Cancer
Breast cancer, the most common cancer in American women, is the leading cause of death in women ages 40 to 44 years and the second most common killer after lung cancer of women of all ages. The incidence of breast cancer has risen steadily since 1950 and is leveling off at about 126 cases per 100,000 women per year. The highest absolute lifetime (calculated to age 85) risk of breast cancer is 1 in 8 for non-Hispanic white women, 1 in 14 for black women, 1 in 21 for New Mexican Hispanics, and 1 in 40 for New Mexican American Indians240 (Table 23-15). In women younger than 50 years of age, blacks experience higher incidence of early onset breast cancer and higher breast cancer mortality.241 Black women in all age groups experience the highest mortality rates for breast cancer although the reason for this disparity is not clearly understood.242 More than two thirds of breast cancer cases occur in women older than 55 years. The median age for breast cancer diagnosis is 61 years of age. The median age at death for breast cancer is 69 years of age.242 Because DCIS is almost exclusively detected by mammography, the large increase in incidence over the past 20 years can be attributed to screening.
Table 23-15
Chance of Being Diagnosed with Breast Cancer
| By Age (years) | By Ratio |
| 30-39 | 1 in 238 |
| 40-49 | 1 in 69 |
| 50-59 | 1 in 38 |
| 60-69 | 1 in 27 |
| Ever∗ | 1 in 8 |
| Never | 7 in 8 |
NOTE: These calculations are averages. An individual’s risk may be higher or lower depending on several factors (e.g., family history, reproductive history, race ethnicity, and others).
Data from Reis LAG et al: Cancer statistics review, 1975-2005, Bethesda, MD, 2008, National Cancer Institute. Available at http://seer.cancer.gov/csr/1975_2005. Based on November 2007 SEER data, posted SEER website, 2008.
Risk factors and possible causes of breast cancer can be classified as reproductive, hormonal, environmental and lifestyle, and familial (Table 23-16). However, two factors emerging as important are postpartum involution of the mammary gland and breast density, which are not as easily classified.
Table 23-16
Factors Associated with Increased Risk of Breast Cancer∗
| Category | Risk Factor | Relative Risk† |
| Race | Blacks have higher incidence up to age 40 yr; whites have higher incidence after age 40 yr | 1.1-1.9 |
| Family history | Breast cancer in first-degree relative before age 60 yr | 2-3 |
| Premenopausal or bilateral breast cancer | >4 | |
| Postmenopausal in first-degree relative | ≤2 | |
| Breast cancer in two first-degree relatives | 4-6 | |
| BRCA1 or BRCA2 | ≤4 | |
| TP53 (Li-Fraumeni syndrome) | ≤4 | |
| Previous medical history | Moderate or florid mammary hyperplasia | 1.5-2 |
| Mammary papilloma | 1.5-2 | |
| Atypical mammary hyperplasia | 4-5 | |
| DCIS, LCIS‡ | 8-10 | |
| Estrogen exposure | Early menarche (before age 12 yr) | 1.1-1.9 |
| Late menopause (after age 55 yr) | 1.1-1.9 | |
| Postmenopausal hormone therapy | 1.4 | |
| Oral contraceptive use | 1.5 | |
| Pregnancy | Nulliparous or late first pregnancy (after age 35 yr) | 1.1-1.9 |
| Radiation | Atomic bomb | 3 |
| Repeated fluoroscopy | 1.5-2§ | |
| Obesity and stature | Postmenopausal | 1.2 |
| Tallness | ≤2 | |
| Dietary/alcohol | High alcohol consumption | 1.4-2 |
| High energy intake | ≤2 | |
| Advanced age | 2-4 | |
| Xenobiotics | ≤2 | |
| Social | Smoking | 2-4 |
| Higher socioeconomic status | ≤2 | |
| Low physical activity | ≤2 | |
| Environmental | Excess radiation to breasts | ??‡ |
| Chemical carcinogens | ≤2-?? | |
| Infectious agents | ≤2-?? |
DCIS, Ductal carcinoma in situ; LCIS, lobular carcinoma in situ.
∗Normal lifetime risk in white non-Hispanic women: 1 in 8.
†Relative risk is defined and discussed in Chapter 5.
‡Data from Lester SC: The breast. In Kumar V, Abbas AK, Fausto N, editors: Robbins and Cotran pathologic basis of disease, ed 7, Philadelphia, 2005, Saunders.
Reproductive Factors: Pregnancy: A clearer understanding of mammary gland structure (morphology) and function from fetal development, to puberty, pregnancy, and aging will help elucidate fundamental changes to breast development and disease. A key element is “branching morphogenesis,” in which the mammary gland produces and delivers copious amounts of milk by forming a rootlike network of branched ducts from a rudimentary epithelial bud.243 Branching morphogenesis begins in fetal development, pauses after birth, starts again in response to estrogens at puberty, and is modified by cyclic ovarian hormonal action. This systemic hormonal action elicits local paracrine interactions between the developing epithelial ducts and their adjacent mesenchyme (embryonic) or postnatal stroma. Then the local cellular crosstalk directs the tissue remodeling, ultimately producing a mature ductal tree.243 The gland is unique as it undergoes most of its branching during adolescence and not fetal development. This allows experimental manipulation of the gland not possible with any other organ.244
A woman’s age when her first child is born affects her risk for developing breast cancer—the younger she is, the lower the risk. A complete pregnancy before age 20 reduces the risk of breast cancer by 20% to 50%.245–247 The protective factor is especially observed in the years of peak incidence, the postmenopausal years.248 Paradoxically, however, a transient increase in breast cancer risk lasting 3 to 5 years after pregnancy is reported in women 25 years of age or older during pregnancy.245,249,250 In addition, pregnancy induces a lifelong, not transient increase, in breast cancer risk in women who are more than 30 at the time of first pregnancy.251 Why age affects this transient increase in breast cancer risk is unknown. A hypothesis for risk at any age is that gland involution after pregnancy and lactation uses some of the same tissue remodeling pathways activated during wound healing (i.e., proinflammatory pathways). The proinflammatory environment, although physiologically normal, promotes tumor progression. The presence of macrophages in the involuting mammary gland may contribute to carcinogenesis.252 (Involution is an important topic discussed on p. 882.)
The main mechanisms for the protective effect of pregnancy are controversial including (1) induction of breast differentiation with lasting protective phenotypic (morphologic) changes; (2) altered cell fate with removal or modification of vulnerable cells; (3) enhancement of the ability for DNA repair or apoptosis, or both; (4) altered systemic hormonal regulation and possible persistent changes in intracellular pathways regulating proliferation; (5) decreased proliferation in the parous involuted gland; and (6) early-life hormonal and dietary exposures.248 Although contradictory evidence exists the majority of data suggest that lasting changes and decreased proliferation are not closely related to hormone-induced protection. In animal models, hormone levels mimicking pregnancy, including estrogen and progesterone combinations and human chorionic gonadotropin, protect against carcinogen-induced cancer253 (Box 23-17). The mechanism whereby protection is caused by an increase in apoptosis has been implicated in the post-pregnancy treatment experimental animal model. Studies of in utero and prepubertal exposures and subsequent breast cancer risk have yielded inconclusive results.
Thus the two prevailing hypotheses, not mutually exclusive, of protective mechanisms induced by pregnancy are (1) the induction of an altered systemic hormonal pattern and (2) an altered cell fate or persistent changes in intracellular regulatory circuits (loops).248 The first hypothesis involves systemic levels of hormones, including growth hormone and prolactin, and their cascading downstream effectors as modifying the chemical-carcinogen–initiated mammary cells (see p. 883). The second hypothesis emphasizes that hormones induce a molecular switch in stem cells that result in cells with long-lasting changes in regulatory circuits regulating proliferation and response to DNA damage.248 Still controversial, stem cells are proposed to be the origin for breast cancer.254–256 Although further research is needed data indicate that reduced stem cells may help explain why early pregnancy reduces the risk of breast cancer. In addition, cancers may result from errant stem or progenitor cells misguided by their microenvironment (stroma) or from mutated somatic cells that interact with stem cells causing dysfunctional signaling that leads to carcinogenesis.257 Much research is devoted to the concept that stem cells are controlled by their microenvironment, systemic hormones, and local growth factors.258 A series of experiments demonstrated that estrogen facilitates epithelial proliferation and morphogenesis by a paracrine mechanism.259 Amphiregulin (AREG) was found to be a major paracrine-mediator of ductal morphogenesis and plays an important role in mammary stem cell self-renewal and differentiation.258 These studies are important because they help elucidate factors in mammary stem differentiation and breast cancer initiation, progression, and protection. Thus hierarchical models are emerging of hormones, growth factors, and transcription factors in which all types of epithelial cells in the mammary gland possibly originate from a single common multipotent stem cell.258 As part of the second hypothesis, investigators are focusing on the TP53 tumor-suppressor gene, which has been demonstrated to be important in pregnancy-related hormone-induced protection. The function of TP53 is required for hormone-mediated protection against the carcinogens dimethylbenz(a)anthracene (DMBA)-induced carcinogenesis in mice.244 Russo and colleagues260 have found that the post-pregnancy involuted mammary gland exhibits elevated expression of genes involved in DNA repair and apoptosis.
Lobular Involution and Age: Part of the uniqueness of the mammary gland is its profound physiologic changes throughout the phases of a woman’s life: puberty, pregnancy, lactation, postlactational involution, and aging. The human breast is organized to 15 to 20 major lobes, each with lobules containing milk-forming acini (see Figure 22-19). With aging, breast lobules regress or involute with a decrease in the number and size of acini per lobule and replacement of the intralobular stroma with the more dense collagen of connective tissue.261 Over time the glandular elements and collagen are replaced with fatty tissue. This process is called lobular involution whereby, over many years, the parenchymal elements progressively atrophy and disappear. A first study of its kind found lobular involution was associated with reduced risk of breast cancer.261–263 Breast cancer risk decreased with increasing extent of involution in both high- and low-risk subgroups defined by family history of breast cancer, epithelial atypia, reproductive history, and age.261 Based on pathologic and epidemiologic factors, these investigators propose that delayed involution (persistent glandular epithelium) is a major risk factor for breast cancer.261–263 Widely appreciated is that as women age, their risk of breast cancer increases. But the rate of increase of breast cancer slows at about 50 years of age.264,265 This slowing has been attributed to a reduction in ovarian hormone production. Milanese and colleagues261 observed a definite increase in the process of involution at about 50 years of age with complete involution present in 5.8% of women ages 40 to 49 years and 21.6% of women ages 50 to 59 years. Investigators propose that involution may contribute to this slowing in the rate of increase of breast cancer among women older than 50 years.263 Importantly, investigators found an inverse association between lobular involution and parity.261 Other investigators have reported that the more children a woman has, the more likely she is to have persistent lobular tissue,266,267 which Milanese and colleagues261 found was associated with increased risk of breast cancer. However, multiparity also has been found to reduce risk of breast cancer.268,269 This apparent contradiction may be explained by studies documenting that full-term pregnancies after 35 years of age are correlated with an increased risk of breast cancer.270 In the Milanese study, age of the mother at each child’s birth was unknown.
Henson and colleagues271 propose that late pregnancy with its concomitant increase in the proliferation of the ductal-alveolar epithelium is likely to interrupt the process of involution, which typically begins between 30 and 40 years of age. The activated stromal environment in the process of involution is similar to that in invasive breast cancer. The long-term protective effects of pregnancy with hormones released during pregnancy affect remodeling of the stromal microenvironment by causing apoptosis and involution. However, a short-term increase in breast cancer risk following pregnancy may be caused by the process of mammary gland involution which returns the tissue back to its prepregnant state and is co-opted by processes of wound healing resulting in a proinflammatory environment that although physiologically normal can promote carcinogenesis.252
Interestingly, oophorectomy, which is associated with a decrease in risk of breast cancer, leads to atrophy of breast parenchyma in young women as is noted in older women. Thus the risk reduction of oophorectomy may be caused by an accelerated involution.271
Risk data are needed on the age of a woman at pregnancy and at breast biopsy to evaluate the relationship of parity, involution, and breast cancer risk. In addition, the Milanese and colleagues261 study had a large number (n = 5197) of women with partial involution only; they propose better quantitative measures of degree of involution are needed. However, they hypothesize that given the inverse association of complete involution and multiparity, the breast cancer risk modification associated with parity is independent of involution. An important finding in their study is that the extent of involution was independent of all known breast cancer risk factors.261
The biologic mechanisms suggested by which involution or lack thereof alters breast cancer risk include that (1) complete involution causes a decrease in epithelial cell number so fewer cells undergo carcinogenesis; (2) aberrant involution or prolonged involution activates tissue remodeling pathways during wound healing, resulting in proinflammatory pathways that although physiologically normal, promote carcinogenesis (see Pregnancy and Breast Cancer, p. 881)252; and (3) failure to involute allows prolonged exposure to intrinsic or extrinsic factors, and genetic, epigenetic, or oxidative stressors. Elucidation on the mechanisms of lobular involution is very important for understanding breast carcinogenesis and factors that reduce breast cancer risk.
Hormonal Factors: The link between breast cancer and hormones is based on six factors that affect risk: (1) the protective effect of an early (i.e., in the 20s) first pregnancy; (2) the protective effect of removal of the ovaries and pituitary gland; (3) the increased risk associated with early menarche, late menopause, and nulliparity; (4) the relationship between types of fat, free estrogen levels, and oxidative changes in estrogen metabolism; (5) the hormone-dependent development and differentiation of mammary gland structures; and (6) the efficacy of antihormone therapies for treatment and prevention of breast cancer. Throughout its existence, the mammary gland epithelium proceeds through critical “exposure periods” of rapid growth or cycles of proliferation, including neonatal growth, pubertal development, pregnancy lactation, and involution (after pregnancy and postmenopause; see p. 882).242,252
Our understanding of the role of systemic hormones as powerful regulators of mammary gland development is shifting. Evidence is pointing to the wide-ranging effects of systemic hormones as possibly not due to their direct hormone action but rather their induced actions from multiple secondary paracrine effectors—thus the term hierarchical.243 Unraveling is a complex model of hormone, paracrine, and adhesion molecule-signaling pathways affecting epithelial and stromal cell fate in development and cancer (Figure 23-46). Despite differences between the organized process of development and the less, even chaotic, environment of invasive cancer, both processes share many identical mechanisms and signaling pathways. Key is tissue remodeling that applies to pubertal growth, immediately after pregnancy and during involution (see previous section).252,272–273
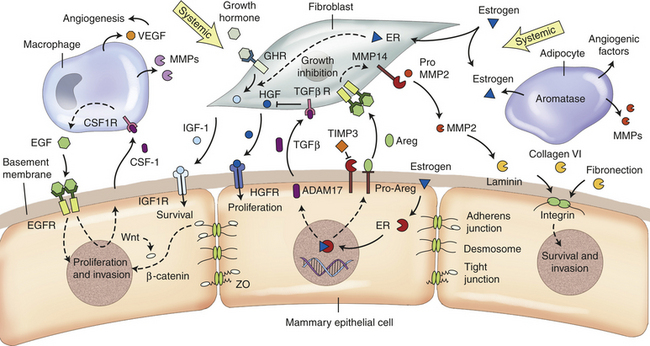
Figure 23-46 Figure of the current hypothesis of mammary gland during development and cancer. A tissue model of interacting endocrine, paracrine, and adhesion signaling pathways that modulate epithelial and stromal cell behavior. Some of the pathways depicted are not exclusive to one type of stromal cell. Dotted arrows indicate indirect interactions. (From Lanigan F et al: Cell Mol Life Sci 64:3165, 2007.)
A vast majority of breast cancers are initially hormone-dependent (estrogen-receptor positive [ER+] and/or progesterone-receptor positive [PR+]), with estrogens playing a crucial role in their development.274 Estrogens control processes critical for cellular functions by regulating activities and expression of key signaling molecules. These processes include regulation of receptor activity, its interaction with other intracellular proteins, and DNA.274 Estrogens thus play prominent roles in cellular proliferation, differentiation, and apoptosis.274–276 It is now appreciated that estrogen has both nuclear (genomic) and non-nuclear (nongenomic) actions (Figure 23-47, A). Studies reveal 58 target genes of estradiol (E2), some of which may be relevant to onset and proliferation of breast cancer (see Figure 23-47, B).275–277 It is well documented that in addition to direct regulation of gene expression (genomic), steroid hormones (e.g., estrogen) regulate cytoplasmic cell-signaling cascades or rapid nongenomic effects.275 In hormone-dependent breast cancers, estrogens, especially the potent 17β-E2 contribute greatly to the development of carcinoma cells, and some carcinomas require estrogen for continued growth.278
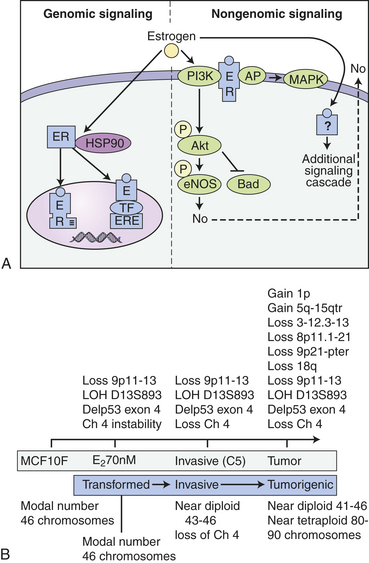
Figure 23-47 Genomic and nongenomic E2 signaling. A, In the classical genomic pathway, E2 binds to cystolic estrogen receptor (ER) and translocates to the nucleus where it binds directly to the estrogen receptor element (ERE) or binds to another transcription factor (TF) tethered to response elements and induces gene transcription. In addition, E2 is able to exert rapid cellular effects through several nongenomic mechanisms. E2 can bind to plasma membrane-bound ER and directly activate signaling cascades, such as the P13K/AKT pathway. Alternatively, on binding E2, ER can recruit adaptor proteins that activate signaling cascades. Lastly, E2 can bind nonmembrane-bound receptors and directly activate signaling pathways. B, Cumulative genomic changes observed in the breast cancer cell line MCF10F transformed with 17 beta estradiol (E1). (A from Mol Med published online 2008 April 20. B from J Steroid Biochem Mol Biol 102[1-5]:89-96, 2006.)
Most of our understanding of carcinogenicity of estrogens is based on animal studies. Two main mechanisms of carcinogenicity of estrogens involve (1) a receptor-mediated hormonal activity shown to stimulate cellular proliferation resulting in increased opportunities for accumulation of genetic damage, and (2) oxidative catabolism of estrogens mediated by various cytochrome P-450 (CYP) complexes that eventually activate and generate reactive oxygen species (ROS) that can cause oxidative stress and genomic damage directly (Figure 23-48). A third mechanism is that of estrogens, and possibly other hormones, functioning as inducers of aneuploidy (gain or loss of chromosomes). LOH, and aneuploidy are crucial events during carcinogenesis. However, it is not clear whether aneuploidy is a result of neoplastic development or a cause.
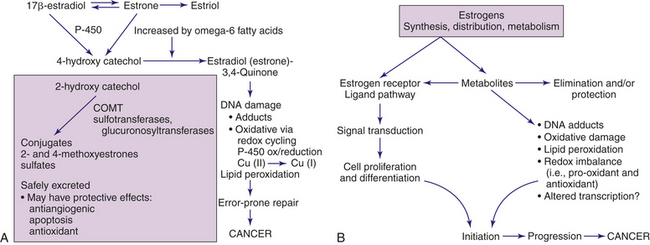
Figure 23-48 Metabolites of estrogen and their associated carcinogenic effects. A, Metabolites of estrogen and carcinogenic pathway. Genotoxicity can be produced by the 4-hydroxy catechol metabolite. A redox cycle catalyzed by microsomal P-450 and cytochrome P-450-reductase can locally generate superoxide (O2•) and hydroxyl radicals to produce additional DNA damage. Catechol estrogens also have been shown to interact with breast tissue nitric oxide, a potent oxidant that induces DNA strand breakage. Unstable polyunsaturated fats (omega-6) can increase the production of quinones. Unstable omega-6 fatty acids can be transformed by the effects of oxygen and heat into carriers of free radicals. 2-Hydroxy catechol, when methylated, may have protective effects against tumor development. Several enzymes are involved with the metabolism of estrogen, including specific cytochrome P-450 isoforms, sulfotransferases, and catechol-O-methyltransferase (COMT). These enzymes may be influenced by environmental factors, including fats, alcohol, and xenobiotic exposures. These enzymes also are polymorphic and their distributions may differ among different ethnic populations. B, Estrogen receptor and estrogen metabolites on cancer initiation and progression. (B adapted from Yager JD: J Natl Cancer Inst Monogr 27:67, 2000; additional data from Russo J, Russo I: Molecular basis of breast cancer: prevention and treatment, Germany, 2004, Springer; Cavalieri EL, Rogan EG: Ann N Y Acad Sci 1028:247-257, 2004.)
Estrogens affect microtubules that are essential for establishing cell shape and cell polarity, processes necessary for epithelial gland organization.274 In addition, the centrosomes, which are necessary for segregating chromosomes into daughter cells, are affected by estrogen. Centrosomes facilitate the coordination of intracellular activities, including cell cycle progression and cell cycle checkpoints (see Chapter 1). Although the mechanisms that promote the formation of abnormal centrosomes are unclear, several possibilities have been proposed in regard to the development of cancer,274 including centrosome amplification in breast tumors and hyperplastic gland development prior to tumor formation,279 progestins may facilitate aneuploidy,280 and bisphenol-A (BPA, found in plastics) induces the same pattern of aneuploidy found by estrogens.281 Women taking hormone replacement therapy (HRT) (estrogen plus progestin) have increased mammographic breast density and increased breast cancer risk compared with women taking only estrogen282–284 (see discussion following).
Evidence is emerging on the roles of estrogen and other hormones as initiators. An experimental system has demonstrated that the natural estrogen 17β-E2, by itself, or its metabolites 2-hydroxy, 4-hydroxy, and 16-a-hydroxy-estradiol induced carcinogenesis of human breast epithelial cells (HBECs).285
Other studies suggest that local (in situ; paracrine) formation of estrogens in breast tumors may be more important than circulating estrogens in plasma for the growth and survival of estrogen-dependent breast cancer in postmenopausal women.274,286,287
One explanation for the estradiol levels (17β-estradiol) in breast cancer tissues being equivalent in premenopausal and postmenopausal women despite significantly decreased plasma levels after menopause is that enzymatic transformation of circulating precursors in peripheral tissues contribute 75% of estrogen in premenopausal women and almost 100% in postmenopausal women.288,289 The specific enzyme complexes involved in the most biologically active estrogen, 17β-estradiol, in breast tissue include (1) aromatase, which converts androstenedione to estrone; (2) estrone sulfatase, which hydrolyzes estrogen sulfate to estrone; and (3) 17β-estradiol hydroxysteroid dehydrogenase (HSD), which reduces estrone to 17β-estradiol in tumor tissues274 (Figure 23-49).
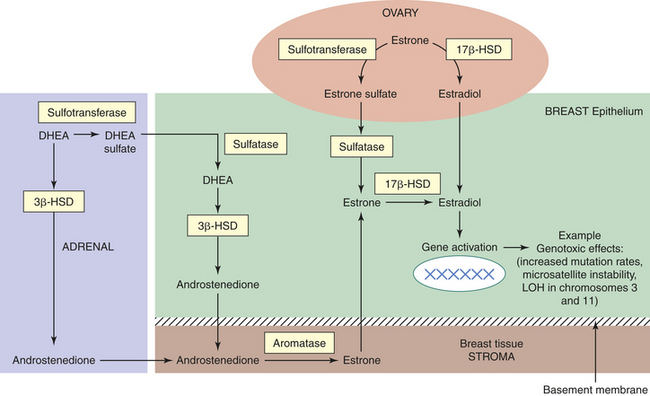
Figure 23-49 Local biosynthesis of estrogens. Three main enzyme complexes (yellow) involved in estrogen formation in breast tissue, including aromatase, sulfatase, and 17β-estradiol hydroxysteroid dehydrogenase (HSD). Thus despite low levels of circulating estrogens in postmenopausal women with breast cancer, the tissue levels are several-fold higher than these in plasma, suggesting tumor accumulation of these estrogens. Data suggest that most abundant is sulfatase in premenopausal and postmenopausal women with breast cancer. Numerous agents can block the aromatase action, exploration of progesterone, and various progestins to inhibit sulfatase and 17β-HSD or stimulate sulfotransferase (i.e., breast cancer cells cannot inactivate estrogens because they lack sulfotransferase) to provide new possibilities for treatment. (Adapted from Russo J, Russo I: Molecular basis of breast cancer: prevention and treatment, Germany, 2004, Springer.)
Breast tissue (endogenous) metabolism of estrogens through the aromatase-mediated pathway is correlated with the risk of breast carcinogenesis.290,291 Evidence suggests that the site of conversion of androgens to estrogens in breast cancer is the stroma and not the malignant epithelial cells. Consistent evidence also suggests that either tumor location is related to areas of high aromatase activity or that tumors themselves induce aromatase expression in surrounding adipose tissue.292
Breast cancer tissues may contain higher sulfatase activity than aromatase activity and produce estrone through the hydrolysis of estrone sulfate274,293–295 (see Figure 23-49). In addition, estrone sulfate has a longer half-life than estrone.295 Thus quantitatively estrone sulfate may be the most important circulating estrogen in women; it increases the reservoir for the production of estrone and, ultimately, estradiol. Furthermore, sulfatase levels have independently predicted breast cancer relapse-free survival.295,296 The association between sulfatase and poor prognosis was significant only in individuals with ER+ tumors. Miyoshi and colleagues found high sulfatase expression was associated with a poorer prognosis in premenopausal and postmenopausal women with ER+ tumors. These results suggest a possibility that, even in premenopausal women, the intratumoral estrogen biosynthesis plays an important role in the growth stimulation of breast tumors.296 Unknown are the relative contributions of estradiol to the intratumoral synthesized estradiol from the ovary versus the total intratumoral quantities. Aromatase inhibitors (anastrozole, letrozole, and exemestane) are extremely effective and very selective (targeted) for this enzyme. Estrogen formation from estrone sulfatase cannot be blocked by aromatase inhibitors.
The first sulfatase inhibitor, 667 COUMATE, was used in a phase 1 clinical trial in postmenopausal women with hormone-dependent breast cancer. Although a small number (five of eight) of women showed evidence of stable disease with 667 COUMATE, all had been unresponsive (refractory) to aromatase inhibition.297
Recent studies in mice illustrate the complex and paradoxical roles of hormones in mammary tissue. On the one hand, short duration of exposure to estrogen and progesterone, or estrogen alone, imparts a protective effect on breast carcinogenesis.298 On the other hand, continuing the same dose of hormones for a prolonged period strongly stimulates the development of tumors in the same mouse models. Different mechanisms have been suggested for the short-term protective effect, including (1) a different cellular developmental fate or (2) a systemic effect involving down-regulation of pituitary hormones (e.g., growth or prolactin hormones, or both). A short duration of hormones or blocking the same hormone pathway (i.e., tamoxifen) produces a similar result or decrease in tumor development. Overwhelming data, however, show that prolonged exposure to estradiol and progesterone (progestins) increase the risk of breast cancer.298
IGFs regulate cellular functions involving cell proliferation, differentiation, and apoptosis. Emerging evidence indicates that members of the IGF family play important roles in the development and progression of cancer. The IGF-1 receptor (IGF-1R), overexpressed in cancer cells, mediates the effects of IGFs and plays a role in cell transformation.299 IGFs are potent mitogens for ER+ breast cell lines. Interruption of IGF action can inhibit estrogenic stimulation of breast cancer cells, evidence of cellular crosstalk between the insulin growth factors and estrogen receptors.300 IGF-binding protein 3 (IGFBP-3) regulates the mitogenic and metabolic effects of IGFs; TP53 may regulate apoptosis in tumor cells through IGFBP-3.274 The protective effects of IGFBP-3 may be modulated by hCG that facilitates differentiation of the mammary gland. Increased levels of IGFBP-3 in the more differentiated Lob 3 compared with Lob 1 is a finding that requires further study.274
During the first trimester of pregnancy hCG increases, then rapidly declines to a slow steady state maintained throughout the rest of pregnancy. The action of hCG is mediated by a G-protein–coupled receptor, which also binds LH. Low levels of these receptors are present in breast tissue. This coupled with the epidemiologic findings of a decreased breast cancer risk in women who complete full-term pregnancy at a young age and a protective effect of hCG against carcinogen-induced mammary tumor development in rats, suggests that hCG may be protective against breast cancer.274,301 Treatment of human breast cancer cells (MCF-7) with hCG resulted in a modest dose-dependent decrease in cell proliferation but a dramatic decrease in cell invasion.301 Experiments showed not only inhibition of genes involved in cell proliferation and invasion but also activation of genes involved in cell differentiation, apoptosis, and DNA repair.302 hCG down-regulates ER levels through an epigenetic mechanism (CpG island methylation, see p. 374) leading to a protective effect.303 Treatment with hCG, however, can stimulate breast cancer growth in animals with overexpressed HER-2/neu oncogene.304 Nonetheless, the antiproliferative and anti-invasive effect may be useful in developing new therapies.
Investigators also have proposed that persistent alteration in the hypothalamic-hypophyseal axis (e.g., occurring during pregnancy) results in reduced circulating levels of mammary hormones, including growth hormone and prolactin that have been identified as important promoters of breast carcinogenesis.298
Hormonal Therapy: Estrogen Only: Data based on an overview of all epidemiologic studies on the effect of menopausal estrogen therapy (ET) show that ET causes a 2% mean increase in breast cancer risk per year of use,305,306 or a 10% increase in risk after 5 years of use.306 Analysis of hormonal therapy risks, however, seems to differ according to body mass index (BMI). The effects of obesity reveal that increases in non–SHBG bound E2 exceeding about 10.2 pg/ml have no further effect on breast cancer risk.306 The increased risk of ET (0.6251 mg/day) is more evident in slender women, estimated at 30% increase in risk in a woman with a BMI of 20 kg/m2 decreasing to an 8% increase in risk in a woman with a BMI of 30 kg/m2.305–308 The equivalent figures for estrogen-progestin therapy (EPT) are 50% in slender women (BMI 20 kg/m2) and 26% in heavier women (i.e., 30 kg/m2). Interestingly, according to these risk estimates, reducing the dose of estrogen in ET and EPT by as much as half has little or no effect on risk.306 These data differ from the randomized Women’s Health Initiative (WHI) with women who had hysterectomy that found a decrease in risk with use of ET.309 These findings are confusing because of other inconsistent data: (1) increased serum levels of estrogen are associated with increased risk310–313; (2) increasing postmenopausal weight increases breast cancer risk, an association between weight and increased serum levels of estrogen311–313; and (3) treating the original breast cancer with aromatase inhibitors sharply reduces risk of contralateral breast cancer. Thus ET increases breast cancer risk; the effect is greatest in slender women and difficult to discern in women with a BMI greater than 30 kg/m2.
Estrogen-Progestin Therapy: To compare risks among several studies Lee and colleagues314 standardized measures to enable data from the WHI randomized trial, cohort studies, and case-control studies to be expressed in the same relative risk terms. The summary of all studies showed a weighted average relative risk at the end of 5 years of use of EPT to be 1.44. The relative risk for the studies from the United States was 1.29 and 1.53 for the Scandinavian studies, a statistically significant difference. The continuous-combined (estrogen-progestin) therapy from the United States studies was associated with a slightly lower risk than the sequential therapy, that is, a 20% increased risk after 5 years. In comparison, there is a 32% increase in risk with sequential therapy. The opposite findings were found for the Scandinavian studies in which the continuous-combined regimens were associated with an 8.8% increase risk compared with a 40% increase with sequential.306 These differences are explained by different continuous-combined progestin doses in the United States and Scandinavia; additionally U.S. women have a greater BMI.306 Importantly, the greater risk in the Scandinavian studies compared to the United States studies is explained by a greater relative effect of EPT on breast cancer risk in leaner women.306,307
In conclusion, ET use is associated with a statistically significant increased risk of breast cancer, especially in slender women.306 An explanation for this risk may be related to the so-called ceiling (effect) to the carcinogenic risk of estrogen on the breast. Slender women may be more vulnerable to the additional exogenous estrogen because endogenously they may have lower serum levels of estrogen than obese women. Thus additional estrogen has no further effect (i.e., ceiling effect) on breast cancer risk in heavier women.306 Heavier women on EPT could likely reduce their breast cancer risk by reducing the progestin dose. In terms of ceiling effect, progestins appear to act independently on estrogen without the estrogen ceiling decreasing the progestin effect.306 A French study showed no increase in breast cancer risk with micronized progesterone instead of progestin (i.e., in EPT therapy),315 and an experimental study in macaques showed no effect of micronized progesterone on breast cell proliferation.316 Micronized progesterone, however, increased mammographic density in a randomized trial.317 An Italian retrospective study found long-term use with EPT using either micronized progesterone or transdermal progesterone were both associated with breast cancer risk; however, transdermal use was associated with a lower risk (RR 1.27 versus 2.14).318
Depending on the tissue, progesterone is classified as either a proliferative or differentiative hormone. Its effects also vary depending on whether it is used in combination with estrogen or alone. The conclusion from studies319,320 is that progesterone is neither inherently proliferative nor antiproliferative but it is capable of stimulating or inhibiting cell growth depending on whether treatment is transient or continuous. Continuous treatment may decrease sensitivity of the cells to the proliferative effects of epidermal growth factor. Investigators reported that different signal transduction pathways are used by natural versus synthetic progestins for the induction of vascular endothelial growth factor (VEGF), which promotes angiogenesis. This distinction may represent the different pathologies reported in progesterone versus synthetic progestin medroxyprogesterone acetate (MPA)–induced breast tumors in mice or the different potencies exhibited by natural and synthetic progestins for inducing proliferation of breast cancer cells in vitro.321 The safe use of progesterone-progestins, in terms of breast cancer, however, is not yet established.
Some studies have shown that OC use increases a young woman’s risk of breast cancer, especially current use.322,323 Other studies have found that the most important variable is the total months of use, with an increase of 38% (relative risk) for 10 years of use.324 A study of women between 35 and 64 years of age that included current and former OC users showed no significant association with increased breast cancer risk.325 A small study (n = 25) showed that baby boomers who took OCs for 10 years and then took hormone therapy for 3 or more years (the first group to do this) had a relative risk of 3.2—more than triple the risk of women who never used either.326 Controversy remains about the relationship between OC use and breast cancer risk; however, the efficacy of OCs in protecting against ovarian cancer and endometrial cancer is well established.
Mammographic Breast Density: Mammographic density (MD) is the radiologic appearance of the breast reflecting variations in breast tissue composition (Figure 23-50). Mammographic density in more than 60% to 75% of the breast is associated with a four- to sixfold greater risk for breast cancer.327 Thus extensive breast density is one of the strongest risk factors for developing breast cancer and second only to age and carrying a BRCA1 or BRCA2 mutation.328 Increased MD is common among 26% to 32% of women in the general population having densities of 50% or greater. Estimates of attributable risk reveal that densities of more than 50% of the breast may account for 16% to 21% of breast cancers, and possibly greater among premenopausal women.329 The strongest correlations of MD with other breast cancer risk factors is with BMI and age. Importantly, MD appears to be an independent risk factor for breast cancer because of its robust association with breast cancer after adjusting for other breast cancer risk factors.330,331
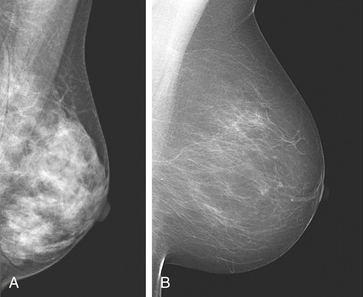
Figure 23-50 Breast density varies among women. The sensitivity of mammography for detecting malignancy is significantly reduced if the breast consists of a high proportion of fibroglandular (dense) breast tissue (A) compared to a breast that is fatty (B). (From O’Malley FP, Pinder SE, editors: Breast pathology, New York, 2006, Churchill Livingstone.)
The Minnesota Breast Cancer Family Study332 demonstrated that the factors of baseline percent density (PD), postmenopausal hormone use, and BMI (inverse relationship) predict changes in MD trends during adult life. Height has been shown to be positively associated with percent of mammographic density and with increased risk of breast cancer. However, these factors all together account for only 20% to 30% of the variation in the population. Investigators detected increased risks of breast cancer in women with MD that persisted for at least 8 years after entry into the study and were greater in younger than in older women. Their data also showed that more extensive mammographic density was strongly associated with greater risk of breast cancer detected by screening and an increased risk of detection of breast cancer between screens. Consistent with other studies, risk of breast cancer was positively correlated with the area of dense tissue but unlike other studies, less so than overall percent density. Because definitive diagnosis by mammography of breast cancer is more difficult in women with dense breasts, the optimal approach for detection remains to be determined.333
Histologic studies of breast biopsy sections and from mastectomy specimens have shown that epithelial and stromal proliferation were associated with mammographic density.333 These data suggest that genetic and environmental factors affecting risk of breast cancer affect the proliferative activity and quantity of epithelial and stromal tissue in the breast, and that these effects are possibly related to differences in mammographic density among women of the same age (see Figure 23-50). It has been shown that the dense area noted by mammogram is related to risk of breast cancer but percent density may be a stronger risk factor. Mammographic density appears to play a large role in explaining variance in the mammographic areas of dense and nondense tissue and, because MD is a continuous trait, is likely to be influenced by multiple genes. Finding the genes involved in MD may help explain why it is a strong risk factor. Evaluation of younger women to understand the pathogenesis of MD holds a promise for improved risk prediction; however, the controversy surrounding mammograms (see Chapter 11 and p. 386) and radiation in younger women speaks for alternative imaging modalities to provide an MD estimate for risk models in these women.329 How MD is related to involution is unknown.
Environmental Factors and Lifestyle: The environmental causes of breast cancer possibly affect the breast the most during critical phases or “windows” of development including early differential stages—that is, undifferentiated cells to alveolar buds and then lobules, puberty, pregnancy and lactation, involution, and menopause. During early phases, mitotic activity and cell division are greater than later in life.
Radiation: Ionizing radiation is a known risk factor for breast cancer. To date, only accidentally or medically induced radiation has been demonstrated to exert a carcinogenic effect on the breast. There are many sources of ionizing radiation, including x-rays, CT scans, fluoroscopy, and other medical radiologic procedures. According to the National Cancer Institute (NCI), CT scans “comprise about 10% of diagnostic radiologic procedures in large U.S. hospitals”; however, they contribute an estimated 65% of the effective radiation dose to the public from all medical x-ray examinations.334 Scientists and clinicians have expressed concern about the increasing number of CT scans performed, particularly for children, because of the high dose radiation exposure and subsequent cancer risk, which has the potential to become a public health issue in the future.335
Between 1950 and 1991 the incidence of breast cancer in the United States increased dramatically as did screening, leading some to suggest that increased exposure may have been a contributing factor.336 However, since the 1980s, screening has resulted in increased detection of small invasive carcinomas and in situ carcinomas. The duration of increased risk from radiation is unknown, but increased risk appears to have lasted at least 35 years in women treated for mastitis, those treated with fluoroscopy, and Atom-bomb survivors.
The type of cancer that can result from radiation exposure depends on the area exposed and the age of the individual at time of exposure. Radiologic exposure of the upper spine, heart, ribs, lungs, shoulders, and esophagus also can expose breast tissue to radiation. X-rays and fluoroscopy of infants may constitute whole body irradiation. The younger the age, the higher the risk. Evidence indicates that childhood exposure to radiation creates the greatest cancer risk whereas exposure after age 40 confers the lowest.334 Breast cancer rates in atomic bomb survivors in Japan were highest among women younger than 20 years of age at time of exposure. An important finding among the Atom-bomb survivors is that those who had early full-term pregnancies were at significantly lower risk than those who had not.337 Therefore, interacting factors can modulate the risks from radiation.
Radiobiologists have long been struggling to estimate the health risks for low doses of radiation (less than 10 cGy). Cancer induction and exposure to low doses of radiation are controversial and the topic of much debate and research. Biologic understanding related to low doses of radiation is presented in Chapters 2 and 12 and a few of these points are relevant here. Data among Japanese A-bomb survivors suggest that for solid tumors the dose response relationship is a linear function of doses between 10 and 250 cGy.338 Estimates of cancer risks at low doses—except for those from direct epidemiologic observations—are obtained by a mathematical model of linear extrapolation from these higher doses. Qualitative and quantitative differences in responses to doses of irradiation are important for understanding whether the biologic effects of low- and high-dose ionizing radiation are linearly distributed. Because cellular responses from low doses may be different than from high doses linear extrapolation may not be accurate.339 Specifically, the concerns of cellular responses from low doses including bystander effects, adaptive response, and potential radiation hypersensitivity responses in certain population subgroups (e.g., hypersensitive to low doses).340 The first study to use global gene expression changes for investigating the effects of extremely low radiation doses and high radiation doses on the cell yielded intriguing results.341 The percentage of total genes responding to the low dose at all experimental times was lower than that for genes responding to high doses. However, the groups of genes responding were different for low- than high-dose exposures. The cellular responses to high doses were apoptosis and cell proliferation. The most dominant response to low-dose irradiation were cell-cell signaling, DNA damage responses, and signal transduction.341 Several types of cellular responses to ionizing radiation, such as the bystander effect or the adaptive response, may distinguish it from the effects from high doses. Further, low-dose-induced alterations are predictive of subsequent genomic damage.342
Considerable attention is being addressed to the low dose (less than 10 cGy) radiation. Glandular doses from screening mammography are low, typically around 2.5 to 4.5 mGy (two-view; 1.76 mGy digital mammography) of about 26 to 30 kVp low energy x-rays.343 Renewed debate has emerged concerning the benefits and harms of routine screening mammography.344–348 The goal of screening is to decrease the death rate from the disease. Thus the debate has centered on whether screening mammography actually saves lives (see Box 11-2). Most study trials covered the age range from 45 to 64 years.345 The U.S. Preventive Services Task Force, however, (ages 40 to 49) and Cochrane Review (ages 45 to 64) both found a 15% relative reduction in the death rate and 0.05% absolute reduction in the death rate in risk from screening for women,345,346 but the Armstrong study estimated that 30 to 200 per 100,000 women ages 40 to 49 will die after annual screening mammography as a result of radiation-induced breast cancer.344 Results of recent studies have led to questioning whether benefits outweigh the harms because risk reduction is low and the potential for overdiagnosis and overtreatment results in risk increase.
It is questionable whether further research on the benefits of screening will enable better estimation of the ratio of benefit to harm in the 40- to 49-year-old group. However, much more research on the harms is needed because they remain too uncertain in this ratio. Toward that end, the rest of this discussion summarizes the experimental biologic data on low-level radiation and breast tissue.
The underlying mechanisms of radiation-induced carcinogenesis are not completely understood. The historical viewpoint has been that the biologic effects of ionizing radiation occur in irradiated cells as a result of DNA damage. This viewpoint implies that (1) these alterations only occur in directly irradiated cells, (2) radiation movement through the cell nucleus is necessary to producing a biologic response, and (3) the target in the cell is DNA.349 Emerging evidence points to radiation-induced non-DNA targeted effects including mutations, chromosomal aberrations, and changes in gene expression (phenotype) in the cells not directly irradiated (non-hit). These phenomena include radiation-induced genomic instability (RIGI) in which the biologic effects include increased frequency of mutations and chromosomal aberrations that occur in descendants of irradiated cells (see Chapter 12 for a discussion of RIGI). Another phenomenon is the “bystander effect,” the so-called innocent cells that did not receive radiation directly but still experience biologic effects. These effects include damaged cell-to-cell signaling, presumably through gap junction channels, and may be the result of oxidative stress350,351 (see Chapters 2 and 12, and Figure 12-11). Together these effects are often called nontargeted effects. A major paradigm shift in radiation biology has occurred because of work involving the bystander effect.351 Clearly, radiation-induced signaling contributes to nontargeted effects.352 Investigators351,353 have shown in vitro and in vivo that radiation activates the inflammatory response and induces activation of multiple signaling pathways. Thus evidence is emerging of a multicellular program of tissue response to damage that also includes surveillance and selective apoptosis of abnormal cells.354–356 Yet normal signaling can be altered by radiation, compromising surveillance and apoptosis of abnormal cells, and causing genomically unstable cells to accumulate and proliferate. Data provide evidence that centrosome deregulation is a mechanism involved in the generation of RIGI352 (Figure 23-51, A; see also Chapter 12).
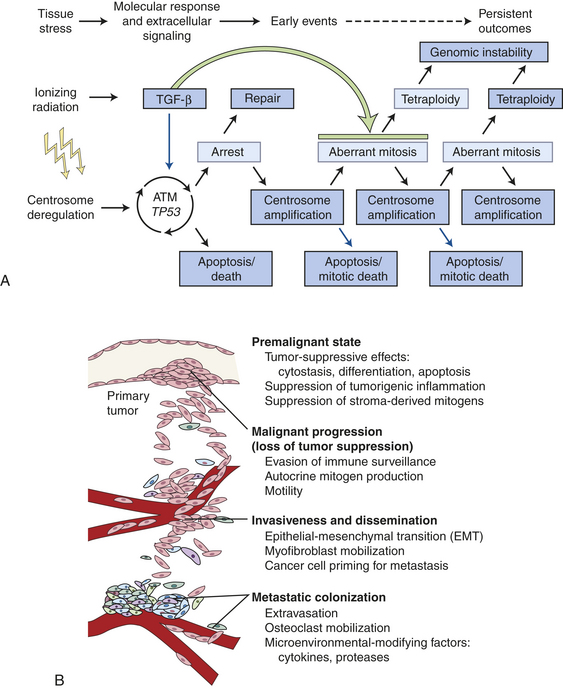
Figure 23-51 Roles of transforming growth factor-beta (TGF-β). A, Roles of TGF-β in response to IR. B, Roles of TGF-β in cancer.
Maxwell and colleagues352 also showed that the cytokine TGF-β has a surveillance role in RIGI. In their experiments using human mammary epithelial cells (HMEC) TGF-β signaling in irradiated tissues could either promote carcinogenesis—causing stromal remodeling from activation of inflammatory responses leading to epithelial tissue transitioning to mesenchymal tissue (EMT)—or reestablish homeostasis by eliminating RIGI cells by activating a TP53-dependent apoptosis, thus inhibiting carcinogenesis. However, with continued chronic exposure to radiation, TGF-β induces the remaining population of stromal cells to undergo tissue transformation or EMT facilitating a neoplastic process (see Figure 23-51, B). Thus, TGF-β, a regulatory cytokine, can exert tumor-suppressor effects or paradoxically can cause tissue changes that facilitate cell invasion and immune regulation, transforming the microenvironment into a malignant environment (see Figure 23-51).357
Other investigators studied low-dose ionizing radiation of the breast stromal microenvironment.358 These studies showed that human mammary stromal fibroblasts respond to protracted (repeated overtime) low-dose ionizing radiation by displaying a “senescence-like phenotype,” thus an epigenetic mechanism. Senescent cells persist overtime, are irreversible, and accumulate in aging tissues.359,360 One hypothesis is that accumulation of senescent cells in the stroma may serve as a means of stromal activation, subsequent inflammation, and stromal remodeling. Fibroblasts respond to a variety of chronic stressors including oxidative stress, UV light, inflammatory cytokines, and numerous genotoxic agents. The combination of these effects may represent a long-term outcome of cellular stress, including genotypic and phenotypic (epigenetic) changes. So in addition to replicative senescence, accumulating evidence indicates that chronic stress to tissue may cause the accumulation of senescent cells in the stroma.358
Ionizing radiation may be only one type of environmental stress creating perturbed mammary stromal changes. Tsai and colleagues358 found that with radiation-induced senescence-like fibroblasts, other disruptions included cytoskeletal alterations, increased extracellular matrix degradation, mammary ductal alterations (e.g., enlarged cystic structures), disorganized cell masses, and changes in cellular death (i.e., apoptosis) pathways. Breast cancer cells growing in this type of stromal environment lead to dysregulated cell-cell and cell-matrix interactions, thereby enabling malignancy.358 Overall, these investigators suggest that protracted, low-dose ionizing radiation exposure fostered an oncogenic environment.
Normal stroma suppresses tumor growth by releasing growth-inhibiting biochemical signals whereas oncogenic stroma promotes tumor growth.358 The tumor effects of stromal fibroblasts have been attributed to their production of matrix metalloproteinases (MMPs; see Chapter 11). MMP-3 has recently been shown to regulate the branching developments of mammary epithelial cells in response to senescent fibroblasts.361 MMPs have been linked in vivo to breast cancer invasion and metastasis.362
Because some human cancers arise from the accumulation of multiple genetic abnormalities, the age at which mammography begins and the total number of mammograms received may be important factors in the development of radiation-induced breast cancer. Women at high risk for developing breast cancer—those with mutations of the BRCA1 or BRCA2 genes or have a first degree relative (parent, sibling, or child) with such a mutation and women who received radiation to the chest for Hodgkin disease—are recommended to start mammography screening with MRI at age 30. However, low-dose ionizing radiation has been shown to increase the risk of breast cancer significantly among BRCA1 and BRCA2 mutation carriers.363 A retrospective cohort study of 1601 female BRCA1 and BRCA2 mutation carriers found an association with reported chest x-ray exposure and significantly increased risk of breast cancer (hazard ratio 1.54). Also a review showed a strong association (odds ratio 3.21) between CHEK2∗1100delC carrier status, history of chest x-rays and breast cancer risk.364 Continuing research will help to determine if high-risk women should be screened with MRI. It is true that MRI can find lesions that mammograms miss, and women are not exposed to ionizing radiation. However, MRIs can increase the detection of cancers that would not be clinically relevant (i.e., not become invasive) and lead to unnecessary treatment, including mastectomies. Thus the role of MRIs is still being investigated and more study is needed to fully understand all its benefits and harms.
In conclusion, the risk of radiation-induced breast cancer depends strongly on when radiation exposure occurs. Exposure before the age of 20 years carries the greatest risk. Biologic mechanisms of low-dose and subsequent cellular effects are emerging. Other factors that also may influence risk include age at first-term pregnancy, parity, possibly a history of benign breast disease and injury, exposure while pregnant, endogenous hormone levels and ratios, and genetic factors.365 Overall, mammography screening, which is convenient and efficient, has a modest effect on breast cancer mortality, in absolute terms. The death rate after 10 years of screening is reduced by 0.05% (i.e., 1 death prevented of 2000 women screened).245 However, the harms include overdiagnosis and overtreatment of healthy women (i.e., 30% more surgery, 20% more mastectomies, and more use of radiotherapy). Unclear from screening and/or overtreatment with radiotherapy are the exact numbers of women that develop radiation-induced breast cancer, heart disease, and lung disturbances. Newer understandings of the biology of breast cancer challenge the widely held view that breast cancer is a uniformly progressive disease not cured unless caught early. It is now understood that breast cancer is a heterogenous disease that may be metastatic from the very start of it and may never metasasize. Women need information on both benefits and harms to make a rational decision about screening (see What’s New: Screening Mammograms: Far from Perfect, p. 386, in Chapter 11).
Diet: Prospective epidemiologic studies on diet and breast cancer risk fail to show an association that is consistent, strong, and statistically significant except for alcohol intake, being overweight, and weight gain after menopause (see discussion following).366 Dietary fat and breast cancer risk is the subject of much study, controversy, and debate. Potential biologic mechanisms between fat intake and breast cancer risk include (1) that fat may stimulate endogenous steroid hormone production (also affect weight gain, age of menarche), (2) they interfere with immune or inflammatory function, and (3) they influence gene expression. Evidence from large, prospective cohort studies has been mostly unsupportive and clinical trials have not supported a strong association with total fat intake.367 Cohort studies, however, suggest a modest positive association between fat intake and the risk of breast cancer,368 but so far more than 70 studies of dietary fat during midlife on risk of breast cancer show the relationship is likely to be small.
Another area of study is how consumption of red meat could increase breast cancer risk. The hypotheses range from available iron content, growth-promoting hormones used in the cattle industry, and carcinogenic heterocyclic amines released from cooking the fatty acid content. Case-control and cohort studies have shown a modest association of red meat intake with breast cancer incidence but no association in a pooled analysis of prospective studies.369 Research has reported an increased risk with red meat consumption.370–372 For other updates on diet and breast cancer risk see Nutrition & Disease: Diet and Breast Cancer Risk Updates.
Studies in animal models and recent observations in humans have provided some evidence that a high intake of omega-6-polyunsaturated fatty acids (omega-6 PUFAs) stimulates several stages in the development of mammary and colon cancer and possibly prostate cancer—from an increase in oxidative DNA damage that affects cell proliferation and increased free estrogen levels that affect hormonal catabolic products.373–376 The prospective Malmo diet and cancer cohort (n = 11,699) found after a 10-year follow-up that omega-6 PUFAs may promote breast cancer.377 Conversely, fish oil–derived omega-3 fatty acids may help to prevent cancer by influencing the activity of enzymes and proteins related to intracellular signaling, inflammation, and eventually cell proliferation.378,379 Studies that show protective effects of fish oil and decreased cancer risk have been confined to countries with high fish intake.
Investigators have identified potential carcinogens in breast fluid in normal women, especially cholesterol derivatives.380 Breast fluid represents secretions from the cells lining the breast ducts, which is where the majority of breast cancers develop.381 These breast secretions have been related to the fat content of the diet. Estrogen levels are also substantially higher in breast secretions than in blood. Thus fat tissue in the breast may be a
source of high concentrations of fat-soluble chemicals (including estrogens), some of which may be carcinogens.381
Further studies are needed to evaluate the benefits of substantially lowering fat intake (20% or less of total calories) and the roles of micronutrient imbalances and childhood nutrition in the development of breast cancer. The role of obesity in breast cancer is complex and seems to be related to fat distribution, type of fatty acids consumed, and sex hormone levels.382
Obesity has been associated with a reduced risk of premenopausal breast cancer. One mechanism suggested is the direct relationship between irregular menstrual cycling, especially obesity and anovulatory cycling, which would result in a decrease in estrogens and progesterone and thus decrease the risk of breast cancer. It is possible that in obese women with hyperinsulinemia the higher insulin levels increase the enzymatic conversion of testosterone to dihydrotestosterone, rather than estradiol, lowering their estrogen levels.381
Obesity, however, is related to increased risk of breast cancer in postmenopausal women. Despite strong links with endogenous estrogen levels, body fat has been consistently but weakly related to increased postmenopausal risk.383 This observation has been surprising because obese postmenopausal women have endogenous estrogen levels (estrone and estradiol) nearly double those of lean women.383,384 This weak association is possibly related to two factors. First, the premenopausal reduction in breast cancer risk related to being overweight possibly persists, opposing the adverse effect of elevated estrogens after menopause. Thus weight gain should be more strongly related to postmenopausal breast cancer risk than attained weight. In two case-control studies and prospective studies, this was indeed true.385–388 A pooled analysis of prospective cohort studies showed women with a BMI of 28 kg/m2 or higher were 26% more likely to develop postmenopausal breast cancer compared with leaner women.389 Premenopausal and postmenopausal weight gain is also associated with higher estradiol and estrone levels and lower SHBG as a transporter protein; low levels cause higher bioavailable estrogen.390 This increase in estrogens, particularly estradiol, is from aromatization in the adipose tissue. Second, use of exogenous hormones postmenopausally obscures the variation in endogenous estrogens caused by adiposity and elevates breast cancer risk regardless of body weight.383 Excess body fat and weight gain are stronger risk factors for women who do not use hormone therapy. A recent prospective study found weight gained at multiple time points throughout adulthood of 20 to 29 kg was associated with a 56% higher risk of breast cancer, and weight gain of 40 to 49 kg doubled the risk of breast cancer among hormone users.391
Weight loss after menopause reduces circulating estrogens and increases SHBG, making weight loss a potentially important prevention strategy especially for those women not on hormone therapy. Weight loss and postmenopausal cancer risk have been examined in prospective studies. In one of the largest studies, women who lost 10 kg or more after menopause and maintained this weight loss halved their risk for breast cancer. This relationship was clearer in nonhormone users.392
Obesity is associated with poor survival among women with breast cancer, and the association of obesity with mortality from breast cancer appears to be stronger than its association with incidence.383,387 Thus the increase in breast cancer risk with increasing BMI among postmenopausal women largely results from increased estrogen, especially estradiol.383
Soy products are a hot topic because of their consumption in Asian countries that have low rates of cancer (see Nutrition & Disease: Diet and Breast Cancer Risks Update, p. 892). These isoflavone compounds, including diadzen and genistein, can bind estrogen receptors but are far less potent than estradiol. Soy may act like other antiestrogens, such as tamoxifen, by blocking the action of endogenous estrogens to reduce breast cancer risk. Thus depending on the estradiol concentration and the timing of administration, soy exhibits weak estrogenic or antiestrogenic activity. Isoflavones can influence transcription and cell proliferation. They modulate enzyme activities, as well as signal transduction, and have antioxidant properties.393 Results of clinical studies on the effects of soy products or isolated isoflavones on vasomotor symptoms are contradictory. Epidemiologic studies, however, have shown a decrease in the prevalence of hot flashes in women from countries with high isoflavone intake, such as Japan, more so than in Western countries. Evidence from epidemiologic, animal, in vitro data, and human clinical trials show isoflavones are promising agents for breast cancer prevention.208,394–399 However, there are concerns that soy or isoflavones may increase proliferating cells. A study of nipple aspirate fluid from women who had ingested high-soy diets and were either premenopausal or took estrogen replacement therapy showed an increase in proliferating cells.400 Controversy has ensued on whether breast cell proliferation can equal breast cancer growth. Soy may cause breast cells to grow; however, in vitro properties of soy for blocking invasion and antiangiogenesis may be more important in preventing breast cancer. In vitro and animal studies show that soy inhibits breast cancer growth, and additional work showed this effect on cancer cells that are both ER+ and ER−.399 In addition, soy may optimize extrarenal 1,25(OH)2 cholecalciferol or vitamin D3 (a prodifferentiating vitamin D metabolite), which could result in growth control and, conceivably, inhibition of tumor progression.394
Environmental Chemicals: Evidence for linking chemicals to the cause of breast cancer is difficult. It is challenging because it is a life history of exposure that is important—not just a single chemical but also complex mixtures of chemicals and their interaction with endogenous hormones and with radiation. The highest rates of breast cancer are found in superindustrialized countries—North America and Europe—and the lowest rates in central Africa and Asia. With industrial development, breast cancer rates increase. An estimated 85,000 synthetic chemicals are registered for use today in the United States, another 1000 or more are added each year, and toxicologic screening for these chemicals is minimal—only about 7%.401 Chemicals persist in the environment, accumulate in adipose tissue, interact with local adipose tissue physiology in an endocrine-paracrine manner, and remain in breast tissue for decades. Some of these chemicals are known human carcinogens and many have been linked to mammary tumors in animals. Women who emigrate to the United States from Asian countries experience an enormous percent increase in risk within one generation. A generation later their daughters’ risk approaches that of women born in the United States. This change in risk suggests that in utero exposures affect subsequent disease risk. However, it is difficult to know whether these changes in risk come from nutritional content, pollutants, cosmetics, food additives, or other factors.
Xenoestrogens are synthetic chemicals that mimic the actions of estrogens and are found in many pesticides, fuels, plastics, detergents, and drugs.206 Because many factors correlated with breast cancer (early menarche, delayed pregnancy and breast-feeding, late menopause, etc.) are associated with lifetime exposure to estrogens, investigators reasoned that environmental chemicals affect estrogen metabolism and contribute to breast cancer. The most significant chemicals may be polychlorinated biphenyls (PCBs), pesticides, BPA (pervasive in polycarbonate plastics), tobacco smoke (active and passive), dioxins, alkyphenols, metals, phthalates, parabens, food additives, HRT, and others (see Table 23-17). Many chemicals are fat soluble with estrogenic effects. Because the amount of these environmental estrogens is presumably minute, their effect may be secondary to an abnormal (e.g., mutagenic) response of the estrogen receptor and DNA or catabolized products of estrogen. (Human studies related to HRT are exhaustive and discussed on p. 883.) Human studies of women exposed to DDT during childhood and early adolescence was associated with a fivefold increase in risk of developing breast cancer.402 Other human studies include heptachlor,403,404 environmental tobacco smoke,405 benzene among enlisted women in the U.S. Army,406 and benzene among women in different professions in Israel related to increased rates of breast cancer.407 Further, a long-term follow-up (30 years) of women who were exposed to DES shows a small increased risk of breast cancer (relative risk 1.35) and no increasing risk over time.381 Table 23-17 contains information on selected studies, chemicals, and risk of breast cancer.
Table 23-17
Selected Chemicals and Risk of Breast Cancer
| Chemical | Comments |
| Bisphenol-A (BPA) | Studies have shown altered reproductive systems and breast tissue when exposed to BPA in utero408 |
| BPA is commonly found in plastics410 | |
| Polyvinyl chloride (PVC) | Used in food packaging, medical products, appliances, cars, toys, credit cards, rain wear409 |
| Has been found in the air near waste sites, landfills, and tobacco smoke | |
| Has been linked to increased mortality from breast and liver cancer among manufacturing workers411, 412 | |
| Pesticides: aldrin and dieldrin (organochlorines) | Used in crops like corn and cotton from 1950s to 1970s |
| Banned by the EPA in 1975 except for termite control; completely banned in 1987 | |
| In vitro assays showed estrogenic activity and dieldrin found in 78% of women diagnosed with breast cancer413 | |
| High incidence of breast cancer in Massachusetts study found associations with higher income and regular use of lawn services, termite treatments, and home pesticides414 | |
| Household products: methylene chloride | Spray paints and paint removers may contain methylene chloride, documented breast cancer in lab animals415 |
| Diethylstilbestrol (DES) | Prescribed for women to avert miscarriages between 1941 and 1971 |
| Exposed daughters known to have higher rates of vaginal cancer, and in the mothers slight increased risk of breast cancer416, 417 | |
| Daughters now known to have slight increased risk of breast cancer418 | |
| Solvents (e.g., benzene, toluene, trichloroethylene, chlorinated organic solvents) | Used in manufacture of computers, also some in cosmetics |
| In 2003 a Taiwanese study documented increased risk of breast cancer among electronic workers exposed to chlorinated organic solvents419 | |
| A Danish study of women 22 to 55 years of age employed in industries (fabricated metal, lumber, furniture, printing, textiles) using solvents doubled the risk of breast cancer420 | |
| Styrene, carbon tetrachloride, formaldehyde | A 1995 study suggested increased risk with occupational exposure—validation in Finland, Sweden, and Italy421–424 |
| Ethylene glycol methyl ether (EGME) | A Duke University study found it acts as hormone sensitizer in vivo and in vitro425,426 |
| Compounds are found in semiconductor industry, varnishes, paints, dyes, and fuel additives | |
| Valproic acid (anticonvulsant medication) | Found to be hormone sensitizing and prescribed for migraines and bipolar disorder425,426 |
| 1,3-butadiene | Air pollutant and synthetic rubber product and some fungicides and tobacco smoke |
| Causes mammary and ovarian tumors in female mice and rats427,428 | |
| Aromatic amines (heterocyclic, polycyclic, moncyclic) | Found in plastics, tobacco smoke, grilled meats and fish, combustion of wood chips and rubber |
| Exposure in adolescence before full-term pregnancy may increase risk429 | |
| Dichlorodiphenyltrichloroethane (DDT) and polychlorinated biphenyls (PCBs) | PCB used in manufacture of electrical equipment430 |
| PCB and DDT are banned in the United States since 1970s but are still found in body fat, as well as breast milk431 | |
| DDT was used as pesticide for insects on farms and swamps | |
| PCB deteriorates slowly in soil | |
| PCB is difficult to study because it is a diverse class of compounds | |
| A 1999 in vitro study showed PCBs proliferate in breast cancer cells432 | |
| Conflicting results; several large studies failed to show relationship with PCBs | |
| Polycystic aromatic hydrocarbons (PAH, including tobacco) | Found in soot and fumes from fuelsIncreased DNA damage (DNA adducts) implicated from the Long Island Breast Cancer Study Project433 |
| Tobacco smoke also contains PAHs | |
| Smokers who began smoking as adolescents have an increased risk of breast cancer434–436 | |
| In 2004 the California EPA concluded that environmental tobacco smoke (ETS) increases the risk of breast cancer, and the association appears stronger for premenopausal women437 | |
| Tobacco smoke also contains the carcinogens polonium-210, vinyl chloride, benzene, and 1-3 butadiene438 | |
| Dioxin | Products containing PVC, PCBs, or other chlorinated compounds release dioxin from incineration |
| Declared a known carcinogen by the EPA in 2000 | |
| It may be the most prevalent of all toxic chemicals | |
| Occurs in meat, poultry, dairy products, and human breast milk | |
| A United Kingdom study linked dioxin to the development of mammary tumors in mice439 | |
| A study in Seveso, Italy, connected dioxin with breast cancer440 | |
| Ethylene oxide | Used to sterilize surgical instruments and in some cosmetics |
| Linked to breast cancer in women exposed to ethylene oxide in commercial sterilization facilities441 |
Physical Activity: Regular physical activity may reduce overall risk of breast cancer, especially in premenopausal or young postmenopausal women.440–442 Yet selection bias in studies related to recreational physical activity during adulthood and random error in the measurements of activity remain concerns.443 A large prospective study found walking for 1 hour per day and additional weekly exercise seemed to be protective against breast cancer regardless of menopausal status.444 Mechanisms for this protective effect are not known but include alterations in endogenous free radical formation and oxidative damage, effects on DNA repair capacity, alteration in carcinogen-metabolizing enzymes, increased intestinal transit times (i.e., reduced exposures to carcinogens), weight loss, and changes in endogenous sex hormone levels.441,442
Familial Factors and Tumor-Related Genes: Genetically, breast cancer can be divided into three main groups: (1) sporadic, the majority or 40% of women with breast cancer have no known family history; (2) inherited dominant cancer gene syndromes, in which the gene is passed to future generations by an autosomal dominant mechanism; and (3) probable polygenic, in which there is family history but it is not passed on to future generations as a dominant gene. Yet to be determined are the number of genes in the polygenic model that could be involved, the nature of the interactions among these genes, and their interaction with environmental factors. The major risk factors for sporadic cancer are related to hormone exposure including age at menarche and menopause, reproductive history, breast-feeding history, and endogenous and exogenous estrogens (see p. 886). Radiation exposure is known to increase risk. Chemical exposures during critical windows of development may also be important (see p. 893). The majority of these cancers occur in postmenopausal women with increased ER expression.
A history of breast cancer in first-degree relatives (mother or sister) increases a woman’s risk two to three times. Risk increases even more if two first-degree relatives are involved, especially if the disease occurred before menopause and was bilateral. In some families, breast cancer occurs at an earlier age and the frequency of bilateral tumors is greater. Women with inherited breast or ovarian cancer have tumors characterized by alterations in particular genes, mainly BRCA1 and BRCA2 breast cancer susceptibility genes, but also CHEK2 (Li-Fraumeni syndrome), ataxia telangiectasia (AT), and STK11 (Peutz-Jeghers syndrome). An obvious hereditary predisposition and strongly penetrant mutations in genes such as BRCA1 and BRCA2 are responsible for 5% to 10% of all breast cancer, or 18,000 individuals per year.445 The probability that a mutation will be present in family kindred increases if the family history includes disease at early ages, clustering of both breast and ovarian cancers (BRCA1), male breast cancer (BRCA2), and other rare cancers, such as sarcomas.445 Even in families with more than four individuals with breast cancer, a germline mutation in BRCA1 or BRCA2 was found in only 65% of people.446 Unexplained familial breast cancer possibly includes other more common lower-penetrant genes.
Investigators estimate that 45% of families with apparent autosomal dominant transmission of breast cancer susceptibility and about 90% of families with dominant inheritance of both breast and ovarian cancer have BRCA1 germline mutations447 (Figure 23-52). The BRCA1 and BRCA2 genes include several “founder” mutations identified in various populations. Most common in the United States are three mutations of the Eastern European population: two in BRCA1 (185delAg and 5382insC) and the 6174delT mutation in BRCA2.445 The penetrance, or lifetime risk, of developing breast cancer and ovarian cancer from BRCA mutations is the subject of intense research. Breast cancer risk associated with mutations in BRCA1 have been estimated in the range of 50% to 80% and in 40% to 70% for BRCA2.446,448 The ovarian cancer risk among BRCA1 carriers is about 40% lifetime, exceeding the risk of 20% for BRCA2 carriers. The risk for ovarian cancer is not the same for all BRCA2 mutations, which depend on the location of the gene mutation.449 In premenopausal women, a modifier of risk for breast cancer has been prophylactic oophorectomy, reducing lifetime risk by 50%.450,451 Thus the risk associated with an inherited predisposition can be reduced significantly by modifying endogenous, and possibly exogenous, hormonal exposures. The risk for other cancers, such as pancreatic, prostate, melanoma, and others, is increased in BRCA2 carriers.445
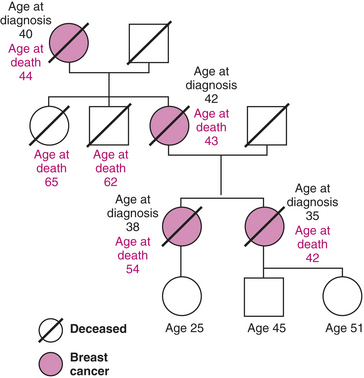
Figure 23-52 Example of family pedigree for breast cancer. Family pedigree showing cases of breast cancer associated with typical dominant transmission of breast cancer. Other possible genetic alterations related to risk of breast cancer include changes in TP53 and alterations in the estrogen receptor. Numerous somatic mutations in the expression of oncogenes in breast cancer cells have been reported.
Genes important to the development of cancer regulate diverse cellular pathways, including the progression of cells through the cell cycle, resistance to apoptosis, and the response to signals that direct cellular differentiation.452 The inactivation of genes (e.g., tumor-suppressor genes) that contribute to the stability of the genome itself can favor errors in other genes that regulate proliferation. The importance of this latter pathway is exemplified by two studies linking the function of the BRCA1 gene with that of the gene for ataxia-telangiectasia mutation (ATM) (Figure 23-53).
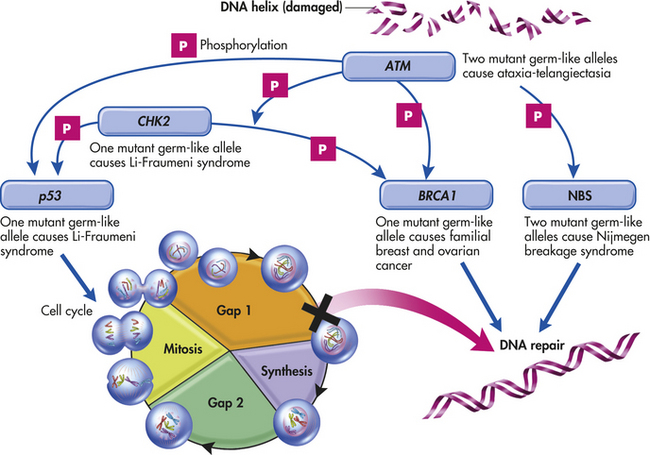
Figure 23-53 Mutations in genes that regulate cellular proliferation and repair of deoxyribonucleic acid (DNA) and lead to breast carcinogenesis. The ataxia-telangiectasia mutated (ATM) gene encodes a protein kinase that activates (through phosphorylation) the tumor-suppressor p53 protein either directly or indirectly by activating CHK2 (a gene that encodes a protein kinase that activates p53 by adding a phosphate group to it) in response to damage to DNA. The p53 protein then triggers the arrest of the cell cycle, increasing time for DNA to be repaired. More simply, the ATM gene is necessary to accomplish DNA repair. The BRCA1 and Nijmegen breakage syndrome (NBS) proteins are also activated by the ATM gene and are thought to be directly involved in the repair of damaged DNA. The inactivating mutations in the genes that encode these proteins increase the risk of breast cancer. (Redrawn from Haber D: N Engl J Med 343[21]:1566, 2000.)
Other tumor-related genes or proteins include p53, Bcl-2, HER-2/neu, and c-myc. About 40% of breast carcinomas reveal high levels of stabilized, often mutant p53 protein in their cells; p53-related defects in tumor cells correlate with a poor prognosis.
Production of the proto-oncogene Bcl-2 decreases or inhibits apoptosis and thereby promotes breast and other cancers. However, the College of American Pathologists has classified it as belonging to category III, meaning there is insufficient evidence to support it as a prognostic factor.453
HER-2/neu, another oncogene, is overexpressed in 25% to 30% of breast cancer cells. It transmits a growth signal to the nucleus. The drug trastuzumab (Herceptin) blocks the signal in about 35% of those affected, thereby decreasing the growth of the tumor.
C-myc is a proto-oncogene expressed in cells and is one of the immediate, early growth response genes that are rapidly induced when quiet cells receive a signal to divide. Mutation of c-myc is amplified in breast, colon, lung, and many other cancers.
PATHOGENESIS Breast cancer is as varied as the breast itself. Table 23-18 lists the different types of breast carcinomas and summarizes their major characteristics. Most breast cancers arise from the ductal epithelium (Figure 23-54). Tumors of the infiltrating ductal type do not become large, but they metastasize early. This type accounts for the majority of breast cancers.

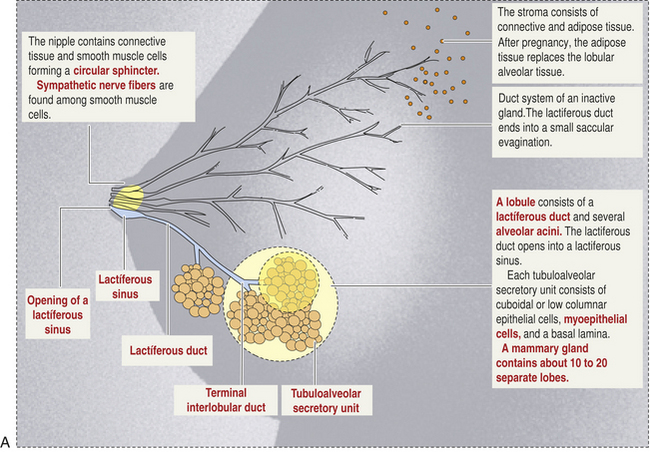
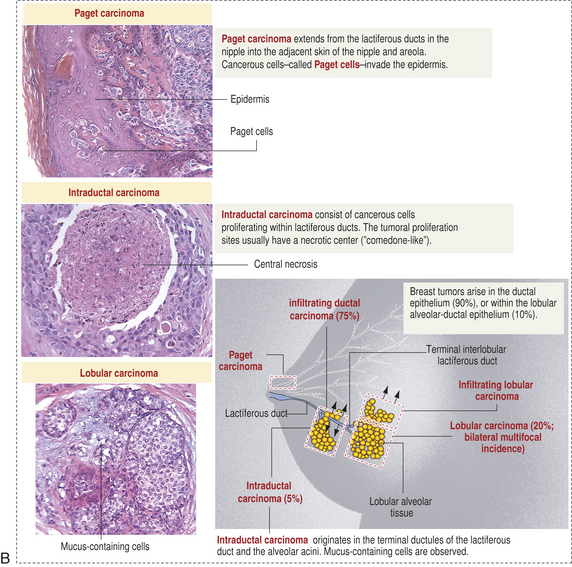
Figure 23-54 Normal breast and breast cancer. A, Normal breast. (From Kierszenbaum AL: Histology and cell biology: an introduction to pathology, ed 2, Philadelphia, 2007, Mosby.) B, Breast cancer.
Breast cancer is a heterogeneous disease with diverse molecular, phenotypic, and pathologic changes. Despite heroic efforts, this diversity has greatly challenged the understanding of breast cancer evolution. Recent research suggests that breast cancer may be heterogeneous from its initial preinvasive stages.454 The historic multistep model of breast carcinogenesis proposing tissue transition through a single pathway of sequential molecular alterations from normal epithelium to invasive carcinoma (e.g., nontypical, atypical hyperplasia, in situ carcinoma) is an important model for understanding colon carcinogenesis (Figure 23-55, A). However, emerging evidence indicates that for breast cancer this model may be oversimplified or flawed, and that other alternative models are possible and have been proposed454,455 (see Figure 23-55). All together three models are proposed for breast carcinogenesis including (1) the multistep (hits and stepwise) sequential acquisition model, (2) the telomere crisis model, and, (3) the imprinted stem cell model. Models are necessary because in humans, carcinogenesis or progression from one lesion to the next cannot be experimentally tested. Nevertheless, several biologic associations are known. DCIS (see p. 898) is associated with invasive cancer at the site of clinical diagnosis.454 This characteristic implies a direct “clonal” progression from DCIS to invasive carcinoma and is the basis for current DCIS treatment.456 Hyperplasia and atypical hyperplasia, however, are not known to have invasive cancer at the site of the lesion (see p. 872). Instead, they are deemed markers of risk throughout the entire breast.454 In addition, from the multistep model where the programmed malignant potential depends on sequential acquisition of genetic alterations with specific “hits” responsible for the transition from DCIS to invasive cancer remain unknown—despite tremendous study (see Figure 23-55, A). Instead, several lines of evidence suggest there may be multiple molecular genetic pathways of complex crosstalking networks that progress toward malignancy.
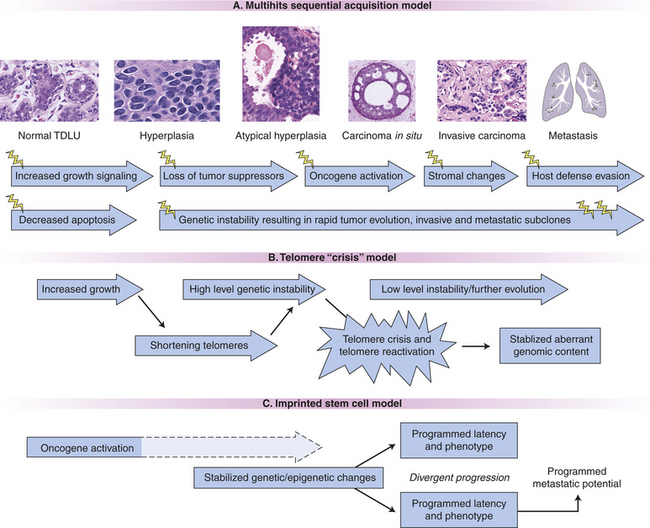
Figure 23-55 Conceptual models of breast carcinogenesis. A, Multihits sequential acquisition model (schematic as lightning bolts) corresponds to structural changes with cancer progression (also see Chapter 11). B, Hyperplasia increases the shortening of telomeres, swiftly increasing genetic instability known as “telomere crisis,” with eventual stability after the enzyme telomerase reactivation, those individual cells reactivating telomerase with this genetic profile give rise to ductal carcinoma in situ (DCIS) and later invasive carcinoma. C, Genetically stable precancer stem cells are turned on through activation of oncogenes with divergent programmed behavior through epigenetic encoding and possible genetic, although not required, content changes. With progression, intermediate structural and molecular events are not required. These cells initiate DCIS and have innate built-in latency to invade with a built-in (innate) metastatic potential. (From Damonte P et al: Breast Cancer Res 10[3]:R50, 2008.)
The telomere crisis model of breast carcinogenesis457 involves the initiation of cancer occurring through telomere shortening and genetic instability (see Figure 23-55, B and Chapter 11). Hyperplasia results in shortening telomeres and increasing genetic instability (telomere crisis). Certain genetic changes may eventually become stabilized with reactivation of the enzyme telomerase, stabilizing the aberrant genomic composition, and immortalizing (cancerizing) the cell. Overall these genetic changes might be considered the origin or “birth” of the cancer-initiating cell or cancer stem cell. This model—imprinted stem cell model—suggests that this “birth” occurs as the tissue becomes DCIS or the cancer stem cell is “born” at the precancer stage. From other experiments, Damonte and colleagues454 modified this model slightly by showing that genetic instability is not required. Instead, these investigators show that overexpression of a single gene is sufficient and that the modifications in the precancer stem cell might be epigenetic as well as genetic. The continuing “low level” genetic changes and epigenetic changes may contribute to heterogeneity and are not required for neoplastic progression (see Figure 23-55, C). Thus this model suggests that the precancer stem cell is programmed from the beginning and is not dependent on multihits as the origin of invasive cancer. The risk for advancing to invasive breast cancer from human DCIS will therefore be predictable at the precancer stage possibly requiring the understanding of the interactions between the DCIS epithelium and stroma.454
Ductal Carcinoma In Situ: Ductal carcinoma in situ (DCIS) refers to a heterogeneous group of proliferations limited to ducts and lobules (Figure 23-56, B). DCIS occurs predominantly in women but can occur in men. The cells sometimes extend to the overlying skin without crossing the basement membrane and mistakenly appear as Paget disease202 (see Table 23-18). Since 1980, with the increased use of mammography, the incidence and presentation have changed dramatically.458 Today DCIS represents at least 15% to 30% of all newly screen-detected breast carcinomas.459 DCIS presents as microcalcifications (low grade) or rod-shaped branching (high grade) on a mammogram (see Figure 23-56, A).
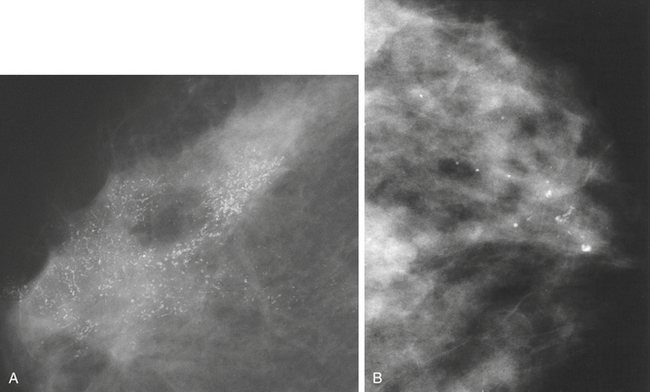
Figure 23-56 Ductal carcinoma in situ (DCIS). A, Malignant microcalcifications. Extensive area of pleomorphic microcalcifications; granular, rod-shaped, and branching microcalcifications can be identified. The appearances are typical of high-grade DCIS. B, Craniocaudal mammography reveals fine and course granular calcifications. Histopathology revealed low-grade DCIS. (A from O’Malley FP, Pinder SE, editors: Breast pathology, New York, 2006, Churchill Livingstone/Elsevier. B from Donegan WL, Spratt JS: Cancer of the breast, ed 5, Philadelphia, 2002, Saunders.)
Molecular genetics has helped define the complexities of DCIS. From genome—wide molecular techniques—the DCIS lesions demonstrate differences in number and type of unbalanced or aberrant chromosome changes as well as intermediate, and poorly differentiated types. These changes seem to mirror the aberrant chromosomal changes observed in invasive breast carcinomas. Still controversial, DCIS does not appear to progress from sequential steps of low grade or risk types to higher grades or risk types en route to cancer or recurrence. This property therefore suggests a stable population. Emerging evidence suggests that DCIS may have programmed potential for phenotype, including progression to invasion, metastasis, hormone receptor expression, and treatment resistance.454 The main issue and challenge revolve around which lesions of the category DCIS become invasive and how soon that happens.458 In a study of 110 autopsies of young and middle-aged women (20 to 54 years), 14% were found to have DCIS,460 suggesting that the preclinical prevalence (subtle histologic distortion and/or nonpalpable mass) is significantly higher than the clinical expression. Other autopsy series show that not all DCIS lesions progress to invasion or become clinically significant.161,167,460,461 DCIS is detected more often in younger women than in older women.
Although there is no universally accepted histopathologic classification, historically most pathologists divided DCIS into five subtypes (papillary, micropapillary, cribriform, solid, and comedo) and often compare the first four types, noncomedo, with comedo. In a single biopsy, however, several types may be mixed and some noncomedo types may express characteristics of the comedo type. Although the term comedo type is widely used it does not specify a grade or an architecture, so newer categories most often use nuclear grade (high, intermediate, or low) and record the architectural pattern separately.459
Lobular Carcinoma in situ: Lobular carcinoma in situ (LCIS) originates from the terminal duct–lobular unit (see Figure 23-54, B). Unlike DCIS, LCIS has a uniform appearance in which the cells occur in noncohesive (discohesive) clusters primarily in lobules. Research, however, suggests that some lobular and ductal carcinomas are closely related.462 LCIS is not associated with calcifications or a stromal involvement that would form a density (lump). Thus it is usually an incidental finding from biopsy for something else. LCIS is fairly uncommon (up to 3.8% of all specimens).463 LCIS is bilateral in 20% to 40% of women, and the majority (80% to 90%) occur before menopause. Although the cells of LCIS and invasive lobular carcinoma are identical,202 whether LCIS is a true neoplasm or a marker of breast cancer risk remains controversial. Evidence to support LCIS as a precursor lesion of invasive carcinoma comes from molecular genetic data highlighting chromosomal alterations and that LOH on chromosome 16q is present in LCIS and invasive carcinoma.43,464 Invasive carcinoma develops in 25% to 35% of women with LCIS, and the contralateral (opposite) breast also is at risk.465
Inflammatory Stroma in Breast Cancer: Dominating the cancer field is the idea that epithelial function depends on the entire tissue, including the stroma or microenvironment. There is compelling evidence that cancer is a tissue-based disease with a possible abnormal aberrant wound healing and inflammatory stromal (reactive stroma) component (see Chapter 11). Early alterations in the stroma that occur with wound healing and inflammation include activation of (1) mesenchymal cells (embryonic) fibroblasts; (2) endothelial cells; and (3) immune cells, including macrophages. Evidence of this type of activation is noted adjacent to tumors and is pathologically known as desmoplastic stroma.
Similar to mesenchymal cells, tumor cells appear to be stimulated, if not initiated, by such proinflammatory microenvironments.466 Inflammation has been correlated with increased cancer incidence and a worse prognosis, or both, for cancers of the prostate, stomach, intestine, liver, and lung. In addition to these clinical data, laboratory data demonstrate how a wound can promote carcinogenesis.467,468 Evidence of the link between wound healing and carcinogenesis is based on the fact that these two distinct pathologies share a common “footprint” or “signature,” a common molecular gene expression signature.466 Furthermore, the genetic expression profile from a wound-healing model of fibroblasts actually predicted metastasis and death in several epithelial cancers. The “activated fibroblast gene signature” also was shown to be an independent marker for local recurrence of breast cancer.469,470
The wound response is based on the theory that “tumors are wounds that do not heal,”471 and that cells involved in angiogenesis and the wound-healing response, including endothelial cells, immune cells, and fibroblasts have a prominent role in carcinogenesis and metastasis.471 Fibroblasts are associated with cancer cells at all stages (Figure 23-58), and their structural and functional roles are emerging. Reactive tumor stroma is associated with an increased number of fibroblasts, increased capillary density, type-I collagen, and fibrin deposition. Thus reactive stroma provides oncogenic signals to promote carcinogenesis (see Figure 23-58).471 Emerging evidence suggests that the development of this wound-reactive pattern and change in tumor tissue architecture create a distinctive phenotype change called epithelial-mesenchymal transition (EMT) (see p. 904).472 EMT is normally activated during early embryogenesis and following injury of some epithelial tissues.473 Therefore, it is possible that cancer cells “reactivate” a latent molecular program usually confined to embryonic development and tissue repair but exaggerated and uncontrolled in cancer.474 The EMT phenotype encourages the “migration step” for tumor invasion, enabling metastasis.475 Interestingly, the tumor cells that undergo EMT have been shown to acquire the characteristics of cancer stem cells.476 Once the metastasizing cancer cells settle in another tissue, they might reestablish the EMT epithelial phenotype.473
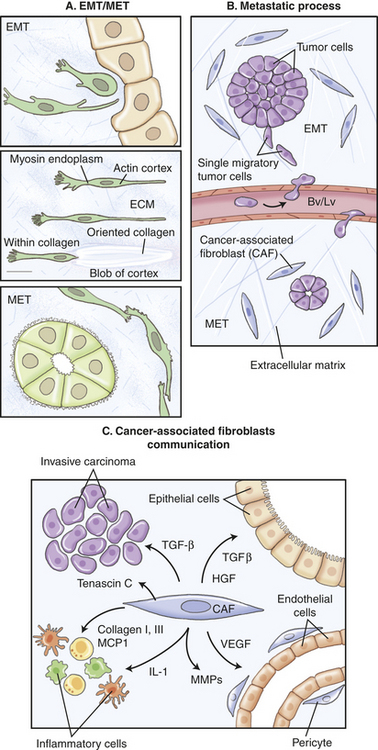
Figure 23-58 The epithelial-mesenchymal transition in cancer (past, present, and future) and functions of activated fibroblasts in the tumor stroma. A, Betty Hey, well known professor of cell biology and embryology, coined the term epithelial-to-mesenchymal (EMT) transformation some 40 years ago and left us beautiful drawings that show not only the transformation to mesenchyme but also the transient nature of the process and the reversion to the epithelial character. This is why the preferred term is now epithelial-to-mesenchymal transition. B, The EMT, as an initial step in the metastatic cascade, has until recently remained a matter of debate; however, powerful imaging tools have convincingly shown that individual cells delaminate (also see Figure 23-59, B) from primary tumors (possibly from decreased cell adhesion) because of dismantling of E-cadherin. C, Fibroblasts communicate with cancer cells, resident epithelial cells, endothelial cells, pericytes, and inflammatory cells through the secretion of growth factors and chemokines. Through the increased deposition of collagen types I and III and de novo expression of tenascin C, fibroblasts induce an altered extracellular-matrix microenvironment that potentially provides additional oncogenic signals. Fibroblasts mediate the inflammatory response by secreting chemokinases such as monocyte chemotactic protein 1 (MCP1) and interleukins such as IL-1. Fibroblasts interact with the microvasculature by secreting matrix metalloproteinases (MMPs) and vascular endothelial growth factor (VEGF). Fibroblasts also provide potentially oncogenic signals such as transforming growth factor-β (TGF-β) and hepatocyte growth factor (HGF) to resident epithelia, and directly stimulate cancer-cell proliferation and invasion by secreting growth factors such as TGF-β and stromal-cell–derived factor 1 (SDF1). Bv, Blood vessels; ECM, extracellular matrix; Lv, lymphatic vessels. (A and B from Acloque H, Thiery JP, Nieto MA: EMBO Rep 9[4]:322-326, 2008. C from Kalluri R, Zeisburg M: Nat Rev Cancer 6:392-401, 2006.)
Another cell that is gaining importance for cancer invasion is the macrophage.477 In one meta-analysis, investigators found that an abundance of macrophages in tumor stroma correlates with a poor prognosis.478 Experimental evidence of the relationship between macrophages and poor prognosis comes from mouse models of breast cancer. In established tumors, tumor-derived cytokines induce the differentiation of the M2 macrophage phenotype. M1 macrophage phenotype is produced mainly in inflamed noncancerous tissue.473 The M2 phenotype stimulates angiogenesis and ECM breakdown from the production of angiogenic growth factors and MMPs, thus promoting carcinogenesis.479 Macrophages may provide a notable “whammy” at the tumor invasive front not only by promoting tumor cell migration, invasion, and intravasation but also by increasing the number of blood vessels as targets for intravasation.477
Invasive Breast Carcinoma: Invasive breast carcinoma is a malignant invasive epithelial lesion derived from the terminal duct lobular unit (Figure 23-54, B, p. 899). It can arise from anywhere in the
Figure 23-57 Schematic figure comparing a wound and invasive tumor. Invasive epithelial tumor (A) and a skin wound (B) in the phase of new tissue formation 3 to 10 days after wounding. Both types of tissue are characterized by the presence of a fibrin clot, inflammatory cells (neutrophils, macrophages, mast cells, and lymphocytes), newly formed vessels, and a large number of fibroblasts and myofibroblasts. These are components of the wound granulation tissue, which strongly resembles the tumor stroma. In addition, migrating and proliferating keratinocytes are present in the wound and in the cancer tissue. The main difference between tumors and wounds is the invasive growth of the transformed keratinocytes (which fill the tumor). (From Schäfer M, Werner S: Nat Rev Mol Cell Biol 9, 628-638, 2008.)
breast parenchyma or accessory breast tissue but it appears to be more common in the upper outer quadrant. The exact molecular events leading to invasion are complex and not completely understood. Unlike the colon cancer multistep model, in which some carcinomas arise sequentially from the preexisting adenoma, breast carcinomas are a group of different disease entities showing parallel progression pathways probably involving crosstalk of multiple signaling pathways.480 From newer data using the stem cell model of breast carcinogenesis, the origin of invasion is hypothesized as the precancer stem cell. Accordingly, the precancer stem cell has an intrinsic programmed latency to invasion and metastases. This newer model suggests that the risk for advancing to invasive breast cancer from human DCIS is predictable at the precancer stage and may depend on the interactions between the DCIS epithelium and the surrounding stroma. It also suggests that these events may involve epigenetic interactions and that genomic alterations may play a role in the programming but their role in neoplastic progression to malignancy and metastasis is unknown.454
Studies of human mammary epithelial cells (HMECs) from healthy individuals are providing insights into how epigenetic and genomic factors fuel cancer progression.481 Hypermethylation of a key cell cycle regulator p16 INK4A, an epigenetic change, creates a previously unknown premalignant lesion (preclonal phase) in healthy disease-free women. These so-called variant HMECs (vHMECs) are thus characterized by lack of p16 INK4A activity and proliferate for an extended period of time with disappearing telomere sequences. These cells exhibit telomere dysfunction and generate chromosomal abnormalities noted in the earliest lesions of breast cancer.482 The existence of this subpopulation of vHMECs with the accompanying telomeric and centrosomal dysfunction may be important early events in breast carcinogenesis.481 Bean and colleagues483 tested this hypothesis in high-risk women and found the frequency of INK4A/ARF promotes hypermethylation and was associated with the combined frequency of other epigenetic hypermethylation changes of the retinoic acid receptor-beta 2 (RARβ), estrogen receptor gene (ESR-1), and BRCA1 genes. In another study (small sample), Bean and colleagues484 found BRCA1 promoter hypermethylation was associated with age and the combined frequency of promoter hypermethylation of the RARβ gene, ESR-1 gene, and p16 (INK4A) gene. Lui and colleagues485 found p16 INKA methylation and γ-tubulin gene amplification had a synergistic effect on promoting tumor progression. Both of these effects were found to increase in atypical ductal hyperplasia and in situ carcinomas. Hypermethylated p16 promoter sequences, and others, could continue to proliferate, thereby generating chromosomal abnormalities that may increase carcinogenesis (Figure 23-59). The continued alterations in the variant cells could occur from microenvironment stromal contributions. Other investigators observed that p16 modulates TP53 in HMEC but not in fibroblasts (see next section).486 How this interaction between signaling pathways is regulated—stroma or intrinsic cell factors—is unknown and potentially important for understanding carcinogenesis. This model differs from the classic view in that it identifies a potentially vulnerable stage of cancer cells.484
Figure 23-59 Model of breast carcinogenesis and breast tissue transformation. A, Molecular mechanisms underlying each of the risk factors for breast cancer are not completely defined. Breast carcinogenesis involves uncontrolled cellular proliferation, alterations in cell signaling pathways, aberrance or loss of apoptosis as a consequence of accumulated genetic damage that lead to activation, and alteration of germline mutation or acquired as somatic mutations as a result of environmental physical (peach) (e.g., ionizing radiation), chemical (blue), lifestyle (purple) (e.g., pregnancy, diet, obesity, physical inactivity), and biologic factors (green) (e.g., aging, endocrine/hormonal milieu); unclassified (pink). The expression of the transformed phenotype requires cumulative genetic alterations and epigenetic alterations. The normal breast is maintained by a complex set of interactions among luminal cells, myoepithelial cells, the basement membrane, and stromal cells. Changes in malignant cells are accompanied or preceded by alterations in the supporting myoepithelial and stromal cells because of genetic and epigenetic events and disruption of normal signaling pathways. The final alteration, invasion of the stroma, is the least understood. Emerging is the possibility that invasion is a result of the loss of myoepithelial and stromal cells to maintain the basement membrane. B, Breast tissue transformation (dark blue cells) from the accumulation of genetic and epigenetic alterations in the epithelium and an altered stromal matrix leads to proliferation and enhanced survival of luminal epithelial cells within the ductal tree compromising normal ductal structure. From proliferation and abnormal survival, the preneoplastic luminal mammary epithelial cells eventually expand to fill the breast ducts. The expanding luminal epithelial mass exerts outward projecting compression forces of increasing magnitude on the basement membrane and adjacent myoepithelium. These forces are countered by an inward projecting resistance force. Importantly, the preneoplastic lesion secretes soluble factors that stimulate immune cell infiltration and activation of fibroblasts to induce a desmoplastic response in the breast stroma. The desmoplastic stroma stiffens over time because of changes in the composition, post-translational modifications, and alterations of the extracellular matrix (ECM). This rigid parenchyma exerts a progressively greater inward projecting resistance force on the expanding preneoplastic duct. Eventually, the number of myoepithelial cells surrounding the prenoeplastic mass decreases and the basement membrane thins, probably owing to increased matrix metalloproteinase (MMP) activity, decreased protein deposition, and compromised assembly. A build-up of interstitial fluid pressure continues, contributed by a leaky vasculature and lymphatic drainage. Responding to their genetic modifications and the altered materials properties of the matrix, the preneoplastic luminal epithelial cells exhibit altered tensional homeostasis and respond to the combination of forces and stromal cues to invade the breast parenchyma. Some fibroblasts transdifferentiate into myofibroblasts and increase tumor migration and invasion by promoting the assembly of collagen fibrils surrounding the distended preneoplastic epithelial ducts. Model of breast carcinogenis and breast tissue transformation. (B from Butcher DT, Alliston T, Weaver VM: Nat Rev Cancer 9:108-122, 2009.) B, Breast tissue transformation.
While all of these changes are occurring, parallel changes also occur because of mutation, epigenetic alterations (see Chapters 11 and 12), or abnormal signaling pathways resulting in altered cellular interactions and tissue structure. Loss of these normal functions also occurs with aging, which may contribute to breast cancer in older women.
The last step in carcinogenesis, the transition of carcinoma in situ to invasive carcinoma, is the subject of emerging evidence. Research shows that tumor-associated stroma undergoes extensive gene expression changes as does the malignant epithelium. Up-regulated genes in the stroma include those of the extracellular matrix, matrix metalloproteinases, and cell cycle-related genes (also see Chapter 11). Important is restoration of normal microenvironment signaling has been shown to revert features of the malignant phenotype despite the mutations in tumor cells.487 Cells of importance in the microenvironment include inflammatory cells, endothelial cells, and stromal fibroblasts. Fibroblasts may play a dominant role in modulating epithelial cell function, promoting tumor progression, and even initiating epithelial cell carcinogenesis.488 Tumor-derived fibroblasts exhibit higher levels of invasion-promoting capacity (IPC) than normal fibroblasts, and this role is enhanced by releasing the matrix metalloproteinases (MMPs) (Figures 23-58 and 23-59).488 Various stimuli within the tumor microenvironment promote EMT of carcinoma cells. Most of the exogenous EMT stimuli are potentially exhibited by cancer-associated fibroblasts.489 The development of this “reactive stromal change” in tumor tissue architecture creates a phenotype normally seen in early embryogenesis that in cancer is reactivated, exaggerated, uncontrolled, and just might acquire the characteristics of cancer stem cells (see Inflammatory Stroma section, p. 900). Numerous signaling pathways (RGKs, Wnt, TGF-β, NF-Kβ) can induce both EMT and invasive cancer by activating master families of transcriptional regulators (Snail, Slug, Twist, ZeB1). These activated regulators inhibit E-cadherin (cell adhesion molecule) causing the dismantling of epithelial cell-cell junctions, which enables the transition of epithelial cells to become motile and invade surrounding tissue (Figure 23-60).490
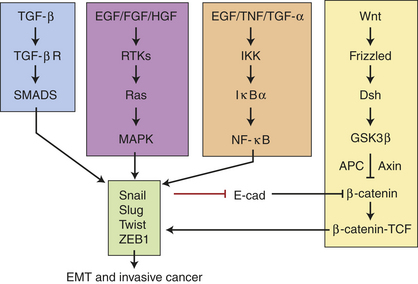
Figure 23-60 Signaling pathways to induce EMT and invasive breast cancer. Numerous signaling pathways including receptor tyrosine kinase (RTKs, Wnt, transforming growth factor-β [TGF-β], NF-kappaKβ can induce EMT and invasive cancer formation by activating a group of master transcriptional regulators of the Snail, Slug, Twist, and ZEB1 families. These regulators inhibit E-cadherin transcription and thereby contribute to the dismantling of epithelial cell-cell junctions and enable the transition of epithelial cells to a more mobile and invasive phenotype. (From Gavert N, Ben-Zé-Ev A: Epithelial-mesenchymal transition and the invasive potential of tumors, Trends Mol Med 14[5]:199-208, 2008.)
Newer techniques, such as microarray technologies (gene chips) that survey many changes in DNA, RNA, and proteins of carcinomas, provide molecular patterns of the overall biologic diversity of invasive breast carcinomas. The four main molecular classes are discussed below.
CLINICAL MANIFESTATIONS Invasive carcinoma of the breast generally presents as a nontender palpable mass or thickened area.202 Lumps caused by breast tumors do not have any classic characteristics. If the local lymphatics are blocked, the subsequent lymphedema and thickening of the skin causes a change called peau d’orange (orange peel skin). Tethering of the skin by Cooper ligaments to the breast mimics the look of an orange peel (Figure 23-61). If the central portion of the breast is involved, retraction of the nipple can occur. Other signs include nipple discharge, palpable nodes in the axilla, or bone pain caused by metastasis to the vertebrae or other skeletal area. Table 23-19 summarizes the clinical manifestations of breast cancers. Manifestations vary according to the type of tumor and stage of disease.
Table 23-19
Clinical Manifestations of Breast Cancer
| Clinical Manifestation | Pathophysiology |
| Chest pain | Metastasis to the lung |
| Dilated blood vessels | Obstruction of venous return by a fast-growing tumor; obstruction dilates superficial veins |
| Dimpling of the skin | Can occur with invasion of the dermal lymphatics because of retraction of Cooper ligament or involvement of the pectoralis fascia |
| Edema | Local inflammation or lymphatic obstruction |
| Edema of the arm | Obstruction of lymphatic drainage in the axilla |
| Hemorrhage | Erosion of blood vessels |
| Local pain | Local obstruction caused by the tumor |
| Nipple/areolar eczema | Paget disease |
| Nipple discharge in a nonlactating woman | Spontaneous and intermittent discharge caused by tumor obstruction |
| Nipple retraction | Shortening of the mammary ducts |
| Pitting of the skin (similar to the surface of an orange [peau d’orange]) | Obstruction of the subcutaneous lymphatics, resulting in the accumulation of fluid |
| Reddened skin, local tenderness, and warmth | Inflammation |
| Skin retraction | Involvement of the suspensory ligaments |
| Ulceration | Tumor necrosis |
Data from Griffiths MJ, Murray KH, Russo PC: Oncology nursing: pathophysiology, assessment, and intervention, New York, 1984, Macmillan.
Figure 23-61 Retraction of nipple caused by carcinoma. (From del Regato JA, Spjut HJ, Cox JD: Ackerman and del Regato’s cancer: diagnosis, treatment, and prognosis, ed 6, St Louis, 1985, Mosby.)
The most common histologic types of breast carcinomas are listed in Table 23-18 (p. 897). Invasive carcinomas of no special type (NST) constitute the majority (70% to 80%) of tumors, however, they cannot be specifically classified. Carcinomas of NST have varying amounts of DCIS. In the future, gene expression profiling may specifically identify subtypes and enable understanding of the clinical relevance (e.g., etiology, presentation, prognosis, or treatment).
EVALUATION AND TREATMENT Approximately 50% of carcinomas of the breast occur in the upper outer quadrant because most of the glandular tissue of the breast is there. Most lymphatic vessels in the breast connect to axillary lymph nodes. Other important lymph nodes are the internal mammary nodes and those close to the collar bone—the infraclavicular and supraclavicular nodes. The lymphatic spread of cancer to the opposite breast, to lymph nodes in the base of the neck, and to the abdominal cavity is caused by obstruction of the normal lymphatic pathways or destruction of lymphatic vessels by surgery or radiation. The less common inner-quadrant tumors may spread to mediastinal nodes or Rotter nodes, which are located between the pectoral muscles. Internal mammary chain nodes are common sites of metastasis. Metastases from the vertebral veins can involve the vertebrae, pelvic bones, ribs, and skull. The brain, lungs, liver, spinal cord, kidneys, adrenal glands, ovaries, and pituitary gland also are sites of metastasis.
Mammography, digital mammography, MRI, ultrasonography, percutaneous needle aspiration, biopsy or minimally invasive biopsy called mammotome, palpation, hormone receptor assays, and gene expression profiling are used to evaluate an individual’s breast cancer, plan treatment, and predict outcome.491 Gene expression profiling results support the hypothesis that estrogen-receptor (ER)−negative (−) and ER-positive (+) cancers originate from distinct cell types and has uncovered biologic processes that govern metastatic progression.492 Four main molecular classes of breast cancer have been identified by profiling, including (1) basal-like (ER−, PR−, HER-2 negative, thus called “triple negative” tumors), (2) luminal-A cancers (mostly ER+ [low grade]), (3) luminal B cancers (mostly ER+ low levels of hormone receptors and often high grade) and (4) HER-2 positive with amplification of the ERBB2 gene and several others of the ERBB2 family. According to Sotiriou and Pusztai492 these subgroups correspond reasonably well to clinical features on the basis of ER and HER2 status. Responding to the need for earlier, more accurate, safer, and cost-effective methods for cancer detection, investigators are studying other methods, including traveling wave and MRI, ultrasound, infrared (thermography), elastography, optical imaging, and others.493,494 Surgical biopsy is the definitive diagnostic test.
Treatment is based on the extent or stage of the cancer (Box 23-18). The extent of the tumor at the primary site, the presence and extent of lymph node metastasis, and the presence of any distant metastases are all evaluated to determine the stage of disease.
Box 23-18 Staging of Breast Cancer—TNM Method

Data from Beahrs OH, Hutter RV, Kennedy BJ, editors: Breast manual for staging of cancer, ed 45, Philadelphia, 1992, Lippincott.
Ovarian ablation in some women younger than 50 years of age with early breast cancer significantly improves long-term survival.495 Radiation therapy is used to prevent metastasis of a small tumor (0.2 cm) or if the cancer is near bone or the edge of the breast that has been surgically excised. Not all women with breast cancer need radiation. The long-term effect of radiotherapy on mortality from breast cancer and other causes remains uncertain. It is especially important to protect the heart and lungs from radiation exposure. Chemotherapy and hormone therapy are most successful as an adjunct to surgery in premenopausal women with hormone-dependent tumors, and are used in individuals with advanced disease. Trastuzumab, the antibody to the Her-2 oncogene, is available and blocks the growth-promoting signal produced by Her-2/neu. Other biologic therapies under investigation are based on discovered relationships between COX-2 and VEGF. Hypoxia upregulates COX-2, which increases proinflammatory prostaglandins (PGE2). Therefore, COX-2 or VEGF inhibitors (e.g., bevacizumab) may be an additional therapeutic choice. Endocrine therapy may be used to prolong survival time and is thought to be most effective in women with ER+ and PR+ tumors. Long-term antiestrogen therapy, such as tamoxifen, has proven efficacy (improves 10-year survival) in individuals with ER+ tumors, and in those with unknown ER status.496 Use of tamoxifen for many years, however, can increase tumor resistance as well as the risk of adverse effects such as blood clots and endometrial cancer. Thus tamoxifen use should not exceed 5 years, and some data support switching to anastrozole or other aromatase inhibitors.497,498 According to a Cochrane Review,436 adjuvant tamoxifen needs further study for women with ER-tumors. Currently, tamoxifen is not recommended as an agent for prevention of breast cancer in those without breast cancer. In early postmenopausal breast cancer, aromatase inhibitors, such as anastrozole, have increased disease-free survival rates more than has tamoxifen499,500 (see p. 883).
Strategies are being studied that have high affinity for breast estrogen receptors but weak affinity for receptors in the uterus, such as the drug raloxifene. Results from the Multiple Outcomes of Raloxifene Evaluation (MORE), a randomized study, found that among postmenopausal women with osteoporosis, raloxifene decreased the risk of invasive breast cancer by 76% (13 cases in 5129 women versus 27 cases in the 2579 placebo group).501 Results from the STAR (study of tamoxifen and raloxifene) trial showed raloxifene to be as effective as tamoxifen in reducing the risk of invasive breast cancer. Raloxifene also has a lower risk of thromboembolic events and cataracts but a nonsignificant higher risk of noninvasive breast cancer. The risk of other cancers, fractures, ischemic heart disease, and stroke were similar for both drugs.501 Coumate 667 (STX 64), a steroid sulfatase inhibitor, has completed a phase 1 trial in postmenopausal women with breast cancer (see Hormones, p. 885). STC 64 is a potent, apparently well-tolerated steroid sulfatase inhibitor, which showed decreases in serum concentrations of steroids with estrogenic properties.502 The effectiveness of sulfatase inhibitors needs further study.