SHOCK, MULTIPLE ORGAN DYSFUNCTION SYNDROME, AND BURNS IN ADULTS



Shock occurs when the cardiovascular system fails to perfuse tissues adequately, resulting in widespread impairment of cellular metabolism. Because tissue perfusion can be disrupted by any factor that alters heart function, blood volume, or blood pressure, shock has many causes and various clinical manifestations. Ultimately, however, shock from any cause progresses to organ failure and death, unless compensatory mechanisms reverse the process or clinical intervention succeeds. Untreated severe shock overwhelms the body’s compensatory mechanisms through positive-feedback loops that initiate and maintain a downward physiologic spiral.
Multiple organ dysfunction syndrome (MODS) is progressive and often involves the ultimate failure of two or more organ systems after a severe illness or injury. The disease process is initiated and perpetuated by uncontrolled systemic inflammatory and stress responses and is characterized by a hypermetabolic and hyperdynamic state that persists as organ dysfunction develops. For many years the syndrome was referred to as multiple organ failure or multiple systems organ failure. Gradually it was recognized that the term organ dysfunction more accurately describes the syndrome as a process of physiologic deterioration.
Major burns result in extensive immediate tissue injury and thus are a form of trauma with wide-reaching effects on all organ systems. The cause of injury may be thermal contact, flame, chemical agents, or electrical agents; each cause requires a different approach in diagnosis and treatment. Closely associated with thermal burns is smoke inhalation injury, which accounts for about 25% of all burn unit admissions. As a multiorgan problem, thermal injuries can have an overwhelming effect on survival of the burned individual. Regardless of the cause of burns, the result is a final common pathway of physiologic response dependent on the extent of burn surface involvement and depth of tissue destruction.
SHOCK
Shock can be classified by type, principal pathophysiologic process, or clinical manifestations. Classification by type is perhaps the most useful because it suggests the cause and pathophysiologic process of the underlying disorder, which must be treated to prevent the irreversible impairment of cellular metabolism. Shock is classified as cardiogenic (caused by heart failure); neurogenic or vasogenic (caused by alterations in vascular smooth muscle tone); anaphylactic (caused by hypersensitivity); septic (caused by infection); or hypovolemic (caused by insufficient intravascular fluid volume). An additional type, traumatic shock, has components of hypovolemic and septic shock.1
Cellular Alterations
Because the body is made up of many cells that may function or malfunction at different stages of metabolic impairment, shock causes many signs and symptoms. Subjective complaints are usually nonspecific and may not be particularly helpful to the clinician attempting diagnosis and treatment. The individual may report feeling sick, weak, cold, hot, nauseated, dizzy, confused, afraid, thirsty, and short of breath.
Impairment of Cellular Metabolism
The common pathway in all types of shock is impairment of cellular metabolism, which is a complex concept. Figure 46-1 illustrates the pathophysiology of shock at the cellular level.
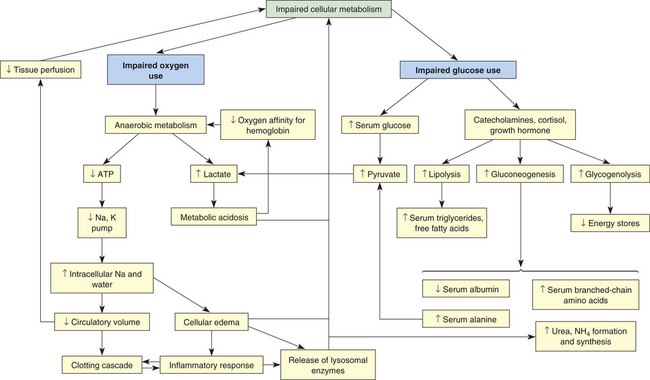
Impairment of Oxygen Use
In all types of shock the cell either is not receiving an adequate amount of oxygen or is unable to use oxygen (see Figure 46-1). In cardiogenic shock, cardiac output is too low to deliver adequate oxygen to the cell. In hypovolemic shock, oxygen delivery is impaired by inadequate numbers of red cells or inadequate volume of intravascular fluid. In neurogenic, anaphylactic, and septic shock, systemic vascular resistance (SVR) is too low and perfusion pressure in the capillaries is inadequate to drive oxygen across cell membranes. In septic shock, hypoxia is made worse by fever, which increases the cell’s oxygen consumption rate, and by endotoxic and inflammatory chemical disruption of cell metabolism, which impairs the cells’ ability to use oxygen.
Without oxygen the cell shifts from aerobic to anaerobic metabolism. Anaerobic metabolism is a less efficient method of extracting energy from carbon bonds, and the cell begins to use adenosine triphosphate (ATP) faster than it can be replaced. Without ATP the cell loses its ability to maintain an electrochemical gradient across its selectively permeable membrane. Specifically, the cell cannot operate the sodium-potassium pump. Sodium and chloride accumulate inside the cell, and potassium exits. Cells of the nervous system and myocardium are profoundly and immediately affected. The resting potentials of these cells are reduced, and action potentials decrease in amplitude (see Chapter 1). Myocardial depressant factor also decreases the contractility of the heart. A variety of clinical manifestations of impaired central nervous system and myocardial function result.
As sodium moves into the cell, water follows. Throughout the body, the water drawn from the interstitium into the cells is “replaced” by water that is in turn drawn out of the vascular space, often called “third spacing” of fluid. This decreases circulatory volume. Within the cells, water causes cellular edema that disrupts cellular membranes, releasing lysosomal enzymes that injure the cells internally and leak into the interstitium.
Three positive-feedback loops then begin that further impair oxygen use: (1) activation of the clotting cascade, (2) decreased circulatory volume, and (3) lysosomal enzyme release. First, enzymatic processes are disrupted by the change in the normal ionic and osmotic levels in the cell, as are those processes governed by the physical laws of diffusion. Diffusion of nutrients and wastes into and out of the cell takes longer, and cellular metabolism is further altered. At the same time, diffusion across capillary membranes occurs more slowly as blood flow in the capillary beds becomes sluggish. Sluggish capillary flow decreases tissue perfusion further and activates the clotting cascade (see Chapter 25). The clotting cascade accounts for common complications of shock, such as acute tubular necrosis, acute respiratory distress syndrome (ARDS), and disseminated intravascular coagulation (DIC). It also may activate or be activated by the inflammatory response.2
Intravascular fluid loss into the intracellular and interstitial spaces, described as “third spacing” earlier, is amplified when serum albumin and other plasma proteins are consumed for fuel, which results in decreased intravascular osmotic pressure, shift of fluid to the interstitial or extracellular spaces, and decreased circulation volume. Decreased circulatory volume magnifies decreased tissue perfusion in all types of shock. Decreased intravascular volume causes decreased cardiac output in septic shock and further decreases cardiac output in cardiogenic shock. In individuals with anaphylactic, neurogenic, or septic shock and an already dilated vasculature, hypotension worsens as a result of decreased circulatory volume. New data on additional mechanisms for hypotension (vasodilation) are illustrated in Figure 46-7 (p. 1706).
Lysosomal enzymes released during shock injure the cell that released them and injure adjacent cells. By damaging the mechanisms of surrounding cells, lysosomal enzymes extend areas of impaired metabolism and cellular injury.
In addition to decreasing ATP stores, anaerobic metabolism affects the pH of the cell, and metabolic acidosis develops. A compensatory mechanism is initiated that enables cardiac and skeletal muscles to use lactic acid as a fuel source, but only for a limited time. The decreasing pH of the cell that is functioning anaerobically has serious consequences. Enzymes necessary for cellular function dissociate under acid conditions. Enzyme dissociation stops cell function, repair, and division. As lactic acid is released systemically, blood pH drops, reducing the oxygen-carrying capacity of the blood (see Chapter 2). Therefore, less oxygen is delivered to the cells. Further acidosis triggers the release of more lysosomal enzymes because the low pH disrupts lysosomal membrane integrity.
Impairment of Glucose Use
Impaired glucose use can be caused by either impaired glucose delivery or impaired glucose uptake by the cells (see Figure 46-1). The reasons for inadequate glucose delivery are the same as those enumerated for inadequate oxygen delivery. In addition, in septic and anaphylactic shock, glucose metabolism may be increased or disrupted because of fever or bacteria, and glucose uptake can be prevented by the presence of vasoactive toxins, endotoxins, histamine, and kinins.
Some of the compensatory mechanisms activated by shock contribute to decreased glucose uptake by the cells. High serum levels of cortisol, growth hormone, and catecholamines account for hyperglycemia and insulin resistance, tachycardia, increased SVR, and increased cardiac contractility. Cells shift to glycogenolysis, gluconeogenesis, and lipolysis to generate fuel for survival (see Chapter 1). Except in the liver, kidneys, and muscles, the body’s cells have extremely limited stores of glycogen. In fact, total body stores can fuel the metabolism for only about 10 hours. The depletion of fat and glycogen stores is not itself a cause of organ failure, but the energy costs of glycogenolysis and lipolysis are considerable and contribute to the cells’ failure.
The depletion of protein is, however, a cause of organ failure. When gluconeogenesis causes proteins to be used for fuel, these proteins are no longer available to maintain cellular structure, function, repair, and replication. The breakdown of protein occurs in starvation states, hyperdynamic metabolic states, and septic shock. Under anaerobic metabolism, protein breakdown liberates alanine, which is converted to pyruvate. In sepsis, pyruvic acid is changed into lactic acid and a positive-feedback loop is formed.
As proteins are broken down anaerobically, ammonia and urea are produced. Ammonia is toxic to living cells. Uremia develops, and uric acid further disrupts cellular metabolism. Proteins are broken down preferentially. Serum albumin and other plasma proteins are consumed for fuel first. Serum protein consumption decreases capillary osmotic pressure and contributes to the development of interstitial edema, creating another positive-feedback loop that decreases circulatory volume. In septic shock, plasma protein breakdown includes breakdown of immunoglobulins, thereby impairing immune system function when it is most needed.
Muscle wasting caused by protein breakdown weakens skeletal and cardiac muscle. Skeletal muscle wasting impairs the muscles that facilitate breathing. Muscle wasting therefore alters the actions of both heart and lungs. The delivery of oxygen and glucose to the cells is directly reduced, as is the removal of waste products, forming another positive-feedback loop.
A final outcome of impaired cellular metabolism is the buildup of metabolic end products in the cell and interstitial spaces. Waste products are toxic to the cells and further disrupt cellular function and membrane integrity. In septic shock, for example, a deficiency in cellular metabolism and the buildup of toxins may precede and cause decreased tissue perfusion.
Types of Shock
Each type of shock (cardiogenic, hypovolemic, neurogenic, anaphylactic, septic) involves numerous clinical manifestations that also characterize many other conditions, making diagnosis difficult. In addition, the body’s many compensatory mechanisms can mask, for a time, many definitive signs of shock.
Cardiogenic Shock
Cardiogenic shock results from the inability of the heart to pump adequate blood to tissues and end organs from any cause, the most common being within hours of an acute myocardial infarction or severe episode of myocardial ischemia. Cardiogenic shock is defined as persistent hypotension and tissue hypoperfusion caused by cardiac dysfunction in the presence of adequate intravascular volume and left ventricular filling pressure.3 Pathologic conditions that reduce contractility, impair diastolic filling, or cause obstruction can lead to cardiogenic shock. The decreased contractility can result from (1) acute myocardial infarction (AMI), cardiomyopathy, sepsis, myocarditis, dysrhythmias, metabolic abnormalities, papillary muscle rupture; (2) impaired diastolic filling related to arrhythmias; and (3) obstruction due to pulmonary embolism, cardiac tamponade, valvular disorders, and wall rupture or defects.4 Although mortality rates have been estimated between 50% and 80%, there is some indication that overall hospital mortality is decreasing because of early recognition and interventional treatment advancements.5 Mortality improves with early revascularization, cardio-supportive drug regimens, and mechanical assistive devices.
Reperfusion and revascularization can be achieved with treatment of fibrinolytic therapies (medications that disintegrate the coronary thrombus), percutaneous interventions (balloon angioplasty, stent placement, and thrombectomies), or surgery (coronary artery bypass, ventriculoplasty or heart transplant) to open the coronary vessels during an acute myocardial infarction (AMI) or replace irreparable heart muscle. Cardio-supportive drug and fluid regimens are initiated to maintain adequate blood pressure, and essential fluid and electrolyte balance, and optimize coronary perfusion to the myocardium. Mechanical assist devices, specifically intra-aortic balloon pumps and percutaneous or ventricular assist devices (VADS), are used to support cardiac output temporarily until the individual improves or transplantation is possible. Implantable VADS, pacemakers, or internal defibrillator devices are sometimes used as permanent treatment for those who survive cardiogenic shock. Continuous hemodynamic monitoring should be used to evaluate vascular volume and pressures, optimize fluid, and monitor drug administration with the goal of improving cardiac output and tissue perfusion.6
As cardiac output decreases, compensatory adaptive responses are activated, such as the renin-angiotensin, neurohormonal, and sympathetic nervous systems, that lead to fluid retention, systemic vasoconstriction, and tachycardia.7 Blood pressure is maintained through vasoconstriction in response to catecholamine release from the adrenals. Catecholamines also increase contractility and heart rate. Increases in blood volume and vascular resistance succeed in normalizing blood pressure and increasing cardiac performance but at the cost of increasing myocardial demands for oxygen and nutrients. Increasing myocardial requirements further strain the already failing heart, which can no longer pump an adequate volume of blood with sufficient force to perfuse the tissues. Thus increased coronary, tissue, and cellular ischemia progressively deteriorate myocardial dysfunction and the shock state (see What’s New? Cardiogenic Shock).
The clinical manifestations of cardiogenic shock are caused by inadequate perfusion to the heart and end organs (Figure 46-2). Subjective complaints of chest pain, dyspnea,
WHAT’s NEW?
Cardiogenic Shock
The “Should We Emergently Revascularize Occluded Coronaries for Cardiogenic Shock” (SHOCK) trial demonstrated that strategies of early revascularization versus initial medical stabilization showed greater reduction in mortality with the latter. However, a relative overall 67% improvement in survival at 6 years was found in the cohort of survivors sustaining acute myocardial infarction with complication of cardiogenic shock who had received early revascularization.
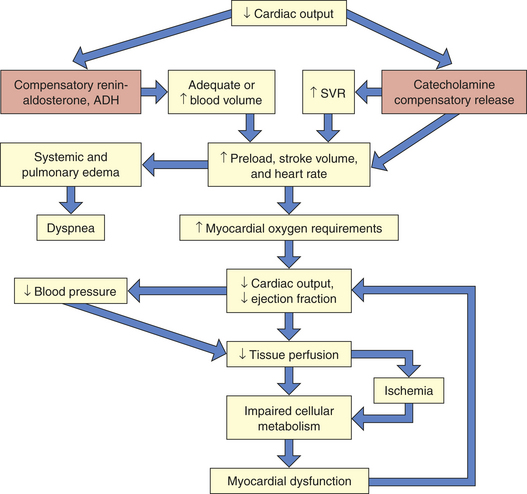
Figure 46-2 Cardiogenic shock. Shock becomes life threatening when compensatory mechanisms (orange boxes) cause increased myocardial oxygen requirements. ADH, Antidiuretic hormone; SVR, systemic vascular resistance.
Data from Hochman J et al: JAMA 295(21):2511-2515, 2006.
and faintness, along with feelings of impending doom, are often revealed. Classic observable signs and symptoms of tachycardia, tachypnea, hypotension, jugular venous distention, and low measured cardiac output are hallmarks. Cyanosis; skin mottling; rapid, faint, or irregular pulses; low urine output; and occasional peripheral edema are additional signs and symptoms of end-organ hypoperfusion. Myocardial dysfunction from fluid overload may result in extra heart sounds and elevated laboratory values. Pulmonary edema is evidenced by audible crackles, wheezes, and abnormal vascular congestion on chest radiography. Metabolic abnormalities involving electrolyte imbalances and elevated inflammatory markers may result from or concur with the cardiac cascade of shock (see What’s New? Interleukin-6 [IL-6]).
Hypovolemic Shock
Hypovolemic shock is caused by loss of whole blood (hemorrhage), plasma (burns), or interstitial fluid (diaphoresis, diabetes mellitus, diabetes insipidus, emesis, or diuresis) in large amounts. Loss of whole blood or plasma causes hypovolemia directly. Loss of interstitial fluid causes an indirect “relative” hypovolemia by promoting diffusion of plasma from the intravascular to the extravascular space. Hypovolemic shock begins to develop when intravascular volume has decreased by about 15%.
Hypovolemia is offset initially by compensatory mechanisms (Figure 46-3). Heart rate and SVR increase as a result of catecholamine release by the adrenals. This boosts cardiac output and tissue perfusion pressures. Compelled by a decrease in capillary hydrostatic pressures, interstitial fluid moves into the vascular compartment. The liver and spleen add to blood volume by disgorging stored red blood cells and plasma. In the kidneys, renin (through several intermediaries) stimulates aldosterone release and the retention of sodium (and hence water), whereas antidiuretic hormone (ADH, or vasopressin) from the posterior pituitary gland increases water retention. Data on the compensation of ADH, however, show that as shock worsens, ADH in plasma decreases. Hypovolemic shock results in compensatory vasoconstriction, increased SVR, and afterload in order to improve blood pressure and perfusion to core organs of the body.
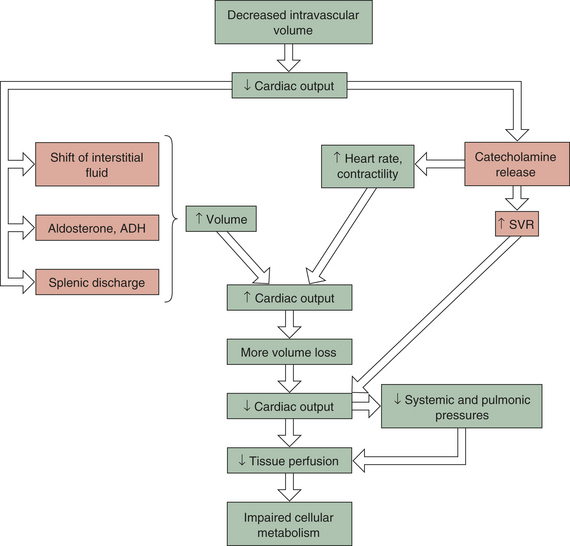
Figure 46-3 Hypovolemic shock. This type of shock becomes life threatening when compensatory mechanisms (orange boxes) are overwhelmed by continued loss of intravascular volume. ADH, Antidiuretic hormone; SVR, systemic vascular resistance.
These compensatory mechanisms are, however, finite. If the initial fluid or blood loss is great or if loss continues, compensation fails, resulting in decreased tissue perfusion. Nutrient delivery to the cells is impaired, and cellular metabolism
fails. Mortality from traumatic hemorrhagic shock ranges from 10% to 31%. Prompt control of hemorrhage is the treatment of choice. Fluid replacement is also important, but the type of fluid to be used and the rate of replacement are controversial.8,9 The clinical manifestations of hypovolemic shock include high SVR, poor skin turgor, thirst, oliguria, low systemic and pulmonary preloads, and rapid heart rates.
Neurogenic Shock
Neurogenic shock is sometimes called vasogenic shock. Both terms refer to a widespread and massive vasodilation that results from an imbalance between parasympathetic and sympathetic stimulation of vascular smooth muscle (see Chapter 29). Occasionally, parasympathetic overstimulation or sympathetic understimulation persists, causing vasodilation for an extended period. Extreme, persistent vasodilation leads to neurogenic shock (Figure 46-4). Neurogenic shock creates “relative hypovolemia.” Blood volume has not changed, but the amount of space containing the blood has increased, so that SVR decreases drastically; thus pressure in the vessels is inadequate to drive nutrients across capillary membranes, and nutrient delivery to the cells is impaired. As with other types of shock, this leads to impaired cellular metabolism.
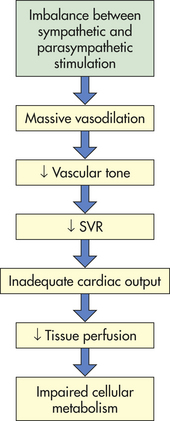
Neurogenic shock can be caused by any factor that stimulates parasympathetic activity or inhibits sympathetic activity of vascular smooth muscle. (Parasympathetic stimulation automatically inhibits sympathetic activity and vice versa; see Chapter 29.) Normally, sympathetic stimulation maintains muscle tone. If sympathetic stimulation is interrupted or inhibited, vasodilation occurs. Therefore, trauma to the spinal cord or medulla, conditions that interrupt the supply of oxygen to the medulla, or conditions that deprive the medulla of glucose (e.g., insulin reactions) can cause neurogenic shock by interrupting sympathetic activity. Depressive drugs, anesthetic agents, and severe emotional stress and pain are other causes of neurogenic shock.
The clinical hallmark of neurogenic shock is a very low SVR, along with other indicators of excessive parasympathetic activity. Bradycardia is the most obvious manifestation, especially in the early stages. Bradycardia may cease when compensatory mechanisms, particularly an increase in sympathetic system activity, have been initiated. The ejection fraction remains high, indicating a healthy myocardium, whereas central venous pressure decreases as the veins dilate. Neurogenic shock causes fainting if blood pressure decreases to the point that cerebral metabolism is not sufficient to support consciousness. Most episodes of fainting are not shock, however; for such episodes to progress to shock is rare. By allowing the blood pressure to equalize from head to toe as the individual becomes prone, fainting can actually prevent shock.
Anaphylactic Shock
Anaphylactic shock is the outcome of a widespread hypersensitivity reaction known as anaphylaxis. The basic physiologic alteration in anaphylactic shock is the same as that in neurogenic shock—that is, vasodilation, peripheral pooling, and relative hypovolemia leading to decreased tissue perfusion and impaired cellular metabolism (Figure 46-5). Anaphylactic shock is often more severe than other types of normovolemic shock because the hypersensitivity reaction that triggers vasodilation has other pathophysiologic effects that rapidly involve the entire body.
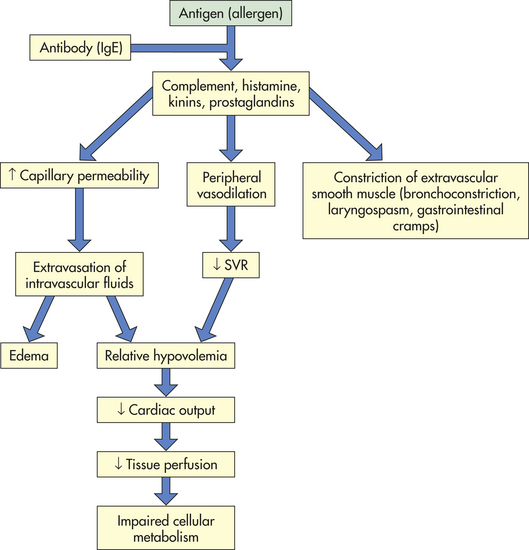
Anaphylactic shock begins as an allergic reaction—an immune and inflammatory response—to an allergen. (An allergen is an antigen to which an individual is hypersensitive; see Chapters 6, 7, and 8 for discussions of immunity, inflammation, and hypersensitivity.) Some allergens known to cause hypersensitivity reactions are snakebite venom, insect venoms, pollens, shellfish, penicillin, and animal sera. Once in the body, the allergen causes an extensive immune and inflammatory response. The vascular effects of this response include vasodilation and increased vascular permeability, resulting in peripheral pooling and tissue edema. The extravascular effects include constriction of extravascular smooth muscle. Constriction often causes respiratory difficulty because it tends to affect smooth muscle layers in the airway walls (e.g., the larynx and bronchioles; see Chapter 32).
The onset of anaphylactic shock is usually sudden, and progression to death can occur within minutes unless emergency treatment is given. The first manifestations of shock may be anxiety, difficulty in breathing, gastrointestinal cramps, edema, hives (urticaria), and sensations of burning or itching of the skin. A precipitous decrease in blood pressure occurs and is followed by impaired mentation. Other signs include decreased SVR (with high or normal cardiac output) and oliguria. Treatment begins with removal of the antigen (if possible). Epinephrine is administered to decrease mast cell and basophil degranulation, cause vasoconstriction, and reverse airway constriction. Volume expanders (e.g., lactated Ringer solution) are given intravenously to reverse the relative hypovolemia, and antihistamines and steroids are given to stop the inflammatory reaction.
Septic Shock
Septic shock is the endpoint of a continuum of progressive dysfunction.10 The syndrome begins with systemic inflammatory response syndrome (SIRS), then sepsis, then severe sepsis, and then septic shock. Consensus on definitions of each component was updated at an international sepsis conference in 2001 (Table 46-1).10,11 The International Sepsis Forum reviewed research for sepsis to identify and define the six most common infection sites (pneumonia, bloodstream, intravascular catheter, intra-abdominal, urosepsis, and surgical wound infection) associated with sepsis in the intensive care setting.12
Table 46-1
Definitions of Septic Shock Components
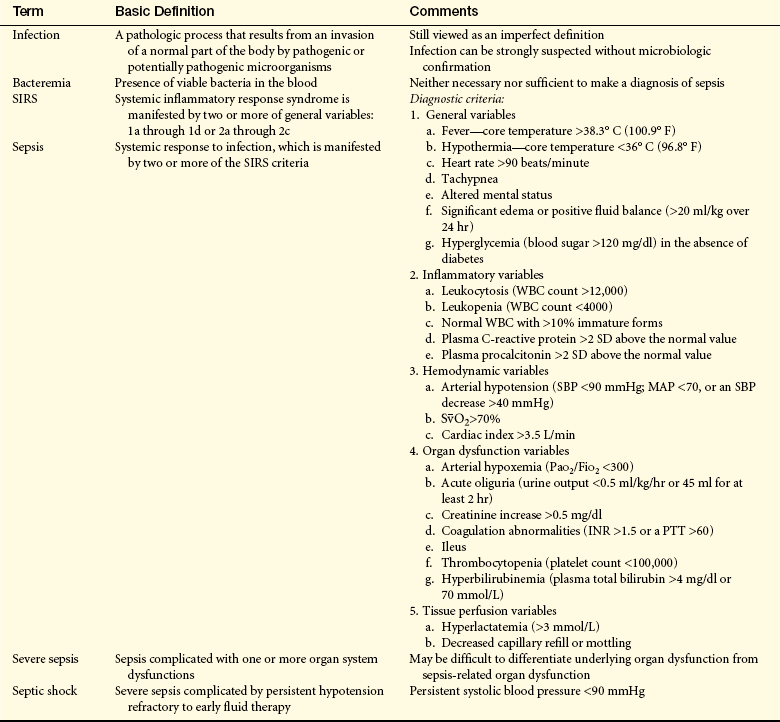
INR, International normalized ratio; MAP, mean arterial pressure; Pao2/Fio2, partial pressure of oxygen in arterial blood/fraction of inspired oxygen;
PTT, partial thromboplastin time; SD, standard deviation; SBP, systolic blood pressure; 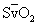 , saturation of hemoglobin with oxygen; WBC, white blood cell.
, saturation of hemoglobin with oxygen; WBC, white blood cell.
Data from Levy MM et al: Crit Care Med 312:1250, 2003; Opal SM, Scand J Infect Dis 35:529, 2003.
Severe sepsis is the eleventh most common cause of death in the United States. Mortality ranges from 28% to 60%.13 Septic shock is caused by gram-negative bacteria, gram-positive bacteria, and fungi. Advances in antibiotic therapy for gram-negative sepsis have made gram-positive bacteria the leading cause of sepsis.14 Even when properly treated with available therapies, it carries a high mortality rate. Prognosis is significantly affected by the source and virulence of the infectious microorganism.
Septic shock begins with a nidus of infection that may be readily discernible or extremely difficult to locate (Figure 46-6). Bacteria then enter the bloodstream to produce bacteremia in one of two ways: (1) directly from the site of infection or (2) from toxic substances released by the bacteria directly into the bloodstream. These toxic substances, which act as triggering molecules in the septic syndrome, include endotoxins released by gram-negative microorganisms, lipoteichoic acids and peptidoglycan released by gram-positive microorganisms, and superantigens.14
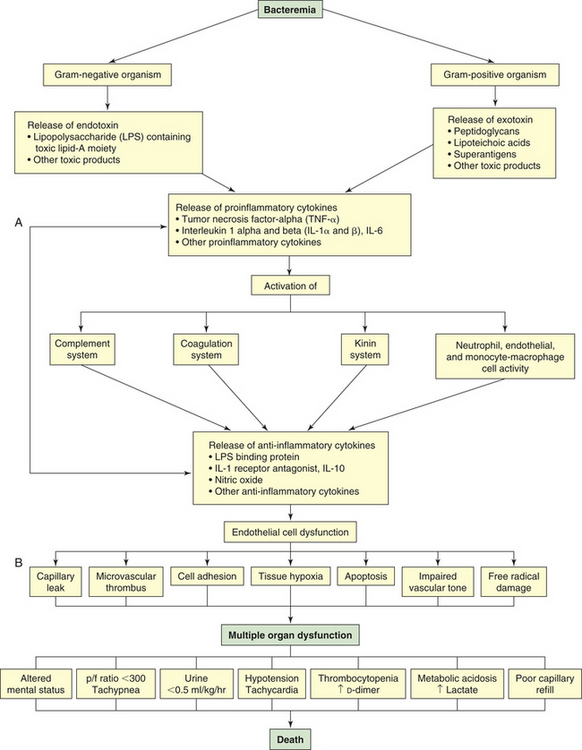
Figure 46-6 Summary of sepsis pathology. p/f (PaO2/FiO2), oxygenation ratio. (A from Larson V, Barke RA: Urol Clin North Am 26[4]:687, 1999. B copyright © 2003, Eli Lilly and Company. All rights reserved. Reprinted with permission from Eli Lilly and Company.)
The triggering molecules cause the host to initiate a proinflammatory response. Proinflammatory cells released include polymorphonuclear leukocytes, macrophages, monocytes, and platelets. Proinflammatory mediators released include cytokines (interleukins [IL]-1, IL-2, IL-6, IL-8, and IL-15; tumor necrosis factor-alpha [TNF-α]; and granulocyte cell–stimulating factor), complement and complement cascade activation, kinins, arachidonic acid metabolites (prostaglandins, prostacyclin, leukotrienes, and thromboxane), soluble adhesion molecules, platelet-activating factor, endorphins, vasoactive neuropeptides, histamine, serotonin, monocyte chemoattractant proteins 1 and 2, proteolytic enzymes (e.g., elastase and lysosomal enzymes), protein kinase, tyrosine kinase, CD14, toxic oxygen metabolites (e.g., superoxide, hydroxyl radical, hydrogen peroxide, peroxynitrite), neopterin, and clotting cascade activation.2,15,16 Proinflammatory cytokines enhance tissue factors, which initiates coagulation. Diminished thrombomodulin (cell surface glycoprotein of endothelial cells) inhibits the conversion of protein C and activated protein C. A compensatory anti-inflammatory response syndrome is presumed to follow this response.2,15,16 Anti-inflammatory mediators released include lipopolysaccharide-binding protein; IL-1 receptor antagonist; soluble CD-14; type 2 IL-1 receptor; leukotriene B4 receptor antagonist; IL-4, IL-10, and IL-13; soluble tumor necrosis factor receptor; transforming growth factor-beta [TGF-β]; epinephrine; and nitric oxide.2,15,16 Presumably the end result is a mixed antagonistic response syndrome as proinflammatory and anti-inflammatory mediators respond, intensify, and lead the host into MODS.
Clinical manifestations of septic shock are persistent low arterial pressure, low SVR from vasodilation, and an alteration in oxygen extraction by all cells. Septic shock and states of prolonged shock causing tissue hypoxia with lactic acidosis increase nitric oxide synthesis, activate ATP-sensitive and calcium-regulated potassium channels (KATP and Kca, respectively) in vascular smooth muscle (see Chapter 29), and lead to depletion of ADH (vasopressin) (Figure 46-7). Tachycardia causes cardiac output to remain normal or become elevated, although myocardial contractility is reduced. Temperature instability is present, ranging from hyperthermia to hypothermia. Effects on other organ systems may result in deranged renal function, gastrointestinal mucosa changes that result in release of bacteria from the gut, jaundice, clotting abnormalities, deterioration of mental status, and tachypnea that often progresses to ARDS.
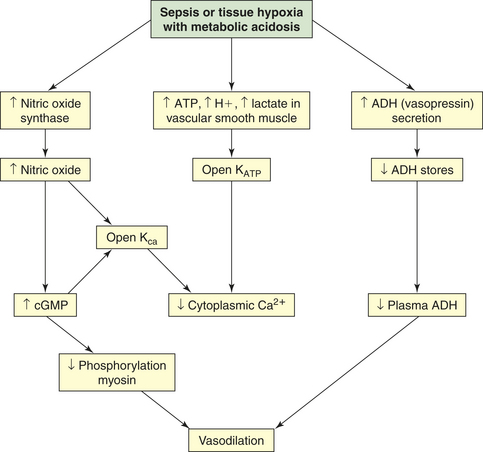
Figure 46-7 Mechanisms of vasodilation in shock. Vasodilatory shock is caused by the inappropriate activation of vasodilatory mechanisms and the failure of constrictor mechanisms. Unregulated nitric oxide, by regulating guanylate cyclase and generating cyclic guanosine monophosphate (cGMP), causes dephosphorylation of myosin and, thus, vasodilation. Nitric oxide synthesis and metabolic acidosis activate the potassium channels (KATP and KCa) in the plasma membrane of vascular smooth muscle. The resulting hyperpolarization (see Chapter 3) of the membrane presents the calcium that mediates norepinephrine and angiotensin II–induced vasoconstriction from entering the cell. Therefore, hypotension and vasodilation stubbornly persist despite high plasma levels of these hormones. In contrast, and unexpectedly, the plasma level of antidiuretic hormone (ADH) (vasopressin) is low despite the presence of hypotension. The early, massive release of ADH may result in future depletion.
Promptly initiated treatment helps reduce mortality and morbidity related to multiple organ dysfunction. Infection and sepsis identified in the early stages and treated with antibiotics and fluid resuscitation may prevent evolution to severe sepsis and shock; however once these are identified, more stringent treatment is recommended. The current guidelines for severe sepsis and septic shock17 recommend initial resuscitation within the first 6 hours to include fluids, either crystalloids or colloids (to maintain CVP 8 to 12 mmHg, mean arterial pressure ≥65 mmHg, urine output 0.5 ml/kg/hr, mixed venous saturation ≥65%); antibiotics; identification of specific anatomic sites of infection with source identification are recommended in the first 6 hours with subsequent control measures taken to eliminate or reduce the spread of infection.17 Continued hemodynamic support and adjunctive therapy, including vasopressors such as norepinephrine or dopamine, as initial vasopressor of choice with epinephrine, phenylephrine and vasopressin as alternatives, inotropic agents, such as dobutamine, if needed because of myocardial dysfunction, and intravenous hydrocortisone for adult septic shock if an individual remains hypotensive despite fluids and vasopressors. Adrenocorticotropic hormone (ACTH or corticotropin) stimulation test is not recommended and administration of recombinant human-activated protein C should be initiated if indicated.17 Other supportive therapies in the treatment of severe sepsis include blood product administration to maintain hemoglobin between 7 and 9 g/dl, targeted tidal volume (6 ml/kg) when mechanically ventilated with plateau pressure less than 30 cm H2O, utilization of sedation and analgesia protocols when needed, and avoidance of neuromuscular blockers if possible. Glucose is to be maintained at less than 150 mg/dl. If renal dialysis is required, intermittent hemodialysis and continuous venovenous hemofiltration are considered equivalent. Bicarbonate therapy is contraindicated for the purpose of treating hypoperfusion lactic acidemia. Use deep vein thrombosis prophylaxis, such as low-dose unfractionated heparin or low-molecular-weight heparin, or mechanical compression prophylaxis if heparin is contraindicated. Stress ulcer prophylaxis with H2-receptor antagonist or proton pump inhibitors is indicated. It is important to discuss advanced directives with the individual and family.17 Gaps in the current understanding of sepsis have led to research using experimental treatment such as immunomodulating therapies, including monoclonal antibodies and vaccines.18–20
Treatment for Shock
The first treatment for shock is to discover and correct or remove the underlying cause. Although this seems a simple tenet, it is one that is not always remembered. Thus treatment for cardiogenic shock begins with treatment of heart failure or at least enhancement of cardiac output. If hypovolemia is the cause of shock, hemorrhage and other causes of fluid loss must be stopped. In neurogenic shock as a result of spinal cord trauma, stabilization of the spine and surrounding tissue is a beginning, and pain usually can be decreased to a level at which neurally mediated decreases of SVR cease. The initial treatment for anaphylactic shock is to remove or neutralize the antigen. Treatment for septic shock begins with eradication of the infective agent, usually with antimicrobials (see preceding discussion).
After the underlying cause or condition is corrected as much as possible, treatment thereafter is supportive. Intravenous fluid is administered to expand intravascular volume, except in cases of cardiogenic shock, which require diuresis to reduce preload. Supplemental oxygen is always given. Cardiotonic drugs are given early in cardiogenic shock and given later in other forms of shock. Steroid use in septic shock remains unproven, although there is evidence that low-dose therapy improves mortality. Stress ulcer prophylaxis and gastric tonometry, to measure splanchnic blood flow, are imperative because the gut is one of the drivers of the septic syndrome.17
Once positive-feedback loops are established, intervention in shock is difficult. Prevention and early treatment offer the best prognosis.
MULTIPLE ORGAN DYSFUNCTION SYNDROME
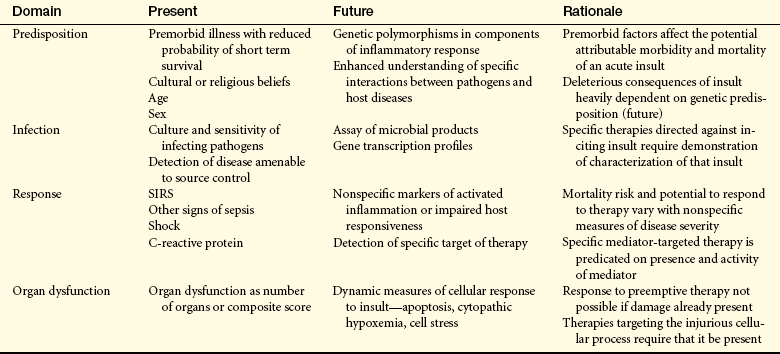
Multiple organ dysfunction syndrome (MODS) is the progressive dysfunction of two or more organ systems resulting from an uncontrolled inflammatory response to a severe illness or injury. The organ dysfunction can progress to organ failure and death. MODS occurs during severe sepsis. In 2001, an international consensus conference developed a set of definitions for sepsis and related disorders (see Table 46-1), and a predisposition-infection-response-organ (PIRO) dysfunction staging system was proposed as a template for staging sepsis (Table 46-2). MODS is the end stage of a variety of injuries that terminate in severe, generalized inflammation.
Table 46-2
Predisposition-Infection-Response-Organ (PIRO) Dysfunction System for Staging Sepsis
SIRS, Systemic inflammatory response syndrome.
From Levy MM et al: 2001 Intensive Care Med 29:530, 2003.
MODS was first recognized as a distinct clinical syndrome in the mid-1970s,21,22 when advances in resuscitation and support technologies allowed many individuals to survive life-threatening illness or trauma only to die of complications of their disease. Today MODS is a leading cause of mortality in surgical intensive care units (ICUs). Mortality for individuals with MODS increases progressively from 54% with two failing organ systems to 100% with five failing organ systems. Moreover, mortality has not improved much over the past 15 to 20 years.23,24
Although sepsis and septic shock are the most common causes, MODS can be initiated by any severe injury or disease process that activates a massive systemic inflammatory response by the host. Documented clinical infection is not necessary for its development. Other common triggers are severe trauma, major surgery, burns, circulatory shock, acute pancreatitis, acute renal failure, ARDS, blood transfusion, heat stroke, liver failure, mesenteric ischemia, Propofol infusion syndrome, persistent inflammatory foci, and necrotic tissue (Box 46-1). MODS is the major cause of death following septic shock, trauma, burn injuries, and ARDS. People at greatest risk for developing MODS are older adults and persons with significant tissue injury or preexisting disease.11
Box 46-1 Other Common Triggers of MODS
Data from Oeckler RA, Hubmayr RD: Eur Resp J 30(6):1216-1226, 2007; Ciesla DJ, Moore EE, Johnson JL, Burch JM, Cothren CC, Sauaia A: Arch Surg 140(5):432-440, 2005; Broessner G, Beer R, Franz G, Lackner P, Engelhard K, Brenneis C, Pfausler B, Schmutzhard E: Crit Care 9(5):R498-R501, 2006; Bouchama A, Knochel JP: N Engl J Med 346:1978-1988, 2002; Varghese GM, John G, Thomas K, Abraham OC, Mathai D: Emerg Med J 22:185–187, 2005; Adukauskienė D, Dockienė I, Naginienė R, Kėvelaitis E, Pundzius J, Kup inskas L: Medicina (Kaunas) 44(7):536-540, 2008; Abboud B, Daher R, Boujaoude J: World J Gastroenterol 14(35):5361-5370, 2008; Beger HG, Rau BM: World J Gastroenterol 13(38):5043-5051, 2007; Carnovale A, Rabitti PG, Manes G, Esposito P, Pacelli L, Uomo G: J Pancreas (Online) 6(5):438-444, 2005; Shaheen MA, Akhtar AJ: J Nat Med Assoc 99(12):1402-1406, 2007; Kam PCA, Cardone D: Anesthesia 62(1):690-701, 2007; Zaccheo MM, Bucher DH: Crit Care Nurse 28(3):18-26, 2008.
inskas L: Medicina (Kaunas) 44(7):536-540, 2008; Abboud B, Daher R, Boujaoude J: World J Gastroenterol 14(35):5361-5370, 2008; Beger HG, Rau BM: World J Gastroenterol 13(38):5043-5051, 2007; Carnovale A, Rabitti PG, Manes G, Esposito P, Pacelli L, Uomo G: J Pancreas (Online) 6(5):438-444, 2005; Shaheen MA, Akhtar AJ: J Nat Med Assoc 99(12):1402-1406, 2007; Kam PCA, Cardone D: Anesthesia 62(1):690-701, 2007; Zaccheo MM, Bucher DH: Crit Care Nurse 28(3):18-26, 2008.
The PIRO system (see Table 46-2), a clinically useful sepsis staging system, stratifies individuals with disease by baseline risk of adverse outcomes and potential to respond to therapy.11
PATHOPHYSIOLOGY In primary MODS the organ injury is directly associated with a specific insult, most often ischemia or impaired perfusion from an episode of shock or trauma, thermal injury, soft tissue necrosis, or invasive infection.25 This decreased perfusion is local (in the injured organs themselves) and generalized. The generalized hypoperfusion in primary MODS usually cannot be detected clinically. As a result of the insult, a stress response is initiated and stress hormones—in particular, catecholamines—are released. The inflammatory and stress responses are not as evident as they are in secondary MODS. In primary MODS during the inflammatory response, presumably neutrophils and macrophages are “primed” by cytokines.25 Any second insult, such as additional tissue injury, infection, or organ ischemia, may then activate the primed cells to produce an exaggerated response of secondary MODS25,26 (Figure 46-8).
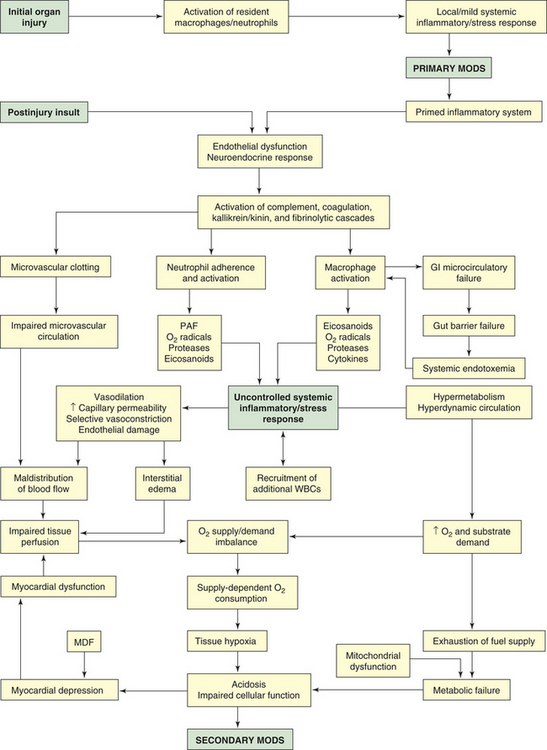
Figure 46-8 Pathogenesis of multiple organ dysfunction syndrome. GI, Gastrointestinal; MDF, myocardial depressant factor; MODS, multiple organ dysfunction syndrome; PAF, platelet-activating factor; WBCs, white blood cells.
The progressive organ dysfunction of secondary MODS is the result of an excessive inflammatory reaction, after a latent period following the initial injury, in organs distant from the site of the original injury. It is postulated that the resulting organ trauma is caused by the host response to a second insult rather than being a direct result of the primary injury. Often the second insult is mild but produces an immense disproportionate response because of the previous priming of leukocytes. The interaction of injured organs then leads to a self-perpetuating inflammation.
Secondary MODS is initiated by the delayed postinjury insult as primed macrophages release a barrage of mediators, particularly the cytokines TNF and IL-1. These mediators damage the endothelium throughout the body. If a gram-negative bacterial infection is present, endotoxin released from the bacteria also causes severe damage to endothelial cells. Normal endothelial cells have little interaction with leukocytes, but when stimulated by TNF, IL-1, IL-6, or endotoxin, they change to a proinflammatory state and express adhesion molecules that mediate adhesion of neutrophils. The adhered neutrophils then migrate through the endothelium, aggregate in the area of damaged tissue, and amplify the inflammation.2 The activated endothelial cells increase production and release of nitric oxide (endothelium), a potent vasodilator that is considered an important factor in the blood flow changes and loss of vascular tone noted in systemic inflammation.27 The injured endothelium also becomes much more permeable, allowing fluid and protein to leak into the interstitial spaces. An important function of normal endothelium is anticoagulation. When damaged, the endothelium loses much of its ability to prevent blood clotting, allowing microvascular thrombi to develop.
The postinjury insult also activates the neuroendocrine system, resulting in a second, more extensive stress response. The normal function of the stress response is to maintain basal and stress-related homeostasis,28 but in MODS homeostasis cannot be maintained. In fact, the endocrine response becomes excessive and injurious. There is an early increase in circulating catecholamines that contributes to many of the clinical manifestations of MODS, such as tachycardia, hypermetabolism, and increased oxygen consumption. Cortisol, glucagon, insulin, human growth hormone, ADH (which may become depleted), and endorphin levels also are increased. Many of these hormones contribute to the extreme catabolic state of MODS, and endorphins, which are vasodilators, decrease SVR. The sympathetic nervous system, to compensate for complications resulting from the injury (e.g., fluid loss, hypotension), also is stimulated. The stimulation persists throughout the period of critical illness.28 The stress response can be amplified by a number of factors, including pain, anxiety, psychosis, and hyperthermia. (Stress response is discussed in detail in Chapter 10.)
Because of endothelial cell dysfunction and the release of mediators, four major plasma cascades are activated: complement, kallikrein-kinin, coagulation, and fibrinolytic.27 Complement components, particularly the anaphylatoxins C3a and C5a, cause vasodilation by stimulating release of histamine from mast cells. They also have strong chemotactic properties. C5a, especially, causes adhesion and the activation and degranulation of neutrophils. Complement is thought to be a powerful trigger for the exaggerated inflammatory response. Activation of the kinin system results in the production of bradykinin, a very potent vasodilator known to decrease SVR. Coagulation mechanisms also are activated, and because tissue injury and endothelial dysfunction are extensive, microvascular thrombosis occurs throughout the body, resulting in impaired microvascular circulation and organ ischemia. Concurrently, fibrinolytic mechanisms are activated. The tendency toward clotting, however, is greater, resulting in a net procoagulant state that can lead to the development of DIC. The overall effect of the activation of the plasma cascades is a hyperinflammatory and hypercoagulant state that contributes to vasodilation, vasopermeability, cardiovascular instability, endothelial dysfunction, and clotting abnormalities.29
Once cytokines and other mediators have been released and the plasma enzyme cascades have been activated, a massive systemic inflammatory response develops. It involves several types of inflammatory cells, particularly neutrophils, macrophages, and mast cells. These cells, having been primed by their response to the initial organ injury, now pour large amounts of chemical mediators into tissues and into the systemic circulation. Neutrophils have tremendous inflammatory potential. The accumulation of activated neutrophils in organs is thought to play a key role in the pathogenesis of MODS.30 When neutrophils adhere to the endothelium, they undergo a “respiratory burst” (oxidative burst) and release oxygen radicals. The respiratory burst occurs as the activated neutrophil experiences a sudden increase in oxidative metabolism, producing large quantities of highly toxic oxygen free radicals. These reactive oxygen species (ROS) cause oxidative stress. The primary ROS produced are superoxide, hydrogen peroxide (H2O2), hydroxyl radical (OH−), and singlet oxygen (O). Oxygen radicals are extremely damaging to vascular endothelium and tissue cells, attacking deoxyribonucleic acid (DNA), cross-linking membrane structures, and inducing membrane peroxidation—reactions that disorganize cell membranes and lead to tissue necrosis2,16 (see also Chapter 2).
Other important mediators released by neutrophils are proteases, particularly collagenase and elastase. Proteases directly damage endothelium and neighboring cells, resulting in increased capillary permeability and organ damage. When activated, neutrophils also release platelet-activating factor (PAF), a mediator that damages endothelium, stimulates clot formation, and activates increasing numbers of phagocytes. Finally, neutrophils release arachidonic acid metabolites (eicosanoids) as a result of lipid peroxidation of their cell membranes. Of the arachidonic acid metabolites (prostaglandins, thromboxanes, leukotrienes), two are particularly important in the pathogenesis of organ hypoperfusion: prostacyclin (PGI2) and thromboxane A2 (TXA2). TXA2 is a powerful vasoconstrictor, and PGI2 is a potent vasodilator. When released in varying amounts in different organ beds, they are largely responsible for the maldistribution of blood flow characteristic of MODS. In total, neutrophils produce at least 50 to 60 toxins.30,31 Collectively, products released by neutrophils cause endothelial dysfunction, systemic vasodilation, selective vasoconstriction (vasoconstriction of certain organ beds or parts of organ beds), increased vascular permeability, and microvascular coagulation.
Macrophages, present in most tissues, are activated by endotoxin, complement, and monocyte chemotactic substances.32 Macrophages share a key role in the development of the unregulated inflammation of secondary MODS with the neutrophils. Like neutrophils, they produce oxygen radicals, proteases, cytokines, nitric oxide, and arachidonic acid metabolites. It has been reported that excessive or prolonged stimulation of macrophages leads to the overproduction of cytokines and nitric oxide that initiate the cycle of harmful effects in MODS.2,16,32 TNF and IL-1, which share many of the same functions and act synergistically, are the major cytokines that mediate inflammation.32,33 TNF has potent metabolic effects, including fever, anorexia, hyperglycemia, hypermetabolism, and weight loss. It activates neutrophils, damages endothelial cells, and potentiates hypotension and shock. IL-1 also has metabolic effects, inducing fever, hypermetabolism, and muscle wasting. Normally, TNF activates cytokines, the coagulation system, fibrinolysis, and neutrophils. With the exception of neutrophil activation, IL-1 causes similar activation in individuals with cancer.34 In the pathogenesis of MODS, the cytokines are linked to all cellular, hemodynamic, and metabolic alterations.
The gastrointestinal mucosa is particularly vulnerable to inflammatory mediators released by macrophages and neutrophils. Under normal circumstances the gut mucosa serves as a barrier to prevent bacteria from the gastrointestinal tract from entering the systemic circulation. Damage to the mucosa results in microcirculatory failure of the gut and consequent loss of the gut barrier function. The loss of intestinal barrier function leads to the systemic spread of bacteria and/or endotoxin from the gut (systemic endotoxemia). This phenomenon is called translocation of bacteria. The idea that the gut acts as a reservoir of bacteria and endotoxin that can initiate or perpetuate the development of MODS is known as the gut hypothesis. The gut hypothesis provides a possible explanation for the fact that an infectious focus is not always found in individuals with MODS. Although this hypothesis has been substantiated by animal studies and has much support, the evidence from human studies is inconclusive.35,36
The numerous inflammatory processes operating in MODS cause maldistribution of blood flow and hypermetabolism. Maldistribution of blood flow refers to the uneven distribution of flow to various organs and between the large vessels and capillary beds of the body. It is caused by generalized vasodilation, increased capillary permeability, selective vasoconstriction, endothelial dysfunction, and impaired microvascular circulation. It is a major factor in the pathophysiology of MODS.27 The alterations in blood flow—which can occur at the cellular, organ, or regional level—lead to impaired tissue perfusion and a decreased supply of oxygen to the cells. The organs most severely affected by hypoperfusion are the lungs, splanchnic bed, liver, and kidneys. Despite supernormal systemic blood flow, oxygen delivery to the tissues decreases. Several factors contribute to the problem. First, blood is shunted past selected regional capillary beds. Shunting, caused by loss of autoregulation in some organs, may be an early indicator of progression of sepsis into MODS.37 This occurs because inflammatory mediators, particularly TXA2, override the normal vascular control to cause selective vasoconstriction and because injured endothelial cells are unable to respond to normal vasodilator mediators. Second, interstitial edema, resulting from microvascular permeability, contributes to decreased oxygen delivery to cells by increasing the distance oxygen must travel to reach the cells. Third, capillary obstruction occurs because of the formation of microvascular thrombi and the aggregation of leukocytes.2,16
Hypermetabolism, with accompanying alterations in carbohydrate, fat, and lipid metabolism, is initially a compensatory measure to meet the body’s increased demands for energy. Eventually, however, hypermetabolism becomes detrimental, placing enormous demands on the heart. Hypermetabolism is the result of (1) the neuroendocrine response to stress with the release of catecholamines and cortisol, and (2) the action of TNF and IL-1. With increased metabolism the calorie requirements are markedly increased,23 and the cardiac output increases 1.5 to 2 times normal.38 The alterations in metabolism affect all aspects of substrate use. Most important is the catabolism of protein, primarily of skeletal muscle and visceral organs. The extreme catabolism of protein can rapidly deplete lean body mass. Hyperglycemia occurs as gluconeogenesis by the liver increases and glucose use by the cells decreases. Fatty acids are mobilized from adipose tissue. The net result of the hypermetabolism is depletion of oxygen and fuel supplies.
Myocardial depression also accompanies MODS. The cause remains unclear, but possible explanations are the effects of myocardial depressant factor (MDF), TNF, and IL-1 on cardiac contractility; alterations in α-adrenergic receptors in the heart; and hypoxia of the myocardium.39,40
The decreased oxygen delivery to the cells (resulting from the maldistribution of blood flow) and the increased oxygen needs of the cells (resulting from hypermetabolism) combine to create an imbalance in oxygen supply and demand. This imbalance is critical in the pathogenesis of MODS because it results in a pathologic condition known as supply-dependent oxygen consumption. Ordinarily the amount of oxygen consumed by the cells depends only on the needs of the cells because there is an adequate reserve of oxygen that can be delivered if required. In MODS, however, the reserve has been exhausted and the amount of oxygen consumed becomes dependent on the amount the circulation is able to deliver. Because the amount is inadequate in MODS, the tissues become hypoxic. Compounding the hypoxic damage to cells is a phenomenon called reperfusion injury (see Chapter 2). Much of the organ damage in MODS occurs with the reestablishment of blood flow after a period of ischemia. During the ischemic episode, energy stores and ATP are depleted and the enzyme xanthine dehydrogenase is converted to xanthine oxidase. With reperfusion of the ischemic tissue, oxygen radicals are formed from oxygen by the action of xanthine oxidase, and they attack the already damaged tissues. Consequently, although reperfusion is necessary to restore oxygen supply to ischemic organs, it can increase the extent of injury. Therefore, because of supply-dependent oxygen consumption and reperfusion injury, tissues become increasingly hypoxic. The result is cellular acidosis, impaired cellular function, and ultimately multiple organ failure.
CLINICAL MANIFESTATIONS In MODS the organs that show clinical manifestations of failure are not always the organs involved as part of the initial injury, and there is usually a lag time between the initial insult and the development of systemic organ failure. The development of primary MODS is difficult to monitor, but there is a well-established general pattern in the clinical development of secondary MODS.25,26 Following the inciting event and aggressive resuscitation of the individual for approximately 24 hours, the individual develops low-grade fever, tachycardia, tachypnea, dyspnea, altered mental status, and a general hyperdynamic and hypermetabolic state (Box 46-2). Following this, the lungs begin to fail and ARDS may appear within 24 to 72 hours (see discussion of ARDS, Chapter 33). Between days 7 and 10, the hypermetabolic and hyperdynamic state intensifies; bacteremia with enteric organisms is common; and signs of hepatic, intestinal, and renal failure develop. During days 14 to 21, the renal failure and liver failure become more severe. Hematologic failure and myocardial failure are usually later manifestations. Encephalopathy, characterized by mental status changes ranging from confusion to deep coma, may occur at any time. This sequence can evolve rapidly, with death occurring between 14 and 21 days later, or it can evolve over weeks. Individuals can recover from either the slowly or rapidly evolving course.
The clinical manifestations of failure of individual organs in MODS are caused by inflammatory mediator damage, tissue hypoxia, and hypermetabolism. Respiratory failure progresses early to ARDS and is characterized by tachypnea, pulmonary edema with crackles and diminished breath sounds, use of accessory muscles, and hypoxemia. Liver failure, although early in its development, is not clinically detectable until the later stages of MODS, when jaundice, abdominal distention, liver tenderness, muscle wasting, and hepatic encephalopathy appear. All aspects of metabolism, substance detoxification, and immune response are impaired. Albumin and clotting factor synthesis decreases, protein wastes accumulate, and liver tissue macrophages (Kupffer cells) no longer function effectively.
The gastrointestinal system is very sensitive to ischemic and inflammatory injury. Clinical manifestations of bowel involvement are hemorrhage, ileus, stress ulcers, malabsorption, diarrhea or constipation, vomiting, anorexia, abdominal pain, and pancreatitis. Intolerance to enteral feeding may develop. Adding to damage caused by injury to the bowel is bacterial translocation into the bloodstream resulting from the loss of the gut barrier function. The overwhelmed liver is unable to clear the bacteria from the systemic circulation. Thus, regardless of whether infection or some other injury was the precipitating cause of MODS, once intestinal bacteria enter the systemic circulation, it is likely that sepsis will be a problem. Renal failure develops at about the same time and is marked by progressive oliguria, azotemia, and edema. If renal shutdown is severe, anuria, hyperkalemia, and metabolic acidosis occur.
The first manifestations of cardiac failure are similar to those of septic shock: tachycardia, bounding pulse, increased cardiac output, fall in SVR, hypotension, warm skin, and supraventricular dysrhythmias. In the terminal stages, profound hypotension and ventricular dysrhythmias may develop. Changes in central nervous system function may be noted. Ischemia and inflammation are responsible for the changes, which include apprehension, confusion, disorientation, restlessness, agitation, headache, decreased cognitive ability and memory, and decreased level of consciousness. When ischemia is severe, seizures and coma can occur.
EVALUATION AND TREATMENT Because there is no specific therapy for MODS, early detection or prevention is extremely important so that supportive measures are initiated instantly.23 Frequent assessment of the clinical status of individuals at known risk is essential. Unfortunately, there is no way to determine with certainty when an organ is failing. Indicators of organ dysfunction are presented in Table 46-1.
Several systems for scoring severity of illness also have been developed. Commonly used systems are the Acute Physiology and Chronic Health Evaluation II and III (APACHE II and APACHE III), sequential organ failure assessment (SOFA), MODS score, and the PIRO staging system.41 Once organ failure develops, monitoring of laboratory values and hemodynamic parameters is necessary to assess the degree of clinical impairment.
The therapeutic management of MODS consists of prevention and support. Prevention of the syndrome is essential! First, if possible, the initial source of inflammation must be eliminated or controlled. Next, a second insult must be avoided. It is paramount to remove any potential site of infection by débriding necrotic tissue, draining abscesses, reducing the numbers of invasive procedures performed, and removing hematomas. Nosocomial infections from contaminated lines and catheters are of concern and must be prevented. Nosocomial infection rates of 15% to 25% have been reported in critically ill individuals.42 Early reduction of long-bone fractures and surgical repair of injured tissues are also important preventive measures.
The goals of therapy are to control infection, provide adequate tissue oxygenation, restore intravascular volume, and support the function of individual organs.43 After the initial injury has been aggressively treated and sources of infection have been removed, antibiotics generally are administered. The choice of agents is based on the individual’s disease process, but the regimen is usually a combination of antibiotics that covers both gram-negative and gram-positive organisms.
Because oxygen is not stored in the tissues, it must be continuously delivered. Maintaining an arterial oxygen saturation of 88% to 92% is recommended,43 and hemoglobin levels should be kept above 9 g/dl.43 Mixed venous oxygen greater than or equal to 70% is recommended. Blood transfusions may be necessary to ensure an adequate hemoglobin level. To deliver oxygen to the organs in the face of profound systemic vasodilation, fluid volume must be restored. Therefore, aggressive fluid therapy is initiated early. Usually large volumes of isotonic crystalloid solutions are administered, although colloids (often albumin) also may be added to maintain adequate preload and circulation volume.43
Finally, support for individual organ systems must be provided. Respiratory failure is treated with mechanical ventilation with low tidal volumes, high oxygen concentrations, and positive end-expiratory pressures (PEEP).44 To provide adequate nutrition and metabolic support, the failing gastrointestinal system is supported with enteral feedings (see Nutrition & Disease: Olive Oil). It is now well recognized that enteral feedings help preserve gut microbial barrier function, thus are preferred to parenteral feedings.36 However, if the individual is unable to tolerate the amount of enteral feeding required to meet the enormous metabolic demands, hyperalimentation may be added. Ideally the feeding formula is carefully calculated to meet the individual’s nutritional requirements. Tight glucose control (80 to 110 mg/dl) is recommended.45 Once renal failure is established, dialysis or continuous hemofiltration may be required to maintain fluid and electrolyte balance. To support the failing cardiovascular system, inotropic drugs, such as low-dose dopamine and dobutamine, or vasopressors, such as norepinephrine, may be required to maximize cardiac contractility and maintain cardiac output. Although steroids have anti-inflammatory effects, use of them is controversial because they have been shown to be effective in adults with septic shock. Obtaining
an ACTH level is not recommended. Deep vein thrombosis prophylaxis is also important.3
Scientific knowledge gained about MODS and inflammatory mediators has led to many investigational therapies. Recombinant human-activated protein C has been found to be effective in the treatment of severe sepsis.17 Novel molecular approaches targeting a variety of interdependent mediators of MODS are being investigated.34
BURNS
Major thermal injury is a source of massive tissue injury and destruction that has wide-reaching effects on virtually all organ systems. Burn is a generic term used to describe cutaneous injury resulting from thermal, chemical, or electrical environmental causes. In addition to cutaneous injury, burns are often associated with smoke inhalation injury or other traumatic injuries that aggravate the local and systemic problems of burns. Pulmonary injury, both primary and secondary, is common and often calls for ventilator support. The use of tracheostomy also has been examined.46,47 This model of multisystem injury provides an opportunity to examine the interaction of shock, inflammation, and immunocompromise in a clinical setting.
Epidemiology and Etiology
The incidence of burns in the United States has dropped from 4.2 cases per 100,000 from 1961 through 1964 to 1.5 per 100,000 from 1993 through 1996.48 Deaths from fire-related injuries are estimated to be 5000 annually in the United States. Deaths from burn injuries decreased more than 50% from 9000 in 197149 to 4000 in 2005.50 This remarkable progress is the result of several factors, including an increased national focus on fire safety and burn prevention, the establishment of regional burn centers, the use of smoke detectors, regulation of consumer product safety, and occupational safety mandates. A decrease in hospitalization reflects a shift to outpatient care and improved prehospital and emergency treatment. Burn assessment and delivery of care can be improved51 to reduce medical transport and treatment costs.
The causes of burn injury may be thermal or nonthermal, such as chemical, electrical, or radioactive. Thermal burns may result from thermal contact, flame, or scald. Adherent materials (e.g., asphalt, tar, or plastic) may likewise produce a serious contact burn. Chemical injuries are a result of contact with substances that are directly toxic to skin or the lining of the respiratory or alimentary tract. Such chemicals are often acid, alkali, or organic agents, termed vesicants, that cause blistering of the epithelial surfaces. Electrical burns may be the result of the conduction of electrical current through the body and the resultant heating of tissue, or flash over the body surface associated with an electrical discharge. Quality of life can be affected but, by self-reports, may exceed normal population averages with proper intervention.52
Burn Wound Depth
The classification of burn wound depth is usually based on the physical appearance and the symptoms associated with the affected skin. The definitive diagnosis is determined by the histologic depth of tissue necrosis. Such evaluation, unfortunately, necessitates a skin biopsy. Because of the invasive nature of biopsy, clinical depth assessment is used, and the ultimate fate of the wound determines final diagnosis. Advances in laser Doppler technology have resulted in extensive exploration of noninvasive means for burn wound depth assessment.53–65
First-degree burns are a partial-thickness injury involving only the epidermis and no injury to the underlying dermal or subcutaneous tissue (Table 46-3). The skin maintains water vapor and bacterial barrier functions. Many sunburns are first-degree injuries caused by exposure of skin to ultraviolet radiation from the sun. Initially there is local pain and erythema, but no blisters appear until after about 24 hours. An extensive first-degree burn may cause systemic responses such as chills, headache, localized edema, and nausea or vomiting. Therapy consists of intravenous hydration until the nausea and vomiting subside 24 to 72 hours after burn injury. Comfort measures for previously healthy children or adults with extensive first-degree burns consist of aspirin for adults or acetaminophen (controversial) for children every 4 hours in age-appropriate doses and frequent application of a water-soluble lotion. First-degree burns heal in 3 to 5 days without scarring.

Second-degree burns describe two categories of burn depth with markedly different characteristics. Both of these are partial-thickness injuries, but they evoke vastly different responses. The hallmark of superficial partial-thickness injury is the appearance of thin-walled, fluid-filled blisters that develop within just a few minutes after injury. Another dominant characteristic of superficial injury is pain. As blisters break or are removed, nerve endings are exposed to air (Figure 46-9). Tactile and pain sensors remain intact throughout healing, with each wound care procedure causing substantial pain. Wounds heal in 3 to 4 weeks if the individual is adequately nourished and no complications develop (Figure 46-10). Scar formation is unusual with this injury. The amount of scarring that develops is a genetically determined trait and is not predictable during the early course of treatment.
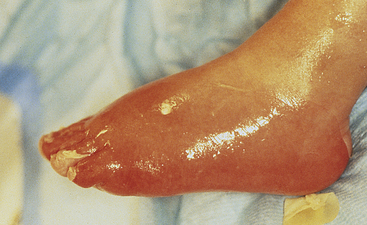
Figure 46-9 Superficial partial-thickness injury. Scald injury following débridement of overlying blister and nonadherent epithelium. (Courtesy Intermountain Burn Center, University of Utah.)
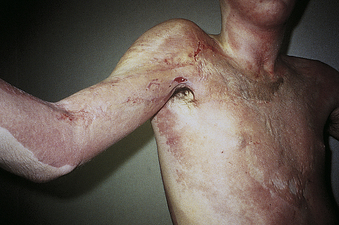
Figure 46-10 Axillary burn scar contracture. Note the blanching of the anterior axillary fold and small ulceration, both indicating the diminished range of motion. (Courtesy Intermountain Burn Center, University of Utah.)
Deep partial-thickness burns involve the entire dermis, sparing skin appendages such as hair follicles and sweat glands (see Table 46-3). The burn often looks waxy white and is surrounded by margins of superficial partial-thickness injury. The injury is often clinically indistinguishable from a full-thickness injury (Figure 46-11), but by 7 to 10 days after burn injury, skin buds and hair will appear from hair follicles, indicating that skin appendages remain. These wounds take weeks to heal, and therapy consists of surgical removal of the burn wound (excision) followed by application of the person’s own unburned skin from another body area (autograft). Wounds that heal slowly produce more scar tissue and continue to be a potential source of infection until closed. In the presence of relative surgical contraindications, such as cardiopulmonary failure, deep partial-thickness wounds are not surgically treated but are allowed to heal from primary intention. The ultimate healing of deep partial-thickness burns commonly results in hypertrophic scarring with poor functional and cosmetic results.
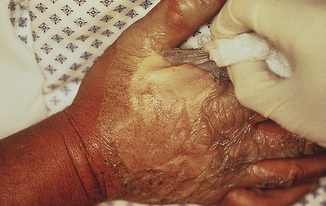
Figure 46-11 Deep partial-thickness wound. Note pale appearance and minimal exudate. (Courtesy Intermountain Burn Center, University of Utah.)
Third-degree burns, or full-thickness injuries, involve destruction of the entire epidermis, dermis, and often the underlying subcutaneous tissue (see Table 46-3). On occasion, all underlying subcutaneous tissue is destroyed and muscle or bone may be involved. Full-thickness wounds often appear relatively innocuous when their color is white and the delineation between normal and burned skin is not accompanied by a marked color change. Elasticity of the dermis is absent, leaving the wound dry and leathery in appearance and texture (Figure 46-12). As marked edema forms, distal circulation may be compromised in areas of circumferential burns. An escharotomy (cutting through burned skin) is performed to release underlying pressure. Full-thickness burns are painless because all nerve endings have been destroyed by the injury.
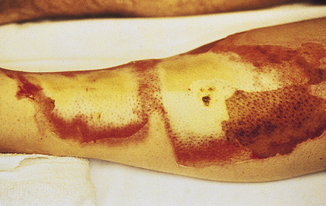
Figure 46-12 Full-thickness thermal injury. The wound is dry and insensate. (Courtesy Intermountain Burn Center, University of Utah.)
The extent of the total body surface area (TBSA) burn is estimated using the “rule of nines” (Figure 46-13). Areas of partial-thickness and full-thickness injury are marked on the diagram in Figure 46-14. First-degree burns are not included in the TBSA estimate. The surface area of the palm, including palmar finger surface, averages 1% of the body surface area over a wide range of ages; thus it can be used to estimate burn areas of irregular size and shape.66
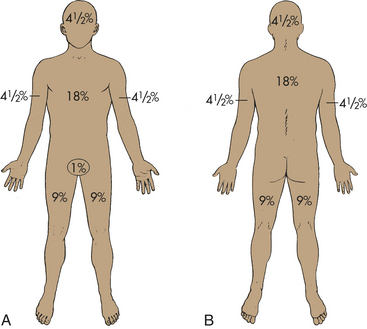
Figure 46-13 Rule of nines. A commonly used assessment tool with estimates of the percentages (in multiples of 9) of the total body surface area burned. A, Adults (anterior view). B, Adults (posterior view).
Severity of burn injury is a combination of many factors, including age, medical history, extent and depth of injury, and body area involved. The American Burn Association has defined criteria to assist healthcare professionals in identifying individuals who require care at a specialized burn center (Box 46-3). The multidisciplinary burn center is recommended for those persons who are at high risk for morbidity, mortality, or permanent functional loss.
PATHOPHYSIOLOGY AND CLINICAL MANIFESTATIONS Burn injury results in dramatic changes in many physiologic functions of the body within the first few minutes after the event. The effect of burn depends on two factors: first, the extent of body surface involved and, second, the depth of cutaneous injury. Body surface burn extent is described by the percentage of TBSA injured. Burns exceeding 20% of TBSA in most adults are major burn injuries and are associated with massive evaporative water losses and flux of large amounts of fluid and electrolytes in the tissues, manifested as generalized edema and circulatory hypovolemia. Depth of cutaneous injury has been categorized in many ways but always depends on the severity of injury of epidermal and dermal elements of the skin and whether the alteration is a permanent or reversible injury.
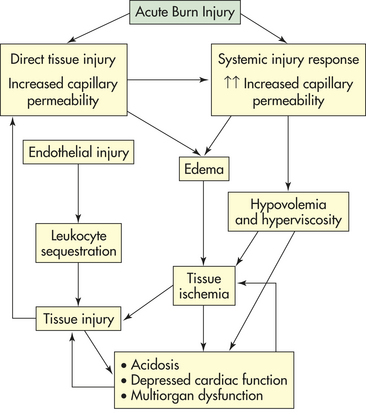
With a major burn injury, a systemic pathophysiology ensues that requires therapeutic intervention to sustain life. The immediate (acute) physiologic consequences of major burn injury centers around the profound, life-threatening hypovolemic shock occurring in conjunction with cellular and immunologic disruption within a few hours of injury (see Figure 46-14). Burn shock consists of a hypovolemic cardiovascular component and a cellular component.
Hypovolemia associated with burn shock results from massive fluid losses from the circulating blood volume. The losses are caused by an increase in capillary permeability that persists for approximately 24 hours after burn injury. Fluid resuscitation is the administration of intravenous fluids, such as lactated Ringer solution, in an effort to restore the circulating blood volume during the period of increasing capillary permeability. In addition to hypovolemia, most other organ systems are affected. Cardiac contractility is diminished during the initial 24-hour resuscitation period with shunting of blood away from the liver, kidney, and gut. This is often termed the ebb phase of the response to trauma and can be seen with other severe injuries. Normal blood volume does not result in restoration of normal cardiac output because of a phenomenon known as myocardial depression (see MODS). The decrease in perfusion of viscera results in a decrease in their function. This may be an explanation for decreased gut barrier function seen in thermal injury.67
There also is evidence that cellular metabolism is disrupted when the burn wound is created resulting in altered cell membrane permeability and loss of normal electrolyte homeostasis. This cellular defect may be the pathophysiologic process responsible for the genesis of burn shock. Numerous circulating factors in burn serum may play a role in these cellular processes. Although the cardiovascular and systemic response is intricately interwoven into the cellular response, these responses are presented here as discrete entities.
Cardiovascular and Systemic Response to Burn Injury
The clinical manifestations of burn shock are the result of more than simple loss of extracellular fluid at the burn wound site. Hypovolemia and numerous local mediators in the burn wound,68 as well as systemic signals, result in alteration of cellular function throughout the body. The restoration of normal intravascular volume with either saline solutions or colloid materials (e.g., albumin, blood, or dextrans) does not reverse changes such as increases in pulmonary vascular resistance or myocardial contractility.69–71 This is reflected in cardiac output with precipitous decreases that often result in inadequate perfusion of most tissues at the capillary level, which is the hallmark of burn shock. Fluid infusion does not return cardiac output to preburn levels.72,73 These findings led to the postulation of a specific MDF.74,75 Other causes also have been suggested, such as reactive oxygen radicals that attack cell membranes and other subcellular organelles as a result of first ischemia and then reperfusion of tissues during burn shock and resuscitation.76 A third factor may be the level of nitric oxide after burn injury, which could have a direct myocardial depressant effect.77,78 The relationship of nitric oxide and myocardial function is not yet totally clear. Gamelli and colleagues79 found nitric oxide production to be significantly depressed in burned individuals who did not survive their injuries. They postulate that nitric oxide may scavenge reactive oxygen radicals and protect tissues from oxidative injury.
Regardless of the contribution of these mechanisms, fluid resuscitation eventually results in improved outcome of a massively burned person. This resuscitation involves infusion of intravenous fluid at a rate faster than the loss of circulation vascular volume for about 24 hours from the time of burn injury and may require up to 30 L in a major burn. Resuscitation from burn shock can be accomplished using any of a number of infusion protocols, most frequently the Parkland formula.80 Lactated Ringer solution is used because it closely approximates extracellular fluid, the repository of fluid leaving the circulatory system during this phase of extensive edema formation (Table 46-4). The use of electrolyte-free fluids, such as D5W, results in life-threatening hypovolemia and hyponatremia. Resuscitation with hypertonic saline has been used in some medical centers but is reserved for special circumstances; its use can result in adverse outcomes.81
Table 46-4
Electrolyte Content of Ringer Lactate Solution and Extracellular Fluid

∗Normal values may vary slightly between laboratories.
†Plus 80-100 ml free water per liter.
The massive edema associated with burn shock is inevitable with fluid resuscitation, and failure to administer resuscitation fluid results in irreversible hypovolemic shock and death. The edema occurs in unburned as well as burned areas (Figure 46-15). Edema often leads to mechanical airway obstruction, necessitating tracheal intubation, and increased severity of the interstitial pulmonary edema associated with inhalation injury.
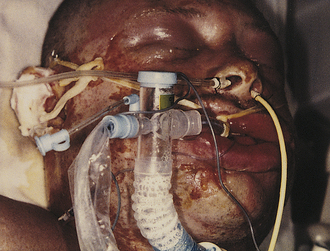
Figure 46-15 Edema related to burn injury. Superficial facial burns can result in marked swelling, requiring prompt endotracheal intubation to maintain the airway. (Courtesy Intermountain Burn Center, University of Utah.)
The most reliable criterion for adequate resuscitation of burn shock is urine output. The individual in hypovolemic shock will, as a compensatory mechanism, decrease or stop urine output in an effort to preserve circulation volume. The adult receiving sufficient intravenous fluids will excrete urine amounting to 30 to 50 ml/hr; children produce 1 ml/kg/hr. If the individual does not have adequate urine output, it often indicates inadequate fluid resuscitation. The massive amount of intravenous fluid required by burned individuals during the shock phase is often intimidating to the person unfamiliar with burns. One common concern is that massive fluid administration will result in pulmonary edema. It should be remembered that the individual is in hypovolemic shock and that fluid is lost dramatically during the resuscitation period from movement to the interstitium, exudation, and evaporation.
The endpoint of burn shock is defined as the state in which the individual is able to maintain adequate urine output for 2 hours with the intravenous fluid administration rate equal to the individual’s calculated maintenance rate (Box 46-4). As burn shock ends, fluid administered remains in the circulating volume and is reflected as an increase in urine output. The mechanism whereby capillary integrity is restored is unknown but usually occurs about 24 hours after burn injury (Figure 46-16). After the individual has reached the endpoint of burn shock, the term used to describe the vascular status of the individual is capillary seal. In individuals with large burns, colloid-containing fluids may be given to help maintain oncotic pressure during the resuscitation phase and afterward to enhance the mobilization of interstitial fluid and diuresis.82
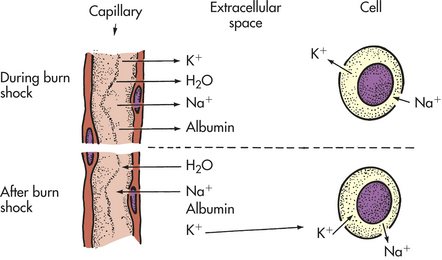
Cellular Response to Burn Injury
In addition to capillary endothelial permeability changes resulting in vascular fluid losses, transmembrane potential changes occur in cells not directly damaged by heat. The normal potential of −90 mV decreases to nearly −70 mV, with an increase in intracellular sodium and water. Such membrane potential changes may be caused by a circulating shock factor.83 Other changes can be categorized as (1) a metabolic response to the burn injury or (2) an immunologic response to the burn injury.
Metabolic Response: The metabolic changes after a burn injury were described in 1967 by Welt and associates as “sick cell syndrome.”84 This was considered to be a cell membrane transport defect related to an alteration in the steady-state composition characterized by high intracellular concentrations of sodium. Trunkey and colleagues85 found a marked decrease in primate muscle extracellular water and an increase in intracellular sodium and water during hypovolemic shock. In addition, other researchers demonstrated an associated decrease in resting membrane potential, a decrease in amplitude of the action potential, and a prolongation of the repolarization and depolarization times in association with a decreased intracellular potassium concentration.86,87 The cellular dysfunction of burn injury extends beyond the transmembrane potential disruption and the sodium-potassium pump impairment to include a loss of intracellular magnesium and phosphate88 and elevated serum lactic dehydrogenase (LDH) levels.89 Thus impairment of basic cellular function may be the underlying cause of the diminished membrane potentials. The data suggest a decrease in the efficiency of membrane pumps. The failure of rapid intravascular volume repletion to restore membrane potential completely suggests other pathways for cellular metabolic derangement.90
Metabolic reactions to the stress of a major burn injury involve the response of the sympathetic nervous system and other homeostatic regulators. Catecholamines are found in elevated amounts in both the serum and urine of burned individuals. Cortisol, glucagon, and insulin levels are elevated, with a corresponding increase in gluconeogenesis, lipolysis, and proteolysis. Changes in lipid metabolism are reflected as an elevation in plasma free fatty acids (FFA) and a decrease in plasma cholesterol and phospholipids.91 Jeschke and colleagues92 found that the use of propranolol, a nonselective beta1- and beta2-blocker, could decrease symptoms of the hypermetabolic response, including a decrease in heart rate and lipolysis. Glucose and lactate kinetics are altered after burn injury. Although tissue hypoxia produces lactic acidosis, its persistence in the presence of adequate tissue perfusion suggests an increased rate of glycogenolysis.93
Burn injury induces a hypermetabolic state that persists until wound closure. Wilmore and colleagues94 described the hypermetabolic state of 20 burned individuals as unrelated to ambient temperatures, with persistent elevation of core body temperatures. The metabolic rate increased with burn size in a curvilinear relationship, with oxygen consumption rarely exceeding two times basal levels. Evaporative water loss and surface cooling are not the primary stimulus for the hypermetabolic state; rather, the hypermetabolism is related to an increase and resetting of the thermal regulatory set point. A core body temperature of 38.5° C (101.3° F) is typical. A reflex arc mobilizes neural or hormonal afferent stimuli to the hypothalamus, producing a catecholamine response clinically manifested as hypermetabolism, hyperthermia, and hyperglycemia.
Evidence also exists that the burn wound itself directly mediates the response to injury at both the local and system levels. Cytokines, oxygen radicals, chemotactic substances, and eicosanoids contribute to the systemic inflammatory response and hypermetabolic state. The inflammatory response to the wound level is magnified into a generalized systemic inflammatory response that is often deleterious.95–97 Vasodilation, increased capillary permeability, and edema occur to facilitate healing of the local area. The distribution of the peripheral circulation after burn injury transports heat and glucose preferentially to the wound. The energy cost of these reparative and transport processes is reflected in the increased metabolism and hyperdynamic circulation.
The extensive evaporative water loss that occurs in burn tissue is a heat-consuming process, and the energy of evaporation is provided by increased visceral heat production. The signal for the response is unknown because individuals whose wounds have been denervated continue to have a posttraumatic hypermetabolic response. Hypothalamic function alterations result in the elevation of human growth hormone (hGH) serum levels in the presence of hyperglycemia, a finding opposite that in normal states.94 Further, the hypermetabolic rate is not decreased during rest, sleep, or warmth.
Evidence of hepatic response to burn injury is characterized by alterations in the clotting factors.98 A hypercoagulable state develops as manifested by an elevated plasma fibrinogen concentration in the presence of shortened prothrombin time (PT) and activated partial thromboplastin time (PTT).99
In summary, extensive burn injury initiates the most marked alterations in body metabolism associated with any illness (Figure 46-17). Much of the work explaining this response has been conducted by Wilmore,100,101 who reported that the persistent tachycardia, hyperpnea, hyperpyrexia, and marked body wasting seen in burn injury reflect heightened metabolic activity and accelerated body catabolism. The development of decreased bone density can last long after discharge from the hospital.102 These system alterations occur as a result of the cutaneous inflammatory process and are thought to facilitate wound repair. The neural component of this alteration is in response to a sympathetic reaction that releases catecholamines in large amounts.
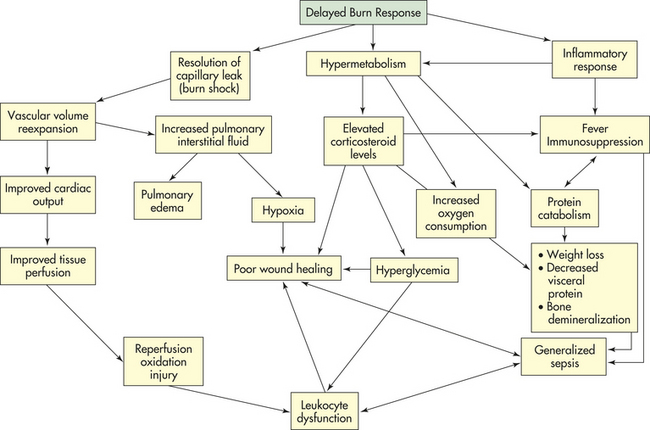
Immunologic Response: The immunologic response to burn injury is immediate, prolonged, and severe. The result in individuals surviving burn shock is immunosuppression with increased susceptibility to potentially fatal systemic burn wound sepsis.
Several cytokines have been identified in the immediate postburn period. IL-1 is detected in the serum of burned individuals. The level of IL-1 correlates inversely with burn survival; low levels may be associated with a higher mortality.103 Fatal burn injury has often shown decreased levels of IL-2, which may result in decreased T helper 1 (Th-1) lymphocytes. Th-1 cells produce IL-2, interferon-gamma, and TNF, which help to initiate cellular immunity and immunoglobulin G (IgG) production. IL-4 is elevated after burn injuries and causes a shift in the T helper cell production from Th-1 to Th-2 lymphocytes. Th-2 cells secrete IL-4, which promotes further conversion of nonspecific Th cells to Th-2 cells. Th-2 cells also produce other cytokines and antibodies.104 IL-6 levels increase quickly after burn injury and remain elevated for several weeks. The level of IL-6 correlates with the extent of burn injury.105 IL-6, together with platelet-activating factor, activates polymorphonuclear neutrophils (PMNs), causing infiltration of neutrophils into burned tissue and adhesion to vascular endothelial surfaces.106,107 IL-8 levels are elevated after burn injury, with significantly greater elevations in individuals with a TBSA burn of 40% or higher. IL-8 activity may play a role in the strong and persistent activation of neutrophils noted in people with large burns.108 Burn blister fluid contains large amounts of IL-6 and IL-8 in addition to substances such as epidermal growth factor, platelet-derived growth factor, and TGF.109
Macrophages, platelets, neutrophils, and vascular endothelial cells release prostaglandins and leukotrienes, which are the byproducts of arachidonic acid metabolism. These chemical mediators cause peripheral vasodilation, pulmonary vasoconstriction, increased capillary permeability, and local tissue ischemia in the burn wound.
A host of chemicals found in burn plasma in altered concentrations also may play a role in burn shock. These include vasoactive amines (histamine, serotonin), products of complement activation (C3a, C5a), prostaglandins, kinins, endotoxin, and metabolic hormones (catecholamines, glucocorticoids). A decrease in complement components C3a and C5a in the circulation after burn injury suggests a nonspecific activation of the complement system.110 Activation of the complement system in injured tissue results in an inflammatory response caused by release of histamine and serotonin by C3a and C5a, because histamine and serotonin alter capillary permeability and participate in the mechanism of burn shock along with kinin polypeptides and other chemical mediators. Prostaglandins function in the inflammatory process by regulating metabolism of cells of inflammation (see Chapter 6).
Burn shock can induce changes in the integrity of the intestinal wall, facilitating bacterial translocation and endotoxemia.111 Bacterial translocation from the gut may be a mechanism of infection leading to septic shock after burn injury and other major trauma.112 Circulating endotoxin is correlated with the development of MODS and death after major burn injury.113
White blood cells are also altered at this time, when their need to inhibit sepsis is vital. Natural resistance to infection in burn wounds is a function of the nonspecific immune system; that is, resistance to microorganisms that infect wounds rests almost solely on the ability of phagocytic cells (i.e., granulocytes, macrophages) to leave the bloodstream, migrate to the site of infection, and ingest and kill microorganisms.114 Normally, opsonins render bacteria susceptible to phagocytosis, but the burn injury triggers a consumptive opsoninopathy. Burn serum contains an inhibitor of C3 conversion that leads to decreased opsonization and PMN dysfunction.115,116
Individuals with altered immunocompetence before burn injury are at additional risk for complications. Opportunistic infections, such as fungal sepsis, can increase hospital stay and ICU costs.117 At risk individuals are those at the extremes of age and those with cardiac disease, malnutrition, immunodeficiency disease, and a history of alcohol or drug abuse.118,119 Additional risk factors include diabetes mellitus and pulmonary or renal dysfunction.
Evaporative Water Loss
One of the major purposes of intact skin is to serve as a barrier to evaporative water loss (EWL) from the body. With major burn injury, this ability of the skin to regulate evaporative water loss is totally disrupted. In a classic study done in 1962, Moncrief and Mason120 attempted to determine the magnitude of such a loss and determined that daily evaporative water loss was in the range of 20 times normal in the early phase of injury, with gradual decreases as wound closure is achieved. Further studies indicated that insensible water loss through burned skin is not from evaporation of water from sweat glands but rather from water vapor formed within the body and lost through the skin.121,122
Calculation of the amount of fluid lost by evaporative water loss includes losses from all sources. Normally the skin is the major source of insensible loss (75%) and the lungs are minor sources of loss (25%), with a total loss of only approximately 600 to 800 ml/day. This changes dramatically with burns, because not only does skin loss increase but also lung loss increases by hypermetabolism and hyperventilation, especially in an intubated individual. Total evaporative losses exceed many liters per day in an adult with large burn wounds. Replacement of the loss is mandatory to prevent volume deficit.
EVALUATION AND TREATMENT Burn recovery is long and stormy, with complications the rule rather than the exception. The goal of burn management is wound closure in a manner that promotes survival. Scar formation with contractures is often a consequence of healing in deep partial-thickness and full-thickness burns (Figure 46-18). Assessment of tissue viability can be difficult in complex extremity injury; pyrophosphate nuclear scanning can assist in evaluation.123
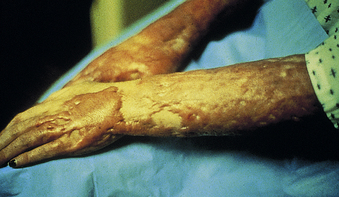
Figure 46-18 Hypertrophic scarring. Deep partial-thickness thermal injury can result in extensive hypertrophic scarring. (Courtesy Intermountain Burn Center, University of Utah.)
The three essential elements of survival of major burn injury are (1) meticulous wound management, (2) adequate fluids and nutrition, and (3) early surgical excision and grafting. Therapy for deep partial- and full-thickness burn injury includes surgical removal of the burn tissue (excision) followed by grafting of the person’s unburned skin (autograft) onto the excised wound. Satisfactory wound closure with cultured epithelial autograft (Figure 46-19) has been
WHAT’s NEW?
Improving Skin Graft Success
Much of the effort in the management of individuals with massive burn wounds focuses on closure of the wound to diminish infection risks and decrease hypermetabolism associated with burns. Cadaveric allograft skin grafting, dermal matrix development, and cultured keratinocyte technology have become tools to promote this strategy. Fibrin sealant has become widely used to improve graft take, decrease blood loss, and improve cosmetic appearance. The cost and efficacy are under clinical evaluation but show promise. The traditional fibrin sealant is an aerosolized fibrin and thrombin that mixes in the aerosol and provides a hemostatic layer that adheres within a few seconds. The more recent applications for skin grafts use a mixture with one tenth the concentration of thrombin, thus affording the surgeon time to position the graft and improve the technical application in the operating room. Such advances may prove cost-effective and provide improved outcomes.
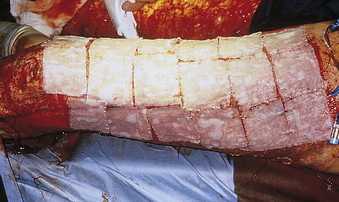
Figure 46-19 Application of cultured epithelial autografts. The thin sheets of keratinocytes are attached to gauze backing to allow application onto the clean, excised thigh. (Courtesy Intermountain Burn Center, University of Utah.)
Data from Achauer BM, Miller SR, Lee TE: J Burn Care Rehabil 15(1):24-28, 1994; Chakravorty RC, Sosnowski KM: Ann Plast Surg 23(6):488-491, 1989; de Moraes AM, Annichino-Bizzacchi JM, Rossi AB: Sao Paulo Med J 116(4):1747-1752, 1998; Dean M, Nicholls M, Wedderburn C: Med J Aust 160(8):526-527, 1994; Foster K: Surgery 142(4 Suppl):S50-S54, 2007; Foster K et al: J Burn Care Res 29(2):293-303, 2008; Jeschke MG et al: Plast Reconstr Surg 113(2):525-530, 2004; McGill V et al: J Burn Care Rehabil 18(5):429-434, 1997; Ofodile FA, Sadana MK: J Natl Med Assoc 83(5):416-418, 1991; Ronfard V et al: Burns 17(3):181-184, 1991; Saltz R et al: J Burn Care Rehabil 10(6):504-507, 1989; Saltz R et al: Plast Reconstr Surg 88(6):1005-1015, 1991; Schumacher J et al: Can J Vet Res 60(2):158-160, 1996.
inconsistent and costly.124,125 Early enthusiasm for synthetic dermal replacement has been tempered by a high rate of dermal graft loss and slow epidermal engraftment.126,127 Such advancements in skin replacement technology include sheets of acellular dermal matrix that can be used with thin, meshed autografts or cultured epithelial autografts.128,129 This concept is also being used on the donor site, and glycosaminoglycan hydrogels may supplement donor site wound dressings130 (see What’s New? Improving Skin Graft Success). Scar reduction or prevention is a challenging problem that is being addressed with pulsed-dye laser treatment.131 Research is also directed toward therapies to modulate hypermetabolic and inflammatory responses. Nutritional therapy is focused on early enteral feeding to reduce the potential of gut-mediated sepsis. Ongoing clinical trials using anabolic agents (e.g., recombinant human growth hormone) and pharmacologic agents that modulate inflammatory and endocrine mediators (e.g., ibuprofen, propranolol) show promise in the treatment of severe burn injuries.80 Burn pain is almost always acute, and treatment strategies usually differ from strategies for chronic pain. In addition to opioid-based agents, newer treatment approaches may include antianxiety agents, hypnosis, and relaxation techniques.132
Acute Physiology and Chronic Health Evaluation II and III (APACHE II and APACHE III) 1713
Anaphylactic shock 1702
Burn 1714
Burn shock 1717
Burn wound depth 1714
Capillary seal 1718
Cardiogenic shock 1699
Deep partial-thickness burn 1714
Escharotomy 1715
First-degree burn 1714
Fluid resuscitation 1717
Full-thickness injury 1715
Gut hypothesis 1711
Hypermetabolism 1711
Hypovolemic shock 1701
Maldistribution of blood flow 1711
Multiple organ dysfunction syndrome (MODS) 1707
Myocardial depression 1711
Neurogenic shock (vasogenic shock) 1702
Nonthermal injury 1714
Partial-thickness injury 1714
Posttraumatic hypermetabolic response 1719
Primary MODS 1707
Secondary MODS 1708
Second-degree burn 1714
Septic shock 1703
Shock 1696
Superficial partial-thickness injury 1714
Supply-dependent oxygen consumption 1711
Thermal injury 1714
Third-degree burn 1715
Total body surface area (TBSA) 1716
Vesicants 1714
REFERENCES
1. Garcia, A. Critical care issue in the early management of severe trauma. Surg Clin North Am. 2006;86(6):1359–1387.
2. Cunneen, J., Cartwright, M. The puzzle of sepsis: fitting the pieces of the inflammatory response with treatment. AACN Clin Issues. 2004;15(1):18–44.
3. Topalian, S., Ginsberg, F., Parrillo, J. Cardiogenic shock. Crit Care Med. 2008;36(1):S66–S77.
4. Jackson, C., Boenau, I.B. How to recognize cardiogenic shock. Emerg Med. 2007;39(10):11–19.
5. Babaev, A., et al. Trends in management and outcomes of patients with acute myocardial infarction complicated by cardiogenic shock. JAMA. 2005;294(4):448–454.
6. Hochman, J., et al. Early revascularization and long-term survival in cardiogenic shock complicating acute myocardial infarction. JAMA. 2006;295(21):2511–2515.
7. Omland, T. Advances in congestive heart failure management in the intensive care unit: b-type natriuretic peptides in evaluation of acute heart failure. Crit Care Med. 2008;36(1):S17–S27.
8. Russell, J. Vasopressin versus norepinephrine infusion in patients with septic shock. N Engl J Med. 2008;358(9):877–887.
9. Holmes, C.L., Walley, K.R. The evaluation and management of shock. Clin Chest Med. 2003;24(4):775–789.
10. Bone, R.C., et al. Definitions for sepsis and organ failure and guidelines for the use of innovative therapies in sepsis. The ACCP/SCCM Consensus Conference Committee. Chest. 1992;101(6):1644–1655.
11. Levy, M.M., et al. 2001 SCCM/ESICM/ACCP/ATS/SIS International Sepsis Definition Conference. Crit Care Med. 2003;31(4):1250–1256.
12. Calandra, T., Cohen, J. The international sepsis forum consensus conference on definitions of infection in the intensive care unit,. Crit Care Med. 2005;33(7):1538–1548.
13. Wenzel, R.P. Health care-associated infections: major issues in the early years of the 21st century. Clin Infect Dis. 2007;15(45-Suppl 1):S85–S88.
14. Albrecht, S.J., et al. Reemergence of gram-negative health care-associated bloodstream infections. Arch Intern Med. 2006;166(12):1289–1294.
15. Kleinpell, R.M., Graves, B.T., Ackerman, M.H. Incidence, pathogenesis, and management of sepsis: an overview. AACN Adv Crit Care. 2006;17(4):385–393.
16. Papathanassoglou, E.D., et al. Association of proinflammatory molecules with apoptotic markers and survival in critically ill multiple organ dysfunction patients. Biol Res Nurs. 2003;5(2):129–141.
17. Dellinger, R.P., et al. Surviving sepsis campaign: international guidelines for management of severe sepsis and septic shock: 2008. Intensive Care Med. 2008;31(1):17–60.
18. Cross, A.S., et al. Development of an anti-core lipopolysaccharide vaccine for the prevention and treatment of sepsis. Vaccine. 2004;22(7):812–817.
19. Cinel, I., Dellinger, R.P. Advances in pathogenesis and management of sepsis. Curr Opin Infect Dis. 2007;20(4):345–352.
20. Honoré, P.M., Joannes-Boyau, O., Gressens, B. Blood and plasma treatments: the rationale of high-volume hemofiltration. Contrib Nephrol. 2007;156:387–395.
21. Baue, A.E. Multiple, progressive or sequential system failure: a syndrome for the 70’s. Arch Surg. 1975;110(7):779–781.
22. Tilney, N.L., Bailey, G.L., Morgan, A.P. Sequential system failure after rupture of abdominal aortic aneurysm: an unsolved problem in postoperative care. Ann Surg. 1973;178(2):117–122.
23. Awad, S.S. State-of-the-art therapy for severe sepsis and multisystem organ dysfunction. Am J Surg. 2003;186(Suppl):23S–30S.
24. Cook, R., et al. Multiple organ dysfunction: baseline and serial component scores. Crit Care Med. 2001;29(11):2046–2050.
25. Rotstein, O.D. Modeling the two-hit hypothesis for evaluating strategies to prevent organ injury after shock/resuscitation. J Trauma. 2003;54(5 Suppl):S203–S206.
26. Brown, M.J., et al. The systemic inflammatory response syndrome, organ failure, and mortality after abdominal aortic aneurysm repair. J Vasc Surg. 2003;37(3):600–606.
27. Aird, W.C. The role of the endothelium in severe sepsis & MODS. Blood. 2003;101:3756–3777.
28. Vanhorebeek, I., Van den Berghe, G. The neuroendocrine response to critical illness is a dynamic process. Crit Care Clin. 2006;22(1):1–15.
29. van Meurs, M., et al. Early organ-specific endothelial activation during hemorrhagic shock and resuscitation. Shock. 2008;29(2):291–299.
30. Brown, K.A., et al. Neutrophils in development of multiple organ failure in sepsis. Lancet. 2006;8(368):157–169.
31. Baue, A.E. Mediators or markers of injury, inflammation, and infection (harbingers of doom or predictors of disaster) and biologic puzzles or ambiguities. Arch Surg. 2007;142(1):89–93.
32. Maier, R.V. Pathogenesis of multiple organ dysfunction syndrome—endotoxin, inflammatory cells, and their mediators: cytokines and reactive oxygen species. Surg Infect (Larchmt). 2000;1(3):197–204.
33. Rankin, J.A. Biological mediators of acute inflammation. AACN Clin Issues. 2004;15(1):3–17.
34. Matsuda, N., Hattori, Y. Systemic inflammatory response syndrome (SIRS): molecular pathophysiology and gene therapy. J Pharmacol Sci. 2006;101(3):189–198.
35. Deitch, E.A., Xu, D., Kaise, V.L. Role of the gut in the development of injury- and shock-induced SIRS and MODS: the gut-lymph hypothesis, a review. Front Biosci. 2006;1(11):520–528.
36. Magnotti, L.J., Deitch, E.A. Burns, bacterial translocation, gut barrier function, and failure. J Burn Care Rehabil. 2005;26(5):383–391.
37. Dixon, B. The role of microvascular thrombosis in sepsis. Anaesth Intensive Care. 2004;32(5):619–629.
38. Di Giantomasso, D., May, C.N., Bellomo, R. Vital organ blood flow during hyperdynamic sepsis. Chest. 2003;124(3):1053–1059.
39. Merx, M.W., Weber, C. Sepsis and the heart. Circulation. 2007;116(7):793–802.
40. Kumar, A., et al. Myocardial dysfunction in septic shock: part II. role of cytokines and nitric oxide. J Cardiothorac Vasc Anesth. 2001;15(4):485–511.
41. Pettila, V., et al. Comparison of multiple organ dysfunction scores in the prediction of hospital mortality in the critically ill. Crit Care Med. 2002;30(8):1705–1711.
42. Blot, S.I., et al. Clinical and economic outcomes in critically ill patients with nosocomial catheter-related bloodstream infections. Clin Infect Dis. 2005;41(11):1591–1598.
43. Rivers, E.P., et al. Early goal-directed therapy in the treatment of severe sepsis and septic shock. N Engl J Med. 2001;345:1368–1377.
44. The Acute Respiratory Distress Syndrome Network. Ventilation with lower tidal volumes as compared with traditional tidal volumes for acute lung injury and the acute respiratory distress syndrome. N Engl J Med. 2000;342:1301–1308.
45. Van den Berghe, G., et al. Intensive insulin therapy in the critically ill patients. N Engl J Med. 2001;345:1359–1367.
46. Caruso, D.M., et al. Rationale for “early” percutaneous dilatational tracheostomy in patients with burn injuries. J Burn Care Rehabil. 1997;18(5):424–428.
47. Saffle, J.R., Morris, S.E., Edelman, L. Early tracheostomy does not improve outcome in burn patients. J Burn Care Rehabil. 2002;23(6):431–438.
48. Clark, D.E., Dainiak, C.N., Reeder, S. Decreasing incidence of burn injury in a rural state. Ing Prev. 2000;6(40):259–262.
49. Brigham, P.A., McLoughlin, E. Burn incidence and medical care use in the United States: estimates, trends, and data sources. J Burn Care Rehabil. 1996;17(2):95–107.
50. American Burn Association: Burn incidence and treatment in the U.S.: 2007 Fact Sheet, 2007. Available at www.ameriburn.org/resources_factsheet.php. Accessed September 28, 2008.
51. Saffle, J.R., Edelman, L., Morris, S.E. Regional air transport of burn patients: a case for telemedicine? J Trauma. 2004;57(1):57–64.
52. Cochran, A., et al. Self-reported quality of life after electrical and thermal injury. J Burn Care Rehabil. 2004;25(1):61–66.
53. Altintas, M.A., et al. Differentiation of superficial-partial vs. deep-partial thickness burn injuries in vivo by confocal-laser-scanning microscopy. Burns. 2009;35(1):80–86.
54. Braue, E.H., Jr., et al. Noninvasive methods for determining lesion depth from vesicant exposure. J Burn Care Res. 2007;28(2):275–285.
55. Chiu, T., Burd, A. Laboratory assessment of burn depth. Plast Reconstr Surg. 2007;119(2):751–752.
56. Cross, K.M., et al. Clinical utilization of near-infrared spectroscopy devices for burn depth assessment. Wound Repair Regen. 2007;15(3):332–340.
57. Devgan, L., et al. Modalities for the assessment of burn wound depth. J Burns Wounds. 2006;5:e2.
58. Hoeksema, H., et al. Accuracy of early burn depth assessment by laser Doppler imaging on different days post burn. Burns. 2009;35(1):36–45.
59. Kassira, W., Namias, N. Outpatient management of pediatric burns. J Craniofac Surg. 2008;19(4):1007–1009.
60. Mandal, A. Burn wound depth assessment—is laser Doppler imaging the best measurement tool available? Int Wound J. 2006;3(2):138–143.
61. McGill, D.J., et al. Assessment of burn depth: a prospective, blinded comparison of laser Doppler imaging and videomicroscopy. Burns. 2007;33(7):833–842.
62. Monstrey, S., et al. Assessment of burn depth and burn wound healing potential. Burns. 2008;34(6):761–769.
63. Ng, D., et al. The use of laser Doppler imaging for burn depth assessment after application of flammacerium. Burns. 2007;33(3):396–397.
64. Renkielska, A., et al. Burn depths evaluation based on active dynamic IR thermal imaging—a preliminary study. Burns. 2006;32(7):867–875.
65. Tehrani, H., et al. Spectrophotometric intracutaneous analysis: a novel imaging technique in the assessment of acute burn depth. Ann Plast Surg. 2008;61(4):437–440.
66. Sheridan, R.L., et al. Planimetry study of the percent of body surface represented by the hand and palm: sizing irregular burns is more accurately done with the palm. J Burn Care Rehabil. 1995;16(6):605–606.
67. Morris, S.E., Navaratnam, N., Herndon, D.N. A comparison of effects of thermal injury and smoke inhalation on bacterial translocation. J Trauma. 1990;30(6):639–643.
68. Arturson, G. Forty years in burns research—the postburn inflammatory response. Burns. 2000;26(7):599–604.
69. Baxter, C.R., Cook, W.A., Shires, G.T. Serum myocardial depressant factor of burn shock. Surg Forum. 1966;17:1–2.
70. Demling, R.H., Will, J.A., Belzer, F.O. Effect of major thermal injury on the pulmonary microcirculation. Surgery. 1978;83(6):746–751.
71. Horton, J.W., et al. Calcium antagonists improve cardiac mechanical performance after thermal trauma. J Surg Res. 1999;87(1):39–50.
72. Aikawa, N., Martyn, J.A., Burke, J.F. Pulmonary artery catheterization and thermodilution cardiac output determination in the management of critically burned patients. Am J Surg. 1978;135(6):811–817.
73. Dobson, E.L., Warner, G.F. Factors concerned in the early stages of thermal shock. Circ Res. 1957;5(1):69–74.
74. Lefer, A.M., Martin, J. Origin of myocardial depressant factor in shock. Am J Physiol. 1970;218(5):1423–1442.
75. Rosenthal, S.R., Hawley, P.L., Hakim, A.A. Purified burn toxic factor and its competition. Surgery. 1972;71(4):527–536.
76. Horton, J.W., Burton, K.P., White, D.J. The role of toxic oxygen metabolites in a young model of thermal injury. J Trauma. 1995;39(3):563–569.
77. Onuoha, G., Alpar, K., Jones, I. Vasoactive intestinal peptide and nitric oxide in the acute phase following burns and trauma. Burns. 2001;27(1):17–21.
78. Ungureanu-Longrois, D., et al. Myocardial contractile dysfunction in the systemic inflammatory response syndrome: role of a cytokine-inducible nitric oxide synthase in cardiac myocytes. J Mol Cell Cardiol. 1995;27(1):155–167.
79. Gamelli, R.L., et al. Burn-induced nitric oxide release in humans. J Trauma. 1995;39(5):869–877. [discussion 77-78].
80. Baxter, C.R. Fluid volume and electrolyte changes of the early postburn period. Clin Plast Surg. 1974;1(4):693–703.
81. Huang, P.P., et al. Hypertonic sodium resuscitation is associated with renal failure and death. Ann Surg. 1995;221(5):543–554.
82. Herndon, D.N., Spies, M. Modern burn care. Semin Pediatr Surg. 2001;10(1):28–31.
83. Evans, J.A., Darlington, D.N., Gann, D.S. A circulating factor(s) mediates cell depolarization in hemorrhagic shock. Ann Surg. 1991;213(6):549–556.
84. Welt, L.G. Membrane transport defect: the sick cell. Trans Assoc Am Physicians. 1967;80:217–226.
85. Trunkey, D.D., et al. The effect of hemorrhagic shock on intracellular muscle action potentials in the primate. Surgery. 1973;74(2):241–250.
86. Cunningham, J.N., Jr., Shires, G.T., Wagner, Y. Changes in intracellular sodium and potassium content of red blood cells in trauma and shock. Am J Surg. 1971;122(5):650–654.
87. Rosenthal, S.R., Tabor, H. Electrolyte changes and chemotherapy in experimental burn and traumatic shock and hemorrhage. Arch Surg. 1945;51:244.
88. Turinsky, J., Gonnerman, W.A., Loose, L.D. Impaired mineral metabolism in postburn muscle. J Trauma. 1981;21(6):417–423.
89. Deets, D.K., Glaviano, V.V. Plasma and cardiac lactic dehydrogenase activity in burn shock. Proc Soc Exp Biol Med. 1973;142(2):412–416.
90. Button, B. Evidence of circulating membrane depolarization factor(s) in hemorrhagic shock. Shock. 1994;1(Suppl):15.
91. Okamoto, R., Glaviano, V.V., Pindok, M. Myocardial lipases and catecholamines in burn shock. Proc Soc Exp Biol Med. 1971;137(1):347–353.
92. Jeschke, M.G., et al. Propranolol does not increase inflammation, sepsis, or infectious episodes in severely burned children. J Trauma. 2007;62(3):676–681.
93. Wilmore, D.W., Aulick, H.L., Goodwin, C.W. Glucose metabolism following severe injury. Acta Chir Scand Suppl. 1980;498:43–47.
94. Wilmore, D.W., et al. Alterations in hypothalamic function following thermal injury. J Trauma. 1975;15(8):697–703.
95. Gump, F.E., Price, J.B., Jr., Kinney, J.M. Blood flow and oxygen consumption in patients with severe burns. Surg Gynecol Obstet. 1970;130(1):23–28.
96. Wilmore, D.W., et al. Influence of the burn wound on local and systemic responses to injury. Ann Surg. 1977;186(4):444–458.
97. Wilmore, D.W., et al. Effect of injury and infection on visceral metabolism and circulation. Ann Surg. 1980;192(4):491–504.
98. Holder, I.A., Neely, A.N. Hageman factor dependent activation and its relationship to lethal Pseudomonas aeruginosa burn wound infections. Agents Actions Suppl. 1992;38(Pt 3):329–342.
99. McManus, W.F., Eurenius, K., Pruitt, B.A., Jr. Disseminated intravascular coagulation in burned patients. J Trauma. 1973;13(5):416–422.
100. Wilmore D.W., ed. The metabolic management of the critically ill, ed 2, New York: Plenum, 1990.
101. Wilmore, D.W., Aulick, L.H. Metabolic changes in burned patients. Surg Clin North Am. 1978;58(6):1173–1187.
102. Edelman, L.S., et al. Sustained bone mineral density changes after burn injury. J Surg Res. 2003;114(2):172–178.
103. Wright, K., et al. Burn-activated neutrophils and tumor necrosis factor-alpha alter endothelial cell actin cytoskeleton and enhance monolayer permeability. Surgery. 2000;128(2):259–265.
104. Goebel, A., et al. Injury induces deficient interleukin-12 production, but interleukin-12 therapy after injury restores resistance to infection. Ann Surg. 2000;231(2):253–261.
105. Nishiura, T., et al. Gene expression and cytokine and enzyme activation in the liver after a burn injury. J Burn Care Rehabil. 2000;21(2):135–141.
106. Biffl, W.L., et al. Interleukin-6 delays neutrophil apoptosis via a mechanism involving platelet-activating factor. J Trauma. 1996;40(4):575–578.
107. Choi, M., et al. Preventing the infiltration of leukocytes by monoclonal antibody blocks the development of progressive ischemia in rat burns. Plast Reconstr Surg. 1995;96(5):1177–1185.
108. Iocono, J.A., et al. Interleukin-8 levels and activity in delayed-healing human thermal wounds. Wound Repair Regen. 2000;8(3):216–225.
109. Ortega, M.R., Ganz, T., Milner, S.M. Human beta defensin is absent in burn blister fluid. Burns. 2000;26(8):724–726.
110. Heideman, M., Kaijser, B., Gelin, L.E. Complement activation and hematologic, hemodynamic, and respiratory reactions early after soft-tissue injury. J Trauma. 1978;18(10):696–700.
111. Grzybowski, J., et al. Antidietary antigen antibodies in the sera of patients with burns as a potential marker of gut mucosa integrity failure. J Burn Care Rehabil. 1992;13(2 Pt 1):194–197.
112. Deitch, E.A., Berg, R. Bacterial translocation from the gut: a mechanism of infection. J Burn Care Rehabil. 1987;8(6):475–482.
113. Yao, Y.M., et al. The association of circulating endotoxaemia with the development of multiple organ failure in burned patients. Burns. 1995;21(4):255–258.
114. Benhaim, P., Hunt, T.K. Natural resistance to infection: leukocyte functions. J Burn Care Rehabil. 1992;13(2 Pt 2):287–292.
115. Alexander, J.W., et al. Consumptive opsoninopathy: possible pathogenesis in lethal and opportunistic infections. Ann Surg. 1976;184(6):672–678.
116. Bjornson, A.B., Altemeier, W.A., Bjornson, H.S. Changes in humoral components of host defense following burn trauma. Ann Surg. 1977;186(1):88–96.
117. Cochran, A., et al. Systemic Candida infection in burn patients: a case-control study of management patterns and outcomes. Surg Infect (Larchmt). 2002;3(4):367–374.
118. Goff, D.R., et al. Cardiac disease and the patient with burns. J Burn Care Rehabil. 1990;11(4):305–307.
119. McGill, V., et al. The impact of substance use on mortality and morbidity from thermal injury. J Trauma. 1995;38(6):931–934.
120. Moncrief, J.A., Mason, A.D., Jr. Water vapor loss in the burned patient. Surg Forum. 1962;13:38–41.
121. Moncrief, J.A. Burns. In Schwartz S.I., ed.: Principles of surgery, ed 2, New York: McGraw-Hill, 1974.
122. Roe, C.F., Kinney, J.M. Water and heat exchange on third-degree burns. Surgery. 1964;56:212–220.
123. Affleck, D.G., et al. Assessment of tissue viability in complex extremity injuries: utility of the pyrophosphate nuclear scan. J Trauma. 2001;50(2):263–269.
124. Ronfard, V., et al. Long-term regeneration of human epidermis on third degree burns transplanted with autologous cultured epithelium grown on a fibrin matrix. Transplantation. 2000;70(11):1588–1598.
125. Williamson, J.S., et al. Cultured epithelial autograft: five years of clinical experience with twenty-eight patients. J Trauma. 1995;39(2):309–319.
126. Fitton, A.R., Drew, P., Dickson, W.A. The use of a bilaminate artificial skin substitute (Integra) in acute resurfacing of burns: an early experience. Br J Plast Surg. 2001;54(3):208–212.
127. Peck, M.D., et al. A trial of the effectiveness of artificial dermis in the treatment of patients with burns greater than 45% total body surface area. J Trauma. 2002;52(5):971–978.
128. Carsin, H., et al. Cultured epithelial autografts in extensive burn coverage of severely traumatized patients: a five year single-center experience with 30 patients. Burns. 2000;26(4):379–387.
129. Wainwright, D., et al. Clinical evaluation of an acellular allograft dermal matrix in full-thickness burns. J Burn Care Rehabil. 1996;17(2):124–136.
130. Kirker, K.R., et al. Glycosaminoglycan hydrogels as supplemental wound dressings for donor sites. J Burn Care Rehabil. 2004;25(3):276–286.
131. Liew, S.H., Murison, M., Dickson, W.A. Prophylactic treatment of deep dermal burn scar to prevent hypertrophic scarring using the pulsed dye laser: a preliminary study. Ann Plast Surg. 2002;49(5):472–475.
132. Jellish, W.S., et al. Effect of topical local anesthetic application to skin harvest sites for pain management in burn patients undergoing skin-grafting procedures. Ann Surg. 1999;229(1):115–120.