STRUCTURE AND FUNCTION OF THE HEMATOLOGIC SYSTEM


All the body’s tissues and organs require oxygen and nutrients to survive. These essential needs are provided by the blood that flows through miles of vessels throughout the human body. The red blood cells provide the oxygen and remove carbon dioxide, and the fluid portion of the blood carries the nutrients and ions for proper acid-base balance. The blood also cleans discarded waste from the tissues, transports hormones, conveys cells (white blood cells), platelets, and other ingredients that are necessary for protecting the entire body from injury and infection and initiating healing, and provides thermal regulation to maintain organs and tissues within an acceptable range of temperatures.
COMPONENTS OF THE HEMATOLOGIC SYSTEM
Blood consists of various cells that circulate in the cardiovascular system suspended in a solution of protein and inorganic materials (plasma), which is approximately 90% water and 10% dissolved substances (solutes). The blood volume amounts to about 6 quarts (5.5 L) in adults. The continuous movement of blood guarantees that critical components are available to all parts of the body to carry out their chief functions: (1) delivery of substances needed for cellular metabolism in the tissues, (2) removal of the wastes of cellular metabolism, (3) defense against invading microorganisms and injury, and (4) maintenance of acid-base balance.
Plasma and Plasma Proteins
In adults, plasma accounts for 50% to 55% of blood volume. Plasma is a complex aqueous liquid containing a variety of organic and inorganic elements (Table 25-1). The concentration of these elements varies depending on diet, metabolic demand, hormones, and vitamins. Plasma differs from serum in that serum is plasma that has been allowed to clot in the laboratory in order to remove fibrinogen and other clotting factors that may interfere with some diagnostic tests.
Table 25-1
Organic and Inorganic Components of Arterial Plasma

Data from Vander AJ, Sherman JH, Luciano DS: Human physiology: the mechanisms of body function, ed 8, New York, 2001, McGraw-Hill.
The plasma contains a large number of proteins (plasma proteins) that constitute about 7% of the total plasma weight. These vary in structure and function and can be classified into two major groups, albumin and globulins. Most plasma proteins are produced by the liver. The major exception is antibodies (immunoglobulins), which are produced by plasma cells in the lymph nodes and other lymphoid tissues (see Chapter 7).
Albumin (about 60% of total plasma protein at a concentration of about 4 g/dl) serves as a carrier molecule for normal components of blood as well as drugs that have low solubility in water (e.g., free fatty acids, lipid-soluble hormones, thyroid hormones, bile salts). Its most essential role is regulation of the passage of water and solutes through the capillaries. Albumin molecules are large and do not diffuse freely through the vascular endothelium, and thus they maintain the critical colloidal osmotic pressure (or oncotic pressure) that regulates the passage of water and solutes into the surrounding tissues (see Chapters 1 and 3). Water and solute particles tend to diffuse out of the arterial portions of the capillaries because the blood pressure is greater in arterial than in venous blood vessels (see Chapter 3). Water and solutes move from tissue cells into the venous portions of the capillaries where the pressures are reversed, oncotic pressure being greater than intravascular pressure or hydrostatic pressure. In the case of decreased production (e.g., cirrhosis, other diffuse liver diseases, protein malnutrition) or excessive loss of albumin (e.g., certain kidney diseases, extensive burns), the reduced oncotic pressure leads to excessive movement of fluid and solutes into the tissue and decreased blood volume.1
The remaining plasma proteins, or globulins, are often classified by their properties in an electric field (serum electrophoresis). Under the normal conditions used to perform serum electrophoresis, albumin is the most rapidly moving protein. The globulins are classified by their movement relative to albumin: alpha (α) globulins (those moving most closely to albumin), beta (β) globulins, and gamma (γ) globulins (those with the least movement). Depending on the electrophoretic procedure the alpha and beta globulins may be subdivided into subregions (α1, α2, β1, or β2-globulins). Fibrinogen is a major plasma protein (about 4% of total plasma protein) that would move between the beta and gamma regions but is removed during the formation of serum. The gamma globulin region consists primarily of immunoglobulin G (IgG) (see Chapter 7).
Plasma proteins can also be classified into groups by function: clotting, defense, transport, or regulation. The clotting factors promote coagulation and stop bleeding from damaged blood vessels. Fibrinogen is the most plentiful of the clotting factors and is the precursor of the fibrin clot. Proteins involved in defense, or protection, against infection include antibodies and complement proteins (see Chapters 6 and 7). Transport proteins specifically bind and carry a variety of inorganic and organic molecules, including iron (transferrin), copper (ceruloplasmin), steroid hormones, and vitamins (e.g., retinol-binding protein). The plasma lipids, triglycerides, phospholipids, cholesterol, and fatty acids are carried through the blood as complexes with plasma proteins; they are known as lipoproteins (see Chapters 1 and 30). Regulatory proteins include a variety of enzymatic inhibitors (e.g., α1-antitrypsin) that protect the tissues from damage, precursor molecules (e.g., kininogen) that are converted into active biologic molecules when needed, and protein hormones (e.g., cytokines) that communicate between cells.
Plasma also contains several charged inorganic ions (electrolytes) that regulate cell function, osmotic pressure, and blood pH. (Electrolytes are described in Chapters 1 and 3.)
Cellular Components of the Blood
The cellular elements of the blood are broadly classified as red blood cells (RBCs) (i.e., erythrocytes), white blood cells (WBCs) (i.e., leukocytes), and platelets (thrombocytes). The components of the blood are listed in Table 25-2.
Table 25-2
Cellular Components of the Blood
∗See bottom row of Figure 25-9 for illustrations of cells.
Erythrocytes: In 1628 Robert Burton described blood as a “hot, temperate red humor whose office is to nourish the whole body, to give it strength and color being dispersed by the veins through every part of it.”2 A few years later, with the invention of the microscope, researchers learned that erythrocytes give blood its red color.
Erythrocytes (red blood cells [RBCs]) are the most abundant cells of the blood, occupying approximately 48% of the blood volume in men and about 42% in women. Erythrocytes are primarily responsible for tissue oxygenation. The erythrocyte contains hemoglobin, which carries the gases, and electrolytes, which regulate diffusion through a cell’s plasma membrane. The mature erythrocyte lacks a nucleus and cytoplasmic organelles (e.g., mitochondria), so it cannot synthesize protein or carry out oxidative reactions. Because it cannot undergo mitotic division, the erythrocyte has a limited life span (approximately 120 days), ages, and is removed from the circulation to be replaced by new erythrocytes.
The erythrocyte’s size and shape are ideally suited to its function as a gas carrier. An RBC is a small disk with two unique properties: (1) a biconcave shape and (2) the capacity to be reversibly deformed (Figure 25-1).3 The flattened, biconcave shape provides a surface area/volume ratio that is optimal for gas diffusion into and out of the cell. During its life span, the erythrocyte, which is 6 to 8 μm in diameter, repeatedly circulates through sinusoids of the spleen and capillaries that are only 2 μm in diameter. Reversible deformity enables the erythrocyte to assume a more compact torpedo-like shape, squeeze through the microcirculation, and return to normal.
Leukocytes: Leukocytes (white blood cells [WBCs]) defend the body against microorganisms that cause infection and remove debris, including dead or injured cells of all kinds (Figure 25-2). The leukocytes act primarily in the tissues but are transported in the circulation. The average adult has approximately 5000 to 10,000 leukocytes/mm3 of blood.
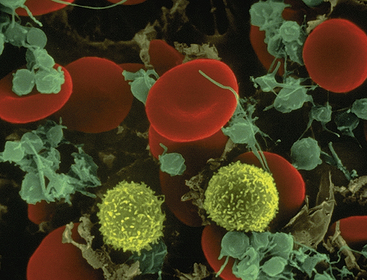
Figure 25-2 Blood cells. Leukocytes are spherical and have irregular surfaces with numerous extending pili (appear as yellow). Erythrocytes are flattened spheres with a depressed center. Activated platelets are green. (Copyright Dennis Kunkel Microscopy, Inc.)
Leukocytes are classified according to structure as either granulocytes or agranulocytes and according to function as either phagocytes or immunocytes. The granulocytes, which include neutrophils, basophils, and eosinophils, are all phagocytes. (Phagocytic action is described in Chapter 6.) Of the agranulocytes, the monocytes and macrophages are phagocytes, whereas the lymphocytes are immunocytes (cells that create immunity; see Chapter 7).
Granulocytes: Granulocytes have many membrane-bound granules in their cytoplasm. These granules contain enzymes capable of killing microorganisms and catabolizing debris ingested during phagocytosis. The granules also contain powerful biochemical mediators with inflammatory and immune functions. These mediators, along with the digestive enzymes, are released from granulocytes in response to specific stimuli. The biochemical mediators have vascular and intercellular effects and the enzymes participate in the breakdown of debris from sites of infection or injury. Granulocytes are capable of amoeboid movement, by which they migrate through vessel walls (diapedesis) and then to sites where their action is needed (see Chapter 6).
The neutrophil (polymorphonuclear neutrophil [PMN]) is the most numerous and best understood of the granulocytes (Figure 25-3, A). Neutrophils constitute about 55% of the total leukocyte count in adults. The cytoplasm of neutrophils contains small lysosomal granules and a central nucleus with two to five distinct lobes. Immature neutrophils are called bands or stabs. Mature neutrophils are called segmented neutrophils because of the characteristic appearance of their nucleus. Neutrophils reach a fully mature state in the bone marrow, and these mature neutrophils are called the marrow neutrophil reserve. Normally it takes about 14 days for neutrophils to develop from early precursors, but this process is accelerated by infection and treatment with colony-stimulating factors.
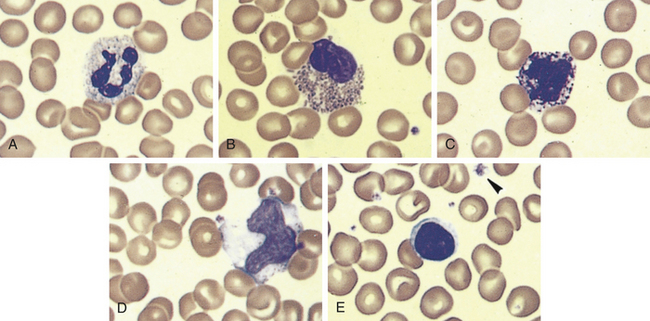
Figure 25-3 Leukocytes. An example of leukocytes in a human blood smear. A, Neutrophil. B, Eosinophil. C, Basophil. D, Monocyte. E, Lymphocyte. (From Erlandsen S, Magney J: Color atlas of histology, St Louis, 1992, Mosby.)
Neutrophils are the chief phagocytes of early inflammation. Soon after bacterial invasion or tissue injury, neutrophils migrate out of the capillaries and into the inflamed site, where they ingest and destroy microorganisms and debris and then die in 1 or 2 days. The dissolution of dead neutrophils releases digestive enzymes from their cytoplasmic granules. These enzymes dissolve cellular debris and prepare the site for healing. (This final function, called débridement, is described in Chapter 6).
Eosinophils: Eosinophils, which have large, coarse granules, constitute only 1% to 4% of the normal leukocyte count in adults (see Figure 25-3, B). Like neutrophils, eosinophils are capable of amoeboid movement and phagocytosis.4 Unlike neutrophils, which ingest cellular debris, eosinophils ingest antigen-antibody complexes, and viruses and are induced by mast cell chemotactic factors to attack parasites. Eosinophil granules contain chemicals (e.g., major basic protein, eosinophil cationic protein, eosinophil peroxidase, eosinophil-derived neurotoxin) that are highly destructive to parasites and viruses. The eosinophil granules contain a variety of enzymes (e.g., histaminase) that help to control inflammatory processes. (Their function in inflammation and defense against parasites is described in Chapters 6 and 7.) Type I hypersensitivity allergic reactions and asthma are characterized by high numbers of circulating eosinophils, which may be involved in a dual role of regulation of inflammation and may contribute to the destructive inflammatory processes observed in the lungs of asthmatics (see Chapter 8).
Basophils, which make up less than 1% (0.01% to 0.3%) of the leukocytes, contain cytoplasmic granules that contain an abundant mixture of biochemical mediators, including histamine, chemotactic factors, proteolytic enzymes, and an anticoagulant (heparin) (see Figure 25-3, C). Stimulation of basophils also induces synthesis of vasoactive lipid molecules (e.g., leukotrienes) and cytokines.5 Basophils are a particularly rich source of the cytokine interleukin-4 (IL-4), which preferentially guides B-cell differentiation toward plasma cells that secrete IgE (see Chapter 7).
The precise function of basophils is poorly understood, but numbers of basophils are often increased at sites of allergic inflammatory reactions and parasitic infection, particularly exoparasites (e.g., ticks). IgE receptors on the basophil would induce degranulation at sites of IgE-mediated hypersensitivity reactions and contribute to the local inflammatory response.
Mast cells are highly similar to basophils, but are generated from a different set of precursor cells in the bone marrow, from which they migrate in an immature form into tissues.5 They reside in vascularized connective tissues just beneath body epithelial surfaces, including the submucosal tissues of the gastrointestinal and respiratory tracts and the dermal layer that lies just below the surface of the skin. Mast cells play a central role in inflammation, and their activation and degranulation affects a great number of cells, including those involved in inflammation (e.g., vascular endothelial cells, smooth muscle cells, circulating platelets and leukocytes, nerves) and healing (e.g., fibroblasts), as well as glandular cells and cells of the immune system. Their activation contributes greatly to increased permeability of blood vessels and smooth muscle contraction (see Figure 6-8).
Agranulocytes: The agranulocytes—monocytes, macrophages, and lymphocytes—differ from the granulocytes in that they contain relatively fewer granules in their cytoplasm. The lymphocytes do not contain any enzyme-filled digestive vacuoles, and the digestive vacuoles of the monocytes and macrophages are larger and fewer than those of the granulocytes.
Lymphocytes constitute approximately 36% of the total leukocyte count and are the primary cells of the immune response (see Figure 25-3, E and Chapter 7). Most lymphocytes transiently circulate in the blood and eventually reside in secondary lymphoid tissues as mature T cells, B cells, or plasma cells. The life span of the lymphocyte can be days, months, or years, depending on its type and subtype. (Lymphocyte function and dysfunction are described in detail in Unit III.)
Natural killer (NK) cells, which resemble large granular lymphocytes, kill some types of tumor cells (in vitro) and some virus-infected cells without being induced by previous exposure to these antigens (see Chapters 6 and 7). Hence they are named natural killer cells to differentiate them from T cytotoxic cells, which are induced by antigen. NK cells also have the capacity to activate T cells and phagocytes and produce a variety of cytokines that can regulate immune responses. The predominant form of NK cells develops in the bone marrow and circulates in the blood, where it accounts for 5% to 10% of the circulating lymphoid pool, and is found mainly in the peripheral blood and spleen. NK cells develop independent of a thymus, although some NK precursors are found in the thymus and may develop into NKT cells that have markers of NK and T cells.
The monocytes and macrophages make up the mononuclear phagocyte system (MPS), formerly called the reticuloendothelial system (RES).6 Monocytes and macrophages are active phagocytes that participate in the immune and inflammatory responses. They also ingest dead or defective host cells, particularly blood cells.
Monocytes are the largest normal blood cell and have a horseshoe-shaped nucleus (see Figure 25-3, D). They are formed and released by the bone marrow into the bloodstream. Monocytes migrate into a variety of tissues and fully mature into tissue macrophages and myeloid dendritic cells (Table 25-3). Other monocytes may mature into macrophages in the circulation and migrate out of the vessels in response to infection or inflammation. Macrophages are generally larger and are more active as phagocytes than monocytes. Dendritic cells frequently extend projections (dendrites) into the tissue and take on a “neuron-like” appearance. The origin and turnover of many of the tissue macrophages are not precisely known. It seems clear that once monocytes leave the circulation, they do not return. They can survive many months or even years.
Table 25-3
Mononuclear Phagocyte System∗
| Name of Cell | Location |
| Committed Stem Cells† | Bone marrow |
| Monoblasts | Bone marrow |
| Promonoblasts | Bone marrow |
| Monocytes | Bone marrow and peripheral blood |
| Macrophages | Tissue |
| Kupffer cells | Liver macrophages |
| Alveolar macrophages | Lung |
| Histiocytes | Connective tissue |
| Macrophages | Bone marrow |
| Fixed and free macrophages | Spleen and lymph nodes |
| Pleural and peritoneal macrophages | Serous cavities |
| Adipose macrophages | Adipose (fat) tissue |
| Microglial cells | Nervous system |
| Mesangial cells | Kidney |
| Osteoclasts | Bone |
| Langerhans cells | Skin |
| Dendritic cells | Lymphoid tissue, lining of respiratory and gastrointestinal tracts |
∗Formerly called the reticuloendothelial system.
†Development of blood cells from stem cells in the marrow is described on this page and illustrated in Figure 25-9.
Modified from Kumar V et al: Robbins and Cotran pathologic basis of disease, ed 7, Philadelphia, 2005, Saunders.
The normal role of macrophages is to remove old and damaged cells and large-molecular substances from the blood. Cellular targets of macrophage phagocytosis include circulating senescent or damaged erythrocytes and platelets (removed primarily in spleen), dead neutrophils (in the circulation and at sites of inflammation), and cells undergoing apoptosis. Noncellular targets include antigen-antibody complexes, cellular debris, products of coagulation, and macromolecules (such as lipids and carbohydrates synthesized by the body as the result of faulty metabolism, as in storage diseases). Macrophages remove and kill contaminating microorganisms in the blood (mostly in the liver and spleen) and at sites of infection. Macrophages and, particularly, dendritic cells are the major “antigen-processing” and “antigen-presenting” cells that initiate immune responses (see Chapter 7). Macrophages initiate wound healing and tissue remodeling and if activated by cytokines from T cells secrete a large array of biologically active chemicals that if uncontrolled result in chronic inflammation and tissue injury (see Chapter 6). Osteoclasts are multinucleated cells specialized for the function of lacunar bone resorptions and remodeling in addition to phagocytosis.
Platelets: Platelets (thrombocytes) are not true cells but disk-shaped cytoplasmic fragments that are essential for blood coagulation and control of bleeding. They are formed by fragmentation of very large (40 to 100 μm in diameter) cells known as megakaryocytes (Figure 25-4). They lack a nucleus, have no deoxyribonucleic acid (DNA), and are incapable of mitotic division. They do, however, contain cytoplasmic granules (i.e., dense granules, alpha granules) capable of releasing biochemical mediators (e.g., adenosine diphosphate [ADP], adenosine triphosphate [ATP], calcium, serotonin from dense granules; coagulation factors, platelet-derived growth factor [PDGF], platelet factor 4 from alpha granules) when stimulated by injury to a blood vessel. Activation also stimulates synthesis of arachidonic acid pathway products (e.g., thromboxane-A2) (see Chapter 6).
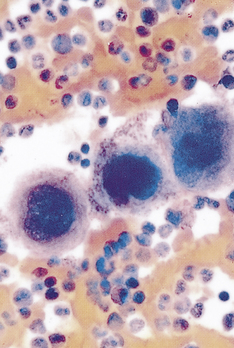
Figure 25-4 Megakaryocyte and platelets. Note the large number of platelets (purple) surrounding the large megakaryocytes in the center. (From Miale JB, Laboratory medicine: hematology, ed 6, St Louis, 1982, Mosby.)
There are approximately 140,000 to 340,000 platelets/mm3 of circulating blood. An additional one third of the body’s available platelets are in a reserve pool in the spleen. A platelet circulates for approximately 10 days, ages, and is removed by macrophages of the MPS, mostly in the spleen.
Lymphoid Organs
The lymphoid system is closely integrated with the circulatory system. The lymphoid organs, some of which are merely aggregations of lymphoid tissue, are classified as primary or secondary. The primary lymphoid organs are the thymus and the bone marrow. The secondary lymphoid organs consist of the spleen, lymph nodes, tonsils, and Peyer patches of the small intestine (see Figure 7-3). All of the lymphoid organs link the hematologic and immune systems in that they are sites of residence, proliferation, differentiation, or function of lymphocytes and mononuclear phagocytes (monocytes and macrophages). (The liver, which also has hematologic functions, is primarily a digestive organ and is described in Chapter 38.)
Spleen
The spleen is the largest of the secondary lymphoid organs. It is a site of fetal hematopoiesis; its mononuclear phagocytes filter and cleanse the blood; its lymphocytes mount an immune response to blood-borne microorganisms; and it serves as a blood reservoir (see Chapter 27).
The spleen is a concave, encapsulated organ that weighs about 150 g and is about the size of a fist. It is located in the left upper abdominal cavity, curved around a portion of the stomach (see Figure 7-3). Strands of connective tissue (trabeculae) extend from the capsule, dividing the spleen into compartments (Figure 25-5). The compartments contain masses of lymphoid tissue called splenic pulp. The spleen is interlaced with many blood vessels, some of which are capable of distending to store blood. Blood that circulates through the spleen comes from the splenic artery, which branches from the descending aorta and reenters the circulatory system through the splenic vein and into the portal vein.
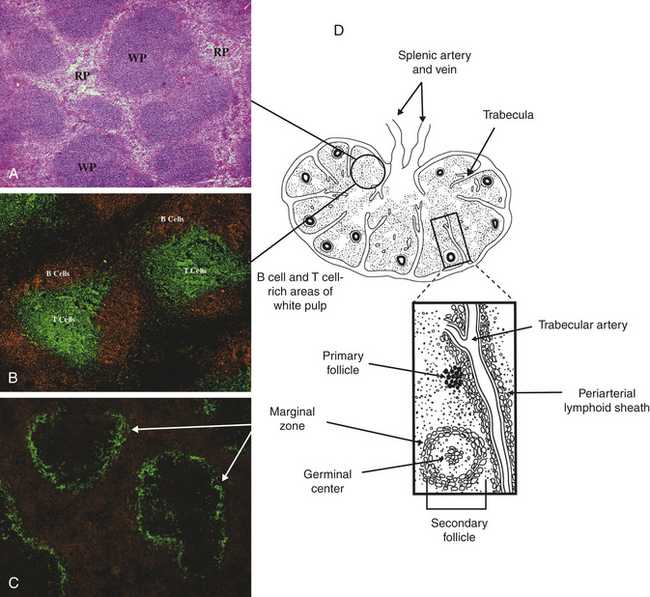
Figure 25-5 Spleen architecture. A, Spleen section stained with hematoxylin and eosin shows areas of densely packed cells, referred to as the white pulp (WP), separated by areas with more dispersed cell populations, referred to as the red pulp (RP). B, Spleen section that has been stained with fluorescently labeled antibodies specific for B cells (orange) and T cells (green) shows the distinct localization of B cells and T cells within the white pulp. C, Staining for macrophages (orange) and the splenic marginal zone (green) shows the density of macrophages and phagocytic cells in the red pulp and marginal zone. D, The spleen is enclosed in a capsule with the interior pulp divided into compartments by strands of connective tissue (trabeculae). The splenic pulp contains regions that are rich in lymphocytes (white pulp) and those containing erythrocytes (red pulp). Arteries residing near the trabeculae (tubercular arteries) are frequently surrounded by a periarterial lymphoid sheath, primarily containing T cells and macrophages, with adjacent lymphoid follicles. (A, B, and C from Mandell G, Bennett J, Dolin R: Principles and practice of infectious diseases, ed 6, Philadelphia, 2005, Churchill Livingstone; D from Hoffman R et al: Hematology: basic principles and practice, ed 4, Philadelphia, 2005, Churchill Livingstone.)
The portion of arterial blood that enters the spleen first encounters the white splenic pulp, which consists of masses of lymphoid tissue containing macrophages and lymphocytes, primarily T lymphocytes in proximity to the arterioles (see Figure 25-5, B and E). Cellular clumps (lymphoid follicles) are formed in the white pulp around the splenic arterioles. The lymphoid follicles consist primarily of B lymphocytes. These are the chief sites of immune function within the spleen. Here blood-borne antigens encounter lymphocytes, initiating the immune response and the conversion of lymphoid follicles into germinal centers (see Chapter 7).7
Some of the blood that enters the terminal capillaries continues through the microcirculation and enters highly distensible storage areas called venous sinuses in the red pulp of the spleen. The venous sinuses are capable of storing more than 300 ml of blood. Passive dilation of the venous sinuses enables the spleen to increase its storage capacity as needed by the body. Sudden reductions in blood pressure cause the sympathetic nervous system to stimulate constriction of the sinuses, resulting in expulsion of as much as 200 ml of blood into the venous circulation, which helps restore blood volume and increases the hematocrit by as much as 4%.
The endothelial lining of the venous sinuses is discontinuous (having gaps between endothelial cells) and therefore extremely permeable so that blood cells are allowed to exit the circulation (Figure 25-6). The red pulp contains a system of loosely interconnected resident macrophages that provide the principal site of splenic filtration. Because of the slow circulation in the sinuses, the macrophages easily phagocytose old, damaged, or dead blood cells of all kinds (but chiefly erythrocytes), microorganisms, macromolecules, and particles of debris. Hemoglobin from phagocytosed erythrocytes is catabolized, and heme (iron) is stored in the cytoplasm of the macrophages or released back into the blood (see Figure 25-16). The macrophages also can remove particulate inclusions containing denatured hemoglobin (Heinz bodies) from erythrocytes without harming the cells themselves. Blood that filters through the red pulp also finds its way into the venous sinuses and hence into the portal circulation.
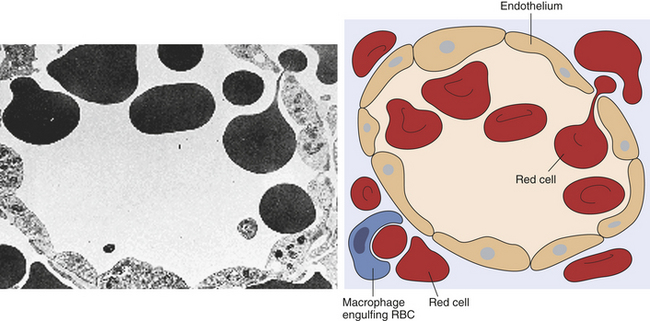
Figure 25-6 Splenic sinus. Transmission electron micrograph and schematic of erythrocytes in the process of squeezing from the red pulp cords into the sinus lumen. Note the degree of deformability required for red cells to pass through the wall of the sinus. (From Damjanov I, Linder J, editors: Anderson’s pathology, ed 10, St Louis, 1996, Mosby. Schematic from Kumar V, Fausto N, Abbas A: Robbins and Cotran pathologic basis of disease, 7th ed, St Louis, 2005, Saunders.)
The spleen is not absolutely necessary for life or for adequate hematologic function. However, splenic absence from any cause (atrophy, traumatic injury, or removal because of disease) has several secondary effects on the body. For example, leukocytosis (high levels of circulating leukocytes) often occurs after splenectomy, suggesting that the spleen exerts some control over the rate of proliferation of leukocyte stem cells in the bone marrow or their release into the bloodstream. Circulating levels of iron may also decrease, reflecting the spleen’s role in the iron cycle. The immune response to encapsulated bacteria (e.g., Streptococcus pneumoniae [pneumococcus], Neisseria meningitidis [meningococcus], Haemophilus influenzae), which is primarily an IgM response, may be severely diminished resulting in increased susceptibility to disseminated infections. Loss of the spleen results in an increase in morphologically defective blood cells in the circulation, confirming the spleen’s role in removing old or damaged cells.
Lymph Nodes
Structurally, lymph nodes are part of the lymphatic system. Lymphatic vessels collect interstitial fluid from the tissues and transport it, as lymph, through vessels of increasing size to the thoracic duct, which drains into the superior vena cava returning the lymph to the circulation. Lymph nodes are distributed throughout the body and provide filtration of the lymph during its journey through the lymphatics. Each lymph node is enclosed in a fibrous capsule, branches of which (trabeculae) extend inward to partition the node into several compartments (Figure 25-7). Reticular fibers of connective tissue divide the compartments into a meshwork throughout the lymph node. The node consists of outer (cortex) and inner (paracortex) cortical areas and an inner medulla. Lymph enters through multiple small afferent lymphatic vessels into the subcapsular sinus, just beneath the capsule, drains into the cortical sinuses to the medullary sinuses, from which the lymph is collected and leaves the node by way of the efferent lymphatic vessel. Blood flows into the lymph nodes through the lymphatic artery, which ends in groups of postcapillary venules disturbed throughout the outer cortex. The blood is drained through the lymphatic vein.
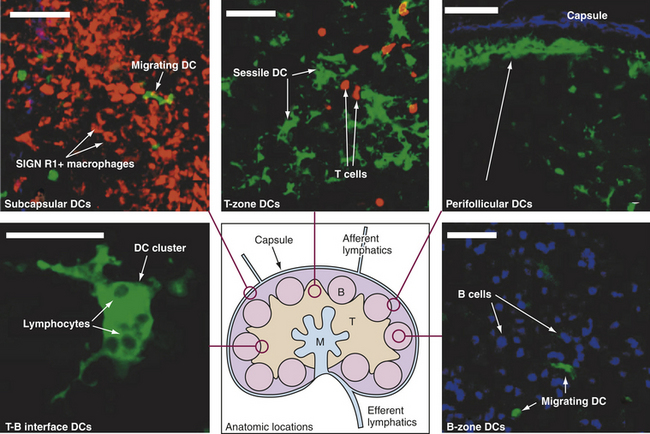
Figure 25-7 Lymph node architecture. Drawing in lower center: Lymph enters via the afferent lymphatics and exits via the efferent lymphatics. T cells enter the lymph nodes via the high endothelial venules and exit via medulla to the efferent lymphatics. B, B-cell follicles; M, medullary cords; T, T-cell zone. Other structures are labeled. Stained microscopic images: all images are linked to the drawing of the lymph node. DC, Dendritic cell. (All scale bars 50 μm.) (From Lindquist RL et al: Nat Immunol 5:1243-1250, 2004.)
Functionally, however, lymph nodes are part of the hematologic and immune systems and are the primary site for the first encounter between antigen and lymphocytes. Lymphocytes enter the lymph node from the blood through the postcapillary venules by means of diapedesis across the endothelial lining. B lymphocytes tend to migrate preferentially to nodes in the cortex and medulla, whereas T lymphocytes predominantly migrate to the paracortex (see Figure 25-7). Macrophages reside in the lymph node, help filter the lymph of debris, foreign substances, and microorganisms, and provide antigen-processing functions. The dendritic cells encounter and process antigens and microorganisms in other tissues, enter the lymph node through the afferent lymph vessels, and migrate throughout the nodes. The reticular network provides adhesive surfaces for trapping large numbers of phagocytes and lymphocytes and facilitating their organization into follicles or primary nodules.8 The presence of antigen, either removed from the lymph by macrophages or presented on the surface of dendritic cells, results in the production of secondary nodules containing germinal centers. In the germinal centers lymphocytes, particularly B cells, respond to antigenic stimulation by undergoing proliferation and further differentiation, including class-switch, into memory cells and plasma cells (see Chapter 7). Plasma cells migrate to the medullary cords. The B lymphocyte proliferation in response to a great deal of antigen (e.g., during infection) may result in lymph node enlargement and tenderness (reactive lymph node).
DEVELOPMENT OF BLOOD CELLS
The typical human requires about 100 billion new blood cells per day. Blood cell production, termed hematopoiesis, is constantly ongoing, occurring in the liver and spleen of the fetus and only in bone marrow (medullary hematopoiesis) after birth (see Chapter 28). This process involves the biochemical stimulation of populations of relatively undifferentiated cells to undergo mitotic division (i.e., proliferation) and maturation (i.e., differentiation) into mature hematologic cells (Table 25-4). Although proliferation and differentiation are usually sequential, certain blood cells proliferate and differentiate simultaneously. Erythrocytes and granulocytes generally differentiate fully before entering the blood, but monocytes and lymphocytes continue to mature in the blood and in secondary lymphatic organs.
Table 25-4
Human Hematopoietic Growth Factors (cytokines, colony-stimulating factors)
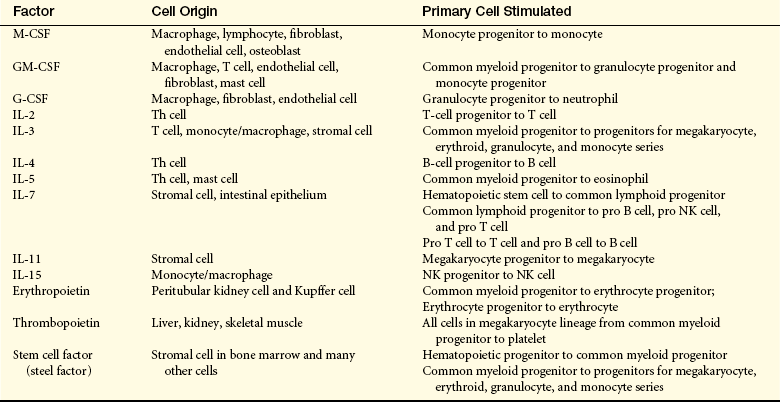
G-CSF, Granulocyte colony-stimulating factor; GM-CSF, granulocyte-macrophage colony-stimulating factor; IL, interleukin; M-CSF, macrophage colony-stimulating factor; NK, natural killer; Th, T helper.
Hematopoiesis continues throughout life, increasing in response to a need to replenish destroyed circulating cells (e.g., during hemorrhage, hemolytic anemia [peripheral destruction of erythrocytes], consumptive thrombocytopenia) or in response to infection. In general, long-term stimuli, such as chronic diseases, cause a greater increase in hematopoiesis than acute conditions, such as hemorrhage. Various abnormalities in medullary hematopoiesis have been identified and is discussed in Chapter 26. Extramedullary hematopoiesis—blood cell production in tissues other than bone marrow—of apparently normal blood cells has been reported in the spleen, liver, and, less frequently, lymph nodes, adrenal glands, cartilage, adipose tissue, intrathoracic areas, and kidneys. In adults, however, extramedullary hematopoiesis is usually a sign of disease, occurring in pernicious anemia, sickle cell anemia, thalassemia, hemolytic disease of the newborn (erythroblastosis fetalis), hereditary spherocytosis, and certain leukemias.
Bone Marrow
Bone marrow is confined to the cavities of bone and is the primary site of residence of hematopoietic stem cells (Figure 25-8). Adults have two kinds of bone marrow: red, or active (hematopoietic), marrow (also called myeloid tissue) and yellow, or inactive, marrow. The large quantity of fat in inactive marrow gives its characteristic yellow color. Not all bones contain active marrow. In adults, active marrow is found primarily in the flat bones of the pelvis (34%), vertebrae (28%), cranium and mandible (13%), sternum and ribs (10%), and in the extreme proximal portions of the humerus and femur (4% to 8%). Inactive marrow predominates in cavities of other bones. (Bones are discussed further in Chapter 41.)
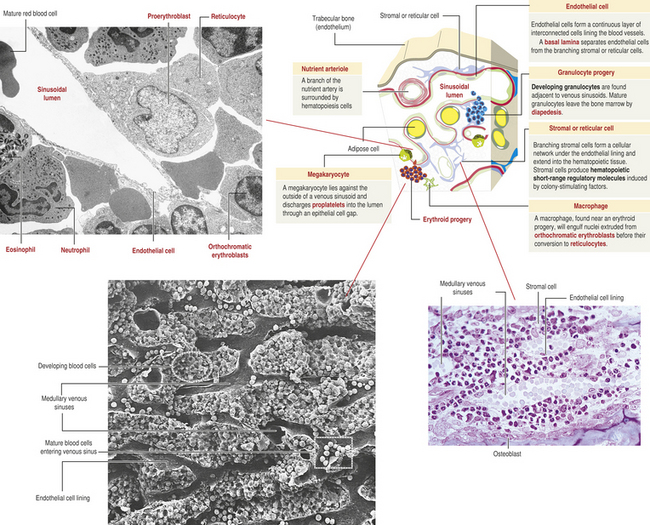
Figure 25-8 Bone marrow: structure and vascularization. (From Kierszenbaum A: Histology and cell biology: an introduction to pathology, 2nd ed, Philadelphia, 2006, Mosby. Scanning electron micrograph from Kessel RG, Kardon RH: Tissues and organs, New York, WH Freeman, 1979.)
Hematopoietic marrow is vascularized by the primary arteries of the bones, which terminate in a capillary network forming large venous sinuses. Hematopoietic marrow and fat fill the spaces surrounding the network of venous sinuses. Newly produced blood cells traverse narrow openings between endothelial cells in the venous sinus walls and thus enter the circulation. Normally, immature cells have not developed the appropriate surface receptors to interact with the endothelium and enter the circulation.
The stromal compartment of the bone marrow contains a variety of cell types, including mesenchymal stem cells, macrophages/osteoclasts, and endothelial-like cells.9 The mesenchymal stem cells can differentiate into fibroblasts, osteoblasts, or adipocytes. Bone marrow fibroblasts secrete a large variety of cytokines (e.g., macrophage colony-stimulating factor [M-CSF], granulocyte-macrophage colony-stimulating factor [GM-CSF], IL-6) that are necessary for hematopoiesis (see Table 25-4). Osteoblasts are responsible for the formation of new bone.10 Additionally, osteoclasts secrete cytokines that drive hematopoietic cell differentiation. Adipocytes are fat cells containing large depositions of lipid. Adipocytes secrete several growth factors, as well as leptin. Leptin preferentially stimulates mesenchymal stem cells to differentiate into osteoblasts.
Macrophages and osteoclasts have common monocytic precursors. Bone marrow macrophages secrete cytokines and chemokines that regulate proliferation of hematopoietic progenitor cells. Other monocytic cells differentiate under the direction of stromal cells and osteoblasts and undergo intercellular fusion into large multinucleate osteoclasts. Osteoclasts remodel bone by resorption and can produce cytokines that affect proliferation of hematopoietic cells.
The hematologic compartment of the bone marrow consists of a variety of cellular niches that favor differentiation of various hematopoietic progenitor cells.11 The niches are distinguished by a variety of locally produced cytokines and growth factors, so that one niche may be more likely to support differentiation of erythroid precursors into erythrocytes, whereas granulocytes may differentiate in a different site.
Cell Differentiation
Each type of blood cell originates from common hematopoietic stem cells that proliferate and differentiate under control of a variety of cytokines and growth factors (Figure 25-9 and see Table 25-4). During this process some hematopoietic stem cells undergo different paths of differentiation into more differentiated stem cells that are committed to a particular line of blood cells.
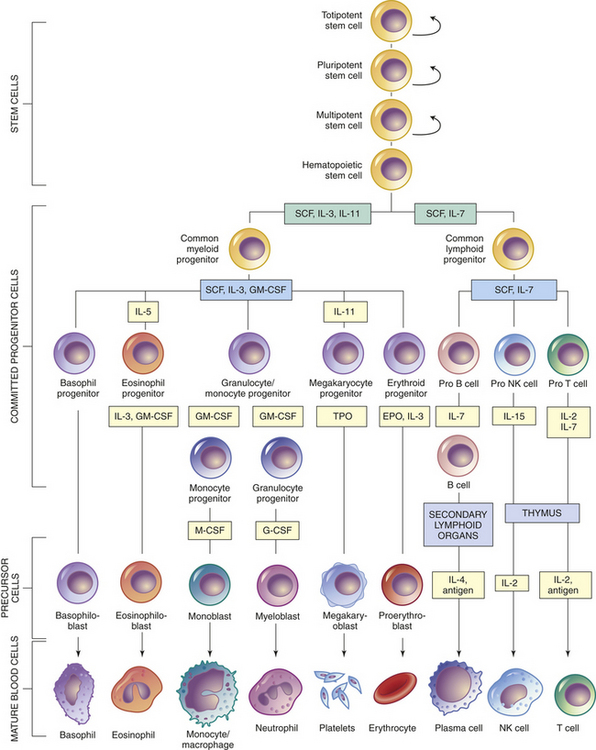
Figure 25-9 Differentiation of hematopoietic cells. Curved arrows indicate proliferation and expansion of pre-hematopoietic stem cell populations. EPO, erythropoietin; G-CSF, granulocyte colony-stimulating factor; GM-CSF, granulocyte-macrophage colony-stimulating factor; IL, interleukin; M-CSF, macrophage colony-stimulating factor; NK, natural killer; SCF, stem cell factor; TPO, thrombopoietin.
All humans originate from a single cell (the fertilized egg) that has the capacity to proliferate and eventually differentiate into the huge diversity of cells of the human body. After fertilization, the egg divides over a 5-day period to form a hollow ball (blastocyst) that implants on the uterus. Until about 3 days after fertilization, each cell (blastomere) is undifferentiated and retains the capacity to differentiate into any cell type. In the 5-day blastocyst, the outer layer cells have undergone differentiation and commitment to become the placenta. Cells of the inner cell mass (embryonic stem cells), however, continue to have unlimited differentiation potential (currently referred to as being pluripotent) and can grow into different kinds of tissue—blood, nerves, heart, bone, and so forth. After implantation, cells of the inner cell mass begin differentiation into other cell types. Differentiation is a multistep process and results in intermediate groups of stem cells (multipotent stem cells) with more limited, but still impressive, abilities to differentiate into many different types of cells (see Figure 25-9).12
The hematopoietic organs contain a population of hematopoietic stem cells that have partially differentiated.13 They have the capacity to differentiate easily into any of the hematologic cell populations but are very difficult to differentiate into other cell types, like nerve or muscle cells.12 The challenge of getting any partially committed multipotent stem cells to differentiate reliably involves coaxing them with identical chemical signals that the body uses naturally for differentiation. This is a daunting task with potentially astonishing clinical implications. For example, bone marrow might become the reservoir from which stem cells are harvested and then stimulated to produce nerve cells to help with the treatments of spinal cord injuries.
As with all stem cells, the hematopoietic stem cells are self-renewing (they have the ability to proliferate without further differentiation) so that a relatively constant population of stem cells is available.12 Some hematopoietic stem cells will continue differentiation into hematopoietic progenitor cells. Progenitor cells retain proliferative capacity but are committed to possible further differentiation into particular types of hematologic cells: lymphoid (lymphocytes, NK cells), granulocyte-monocyte (granulocytes, monocytes, macrophages), and megakaryocyte-erythroid (platelets, erythrocytes) progenitor cells (see Figure 25-9).
As with all other forms of cellular differentiation, successful hematopoiesis requires that progenitor cells interact with neighboring cells (e.g., stromal cells of the bone marrow) through a variety of adhesion molecules and are exposed to particular signaling molecules (e.g., cytokines) (see Table 25-4).14 Stromal cells apparently express steel factor, a stem cell factor, which activates stem cells to develop. Several cytokines participate in hematopoiesis, particularly colony-stimulating factors (CSFs or hematopoietic growth factors), which stimulate the proliferation of progenitor cells and their progeny and initiate the maturation events necessary to produce fully mature cells (see Figure 25-9).15 Multiple cell types in the hematopoietic organs, including endothelial cells, fibroblasts, and lymphocytes, produce CSFs.
Hematopoiesis in the bone marrow occurs in two separate pools, the stem cell pool and the bone marrow pool (Figure 25-10). The stem cell pool is responsible for maintaining the number of pluripotent stem cells and partially committed progenitor cells. The bone marrow pool contains cells that are proliferating and maturing in preparation for release into the circulation and mature cells that are stored for later release into the peripheral blood. The peripheral blood also contains two pools of cells; those in the circulation and those stored around the walls of the blood vessels (often called the marginating storage pool). The marginating storage pool primarily consists of neutrophils that adhere to the endothelium in vessels where the blood flow is relatively slow. These cells can rapidly move into tissues and mucous membranes when needed in an inflammatory response. The infiltrating cells are replenished from the circulating pool.
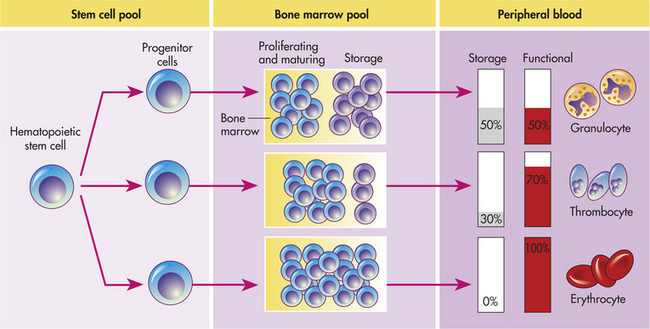
Figure 25-10 Hematopoiesis. Hematopoiesis from the stem cell pool; activity mainly in the bone marrow and in the peripheral blood. (Modified from Harmening DM, editor: Clinical hematology and fundamentals of hemostasis, ed 3, Philadelphia, 1997, FA Davis.)
Under certain conditions of rapid depletion of the circulating pool, the circulating hematologic cells need to be rapidly replenished. Medullary hematopoiesis can be accelerated by any or all of three mechanisms: (1) conversion of yellow bone marrow, which does not produce blood cells, to hematopoietic red marrow by the actions of erythropoietin (a hormone that stimulates erythrocyte production); (2) faster differentiation of progenitor cells; and (3) faster proliferation of stem cells into progenitor cells (see Table 25-4).
Clinical Uses of Colony-Stimulating Factors
Neutrophils are normally present in the blood in the range of 4000 to 6000 cells/μL, and in response to a bacterial infection, numbers usually increase to 10,000 to 20,000 cells/μL. Susceptibility to infection develops when normal levels drop below 1000 cells/μL, such as during congenital neutropenia or as a consequence of cytotoxic therapy for cancer. Similarly normal levels of other hematologic cells may be suppressed, e.g., congenital or acquired immune deficiencies and anemia (see Chapters 8 and 9).
The numbers of circulating hematologic cells are under the control of CSFs (see Table 25-4). Administration of CSFs can raise white cell numbers to extremely high levels in healthy individuals. These excessive levels of white blood cells may result in production of toxic products and tissue damage. Therapy with CSFs has been tested in individuals with subnormal levels of circulating blood cells, such as acquired immunodeficiency syndrome (AIDS), aplastic anemia, or congenital neutropenia or as a consequence of cytotoxic therapy lymphoma or leukemia (Figure 25-11). CSF therapy can stimulate increases in circulating granulocyte-monocyte populations, but the degree of response depends on the available numbers of stem and progenitor cells that have survived chemotherapy or the effects of disease. CSF treatment has corrected some cases of congenital neutropenia and resulted in reconstitution of hematopoiesis after bone marrow transplantation.16 CSF treatment can result in shorter periods of intensive nursing and hospitalization. Recombinant CSFs (e.g., granulocyte colony-stimulating factor [G-CSF], GM-CSF, erythropoietin) are being mass-produced for therapeutic use.
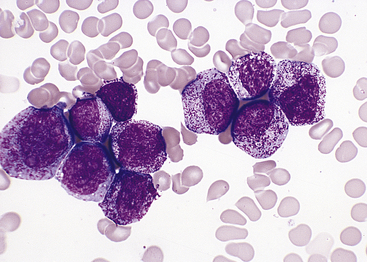
Figure 25-11 Colony-stimulating factor (CSF) effects. Morphologic effects of growth factor. Marrow aspirate from a patient receiving granulocyte colony-stimulating factor (G-CSF) showing an early neutrophil response. There is a marked shift toward immaturity in the neutrophils with the majority at the promyelocyte and early myelocyte stages of maturation (Wright-Giemsa stain). (From Damjanov I, Linder J, editors: Anderson’s pathology, ed 10, St Louis, 1996, Mosby.)
Development of Erythrocytes
For almost 100 years it was believed that erythrocytes developed from lymphocytes that were transformed in the spleen. It was not until the 1850s that the bone marrow was identified as the site of erythropoiesis, or development of red blood cells.
Erythropoiesis
In the confines of the bone marrow erythroid progenitor cells proliferate and differentiate into large, nucleated proerythroblasts, which are committed into producing cells of the erythroid series (Figure 25-12). Erythroid development from the proerythroblast onward is contained in a compartment referred to as the erythron.17 The proerythroblast, which has ribosomes and can produce protein, differentiates through several intermediate forms of erythroblast while synthesizing hemoglobin and progressively eliminating most intracellular structures, including the nucleus. Thus the maturing erythroblast becomes more compact and progressively assumes the shape and characteristics of an erythrocyte. Hemoglobin is readily apparent and increases in quantity as nuclear size shrinks throughout the basophilic and polychromatophilic stages. The orthochromatic erythroblast (normoblast) is the smallest of the nucleated erythrocyte precursors.
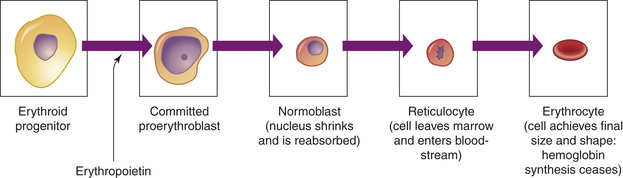
Figure 25-12 Erythrocyte differentiation. Erythrocyte differentiation from large nucleated progenitor cells to small nonnucleated erythrocytes.
The last immature form of erythroblast is the reticulocyte, which is anucleate and contains a meshlike (reticular) network of ribosomal ribonucleic acid (RNA) that is visible microscopically after staining with certain dyes. The reticulocyte contains polyribosomes (for globin synthesis) and mitochondria (for oxidative metabolism and heme synthesis). The reticulocyte matures into an erythrocyte within 24 to 48 hours. During this period, mitochondria and ribosomes disappear and the cell becomes smaller and more disk like. With these final changes, the erythrocyte loses its capacity for hemoglobin synthesis and oxidative metabolism. Reticulocytes remain in the marrow approximately 1 day and are released into the venous sinuses. They continue to mature in the bloodstream and may travel to the spleen for several days of additional maturation. The normal reticulocyte count is 1% of the total red blood cell count. Approximately 1% of the body’s circulating erythrocyte mass normally is generated every 24 hours. Therefore, the reticulocyte count is a useful clinical index of erythropoietic activity and indicates whether new red cells are being produced.
Regulation of Erythropoiesis
In healthy individuals, the total volume of circulating erythrocytes remains surprisingly constant. Most steps of erythropoiesis are primarily under the control of a feedback loop involving the glycoprotein erythropoietin (see Table 25-4). In conditions of tissue hypoxia, erythropoietin is secreted by the liver and, primarily, by the peritubular cells of the kidney (Figure 25-13). Rising levels of circulating erythropoietin cause a compensatory increase in proliferation and differentiation of proerythroblasts in the bone marrow. The density of cellular erythropoietin receptor decreases progressively during erythroid maturation to almost undetectable levels on reticulocytes. The normal steady-state rate of production of approximately 2.5 million erythrocytes per second can increase to 17 million per second during anemia or under conditions of low oxygen, such as high-altitude or pulmonary disease. Thus the body responds to reduced oxygenation of blood in two ways: (1) stimulation of chemoreceptors of the carotid body and aortic arch that signal the brain to increase respiration and (2) stimulation of receptors on the kidney peritubular cells to increase erythropoietin synthesis and release.
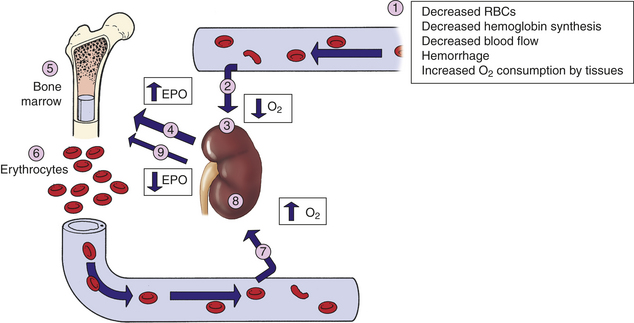
Figure 25-13 Role of erythropoietin in regulation of erythropoiesis. (1) Decreased arterial oxygen levels result in (2) decreased tissue oxygen (hypoxia) that (3) stimulates the kidney to increase (4) production of erythropoietin. Erythropoietin is carried to the bone marrow (5) and binds to erythropoietin receptors on proerythroblasts, resulting in increased red cell production and maturation and expansion of the erythron (6). The increased release of red cells into the circulation frequently corrects the hypoxia in the tissues (7). (8) Perception of normal oxygen levels by the kidney causes (9) diminished production of erythropoietin (negative feedback) and return to normal levels of erythrocyte production. EPO, Erythropoietin; O2, oxygen in the blood and tissue; RBCs, red blood cells.
One of the most significant advances in the study of hematopoietic growth factors has been the development of erythropoietin for use in individuals with chronic renal failure. In 1986 large amounts of recombinant human erythropoietin (r-HuEPO) became widely available for clinical research. Erythropoietin is administered intravenously or subcutaneously for the treatment of anemia caused by decreased production of erythropoietin. An immediate effect of increased endogenous or exogenous erythropoietin is an increase in the blood reticulocyte count, followed by increasing levels of erythrocytes. The most significant side effect associated with r-HuEPO is increased blood pressure.
Hemoglobin Synthesis
Hemoglobin (Hb), the oxygen-carrying protein of the erythrocyte, constitutes approximately 90% of the cell’s dry weight. Hemoglobin-packed blood cells take up oxygen in the lungs and exchange it for carbon dioxide in the tissues. A single erythrocyte can contain as many as 300 hemoglobin molecules. Hemoglobin increases the oxygen carrying capacity of blood by 100-fold. Each hemoglobin molecule is composed of two pairs of polypeptide chains (the globins) and four colorful complexes of iron plus protoporphyrin (the hemes) (Figure 25-14).18 Hemoglobin is responsible for blood’s ruby-red color.
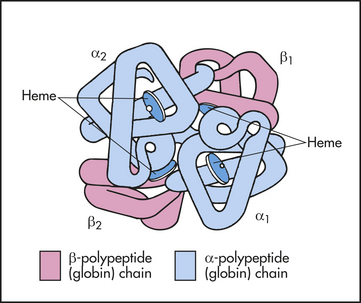
Figure 25-14 Molecular structure of hemoglobin. Molecule is a spherical tetramer weighing approximately 64,500 daltons. It contains a pair of α-polypeptide and a pair of β-polypeptide chains and several heme groups.
Several variants of hemoglobin exist, but they differ only slightly in primary structure based on the use of different polypeptide chains; alpha, beta, gamma, delta, epsilon, or zeta (α, β, γ, δ, ε, or ζ) (Table 25-5). Each polypeptide chain contains approximately 150 amino acids and is arranged in the knotted-sausage configuration shown in Figure 25-14. The chains assemble to form a tetrahedron containing two pairs of identical chains. Hemoglobin A, the most common type in adults, is composed of two α- and two β-polypeptide chains (α2β2). A normal variant, fetal hemoglobin (hemoglobin F) is a complex of two α- and two γ-polypeptide chains (α2γ2) that binds oxygen with a much greater affinity than adult hemoglobin.
Table 25-5
Structure of Normal Hemoglobin Molecules
| Type of Hemoglobin (Hb) | Identity of Polypeptide Chain | Significance |
| HbA | α2β2 | 92% of adult Hb |
| HbA1c | α2 (β-NH-glucose) | 5% of adult Hb; increased in diabetes (see Chapter 21) |
| HbA2 | α2δ2 | 2% of adult Hb; increased in beta-thalassemia (see Chapter 28) |
| HbF | α2γ2 | Major fetal Hb from the third through ninth month of gestation; promotes oxygen transfer across platelets; increase in beta-thalassemia |
| Hb Gower I | ε4 or ζ2ε2 | Present in early embryo; function unknown |
| Hb Gower II | α2ε2 | Present in early embryo; function unknown |
| Hb Portland | ζ2γ2 | Present in early embryo; function unknown |
Heme is a large, flat, iron-protoporphyrin disk that is synthesized in the mitochondria and can carry one molecule of oxygen (O2). Thus an individual hemoglobin molecule with its four hemes can carry four oxygen molecules. If all four oxygen-binding sites are occupied by oxygen, the molecule is said to be saturated. Through a series of complex biochemical reactions, protoporphyrin, a complex four-ringed molecule, is produced and bound with ferrous iron. It is crucial that the iron be correctly charged; reduced ferrous iron (Fe2+) can bind oxygen in the lungs and release it in the tissues, where oxygen concentration is less, whereas ferric iron (Fe3+) cannot. Binding of oxygen to ferrous iron (oxyhemoglobin) temporally oxidizes Fe2+ to Fe3+, but after the release of oxygen the body reduces the iron to Fe2+ (deoxyhemoglobin [reduced hemoglobin]) and reactivates the hemoglobin’s capacity to bind oxygen. Without reactivation by methemoglobin reductase, the Fe3+-containing hemoglobin (methemoglobin) cannot bind oxygen.
Several other molecules can competitively bind to deoxyhemoglobin. Carbon monoxide (CO) directly competes with oxygen for binding to ferrous ion with an affinity that is about 200-fold greater than oxygen. Thus even a small amount of CO can dramatically decrease the ability of hemoglobin to bind and transport oxygen. Hemoglobin also binds carbon dioxide (CO2), but at a binding site separate from where oxygen binds. In the lungs, CO2 is released allowing hemoglobin to bind oxygen.
Erythrocytes may play a role in the maintenance of vascular relaxation. Nitric oxide (NO) produced by blood vessels is a major mediator of relaxation and dilation of the vessel walls. In the lungs, hemoglobin can concurrently bind oxygen to the ferrous ion and NO to cysteine residues in the globins. As hemoglobin transfers its oxygen to tissue, it may also shed small amounts of nitric oxide, contributing to dilation of the blood vessels and helping get the oxygen into tissues.
Nutritional Requirements for Erythropoiesis
Normal development of erythrocytes and synthesis of hemoglobin depends on an optimal biochemical milieu and adequate supplies of the necessary building blocks, including protein, vitamins, and minerals (Table 25-6). If these components are lacking for a prolonged time, erythrocyte production slows and anemia (insufficient numbers of functional erythrocytes) may result (see Chapter 26).
Table 25-6
Nutritional Requirements for Erythropoiesis
| Nutrient | Role in Erythropoiesis | Consequence of Deficiency |
| Protein (amino acids) | Structural component of plasma membrane | Decreased strength, elasticity, and flexibility of membrane; hemolytic anemia |
| Synthesis of hemoglobin | Decreased erythropoiesis and life span of erythrocytes | |
| Cobalamin (vitamin B12) | Synthesis of DNA, maturation of erythrocytes, facilitator of folate metabolism | Macrocytic (megaloblastic) anemia |
| Folate (folic acid) | Synthesis of DNA and RNA, maturation of erythrocytes | Macrocytic (megaloblastic) anemia |
| Vitamin B6 (pyridoxine) | Heme synthesis | Microcytic-hypochromic anemia |
| Vitamin B2 (riboflavin) | Oxidative reactions | Normocytic-normochromic anemia |
| Vitamin C (ascorbic acid) | Iron metabolism, acts as a reducing agent to maintain iron in its ferrous (Fe2+) form | Normocytic-normochromic anemia |
| Pantothenic acid | Heme synthesis | Unknown in humans∗ |
| Niacin | None, but needed for respiration in mature erythrocytes | Unknown in humans |
| Vitamin E | Heme synthesis (?); protection against oxidative damage in mature erythrocytes | Hemolytic anemia with increased cell membrane fragility; shortens life span of erythrocytes in individuals with cystic fibrosis |
| Iron | Hemoglobin synthesis | Iron deficiency anemia |
| Copper | Required for optimal mobilization of iron from tissues to plasma | Microcytic-hypochromic anemia |
DNA, Deoxyribonucleic acid; RNA, ribonucleic acid.
∗Although pantothenic acid is important for optimal synthesis of heme, experimentally induced deficiency failed to produce anemia or other hematopoietic disturbances.
Data from Strine-Martin EA, Lotspeich-Steininger CA, Koepke JA: Clinical hematology: principles, procedures, correlations, ed 2, Philadelphia, 1998, Lippincott.
Erythropoiesis cannot proceed in the absence of vitamins, especially B12, folate (folic acid), B6, riboflavin, pantothenic acid, niacin, ascorbic acid, and vitamin E. Dietary vitamin B12 is a large molecule that requires a protein secreted by parietal cells into the stomach (intrinsic factor [IF]) for transport across the ileum. Once absorbed, vitamin B12 is stored in the liver and used as needed in erythropoiesis. Defects in IF production lead to decreased B12 absorption and pernicious anemia.
Folate is the second most important vitamin for erythrocyte production and maturation. Folate is necessary for DNA synthesis, being a component of three of the four DNA bases (thymine, adenine, and guanine), and RNA synthesis. Folate absorption occurs principally in the upper small intestine and is stored in the liver. Folate deficiency is more common than vitamin B12 deficiency and occurs more rapidly. Folate stores can be depleted within a few months, whereas vitamin B12 depletion can take years. Folate supplements are prescribed for pregnant women because pregnancy increases the demand for folate and may cause anemia.
Normal Destruction of Senescent Erythrocytes
After about 100 to 120 days in the circulation, old erythrocytes are removed by tissue macrophages, primarily in the spleen. Although mature erythrocytes lack nuclei, mitochondria, and endoplasmic reticulum, they do have cytoplasmic enzymes capable of glycolysis (anaerobic glucose metabolism) and production of small quantities of ATP, which provides the energy needed to maintain cell function and membrane pliability. Metabolic processes diminish as the erythrocyte ages, so less ATP is available to maintain plasma membrane function. The senescent red cell becomes increasingly fragile and loses its reversible deformability, becoming susceptible to rupture while passing through narrowed regions of the microcirculation.
Additionally, the plasma membrane of senescent red cells undergoes phospholipid rearrangement that is recognized by receptors on macrophages (primarily in the spleen) that selectively remove and sequester the red cells. If the spleen is dysfunctional or absent, macrophages in the liver (Kupffer cells) take over.
The erythrocytes are digested by proteolytic and lipolytic enzymes in the phagolysosomes (digestive vacuoles) of the macrophage. The heme and globin of methemoglobin dissociate easily, and the globin is broken down into its component amino acids. The iron in hemoglobin is oxidized, forming Fe+3 (methemoglobin), and recycled (see following section).
Porphyrin is reduced to bilirubin, which is transported to the liver, conjugated, and finally excreted in the bile as glucuronide (Figure 25-15). Approximately 6 g of hemoglobin is catabolized daily, producing 200 mg of bilirubin. Bacteria in the intestinal lumen transform conjugated bilirubin into urobilinogen. Although a small portion is reabsorbed to be either metabolized further by the liver or excreted by the kidney into the urine, most urobilinogen is excreted in feces. Conditions causing accelerated erythrocyte destruction increase the load of bilirubin for hepatic clearance, leading to increased serum levels of unconjugated bilirubin and increased urinary excretion of urobilinogen. Gallstones (cholelithiasis) can result from a chronically elevated rate of bilirubin excretion.
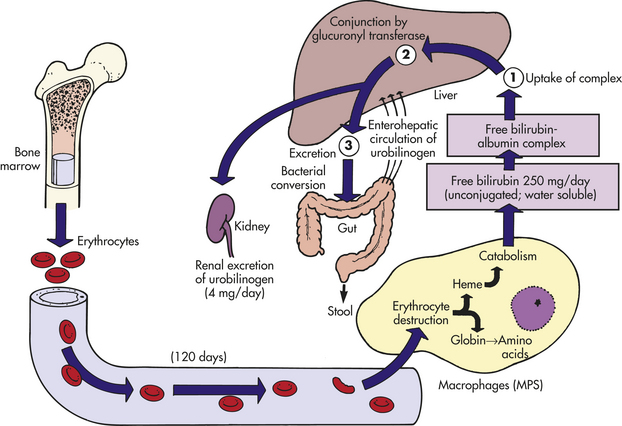
Iron Cycle
Approximately 67% of total body iron is bound to heme in erythrocytes (hemoglobin) and muscle cells (myoglobin), and approximately 30% is stored in mononuclear phagocytes (i.e., macrophages) and hepatic parenchymal cells as either ferritin or hemosiderin. The remaining 3% (less than 1 mg) is lost daily in urine, sweat, bile, and epithelial cells in the gut.
Iron is continually recycled.19 The methemoglobin released from the breakdown of senescent or damaged erythrocytes (see preceding section) is dissociated by the enzyme heme oxygenase, and the iron released into the bloodstream, where it is free to bind again to transferrin, or stored in the macrophage’s cytoplasm as ferritin or hemosiderin (Figure 25-16). A minute amount of iron is stored in muscle cells by the heme-containing protein myoglobin. Unavailable stores of iron are present in cytochromes, catalases, and peroxidase enzymes.
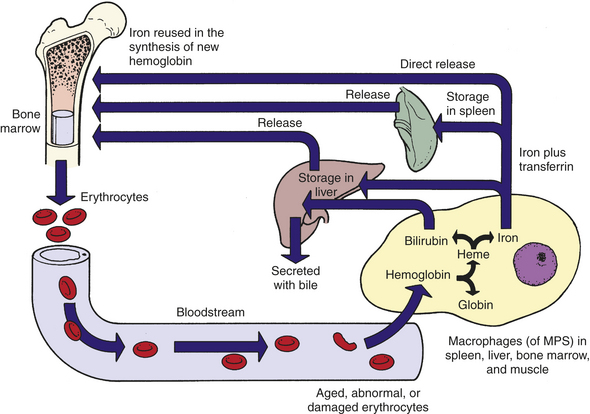
Figure 25-16 Iron cycle. Iron (Fe) released from gastrointestinal epithelial cells circulates in the bloodstream associated with its plasma carrier, transferrin. It is delivered to erythroblasts in bone marrow, where most of it is incorporated into hemoglobin. Mature erythrocytes circulate for approximately 100 to 120 days, after which they become senescent and are removed by the mononuclear phagocyte system (MPS). Tissue macrophages (mostly in spleen) break down ingested erythrocytes and return iron to the bloodstream directly or after storing it as a ferritin or hemosiderin.
The protein ferritin is the major intracellular iron storage protein. Apoferritin, which is ferritin without attached iron, can store thousands of atoms of iron. Several (24) apoferritin complexes combine to form the micelle ferritin. Large aggregates of micelles (if a large amount of iron is present) produce numerous ferritin micelles, known as hemosiderin. Hemosiderin is visible as an iron-based pigment under a light microscope as cell inclusions. The iron within deposits of hemosiderin is poorly available to supply iron when needed. Conditions leading to large amounts of iron include hemolysis, severe congestion, unusual increases in dietary iron consumption, increased absorption, or decreased loss. The most common cause of hemosiderin deposition is simple bruising. Hemosiderin in small amounts within iron-rich tissues (i.e., spleen, liver, bone marrow) is considered normal. Large aggregates or its presence in tissue such as the lungs or subcutaneous tissue suggest a pathologic condition.
Iron balance is maintained through controlled absorption rather than excretion. Dietary iron (primarily as Fe2+) is transported by a divalent metal ion transporter directly across the membranes of epithelial cells in the duodenum and proximal jejunum.20 (Transport mechanisms are described in Chapter 1.) Regulation of iron transport across the plasma membrane of gastrointestinal epithelial cells is related to the cells’ iron content and the overall rate of erythropoiesis. If the body’s iron stores are low or the demand for erythropoiesis increases, iron is transported rapidly through the epithelial cell and into the plasma. If body stores are high and erythropoiesis is not increased, iron transport is shut down, although iron can cross the epithelial cells’ plasma membrane passively and is stored as ferritin. Excretion of iron occurs when the epithelial cells of the intestinal mucosa slough off.
Iron from either dietary sources or erythrocyte catabolism is transported in the blood bound to apotransferrin, which is then called transferrin; under normal conditions, only one third of the iron-binding sites on transferrin molecules are occupied. Apotransferrin is a glycoprotein synthesized primarily by hepatocytes in the liver but also produced in small quantities by tissue macrophages, submaxillary and mammary glands, and ovaries or testes. Iron for hemoglobin production is carried by transferrin to the bone marrow, where it binds to transferrin receptors on erythroblasts. Transferrin receptors are on the plasma membrane of all nucleated cells, although at particularly high levels on erythroid precursors and rapidly proliferating cells (e.g., lymphocytes), and are thought to be the only route of cellular entry for transferrin-attached iron. Transferrin is recycled (transferrin cycle) in the following manner:
1. The transferrin-iron complex binds to a transferrin receptor on the erythroblast’s plasma membrane.
2. The complex moves into the cell by receptor-mediated endocytosis.
3. Iron is released (dissociated) from transferrin.
4. The dissociated transferrin is returned to the bloodstream for reuse.
The iron is transported to the erythroblast’s mitochondria (the site of hemoglobin production), where the enzyme heme synthetase inserts ferrous iron into protoporphyrin to form heme. Heme then is bound to globin to form hemoglobin. Iron not used in erythropoiesis is stored temporarily as ferritin or hemosiderin and later excreted.
Development of Leukocytes
Leukocytes consist of lymphocytes, granulocytes, and monocytes. Most of the leukocytes arise from stem cells in the bone marrow (their pathways of differentiation are shown in Figure 25-9). Hematopoietic stem cells differentiate into two populations of progenitor cells: common lymphoid progenitors and common myeloid progenitors. Lymphoid progenitors that remain in the bone marrow undergo differentiation into the B-cell lineage, after which they are released into the circulation and undergo further maturation in the secondary lymphoid organs (described in Chapter 7 [see Figure 7-12]). The common myeloid progenitors futher differentiate into progenitors for basophils, mast cells, eosinophils, and megakaryocytes, and granulocyte/monocyte progenitors. The granulocyte/monocyte progenitors further differentiate into monocyte progenitors and granulocyte progenitors, which develop into monocytes/macrophages and neutrophils, respectively. Development from hematopoietic stem cell to common granulocyte-monocyte progenitors primarily is under the control of stem cell factor, IL-3, and GM-CSF, whereas further differentiation into granulocytic and monocytic progenitors is controlled by G-CSF and M-CSF, respectively (see Table 25-4).
Monocytic progenitors undergo development into monocytes within 24 hours and are released into the circulation. Monocytes mature into various forms of macrophages, which is usually complete within 1 or 2 days after release (see Table 25-3).21
Progenitor cells for granulocytes normally fully mature in the bone marrow into neutrophils, eosinophils, and basophils. The ultimate phenotype is determined by relative local bone marrow concentrations of early and late-acting cytokines, including GM-CSF, G-CSF, IL-3, IL-5, stem cell factor, and others (see Table 25-4). Granulocytes are released into the blood within 14 days of development. The bone marrow selectively retains immature granulocytes as a reserve pool that can be rapidly mobilized in response to the body’s needs.
Most leukocytes exist in the body from days to years, depending on type. Maintenance of optimal levels of granulocytes and monocytes in the blood depends on the availability of pluripotent stem cells in the marrow, induction of these into committed stem cells, timely release of new cells from the marrow, and mobilization of the granulocyte reserve pool. Leukocyte production increases in response to infection, to the presence of steroids, and to reduction or depletion of reserves in the marrow. It is also associated with strenuous exercise, convulsive seizures, heat, intense radiation, paroxysmal tachycardias, pain, nausea and vomiting, and anxiety.
Development of Platelets
Platelets (thrombocytes) are derived from stem cells and progenitor cells that differentiate into megakaryocytes.22 During thrombopoiesis, the megakaryocyte progenitor is programmed to undergo an endomitotic cell cycle (endomitosis) during which DNA replication occurs, but anaphase and cytokinesis are blocked (see Chapter 1). Thus the megakaryocyte nucleus enlarges and become extremely polyploidy (up to 100-fold or more of the normal amount of DNA) without cellular division. Concurrently, the numbers of cytoplasmic organelles (e.g., internal membranes, granules) increase, and the cell develops cell surface elongations and branches that progressively fragment into platelets. A single megakaryocyte may produce thousands of platelets. Like erythrocytes, platelets released from the bone marrow lack nuclei.
About two thirds of platelets enter the circulation, and the remainder reside in the splenic pool. Platelets circulate in the bloodstream for about 10 days before losing their ability to carry out thrombogenic activity. Senescent platelets are sequestered and destroyed in the spleen by mononuclear cell phagocytosis.
An adequate level of committed platelet precursors (megakaryoblasts) in the bone marrow and differentiation into circulating platelets are controlled by specific interactions between megakaryocyte progenitors and stromal cells in the bone marrow as well as thrombopoietin (TPO), a hormonal growth factor primarily produced by the liver, and various cytokines and colony-stimulating factors and interleukins (see Table 25-4). Platelets express high affinity receptors for TPO, and when circulating platelet levels are normal, TPO is adsorbed onto the platelet surface and prevented from accessing the bone marrow and initiating further platelet production.23 TPO stimulates committed cells at further stages of differentiation to differentiate faster so that rates of megakaryocyte development, endomitosis, and platelet release are increased. During inflammation IL-6 induces increased production of TPO, which increases production of newly formed platelets, which are more thrombogenic.
MECHANISMS OF HEMOSTASIS
Hemostasis is defined as arrest of bleeding (Figure 25-17). As a result of hemostasis, damaged blood vessels maintain a relatively steady state of blood volume, pressure, and flow. The importance of hemostasis clearly varies with vessel size. Damage to large vessels cannot easily be controlled by hemostasis but requires vascular contraction and dramatically decreased blood flow into the damaged vessels.
Three equally important components of hemostasis are the vasculature (endothelial cells and subendothelial matrix), platelets, and blood proteins (clotting factors). The general sequence of events in hemostasis are (1) vascular injury leads to a transient arteriolar vasoconstriction to limit blood flow to the affected site; (2) damage to the endothelial cell lining of the vessel exposes prothrombogenic subendothelial connective tissue matrix leading to platelet adherence and activation and formation of a hemostatic plug to prevent further bleeding (primary hemostasis); (3) tissue factor, produced by the endothelium, collaborates with secreted platelet factors and activated platelets to activate the clotting (coagulation) system to form fibrin clots and further prevent bleeding (secondary hemostasis); and (4) the fibrin/platelet clot contracts to form a more permanent plug, and regulatory pathways are activated (fibrinolysis) to limit the size of the plug and begin the healing process.
Function of Blood Vessels
The vessel walls consist of a layer of endothelial cells that adhere to an underlying matrix of connective tissue. The matrix contains a variety of proteins, including collagen, fibronectin, and laminins. Endothelial cells adhere to the matrix and to each other through receptors (e.g., vascular endothelial cell-specific cadherin [VE-cadherin], platelet-endothelial cell adhesion molecule-1 [PECAM-1], integrins [especially α2β1 and α5β1]) that are expressed only on the intercellular and basal surfaces.
Under normal conditions the endothelium actively regulates blood flow and prevents spontaneous activation of platelets and the clotting system (see Figure 25-17). Endothelial cells produce nitric oxide (NO) from L-arginine and prostacyclin (PGI2) from arachidonic acid. Both NO, via cGMP, and PGI2 are vasodilators that work in concert with endothelin (a vasoconstrictor) to maintain blood flow and pressure.24 NO and PGI2 also inhibit platelet adhesion and aggregation. Synergism between PGI2 and NO is significant. PGI2 production varies a great deal in response to stimuli, whereas NO is released continually to regulate vascular tone. NO has other biologic functions including cell signaling, free radical production, and possibly others. Endothelium also produces adenosine diphosphatase, which degrades ADP (a potent activator of platelets).
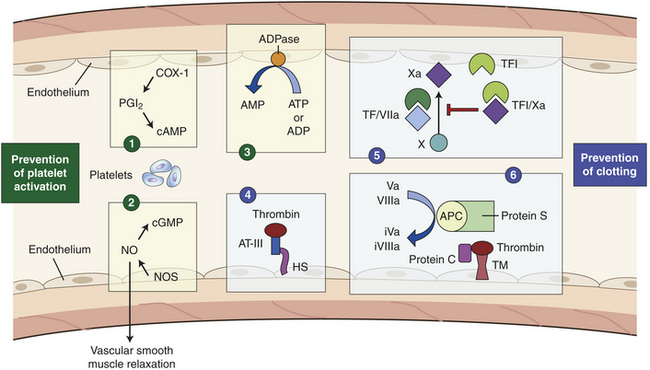
Figure 25-17 Hemostasis. Endothelium controls hemostasis by preventing platelet activation (1-3) and preventing activation of the clotting system (4-6). (1) Prostacyclin production. Injury activates inflammation (COX-1 arachidonic acid). Enzymes convert arachidonic acid into prostacylin I2 (PGI2) in endothelial cells. PGI2 eventually increases intracellular cyclic adenosine monophosphate (cAMP); cAMP inhibits platelet aggregation and induces vasodilation. Nitric oxide (NO) formation is induced by NO synthases (NOS) and NO causes increased cyclic guanosine monophosphate (cGMP). (2) Nitric oxide system. Endothelial cell NOS produces nitric oxide, which controls platelet activation through cGMP-mediated signaling. (3) ADPase. Endothelial cells express a surface bound ADPase (CD39) that converts circulating ADP and ATP to AMP. (4) Antithrombin III–heparan sulfate system. Antithrombin III (AT-III) inhibits thrombin slowly when heparan sulfate (HS) is absent. When HS is present, it quickly activates thrombin because it binds to a specific site on AT-III that causes an instant conformational change in AT-III, allowing it to quickly activate thrombin. (5) Tissue factor inhibitor (TFI) system. Expression of TFI on the endothelial cells and secreted into the circulation complexes with factor IXa to form a competitive inhibitor of the tissue factor/factor VIIa complex (TF/VIIa) and prevent further activation of factor X to Xa. (6) Protein C/protein S pathway (thrombomodulin). Thrombin in the circulation binds to thrombomodulin on the endothelial cell creating a complex that can bind and activate protein C to activated protein C (APC) that complexes in the blood or on the surface of active platelets with protein S. This complex degrades circulating clotting factors Va and VIIIa to inactive forms (iVa, iVIIIa) to prevent further activation of clotting.
The endothelial cell surface contains antithrombotic molecules, such as glycosaminoglycans (e.g., heparan sulfate), thrombomodulin, and plasminogen activators. These limit platelet activation and fibrin deposition. Although thrombomodulin and plasminogen activators help control hemostasis in normal vessels, their effects are magnified during vascular damage and clot formation; therefore, further information is provided on these molecules in the following section on control of hemostatic mechanisms.
As a result of damage to the vessels, the endothelial cell barrier is frequently compromised, remaining endothelial cells activated by product of tissue damage, and the underlying matrix exposed. Endothelial cells contain intracellular structures (Weibel-Palade bodies) that contain von Willebrand factor (vWF) that is released during damage. The matrix, in addition to collagen and other connective tissue, contains vWF and can bind additional vWF released by the endothelium.25 The matrix itself and vWF are potent activators of platelets.
Function of Platelets
Platelets normally circulate freely, suspended in plasma, in an unactivated state. The role of platelets is to (1) contribute to regulation of blood flow into a damaged site by induction of vasoconstriction (vasospasm), (2) initiate platelet-to-platelet interactions resulting in formation of a platelet plug to stop further bleeding, (3) activate the coagulation (or clotting) cascade to stabilize the platelet plug, and (4) initiate repair processes including clot retraction and clot dissolution (fibrinolysis). The normal platelet count ranges from 140,000 to 340,000/mm3. If platelet counts drop below 100,000/mm3 an individual is usually considered thrombocytopenic (abnormally low numbers of platelets) and may experience prolongation of normal clotting but is usually not at risk for spontaneous major bleeding episodes unless the platelet count falls below 20,000/mm3. If platelet numbers are elevated (thrombocytosis) the risk for spontaneous blood clots (thrombosis), stroke, or heart attack is increased.
The state of platelet activation is primarily under the control of endothelial cells lining the vessels. Damage to the vessel initiates a process of platelet activation; (1) increased platelet adhesion to the damaged vascular wall; (2) activation leading to secretion of chemicals from platelet granules, which stimulate changes in platelet shape and biochemistry; and (3) aggregation as platelet-vascular wall and platelet-platelet adherence increases.26 This process leads to activation of the clotting system and development of an immobilizing meshwork of platelets and fibrin (Figure 25-18).
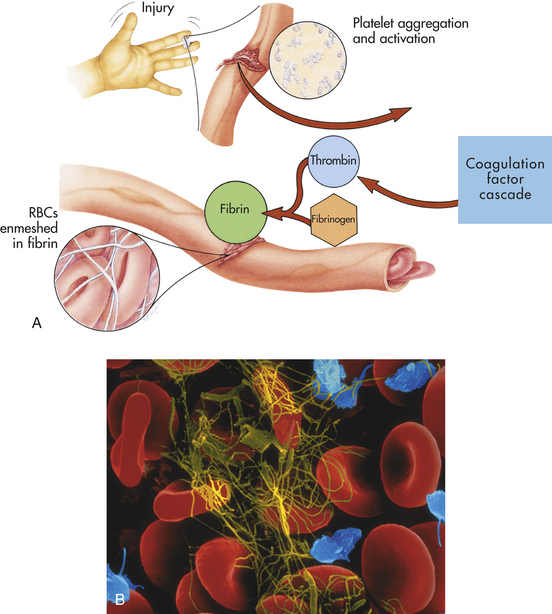
Figure 25-18 Blood clotting mechanism. A, The clotting mechanism involves release of platelet factors at the injury site, formation of thrombin and trapping of red blood cells (RBCs) in fibrin to form a clot. B, An electron micrograph showing entrapped RBCs in a fibrin clot. (A from Thibodeau GA, Patton KT: Anatomy & physiology, ed 5, St Louis, 2003, Mosby; B copyright Dennis Kunkel Microscopy, Inc.)
Adhesion
Normally, platelets are generally observed “rolling” along the margins of vessels. At sites of vessel injury, however, platelets become adherent to the site of endothelial damage where the subendothelial matrix is exposed and endothelial cells have released vWF and decreased their antithrombotic activities (Figure 25-19).25 Platelet adhesion is mostly mediated by the binding of platelet surface receptor glycoprotein-Ib (GPIb) (in a complex with clotting factors IX and V) to von Willebrand factor (vWF) (Figure 25-20).27 The vWF protein is found in the subendothelial matrix and is released by endothelial cells and platelets. Deficiencies in GPIb (Bernard-Soulier syndrome) or of vWF (von Willebrand disease) lead to highly defective hemostasis and congenital bleeding disorders. Platelet adhesion narrows the diameter of the blood vessel resulting in increasing shear forces that could strip platelets off the vessel surface. However, those same forces induce conformational changes in the vWF molecule that result in increased affinity with GPIb, thus stabilizing the adherent platelet.28
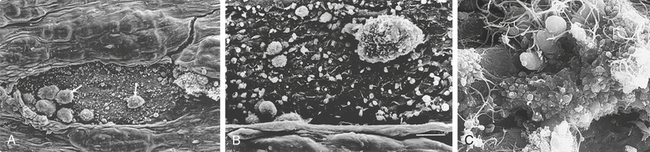
Figure 25-19 Platelet activation. A, After endothelial denudation, platelets and leukocytes adhere to the subendothelium in a monolayer fashion. B, Higher-power view showing leukocytes and platelets adherent to the subendothelium. C, High magnification of a thrombus showing a mixture of red cells and platelets incorporated into the fibrin meshwork. (A and B from Libby P, et al: Braunwald’s heart disease: a textbook of cardiovascular medicine, ed 8, Philadelphia, 2007, Saunders, as reproduced from Faggiotto A, Ross R: Studies of hypercholesterolemia in the nonhuman primate. II. Fatty streak conversion to fibrous plaque. Arteriosclerosis 341-356, 1984; C from Damjanov I, Linder J, editors: Anderson’s pathology, ed 10, St Louis, 1996, Mosby.)
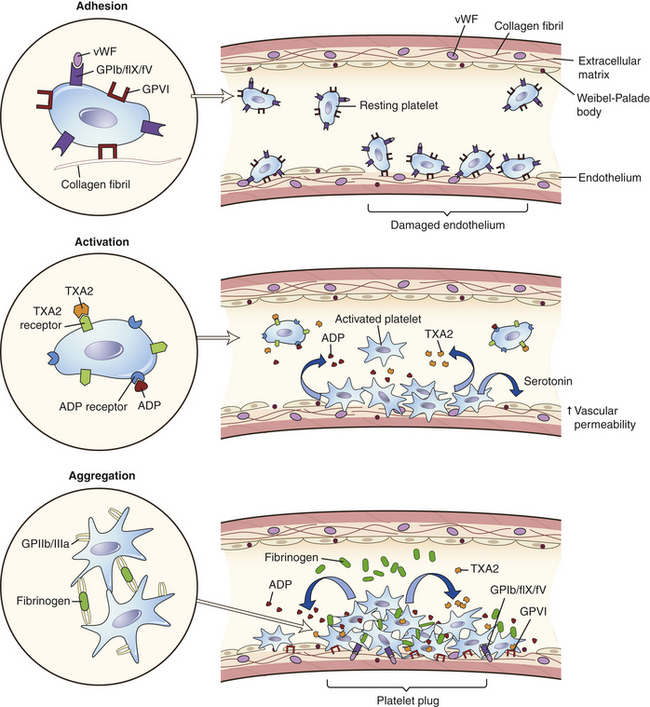
Figure 25-20 Platelet adhesion, activation, aggregation. Adhesion. Damage to a vessel may result in the removal of endothelial cells covering the extracellular matrix. The matrix contains collagen and von Willebrand factor (vWF) that was stored in the Weibel-Palade bodies of endothelial cells and secreted. Resting platelets express receptors for collagen (GPVI) and for vWF (GPIb in a complex with factor IX and factor V, GPIb/fIX/fV) that mediate adherence to the exposed matrix. Activation. Adhesion leads to activation of the platelets with a change in morphology and release of mediators contained in alpha and dense granules; adenosine diphosphate (ADP) and thromboxane (TXA2) acting through specific receptors are primarily responsible for recruitment and activation of more platelets to the clot, as well as platelet spreading and complete coverage of the exposed matrix. Mediators such as serotonin enhance the inflammatory response by binding to neighboring endothelial cells and inducing further vascular permeability. Aggregation. The platelet plug is consolidated by further production of ADP and TXA2, which induce expression of platelet receptors (GPIIb-IIIa) for fibrinogen. The binding of fibrinogen to the fibrinogen receptors on adjacent facilitates platelet aggregation by triggering further platelet spreading and clot retraction. Aggregation is further facilitated by continuing interactions between platelet receptors and collagen and vWF in the matrix.
Platelet adhesion is also facilitated by other interactions between platelet receptors and exposed molecules of the subendothelial matrix. For instance, adhesion is increased through binding of the platelet collagen receptor GPVI to exposed collagen in the matrix.
Activation
As a result of interactions with the endothelium or the subendothelial matrix, as well as exposure to inflammatory mediators produced by the endothelium and other cells, the platelets are activated. Activation results in dynamic changes in platelet shape from smooth spheres to those with spiny projections and degranulation (also called the platelet-release reaction) resulting in the release of various potent biochemicals (Figure 25-21).29
Figure 25-21 Micrograph of an active and moderately active platelet. (Copyright Dennis Kunkel Microscopy, Inc.)
Platelets contain three types of granules: lysosomes, dense bodies, and alpha granules.30 The contents of the dense bodies and alpha granules are particularly important in hemostasis. The dense bodies contain ADP, serotonin, and calcium. ADP recruits and activates other platelets through specific receptors. During activation the platelet plasma membrane undergoes several important changes, including becoming ruffled and sticky, cellular spreading to make tight contacts between neighboring platelets causing the platelet plug to seal the injured endothelium, and externalization of the phospholipid phosphatidylserine, which provides a matrix for activation of clotting factors (see Figure 25-20). Serotonin is a vasoactive amine that functions like histamine and increases vasodilation and vascular permeability. Calcium is necessary for many of the adhesive interactions as well as intracellular signaling mechanisms that control platelet activation.
Alpha granules contain a large number of clotting factors (e.g., fibrinogen, factor V), growth factors (e.g., PDGF), and heparin-binding proteins (e.g., platelet factor 4). Many of these mediators either promote or inhibit platelet activity and the eventual process of clot formation. PDGF stimulates smooth muscle cells and promotes tissue repair. Heparin-binding proteins enhance clot formation at the site of injury.
Platelets also initiate production of the prostaglandin derivative thromboxane-A2 (TXA2), which counters the effects of PGI2 that is produced by endothelial cells (see Figure 25-20). TXA2 promotes the degranulation of platelets, increases expression of platelet fibrinogen receptors, and stimulates platelet aggregation. The balance between TXA2 and PGI2 affects platelet aggregation, which is favored by TXA2 excess and inhibited by PGI2 excess. An isoform of cyclooxygenase (COX-1) converts arachidonic acid to TXA2 in platelets. Aspirin at low doses specifically and irreversibly inactivates COX-1, decreasing production of TXA2 and decreasing platelet activation.31 A few days of low doses of aspirin lead to more than 95% inhibition of TXA2.
Other stimuli of platelet activation include epinephrine, thrombin, and collagen. Thrombin and collagen are particularly strong stimuli. Thrombin cleaves the extracellular domain of G-protein–coupled protease-activated receptors (PARs), thereby initiating transmembrane signaling.
Aggregation
Platelet aggregation is stimulated primarily by TXA2 and ADP, which induce functional fibrinogen receptors on the platelet. The GPIIb-IIIa complex (also called integrin αIIbβ3) undergoes a conformational change during activation to become a calcium-dependent receptor for fibrinogen (see Figure 25-20). It is a member of the integrin receptor family and binds other matrix proteins (e.g., fibronectin, fibrinogen, thrombospondin). Although the GPIIb-IIIa complex is the most abundant aggregation receptor on the platelet, receptors for vWF (GPIb) and collagen (GPVI) also contribute to the process. Interplatelet aggregation and clot retraction that form the primary hemostatic plug are facilitated by fibrinogen bridges between receptors on the platelets. The GPIIb-IIIa–fibrinogen pathway is essential for the formation of a thrombus and as such is an important therapeutic target for blockage by antiplatelet drugs.31 In addition, fibrin strands within the clot shorten, becoming denser and stronger, helping the clot to approximate the edges of the injured vessel wall and sealing the site of injury. Contraction of myosin and actin filaments in the platelet cytoskeleton mediates platelet contraction and fusing of the platelet mass into a secondary hemostatic plug. Contraction expels serum from the fibrin meshwork, resulting in greater packing and increased strength. This process usually begins within a few minutes after a clot has formed, and most of the serum is expelled within 20 to 60 minutes.32
If blood vessel injury is minor, primary hemostasis is achieved by formation of the platelet plug within 3 to 5 minutes of injury. Platelet plugs seal the many minute ruptures that occur daily in the microcirculation, particularly in capillaries. With too few platelets, numerous small hemorrhagic areas called purpuras develop under the skin and throughout the tissues (see Chapter 27). If primary hemostasis is inadequate to prevent further bleeding, the process proceeds through secondary hemostasis to create a larger complex of more tightly interactive platelets within a matrix created by activation of the clotting system.
Function of Clotting Factors
A blood clot is a meshwork of protein strands that stabilizes the platelet plug and traps other cells, such as erythrocytes, phagocytes, and microorganisms. The strands are made of fibrin, which is produced by the clotting (coagulation) system. The clotting system was described in Chapter 6 and consists of a family of proteins that circulate in the blood in inactive forms (proenzymes). Initiation of the system results in sequential enzymatic activation (cascade) of multiple members of the system until a fibrin clot is created (Table 25-7).
Table 25-7
Coagulation Factors and Synonyms
| Factor | Synonym | Primary Function |
| I | Fibrinogen | Source of fibrin to form clot |
| II | Prothrombin | Source of thrombin that activates fibrinogen, V, VII, VIII, XI, XIII, protein C, platelets |
| Tissue factor | Previously called factor III | Cofactor for factor VIIa |
| Calcium | Previously called factor IV | Cofactor for clotting factor binding to phosphatidylserine |
| V | Labile factor | Va is cofactor in the prothrombinase complex |
| VII | Stable factor, proconvertin | VIIa forms a complex with tissue factor and activates factors IX and X |
| VIII | Antihemophilic factor | VIIIa is a component of tenase complex |
| IX | Christmas factor | IXa is a component of tenase complex, activates factor X |
| X | Stuart-Prower factor | Xa is component of prothrombinase complex, activates prothrombin |
| XI | Plasma thromboplastin antecedent | XIa activates factor IX |
| XII | Hageman (contact) factor | XIIa activates factor XI |
| XIII | Fibrin-stabilizing factor | XIIIa cross-links fibrin |
The clotting system is usually presented as two pathways of initiation (intrinsic and extrinsic pathways) that join in a common pathway (Figure 25-22).26 The intrinsic pathway is activated when Hageman factor (factor XII) in plasma contacts negatively charged subendothelial substances exposed by vascular injury. The extrinsic pathway is activated when membrane-bound or soluble tissue factor (TF) (also called tissue thromboplastin), a substance released by damaged endothelial cells, reacts with a high affinity with activated factor VII (TF/VIIa).33 The resultant complexes of both pathways are enzymatically active with factor X as the substrate.34
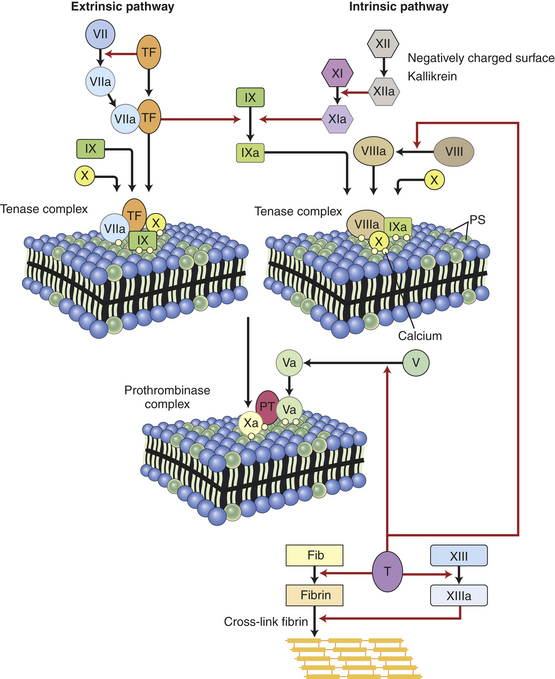
Figure 25-22 The clotting system. The clotting system is frequently presented with two routes of initiation: the intrinsic and extrinsic pathways. The intrinsic pathway is initiated by activation of factor XII to XIIa by negatively charged molecules or surfaces or by the action of kallikrein, a member of the kinin system. Factor XIIa induces enzymatic activation of factor XI to XIa, which activates IX to IXa. A complex of factors IXa/VIIIa/X and calcium aggregate on the surface of activated platelets that have externalized the plasma membrane phospholipid phosphatidylserine (PS). The IXa/VIIIa complex is an active enzyme (“tenase”) that can activate factor X to Xa. The extrinsic pathway is the primary physiologic route of activation and is activated by exposure of tissue factor (TF) on endothelial cells or in the circulation. TF activates factor VII in the blood to factor VIIa. The TF/VIIa complex associates with circulating factors IX and X and calcium on the PS-rich surface of activated platelets. The TF/VIIa/IX complex is a tenase that produces Xa. The VIIa/TF complex also can activate factor IX to IXa and facilitate the formation of the VIIIa/IXa tenase. Factor Xa associates with activated factor V (Va) and prothrombin (PT) in a PS and calcium-dependent complex that is a “prothrombinase,” which converts PT to thrombin (T). Thrombin is an active enzyme that converts fibrinogen (Fib) to fibrin. Thrombin also activates factor XIII to XIIIa, which cross-links fibrin into a matrix that seals clots. Thrombin amplifies the clotting process by activating factor VIII to VIIIa and facilitating formation of the VIIIa/IXa tenase and by activating factor V to Va to create further prothrombinase complexes.
Activated platelets are important participants in clotting. The phosphatidylserine-rich surface produced during activation provides a matrix on which several important complexes of clotting factors are formed. These include the intrinsic pathway’s tenase complex (factor X and activated factors VIII and IX) that activates factor X and the prothrombinase complex (prothrombin and activated factors X and V) that activates prothrombin into thrombin (see Figure 25-22). Thrombin then converts fibrinogen into fibrin, which polymerizes into a fibrin clot. Thrombin has broad reactivity in the inflammatory response.35 In addition to producing fibrin, thrombin is an activator of other coagulation proteins (e.g., factors V, VIII, XI, XIII), platelets (e.g., aggregation, degranulation), endothelial cells (e.g., up-regulation of adhesion molecules for leukocytes, increased NO, PGI2, PDGF), and monocytes (e.g., cytokine secretion, increased receptors for endothelial cells).
The extrinsic pathway is clearly predominant; individuals with deficiencies in intrinsic pathway components (i.e., factor XI, factor XII) do not have prolonged bleeding.36 As with the complement cascade, the clotting system is complex with a large number of alternative activators and inhibitors, and the relative importance of particular factors may differ between in vivo hemostasis and in vitro testing of clotting or may depend on the particular mechanism by which the pathway is activated.37 Also there is interaction between components of the intrinsic and extrinsic pathways so that an activated member of one pathway may activate a member of the other pathway (e.g., factor VIIa of the extrinsic pathway can directly activate factor IX of the intrinsic pathway).
Another similarity with the complement system is that some complexes may have biologic activities outside the predominantly described pathway. For instance, in vitro studies have showed that TF/VIIa complex activates protease-activated receptors (PARs); thus TF may contribute to other biologic processes by facilitating signaling in vascular cells (Figure 25-23). Abnormal TF expression in the vessel wall and/or low circulating cells initiates life-threatening thrombosis in various diseases. TF also contributes to inflammation, tumor angiogenesis and metastasis, and cell migration (Figure 25-24).
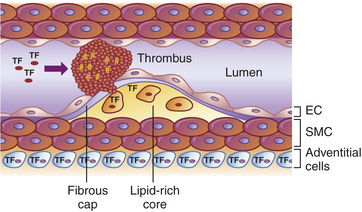
Figure 25-23 Tissue factor (TF) in thrombus formation after rupture of an atherosclerotic plaque. In atherosclerosis TF is expressed by macrophage-derived foam cells and within atherosclerotic plaque. High levels of TF exposed on rupture trigger thrombosis and myocardial infarction. In addition, blood-borne TF may contribute to thrombus propagation. TF is also expressed by adventitial cells (blue). EC, Endothelial cells; SMC, smooth muscle cells. (Modified from Mackman N: Arterioscler Thromb Vasc Biol 24[6]:1015-1022, 2004.)
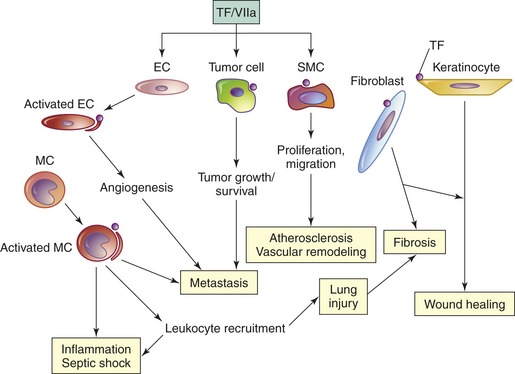
Figure 25-24 Factor VIIa signaling through tissue factor (TF) expressed on the surface of cells mediates changes in disease-related cellular activities, EC, Endothelial cell; MC, monocyte/macrophage; SMC, smooth muscle cell. (Modified from Rao LV, Pendurthi UR: Arterioscler Thromb Vasc Biol 25[1]:47-56, 2005.)
Control of Hemostatic Mechanisms
The endothelium is the major site of hemostasis. Despite the continual presence of clotting factors and platelets in the circulation, blood normally remains fluid. Thus the major regulatory factors that control hemostasis reside where the greatest probability of clotting would occur: on the endothelial cell surface (see Figure 25-17). The primary anticoagulant mechanisms include thrombin inhibitors (e.g., antithrombin III), tissue factor inhibitors (e.g., tissue factor pathway inhibitor), and mechanisms for degrading activated clotting factors (e.g., protein C).38 Antithrombotic mechanisms are listed in Table 25-8.
Table 25-8
Antithrombotic Mechanisms of Endothelial Cells
| Function Regulated | Substances Involved |
| Clotting cascade | Tissue factor pathway inhibitor |
| Antithrombin III | |
| Heparan sulfate | |
| Thrombomodulin/protein C/protein S | |
| Vessel and platelet activity | Cover prothrombotic intercellular matrix molecules |
| Prostacyclin (PGI2) | |
| Nitric oxide (NO) | |
| Adenosine diphosphate | |
| Eliminate fibrin clot | Plasminogen activators |
Antithrombin III (AT-III) is a circulating plasma serine protease inhibitor produced by the liver. Specifically, it inhibits thrombin and several activated clotting factors (e.g., VIIa, IXa, Xa, XIa, XIIa). Clinically administered heparin or heparan sulfate (on the surface of endothelial cells) binds to AT-III and induces a conformational change that greatly enhances its activity. Under normal conditions the presence of endothelial cell heparan sulfate and available AT-III in the circulation cooperate to protect the vessels from the effects of spontaneously activated thrombin (see Figure 25-17). Acquired AT-III deficiencies can result from infection with bacteria that produce AT-III inhibitors, sepsis, liver disease, and nephrotic syndrome and lead to venous thrombosis and pulmonary embolism.
Tissue factor pathway inhibitor (TFPI) is produced by endothelial cells and complexes to, and reversibly inhibits, factor Xa. The resultant TFPI/Xa complex inhibits TF/VIIa, which mediates feedback inhibition of tissue factor as well as factor VIIa (see Figure 25-17). Although the majority of TFPI remains associated with endothelial surfaces, about 20% circulates in plasma with lipoproteins. Heparin increases plasma levels of TFPI, which may contribute to heparin’s antithrombotic effects.
Thrombomodulin is a membrane thrombin-binding protein on the surface of endothelial cells. Protein C in the circulation binds to thrombomodulin in a thrombin-dependent manner and is converted to activated protein C (see Figure 25-17). Activated protein C, in association with a cofactor (protein S), degrades factors Va and VIIIa. Deficiencies of AT-III, protein C, or protein S are important causes of hypercoagulation (increased clotting).39 Expression of thrombomodulin and the endothelial cell protein C receptor is down-regulated by cytokines and other products of inflammation (e.g., IL-1α, tumor necrosis factor-alpha [TNF-α], endotoxin). Decreased expression prevents protein C activation, thereby enhancing clot formation. Activated protein C inhibits the adhesion of neutrophils to the endothelium, but during inflammation the neutrophil enzyme elastase enzymatically removes thrombomodulin from the endothelial cell surface.40
Lysis of Blood Clots
Concurrent with activation of coagulation is the activation of pathways that limit the size of the clot and remove the clot after bleeding has ceased and repair has begun. The primary mechanism for lysis (breakdown) of blood clots is the fibrinolytic system (plasminogen-plasmin system) that produces plasmin. Plasmin (also called fibrinase or fibrinolysin) is a serine protease that degrades fibrin polymers in clots.41
The inactive precursor of plasmin is plasminogen, which is produced in the liver (Figure 25-25). Plasminogen activation may occur by several means, although the most physiologically important is by the action of tissue plasminogen activator (t-PA).42 Endothelial cells at a site of vascular injury express t-PA, which is also a serine protease that reaches maximum enzymatic activity after binding to fibrin and proteolytically activates plasminogen to plasmin. Another activator of plasminogen is urokinase-like plasminogen activator (u-PA). The u-PA is a serine protease that can bind to a specific cellular u-PA receptor (u-PAR) causing activation of plasminogen resulting in plasmin generation. This urokinase is the major activator of fibrinolysis in the extravascular or tissue compartment, whereas t-PA is largely involved in intravascular fibrinolysis. Several cancers appear to use membrane-bound u-PA to digest intercellular matrix and greatly facilitate tumor invasion and metastasis. Both t-PA and u-PA (see following) have been used clinically to treat diseases associated with a blood clot (e.g., pulmonary embolism, myocardial infarction, stroke).
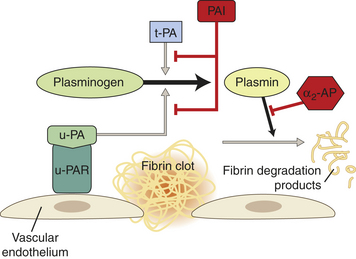
Figure 25-25 The fibrinolytic system. Fibrinolysis is initiated by the binding of plasminogen to fibrin. Although tissue plasminogen activator (t-PA) initiates intravascular fibrinolysis, urokinase plasminogen activator (u-PA) is the major activator of fibrinolysis in tissue (extravascular). Inhibitors (indicated by red lines) of fibrinolysis include α2-antiplasmin (α2-AP) that inhibits plasmin and plasminogen activator inhibitor-1 (PAI-1) that inhibits t-PA. Plasmin digests the fibrin into smaller soluble pieces (fibrin degradation products). u-PAR, urokinase-like plasminogen activator receptor.
As with most components of inflammation, plasmin interacts greatly with other factors. In addition to activation by t-PA and u-PA, plasminogen is activated to plasmin by thrombin, fibrin, factor XIIa, factor XIa, and kallikrein. Plasmin is proteolytic to several substrates, including activation of collagenases, complement components C1 and C3, and factor XII and cleaves fibronectin, fibrin, thrombospondin, laminin, and vWF. Plasmin activity is usually controlled directly by the serine protease inhibitor α2-antiplasmin or by inhibiting plasminogen activation by specific plasminogen activator inhibitors (PAIs).
Congenital defects in the fibrinolytic system are very rare and variable in effects. Congenital plasminogen deficiency is associated with venous thrombosis if the plasminogen level is decreased by more than 50%. Apparent defects in plasminogen may result from dysfunctional plasminogen or an absence of fibrinogen. Congenital deficiencies of plasminogen activator or abnormal increases in plasminogen activator inhibitor also may increase the chance for spontaneous thrombosis. Alternative routes of fibrin degradation may mitigate the severity of symptoms in these deficiencies. Other enzymes released during inflammation (e.g., leukocyte elastase, cathepsin G, metalloproteinases [MMPs]) are fibrinolytic.
Cross-linked fibrin is deposited in tissues around wounds, inflammatory sites, and tumors. Fibrin removal is an important biologic process, for intravascular and extravascular spaces, with various controlling mechanisms that can lead to abnormalities of fibrin accumulation, and thrombotic events and can be a structural barrier to tumor invasion (see Chapter 12).
Products of fibrinolysis include fibrin degradation products (FDPs) (see Figure 25-25). A major FDP is D-dimer. D-dimer is two D domains from adjacent fibrin monomers that are cross-linked by factor XIIIa. Measurement of levels of circulating D-dimer has been used for diagnosis of deep venous thrombosis (DVT) or pulmonary embolism (PE).43 Despite extensive literature, the diagnostic role of D-dimer is unclear because of the use of multiple D-dimer assays with different sensitivities and variabilities.
CLINICAL EVALUATION OF THE HEMATOLOGIC SYSTEM
The bone marrow is the soft spongy tissue found within bones, especially the sternum, pelvis, and femur. Several abnormal conditions in the numbers or morphology of circulating blood cells or suspected infection of the marrow may justify further investigation of the bone marrow.
Usually bone marrow is aspirated from the sternum or pelvis using a needle. In children a bone marrow aspirate can be obtained from the vertebrae or the femur. The aspirate is examined microscopically and may be cultured if infection (e.g., fungi, mycobacteria, brucellosis, typhoid fever) is suspected. Microscopic evaluation may also include flow cytometry, chromosome analysis, or polymerase chain reaction (PCR) related to the presence of atypical cells, atypical numbers of normal cells, and the absence of particular cell types. A normal bone marrow aspirate contains stromal cells (fibroblasts, macrophages, osteoblasts, adipocytes), stem cells (hematopoietic, mesenchymal, and endothelial), and immature and mature forms of blood cells (erythrocytes, leukocytes, platelets) (Figure 25-26). The differential cell count of a bone marrow aspirate involves examining approximately 400 nucleated cells under oil-immersion magnification and counting populations of cells differentiated based on morphology. The relative number of each type of stem cell is expressed as a fraction of 400 (Table 25-9).
Table 25-9
Differential Cell Counts in Bone Marrow with Age
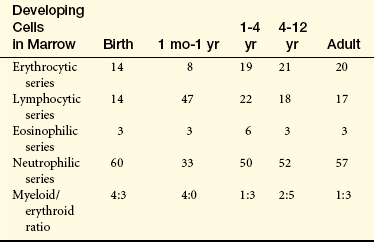
NOTE: Values are percentages of cell types counted during examination of a marrow specimen containing approximately 400 nucleated cells.
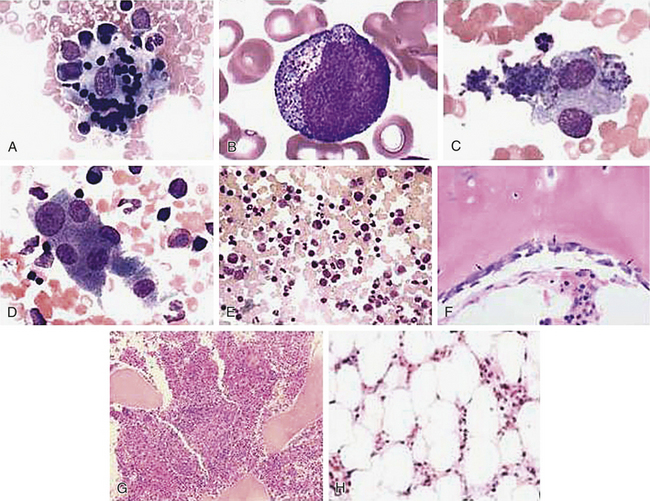
Figure 25-26 Bone marrow samples. Images A through D contain bone marrow aspirates; images E through H contain bone marrow biopsy specimens; images A through F are examples of normal bone marrow. A, An erythroblastic island in which several erythroid precursors cluster around a central “nursing” histiocyte. B, An early promyelocyte within which primary granules are prominent and a centrosome is present. C, A megakaryocyte with clusters or chains of platelets (small, darkly staining objects) being shed. D, Osteoclasts containing multiple nuclei that are separated by cytoplasm. These cells may be confused with megakaryocytes. E, Low-power scan of a normal bone marrow with a heterogeneous population of cells. F, Bone marrow biopsy illustrating the linear distribution of the osteoblasts along the bony trabecula. G, Bone marrow biopsy of a 63-year-old male who was being evaluated for anemia. A discrete germinal center is present (left-center of image) in this otherwise normal marrow biopsy. H, High-power view of a bone marrow biopsy from an individual with aplastic anemia showing markedly hypocellular marrow containing mainly fat cells. (Sources for photographs: A, American Society of Hematology Image Bank (ASHIB); 2004:101123; B, ASHIB; 2005:101355; C, ASHIB; 2005:101324; D, ASHIB; 2005:101358; E, ASHIB; 2005:101401; F, ASHIB; 2002:100504; G, ASHIB; 2004:101190; H from Kumar V et al: Robbins and Cotran pathologic basis of disease, ed 7, Philadelphia, 2005, Saunders.)
Bone marrow iron stores, primarily in macrophages, can be examined using special stains (e.g., Prussian blue) for iron-containing granules. A direct measure of iron stores also can be obtained only from liver biopsy specimens, although bone marrow is preferred as a safer procedure and because the bone marrow is the immediate source of iron destined for erythrocyte production.
Bone marrow aspiration is an important diagnostic test for severe central defects in hematopoiesis (e.g., aplastic anemia, metabolic anemias arising from insufficient iron or inadequate erythropoietin, thrombocytopenia, neutropenia; see Chapters 26 and 27). Examination of the bone marrow is also useful to diagnosis B lymphocyte immune deficiencies (see Chapter 8), nonmalignant myeloproliferative disorders (e.g., polycythemia vera), lymphoid/monocytic malignancies (e.g., leukemias, myelomas, lymphomas; see Chapters 27 and 28). This test can also be used to monitor the effects of chemotherapy on malignancies that have invaded the bone marrow (see Figure 25-26). A marrow aspirate that is richly cellular implies normal or increased hematopoiesis but does not indicate whether marrow activity is effective.
Results from bone marrow aspiration are sometimes limited because this technique disturbs the architecture of the marrow and only provides an analysis of the general cellularity (numbers of constituent cells) of the marrow. On occasion, analysis of an aspirate may only suggest the presence of a malignancy or a central defect in hematopoiesis without being clearly confirmatory, or the sample may be inadequate for diagnosis of bone marrow fibrosis. In these cases the need for a bone marrow biopsy may be indicated. During a biopsy, a special needle is used to obtain a “core” or cylindrical sample of bone and marrow in which the three dimensional structure of the marrow is preserved. The biopsy specimens provide the most reliable and complete information about marrow cellularity (see Figure 25-26). Obtaining a bone marrow biopsy is, however, usually more painful and expensive than aspiration. Therefore, biopsy is not performed unless insufficient information is obtained from aspiration.
Blood Tests
Blood tests provide information about the absolute and relative numbers of blood cells in a specimen of blood, as well as various structural and functional characteristics of the cells, and usually provide the initial justification for performing a bone marrow aspiration. Deviations from the normal differential distribution and the presence of abnormal or immature cells can reflect disease, physiologic states (e.g., pregnancy, infancy, old age), injury, or dysfunction in almost any part of the body. Blood tests that reflect chiefly hematologic disorders are listed in Table 25-10.
Table 25-10
Blood Tests for Hematologic Disorders
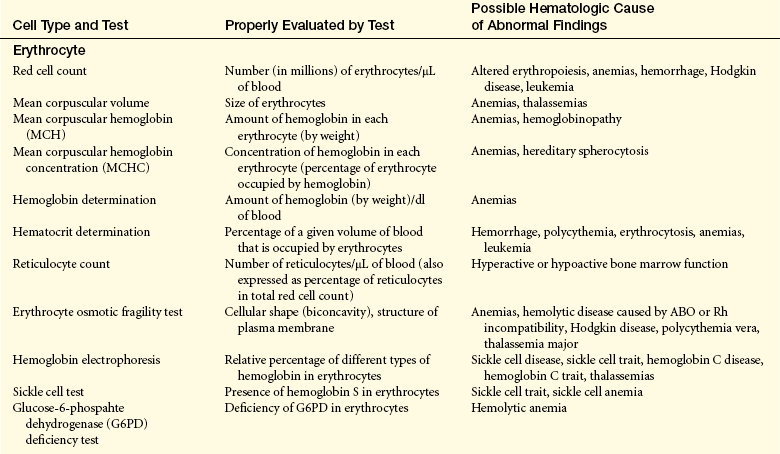




NOTE: See Figure 25-23 and Table 25-7 for information about clotting factors and their sequence of activation in the coagulation cascade.
Data from Byrne CJ et al: Laboratory tests: implications for nursing care, Menlo Park, CA, 1986, Addison-Wesley; Bick RL et al: Hematology: clinical and laboratory practice, St Louis, 1993, Mosby.
Pediatrics and the Hematologic System
Blood cell counts tend to rise above adult levels at birth and then decline gradually throughout childhood. Table 25-11 lists normal ranges during infancy and childhood. The immediate rise in values is the result of accelerated hematopoiesis during fetal life, increased numbers of cells that result from the trauma of birth, and cutting of the umbilical cord.
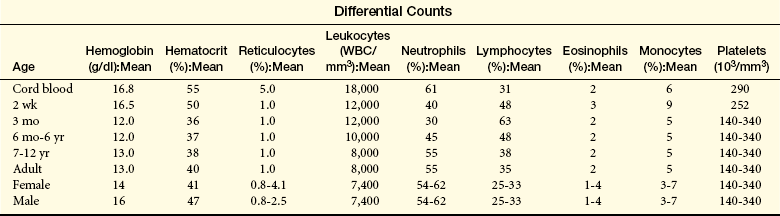
Average blood volume in the full-term neonate is 85 ml per kilogram of body weight. The premature infant has a slightly larger blood volume of 90 ml per kilogram of body weight, with the mean increasing to 150 mg/kg during the first few days after birth. In both full-term and premature infants, blood volume decreases during the first few months. Thereafter the average blood volume is 75 to 77 ml/kg, which is similar to that of older children and adults.
The hypoxic intrauterine environment stimulates erythropoietin production in the fetus and accelerates fetal erythropoiesis, producing polycythemia (excessive proliferation of erythrocyte precursors) of the newborn. After birth the oxygen from the lungs saturates arterial blood, and more oxygen is delivered to the tissues. In response to the change from a placental to a pulmonary oxygen supply during the first few days of life, levels of erythropoietin and the rate of blood cell formation decrease.
The very active rate of fetal erythropoiesis results in a large number of immature erythrocytes (reticulocytes) in the peripheral blood of full-term neonates. After birth the number of reticulocytes decreases about 50% every 12 hours so that it is rare to find an elevated reticulocyte count after the first week of life. During this period of rapid growth, the rate of erythrocyte destruction is greater than that in later childhood and adulthood. In full-term infants, normal erythrocyte life span is 60 to 80 days; in premature infants it may be as short as 20 to 30 days; and in children and adolescents it is the same as that in adults—120 days.
The postnatal fall in hemoglobin and hematocrit values is more marked in premature infants than in the full-term infant. In the preschool and school-age child, hemoglobin, hematocrit, and red blood cell counts gradually rise. Metabolic processes within the erythrocytes of neonates differ significantly from those of erythrocytes in the normal adult. Among other differences, the relatively young population of erythrocytes in the newborn consumes greater quantities of glucose than do erythrocytes in adults.
At birth, the lymphocyte count is high and continues to rise during the first year of life, and then steadily declines until lower adult values are reached. The lymphocytes of children also tend to have more cytoplasm and less compact nuclear chromatin than do the lymphocytes of adults. A possible explanation is that children tend to have more frequent viral infections, some of which are subclinical, and are receiving vaccinations, both of which are associated with atypical lymphocytes.
The neutrophil count, like the lymphocyte count, is high at birth and rises during the first days of life. After 2 weeks the neutrophil count falls to within or below the normal adult range. By approximately 4 years of age, the neutrophil count is the same as that of an adult. The eosinophil count is high in the first year of life and higher in children than in teenagers or adults. Monocyte counts also are high in the first year of life but then decrease to adult levels. Platelet counts in full-term neonates are comparable with platelet counts in adults and remain so throughout infancy and childhood.
Aging and the Hematologic System
Blood composition changes little with age. Erythrocyte life span in older adults is normal, although erythrocytes are replenished more slowly after bleeding, probably because of iron depletion. Total serum iron, total iron-binding capacity, and intestinal iron absorption are all decreased in older adults. Iron deficiency is often responsible for the low hemoglobin levels noted in older adults. The plasma membranes of erythrocytes become increasingly fragile, presumably because of physical trauma inflicted during circulation.
Lymphocyte function decreases with age (see Chapters 7 and 8), causing changes in cellular immunity with some decline in T cell function. The humoral immune system is less able to respond to antigenic challenge. No changes in platelet numbers or structure have been observed in elderly persons, yet platelet adhesiveness probably increases. Although fibrinogen levels and factors V, VII, and IX tend to be increased in older adults, evidence concerning hypercoagulability is inconclusive.
Albumin 953
Antithrombin III (AT-III) 978
Apoferritin 969
Apotransferrin 971
Basophil 956
Blood clot 976
Bone marrow 962
Clotting (coagulation) system 976
Clotting factor 954
Colony-stimulating factor (CSF, hematopoietic growth factor) 963
Cyclooxygenase (COX-1) 976
D-dimer 980
Dendritic cell 957
Deoxyhemoglobin 968
Endomitosis 971
Eosinophil 956
Erythroblast 966
Erythrocyte (red blood cell [RBC]) 954
Erythropoiesis 965
Erythropoietin 965
Fibrin degradation product (FDP) 980
Fibrinolysis 972
Fibrinolytic system (plasminogen-plasmin system) 979
Globin 967
Globulin 953
GPIIb-IIIa complex 976
Granulocyte 955
Hematopoiesis 961
Hematopoietic stem cell 962
Heme 967
Hemoglobin (Hb) 967
Hemosiderin 969
Hemostasis 972
Immunocyte 955
Leukocyte (white blood cell [WBC]) 955
Lymphocyte 956
Macrophage 957
Marginating storage pool 963
Mast cell 956
Megakaryocyte 957
Mesenchymal stem cell 961
Methemoglobin 968
Monocyte 957
Mononuclear phagocyte system (MPS) 957
Myeloid tissue 962
Myoglobin 969
Natural killer (NK) cell 957
Neutrophil (polymorphonuclear neutrophil [PMN]) 955
Nitric oxide (NO) 972
Normoblast 966
Oxyhemoglobin 968
Phagocyte 955
Plasma 952
Plasma protein 952
Plasmin 979
Plasminogen 979
Platelet (thrombocyte) 957
Platelet adhesion 973
Platelet aggregation 976
Platelet-release reaction 973
Proerythroblast 965
Prostacyclin (PGI) 972
Protein C 979
Protein S 979
Protoporphyrin 968
Reticulocyte 966
Serum 952
Steel factor 963
Stromal cell 963
Thrombomodulin 979
Thromboxane A2 (TXA2) 976
Tissue factor (TF, tissue thromboplastin) 977
Tissue factor pathway inhibitor (TFPI) 979
Tissue plasminogen activator (t-PA) 980
Transferrin 971
Urokinase-like plasminogen activator (u-PA) 980
von Willebrand factor (vWF) 973
REFERENCES
1. Chuang, V.T., Otagiri, M. Recombinant human serum albumin. Drugs Today. 2007;43(8):547–561.
2. Babior, B.M., Stossel, T.P. Hematology: a pathophysiological approach. New York: Churchill Livingstone; 1984.
3. Mohandas, N., Gallagher, P.G. Red cell membrane: past, present, and future. Blood. 2008;112(10):3939–3948.
4. Karikyawasam, H.H., Robinson, D.S. The eosinophil: the cell and its weapons, the cytokines, its locations,. Semin Respir Crit Care Med. 2006;27(2):117–127.
5. Prussin, C., Metcalfe, D.D. IgE, mast cells, basophils, and eosinophils. J Allergy Clin Immunol. 2006;117(2 Suppl):S450–S456.
6. Hume, D.A. The mononuclear phagocyte system,. Curr Opin Immunol. 2005;18(1):49–53.
7. Junt, T., Scandella, E., Ludewig, B. Form follows function: lymphoid tissue microarchitecture in antimicrobial immune defense. Nat Rev Immunol. 2008;8(10):764–775.
8. Mueller, S.N., Ahmed, R. Lymphoid stroma in the initiation and control of immune responses. Immunol Rev. 2008;224(1):284–294.
9. Ross, F.P., Christiano, A.M. Nothing but skin and bones. J Clin Invest. 2006;116(5):1140–1149.
10. Bar-Shavit, Z. The osteoclast: a multinucleated, hematopoietic-origin, bone-resorbing osteoimmune cell,. J Cell Biochem. 2007;102(5):1130–1139.
11. Yin, T., Li, L. The stem cell niches in bone,. J Clin Invest. 2006;116(5):1195–1201.
12. Stem cell information, Bethesda MD, 2009. National Institutes of Health, U.S. Department of Health and Human Services [cited, January 17, 2009]. Available at http://stemcells.nih.gov/info.
13. Ratajczak, M.Z. Phenotypic and functional characterization of hematopoietic stem cells. Curr Opin Hematol. 2008;15(4):293–300.
14. Valtieri, M., Sorrentino, A. The mesenchymal stromal cell contribution to homeostasis,. J Cell Physiol. 2008;217(2):296–300.
15. Möhle, R., Kanz, L. Hematopoietic growth factors for hematopoietic stem cell mobilization and expansion. Semin Hematol. 2007;44(3):193–202.
16. Pusic, I., DiPersio, J.F. The use of growth factors in hematopoietic stem cell transplantation,. Curr Pharm Des. 2008;14(20):1950–1961.
17. Chasis, J.A., Mohandas, N. Erythroblastic islands: niches for erythropoiesis. Blood. 2008;112(3):470–478.
18. Schechter, A.N. Hemoglobin research and the origins of molecular medicine. Blood. 2008;112(10):3927–3938.
19. Edison, E.S., Bajel, A., Chandy, M. Iron homeostasis: new players, newer insights. Eur J Haematol. 2008;81(6):411–424.
20. West, A.R., Oates, P.S. Mechanisms of heme iron absorption: current questions and controversies. World J Gastroenterol. 2008;14(26):4101–4110.
21. Naito, M. Macrophage differentiation and function in health and disease. Path Int. 2008;58(3):143–155.
22. Kaushansky, K. Historical review: megakaryopoiesis and thrombopoiesis. Blood. 2008;111(3):981–986.
23. Marcucci, R., Romano, M. Thrombopoietin and its splicing variants: structure and functions in thrombopoiesis and beyond. Biochim Biophys Acta. 2008;1782(7-8):427–432.
24. Freestone, B., Lip, G.Y. The endothelium and atrial fibrillation. The prothrombotic state revisited,. Hamostaseologie. 2008;28(4):207–212.
25. Reininger, A.J. Function of von Willebrand factor in haemostasis and thrombosis. Haemophilia. 2008;14(Suppl 5):11–26.
26. Furie, B., Furie, B.C. Mechanisms of thrombus formation. N Engl J Med. 2008;359(9):938–949.
27. Reininger, A.J. VWF attributes—impact on thrombus formation. Thromb Res. 2008;122(Suppl 4):S9–S13.
28. Andrews, R.K., Berndt, M.C. Platelet adhesion: a game of catch and release. J Clin Invest. 2008;118(9):3009–3011.
29. Ren, Q., Ye, S., Whiteheart, S.W. The platelet release reaction: just when you thought platelet secretion was simple,. Curr Opin Hematol. 2008;15(5):537–541.
30. Gleissner, C.A., von Hundelshausen, P., Ley, K. Platelet chemokines in vascular disease. Arterioscler Thromb Vasc Biol. 2008;28(11):1920–1927.
31. Krötz, F., Sohn, H.-Y., Klauss, V. Antiplatelet drugs in cardiological practice: established strategies and new developments. Vasc Health Risk Manag. 2008;4(3):637–645.
32. De Gaetano, G., Crescente, M., Cerletti, C. Current concepts about inhibition of platelet aggregation. Platelets. 2008;19(8):565–570.
33. Hoffman, M. Some things I thought I knew about tissue factor that turn out to be wrong. Thromb Res. 2008;122(Suppl 1):S73–S77.
34. Pendurthi, U.R., Rao, L.V. Factor VIIa interaction with tissue factor and endothelial cell protein C receptor on cell surfaces. Semin Hematol. 2008;45(2 Suppl 1):S21–S24.
35. Di Cera, E. Thrombin. Mol Aspects Med. 2008;29(4):203–254.
36. Müller, F., Renné, T. Novel roles for factor XII-driven plasma contact activation system. Curr Opin Hematol. 2008;15(5):516–521.
37. Henry, B.L., Desai, U.R. Recent research developments in the direct inhibition of coagulation proteinases—inhibitors of the initiation phase. Cardiovasc Hematol Agents Med Chem. 2008;6(4):323–336.
38. Wang, L., Bastarache, J.A., Ware, L.B. The coagulation cascade in sepsis,. Curr Pharm Des. 2008;14(19):1860–1869.
39. Castoldi, E., Hackeng, T.M. Regulation of coagulation by protein S. Curr Opin Hematol. 2008;15(5):529–536.
40. Jackson, C.J., Xue, M. Activated protein C—an anticoagulant that does more than stop clots. Int J Biochem Cell Biol. 2008;40(12):2692–2697.
41. Weisel, J.W., Litvinov, R.I. The biochemical and physical process of fibrinolysis and effects of clot structure and stability on the lysis rate,. Cardiovasc Hematol Agents Med Chem. 2008;6(3):161–180.
42. Zorio, E., et al. Fibrinolysis: the key to new pathogenetic mechanisms. Curr Med Chem. 2008;15(9):923–929.
43. Righini, M., et al. D-dimer for venous thromboembolism diagnosis: 20 years later. J Thromb Haemost. 2008;6(7):1059–1071.