ALTERATIONS OF ERYTHROCYTE FUNCTION



Alterations of erythrocyte function involve either insufficient or excessive numbers of erythrocytes in the circulation or normal numbers of cells with abnormal components. Anemias are conditions in which there are too few erythrocytes or an insufficient volume of erythrocytes in the blood. Polycythemias are conditions in which erythrocyte numbers or volume is excessive. Each of these conditions has many causes and is a pathophysiologic manifestation of a variety of disease states.
ANEMIA
Strictly speaking, anemia is a reduction in the total number of erythrocytes in the circulating blood or a decrease in the quality or quantity of hemoglobin. Anemias commonly result from (1) impaired erythrocyte production, (2) blood loss (acute or chronic), (3) increased erythrocyte destruction, or (4) a combination of these three.
Classification
Anemias are classified by their causes or to changes in their morphology (size, shape, or hemoglobin content) (Box 26-1). The most common classification is based on changes that affect the size or hemoglobin content of the erythrocyte (Table 26-1). The terminology reflects these characteristics; terms that end in “-cytic” refer to cell size, whereas “-chromic” refers to hemoglobin content (Table 26-2). Additional descriptors of erythrocytes associated with some anemias include anisocytosis (assuming various sizes) or poikilocytosis (assuming various shapes) (Figure 26-1).
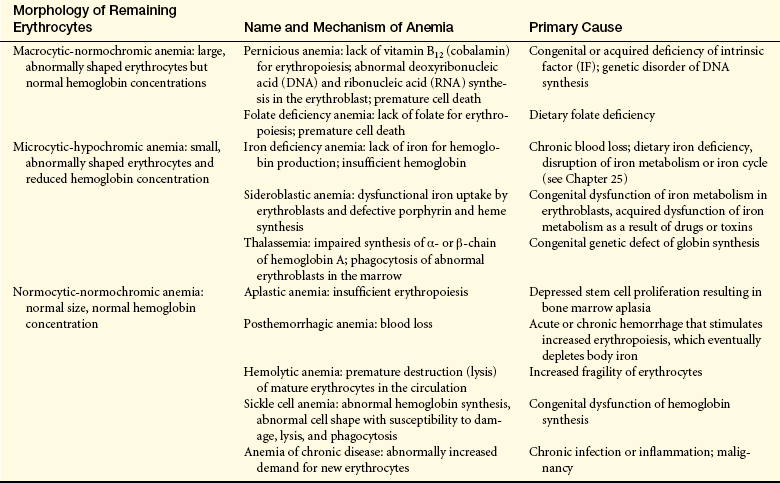
Table 26-2
Terms Used in Assessment of Erythrocytes
| Erythrocyte Volume | Hemoglobin Content | |
| Normal | Normocytic | Normochromic |
| Increased | Macrocytic (higher mean corpuscular volume [MCV]) | Hyperchromic (higher mean corpuscular hemoglobin concentration [MCHC]) |
| Decreased | Microcytic (lower MCV) | Hypochromic (lower MCHC) |
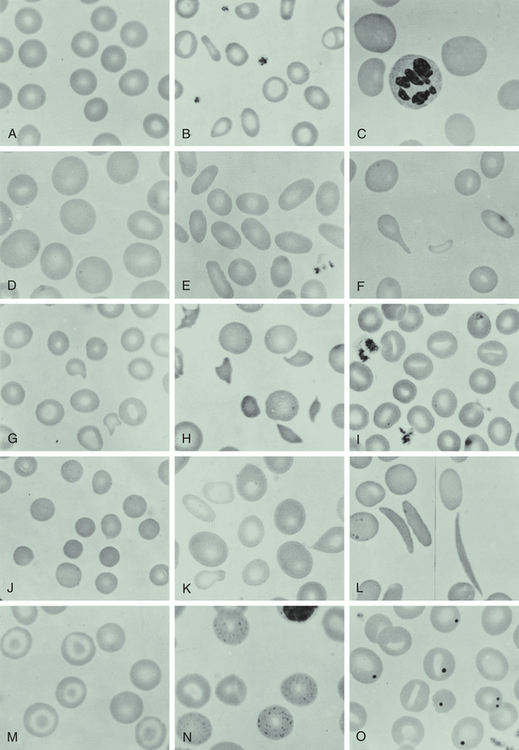
Figure 26-1 Appearance of red blood cells in various disorders. A, Normal blood smear. B, Microcytic-hypochromic anemia (iron deficiency). C, Macrocytic anemia (pernicious anemia). D, Macrocytic anemia in pregnancy. E, Hereditary elliptocytosis. F, Myelofibrosis (teardrop). G, Hemolytic anemia associated with prosthetic heart valve. H, Microangiopathic anemia. I, Stomatocytes. J, Spherocytes (hereditary spherocytosis). K, Sideroblastic anemia; note the double population of red blood cells. L, Sickle cell anemia. M, Target cells (after splenectomy). N, Basophil stippling in case of unexplained anemia. O, Howell-Jolly bodies (after splenectomy). (From Wintrobe MM et al: Clinical hematology, ed 8, Philadelphia, 1981, Lea & Febiger.)
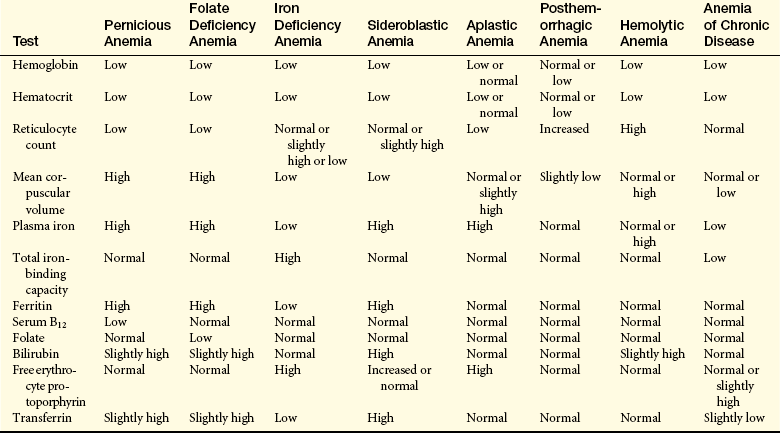
CLINICAL MANIFESTATIONS The fundamental physiologic manifestation of anemia is a reduced oxygen-carrying capacity of the blood resulting in tissue hypoxia. Symptoms of anemia vary, depending on the body’s ability to compensate for hypoxia (Figure 26-2). Anemia that is mild and develops gradually, so-called asymptomatic anemia, is usually easier to compensate for and may cause problems for the individual only during physical exertion. As the reduction in red blood cells (RBCs) continues, symptoms become more pronounced and alterations of specific organs and compensatory effects become more apparent. Compensation generally involves the cardiovascular, respiratory, and hematologic systems. (Hematologic findings associated with various anemias are listed in Table 26-3 and progression and manifestations of anemias are shown in Figure 26-2.)
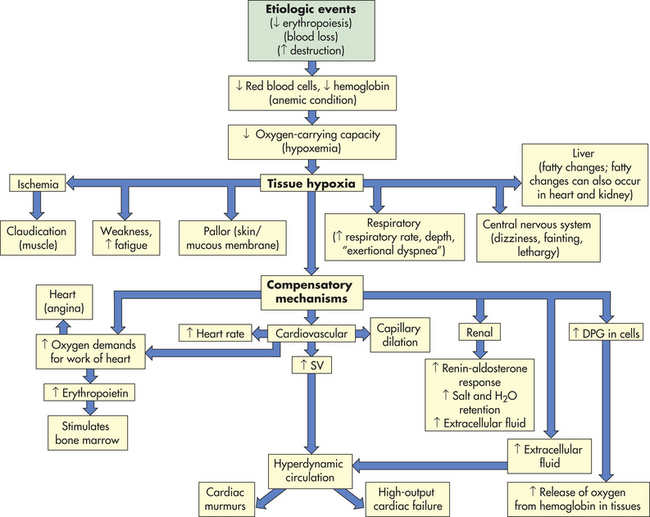
The initial manifestations of anemia are apparent in the cardiovascular system. With hemorrhage, a reduction in the number of RBCs results in reduced blood volume. Compensation for a reduced blood volume causes fluids to move from the interstitium into the intravascular space (osmotic gradient), expanding plasma volume. This compensatory mechanism maintains adequate blood volume, increasing venous return, preload, and stroke volume, but the viscosity (thickness) decreases causing the blood to become diluted. The diluted blood flows faster and more turbulently than normal blood.
Hypoxemia, reduced oxygen levels in the blood, further contributes to cardiovascular dysfunction by causing systemic arterial dilation leading to decreased vascular resistance, which effectively reduces afterload (the pressure necessary to eject blood from the left ventricle into the aorta). Additionally, anemia activates the sympathetic nervous system, causing the heart rate to increase.
These hemodynamic alterations—increased preload, heart rate, and stroke volume, and a reduced afterload—all contribute to increased cardiac output in an effort to maintain adequate oxygen delivery. Without timely interventions, cardiac compensatory mechanisms fail and precipitate the development of congestive heart failure. (Mechanisms of congestive heart failure are described in Chapter 30.)
Tissue hypoxia creates additional demands and compensatory actions on the pulmonary and hematologic systems. The rate and depth of breathing increase in an attempt to increase the availability of oxygen. These demands are accompanied by an increase in the release of oxygen from hemoglobin because of an increase in 2,3-diphosphoglycerate (DPG) in the erythrocytes. (Mechanisms of oxygen transport and release by hemoglobin are described in Chapter 32.) When compensatory mechanisms fail, individuals may experience shortness of breath (dyspnea), a rapid, pounding heartbeat (palpitations), dizziness, and fatigue even at rest. In mild, chronic conditions, these symptoms might be experienced only when demand for oxygen is increased (i.e., during physical exertion), but in severe conditions they may be experienced at rest. Decreased blood supply to skeletal and cardiac muscle also may contribute to the development of muscle pain (claudication) and cardiac angina.
Manifestations of anemia may be observed in other parts of the body. The skin, mucous membranes, lips, nail beds, and conjunctivae become pale as a result of reduced hemoglobin concentration. If anemia is caused by RBC destruction (hemolysis), the skin may become yellowish because of accumulation of the products of hemolysis. Tissue hypoxia also affects the skin causing impaired healing and loss of elasticity, as well as thinning and early graying of the hair.
Affects on the nervous system can occur if the anemia is caused by a vitamin B12 deficiency. Myelin degeneration may occur with the resultant loss of fibers in the spinal cord, producing paresthesias (numbness), gait disturbances, extreme weakness, spasticity, and reflex abnormalities. Decreased oxygen supply to the gastrointestinal (GI) tract often produces abdominal pain, nausea, vomiting, and anorexia. A low-grade fever of less than 38.5° C (less than about 101° F) occurs in some anemic individuals and may be the result of leukocyte pyrogens released from ischemic tissues.
When the anemia is severe or rapid in onset (i.e., hemorrhage), peripheral blood vessels constrict, diverting blood flow to vital organs. Decreased blood flow detected by the kidneys activates the renal renin-angiotensin response. This lifesaving maneuver causes vasoconstriction and increases salt and water retention to increase blood volume and improve kidney perfusion. These situations are emergencies and require immediate intervention to stop the acute loss of blood; consequently, long-term compensatory mechanisms do not develop.
Interventions for slowly developing anemic conditions require treatment of the underlying disorder and palliation of associated symptoms. Therapeutic interventions include control of bleeding, transfusions, dietary correction, and administration of supplemental vitamins or iron.
Macrocytic-Normochromic Anemias
The macrocytic (megaloblastic) anemias are characterized by unusually large stem cells (megaloblasts) in the marrow that mature into erythrocytes that are unusually large in size (macrocytes), thickness, and volume.1 The hemoglobin content is normal (normochromic). These anemias are the result of defective erythrocyte precursor DNA synthesis commonly caused by deficiencies of vitamin B12 (cobalamin) or folate, coenzymes that are required for nuclear maturation and the DNA synthesis pathway. More than half the residents in nursing homes are anemic, and about a third of those cases are caused by deficiencies in vitamin B12, folate, or iron.2,3 Vitamin B12 deficiency in older adults often goes unrecognized because of the subtle nature of manifestations that are potentially serious, particularly hematologically and neurologically.
In spite of defective DNA synthesis in megaloblasts, ribonucleic acid (RNA)–controlled processes (RNA replication and hemoglobin synthesis) occur at a normal rate, resulting in the unequal growth and development of the cytoplasm and nucleus. Asynchronous development leads to a larger than normal normoblast with a disproportionally small nucleus. With each cell division the disproportion between RNA and DNA becomes more obvious. The chromatin in the nucleus fails to clump normally, resulting in finely distributed chromatin throughout the nucleus. The altered pattern of chromatin deposition allows for microscopic differentiation of normoblasts from megaloblasts. Hemoglobin increases in proportion to the size of the cell; thus the mean corpuscular hemoglobin concentration (MCHC) remains normal and the megaloblastic anemias, in the absence of complications, are normochromic.
Immature precursors of the megaloblastic RBCs have a greater chance of dying during maturation than do normoblastic precursors. Phagocytosis of these cells occurs within the bone marrow, resulting in a reduction of reticulocytes and erythrocytes in the circulation. Additionally there is an increase in lactic dehydrogenase, reflecting cellular destruction, and indirect bilirubin, from the breakdown of heme. Both of these substances may be measured in the blood, providing biochemical evidence of ineffective erythropoiesis.
Defective DNA synthesis also may result in significant enlargement of neutrophil precursors creating giant metamyelocytes with a tendency to have more nuclear lobes than normal. Other cells throughout the body also may demonstrate enlargement and nuclear abnormalities. Cells lining epithelium and those with high turnover rates are most affected.
Pernicious Anemia
Pernicious anemia (PA), the most common type of megaloblastic anemia, is caused by vitamin B12 deficiency, which is often associated with the end stage of type A chronic atrophic (congenital or autoimmune) gastritis (see Figure 26-1, C; Figure 26-3). Pernicious means highly injurious or destructive and reflects the fact that this condition was once fatal. It most commonly affects individuals older than the age of 50 who are of Northern European descent, as well as blacks and Hispanics. Females are more prone to develop PA, with black females having an earlier onset.
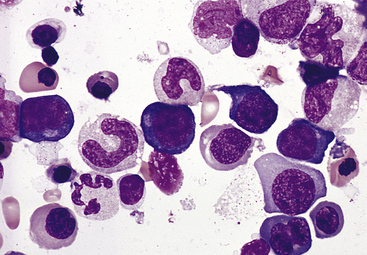
Figure 26-3 Bone marrow aspirate from individual with pernicious anemia. Bone marrow aspirate smear from an individual with megaloblastic red blood cell precursors and giant metamyelocytes. The chromatin in the red blood cell nuclei is more dispersed than in normal red blood cell precursors at comparable stages of maturation; the giant metamyelocytes have dispersed nuclear chromatin in contrast to a normal metamyelocyte, which has condensed chromatin (Wright-Giemsa stain). (From Damjanov I, Linder J, editors: Anderson’s pathology, ed 10, St Louis, 1996, Mosby.)
PATHOPHYSIOLOGY The principal disorder in PA is an absence of intrinsic factor (IF), a transporter required for absorption of dietary vitamin B12. Vitamin B12 catalyzes the action of methionine synthase and R-methylmalonyl-coenzyme A (CoA) mutase, which acts to promote nuclear maturation and DNA synthesis in erythrocytes. IF, along with hydrochloric acid, is secreted by gastric parietal cells and complexes with dietary vitamin B12 in the small intestine. The B12-IF complex binds to cell surface receptors in the ileum and is transported across the intestinal mucosa. Deficiency in IF secretion may be congenital or result from adult onset gastric mucosal atrophy and destruction of parietal cells. In older adults, virtually all the vitamin B12-deficiency anemia is caused by a failure of IF-related absorption.4 Congenital IF deficiency is a genetic disorder that demonstrates an autosomal recessive inheritance pattern. Gastric atrophy commonly occurs in the presence of type A chronic gastritis and may be autoimmune. Autoantibodies against gastric parietal cells are frequently observed.5 The autoantibodies are directed against H+, K+-ATPase, an enzyme responsible for secretion of hydrogen ions by parietal cells in exchange for potassium ions. Other characteristics associated with chronic gastric atrophy include achlorhydria, low serum levels of pepsinogen I, hypergastrinemia, and gastric carcinoids.
Early in the disease process the gastric submucosa becomes infiltrated with inflammatory cells, eventually extending into the lamina propria and causing degeneration of the parietal and zymogenic cells. Late in the course of the disease, the parietal and zymogenic cells are destroyed and replaced by mucus-containing cells (intestinal metaplasia). The mechanism of cellular destruction in autoimmune gastritis is unknown, but it is thought to involve signaling through death-inducing pathways (e.g., Fas ligand [Fas/FasL] and tumor necrosis factor/tumor necrosis factor receptor [TNF/TNFR] pathways). Loss of parietal cells results in IF deficiency. A direct correlation exists between the severity of the gastric lesion and the degree of malabsorption of vitamin B12. Malabsorption of vitamin B12 also may be secondary to secreted autoantibodies against IF, which prevents the formation of the B12-IF complex.5
As with most autoimmune diseases, genetic factors increase the risk for developing chronic gastritis and PA. Family clusters have been identified; 20% to 30% of individuals related to persons with PA also have PA. These relatives, particularly first-degree female relatives, also demonstrate a higher frequency of the presence of gastric autoantibodies.
PA also is associated with other autoimmune conditions, particularly those affecting the endocrine system, including chronic autoimmune thyroiditis (Hashimoto thyroiditis), type 1 diabetes mellitus, Addison disease, primary hypoparathyroidism, Graves disease, and myasthenia gravis.
Environmental conditions also may lead to chronic gastritis. These include excessive alcohol ingestion, hot tea, and smoking. Complete or partial gastrectomy causes IF deficiency. Helicobacter pylori has been identified as a causative agent in the development of vitamin B12 deficiency.6 Drugs known as proton pump inhibitors (PPIs) are used to decrease gastric acidity, but also may decrease cobalamin absorption, although it is not thought that they actually cause PA. Individuals with type A chronic gastritis PA are at risk for developing gastric adenocarcinoma of the noncardia stomach from intestinal metaplasia and esophageal squamous cell carcinoma. The incidence of carcinoma in these individuals is 2% to 3%. Type B gastritis is caused by H. pylori with a decreased risk for development of cancer.
CLINICAL MANIFESTATIONS PA develops slowly—possibly from 20 to 30 years; 60 years of age is the median age at time of diagnosis. Because of the slow onset of symptoms, PA is usually severe by the time treatment is sought. Early symptoms are often ignored because they are vague and include infections, mood swings, and gastrointestinal, cardiac, or kidney ailments. When the hemoglobin level in the blood has decreased significantly (7 to 8 g/dl), the individual experiences the classic symptoms of anemia—weakness, fatigue, paresthesias of the feet and fingers, difficulty in walking, loss of appetite, abdominal pains, and weight loss. The tongue may become sore, smooth, and beefy red secondary to atrophic glossitis. The skin may become “lemon yellow” (sallow) as a result of a combination of pallor and icterus. The liver may be enlarged, especially in older adults, indicating right-sided heart failure. The spleen also may enlarge but remains nonpalpable.
Neurologic manifestations result from nerve demyelination that may produce neuronal death. The posterior and lateral columns of the spinal cord also may be affected, causing a loss of position and vibration sense, ataxia, and spasticity. These complications pose a serious threat because they are not reversible, even with appropriate treatment. The cerebrum also may be involved with manifestations of affective disorders, most commonly of the depressive types. An increased prevalence of serum vitamin B12 deficiency has been reported among individuals with Alzheimer disease.
EVALUATION AND TREATMENT Diagnosis of PA is based on a variety of tests (see Table 26-3), which include blood tests, bone marrow aspiration, serologic studies, gastric biopsy, clinical manifestations, and the Schilling test. The Schilling test indirectly evaluates vitamin B12 absorption by administering radioactive B12 and measuring excretion in the urine. Low urinary excretion is significant for PA. A second test often is done to confirm the diagnosis. In the second test, IF may be administered to see whether urinary excretion increases. If urinary excretion does not increase, other causes of PA must be considered.
Serologic studies, however, have replaced the Schilling test for diagnosing PA. Measuring methylmalonic acid and homocysteine levels, which are elevated early in PA, is more sensitive. The presence of circulating antibodies against parietal cells and intrinsic factor is also useful in diagnosis.5 Gastric biopsy reveals total achlorhydria (absence of hydrochloric acid), which is diagnostic for PA because it occurs only in the presence of this gastric lesion.
Replacement of vitamin B12 (cobalamin) is the treatment of choice. Cyanocobalamin or hydroxocobalamin (1000 mcg) is administered parenterally on a monthly schedule. Initial injections are administered weekly until the deficiency is corrected. Conventional wisdom and practice assumed that oral preparations were ineffective because there was no IF to facilitate absorption. Recent experience, however, has determined that vitamin B12 will be absorbed across the small bowel so that oral administration is beneficial in dosages higher than parenteral dosages.
The effectiveness of cobalamin replacement therapy is determined by a rising reticulocyte count. Within 5 to 6 weeks, blood counts return to normal. PA cannot be cured, so maintenance therapy is lifelong. Blood transfusions are given if the individual shows signs of circulatory collapse, heart failure, or severe angina pectoris.
Untreated PA is fatal, usually because of heart failure. Death occurs after a course of remissions and exacerbations lasting from 1 to 3 years. Since 1926, when replacement therapy began, mortality has been reduced significantly. Today, death from PA is rare, and any relapses that occur are usually the result of noncompliance with therapy.
Folate Deficiency Anemia
Folate (folic acid) is an essential vitamin for erythrocyte production and maturation. Humans totally depend on dietary intake of folate, requiring 50 to 200 mcg/day, with pregnant and lactating females requiring increased amounts. Folate synthesis takes place in the human intestine, although not in quantities sufficient to make any significant contribution.
Absorption of folate occurs primarily in the upper small intestine and does not depend on the presence of any other facilitating factor. From the small intestine it is circulated to and through the liver where it is stored. Folate deficiency is more common than cobalamin deficiency, particularly in alcoholics and individuals with chronic malnourishment. Alcohol interferes with folate metabolism in the liver, causing a profound depletion of folate stores. Fad diets and diets low in vegetables also may cause folate deficiency because of the absence of plant sources of folate. At least 10% of North Americans have a folate deficiency, although the incidence has been on the decrease in the United States since the fortification of food with folate and the increased use of folate supplements.
Folates are coenzymes required for the synthesis of thymine and purines (adenine and guanine) and the conversion of homocysteine to methionine. Deficient production of thymine, in particular, affects cells undergoing rapid division (e.g., bone marrow cells undergoing erythropoiesis). The clinical manifestations of folate deficiency become apparent when the synthesis of thymidylate is critically impaired and progresses to the development of megaloblastic anemia.
PATHOPHYSIOLOGY Impaired DNA synthesis secondary to a folate deficiency results in megaloblastic cells with clumped nuclear chromatin. Anemia may result from apoptosis of erythroblasts in the late stages of erythropoiesis. In addition to anemia, folate deficiency in pregnant women is associated with neural tube defects of the fetus. Folate is necessary for the reduction of circulating levels of homocysteine, a risk factor for the development of atherosclerosis (see Chapter 30), thus a folate deficiency increases the risk for developing coronary artery disease. A deficiency of folate also is implicated in the development of cancers, specifically colorectal cancers.
CLINICAL MANIFESTATIONS Clinical manifestations of folate deficiency anemia are similar to the cachectic, malnourished appearance of individuals with PA. Specific symptoms include severe cheilosis (scales and fissures of the lips and corners of the mouth), stomatitis (inflammation of the mouth), and painful ulcerations of the buccal mucosa and tongue. Gastrointestinal symptoms may be present and include dysphagia (difficulty swallowing), flatulence, and watery diarrhea, as well as histologic and roentgenographic changes of the GI tract suggestive of the chronic malabsorption syndrome, sprue. Neurologic manifestations, such as those that occur in PA, are generally not seen in folate deficiency anemia. Any neurologic symptoms are usually caused by a thiamine deficiency, which often accompanies folate deficiency.
EVALUATION AND TREATMENT Determination of a folate deficiency is based on measurement of serum folate levels and symptoms. Successful treatment requires daily oral administration of folate preparations until adequate blood levels are obtained and clinical symptoms are reduced or eliminated. One milligram per day is sufficient for most individuals, although persons with alcoholism may require 5 mg. Prophylactic dosages of 0.1 to 0.4 mg/day are sometimes given during pregnancy. Parenteral administration of folic acid (citrovorum factor or leucovorin) generally is not used except in situations in which an individual has been using drugs that inhibit dihydrofolate reductase. After administration of folate, the manifestations of anemia disappear within 1 to 2 weeks.
After the folate deficiency has been corrected, long-term treatment with folate is not necessary if the appropriate dietary adjustments are made to maintain adequate intake. An intake of folate (400 mcg/day) is recommended as a measure to prevent heart disease.
Microcytic-Hypochromic Anemias
The microcytic-hypochromic anemias are characterized by erythrocytes that are abnormally small and contain abnormally reduced amounts of hemoglobin (see Figure 26-1, B). Hypochromia occurs even in cells of normal size.
Microcytic-hypochromic anemia results from a wide variety of conditions that are related to (1) disorders of iron metabolism, (2) disorders of porphyrin and heme synthesis, or (3) disorders of globin synthesis. Specific disorders include iron deficiency anemia, sideroblastic anemia, and thalassemia (thalassemia is discussed in Chapter 28).
Iron Deficiency Anemia
Iron deficiency anemia (IDA) is the most common type of anemia worldwide, occurring in both developing and developed countries and affecting as many as one fifth of the world population. Those at greatest risk for developing hypoferremia and IDA are the chronic poor, women of childbearing age, and children. Iron deficiency in children is associated with numerous adverse health-related manifestations, especially cognitive impairment, which may be irreversible. Teens who had iron deficiency as infants are likely to score lower on cognitive and motor tests, even if the iron deficiency was identified and treated in infancy.
Children in developing countries often are affected by chronic parasite infestations that result in intestinal blood and iron loss that outpaces dietary intake.7 Treatment of helminth infections results in an improvement in the anemia as well as in appetite and growth. Iron deficiency also occurs in individuals with lead poisoning. Treatment of the iron deficiency is associated with a decrease in lead levels.
In the United States, 720,000 children (9%) ages 1 to 2 years are estimated to be iron deficient, of whom 240,000 (3%) are anemic, which may be a result of increased iron requirements with growth. Females demonstrate a higher incidence of hypoferremia (13.9%) than do males (8.3%), as well as IDA; 4% to 6% in females and 4% in males. The incidence peaks in females during their reproductive years and decreases after menopause. In females, menorrhagia (excessive bleeding during menstruation) is a common cause of primary IDA. Those at highest risk are black females living in urban poverty.8 Males demonstrate a higher incidence during childhood and adolescence, a decrease occurring during young adulthood, and an upswing during late adulthood. An increased prevalence of iron deficiency has been demonstrated in overweight children. The most common cause of IDA in well-developed countries is pregnancy and chronic blood loss.4 Blood loss of 2 to 4 ml/day (1 to 2 mg of iron) is sufficient to cause iron deficiency and may result from erosive esophagitis, gastric and duodenal ulcers, colon adenomas, and cancers. H. pylori infections also have been found to cause IDA of unknown origin, although H. pylori impairs iron uptake.
Other causes of IDA are (1) medications that cause gastrointestinal bleeding (aspirin, nonsteroidal anti-inflammatory drugs [NSAIDs]); (2) surgical procedures that decrease stomach acidity, intestinal transit time, and absorption; (3) insufficient dietary intake of iron; and (4) eating disorders, such as pica, which is the craving and eating of nonnutritional substances.
Iron is the essential for several biologic processes. As a component of hemoglobin, iron is in constant demand for use in normal erythropoiesis. Iron is recyclable; therefore, the body maintains a balance between iron that is contained in hemoglobin and iron that is in storage and available for future hemoglobin synthesis. Iron metabolism for erythropoiesis is complex and not well understood. Sources of iron include a small portion absorbed from the duodenum and, to a lesser extent, from the stomach, ileum, and colon. A much larger portion is available through recycling of iron from senescent RBCs (see Chapter 25).
Iron also contributes to immune function by regulating immune effector mechanisms (i.e., cytokine activities [interferon-gamma (INF-γ)], nitric oxide formation, and T-cell proliferation).
Acquired hypoferremia may be part of the body’s response to infection. Anemia can be part of the nonspecific acute phase response to any type of inflammation of sufficient degree. Many pathogens require iron for survival; thus hypoferremia would hamper their growth. However, the precise benefits or detriments of iron deficiency and immunity are still controversial.
PATHOPHYSIOLOGY IDA can be classified as arising from one of two different etiologies or a combination of both. Nutritional iron deficiency results from inadequate dietary intake or excessive blood loss. In both instances there is no intrinsic dysfunction in iron metabolism; however, both deplete iron stores and result in IDA caused by reduced hemoglobin synthesis. A second category is a metabolic or functional iron deficiency in which various metabolic disorders lead to either insufficient iron delivery to bone marrow or impaired iron use within the marrow. Paradoxically, iron stores may be sufficient but delivery is inadequate to maintain heme synthesis, thus producing a functional or relative iron deficiency.
IDA occurs when the demand for iron exceeds the supply and develops slowly through three overlapping stages. In stage I, the body’s iron stores are depleted. Erythropoiesis proceeds normally, with the hemoglobin content of RBCs remaining normal. In stage II, iron transportation to bone marrow is diminished, resulting in iron deficiency erythropoiesis. Stage III begins when the small hemoglobin-deficient cells enter the circulation in sufficient numbers to replace the normal mature erythrocytes that have been removed from the circulation. Manifestations of IDA appear in stage III when iron stores are depleted and there is diminished hemoglobin production.
CLINICAL MANIFESTATIONS Symptoms of IDA begin gradually, and the symptoms are usually not severe enough for individuals to seek medical attention until hemoglobin levels have decreased to about 7 to 8 g/dl. Early symptoms include fatigue, weakness, and shortness of breath. Pale earlobes, palms, and conjunctivae (Figure 26-4) are also common signs.
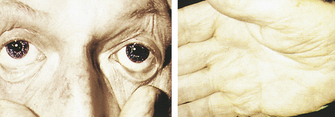
Figure 26-4 Pallor and iron deficiency. Pallor of the skin, mucous membranes, and palmar creases in an individual with hemoglobin of 9 g/dl. Palmar creases become as pale as the surrounding skin when the hemoglobin level approaches 7 g/dl. (Courtesy Hoffbrand AV, Pettit JE, editors: Sandoz atlas of clinical hematology, London, 1988, Gower Medical.)
Progressive IDA causes more severe alterations, with structural and functional changes apparent in epithelial tissue (see Figure 26-4). The nails become brittle, thin, coarsely ridged, and spoon-shaped or concave (koilonychia) as a result of impaired capillary circulation (Figure 26-5). The tongue becomes red, sore, and painful, which is caused by atrophy of the papillae (glossitis) (Figure 26-6). The degree of pain experienced is directly associated with the amount of iron deficiency. Individuals also experience dryness and soreness in the epithelium at the corners of the mouth, known as angular stomatitis. Difficulty in swallowing is associated with an esophageal “web,” a thin, concentric, smooth extension of normal esophageal tissue consisting of mucosa and submucosa at the juncture between the hypopharynx and esophagus. The duration of iron deficiency required for web formation is uncertain. Dysphagia also is exacerbated by hyposalivation. The pathophysiology associated with these epithelial lesions is not well understood, but the lesions have the potential to become cancerous.
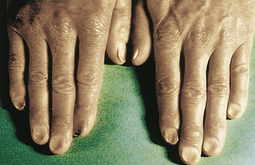
Figure 26-5 Koilonychia. The nails are concave, ridged, and brittle. (Courtesy Hoffbrand AV, Pettit JE, editors: Sandoz atlas of clinical hematology, London, 1988, Gower Medical.)
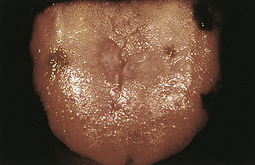
Figure 26-6 Glossitis. Tongue of individual with iron deficiency anemia has bald, fissured appearance caused by loss of papillae and flattening. (Courtesy Hoffbrand AV, Pettit JE, editors: Sandoz atlas of clinical hematology, London, 1988, Gower Medical.)
Nonheme iron is a component of many enzymes in the body (e.g., cytochromes, myoglobin, catalases, peroxidases), particularly those involved in the metabolism of amine neurotransmitters, reduction of nucleotides, and biosynthesis of methionine. Abnormalities and deficiencies of iron-dependent enzymes may account for many of the clinical manifestations of IDA. Individuals with IDA also exhibit gastritis, neuromuscular alterations, irritability, headache, numbness, tingling, and vasomotor disturbances. The pathogenesis of neurologic symptoms is unknown but may be caused by hypoxia in already compromised cerebral vessels. Gait disturbances are rare. Mental confusion, memory loss, and disorientation often are associated with anemia in older adults and may be wrongly perceived as “normal” events related to aging.
EVALUATION AND TREATMENT Initial evaluation is based on the presence of a decreased hemoglobin and hematocrit. Additional measurements, however, are needed to determine the cause of the anemia (see Table 26-3). Iron stores may be measured directly by bone marrow biopsy and iron staining or indirectly by laboratory tests for serum ferritin, transferrin saturation, or total iron-binding capacity. Serum ferritin is a widely accepted and available measurement of iron status that has been used for the past 25 years; 1 mcg/L serum ferritin corresponds to 8 to 10 mg or 120 mcg of storage iron/kg body weight.8 Serum ferritin level has demonstrated its superiority over other measures (i.e., mean corpuscular volume [MCV], transferrin saturation). One limit to the serum ferritin is the elevation of values independent of iron status that accompanies acute or chronic inflammation, malignancy, liver disease, or alcoholism.
An indicator of iron levels is the level of serum transferrin receptor (sTfR). Transferrin receptors are membrane glycoproteins that bind circulating transferrin for transport into cells. Soluble forms of the receptor are found in serum. The ratio of serum levels of transferrin receptor to ferritin (R/F) reliably and accurately estimate body iron stores and differentiate primary IDA from anemia secondary to chronic disease. A major drawback, however, is the lack of proper standardization for the sTfR assay.
The first step in treatment of IDA is to identify and eliminate sources of blood loss.9 With ongoing bleeding, any pharmacologic therapy is likely to be ineffective. Iron replacement therapy is very effective in the treatment of nutritional deficient anemia. In fact, the most conclusive evidence for the diagnosis of IDA is an increase in hemoglobin of 1 to 2 g/dl after iron therapy is initiated. Iron is available in ferrous or ferric forms; however, ferrous is preferable because it is more readily absorbed. The ferrous form is available as sulfate, gluconate, or fumarate. Ferrous sulfate is the cheapest and most commonly used.
Initial iron replacement therapy is 150 to 200 mg/day; however, recent studies have found that dosages as low as 60 mg/day are effective in certain individuals. Once therapy has begun, individuals demonstrate a rapid decrease in fatigue, lethargy, and other associated symptoms. Hematocrit levels should improve within 1 to 2 months of therapy; however, the serum ferritin level is a more precise measurement of improvement and total body stores of iron. Once the serum ferritin level reaches 50 mcg/L, adequate replacement of iron has occurred. Replacement therapy is usually continued for 3 to 6 months after bleeding has been contained; however, therapy may continue for as long as 24 months. Daily therapy (60–120 mg/day) for menstruating females may be required until menopause.
Parenteral iron replacement is used in instances of uncontrolled blood loss, intolerance to oral iron, intestinal malabsorption, and poor adherence to oral therapy. Iron dextran has been the only parenteral agent available in the United States. Intramuscular injection is the recommended method; however, intravenous administration is generally preferred because of the ability to administer larger doses. A significant concern in the use of IV dextran is the potential for severe anaphylactic reaction. Delayed allergic reactions are also major concerns.
Newer medications that have recently been approved for parenteral therapy in treating IDA are sodium ferric gluconate complex in sucrose (Ferrlecit) and iron sucrose injection (Venofer). Iron dextran is recommended as the first choice in spite of its higher rate of adverse reactions. For individuals who are intolerant of iron dextran, the two newer agents are safe and effective alternatives. Drawbacks to their use include higher cost and the need for multiple infusions.
Sideroblastic Anemia
Sideroblastic anemias (SAs) are a heterogeneous group of disorders characterized by anemia of varying severity caused by a deviation in mitochondrial metabolism leading to ineffective iron uptake and dysfunctional heme synthesis.10 Ringed sideroblasts within the bone marrow are diagnostic of SA. Ringed sideroblasts are erythroblasts that contain iron granules that have not been synthesized into hemoglobin, but instead are arranged in a perinuclear collar around one third or more of the nucleus (see Figure 26-1, K). Individuals with SA also have increased levels of iron in their tissue. The blood contains hypochromic erythrocytes, either microcytic or macrocytic depending on the form of the disease.
PATHOPHYSIOLOGY SAs have multiple etiologies but all share the commonality of altered mitochondrial heme synthesis in the erythroid cells in bone marrow. Mitochondrial aminolevulinic acid (ALA) synthase uses glycine to convert succinyl CoA into ALA.11 ALA undergoes further enzymatic modification in the cytoplasm to the porphyrin structure becoming coproporphyrinogen III, which reenters the mitochondria. Within the mitochondria the molecule is progressively converted to protophorphyrin IX, which has ferrous iron (Fe2+) inserted by the enzyme ferrochelatase. Disruptions to this pathway lead to the accumulation of iron in the mitochondria and the characteristic sideroblasts.
SAs are either hereditary or acquired. Hereditary SAs are rare and occur almost exclusively in males, suggesting a predominant recessive X-linked transmission. An occasional autosomal recessive transmission occurs with mitochondrial mutations and deficiencies of ferrochelatase.12 The anemia of hereditary SA is usually present in infancy or childhood, but may remain undetected until midlife. In some instances, other symptoms (e.g., diabetes or cardiac failure resulting from tissue iron overload) may be the first manifestation of SA. Differentiation of SA from idiopathic hemachromatosis needs to be confirmed because both are characterized by tissue iron deposition.
The severity of the anemia is quite variable, and qualitative alterations of the erythrocytes (e.g., decreased MCV and increased RBC volume distribution width) may be evident even when anemia is not present. Dimorphism, in which normocytic and normochromic cells are seen concomitantly with microcytic-hypochromic cells, may be present and is seen more commonly in individuals with mild anemia, female carriers, or those receiving treatment with pyridoxine. Anisocytosis and poikilocytosis also are seen on examination of the blood smear.
Hereditary SA (X-linked sideroblastic anemia [XLSA]) has been linked to missense mutations in the erythroid-specific ALAS-E gene Xp11.21.12 More than 25 missense mutations have been identified. ALAS is the first and rate-limiting enzyme in the heme biosynthesis pathway, and mutations lead to reduced synthesis of protoporphyrin IX and the characteristic accumulation of iron in the erythrocyte.
Acquired sideroblastic anemias (ASAs) are the most common SAs. The causes of primary forms of ASA are unknown (idiopathic) or associated with other myeloproliferative or myeloplastic disorders. Another form, reversible SAs, is secondary to various conditions, such as alcoholism, drug reactions, copper deficiency, and hypothermia, with drugs and toxins being the leading cause.
The leading known cause of primary ASA, myelodysplastic syndrome (MDS), is a group of disorders of hematopoietic stem cells, with all three stem cell lines demonstrating dysplastic characteristics.13 Initially, all ASAs associated with myelodysplastic syndrome were considered to be one and the same and identified as refractory anemia with ringed sideroblasts. This classification proved unsatisfactory because different outcomes were observed in individuals who had the same apparent disease. Further investigations discovered morphologic and chromosomal characteristics that predicted different clinical courses. Two subsets of myelodysplastic ringed sideroblasts were identified based on which cell lines were affected. In one subset dysplastic features were limited to the erythroid line and was classified as pure SA. Individuals with pure SA require transfusions, which may produce iron overload.14 With adequate chelation therapy, they are able to survive and thrive for many years. A significant outcome of this condition is the rare occurrence of conversion to leukemia.
The second subset was characterized by abnormalities of multiple cell lineages. In addition to SA, major alterations of neutrophil and platelet were observed. Infections, frequently fatal, are common secondary to neutropenia and neutrophil dysfunction. Bleeding from thrombocytopenia and platelet dysfunction also are prevalent. Of those who survive, 40% develop acute (myeloblastic) leukemia.
Reversible SA, the most prevalent form of ASA, is a result of several factors (predominantly drugs or toxins [e.g., lead, zinc]) that affect heme biosynthesis, and anemia related to these causes is reversible with reduced exposure to the toxin or drug. The most frequent cause of reversible SA is alcohol abuse. Excessive alcohol inhibits pyridoxal phosphate, which is a cofactor for ALA-synthase in mitochondria. Alcohol abuse may also secondarily result in nutritional deficiencies that may affect heme biosynthesis, such as pyridoxine and copper. Copper is a cofactor for mitochondrial ferrochelatase, which controls the insertion of iron into protoporphyrin IX to form heme. Other drugs that are copper chelators (e.g., penicillamine) have similar effects leading to SA.
Other drugs that cause ASA include antituberculous agents (isoniazid [INH], pyrazinamide, and cycloserine) and chloramphenicol. Antituberculous agents interfere with vitamin B6 metabolism, which reduces ALA synthesis, thus decreasing heme generation. Chloramphenicol causes direct mitochondrial injury by inhibiting mitochondrial membrane proteins and thus mitochondrial respiration. Additionally, therapeutic drug levels are known to inhibit erythroid colony growth but not granulocyte colony growth.
CLINICAL MANIFESTATIONS The anemias of SA are generally moderate to severe, with hemoglobin levels varying from 4 to 10 g/dl. In addition to the cardiovascular and respiratory manifestations common to all anemias, individuals with SA demonstrate signs of iron overload known as erythropoietic hemochromatosis. Mild to moderate enlargement of the spleen (splenomegaly) and liver (hepatomegaly) occurs; however, liver function remains normal or only slightly impaired. Occasionally abnormal skin pigmentation (bronze colored) is seen. Neurologic and epithelial alterations commonly associated with other anemias are nonexistent. Heart rhythm disturbances, along with congestive heart failure, are major life-threatening complications related to cardiac iron overload. These manifestations are fortunately rare and occur late in the progression of the disease. Young children and infants who are severely affected may demonstrate growth and developmental impairment.
EVALUATION AND TREATMENT Initially, SA may be mistaken for deficiency of stem cells in the marrow (hypoplastic anemia) or IDA (laboratory findings are listed in Table 26-3). Bone marrow examination establishes the diagnosis. The marrow is packed with erythrocyte stem cells, and mononuclear phagocytes in the marrow are loaded with iron in the form of hemosiderin. Platelet and leukocyte values are generally normal; however, they may be reduced if splenomegaly is evident. The presence of sideroblasts confirms the diagnosis of SA.
Initial treatment of SA is directed toward identification of a causative agent (i.e., drugs or toxins).15 Treatment is supportive, with transfusions being the primary intervention. Following removal of the agent, oral pyridoxine (100 mg/day) may be administered on a trial basis. Acquired SA related to alcohol abuse and pyridoxine antagonists often demonstrates a complete response to pyridoxine. SA caused by other etiologies does not demonstrate the same improvement.
Individuals with hereditary XLSA also are initially treated with pyridoxine therapy in doses of 50 to 200 mg/day. Approximately one third of individuals with hereditary SA respond to this therapy. An optimal response is related to reticulocytosis with blood hemoglobin levels returning to normal within 1 to 2 months and low free erythrocyte protoporphyrin levels also returning to normal. Morphologic abnormalities of cells (microcytosis), however, do not disappear, even in the presence of normal ALA synthase activity and hemoglobin. Hemoglobin levels also may increase in response to therapy but stabilize at less than normal levels. When a response to pyridoxine therapy is observed, lifelong maintenance therapy at a lowered dosage is instituted. Discontinuing therapy initiates a relapse. Individuals not responding to pyridoxine require blood transfusions to relieve symptoms and permit growth and development.
Individuals who demonstrate evidence of iron overload require iron depletion therapy to prevent or minimize organ damage. Phlebotomies are generally well tolerated and preferable for individuals who have a mild to moderate anemia without other complications, such as heart disease. Once all the stored iron is removed, maintenance phlebotomies are performed on a continuing basis. Individuals who have severe anemia and/or depend on transfusions become extremely overloaded with iron. When this occurs, iron chelation therapy with desferrioxamine is necessary to eliminate excess iron.
As stated, individuals with acquired SA infrequently respond to pyridoxine. Fortunately, these individuals are rarely incapacitated by SA. In the absence of abnormalities of other blood cells and without iron overload, progression takes place over many years. Transfusion and chelation therapy are the same as for hereditary SA when indicated.
Recent advances in treatment for SAs include prolonged administration of erythropoietin and stem cell transplant. Treatment with recombinant human erythropoietin improves anemia in 30% of those with myelodysplastic syndrome.16 Those with the subset of MDS identified as refractory anemia have the overall best response rate. Stem cell transplant has been found to successfully treat congenital SA; however, this treatment is in the early stages of use, and long-term efficacy has not yet been established. Death from SA is relatively rare and often secondary to complications, such as infection, bone marrow failure, liver failure, or cardiac failure or arrhythmias, or both.
Normocytic-Normochromic Anemias
Normocytic-normochromic anemias are characterized by erythrocytes that are relatively normal in size and hemoglobin content but insufficient in number. These anemias have no common etiology, pathologic mechanisms, or morphologic characteristics. They are less frequent than macrocytic-normochromic and microcytic-hypochromic anemias. NNAs include five distinct groups: aplastic, posthemorrhagic (acute blood loss), hemolytic, sickle cell, and anemia of chronic inflammation. (Sickle cell anemia is discussed in Chapter 28).
Aplastic Anemia
Aplastic anemia (AA) is a critical condition characterized by pancytopenia, a reduction or absence of all three blood cell types, resulting from failure or suppression of bone marrow to produce adequate amounts of blood cells (Figure 26-7). The rate or decline in the quantity of blood cells is related to their respective life span; thus RBCs (life span about 120 days) are last to demonstrate a reduction in numbers.
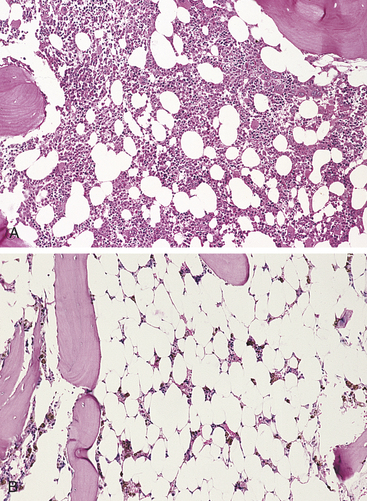
Figure 26-7 Aplastic anemia. A, Normal bone marrow of an adult. Hematopoietic cells account for approximately 40% of marrow’s cellularity. B, There is a marked reduction in hematopoietic cells with expansion of fat cells. (From Damjanov I, Linder J: Pathology: a color atlas, St Louis, 2000, Mosby.)
The incidence of AA is relatively rare (annual rate of 2 to 5 new cases per million per year). The incidence in developing countries is somewhat higher and is thought to be caused by unregulated use of and exposure to certain chemicals known to cause AA. The incidence is bimodal, with one peak occurring between 15 and 25 years of age and a second peak occurring in individuals older than age 60. AA is equally distributed between genders.
AAs are the most common type, with idiopathic AA (primary acquired) accounting for approximately 75% of all confirmed cases. Secondary AA, which accounts for approximately 15% of cases, is caused by a variety of known chemical agents and ionizing radiation. Chemical agents include benzene, arsenic, and multiple drugs, including chloramphenicol and alkylating and antimetabolite chemotherapeutic drugs (6-mercaptopurine, vincristine, and busulfan).17 Other drugs known to cause AA are identified in Table 26-4. The development of AA with use of these agents is generally dose related, and the effect can be controlled with diminished dosages. In other instances, AA might develop after the use of small amounts of these drugs (idiosyncratic), with the anemia following a severe, rapid, irreversible progression. Liver disease is also recognized as a cause of AA.
Table 26-4
Anemias Secondary to Drug Effects


X, Rare number of reported cases; XXXX, substantial number of reported cases; XX, XXX, intermediate number of reported cases; X∗, “pure red cell” aplasia.
AA is constitutional or familial in origin or is associated with one or more somatic abnormalities in approximately 5% to 10% of affected individuals. A subset of these is found to have defective telomerase RNA resulting in shortened telomeres. This abnormality also is found in some individuals with idiopathic AA.
Total body irradiation also causes AA and in certain instances may be used therapeutically for this effect. Infections are also known to cause AA, with viruses being the most common agent. Viral infections identified as causing AA include the human immunodeficiency virus (HIV) infections, Epstein-Barr virus, and hepatitis (non-A, non-B, non-C, and non-G virus). Persistent parvovirus B19 infection also has been identified as producing bone marrow failure resulting in AA. Parvovirus B19 has been identified as the cause of aplastic crisis in children who have sickle cell hemoglobinopathies and hereditary spherocytosis.
Another condition associated with AA is pure red cell aplasia (PRCA), in which only the RBCs are affected. PRCA is a rare disorder and has been associated with autoimmune, viral, and neoplastic (leukemias) disorders; infiltrative disorders of the bone marrow (myelofibrosis); renal failure; hepatitis; mononucleosis; and systemic lupus erythematosus.17 It also is a well recognized but infrequent complication of allogeneic bone marrow transplantation, particularly when there is donor-recipient ABO mismatch. A thymoma often is found in association with PRCA and is also present in Diamond-Blackfan syndrome, a congenital disorder.
A very small percentage of AA cases is linked to genetic alterations or predisposition. Fanconi anemia is a rare genetic anemia characterized by pancytopenia resulting from defects in DNA repair. This anemia develops early in life and is accompanied by multiple congenital anomalies.
PATHOPHYSIOLOGY The characteristic lesion of AA is a hypocellular bone marrow that has been replaced with fat. Most cases of AA result from an autoimmune disease directed against hematopoietic stem cells.18 The evidence supporting an autoimmune process includes the response of AA to immunosuppressive therapy including depletion of T-cells by antithymocyte antibodies. Cytotoxic T cells (Tc cells) appear to be the main culprits, although the causative antigen has yet to be identified. Th1 cytokines (involved in the differentiation of Tc cells), such as INF-γ and TNF-α, as well as cellular contact with Tc cells through FasL, induce apoptosis of CD34+ target cells, which includes most of the hematopoietic progenitors.
CLINICAL MANIFESTATIONS The onset of symptoms is insidious and related to the rapidity with which the bone marrow is destroyed and replaced.17 Approximately 50% of AA cases progress rapidly, with a high risk of death from overwhelming infection or bleeding. In some cases the rate of decline is slow and the individual may adapt progressively to a new level of hematologic function. This condition is referred to as hypoplastic anemia rather than aplastic anemia.
Initial symptoms depend on which cell line is affected. Rapidly progressing disease is usually associated with hypoxemia, pallor (occasionally with a brownish pigmentation of the skin), and weakness along with fever and dyspnea with rapidly developing signs of hemorrhaging if platelets are affected (e.g., unexplained bruising, nosebleeds, bleeding gums, bleeding in the GI tract, prolonged bleeding at sites of minor injury). A slower onset over weeks or months is characterized by progressive weakness and fatigue with developing signs of hemorrhaging. Major hemorrhage may occur from any organ; however, it is generally observed in the late stages and is often secondary to other events. Menorrhagia and purpura also may be evident; however, purpura is not necessarily a classic indication of AA and may not be representative of the degree of thrombocytopenia. In both rapid and slow onset AA, diminished leukocyte production may result in a progressive frequency and prolongation of infections.
Late manifestations of the condition include ulcerations of the mouth and pharynx or a low-grade cellulitis in the neck. Splenomegaly is extremely rare, and if present, other conditions that may imitate AA should be ruled out. Neurologic changes are only evident when hemorrhages have occurred within the system; however, some individuals have complained of paresthesias.
EVALUATION AND TREATMENT Diagnosis is made by blood tests and bone marrow biopsy. AA is suspected if levels of circulating erythrocytes, leukocytes, and platelets are diminished; a granulocyte count less than 500/μL, a platelet count less than 20,000/μL, and an absolute reticulocyte count less than or equal to 40 × 109/L. The diagnosis is confirmed by a bone marrow biopsy. The bone marrow usually has reduced cellularity (i.e., less than 25% normal cellularity). The morphology of the few remaining hematopoietic cells is usually normal. Occasionally the RBCs are macrocytic, with anisocytosis and poikilocytosis, and may appear immature.
Marrow biopsied from individuals with typical AA contains yellowish white material consisting mainly of fat, fibrous tissue, and lymphocytes. Pancytopenia is usually characterized by decreased stem cell and progenitor cell populations to approximately 1% or less of normal.
Up until the past 20 years, treatment involved determining the cause, removal of exposure to the potential causative agent, transfusion, and prevention and treatment of infection and hemorrhage. Stimulation of blood cell production also was used, and in some instances splenectomy was recommended. The prognosis with these forms of treatment was extremely poor. In acute cases, 25% of individuals succumbed within 4 months, and approximately 70% died within 5 years; only about 10% experienced complete recovery. Newer forms of treatment, such as bone marrow transplant (BMT), immunosuppression, and identification of high-risk individuals, has decreased mortality significantly.18
Bone marrow and, most recently, peripheral blood stem cell transplantation from a histocompatible sibling often cures the underlying bone marrow failure.19 Survival rates of 75% to 80% have been reported, and mortality rates within the first 100 days have decreased. Before transplantation the recipient usually received radiation or chemotherapy to deplete the bone marrow of disease-causing lymphocytes. Thus an unsuccessful transplantation may leave the recipient with a depleted immune system and an increased vulnerability to infection. Graft-versus-host (GVH) disease remains a risk and is a major contributor to premature death.20 Children demonstrate higher survival than adults.
For those individuals unable to undergo bone marrow transplantation or who lack a suitable sibling donor, immunosuppression remains the treatment of choice.20 Antithymocyte globulin (ATG) specifically suppresses lymphocytes, including those autoreactive lymphocytes destroying the bone marrow cells. Drugs like cyclosporine, which is often used in combination with ATG, broadly suppress the activity of immune cells. Response rates, that is, increased blood cell counts, of 40% to 50% may occur in individuals who receive ATG. The addition of cyclosporine has increased the response and survival rates to as much as 70% to 80%, with a 5-year survival rate between 80% and 90%. Cyclosporine as a single therapeutic agent is not as effective. Corticosteroids are often used concurrently with ATG and cyclosporine. Cyclophosphamide also has been used as an immunosuppressive agent and has produced the same effects as ATG; however, its use has been discontinued because of its toxicity. The addition of recombinant hematopoietic growth factors, such as granulocyte-macrophage colony-stimulating factor (GM-CSF), IL-6, and epoetin, to immunosuppressive therapy has produced significant additional benefit in both children and adults.
Immunosuppressive therapy is not without risk. Individuals receiving immunosuppressive therapy are at risk of experiencing treatment failure or late clonal/malignant conditions or both. Late clonal/malignant conditions include paroxysmal nocturnal hemoglobinuria (PNH), MDS, acute leukemia, or solid tumor. Although quite rare (less than 3%), administration of ATG may cause an anaphylactic reaction in some individuals.
Posthemorrhagic Anemia (Acute Blood Loss)
Posthemorrhagic anemia is a normocytic-normochromic anemia caused by acute blood loss. Initial manifestations of this event depend on the severity of blood loss. If blood loss is severe, the significant manifestations are related to loss of blood volume rather than loss of hemoglobin.
A normal, healthy young adult can tolerate a blood loss of 500 to 1000 ml (10% to 20% of volume) without experiencing any symptoms. Additional losses up to 1500 ml do not cause obvious symptoms if the individual is recumbent—symptoms appear only when assuming an upright position. When blood loss exceeds 1500 ml, symptoms are apparent even in a recumbent position (Table 26-5).
Table 26-5
Clinical Manifestations of Acute Blood Loss of Increasing Severity
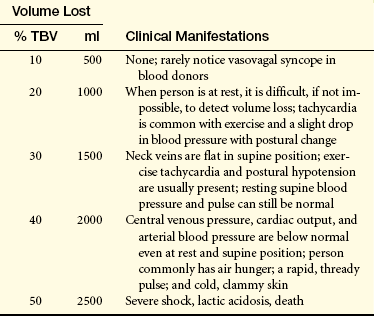
Adapted from Hillman RS: Acute blood loss anemia. In Beutler E et al, editors: Williams hematology, ed 5, New York, 1995, McGraw-Hill.Data based on a 70-kg person with a total blood volume of 5000 ml.
Volume loss reduces mean systemic filling pressure, resulting in decreased venous return. The initial manifestations (increased sympathetic nerve activation and a reduction in blood pressure, cardiac output, and central venous pressure) are caused by cardiovascular adaptations to blood volume depletion. If blood loss exceeds 2000 ml, severe shock, lactic acidosis, and death occur. (Shock is discussed in Chapter 46.)
If the acute blood loss is not severe (does not cause the preceding manifestations), complete recovery is possible. Within 24 hours of blood loss, lost plasma is replaced by mobilizing water and electrolytes from tissues and interstitial spaces into the vascular system. The hemodilution that results lowers the hematocrit; concurrently, there is often a rapid elevation of circulating neutrophils and platelets. Neutrophils can rise to levels between 10,000 and 30,000/μL within a few hours as a result of a shift of marginated leukocytes into the circulation and a release of leukocytes from the bone marrow. The platelet count can rise to levels of about 1million/μL. In severe blood loss, more immature cells—metamyelocytes, myelocytes, and nucleated red blood cells—may enter the circulation. Reduction in tissue oxygenation stimulates production of erythropoietin and increasing production of RBCs (reticulocytes) in the bone marrow. Iron recovery from destroyed RBCs may occur if the acute blood loss is internal; however, if blood is lost externally, iron stores may be depleted and erythropoiesis may be impeded. Hemorrhage that is chronic (occult [i.e., bleeding ulcer or neoplasm]) produces adaptations that are less prominent, but the individual may experience an IDA when iron reserves become depleted.
Initial treatment for acute blood loss is restoration of blood volume by intravenous administration of saline, dextran, albumin, or plasma. Large volume losses may require transfusion of fresh whole blood.
Successful therapy is first indicated by a return of erythrocytes to their normal size and shape. As the bone marrow begins to produce more erythrocytes, an increase in reticulocytes (10% to 15% after 7 days) is seen. Changes in the appearance of RBCs (polychromatophilia and macrocytosis) associated with reticulocytosis may give the impression that an underlying hemolytic process is occurring. A normal erythrocyte count is usually noted in 4 to 6 weeks, but hemoglobin restoration may take 6 to 8 weeks.
Hemolytic Anemia
The predominant event in hemolytic anemias is premature accelerated destruction of erythrocytes, either episodically or continuous. The consequences of the anemia are elevated levels of erythropoietin to induce accelerated production of erythrocytes and an increase in the products of hemoglobin catabolism.
Hemolytic anemias may be either congenital or acquired. Congenital hemolytic anemias result from intrinsic defects in erythrocytes, including the red cell membrane (e.g., hereditary spherocytosis, paroxysmal nocturnal hemoglobinuria), enzymatic pathways (e.g., glucose-6-phosphate dehydrogenase deficiency), and hemoglobin synthesis (e.g., the thalassemia syndromes, sickle cell anemia). (Glucose-6-phosphate dehydrogenase deficiency, thalassemia, and sickle cell disease are discussed in Chapter 28.) Acquired hemolytic anemias are usually immunologic (immune hemolytic anemias), such as RBC destruction caused by autoantibodies against erythrocyte antigens (e.g., autoimmune hemolytic anemia), isohemagglutinins (e.g., mismatched RBC transfusions), or allergic reactions against drug antigens adsorbed onto the erythrocyte surface (drug-induced hemolytic anemia). (Isohemagglutinins, erythrocyte antigens, autoantibodies, and allergic reactions are discussed in Chapter 8.) Acquired hemolytic anemia may also be secondary to erythrocyte damage caused by cardiac valve prostheses or by increased shear stresses in narrowed small vessels (e.g., during disseminated intravascular coagulation). Causes of acquired and hereditary hemolytic anemias are listed in Table 26-6.
Table 26-6
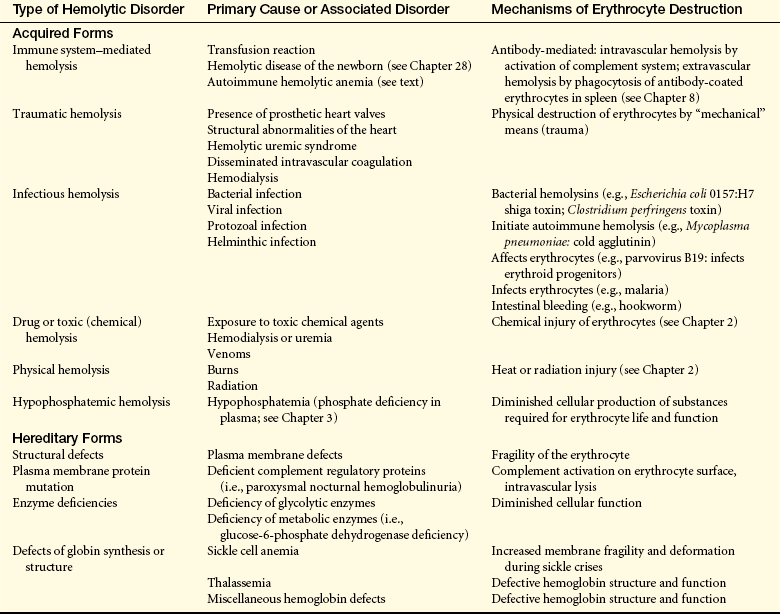
From Lee GR et al: Wintrobe’s clinical hematology, ed 9, Philadelphia, 1993, Lea & Febiger.
PATHOPHYSIOLOGY Hemolytic anemias can be classified by a variety of parameters, although no system is entirely satisfactory. Dividing these anemias into inherited or acquired is the preferred and most useful method. Pathophysiologic mechanisms also can be discussed in the context of where hemolysis occurs. Hemolysis occurs within blood vessels (intravascular) or lymphoid tissues (extravascular) that filter blood—that is, spleen and liver. Intravascular hemolysis is the least common and typically caused by physical destruction of RBCs in the circulation, frequently by antibody and complement. Extravascular hemolysis results from removal of damaged or opsonized erythrocytes by cells of the mononuclear phagocyte system (MPS). Erythrocytes continuously circulate through the spleen, passing through the thin-walled splenic cords into the splenic sinusoids, a sponge-like labyrinth of macrophages with long dendritic processes. Normally, RBCs are able to alter their shape to allow passage through openings in the splenic cords. Macrophages will phagocytose RBCs with structure alterations of the membrane surface or that have become more rigid are incapable of maneuvering through this network. In some cases, IgG antibodies or complement component C3b can coat erythrocytes without causing hemolysis, but can function as opsonins that are recognized by macrophages.
Paroxysmal nocturnal hemoglobinuria may be congenital or acquired secondarily to acquired aplastic anemia. The disease results from a mutation in the X-linked gene for phosphatidylinositol glycan—class A (PIG-A), which results in a defect in expression of glycosylphosphatidylinositol (GPI) in hematologic stem cells.21 GPI is a lipid anchor that is necessary for attachment of a large number of proteins to the plasma membrane. Several GPI-anchored proteins on erythrocytes are complement regulatory proteins, including CD55 and CD59. Normally low levels of complement are activated on cell surfaces through the alternative pathway (see Chapter 6). Erythrocytes are protected from complement-mediated damage by CD55, which accelerates the degradation of any C3 convertase that forms on the cell surface, and by CD59, which prevents C9 aggregation and pore formation by the the membrane attack complex. Thus RBCs deficient in CD55 and CD59 undergo complement-mediated intravascular lysis and release of hemoglobin. In addition to anemia and hemoglobinuria, affected individuals also present with severe fatigue, abdominal pain, and thrombosis.22 The cause of death is usually thrombosis of the abdominal or cerebral veins.23 Thrombosis most likely results from a depletion of vascular nitric oxide (NO) by free hemoglobin, which has a high affinity for NO. The result is dysregulation of normal hemostasis and increased platelet vascular adherence and clot formation (see Chapter 25).
Autoimmune hemolytic anemias (AIHAs) are acquired disorders caused by autoantibodies against antigens normally on the surface of erythrocytes.24 Three types of AIHAs have been described: (1) warm reactive antibody type, (2) cold agglutinin type, and (3) cold hemolysin type (paroxysmal cold hemoglobinuria). This classification is based on the optimal temperature at which the antibody binds to erythrocytes and the mechanism of RBC destruction.
Warm autoimmune hemolytic anemia is uncommon (incidence of about 1 per 80,000 population annually), although it is the most common form of AIHA (80 to 90% of cases), and generally occurs in individuals older than the age of 40.25 Approximately half of the cases are secondary to other diseases, especially lymphomas but also chronic lymphocytic leukemia, other neoplastic disorders, or systemic lupus erythematosus (SLE). The anemia is caused by IgG that binds optimally to erythrocytes at normal body temperature (98.6° F, 37° C). Most cases are related to antibody against Rh-related antigens other than the D epitope (Rh antigens are discussed in Chapter 8). The spectrum of antibody specificities includes antibodies against the e, E, or c antigens of the Rh complex. Other cases are caused by IgG antibodies against erythrocyte antigens outside the Rh complex and include antibodies against Wrb, Ena, the Kell blood group, and many others. The warm reactive IgG antibodies usually do not activate complement because of the rather sparse distribution of antigens on the RBC surface. (Activation of complement by antibody is discussed in Chapters 6, 7, and 8.) RBC destruction is caused by extravascular processes. The IgG-coated RBCs bind to the Fc receptors on monocytes and splenic macrophages and are removed by phagocytosis.
Cold agglutinin autoimmune hemolytic anemia is mediated by immunoglobulin M (IgM) antibodies and occurs less often than warm antibody hemolysis, affecting mostly the middle-aged and older adults.26 Cold antibodies optimally bind to RBCs at colder temperatures (lower than 31° C [87.8° F])with maximal binding capacity at 4° C (39.2° F). Cold agglutinin autoantibodies may appear acutely during recovery of certain infectious disorders, particularly infectious mononucleosis and mycoplasma pneumonia. With these conditions, the individuals are usually younger than those with primary disease; the anemia may be severe but may be self-limiting. Chronic cold agglutinin AIHAs also can occur in association with lymphoid neoplasm and other unknown or idiopathic conditions.27
The IgM autoantibody is usually monoclonal and directed against erythrocyte carbohydrate antigens of the I system (i.e., i, I) or the P system (i.e., Pr).26 In the colder areas of the body, particularly during cold weather (e.g., fingers, toes, nose, ears, exposed skin), the IgM autoantibodies bind to circulating erythrocytes. The IgM is rapidly released when the blood recirculates and warms. IgM is an extremely efficient activator of complement, resulting in the stable deposition of C3b on the cell surface. If an adequate amount of complement is deposited, the erythrocytes become vulnerable to recognition and rapid phagocytosis by mononuclear phagocytes in the liver and spleen (also see Chapter 8). The severity of hemolysis is variable and may result in a progressive chronic anemia. If the level of antibody is high or has particularly strong binding, hemagglutination may occur in the capillaries of exposed sites, such as fingers, toes, and ears, when temperatures are below 30° C (86° F). Obstruction of blood flow caused by RBC agglutination may lead to a bluish discoloration of the skin (acrocyanosis) that resolves as the skin is warmed. Prolonged exposure to the cold may lead to gangrene. Cold hemolysin autoimmune hemolytic anemia (paroxysmal cold hemoglobinuria) is a disorder in which exposure to cold initiates acute and severe intravascular hemolysis that unlike cold agglutinin anemia results in hemoglobulinuria.26 The chronic form of this anemia is extremely rare, but an acute form of paroxysmal cold hemoglobinuria is frequently observed (30% to 40% of cases) in AIHA of childhood. The acute form occurs primarily in young children younger than the age of 10 and is usually preceded by an upper respiratory tract infection or flulike symptoms. Infections with measles, mumps, Mycoplasma (pneumonia), and Varicella have also been linked to an onset of paroxysmal cold hemoglobinuria. The anemia may be rapidly progressing and severe and associated with fever, reddish brown urine, hemoglobinuria, jaundice, abdominal pains, and pallor, with about 25% of individuals presenting with hepatomegaly and splenomegaly.
Paroxysmal cold hemoglobinuria is generally caused by IgG autoantibodies against the P blood group antigen.26 Antibody binding occurs in the colder portions of the body. As the erythrocyte recirculates, enzymes of the complement cascade are activated, and cells are destroyed in the vasculature by complement-mediated lysis. The involved antibody, also called Donath-Landsteiner antibody, was first recognized in individuals with anemia secondary to chronic syphilis infection. A transfusion reaction is an example of alloimmune hemolytic anemia (also see Chapter 8). Transfused blood that is mismatched for ABO antigens is destroyed by preexisting isohemagglutinins in the recipient. Isohemagglutinins, which are generally IgM antibodies, activate complement, resulting in a rapid intravascular hemolysis. The individual may immediately experience fever, chills, dyspnea, and hypotension and may progress to shock. In some cases the hemolytic reaction may be delayed and develop 3 to 10 days after transfusion. The delayed reaction is caused by a low titer of preexisting antibodies to minor RBC antigens.
Drug-induced hemolytic anemia is a form of immune hemolytic anemia usually resulting from an allergic reaction against foreign antigens (e.g., antibiotics) (also see Chapter 8). Usually the drug is small molecular weight and functions as a hapten and binds to proteins on the surface of erythrocytes. This is sometimes called the hapten model and is based on anemia caused by penicillin, cephalosporins (more than 90% of cases), and, very recently, hydrocortisone (Figure 26-8).28 IgG antibody against the drug or against the unique antigen formed by the interface of the drug and erythrocyte protein is formed and binds to the erythrocyte at normal body temperature. Hemolysis is usually extravascular because the opsonized RBCs are removed by phagocytes in the spleen and liver, although complement-dependent intravascular hemolysis may occur in some individuals. This form of drug-induced anemia usually follows a large intravenous infusion of an antibiotic and occurs 1 to 2 weeks after the initiation of therapy. Cessation of administration of the drug results in rapid resolution of the anemia.
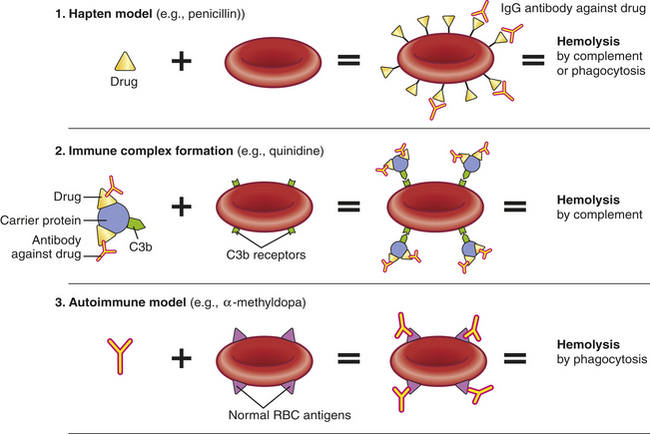
Figure 26-8 Models of drug-induced hemolytic anemia. (See discussion in text). IgG, Immunoglobulin G; RBC, red blood cell.
The erythrocyte plasma membrane contains receptors of components of the complement system, such as C3b, and can bind circulating immune complexes that have activated the complement cascade (see Chapter 8 for a discussion of immune complexes). This forms the basis for the immune complex model of drug-induced hemolytic anemia and was first described for anemia resulting from administration of the drug quinidine. The drug or a metabolite of the drug, both of which are haptens, initially binds to plasma proteins and becomes immunogenic (see Chapter 7). The circulating drug/protein complex reacts with the resultant antibody (usually IgM, although IgG complexes have also been described) and activate the complement system resulting in the deposition of C3b into the complex. Binding of the immune complexes to the erythrocyte surface results in further complement activation and intravascular hemolysis. This mechanism also may explain some of the anemia observed in other immune complex conditions, such as SLE.
In at least one instance, administration of the drug α-methyldopa induces an immune response against normal erythrocyte antigens and thus initiates a true AIHA (autoantibody model). The autoantibody is usually against Rh blood group antigens. It is estimated that 20% of individuals taking α-methyldopa develop detectable antibodies, but only 1% actually develop clinically significant anemia. The mechanism by which α-methyldopa induces autoantibodies against the erythrocytes is unknown.
CLINICAL MANIFESTATIONS The presence and severity of signs and symptoms of hemolytic anemia depend on the degree of anemia and hemolysis and the success of compensatory erythropoiesis. Adaptation to red cell destruction is facilitated by increased red cell production. Bone marrow is capable of increasing red cell production up to eight times its normal rate. Accelerated RBC production that is incapable of keeping up with destruction develops into a true hemolytic anemia.
The severity of anemia varies widely from individual to individual, even in individuals who have the same illness. Severe disease is commonly diagnosed shortly after birth or within the first year of life. Mild to moderate anemia is more common because the shortened erythrocyte survival time is offset by increased erythropoiesis. Some individuals have no symptoms of anemia, and the underlying hemolytic process remains undetected unless some other complication develops during the course of the disease.
Jaundice (icterus) is present when heme destruction exceeds the liver’s ability to conjugate and excrete bilirubin. Jaundice is first noticed in the neonatal period. Children and adults with congenital hemolytic anemia may not have icterus, or it may be mild enough that it goes unnoticed. In some individuals, faint scleral icterus may be the only indication of hemolytic disease.
Acute conditions that disrupt the delicate equilibrium of accelerated erythropoiesis and RBC destruction may precipitate a crisis. The most common type of crisis is aplastic and results from failure of bone marrow RBC production. The most common cause of aplastic crisis is human parvovirus B19 infection.
Commonly individuals with congenital hemolytic disorders demonstrate splenomegaly, which is often only mild in nature. In some cases the spleen may become quite enlarged and may cause discovery of the underlying hemolytic disorder. Another underlying condition that may be the cause of inadvertently determining the presence of the anemic disorder is the development of gallstones.
Children who have hemolytic anemia often demonstrate skeletal abnormalities caused by expansion of erythroid bone marrow during the active phase of growth and development. These alterations are more pronounced in the bony structures of the face and skull and may result in pathologic fractures (see Chapter 28). Cardiovascular and respiratory manifestations vary with the degree of anemia. In spite of the disorder being characterized as hemolytic in nature, thromboembolism may occur. Pulmonary embolism is a common finding during autopsies of individuals with immune hemolytic anemia.
EVALUATION AND TREATMENT Diagnosis is based on clinical manifestations, bone marrow studies, and blood tests (see Table 26-3). Abnormally increased numbers of erythrocyte stem cells are found in the marrow, a finding termed erythroid hyperplasia. Accelerated erythropoiesis causes large numbers of fragile and immature erythrocytes (stem cells and reticulocytes) to be released prematurely into the circulation. These cells are observed in blood smears. If the bone marrow is able to consistently maintain adequate compensation, the hemoglobin may remain stable. The mean corpuscular volume, however, may be decreased in the presence of reticulocytes. A blood smear is helpful in determining the presence of spherocytes or schistocytes, as well as examining white blood cells and platelets for coexisting hematologic or malignant conditions.
Acquired hemolytic anemias are treated by removing the cause or treating the underlying disorder. Acute fulminating hemolytic anemia (hemolytic crisis) is treated with fluid and electrolyte replacement to prevent shock and renal damage, which may be caused by RBC debris clogging the kidney tubules. Transfusions of blood products sometimes are given. Splenectomy is performed if the spleen is the major site of hemolysis and splenomegaly is significant.
Folate also is used in treating chronic hemolytic disease to prevent megaloblastic crisis because long-term erythrocyte turnover increases folate requirements. The use of monoclonal antibody therapy has proven beneficial. Rituximab is a monoclonal antibody directed against the CD20 antigen and specifically depletes or suppresses B cells throughout the body. CD20 is expressed on most cells in the B-cell lineage, except hematopoietic stem cells and plasma cells. Rituximab is used to treat a variety of leukemias and lymphomas and autoimmune diseases (e.g., rheumatoid arthritis, idiopathic thrombocytopenia, multiple sclerosis, type 1 diabetes mellitus, systemic lupus erythematosus). It is used successfully in several types of immune hemolytic anemias.29 Eculizumab is a monoclonal antibody against complement protein C5, which blocks the enzymatic activation of C5 to C5a and C5b.23 Thus treatment with eculizumab prevents the formation of the membrane attack complex and complement-mediated cell lysis (see Chapter 6). Eculizumab has been used in multiple trials to treat paroxysmal nocturnal hemoglobinuria.30 Treatment has rapidly decreased serum markers for intravascular hemolysis, the need for transfusions, and the risk for thrombosis and diminished many other symptoms, including fatigue and abdominal pains.30,31 Blockage of C5 activation mimics individuals who have congenital deficiencies in C5 (see Chapter 8). Lack of C5 increases the risk for disseminated infections with Neisseria sp., particularly Neisseria meningitidis. Thus immunization with the meningococcal vaccine is recommended before treatment with eculizumab.31
Anemia of Chronic Disease
Anemia of chronic disease (ACD) is a mild to moderate anemia resulting from decreased erythropoiesis in individuals with conditions of chronic systemic disease or inflammation (e.g., cancer, infections, autoimmune diseases).11 These conditions include acquired immunodeficiency disease (AIDS), rheumatoid arthritis, SLE, malaria, acute and chronic hepatitis, and chronic renal failure. This form of anemia also is commonly noted in the presence of congestive heart failure (CHF). The anemia develops after 1 to 2 months of disease activity. The severity of anemia is related to that of the underlying disorder. Individuals may be asymptomatic, or the anemia may be a coincidental clinical finding. Morphologically, ACD is initially normocytic-normochromic, but as the condition progresses it becomes hypochromic and microcytic. ACD is characterized by normal iron stores with low circulating iron (less than 60 mcg/dl).
ACD is one of the most common conditions encountered in medicine and is probably only secondary to IDA in overall incidence. In individuals older than age 65 anemia is present in 10% of those who live in the community and more than 50% of those who reside in nursing homes3; two thirds of which is ACD or unexplained anemia. Older adults may be predisposed to ACD related to age-associate hematopoietic restriction. Additionally, older adults demonstrate increased concentrations of inflammatory cytokines, which play a significant role in the development of ACD. Older adult who present with characteristics of ACD without an underlying malignancy or inflammatory condition are described as having primary defective iron-utilization syndrome.
PATHOPHYSIOLOGY ACD results from a combination of (1) decreased erythrocyte life span, (2) suppressed production of erythropoietin, (3) ineffective bone marrow erythroid progenitor response to erythropoietin, and (4) altered iron metabolism and iron sequestration in macrophages.32–35 During chronic inflammation a large variety of cytokines are released by lymphocytes and macrophages, including TNF-α, INF-γ, interleukin-1β (IL-1β), (IL-3, and IL-6) (also see Chapter 7).32,35
The precise mechanism by which RBC destruction is mediated remains unclear, but it is thought to result from activation of macrophages by TNF-α. Macrophages are responsible for removal of senescent or damaged erythrocytes. Increased erythrocyte sensitivity to phagocytosis may be a result of bacterial toxins or factors released from tumors. The erythropoietic defect in ACD is failure to increase erythropoiesis in response to decreased numbers of RBCs. In part, decreased erythropoiesis results from diminished production of erythropoietin by the kidneys. The kidney is frequently affected by chronic inflammatory processes through the production of circulating immune complexes and other factors that deposit in the kidney and activate secondary inflammatory mechanisms. In addition, the failure in erythropoiesis may reflect decreased responsiveness of erythroid progenitors to erythropoietin. Decreased availability of iron (discussed following) would diminish the rate of erythropoiesis. Proliferation of erythroid cells is also inhibited by proinflammatory cytokines, especially TNF-α, INF-γ, and IL-1β. TNF-α also directly induces apoptosis of erythroid progenitors, thus diminishing the number of responsive cells. In individuals who had anemia secondary to rheumatoid arthritis, the bone marrow contained elevated levels of IL-3, which correlated with diminished expression of integrins on the surface of cells of the erythroid series.33 Loss of integrins may prevent adequate interaction with stromal cells and matrix proteins and inhibit erythropoiesis.
Impaired iron metabolism is partially the result of iron sequestration. IL-6 in particular affects hepatocytes and increases the release of the peptide hepcidin, which regulates the activity of ferroportin. Ferroportin is the primary transporter for the export of iron from macrophages to the plasma. Increased levels of hepcidin result in decreased ferroportin activity and suppression of iron release11 (Figure 26-9). Additionally, normal iron transport by transferrin may be decreased as a result of competitive iron binding by inflammation-related increases in circulating lactoferrin and apoferritin. Lactoferrin is a member of the transferring family of nonheme iron-binding glycoproteins, and under normal conditions is present in the blood in only small amounts. During inflammation neutrophils release lactoferrin to bind iron and reduce its availability for bacteria. However, the affinity of iron for lactoferrin is 260 times greater than for transferrin. Lactoferrin-bound iron is removed by the mononuclear-phagocyte system and converted into ferritin, the storage form of iron. Apoferritin also has a higher affinity for iron and affects available iron in a similar manner.
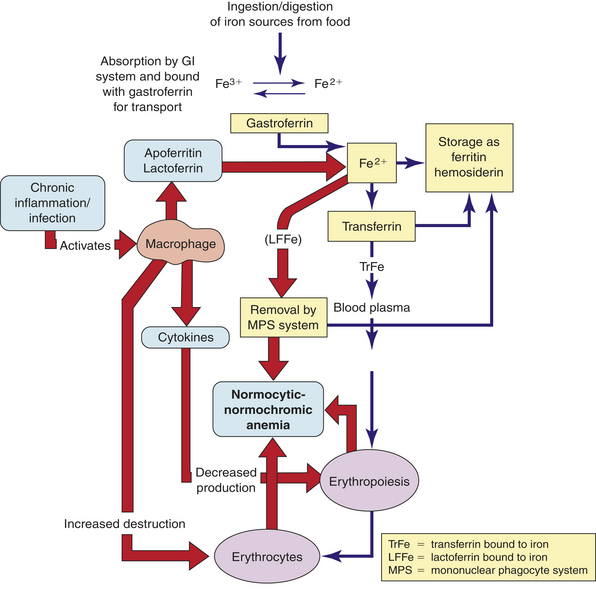
Figure 26-9 Pathophysiology of anemia of chronic disease. Normal iron metabolism is indicated by the narrow arrows. Abnormal mechanisms that are instrumental in the development of anemia of chronic inflammation are indicated by thick arrows. (See discussion in text.) GI, Gastrointestinal.
CLINICAL MANIFESTATIONS ACD has fewer and milder manifestations than most other anemias. The anemia is usually in the mild to moderate range. If hemoglobin levels drop significantly, clinical manifestations of IDA appear.
EVALUATION AND TREATMENT The most significant finding of ACD is a very-high total body iron storage, although inadequate iron is available in the bone marrow for erythropoiesis. Very often the first indication of ACD is a failure to respond to conventional iron replacement therapy. Levels of erythropoietin are generally lower than expected for the degree of anemia. The affected individuals also frequently present with low or normal total iron binding capacity (TIBC), normal or high serum ferritin levels, and low concentrations of soluble transferrin receptor (blood test findings are listed in Table 26-3). Occasionally it may be difficult to differentiate ACD from IDA; however, measurement of sTfR may be useful. Levels of sTfR do not respond to iron supplementation in ACD but do so in IDA.
Use of erythropoietin in treatment of ACD associated with arthritis, malignancies, and AIDS has met with limited success. Transfusion of critically ill individuals may worsen the outcome and increase morbidity and mortality.36 The principal treatment is alleviation of the underlying disorder. Individuals who have ACD but demonstrate no evidence of inflammatory or infectious conditions are screened for the presence of malignancies.
MYELOPROLIFERATIVE RED BLOOD CELL DISORDERS (POLYCYTHEMIA)
Hematologic dysfunction results from an overproduction of cells as well as a deficiency. One or more hematopoietic lines may be overproduced in the marrow in response to exogenous (e.g., radiation, drugs) or endogenous (physiologic compensatory responses, immune disorders) signals. Excessive RBC production is classified as polycythemia. Polycythemia exists in two forms: relative and absolute. Relative polycythemia results from hemoconcentration of the blood associated with dehydration that may be caused by decreased water intake, diarrhea, excessive vomiting, or increased use of diuretics. It is usually of minor consequence development and resolves with fluid administration or treatment of the underlying condition.
Absolute polycythemia consists of two forms: primary or secondary. Secondary polycythemia, the more common type, is caused by an increase in erythropoietin as a physiologic response to chronic hypoxia. This hypoxia is noted in individuals who live at higher altitudes (i.e., above 10,000 ft), smokers with increased levels of carbon monoxide (CO), and individuals with chronic obstructive pulmonary disease and congestive heart failure. Abnormal types of hemoglobin (e.g., HbSan Diego or HbChesapeake), which have a greater affinity for oxygen, also cause secondary polycythemia, as does secretion of erythropoietin by certain tumors (e.g., renal cell carcinoma, hepatoma, cerebral hemiangioblastoma).
Primary polycythemia (known as polycythemia vera) is one of several disorders collectively known as chronic myeloproliferative disorders (CMPDs).37 Others in this group include essential thrombocytosis, chronic idiopathic myelofibrosis, chronic myeloid leukemia, chronic neutrophilic leukemia, and chronic eosinophilic leukemia.38 All of these disorders result from abnormal regulation of the multipotent hematopoietic stem cells. The major characteristics shared by these disorders are (1) involvement of a multipotent hematopoietic progenitor cell; (2) overproduction of one or more of the formed elements of the blood in the absence of a defined stimulus; (3) dominance of a transformed progenitor cell over the nontransformed progenitor cells; (4) marrow hypercellularity or fibrosis; (5) cytogenetic abnormalities; (6) predisposition to thrombus formation and hemorrhage; and (7) spontaneous transformation to acute leukemia. Determining a precise distinction between the CMPDs is difficult if not impossible because of overlapping clinical features and a lack of specific molecular markers. As a result, diagnosis is quite challenging.
PATHOPHYSIOLOGY Polycythemia vera (PV) is a neoplastic, nonmalignant condition characterized by an increase in red blood cells, with increased levels of white blood cells (leukocytosis) and platelets (thrombocytosis), and splenomegaly. Erythrocytosis is the essential component of PV. Clonal proliferation of erythroid progenitors occurs in the bone marrow independent of erythropoietin, although the cells express a normal erythropoietin receptor. However, more than 95% of PV-affected individuals possess an acquired mutation in Janus kinase 2 (JAK2).39 JAK2 increases the activity of the erythropoietin receptor and is self-regulatory so that JAK2 activity diminishes over time. The mutation associated with PV negates the self-regulatory activity of JAK2 so that the erythropoietin receptor is constitutively active regardless of the level of erythropoietin.40
CLINICAL MANIFESTATIONS PV is relatively rare, with an estimated incidence of 2.3 per 100,000 individuals, peak incidence is between the ages of 60 and 80 years of age, with a median incidence of 55 to 60. However, PV has been observed in individuals younger than the age of 40. Males are twice as likely to develop PV. PV is more common in whites, particularly those of Northern European Jewish ancestry, than in blacks. PV is rarely found in children or in multiple members of a single family; however, an autosomal dominant form exists that is characterized by increased production of erythropoietin.
Almost every individual initially presents with an enlarged spleen, frequently with abdominal pain and discomfort. As the disease progresses many of the symptoms are related to the increased blood cellularity and viscosity. Increased viscosity, as well as thrombocythemia and increased platelet dysfunction, leads to a hypercoagulable state with formation of venous and arterial thrombosis and vessel occlusion.41 Thrombi with occlusion of major and minor blood vessels lead to tissue and/or organ ischemia or infarction (tissue injury and/or death). Extreme thrombocythemia (greater than 1,5000,000/mm3 of blood) increases the risk for excessive bleeding, rather than thrombosis.
Increased blood viscosity results in a variety of circulatory alterations in PV, such as plethora (ruddy, red color of the face, hands, feet, ears, and mucous membranes) and engorgement of the retinal and cerebral vessels. Individuals also may experience headache, drowsiness, delirium, mania, psychotic depression, chorea, and visual disturbances. The risk for death from cerebral thrombosis is increased approximately fivefold in individuals with PV.
Cardiac workload and output remain essentially unchanged; however, increased blood volume may lead to elevated blood pressure. Coronary blood flow may be affected and lead to the onset of angina, although myocardial infarctions caused by PV are relatively rare. Other evidence of cardiovascular involvement is the development of Raynaud phenomenon and thromboangiitis obliterans.
Additionally, gastrointestinal gastric and duodenal thrombosis may occur with resultant hemorrhaging. The development of mesenteric thrombosis requires immediate medical intervention. Splenomegaly and hepatomegaly result from pooling of blood in these organs; consequently individuals may develop portal hypertension. The respiratory system, generally not affected by PV, becomes involved if thrombosis and embolization occur.
A unique feature of PV, one that is potentially instrumental in diagnosis, is extreme, painful itching that is intensified on exposure to water (aquagenic pruritus). The intensity of the itching is related to the concentration of mast cells in the skin, but is generally not responsive to antihistamines or topical lotions.
EVALUATION AND TREATMENT PV is frequently suspected based on clinical features, such as a thrombotic event, splenomegaly, or aquagenic pruritus. Diagnosis of PV is made from blood and laboratory findings (Box 26-2). Blood manifestations include an absolute increase in RBCs, with hematocrits ranging from 18 to 24 g/dl and RBC counts of 7 to 10 × 1012. Absolute total blood volume also is increased as well as possible moderate increases of white blood cells and platelets. Erythrocytes appear normal, but anisocytosis may occur occasionally. Bone marrow examination may be done, but is not very valuable unless performed in association with cytogenetic and molecular studies for relevant mutations in JAK2.42 Typically the marrow is hypercellular but not in such a manner as to differentiate it from other myeloproliferative disorders. Additional observations on abnormal megakaryocyte morphologies, formation of clusters of abnormal cells, and increased fibrosis add more specificity and usefulness to bone marrow analysis. Elevated serum levels of erythropoietin are not helpful because most individuals with PV have normal or low levels.
Treatment of PV is challenging and directed toward minimizing the risk of thrombosis and preventing progression to myelofibrosis and acute leukemia. In low-risk individuals (e.g., those younger than age 60 or with no history of thrombosis and without risk factors for cardiovascular disease), the recommended therapy is phlebotomy and low-dose aspirin.39 The guideline for phlebotomy (300 to 500 ml at a time to reduce erythrocytosis and blood volume) is maintenance of the hematocrit at less than 45%. The initial phlebotomies are performed two or three times a week until the hematocrit drops, and are repeated every 3 to 4 months to maintain a safe hematocrit. Aspirin is used for its antithrombotic (decrease in thromboxane) properties.
The recommended therapy for those at high risk includes use of hydroxyurea, an antimetabolite that blocks DNA synthesis and reduces vascular cellularity. Unlike other similar drugs, hydroxyurea reduces the risk of thrombotic complications, but does not increase the risk for developing leukemia.39 INF-α has been used when other forms of treatment have failed. Interferon inhibits the growth of the abnormal progenitors and inhibits the actions of cytokines that may lead to the development of myelofibrosis. Therapy with INF-α is complicated by its proinflammatory activities, thus fever, flulike symptoms, and more severe complications are common. INF-α is not consistently effective in high-risk adult cases, but is considered in individuals who are intolerant to hydroxyurea, younger individuals, and pregnancy.
Radioactive phosphorus (32P) has been used to suppress erythropoiesis. It is generally effective for an extended time, and as many as 18 months may elapse between treatments. Side effects of 32P treatment include suppression of hematopoiesis resulting in anemia, leukopenia, or thrombocytopenia. Development of acute leukemia is also a major side effect of 32P, occurring after 7 or more years of treatment, making this therapy more useful in older adults.
Without proper treatment, 50% of individuals with PV die within 18 months of the onset of initial symptoms. The primary cause of death is thrombosis, which is more prevalent in older individuals and those with prior vascular complications. Hemorrhage is rare but more common in individuals with high platelet counts and those taking antiplatelet drugs. Conversion to acute myeloid leukemia (AML) occurs in 10% of individuals within 15 years, increasing to 50% within 20 years. This leukemia is generally refractory to conventional treatment and remains a significant potential adverse outcome of PV. Conversion to AML is more common in individuals who were treated with alkylating agents, whereas those treated only with INF or hydroxyurea had the same incidence of conversion as those who received no treatment. Although PV is a chronic disorder and remissions occur with appropriate therapy, the prevention of significant morbidity and mortality is possible, and survival for 10 to 15 years is common.
Absolute polycythemia 1009
Anemia 989
Anemia of chronic disease (ACD) 1007
Anisocytosis 989
Aplastic anemia (AA) 1000
Apoferritin 1008
Autoimmune hemolytic anemia (AIHA) 1005
Cold agglutinin autoimmune hemolytic anemia 1005
Cold hemolysin autoimmune hemolytic anemia (paroxysmal cold hemoglobinuria) 1005
Dimorphism 998
Drug-induced hemolytic anemia 1006
Erythropoietic hemochromatosis 999
Fanconi anemia 1002
Folate (folic acid) 995
Hemolytic anemia 1003
Hypoplastic anemia 999
Hypoxemia 990
Intrinsic factor (IF) 993
Iron deficiency anemia (IDA) 995
Lactoferrin 1008
Macrocytic anemia (megaloblastic anemia) 990
Microcytic-hypochromic anemia 995
Myelodysplastic syndrome (MDS) 998
Normocytic-normochromic anemia 1000
Pancytopenia 1000
Paroxysmal nocturnal hemoglobinuria 1005
Pernicious anemia (PA) 991
Poikilocytosis 989
Polycythemia 1008
Polycythemia vera (PV) 1009
Posthemorrhagic anemia 1003
Pure red cell aplasia (PRCA) 1001
Relative polycythemia 1008
Ringed sideroblast 998
Sideroblastic anemia (SA) 998
Warm autoimmune hemolytic anemia 1005
REFERENCES
1. Aslinia, F., Mazza, J.J., Yale, S.H. Megaloblastic anemia and other causes of macrocytosis. Clin Med Res. 2006;4(3):236–241.
2. Gaskell, H., et al. Prevalence of anaemia in older persons: systemic review. BMC Geriat. 2008;8(January 14):1–8.
3. Patel, K.V. Epidemiology of anemia in older adults. Semin Hematol. 2008;45(4):210–217.
4. Carmel, R. Nutritional anemias and the elderly. Semin Hematol. 2008;45(4):225–234.
5. Vojdani, A. Antibodies as predictors of complex autoimmune diseases. Int J Immunopathol Pharmacol. 2008;21(2):267–278.
6. Desai, H.G., Gupte, P.A. Helicobacter pylori link to pernicious anaemia. J Assoc Physicians India. 2007;55(12):857–859.
7. West, A.R., Oates, P.S. Mechanisms of heme iron absorption: current questions and controversies. World J Gastroenterol. 2008;14(26):4101–4110.
8. Killip, S., Bennett, J.M., Chambers, M.D. Iron deficiency anemia. Am Fam Physician. 2007;75(5):671–678.
9. Alleyne, M., Horne, M.K., Miller, J.L. Individualized treatment for iron-deficiency anemia in adults. Am J Med. 2008;121(11):943–948.
10. Finsterer, J. Hematological manifestations of primary mitochondrial disorders. Acta Haematol. 2007;118(2):88–98.
11. Edison, E.S., Bajel, A., Chandy, M. Iron homeostasis: new players, newer insights. Eur J Haematol. 2008;81(6):411–424.
12. Camaschella, C. Recent advances in the understanding of inherited sideroblastic anaemia. Brit J Haematol. 2008;143(1):27–38.
13. Koppel, A., Schiller, G. Myelodysplastic syndrome: an update on diagnosis and therapy. Curr Oncol Rep. 2008;10(5):372–378.
14. Dreyfus, F. The deleterious effects of iron overload in patients with myelodysplastic syndromes. Blood Rev. 2008;22(Suppl 2):S29–S34.
15. Malcovati, L., Nimer, S.D. Myelodysplastic syndromes: diagnosis and staging. Cancer Contr. 2008;15(Supp):4–13.
16. Moyo, V., et al. Erythropoiesis-stimulating agents in the treatment of anemia in myeodysplastic syndromes: a meta-analysis. Ann Hematol. 2008;87(7):527–536.
17. Young, N.S., Calado, R.T., Scheinberg, P. Current concepts in the pathophysiology and treatment of aplastic anemia. Blood. 2006;108(8):2509–2519.
18. Bacigalupo, A. Aplastic anemia: pathogenesis and treatment. Hematol 2007. 2007;2007:23–28.
19. Armand, P., Antin, J.H. Allogeneic stem cell transplantation for aplastic anemia. Biol Blood Marrow Transplant. 2007;13(5):505–516.
20. Davies, J.K., Guinan, E.C. An update on the management of severe idiopathic aplastic anaemia in children. Brit J Haematol. 2007;136(4):549–564.
21. Brodsky, R.A. Narrative review: paroxysmal nocturnal hemoglobinuria: the physiology of complement-related hemolytic anemia. Ann Intern Med. 2008;148(8):587–595.
22. Ziakas, P.D., Poulou, L.S., Pomont, A. Thrombosis in paroxysmal nocturnal hemoglobinuria at a glance: a clinical review. Curr Vasc Pharmacol. 2008;6(4):347–353.
23. Brodsky, R. Advances in the diagnosis and therapy of paroxysmal nocturnal hemoglobinuria. Blood Rev. 2008;22(2):65–74.
24. Valent, P., Lechner, K. Diagnosis and treatment of autoimmune haemolytic anaemias in adults: a clinical review. Wien Klin Wochenschr. 120(5–6), 2008.
25. Packman, C.H. Hemolytic anemia due to warm autoantibodies. Blood Rev. 2008;22(1):17–31.
26. Petz, L.D. Cold antibody autoimmune hemolytic anemias. Blood Rev. 2008;22(1):1–15.
27. Berentsen, S., Belske, K., Tjønnfjord, G.E. Primary chronic cold agglutinin disease: an update on pathogenesis, clinical features and therapy. Hematology. 2007;12(5):361–370.
28. Martinengo, M., et al. The first case of drug-induced immune hemolytic anemia due to hydrocortisone. Transfusion. 2008;48(9):1925–1929.
29. Garvey, B. Rituximab in the treatment of autoimmune haematological disorders. Brit J Haematol. 2008;141(2):149–169.
30. Charneski, L., Patel, P.N. Eculizumab in paroxysmal nocturnal haemoglobinuria. Drugs. 2008;68(10):1341–1346.
31. Hill, A. Update on eculizumab for the treatment of paroxysmal nocturnal hemoglobinuria. Clin Adv Hematol Oncol. 2008;6(7):499–500.
32. Handelman, G.J., Levin, N.W. Iron and anemia in human biology: a review of mechanisms. Heart Fail Rev. 2008;13(4):393–404.
33. Jaworski J et al: Decreased expression of integrins by hematopoietic cells in patients with rheumatoid arthritis and anemia: relationship with bone marrow cytokine levels, J Investig Allerg Clin Immunol 18(1):17-21.
34. Foley, R.N. Erythropoietin: physiology and molecular mechanisms. Heart Fail Rev. 2008;13(4):405–414.
35. Zarychanski, R., Houston, D.S. Anemia of chronic disease: a harmful disorder or an adaptive, beneficial response? Can Med Assoc J. 2008;179(4):333–337.
36. Asare, K. Anemia of critical illness. Pharmacotherapy. 2008;28(10):1267–1282.
37. Levine, R.L., Gilliland, D.G. Myeloproliferative disorders. Blood. 2008;112(6):2190–2198.
38. National Institutes of Health, U.S. Department of Health and Human Services. Chronic myeloproliferative disorders treatment: health professional version, Bethesda, MD, 2008, Available at http://cancer.gov/cancertopics/pdq/treatment/myeloproliferative/healthprofessoinal/. Accessed January 22, 2009.
39. Finazzi, G., Barbui, T. Evidence and expertise in the management of polycythemia vera and essential thrombocythemia. Leukemia. 2008;22(8):1494–1502.
40. Chen, G., Prchal, J.T. Polycythemia vera and its molecular basis: an update. Best Pract Res Clin Haemotol. 2006;19(3):387–397.
41. Landolfi, R., Cipriani, M.C., Novarese, L. Thrombosis and bleeding in polycythemia vera and essential thrombocythemia: pathogenetic mechanisms and prevention. Best Pract Res Clin Haematol. 2006;19(3):617–633.
42. Tefferi, A. The diagnosis of polycythemia vera: new tests and old dictums. Best Pract Res Clin Haematol. 2006;19(3):455–469.