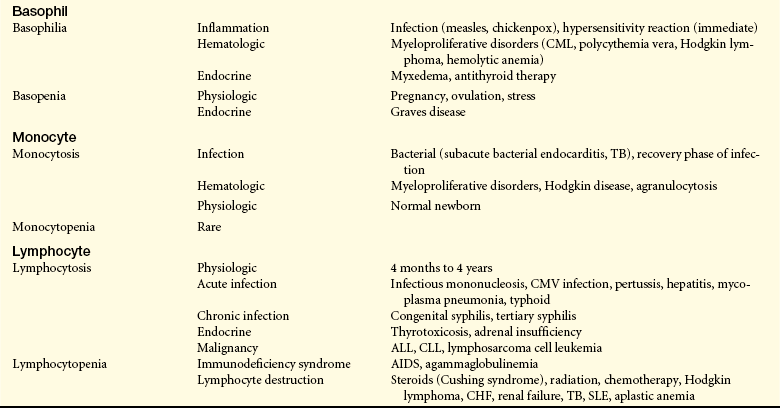
AIDS, Acquired immunodeficiency syndrome; ALL, acute lymphocytic leukemia; CHF, congestive (left) heart failure; CLL, chronic lymphocytic leukemia; CML, chronic myelogenous leukemia; CMV, cytomegalovirus; GI, gastrointestinal; MI, myocardial infarction, SLE, systemic lupus erythematosus; TB, tuberculosis.
When the demand for circulating mature neutrophils exceeds the supply, the marrow begins to release immature neutrophils (and other leukocytes) into the blood. Premature release of the immature white cells is responsible for the phenomenon known as a shift-to-the-left or leukemoid reaction. This refers to the microscopic detection of disproportionate numbers of immature leukocytes in peripheral blood smears. Many diagrams present cellular differentiation and maturation progressing from left to right within the drawing, instead of vertically as shown in Figure 25-9. An early release of immature leukocytes would shift the distribution of cells in the blood toward those on the left side of the diagram. This phenomenon is also seen in the blood smear of individuals with leukemia, hence the term leukemoid reaction. As infection or inflammation diminishes and as granulopoiesis replenishes circulating granulocytes, a return to normal occurs.
Neutropenia is a condition associated with reduction in circulating neutrophils. Clinically, neutropenia exists when the neutrophil count is less than 2000/μL.1 A reduction in neutrophils occurs in severe prolonged infections when production of granulocytes cannot keep up with demand. Neutropenia is considered mild with a neutrophil count between 1000 and 1500/μL. Moderate neutropenia is a neutrophil count between 500 and 1000/μL, and severe neutropenia is a count less than 500/μL. Neutrophil reduction results from severe or prolonged infections when granulocyte production does not keep up with demand.
Other causes of neutropenia, in the absence of infection, may be (1) decreased neutrophil production or ineffective granulopoiesis, (2) reduced neutrophil survival, and (3) abnormal neutrophil distribution and sequestration. Neutropenia also is categorized as primary or secondary; primary disorders are further identified as congenital or acquired.
Congenital defects in neutrophil production include cyclic neutropenia and neutropenia with congenital immunodeficiency diseases, as well as multiple syndromes (e.g., Kostmann, Shwachman-Diamond, Diamond-Blackfan, Griscelli, Chédiak-Higashi, and Barth syndromes). Primary acquired neutropenia is associated with multiple conditions, for example, hypoplastic anemia or aplastic anemia, leukemia (acute myelogenous leukemia [AML]/chronic lymphocytic leukemia [CLL]), lymphomas (Hodgkin, non-Hodgkin), and myelodysplastic syndrome (MDS). The megaloblastic anemias (vitamin B12 and folate deficiency) as well as starvation and anorexia nervosa cause neutropenia because of an inadequate supply of vitamins and nutrients for protein production.
Reduced neutrophil survival and abnormal distribution and sequestration are usually secondary to other disorders. Neutropenia occurs in a variety of immunologic disorders, particularly systemic lupus erythematosus, rheumatoid arthritis, Felty and Sjögren syndromes, splenomegaly, and drug-related causes.
Severe granulocytopenia (less than 500/μL) or agranulocytosis (complete absence of granulocytes in blood) is usually secondary to arrested hematopoiesis in the bone marrow or massive cell destruction in the circulation. Chemotherapeutic agents used to treat hematologic and other malignancies cause generalized bone marrow suppression. Several other drugs and large doses of ionizing radiation cause agranulocytosis, which occurs rarely but carries a high mortality rate (10% to 50%). Clinical manifestations of agranulocytosis include recurrent and persistent life-threatening infection (particularly of the respiratory system) leading to septicemia, general malaise, fever, tachycardia, and ulcers in the mouth and colon. If untreated, sepsis caused by agranulocytosis results in death within 3 to 6 days.
Eosinophilia is an absolute increase (more than 450/μL) in the total numbers of circulating eosinophils. Allergic disorders (type I hypersensitivity) associated with asthma, hay fever, and drug reactions, as well as parasitic infections (particularly with metazoal parasites) are often cited as causes. Hypersensitivity reactions and the normal defense against parasites trigger the release of eosinophil chemotactic factor of anaphylaxis (ECF-A) from mast cells, attracting eosinophils to the area. (These processes are described and illustrated in Chapters 7 and 8.) Tissues with abundant mast cells, such as the respiratory and gastrointestinal tracts, are particularly common sites for eosinophil invasion. Mast cells also release interleukin-5 (IL-5), which stimulates the bone marrow to produce and release more eosinophils into the blood. Eosinophilia may also be associated with dermatologic disorders, such as atopic dermatitis, eczema, and pemphigus. Various types of eosinophilic scleroderma-like diseases also have been reported to occur in association with hemato-oncogenic disorders (i.e., eosinophilic cellulitis [Wells syndrome] and eosinophilic fasciitis [Schulman syndrome]). Increased numbers of eosinophils have been observed in individuals with eosinophilia-myalgia syndrome (EMS), which is associated with ingestion of tryptophan, and a relationship between EMS and fibromyalgia syndrome (FMS) has been suggested.
Eosinopenia, a decrease in circulating numbers of eosinophils, generally is caused by migration of eosinophils into inflammatory sites. It also may be seen in Cushing syndrome and as a result of stress caused by surgery, shock, trauma, burns, or mental distress. Other conditions causing eosinopenia are detailed in Table 27-1.
Basophilia, an increase in circulating numbers of basophils, is rare and generally is a response to inflammation and immediate hypersensitivity reactions. Basophils contain histamine that is released during an allergic reaction. An increase in levels of basophils is seen also in myeloproliferative disorders, such as chronic myeloid leukemia and myeloid metaplasia. Other conditions associated with basophilia are listed in Table 27-1.
Basopenia (also known as basophilic leukopenia), a decrease in circulating numbers of basophils, is seen in hyperthyroidism, acute infection, and long-term therapy with steroids. A decrease in basophils may be seen during ovulation and pregnancy. Other conditions associated with basopenia are listed in Table 27-1.
Monocytosis is an increase (generally greater than 800/μL) in numbers of circulating monocytes. The condition is often transient and not related to a dysfunction of monocyte production. When present, it most commonly occurs with neutropenia associated with bacterial infections, particularly in the late stages or recovery stage, when monocytes are needed to phagocytize surviving microorganisms and debris. Monocytosis often is seen in chronic infections, usually with intracellular bacteria, such as tuberculosis (TB), brucellosis, and listeriosis, and subacute bacterial endocarditis (SBE). Peripheral monocytosis has been found to correlate with the extent of myocardial damage following myocardial infarction. Increased numbers of monocytes also may indicate marrow recovery from agranulocytosis. Other conditions associated with monocytosis are identified in Table 27-1.
Monocytopenia, a decrease in numbers of circulating monocytes, is rare, and not much is known about this condition because of the small numbers of monocytes generally present in the blood. Monocytopenia, however, has been identified with hairy cell leukemia and prednisone therapy.
Lymphocytes
Quantitative alteration of lymphocytes occurs when lymphocytes are activated by antigenic stimuli, usually microorganisms (see Chapter 7). A lymphocytosis is rare in acute bacterial infections and occurs most commonly in acute viral infections, particularly those caused by the Epstein-Barr virus (EBV), a causative agent in infectious mononucleosis. Other specific disorders associated with lymphocytosis are listed in Table 27-1.
Lymphocytopenia may be attributable to (1) abnormalities of lymphocyte production associated with neoplasias and immune deficiencies, and (2) destruction by drugs, viruses, or radiation. It also can occur in individuals for no apparent reason. Other conditions associated with lymphocytopenia are identified in Table 27-1. The lymphocytopenia associated with heart failure and other acute illnesses may be caused by elevated levels of cortisol. Lymphocytopenia is a major problem in acquired immunodeficiency syndrome (AIDS) in which the human immunodeficiency virus (HIV) is cytopathic for T helper lymphocytes. (For a more detailed discussion of AIDS, see Chapter 9.)
Infectious Mononucleosis
Infectious mononucleosis (IM) is an acute, self-limiting, neoplastic lymphoproliferative clinical syndrome characterized by acute viral infection of B lymphocytes (B cells). The most common etiologic agent is EBV, a ubiquitous, lymphotrophic, gamma-group herpesvirus, which was first recognized as the causative agent in IM in the late 1960s. EBV accounts for approximately 85% of all IM cases. Other etiologic agents that may cause symptoms resembling IM are viruses (cytomegalovirus [CMV], adenovirus, HIV, hepatitis A, influenza A and B, and rubella), as well as the bacteria Toxoplasma gondii, Corynebacterium diphtheriae, and Coxiella burnetii. IM caused by CMV is generally noted in older individuals, with fever and malaise the major complaints; the major manifestations of EBV-induced IM are the classic triad of symptoms of pharyngitis, lymphadenopathy, and fever.
Approximately 50% to 85% of children are infected with EBV by age 4, and more than 90% of adults have indications of subclinical EBV infections. These early infections are usually asymptomatic and provide immunity to EBV, thus early EBV infections rarely develop into IM. IM may arise when the initial infection occurs during adolescence or later, but still only results in IM in 35% to 50% of these individuals. Symptomatic IM usually affects young adults between ages 15 and 35 years, with the peak incidences occurring between 15 and 19 years; males have a later peak (18 to 23 years) than females. The overall incidence rate for this age group is 6 to 8 cases per 1000 persons per year. Children from low socioeconomic environments are particularly susceptible to infections with EBV. IM is uncommon in individuals over age 40 years, but if it does occur, it is more commonly caused by CMV.
Transmission of EBV is usually by saliva through personal contact (e.g., kissing, hence the term “kissing disease”). The virus also may be present in other mucosal secretions of the genital, rectal, and respiratory tract, as well as blood. No evidence of aerosol transmission through sneezing or coughing has been documented. The disease begins with widespread infection of B lymphocytes, all of which possess receptors for EBV. The virus initially infects the oropharynx, nasopharynx, and salivary epithelial cells with later spread to the lymphoid tissue and B cells. Infection of B cells permits the virus to enter the bloodstream, which spreads the infection systemically.
In the immunocompetent individual, unaffected B cells produce antibodies (IgG, IgM, IgA) against the virus. Concomitantly, there is a massive activation and proliferation of cytotoxic T cells (CD8) directed against EBV-infected cells; CD8 lymphocytes can account for greater than 50% of the total circulating lymphocytes. The immune response against EBV-infected cells (cellular infiltration, production of cytokines) is largely responsible for the cellular proliferation in the lymphoid tissues (lymph nodes, spleen, tonsils, occasionally liver). Sore throat and fever, two of the earliest manifestations, are caused by inflammation at the site of viral entry and initial infection (the mouth and throat).
CLINICAL MANIFESTATIONS The incubation period of IM is approximately 30 to 50 days (4 to 8 weeks). Flulike symptoms such as headache, malaise, fatigue, arthralgia, fever, chills, and dysphagia, may appear within the first 3 to 5 days, although some individuals remain asymptomatic. These symptoms may vary in severity for the next 7 to 20 days. At the time of diagnosis the individual usually has the classic triad of symptoms: fever, pharyngitis, and lymphadenopathy of the cervical lymph nodes. The pharyngitis is usually diffuse and often accompanied by a whitish or grayish green, thick exudate. It also is quite painful and is the symptom that most often causes the individual to seek treatment. IM is usually self-limiting, and recovery occurs in a few weeks. Fatigue may last for 1 to 2 months after resolution of the infection.
Although severe clinical complications are rare, as the condition progresses, generalize lymph node enlargement may develop and enlargement of the spleen and liver also may occur. Splenomegaly is clinically evident 50% of the time and is demonstrated radiologically 100% of the time. Difficulty in detecting splenomegaly with physical examination contributes to the underestimation of actual enlargement. Splenic rupture is rare (only 0.1% to 0.15% of all cases) and can occur spontaneously as a result of mild trauma, occurring primarily in males between days 4 and 21 after the onset of symptoms. It is the most common cause of death related to IM. Other causes of fatalities are hepatic failure, extensive bacterial infection, or viral myocarditis.
Other organ systems are rarely involved, but such involvement may result in additional symptoms, such as meningitis, encephalitis, Guillain-Barré syndrome, Bell palsy, optic neuritis, mental impairment, transverse myelitis, cerebellar ataxia, and demyelinating diseases. Ocular manifestations may include eyelid and periorbital edema, dry eyes, keratitis, uveitis, conjunctivitis, retinitis, oculoglandular syndrome, choroiditis, papillitis, and ophthalmoplegia. In children, Reye syndrome also has been associated with EBV infection.
Pulmonary involvement is rare, but when present may include hilar and mediastinal lymphadenopathy, interstitial pneumonitis, and pleural effusion. Pneumonia and respiratory failure have been documented; however, they are more likely to develop in immunocompromised individuals. Approximately 3% to 10% of adults older than 40 years of age have never been infected with EBV and are susceptible to IM later in life. In these individuals the classic symptoms are not generally present, making diagnosis more difficult. If an older individual has an elevated temperature that cannot be explained and persists for more than 2 weeks, EBV infection should be suspected, particularly in the presence of abnormal liver function tests with hepatomegaly and jaundice. Other neurologic manifestations that may be present include peripheral neuropathy and Guillain-Barré syndrome.
EVALUATION AND TREATMENT The blood of affected individuals contains an increased number of atypical lymphocytes (Figure 27-1). Diagnosis of IM is commonly based on Hoagland’s criteria of at least 50% lymphocytes and at least 10% atypical lymphocytes in the blood in the presence of fever, pharyngitis, and adenopathy confirmed by a positive serologic test. Serologic tests are used to determine a heterophile antibody response.2 Heterophile antibodies are a heterogeneous group of immunoglobulin M (IgM) antibodies that are agglutinins against nonhuman red blood cells (e.g., sheep, horse) and are detected by qualitative (Monospot) or qualitative methods (heterophile antibody test).
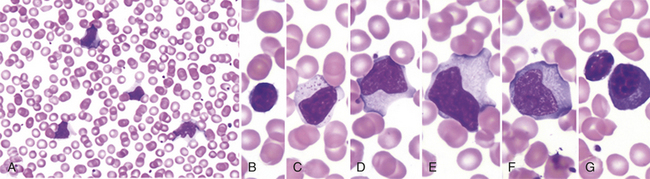
Figure 27-1 Peripheral blood smear in infectious mononucleosis. Low power (A) shows moderately high white blood cell count and high number of reactive, or “atypical” lymphocytes. Higher power (B-G) illustrates spectrum of lymphoid morphology, including small resting lymphocyte (B) for comparison, large granular lymphocyte (C), atypical forms (D-F), also referred to as “reactive” lymphs, and circulating plasma cell (G). (From Hoffman R, et al: Hematology: basic principles and practice, ed 5, Philadelphia, 2009, Churchill Livingstone.)
The Monospot test is limited because other infections (e.g., CMV, adenovirus) and toxoplasmosis also produce heterophilic antibodies. Thus 5% to 15% of Monospot tests yield false-positive results. Levels of heterophilic antibodies in the blood increase as the condition progresses, although some individuals and children younger than age 4 years do not produce them. These individuals give a false-negative result. Specificity for diagnosis of EBV infection may be increased with viral-specific serology tests that identify EBV-specific antibodies (e.g., IgG or IgM against the viral capsid antigen [VCA], or IgG against the EBV nuclear antigen [EBNA]). These tests are more expensive and labor intensive so are reserved for instances in which the Monospot test is not appropriate.
Because IM is usually self-limiting, medical intervention is rarely required. Treatment of IM is supportive and includes rest and alleviation of symptoms with analgesics and antipyretics. Ibuprofen, not aspirin, is used with children and adolescents because of the reported incidence of Reye syndrome associated with EBV infection. Pharyngitis of streptococcal origin, which occurs in 20% to 30% of cases, is treated with penicillin or erythromycin. Ampicillin is contraindicated because it causes a rash in most individuals with IM.
Bed rest and avoidance of strenuous activity should be included in the therapy. Steroids may be used, but only in the presence of severe complications (e.g., impending airway obstruction) or other organ system involvement (e.g., nervous system manifestations, thrombocytopenic purpura, myocarditis, pericarditis). Acyclovir has been used with immunosuppressed individuals; however, clinical improvement has been minimal and therefore it is not recommended for standard treatment.
In the rare event of splenic rupture, the treatment has been removal of the spleen and continues to be the choice in hemodynamically unstable individuals. More recent practice has been to repair the spleen to avoid overwhelming postoperative infection (OPSI). Children are at greater risk of OPSI than adults. Postsplenectomy vaccinations for Streptococcus pneumoniae, Haemophilus influenzae, and Meningococcus are essential because these microorganisms are responsible for 92% of fatal infections. Treatment may also be necessary for airway obstruction from massive edema of the Waldeyer ring or for autoimmune hemolytic anemia, which occurs in approximately 3% to 5% of cases.
Fatal IM also is expressed with the inherited X-linked lymphoproliferative (XLP) syndrome. The underlying cause leading to death is the absence of a functional SAP protein that allows for the unregulated proliferation of cytotoxic T cells and the concomitant production and release of cytokines.
Leukemias
Leukemia is a clonal malignant disorder of leukocytes in the blood and blood-forming organs. The common feature of all forms of leukemia is an uncontrolled proliferation of malignant leukocytes, causing an overcrowding of bone marrow and decreased production and function of normal hematopoietic cells. The first description of a “leukemic” individual was written by Velpeauin 1827.3 Virchow, a pathologist, coined the term white blood (Weissus blut) and later originated the term leukemia. Since Virchow’s initial discovery, the overall classification of leukemia has become increasingly complex and undergone several permutations. The current classification of leukemia is based on (1) the predominant cell of origin (either myeloid or lymphoid) and (2) the rate of progression, which usually reflects the degree at which cell differentiation was arrested when the cell became malignant (acute or chronic) (Figure 27-2). Acute leukemia is characterized by undifferentiated or immature cells, usually a blast cell, and the onset of disease is abrupt and rapid with a short survival time. In chronic leukemia the predominant cell is more differentiated but does not function normally, with a relatively slow progression. Thus there are four types of leukemia: acute lymphocytic (ALL), acute myelogenous (AML), chronic lymphocytic (CLL), and chronic myelogenous (CML). In 1976 the French-American-British Cooperative Group developed more extensive criteria for the classification of acute leukemias. This system is based on characteristics that may provide significant therapeutic prognostic information, such as structure, number of cells, genetics, identification of surface markers, and histochemical staining.
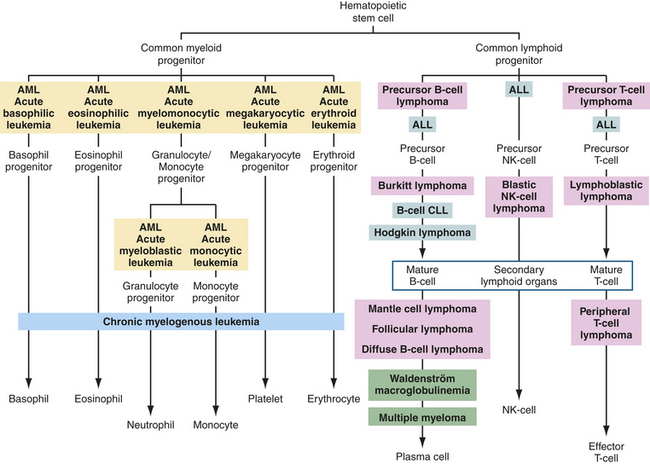
Figure 27-2 Origins of leukemias and lymphomas. Differentiation pathways of blood-forming cells and reported sites from which specific leukemias and lymphomas originate. Tumors of similar types are given the same background coloring. ALL, acute lymphocytic leukemia; AML, acute myelogenous leukemia; CLL, chronic lymphocytic leukemia; NK, natural killer.
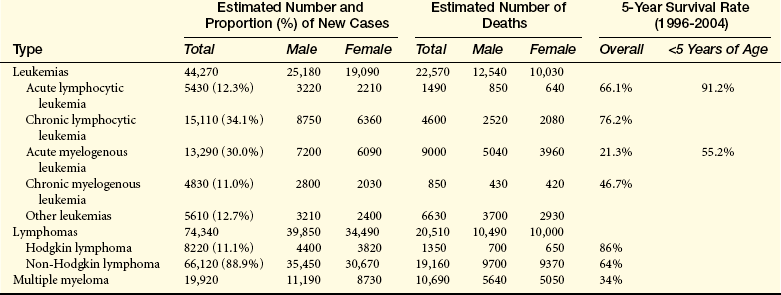
Leukemia occurs with varying frequencies at different ages and is more common in adults than children (Figure 27-3). It is estimated that more than 44,000 cases of leukemia were newly diagnosed in 2008, with males having a slightly higher incidence than females (Table 27-2).4 In all types of leukemia males have a higher incidence rate (56%) as do Americans of European descent. White children have higher rates of leukemia than children of other groups. ALL is the least common type overall, but is the most common in children (approximately 61% of ALL cases are diagnosed before the age of 20). Leukemia accounts for about 30% of all childhood cancers, and ALL accounts for almost 78% of all new cases of leukemia in children. CLL and AML are the most common types in adults. CML is found mostly in adults.
Table 27-2
Estimated New Cases and Deaths: Leukemia and Lymphoma in the United States in 2008
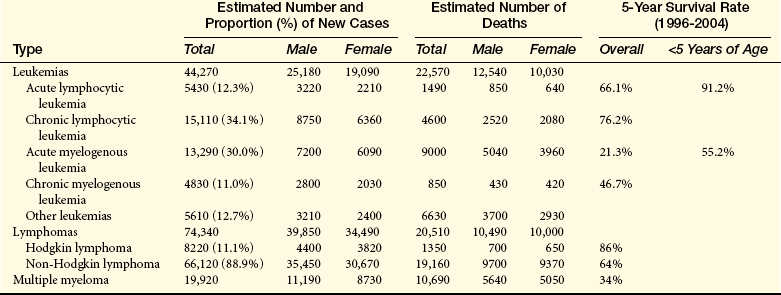
Data from Cancer Facts and Figures 2008, American Cancer Society.
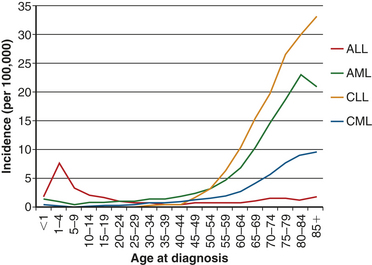
Figure 27-3 Age-related incidence at diagnosis of leukemias. The incidences of acute myelogenous leukemia (AML), chronic lymphocytic leukemia (CLL), and chronic myelogenous leukemia (CML) are relatively stable until middle age and then increase dramatically. The incidence of acute lymphocytic leukemia (ALL) peaks in childhood, then diminishes until middle age when the incidence begins rising slowly with age. Data obtained from http://seer.cancer.gov/csr/1975_2005/results_merged/sect_13_leukemia.pdf.
Over the past two decades the rates of induced remission and survival in most forms of leukemia have increased. Current survival rates range from 25% for AML to 75% for CLL. This progress is the result of more effective chemotherapeutic agents, improved blood product and antimicrobial support, and specialized nursing care. Chemotherapy and bone marrow transplants have significantly increased the survival time for individuals with acute leukemia.
PATHOPHYSIOLOGY All leukemias have certain pathophysiologic features in common. Although the exact cause of leukemia is unknown, several risk factors and related genetic aberrations are associated with the onset of malignancy. There is a statistically significant tendency for leukemia to reappear in families. There is also an increased incidence of leukemia in association with other hereditary abnormalities such as Down syndrome, Fanconi aplastic anemia, Bloom syndrome, trisomy 13, Patau syndrome, and some immune deficiencies (i.e., ataxia-telangiectasia, Wiskott-Aldrich syndrome, and congenital X-linked agammaglobulinemia; see Chapter 8).
Genetic translocations (mitotic errors) are observed in leukemic cells. The most common genetic abnormality is the reciprocal translocation between chromosomes 9 and 22 t(9;22)(q34;q11), the Philadelphia chromosome.5 The Philadelphia chromosome was first observed in persons with CML, and is present in 95% of those with CML, 3% of individuals with AML, and 20% of those with ALL (primarily adults).6 This translocation results in the novel fusion of the BCR1 gene region from chromosome 22 and the proto-oncogene ABL1 from chromosome 9 (Figure 27-4). The BCR-ABL1 joining results in the expression of a unique fused oncoprotein BCR-ABL1.5 The ABL1 protein is a tyrosine kinase in the signaling pathway that promotes cell proliferation. The BCR-ABL1 variant possessed greater tyrosine kinase activity and proved to be essential for transformation into leukemic cells. BCR-ABL1 appears to excessively activate intracellular pathways leading to increased proliferation, decreased sensitivity to apoptosis, and premature release of immature cells into the circulation. In most leukemias and lymphomas a single major genetic abnormality, such as the t(9;22) translocation, does not lead to an aggressive malignancy. The initial event is usually followed by a series of secondary genetic changes.7 Thus the original tumor becomes genetically unstable and diverse.
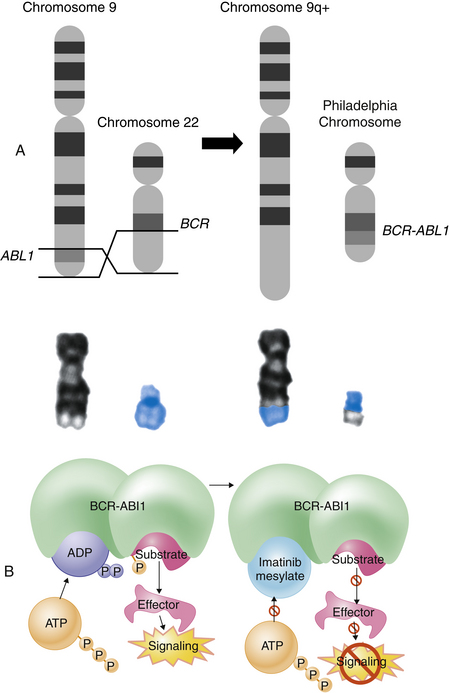
Figure 27-4 Philadelphia chromosome. The Philadelphia chromosome is an example of a reciprocal chromosomal translocation that results in an abnormal gene product responsible for a clinical disorder. A, An exchange occurs between the long arm of chromosome 9 (black chromosome) and the long arm of chromosome 22 (blue chromosome); t(9;22)(q34;q11). B, Mechanism of action of imatinib. By occupying the ATP-binding pocket of the ABL kinase domain, imatinib prevents substrate phosphorylation and downstream activation of signals, thus inhibiting the leukemogenic effects of BCR-ABL1 on cells in chronic myelogenous leukemia. ADP, adenosine diphosphate; ATP, adenosine triphosphate; P, phosphate group. (A, Top portion from Rakel R, Bope E: Conn’s current therapy 2008, Philadelphia, 2008, Saunders. A, Lower portion from Yanoff M, Duker J: Ophthalmology, ed 3, Edinburgh, 2009, Mosby. B from Goldman L, Ausiello D: Cecil medicine, ed 23, Philadelphia, 2008, Saunders.)
Risk factors for the onset of leukemia include environmental factors as well as other diseases. Increased risk in adults has been linked to cigarette smoke, exposure to benzene, and ionizing radiation. Large doses of ionizing radiation particularly result in an increased incidence of myelogenous leukemia. Infections with HIV or hepatitis C virus increase the risk for leukemia, and it is now widely accepted that some types of leukemia are caused by infection with the human T-cell leukemia/lymphoma virus-1 (HTLV-1). Drugs that cause bone marrow depression (e.g., chloramphenicol, phenylbutazone, and certain alkylating agents, such as cytoxan) also can predispose an individual to leukemia. AML is the most frequently reported secondary cancer after high doses of chemotherapy for Hodgkin lymphoma, non-Hodgkin lymphoma, multiple myeloma, ovarian cancer, and breast cancer. Acute leukemia also may develop secondary to certain acquired disorders, including CML, CLL, polycythemia vera, myelofibrosis, Hodgkin lymphoma, multiple myeloma, ovarian cancer, and sideroblastic anemia.
Leukemias are considered clonal disorders in that a single progenitor cell undergoes malignant transformation. The leukemia blasts literally “crowd out” the marrow and cause cellular proliferation of the other cell lines to cease. Normal granulocytic-monocytic, lymphocytic, erythrocytic, and megakaryocytic progenitor cells cease to function, resulting in pancytopenia (a reduction in all cellular components of the blood). An interesting observation is that leukemic cells apparently divide more slowly and take longer to synthesize deoxyribonucleic acid (DNA) than other blood precursors. Leukemic cells accumulate relentlessly in the bone marrow causing overcrowding of the marrow, and they compete with cellular proliferation and function of normal hematopoietic cells. Thus leukemia has been termed an accumulation disorder, as well as a proliferation disorder. In the majority of cases, leukemic cells are ejected into the blood, where they accumulate. These cells also may infiltrate and accumulate in the liver, spleen, lymph nodes, and other organs throughout the body. The presentation of large numbers of leukemic cells in the blood may be one of the most dramatic indicators of leukemia; however, leukemia is still a primary disruption of the bone marrow.
Acute Leukemias
Acute leukemias consist of two types: acute lymphocytic leukemia (ALL) and acute myelogenous leukemia (AML). Acute leukemias are seen in both genders and in all ages, with the incidence increasing dramatically in individuals older than 50 years. Mortality for all acute leukemias in the United States is about 7 per 100,000. In children younger than 15 years, leukemia accounts for a third of all deaths from cancer. North American and Scandinavian countries have the highest mortality; Eastern European countries, Asia (except Japan), and Central America have the lowest mortality. Japan’s higher mortality is the result of the atomic bombs dropped in World War II. Blacks have consistently shown a lower mortality than whites. More than 5400 new cases of ALL and 4800 cases of AML occurred in 2008, with more than 1400 deaths attributed to ALL and 450 to AML.4,8
PATHOPHYSIOLOGY ALL is a progressive neoplasm defined by the presence of greater than 30% lymphoblasts in the bone marrow or blood. Most cases of ALL occur in children (80% of ALL), and it is the most common leukemia in children, most often occurring in the first decade. The median age of diagnosis of ALL is age 13. Although adults with ALL account for only 20% of all cases, their mortality rate is significantly higher (see Table 27-2). The significant difference between the incidence of ALL in adults and children is thought to be determined by differences in the biology of the disease.
Immunotyping of leukemic blast cells allows for the identification of subtypes of ALL. Approximately 75% of ALL in children originate from transformed precursor B cells, whereas adult ALL is a mixture of cancers of precursor B-cell or precursor T-cell origin. A small percentage of ALL cases have neither B- nor T-cell origination and are called null cell (Table 27-3). Precursor B-cell ALL can be subdivided into different phenotypes, depending on their progression through the B-cell maturation process before becoming malignant.9,10 The general phenotype of precursor B-cell ALL expresses CD19, human leukocyte antigen DR (HLA-DR), and other B-cell–associated antigens in the cytoplasm. The most immature form (pro-B ALL) occurs in about 5% of precursor B-cell ALL and is characterized by lack of expression of CD10. CD10 (common acute lymphocytic leukemia antigen [CALLA]) is a cell surface metalloprotease. Lack of CD10 is frequently associated with translocation of the myeloid/lymphoid leukemia (MLL) gene and a poor prognosis. The common precursor B-cell ALL makes up approximately 80% of precursor B-cell ALL cases; these express surface CD10, but have not yet undergone rearrangement of the immunoglobulin genes. The remaining individuals have a more mature form of precursor B-cell ALL (pre–B-cell ALL) in which the cells express immunoglobulin molecules in the cytoplasm. Less common variations include cells that are intermediate between the common precursor and pre–B-cell phenotypes and express immunoglobulin heavy chain, but no light chain, and cells that are more mature than the pre–B-cell ALL and express surface immunoglobulin and the absence of staining for the enzyme—terminal deoxynucleotidyl transferase (TdT).
Table 27-3
Immunophenotype of Adult Acute Lymphocytic Leukemia
ALL, Acute lymphoblastic leukemia; cALL, common acute lymphoblastic leukemia; cy, cytoplasmic; IgH,immunoglobulin heavy chain;
Igl, immunoglobulin light chain; TdT, terminal deoxynucleotidyl transferase.
∗Usually no surface light chain (L) expression.
From Faderl S et al: Cancer 98:1337-1354, 2003.
The T-cell lineage ALL (precursor T-cell ALL) is distinguished by T-cell–associated markers.9,10 Cytoplasmic CD3 is the most common T-cell lineage specific marker, but CD7, CD2, and CD5 are frequently used. In addition to lymphoid markers, T-cell receptor (TCR) gene rearrangements are the most common genetic alteration in T-cell ALL. No specific cytogenetic abnormality, however, has been linked to the subtype of T-cell ALL. ALL blast cells also can express myeloid markers in 15% to 50% of adults and 5% to 35% of children.
Precursor B-cell ALL is strongly associated with aneuploidy of various types, ranging from hypodiploid to hyperdiploid with more than 50 chromosomes.6,9,10 Individuals with hyperdiploid ALL usually have a better prognosis than those with fewer than 46 chromosomes. Precursor T-cell ALL generally have fewer cytogenetic abnormalities, and the majority involve deletions. Genetic translocations between the MYC locus on chromosome 8 and one of the loci for the Ig heavy or light-chain genes (14q32, 2p12, and 22q11) are characteristic (also see Chapters 7 and 11). Several other translocations are commonly observed in ALL, including the Philadelphia chromosome and translocations involving the ETV6 (formerly TEL) and MLL genes (Figure 27-5).6 Philadelphia chromosome–positive ALL carries the worst prognosis of all types of ALL and is found in 25% to 30% of adult ALL cases but less than 5% of childhood ALL cases. A translocation between chromosomes 12 and 21 (t[12;21]) results in fusion of the ETV6 oncogene from chromosome 12 with the AML1 (acute myeloid leukemia 1) gene on chromosome 21 to produce a fusion protein, ETV6-AML1. AML1 is a transcription factor for several genes important in hematopoiesis (e.g., IL-3, granulocyte-macrophage colony-stimulating factor [GM-CSF], CSF1 receptor).6 The t(12;21) translocation occurs in 25% to 30% of childhood pre–B-cell ALL cases but in only 2% of adult ALL cases. This translocation significantly affects the prognosis of childhood ALL; children younger than 10 years with pre–B-cell ALL and the ETV6-AML1 translocation have a 5-year cure rate of 90%, compared with 60% to 65% in those without the translocation.
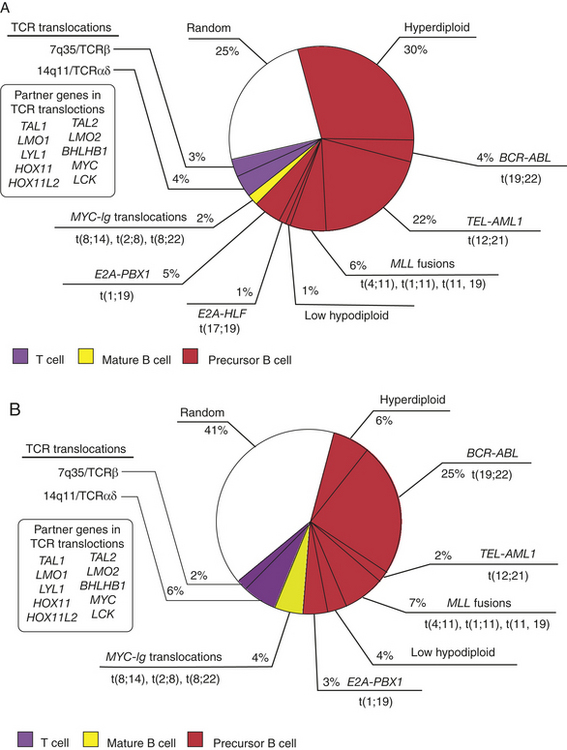
Figure 27-5 Frequency of the major chromosomal translocations in (A) pediatric and (B) adult acute lymphocytic leukemia (ALL). The genes affected by chromosomal translocation are shown in boldface type. TCR translocations in T-ALL can activate a number of different proto-oncogenes as shown in the insert, including TAL1, LMO1/2, TLX1, TLX3, and MYC. (Modified From Hoffman R, et al: Hematology: basic principles and practice, ed 5, Philadelphia, 2009, Churchill Livingstone.)
Translocations of the MLL gene on chromosome 11 occur in about 10% of individuals with ALL and in 70% of infants with AML or ALL.6 Infants and adults with this translocation develop a very aggressive form of leukemia with a very poor prognosis and frequent treatment failure, although children with this abnormality have better outcomes. The most common translocations involving MLL are t(4;11) and t(11;19). The t(4;11) translocation results in a fusion of MLL with the AFF1 (ALL1 fused gene from chromosome 4) gene, and the t(11;19) translocation fuses MLL with the MLLT1(formerly ENL) gene.
Specific causes of ALL are unknown, but multiple factors may contribute to its development.9,10 Risk factors for childhood ALL include prenatal exposure to x-rays and postnatal exposure to high-dose radiation. Individuals with Down syndrome have an increased risk for developing ALL and AML. Increased risk for ALL is also seen in individuals with other genetic conditions, including neurofibromatosis, Shwachman syndrome, Bloom syndrome, and ataxia telangiectasis (see Chapter 8). A unique characteristic of ALL, unlike other forms, is that ALL develops at different rates in different locations. Individuals in developed countries and in higher socioeconomic categories have an increased incidence of ALL. Prevention is almost impossible because there are no known causes.
AML is the most common adult leukemia; the mean age of diagnosis is 67 years of age. It results from an abnormal proliferation of myeloid precursor cells, decreased rate of apoptosis, and an arrest in cellular differentiation.11 Therefore, the bone marrow and peripheral blood are characterized by leukocytosis and a predominance of blast cells. As the immature blasts increase, they replace normal myelocytic cells, megakaryocytes, and erythrocytes. This displacement eventually leads to complications of bleeding, anemia, and infection. AML increases with age, peaking in the sixth decade of life. Certain risk factors have been identified as possible causes, including exposure to radiation, benzene, and chemotherapy. Hereditary conditions, such as Down syndrome, Fanconi aplastic anemia, Bloom syndrome, ataxia telangiectasis, trisomy 13 (Patau syndrome), Wiskott-Aldrich syndrome, and congenital X-linked agammaglobulinemia, are known to be associated with a higher risk for AML (see Table 27-2). AML subtypes are classified based on the stage of development myeloblasts have reached at the time of diagnosis. These subtypes are included in Box 27-1.
More than 150 structural chromosomal abnormalities and several duplications or deletions within genes have been identified in AML.6 The most common abnormalities are balanced translocations or inversions that disrupt genes critical to hematopoiesis of myeloid cells. The most common translocation is between chromosomes 8 and 21 in which the RUNX1T1(formerely ETO) (encodes a transcription factor) gene on chromosome 8 is fused with the AML1 gene on chromosome 21 resulting in an AML1-RUNX1T1 fusion gene and a fusion gene product, AML1-RUNX1T1. Production of AML1-RUNX1T1 disrupts the normal hematopoiesis process for myeloid cells and directly leads to the AML malignant phenotype.
Many kinds of mutations have been found in AML; however, a mutation in the receptor tyrosine kinase FLT3 occurs in about one third of AML persons. FLT3 conveys a proliferation signal normally expressed early in the development of bone marrow stem cells, but mutated FLT3 remains active and promotes blast cell proliferation. Several FLT3 inhibitors are in various stages of clinical development. Another mutation in receptor tyrosine kinases is c-KIT, which also provides a proliferative and/or survival signal to progenitor cells. Together these mutations result in proliferation but not differentiation.
CLINICAL MANIFESTATIONS The clinical manifestations of all the varieties of acute leukemia are generally similar. (Mechanisms associated with common manifestations are summarized in Table 27-4.) Signs and symptoms related to bone marrow depression include fatigue caused by anemia, bleeding resulting from thrombocytopenia (reduced numbers of circulating platelets), and fever caused by infection. Sites of infection include the oral cavity, throat, respiratory tract, lower colon, urinary tract, and skin. Common organisms include the gram-negative bacilli Escherichia coli, Pseudomonas aeruginosa, and Klebsiella pneumoniae. Fever is an early sign, often accompanied by chills. Bleeding can occur in skin, gums, mucous membranes, and gastrointestinal and genitourinary tracts. Visible signs of bleeding include petechiae and ecchymosis, as well as discoloration of the skin, gingival bleeding, hematuria, and midcycle or heavy menstrual bleeding.
Table 27-4
Clinical Manifestations and Related Pathophysiology in Leukemia

CLL, Chronic lymphocytic leukemia; CML, chronic myelogenous leukemia; TNF, tumor necrosis factor.
Anorexia can occur in all varieties of acute leukemia and is associated with weight loss, diminished sensitivity to sour and sweet tastes, wasting away of muscle, and difficulty in swallowing. Liver, spleen, and lymph node enlargement is more common in ALL than in AML (Figure 27-6). Splenomegaly and hepatomegaly usually occur together. The leukemic individual often experiences abdominal pain and tenderness and breast tenderness. Pain in the bones and joints is thought to result from leukemia infiltration with secondary stretching of the periosteum.
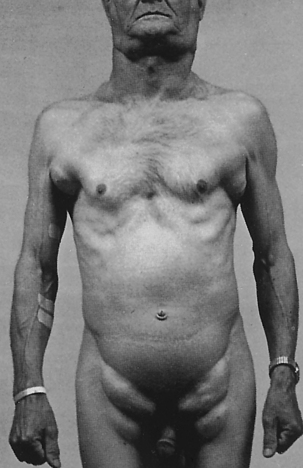
Figure 27-6 Lymphadenopathy. Individual with lymphocyte leukemia with extreme but symmetric lymphadenopathy. (Courtesy Dr. A.R. Kagan, Los Angeles. From del Regato JA, Spjut HJ, Cox JD: Ackerman and del Regato’s cancer, ed 2, St Louis, 1985, Mosby.)
Central nervous system (CNS) involvement is common and may be caused by either leukemic infiltration or cerebral bleeding. Headache, vomiting, papilledema, facial palsy, blurred vision, auditory disturbances, and meningeal irritation can occur if leukemic cells infiltrate the cerebral or spinal meninges. CNS involvement at the time of diagnosis is rare, and less than 5% of children and less than 10% of adults are affected. Without CNS prophylaxis, approximately one third of individuals will develop CNS complications. Interventions associated with CNS prophylaxis include cranial irradiation, chemotherapy, and high doses of systemic chemotherapy. Specific treatment modalities or combinations of treatment vary and are determined by age and risk status.
EVALUATION AND TREATMENT Leukemia is often confused with other conditions, making early detection difficult. Persistent symptoms need intensive medical investigation. The diagnosis is made through examination of blood cells and bone marrow. A stained peripheral blood smear will exhibit low red blood cell and platelet counts along with the presence of leukemic blast cells (Figure 27-7). Examination of bone marrow demonstrates hypercellularity with 60% to 100% blast cells, an occasional normal myeloid, and erythroid precursors and rare to no megakaryocytes.
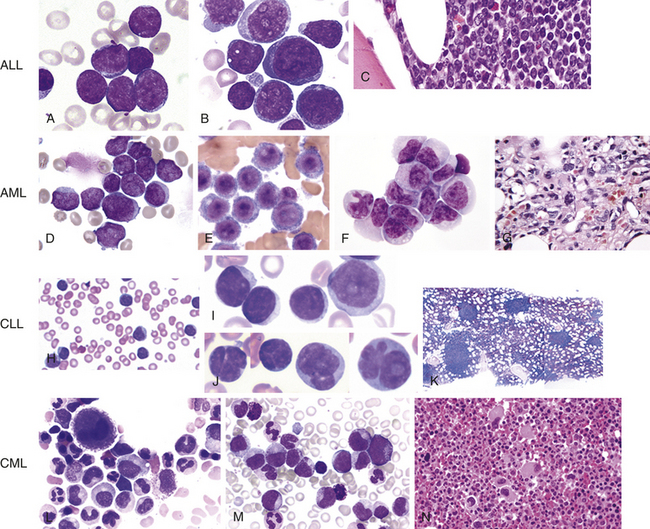
Figure 27-7 Morphologic aspects of leukemia cells. Acute lymphoblastic leukemia (ALL) (A-C). A, Typical uniform lymphoblasts with intermediate-sized nuclei, fine but “smudgy” chromatin, absence of nucleoli, and scant cytoplasm. B, Lymphoblasts with more cytologic variation, including variability in size, number of nucleoli, and amount of cytoplasm. C, Histologic features of ALL in bone core biopsy. Acute myeloid leukemia (AML) (D-G)D, Acute myeloblastic leukemia with minimal or no maturation. The cells are myeloblasts with dispersed chromatin and variable amounts of agranular cytoplasm. Some display medium-sized, poorly defined nucleoli. E, Acute monoblastic leukemia; characteristic monoblasts with round nuclei and delicate chromatin and prominent nucleoli. Cytoplasm is abundant. F, Acute monocytic leukemia with most of the cells in this field being promonocytes. Monoblasts and an abnormal monocyte also are present. G, Marrow biopsy of acute megakaryoblastic leukemia containing large and small blasts and atypical megakaryocytes. Chronic lymphocytic leukemia (CLL) (H-K)H, Peripheral blood smear typically shows lymphocytosis. Cytologic features of CLL cells differ. I, Classic cells have a small nucleus with a “soccer ball” chromatin pattern. J, Some cases have increased large cells, or prolymphocytes, with more open chromatin and prominent “punched-out” nucleoli (prolymphocyte, right side). K, The bone marrow can show nodular infiltrates of CLL cells. Chronic myelogenous leukemia (CML) (L-N)L, Peripheral smear shows marked leukocytosis due to a granulocytic proliferation of all stages with particularly increased myelocytes and absolute basophilia. M, Bone core biopsy illustrates markedly hypercellular marrow due to granulocytic proliferation and increased small hypolobated megakaryocytes. N, Bone marrow aspirate shows granulocytic proliferation and small, “dwarf” megakaryocyte. (A-C, H-N from Hoffman R, et al: Hematology: basic principles and practice, ed 5, Philadelphia, 2009, Churchill Livingstone. D-G from Abeloff M, et al: Abeloff’s clinical oncology, ed 4, Philadelphia, 2008, Churchill Livingstone.)
Chemotherapy, used in varying combinations, is the treatment of choice for leukemia.9,10,12 Supportive measures include blood transfusions, antibiotics, antifungals, and antivirals. Allopurinol is used for preventing production of uric acid (which is elevated from cellular death because of treatment). Stem cell transplantation is now considered standard therapy for selected individuals with leukemia.
Bone marrow transplantation as a treatment has been increasing during the past two decades. Two controversial treatments are immunotherapy agents that induce differentiation of immature granulocytes (i.e., cis-retinoic acid) and marrow transplants. Although there has not been a marked improvement in response or survival of AML, dramatic improvements in survival and response of people with ALL have occurred.
The 5-year survival rate for those with leukemia is 38%, largely because of poor survival rates of individuals with certain types of leukemia (e.g., acute myelogenous). Since the 1970s, 5-year survival rates for those with ALL have increased from 38% to 65% for adults and from 53% to 85% for children. Factors influencing increased survival rate include the use of combined and multimodality treatment methods, improved supportive services such as blood banking and nutritional support, and antimicrobial treatment. The presence of the Philadelphia chromosome (observed in about 5% of children with ALL, in 30% of adults with ALL, and occasionally in AML) is a poor prognostic indicator.
Stimulation of blood cell growth and development with hematopoietic drugs has increased neutrophil recovery during chemotherapy and bone marrow transplant. Blood granulocyte numbers (e.g., eosinophils, neutrophils, basophils or mast cells) are normally in the range of 4000 to 6000 cells/μL, and susceptibility to infection develops below 1000 cells/μL. During a natural response to a bacterial infection, granulocytes usually rise in number to 10,000 to 20,000 cells/μL. Leukemia itself as well as the chemotherapeutic agents used to treat the disease can result in dramatic decreases in circulating granulocytes. The administration of colony-stimulating factors (CSFs) can raise white cell numbers and afford protection from infections.
Chronic Leukemias
The two main types of chronic leukemia are (1) chronic myelogenous leukemia (CML) and (2) chronic lymphocytic leukemia (CLL) (see Table 27-2). Several forms of CML can occur, depending on the lineage of the malignant cells (e.g., chronic neutrophilic leukemia [CNL], chronic eosinophilic leukemia [CEL]). Unlike cells in acute leukemia, chronic leukemic cells are well differentiated and can be readily identified. Individuals with chronic leukemia have a longer life expectancy, usually extending several years from the time of diagnosis.
The chronic leukemias account for the majority of cases in adults, accounting for approximately 30% of leukemias in the Western world. It is estimated that in 2008 more than 15,000 cases of CLL and 4500 cases of CML will be newly diagnosed in the United States.13,14 The incidences of CLL and CML increase significantly in individuals older than 40 years, with prevalence in the sixth through eighth decades. CML is a group of diseases called myeloproliferative disorders, which also include polycythemia vera, primary thrombocytosis, and idiopathic myelofibrosis (invasion of bone marrow by fibrous tissue).
PATHOPHYSIOLOGY CLL involves malignant transformation and progressive accumulation of monoclonal B lymphocytes; rarely (less than 5%) are CLL malignancies of T-cell origin. The characteristic immunophenotype is expression of CD5, CD19, and CD23 molecules and low amounts of surface membrane Ig and CD20 molecules.15 CD5 is a signal transduction molecule linked to the B-cell receptor (BCR), CD19 is a low-affinity antigen receptor expressed on maturing B cells, but is lost in plasma cells, and CD23 is a low-affinity receptor for the Fc portion of IgE. Thus CLL is derived from transformation of a partially mature B cell that has not yet encountered antigen. The gene for the variable region of the antibody heavy chain (IGHV) is frequently mutated (30% to 40% of persons). (See Chapter 7 concerning immunoglobulin heavy-chain structure.) Clients with a mutated IGHV tend to have a more benign condition with a more slowly developing and less malignant disease.
The etiology of CLL is unknown. A familial tendency suggests a genetic linkage; first-degree relatives have a three times greater risk of developing the disease. It is rare in individuals less than 45 years of age, and when diagnosed, 95% of individuals are older than age 50. Genetic anomalies occur in approximately 90% of cases, frequently as deletions, although none has been linked to the etiology of CLL.
CLL cells that accumulate in the marrow do not interfere with normal blood cell production to the extent found in acute leukemias. This is a significant feature explaining the reduced severity in the beginning stage of disease. Accumulation of malignant B cells is the result of cell cycle arrest in the G0/G1 phase. CLL cells tend to express increased levels of proapoptotic proteins (e.g., BCL2) and suppress antiapoptotic proteins (e.g., BCL2L.1), which reduces their sensitivity to apoptosis. Because the major pathophysiologic deficit in CLL is the failure of B cells to mature into plasma cells that synthesize immunoglobulin, this often results in hypogammaglobulinemia (60% of clients).
CML is a member of the family of myeloproliferative disorder that also includes polycythemia vera (see Chapter 26), essential thrombocythemia, chronic idiopathic myelofibrosis (invasion of bone marrow by fibrous tissue), chronic neutrophilic leukemia, and chronic eosinophilic leukemia. CML is clonal and thought to arise from a hematopoietic stem cell. The cells observed in CML are heterogeneous in differentiation, depending on the stage of the disease.16 During the chronic phase the predominant cell is a long-lasting hematopoietic stem cell. A leukemic granulocyte-monocyte progenitor cell is seen. The Philadelphia chromosome is present in more than 95% of CML, and the presence of the BCR-ABL1 protein is responsible for initiation of CML. In advanced disease, the accumulation of additional mutations leads to the more aggressive leukemic phenotype.
CLINICAL MANIFESTATIONS Chronic leukemia advances slowly and insidiously. Approximately 70% of individuals with CLL are asymptomatic at the time of diagnosis. When symptoms do appear, the most common finding is lymphadenopathy. The most significant effect of CLL is suppression of humoral immunity and increased infection with encapsulated bacteria. Frequently the level of neutrophils is depressed, which adds to the risk of infection. Invasion of most organ cells is uncommon but infiltration does occur in lymph nodes, liver, spleen, and salivary glands. CNS involvement is rare. Approximately 10% of individuals develop a more aggressive malignancy, usually a diffuse large B-cell lymphoma. In these individuals, extreme fatigue, weight loss, night sweats, low-grade fever, and elevated levels of the enzyme lactic dehydrogenase, hypercalcemia, anemia, and thrombocytopenia are common.
Individuals with CML may progress through three phases of the disease; a chronic phase lasting 2 to 5 years during which symptoms may not be apparent, an accelerated phase of 6 to 18 months during which the primary symptoms develop, and a terminal blast phase (“blast crisis”) with a survival of only 3 to 6 months. The accelerated phase is characterized by excessive proliferation and accumulation of malignant cells. Splenomegaly is the most common finding, which is prominent and painful, but lymphadenopathy generally is not present. Liver enlargement also occurs, but liver function is rarely altered. Hyperuricemia is common and produces gouty arthritis. Infections, fever, and weight loss also are seen often. The terminal blast phase is characterized by rapid and progressive leukocytosis with an increase in basophils. In the later stages of the terminal phase, which then resembles AML, blast cells or promyelocytes predominate, and the individual experiences a blast crisis.
The acute effects of CML resemble those of acute leukemia but with more prominent and painful splenomegaly. Liver function rarely is altered despite enlargement, and lymphadenopathy generally is found only in the acute phase of the disease. Hyperuricemia invariably is present and produces gouty arthritis. Infections, fever, and weight loss are common findings in clients with CML.
EVALUATION AND TREATMENT Diagnosis of chronic leukemia depends on laboratory analyses of peripheral blood and bone marrow. Diagnosis of CLL is based on detection of a monoclonal B-cell lymphocytosis in the blood. The cells must have the immunophenotype characteristic of CLL (CD5+, CD19+, CD20 [weak], CD23+), at levels in excess of 5000 cells/μL, over a sustained period of time (usually 4 weeks). Bone marrow may contain more than 30% lymphocytes and be normocellular or hypercellular.
Treatment is frequently based on prognostic indicators. Typically, individuals with CLL survive 10 years or more. However, those with certain risk markers have a more aggressive disease that shortens survival to less than 3 years. Markers of high risk include anemia, thrombocytopenia, and no mutations in the IGHV gene. Mutations in IGHV correlate very closely with levels of intracellular ZAP-70, detection of which may be substituted for tests of IGHV mutation. ZAP-70 is a tyrosine kinase that is linked to the T-cell receptor (see Chapter 7). It is not normally detected in CLL cells with mutated IGHV, but is easily detectable by immunohistology in cells with an unmutated IGHV.
Chlorambucil, administered with or without corticosteroids, on a daily or intermittent schedule is the most common treatment for individuals with the most aggressive disease. Relief of symptoms is often achieved, but there is no substantial effect on survival. Combination therapy (CHOP) that includes cyclophosphamide, hydroxydaunomycin (Adriamycin), vincristine (Oncovin), and prednisone has an improved response rate but still does not demonstrate improved survival. Fludarabine, a purine analog, has a higher response rate and disease-free intervals, although survival is not affected. Promising results also have been obtained with the use of monoclonal antibodies (rituximab and alemtuzumab). Stem cell (autogenic and allogeneic) transplant also is being investigated as treatment; however, the advanced age at which individuals contract CLL makes its use less desirable.
Present treatment modalities for CML do not cure the disease, prevent blastic transformation, or prolong the average survival time. Standard treatment consists of combined chemotherapy, biologic response modifiers, and allogeneic stem cell transplant. Although transplantation is potentially curative, its use is limited by donor availability and high toxicity in older adults thus limiting use to those older than 65 years. Allogeneic bone marrow transplantation had increased survival time significantly (20% to 30%) when used after high-dose radiation and chemotherapy and with concurrent treatment with interferon. Traditional chemotherapy agents used are hydroxyurea and busulfan. The development and introduction of the tyrosine kinase inhibitor imatinib mesylate (Gleevec) as a treatment modality have changed current management of CML. Imatinib mesylate is highly specific for CML and suppression of BCR-ABL kinase activity. Suppression of hematologic symptoms occurs in 97% of treated individuals, and the use of imatinib mesylate has become the standard of care for CML. A small percentage of clients develop additional mutations in BCR-ABL that confer resistance to imatinib mesylate.17 Several new tyrosine kinase inhibitors are under investigation as treatments for CML.
ALTERATIONS OF LYMPHOID FUNCTION
Lymphadenopathy is characterized by enlarged lymph nodes. Lymph node enlargement is caused by an increase in size and number of its germinal centers caused by proliferation of lymphocytes and monocytes or invasion by malignant cells. Normally, lymph nodes are not palpable or are barely palpable. Enlarged lymph nodes are characterized by being palpable and often also may be tender or painful to touch, although not in all situations (see Figure 27-6).
Localized lymphadenopathy usually indicates drainage of an area associated with an inflammatory process or infection (reactive lymph nodes). Generalized lymphadenopathy is generally a result of malignant or nonmalignant disease, particularly in adults. Palpable nodes, however, do not always indicate serious disease and may indicate only a reaction to minor trauma or infection of a specific structure. The location and size of the enlarged nodes are important factors in diagnosing the cause of the lymphadenopathy, as are the individual’s age, gender, and geographic location. Generalized lymphadenopathy occurs with non-Hodgkin lymphomas, chronic lymphocytic leukemia, histiocytosis, and disorders that produce lymphocytosis. In general, lymphadenopathy results from one of four types of conditions: (1) neoplastic disease, (2) immunologic or inflammatory conditions, (3) endocrine disorders, or (4) lipid storage diseases. Diseases of unknown cause, including reactions to drugs, also may lead to generalized lymphadenopathy.
Malignant Lymphomas
Lymphomas consist of a diverse group of neoplasms that develop from the proliferation of malignant lymphocytes in the lymphoid system. The classification of lymphomas was published by the World Health Organization (WHO) and is derived from the Revised European-American Lymphoma (REAL) classification. This classification is based on the cell type from which the lymphoma probably originated (Box 27-2).18 The groups include Hodgkin lymphoma and two that were previously classified as non-Hodgkin lymphoma (B-cell neoplasms, T-cell and natural killer [NK] cell neoplasms). With the new classification, multiple myeloma, which was previously classified independently, is included as a B-cell lymphoma.
Incidence rates of lymphoma differ with respect to age, gender, geographic location, and socioeconomic class. The estimated number of new cases of lymphoma for 2008 is almost 75,000 individuals (see Table 27-2). It is estimated that more than 20,000 individuals will have died from lymphoma in 2008. Since the early 1970s, the incidence of non-Hodgkin lymphoma has nearly doubled. The exact reason for this increase remains a mystery; however, a modest portion of the increase had been attributed to lymphomas developing in association with immune deficiencies, including AIDS and organ transplants. Conversely, the incidence of Hodgkin lymphoma has declined over the same time period, especially among older adults.
In general, lymphomas are the result of genetic mutations or viral infection. Malignant transformation produces a cell with uncontrolled and excessive growth that accumulates in the lymph nodes and other sites, producing tumor masses. Lymphomas usually start in the lymph nodes or lymphoid tissues of the stomach or intestines.
Hodgkin Lymphoma
Hodgkin lymphoma (HL) is a malignant lymphoma first characterized by Thomas Hodgkin in 1832. It is estimated that more than 8000 individuals will be newly diagnosed with HL in 2008 (see Table 27-2). The incidence of HL is approximately 3.1 per 100,000 men and 2.5 per 100,000 women.19 The median age of diagnosis is age 38. Incidence rates for HL have declined, especially among older adults. The decrease in incidence in older adults is attributed to improved diagnostic accuracy. The incidence is greater in whites than blacks. Denmark, the Netherlands, and the United States have the highest incidence of HL, and Japan and Australia have the lowest incidence. HL peaks at two different ages: early in life in the second and third decades and later in life during the sixth and seventh decades.
PATHOPHYSIOLOGY HL is characterized by its progression from one group of lymph nodes to another, the development of systemic symptoms, and the presence of Reed-Sternberg (RS) cells (Figure 27-8). It is widely accepted that the RS cell represents the malignant transformed lymphocyte. The RS cells are often large and binucleate, with occasional mononuclear variants. The RS cells are necessary for the diagnosis of HL; however, they are not specific to HL. In rare instances, cells resembling them can be found in benign illnesses, as well as in other forms of cancer, including non-Hodgkin lymphomas and solid tissue cancers and in infectious mononucleosis.
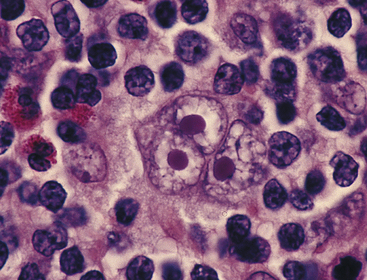
Figure 27-8 Reed-Sternberg cell. A large multinucleated or multilobed cell (center of photograph) with inclusion body–like nucleoli surrounded by a halo of clear nucleoplasm. (From Damjanov I, Linder J, editors: Anderson’s pathology, ed 10, St Louis, 1996, Mosby.)
The triggering mechanism for the malignant transformation of cells remains unknown. Classical HL appears to be derived from a B cell in the germinal center that has not undergone successful immunoglobulin gene rearrangement (see Chapter 7) and would normally be induced to undergo apoptosis. Survival of this cell may be linked to infection with EBV. Laboratory and epidemiologic studies have linked HL with EBV infections and EBV DNA. RNA, and proteins are frequently observed in HL cells. The RS cells secrete and release cytokines (e.g., IL-10, transforming growth factor-beta (TGF-β) that result in the accumulation of inflammatory cells that produces the local and systemic effects. HL is subcategorized into two main types: classical Hodgkin and nodular lymphocyte–predominant Hodgkin. Classical HL is subclassified into four types (Table 27-5) based on the morphology of RS cells, and the characteristics of the inflammatory cell infiltrate in the tumor.

The molecular events causing malignant transformation remain controversial; although RS cells are apparently from B-cell lineage, they express very few B-cell markers and express markers normally not found on B cells. For instance, RS cells do not express immunoglobulin, but do express CD15 (a carbohydrate adhesion molecule found on neutrophils), TARC (a Th2-cell specific chemokine), and T-cell–associated antigens (e.g., β-chain of the T-cell receptor).18 The precise genetic defects leading to development of HL are unknown, although several have been suggested. These generally include defects in immunoglobulin variable region gene rearrangement or defects in other B-cell–specific differentiation genes.
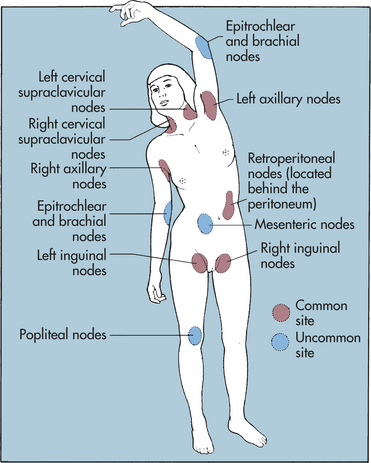
CLINICAL MANIFESTATIONS Many of the characteristic clinical features (Box 27-3) of HL can be explained by the complex action of cytokines and other growth factors that are secreted by the malignant cells. These substances induce infiltration and proliferation of inflammatory cells, resulting in an enlarged, painless lymph node in the neck (often the first sign of HL) (Figure 27-9). The discovery of an asymptomatic mediastinal mass on routine chest x-ray is not uncommon and is often an initial sign of HL. The cervical, axillary, inguinal, and retroperitoneal lymph nodes are commonly affected in HL (Figure 27-10). Local symptoms caused by pressure and obstruction of the lymph nodes are the result of the lymphadenopathy.

Figure 27-9 Hodgkin lymphoma and enlarged cervical lymph node. Typical enlarged cervical lymph node in the neck of a 35-year-old woman with Hodgkin lymphoma. (From del Regato JA, Spjut HJ, Cox JD: Ackerman and del Regato’s cancer, ed 2, St Louis, 1985, Mosby.)
About one third of individuals will have some degree of systemic symptoms.20 Intermittent fever, without other symptoms of infection, drenching night sweats, itchy skin (pruritus), and fatigue are relatively common. These constitutional symptoms accompanied by weight loss are associated with a poor prognosis. The Cotswold staging classification system used for HL is able to establish a correlation between the anatomic extent of the disease and prognosis (Table 27-6). This classification system is based on the individual’s medical history, examination (presence of symptoms and palpable lymph nodes), and other radiologic and hematologic results. Prognostic indicators include clinical stage, histologic type, tumor cell concentration and tumor burden, constitutional symptoms, and age.
Table 27-6
Cotswold Staging Classification System

Data from Lister TA, Crowther D: Staging for Hodgkin’s disease, Semin Oncol 17:696, 1990.
Although HL rarely arises in the lung, mediastinal and hilar node adenopathy can cause secondary involvement of the trachea, bronchi, pleura, or lungs. Retroperitoneal nodes can involve vertebral bodies and nerves, causing displacement of ureters. Spinal cord involvement is more common in the dorsal and lumbar regions than in the cervical region. Although uncommon, skin manifestations include psoriasis and eczematoid lesions, causing itching and scratching.
As a result of direct invasion from mediastinal lymph nodes, pericardial involvement can cause pericardial friction rub, pericardial effusion, and engorgement of the neck veins. The gastrointestinal (GI) tract and urinary tract rarely are involved. Anemia often is found in individuals with HL, accompanied by a low serum iron and iron-binding capacity. Other laboratory findings include elevated sedimentation rate, leukocytosis, and eosinophilia. Leukopenia occurs in advanced states of HL.
Splenic involvement of HL depends on histopathologic type (see Table 27-5). The spleen is involved in 60% of cases of mixed cellularity and lymphocytic depletion types. With lymphocyte predominance and nodular sclerosis types, only 34% of cases reveal splenic involvement.
EVALUATION AND TREATMENT Because of the variability in symptoms, early definitive detection may be difficult. Asymptomatic lymphadenopathy can progress undetected for several years. Careful evaluation, including chest x-rays, lymphangiography, and biopsy, should be carried out for individuals with fever of unknown origin and peripheral lymphadenopathy.20 A lymph node biopsy with scattered RS cells and a cellular infiltrate is highly indicative of HL. The effectiveness of treatment is related to the age of the individual and the extent of the disease. Approximately 75% of individuals diagnosed with HL are cured, largely because of successful treatment with irradiation and chemotherapy (Figure 27-11). More recent treatments include high-dose chemotherapy with bone marrow or stem cell transplant. Monoclonal antibodies also are being developed and nonmyeloablative allogeneic stem cell transplant has been found to help certain individuals even though this treatment is still under development.
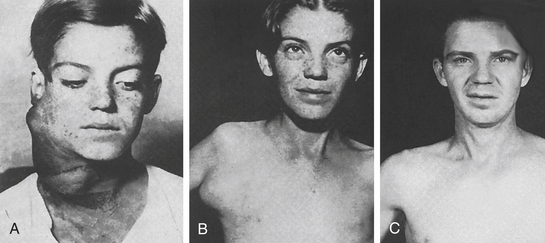
Figure 27-11 Cervical Hodgkin lymphoma. A, Young boy with extensive cervical Hodgkin lymphoma. B, Appearance several years later, when axillary manifestations developed. C, Appearance 23 years after initial treatment with radiation. (From del Regato JA, Spjut HJ, Cox JD: Ackerman and del Regato’s cancer, ed 2, St Louis, 1985, Mosby.)
The 5-year survival rate varies depending on which stage is identified at diagnosis.20 The 5-year survival rate for stage I and II is 90% to 95%, 80% to 85% for stage III, and 75% for stage IV. Those with stage I or II disease are candidates for chemotherapy, combined, or radiation therapy alone. Individuals with stage III or IV disease, bulky disease (more than 10-cm mass or mediastinal disease with a transverse diameter exceeding 33% of the transthoracic diameter), or presence of B symptoms require combined chemotherapy with or without additional radiation treatment. Other factors, if present, have an influence on survival. Poorer survival is related to a high white blood cell count (greater than 15,000) or low hemoglobin (Hb) (less than 10.5); low lymphocyte count (less than 600); and being male. Cure for HL can be achieved in 70% of cases with current therapies.
Non-Hodgkin Lymphoma
The previously used generic classification of non-Hodgkin lymphoma (NHL) has been reclassified in the WHO/REAL scheme into B-cell neoplasms, which includes a variety of lymphomas including myelomas that originate from B cells at various stages of differentiation, and T-cell and NK-cell neoplasms, which includes lymphomas that originate from either T or NK cells. These cancers are differentiated from HL by lack of RS cells and other cellular changes not characteristic of HL.
More than 66,000 cases of NHL and 19,000 deaths are predicted for 2008 (see Table 27-2).21 The median age of diagnosis is 67 years of age. The incidence of NHL has increased from 8 persons per 100,000 in 1973 to 23.5 per 100,000 in 2006. Lymphomas from HIV and EBV have accounted for some of the increase but an actual cause has yet to be determined. Conversely, the mortality rate has risen at a slower rate. It is thought that newer treatment modalities are improving survival rates.
PATHOPHYSIOLOGY NHL is best described as a progressive clonal expansion of B cells, T cells, or NK cells. The genetic lesions affecting proto-oncogenes or tumor-suppressor genes result in cell immortalization and the resultant increase in malignant cells. Oncogenes may be activated by chromosomal translocations or the tumor-suppressor loci may be inactivated by deletion or mutation of chromosomes. Oncogenic viruses also may alter the genome of certain subtypes. The various subtypes of NHL may be identified by specific diagnostic markers related to various cytogenic lesions.
Lymphomas most likely originate from mutations in cellular genes (many of which are environmentally induced) in a single cell that lead to loss of control of proliferation and other aspects of cell growth. The most common type of chromosomal alteration in NHL is translocation, which disrupts the genes encoded at the breakpoints. Risk factors include a family history, exposure to a variety of mutagenic chemicals, irradiation, infection with certain cancer-related viruses (e.g., EBV, human herpesvirus-8, HIV, HTLV-1, hepatitis C), and immune suppression related to organ transplantation. Gastric infection with Helicobacter pylori increases the risk for gastric lymphomas. NHL is a disease of middle age, usually found in individuals more than 50 years old.
B cells account for approximately 85% of NHLs, with T cells and NK cells accounting for the remaining 15%. A very small percent originates from macrophages. NHL tumors are categorized by the level of differentiation, cell of origin, and rate of cellular proliferation. Tumor aggressiveness of many B-cell NHLs may be predicted by the pattern of cell growth and size. Tumors with a characteristic nodular pattern, vaguely resembling lymphoid follicular structures, are generally less aggressive than lymphomas with a diffuse pattern of proliferation. Small lymphocyte lymphomas are less aggressive than large cell lymphomas, which are generally intermediate to high grade in aggressiveness. However, small cells are characteristic of some subtypes of high-grade lymphomas.
CLINICAL MANIFESTATIONS Clinical manifestations of NHL usually start out as localized or generalized lymphadenopathy, similar to HL. The cervical, axillary, inguinal, and femoral chains are the most commonly affected sites. Generally, the swelling is painless and the nodes have enlarged and transformed over a period of months or years. Other sites of involvement are the nasopharynx, GI tract, bone, thyroid, testes, and soft tissue. Some individuals have retroperitoneal and abdominal masses with symptoms of abdominal fullness, back pain, ascites (fluid in the peritoneal cavity), and leg swelling.
Lymphomas are classified as low, intermediate, or high grade. A low-grade lymphoma, which also may be termed indolent, has a slow progression. Individuals with low-grade lymphoma commonly present with a painless, peripheral adenopathy. Spontaneous regression of these nodes may occur, mimicking the presence of an infection. Night sweats with an elevated temperature (more than 38° C [100.4° F]) and weight loss, as well as extranodular involvement, are not commonly present in the early stages but are common in advanced or end stage. Cytopenia, reflective of bone marrow involvement, is often observed. Hepatomegaly is common; however, splenomegaly is present in approximately 40% of individuals. Fatigue and weakness are more prevalent with advanced stages.
Immediate and high-grade lymphomas, which are more progressive, have a more varied clinical presentation. A high-grade lymphoma also may be termed aggressive. Adenopathy is common with more than one third of individuals having extranodal involvement. Common sites are the GI tract, skin, bone marrow, sinuses, genitourinary (GU) tract, thyroid, and CNS. Night sweats, with an increased temperature (more than 38° C [100.4° F]), as well as weight loss (more than 10% from baseline within 6 months) are present in approximately 30% to 40% of individuals. Some individuals have retroperitoneal and abdominal masses with symptoms of abdominal fullness, back pain, ascites (fluid in the peritoneal cavity), and leg swelling. Hepatomegaly and splenomegaly are often present. Differences in clinical features are noted in Table 27-7.
Table 27-7
Clinical Differences Between Non-Hodgkin Lymphoma and Hodgkin Lymphoma
| Characteristic | Non-Hodgkin Lymphoma | Hodgkin Lymphoma |
| Nodal involvement | Multiple peripheral nodes | Localized to single axial group of nodes (i.e., cervical, mediastinal, para-aortic) |
| Mesenteric nodes and Waldeyer ring commonly involved | Mesenteric nodes and Waldeyer ring rarely involved | |
| Spread | Noncontiguous | Orderly spread by contiguity |
| B symptoms∗ | Uncommon | Common |
| Extranodal involvement | Common | Rare |
| Extent of disease | Rarely localized | Often localized |
EVALUATION AND TREATMENT Biopsy is considered the primary means for diagnosis of NHL. Staging of NHL is necessary to identify treatment and make a prognosis. In addition to biopsy, computed tomography (CT) scans of the neck, chest, abdomen, and pelvis, as well as bilateral bone marrow aspirate, are performed. Data from all three procedures is necessary for appropriate staging. A common finding in NHL is noncontiguous lymph node involvement, which is not common in HL. The Ann Arbor staging system is most commonly used to stage NHL (Table 27-8). Treatment for NHL is quite diverse and depends on type (B cell or T cell) of tumor stage, histologic status (low, intermediate, or high grade), symptoms, age, and any comorbidities.

In general, treatment is initiated at the time of diagnosis; however, some low-grade lymphomas are widely disseminated at diagnosis, and because current therapy is not curative, observation without treatment may be the most appropriate choice. A partial remission may be achieved in some cases in which evidence of the disease remains but it does not progress.
Success of treatment is dependent on several parameters, including the type of lymphoma, stage of disease, cell type, involvement of organs outside the lymph nodes, age of the person, and the severity of the body’s reaction to the disease (e.g., fever, night sweats, weight loss).20,22 Treatment with chemotherapy alone may be adequate in many cases, although radiation therapy is frequently included. Low-dose chemotherapy has been followed by autologous stem cell transplantation in some NHLs or for recurrent disease. Treatment of B-cell lymphomas with rituximab has proven effective. Rituximab is a commercial monoclonal antibody against antigen CD20, which is expressed on the surface of all B cells, including those that are malignant. Administration of rituximab depletes most B cells and allows the replenishment of normal B cells from the lymphoid stem cell pool. It has also proven useful in a variety of autoimmune diseases, including immune thrombocytopenia purpura, autoimmune anemias, systemic lupus erythematosus, and rheumatoid arthritis.
Individuals with NHL can survive for extended periods.20,22 Survival with nodular lymphoma ranges up to 15 years, but those with diffuse disease generally do not survive as long. Overall, the survival rates of NHL are less than for Hodgkin lymphoma. For NHL, the survival rates are 1 year, 77%; 5 years, 59%; and 10 years, 42%. Many investigators believe that more aggressive treatment increases the cure rate. High-grade NHL is seen with increasing frequency in persons with AIDS and has an extremely poor prognosis.
Burkitt Lymphoma
Burkitt lymphoma is a B-cell tumor with unique clinical and epidemiologic features that accounts for 30% of childhood lymphomas worldwide. It occurs in children from east-central Africa and New Guinea and is characterized by a rapidly growing tumor primarily in the jaw and facial bones (Figure 27-12). In the United States, Burkitt lymphoma is rare, usually involves the abdomen, and is characterized by extensive bone marrow invasion and replacement. EBV, found in nasopharyngeal secretions, is associated with Burkitt lymphoma in African children.

Figure 27-12 Burkitt lymphoma. Burkitt lymphoma involving the jaw in a young African boy. (Courtesy Dr. JNP Davies, Albany, NY. From del Regato JA, Spjut HJ, Cox JD: Ackerman and del Regato’s cancer, ed 2, St Louis, 1985, Mosby.)
PATHOPHYSIOLOGY EBV is associated with almost all cases (more than 90%) of Burkitt lymphoma. It is suspected that suppression of the immune system by other illnesses (e.g., HIV infection, chronic malaria) increases the individual’s susceptibility to EBV. B cells are particularly sensitive because of specific surface receptors for EBV. As a result, the B cell undergoes chromosomal translocations that result in overexpression of the C-MYC proto-oncogene and loss of control of cell growth (Figure 27-13). The most common translocation (75% of individuals) is between chromosomes 8 (containing the C-MYC gene) and 14 (containing the immunoglobulin heavy-chain genes). Other translocations have been reported between chromosome 8 and chromosomes 2 or 22, which contain genes for immunoglobulin light chains.
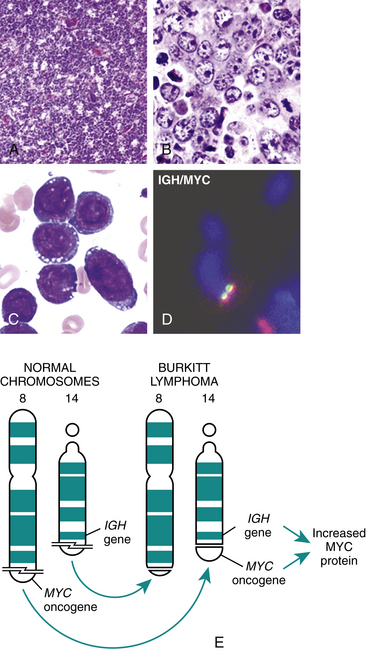
Figure 27-13 Burkitt lymphoma cells. A, A case of Burkitt lymphoma illustrated at low power showing the “starry sky” appearance. This is due to the dense proliferating cells producing the dark sky, and the scattered lighter-staining tingible body macrophages (stars) phagocytizing dying cells. B, Higher magnification illustrating the syncytia of intermediate-sized cells with coarse chromatin and multiple nucleoli. Note the tingible body macrophage with abundant light cytoplasm and ingested debris (center bottom). C, Burkitt cells as seen on a Wright-stained bone marrow aspirate in a person with Burkitt leukemia. Notice deep blue cytoplasm with numerous vacuoles. D, Fluorescence in situ hybridization (FISH) with probes to MYC and IGH illustrate the IGH/MYC fusion. E, The 8, 14 chromosomal translocation and associated oncogenes in Burkitt lymphoma. (A-D, from Hoffman R, et al: Hematology: basic principles and practice, ed 5, Philadelphia, 2009, Churchill Livingstone. D, courtesy of Dr. Yanming Zhang, University of Chicago. E, from Kumar: Robbins and Cotran pathologic basis of disease, ed 7, Philadelphia, 2005, Saunders.)
CLINICAL MANIFESTATIONS In non-African Burkitt lymphoma the most common presentation is abdominal swelling. More advanced disease may involve other organs—eye, ovaries, kidneys, glandular tissue (breast, thyroid, tonsil)—and presents with type B symptoms (night sweats, fever, weight loss).
EVALUATION AND TREATMENT The distribution of tumors and biopsies of enlarged lymph nodes or the bone marrow containing malignant B cells are usually indicative of Burkitt lymphoma. It is one of the most aggressive and quickly growing malignancies. However, the African variety in children has been successfully treated with radiotherapy and cyclophosphamide (60% survival overall; 90% survival with limited disease). The American type is more resistant to treatment.
Lymphoblastic lymphoma
Lymphoblastic lymphoma (LL) is a relatively rare variant of NHL (2% to 4%) but accounts for almost one third of cases of NHL in children and adolescents, with a male predominance. The vast majority of LL (more than 85%) is of T-cell origin, and the remainder arises from B cells. LL is similar to acute lymphoblastic leukemia and may be considered a variant of that disease.
PATHOPHYSIOLOGY The disease arises from a clone of relatively immature T cells that becomes malignant in the thymus. As with most lymphoid tumors, LL is frequently associated with translocations, primarily of the chromosomes that encode for the T-cell receptor (chromosomes 7 and 14). These aberrations result in increased expression of a variety of transcription factors and loss of growth control.
CLINICAL MANIFESTATIONS The first sign of LL is usually a painless lymphadenopathy in the neck. Peripheral lymph nodes in the chest become involved in about 70% of individuals, mostly above the diaphragm. LL is a very aggressive tumor that presents as stage IV in most people. T-cell LL is associated with a unique mediastinal mass (up to 75%) because of the apparent origin of the tumor in the thymus. The mass results in chest pain and may cause compression of bronchi or superior vena cava. The tumor may infiltrate the bone marrow in about half of those affected, and suppression of bone marrow hematopoiesis leads to increased susceptibility to infections. Other organs, including the liver, kidney, spleen, and brain, may also be affected. Many individuals express type B symptoms: fever, night sweats, and significant weight loss.
EVALUATION AND TREATMENT The most common therapeutic approach is combined chemotherapy with multiple drugs. In early disease, the response rate is high with increased survival; the 5-year survival in children is 80% to 90%, and it is 45% to 55% in adults. Although LL is easily treated, there is a high relapse rate: 40% to 60% of adults.
Conditions That Mimic Lymphomas
Certain other clinical conditions mimic the malignant lymphomas. These conditions include TB, syphilis, systemic lupus erythematosus, lung cancer, and bone cancer. An important distinction between lymphomas and other conditions is that lymphomas usually involve localized lymphadenopathy. Infectious precursors of malignant lymphomas are characterized by more generalized lymphadenopathy with systemic signs and symptoms.
Plasma Cell Malignancies
The plasma cell is the end-stage cell of the humoral immune response (see Chapter 7). Immunocompetent B cells presented with antigen and stimulated with cytokines from T helper cells will undergo proliferation and differentiation into antibody-producing plasma cells. Antigen-reactive B cells have undergone rearrangement of immunoglobulin heavy-chain variable region genes (V, D, J) and express surface IgM or IgD, or both. After simulation with antigen, the B cells may not undergo any further genetic rearrangement and develop into plasma cells that secrete IgM or selectively rearrange the immunoglobulin heavy-chain genes to irreversibly switch to secreting IgG, IgA, or IgE. During this process some cells may undergo malignant transformation, leading to one of several types of plasma cell malignancies (Figure 27-14). The most common and most aggressive plasma cell tumor is multiple myeloma. Other diseases in this classification include precursors to malignant myeloma (smoldering myeloma, monoclonal gammopathy of undetermined significance [MGUS]), solitary plasmacytoma of the bone, and Waldenström macroglobulinemia.23 A common characteristic of these tumors is secretion of complete or partial immunoglobulin molecules.
Figure 27-14 Distribution of monoclonal gammamopathy types. A, Distribution of serum monoclonal proteins in 1311 clients seen at the Mayo Clinic during 2004. B, Diagnoses in 1423 cases of monoclonal gammopathy seen at the Mayo Clinic during 2004. Ig, Immunoglobulin; MGUS, monoclonal gammopathy of undetermined significance; PC, plasmacytoma; SMM, smoldering multiple myeloma; WM, Waldenström’s macroglobulinemia. (From Goldman L, Ausiello D: Cecil medicine, ed 23, Philadelphia, 2008, Saunders.)
Multiple Myeloma
Multiple myeloma (MM) is a clonal plasma cell cancer characterized by the slow proliferation of malignant cells as tumor cell masses in the bone marrow that usually results in destruction of the bone. Most MMs secrete large amounts of monoclonal proteins that resemble intact immunoglobulins. The reported incidence of myeloma has doubled in the past two decades, possibly as a result of more sensitive testing used for diagnosis. The annual incidence rate in the United States is 5.6 per 100,000, with almost 20,000 new cases and almost 11,000 deaths estimated for 2008.24 Multiple myeloma occurs in all races, but the incidence in blacks is about twice that of whites. It rarely occurs before the age of 40 years—peak age of incidence is about 70 years. It is slightly more common in men than women. Neoplastic cells of multiple myeloma reside in the bone marrow and are usually not found in the peripheral blood. Occasionally it may spread to other tissues, especially in very advanced disease.
PATHOPHYSIOLOGY Many myelomas are aneuploidy, with chromosomal numbers ranging from 44 chromosomes to near tetraploid. Chromosomal translocations (break points) are responsible for development of myeloma in most individuals. The primary translocation involves the immunoglobulin heavy chain on chromosome 14 that relocates to sites of containing genes that cell cycle (cyclins) on chromosomes 11(q13), 12(p13), and 6(p21); oncogenes on chromosomes 16(q23), 8(q24), and 20; and fibroblast growth factor receptor on chromosome 4(p16).6 A progression of further secondary genetic alterations causes progression to an aggressive MM (Figure 27-15). The molecular pathogenesis of multiple myeloma also involves proto-oncogene mutations and, more rarely, inactivation of tumor-suppressor genes. The precise timing and reason for the genetic alteration and accumulation are unknown, but probably occur initially late in B-cell development after exposure to antigen.
Figure 27-15 Myeloma cell proliferation and disease progression. Development of the malignant myeloma results from multiple genetic changes; initially a translocation involving the immunoglobulin heavy-chain genes on chromosome 14 or a deletion in chromosome 13. The intermediate phenotype is frequently genetically unstable, leading to further mutations that result in a myeloma. Interactions between myeloma cells and extracellular matrix proteins further increase adhesion molecule expression, antiapoptotic pathways, angiogenesis, bone resorption, and cytokine secretion. IL-6, Interleukin-6; MGUS, monoclonal gammopathy of undetermined significance; MIP-1α, macrophage inflammatory protein-1α NRAS, neuroblastoma RAS viral (v-ras) oncogene hemolog; OPG, osteoprotegerin; RANKL, receptor activator of nuclear factor-κB ligand; VEGF, vascular endothelial growth factor. (From Abeloff M, et al: Abeloff’s clinical oncology, ed 4, Philadelphia, 2008, Churchill Livingstone.)
Malignant plasma cells arise from one clone of B cells that produce abnormally large amounts of one class of immunoglobulin (usually IgG, occasionally IgA, and rarely IgD or IgE). The malignant transformation may begin early in B-cell development, possibly before encountering antigen in the secondary lymphoid organs. The myeloma cells return to either the bone marrow or other soft tissue sites. Their return is aided by cell adhesion molecules that help them target favorable sites that promote continued expansion and maturation.
Myeloma cells in the bone marrow directly secrete hepatocyte growth factor and parathyroid hormone-related peptide and adhere to stromal cells inducing their production of several cytokines (e.g., IL-6, IL-1, tumor necrosis factor-alpha (TNF-α), IL-11, macrophage inflammatory protein). (Lymphocytes and cytokines are described in Chapter 7.) All of these factors, IL-6 in particular, act as an osteoclast-activating factor and stimulate osteoclasts to reabsorb bone. This process results in bone lesions and hypercalcemia (high calcium levels in the blood) resulting from release of calcium from the breakdown of bone.
The antibody produced by the transformed plasma cell is usually defective, containing truncations, deletions, and other abnormalities, and is frequently referred to as a paraprotein (abnormal protein in the blood). Because of the large number of malignant plasma cells, the abnormal antibody, called the M protein, becomes the most prominent protein in the blood in 80% of myeloma clients (Figure 27-16). Suppression of normal plasma cells by the myeloma results in diminished or absent normal antibodies. The excessive amount of M protein may also contribute to many of the clinical manifestations of the disease. The myeloma may produce free immunoglobulin light chain (Bence Jones protein) that is present in the blood and urine in approximately 80% of clients and contributes to damage of renal tubular cells.
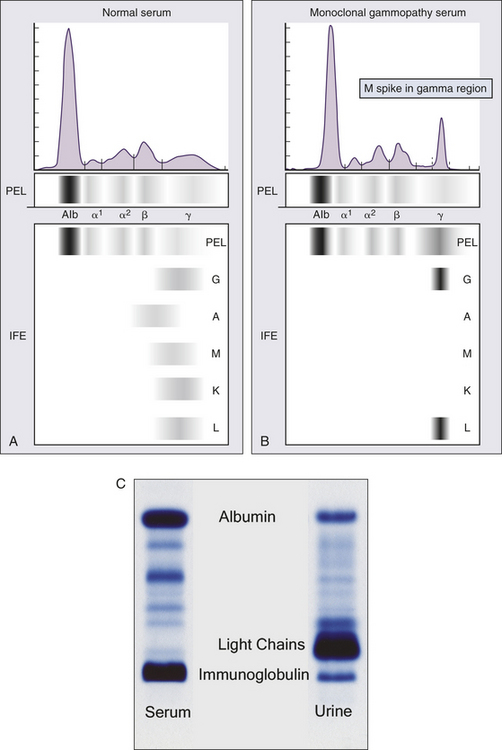
Figure 27-16 M protein. Serum protein electrophoresis (PEL) is used to screen for M proteins in multiple myeloma. A, In normal serum the proteins separate into several regions between albumin (Alb) and a broad band in the gamma (γ) region, where most antibodies (gamma globulins) are found. Immunofixation (IFE) can identify the location of IgG (G), IgA (A), IgM (M), and kappa (K) and lambda (L) light chains. B, Serum from an individual with multiple myeloma contains a sharp M protein (M spike). The M protein is monoclonal and contains only one heavy chain and one light chain. In this instance the IFE identifies the M protein as an IgG containing a lambda light chain. C, Serum and urine protein electrophoretic patterns in a client with multiple myeloma. Serum demonstrates an M protein (immunoglobulin) in the gamma region, and the urine has a large amount of the smaller-sized light chains with only a small amount of the intact immunoglobulin. (A and B, from Abeloff M, et al: Abeloff’s clinical oncology, ed 4, Philadelphia, 2008, Churchill Livingstone. C, From McPherson R, Pincus M: Henry’s clinical diagnosis and management by laboratory methods, ed 21, Edinburgh, 2006, Saunders.)
CLINICAL MANIFESTATIONS The common presentation of MM is characterized by elevated levels of calcium in the blood (hypercalcemia) (13% of persons), renal failure (19%), anemia (72% of persons), and bone lesions (80% of persons).25 The hypercalcemia and bone lesions result from infiltration of the bone by malignant plasma cells and stimulation of osteoclasts to reabsorb bone. This process results in the release of calcium (hypercalcemia) and development of “lytic lesions” (round, “punched out” regions of bone) (Figure 27-17). Destruction of bone tissue causes pain, the most common presenting symptom, and pathologic fractures. The pain may be felt in a single bone of the entire skeleton, and the bones most commonly involved, in decreasing order of frequency, are the vertebrae, ribs, skull, pelvis, femur, clavicle, and scapula. Spinal cord compression, because of the weakened vertebrae, occurs in about 10% of individuals. The pain is initially aching, intermittent, and aggravated by weight-bearing. As the disease progresses, pain becomes severe and prolonged. It is common for the individual with myeloma to be treated for a slipped disk or arthritis before the correct diagnosis of myeloma is established. The individual may complain of weakness, fatigue, weight loss, and anorexia in addition to pain.
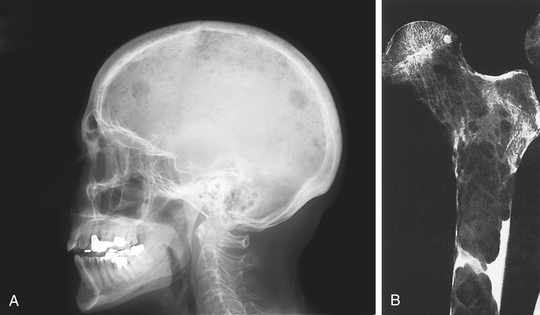
Figure 27-17 Osteolytic lesions in individuals with multiple myeloma. A, Lesions in the skull on radiograph in a client with myeloma. B, Roentgenogram of femur showing extensive bone destruction caused by tumor. Note absence of reactive bone formation. (A from Abeloff M, et al: Abeloff’s clinical oncology, ed 4, Philadelphia, 2008, Churchill Livingstone. B from Kissane JM, editor: Anderson’s pathology, ed 9, St Louis, 1990, Mosby.)
Proteinuria is observed in 90% of individuals. Renal failure may be either acute or chronic and is usually secondary to the hypercalcemia. Bence Jones protein may lead to damage of the proximal tubules. Anemia is usually normocytic and normochromic and results from inhibited erythropoiesis caused by tumor cell infiltration of the bone marrow.
The high concentration of paraprotein in the blood may lead to hyperviscosity syndrome. The increased viscosity interferes with blood circulation to various sites (brain, kidneys, extremities). Hyperviscosity syndrome is observed in up to 20% of individuals with MM. Additional neurologic symptoms (e.g., confusion, headaches, blurred vision) may occur secondary to hypercalcemia or hyperviscosity.
Suppression of the humoral (antibody-mediated) immune response results in repeated infections, primarily pneumonias and pyelonephritis. The most commonly involved organisms are encapsulated bacteria that are particularly sensitive to the effects of antibody; pneumonia caused by S. pneumoniae, Staphylococcus aureus, or K. pneumoniae or pyelonephritis caused by E. coli or other gram-negative organisms. Cell-mediated (T cell) function is relatively normal. Overwhelming infection is the leading cause of death from MM.
MM is a progressive disorder and is often preceded by a condition known as monoclonal gammopathy of undetermined significance (MGUS). MGUS is diagnosed by the presence of an M protein in the blood or urine without additional evidence of MM.25 MGUS is present in approximately 1% of the general population and in 3% of individuals older than 70 years. Although MGUS is considered nonpathologic and requires no treatment, about 16% of individuals with MGUS progress to malignant plasma cell disorders. Progression of MM following MGUS advances to asymptomatic MM and finally symptomatic MM. Asymptomatic MM also may be referred to as smoldering myeloma and indolent myeloma.25 Smoldering myeloma is usually characterized by the presence of an M protein and clonal bone marrow plasma cells, but with no indication of end-organ damage.
Most cases of symptomatic plasma cell tumors are multiple myeloma (about 94%). The remaining 6% is divided equally between solitary plasmacytomas and extramedullary plasmacytomas.23 Solitary plasmacytoma is characterized by a solitary tumor of malignant plasma cells that may result in a single bone lesion or may be in the tissues (extramedullary plasmacytoma).25 Extramedullary plasmacytoma can be found in a variety of soft tissues, but commonly in those of the upper respiratory tract (e.g., tonsils, nasopharynx, sinuses). Additionally, MM is staged to help determine prognosis and appropriate treatment (Table 27-9).
Table 27-9
International Staging System for Multiple Myeloma
| Stage | Criteria |
| I | Serum β2-microglobulin <3.5 mg/L |
| Serum albumin ≥3.5 g/dl | |
| II | Not stage I or III∗ |
| III | Serum β2-microglobulin ≥5.5 mg/L |
∗There are two categories for stage II: serum β2-microglobulin <3.5 mg/L but serum albumin <3.5 g/dl; or serum β2-microglobulin 3.5 to <5.5 mg/L irrespective of the serum albumin level.
From Greipp PR et al: J Clin Oncology 23(15):3412-3420, 2005.
EVALUATION AND TREATMENT Diagnosis of MM is made by symptoms, radiographic and laboratory studies, and a bone marrow biopsy. Quantitative measurements of immunoglobulins (IgG, IgM, IgA) are usually performed. Typically, one class of immunoglobulin (the M protein produced by the myeloma cell) is greatly increased, whereas the others are suppressed. Serum electrophoretic analysis reveals increased levels of M protein. Because the M protein is monoclonal, each molecule has the same electric change and migrates at about the same site on electrophoresis, resulting in a highly concentrated protein (M spike). Bence Jones protein is observed in the urine or serum by immunoelectrophoresis or in the serum using enzyme-linked immunosorbent assay (ELISA) assays. Usually individuals with Bence Jones protein also have M protein in their blood. However, variants of MM include individuals in which free light chain only is produced and a rare variant that produces only free heavy chain, and approximately 1% are nonsecretory so that neither an M protein nor Bence Jones protein is produced. The amount of M protein in the blood may be used as a measure of the extent of the disease or as a measure of response to therapy. The serum level of another protein, free β2-microglobulin, is a useful indicator of prognosis or effectiveness of therapy.
A bone marrow biopsy is performed to confirm the presence of myeloma cells in the marrow (Figure 27-18). Radiographic studies include x-ray, CT scans, and magnetic resonance imaging (MRI) to document the presence of bone lesions and areas of destruction. Diagnosis is based on findings and the degree of involvement. The individual must have all three major criteria (Box 27-4).
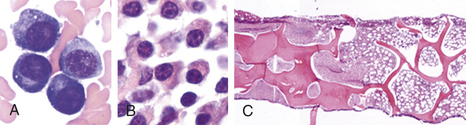
Figure 27-18 Myeloma cells. The typical myeloma type has fairly mature-appearing plasma cells with eccentric nuclei on bone marrow aspirate (A) and biopsy (B). C, Osteosclerotic myeloma in which the left side of the photograph shows bone sclerosis and the marrow cavity replaced by myeloma. (From Hoffman R, et al: Hematology: basic principles and practice, ed 5, Philadelphia, 2009, Churchill Livingstone.)
Combinations of chemotherapy, radiation therapy, and plasmapheresis (exchange), and marrow transplantation have been the standards of treatment.23 Conventional combinations of chemotherapeutic agents have included melphalan and prednisone (MP); MP with vincristine, carmustine, and cyclophosphamide; vincristine, doxorubicin, and dexamethasone; and thalidomide and dexamethasone. The drug thalidomide disrupts the stromal marrow–MM cell interaction by modulating cell surface adhesion molecules and inhibiting angiogenesis. In addition, it increases apoptosis and G1 growth arrest (i.e., the cell cycle gap 1; see Chapter 1) of MM cells.
Dose intensification improves the outcomes in younger clients; however, long-term remissions are obtained in a minority of clients. Thus intensive research measuring the effect of novel new therapies is the objective of ongoing trials. Gene expression profiling (GEP) helps improve the treatment of MM because it identifies prognostic subgroups and defines the molecular pathways associated with these subgroups. Newer agents (e.g., bortezomib, lenalidomide) have broadened the therapeutic regimens for end-stage myeloma.
High-dose chemotherapy followed by blood-forming stem cell transplantation (SCT) has become standard treatment for younger individuals (up to 70 years old in some trials).23,26 Survival is increased with SCT compared with chemotherapy alone. SCT uses the client’s own blood-forming stem cells (autologous) or a donor’s cells (allogeneic). Survival may be prolonged by performing a second autologous transplant within 6 to 12 months from the first transplant.
Radiation with high-energy x-rays is used more for a localized effect rather than systemic. It is most often used to treat areas of the bone that have been damaged and are not responding to chemotherapy. In addition, it may be used to treat sites where tumor has led to collapse of vertebrae and spinal cord compression.
Additional interventions are used to prevent and treat complications arising from progression of the disease. Drugs that inhibit bone resorption—bisphosphonates—reduce the incidence of skeletal damage, which also reduces hypercalcemia and decreases bone pain. Hydration and diuretics may be used to maintain a high urine output, and antibiotics to treat recurring infections.
The prognosis for persons with MM remains poor. The median survival for all states of MM is 3 years. Individuals with multiple bone lesions, if untreated, rarely survive more than 6 to 12 months. Individuals with inactive (indolent) myeloma, however, can survive for many years. With chemotherapy and aggressive management of complications, median survival may increase to 24 to 30 months, with a 10-year survival rate of 3%.
Waldenström Macroglobulinemia
Waldenström macroglobulinemia (WM), also called lymphoplasmacytic lymphoma, is a rare type of slow-growing plasma cell tumor that secretes a monoclonal IgM molecule. Approximately 1500 new cases are diagnosed yearly in the United States; the median age of diagnosis is 63 years of age.27 WM shares a great deal of similarity with multiple myeloma regarding its plasma cell origin, diagnosis, and treatment. However, the overproduction of the macromolecule IgM leads to certain unique clinical characteristics.
PATHOPHYSIOLOGY WM arises from plasma cells that have undergone genetic rearrangement of the variable region genes (V, D, J), but have not undergone class switch. Therefore, the principal secretory product of the tumor is IgM. Although no definitive genetic defect has been identified, WM may originate from aberrant B cell maturation and class switch.
Most of the pathology is associated with the production of large amounts of IgM, a large-molecular-weight protein (about 900,000 daltons). Excessive production leads to thickening of the blood and abnormally high blood viscosity (hyperviscosity syndrome). The increased viscosity interferes with circulation to various sites (e.g., eyes, brain, kidneys, extremities). IgM paraprotein may also result in cryoglobulins (proteins that precipitate from the blood at lower than body temperature). Hyperviscosity syndrome is observed in up to 20% of individuals with WM.
CLINICAL MANIFESTATIONS Many clients with WM are asymptomatic. The most common symptoms include weakness and fatigue, bleeding (from gums and nose), weight loss, and bruising. Bleeding may result secondary to formation of complexes among the macroglobulin, clotting factors, and platelets that diminish hemostatic capacity. If hyperviscosity syndrome occurs, the individual may develop neurologic problems (e.g., blurred vision, loss of vision, headaches, dizziness, vertigo), The macromolecules may also precipitate in colder regions of the body (cryoglobulins) leading to Raynaud phenomenon.
Although the malignant plasma cells invade the bone marrow, erosion of the bone is not commonly observed; less than 5% of individuals have lytic bone lesions. The tumor often disseminates to other organs, including the spleen, lymph nodes, and liver. Anemia occurs in about 10% of clients, secondary to tumor infiltration of the bone marrow. Peripheral hemolysis can also result from production of cold agglutinins.
EVALUATION AND TREATMENT Diagnosis is made based on high levels of monoclonal IgM in the blood and the identification of malignant cells in bone marrow aspirates. Other hematologic abnormalities may be observed, especially anemia (80% of clients with symptomatic WM) but also thrombocytopenia and leukopenia. Bence Jones protein may be observed in almost half of individuals with WM.
Treatment is similar to that described for multiple myeloma.28 First-line therapy includes combined chemotherapy with nucleoside analogs, alkylating agents, and monoclonal antibody (e.g., rituximab). Bone marrow stem cell transplantation has also proven effective in some individuals. The current recommendations include treatment with a combination of dexamethasone, rituximab, and cyclophosphamide, with the use of other drugs (e.g., doxorubicin, vincristine, and nucleoside analogs) in individuals with very high levels of M protein.
ALTERATIONS OF SPLENIC FUNCTION
The spleen has been an organ of mystery and perplexity in the study of medicine. Its relationship to other organs and disease processes, particularly the immune and hematologic systems, was not identified until the eighteenth century. The complexities of splenic function are not totally understood, and its mysteries are still being explored. The spleen is a useful organ, but its functions overlap those of other organs so that one is capable of living a normal, healthy life without the spleen. The relationship between asplenia and a higher risk for infection was not recognized until the early 1950s.
In the past, splenomegaly (enlargement of the spleen) was associated with various disease states. It is now recognized that splenomegaly is not necessarily pathologic; an enlarged spleen may be present in certain individuals without any evidence of disease. Splenomegaly may be, however, one of the first physical signs of underlying conditions, and its presence should not be ignored. In conditions in which splenomegaly is present, the normal functions of the spleen may become overactive, producing a condition known as hypersplenism.
Current criteria indicating the presence of hypersplenism include (1) anemia, leukopenia, thrombocytopenia, or combinations of these; (2) cellular bone marrow; (3) splenomegaly; and (4) improvement after splenectomy. Some individuals may seek treatment for problems even though they have not met all these clinical criteria; therefore, the relevance and significance of hypersplenism are still uncertain. Primary hypersplenism is recognized when no etiologic factor has been identified; secondary hypersplenism occurs in the presence of another condition.
PATHOPHYSIOLOGY Splenomegaly without a specific etiology is seen in 7% to 15% of individuals who are being evaluated for primary splenomegaly and is generally a diagnosis of exclusion. Specific conditions causing secondary splenomegaly and resulting hypersplenism are many and are related to all other categories of disease that affect individuals. Secondary splenomegaly may be classified according to the underlying cause. Specific conditions related to these various classifications of splenomegaly are detailed in Box 27-5. Different pathologic processes that produce splenomegaly are described briefly.
Acute inflammatory or infectious processes cause splenomegaly because of an increased demand for defensive activities. Acutely enlarged spleens secondary to infection may become so filled with erythrocytes that their natural rubbery resilience is lost and become fragile and vulnerable to blunt trauma. Splenic rupture is a complication associated with infectious mononucleosis; rupture occurs mostly in males between the fourth and twenty-first day of acute illness.
Congestive splenomegaly is accompanied by ascites, portal hypertension, and esophageal varices and is most commonly seen in those with hepatic cirrhosis. Splenic hyperplasia develops in disorders that increase splenic workload and is associated most commonly with various types of anemia (hemolytic) and chronic myeloproliferative disorders (i.e., polycythemia vera).
Infiltrative splenomegaly is caused by engorgement by the macrophages with indigestible materials associated with various “storage diseases.” Tumors and cysts cause actual growth of the spleen. Metastatic tumors in the spleen are rare and may result from primary tumors of the skin, lung, breast, and cervix.
CLINICAL MANIFESTATIONS Overactivity of the spleen results in hematologic alterations that affect all blood components. Sequestering of red blood cells, granulocytes, and platelets results in a reduction of all circulating blood cells. The spleen may sequester up to 50% of the red blood cell population, thereby upsetting the normal physiologic concentration of red blood cells in the circulation. The rate of splenic pooling is directly related to spleen size and the degree of increased blood flow through it. Sequestering exposes the red blood cells to splenic conditions that accelerate destruction, further contributing to the decreased red blood cell concentration. Anemia is the result of these combined activities. Anemia may be further potentiated by an increase in blood volume, which produces a dilutional effect on the already reduced concentration of red blood cells. The dilutional effect, as well as the removal and destruction of red blood cells, depends primarily on the degree of splenomegaly.
White blood cells and platelets also are affected by sequestering, although not to the same degree as the red blood cell. Again, the size of the spleen is the determining factor in the number of cells sequestered.
EVALUATION AND TREATMENT Treatment for hypersplenism is splenectomy; however, it is not always the treatment of choice. A splenectomy should be performed when its removal is considered necessary to alleviate the destructive effects on red blood cells. Clinical indicators should determine the need for splenectomy, not necessarily the specific condition. Splenectomy for splenic rupture no longer is considered mandatory in light of the possibility of overwhelming sepsis after removal. Repair and preservation of the ruptured spleen are now considered before the decision to remove the spleen. Splenectomy also may be performed as treatment for hairy cell leukemia, Felty syndrome, agnogenic myeloid metaplasia, thalassemia major, Gaucher disease, hemodialysis, splenomegaly, splenic venous thrombosis, and thrombotic thrombocytopenia purpura (TTP).
Individuals are able to lead normal lives after splenectomy, but hematologic abnormalities often exist after removal of the spleen. The red blood cells become thinner, broader, and wrinkled as a result of increases in surface area and membrane lipids. The white blood cell count increases dramatically 1 week after removal and then levels off to approximately 40% above normal. Platelets also rise immediately after surgery and then level off to above-normal levels for the duration of the individual’s life. Increased platelet levels have been implicated in ischemic heart disease in males because of increased thrombocytosis and hypercoagulability.
A major postoperative complication following splenectomy is OPSI. Unless treated in time, OPSI may rapidly progress to septic shock and possibly disseminated intravascular coagulation (DIC). Initial statistics indicate a mortality rate of 50% to 70%, with most deaths occurring within the first 48 hours after hospitalization. Prompt medical attention can reduce the mortality rate to 10%.
ALTERATIONS OF PLATELETS AND COAGULATION
Hemostasis is dependent on adequate numbers of platelets and levels of coagulation factors. Diminished or excessive levels may lead to defective hemostasis or spontaneous and unnecessary activation of clotting. (Hemostasis is described in Chapter 25.) Diminished hemostasis results in either internal or external hemorrhage. Diffuse hemorrhage into skin tissues that is visible through the skin causes a red-purple discoloration identified as a purpura. Purpuric disorders occur when there are not enough normal platelets to plug damaged vessels or prevent leakage from the many minute tears that occur daily in normal capillaries. Disorders of the clotting system tend to result in more serious internal bleeding than platelet defects and usually are caused by a deficiency of one or several clotting factors. Disorders that result in spontaneous clotting can result from genetic disorders of clotting system components or from acquired diseases that activate clotting. These disorders are known collectively as thromboembolic disease.
Disorders of Platelets
Quantitative or qualitative abnormalities of platelets can interrupt normal blood coagulation and prevent hemostasis.29 The quantitative abnormalities are thrombocytopenia, a decrease in the number of circulating platelets, and thrombocythemia, an increase in the number of platelets. Qualitative disorders affect the structure or function of individual platelets and can coexist with the quantitative disorders. Qualitative disorders usually prevent platelet adherence and aggregation, preventing formation of a platelet plug.
Thrombocytopenia
Thrombocytopenia is defined as a platelet count less than 150,000 platelets/mm3 of blood, although most health care providers do not consider the decrease of significance unless the count falls to less than 100,000 platelets/mm3 of blood.30 Hemorrhage resulting from minor trauma does not usually occur until the count falls below 50,000/mm3. Spontaneous bleeding without apparent trauma can occur with counts between 10,000 and 15,000/mm3, resulting in petechiae, ecchymoses, larger purpuric spots, or frank bleeding from mucous membranes. Severe spontaneous bleeding may result if the count is less than 10,000/mm3 and can be fatal if it occurs in the gastrointestinal tract, respiratory system, or CNS.
Before the diagnosis of thrombocytopenia is made, pseudothrombocytopenia must be ruled out. This phenomenon occurs in approximately 1 in 1000 to 1 in 10,000 laboratory samples and is an in vitro artifact that may occur when a blood sample is analyzed by an automated cell counter. Platelets in the sample may become nonspecifically agglutinated by immunoglobulins in the presence of ethylenediaminetetraacetic acid (EDTA), a preservative in banked blood. The agglutinated platelets are not counted, thus giving an apparent, but false, thrombocytopenia. Thrombocytopenia also may be falsely diagnosed because of a dilutional effect observed after massive transfusion of platelet-poor packed cells to treat a hemorrhage. This occurs when more than 10 units of blood have been transfused within a 24-hour period. The hemorrhage that necessitated the transfusion also accelerates the loss of platelets, which further contributes to the pseudothrombocytopenic state. Splenic sequestering of platelets secondary to hypersplenism (congestive) induces an apparent thrombocytopenia, as does hypothermia (less than 25° C [77° F]), which is reversed when temperatures return to normal, suggesting an increased platelet sequestration in response to chilling.
PATHOPHYSIOLOGY Thrombocytopenia results from decreased platelet production, increased consumption, or both. The condition also may be congenital or acquired and primary or secondary to other acquired or congenital conditions. Thrombocytopenia secondary to congenital conditions occurs in a large number of different diseases, although each is relatively rare. These include thrombocytopenia with absence of radius (TAR) syndrome, Wiskott-Aldrich syndrome (see Chapter 8), various forms of MYH9 gene mutation (e.g., May-Hegglin syndrome), X-linked thrombocytopenia, and many other examples.
Acquired thrombocytopenia is more common and may occur as a result of decreased platelet production secondary to viral infections (e.g., EBV, rubella, CMV, HIV), drugs (e.g., thiazides, estrogens, quinine-containing medications, chemotherapeutic agents, ethanol), nutritional deficiencies (vitamin B12 or folic acid in particular), chronic renal failure, bone marrow hypoplasia (e.g., aplastic anemia), radiation therapy, or bone marrow infiltration by cancer. Most common forms of thrombocytopenia are the result of increased platelet consumption. Examples include heparin-induced thrombocytopenia, idiopathic (immune) thrombocytopenic purpura, thrombotic thrombocytopenic purpura.
Heparin-Induced Thrombocytopenia: Heparin is a common cause of drug-induced thrombocytopenia. Approximately 4% of individuals treated with unfractionated heparin develop heparin-induced thrombocytopenia (HIT). The incidence is lower (about 0.1%) with the use of low-molecular-weight heparin. HIT is an immune-mediated, adverse drug reaction caused by IgG antibodies against the heparin-platelet factor 4 complex leading to platelet activation through platelet Fc γIIa receptors (Figure 27-19).31 The release of additional platelet factor 4 from activated platelets and activation of thrombin lead to increased platelet consumption and a decrease in platelet counts beginning 5 to 10 days after administration of heparin.
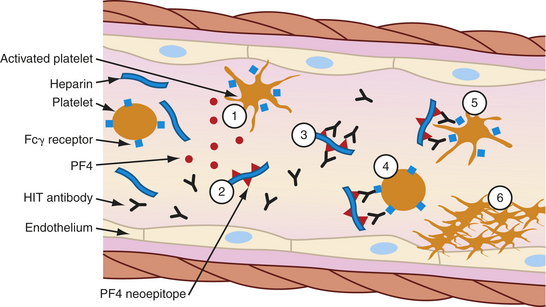
Figure 27-19 Pathogenesis of heparin-induced thrombocytopenia (HIT). (1) Activated platelets release procoagulant proteins from α-granules, including platelet factor 4 (PF4). Administered heparin binds PF4 (2), which undergoes a conformation change and expresses a new antigen (neoepitope). Individuals with HIT produce an immunoglobulin G (IgG) antibody that specifically reacts (3) with multiple identical neoepitopes on the heparin-PF4 complex. The reaction forms heparin-PF4-IgG immune complexes. Platelets express FcγRIIa receptors (Fcγ receptor) that react (4) with the Fc portion of IgG in immune complexes. Cross-linking of Fc receptors (5) results in FcγRlIa-dependent platelet activation. The activated platelets mediate a series of events that lead to further activation of the coagulation cascade, resulting in thrombin generation. Further release of PF4 from newly activated platelets leads to a cycle of continuing platelet activation and (6) formation of a primary clot. The reaction can be enhanced by the release of platelet-derived microparticles that are rich in surface phosphatidylserine and increase activation of coagulation and by the binding of heparin-PF4 complexes and HIT-IgG to the vascular endothelium (not shown.)
CLINICAL MANIFESTATIONS The hallmark of HIT is thrombocytopenia. A decrease of approximately 50% in the platelet count is seen in more than 95% of individuals. However, 30% or more of those with thrombocytopenia are also at risk for venous or arterial thrombosis.31 Venous thrombosis is most common and results in deep venous thrombosis and pulmonary emboli. Arterial thromboses affect the large arteries of the lower extremities, causing acute limb ischemia. Arterial thrombosis also may lead to cerebrovascular accidents and myocardial infarctions. Other major arteries (renal, mesenteric, upper limb) also may be affected. Bleeding is uncommon in HIT, even with low platelet counts.
EVALUATION AND TREATMENT Diagnosis is primarily based on clinical observations.31 The individual presents with dropping platelet counts after 5 days or longer of heparin treatment. On average, platelet counts may reach 60,000/mm3. Because most clients are postsurgery, and the onset of symptoms, including thrombosis, may be delayed until after release from the hospital, other possible causes of thrombocytopenia (e.g., infection, other drug reactions) must be considered.
Tests are available to measure antibodies against heparin-platelet factor 4.32 The test sensitivity is extremely high (more than 90%), but the specificity is less because of false-positive reactions (e.g., those on dialysis). HIT antibody titers may be measured, but the titers must be evaluated in the context of the clinical presentation. If HIT is not recognized and treated, intravascular aggregation of platelets causes rapid development of arterial and venous thrombosis. Although rare, heparin antibodies have caused anaphylactic shock.
Treatment is the withdrawal of heparin and use of alternative anticoagulants. A switch to low-molecular-weight heparin is not indicated, and warfarin should not be used until the symptoms of HIT have resolved because of an increased risk of initiating skin necrosis. The thrombocytopenia should progressively resolve. The chance of spontaneous blood clots can be diminished using thrombin inhibitors (e.g., lepirudin, argatroban).31
Immune Thrombocytopenic Purpura: The most common cause of thrombocytopenia secondary to increased platelet destruction is immune thrombocytopenic purpura (ITP). The incidence of ITP is estimated to be 5.8 to 6.6 per 100,000 in the general population. ITP was formerly known as idiopathic thrombocytopenic purpura; however, it is widely recognized now as an immune process, hence the change from idiopathic to immune.33 ITP may be acute or chronic. The acute form is frequently observed in children and typically lasts 1 to 2 months with a complete remission. In some instances it may last for up to 6 months, and some children (7% to 28%) may progress to the chronic condition. Acute ITP is usually secondary to infections (particularly viral) or other conditions that lead to large amounts of antigen in the blood, such as drug allergies or systemic lupus erythematosus (SLE) (see Chapter 8). Under these conditions the antigen usually forms immune complexes with circulating antibody, and it is thought that the immune complexes bind to Fc receptors on platelets, leading to their destruction in the spleen. The acute form of ITP usually resolves as the source of antigen is removed (e.g., the viral infection resolves).
Chronic ITP is associated with autoantibodies against platelet-specific antigens. This form is more commonly observed in adults, with highest prevalence in women between 20 and 40 years old, although it can develop at most any age. The chronic form tends to get progressively worse.
The autoantibodies are generally of the IgG class, although IgA and IgM antibodies also have been identified. They react against one or more of several platelet glycoproteins (e.g., GPIIb-IIIa, GPIb-IX, GPIa-IIa) (see Chapter 25).34 The antibody-coated platelets are removed from the circulation by mononuclear phagocytes in the spleen through the Fc receptor.
CLINICAL MANIFESTATIONS Initial manifestations are usually minor bleeding problems (development of petechiae and purpura) over the course of several days, that progress to major hemorrhage from mucosal sites (epistaxis, hematuria, menorrhagia, bleeding gums). Rarely will an individual present with intracranial bleeding or internal bleeding at other sites.
During pregnancy, an individual with ITP may have a newborn that is also thrombocytopenic. In most individuals the antiplatelet antibody is an IgG that readily crosses the placenta (see Chapters 7 and 8). If the fetal platelets express the same antigen as the mother, the maternal antibody will coat the platelets potentially resulting in thrombocytopenia in utero. A variant of neonatal thrombocytopenia (neonatal alloimmune thrombocytopenia) occurs when the mother does not have ITP, but makes IgG antibodies against an antigen inherited from the father and found on fetal platelets but not on maternal platelets.35 Alloimmune neonatal thrombocytopenia occurs in 1 of 2000 pregnancies. The most common antibody in this condition is against the human platelet antigen-a (HPA-a) antigen on the GPIIIa protein. Neonatal thrombocytopenia, either secondary to maternal autoimmune thrombocytopenia or as alloimmune thrombocytopenia, may occur to various degrees. The most severe form results in fetal platelet counts below 20,000/mm3 with a high associated risk of intracranial hemorrhage.
EVALUATION AND TREATMENT Diagnosis of ITP is based on a history of bleeding and associated symptoms, such as weight loss, fever, and headache. Physical examination includes notations on the types of bleeding, location, and severity. Evidence of infections (bacterial, HIV and other viral), medication history, family history, and evidence of thrombosis are also assessed. Other diagnostic tests include complete blood count (CBC) and peripheral blood smear. Unlike some other forms of thrombocytopenia, splenectomy is rarely observed. Testing for antiplatelet antibodies is usually not helpful. Although most cases of ITP are associated with elevated levels of IgG on platelets, other forms of thrombocytopenia also have a high incidence of platelet-associated antibodies; thus the specificity is low (50% to 65%).36 In addition, some cases of ITP will not present with elevated platelet-associated antibodies; the sensitivity is 75% to 94%, so that a negative test does not completely rule out ITP.
The acute form of ITP usually resolves without major clinical consequences. As with most autoimmune diseases, the course of the chronic form is variable, with multiple remissions and exacerbations. For many individuals the platelet count may remain adequate enough to avoid clinically serious bleeding. However, the presence of spontaneous bleeding suggests more severe disease and requires immediate attention. Treatment is initiated when platelet counts are less than 30,000 or less than 50,000 with evidence of bleeding from mucous membranes or when the individual is at high risk to develop bleeding.
Treatment is palliative, not curative, focusing on prevention of platelet destruction. Initial therapy for ITP is infusion of glucocorticoids (e.g., prednisone), which suppresses the production of antiplatelet antibodies and prevents sequestering and further destruction of platelets. If platelet counts do not increase appropriately, other medications may be tried. Treatment with intravenous immunoglobulin (IVIG) is used to prevent major bleeding. The response rate is 80%, but the effects are transient, lasting only days or a few weeks. Anti-(Rho)D (RhoGAM), which is a preparation of antibody against the D antigen of the Rh blood group, has been used with limited success to treat individuals who are Rh-positive.
If all other therapies are ineffective, splenectomy is considered to remove the primary site of platelet destruction.37 The response rate (resolution of the thrombocytopenia) is 60% to 70%; however, the procedure is not without risk. Approximately 10% to 20% of individuals who undergo splenectomy suffer a relapse and require further treatment. It is thought that other reticuloendothelial organs, particularly the liver, can become major sites for platelet destruction. If splenectomy is unsuccessful and life-threatening thrombocytopenia persists, more aggressive immunosuppressive medications (e.g., azathioprine, cyclophosphamide) may be used. Because of potential major complications, these medications are reserved for individuals who are severely thrombocytopenic and refractive to other therapies.
Thrombotic Thrombocytopenic Purpura: Thrombotic thrombocytopenic purpura (TTP) is characterized by thrombotic microangiopathy in which platelets aggregate and cause occlusion of arterioles and capillaries within the microcirculation.38 Aggregation may lead to increased platelet consumption and organ ischemia. TTP is relatively uncommon, occurring in about 5 per 1 million individuals per year. The incidence is increasing, which appears to be an actual increase in the number of affected individuals rather than a result of improved recognition. There are two forms of TTP: familial or acquired idiopathic. The familial form is the more rare and is usually chronic, relapsing, and seen in children. The child experiences predictable recurring episodes at approximately 3-week intervals and are responsive to treatment. Acquired TTP is more common, as well as more acute and severe. It occurs mostly in females in their 30s and is rarely observed in infants or older adults.
Platelet aggregation and microthrombi formation are found throughout the entire vascular system, causing damage to multiple organs. Organs most susceptible to damage are the kidney, brain, and heart. Other organs often affected are the pancreas, spleen, and adrenal glands. The thrombi are primarily composed of platelets with minimal fibrin and red cells, differentiating them from thrombi secondary to intravascular coagulation. Most cases of TTP are related to a dysfunction of the plasma metalloprotease ADAMTS13. This enzyme is responsible for digesting large precursor molecules of von Willebrand factor (vWF) produced by endothelial cells into smaller molecules. Defects in ADAMTS13 result in expression of large-molecular-weight vWF on the endothelial cell surface and the formation of large aggregates of platelets. The aggregates may break off and form occlusions in smaller vessels. Most individuals with TTP (about 80%) have less than 5% of normal plasma ADAMTS13 levels. TTP also is commonly associated with an IgG autoantibody against ADAMTS13 that is able to neutralize the enzyme’s activity and accelerate its clearance from the plasma.
CLINICAL MANIFESTATIONS The rare familial chronic relapsing TTP observed in children is usually recognized and successfully treated. The acquired acute idiopathic TTP is much more common and more severe.38 Early diagnosis and treatment is important because the disease may be fatal within 90 days of onset. TTP is clinically related to and must be distinguished from other thrombotic microangiopathic conditions, including hemolytic uremic syndrome, malignant hypertension, preeclampsia, or the pregnancy-induced HELLP (hemolysis, elevated liver enzymes, low platelet count) syndrome. Hemolytic uremic syndrome (HUS) shares many of the clinical characteristics of TTP; however, HUS often follows a hemorrhagic, diarrheal illness.
EVALUATION AND TREATMENT Acute idiopathic TTP is characterized by a “pathognomonic pentad” of symptoms. These include extreme thrombocytopenia (less than 20,000 platelets/mm3), intravascular hemolytic anemia, ischemic signs and symptoms most often involving the CNS (about 65% present with memory disturbances, behavioral irregularities, headaches, or coma), kidney failure (affecting about 65% of individuals), and fever (present in about 33%).38 It is not mandatory that all five be present to begin treatment. A routine blood smear usually reveals fragmented red cells (schizocytes) produced by shear forces when red cells are in contact with the fibrin mesh in clots that form in the vessels. As a result of tissue injury, serum levels of lactate dehydrogenase (LDH) may be very high, and low-density lipoprotein (LDL) levels may be elevated. Tests for antibody on red cells are negative, excluding immune hemolytic anemia.
Untreated acute TTP has a mortality rate of 90%, which can be reduced to 12% to 20% with prompt treatment. Plasma exchange with fresh frozen plasma replenishes functional ADAMTS13 and is the treatment of choice achieving a response rate of 70% to 85%. Additionally, steroids (glucocorticoids) are administered. In the absence of major organ damage, this approach may lead to complete recovery with no long-term complications. Relapses do occur at a rate of 13% to 36%, and recurrences have been reported, some as far out as 9 years. Individuals who do not respond to conventional treatment may be candidates for splenectomy; however, postoperative hemorrhage remains a dangerous complication. Immunosuppressive (azathioprine) therapy has been successful in some individuals.
Thrombocythemia
Thrombocythemia (also called thrombocytosis) is defined as a platelet count greater than 400,000/mm3 of blood.39 Thrombocythemia may be primary or secondary (reactive) and is usually asymptomatic until the count exceeds 1 million/mm3 of blood. Then intravascular clot formation (thrombosis), hemorrhage, or other abnormalities can occur.
PATHOPHYSIOLOGY Secondary thrombocythemia may occur after splenectomy because platelets that normally would be stored in the spleen remain in circulating blood. The increase in platelets may be gradual, with thrombocythemia not occurring for up to 3 weeks after splenectomy. Reactive thrombocythemia may occur during some inflammatory conditions, such as rheumatoid arthritis and cancers. In these conditions, excessive production of some cytokines (e.g., IL-6, IL-11) may induce increased production of thrombopoietin in the liver, resulting in increased megakaryocyte proliferation. Reactive thrombocythemia may also occur during a variety of physiologic conditions, such as after exercise. Because of the relatively self-resolving nature of secondary thrombocythemia, the remaining discussion will focus on the more severe primary form.
Essential (primary) thrombocythemia (ET) is a chronic myeloproliferative disorder characterized by excessive platelet production resulting from a defect in bone marrow megakaryocyte progenitor cells.40 The overall incidence of ET is 0.8 per 100,000 in the United Kingdom, 2.53 in the United States, and 0.59 in Denmark. It is more common in middle-age individuals, with the majority of cases occurring between ages 50 and 60 years. There is no known gender preference. There also is a rare hereditary type of ET called familial essential thrombocythemia (FET) that is inherited in an autosomal dominant pattern.
The thrombocythemia is secondary to increased plasma thrombopoietin levels resulting from defects in the thrombopoietin receptor. The defective receptor cannot adequately bind and remove thrombopoietin from the blood, thus circulating levels remain high. Along with increased platelet levels, there may be a concomitant increase in red cells, indicating a myeloproliferative disorder; however, the increase in red cells is not to the extent seen in polycythemia vera (see Chapter 26). The bone marrow of affected individuals with ET is characterized by hyperplasia of megakaryocytes. The platelets of affected individuals appear to have a normal survival time, compatible with a defect in production rather than an increase in platelet life span.
RBCs in ET tend to aggregate and contribute to the blockage of flow in the microvasculature and altered interactions between platelets and the vascular endothelium.41 Increased adherence of erythrocytes to the endothelium appears to result from a mutation in an erythrocyte Janus kinase 2 (JAK2) that is responsible for phosphorylation of the erythrocyte receptor for endothelial laminin.42,43 The frequency of JAK2 mutations in ET is about 30%. Increased platelet aggregation arises from several mutations that result in altered platelet membrane glycoproteins, particularly resulting in increased expression of GPIV, and increased secretion of thromboxane.
CLINICAL MANIFESTATIONS Clinical manifestations vary significantly among individuals. Those with ET are at risk for large-vessel arterial or venous thrombosis, and, particularly, microvasculature thrombosis leading to ischemia in the fingers, toes, or cerebrovascular regions.41 The primary presenting symptoms of microvasculature thrombosis are erythromyalgia, headache, and paresthesias. Erythromyalgia is characterized by unilateral or bilateral warm, congested, red hands and feet with painful burning sensations, particularly in the forefoot sole and one or more toes. The lower extremities are affected more often, and only one side may be involved. The pain is initiated by standing, exercise, or warmth and relieved by elevation and cooling. In extreme situations, acrocyanosis and gangrene may result.
Arterial thrombosis is more common than venous thrombosis and may involve the coronary and renal arteries. The carotid, mesenteric, and subclavian arteries also may be affected. Myocardial ischemia and infarction have occurred without clear evidence of coronary artery disease. Deep venous thrombosis of the lower extremities and pulmonary embolism are the major sites for venous involvement. Intra-abdominal venous thrombosis (portal and hepatic) also are common sites of venous thrombosis.
Microvascular thrombosis in the CNS is usually associated with headache and dizziness, with paresthesias, transient ischemic attacks, strokes, visual disturbances, and seizures also being reported. Major thrombotic events, not directly related to the platelet count, occur in about 20% to 30% of individuals with ET. Prior history of thrombotic events, advanced age, and duration of thrombocytosis are predictors of future thrombotic complications. Individuals older than age 60 are at greatest risk.
Although thrombosis is the most common symptom, hemorrhage can also occur. Sites for bleeding include the GI tract, skin, mucous membranes, urinary tract, gums, tooth sockets (after extraction), joints, eyes, and brain. GI bleeding may be mistaken for a duodenal ulcer. Hemorrhage is not severe, and generally occurs in the presence of very high platelet counts, and occasionally requires transfusion. Important is recognition that bleeding and clotting may exist simultaneously and individuals will not necessarily be “bleeders” or “clotters.”
EVALUATION AND TREATMENT Initial diagnosis is not difficult; as many as two thirds of affected individuals are diagnosed from a routine CBC. Secondary thrombocytosis may present as a moderate rise in the platelet count that resolves with treatment or resolution of the underlying condition. ET is diagnosed by a platelet count greater than 600,000/mm3 and remains elevated, with no other indicated cause, such as arthritis, iron deficiency anemia, cancer, or splenectomy. Many individuals present with a mild anemia and a slightly elevated white blood cell count.
After diagnosis, these individuals may recall events related to thrombosis or hemorrhage. Manifestations of ET may be mistaken for CML; therefore, differentiation of the two is important because treatment varies significantly. Identification of the Philadelphia chromosome is recommended in all cases of ET.
Treatment of ET is directed toward preventing thrombosis or hemorrhage.44 Whether to reduce platelet count remains a significant treatment issue. Historically treatment of ET relied on the use of alkylating agents (busulfan) or radiophosphorus (32P) to suppress platelet production. Hydroxyurea, a nonalkylating myelosuppressive agent, has been the drug of choice to suppress platelet production: however, long-term use may cause progression to other myelodysplastic disorders, particularly AML or myelofibrosis.44 Conversion to myelofibrosis occurs approximately 8% of the time and conversion to AML occurs approximately 3.5% of the time when treated with Vhydroxyurea as a single cytotoxic agent, but increases to 14% when more than one cytotoxic agent is used.
Interferon (TFN) also may be used and has a response rate of 80%. TFN may not work for everyone because it has many side effects and 20% of individuals may be intolerant. Anagrelide is now considered to be the drug of choice. Anagrelide specifically interferes with platelet maturation rather than production, thus not affecting erythropoiesis or leukopoiesis.
Aspirin also is used in the treatment of ET; however, its action is not to reduce the platelet count but to prevent adherence of platelets to each other and prevent thrombus formation. Early studies with aspirin found hemorrhage to be a major contraindication for its use; however, in lower doses (80 to 160 mg/daily) it effectively alleviates erythromyalgia and transient neurologic manifestations.
Prognosis and survival of individuals with ET have been somewhat difficult to establish. ET is not necessarily considered life threatening, but in those older than age 60 and who have had previous incidences of thrombosis, complications are more common and have a higher risk of mortality.
Alterations of Platelet Function
Qualitative alterations in platelet function occur with an increased bleeding time in the presence of a normal platelet count. Associated clinical manifestations include spontaneous petechiae and purpura, bleeding from the GI tract, genitourinary tract, pulmonary mucosa, and gums. Congenital alterations in platelet function (thrombocytopathies) are quite rare and may be categorized into several types of disorders: (1) platelet-vessel wall adhesion, (2) platelet-platelet interactions, (3) platelet granules and secretion, (4) arachidonic acid pathways, and (5) membrane phospholipid regulation (coagulation protein-platelet interactions).45
Disorders of platelet-vascular wall adhesion result from aberrations of the platelet membrane glycoprotein GPIb-IX-V (Bernard-Soulier syndrome), the collagen receptor (GPVI) or deficiencies of vWF. The GPIb protein is the most commonly mutated in individuals with Bernard-Soulier syndrome. Lack of these proteins prevents platelets from adhering to collagen, resulting in impaired hemostasis and clinical hemorrhage.
Disorders of platelet-platelet interactions result in failure of platelets to aggregate in response to adenosine diphosphate (ADP), collagen, epinephrine, or thrombin because of a deficiency in the glycoprotein (αIIbβ3) that acts as a receptor for fibrinogen, vWF, and fibronectin (Glanzmann thrombasthenia). Lack of this protein results in a failure to build “fibrinogen bridges” between platelets (see Figure 25-20). Defects also can occur in platelet receptors for platelet activators. These include mutations in the receptors for thromboxane or ADP.
Disorders of platelet granules and secretion and arachidonic pathways are characterized by initial normal platelet aggregation with collagen or ADP; however, there is failure of subsequent processes, specifically secretion of prostaglandins and release of granules. Defective α-granule numbers or release (gray platelet syndrome) results from mutations in several aspects of granule function, including biosynthesis or loading of proteins normally found in these granules. Defects in dense granules include Hermansky-Pudlak syndrome, Chédiak-Higashi syndrome, and delta-storage pool disease. These usually result from mutations in proteins involved in formation of dense granules or their movement to the plasma membrane. Defects in the thromboxane pathway prevent the release of this mediator.
Externalization of plasma membrane phosphatidylserine (PS) is necessary for effective platelet function. In Scott syndrome, the enzyme responsible for PS efflux is defective, thus platelets are unable to support the activation of factor X and prothrombin. The inreverse of Scott syndrome is Stormorken syndrome, in which platelets constitutively externalize PS.
Acquired disorders of platelet function are more common than the congenital disorders and may be categorized into three principal causes: (1) drug effects, (2) systemic inflammatory conditions, and (3) hematologic conditions.
Multiple drugs are known to affect platelet function in several ways: inhibition of platelet membrane receptors, inhibition of prostaglandin pathways, and inhibition of phosphodiesterase activity. Aspirin is the most commonly used drug that affects platelets and the only drug specifically used for its platelet effects. It irreversibly inhibits cyclooxygenase function for several days after administration. Nonsteroidal anti-inflammatory drugs also affect cyclooxygenase, although in a reversible fashion.
Systemic disorders that affect platelet function are chronic renal disease, liver disease, cardiopulmonary bypass surgery, severe deficiencies of iron or folate, and antiplatelet antibodies associated with autoimmune disorders. Hematologic disorders that cause platelet dysfunction are chronic myeloproliferative disorders, multiple myeloma, leukemias, myelodysplastic syndromes, and dysproteinemias.
Disorders of Coagulation
Disorders of coagulation usually are caused by defects or deficiencies of one or more of the clotting factors. (Normal function of the clotting factors is described in Chapter 25.) Qualitative or quantitative abnormalities of clotting factors interfere with or prevent the enzymatic reactions that transform circulating clotting proteins into a stable fibrin clot (see Figure 25-22).
Some clotting factor defects are inherited and usually involve a single factor, such as hemophilias and von Willebrand disease, caused by deficiencies of specific clotting factors (see Chapter 28). Other coagulation defects are acquired and tend to result from deficient synthesis of clotting factors by the liver. Causes include liver disease and dietary deficiency of vitamin K.
Other coagulation disorders are attributed to pathologic conditions that trigger coagulation inappropriately. For example, any cardiovascular abnormality that alters normal blood flow by speeding it up, slowing it down, or obstructing it can result in spontaneous coagulation within the vessels. Coagulation is also stimulated by the presence of tissue factor, which is released by damaged or dead tissues. Vasculitis, or inflammation of the blood vessels, as well as vessel damage, activates platelets, which in turn activates the coagulation cascade. In extensive or prolonged vasculitis, blood clot formation can suppress mechanisms that normally control clot formation and breakdown, leading to clogging of the vessels. In each of these acquired conditions, normal hemostatic function proves detrimental to the body by consuming coagulation factors excessively or by overwhelming the normal control of clot formation and breakdown (fibrinolysis).
Impaired Hemostasis
Impaired hemostasis, or the inability to promote coagulation and the development of a stable fibrin clot, is commonly associated with liver disorders, either resulting from the lack of vitamin K or specific diseases of the liver.
Vitamin K Deficiency: Vitamin K, a fat-soluble vitamin, is necessary for synthesis and regulation of prothrombin, procoagulant factors (VII, IX, X), and anticoagulant regulators (proteins C and S) within the liver.46 Vitamin K is found in green leafy vegetables and is the primary dietary source. Vitamin K also is synthesized by intestinal flora, but its contribution to the overall supply of vitamin K is uncertain. The most common cause of vitamin K deficiency is parenteral nutrition in combination with broad-spectrum antibiotics that destroy normal gut flora. Rarely is a deficiency caused by lack of dietary intake; however, bulimia can suppress vitamin K–dependent activity. Clinical manifestations of vitamin K deficiency are caused by a reduction of vitamin K–dependent proteins. The severity of manifestations is related to the degree of deficiency and ranges from laboratory abnormalities to significant hemorrhage.
Parenteral administration of vitamin K is the treatment of choice and usually results in correction of the deficiency. Improvement of clotting tests is usually noted within 8 to 12 hours. Fresh frozen plasma may be administered but usually is reserved for individuals with life-threatening hemorrhages or who require emergency surgery.
Liver Disease: Individuals who have liver disease (e.g., acute or chronic hepatocellular diseases, cirrhosis, vitamin K deficiency) or major liver surgery present with a broad range of hemostasis derangements that may be characterized by defects in the clotting or fibrinolytic systems or platelet function.46 The hepatic parenchymal cells produce most of the factors involved in hemostasis. Thus damage to the liver frequently results in diminished production of factors involved in clotting, usually in proportion to the degree of hepatic parenchymal cell damage. For instance, factor VII is most sensitive to liver damage because of its rapid turnover. Factor IX levels are less affected and do not decline until liver destruction is well advanced. The liver is also a major site for production of plasminogen and α2-antiplasmin of the fibrinolytic system, as well as thrombopoietin and the metalloprotease ADAMTS13. Diminished thrombopoietin may lead to thrombocytopenia from decreased platelet production. Decreased production of ADAMTS13 results in increased levels of large precursor molecules of vWF, which leads to the formation of large aggregates of platelets.
In conditions of severe liver disease (e.g., cirrhosis) circulating levels of most clotting factors are significantly depressed. Concurrently production of clotting system regulators (e.g., antithrombin, protein C, protein S) and of fibrinogen is diminished. The fibrinolytic system is commonly active due to decreased levels of plasmin inhibitor and unaffected levels of fibrinolytic activators (e.g., tPA, uPA). The affected individuals also are thrombocytopenic because of diminished thrombopoietin and ADAMTS13, as well as increased platelet sequestration in the spleen, which is frequently enlarged in cirrhosis and is associated with portal hypertension. Thus the individuals with cirrhosis may appear to have a condition similar to DIC (see next section).
Treatment of hemostatic alterations in liver disease must be comprehensive to cover all aspects related to platelet, clotting, and fibrinolytic dysfunctions. Fresh frozen plasma administration is the treatment of choice, but not all individuals tolerate the volume needed to adequately replace all deficient factors. Alternative modalities include the addition of exchange transfusions and platelet concentration to plasma administration.
Consumptive Thrombohemorrhagic Disorders
Consumptive thrombohemorrhagic disorders are a heterogeneous group of conditions that demonstrate the entire range of hemorrhagic and thrombotic pathologic conditions. Symptoms range from subtle to devastating and generally are considered to be intermediary disease processes that complicate many primary disease states. These disorders also are characterized by confusion and controversy regarding diagnosis, treatment, and management. No one definition can cover all possible varieties of these disorders; however, disseminated intravascular coagulation is the most common term used in the clinical setting to describe a pathologic condition associated with hemorrhage and thrombosis.
Disseminated Intravascular Coagulation: Disseminated intravascular coagulation (DIC) is an acquired clinical syndrome characterized by widespread activation of coagulation resulting in formation of fibrin clots in medium and small vessels throughout the body. Disseminated clotting may lead to blockage of blood flow to organs, resulting in multiple organ failure. The magnitude of clotting may result in consumption of platelets and clotting factors leading to severe bleeding. The Subcommittee on DIC of the International Society on Thrombosis and Hemostasis defined DIC as, “An acquired syndrome characterized by the intravascular activation of coagulation with loss of localization arising from different causes. It can originate from and cause damage to the microvasculature, which if sufficiently severe, can produce organ dysfunction.”47,48
The clinical course of DIC largely is determined by the intensity of the stimulus, host response, and comorbidities and ranges from an acute, severe, life-threatening process that is characterized by massive hemorrhage and thrombosis to a chronic low-grade condition. The chronic condition is characterized by subacute hemorrhage and diffuse microcirculatory thrombosis. DIC may be localized to one specific organ or generalized, involving multiple organs.
Because of the complexity and wide variations in manifestations of DIC, diagnosis has been confusing and difficult. Minimally acceptable diagnostic criteria have been established and include a systemic thrombohemorrhagic disorder with laboratory evidence of (1) clotting activation, (2) fibrinolytic activation, (3) coagulation inhibitor consumption, and (4) biochemical evidence of end-organ damage or failure.
DIC is secondary to a wide variety of well-defined clinical conditions, specifically those capable of activating the clotting cascade (Box 27-6). These include (1) arterial hypotension, frequently accompanying shock; (2) hypoxemia; (3) acidemia; and (4) stasis of capillary blood flow.
Sepsis is the most common condition associated with DIC. Gram-negative microorganisms, as well as some gram-positive microorganisms, fungi, protozoa (malaria), and viruses (influenza, herpes) are capable of precipitating DIC by causing damage to vascular endothelium. Gram-negative endotoxins are the primary cause of endothelial damage; DIC may occur in up to 50% of individuals with gram-negative sepsis. DIC occurs in approximately 10% to 20% of individuals with metastatic cancer or acute leukemia. Direct tissue damage (ischemia and necrosis, surgical manipulation, crushing injury) also result in release of tissue factor (TF) by the endothelium. Severe trauma, especially to the brain, can induce DIC. DIC occurs in about two thirds of individuals with a systemic inflammatory response to trauma. Some complications of pregnancy also are associated with DIC; incidences range from 50% for women with placental abruptions to less than 10% for severe preeclampsia. Other causes of DIC have been identified, most notably blood transfusion. Transfused blood dilutes the clotting factors, as well as circulating naturally occurring antithrombins. In hemolytic transfusion reactions, the endothelium is damaged by complement-mediated reactions.
PATHOPHYSIOLOGY The coagulation system is designed to function at local areas of vascular damage, resulting in cessation of bleeding and activation of repair to the vessels. DIC results from abnormally widespread and ongoing activation of clotting (Figure 27-20). A variety of conditions are associated with DIC (see Box 27-6), primarily by activating the extrinsic clotting cascade. The common pathway for DIC appears to be excessive and widespread exposure of TF. This may occur by several mechanisms. Widespread damage to vascular endothelium results in exposure of subendothelial TF. Several types of cells, either after stimulation by cytokines or constitutively, express TF on their surfaces. Endothelial cells and monocytes do not normally express surface TF unless stimulated by inflammatory cytokines (particularly IL-6 and TNF-α).49 Many tumors express surface TF or produce cytokines that stimulate TF expression by endothelium or monocytes or both.50 These cytokines are abundantly produced during many of the conditions listed in Box 27-6. Endotoxin, in particular, triggers the release of multiple cytokines that play a significant role in the development and maintenance of DIC. Proinflammatory cytokines (TNF-α, interleukins [IL-1, IL-6, IL-8], and platelet activating factor [PAF]) are responsible for the clinical signs and symptoms associated with sepsis. They also contribute to the development of DIC by activating endothelial cells, causing release of TF and vWF, increasing plasminogen activator inhibitor-1 (PAI-1) synthesis and tissue factor activity, and decreasing thrombomodulin expression, thereby promoting development of thrombi. TF also may be released directly into the bloodstream from circulating white blood cells (monocyte/endotoxin interaction).
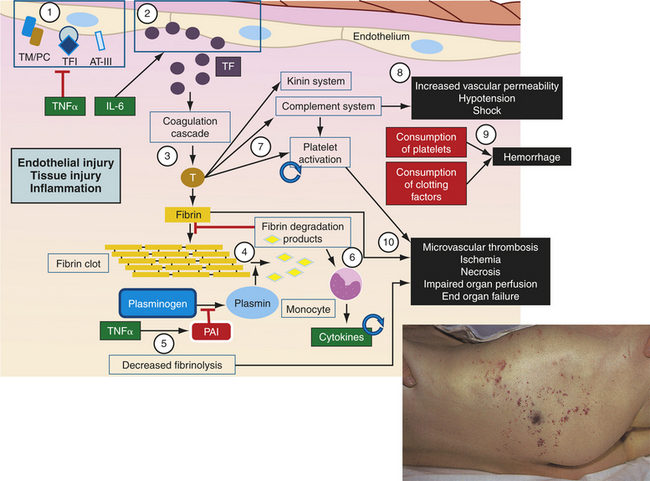
Figure 27-20 Pathophysiology of disseminated intravascular coagulation (DIC). DIC is initiated by a variety of factors (endothelial injury, tissue injury, inflammation, and others), most of which either directly or indirectly result in release of large amounts of tissue factor. Many cytokines create a procoagulant environment by concurrently (1) suppressing normal control of homeostasis and (2) inducing tissue factor release by endothelium or monocytes. Tissue factor initiates the coagulation cascade (3) leading to the activation of thrombin, production of fibrin, and polymerization into a fibrin clot. Fibrinolysis normally digests clots (4) through the activity of plasmin, resulting in the production of various fibrin degradation products. However, during DIC, factors, such as TNF-α, induce (5) inhibitors of plasmin generation, thus leading to diminished fibrinolysis. Fibrin split products possess several biological activities that affect DIC, including (6) the induction of further cytokine release by monocytes. Enzymatically active products of the coagulation cascade, including thrombin, activate (7) other inflammatory systems, including platelets and the kinin and complement systems. Activation of platelets and monocytes continue the procoagulant cycle (indicated by circular arrows) by inducing additional tissue factor and cytokines. Mediators produced from the kinin and complement system (8) affect vascular endothelium leading to increased vascular permeability that contributes to hypotension and potential shock. The uncontrolled consumption of platelets and clotting factors (9) compromises the normal hemostatic mechanisms resulting in potential systemic hemorrhages. Excess activation of the coagulation cascade and platelets, with decreased fibrinolysis, leads to systemic microvascular thrombosis (10) and blockage of the vessels with progressive ischemia. Uncontrolled DIC will eventually lead to multiple end-organ failure. For further details of these mechanisms see Chapters 6 and 25. AT III, Antithrombin III; IL-6, interleukin-6; PAI, plasminogen activator inhibitor; T, thrombin; TF, tissue factor; TFI, tissue factor inhibitor; TM/PC, thrombomodulin/protein C complex; TNF-α, tumor necrosis factor-alpha. Insert is an example of DIC resulting from staphylococcal septicemia. Note the characteristic skin hemorrhage ranging from small purpuric lesions to larger ecchymoses.
TF binds clotting factor VII, which leads to conversion of prothrombin to thrombin and formation of fibrin clots (see Figure 25-22).This pathway appears to be the primary route by which DIC is initiated; inhibition of TF or factor VIIa completely prevents the generation of thrombi by gram-negative bacterial endotoxin in animal models of DIC.
Not only is the clotting system extensively activated in DIC, but the predominant natural anticoagulants (tissue factor inhibitor, antithrombin III [AT III], protein C) are also greatly diminished (see Figure 25-17). During DIC the activation of clotting is prolonged by the increased rate of consumption because of persistent thrombin production, as well as decreased synthesis, of these inhibitors and protein S and by cytokine-mediated decreased expression of thrombomodulin on the endothelial cell surface. Hepatic dysfunction in sepsis results in decreased antithrombin synthesis and extravascular leakage of this protease inhibitor because of capillary leakage. Additionally, antithrombin is degraded by elastase released by activated neutrophils, and clotting is initiated concurrently with loss of regulation of the extent of thrombosis, thus the amount of thrombin produced during DIC exceeds the ability of the body’s naturally occurring anticoagulants to regulate it.
The rate of fibrinolysis is also diminished in DIC. The primary component of the fibrinolysis is plasmin, which exists in the circulation as an inactive precursor, plasminogen (see Figure 25-25). Plasminogen is activated to plasmin that digests fibrin clots, thus controlling the extent of fibrin deposition in the vessels. During DIC the activity of plasmin is diminished by increased production of its natural inhibitor, PAI-1. Although some fibrinolytic activity remains, the level is inadequate to control the systemic deposition of fibrin. The slow breakdown of fibrin by plasmin produces FDPs that are released into the blood. These are potent anticoagulants that are normally removed from blood by fibronectin and macrophages. FDPs, along with thrombin, induce further cytokine release from monocytes, contributing to endothelial damage and TF release. During DIC the presence of fibrin degradation products is prolonged, probably because of diminished production of fibronectin. Fibronectin is a glycoprotein with adhesive properties that mediate removal of particulate matter (e.g., fibrin clumps). Low levels of fibronectin suggest a poor prognosis.
Although thrombosis is generalized and widespread, individuals with DIC are paradoxically at risk for hemorrhage. Hemorrhage is secondary to the abnormally high consumption of clotting factors and platelets, as well as the anticoagulant properties of FDPs, which interfere with polymerization of fibrin monomers. Both thrombin and FDPs have a high affinity for platelets and cause platelet activation and aggregation—an event that occurs early in the development of DIC—which facilitates microcirculatory coagulation and obstruction in the initial phase. However, platelet consumption exceeds production, resulting in a thrombocytopenia that increases bleeding. (Box 27-7).
Activation of clotting also leads to activation of other inflammatory pathways, including the kallikrein-kinin and complement systems (see Chapter 6). Factor XIIa, generated in DIC, converts prekallikrein to kallikrein, ultimately resulting in conversion to circulating kinins. Activation of these systems contributes to increased vascular permeability, hypotension, and shock. Activated complement components also induce platelet destruction, further contributing initially to the thrombosis and later to the thrombocytopenia.
The deposition of fibrin clots in the circulation interferes with blood flow, causing widespread organ hypoperfusion. This condition may lead to ischemia, infarction, and necrosis, further potentiating and complicating the existing DIC process by causing further release of TF and eventually organ failure.
In addition to initiation of clotting by TF, DIC may be precipitated by direct proteolytic activation of factor X. This has been described as “thrombin mimicry” and is the result of activated factor X directly converting fibrinogen to fibrin. The proteases that activate factor X may come from snake venom, some tumor cells, or the pancreas and liver, where they are released during episodes of pancreatitis and various stages of liver disease. Direct proteolytic activity appears to be independent of any type of damage to the endothelium or tissue.
Vascular obstruction results from circulatory deposition of thrombin and clot formation that impedes blood flow, causing widespread organ hypoperfusion that can lead to tissue ischemia, infarction, and necrosis. The resulting tissue damage further potentiates and complicates the existing DIC process. Because organ perfusion is drastically impaired, manifestations of multisystem organ dysfunction and failure ultimately result. Multisystem organ dysfunction and failure are discussed in Chapter 46. Whatever initiates the process of DIC, the cycle of thrombosis and hemorrhage persists until the underlying cause of the DIC is removed or appropriate therapeutic interventions are used.
CLINICAL MANIFESTATIONS Clinical signs and symptoms of DIC present a wide spectrum of possibilities, depending on the underlying disease process that initiates DIC and whether the DIC is acute or chronic (see Box 27-7). Most symptoms are the result of either hemorrhage or thrombosis. Acute DIC presents with rapid development of hemorrhaging, such as oozing from venipuncture sites, arterial lines, and surgical wounds, or development of ecchymotic lesions (purpura, petechiae) and hematomas. Other sites of bleeding include the eyes (sclera and conjunctiva), the nose (epistaxis), and the gums. Most individuals with DIC demonstrate bleeding at three or more unrelated sites, and any combination may be observed. Shock of variable intensity, out of proportion to the amount of apparent blood loss, also may be observed. Hemorrhaging into closed compartments of the body also can occur and may precede the development of shock.
DIC has been conceptualized as a systemic hemorrhagic disorder because bleeding, sometimes very extensive, is usually the initial observation. Symptoms of thrombosis are not always as evident, even though it is often the first pathologic alteration to occur and ultimately determines the degree of morbidity and risk for death. A large amount of microvascular and macrovascular occlusion may occur that is not clinically obvious. Several organ systems are susceptible to microvascular thrombosis that affects their function; these include the cardiovascular, pulmonary, central nervous, renal, and hepatic systems. Quick and accurate clinical diagnosis is critical to preventing further progression of DIC that may lead to multisystem organ dysfunction or failure. Indicators of multisystem failure include changes in level of consciousness, behavior, and mentation; confusion; seizure activity; oliguria; hematuria; hypoxia; hypotension; hemoptysis; chest pain; and tachycardia. Symmetric cyanosis of the fingers and toes (“blue finger/toe syndrome”) and, in some instances, of the nose and breasts may be present. Symmetric parts are often affected and are indicative of microvascular thrombosis. This may progress to infarction and gangrene, requiring amputation. Jaundice also may be present and is believed to result from red blood cell destruction rather than hepatic dysfunction.
Individuals with chronic or low-grade DIC do not present with overt manifestations of hemorrhaging and thrombosis but instead have subacute bleeding and diffuse thrombosis and are described as having a compensated DIC, or non-overt DIC. The major characteristic of this state is an increased turnover and decreased survival time of the components of hemostasis: platelets and clotting factors. On occasion diffuse or localized thrombosis develops, but this is infrequent.
EVALUATION AND TREATMENT No single laboratory test can be used to effectively diagnosis DIC. Diagnosis is based primarily on clinical symptoms and confirmed by a combination of laboratory tests. The individual must present with a clinical condition that is known to be associated with DIC. The most commonly used combination of laboratory tests usually confirms thrombocytopenia or a rapidly decreasing platelet count on repeated testing, prolongation of clotting times, the presence of fibrin degradation products, and decreased levels of coagulation inhibitors. The relationships among these criteria are summarized in Box 27-8. Platelet counts less than 100,000/mm3 or a progressive decrease in platelet counts is very sensitive for DIC, although not greatly specific. These changes usually indicate consumption of platelets.
The standard coagulation tests (e.g., prothrombin time [PT], activated partial thromboplastin time [aPTT]) also have a high degree of sensitivity, but they are not highly specific for DIC. As a result of consumption of circulating clotting factors, these tests are usually abnormal, ranging from shortened to prolonged times. However, conditions other than DIC may prolong clotting times. Assays of specific clotting factors do not contribute meaningful diagnostic information.
Detection of various fibrin degradation products is more specific for DIC; of these tests the detection of D-dimers is the most widely used, reliable and specific test.51 A D-dimer is a molecule produced by plasmin degradation of cross-linked fibrin in clots. D-dimers in the blood can be quantified using enzyme-linked immunosorbent assay (ELISA) tests that include commercially available and highly specific monoclonal antibody against the D-dimer. Agglutination tests for other fibrin degradation products are available. Fibrin degradation products, in general, are elevated in the plasma in 95% to 100% of cases; however, they are less specific and only document the presence of plasmin and its action of fibrin, whereas detection of D-dimers measures a specific DIC-related product.
ELISAs for markers of thrombin activity are sometimes used. Normal conversion of prothrombin to thrombin produces an inactive prothrombin fragment 1.2 (PF1+2).51 This fragment is released from the prothrombin molecule generating an intermediate factor, prethrombin 2. Once generated, prethrombin 2 can be split to produce thrombin that can then proteolyze fibrinogen, liberating fibrinopeptide A (FPA) or combine with its major antagonist, antithrombin, and form a stable inactive enzyme inhibitor complex, the thrombin-antithrombin (TAT) complex. Assays of these factors (PF1+2, FPA, TAT) are now generally available to quantify their blood levels, providing evidence of excessive factor Xa (PF1+2) and thrombin (FPA) generation.
Levels of coagulation inhibitors (e.g., AT III, protein C) can be measured by assays that rely on function or by ELISAs that quantify the amount of the specific inhibitor. AT III levels can provide key information for diagnosing and monitoring therapy of DIC. Initial levels of functional AT III are low in DIC because thrombin is irreversibly complexed with activated clotting factors and AT III.
Treatment of DIC is directed toward (1) eliminating the underlying pathology, (2) controlling ongoing thrombosis, and (3) maintaining organ function. Elimination of the underlying pathology is the initial intervention in the treatment phase in order to eliminate the trigger for activation of clotting. Once the stimulus is gone, production of coagulation factors in the liver leads to restoration of normal plasma levels within 24 to 48 hours.
Control of thrombosis is more difficult to attain. Heparin has been used for this; however, its use is controversial because its mechanism of action is binding to and activating AT III, which is deficient in many types of DIC. Currently heparin is indicated only in certain situations related to DIC. For instance, heparin seems to be effective in DIC caused by a retained dead fetus or associated with acute promyelocytic leukemia. Organ function is compromised by microthrombi, and there is a risk of losing an extremity because of vascular occlusion, thus heparin is also indicated in these conditions. Heparin’s usefulness, however, for DIC that is precipitated by septic shock has not been established and so is contraindicated in that instance; heparin is also contraindicated when there is evidence of postoperative bleeding, peptic ulcer, or CNS bleeding.
Replacement therapy (interventions based on restoring the balance of coagulation factors, deficient coagulation factors, platelets, and other coagulation elements) is gaining recognition as an effective treatment modality. Components used in replacement therapy include platelets, fresh frozen plasma, and cryoprecipitate. Platelets are given for thrombocytopenia, plasma provides volume and replaces clotting factors, and cryoprecipitate replaces fibrinogen. Their use is not without controversy, however, because of the possible risk of adding components that will increase the rate of thrombosis. Clinical judgment is the key factor in determining whether replacement is to be used as a treatment modality.
Several clinical trials are evaluating replacement of anticoagulants (i.e., AT III, protein C). Replacement of AT III appears to be effective in DIC caused by sepsis. Low levels of AT III correlate with sepsis-initiated DIC, which makes a case for its use. AT III is an α2-globulin that inactivates thrombin, factor Xa, factor IXa, and other activated components of the clotting system. Heparin augments AT III, but the increased benefit of a combination of heparin with AT III replacement has not been established. Antifibrinolytic drugs also are used in treatment but are limited to instances of life-threatening bleeding that have not been controlled by blood component replacement therapy.
Maintenance of organ function is achieved by fluid replacement to sustain adequate circulating blood volume and to maintain optimal tissue and organ perfusion. Fluids may be required to restore blood pressure, cardiac output, and urine output to normal parameters.
Thromboembolic Disease
Certain conditions within the blood vessels predispose an individual to develop clots spontaneously.52 A stationary clot attached to the vessel wall is called a thrombus (Figure 27-21). A thrombus is composed of fibrin and blood cells and can develop in either the arterial or venous system. Arterial thrombi form under conditions of high blood flow and are composed mostly of platelet aggregates held together by fibrin strands. Venous thrombi form in conditions of low flow and are composed mostly of red cells with larger amounts of fibrin and few platelets.
Figure 27-21 Thrombus. Thrombus arising in valve pocket at upper end of superficial femoral vein. Postmortem clot on the right is shown for comparison. (From McLachlin J, Paterson JC: Surg Gynecol Obstet 93:1, 1951.)
A thrombus may eventually grow large enough to reduce or obstruct blood flow to tissues or organs, such as the heart, brain, or lungs, depriving them of essential nutrients critical to survival. A thrombus also has the potential of detaching from the vessel wall and circulating within the bloodstream (referred to as an embolus). The embolus may become lodged in smaller blood vessels, blocking blood flow into the local tissue or organ and leading to ischemia. Whether episodes of thromboembolism are life threatening depends on the site of vessel occlusion.
Therapy consists of removal or breakdown of the clot and supportive measures. Anticoagulant therapy is effective in treating or preventing venous thrombosis; it is not as useful in treating or preventing arterial thrombosis. Parenteral heparin is the major anticoagulant used to treat thromboembolism. Oral coumarin drugs also are widely used, particularly for individuals not hospitalized. More aggressive therapy may be indicated for such conditions as pulmonary embolism, coronary thrombosis, or thrombophlebitis. Streptokinase and urokinase activate the fibrinolytic system and are administered to accelerate the lysis of known thrombi. Thrombolytic therapy has limited uses and is prescribed with a high degree of caution because it can cause hemorrhagic complications.
The risk for developing spontaneous thrombi is related to several factors, referred to as the Virchow triad: (1) injury to the blood vessel endothelium, (2) abnormalities of blood flow, and (3) hypercoagulability of the blood.
Vascular endothelial injury can result from atherosclerosis (plaque deposits on arterial walls). Atherosclerosis initiates platelet adhesion and aggregation, promoting the development of atherosclerotic plaques that enlarge, causing further damage and occlusion. Other causes of vessel endothelial injury may be related to hemodynamic alterations associated with hypertension and turbulent blood flow. Injury also is caused by radiation injury, exogenous chemical agents (toxins from cigarette smoke), endogenous agents (cholesterol), bacterial toxins or endotoxins, or immunologic mechanisms. Whatever the precipitating cause of endothelial injury, it is a potent thrombogenic agent.
Sites of turbulent blood flow in the arteries and stasis of blood flow in the veins are at risk for thrombus formation. In areas of turbulence, platelets and endothelial cells may be activated, leading to thrombosis. In sites of stasis, platelets may remain in contact with the endothelium for prolonged times, and clotting factors that would normally be diluted with fresh-flowing blood are not diluted and may become activated. The most common clinical conditions that predispose to venous stasis and subsequent thromboembolic phenomena are major surgery (e.g., orthopedic surgery), acute myocardial infarction, congestive heart failure, limb paralysis, spinal injury, malignancy, advanced age, the postpartum period, and bed rest longer than 1 week. Turbulence and stasis occur with ulcerated atherosclerotic plaques (myocardial infarction), hyperviscosity (polycythemia), and conditions with deformed red cells (sickle cell anemia).
Hypercoagulability, or thrombophilia, is the condition in which an individual is at risk for thrombosis. Hypercoagulability is differentiated according to whether it results from primary (hereditary) or secondary (acquired) causes. Primary causes include defects in proteins involved in hemostasis. Secondary causes include a variety of clinical disorders or conditions (Box 27-9). It is not well understood why there is not a greater incidence of thrombosis formation in hypercoagulable states associated with various disease states and conditions.
Hereditary Thrombophilias: A large number of inherited conditions have been identified that increase the risk to develop thrombosis (Box 27-10).53 Most are autosomal dominant, thus individuals who are homozygous for the mutation are at greatest risk for thrombosis. These include mutations in coagulation proteins, fibrinolytic proteins, platelet receptors, and other factors. The particular mutations that have been most strongly linked as risk factors for venous thrombosis or for arterial thrombosis leading to coronary artery disease or stroke include those that affect fibrinogen, prothrombin (G20210A variant), factor V (factor V Leiden) of the coagulation system, PAI-1 of the fibrinolytic system, the platelet receptor GPIIIa, and methylenetetrahydrofolate reductase (MTHFR), as well as mutations that result in excessive levels of homocysteine (hyperhomocysteinemia). Other inherited thrombophilias are risk factors primarily for venous thrombosis.54 These include deficiencies in protein C, protein S, and AT III.55
Factor V Leiden results from a single nucleotide mutation of guanine to adenine at nucleotide 1691 (G1691A). Activated factor V (Va) is usually inactivated by protein C, but this single mutation results in a change in amino acid 506 from arginine to glutamine. The change alters the site where protein C would cleave factor Va and confers partial resistance, resulting in prolonged high levels of Va and prolongation of clot formation.56 Although this mutation increases the risk for thrombosis, most individuals with factor V Leiden do not have clinically relevant thrombotic events. It is the most common hereditary thrombophilia and is found in about 30% of individuals presenting with deep venous thrombosis (DVT) or pulmonary embolism. It is primarily observed in individuals of European ancestry and in about 5% of whites in the United States and Europe.
The second most common inherited thrombophilia is a mutation in the prothrombin gene, resulting in a replacement of guanine at nucleotide 20210 with an adenine (G20210A variant).54 This mutation is observed in about 2% to 5% of individuals of European ancestry, but is found in 5% to 10% of individuals presenting with venous thrombosis. The G20210A variant leads to overproduction of prothrombin and prolongation of clot formation.
MTHFR mutation leads to alterations in the metabolism of the amino acid homocysteine into methionine and abnormally elevated levels of that amino acid in the blood (hyperhomocysteinemia).57 Acquired hyperhomocysteinemia may result from deficiencies in vitamins B6 or B12, endocrine diseases (e.g., diabetes mellitus, hypothyroidism), pernicious anemia, inflammatory bowel disease, renal failure, and therapy with some drugs. Individuals with homocysteine levels above the 95th percentile are 2.5 times more likely to experience an episode of DVT.
More than 100 different known mutations lead to defects of proteins C, protein S, and AT III and increase the risk of venous thrombosis. Mutations may lead to either quantitative (low levels of protein) or qualitative (production of defective protein) changes.
Tests to diagnose inherited thrombophilias include prothrombin time, partial thromboplastin time, levels of protein C, protein S, and AT III. More elaborate tests to detect precise mutations in factor V, prothrombin, or MTHFR may be indicated.
Acquired Hypercoagulability: Deficiencies in protein S and C and AT III may be acquired and contribute to a hypercoagulable state.56 Conditions associated with an acquired protein deficiency include DIC, liver disease, infection, DVT, acute respiratory distress syndrome, L-asparaginase therapy, HUS, and TTP. The postoperative state also predisposes an individual to protein C or S deficiency; however, its role in contributing to DVT remains unclear.
Acquired hypercoagulable states include the antiphospholipid syndrome (APS), an autoimmune syndrome characterized by autoantibodies against plasma membrane phospholipids and phospholipid-binding proteins. As with most autoimmune diseases, the predominantly affected individual is female and of reproductive age. Those with APS are at risk for arterial and venous thrombosis and a variety of obstetric complications, including pregnancy loss and preeclampsia or eclampsia (Figure 27-22).58 In severe cases the patients may die from recurrent major thrombus formation.59 The pathophysiology is related to autoantibodies directly reacting with platelets or endothelial cells (increasing the risk for thrombosis) or the placental surface (resulting in damage to the placenta). The predominant diagnostic tests measure prolongation of laboratory blood coagulation tests related to an antibody inhibitor (lupus anticoagulant) and specific ELISAs for antibodies against phospholipids (e.g., anticardiolipin antibody) or proteins that bind to phospholipids (e.g., β2-glycoprotein I).60 Highly effective therapy (i.e., unfractionated or low-molecular-weight heparin with low-dose aspirin) is available to prevent the obstetric complications.61
SUMMARY REVIEW
Alterations of Leukocyte Function
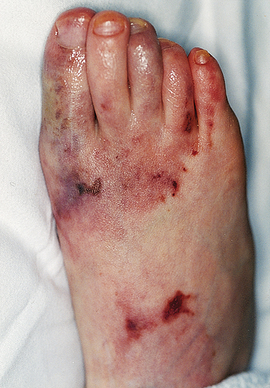
Figure 27-22 Arterial thrombosis associated with antiphospholipid antibodies. A 12-year-old girl with systemic lupus erythematosus and antiphospholipid antibodies with painful cutaneous vasculitis of the right foot. Arterial thrombosis documented by angiography resulted in cyanosis of the large toe. Symptoms resolved with treatment with heparin and corticosteroids. (From Kliegman R, et al: Nelson textbook of pediatrics, ed 18, Philadelphia, 2007, Saunders.)
1. Quantitative alterations of leukocytes (too many or too few) can be caused by bone marrow dysfunction or premature destruction of cells in the circulation. Many quantitative changes in leukocytes occur in response to invasion by microorganisms.
2. Leukocytosis is a condition in which the leukocyte count is higher than normal and is usually a response to stress and invasion of microorganisms.
3. Leukopenia is a condition in which the leukocyte count is lower than normal and is caused by pathologic conditions such as malignancies and hematologic disorders.
4. Granulocytosis (particularly as a result of an increase in neutrophils) occurs in response to infection. The marrow releases immature cells, causing a shift-to-the-left, when responding to an infection that has created a demand for neutrophils that exceeds the supply in the circulation.
5. Eosinophilia results most commonly from parasitic invasion and ingestion or inhalation of toxic foreign particles.
6. Basophilia is seen in hypersensitivity reactions because of the high content of histamine and subsequent release.
7. Monocytosis occurs during the late or recuperative phase of infection when macrophages (mature monocytes) phagocytose surviving microorganisms and debris.
8. Granulocytopenia, a significant decrease in neutrophils, can be a life-threatening condition if sepsis occurs; it is often caused by chemotherapeutic agents, severe infection, and radiation.
9. Infectious mononucleosis is an acute infection of B lymphocytes most commonly associated with EBV, a type of herpesvirus. Transmission of EBV is by personal contact, commonly through saliva, thus its nickname, the kissing disease.
10. Two of the earliest manifestations of momonucleosis are sore throat and fever caused by inflammation at the primary site of viral entry.
11. Most causes of EBV mononucleosis include fever lasting 7 to 10 days, sore throat, and enlargement and tenderness of the cervical lymph nodes. It is self-limiting, and treatment consists of rest and relief of symptoms.
12. The common pathologic feature of all forms of leukemia is an uncontrolled proliferation of leukocytes, overcrowding the bone marrow and resulting in decreased production and function of the other blood cell lines.
13. All leukemias are classified by the cell type involved, lymphocytic or myelogenous, and are differentiated by onset, acute or chronic. Thus there are four major types of leukemia: ALL, CLL, AML, and CML.
14. Although the exact cause of leukemia is unknown, it is considered a clonal disorder. A high incidence of acute leukemias and CLL is reported in certain families, suggesting a genetic predisposition.
15. The most common genetic abnormality in adult ALL is the Philadelphia chromosome. In about a third of clients with AML there is a mutation in the receptor tyrosine kinase FLT3.
16. In leukemia, blasts (precursor cells) “crowd out” the marrow and cause cellular proliferation of the other cell lines to cease.
17. The major clinical manifestation of leukemia includes fatigue caused by anemia, bleeding caused by thrombocytopenia, fever secondary to infection, anorexia, and weight loss.
18. Chemotherapy is the treatment of choice for leukemia. Acute leukemias are associated with an increasing survival rate of 80% to 90%, with long-term survival of 30% to 40%. Chronic leukemias are associated with a longer life expectancy than are acute leukemias.
19. Chronic leukemias progress differently than acute leukemias, advancing slowly and without warning. The presence of the Philadelphia chromosome is a diagnostic marker for CML.
Alterations of Lymphoid Function
1. The number of lymphocytes is decreased (lymphocytopenia) in most acute infections and in some immunodeficiency syndromes.
2. Lymphocytosis occurs in viral infections (IM and infectious hepatitis, in particular), leukemia, lymphomas, and some chronic infections.
3. Lymphomas are tumors of primary lymphoid tissue (thymus, bone marrow) or secondary lymphoid tissue (lymph nodes, spleen, tonsils, intestinal lymphoid tissue). The two major types of malignant lymphomas are Hodgkin lymphoma (HL) and non-Hodgkin lymphoma (NHL).
4. HL is associated with a highly distinctive cell, the Reed-Sternberg cell (RS), in the lymph nodes. The RS cell is derived from a malignant B cell that usually becomes binucleate.
5. A virus might be involved in the pathogenesis of HL. Some familial clustering suggests an unknown genetic mechanism.
6. An enlarged painless mass or swelling, most commonly in the neck, is an initial sign of HL. Local symptoms are produced by lymphadenopathy, usually caused by pressure or obstruction.
7. Treatment of HL includes radiation therapy and chemotherapy. A cure is possible regardless of the stage of HL; however, individuals treated with chemotherapy who relapse in less than 2 years have a poor prognosis.
8. The cause of lymph node enlargement and cancerous transformation in NHL is unknown. Immunosuppressed persons have a higher incidence of NHL, suggesting an immune mechanism.
9. Generally, with NHL, the swelling of lymph nodes is painless, and the nodes enlarge and transform over months or years.
10. Individuals with NHL can survive for long periods. Treatment is chemotherapy.
11. Burkitt lymphoma involves the jaw and facial bones and occurs in children from east-central Africa and New Guinea.
12. Multiple myeloma (MM) is a neoplasm of B cells (immature plasma cells) and mature plasma cells. It is characterized by multiple malignant tumor masses of plasma cells scattered throughout the skeletal system and sometimes found in soft tissue.
13. Myeloma cells usually secrete monoclonal protein (M protein) that is an abnormal antibody molecule. The myeloma cell may also secrete free antibody light chain that is excreted in the urine (Bence Jones protein).
14. The exact cause of MM is unknown, but genetic factors and chronic stimulation of the mononuclear phagocyte system by bacteria, viral agents, and chemicals have been suggested.
15. The major clinical manifestations for MM include recurrent infections caused by suppression of the humoral immune response and renal disease as a result of Bence Jones proteinuria.
16. Chemotherapy is the treatment of choice for MM. Survival is still only 2 to 3 years with chemotherapy, however. Treatment with thalidomide is showing promise as an effective therapeutic agent in producing long-term remissions.
17. Waldenström macroglobulinemia is a rare type of slow-growing plasma cell tumor that secretes a monoclonal IgM molecule.
Alterations of Splenic Function
1. Splenomegaly (enlargement of the spleen) may be considered normal in certain individuals, but its presence should not be ignored.
2. Splenomegaly results from (a) acute inflammatory or infectious processes, (b) congestive disorders, (c) infiltrative processes, and (d) tumors or cysts.
3. Hypersplenism (overactivity of the spleen) results from splenomegaly. Hypersplenism results in sequestering of the blood cells, causing increased destruction of red blood cells, which leads to the development of anemia.
Alterations of Platelets and Coagulation
1. Thrombocytopenia is characterized by a platelet count less than 100,000 platelets/mm3 of blood; a count less than 50,000/mm3 increases the potential for hemorrhage associated with minor trauma.
2. Thrombocytopenia exists in primary or secondary forms and is commonly associated with autoimmune diseases and viral infections; bacterial sepsis with DIC also results in thrombocytopenia.
3. Heparin-induced thrombocytopenia develops in approximately 4% of individuals receiving unfractionated heparin.
4. Immune thrombocytopenic purpura (ITP) is a major cause of platelet destruction, often affecting females, and results in hemorrhaging that ranges from minor development of petechiae to major bleeding from mucosal sites.
5. Thrombotic thrombycytopenic purpura (TTP) causes platelet aggregation leading to microcirculatory occlusion.
6. Thrombocythemia is characterized by a platelet count more than 400,000 platelets/mm3 of blood and is symptomatic when the count exceeds 1 million/mm3, at which time the risk for intravascular clotting (thrombosis) is high.
7. Thrombocythemia is caused by accelerated platelet production in the bone marrow.
8. Qualitative alterations in normal platelet adherence or aggregation prevent platelet plug formation and may result in prolonged bleeding times.
9. Prolonged bleeding can result from alterations in platelet function, including adhesion between platelets and the vessel wall, platelet-platelet adhesion, platelet granule secretion, arachidonic acid pathway activity, and membrane phospholipid regulation.
10. Disorders of coagulation are usually caused by defects or deficiencies of one or more clotting factors.
11. Coagulation is impaired when there is a deficiency of vitamin K because of insufficient production of prothrombin and synthesis of clotting factors II, VII, IX, and X, often associated with liver diseases.
12. DIC is a complex syndrome that results from a variety of clinical conditions that release tissue factor causing an increase in fibrin and thrombin activity in the blood and producing augmented clot formation and accelerated fibrinolysis. Sepsis is a condition that is often associated with DIC.
13. DIC is characterized by a cycle of intravascular clotting followed by active bleeding caused by the initial consumption of coagulation factors and platelets and diffuse fibrinolysis.
14. Diagnosis of DIC is based on measurement in the blood of end products characteristic of dysfunctional coagulation activity. Treatment is complex and nonstandardized and focused on removing the primary cause, restoring hemostasis, and preventing further organ damage.
15. Thromboembolic disease results from a fixed (thrombus) or moving (embolus) clot that blocks flow within a vessel, denying nutrients to tissues distal to the occlusion; death can result when clots obstruct blood flow to the heart, brain, or lungs.
16. Hypercoagulability is the result of deficient anticoagulation proteins. Secondary causes are conditions that promote venous stasis.
17. The term Virchow triad refers to three factors that can cause thrombus formation: (1) injury to the vessel wall, (2) abnormalities of blood flow, and (3) alterations in the blood constituents leading to hypercoagulability.
18. Autoantibodies against phospholipids result in a state of acquired hypercoagulability, an increased risk for venous or arterial thrombosis, and a high incidence of pregnancy complications.
Acute idiopathic TTP 1047
Acute leukemia 1019
Acute lymphocytic leukemia (ALL) 1021
Acute myelogenous leukemia (AML) 1021
Agranulocytosis 1015
Arterial thrombus(pl., thrombi) 1055
Basopenia 1017
Basophilia 1017
B-cell neoplasms 1033
Bence Jones protein 1038
β2-microglobulin 1041
Burkitt lymphoma 1035
Chronic leukemia 1019
Chronic lymphocytic leukemia (CLL) 1025
Chronic myelogenous leukemia (CML) 1025
Chronic relapsing TTP 1047
Compensated DIC 1053
Congestive splenomegaly 1043
Consumptive thrombohemorrhagic disorders 1050
d-dimer 1054
Disseminated intravascular coagulation (DIC) 1050
Embolus 1055
Eosinopenia 1017
Eosinophilia 1015
Erythromyalgia 1048
Essential (primary) thrombocythemia (ET) 1047
Granulocytopenia 1015
Granulocytosis 1015
Heparin-induced thrombocytopenia (HIT) 1044
Heterophile antibodies 1018
Hodgkin lymphoma (HL) 1031
Hypercoagulability 1055
Hypersplenism 1042
Immune thrombocytopenic purpura (ITP) 1045
Impaired hemostasis 1049
Infectious mononucleosis (IM) 1017
Infiltrative splenomegaly 1043
Leukemia 1019
Leukemoid reaction 1015
Leukocytosis 1014
Leukopenia 1014
Lymphadenopathy 1030
Lymphoblastic lymphoma (LL) 1036
Lymphocytopenia 1017
Lymphocytosis 1017
Lymphoplasmacytic lymphoma 1042
Microvasculature thrombosis 1048
Monoclonal gammopathy of undetermined significance (MGUS) 1040
Monocytopenia 1017
Monocytosis 1017
M protein 1038
Multiple myeloma, (MM 1037
Neutropenia 1015
Neutrophilia 1015
NK-cell neoplasms 1033
Non-Hodgkin lymphoma (NHL) 1033
Pancytopenia 1021
Philadelphia chromosome 1020
Pseudothrombocytopenia 1044
Purpura 1044
Qualitative leukocyte disorder 1014
Quantitative leukocyte disorder 1014
Reed-Sternberg (RS) cell 1031
Shift-to-the-left 1015
Smoldering myeloma 1040
Solitary plasmacytoma 1040
Splenomegaly 1042
T-cell neoplasms 1033
Thrombocythemia 1047
Thrombocytopenia 1044
Thrombocytosis 1047
Thromboembolic disease 1044
Thrombophilia 1055
Thrombotic thrombocytopenic purpura (TTP) 1046
Thrombus 1055
Vasculitis 1049
Venous thrombus 1055
Virchow triad 1055
Waldenström macroglobulinemia 1042
REFERENCES
1. Marris, J.A. Care of patients with neutropenia. Clin J Oncol Nurs. 2006;10(2):164–166.
2. Bell, A.T., Fortune, B., Sheeler, R. Clinical inquiries: what test is the best for diagnosing infectious mononucleosis? J Fam Pract. 2006;55(9):799–802.
3. Gunz, F.W. The dreaded leukemias and the lymphomas: their nature and their prospects. In: Wintrobe M.M., ed. Blood, pure and eloquent: a story of discovery, of people, and of ideas. New York: McGraw-Hill, 1980.
4. , SEER stat fact sheets—acute lymphocytic leukemia. Bethesda, MD: National Institutes of Health, U.S. Department of Health and Human Services; 2008. Available at. http://seer.cancer.gov/statfacts/html/alyl.html [Accessed February 3, 2009].
5. Druker, B.J. Translation of the Philadelphia chromosome into therapy for CML. Blood. 2008;112(13):4808–4817.
6. Keen-Kim, D., Nooraie, F., Rao, P.N. Cytogenetic biomarkers for human cancer. Front Biosci. 2008;13(May 1):5928–5949.
7. Dick, J.E. Stem cell concepts renew cancer research. Blood. 2008;112(13):4793–4807.
8. , SEER stat fact sheets—acute myeloid leukemia. Bethesda, MD: National Institutes of Health, U.S. Department of Health and Human Services; 2008. Available at. http://seer.cancer.gov/statfacts/html/amyl.html [Accessed February 3, 2009].
9. , Childhood acute lymphoblastic leukemia treatment: health professional version. Bethesda, MD: National Institutes of Health, U.S. Department of Health and Human Services; 2008. Available at. www.cancer.gov/cancertopics/pdq/treatment/childALL/healthprofessional [Accessed February 3, 2009].
10. , Adult acute lymphoblastic leukemia treatment: health professional version. Bethesda, MD: National Institutes of Health, U.S. Department of Health and Human Services; 2008. Available at. www.cancer.gov/cancertopics/pdq/treatment/adultALL/healthprofessional [Accessed February 3, 2009].
11. Estey, E., Dohner, H. Acute myeloid leukemia. Lancet. 2006;368(9550):1894–1907.
12. , Adult acute myeloid leukemia treatment: health professional version. Bethesda, MD: National Institutes of Health, U.S. Department of Health and Human Services; 2008. Available at. www.cancer.gov/cancertopics/pdq/treatment/adultAML/healthprofessional [Accessed February 3, 2009].
13. , SEER stat fact sheets—chronic lymphoblastic leukemia. Bethesda, MD: National Institutes of Health, U.S. Department of Health and Human Services; 2008. Available at. http://seer.cancer.gov/statfacts/html/clyl.html [Accessed February 3, 2009].
14. , SEER stat fact sheets—chronic myeloid leukemia. Bethesda, MD: National Institutes of Health, U.S. Department of Health and Human Services; 2008. Available at. http://seer.cancer.gov/statfacts/html/alyl.html [Accessed February 3, 2009].
15. Montserrat, E., Moreno, C. Chronic lymphocytic leukemia: a short overview. Ann Oncol. 2008;19(Suppl 7):vii320–vii325.
16. Savona, M., Talpaz, M. Getting to the stem of chronic myeloid leukemia. Nat Rev Cancer. 2008;8(5):341–350.
17. Kantarjian, H.M., et al. New insights into the pathophysiology of chronic myeloid leukemia and imatinib resistance. Ann Intern Med. 2006;145(12):913–923.
18. Jaffe, E.S., et al. Classification of lymphoid neoplasms: the microscope as a tool for disease discovery. Blood. 2008;112(12):4384–4399.
19. , SEER stat fact sheets—Hodgkin lymphoma. Bethesda, MD: National Institutes of Health, U.S. Department of Health and Human Services; 2008. Available at. http://seer.cancer.gov/statfacts/html/hodg.html [Accessed February 3, 2009].
20. , What you need to know about Hodgkin lymphoma. Bethesda, MD: National Institutes of Health, U.S. Department of Health and Human Services; 2008. Available at. http://cancer.gov/cancertopics/wyntk/hodgkin [Accessed February 12, 2009].
21. , SEER stat fact sheets—non-Hodgkin lymphoma. Bethesda, MD: National Institutes of Health, U.S. Department of Health and Human Services; 2008. Available at. http://seer.cancer.gov/statfacts/html/nhl.html [Accessed February 3, 2009].
22. , Adult non-Hodgkin lymphoma treatment: health professional version. Bethesda, MD: National Institutes of Health, U.S. Department of Health and Human Services; 2008. Available at. www.cancer.gov/cancertopics/pdq/treatment/myeloma/healthprofessional [Accessed February 3, 2009].
23. , Multiple myeloma and other plasma cell neoplasms treatment: health professional version. Bethesda, MD: National Institutes of Health, U.S. Department of Health and Human Services; 2008. Available at. www.cancer.gov/cancertopics/pdq/treatment/adult-non-hodgkins/healthprofessional [Accessed February 3, 2009].
24. , SEER stat fact sheets—myeloma. Bethesda, MD: National Institutes of Health, U.S. Department of Health and Human Services; 2008. Available at. http://seer.cancer.gov/statfacts/html/mulmy.html [Accessed February 3, 2009].
25. Kyle, R.A., Rajkumar, S.V. Criteria for diagnosis, staging, risk stratification and response assessment of multiple myeloma. Leukemia. 2009;23(1):3–9.
26. Yasui, H., et al. Recent advances in the treatment of multiple myeloma. Curr Pharm Biotechnol. 2006;7(5):381–393.
27. , Waldenström macroglobulinemia: questions and answers: fact sheet. Bethesda MD: National Institutes of Health, U.S. Department of Health and Human Services; 2008. Available at. http://cancer.gov/cancertopics/factsheet/Sites-Types/WM [Accessed February 12, 2009].
28. Dimopoulos, M.A., et al. Update on treatment recommendations from the fourth international workshop on Waldenström’s macroglobulinemia. J Clin Oncol. 2009;27(1):120–126.
29. Nachman, R.L., Rafii, S. Platelets, petechiae, and preservation of the vascular wall. N Eng J Med. 2008;359(12):1261–1270.
30. , Thrombocytopenia. Bethesda, MD: National Institutes of Health, U.S. Department of Health and Human Services; 2008. Available at. www.nhlbi.nih.gov/health/dci/Diseases/thcp/thcp_all.html [Accessed February 3, 2009].
31. Selleng, K., Selleng, S., Greinacher, A. Heparin-induced thrombocytopenia in intensive care patients. Semin Thromb Hemost. 2008;34(5):425–438.
32. Warkentin, T.E., Sheppard J-, A.I. Testing for heparin-induced thrombocytopenia antibodies. Transfus Med Rev. 2006;20(4):259–272.
33. , Idiopathic thrombocytopenic purpura. Bethesda, MD: National Institutes of Health, U.S. Department of Health and Human Services; 2008. Available at. www.nhlbi.nih.gov/health/dci/Diseases/Itp/ITP.all.html [Accessed February 3, 2009].
34. Curtis, B.R. Genotyping for human platelet alloantigen polymorphisms: applications in the diagnosis of alloimmune platelet disorders. Semin Thromb Hemost. 2008;34(6):539–548.
35. Arnold, D.M., Smith, J.W., Kelton, J.G. Diagnosis and management of neonatal alloimmune thrombocytopenia. Transfus Med Rev. 2008;22(4):255–267.
36. Bennett, C.M., de Jong, J.L.O., Neufeld, E.J. Targeted ITP strategies: do they elucidate the biology of ITP and related disorders? Pediatr Blood Cancer. 2006;47(5 Suppl):706–709.
37. Psaila, B., Bussel, J.B. Refractory immune thrombocytopenic purpura: current strategies for investigation and management. Brit J Haematol. 2008;143(1):16–26.
38. , Thrombotic thrombocytopenic purpura. Bethesda, MD: National Institutes of Health, U.S. Department of Health and Human Services; 2008. Available at. www.nhlbi.nih.gov/health/dci/Diseases/Itp/ITP.all.html [Accessed February 16, 2009].
39. , Thrombocytopenia & thrombocytosis. Bethesda, MD: National Institutes of Health, U.S. Department of Health and Human Services; 2008. Available at. www.nhlbi.nih.gov/health/dci/Diseases/thrm/thrm_all.html [Accessed February 3, 2009].
40. Harrison, C.N., Green, A.R. Essential thrombocythaemia. Best Pract Res Clin Haematol. 2006;19(3):439–453.
41. Landolfi, R., Di Gennaro, L., Falanga, A. Thrombosis in myeloproliferative disorders: pathogenetic facts and speculation. Leukemia. 2008;22(11):2020–2028.
42. Kota, J., Caceres, N., Constantinescu, S.N. Aberrant signal transduction pathways in myeloproliferative neoplasms. Leukemia. 2008;22(10):1828–1840.
43. Tefferi, A., Elliott, M. Thrombosis in myeloproliferative disorders: prevalence, prognostic factors, and the role of leukocytes and JAK2V617F. Semin Thromb Hemost. 2007;33(4):313–320.
44. Barbui, T., Finazzi, G. Therapy for polycythemia vera and essential thrombocythemia is driven by the cardiovascular risk. Semin Thromb Hemost. 2007;33(4):321–329.
45. Salles, I.I., et al. Inherited traits affecting platelet function. Blood Rev. 2008;22(3):155–172.
46. Wada, H., Usui, M., Sakuragawa, N. Hemostatic abnormalities and liver disease. Semin Thromb Hemost. 2008;34(8):772–778.
47. Taylor, F.B., Jr., et al. Towards definition, clinical and laboratory criteria, and a scoring system for disseminated intravascular coagulation. Thromb Hemost. 2001;86(5):1327–1330.
48. Furlong MA, Furlong BR: Disseminated intravascular coagulation. Available at www.emedicine.com/emerg/topic150.htm. Accessed March 16, 2005.
49. Kwaan, H.C., Vicuna, B. Incidence and pathogenesis of thrombosis in hematologic malignancies. Semin Thromb Hemost. 2007;33(4):303–312.
50. Palumbo, J.S. Mechanisms linking tumor cell-associated procoagulant function to tumor dissemination. Semin Thromb Hemost. 2008;34(2):154–160.
51. Wada, H., Sakuragawa, N. Are fibrin-related markers useful for the diagnosis of thrombosis. Semin Thromb Hemost. 2008;34(1):33–38.
52. , Excessive blood clotting. Bethesda, MD: National Institutes of Health, U.S. Department of Health and Human Services; 2008. Available at. www.nhlbi.nih.gov/health/dci/Diseases/ebc/ebc_all.html [Accessed February 3, 2009].
53. Chan, M.Y., Andreotti, F., Becker, R.C. Hypercoagulable states in cardiovascular disease. Circulation. 2008;118(22):2286–2297.
54. Lippi, G., Franchini, M. Pathogenesis of venous thromboembolism: when the cup runneth over. Semin Thromb Hemost. 2008;34(8):747–761.
55. Middeldorp, S., Vlieg, A.H. Does thrombophilia testing help in the clinical management of patients? Brit J Haematol. 2008;143(3):321–335.
56. Boekholdt, S.M., Kramer, M.H.H. Arterial thrombosis and the role of thrombophilia. Semin Thromb Hemost. 2007;33(6):588–596.
57. Cohn, D.M., Roshani, S., Middeldorp, S. Thrombophilia and venous thromboembolism: implications for testing. Semin Thromb Hemost. 2007;33(6):573–581.
58. Tincani, A., et al. Lupus and the antiphospholipid syndrome in pregnancy and obstetrics: clinical characteristics, diagnosis, pathogenesis, and treatment. Semin Thromb Hemost. 2008;34(3):267–273.
59. Merrill, J.T., Asherson, R.A. Catastrophic antiphosholipid syndrome. Nat Clin Pract Rheumatol. 2006;2(2):81–89.
60. Pierangeli, S.S., Harris, E.N. A quarter of a century in anticardiolipin antibody testing and attempted standarization has led us to here, which is? Semin Thromb Hemost. 2008;34(4):313–328.
61. Heilmann, L., et al. Pregnancy outcome in women with antiphospholipid antibodies: report on a retrospective study. Semin Thromb Hemost. 2008;34(8):794–802.