ALTERATIONS OF HEMATOLOGIC FUNCTION IN CHILDREN


This chapter briefly explains fetal and neonatal hematopoiesis and postnatal changes in blood as a foundation for understanding the pathophysiology of specific blood disorders in childhood. Among the diseases that affect erythrocytes are acquired disorders, such as iron deficiency anemia, hemolytic disease of the newborn, and anemia of infectious disease; and inherited disorders, such as glucose-6-phosphate dehydrogenase (G6PD) deficiency, hereditary spherocytosis, sickle cell disease, and the thalassemias. Disorders of coagulation and platelets include inherited hemorrhagic diseases, such as the hemophilias, and antibody-mediated hemorrhagic diseases, which include idiopathic thrombocytopenic purpura, autoimmune neonatal thrombocytopenias, and autoimmune vascular purpuras. Finally, leukocyte disorders, such as leukemia and the lymphomas (non-Hodgkin lymphoma as well as Hodgkin disease), are discussed.
FETAL AND NEONATAL HEMATOPOIESIS
As the developing embryo becomes too large for oxygenation of tissues by simple diffusion, the production of erythrocytes begins within the vessels of the yolk sac. Shortly after 2 weeks of gestation, circulating erythrocytes play a major role in delivering oxygen to the tissues. At approximately the eighth week of gestation, the site of erythrocyte production shifts from the vessels to the liver sinusoids and the production of leukocytes and platelets begins in the liver and spleen. Erythropoiesis in the liver and, to a lesser extent, in the spleen and lymph nodes, reaches a peak at approximately 4 months. Hepatic blood formation declines steadily thereafter but does not disappear entirely during the remainder of gestation. By the fifth month of gestation, hematopoiesis begins to occur in the bone marrow and increases rapidly until hematopoietic (red) marrow fills the entire bone marrow space. By the time of delivery, the marrow is the only significant site of hematopoiesis.
In neonates and young infants, hematopoietic marrow progressively fills the bony cavities of the entire axial skeleton (skull, vertebrae, ribs, sternum), the long bones of the limbs, and many intramembranous bones. (These structures are described in Chapter 43.) Fatty (yellow) marrow gradually replaces hematopoietic marrow in some bones. During childhood, hematopoietic tissue retreats centrally to the vertebrae, ribs, sternum, pelvis, scapulae, skull, and proximal ends of the femur and humerus.
In diseases characterized by hemolysis, erythrocyte production can increase as much as eight times the normal because erythropoietin causes hematopoietic marrow to increase in volume. Initially, hematopoietic marrow expands from the ends of the long bones toward the middle of the shafts, replacing fatty marrow. Next, blood cell production begins to occur outside the marrow cavities, especially in the liver and spleen. Extramedullary hematopoiesis is more likely to occur in children than in adults because the bony cavities of children already are filled with red marrow (Figure 28-1). This is why hemolytic disease causes especially pronounced enlargement of the spleen and liver in children.
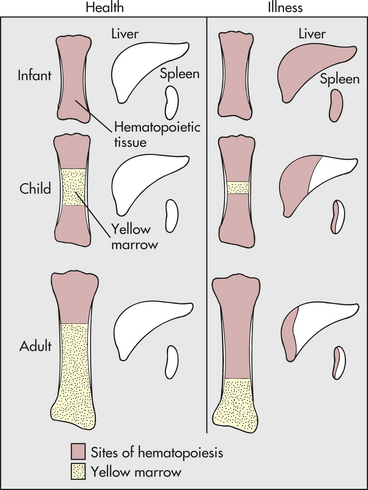
Figure 28-1 Sites of hematopoiesis in health and illness. With normal maturation, red marrow is partly replaced by yellow marrow in the shafts of the long bones. In adults, red marrow is largely restricted to the proximal ends of the femur and humerus. In response to hemolysis, red marrow replaces yellow marrow in the long bones. In infants, whose long bones already are filled with red marrow, additional hematopoiesis takes place in the liver and spleen. In children and adults, red marrow can replace yellow marrow in response to hemolysis, necessitating less hematopoiesis in the liver and spleen.
The erythrocytes undergo striking changes during gestation, particularly during the first two trimesters, at which time they nearly double in numbers and in hemoglobin content. A proportionate increase in hematocrit also occurs. By the end of gestation the erythrocyte count has more than tripled but the size of each erythrocyte has decreased.
A biochemically distinct type of hemoglobin is synthesized during fetal life. The three embryonic hemoglobins (Gower 1, Gower 2, and Portland) and the fetal hemoglobin (Hb F) are composed of two α and two γ-chains of polypeptides, whereas the adult hemoglobins (Hb A and Hb A2) are composed of two α-chains and two β-chains. (The structure of an adult hemoglobin molecule is illustrated in Figure 25-14, and types of hemoglobin are defined in Table 25-5.) Some unknown regulatory mechanism promotes γ-chain synthesis and inhibits β- and δ-chain synthesis in utero. This results in production of embryonic or fetal hemoglobin. After birth, γ-chain synthesis is inhibited, whereas β- and δ-chain synthesis is facilitated, resulting in production of adult hemoglobins.
Fetal hemoglobin has greater affinity for oxygen than does adult hemoglobin because it interacts less readily with an enzyme (2,3-diphosphoglycerate [DPG]) that inhibits hemoglobin-oxygen binding. The decreased inhibitory effects of 2,3-DPG enable fetal blood to transport oxygen despite the relative lack of oxygen in the uterine environment. The increased affinity for oxygen enables Hb F to bind with maternal oxygen in the placental circulation.
During the first trimester, nearly all of the hemoglobin in the fetus is embryonic, but some Hb A can be detected. Therefore, it is possible to identify as early as 16 to 20 weeks of gestation some disorders of adult hemoglobin, such as sickle cell anemia and thalassemia major. In the 6-month fetus, Hb F constitutes 90% of the total. This percentage then begins to decline. At birth, neonatal hemoglobin consists of 70% Hb F, 29% Hb A, and 1% Hb A2. Between 6 and 12 months of age, normal adult hemoglobin percentages are established (see Chapter 25).
POSTNATAL CHANGES IN THE BLOOD
Blood cell counts tend to rise above adult levels at birth and then decline gradually throughout childhood. Table 28-1 lists normal ranges during infancy and childhood. The immediate rise in values is the result of accelerated hematopoiesis during fetal life, increased numbers of cells that result from the trauma of birth, and cutting of the umbilical cord. These events surrounding the birth also are accompanied by a “shift to the left,” that is, the presence of large numbers of immature erythrocytes and leukocytes (particularly granulocytes) in peripheral blood (see Chapter 25). The shift to the left disappears as the infant develops, usually within the first 2 to 3 months of life. Other unique postnatal characteristics, particularly of lymphocytes, may be caused by exogenous factors, such as viral infections.
Table 28-1
Hematologic Values During Infancy and Childhood
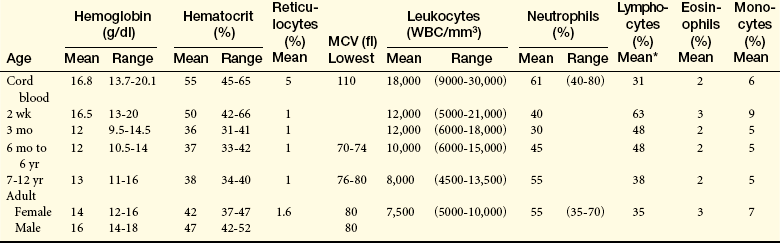
fl, Femtoliters; MCV, mean corpuscular volume; WBC, white blood cells.
∗Relatively wide range.
From Behrman R et al, editors: Nelson textbook of pediatrics, 17th ed, Philadelphia, 2004, Saunders.
Average blood volume in the full-term neonate is 85 ml/kg of body weight. The premature infant has a slightly larger blood volume of 90 ml/kg of body weight, with the mean increasing to 150 mg/kg during the first few days after birth. In full-term and premature infants, blood volume decreases during the first few months. Thereafter the average blood volume is 75 to 77 ml/kg, which is similar to that of older children and adults.
Erythrocytes
The hypoxic intrauterine environment stimulates erythropoietin production in the fetus. This accelerates fetal erythropoiesis, producing polycythemia (excessive proliferation of erythrocyte precursors) of the newborn. After birth the oxygen from the lungs saturates arterial blood and the amount of oxygen delivered to the tissues increases. In response to the change from a placental to a pulmonary oxygen supply during the first few days of life, levels of erythropoietin and the rate of blood cell formation decrease. The very active rate of fetal erythropoiesis is reflected by the large numbers of immature erythrocytes (reticulocytes) in the peripheral blood of full-term neonates. After birth the number of reticulocytes decreases about 50% every 12 hours so it is rare to find an elevated reticulocyte count after the first week of life. A decrease in extramedullary hematopoiesis also occurs at this time. In the peripheral blood the erythrocyte count drops for 6 to 8 weeks after birth. During this period of rapid growth the rate of erythrocyte destruction is greater than that in later childhood and adulthood. In full-term infants, normal erythrocyte life span is 60 to 80 days; in premature infants it may be as short as 20 to 30 days; and in children and adolescents, it is the same as that in adults—120 days. (Mechanisms of hemolysis are described in Chapter 25.)
In the premature infant the postnatal fall in hemoglobin and hematocrit values is more marked than in the full-term infant. In the preschool and school-age child, there is a gradual rise in hemoglobin, hematocrit, and red blood cell(RBC) count. Values in males and females first begin to diverge in adolescence. In the female the gradual hemoglobin increase continues into early puberty, at which time it stabilizes. In the male the hemoglobin increase keeps pace with growth and maturation and eventually surpasses that of the female. This higher value in the mature male is related to androgen secretion.
Metabolic processes within the erythrocytes of neonates differ significantly from those of erythrocytes in the normal adult. The relatively young population of erythrocytes in the newborn consumes greater quantities of glucose than do erythrocytes in adults. Several enzymes that regulate glucose consumption are increased in the erythrocytes of neonates, with a subsequent increase in the rate of glycolysis.
Leukocytes and Platelets
The lymphocytes of children tend to have more cytoplasm and less compact nuclear chromatin than do the lymphocytes of adults. The significance of these differences is unknown. One possible explanation is that children tend to have more frequent viral infections, which are associated with atypical lymphocytes. Even minor infections, in which the child fails to exhibit clinical manifestations of illness, and administration of immunizations may result in lymphocyte changes.1
The lymphocyte count is high at birth and continues to rise in some healthy infants during the first year of life. Then a steady decline occurs throughout childhood and adolescence until lower adult values are reached. It is unknown whether these developmental variations are physiologic or are a pathologic response to frequent viral infections and immunizations in children.
At birth the neutrophil count is very high and rises further during the early days of life.2 After 2 weeks, neutrophil counts fall to within or below normal adult ranges. By approximately 4 years of age, the neutrophil count is the same as that of an adult. White children have slightly higher counts than black children.3
Eosinophil count is high in the first year of life and is higher in children than in teenagers or adults.4 Monocyte counts are high in the first year of life and then decrease to adult levels. No relationship between age and basophil count has been found. Platelet counts in full-term neonates are comparable to platelet counts in adults and remain so throughout infancy and childhood.5
DISORDERS OF ERYTHROCYTES
Anemia is the most common blood disorder in children. Like the anemias of adulthood, the anemias of childhood are caused by ineffective erythropoiesis or premature destruction of erythrocytes. The most common cause of insufficient erythropoiesis is iron deficiency, which may result from insufficient dietary intake or chronic loss of iron caused by bleeding. The hemolytic anemias of childhood may be divided into two large categories. The first category consists of disorders that result from premature destruction caused by intrinsic abnormalities of the erythrocytes, and the second category consists of disorders that result from damaging extraerythrocytic factors. The hemolytic anemias are inherited, congenital, or both.
The most dramatic form of acquired congenital hemolytic anemia is hemolytic disease of the newborn (HDN), also termed erythroblastosis fetalis. HDN is an alloimmune disease in which maternal blood and fetal blood are antigenically incompatible, causing the mother’s immune system to produce antibodies against fetal erythrocytes. Fetal erythrocytes that have been attacked by (i.e., bound to) maternal antibodies are recognized as foreign or defective by the fetal mononuclear phagocyte system and are removed from the circulation by phagocytosis, usually in the fetal spleen. (For a complete discussion of HDN, see p. 1066.) Other acquired hemolytic anemias—some of which begin in utero—include those caused by infections or the presence of toxic chemicals.
The inherited forms of hemolytic anemia result from intrinsic defects of the child’s erythrocytes, any of which can lead to erythrocyte removal by the mononuclear phagocyte system. Structural defects include abnormal cellular size and abnormalities of plasma membrane structure (spherocytosis). Intracellular defects include enzyme deficiencies, the most common of which is G6PD deficiency, and defects of hemoglobin synthesis, which manifest as sickle cell disease or thalassemia, depending on which component of hemoglobin is defective. These and other causes of childhood anemia, some more common than others, are listed in Table 28-2.
Table 28-2
| Cause | Anemic Condition |
| Deficient Erythropoiesis or Hemoglobin Synthesis | |
| Decreased stem cell population in marrow (congenital or acquired pure red cell aplasia) | Normocytic-normochromic anemia |
| Decreased erythropoiesis despite normal stem cell population in marrow (infection, inflammation, cancer, chronic renal disease, congenital dyserythropoiesis) | Normocytic-normochromic anemia |
| Deficiency of a factor or nutrient needed for erythropoiesis | |
| Cobalamin (vitamin B12), folate | Megaloblastic anemia |
| Iron | Microcytic-hypochromic anemia |
| Increased or Premature Hemolysis | |
| Alloimmune disease (maternal-fetal Rh, ABO, or minor blood group incompatibility) | Hemolytic disease of the newborn (HDN) |
| Autoimmune disease (idiopathic autoimmune hemolytic anemia, symptomatic systemic lupus erythematosus, lymphoma, drug-induced autoimmune processes) | Autoimmune hemolytic anemia |
| Inherited defects of plasma membrane structure (spherocytosis, elliptocytosis, stomatocytosis) or cellular size or both (pyknocytosis) | Hemolytic anemia |
| Infection (bacterial sepsis, congenital syphilis, malaria, cytomegalovirus infection, rubella, toxoplasmosis, disseminated herpes) | Hemolytic anemia |
| Intrinsic and inherited enzymatic defects (deficiencies of glucose-6-phosphate dehydrogenase [G6PD], pyruvate kinase, 5′-nucleotidase, glucose phosphate isomerase) | Hemolytic anemia |
| Inherited defects of hemoglobin synthesis | Sickle cell anemia Thalassemia |
| Disseminated intravascular coagulation (see Chapter 27) | Hemolytic anemia |
| Galactosemia | Hemolytic anemia |
| Prolonged or recurrent respiratory or metabolic acidosis | Hemolytic anemia |
| Blood vessel disorders (cavernous hemangioma, large vessel thrombus, renal artery stenosis, severe coarctation of the aorta) (see Chapter 31) | Hemolytic anemia |
Acquired Disorders
Iron deficiency anemia is the most common blood disorder of infancy and childhood, with the highest incidence occurring between 6 months and 2 years of age. Incidence is not related to gender or race, but socioeconomic factors are important because they affect nutrition, for example, the risk of iron deficiency anemia in children of single, homeless women.6 However, greater use of iron-fortified products has decreased the prevalence of anemia in low-income infants.7 Iron deficiency anemia is a common disorder in children because of their extremely high need for iron for normal growth to occur.
Between 4 years of age and the onset of puberty, dietary iron deficiency is uncommon. During adolescence, however, it is relatively common, especially in menstruating females. Rapid growth, together with the average teenager’s dietary habits, causes iron depletion. (Mechanisms of iron depletion are described in Chapter 25.)
PATHOPHYSIOLOGY Although inadequate intake of iron is the most common cause of iron deficiency anemia during the first few years of life and during adolescence, blood loss is the most common cause in childhood. Chronic iron deficiency anemia from occult (hidden) blood loss may be caused by a gastrointestinal lesion, parasitic infestation, or hemorrhagic disease. As many as one third of infants with severe iron deficiency anemia have chronic intestinal blood loss induced by exposure to a heat-labile protein in cow’s milk. Such exposure causes an inflammatory gastrointestinal reaction that damages the mucosa and results in diffuse hemorrhage.
The amount of iron available for hemoglobin synthesis in the infant depends on iron stores present at birth, rate of growth, the amount of dietary iron absorbed, and physiologic or pathologic loss of iron. During the period of inactive erythropoiesis immediately after birth, iron from erythrocytes that die at the end of their normal life span is stored, as hemosiderin, in bone marrow and liver tissue. This creates an iron reserve that can be used in lieu of dietary intake. The greatest stores are present 4 to 8 weeks after birth. Until erythropoiesis resumes, these iron stores are mobilized. In the premature infant, resumption of erythropoiesis depletes iron stores within 6 to 12 weeks; in the full-term infant, depletion takes longer—about 16 to 20 weeks. Once iron stores have been used, the infant depends on dietary iron.
The amount of dietary iron available for erythropoiesis depends on which foods are consumed. Iron-fortified cereals, green and yellow vegetables, fruits, and milk are common in the average 6-month-old infant’s diet and provide iron in the amount of 0.9 to 1.5 mg/kg/day, amounts that satisfy the normal average daily requirement. Iron-fortified formulas are available commercially.
CLINICAL MANIFESTATIONS The symptoms of mild anemia—lethargy and lassitude—usually are not present or detectable in infants and young children, who are unable to describe these symptoms. Therefore, parents usually do not notice any change in the child’s behavior or appearance until moderate anemia has developed. General irritability, decreased activity tolerance, weakness, and lack of interest in play are nonspecific indications of anemia. In mild to moderate iron deficiency anemia (hemoglobin of 6 to 10 g/dl), compensatory mechanisms of tissue oxygenation, such as increased amounts of 2,3-DPG within erythrocytes and a shift of the oxyhemoglobin dissociation curve, may be so effective that few clinical manifestations are apparent. When the hemoglobin falls below 5 g/dl, however, pallor, tachycardia, and systolic murmurs may occur.
Splenomegaly is evident in 10% to 15% of children with iron deficiency anemia, and if the condition is long-standing, the sutures of the skull may be widened. Chronic anemia also may result in decreased physical growth and developmental delays. Some children exhibit pica, a behavior in which nonfood substances are eaten. Because children with iron deficiency anemia may be obese, underweight, or of normal weight, other manifestations of undernutrition must be identified.
Iron deficiency anemia may affect neurologic and intellectual function. Some research findings indicate that low iron in the blood affects attention span, alertness, and learning ability, even when anemia is not severe.
EVALUATION AND TREATMENT The most definitive test for differentiating iron deficiency from other microcytic states is the absence of iron stores in the bone marrow. However, measurement of serum ferritin iron concentration, transferrin saturation, iron-binding capacity, and, more recently, serum transferrin receptors may prevent proceeding to actual bone marrow evaluation. Evaluation and treatment of iron deficiency anemia in children are similar to evaluation and treatment in adults (see Chapter 26). Oral administration of simple ferrous salts usually is satisfactory, but additional vitamin C may be needed to promote absorption.8 Administration of supplementary trace metals or other vitamins is not necessary. If malabsorption is the cause of the anemia (or if oral administration has not been successful), iron dextran (Imferon) is given intravenously. Iron therapy is continued for at least 2 months after erythrocyte indexes have returned to normal in order to replenish iron stores.9
Dietary modification is required to prevent recurrences of iron deficiency anemia. The child’s intake of iron-rich foods is increased, and the intake of cow’s milk may be restricted, with the exact amount depending on the child’s age (from 16 to 32 ounces). Limiting milk intake makes the child hungrier for other iron-rich foods and prevents gastrointestinal blood loss in children whose anemia is aggravated or caused by inflammatory reactions to proteins in cow’s milk.
Hemolytic Disease of the Newborn
HDN can occur only if antigens on fetal erythrocytes differ from antigens on maternal erythrocytes. The antigenic properties of erythrocytes are determined genetically: they may be type A, B, or O and may or may not include Rh antigen D. Erythrocytes that express Rh antigen D are Rh-positive; those that do not are Rh-negative. The frequency of Rh negativity is higher in whites (15%) than in blacks (5%), and is rare in Asians. Maternal-fetal incompatibility exists if mother and fetus differ in ABO blood type or if the fetus is Rh-positive and the mother is Rh-negative. (The antigenic properties of erythrocytes are described in Chapter 8.)
ABO incompatibility occurs in about 20% to 25% of all pregnancies, but only 1 in 10 cases of ABO incompatibility results in HDN. Rh incompatibility occurs in less than 10% of pregnancies and rarely causes HDN in the first incompatible fetus. Even after five or more pregnancies, only 5% of women have babies with hemolytic disease. Usually erythrocytes from the first incompatible fetus cause the mother’s immune system to produce antibodies that affect the fetuses of subsequent incompatible pregnancies. Only one in three cases of HDN is caused by Rh incompatibility; most cases are caused by ABO incompatibility.
PATHOPHYSIOLOGY If the mother and fetus have antigenically incompatible erythrocytes, HDN will result (1) if the mother’s blood contains preformed antibodies against fetal erythrocytes or produces them on exposure to fetal erythrocytes, (2) if sufficient amounts of antibody (usually immunoglobulin G [IgG]) cross the placenta and enter fetal blood, and (3) if IgG binds with sufficient numbers of fetal erythrocytes to cause widespread antibody-mediated hemolysis or splenic removal. (Antibody-mediated cellular destruction is discussed in Chapter 7.)
Individuals usually form IgM antibodies against the ABO antigen they do not express; against the A antigen if the mother is blood type O or B or against the B antigen if the mother is type O or A. These are produced early in life against gastrointestinal bacteria that make antigens similar to A and B. IgM antibodies do not cross the placenta or cause HDN. Occasionally, a blood type O individual may have also produced IgG against the A or B antigen. If the fetus is blood group A or B, the ABO incompatibility can cause HDN in the first pregnancy. The severity of HDN is usually not severe because A and B antigens are expressed on most cells, including the placenta, and much of the IgG against A or B will be absorbed before encountering fetal blood cells.
Anti-Rh antibodies, on the other hand, are formed only in response to the presence of incompatible (Rh-positive) erythrocytes in the blood of an Rh-negative mother. Sources of exposure include fetal blood that is mixed with the mother’s blood at the time of delivery, transfused blood, and, rarely, previous sensitization of the mother by her own mother’s incompatible blood.
The first Rh-incompatible pregnancy usually presents no difficulties because very few fetal erythrocytes cross the placental barrier during gestation. When the placenta detaches at birth, however, large numbers of fetal erythrocytes usually enter the mother’s bloodstream. If the mother is Rh-negative and the fetus is Rh-positive, the mother produces anti-Rh antibodies. The capacity of the mother’s immune system to produce anti-Rh antibodies depends on many factors, including her genetic capacity to make antibodies against the Rh antigen D, the amount of fetal-to-maternal bleeding, and the occurrence of any bleeding earlier in the pregnancy. Anti-Rh antibodies persist in the bloodstream for a very long time, and if the next offspring is Rh-positive, the mother’s anti-Rh antibodies can enter the fetus’s bloodstream and destroy the erythrocytes. Antibodies against Rh antigen D are of the IgG class and easily cross the placenta.
IgG-coated fetal erythrocytes are destroyed extravascularly, primarily by mononuclear phagocytes in the spleen. As hemolysis proceeds, the fetus becomes anemic. Erythropoiesis accelerates, particularly in the liver and spleen, and immature nucleated cells (erythroblasts) are released into the bloodstream (hence the name erythroblastosis fetalis) (Figure 28-2). The degree of anemia depends on the length of time the antibody has been in the fetal circulation, antibody concentration, and the ability of the fetus to compensate for increased hemolysis. Unconjugated (indirect) bilirubin, which is formed during breakdown of hemoglobin, is transported across the placental barrier into the maternal circulation and is excreted by the mother. Hyperbilirubinemia occurs in the neonate after birth because excretion of lipid-soluble unconjugated bilirubin through the placenta no longer is possible.
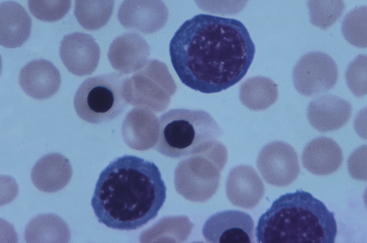
Figure 28-2 Rh incompatibility in hemolytic disease of the newborn. This micrograph shows immature red blood cells not normally found in blood. Large purple cells are erythroblasts; nucleated red blood cells are normoblasts. Normal red blood cells also are shown (× 500). (Copyright Ed Reschke.)
The pathophysiologic effects of HDN are more severe in Rh incompatibility than in ABO incompatibility. ABO incompatibility may resolve after birth without life-threatening complications. Maternal-fetal incompatibility in which a mother with type O blood has a child with type A or B blood usually is so mild that it does not require treatment.
Rh incompatibility is more likely than ABO incompatibility to cause severe or even life-threatening anemia, death in utero, or damage to the central nervous system (CNS). Severe anemia alone can cause death as a result of cardiovascular complications (see Chapter 26). Extensive hemolysis also results in increased levels of unconjugated bilirubin in the neonate’s circulation. If bilirubin levels exceed the liver’s ability to conjugate and excrete bilirubin, some of it is deposited in the brain, causing cellular damage and eventually, if the neonate does not receive exchange transfusions, death.
Fetuses that do not survive anemia in utero usually are stillborn, with gross edema in the entire body, a condition called hydrops fetalis. Death can occur as early as 17 weeks of gestation and results in spontaneous abortion.
CLINICAL MANIFESTATIONS Neonates with mild HDN may appear healthy or slightly pale, with slight enlargement of the liver and spleen. Pronounced pallor, splenomegaly, and hepatomegaly indicate severe anemia, which predisposes the neonate to cardiovascular failure and shock. Life-threatening Rh incompatibility is rare today, largely because of the routine use of Rh immune globulin.
Because the maternal antibodies remain in the neonate’s circulatory system after birth, erythrocyte destruction can continue. This causes hyperbilirubinemia and icterus neonatorum (neonatal jaundice) shortly after birth. Without replacement transfusions, in which the child receives Rh-negative erythrocytes, the bilirubin is deposited in the brain, a condition termed kernicterus. Kernicterus produces cerebral damage and usually causes death (icterus gravis neonatorum). Infants who do not die may have mental retardation, cerebral palsy, or high-frequency deafness.
EVALUATION AND TREATMENT Routine evaluation of fetuses at risk for HDN (i.e., fetuses resulting from Rh- or ABO-incompatible matings) include the Coombs test. The indirect Coombs test measures antibody in the mother’s circulation and indicates whether the fetus is at risk for HDN. The direct Coombs test measures antibody already bound to the surfaces of fetal erythrocytes and is used primarily to confirm the diagnosis of antibody-mediated HDN. Determining prior history of fetal hemolytic disease, as well as diagnostic tests, may help predict the severity of the disorder. Diagnostic measures include maternal antibody titers, fetal blood sampling, amniotic fluid spectrophotometry, and ultrasound fetal assessment.10
The key to treatment of HDN resulting from Rh incompatibility lies in prevention (immunoprophylaxis). One of the success stories of immunology has been the spectacular results obtained through the use of Rh immune globulin (RhoGAM), a preparation of antibody against Rh antigen D. If an Rh-negative woman is given Rh immune globulin within 72 hours of exposure to Rh-positive erythrocytes, she will not produce antibody against the D antigen and the next Rh-positive baby will be protected (Figure 28-3). The injected antibodies remain in the mother’s bloodstream long enough to prevent her immune system from producing its own anti-Rh antibodies but not long enough to affect subsequent offspring. The mother must be given Rh immune globulin injections after the birth of each Rh-positive baby and after an miscarriage. Also the mother must be especially careful not to receive a transfusion containing Rh-positive blood, because this also would stimulate production of anti-Rh antibodies. In many hospitals, Rh immune globulin is given prophylactically at 28 weeks to all pregnant Rh-negative women with Rh-positive partners. Immunoprophylaxis with Rh immune globulin, unfortunately, appears to be underused in the United States. Failure to use immunoprophylaxis, such as in cases of unrecognized miscarriage, has led to a small increase in mothers who will require comprehensive treatment during subsequent pregnancies.11
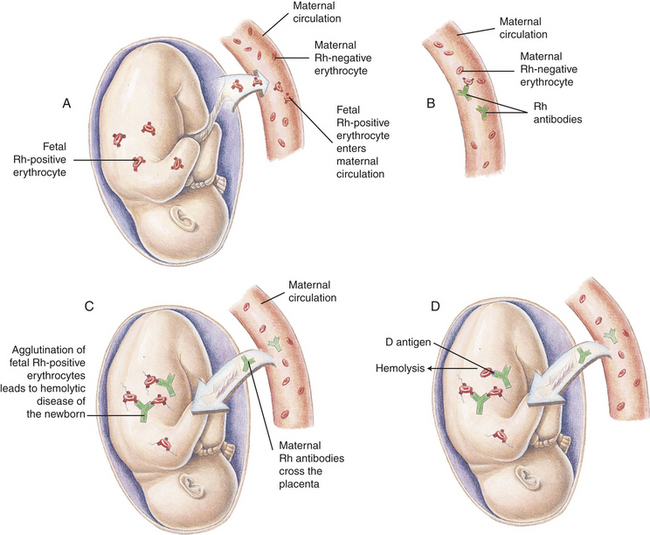
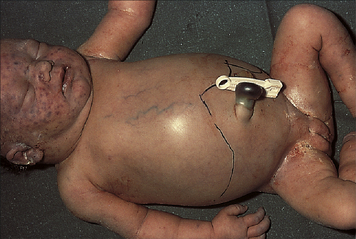
Figure 28-3 Hemolytic disease of the newborn (HDN) A, Before or during delivery, Rh-positive erythrocytes from the fetus enter the blood of an Rh-negative woman through a tear in the placenta. B, The mother is sensitized to the Rh antigen and produces Rh antibodies. Because this usually happens after delivery, there is no effect on the fetus in the first pregnancy. C, During a subsequent pregnancy with an Rh-positive fetus, Rh-positive erythrocytes cross the placenta, enter the maternal circulation, and(D) stimulate the mother to produce antibodies against the Rh antigen. The Rh antibodies from the mother cross the placenta, using agglutination and hemolysis of fetal erythrocytes, and HDN develops (E). (Modified from Seeley RR, Stephens TD, Tate P: Anatomy and physiology, ed 3, St Louis, 1995, Mosby.)
If antigenic incompatibility of the mother’s erythrocytes is not discovered in time to administer Rh immune globulin and a child is born with HDN, treatment consists of exchange transfusions in which the neonate’s blood is replaced with new Rh-positive blood that is not contaminated with anti-Rh antibodies. This treatment is instituted during the first 24 hours of extrauterine life to prevent kernicterus. Phototherapy also is used to reduce the toxic effects of unconjugated bilirubin.
Jaundice and indirect hyperbilirubinemia are reduced when the infant is exposed to high-intensity light in the visible spectrum, the most effective being the blue range (from 420 to 470 nm). Bilirubin in the skin absorbs light energy, which, by photoisomerization, converts the toxic unconjugated bilirubin into conjugated isomers that are excreted in the bile. Phototherapy also causes autosensitization that results in oxidation reactions. Breakdown products from the oxidation reactions are excreted by the liver and kidney without need for conjugation. The therapeutic effect of phototherapy depends on the light energy emitted in the effective wavelengths, the distance between the infant and the light source, and the amount of skin exposed; the rate of hemolysis and the infant’s ability to excrete bilirubin also are factors in determining the effectiveness of phototherapy in lowering serum bilirubin levels.
Anemia of Infectious Disease
Infections of the newborn, often initially acquired by the mother and transmitted to the fetus, may result in a hemolytic anemia with clinical manifestations similar to those of HDN. Congenital syphilis, toxoplasmosis, cytomegalic inclusion disease, rubella, coxsackievirus B infection, herpesvirus infection, and bacterial sepsis can cause hemolytic anemia in the neonate.
The exact mechanism of anemia caused by congenital infections is unclear. In some instances it is related to direct injury of erythrocyte membranes or erythrocyte precursors by the infectious microorganism. In other instances it results from traumatic destruction of erythrocytes during their passage through inflamed capillaries.
Inherited Disorders
A number of inherited and intrinsic erythrocyte defects are known to cause increased hemolysis (see Table 28-2). These defects may be associated with enzymatic abnormalities that disrupt metabolic processes and prevent normal biochemical balance within the cell, with alterations of hemoglobin structure or synthesis, or with plasma membrane defects accompanied by changes in erythrocyte size or shape.
Glucose-6-Phosphate Dehydrogenase Deficiency
Glucose-6-phosphate dehydrogenase (G6PD) deficiency is an inherited, X-linked recessive disorder, most fully expressed in homozygous males, although partial expression and a carrier state are possible in heterozygous females. (X-linked inheritance is discussed in Chapter 4.) The deficiency is present in 10% of blacks and tends to occur in Sephardic Jews, Greeks, Iranians, Chinese, Filipinos, and Indonesians, with a frequency ranging from 5% to 40%.
PATHOPHYSIOLOGY G6PD is an enzyme that normally enables erythrocytes to maintain metabolic processes despite injurious conditions, such as the presence of certain drugs (sulfonamides, antimalarial agents, salicylates, or naphthaquinolones); ingestion of fava beans (a dietary staple in some Mediterranean areas); hypoxemia; infection; fever; or acidosis. Therefore, G6PD deficiency is usually asymptomatic unless one of these stressors is present. Erythrocyte damage in affected children begins after intense or prolonged exposure to one of these substances or conditions, and it ceases when they are removed. In black males the G6PD defect becomes more pronounced as the erythrocyte ages; in other populations the defect is profound even in young erythrocytes. By ingesting a substance with oxidant properties, such as a salicylate (aspirin), a pregnant woman may precipitate an episode of hemolysis in a fetus with G6PD deficiency.
In the absence of G6PD, oxidative stressors damage hemoglobin and the plasma membranes of erythrocytes and possibly interfere with the activities of other enzymes within the cell. Hemoglobin is oxidized progressively to methemoglobin, sulfmethemoglobin, and denatured globin-glutathione complexes. Eventually, exposure to oxidating substances results in the precipitation of insoluble hemoglobin inclusions, called Heinz bodies, within the cell. Plasma damage and the presence of Heinz bodies cause hemolysis, chiefly in the spleen.
CLINICAL MANIFESTATIONS In Asian and Mediterranean infants, G6PD deficiency is likely to be associated with icterus neonatorum. The most common clinical manifestation of G6PD deficiency is acute hemolytic anemia, usually after infections or the ingestion of certain oxidative drugs. The fava bean produces a severe hemolytic reaction called favism in infants with G6PD deficiency.12
Hemolytic episodes are characterized by pallor, icterus, dark urine, back pain, and, in severe cases, shock, cardiovascular collapse, and death. Between hemolytic episodes, anemia is absent and erythrocyte survival is normal.
EVALUATION AND TREATMENT Direct or indirect demonstration of reduced G6PD activity in erythrocytes is required for evaluation. Satisfactory screening test results are based on discoloration of methylene blue and reduction of methemoglobin. Immediately after a hemolytic episode, reticulocytes and young erythrocytes predominate. Because young erythrocytes have significantly higher enzyme activity than do older cells, testing should be performed a few weeks after a crisis so that a low level of enzyme activity can be demonstrated. G6PD activity that is within the low normal range in the presence of a high reticulocyte count suggests G6PD deficiency. G6PD deficiency also can be detected by electrophoretic analysis.
Prevention of hemolysis is the most important therapeutic measure. Males belonging to high-risk groups (Greeks, southern Italians, Sephardic Jews, Filipinos, Chinese, Africans, Thais) should be tested for the defect before being given drugs known to be oxidative. When hemolysis has occurred, supportive treatment may include blood transfusions and oral iron therapy. Spontaneous recovery generally follows treatment.
Hereditary Spherocytosis
Hereditary spherocytosis (HS), also known as congenital hemolytic anemia or congenital acholuric jaundice, is the most common of the hemolytic disorders in which there is no abnormality of hemoglobin.
PATHOPHYSIOLOGY Transmitted as an autosomal dominant trait, HS is presumed to represent new mutations in about 25% of cases. The defect is believed to be caused by an undefined abnormality of proteins or spectrins of the erythrocyte membrane. Affected cells are unduly permeable to sodium and acquire a particular characteristic structure (Figure 28-4). An increased concentration of intracellular sodium is believed to lead to increased use of adenosine triphosphate (ATP) to drive the so-called cation pump. Early aging and destruction of erythrocytes are believed to result from metabolic overwork and loss of erythrocyte membrane.13
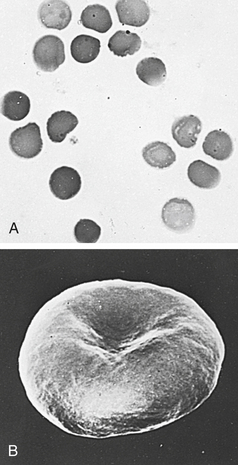
Figure 28-4 The microspherocyte A, Blood smear from individual with hereditary spherocytosis (Wright stain). B, Scanning electron micrograph. (Courtesy Dr. M Bessis. From Miale JB: Laboratory medicine: hematology, ed 6, St Louis, 1982, Mosby.)
Circulation of blood to the spleen creates a metabolic environment that is stressful to spherocyte cells, and repeated passages through this stressful environment result in their sequestration and destruction. The spherocyte is relatively rigid and passes with difficulty through the small openings between the splenic cords and sinuses. Thus the spleen is intimately involved in the hemolytic process.
CLINICAL MANIFESTATIONS The presenting signs of HS are anemia, jaundice, and splenomegaly. Anemia may be mild of even absent in some cases depending on whether the hemolysis is well compensated. If this is the case, the reticulocyte count will be elevated. Splenomegaly is usually mild. HS can present at any age, from the neonatal period until older adulthood. More severe types of HS present with signs of hemolytic anemia and hyperbilirubinemia during the newborn period.14 Children who suffer from HS may have values considerably lower than children who do not. These children therefore may have life-threatening anemia with clinical symptoms ranging from difficulty tolerating feeding, circumoral pallor, tachycardia, nasal flaring, diaphoretic episodes, and lethargy. These children also are at increased risk for gallstones because their bodies make extra bile pigment. Infection (specifically parvovirus), fever, and stress can stimulate the spleen to destroy more red blood cells than usual, leading to a worsening anemia in an already baseline anemic child.
EVALUATION AND TREATMENT It is important to ascertain family history of spherocytosis. Laboratory findings include spherocytes in the peripheral blood smear, elevated reticulocyte count with or without anemia, indirect hyperbilirubinemia, and a positive osmotic fragility test. An osmotic fragility test is performed by placing the individual’s red blood cells in a saline solution for 24 hours. Spherocytes do not tolerate weak saline solutions, thus causing them to burst more readily than normal cells. Treatment of HS is based on disease severity. Although some children with severe HS will have severe anemia, blood transfusions are rarely required. Treatment before the age of 5 years consists of daily folic acid supplementation to help with production of healthy red blood cells. In the past, splenectomy was the first line of treatment. Currently, however, splenectomy is only recommended for those children older than 5 years of age with severe disease or those who develop symptomatic gallstones. Partial splenectomy, in which only a portion of the spleen is removed, is being performed on children with HS in an attempt to decrease the risk of postsplenectomy complications.15
Sickle Cell Disease
Sickle cell disease (SCD) is a group of disorders characterized by the presence of an abnormal form of hemoglobin—hemoglobin S (Hb S)—within the erythrocytes. Hb S is formed by a genetic mutation in which one amino acid (valine) replaces another (glutamic acid) (Figure 28-5 A). Hb S, the so-called sickle hemoglobin, reacts to deoxygenation and dehydration by solidifying and stretching the erythrocyte into an elongated sickle shape. This change has a variety of pathologic consequences, including hemolytic anemia.
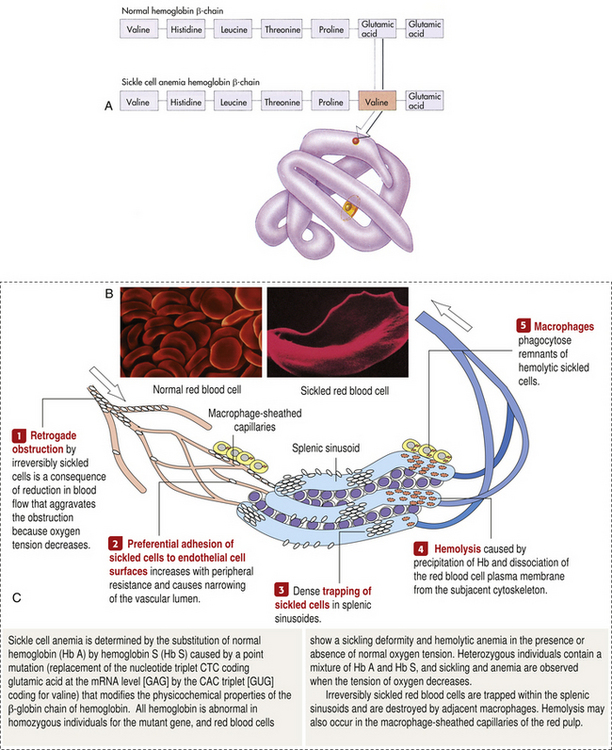
Figure 28-5 Sickle cell hemoglobin A, Sickle cell hemoglobin is produced by a recessive allele of the gene encoding the β-chain of the protein hemoglobin. It represents a single amino acid change—from glutamic acid to valine at the sixth position on the chain. In this model of a hemoglobin molecule, the position of the mutation can be seen near the end of the upper arm. B, Color-enhanced electron micrograph shows normal erythrocytes. C, Illustration of the characteristic shape of a red blood cell containing the abnormal hemoglobin. (A from Raven PH, Johnson GB: Biology, ed 3, St Louis, 1992, Mosby. B copyright Dennis Kunkel Microscopy, Inc. C from Miale JB: Laboratory medicine: hematology, ed 6, St Louis, 1982, Mosby.)
SCD is an inherited autosomal recessive disorder that is expressed as sickle cell anemia, sickle cell–thalassemia disease, or sickle cell–Hb C disease, depending on mode of inheritance (Table 28-3). (See Chapter 4 for a discussion of genetic inheritance of disease.) Sickle cell anemia, a homozygous form, is the most severe. Sickle cell–thalassemia disease and sickle cell–Hb C disease are heterozygous forms in which the child simultaneously inherits another type of abnormal hemoglobin from one parent. Sickle cell trait, in which the child inherits Hb S from one parent and normal hemoglobin (Hb A) from the other, is a heterozygous carrier state that rarely has clinical manifestations. All forms of SCD are lifelong conditions and have no known cure.
Table 28-3
Inheritance of Sickle Cell Disease
| Hemoglobin (Hb) Inherited from First Parent | Hemoglobin Inherited from Second Parent | Form of Sickle Cell Disease in Child |
| Hb S (an abnormal Hb) | Hb S | Sickle cell anemia: homozygous inheritance in which the child’s Hb is mostly Hb S, with the remainder fetal hemoglobin (Hb F) |
| Hb S | Defective or insufficient α- or β-chains of Hb A (alpha- or beta-thalassemia) | Sickle cell: thalassemia disease (heterozygous inheritance of Hb S and alpha- or beta-thalassemia) |
| Hb S | Hb C or D (both abnormal Hb) | Sickle cell: Hb C (or D) disease (heterozygous inheritance of Hb S and either Hb C or D) |
| Hb S | Normal Hb (mostly Hb A) | Sickle cell trait, the carrier state (heterozygous inheritance of Hb S and normal Hb) |
NOTE: See Chapter 25 for a description of normal fetal and adult hemoglobins.
SCD tends to occur in people with origins in equatorial countries, particularly central Africa, the Near East, the Mediterranean area, and parts of India. In the United States, SCD is most common in blacks, with a reported incidence ranging from 1 in 400 to 1 in 500 live births. In the general population the risk of two black parents having a child with sickle cell anemia is 0.7%. Sickle cell–Hb C disease is less common (1 in 800 births), and sickle cell–thalassemia disease occurs in 1 in 1700 births.
Sickle cell trait occurs in 7% to 13% of blacks, whereas its incidence among East Africans may be as high as 45%. The sickle cell trait may provide protection against lethal forms of malaria, a genetic advantage to carriers who reside in endemic regions for malaria (Mediterranean and African zones) but no advantage to carriers living in the United States.
PATHOPHYSIOLOGY Deoxygenation is probably the most important variable in determining the occurrence of sickling.16 The degree of deoxygenation required to produce sickling varies with the percentage of Hb S in the cells. Sickle trait cells will sickle at oxygen tensions of about 15 mm Hg, whereas those from an individual with SCD will begin to sickle at about 40 mmHg. Hb S that is not bound with oxygen forms aggregates of semisolid gel that become stacked within the erythrocyte, stretching it into an elongated crescent (Figures 28-5, C and 28-6). Sickled erythrocytes are stiff and cannot change shape as easily as normal cells when they pass through the microcirculation. (The reversible deformability of erythrocytes is described in Chapter 25.) As a result, sickled erythrocytes tend to plug the blood vessels, causing vascular occlusion, pain, and organ infarction. Sickled cells undergo hemolysis in the spleen or become sequestered there, causing blood pooling and infarction of splenic vessels. The anemia that follows triggers erythropoiesis in the marrow and, in extreme cases, in the liver.
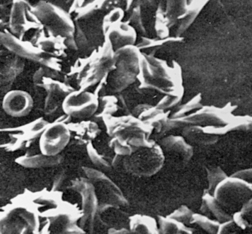
Figure 28-6 Normal and sickle-shaped blood cells. Scanning electron micrograph of normal and sickle-shaped red blood cells. The irregularly shaped cells are the sickle cells; the circular cells are the normal blood cells. (From Raven PH, Johnson GB: Biology, ed 3, St Louis, 1992, Mosby.)
Sickling usually is not permanent; most sickled erythrocytes regain a normal shape after reoxygenation and rehydration. Irreversible sickling is not caused by irreversible hemoglobin changes but rather by irreversible plasma membrane damage caused by sickling. The precise nature of the permanent membrane injury is not known, but it is known that while in the sickled state, the plasma membrane loses some of its capacity for active transport, permitting an influx of calcium ions. (Membrane transport and the effects of calcium influx are described in Chapters 1 and 2.) In people with sickle cell anemia, in which the erythrocytes contain a high percentage of Hb S (75% to 95%), up to 30% of the erythrocytes can become irreversibly sickled. Occasionally, irreversible sickling occurs in SCD but never in the carrier state (sickle cell trait).
Sickling is an occasional, intermittent phenomenon that can be triggered or sustained by one or more of the following stressors: decreased oxygen tension (PO2) of the blood (i.e., hypoxemia), increased hydrogen ion concentration in the blood (decreased pH), increased plasma osmolality, decreased plasma volume, and low temperature (Figure 28-7). The same decrease in PO2 will cause the most sickling in persons with sickle cell anemia (high concentrations of Hb S), the second most in children with sickle cell thalassemia, the third most in those with sickle cell–Hb C disease, and the least or none in those with sickle cell trait. The duration of the PO2 decrease also is important, because sickling tends to occur only after the inciting stimulus has been present for some time.
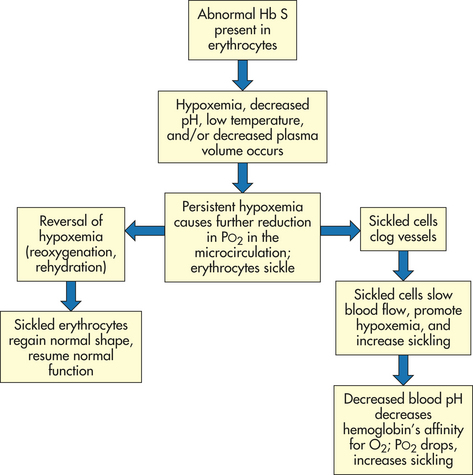
The level of PO2 in the microcirculation also affects sickling because hemoglobin releases whatever oxygen it is carrying to tissues. The PO2 normally is lower in the microcirculation. The added reduction in PO2 caused by persistent hypoxemia—induced by stressors—eventually results in sickling in the microcirculation of all cells that contain Hb S in that site (not throughout the body). Sickling within the microcirculation decreases blood flow as sickled cells clog the vessels. Slow blood flow promotes hypoxemia and perpetuates sickling. Finally, decreased blood pH decreases hemoglobin’s affinity for oxygen. As less oxygen is taken up by hemoglobin in the lungs, PO2 drops, promoting sickling further.
Polymerization of sickle hemoglobin is central to the disorder. Polymerization stiffens the sickle erythrocyte, changing it from a flexible, nourishing cell to an inflexible obstacle that starves and damages tissues.
Increased osmolality of the plasma (increased concentration of solutes; see Chapters 1 and 3) draws water out of the erythrocytes. This promotes sickling by raising the relative Hb S content in erythrocytes. Decreased plasma volume, which occurs in states of dehydration, causes the blood to become viscous (thick and sticky). Increased viscosity of the blood is the final common pathway leading to many pathologic effects. Viscous blood flows slowly and promotes vascular obstruction by increasing opportunities for sickling while decreasing opportunities for reoxygenation in the lungs. This is an example of positive feedback in a vicious cycle of events. Low temperatures precipitate sickle crisis, presumably because of vasoconstriction.16
Once sickling begins, it tends to perpetuate itself until PO2 returns to normal; then it ceases spontaneously. The extent, severity, and clinical manifestations of sickling depend to a great extent on the percentage of hemoglobin that is Hb S. That is why homozygous inheritance of Hb S produces the severest form of SCD—sickle cell anemia. Heterozygous inheritance of SCD results in less sickling because the individual’s erythrocytes contain other forms of abnormal hemoglobin that although defective, do not participate in sickling to any great degree. Heterozygous inheritance (sickle cell trait), in which abnormal hemoglobin is inherited from one parent and normal hemoglobin from the other, rarely results in sickling because normal Hb F and Hb A do not participate in sickling at all. Anemia persists because Hb F does not live 120 days.
CLINICAL MANIFESTATIONS Clinical manifestations of SCD may first be seen at 6 to 12 months of age as fetal hemoglobin is replaced by Hb S. There are two characteristics of SCD that determine presentation. The first is its nature to be a chronic disease with acute exacerbations. The second is that it is a condition affecting red blood cells that supply oxygen to all cells of the body. Therefore, SCD can affect any part of the body. When sickling occurs, the general manifestations of hemolytic anemia—pallor, fatigue, jaundice, and irritability—sometimes are accompanied by acute manifestations called crises. Extensive sickling can precipitate four types of crises: (1) vasoocclusive (or thrombotic) crisis, (2) aplastic crisis, (3) sequestration crisis, or rarely (4) hyperhemolytic crisis. Sites of specific dysfunction are shown in Figure 28-8.
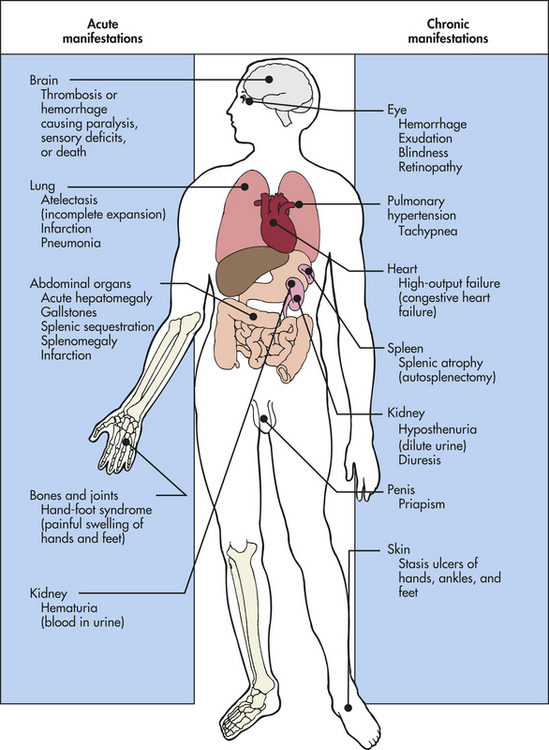
Vasoocclusive crisis (thrombotic crisis) begins with sickling in the microcirculation. As blood flow is obstructed by tangled masses of rigid, sickled cells, vasospasm occurs and a “logjam” effect brings all blood flow through the vessel to a halt. Unless the process is reversed, thrombosis and infarction (death caused by lack of oxygen) of local tissue follow. Vasoocclusive crisis is extremely painful and may last for days or even weeks, with an average duration of 4 to 6 days. The frequency of this type of crisis is variable and unpredictable.
Vasoocclusive crises may develop spontaneously or be precipitated by infection, exposure to cold, dehydration, low PO2, acidosis (low pH), or localized hypoxemia. Symmetric, painful swelling of the hands and feet (hand-foot syndrome) caused by infarction in the small vessels of the extremities often is the initial manifestation of SCD in infancy. In older children and adults the large joints and surrounding tissue become painful and swollen. Priapism (persistent erection of the penis) may occur if penile veins become obstructed. Severe abdominal pains often are caused by infarction in abdominal structures. Strokes resulting from cerebral occlusion may leave the child with paralysis (usually hemiplegia) or other CNS deficits.
Aplastic crisis, a transient cessation in red blood cell production resulting in acute anemia, occurs as a result of a viral infection, almost always infection with parvovirus B19 which is the virus responsible for the common childhood infection known as fifth disease. The virus causes temporary shutdown of red blood cell production in the bone marrow, or reticulocytosis. However, hemolysis, a component of SCD, continues. The outcome is a severe drop in hemoglobin with an extremely low reticulocyte count.
In sequestration crisis large amounts of blood become acutely pooled in the liver and spleen. This type of crisis is seen only in a young child. Because the spleen can hold as much as one fifth of the body’s blood supply at one time, mortality rates up to 50% have been reported, with death caused by cardiovascular collapse. If blood volume and pressure are maintained by hydration and blood transfusion, much of the sequestered blood eventually is remobilized. Removal of the spleen is the treatment for recurrent sequestration crises and may be performed after the child reaches 5 years of age.17
Hyperhemolytic crisis, an accelerated rate of red blood cell destruction, is unusual but may occur in association with certain drugs or infections. It is characterized by anemia, jaundice, and reticulocytosis. The concomitant presence of G6PD deficiency (see p. 1068) contributes to hyperhemolytic episodes, especially when combined with infections.
Acute chest syndrome is the presence of a new pulmonary infiltrate (involving at least one complete lung segment—not atelectasis) with chest pain, a temperature of more than 38.5° C (101.3° F) increased respiratory rate (tachypnea), wheezing, or cough in an individual with SCD. An injured, underventilated, and inflamed lung becomes “spleenlike” as sickled red cells attach to its endothelium, fails to be reoxygenated, and eventually undergoes more inflammation and lung infarction. The prognosis is poor, and infarction is a leading cause of morbidity. Acute chest syndrome is the cause of death in approximately 25% of all deaths in persons with SCD.17
Infection is the most common cause of death resulting from SCD. Sepsis and meningitis develop in as many as 10% of children with sickle cell anemia during the first 5 years of life, with a mortality rate of 25%. Survival time is unpredictable, and many young adults die in their 20s.
Glomerular disease, characterized by damage to the glomeruli allowing protein and often red blood cells to leak into the urine, can be caused by sickling of red blood cells in the kidneys, which also can damage the glomeruli. The earliest manifestation of SCD in the kidney is hyposthenuria, or the ability to concentrate urine. In young children this often results in bed-wetting. Extensive damage to the glomeruli will result in nephropathy that may progress to renal failure. Proteinuria is an early manifestation of sickle nephropathy.
Cholecystitis, inflammation of the gallbladder that occurs when a gallstone blocks the cystic duct, can be caused by hemolysis resulting in an increase of bilirubin, which in turn causes the formation of gallstones in the gallbladder. The presence of gallstones can cause right upper quadrant pain, nausea, vomiting, and an elevated white blood cell count and alkaline phosphatase. Cholecystectomy may be required.
Sickle cell–Hb C disease is usually milder than sickle cell anemia. The peripheral blood smear reveals many target cells resulting from the presence of Hb C. The main clinical problems are related to vasoocclusive crises and are believed to result from higher hematocrit values and viscosity. In older children, sickle cell retinopathy, renal necrosis, and aseptic necrosis of the femoral heads occur along with obstructive crises.
Sickle cell–thalassemia has the mildest clinical manifestations of all the SCDs. Even though most of the child’s hemoglobin is Hb S (60% to 90%), normal hemoglobins (Hb A and Hb F) also are present. The normal hemoglobins, particularly Hb F, inhibit sickling. In addition, the erythrocytes tend to be small (microcytic) and to contain relatively little hemoglobin (hypochromic). Their small size makes them less likely than normal-size cells to clog the microcirculation, even when in a sickled state.
The sickle cell trait does not affect life expectancy or interfere with daily activities. However, on rare occasions, severe hypoxia caused by shock, vigorous exercising at high altitudes, flying at high altitudes in unpressurized aircraft, or undergoing anesthesia is associated with vasoocclusive episodes in persons with sickle cell trait. These cells form an ivy shape instead of a sickle shape.
EVALUATION AND TREATMENT The parents’ hematologic history and clinical manifestations may suggest that a child has SCD, but hematologic tests are necessary for diagnosis. If the sickle solubility test confirms the presence of Hb S in peripheral blood, hemoglobin electrophoresis provides information about the amount of Hb S in erythrocytes. Prenatal diagnosis can be made after chorionic villus sampling as early as 8 to 10 weeks of gestation or amniotic fluid analysis at 15 weeks of gestation. Newborn screening for SCD should be performed according to state law.
Treatment advances over the past 25 years have significantly decreased morbidity and mortality in children with SCD. Aggressive management of fever, early diagnosis of acute chest syndrome (hypoxia, decreased hemoglobin, progressive multilobar pneumonia, fat emboli), judicious use of transfusions, and proper treatment of pain can improve quality of life and prognosis for these children. Treatment of SCD consists of supportive care aimed at preventing consequences of anemia and avoiding crises. Crises can be prevented by avoiding fever, infection, acidosis, dehydration, constricting clothes, and exposure to cold. Immediate correction of acidosis and dehydration with appropriate intravenous fluids is imperative. Infections require aggressive antibiotic therapy. Oxygen is not needed unless the child becomes hypoxic. Pain associated with SCD is very complex, requiring continuous adjustment of analgesics.
Protocols to implement individual-controlled analgesia in the emergency department shorten the time of initiation of narcotic therapy and are preferred by individuals.18 Therapeutic use of antisickling agents (urea, cyanate, carbamoyl phosphate) currently is regarded as unsafe and ineffective. Hydroxyurea increases Hb F synthesis in individuals with sickle cell anemia and increases hemoglobin and mean corpuscular volume while decreasing reticulocytes and bilirubin. It is safe and well tolerated and has been used successfully in children for more than 10 years.19 To avoid increased acidosis, acetaminophen is preferable to salicylates for antipyretic therapy. Immunization against influenza and pneumococcal microorganisms should be administered. Blood transfusion, including hypertransfusion therapy (e.g., packed red blood cells to raise the hematocrit to a level of 35% for a period of time), can be effective but must be weighed against the risks of hemosiderosis and iron and splenic overload. Oral maintenance therapy with folic acid is needed to meet the increased demands of chronic hemolytic anemia. Splenectomy may be performed if sequestration crises recur (Box 28-1). The most definitive approach to the treatment of SCD requires a permanent alteration in the hemoglobin phenotype. This can be accomplished through stem cell transplantation.
Genetic counseling and psychologic support are important for the child and family. Recently, a genetic technique called preimplantation genetic diagnosis has been performed on parents to diagnose whether their offspring will carry the gene for SCD. Figure 28-9 summarizes this prepregnancy sickle cell test. Genetic counseling enables people with SCD or trait to make informed decisions about transmitting this genetic disorder to their offspring because there is a 25% chance with each pregnancy that a child born to two parents with sickle cell trait will have SCD.
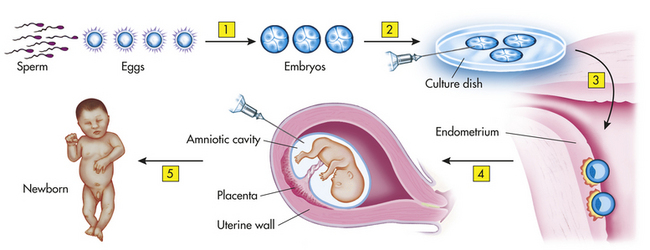
Figure 28-9 Prepregnancy sickle cell test. (This technique has potential for other inherited diseases.) 1, Fertilization produces several embryos. 2, The embryos are tested for the presence of the gene. 3, The embryo(s) without the gene are implanted. 4, Amniocentesis confirms whether the fetus (or fetuses) has the sickle cell gene. 5, Woman has a normal child.
Thalassemias
The alpha- and beta-thalassemias are inherited autosomal recessive disorders that cause an impaired rate of synthesis of one of the two chains—α or β—of Hb A. The disorder was named thalassemia, which is derived from the Greek word for sea, because it was defined initially in persons with origins near the Mediterranean Sea. Beta-thalassemia, in which synthesis of the β-globin chain is slowed or defective, is prevalent among Greeks, Italians, and some Arabs and Sephardic Jews. Alpha-thalassemia, in which the α-chain is affected, is most common among Chinese, Vietnamese, Cambodians, and Laotians. Both alpha- and beta-thalassemia are common among blacks.
Alpha- and beta-thalassemia can be major or minor, depending on how many of the genes that control α- or β-chain synthesis are defective and whether the defects are inherited homozygously (thalassemia major) or heterozygously (thalassemia minor). Pathophysiologic effects range from mild microcytosis to death in utero, depending on the number of defective genes and mode of inheritance. The anemic manifestation of thalassemia is microcytic-hypochromic hemolytic anemia.
PATHOPHYSIOLOGY Normally two genes control β-chain synthesis and four genes control α-chain synthesis. The number of genetic defects in the controlling genes determines the severity of the disorder. As in SCD the hemoglobin abnormality usually consists of the substitution of a single amino acid for another amino acid. Other molecular abnormalities that cause thalassemia are two amino acid substitutions, amino acid deletions or fusions, and synthesis of elongated chains.
The fundamental defect in beta-thalassemia is the uncoupling of α- and β-chain synthesis. β-Chain production is depressed—moderately in the heterozygous form, beta-thalassemia minor, and severely in the homozygous form, beta-thalassemia major (also called Cooley anemia). Depression of β-chain synthesis results in erythrocytes having a reduced amount of hemoglobin and accumulations of free α-chains. The free α-chains are unstable and easily precipitate in the cell. Most erythroblasts that contain precipitates are destroyed by mononuclear phagocytes in the marrow, resulting in ineffective erythropoiesis and anemia. Some of the precipitate-carrying cells do mature and enter the bloodstream, but they are destroyed prematurely in the spleen, resulting in mild hemolytic anemia.
There are four forms of alpha-thalassemia:
1. Alpha trait (the carrier state), in which a single α-chain–forming gene is defective
2. Alpha-thalassemia minor, in which two genes are defective
3. Hemoglobin H disease, in which three genes are defective
4. Alpha-thalassemia major, a fatal condition in which all four alpha-forming genes are defective; death is inevitable because α-chains are absent and oxygen cannot be released to the tissues
Beta-thalassemia occurs more commonly than does alpha-thalassemia. Occasionally synthesis of γ- or δ-polypeptide chains is defective, resulting in gamma- or delta-thalassemia. (Hemoglobin chains are described in Chapter 25.)
CLINICAL MANIFESTATIONS Beta-thalassemia minor causes mild to moderate microcytic-hypochromic anemia, mild splenomegaly, bronze coloring of the skin, and hyperplasia of the bone marrow. The degree of reticulocytosis depends on the severity of the anemia, resulting in skeletal changes. Hemolysis of immature (and therefore fragile) erythrocytes may cause a slight elevation in serum iron and indirect bilirubin levels. Persons with beta-thalassemia minor usually are asymptomatic.
Persons with beta-thalassemia major may become quite ill. Anemia is severe and results in a significant cardiovascular burden, with high-output congestive heart failure. In the past, death resulted from cardiac failure. Today, blood transfusions can increase life span by one to two decades, and death usually is caused by hemochromatosis (from transfusions). (Hemosiderosis and hematochromatosis are described in Chapter 26.) Liver enlargement occurs as a result of progressive hemosiderosis, whereas enlargement of the spleen is caused by extramedullary hematopoiesis and increased destruction of red blood cells. Spinal impairment that starts in infancy retards linear growth. Bone marrow hyperplasia causes a characteristic deformity of the facial bones, as the nasal bridge, mandible, and maxilla widen.
People who inherit the mildest form of alpha-thalassemia, the alpha trait, usually are symptom free, having, at most, mild microcytosis. Alpha-thalassemia minor has clinical manifestations that are virtually identical to those of beta-thalassemia minor: mild microcytic-hypochromic reticulocytosis, bone marrow hyperplasia, increased serum iron concentrations, and moderate splenomegaly.
Signs and symptoms of alpha-thalassemia are similar to those of beta-thalassemia major but milder. Moderate microcytic-hypochromic anemia, enlargement of the liver and spleen, and bone marrow hyperplasia are evident.
Alpha-thalassemia major causes hydrops fetalis and fulminant intrauterine congestive heart failure. In addition to edema and massive ascites, the fetus has a grossly enlarged heart and liver. Diagnosis usually is made postmortem. Prenatal screening for this disorder can be performed by use of chorionic villus sampling. These cells can be analyzed, and a deoxyribonucleic acid (DNA) genetic map can be constructed and evaluated for the abnormalities characteristic of hydrops fetalis.
Alpha-thalassemia major and beta-thalassemia major are life threatening. Children with thalassemia major generally are weak, fail to thrive, show poor development, and experience cardiovascular compromise with high-output failure secondary to anemia. Untreated, they will die by 5 to 6 years of age.
EVALUATION AND TREATMENT Evaluation of thalassemia is based on familial disease history, clinical manifestations, and blood tests. Peripheral blood smears that show microcytosis and hemoglobin electrophoresis that demonstrates diminished amounts of α- or β-chains are used to make the diagnosis. Analysis of fetal DNA from withdrawn amniotic fluid is used as a screening test to detect hydrops fetalis (alpha-thalassemia major). Newborn screening for thalassemia should be done according to state law.
“Silent” carriers or those who have thalassemia minor generally have few if any symptoms and require no specific treatment. Therapies to support and prolong life are necessary, however, for thalassemia major. There is no cure for either condition. Prenatal diagnosis and genetic counseling may be the most important therapeutic measures offered.
At present, thalassemia major is treated with the following therapies:
1. Blood transfusions, which can return hemoglobin and hematocrit levels to normal thus alleviating the anemia-induced cardiac failure; iron overload and hemochromatosis are complications of transfusion therapy
2. Iron chelation therapy in combination with hypertransfusion (transfusion to a hematocrit of 35 ml/dl)
3. Splenectomy, which can reduce the need for transfusions by eliminating the site of hemolysis, thus prolonging erythrocyte survival
4. Bone marrow, cord blood, and stem cell transplantation is currently the only cure for thalassemia (Box 28-2)
DISORDERS OF COAGULATION AND PLATELETS
Hemophilias
Awareness of a serious bleeding disorder in males was documented nearly 2000 years ago in the Babylonian Talmud, which exempted from the rite of circumcision those boys having male relatives prone to excessive bleeding. In 1803 the first description of this disorder appeared in the medical literature, where it was noted to be X linked in nature and associated with joint bleeding and crippling.
Table 28-4 lists the coagulation factors. Until 1952 the term hemophilia was reserved for deficiency of factor VIII (antihemophilic factor). Since then two additional coagulation proteins, factor IX (plasma thromboplastin component [PTC]) and factor XI (plasma thromboplastin antecedent [PTA]), have been identified and their deficiency associated with similar clinical manifestations. Congenital deficiencies of these three plasma clotting factors—VIII, IX, XI—account for 90% to 95% of the hemorrhagic bleeding disorders collectively called hemophilia.
Table 28-4
| Clotting Factors | Synonym | Disorder |
| I | Fibrinogen | Congenital deficiency (afibrinogenemia) and dysfunction (dysfibrinogenemia) |
| II | Prothrombin | Congenital deficiency or dysfunction |
| V | Labile factor, proaccelerin | Congenital deficiency (parahemophilia) |
| VII | Stable factor or proconvertin | Congenital deficiency |
| VIII | Antihemophilic factor (AHF) | Congenital deficiency is hemophilia A (classic hemophilia) |
| IX | Christmas factor | Congenital deficiency is hemophilia B |
| X | Stuart-Power factor | Congenital deficiency |
| XI | Plasma thromboplastin antecedent | Congenital deficiency, sometimes referred to as hemophilia C |
| XII | Hageman factor | Congenital deficiency is not associated with clinical symptoms |
| XIII | Fibrin-stabilizing factor | Congenital deficiency |
Table 28-5
Prognostic Factors in Acute Lymphoblastic Leukemia (ALL)
| Prognostic Factor | Better Prognosis | Worse Prognosis |
| Age∗ | ||
| <2 yr or >10 yr | X | |
| 2-7 yr | X | |
| Gender∗ | ||
| Male | X | |
| Female | X | |
| Initial white blood count∗ | ||
| >50,000/mm3 | X | |
| <10,000/mm3 | X | |
| Race | ||
| Black | X | |
| White | X | |
| Immunology∗ | ||
| T- or B-cell ALL | X | |
| Early pre–B-cell or common acute lymphocytic leukemia antigen (CALLA) | X | |
| Leukemic involvement | ||
| Mediastinal mass | X | |
| Central nervous system involvement at diagnosis | X | |
| Splenic enlargement | X |
∗The four most reliable prognostic factors. Initial prognostic factors become less effective predictors with increasing length of remission. Age and gender are not significant after 15 months of continuous remission, and white blood cell count is not significant after 24 months of continuous remission.
Types of Hemophilia: It is estimated that hemophilia occurs in 1 in 5000 male births. Eighty percent to 85% of those with hemophilia have hemophilia A and 10% to 15% have hemophilia B.20
Hemophilia A (classic hemophilia) is caused by factor VIII deficiency. It is the most common of the hemophilias. Hemophilia A is inherited as an X-linked recessive disorder that affects men and is transmitted by women.
Hemophilia B (Christmas disease), caused by factor IX deficiency, also is transmitted as an X-linked recessive trait and is clinically indistinguishable from factor VIII deficiency. Hemophilia A and hemophilia B occur with varying degrees of clinical severity, depending on concentrations of clotting factor VIII or IX in the blood. Severe hemophilia (concentration of clotting factors less than 1% of normal) is associated with spontaneous bleeding. In moderate hemophilia (1% to 5% of normal), bleeding usually occurs only after trauma; in the mild form (5% to 35% of normal), bleeding occurs only after severe trauma or surgery. The severity of hemophilia is similar in all affected members of a family.
Hemophilia C (factor XI deficiency) occurs as an autosomal recessive disease and occurs equally in men and women. Bleeding usually is less severe than in hemophilia A or B.
von Willebrand disease results from an inherited autosomal dominant trait with variable clinical manifestations and hematologic findings. The factor VIII deficiency differs from that of hemophilia A in mode of inheritance and response to treatment. In hemophilia A the deficiency is inherited as an X-linked recessive trait, whereas in von Willebrand disease, it is inherited as an autosomal dominant trait. The most important difference, however, is in responses to the infusion of plasma. In von Willebrand disease, infusion of plasma causes factor VIII activity to increase for several days because infusion of factor VIII temporarily induces endogenous synthesis of factor VIII.
PATHOPHYSIOLOGY Two types of defects dominate the hereditary defects of hemophilia to date: gene deletions and point mutations. Both types of genetic defects are associated with severe hemophilia A, in which no factor VIII circulates in the blood. Many different gene deletion mutations are associated with factor VIII and factor IX disease. The molecular defect that leads to the deletional mutation is identical among members of a given family.21
Point mutations, in which a single base in the DNA is mutated to another base, represent a second type of mutation that causes hemophilia. When a point mutation gives rise to a de novo stop codon (nonsense mutation), translation of the protein ceases and a shortened version of the protein is synthesized. Usually the protein is destroyed intracellularly and never reaches the plasma. This type of defect is associated with severe hemophilia, that is, with coagulant activity levels below 1%. Point mutations in which one amino acid is substituted for another can cause phenotypes of varying severity. The mutation of an important amino acid can destroy protein function, activation, or folding; inhibit intracellular processing; or cause protein clearance.22 Unlike deletional mutations, point mutations at the same site have been recorded in different families with hemophilia.
Table 28-4 summarizes the types of coagulation disorders. Not all the disorders are discussed in this chapter because some are extremely rare (congenital dysfibrinogenemias) and others have no clinical significance (e.g., Hageman factor deficiency, a condition in which profound laboratory deficiency of factor XII is associated with absolutely no clinical defects).
CLINICAL MANIFESTATIONS Children with severe hemophilia start to bleed at different ages. Although there is no transfer of maternal clotting factor to the fetus, many boys with hemophilia are circumcised without excessive bleeding. Normal hemostasis is achieved in these infants because clotting is activated through the extrinsic coagulation cascade, which does not involve factors VII, IX, or XI.
During the first year, spontaneous bleeding often is minimal, but hematoma formation may result from injections and from firm holding (e.g., under the arms). Many children present when reaching developmental milestones (i.e., crawling, pulling to stand) and become easily injured (i.e., increased bruising, swelling, redness at joints, mouth bleeding). By 3 to 4 years of age, 90% of children with hemophilia have had episodes of persistent bleeding from relatively minor traumatic lacerations (e.g., to the lip or tongue). This usually is the first clinical manifestation of hemophilia. Hemorrhage into the elbows, knees, and ankles cause pain, limit joint movement, and predisposes the child to degenerative joint changes. Spontaneous hematuria and epistaxis are troublesome but minor complications.
Recurrent bleeding—spontaneous and after minor trauma—is a lifelong problem. Many affected individuals experience phases or cycles of spontaneous bleeding episodes. Mechanisms that cause this phenomenon are unknown. Intracranial hemorrhage and bleeding into the neck or abdomen constitute life-threatening emergencies.
EVALUATION AND TREATMENT Although laboratory tests are of primary value in the evaluation of hemorrhagic disorders, the history and physical assessment also should be given careful consideration. The three phases of coagulation can be assessed individually by simple, reliable tests. In any hemorrhagic condition, the adequacy of phase III should be determined first. Unless adequate fibrinogen is present, the blood is incapable of coagulation; thus other laboratory tests that require formation of a visible clot will be invalid. Phase III can be evaluated by the thrombin time, the time required for plasma to clot after the addition of bovine thrombin. Fibrinogen can be measured by chemical or immunologic methods.
Phase II is assessed by the prothrombin time (PT), the time required for plasma to clot after the addition of thromboplastin and calcium. If phase III is intact, a prolonged prothrombin time indicates a deficiency involving factors II, V, VII, or X, alone or in combination. Specific assays for each of the factors are available.
Phase I, the most complex part of coagulation, can be evaluated by several tests. The activated partial thromboplastin time (aPTT) is the time required for clotting of plasma that has been activated by incubation with kaolin when calcium and platelets (or partial thromboplastin) are added. aPTT assesses the adequacy of factors XII, XI, IX, and VII. The prothrombin consumption time is a standard prothrombin test of serum instead of plasma. Because prothrombin is used up during coagulation, the serum normally contains little prothrombin and the serum prothrombin time is prolonged. Deficiencies of the phase I factors are associated with poor use of prothrombin. If the serum and plasma prothrombin times are similar, deficiency of a phase I factor is likely. The thromboplastin generation test is the most sensitive of all phase I tests. The test can precisely identify deficiencies of factors VIII and IX. If the aPTT, prothrombin consumption, or thromboplastin generation test results are abnormal, the way in which they can be corrected identifies the specific deficiency.
The treatment of hemophilia has advanced during the past 70 years. Plasma first was used in the 1920s, and by the 1940s it was used routinely to treat persons with hemophilia. The disadvantages of fresh frozen plasma (FFP), which is low in factor VIII per volume of plasma, led to the development of cryoprecipitate. In 1964 cryoprecipitate (quick-frozen precipitate), which is rich in factor VIII per volume, was used to treat persons with hemophilia. Although cryoprecipitate advanced the treatment of hemophilia A, it has several disadvantages. The most notable complication is the possibility of transmission of viral diseases. Factor VIII concentrates were first introduced in 1965. In addition to the predictable factor VIII content, other advantages of the early factor VIII concentrates included greater purity than cryoprecipitate and less contamination with other plasma proteins. The development of recombinant factor VIII resulted in new factor VIII products that minimize the risk of transmission of viral infection (e.g., HIV and hepatitis) and are potentially less expensive than plasma-derived factor VIII.
Recombinant antihemolytic factor plasma/albumin-free method (rAHF-PFM, Advate) is a product used for the prevention and control of bleeding episodes in individuals with hemophilia A, and in the perioperative management of those with hemophilia A. By excluding proteins or raw materials derived from human or animal sources in the final product, the risk of transmission of potentially infectious agents is removed.23
Primary prophylaxis consists of regular infusions of factor VIII or IX with the goal of preventing joint bleeding. It is usually given to children with severe hemophilia. A 5-year, multicenter trial found prophylaxis initiated in children between 6 and 30 months of age to be effective in the prevention of joint bleeding, structural joint damage, and frequency of bleeding in boys with factor VIII deficiency.24
Congenital Hypercoagulability and Thrombosis: Hereditary bleeding disorders, such as hemophilia, have been recognized and treated for centuries; however, the counterpart of these disorders, thrombophilia, has not been recognized until very recently. The inherited thrombophilic conditions generally are caused by defects in the clotting factors that inhibit clot formation; thus the balance between bleeding and clotting is directed toward the clotting aspects of hemostasis. Defects in specific proteins (C and S) and antithrombin (AT), as well as resistance to activated protein C (APC) and hyperhomocystinemia, are the major recognized causes of inherited thrombophilia.
Both proteins C and S are inhibitors of coagulation and depend on vitamin K for synthesis in the hepatocytes of the liver. Decreased levels of either of these proteins interfere with the normal homeostatic balance of procoagulant and anticoagulant activity at the endothelial level. Protein C and S deficiency states predispose affected individuals to thrombosis, especially venous thrombosis of the lower extremities.
Inheritance of protein C deficiency is autosomal dominant. Heterozygotes have protein C levels 50% to 60% of normal and may develop superficial thrombophlebitis, deep venous thrombosis, or pulmonary embolism in their late teens and early 20s. The majority of these thrombotic events (75%) occur spontaneously, whereas only 25% are the result of predisposing conditions.25 Homozygotes have less than 1% of normal levels of protein C and tend to develop thrombosis of the cutaneous vessels with large areas of skin necrosis. It is rare for individuals with protein C deficiency to develop arterial thrombosis.
Protein C deficiency exists in two forms: types I and II. Type I, the more common form, involves a reduction in both biologic and immunologic activity of protein C. Type I is caused by deletion of the entire gene. In type II, the less common form, there is a normal level of protein C antigen but decreased functional levels of activity.
Neonatal purpura fulminans is a fatal syndrome found in infants who are homozygous or double heterozygous for types I and II protein deficiency. Manifestations of this syndrome are ecchymosis that becomes apparent on the first day of life and develops around the head, trunk, and extremities. These cutaneous manifestations often are accompanied by cerebral thrombosis and infarction. The lesions apparent on the skin often coalesce and demonstrate ulceration and necrosis. The condition is treated with fresh frozen plasma and heparinization, although the infant rarely survives.25
Treatment for protein C deficiency is heparin for acute episodes of thrombosis. Long-term therapy is required and consists of either oral warfarin sodium (Coumadin) or subcutaneous heparin (2500 to 5000 units) every 12 hours. Protein C concentrates have been developed; however, they have not yet been formally approved for treatment.
Protein S deficiency is similar to protein C deficiency, and the inheritance pattern (autosomal dominant) is also similar. Heterozygotes demonstrate a strong tendency for deep venous thrombosis, with the first incidence often occurring before age 25 years. Other manifestations include superficial thrombophlebitis and pulmonary emboli. There are predisposing conditions for thrombi development in some cases, with evidence of spontaneous thrombi development in most cases.
Protein S deficiency exists in two forms: type I and type II. Type I is identified as a quantitative deficiency and manifests as low levels of protein S antigen and activity, and type II is identified as a qualitative deficiency with low levels of free protein S and normal levels of free and total protein S antigen.25
Homozygotes demonstrate severe manifestations of the condition and may develop a form of purpura fulminans in the neonatal period. It also is possible that the homozygous state may lead to uterine death. Treatment with heparin and warfarin (Coumadin) is similar to that of protein C deficiency.
Antithrombin III (AT III) deficiency is inherited as an autosomal dominant condition, with the heterozygote state being the most common. AT III also exists in two forms, type I and type II, with type I being a quantitative deficiency of the AT III antigen. Type II is characterized as a dysfunctional form: normal levels of AT III are present but with reduced activity.
Individuals with AT III deficiency are at risk for early development of venous thrombosis and pulmonary embolism. These events often occur in the middle to late teens, and can occur as early as 10 years of age. The deep veins of the lower extremities most commonly are involved, with the iliofemoral vein being the most common site of involvement. Other sites include the mesenteric veins, vena cava, renal veins, and retinal veins. Cerebral thromboses also have been described. Arterial thrombotic events are rare. In some cases, thrombosis is precipitated by predisposing conditions, such as surgery, trauma, pregnancy, oral contraceptives, and infection.
The treatment of choice for AT III deficiency is heparin. Antiplatelet agents (e.g., aspirin, dipyridamole) may be used, as well as AT III concentrates.
Antibody-Mediated Hemorrhagic Disease
The antibody-mediated hemorrhagic diseases are a group of disorders caused by the immune response. Antibody-mediated destruction of platelets or antibody-mediated inflammatory reactions to allergens damage blood vessels and cause seepage into tissues. The thrombocytopenic purpuras may be intrinsic or idiopathic, or they may be transient phenomena transmitted from mother to fetus. The inflammatory, or “allergic,” purpuras occur in response to allergens in the blood. All these disorders first appear during infancy or childhood.
Idiopathic Thrombocytopenic Purpura: Acute idiopathic thrombocytopenic purpura (ITP) (autoimmune or primary thrombocytopenic purpura) is the most common of the thrombocytopenic purpuras of childhood. It is a disorder of platelet consumption in which antiplatelet antibodies bind to the plasma membranes of platelets, causing platelet sequestration and destruction by mononuclear phagocytes in the spleen and other lymphoid tissues at a rate that exceeds the ability of the bone marrow to produce them.
PATHOPHYSIOLOGY Platelets have several tissue-specific antigens on their plasma markers that may be targets for antiplatelet antibody. In approximately 70% of cases of ITP, there is an antecedent viral disease (e.g., cytomegalovirus [CMV], Epstein-Barr virus [EBV], human immunodeficiency virus [HIV], parvovirus, or viral respiratory infection), thus suggesting that viral sensitization has occurred. The interval between infection and onset of purpura is 1 to 4 weeks. A comparison with purpura seen in adults has identified an immune mechanism as the basis for ITP. High levels of IgG have been found bound to platelets and may represent immune complexes on the platelet surface.26
CLINICAL MANIFESTATIONS One to 4 weeks after a viral infection, bruising and a generalized petechial rash often occur with acute onset. Asymmetric bleeding is typical and is found most often on the legs and trunk. Hemorrhagic bullae of the gums, lips, and other mucous membranes may be prominent. Epistaxis (nose bleeding) may be severe and difficult to control. Except for the signs of bleeding, the child appears well. The acute phase of the disease associated with spontaneous hemorrhages lasts 1 to 2 weeks, but thrombocytopenia often persists. Although its incidence is less than 1%, intracranial hemorrhage is the most serious complication of ITP. In some cases the onset is more gradual and clinical manifestations consist of moderate bruising and a few petechiae.
EVALUATION AND TREATMENT Laboratory examination reveals a reduced platelet count, and the few platelets observed on a peripheral blood smear are large in size, reflecting increased bone marrow production. The Ivy bleeding time is prolonged. Bone marrow aspiration reveals megakaryocytes in normal or increased numbers and normal erythrocytes and granulocytes.
Even without treatment, the prognosis for children with ITP is excellent—75% recover completely within 3 months. After the initial acute phase, spontaneous clinical manifestations subside. By 6 months after onset, 80% to 90% of affected children have regained normal platelet counts.27
Because of the short life span of platelets (10 days), fresh blood or platelets are of no value or of transient benefit; however, their use is indicated when life-threatening hemorrhage occurs. Corticosteroid therapy reduces the severity and shortens the duration of the initial phase by suppressing the immune attack on platelets. Recent evidence indicates that the use of high-dose methylprednisolone increases bone resorption and may cause osteonecrosis in children with ITP.28 Intravenous IgG has been demonstrated to increase the platelet count in some children with ITP, but it is quite costly (see What’s New? Monoclonal Antibody Therapy for Treatment of Idiopathic Thrombocytopenic Purpura [ITP]). A newer product, anti-D, is a gamma globulin fraction containing a high proportion of antibodies to the RhO(D) antigen of the red blood cells. Intravenous anti-D is a safe and effective treatment for Rh-positive, nonsplenectomized individuals with ITP, although it is expensive. Intravenous anti-D is an effective treatment for Rh-positive nonsplenectomized children with ITP, although it is associated with side effects including chills, fever, headache, and a decrease in hemoglobin levels. Administration of steroids and antipyretics prior to the anti-D treatment may prevent side effects.27
Parents should be instructed to protect the child from falls or other trauma that might result in bleeding. Splenectomy should be reserved for chronic cases that fail to respond to nonsurgical intervention.
Autoimmune Neonatal Thrombocytopenias: Antibody-mediated thrombocytopenic purpura occurs in neonates in either autoimmune or alloimmune form. Both forms are characterized by the immunologic destruction of platelets by antibodies (IgG) against tissue-specific antigens expressed by the platelets (i.e., platelet-specific antigens).
Autoimmune neonatal thrombocytopenia was first noted in the early 1950s when it was observed that mothers with ITP often delivered infants who were transiently thrombocytopenic. Neonatal thrombocytopenia was observed in approximately 50% of infants at risk and lasted an average of 1 month. As platelet counts returned to normal, a concomitant drop in the level of maternal antiplatelet antibody on the child’s platelets occurred. The antibody is directed against antigens common to maternal and neonatal platelets. The prognosis generally is favorable. The frequency of intracranial
hemorrhage has been estimated to be 1% to 3% of cases. The principal aim of the management of affected infants is to prevent the deleterious consequences of severe thrombocytopenia by administering intravenous immunoglobulins.29
Neonatal alloimmune thrombocytopenic purpura (NATP) is less common, estimated to occur in 1 in 800 to 1000 live births. NATP is suspected in thrombocytopenic infants of mothers with normal platelet counts and no history of purpura. The disorder is caused by the production of a maternal antibody against a fetal platelet-specific antigen inherited from the father and not shared by the mother. More than 50% of NATP cases are associated with the presence of the P1A1 antigen on neonatal and paternal platelets but not on maternal platelets.
It is not known why NATP occurs in only half of the neonates genetically at risk for NATP. Because 98% of the population show P1A1 positivity, approximately 1 in 50 pregnancies would be expected to show maternal-fetal incompatibility, but the incidence of NATP is 100 times less. NATP does not develop in neonates born to some mothers with high antiplatelet antibody levels.
The diagnosis of NATP is confirmed by detection in the maternal serum of antibody that reacts with platelets from the infant and father but not with platelets from the mother. In approximately 75% to 85% of cases, NATP recurs in subsequent pregnancies. Purpura usually develops in the affected infant shortly after delivery, and intracranial, renal, and gastrointestinal hemorrhages are possible. The mortality rate from intracranial hemorrhage has been estimated at 10% to 15%. Following birth, maternal platelet transfusion (mother to infant) is the treatment of choice.29
Most of the life-threatening clinical manifestations of transient neonatal thrombocytopenia and NATP can be avoided through cesarean delivery. If the mother has antiplatelet disease, however, surgery can result in hemorrhage and serious maternal morbidity. Maternal morbidity resulting from NATP during pregnancy is low (less than 5%): the principal maternal risk is bleeding from surgical incisions during cesarean delivery. This poses a problem for the obstetrician. The incidence of transient thrombocytopenia in infants born to mothers with NATP is about 50%. If all deliveries were cesarean, half the mothers would undergo cesarean delivery unnecessarily. Conversely, if all deliveries were vaginal, half the infants—those with thrombocytopenia—would be at risk for intracranial bleeding.
A considerable amount of research has focused on methods of predicting whether the fetus is thrombocytopenic so that the route of delivery can be chosen to minimize the risks for both mother and child. No satisfactory method has been found, despite reports from many laboratories that fetal platelet counts correlate closely with levels of antiplatelet antibody on maternal platelets or in the maternal circulation. Equally unreliable are predictions of neonatal thrombocytopenia based on immunosuppression with corticosteroids. Research continues in areas such as the identification of specific subclasses of antiplatelet antibodies.
Autoimmune Vascular Purpura: Autoimmune vascular purpura (allergic purpura) is caused by antibody-mediated injury of blood vessel walls, typically arterioles and capillaries. The inflammatory reaction is to foreign proteins or chemicals in the blood (microorganisms, drugs, or other chemicals).
Autoimmune vascular purpura usually is seen in children, with the incidence decreasing in adolescents and adults and occurring only rarely in older adults. The average age at onset is 5 years, with a slightly higher proportion of males affected. Purpura occurs as vessel integrity is disrupted by inflammatory processes, causing effusion of serosanguineous exudate to perivascular tissues.
Clinical manifestations vary and include headache, anorexia, fever, abdominal pain, arthralgias, and skin lesions (urticaria and erythema). The lesions usually are located symmetrically on the proximal portions of the extremities, particularly on the legs and buttocks, and may be accompanied by itching or paresthesias. Abdominal pain results from hemorrhage into the bowel, which may lead to colic, nausea, and vomiting. These symptoms may precede the appearance of skin lesions. The pain usually is midabdominal but may radiate to other parts of the abdomen. Constipation may occur.
Some forms of autoimmune vascular purpura may produce joint pain and tenderness. Periarticular swelling and edema of the hands and feet are common, but hemarthrosis does not occur. These symptoms may precede the onset of symptoms associated with abdominal pain and purpura. Subacute glomerulonephritis occurs in some cases but usually is reversible.
The characteristic skin lesions (purpura and cutaneous manifestations of allergy), accompanied by a history of joint and abdominal pain, are clues for diagnosis. Laboratory test results often reveal no major abnormalities. Attacks may last several weeks and may recur at odd intervals and with changing manifestations with each episode. Treatment, if necessary, consists of the alleviation of symptoms.
LEUKEMIA AND LYMPHOMA
Leukemia, the most common malignancy of childhood, represents approximately 40% of all childhood cancers. Childhood lymphoma is the third most common malignant neoplasm of children in the United States, representing approximately 11% of all childhood cancers. (See Chapter 27 for a discussion of leukemia in adults.)
Leukemia
Of the varieties of childhood leukemia, 80% to 85% of leukemias in children are acute lymphocytic leukemia (ALL) or acute undifferentiated leukemia (AUL). The remaining 15% to 20% are acute nonlymphocytic leukemias (ANLLs) (which include myeloblastic, promyelocytic, monocytic, and myelomonoblastic leukemias) and the very rare red blood cell leukemia, erythroleukemia. Because the vast majority of ANLLs involve the myeloblastic cell, many experts refer to the disease as acute myelogenous leukemia (AML). Leukemia accounts for 25% of cases of cancer in black children and 34% of cases of cancer in white children. Approximately 4900 new cases are diagnosed each year in the United States. Of those 4900 children, 75% to 80% are diagnosed with ALL.30
The peak incidence for childhood ALL is between 2 and 6 years of age. ALL affects more white and Hispanic than black children and more males than females. Male predominance is greater in T-cell disease. There also is a higher incidence of ALL in Western and industrialized nations.30
Types of Leukemia
A number of different classifications are used for the leukemias. First, acute leukemia is differentiated from chronic leukemia. Second, the cell line determines whether lymphoid cells or myeloid cells are involved. In acute leukemia this difference separates ALL from AML and vice versa. Within each of these categories, further subdivisions have been developed. (See Chapter 27 for a discussion of leukemias in adults.)
Cytogenic studies of leukemic cells are performed routinely at most major treatment centers during the diagnostic process. Abnormal morphologic characteristics, as well as abnormalities in the number of copies of chromosomes, are found in leukemic cells. Hyperdiploidy (increased number of chromosome copies) is associated with a good prognosis. Common translocations associated with ALL are TEL-AML1, BCR-ABL, and MLL. TEL-AML1 is the most common abnormality (in 20% to 30% of cases) and occurs when the TEL gene on chromosome 12p13 fuses with the AML1 gene on chromosome 21q22. TEL-AML1 is associated with a favorable outcome. MLL arrangement, t(4;11), is located on chromosome 11q23. This translocation, the most common within this subtype, is found in infant ALL and is associated with a poor prognosis despite intensive therapy. Philadelphia chromosome positive (Ph+) leukemia expresses the BCR-ABL protein and is characterized by the presence of t(9;22)(q34;q11) translocation. Ph+ leukemia can be ALL or chronic myelocytic leukemia (CML), depending where the breakpoint on chromosome 22 occurs. In CML, the translocation can be detected in multiple cell lines. Ph+ ALL occurs in 2% to 3% of cases and responds poorly to conventional chemotherapy. In CML, 99% of cases are characterized by the presence of t(9;22).31
Classification of childhood leukemia has become a complex but essential process that determines treatment. Previous classification by the French-American-British Cooperative Group (FAB) was based primarily on the morphologic and biochemical system. A classification scheme developed by the World Health Organization (WHO) is based on a more comprehensive system that uses morphology, immunophenotyping, and cytogenic and clinical features. The three major classifications of childhood leukemia are ALL (75% to 80%), AML (15% to 20%), and CML (less than 5%).
Flow cytometric immunophenotyping has made distinguishing between lymphoblastic and nonlymphoblastic leukemia much easier than in the past, when the degree of immaturity of the cell sometimes made such distinction difficult.
Immunologic classification has been used on identification of various surface markers. Five categories of ALL have been identified on the basis of their presumed origin from thymic cells (T cells) and bursa-equivalent cells (B cells) of normal lymphocytes:
1. T-cell ALL—characterized by the presence of abnormal T lymphocytes and found more commonly in older boys whose diagnosis includes mediastinal masses, high white blood cell counts, and hepatosplenomegaly (20% of ALL)
2. B-cell ALL—characterized by the presence of abnormal B lymphoblasts and associated with a poor prognosis (5% of ALL)
3. Pre–B-cell ALL—characterized by the presence of pre-B lymphoblasts (20% of ALL)
4. Unclassified ALL—also known as null cell (meaning neither T nor B lymphoblasts), and now classified as early B-cell lineage (15% of ALL)
5. Common ALL—characterized by the presence of a specific antigen known as common ALL antigen, or common acute lymphocytic leukemia antigen (CALLA), recently designated cellular differentiation 10 or CD10, in which the actual cell usually is considered to be of the B lineage (39% of ALL)
PATHOGENESIS The exact cause of childhood leukemia is unclear. Investigations have focused on genetic susceptibility, environmental factors, and viral infections32 (see Chapter 13). Observations of a familial tendency and links with a number of inherited disorders have implicated genetic factors in the origin of leukemia. Analysis of leukemia in twins has revealed a frequent prenatal origin and an early or initiating role for chromosome translocations. In addition, twin studies also suggest that there is a protracted latency and the need in ALL and AML for postnatal exposures or genetic events to produce clinical disease.33 A positive family history of hematopoietic malignancies among first- or second-degree relatives has been associated with a slight increase for risk for childhood ALL, although it is modest (odds ratio 2.06).34
Inherited diseases that predispose a child to leukemia (ALL and AML) include Down syndrome, Fanconi anemia, Bloom syndrome, Diamond-Blackfan anemia, Klinefelter syndrome, Shwachman-Diamond syndrome, and ataxia-telangiectasia. Leukemia also has been associated with known genetic diseases, such as congenital agammaglobulinemia. AML in children sometimes is associated with loss or deletion of chromosome 7. AML can develop from preexisting myeloproliferative disorders that also are preleukemia syndromes (i.e., myelodysplastic syndrome).
Childhood exposure to ionizing radiation, drugs, or viruses has been associated with the risk of developing cancer. Retrospective research has shown a significant correlation between radiation-induced malignancies from radiotherapy (as cancer treatment) or from radiation exposure from diagnostic imaging. The relationship between childhood cancer and electromagnetic fields, small appliances, radon, and other sources has been the focus of many epidemiologic studies; however, no conclusive evidence has been observed.32
Although chemicals such as benzene have been associated with the development of AML in adults, no evidence suggests a similar chemical or drug association in childhood leukemia. Leukemia (primarily AML) has been reported as a secondary malignancy (development of a second cancer after the first) in children treated for Hodgkin disease and Wilms tumor, although such cases are rare. In most cases the children received chemotherapy (alkylating agents or dactinomycin) and radiation therapy for the primary cancer, perhaps accounting for the subsequent development of another cancer.
Leukemic “clusters” that represent a greater number of leukemia cases occurring in a particular geographic location have raised speculation about environmental factors or infectious patterns of transmission. Careful follow-up, however, has failed to document the abnormal clustering.35
The strongest association between viruses and the development of cancer in children has been EBV and Burkitt lymphoma, and nasopharyngeal carcinoma and Hodgkin disease. Children with AIDS have an increased risk of developing non-Hodgkin lymphoma and Kaposi sarcoma. However, with the use of highly active antiretroviral therapy in the developed world, the incidence of acquired immunodeficiency syndrome (AIDS)–related malignancies has declined dramatically.36 Retroviruses have not been linked with childhood leukemia.
CLINICAL MANIFESTATIONS Few variations appear in the presenting symptoms of the various cell types of acute leukemias. The onset may be abrupt or insidious, but the most common symptoms reflect the consequence of bone marrow failure, which results in decreased red blood cells and platelets and changes in white blood cells. Pallor, fatigue, petechiae, purpura, bleeding, and fever generally are present. Approximately 45% of children have a hemoglobin level below 7 g/dl; in contrast to adults, children seem to demonstrate fewer symptoms. If acute blood loss occurs, however, characteristic symptoms of tachycardia, air hunger, restlessness, and thirst may be present. Epistaxis, excessive bruising, and hematuria often occur in children with severe thrombocytopenia. Three fourths of children with ALL have platelet counts less than 100,000/mm3 at diagnosis, and 28% have platelet counts below 20,000/mm3. Half of all children newly diagnosed with AML have platelet counts less than 50,000/mm3. Disseminated intravascular coagulation occurs more commonly with AML, particularly with promyelocytic leukemia. The granules in the leukemic promyelocytes may then indicate thromboplastin activity.
Fever usually is present as a result of two causes: (1) infection associated with the decrease in functional neutrophils and (2) hypermetabolism associated with the ongoing rapid growth and destruction of leukemic cells. In most children with ALL, the total white blood count is less than 10,000/mm3, and with AML most have white cell counts below 50,000/mm3. In a few children, however, the peripheral white blood count can go well above 100,000/mm3. White blood cell counts greater than 200,000/mm3 can cause leukostasis, an intravascular clumping of cells that result in infarction and hemorrhage, usually in the brain and lung. The three most important favorable prognostic factors are age at diagnosis (2 to 10 years), initial leukocyte count (less than 50,000/mm3), and initial response to treatment.
Renal failure as a result of hyperuremia (high uric acid levels) can be associated with ALL, particularly at diagnosis. Cell breakdown results as a natural process in the presence of a high white blood cell count or as a result of cellular breakdown caused by chemotherapy. Uric acid levels rise as an end product of purine metabolism from cellular destruction. Because the major excretory pathway is through the kidney, urates can precipitate in renal tubules or ureters and can lead to oliguria and acute renal failure. Renal failure is preventable if uric acid levels are monitored and treatment is aimed at optimal hydration, alkalinization of urine to assist with the excretion of soluble urates, and blockage of further uric acid formation by administration of the drug allopurinol.
Extramedullary invasion with leukemic cells can occur in nearly all body tissue. Most children with ALL have some extramedullary involvement at diagnosis. Leukemic invasion of tissue other than bone marrow is believed to represent metastatic infiltration. Hepatosplenomegaly and lymphadenopathy, resulting from extramedullary hematopoiesis, occur in nearly one half of children with ALL, but they are less common in children with AML.
The CNS is a common site of infiltration of extramedullary leukemias, although less than 10% of children with ALL have CNS involvement at diagnosis. CNS infiltration manifests later in the course of the disease. Because successful chemotherapy prolongs the time of remission, the incidence of CNS involvement has increased. The most common symptoms of CNS involvement relate to increased intracranial pressure, causing early morning headaches, nausea, vomiting, irritability, and lethargy. Prophylactic CNS treatment therefore is necessary because systemic treatment with chemotherapy does not cross the blood-brain barrier.
Gonadal involvement, with testicular and ovarian infiltration, has been demonstrated in postmortem examination in 57% and 35% of children, respectively. Clinical detection of gonadal involvement is much less frequent. The incidence of testicular involvement, like CNS involvement, has increased with lengthened duration of remission. Prophylactic treatment has not been successful and currently is not recommended.
Leukemic infiltration into bones and joints is common in children. Reports of bone or joint pain actually lead to the diagnosis of leukemia in some children. In most children, bone pain is characterized as migratory, vague, and without areas of swelling or inflammation. If joint pain is the primary symptom and some swelling is associated with the pain, however, misdiagnoses of rheumatoid arthritis and rheumatic fever may occur.
Other organs reported to be sites of leukemic invasion include the kidneys, heart, lungs, thymus, eyes, skin, and gastrointestinal tract. Of these, the kidneys, lungs, and gastrointestinal tract are the most frequently reported sites. Skin involvement is more common in AML than in ALL.
EVALUATION AND TREATMENT Although blood test results can raise the clinician’s suspicion of leukemia, a bone marrow aspiration is required to establish the diagnosis. The blast cell is the hallmark of acute leukemia (Figure 28-10). The blast cell is a relatively undifferentiated cell characterized by diffusely distributed nuclear chromatin, with one or more nucleoli and basophilic cytoplasm (Figure 28-11).
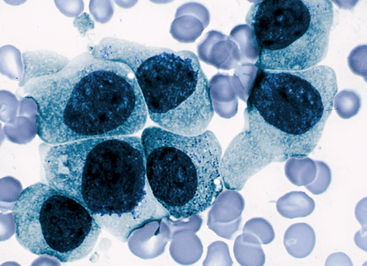
Figure 28-10 Monoblasts from acute monoblastic leukemia. Monoblasts in a marrow smear from an individual with acute monoblastic leukemia (M5A). The monoblasts are larger than myeloblasts and usually have abundant cytoplasm, often with delicate scattered azurophilic granules (an element that stains well with blue aniline dyes). (From Damjanov I, Linder J, editors: Anderson’s pathology, ed 10, St Louis, 1996, Mosby.)
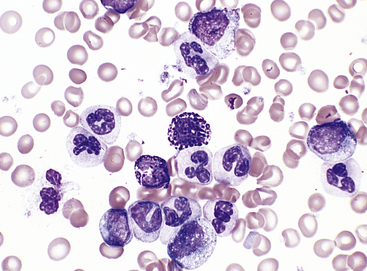
Figure 28-11 Leukocytosis and basophilia in chronic myeloid leukemia. Blood smear from a child with chronic myeloid leukemia (blasts) showing marked leukocytosis and basophilia. Karyotype analysis identified a Philadelphia chromosome (Wright-Giemsa stain). (From Damjanov I, Linder J, editors: Anderson’s pathology, ed 10, St Louis, 1996, Mosby.)
Healthy children have less than 5% blast cells in the bone marrow and none in the peripheral blood. The bone marrow is categorized on the basis of blast percentage. Normal bone marrow is called M1 marrow; M2 and M3 represent an increased percentage of blasts in the sample. This categorization system should not be confused with the similar terminology used to denote subtypes of AML. In ALL the bone marrow often is replaced by 80% to 100% blast cells, with a reduction in normally developing red blood cells and granulocytes. The marrow, which is considered hypercellular, is composed of a homogeneous population of cells. Occasionally, however, the marrow appears hypocellular, making the diagnosis difficult to differentiate from aplastic anemia. When this occurs, bone marrow biopsy or biopsy of extramedullary sites is necessary to confirm the diagnosis.
Combination chemotherapy, with or without radiation therapy to localized sites, such as the CNS, is the treatment of choice for acute leukemia. In ALL, identification of various risk groups has led to the development of different intensities of drug protocols. Thus treatment is tailored specifically for a particular risk group. (Table 28-5 outlines the various prognostic factors for ALL that are considered in determining the degree of risk.).
Most ALL treatment programs have four distinct phases: (1) induction of remission, (2) preventive therapy for the CNS, (3) intensification (also called consolidation), and (4) maintenance. In remission induction, the goal is no clinical evidence of disease and a normal bone marrow biopsy result, which is achieved in 95% of children with ALL. Children with persistent leukemia at the end of 1 month of induction therapy have a dismal prognosis. Prophylactic CNS treatment historically included both chemotherapy and radiation, but therapy, although effective in preventing CNS leukemia, adversely affects neurologic and intellectual function. A marked incidence of learning disabilities has been identified in children previously treated to prevent CNS disease.37 CNS radiation is no longer given and intrathecal regimens are less toxic. Once remission is achieved, an intensification phase of treatment begins. This treatment is necessary because leukemic cells will continue to be present despite successful remission. Thus the goal of the intensification phase is to further decrease and eliminate the remaining leukemic cells. Intensification therapy often overlaps prophylactic CNS treatment. The final phase of initial treatment is called maintenance therapy. The goal of this phase is to maintain disease control. The optimal duration of maintenance therapy is not well defined, but it usually continues for 2.5 to 3 years. During maintenance therapy, intermittent “pulses” of new drugs may be given. Periods of intensified therapy are believed to minimize development of drug-resistant leukemic cells.
ALL is a curable disease. This prognosis is a dramatic reversal of the outlook for a child diagnosed with this disease 40 years ago, when ALL was uniformly fatal and the average survival time was only 2 to 3 months. Today, with prompt and appropriate treatment, 75% to 85% of children with ALL are cured. Those children with the more favorable early pre–B-cell or CALLA-positive ALL have a survival rate of 90%.30
Prognostic factors in AML are not as well defined as they are for ALL because of the small number of affected children and their overall poor prognosis. The goal of treatment for AML is similar to that of ALL except that much more aggressive chemotherapy is administered. With intensive chemotherapy, significant bone marrow suppression is necessary but predisposes children to infection, bleeding, and anemia. The use of colony-stimulating factor (CSF), which stimulates the rapid proliferation of specific blood cell lines, is an advance that shortens this period of bone marrow aplasia (CSFs are discussed in Chapter 25). Although initial remission is achieved relatively easily in all cases of ALL, successful and lasting remission can be achieved in only 70% to 80% of children with AML.30 If remission is achieved, further treatment, called continuation therapy, is required. The specific intensity, timing, and length of continuation therapy are controversial. The use of either a stem cell or bone marrow transplantation (BMT) is an important treatment consideration in AML. Because long-term remission and cure of AML are difficult to achieve with chemotherapy alone, transplant often is recommended after the first remission is achieved. Transplant is the treatment of choice after relapse of AML. The long-term survival rate for children with AML, whether treated with chemotherapy or chemotherapy and BMT, is approximately 45% to 50%.30
Lymphomas
Non-Hodgkin lymphoma (NHL) and Hodgkin disease make up approximately 15% of all childhood cancer. Approximately 800 cases of childhood lymphoma are diagnosed in the United States annually.30 Either group of diseases is rare before age 5 years, and the relative incidence increases throughout childhood. NHL is 1.5 times more common than Hodgkin disease in children. Boys are more likely to be diagnosed with a malignant lymphoma than are girls. There is an increased incidence of lymphoma in children with congenital immunodeficiency syndromes such as Wiskott-Aldrich syndrome, severe combined immunodeficiency (SCID), X-linked lymphoproliferative disease, and ataxia-telangiectasia, as well as those with AIDS. Increased incidence of NHL also is associated with immunosuppression after solid organ and stem cell transplants, particularly T-cell–depleted stem cell transplantation.
Non-Hodgkin Lymphoma
The classification of non-Hodgkin lymphoma (NHL) has been confusing because of the heterogeneity of this group of diseases. Generally most classification systems divide NHL into two categories—nodular or diffuse—on the basis of cellular pattern. Whereas one half of all adults with NHL have a nodular form of the disease, children rarely demonstrate this pattern. Nodular disease represents a less aggressive form of lymphoma. Almost without exception, childhood NHL becomes evident as a diffuse disease and can be further subdivided into three groups: (1) large cell (histiocytic), (2) lymphoblastic, and (3) small noncleaved cell (Burkitt or non-Burkitt lymphoma). Large cell NHL often involves chromosomal translocations. Disease sites commonly involve extranodal sites, such as brain, lung, bone, and skin. Lymphoblastic NHL also shows chromosomal translocations, particularly chromosomes 7 and 14. Disease sites commonly include the mediastinum and peripheral lymph nodes. Small noncleaved cell NHL involves chromosome translocations of 8 and 14. It is believed that this translocation triggers the c-myc oncogene. Children with small noncleaved cell NHL commonly have intra-abdominal disease at diagnosis.
An area of intensive study concerns the apparent biologic similarities of NHL and ALL in children. These two diseases are cytologically identical, and the histologic distinction between them is indicated by the degree of infiltration in the blood and bone marrow. The more bone marrow involvement and the less nodal and organ infiltration present, the more likely the disease is to be classified as ALL. Childhood NHL also is much more like ALL in its clinical manifestations and much less like Hodgkin disease or adult NHL.
As in ALL, immunophenotyping is an important part of the classification of childhood NHL. Almost 45% of the disease in children originates from T cells; an equal number originates from B cells. The remaining group, which represents 10% of childhood NHLs, is classified as non-T, non-B.
PATHOGENESIS The origin of NHL in childhood is still elusive. Although defective host immunity is implicated in most children in whom NHL develops, an immune deficit cannot be identified. Viral etiology is suggested, but the role in development of human lymphoma is still unclear. The strongest correlation exists between EBV and African Burkitt lymphoma. This form of NHL is associated with a breakpoint on chromosome 8 that is located near the c-myc oncogene.38 The relationship between EBV infection and Burkitt lymphoma outside Africa is weak, however, even though the tumor is histopathologically and clinically indistinguishable. Chronic immunostimulation also has been suggested as a factor in the development of lymphomas because these diseases are seen more often when chronic persistent antigenic stimulation occurs from infection, such as malaria or intestinal parasites. Genetic susceptibility also may play a role in the process of malignant transformation.
CLINICAL MANIFESTATIONS In children, NHL has been found to arise from any lymphoid tissue. Signs and symptoms therefore are specific for the site involved. Some children have such widespread involvement that no original site can be determined. Because childhood NHL is a rapidly progressive disease, symptoms generally are present only a few weeks before diagnosis is made. Rapidly enlarging lymphoid tissue and painless lymphadenopathy are common in about one third of children with abdominal sites of involvement, usually representing a gastrointestinal origin for the disease. Symptoms often include abdominal pain and vomiting, but a palpable mass is not always present. Most children with abdominal symptoms have diffuse, small noncleaved cell NHL (Burkitt or non-Burkitt) of B-cell origin. If the tumor recurs, it appears again in the abdomen before distant spread.
The other common site of childhood NHL is the chest region. An anterior mediastinal mass, with or without pleural effusion, often is present. If the mass is large enough, respiratory compromise, tracheal compression, and superior vena cava syndrome may arise, which constitute a medical emergency. Children with anterior mediastinal involvement often are male adolescents and usually have diffuse lymphoblastic lymphoma of T-cell origin. This form of diffuse lymphoblastic lymphoma often evolves into extensive bone marrow involvement and is considered to be an overt leukemic phase (Figure 28-12); therefore, it is referred to as leukemic transformation. CNS involvement and testicular infiltration often then occur. CNS involvement occurs in about 30% of individuals with NHL, usually causing multiple deep-seated lesions within the brain parenchyma. In children with AIDS, NHL is the most common mass lesion found in the brain.
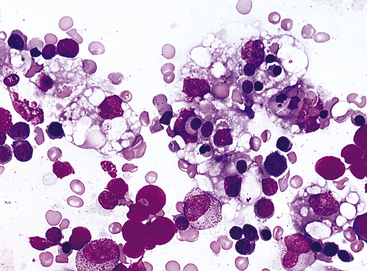
Figure 28-12 Bone marrow aspirate from a child with T-cell lymphoma in a lymph node biopsy. There is marked histiocytic hyperplasia. Two of the histiocytes contain phagocytosed red cells. The histiocytic hyperplasia regressed with disease remission and recurred with relapse of the lymphoma (Wright-Giemsa stain). (From Damjanov I, Linder J, editors: Anderson’s pathology, ed 10, St Louis, 1996, Mosby.)
Bone marrow involvement is less common than other primary sites, whereas CNS involvement is common. Relatively few children (10% to 20%) with NHL have lymphoid tissue involvement of the head and neck (Waldeyer ring, nasopharynx, sinuses). Signs and symptoms include tonsillitis, sinusitis, and a painless nasopharyngeal mass. In African Burkitt lymphoma, involvement of facial bones, particularly the jaw, is common, although this occurs infrequently in non-African cases.
EVALUATION AND TREATMENT Diagnosis is made by biopsy of disease sites, usually the involved lymph nodes. Other sites of biopsy include the tonsils, bone marrow, spleen, liver, bowel, or skin. Advances in understanding the disease and progress in treatment strategies have meant that most children with NHL are cured of the disease. The primary therapeutic modality for childhood NHL is chemotherapy, regardless of stage or site(s) of the disease. The tumor responds to many different agents. Many children with NHL receive intrathecal agents (methotrexate [Mexate] or cytarabine [cytosine arabinoside], or both) for CNS prophylaxis. Radiation is not routinely used to treat childhood NHL, with the exception of emergent situations when airway, intestinal, or spinal obstruction requires immediate reduction in tumor size. Cranial radiation therapy (XRT) may be used for children with T-cell lymphoblastic lymphoma.
Children with advanced small noncleaved cell lymphoma of the abdomen have the poorest prognosis. Although remission occurs in more than 90% of these children, most experience subsequent relapses. Even in the presence of advanced lymphoblastic lymphoma, however, 70% to 80% of children can be cured. Children with localized disease in more easily treated sites are likely to be cured with prompt and appropriate treatment. Overall, children with localized diseases have a 90% survival rate and those with advanced disease have a 70% to 80% survival rate.39
Hodgkin Lymphoma
Hodgkin lymphoma accounts for 6% of all childhood cancers with a significant male-to-female dominance of 4:1 in young children. There is an increased incidence in children with immunologic disorders, whether caused by genetics, infection, or iatrogenic agents. EBV has been associated with Hodgkin lymphoma. Approximately 15% to 25% of adolescents and young adults have EBV-positive Hodgkin lymphoma.40
Clustering of cases within families may suggest a genetic predisposition to the disease or common exposure to a causative agent.41
Hodgkin lymphoma accounts for 6% of all childhood cancers. It occurs infrequently in children younger than 2 years, and few cases are observed before age 5 years. A gradual rise in incidence occurs through age 11 years, with a marked increase through adolescence that continues into the 30s.
Individuals typically have painless supraclavicular or cervical adenopathy. These nodes are firm and rubbery and may be sensitive to palpation if they have grown rapidly. At least two thirds of individuals have mediastinal involvement that may cause symptoms ranging from a nonproductive cough to tracheal or bronchial compression leading to airway obstruction. Systemic symptoms may include fatigue, anorexia, weight loss, fever, drenching night sweats, and pruritus.
The Ann Arbor staging system considers extent and location of disease, as well as substage classifications that consider systemic symptoms (presence of fever of 38° C [100.4° F] for three consecutive days, drenching night sweats, or unexplained loss of 10% or more of body weight in the 6 months preceding diagnosis). Combination chemotherapy used in conjunction with involved field low-dose radiation has been shown to be an effective treatment, with long-term cure rates reported from 90% to 95%.40
Activated partial thromboplastin time (aPTT) 1080
Acute chest syndrome 1074
Alpha-thalassemia major 1077
Alpha-thalassemia minor 1077
Alpha trait 1077
Antithrombin III (AT III) deficiency 1081
Aplastic crisis 1074
Autoimmune neonatal thrombocytopenia 1082
Autoimmune vascular purpura (allergic purpura) 1083
Beta-thalassemia major (Cooley anemia) 1077
Beta-thalassemia minor 1077
Blast cell 1085
Cholecystitis 1076
Embryonic hemoglobin (Gower 1, Gower 2, and Portland) 1063
Fetal hemoglobin (Hb F) 1063
Glomerular disease 1075
Glucose-6-phosphate dehydrogenase (G6PD) deficiency 1068
Hemoglobin H disease 1077
Hemoglobin S (Hb S) 1071
Hemolytic disease of the newborn (HDN) (erythroblastosis fetalis) 1065
Hemophilia A (classic hemophilia) 1078
Hemophilia B (Christmas disease) 1078
Hemophilia C (factor XI deficiency) 1079
Hereditary spherocytosis (HS) 1070
Hodgkin lymphoma 1088
Hydrops fetalis 1067
Hyperbilirubinemia 1067
Hyperhemolytic crisis 1074
Icterus gravis neonatorum 1068
Icterus neonatorum (neonatal jaundice) 1068
Idiopathic thrombocytopenic purpura (ITP) (autoimmune or primary thrombocytopenic purpura) 1081
Kernicterus 1068
Neonatal alloimmune thrombocytopenic purpura (NATP) 1082
Neonatal purpura fulminans 1080
Non-Hodgkin lymphoma (NHL) 1087
Polymerization 1073
Preimplantation genetic diagnosis 1076
Protein C deficiency 1080
Protein S deficiency 1081
Prothrombin consumption time 1080
Prothrombin time (PT) 1080
Sequestration crisis 1074
Sickle cell anemia 1071
Sickle cell disease (SCD) 1071
Sickle cell trait 1071
Sickle cell–Hb C disease 1071
Sickle cell–thalassemia disease 1071
Thalassemia 1077
Thrombin time 1080
Thrombophilia 1080
Thromboplastin generation test 1080
Vasoocclusive crisis (thrombotic crisis) 1074
von Willebrand disease 1079
REFERENCES
1. Graham, B.S. Pathogenesis of respiratory syncytial virus vaccine-augmented pathology. Am J Resp Crit Care Med. 1995;152:S63–S66.
2. Schelonka, R.L., et al. Differentiation of segmented and band neutrophils during the newborn period. J Pediatr. 1995;127:298–300.
3. Bartlett, J.A., et al. Immune function in healthy inner-city children. Clin Diag Lab Immunol. 2001;8:740–746.
4. Cunningham, A.S. Eosinophil counts—age and sex differences. J Pediatr. 1975;87:426–427.
5. Kühne, T., Imbach, P. Neonatal platelet physiology and pathophysiology. Eur J Pediatr. 1998;157:87–94.
6. Khan, J.L., et al. Persistence and emergence of anemia in children during participation in the Special Supplemental Nutrition Program for Women, Infants, and Children. Arch Pediatr Adolesc Med. 2002;156(10):1028–1032.
7. Sherry, B., Mei, Z., Yip, R. Continuation of the decline in prevalence of anemia in low-income infants and children in 5 states. Pediatrics. 2001;107:677–682.
8. Shah, M., et al. Effect of orange and apple juice on iron absorption in children. Arch Pediatr Adolesc Med. 2003;157:1232–1236.
9. Cook, J.D. Diagnosis and management of iron-deficiency anemia. Bailliere’s Best Pract Clin Hematol. 2005;18:319–322.
10. Koelewijn, J.M., et al. Effect of screening for red cell antibodies, other than anti-D, to detect hemolytic disease of the fetus and newborn: a population study in the Netherlands. Transfusion. 2008;48:941–952.
11. Chilcott, J., et al. The economics of routine antenatal anti-D prophylaxis for pregnant women who are rhesus negative. Br J Obstet Gynecol. 2004;111:903–907.
12. Luzzatto, L. Glucose-6-phosphate dehydrogenase deficiency and hemolytic anemia. In: Nathan D.G., et al, eds. Hematology of infancy and childhood. ed 6. Philadelphia: Saunders; 2003:704–725.
13. Lanzkowsky, P. Glucose-6-phosphate dehydrogenase deficiency. In: Lanzkowsky P., ed. Manual of pediatric hematology oncology. ed 4. Burlington, Mass: Elsevier; 2005:153–157.
14. Delhommeau, F., et al. Natural history of hereditary spherocytosis during the first year of life. Blood. 2000;95:393–397.
15. Bolten-Maggs, P.H. Hereditary spherocytosis: new guidelines. Arch Dis Childhood. 2004;89:809–812.
16. Smith, W.R., et al. Temperature changes, temperature extremes and their relationship to emergency department visits and hospitalizations for sickle cell disease. Pain Manag Nurs. 2003;4:106–111.
17. Driscoll, C.M. Sickle cell disease. Pediatr Rev. 2007;28:259–268.
18. Melzer-Lange, M.D., et al. Patient-controlled analgesia for sickle-cell pain crisis in a pediatric emergency department. Pediatr Emerg Care. 2004;20:2–4.
19. Thornburg, C.D., et al. A pilot study of hydroxyurea to prevent chronic organ damage in young children with sickle cell anemia. Pediatr Blood Cancer. 2008 Dec 5. [(Epub ahead of print)].
20. Ross J., ed. Perspectives of hemophilia carriers. Montreal, Canada: World Federation of Hemophilia, 2004.
21. Peyvandi, F., et al. Genetic diagnosis of haemophilia and other inherited bleeding disorders. Haemophilia. 2006;12(Suppl 3):82–89.
22. Bowen, D.J. Haemophilia A and haemophilia B: molecular insights. Mol Pathol. 2002;55(2):127–144.
23. Ananyeva, N., et al. Treating hemophilia A with recombinant blood factors: a comparison. Exp Opin Pharmacother. 2004;5:1061–1070.
24. Manco-Johnson, M.J., et al. Prophylaxis versus episodic treatment to prevent joint disease in boys with severe hemophilia. N Engl J Med. 2007;357:535–544.
25. Young, G. Diagnosis and treatment of thrombosis in children: general principles. Pediatr Blood Cancer. 2006;46:540–546.
26. Kubota, M., et al. Serum immunoglobulin levels at onset: association with the prognosis of childhood idiopathic thrombocytopenic purpura. Internat J Hematol. 77, 2003. [304–304].
27. Gupta, V., Tilak, V., Bhatia, B.D. Immune thrombocytopenic purpura. Indian J Pediatr. 2008;75:723–728.
28. Yldrm, Z.K., et al. Late side effects of high-dose steroid therapy on skeletal system in children with idiopathic thrombocytopenic purpura. J Pediatr Hematol Oncol. 2008;30:749–753.
29. Nugent, D.J. Immune thrombocytopenic purpura of childhood, Hematology Am Soc Hematol Educ Program. 2006:97–103.
30. American Cancer Society. Cancer facts and figures 2008. Atlanta: American Cancer Society; 2008.
31. Plon, S.E., Malkin, D. Childhood cancer and heredity. In: Pizzo P.A., Poplack D.G., eds. Principles and practice of pediatric oncology. Philadelphia: Lippincott Williams & Wilkins; 2006:14–37.
32. Buka, I., Koranteng, S., Osornio-Vargas, A.R. Trends in childhood cancer incidence: review of environmental linkages. Pediatr Clin North Am. 2007;54:177–203.
33. Greaves, M.F., et al. Leukemia in twins: lessons in natural history. Blood. 2003;102:2321–2333.
34. Infante-Rivard, C., Guiguet, M. Family history of hematopoietic and other cancers in children with acute lymphoblastic leukemia. Cancer Detect Prev. 2004;28:83–87.
35. Laurier, D., et al. Epidemiological studies of leukaemia in children and young adults around nuclear facilities: a critical review. Radiat Prot Dosimetry. Oct 15, 2008. [(Epub ahead of print)].
36. Powles, T., et al. Head and neck cancer in patients with human immunodeficiency virus-1 infection: incidence, outcome and association with Epstein-Barr virus. J Laryngol Otol. 2004;118(3):207–212.
37. Koh, S., et al. Anterior lumbosacral radiculopathy after intrathecal methotrexate treatment. Pediatr Neurol. 1999;21:576–578.
38. Link, M.P., Weinstein, H.J. Malignant non-Hodkgin lymphoma in children. In: Pizzo P.A., Poplack D.G., eds. Principles and practice of pediatric oncology. Philadelphia: Lippincott Williams & Wilkins; 2006:722–747.
39. Pinkerton, R. Continuing challenges in childhood non-Hodgkin’s lymphoma. Br J Hematol. 2005;130:480–488.
40. Hudson, M.M., Donaldson, S.S., Onciu, M. Malignant non-Hodkgin lymphoma in children. In: Pizzo P.A., Poplack D.G., eds. Principles and practice of pediatric oncology. Philadelphia: Lippincott Williams & Wilkins; 2006:695–721.
41. Oliapuram Jose, B., et al. Pediatric Hodgkin’s disease. J Ky Med Assoc. 2004;102(3):104–106.