DISORDERS OF THE HEART WALL
Pericardial disease is often a localized manifestation of another disorder, such as infection (bacterial, viral, fungal, rickettsial, parasitic); trauma or surgery; neoplasm; or a metabolic, immunologic, or vascular disorder (uremia, rheumatoid arthritis, systemic lupus erythematosus, periarteritis nodosa). The pericardial response to injury from these diverse causes may consist of acute pericarditis, pericardial effusion, or constrictive pericarditis.
Acute Pericarditis
Acute pericarditis is acute inflammation of the pericardium. It is estimated that up to 5% of emergency department visits for nonischemic chest pain are caused by acute pericarditis. The etiology of acute pericarditis is most often idiopathic or caused by viral infection by coxsackievirus, influenzavirus, hepatitis, measles, mumps, or varicella viruses.170 It is also the most common cardiovascular complication of human immunodeficiency virus (HIV) infection. Other causes include myocardial infarction, trauma, neoplasm, surgery, bacterial infection (especially tuberculosis), connective tissue disease, or radiation therapy. The pericardial membranes become inflamed and roughened, and a pericardial effusion may develop that can be serous, purulent, or fibrinous (Figure 30-25). In some individuals, a large effusion can develop rapidly causing cardiac tamponade. In most individuals an effusion accumulates slowly and in lesser amounts that can recur in up to 30% of individuals, and can rarely progress to constrictive pericarditis.170–172
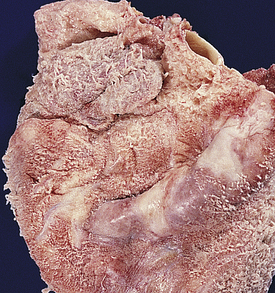
Figure 30-25 Acute pericarditis. Note shaggy coat of fibers covering surface of heart. (From Damjanov I, Linder J: Pathology: a color atlas, St Louis, 2000, Mosby.)
Most individuals with acute pericarditis describe several days of fever, myalgias, and malaise followed by the sudden onset of severe chest pain that worsens with respiratory movements and with lying down. Although the pain may radiate to the back, it is generally felt in the anterior chest and may be confused initially with the pain of acute myocardial infarction. Individuals with acute pericarditis also may report dysphagia, restlessness, irritability, anxiety, and weakness.
Physical examination often discloses low-grade fever and sinus tachycardia. A pericardial friction rub—a short, scratchy, grating sensation similar to the sound of sandpaper—may be heard at the cardiac apex and left sternal border and is highly specific for pericarditis. The rub is caused by the roughened pericardial membranes rubbing against each other. Friction rubs are not always present and may be intermittently heard. ECG changes may reflect inflammatory processes through diffuse ST segment elevation that is concaved upward without Q waves.173 The ECG may remain abnormal for days or even weeks. Echocardiography may reveal a pericardial effusion.
Treatment for uncomplicated acute pericarditis consists of relieving symptoms. Rest is helpful during episodes of acute pain. Salicylates and nonsteroidal anti-inflammatory drugs reduce inflammation.170,174 Combined nonsteroidals and colchicine (prevents fibrosis) is a highly effective regimen.170,174,175 Additional analgesics may be given to relieve pain. Exploration of the underlying cause is important. If pericardial effusion develops, aspiration of the excessive fluid may be necessary.
Pericardial Effusion
Pericardial effusion, the accumulation of fluid in the pericardial cavity, can occur in all forms of pericarditis.170,171 The fluid may be a transudate, such as the serous effusion that develops with left heart failure, overhydration, or hypoproteinemia. More often, however, the fluid is an exudate, which indicates pericardial inflammation like that seen with acute pericarditis, heart surgery, some chemotherapeutic agents, infections, and autoimmune disorders, such as systemic lupus erythematosus. (Types of exudate are described in Chapter 6.) If the fluid is serosanguineous, the underlying cause is likely to be tuberculosis, neoplasm, uremia, or radiation. Idiopathic serosanguineous (cause unknown) effusion is possible, however. Effusions of frank blood are generally related to aneurysms, trauma, or coagulation defects. If chyle leaks from the thoracic duct, it may enter the pericardium and lead to cholesterol pericarditis.
Pericardial effusion, even in large amounts, is not necessarily clinically significant, except that it indicates an underlying disorder. The important consideration is whether the fluid creates sufficient pressure to cause cardiac compression, which is a serious condition known as tamponade.171 If an effusion develops gradually, the pericardium can stretch to accommodate large quantities of fluid without compressing the heart. If the fluid accumulates rapidly, however, even a small amount (50 to 100 ml) may cause serious tamponade. The danger is that pressure exerted by the pericardial fluid eventually will equal diastolic pressure within the heart chambers, thus preventing chamber filling.171 The first structures to be affected by tamponade are the right atrium and ventricle, where diastolic pressures are normally lowest. Compression by pericardial fluid interferes with right atrial filling during diastole, resulting in increased venous pressure, systemic venous congestion, and signs and symptoms of right heart failure (distention of the jugular veins, edema, hepatomegaly). Decreased atrial filling leads to decreased ventricular filling, decreased stroke volume, and reduced cardiac output. If the left atrium collapses because of lack of filling, life-threatening circulatory collapse may occur.176
Individuals with cardiac tamponade most often present with dyspnea, tachycardia, jugular venous distention, cardiomegaly, and pulsus paradoxus.177 Pulsus paradoxus means that the arterial blood pressure during expiration exceeds arterial pressure during inspiration by more than 10 mmHg. This clinical finding reflects impairment of diastolic filling of the left ventricle plus reduction of blood volume within all four cardiac chambers. Presence of a large pericardial effusion or tamponade magnifies the normally insignificant effect of inspiration on intracardiac flow and volume.
Other clinical manifestations of pericardial effusion are distant or muffled heart sounds, poorly palpable apical pulse, dyspnea on exertion, and dull chest pain. A chest roentgenogram may disclose a “water-bottle” configuration of the cardiac silhouette. An echocardiogram can detect an effusion as small as 20 ml and is considered the most accurate and reliable method of diagnosis, although CT also is commonly used.178
Treatment of pericardial effusion or tamponade generally consists of pericardiocentesis (aspiration of excessive pericardial fluid). Pericardiocentesis is diagnostic and therapeutic: the fluid is analyzed to identify the cause of the effusion, and its removal alone may bring dramatic relief from symptoms. Persistent pain may be treated with analgesics, anti-inflammatory medications, or steroids. Surgery may be required if the underlying cause of tamponade is trauma or aneurysm. If an effusion is neoplasm induced, chemotherapeutic agents may be injected into the pericardial space.179 If the effusion recurs, a pericardial “window” can be created or the individual may require pericardectomy.180
Constrictive Pericarditis
Constrictive pericarditis, or restrictive pericarditis (chronic pericarditis), was synonymous with tuberculosis years ago; tuberculosis continues to be an important cause of pericarditis in immunocompromised individuals.181 In the United States, this form of pericardial disease is more often idiopathic or associated with radiation exposure, rheumatoid arthritis, uremia, or CABG. In constrictive pericarditis, fibrous scarring with occasional calcification of the pericardium causes the visceral and parietal pericardial layers to adhere, obliterating the pericardial cavity. The fibrotic lesions encase the heart in a rigid shell (Figure 30-26). Like tamponade, constrictive pericarditis compresses the heart and eventually reduces cardiac output. Unlike tamponade, however, constrictive pericarditis never develops suddenly.
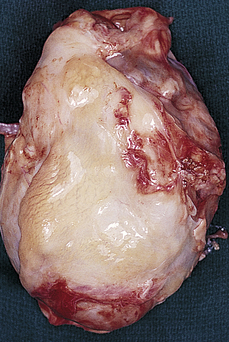
Figure 30-26 Constrictive pericarditis. The fibrotic pericardium encases the heart in a rigid shell. (From Damjanov I, Linder J: Pathology: a color atlas, St Louis, 2000, Mosby.)
Because the onset of constrictive pericarditis is gradual, clinical manifestations seldom include pulsus paradoxus. Symptoms tend to be exercise intolerance, dyspnea on exertion, fatigue, and anorexia. Clinical assessment shows weight loss, edema, distention of the jugular vein, and hepatic congestion. Restricted ventricular filling may cause a pericardial knock (early diastolic sound).
ECG findings include T wave inversions and atrial fibrillation. Chest roentgenograms often disclose prominent pulmonary vessels and calcification of the pericardium. An echocardiogram may suggest evidence of nonspecific pericardial thickening.171 CT or MRI is best able to detect constrictive processes. Some individuals require diagnostic thoracotomy in order to make the diagnosis, especially in cases of postoperative restrictive pericarditis.
Initial treatment for constrictive pericarditis consists of dietary sodium restriction, digitalis glycosides, and diuretics to improve cardiac output. If these modalities are not successful, surgical excision of the restrictive pericardium is indicated.
Disorders of the Myocardium: The Cardiomyopathies
The cardiomyopathies are a diverse group of diseases that primarily affect the myocardium. Most are the result of underlying cardiovascular disorders, such as ischemic heart disease or hypertension. Cardiomyopathies also can be secondary to infectious disease, exposure to toxins, systemic connective tissue disease, infiltrative and proliferative disorders, or nutritional deficiencies. Despite this large number of possible causes, most cases of cardiomyopathy are idiopathic; that is, their cause is unknown. The cardiomyopathies are categorized as dilated, hypertrophic, or restrictive depending on their tissue characteristics, genomics, and hemodynamic effects182 (Figure 30-27 and Table 30-8). An individual may display characteristics of more than one type.
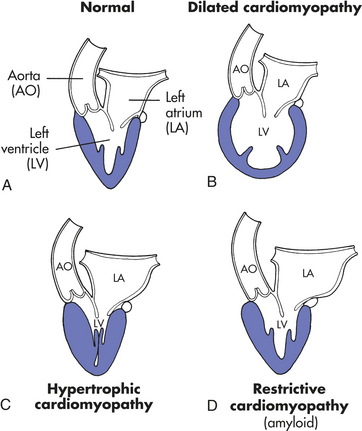
Figure 30-27 Diagram showing major distinguishing pathophysiologic features of the types of cardiomyopathy. A, The normal heart. B, In the dilated type of cardiomyopathy, the heart has a globular shape and the largest circumference of the left ventricle is not at its base but midway between apex and base. C, In the hypertrophic type of cardiomyopathy the wall of the left ventricle is greatly thickened; the left ventricular cavity is small, but the left atrium may be dilated because of poor diastolic relaxation of the ventricle. D, In the restrictive type the left ventricular cavity is of normal size, but again, the left atrium is dilated because of the reduced diastolic compliance of the ventricle. (From Kissane JM, editor: Anderson’s pathology, ed 9, St Louis, 1990, Mosby.)
Dilated Cardiomyopathy
Dilated cardiomyopathy (congestive cardiomyopathy) is characterized by ventricular dilation and grossly impaired systolic function, leading to dilated heart failure (Figure 30-28). The most common causes are ischemic heart disease or valvular heart disease. The basic problem is diminished myocardial contractility, which is reflected in diminished systolic performance of the heart. Abnormalities in myocardial energy metabolism are implicated.183 Dilated cardiomyopathy causes decreased ejection fraction, increased end-diastolic and residual volumes, decreased ventricular stroke volume, and biventricular failure.
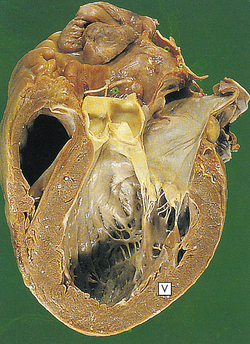
Figure 30-28 Dilated cardiomyopathy. The dilated left ventricle has a thin wall (V). (From Stevens A, Lowe J: Pathology, St Louis, 1995, Mosby.)
About two thirds of the cases of dilated cardiomyopathy are idiopathic; the remainder result from some underlying disease process. Secondary causes of dilated cardiomyopathy include ischemic heart disease, valvular heart disease, diabetes, renal failure, alcohol use, drug toxicity, nutritional deficiencies postpartum, postinfectious, and hyperthyroidism.
Idiopathic dilated cardiomyopathy has a familial origin in 20% to 30% of cases and genes coding for contractile proteins are implicated. In the majority of familial and sporadic idiopathic cases, cardiac-specific autoantibodies can be detected.184
Ischemic heart disease damages the ventricular myocardium both through necrosis and peri-infarct remodeling (see p. 1165). Valvular heart disease causes cardiac chamber volume and pressure overload that can result in long-term myocardial dysfunction (see p. 1181). Diabetes and uremia are associated with decreased myocardial contractility and dilated cardiomyopathy. Alcohol can be directly toxic to the myocardium,185 as can many drugs such as some chemotherapeutic, inotropic, and antidysrhythmic agents. Many nutritional deficiencies can cause cardiomyopathy including niacin, vitamin D, and selenium. Peripartum cardiomyopathy usually develops in the first 3 to 4 months after completion of a pregnancy, after the period of maximum physiologic stress is thought to have ended. Dilated cardiomyopathies also may be the late consequences of previous viral (especially coxsackievirus), bacterial, or parasitic infections or an autoimmune process. Inflammatory and immune responses include release of cytokines and interleukins resulting in significant myocarditis and contractile dysfunction.186 Hyperthyroidism may present with atrial fibrillation as well as dilated cardiomyopathy, which may be reversible with treatment of the thyroid disorder. (Pathophysiologic effects of the cardiomyopathies are summarized in Table 30-8.)
The most common symptoms of dilated cardiomyopathy are dyspnea and fatigue. Pulmonary congestion is expected, although fulminant pulmonary edema is uncommon. Palpitations and associated dysrhythmias may cause dizziness (syncope). Systemic and pulmonary emboli are common complications. Chest pain may be present but it is usually nonspecific and unlike anginal pain.
In the presence of dilated heart failure, blood pressure may be elevated initially; however, hypotension indicates progressive decreases in contractility. Extra heart sounds and cardiac murmurs may be present as well. Dilated cardiomyopathy may be difficult to distinguish from acute myocarditis, valvular heart disease, CAD, and hypertensive heart disease. Echocardiography and MRI can confirm the diagnosis; however, careful evaluation for potentially reversible underlying causes is essential.
General treatment for dilated cardiomyopathy consists of salt restriction and the careful use of vasodilators, diuretics, and inotropic agents. Anticoagulants are given to prevent pulmonary and systemic embolism. Corticosteroids and immunosuppressants can benefit individuals with documented inflammatory disease. Myocardial pacemakers (pacing) can improve cardiac output in many individuals. Cardiac transplantation may be lifesaving. The use of cardiac stem cells to restore myocardial contractility is an area of promising research.187
Hypertrophic Cardiomyopathy
Hypertrophic cardiomyopathy refers to two major categories of thickening of the myocardium: (1) hypertrophic obstructive cardiomyopathy (asymmetric septal hypertrophic cardiomyopathy or subaortic stenosis) and (2) hypertensive or valvular hypertrophic cardiomyopathy. These two categories are very different in their etiology, pathophysiology, and clinical presentation.
Hypertrophic obstructive cardiomyopathy is the most commonly inherited cardiac disorder and is one of an autosomal dominant inheritance.188,189 It is characterized by thickening of the septal wall (Figure 30-29), which may cause outflow obstruction to the left ventricle outflow tract.190 Additional changes include abnormalities of collagen deposition and altered contractile proteins in the myocytes. The thickening of the septum results in a hyperdynamic state, especially with exercise. Diastolic relaxation also is impaired and ventricular compliance is decreased. Obstruction of left ventricular outflow can occur when heart rate is increased and intravascular volume is decreased. Individuals complain of angina, syncope, palpitations, and symptoms of myocardial infarction and left heart failure. Examination may reveal extra heart sounds and murmurs. Echocardiography and cardiac catheterization can confirm the diagnosis. This type of hypertrophic cardiomyopathy is a significant risk for serious ventricular arrhythmias and sudden death. Management includes beta-blockers to slow the heart rate, surgical resection of the hypertrophied myocardium, septal ablation, and prophylactic placement of an ICD in high-risk individuals.190
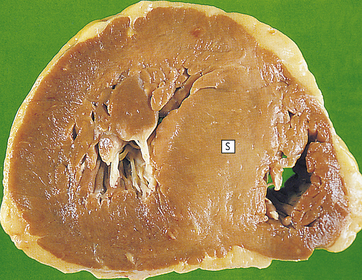
Figure 30-29 Hypertrophic cardiomyopathy. There is marked left ventricular hypertrophy. This often affects the septum (S). (From Stevens A, Lowe J: Pathology, St Louis, 1995, Mosby.)
Hypertensive, or valvular hypertrophic, cardiomyopathy occurs because of increased resistance to ventricular ejection commonly seen in hypertension or in valvular stenosis (usually aortic). In this case, hypertrophy of the myocytes is an attempt to compensate for increased workload; however, long-term dysfunction of the myocytes develops over time, with diastolic dysfunction leading eventually to systolic dysfunction of the ventricle (see Heart Failure, p. 1189).
Restrictive Cardiomyopathies
Restrictive cardiomyopathy is characterized by restrictive filling and reduced diastolic volume of either or both ventricles with normal or near-normal systolic function and wall thickness.191 It may occur idiopathically or as a cardiac manifestation of systemic diseases, such as scleroderma, amyloidosis, sarcoidosis, lymphoma, and hemochromatosis, or a number of inherited storage diseases.191 The myocardium becomes rigid and noncompliant, impeding ventricular filling and raising filling pressures during diastole. The overall clinical and hemodynamic picture mimics and may be confused with that of constrictive pericarditis.
The most common clinical manifestation of restrictive cardiomyopathy is right heart failure with systemic venous congestion. Cardiomegaly and dysrhythmias are common. A thorough evaluation for the underlying cause should be initiated (and may include myocardial biopsy) because there is no effective therapy for restrictive cardiomyopathy other than treating the underlying disease process.191 Death occurs as a result of heart failure or dysrhythmias.
Disorders of the Endocardium
Disorders of the endocardium, the innermost lining of the heart wall, all damage the heart valves, which are made up of endocardial tissue. Endocardial damage can be either congenital or acquired. The acquired forms cause inflammatory, ischemic, traumatic, degenerative, or infectious alterations of valvular structure and function.192 Structural alterations of the heart valves lead to stenosis, incompetence, or both. Although all four heart valves may be affected, those of the left heart (mitral and aortic semilunar valves) are far more commonly affected than those of the right heart (tricuspid and pulmonic semilunar valves).
In valvular stenosis the valve orifice is constricted and narrowed, impeding the forward flow of blood and increasing the workload of the cardiac chamber proximal to the diseased valve (Figure 30-30). Intraventricular or atrial pressure increases in the chamber to overcome resistance to flow through the valve. Increased pressure causes the myocardium to work harder, causing myocardial hypertrophy. In valvular regurgitation (also called insufficiency or incompetence) the valve leaflets, or cusps, fail to shut completely, permitting blood flow to continue even when the valve is supposed to be closed (see Figure 30-30). During systole or diastole some blood leaks back into the chamber proximal to the incompetent valve. Valvular regurgitation increases the volume of blood the heart must pump and increases the workload of the affected heart chamber. Increased volume leads to chamber dilation, and increased workload leads to hypertrophy.
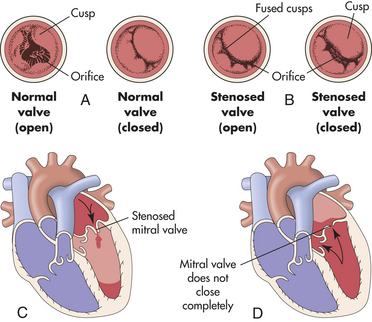
Figure 30-30 Valvular stenosis and regurgitation. A, Normal position of the valve leaflets, or cusps, when the valve is open and closed. B, Open position of a stenosed valve (left) and open position of a closed regurgitant valve (right). C, Hemodynamic effect of mitral stenosis. The stenosed valve is unable to open sufficiently during left atrial systole, inhibiting left ventricular filling. D, Hemodynamic effect of mitral regurgitation. The mitral valve does not close completely during left ventricular systole, permitting blood to reenter the left atrium.
Valvular dysfunction stimulates chamber dilation and/or myocardial hypertrophy, both of which are compensatory mechanisms intended to increase the pumping capability of the heart. Eventually, myocardial contractility is diminished, the ejection fraction is reduced, diastolic pressure increases, and the ventricles fail from overwork. Depending on the severity of the valvular dysfunction and the capacity of the heart to compensate, valvular alterations cause a range of symptoms and some degree of incapacitation (Table 30-9). The effects of valvular dysfunction are treated with medications until surgical prosthetic valve replacement becomes necessary.
Table 30-9
Clinical Manifestations of Valvular Stenosis and Regurgitation
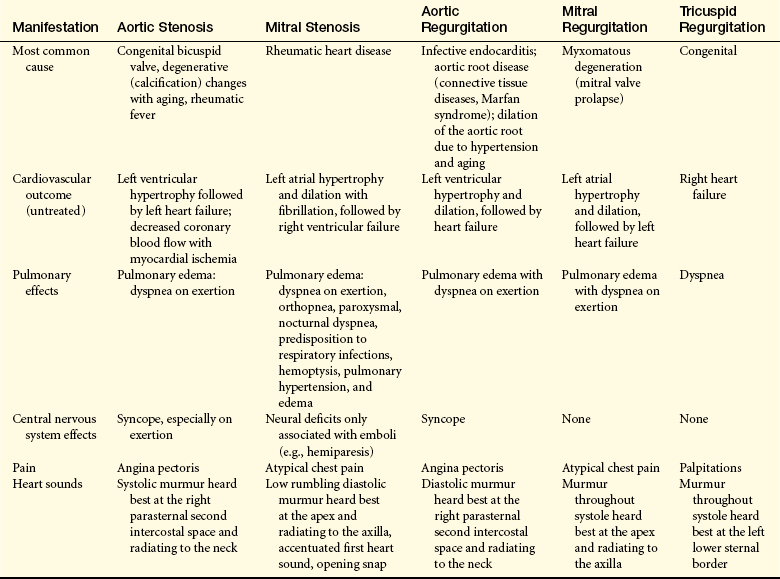
Data from Braunwald E, editor: Heart disease: a textbook of cardiovascular medicine, ed 7, Philadelphia, 2005, Saunders; Carabello BA, Paulus WJ: Valvular heart disease. In Crawford MH, DiMarco JP, editors: Cardiology, London, 2001, Mosby-Wolfe.
Aortic Stenosis: Aortic stenosis is the most common valvular abnormality affecting nearly 2% of adults older than 65 years of age. The three common causes are (1) congenital bicuspid valve, (2) degeneration with aging, and (3) inflammatory damage caused by rheumatic heart disease (less than 10% of cases). Numerous gene abnormalities have been associated with aortic stenosis.192–194 Aortic stenosis is also associated with many risk factors for coronary artery disease.195 Evidence suggests that degenerative aortic stenosis is linked to hyperlipidemia and that its prevalence might be decreased by more aggressive lipid lowering in adults.193,196 Disorders in calcium transport, apoptosis of endocardial cells, and decreased nitric oxide synthesis also have been implicated.193 Aortic valve degeneration with aging is associated with lipoprotein deposition in the tissue with chronic inflammation and leaflet calcification.197 The orifice of the aortic semilunar valve narrows, causing diminished blood flow from the left ventricle into the aorta (see Figures 30-30 and 30-31). Outflow obstruction increases pressure within the left ventricle as it tries to eject blood through the narrowed opening. Left ventricular hypertrophy develops to compensate for the increased workload. Eventually, hypertrophy increases myocardial oxygen demand that the coronary arteries may not be able to supply. If this occurs, ischemia may cause attacks of angina. Untreated aortic stenosis can lead to dysrhythmias, myocardial infarction, and heart failure.195
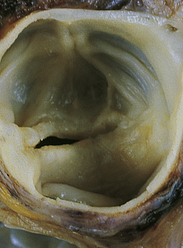
Figure 30-31 Aortic stenosis. Mild stenosis in valve leaflets of a young adult. (From Damjanov I, Linder J: Pathophysiology: a color atlas, St Louis, 2000, Mosby.)
Aortic stenosis tends to develop gradually. The classic manifestations of aortic stenosis are angina, syncope, and heart failure.192,195 These manifestations are attributable to diminished stroke volume that results in diminished tissue perfusion. Clinical manifestations include decreased stroke volume, reduced systolic blood pressure, and narrowed pulse pressure (difference between systolic and diastolic pressure). Heart rate is often slow, and pulses are faint. Resistance to flow through the stenotic valve gives rise to a crescendo-decrescendo systolic heart murmur heard best at the second intercostal space and may radiate to the neck.
Mitral Stenosis: Mitral stenosis impairs the flow of blood from the left atrium to the left ventricle. Mitral stenosis is most commonly caused by acute rheumatic fever (see p. 1185) and is two to three times more common in women than in men.192 Autoimmunity in response to group A beta-hemolytic streptococcal M protein antigens leads to inflammation and scarring of the valvular leaflets (Figure 30-32). Scarring causes the leaflets to become fibrous and fused and the chordae tendineae cordis becomes shortened.
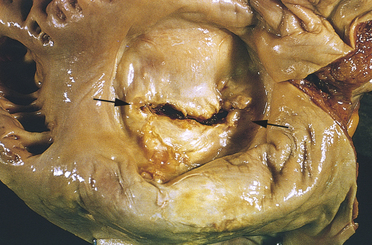
Figure 30-32 Mitral stenosis with classic “fish mouth” (arrows) orifice. (From Kumar: Pathologic basis of disease, ed 8, St Louis, 2010, Mosby.)
Clinical manifestations depend on the size of the valvular orifice. Impedance to blood flow results in incomplete emptying of the left atrium and elevated atrial pressure as the chamber tries to force blood through the stenotic valve. Continued increases in left atrial volume and pressure cause chamber dilation and hypertrophy. The risk of developing atrial dysrhythmias (especially fibrillation) and dysrhythmia-induced thrombi is high. As mitral stenosis progresses, symptoms of decreased cardiac output occur, especially during exertion. Continued elevation of left atrial pressure and volume causes pressure to rise in the pulmonary circulation. The outcomes of untreated chronic mitral stenosis are pulmonary hypertension, edema, and right ventricular failure.
Blood flow through the stenotic valve gives rise to a rumbling decrescendo diastolic murmur heard best over the cardiac apex and radiating to the left axilla. If the mitral valve is forced open during diastole, it may make a sharp noise called an opening snap. The first heart sound (S1) is often accentuated and somewhat delayed because of increased left atrial pressure. Other signs and symptoms result from pulmonary congestion and right heart failure. Atrial enlargement is demonstrated by chest roentgenograms and electrocardiography.
Aortic Regurgitation: Aortic regurgitation results from an inability of the aortic valve leaflets to close properly during diastole resulting from abnormalities of the leaflets or the aortic root and annulus, or both. It can be congenital (bicuspid valve) or acquired. Acquired aortic regurgitation can be caused by rheumatic heart disease, bacterial endocarditis, syphilis, hypertension, connective tissue disorders (e.g., Marfan syndrome and ankylosing spondylitis), appetite suppressing medications, trauma, or atherosclerosis.192 In many cases dilation of the aortic root as a cause of regurgitation is idiopathic and more than a third of cases of aortic regurgitation have no known cause. The hemodynamic repercussions depend on the size of the “leak.” During systole, blood is ejected from the left ventricle into the aorta. If the aortic semilunar valve fails to close completely, some of the ejected blood flows back into the left ventricle during diastole. Volume overload occurs in the ventricle because it receives blood from the left atrium and the aorta during diastole. Over time, the end-diastolic volume of the left ventricle increases and myocardial fibers stretch to accommodate the extra fluid. Compensatory dilation permits the left ventricle to increase its stroke volume and maintain cardiac output. Ventricular hypertrophy also occurs as an adaptation to the increased volume and increased afterload created by the high stroke volume and resultant systolic hypertension.192 Ventricular dilation and hypertrophy eventually cease to compensate for aortic incompetence, and heart failure develops.198
Clinical manifestations include widened pulse pressure resulting from increased stroke volume and diastolic backflow. Turbulence across the aortic valve during diastole produces a decrescendo murmur heard best in the second, third, or fourth intercostal spaces parasternally and may radiate to the neck. Large stroke volume and rapid runoff of blood from the aorta cause prominent carotid pulsations and bounding peripheral pulses (Corrigan pulse). Other symptoms are usually associated with heart failure that occurs when the ventricle can no longer pump adequately. Dysrhythmias and endocarditis are common complications of aortic regurgitation.
Mitral Regurgitation: Mitral regurgitation has a variety of causes. The most common are mitral valve prolapse and rheumatic heart disease. Other causes include infective endocarditis, CAD, connective tissue diseases (Marfan syndrome), and congestive cardiomyopathy.192 Mitral regurgitation permits backflow of blood from the left ventricle into the left atrium during ventricular systole, giving rise to a loud pansystolic (throughout systole) murmur heard best at the apex that radiates into the back and axilla. Because of increased volume in the left atrium entering the ventricle, the left ventricle becomes dilated and hypertrophied to maintain adequate cardiac output. The volume of backflow reentering the left atrium gradually increases, causing atrial dilation and associated atrial fibrillation. As the left atrium enlarges, the valve structures stretch and become deformed, leading to further backflow. As mitral valve regurgitation progresses, left ventricular function may become impaired to the point of failure. Eventually, increased atrial pressure also causes pulmonary hypertension and failure of the right ventricle. Mitral incompetence is usually well tolerated—often for years—until ventricular failure occurs. Most clinical manifestations are caused by heart failure.
Tricuspid Regurgitation: Tricuspid regurgitation is more common than tricuspid stenosis and usually is associated with cardiac failure and dilation of the right ventricle secondary to pulmonary hypertension. Rheumatic heart disease and infective endocarditis are less common causes. Tricuspid valve incompetence leads to volume overload in the right ventricle, increased systemic venous blood pressure, and right heart failure. Pulmonic semilunar valve dysfunction can have the same consequences as tricuspid valve dysfunction.
Mitral Valve Prolapse Syndrome: Mitral valve prolapse syndrome is a condition in which the anterior and posterior cusps of the mitral valve billow upward (prolapse) into the atrium during systole (Figure 30-33). The most common cause of mitral valve prolapse is myxomatous degeneration of the leaflets in which the cusps are redundant, thickened, and scalloped because of changes in tissue proteoglycans, increased proteinases, and infiltration by myofibroblasts.192 The chordae tendineae may be elongated, permitting the valve cusps to stretch upward. Mitral regurgitation occurs if the ballooning valve permits blood to leak into the atrium.
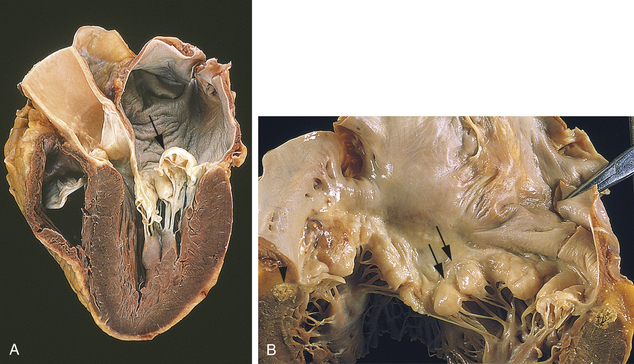
Figure 30-33 Mitral valve prolapse. A, Prolapsed mitral valve. Prolapse permits the valve leaflets to billow back (arrow) into the atrium during left ventricular systole. The billowing causes the leaflets to part slightly, permitting regurgitation into the atrium. B, Looking down into the mitral valve, the ballooning (arrows) of the leaflets is seen. (From Kumar V: Pathologic basis of disease, ed 8, St Louis, 2010, Mosby.)
Mitral valve prolapse is the most common valve disorder in the United States, with a prevalence of 1% to 3% in adults.192,199 Mitral valve prolapse tends to be most prevalent in young women. Studies suggest an autosomal dominant and X-linked inheritance pattern.199 Because mitral valve prolapse often is associated with other inherited connective tissue disorders (Marfan syndrome, Ehlers-Danlos syndrome, osteogenesis imperfecta), it is thought to result from a genetic or environmental disruption of valvular development during the fifth or sixth week of gestation. There may be a relationship between symptomatic mitral valve prolapse and hyperthyroidism. Other neuroendocrine abnormalities have been suggested, including polymorphisms of the angiotensin II type 1 (AT1) receptor and alterations in ANS function.
Many cases of mitral valve prolapse are completely asymptomatic. Cardiac auscultation on routine physical examination may disclose a regurgitant murmur or midsystolic click in an otherwise healthy individual, or echocardiography may demonstrate the condition in the absence of auscultatory findings. Symptomatic mitral valve prolapse can cause palpitations related to dysrhythmias, tachycardia, lightheadedness, syncope, fatigue (especially in the morning), lethargy, weakness, dyspnea, chest tightness, hyperventilation, anxiety, depression, panic attacks, and atypical chest pain. Many symptoms are vague and puzzling and are unrelated to the degree of prolapse. Although severe sequelae—such as chordae rupture, ventricular failure, systemic emboli, and sudden death—are possible, the disorder is actually associated with minimal mortality and morbidity. Most individuals with mitral valve prolapse have an excellent prognosis, do not develop symptoms, and do not require any restriction in activity or medical management. However, a subset of individuals have an increased risk for complications such as infective endocarditis, cardioembolic stroke, and sudden death. These high-risk individuals can be identified by clinical and echocardiographic findings. Overall, the most common complication of mitral valve prolapse is infective endocarditis.
EVALUATION AND TREATMENT The diagnosis of valvular heart disease is most often made by echocardiography. Cardiac catheterization is done prior to surgery to more directly evaluate valve structure and function as well as cardiac output.200 Valvular heart disease can often be managed temporarily with careful fluid management and medications, such as diuretics and vasodilators. However, most significant valvular abnormalities eventually require surgical intervention either by repair of the valve or replacement with either a porcine or mechanical valve.200 In the case of mechanical valve replacement, lifelong antibiotic prophylaxis prior to invasive procedures is required, as is lifelong anticoagulation to prevent clot formation on the valve with the possibility of embolization.
The majority of individuals with mitral valve prolapse have very few complications and require no treatment. Management is matched to the degree of mitral regurgitation. If regurgitation is present, antibiotic prophylaxis for infective endocarditis may be indicated before invasive procedures but physical activities are not restricted. Occasionally, beta-blockers are required to alleviate syncope, severe chest pain, or palpitations. Hypovolemia (resulting from diuretics or donating blood) is avoided because it can decrease ventricular volume, thereby increasing stress on the prolapsed mitral valve. Because surgical repair of redundant mitral valve tissue is safe and effective, some surgeons recommend operative treatment even in asymptomatic individuals who have prolapse and regurgitation to reduce the risk of stroke or sudden death.201
Acute Rheumatic Fever and Rheumatic Heart Disease
Rheumatic fever is a diffuse, inflammatory disease caused by a delayed immune response to infection by group A beta-hemolytic streptococci. In its acute form, rheumatic fever is a febrile illness characterized by inflammation of the joints, skin, nervous system, and heart.202 If untreated, rheumatic fever can cause scarring and deformity of cardiac structures, resulting in rheumatic heart disease (RHD).
The incidence of acute rheumatic fever declined in the United States during the 1960s, 1970s, and early 1980s because of medical and socioeconomic improvements, as well as changes in the virulence of group A streptococci. More recent outbreaks in the United States and abroad corresponded to the reappearance of highly virulent strains. These virulent microorganisms have different M protein serotypes than the less pathogenic strains and can be identified as nephritogenic or rheumatogenic.203 Because crowding and poor hygiene are environmental risk factors for acute rheumatic fever, the disease continues to be a major cause of death and disability for underprivileged populations.
The acute disease occurs most often in children between 5 and 15 years of age. Only 3% of those in whom pharyngeal streptococcal infection develops acquire acute rheumatic fever. Because beta-hemolytic streptococcus infection must persist for some time to cause acute rheumatic fever, appropriate antibiotic therapy given within the first 9 days of infection usually prevents rheumatic fever. Initiation of antibiotic therapy 2 weeks after the start of streptococcal infection does not prevent rheumatic fever in susceptible individuals.
Rheumatic fever tends to run in families, lending support to the concept of genetic predisposition, perhaps involving an abnormal immune response to antigens expressed by the bacterial membrane. Individuals who have experienced one attack of acute rheumatic fever are more susceptible than the general population to recurrent attacks.
PATHOPHYSIOLOGY Acute rheumatic fever can develop only as a sequel to pharyngeal infection by group A beta-hemolytic streptococci. Streptococcal skin infections do not progress to acute rheumatic fever because the strains of the microorganism that infect the skin do not have the same antigenic molecules in their cell membranes as do those that cause pharyngitis and therefore do not elicit the same kind of immune response. However, skin infections and pharyngeal infections can cause acute glomerulonephritis. Acute rheumatic fever affects the heart, joints, CNS, and skin through an abnormal humoral and cell-mediated immune response to the M proteins on the microorganisms that cross react with normal tissues203,204 (Figure 30-34). These antigens can bind to receptors on cells in the heart, muscle, brain, and synovial joints.
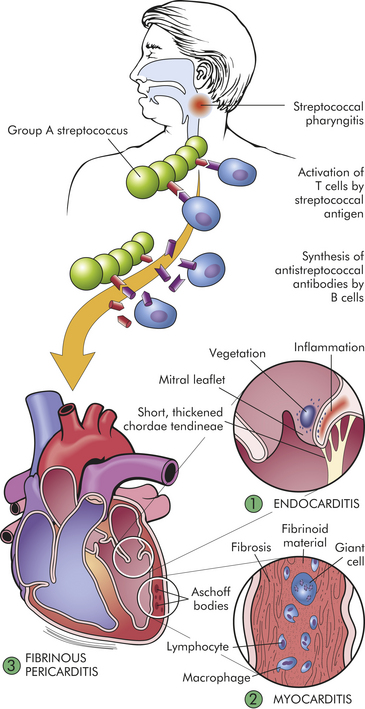
Figure 30-34 Pathogenesis and structural alterations of acute rheumatic heart disease. Beginning usually with a sore throat, rheumatic fever can develop only as a sequel to pharyngeal infection by group A beta-hemolytic streptococcus. Suspected as a hypersensitivity reaction, it is proposed that antibodies directed against the M proteins of certain strains of streptococci cross-react with tissue glycoproteins in the heart, joints, and other tissues. The exact nature of cross-reacting antigens has been difficult to define, but it appears that the streptococcal infection causes an autoimmune response against self-antigens. Inflammation is found in various sites including (1) endocardium, (2) myocardium, and (3) pericardium. The most distinctive inflammatory lesions within the heart are called Aschoff bodies. The chronic sequelae result from progressive fibrosis because of healing of the inflammatory lesions and the changes induced by valvular deformities. (From Damjanov I: Pathology for the health-related professions, ed 2, Philadelphia, 2000, Saunders.)
Diffuse, proliferative, and exudative inflammatory lesions develop in the connective tissues, especially in the heart, joints, and skin. The inflammation may subside before treatment, leaving behind damage to the heart valves and increasing the individual’s susceptibility to recurrent acute rheumatic fever after any subsequent streptococcal infections. Repeated attacks of acute rheumatic fever cause chronic proliferative changes in the previously mentioned organs as a result of scarring, granulomas, and thromboses.
Approximately 10% of cases of rheumatic fever develop rheumatic heart disease. Rheumatic heart disease begins as carditis, or inflammation of the heart. Even mild cases of rheumatic fever can cause carditis in all three layers of the heart wall (endocardium, myocardium, pericardium) (see Chapter 29, Figure 29-2). The primary lesion usually involves the endocardium, which lines the heart chambers and includes the heart valves. Endocardial inflammation causes swelling of the valve leaflets, with secondary erosion along the lines of leaflet contact. Small beadlike clumps of vegetation containing platelets and fibrin are deposited on eroded valvular tissue and on the chordae tendineae (Figure 30-35). (The chordae tendineae anchor the valve leaflets; see Chapter 29, Figure 29-3). The valves lose their elasticity, and the leaflets may adhere to each other. Scarring and shortening of the involved structures occur over time.
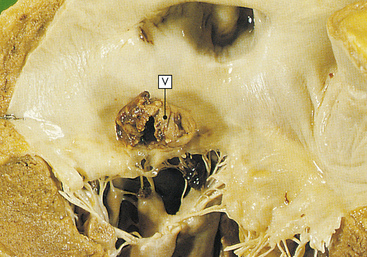
Figure 30-35 Mitral stenosis. Mitral stenosis and clumps of vegetation (V) containing platelets and fibrin. Mitral leaflets are thickened and fused and have clumps of vegetation containing platelets and fibrin. (From Stevens A, Lowe J: Pathology, St Louis, 1995, Mosby.)
If inflammation penetrates the myocardium, localized fibrin deposits develop that are surrounded by areas of necrosis. These fibrinoid necrotic deposits are called Aschoff bodies. Pericardial inflammation is usually characterized by serofibrinous effusion within the pericardial cavity. Cardiomegaly and left heart failure may occur during episodes of untreated acute or recurrent rheumatic fever. Conduction defects and atrial fibrillation are often associated with rheumatic heart disease.
CLINICAL MANIFESTATIONS Many common clinical manifestations of acute rheumatic fever—fever, lymphadenopathy, arthralgia, nausea, vomiting, epistaxis, abdominal pain, and tachycardia—are associated with other disorders as well and are therefore not diagnostic of the disease. The major specific manifestations of acute rheumatic fever are carditis, acute migratory polyarthritis, chorea, and erythema marginatum, which may occur singly or in combination after a latent period of 1 to 5 weeks after streptococcal infection of the pharynx.
Carditis: The earliest cardiac manifestation of acute rheumatic fever may be a previously undetected murmur caused by mitral or aortic semilunar valve dysfunction. Chest pain is caused by pericardial inflammation. Pericardial effusion produces an audible friction rub. Extra heart sounds, heart block (see p. 1199), atrial fibrillation, and a prolonged PR interval are often associated with chronic rheumatic heart disease. Endocardial inflammation may be manifested years later with serious valvular diseases (stenosis and regurgitation) and recurrent infective endocarditis.
Polyarthritis: The classic presenting manifestation of acute rheumatic fever is acute migratory polyarthritis (inflammation of more than one joint). Although all of the synovial joints may be involved, the large joints of the extremities are most often affected. Two or more joints are usually involved simultaneously or in succession, with each joint being symptomatic for approximately 2 to 3 days while the overall polyarthritis continues for up to 3 weeks. Exudative synovitis causes heat, redness, swelling, severe pain, and tenderness but no permanent disability. Palpable subcutaneous nodes often develop over bony prominences and along extensor tendons. They do not interfere with joint function and often go unnoticed.
Chorea: Sydenham chorea, or St. Vitus dance, is a disorder of the CNS characterized by sudden, aimless, irregular, involuntary movements. (Chorea is described in Chapter 16.) It is the most common acquired chorea in children and is more common in girls than in boys. It consists of psychologic and neurologic changes that occur 1 to 6 months after a streptococcal infection. The chorea is self-limiting, although severe cases may require the use of dopamine receptor blockers and antiepileptic medications.205 It resolves within 1 to 6 months and has no permanent neural sequelae.
Erythema Marginatum: Erythema marginatum is a distinctive truncal rash that often accompanies acute rheumatic fever. It consists of nonpruritic, pink, erythematous macules that never occur on the face or hands. The rash is transitory and may change in appearance within minutes or hours. Heat (e.g., bathing) darkens the rash. The macules may fade in the center and be mistaken for ringworm.
EVALUATION AND TREATMENT Criteria for the diagnosis of rheumatic fever have been developed and updated by both the American Heart Association and the World Health Organization (Table 30-10).206–208 No single laboratory test, sign, or symptom is pathognomonic of acute rheumatic fever but certain combinations of criteria indicate that acute disease is probably present.
Table 30-10
Jones Criteria (Revised) for Diagnosis of Rheumatic Fever
| Criteria | Description |
| Essential | Evidence of streptococcal infection (increased titer of streptococcal antibodies: antistreptolysin-O [ASO]; positive throat culture for group A streptococci; recent scarlet fever) |
| Major | Carditis, arthritis, chorea, erythema marginatum, subcutaneous nodules |
| Minor | Clinical: arthralgia, fever |
| Laboratory: increased C-reactive protein, increased white blood cell count, increased erythrocyte sedimentation rate | |
| Electrocardiographic: prolonged PR interval |
From Dajani AS, et al: Guidelines for the diagnosis of rheumatic fever: Jones criteria, updated 1992, Circulation 87: 302-307, 1993.
When correlated with findings from physical assessment, laboratory values lend significant support to the diagnosis of acute rheumatic fever. A throat culture positive for group A beta-hemolytic streptococci can be an important finding when associated with certain physical signs. Cultures may be negative when the rheumatic attack begins, however. Documented recent scarlet fever is another potentially strong diagnostic aid to acute rheumatic fever, but diagnosis of scarlet fever also depends on a positive throat culture and may be difficult to distinguish from other disorders associated with a similar rash. Most strains of group A beta-hemolytic streptococcus produces a hemolytic factor called streptolysin-O. Antibodies against this hemolytic factor increase as an individual’s immune system fights the disease. A high or rising antistreptolysin-O (ASO) antibody titer is an accurate means of diagnosing the presence of a streptococcal infection. ASO antibody titers higher than 250 Todd units in adults and 333 Todd units in children are considered elevated. Several other antibody tests are sensitive indicators of streptococcal infection. These include antideoxyribonucleotidase (anti-DNase B), antihyaluronidase, and antistreptozyme (ASTZ).
Elevated white blood cell count, erythrocyte sedimentation rate, and CRP indicate inflammation. All three are usually increased at the time cardiac or joint symptoms begin to appear. They are more useful in identifying an acute inflammatory process and suggesting prognosis than in diagnosing acute rheumatic fever. The levels of these tests decrease as the inflammatory process resolves.
Therapy for acute rheumatic fever is aimed at eradicating the streptococcal infection using a 10-day regimen of antibiotics. Nonsteroidal anti-inflammatory drugs (NSAIDs) are used as anti-inflammatory agents for rheumatic carditis and arthritis. Serious carditis may require that cardiac glycosides, corticosteroids, diuretics, and bed rest be added to the regimen. Surgical repair of damaged valves may be necessary in cases of chronic recurrent rheumatic fever or carditis. Active disease is considered resolved when (1) the murmur has disappeared or cardiac status becomes stable, (2) major manifestations are no longer present, (3) the individual is afebrile, and (4) the erythrocyte sedimentation rate is normal or stabilized. This may take 1 to 6 months.
A rheumatic recurrence will develop in 50% to 65% of children with known rheumatic fever if they have another group A streptococcal infection. Recurrence rates decline with the length of time elapsed since the last infection. Continuous prophylactic antibiotic therapy for as long as 5 years is necessary to prevent recurrence of acute rheumatic fever. Several group A streptococcus vaccines are in development.209
Infective Endocarditis
Infective endocarditis is a general term used to describe infection and inflammation of the endocardium—especially the cardiac valves. Bacteria are the most common cause of infective endocarditis, especially streptococci, staphylococci, or enterococci.210,211 Other causes include viruses, fungi, rickettsiae, and parasites. Recognizing the likely mode of exposure is helpful in identifying the microorganism involved.211 Untreated, infective endocarditis is a lethal disease but morbidity and mortality diminish significantly with the use of antibiotics and improved diagnostic techniques.
The American Heart Association has identified the cardiac disorders at highest risk for infective endocarditis.212 These disorders are (1) the presence of prosthetic heart valves, (2) a history of infective endocarditis, (3) unrepaired or incompletely repaired congenital heart disease, (4) congenital heart disease repaired with prosthetic materials, and (5) cardiac transplant recipients who develop valvular disease. Other risk factors for infective endocarditis include acquired valvular heart disease, male gender, intravenous drug abuse, long-term indwelling catheterization (e.g., for pressure monitoring, hyperalimentation, or hemodialysis), and recent cardiac surgery.212
PATHOPHYSIOLOGY The pathogenesis of infective endocarditis is a complex process that requires at least three critical elements (Figure 30-36). First, the endocardium (e.g., heart valve) must be “prepared” (usually by endothelial damage) for microorganism colonization. Second, blood-borne microorganisms must adhere to the damaged endocardial surface. Third, the adherent microorganisms must proliferate and promote the propagation of infective endocardial vegetation.
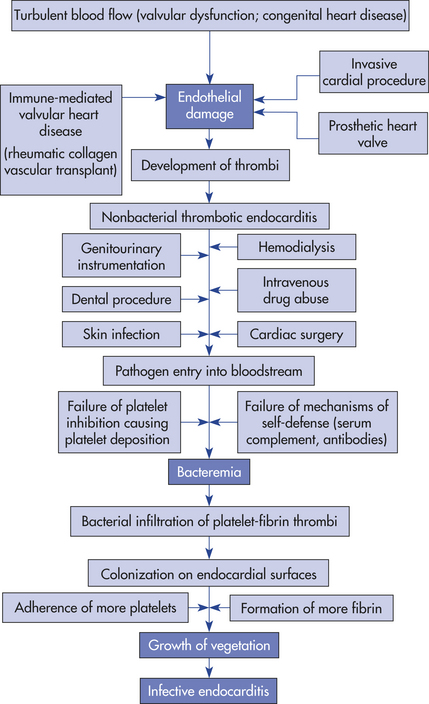
The first critical element, endocardial damage, exposes the endothelial basement membrane. The basement membrane contains a type of collagen that attracts platelets and thereby stimulates thrombus formation on the membrane. Platelet activation and thrombus formation can cause an inflammatory reaction termed nonbacterial thrombotic endocarditis.211,212
Bacteremia and adherence constitute the second critical element. Infective endocarditis cannot develop unless microorganisms gain access to the bloodstream. Microorganisms may enter the bloodstream as a result of minor procedures, such as dental cleaning or bladder catheterization, or they may spread from uncomplicated upper respiratory or skin infections. Any time pathogens gain access to the bloodstream, the potential for endocardial infection exists. Adherence of microorganisms to the endocardial surface is facilitated by the coexistence of nonbacterial thrombotic endocarditis. It should be noted, however, that highly invasive organisms can cause infective endocarditis even on the healthy intact endocardium through bacterial virulence factors called adhesins.212 Some bacteria are able to synthesize extracellular polysaccharides, such as dextran or fibronectin that promote stickiness on endocardial surfaces.
The third critical element, bacterial proliferation and vegetation formation, also is promoted by coexistent nonbacterial thrombotic endocarditis. Once the endocardial surface is colonized, formation of infected vegetation proceeds by a series of complex steps (Figure 30-37). Within 3 to 6 hours after infection, microbial replication occurs and bacterial colonies form within aggregates of fibrin and platelets. Within 24 hours, infected vegetation has increased in size, with colonies of microorganisms sandwiched between layers of fibrin and platelets. Bacteria may accelerate fibrin formation by activating the clotting cascade. As the growing bacterial colonies become progressively enmeshed in the tight fibrin network, which contains few phagocytic cells, they become less and less susceptible to the host’s mechanisms of self-defense. Although endocardial tissue is constantly bathed in antibody-containing blood and is surrounded by scavenging monocytes and polymorphonuclear leukocytes, bacterial colonies are inaccessible to host defenses because they are embedded in the protective fibrin clots. The lesions can form anywhere on the endocardium but usually occur on the endocardial surfaces of heart valves and surrounding structures (see Figure 30-37).
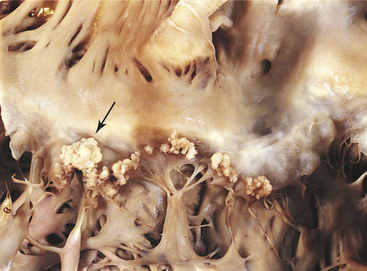
Figure 30-37 Bacterial endocarditis of mitral valve. Lesion (see arrow) in combination with old rheumatic valvulitis. (From Damjanov I, Linder J: Pathology: a color atlas, St Louis, 2000, Mosby.)
CLINICAL MANIFESTATIONS Infective endocarditis may be acute, subacute, or chronic. It causes varying degrees of valvular dysfunction and may be associated with manifestations involving several organ systems (lungs, eyes, kidneys, bones, joints, CNS), making diagnosis exceedingly difficult. Signs and symptoms of infective endocarditis are caused by infection and inflammation, systemic spread of microemboli, and immune complex deposition. The “classic” findings are fever; new or changed cardiac murmur; and petechial lesions of the skin, conjunctiva, and oral mucosa. Characteristic physical findings include Osler nodes (painful erythematous nodules on the pads of the fingers and toes) and Janeway lesions (nonpainful hemorrhagic lesions on the palms and soles).213 Other manifestations include weight loss, back pain, night sweats, and heart failure. CNS, splenic, renal, pulmonary peripheral arterial, coronary and ocular emboli may lead to a wide variety of signs and symptoms. Sudden onset of severely debilitating symptoms indicates acute disease.
EVALUATION AND TREATMENT The criteria for the diagnosis of infective endocarditis include persistent bacteremia, new heart murmurs, vascular complications, and appropriate echocardiographic findings.211,214 If infective endocarditis extends into the heart wall and invades the conduction system, electrocardiography may show a prolonged PR interval, left bundle branch block, or complete heart block (see Table 30-12). If emboli are suggested, organ scans can be performed to confirm their presence. Antimicrobial therapy is generally given for 4 to 6 weeks, beginning with intravenous and ending with oral administration.215 In some cases two different antibiotics are given simultaneously to eliminate the offending microorganism and prevent the development of drug resistance. Other drugs may be necessary to treat left heart failure secondary to valvular dysfunction, and surgical intervention to repair or replace the valve may be required.193
Antibiotic prophylaxis is indicated for high-risk individuals (prosthetic valve, congenital heart disease, cardiac transplant recipients with valvular disease) prior to dental procedures that involve manipulation of gingival tissue or the periapical region of teeth or perforation of the oral mucosa.212 Prophylaxis is no longer recommended for genitourinary or gastrointestinal procedures, although this has sparked controversy.212,216
Cardiac Complications in Acquired Immunodeficiency Syndrome
Individuals infected with the HIV and resultant acquired immunodeficiency syndrome (AIDS) are at risk for numerous cardiac complications. Pericardial effusion and left heart failure are the most common complications of HIV infection. Other conditions include cardiomyopathy, myocarditis, tuberculous pericarditis, infective and nonbacterial endocarditis, heart block, pulmonary hypertension, and non-antiretroviral drug-related cardiotoxicity. Malignancies, such as lymphoma and Kaposi sarcoma, are often seen in individuals with AIDS and can affect the heart. Furthermore, treatment with highly active antiretroviral therapy (HAART) can cause hyperlipidemia and atherosclerotic disease.217,218
MANIFESTATIONS OF HEART DISEASE
Heart failure is defined as the pathophysiologic condition in which the heart is unable to generate an adequate cardiac output such that there is inadequate perfusion of tissues or increased diastolic filling pressure of the left ventricle, or both, so that pulmonary capillary pressures are increased. It is estimated that nearly 10% of Americans older than age 65 have symptomatic heart failure and approximately 20% of asymptomatic individuals older than age 40 have some evidence of myocardial dysfunction.36 In the past three decades, hospitalizations for heart failure have increased 171%.36 Mortality remains high with an estimated 8-year survival rate of only 15%, overall.36 The most common risk factors for heart failure are increasing age, hypertension, ischemic heart disease, obesity, diabetes, and renal failure. Others include valvular heart disease, cardiomyopathies, myocarditis, congenital heart disease, and excessive alcohol use. Numerous genetic polymorphisms have been linked to an increased risk for heart failure, including genes for cardiomyopathies, myocyte contractility, and neurohumoral receptors. Genetic changes in kinases, phosphatases, and cellular calcium cycling are being explored.219 Most causes of heart failure result in dysfunction of the left ventricle (systolic and diastolic heart failure). The right ventricle also may be dysfunctional, especially in pulmonary disease (right ventricular failure). Finally, some conditions cause inadequate perfusion despite normal or elevated cardiac output (high-output failure).
Types
Left Heart Failure (Congestive Heart Failure): Left heart failure, commonly called congestive heart failure, is categorized as systolic heart failure or diastolic heart failure. Synonyms for these terms are systolic ventricular dysfunction and diastolic ventricular dysfunction. These two types of heart failure can occur together in one individual or singly.
Systolic Heart Failure: Systolic heart failure is defined as an inability of the heart to generate an adequate cardiac output to perfuse vital tissues. Cardiac output depends on the heart rate and stroke volume. Stroke volume is influenced by three major factors: contractility, preload, and afterload (see Chapter 29).
Contractility is reduced by diseases that disrupt myocyte activity. Myocardial infarction is the most common cause of decreased contractility; other causes include myocarditis and cardiomyopathies. Secondary causes of decreased contractility, such as myocardial ischemia and increased myocardial workload, contribute to inflammatory, immune, and neurohumoral changes that mediate a process called ventricular remodeling. Ventricular remodeling results in hypertrophy and dilation of the myocardium and causes progressive myocyte contractile dysfunction over time220 (Figure 30-38). When contractility is decreased, stroke volume falls, and left ventricular end-diastolic volume (LVEDV) increases. This causes dilation of the heart and an increase in preload.
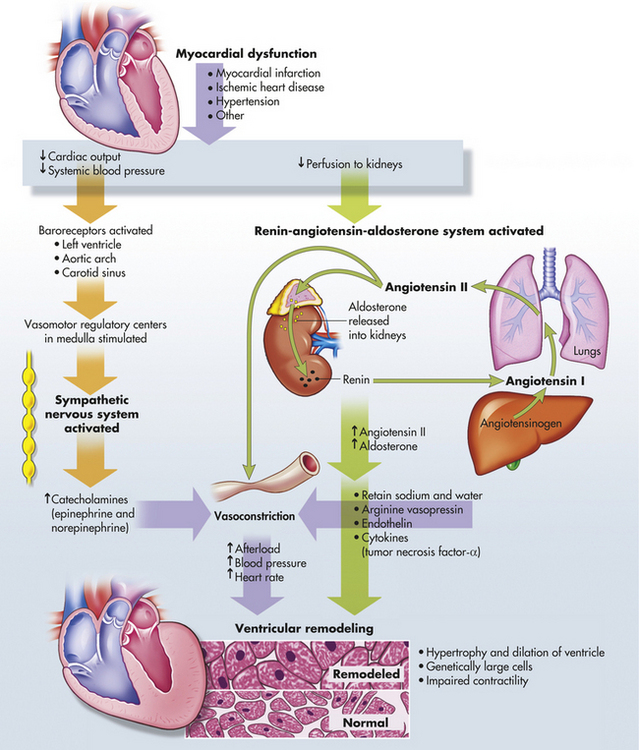
Figure 30-38 Pathophysiology of ventricular remodeling. Myocardial dysfunction activates the renin-angiotensin-aldosterone and sympathetic nervous systems, releasing neurohormones (angiotensin II, aldosterone, catecholamines, and cytokines). These neurohormones contribute to ventricular remodeling. (Redrawn from Carelock J, Clark AP: Am J Nurs 101[12]:27, 2001.)
Preload, or LVEDV, increases with decreased contractility (see earlier) or when there is an excess of plasma volume (intravenous fluid administration, renal failure, mitral valvular disease). Increases in LVEDV can actually improve cardiac output to a certain point, but as preload continues to rise, it causes a stretching of the myocardium that eventually can lead to dysfunction of the sarcomeres and decreased contractility (Figure 30-39).
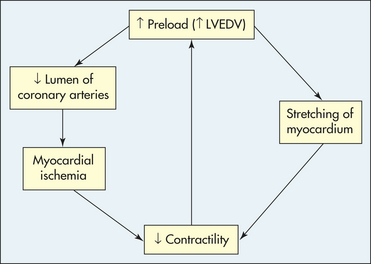
Figure 30-39 The effect of elevated preload on myocardial oxygen supply and demand. LVEDV, Left ventricular end-diastolic volume.
Increased afterload is most commonly a result of increased peripheral vascular resistance (PVR), such as that seen with hypertension (Figure 30-40). Although much less common, it also can be the result of aortic valvular disease. With increased PVR, there is resistance to ventricular emptying and more workload for the left ventricle, which responds with hypertrophy of the myocardium. Hypertrophy is mediated by angiotensin II and catecholamines and results in an increase in oxygen and energy demand by the thickened myocardium. The myocardium consumes a huge amount of metabolic energy and relies on the efficient production of ATP. This production of ATP depends on the myocytes getting enough fuel, having adequate mitochondrial function, and using an effective creatine kinase system. When demand for energy is greater than the ability of these systems to supply the necessary ATP, contractility of the myocardium is compromised.221–223 An energy-starved state develops that further contributes to changes in the myocytes themselves and ventricular remodeling that significantly impairs contractility and, therefore, ventricular function (see Figure 30-38). Remodeling also results in the deposition of collagen between the myocytes, which can disrupt the integrity of the muscle, decrease contractility, and make the ventricle more likely to dilate and fail. Weakness of the cardiac muscle due to hypertension-induced hypertrophy is called hypertensive hypertrophic cardiomyopathy.
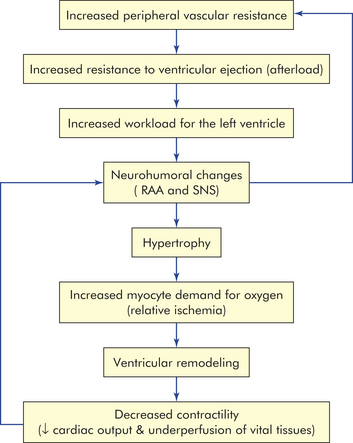
As cardiac output falls, renal perfusion diminishes with activation of the RAAS, which acts to increase PVR and plasma volume, thus increasing afterload and preload further. In addition, baroreceptors in the central circulation detect the decrease in perfusion and stimulate the SNS to cause yet more vasoconstriction and to cause the hypothalamus to produce antidiuretic hormone. This vicious cycle of decreasing contractility, increasing preload, and increasing afterload causes progressive worsening of left heart failure (Figure 30-41).
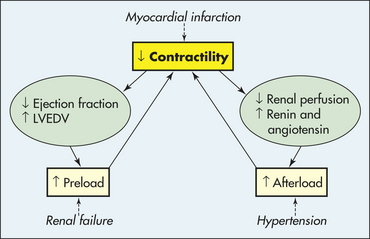
Figure 30-41 The vicious cycle of systolic heart failure. Although the initial insult may be one of primary decreased contractility (e.g., myocardial infarction), increased preload (e.g., renal failure), or increased afterload (e.g., hypertension), all three factors play a role in the progression of left heart failure. LVEDV, Left ventricular end-diastolic volume.
In addition to these hemodynamic interactions, systolic congestive heart failure is characterized by a complex constellation of neurohumoral, inflammatory, and metabolic processes:
1. Catecholamines. Sympathetic nervous system activation initially compensates for a decrease in cardiac output by increasing heart rate and peripheral vascular resistance. However, catecholamines cause numerous deleterious effects on the myocardium, including direct toxicity to myocytes, induction of myocyte apoptosis, myocardial remodeling, down-regulation of adrenergic receptors, facilitation of dysrhythmias, and potentiation of autoimmune effects on the heart muscle.223–225
a. Angiotensin II (Ang II). Activation of the RAAS causes not only increases in preload and afterload but also direct toxicity to the myocardium (see Figure 30-38). Ang II mediates remodeling of the ventricular wall, contributing to sarcomere death, loss of the normal collagen matrix, and interstitial fibrosis. This leads to decreased contractility, changes in myocardial compliance, and ventricular dilation.
b. Aldosterone. Aldosterone not only causes salt and water retention by the kidney but also contributes to myocardial fibrosis, autonomic dysfunction, and dysrhythmias. It also has been implicated in endothelial dysfunction and prothrombotic effects.226,227
3. Arginine vasopressin. Arginine vasopressin is also known as antidiuretic hormone and causes both peripheral vasoconstriction and renal fluid retention. These actions exacerbate hyponatremia and edema in heart failure.228
4. Natriuretic peptides. Atrial and brain natriuretic peptides (BNPs) are increased and may have some protective effect by decreasing preload; however, their compensatory mechanisms are inadequate in heart failure.229–231
a. Endothelial hormones. Endothelin is a potent vasoconstrictor and is associated with a poor prognosis in individuals with heart failure.231
b. TNF-α and IL-6. TNF-α is elevated in heart failure and contributes to myocardial remodeling.232 It down-regulates the synthesis of the vasodilator nitric oxide (NO), induces myocyte apoptosis, and may contribute to weight loss and weakness in individuals with heart failure (cardiac cachexia).233,234 IL-6 also is elevated in individuals with severe heart failure and cardiogenic shock and may contribute to further deleterious immune activation.232
6. Myocyte calcium transport. Calcium transport into, out of, and within myocytes is critical to normal contractile function. Changes in calcium ion channels, intracellular transport mechanisms in the sarcoplasmic reticulum, and calcium cycling have all been implicated in decreased myocardial contractility and heart failure.221,223,231
7. Insulin resistance and diabetes. Insulin resistance is a likely contributor to, as well as complication of, heart failure. Insulin resistance causes abnormal myocyte fatty acid metabolism and generation of ATP, which contributes to decreased myocardial contractility and remodeling.221 (see What’s New? Metabolic Changes in Heart Failure). Heart failure activates the sympathetic nervous system and RAAS, which contribute to insulin resistance. Diabetes contributes to heart failure through disturbed calcium metabolism, oxidative stress, changes in fatty acid and glucose metabolism, and mitochondrial dysfunction. Unfortunately, many of the new medications used to treat diabetes and insulin resistance have deleterious side effects on cardiac functioning; however, newer agents that modify fatty acid metabolism and insulin activity are being explored.221
The interaction of these metabolic, neurohumoral, and inflammatory processes results in a gradual decline in myocardial function. Pathologically, the heart muscle exhibits progressive changes in myocyte myofilaments, decreased contractility, myocyte apoptosis and necrosis, abnormal fibrin deposition in the ventricle wall, myocardial hypertrophy, and changes in the ventricular chamber geometry. These changes reduce myocardial function and cardiac output and lead to increased morbidity and mortality. These discoveries have led to the routine use of ACE inhibitors or Ang II receptor blockers plus beta-blockers in the management of heart failure, which has resulted in significant decreases in
morbidity and mortality.235–237 Individuals selected for use of these medications may soon be facilitated through the use of pharmacogenetics that can identify those genotypes most likely to respond favorably to specific treatment options.238 Aldosterone blockade with spironolactone is associated with a significant improvement in cardiac function and vasopressin blockade (e.g., tolvaptan) improve fluid balance.226,227 Unfortunately, endothelin blockers and inflammatory cytokine blockers (e.g., etanercept) have not been effective.235
The clinical manifestations of left heart failure are the result of pulmonary vascular congestion and inadequate perfusion of the systemic circulation. Individuals experience dyspnea, orthopnea, cough of frothy sputum, fatigue, decreased urine output, and edema. Physical examination often reveals pulmonary edema (cyanosis, inspiratory crackles, pleural effusions), hypotension or hypertension, an S3 gallop, and evidence of underlying CAD or hypertension. The diagnosis is made with echocardiography, revealing decreased cardiac output and cardiomegaly; some people may require invasive catheterization to document underlying coronary disease. Serum BNP levels should be measured to assist in diagnosing heart failure and to give some insight into its severity
and response to treatment239–241 (see What’s New? Brain Natriuretic Peptide and Heart Failure).
Management of systolic left heart failure is aimed at interrupting the worsening cycle of decreasing contractility, increasing preload, and increasing afterload, as well as blocking the neurohormonal mediators of myocardial toxicity. The acute onset of left heart failure is most often the result of acute myocardial ischemia and must be managed in conjunction with the underlying coronary disease. Oxygen, nitrates, and morphine administration improve myocardial oxygenation and help relieve coronary spasm while lowering preload through systemic venodilation.237 Diuretics reduce preload and are the mainstay of therapy.237,242 Intravenous inotropic drugs, such as dopamine or dobutamine, increase contractility and can help raise the blood pressure in hypotensive individuals. New calcium-sensitizing inotropic drugs (e.g., levosimendan) have shown promise for acute heart failure in selected individuals.231,243 ACE inhibitors (which reduce preload and afterload) and intravenous beta-blockers (which reduce myocardial demand) have been found to reduce mortality but must be used with caution in hypotensive individuals.220,236,237,242,244 Intravenous administration of nesiritide (recombinant BNP) also improves preload and contractility; however, results of this therapy have been mixed.231,237,244 Individuals with severe systolic failure because of myocardial ischemia may benefit from acute coronary bypass or PCI. Those with refractory hypotension may be supported with the intra-aortic balloon pump (IABP) until they can be taken safely to the operating room; the IABP is positioned in the aorta just distal to the aortic valve and is inflated during diastole to improve coronary perfusion and deflated during systole to reduce afterload.
Management of chronic left heart failure also relies on increasing contractility and reducing preload and afterload. The current standard of care for chronic heart failure includes diuretics, ACE inhibitors, and beta-blockers for all clinical stages. Salt restriction and diuretics (especially aldosterone-blockers such as spironolactone) are effective in reducing preload and improving outcomes.226,235,236 ACE inhibitors (or Ang II receptor blockers) reduce preload and afterload and have been shown to significantly reduce mortality in chronic left heart failure.235,236,245 Beta-blockers, especially some of the newer drugs such as bisoprolol, improve symptoms and increase survival.235,236,246 The ionotropic drug digoxin may be considered in some individuals, especially those with atrial fibrillation.236,247 Interestingly, statins have been associated with improved outcomes in some heart failure trials; however, their mechanism of action in heart failure is still being explored.235,248,249 Anticoagulants and antithrombotics may be indicated in selected individuals, particularly those with intracardiac thrombi or atrial fibrillation.236,248 Although many individuals with left heart failure die suddenly from dysrhythmias, prophylactic administration of antidysrhythmics has not been shown to improve survival. In individuals with sustained ventricular tachycardia, amiodarone or ICDs are indicated. Cardiac resynchronization therapy is proving to be an important modality in selected individuals.250 Coronary bypass surgery or PCI may improve perfusion to ischemic myocardium (hibernating myocardium) and improve cardiac output. Other types of surgical intervention that improve ventricular geometry may be considered.251,252 Finally, heart transplant may be the only remaining option. Experimental therapies, including gene and stem cell therapies, are being explored.253,254
Diastolic Heart Failure: Diastolic heart failure can occur singly or along with systolic heart failure. Isolated diastolic heart failure is defined as pulmonary congestion despite a normal stroke volume and cardiac output. It is the cause of approximately 50% of all cases of left heart failure and is more common in women.255 The major causes of diastolic dysfunction include hypertension-induced myocardial hypertrophy and myocardial ischemia with resultant ventricular remodeling. Hypertrophy and ischemia cause a decreased ability of the myocytes to actively pump calcium from the cytosol, resulting in impaired relaxation.255,256 Other causes include aortic valvular disease, mitral valve disease, pericardial diseases, and cardiomyopathies. Diabetes also increases the risk for diastolic dysfunction.
Two areas of pathophysiologic changes in the ventricle have been identified in diastolic dysfunction: decreased compliance of the left ventricle and abnormal diastolic relaxation (lusitropy). Decreased ventricular compliance has been linked to changes in myocardial structure such as that seen with molecular alterations in collagen, which forms the extracellular matrix for myocytes. Another recently identified structural change is because of abnormalities in an intracellular protein component of the myocyte cytoskeleton called titin.257 Abnormal lusitropy is caused by changes in calcium transport from myocytes and may be related to the activity of sarcoplasmic reticulum-calcium adenosine triphosphatase (ATPase).257,258 The resultant noncompliant and poorly lusitropic ventricle cannot accept filling with blood without significant resistance and an increase in wall tension. Thus diastolic failure occurs because a normal LVEDV is associated with an increased left ventricular end-diastolic pressure (LVEDP), which is then reflected back into the pulmonary circulation and results in pulmonary edema. The increase in pressure is made worse when ventricular filling is rapid so symptoms worsen with tachycardia (e.g., with exercise).
Individuals with diastolic dysfunction most often present with dyspnea on exertion and fatigue. If diastolic dysfunction is severe, there may be evidence of pulmonary edema (crackles on auscultation, pleural effusions). Late in diastole, atrial contraction with rapid ejection of blood into the noncompliant ventricle may give rise to an S4 gallop. Electrocardiography will often reveal evidence of left ventricular hypertrophy, and chest x-ray will show pulmonary congestion without cardiomegaly (Table 30-11).There also may be evidence of underlying coronary disease, hypertension, or valvular disease. Diagnosis is made initially by echocardiography, which demonstrates poor ventricular filling with normal ejection fractions. Management is aimed at improving ventricular relaxation and prolonging diastolic filling times to reduce diastolic pressure. Beta-blockers, ACE inhibitors, ARBs, and aldosterone blockers have been used with varying success.256,257 Inotropic drugs are not indicated in isolated diastolic heart failure because contractility and ejection fraction are not affected.
Table 30-11
Comparison of Systolic and Diastolic Heart Failure
| Characteristic | Systolic Heart Failure | Diastolic Heart Failure |
| Gender | Male>female | Female>male |
| Left ventricular ejection fraction | Decreased | Normal |
| Left ventricular chamber size | Increased | Decreased |
| Left ventricular hypertrophy on electrocardiogram | Possible | Probable |
| Chest radiography | Pulmonary congestion with cardiomegaly | Pulmonary congestion without cardiomegaly |
| Gallop | S3 | S4 |
Adapted from Jessup M, Brozena S: N Engl J Med 348(20):2007-2018, 2003.
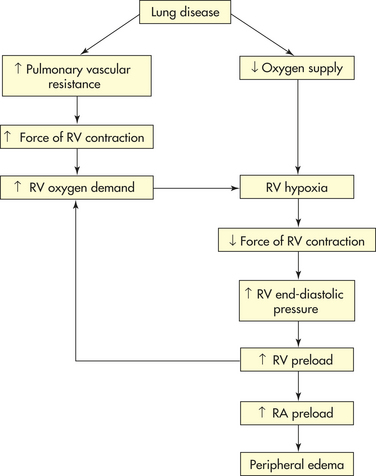
Right Heart Failure: Right heart failure is defined as the inability of the right ventricle to provide adequate blood flow into the pulmonary circulation at a normal central venous pressure.259 It most often results from left heart failure when the increase in left ventricular filling pressure that is reflected back into the pulmonary circulation is severe enough. As pressure in the pulmonary circulation rises, the resistance to right ventricular emptying increases.259,260 The right ventricle is poorly prepared to compensate for this increased workload and will dilate and fail. When this happens, pressure will rise in the systemic venous circulation, resulting in jugular venous distension, peripheral edema, and hepatosplenomegaly. Treatment relies on management of the left ventricular dysfunction as just outlined. When right heart failure occurs in the absence of left heart failure, it is caused most commonly by diffuse hypoxic pulmonary disease such as COPD, cystic fibrosis, and ARDS (Figure 30-42). The mechanisms for this type of right ventricular dysfunction (cor pulmonale) are discussed in Chapter 33. Finally, right heart failure can result from right ventricular MI, cardiomyopathies, and pulmonic valvular disease.
High-Output Failure: High-output failure is the inability of the heart to adequately supply the body with blood-borne nutrients, despite adequate blood volume and normal or elevated myocardial contractility. In high-output failure the heart increases its output but the body’s metabolic needs are still not met. Common causes of high-output failure are anemia, septicemia, hyperthyroidism, and beriberi (Figure 30-43).
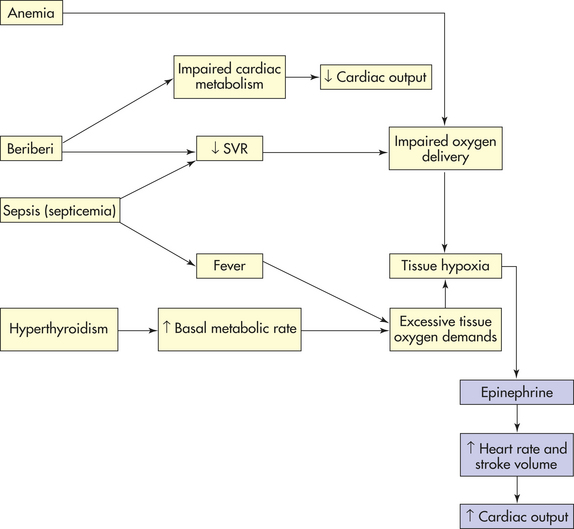
Anemia decreases the oxygen-carrying capacity of the blood (see Chapter 26). Metabolic acidosis occurs as the body’s cells switch to anaerobic metabolism (see Chapter 3). In response to metabolic acidosis, heart rate and stroke volume increase in an attempt to circulate blood faster. If anemia is severe, however, even maximum cardiac output does not supply the cells with enough oxygen for metabolism.
In septicemia, disturbed metabolism, bacterial toxins, and the inflammatory process cause systemic vasodilation and fever. Faced with a lowered systemic vascular resistance (SVR) and an elevated metabolic rate, cardiac output increases to maintain blood pressure and prevent metabolic acidosis. In overwhelming septicemia, however, the heart may not be able to raise its output enough to compensate for vasodilation. Body tissues show signs of inadequate blood supply despite a very high cardiac output.
Hyperthyroidism accelerates cellular metabolism through the actions of elevated levels of thyroxine from the thyroid gland. This may occur chronically (thyrotoxicosis) or acutely (thyroid storm). Because the body’s demand for oxygen threatens to cause metabolic acidosis, cardiac output increases. If blood levels of thyroxine are high and the metabolic response to thyroxine is quite vigorous, even an abnormally elevated cardiac output may be inadequate.
In the United States, beriberi (thiamine deficiency) usually is caused by malnutrition secondary to chronic alcoholism. Beriberi actually causes a mixed type of heart failure. Thiamine deficiency impairs cellular metabolism in all tissues, including the myocardium. In the heart, impaired cardiac metabolism leads to insufficient contractile strength. In blood vessels, thiamine deficiency leads mainly to peripheral vasodilation, which decreases SVR. Heart failure ensues as decreased SVR triggers increased cardiac output, which the impaired myocardium is unable to deliver. The strain of demands for increased output in the face of impaired metabolism may deplete cardiac reserves until low-output failure begins.
Dysrhythmias
A dysrhythmia, or arrhythmia, is a disturbance of heart rhythm. Normal heart rhythms are generated by the SA node and travel through the heart’s conduction system, causing the atrial and ventricular myocardium to contract and relax at a regular rate that is appropriate to maintain circulation at various levels of physical activity (see Chapter 29). Dysrhythmias range in severity from occasional “missed” or rapid beats to serious disturbances that impair the pumping ability of the heart, contributing to heart failure and death. Dysrhythmias can be caused by either an abnormal rate of impulse generation (Table 30-12) by the SA node or other pacemaker or the abnormal conduction of impulses (Table 30-13) through the heart’s conduction system, including the myocardial cells themselves.
SUMMARY REVIEW







1. Varicosities are areas of veins in which blood has pooled, usually in the saphenous veins. Varicosities may be caused by damaged valves as a result of trauma to the valve or by chronic venous distention involving gravity and venous constriction.
2. Chronic venous insufficiency is inadequate venous return over a long period that causes pathologic ischemic changes in the vasculature, skin, and supporting tissues.
3. Venous stasis ulcers follow the development of chronic venous insufficiency and probably develop as a result of the borderline metabolic state of the cells in the affected extremities.
4. DVT occurs in individuals who have venous stasis (immobility, age, left heart failure), spinal cord injury, vein wall damage (trauma, intravenous medications), or hypercoagulable states (pregnancy, oral contraceptives, malignancy, genetic coagulopathies).
5. DVT is often asymptomatic but may lead to fatal pulmonary emboli; prevention and careful assessment in individuals at risk are crucial.
1. An aneurysm is a localized dilation of a vessel wall to which the aorta is particularly susceptible.
2. A thrombus is a clot that remains attached to a vascular wall.
3. An embolus is a mobile aggregate of a variety of substances that occludes the vasculature. Sources of emboli include thrombi, air, amniotic fluid, bacteria, fat, and foreign matter.
4. The most common sources of arterial thrombotic emboli from the heart are mitral and aortic valvular disease and atrial fibrillation. Tissues affected include the lower extremities, the brain, and the heart.
5. Emboli to the central organs cause tissue death in lungs, kidneys, and mesentery.
6. The generation of air emboli requires a connection between the vascular compartment and a source of air. These emboli cause ischemia and necrosis when a vessel is totally blocked.
7. Amniotic fluid may be forced into the bloodstream and generate an embolus during the labor and delivery of pregnancy.
8. Aggregates of bacteria in the vasculature may be large enough to form an embolus.
9. Fat emboli are caused mainly by trauma to the long bones, either through defective fat metabolism after trauma or through the release of fat globules from bone marrow exposed by fracture.
10. The introduction of foreign matter into the vasculature can occur with trauma and also can occur in a hospital setting in which intravenous and intra-arterial lines are being used.
11. Vasospastic disorders include Raynaud disease, involving arterioles of the extremities; variant angina, involving coronary arteries; and Buerger disease, involving arteries of the hands and feet.
12. Hypertension is a sustained elevation of the systemic arterial blood pressure resulting from increases in cardiac output or total peripheral resistance or both. Hypertension can be primary (without known cause) or secondary (caused by disease or drugs). Systolic hypertension is the most significant factor in causing target organ damage.
13. The risk factors for hypertension include a positive family history; male gender; advanced age; black race; obesity; high sodium intake; low potassium, calcium, and magnesium intake; diabetes mellitus; labile blood pressure; cigarette smoking; and heavy alcohol consumption.
14. Primary hypertension is the result of extremely complicated interactions of genetics and the environment mediated by a host of neurohumoral effects. These genes interact with diet, smoking, age, and the other risk factors to cause chronic changes in vasomotor tone and blood volume.
15. The most frequently cited theories of the pathogenesis of primary hypertension include overactivity of the SNS; overactivity of the RAAS; alterations in other neurohumoral mediators of blood volume and vasomotor tone such as ANP, BNP, and adrenomedullin; inflammation; a complex interaction involving insulin resistance and endothelial function; and obesity-related hormonal changes.
16. Clinical manifestations of hypertension result from damage of organs and tissues outside the vascular system. These include heart disease, renal disease, CNS problems, and retinal changes.
17. Hypertension is managed pharmacologically, using diuretics, adrenergic blockers, calcium channel blockers, ACE inhibitors, and Ang II receptor blockers. Nonpharmacologic methods include cessation of smoking, dietary modifications, and exercise.
18. Orthostatic hypotension is a drop in blood pressure that occurs on standing. The compensatory vasoconstriction response to standing is altered by a marked vasodilation and blood pooling in the muscle vasculature.
19. Orthostatic hypotension may be acute or chronic. The acute form is caused by a delay in the normal regulatory mechanisms. The chronic forms are secondary to a specific disease or are idiopathic in nature.
20. The clinical manifestations of orthostatic hypotension include fainting and may involve cardiovascular symptoms, as well as impotence and bowel and bladder dysfunction.
21. Atherosclerosis is a form of arteriosclerosis and is the leading cause of coronary artery and cerebrovascular disease.
22. Atherosclerosis is an inflammatory disease that begins with endothelial tissue and progresses through several stages to become a fibrotic plaque.
23. Traditional risk factors include age, family history, gender, smoking, dyslipidemia, hypertension, and diabetes.
24. Novel risk factors include elevated CRP, increased serum fibrinogen, oxidative stress, infection, and periodontal disease.
25. Once a plaque has formed, it can rupture resulting in thrombosis and vasoconstriction leading to obstruction of the lumen and inadequate perfusion of distal tissues.
26. PAD is atherosclerosis of arteries that perfuse the limbs, especially the lower extremities.
27. PAD is often asymptomatic but can present with intermittent claudication (pain in leg on walking). Treatment includes risk factor reduction and antiplatelet therapy.
28. CAD is spasm or occlusion of the coronary arteries and is most often the result of atherosclerotic lesions that limit the flow of blood to the heart.
29. Many risk factors contribute to the onset and escalation of CAD, including advanced age, male gender (younger than age 60), hypertension, dyslipidemia (including elevated Lp[a]), diabetes mellitus, smoking, obesity, sedentary lifestyle, psychosocial factors, elevated CRP, and possibly infectious agents.
30. CAD results in an imbalance between coronary supply of blood and myocardial demand for oxygen and nutrients such that reversible myocardial ischemia or irreversible infarction may result.
31. Reversible myocardial ischemia presents clinically in several ways. Chronic coronary obstruction results in recurrent predictable chest pain called stable angina. Abnormal vasospasm of coronary vessels results in unpredictable chest pain called Prinzmetal angina. Myocardial ischemia that does not cause detectable symptoms is called silent ischemia.
32. Stable angina is evaluated by noninvasive techniques of assessing coronary flow with or without exercise (stress ECG, thallium, or SPECT). Management may include lifestyle changes, vasodilators, antithrombotics, PCI, or CABG surgery.
33. When there is sudden coronary obstruction because of thrombosis formation over a ruptured atherosclerotic plaque, the acute coronary syndromes result. Unstable angina causes reversible myocardial ischemia and is a harbinger of impending infarction. Myocardial infarction results when prolonged ischemia causes irreversible damage to the heart muscle. Sudden cardiac death can occur in any of the acute coronary syndromes.
34. Unstable angina occurs because of transient episodes of thrombotic vessel occlusion and vasoconstriction at the site of plaque damage, with return of perfusion before significant myocardial necrosis occurs. This must be managed aggressively with antithrombotic agents to prevent myocardial infarction.
35. When coronary blood flow is interrupted for an extended period, myocyte necrosis occurs; this is called MI. Pathologically, there are two major types of myocardial infarction: subendocardial infarction and transmural infarction. In addition to myocyte necrosis, other changes in the heart with MI include hibernating, stunning, and remodeling of the myocardium.
36. Acute coronary syndromes are assessed by measuring serum enzymes, such as creatinine kinase and troponins, as well as looking for characteristic changes in the ECG. Those individuals at highest risk for complications present with ST segment elevations on the ECG (STEMI) and require immediate intervention. Smaller subendocardial infarctions are not associated with ST segment elevations (non-STEMI) but suggest that additional myocardium is still at risk for recurrent ischemia and infarction. Management may include thrombolytic drugs, antithrombotic drugs, vasodilators, PCI, or immediate surgery.
37. Dysrhythmias, congestive heart failure, and sudden death are the most common complications of the acute coronary syndromes.
1. Inflammation of the pericardium (pericarditis) may result from innumerable sources (infection, drug therapy, tumors). Pericarditis presents with symptoms that are physically troublesome, but in and of themselves they are not life threatening.
2. Fluid may collect within the pericardial sac (pericardial effusion). Cardiac function may be severely impaired if a large volume of fluid accumulates rapidly.
3. Cardiomyopathies are a diverse group of primary myocardial disorders that are poorly understood. The cardiomyopathies are categorized as dilated (congestive), restrictive (rigid and noncompliant), and hypertrophic (asymmetric). Size of the cardiac muscle walls and chambers may increase or decrease, depending on the type of cardiomyopathy, thereby altering contractile activity.
4. Hemodynamic integrity of the cardiovascular system depends to a great extent on properly functioning cardiac valves. Congenital or acquired disorders that result in stenosis or incompetence or both can structurally alter the valves.
5. Characteristic heart sounds, cardiac murmurs, and systemic complaints assist in determination of which valve is abnormal. If severely compromised function exists, a prosthetic heart valve may be surgically implanted to replace the faulty one.
6. Mitral valve prolapse is a common finding, especially in young women. Although not grossly abnormal, the mitral valve leaflets do not position themselves properly during systole. MVP may be a completely asymptomatic condition, or it may result in severe subjective symptoms. Afflicted valves may be at greater risk for developing infective endocarditis.
7. Rheumatic fever is an inflammatory disease that results from a delayed autoimmune response to a streptococcal infection. The disorder usually resolves without sequelae if treated early.
8. Severe or untreated cases of rheumatic fever may progress to rheumatic heart disease, a potentially disabling cardiovascular disorder.
9. Infective endocarditis is a general term for infection and inflammation of the endocardium, especially the cardiac valves. A wide range of conditions predisposes one to the development of this disorder. In the mildest cases, valvular function may be slightly impaired by vegetations that collect on the valve leaflets. If infective endocarditis is left unchecked, severe valve abnormalities, chronic bacteremia, and systemic emboli may occur as vegetations break off the valve surface and travel through the bloodstream. Antibiotic therapy can limit the extent of this disease.
10. HIV is associated with cardiac abnormalities, including myocarditis, endocarditis, pericarditis, and cardiomyopathy. Left heart failure is the most common clinical manifestation.
Manifestations of Heart Disease
1. Heart failure is an inability of the heart to supply the metabolism with adequate circulatory volume and pressure.
2. Left heart failure can be categorized as systolic heart failure or diastolic heart failure.
3. Systolic heart failure is defined as an inability of the heart to generate an adequate cardiac output to perfuse vital tissue.
4. Cardiac output depends on the heart rate and stroke volume. Stroke volume is influenced by contractility, preload, and afterload. Myocardial infarction is the most common cause of decreased contractility. Myocardial ischemia results in ventricular remodeling that causes progressive myocyte contractile dysfunction over time.
5. Preload LVEDV is increased when there is decreased contractility or excess plasma volume.
6. Increased afterload is most commonly the result of increased peripheral vascular resistance. This increase in resistance decreases ventricular emptying and makes more workload for the left ventricle, resulting in hypertrophy and ventricular remodeling. The vicious cycle of decreasing contractility, increasing preload, and increasing afterload causes progressive worsening.
7. Neurohumoral mechanisms of CHF include abnormalities in the SNS, RAAS, arginine vasopressin, natriuretic peptides, inflammatory cytokines, and in myocyte metabolism.
8. The clinical manifestations of left heart failure are the result of pulmonary vascular congestion and inadequate systemic perfusion.
9. Management of left heart failure relies on increasing contractility and reducing preload and afterload.
10. Diastolic heart failure can occur singly or with systolic heart failure. The major causes of diastolic dysfunction include hypertension-induced myocardial hypertrophy and ischemia with resultant ventricular remodeling.
11. Right heart failure can result from left heart failure and/or diffuse hypoxic pulmonary disease, such as COPD, cystic fibrosis, and ARDS. These mechanisms are discussed in Chapter 33.
12. High output failure is the inability of the heart to adequately supply the body with blood-borne nutrients despite adequate volume and normal or elevated myocardial contractility. Common causes are anemia, septicemia, hyperthyroidism, and beriberi.
13. A dysrhythmia (arrhythmia) is a disturbance of heart rhythm. Dysrhythmias range in severity from occasional missed beats or rapid beats to disturbances that impair myocardial contractility and are life threatening.
14. Dysrhythmias can occur because of an abnormal rate of impulse generation or the abnormal conduction of impulses.
Acute coronary syndrome 1160
Acute orthostatic hypotension 1157
Acute pericarditis 1176
Aneurysm 1144
Angina pectoris 1166
Aortic dissection 1146
Aortic regurgitation 1183
Aortic stenosis 1181
Arcus senilis 1167
Atherosclerosis 1157
Cardiomyopathy 1178
Carditis 1186
Chronic left heart failure 1194
Chronic orthostatic hypotension 1157
Chronic venous insufficiency (CVI) 1143
Chylomicrons 1161
Complicated plaques 1159
Constrictive pericarditis (restrictive pericarditis, chronic pericarditis) 1177
Coronary artery bypass graft 1169
Coronary artery disease (CAD) 1160
Deep venous thrombosis (DVT) 1143
Diastolic heart failure 1194
Dilated cardiomyopathy (congestive cardiomyopathy) 1178
Dressler postinfection syndrome 1176
Dyslipidemia (dyslipoproteinemia) 1162
Dysrhythmia (arrhythmia) 1175
Embolism 1147
Embolus 1147
Erythema marginatum 1186
Essential (or idiopathic) hypertension 1149
False aneurysm 1146
Fatty streak 1159
Fibrous plaque 1159
Foam cell 1159
Heart failure 1189
Hibernating myocardium 1172
High-density lipoprotein (HDL) 1162
Highly sensitive C-reactive protein (hs-CRP) 1165
High-output failure 1195
Hyperhomocysteinemia 1165
Hypertension 1149
Hypertensive (valvular hypertrophic) cardiomyopathy 1180
Hypertrophic cardiomyopathy 1180
Hypertrophic obstructive cardiomyopathy 1180
Infarction 1160
Infective endocarditis 1187
Intermittent claudication 1160
Ischemia 1160
Ischemic preconditioning 1172
Isolated systolic hypertension 1149
Lanthanic (silent) disease 1155
Left heart failure (congestive heart failure) 1190
Lipoprotein 1161
Lipoprotein (a) (Lp[a]) 1164
Low-density lipoprotein (LDL) 1162
Malignant hypertension 1154
Metabolic syndrome 1164
Minimally invasive direct coronary artery bypass (MIDCAB) 1169
Mitral regurgitation 1183
Mitral stenosis 1181
Mitral valve prolapse syndrome 1183
Myocardial infarction (MI) 1169
Myocardial remodeling 1172
Myocardial stunning 1172
Nonbacterial thrombotic endocarditis 1187
Non–ST elevation MI (non-STEMI) 1169
Organic brain syndrome 1176
Orthostatic (postural) hypotension 1156
Percutaneous coronary intervention (PCI) 1169
Pericardial effusion 1177
Pericarditis 1176
Peripheral artery disease (PAD) 1160
Plaque 1157
Post-thrombotic syndrome (PTS) 1144
Primary hypertension (essential hypertension, idiopathic hypertension) 1149
Prinzmetal angina 1165
Raynaud disease 1149
Raynaud phenomenon 1148
Restrictive cardiomyopathy 1180
Rheumatic fever 1185
Rheumatic heart disease (RHD) 1185
Right heart failure 1194
Saccular aneurysm 1146
Secondary hypertension 1149
Silent ischemia 1165
Stable angina 1165
ST elevation MI (STEMI) 1169
Superior vena cava syndrome (SVCS) 1144
Sydenham chorea (St. Vitus dance) 1186
Systolic heart failure 1190
Tamponade 1177
Thromboangiitis obliterans (Buerger disease) 1148
Thromboembolism 1147
Thromboembolization 1144
Thromboembolus 1143
Thrombus 1143
Tricuspid regurgitation 1183
True aneurysm 1146
Unstable angina 1169
Valvular regurgitation 1181
Valvular stenosis 1181
Varicose vein 1142
Venous stasis ulcer 1143
Ventricular aneurysm 1176
Ventricular remodeling 1190
Very-low-density lipoprotein (VLDL) 1162
Xanthelasma 1167
REFERENCES
1. World Health Organization: Fact sheet No 317, cardiovascular diseases, February 2007. Available at www.who.int/mediacentre/factsheets/fs317/en/index.html. Accessed February 15, 2009.
2. Eberhardt, R.T., Raffetto, J.D. Chronic venous insufficiency. Circulation. 2005;111(18):2398–2409.
3. Meissner, M.H., et al. The hemodynamics and diagnosis of venous disease. J Vasc Surg. 2007;46(Suppl S):4S–24S.
4. Dietzek, A.M. Endovenous radiofrequency ablation for the treatment of varicose veins. Vascular. 2007;15(5):255–261.
5. Cushman, M. Epidemiology and risk factors for venous thrombosis. Semin Hematol. 2007;44(2):62–69.
6. Mackman, N. Triggers, targets and treatments for thrombosis. Nature. 2008;451(7181):914–918.
7. Philbrick, J.T., et al. Air travel and venous thromboembolism: a systematic review. J Gen Intern Med. 2007;22(1):107–114.
8. Baker, W.F., Jr., Bick, R.L. The clinical spectrum of antiphospholipid syndrome. Hematol Oncol Clin North Am. 2008;22(1):33–52. [v-vi].
9. Gatt, A., Makris, M. Hyperhomocysteinemia and venous thrombosis. Semin Hematol. 2007;44(2):70–76.
10. Lim, W., Eikelboom, J.W., Ginsberg, J.S. Inherited thrombophilia and pregnancy associated venous thromboembolism. BMJ. 2007;334(7607):1318–1321.
11. Bezemer, I.D., et al. Gene variants associated with deep vein thrombosis. JAMA. 2008;299(11):1306–1314.
12. Wakefield, T.W., Myers, D.D., Henke, P.K. Mechanisms of venous thrombosis and resolution. Arterioscler Thromb Vasc Biol. 2008;28(3):387–391.
13. Tapson, V.F. Acute pulmonary embolism. N Engl J Med. 2008;358(10):1037–1052.
14. Meissner, M.H., et al. Secondary chronic venous disorders. J Vasc Surg. 2007;46(Suppl S):68S–83S.
15. Frances, C. Prophylaxis for thromboembolism in hospitalized medical patients. N Engl J Med. 2007;356:1438–1444.
16. Sjalander, A., et al. Efficacy and safety of anticoagulant prophylaxis to prevent venous thromboembolism in acutely ill medical inpatients: a meta-analysis. J Intern Med. 2008;263(1):52–60.
17. Hill, J., Treasure, T. Reducing the risk of venous thromboembolism (deep vein thrombosis and pulmonary embolism) in inpatients having surgery: summary of NICE guidance. BMJ. 2007;334(7602):1053–1054.
18. Blaivas, M. Ultrasound in the detection of venous thromboembolism. Crit Care Med. 2007;35(5 Suppl):S224–S234.
19. Di Nisio, M., et al. Diagnostic accuracy of D-dimer test for exclusion of venous thromboembolism: a systematic review. J Thromb Haemost. 2007;5(2):296–304.
20. Thomas, S.M., et al. Diagnostic value of CT for deep vein thrombosis: results of a systematic review and meta-analysis. Clin Radiol. 2008;63(3):299–304.
21. Sampson, F.C., et al. The accuracy of MRI in diagnosis of suspected deep vein thrombosis: systematic review and meta-analysis. Eur Radiol. 2007;17(1):175–181.
22. McRae, S.J., Eikelboom, J.W. Latest medical treatment strategies for venous thromboembolism. Exp Opin Pharmacother. 2007;8(9):1221–1233.
23. Agnelli, G., Becattini, C. Treatment of DVT: how long is enough and how do you predict recurrence. J Thromb Thrombolysis. 2008;25(1):37–44.
24. Wilson, L.D., Detterbeck, F.C., Yahalom, J. Clinical practice. Superior vena cava syndrome with malignant causes. N Engl J Med. 2007;356(18):1862–1869.
25. Schifferdecker, B., et al. Nonmalignant superior vena cava syndrome: pathophysiology and management. Cath Cardiovasc Interven. 2005;65(3):416–423.
26. Kuivaniemi, H., Platsoucas, C.D., Tilson, M.D., 3rd. Aortic aneurysms: an immune disease with a strong genetic component. Circulation. 2008;117(2):242–252.
27. Thompson, A.R., et al. Candidate gene association studies in abdominal aortic aneurysm disease: a review and meta-analysis. Eur J Vasc Endovasc Surg. 2008;35(1):19–30.
28. Raffetto, J.D., Khalil, R.A. Matrix metalloproteinases and their inhibitors in vascular remodeling and vascular disease. Biochem Pharmacol. 2008;75(2):346–359.
29. Rentschler, M.E., Baxter, B.T. Medical therapy approach for treating abdominal aortic aneurysm. Vascular. 2007;15(6):361–365.
30. Eliason, J.L., Upchurch, G.R., Jr. Endovascular abdominal aortic aneurysm repair. Circulation. 2008;117(13):1738–1744.
31. Kamalakannan, D., Rosman, H.S., Eagle, K.A. Acute aortic dissection. Crit Care Clin. 2007;23(4):779–800. [vi].
32. Puechal, X., Fiessinger, J.N. Thromboangiitis obliterans or Buerger’s disease: challenges for the rheumatologist. Rheumatol. 2007;46(2):192–199.
33. Paraskevas, K.I., et al. Thromboangiitis obliterans (Buerger’s disease): searching for a therapeutic strategy. Angiology. 2007;58(1):75–84.
34. Gayraud, M. Raynaud’s phenomenon. Joint Bone Spine. 2007;74(1):e1–8.
35. Pope, J.E. The diagnosis and treatment of Raynaud’s phenomenon: a practical approach. Drugs. 2007;67(4):517–525.
36. Rosamond, W., et al. Heart disease and stroke statistics—2008 update: a report from the American Heart Association Statistics Committee and Stroke Statistics Subcommittee. Circulation. 2008;117(4):e25–146.
37. Chobanian, A.V. The Seventh Report of the Joint National Committee on Prevention, Detection, Evaluation, and Treatment of High Blood Pressure. JNC Report. JAMA. 2003;289(19):2560–2572.
38. Elliott, W.J. Systemic hypertension. Curr Probl Cardiol. 2007;32(4):201–259.
39. Puddu, P., et al. The genetic basis of essential hypertension. Acta Cardiol. 2007;62(3):281–293.
40. Weder, A.B. Genetics and hypertension. J Clin Hypertens. 2007;9(3):217–223.
41. Adrogue, H.J., Madias, N.E. Sodium and potassium in the pathogenesis of hypertension. N Engl J Med. 2007;356(19):1966–1978.
42. Frohlich, E.D. The salt conundrum: a hypothesis. Hypertension. 2007;50(1):161–166.
43. Rodriguez-Iturbe, B., Vaziri, N.D. Salt-sensitive hypertension—update on novel findings. Nephrol Dial Transpl. 2007;22(4):992–995.
44. Harding, S., et al. Overweight, obesity and high blood pressure in an ethnically diverse sample of adolescents in Britain: the Medical Research Council DASH study. Int J Obesity. 2008;32(1):82–90.
45. Schultz, H.D., Li, Y.L., Ding, Y. Arterial chemoreceptors and sympathetic nerve activity: implications for hypertension and heart failure. Hypertension. 2007;50(1):6–13.
46. Coffman, T.M., Crowley, S.D. Kidney in hypertension: Guyton redux. Hypertension. 2008;51(4):811–816.
47. Johnson, R.J., et al. Pathogenesis of essential hypertension: historical paradigms and modern insights. J Hypertens. 2008;26(3):381–391.
48. Hamming, I., et al. The emerging role of ACE2 in physiology and disease. J Pathol. 2007;212(1):1–11.
49. Wysocki, J., Gonzalez-Pacheco, F.R., Batlle, D. Angiotensin-converting enzyme 2: possible role in hypertension and kidney disease. Curr Hypertens Rep. 2008;10(1):70–77.
50. Hernandez Schulman, I., Zhou, M.S., Raij, L. Cross-talk between angiotensin II receptor types 1 and 2: potential role in vascular remodeling in humans [comment]. Hypertension. 2007;49(2):270–271.
51. Barri, Y.M. Hypertension and kidney disease: a deadly connection. Curr Hypertens Rep. 2008;10(1):39–45.
52. Stowasser, M., Gordon, R.D. Aldosterone excess, hypertension, and chromosome 7p22: Evidence continues to mount [comment]. Hypertension. 2007;49(4):761–762.
53. Perkins, J.M., Davis, S.N. The renin-angiotensin-aldosterone system: a pivotal role in insulin sensitivity and glycemic control. Curr Opin Endocrinol Diabetes Obes. 2008;15(2):147–152.
54. Matchar, D.B., et al. Systematic review: comparative effectiveness of angiotensin-converting enzyme inhibitors and angiotensin II receptor blockers for treating essential hypertension. Ann Intern Med. 2008;148(1):16–29.
55. Ostergren, J. Renin-angiotensin-system blockade in the prevention of diabetes. Diabetes Res Clin Pract. 2007;76(Suppl 1):S13–S21.
56. Voors, A.A. Vascular benefits of angiotensin receptor blockers. Exp Opin Invest Drugs. 2007;16(7):987–997.
57. Woodard, G.E., Rosado, J.A. Natriuretic peptides in vascular physiology and pathology. Rev Cell Mol Biol. 2008;268:59–93.
58. Gardner, D.G., et al. Molecular biology of the natriuretic peptide system: implications for physiology and hypertension. Hypertension. 2007;49(3):419–426.
59. de Sa, D.D., Chen, H.H. The role of natriuretic peptides in heart failure. Curr Cardiol Rep. 2008;10(3):182–189.
60. Xue, H., et al. Atrial natriuretic peptide gene promoter polymorphism is associated with left ventricular hypertrophy in hypertension. Clin Sci. 2008;114(2):131–137.
61. Irzmanski, R., et al. The concentration of atrial and brain natriuretic peptide in patients with idiopathic hypertension. Med Sci Monit. 2007;13(10):CR449–CR456.
62. Hildebrandt, P. Richards AM: Amino-terminal pro-B-type natriuretic peptide testing in patients with diabetes mellitus and with systemic hypertension. Am J Cardiol. 2008;101(3A):21–24.
63. Savoia, C., Schiffrin, E.L. Vascular inflammation in hypertension and diabetes: molecular mechanisms and therapeutic interventions. Clin Sci. 2007;112(7):375–384.
64. Virdis, A., et al. C-reactive protein and hypertension: Is there a causal relationship? Curr Pharm Design. 2007;13(16):1693–1698.
65. Feldstein, C., Romero, C. Role of endothelins in hypertension. Am J Ther. 2007;14(2):147–153.
66. Kohan, D.E. Endothelin-1 and hypertension: from bench to bedside. Curr Hypertens Rep. 2008;10(1):65–69.
67. Lapu-Bula, R., Ofili, E. From hypertension to heart failure: role of nitric oxide-mediated endothelial dysfunction and emerging insights from myocardial contrast echocardiography. Am J Cardiol. 2007;99(6B):714.
68. Bian, K., et al. Vascular system: role of nitric oxide in cardiovascular diseases. J Clin Hypertens. 2008;10(4):304–310.
69. Heerkens, E.H., Izzard, A.S., Heagerty, A.M. Integrins, vascular remodeling, and hypertension. Hypertension. 2007;49(1):1–4.
70. Kakar, P., Lip, G.Y. Hypertension: endothelial dysfunction, the prothrombotic state and antithrombotic therapy. Exp Rev Cardiovasc Ther. 2007;5(3):441–450.
71. Boban, M., et al. Obesity-hypertension: emerging concepts in pathophysiology and treatment. Am J Med Sci. 2007;334(1):23–30.
72. Deedwania, P., Srikanth, S. Diabetes and vascular disease. Exp Rev Cardiovasc Ther. 2008;6(1):127–138.
73. Meeuwisse-Pasterkamp, S.H., van der Klauw, M.M., Wolffenbuttel, B.H. Type 2 diabetes mellitus: prevention of macrovascular complications. Exp Rev Cardiovasc Ther. 2008;6(3):323–341.
74. Grossman, E., Messerli, F.H. Hypertension and diabetes. Adv Cardiol. 2008;45:82–106.
75. Sarafidis, P.A., McFarlane, S.I., Bakris, G.L. Antihypertensive agents, insulin sensitivity, and new-onset diabetes. Curr Diabetes Rep. 2007;7(3):191–199.
76. Rao, M.V., et al. Hypertension and CKD: Kidney Early Evaluation Program (KEEP) and National Health and Nutrition Examination Survey (NHANES):1999-2004. Am J Kidney Dis. 2008;51(4 Suppl 2):S30–S37.
77. Takeda, S., et al. The renin-angiotensin system, hypertension and cognitive dysfunction in Alzheimer’s disease: new therapeutic potential. Front Biosci. 2008;13:2253–2265.
78. van den Born, B.J., Koopmans, R.P., van Montfrans, G.A. The renin-angiotensin system in malignant hypertension revisited: plasma renin activity, microangiopathic hemolysis, and renal failure in malignant hypertension. Am J Hypertens. 2007;20(8):900–906.
79. Feldstein, C. Management of hypertensive crises. Am J Ther. 2007;14(2):135–139.
80. Pickering, T., et al. AHA scientific statement: recommendations for blood pressure measurement in humans and experimental animals part 1: blood pressure measurement in humans: a statement for professionals from the Subcommittee of Professional and Public Education of the American Heart Association Council on High Blood Pressure Research. Hypertension. 2005;45:142–161.
81. Ogedegbe, G. White-coat effect: unraveling its mechanisms. Am J Hypertens. 2008;21(2):135.
82. Rosendorff, C., et al. Treatment of hypertension in the prevention and management of ischemic heart disease: a scientific statement from the American Heart Association Council for High Blood Pressure Research and the Councils on Clinical Cardiology and Epidemiology and Prevention. Circulation. 2007;115(21):2761–2788.
83. The Consensus Committee of the American Autonomic Society and the American Academy of Neurology. Consensus statement on the definition of orthostatic hypotension, pure autonomic failure, and multiple system atrophy. Neurol. 1996;46(5):1470.
84. Medow, M.S., et al. Pathophysiology, diagnosis, and treatment of orthostatic hypotension and vasovagal syncope. Cardiol Rev. 2008;16(1):4–20.
85. Ejaz, A.A., Kazory, A., Heinig, M.E. 24-hour blood pressure monitoring in the evaluation of supine hypertension and orthostatic hypotension. J Clin Hypertens. 2007;9(12):952–955.
86. Hansson, G.K. Inflammation, atherosclerosis, and coronary artery disease. N Engl J Med. 2005;352:1685–1695.
87. Packard, R.R., Libby, P. Inflammation in atherosclerosis: from vascular biology to biomarker discovery and risk prediction. Clin Chem. 2008;54(1):24–38.
88. VanEpps, J., Vorp, D. Mechanopathobiology of atherogeneisis: a review. J Surg Res. 2007;142:202–217.
89. Jevon, M., Dorling, A., Hornick, P.I. Progenitor cells and vascular disease. Cell Prolif. 2008;41(Suppl 1):146–164.
90. Deanfield, J., Hlacox, J., Rabelink, T. Endothelial function and dysfunction: testing and clinical relevance. Circulation. 2007;106:1285–1295.
91. Schafer, A., Bauersachs, J. Endothelial dysfunction, impaired endogenous platelet inhibition and platelet activation in diabetes and atherosclerosis. Curr Vasc Pharmacol. 2008;6(1):52–60.
92. Langer, H.F., Gawaz, M. Platelet-vessel wall interactions in atherosclerotic disease. Thromb Haemost. 2008;99(3):480–486.
93. Liang, C.P., et al. The macrophage at the crossroads of insulin resistance and atherosclerosis. Circ Res. 2007;100(11):1546–1555.
94. Doran, A.C., Meller, N., McNamara, C.A. Role of smooth muscle cells in the initiation and early progression of atherosclerosis. Arterioscler Thromb Vasc Biol. 2008;28(5):812–819.
95. Dunn, S., et al. The lectin-like oxidized low-density-lipoprotein receptor: a pro-inflammatory factor in vascular disease. Biochem J. 2008;409(2):349–355.
96. Agius, L.M. Complicated atheromatous plaque as integral atherogenesis. J Clin Pathol. 2007;60(6):589–592.
97. Rodriguez, J.A., et al. Metalloproteinases and atherothrombosis: MMP-10 mediates vascular remodeling promoted by inflammatory stimuli. Front Biosci. 2008;13:2916–2921.
98. Parahuleva, M.S., et al. Factor seven activating protease (FSAP) expression in human monocytes and accumulation in unstable coronary atherosclerotic plaques. Atherosclerosis. 2008;196(1):164–171.
99. Ellahham, S. Role of antiplatelet agents in the primary and secondary prevention of atherothrombotic events in high risk-patients. South Med J. 2008;101(3):273–283.
100. Stern, S. Are we getting nearer to screening for atherosclerosis? Circulation. 2008;117(1):122–126.
101. Sanz, J., Moreno, P.R., Fuster, V. The year in atherothrombosis. J Am Coll Cardiol. 2007;49(16):1740–1749.
102. Schaar, J.A., et al. Current diagnostic modalities for vulnerable plaque detection. Curr Pharm Des. 2007;13(10):995–1001.
103. Shamoun, F., Sural, N., Abela, G. Peripheral artery disease: therapeutic advances. Exp Rev Cardiovasc Ther. 2008;6(4):539–553.
104. Damani, S.B., Topol, E.J. Future use of genomics in coronary artery disease. J Am Coll Cardiol. 2007;50(20):1933–1940.
105. McGill, H.C., Jr., McMahan, C.A., Gidding, S.S. Preventing heart disease in the 21st century: implications of the Pathobiological Determinants of Atherosclerosis in Youth (PDAY) study. Circulation. 2008;117(9):1216–1227.
106. Peng, R., et al. Course particulate matter air pollution and hospital admissions for cardiovascular and respiratory diseases among Medicare patients. JAMA. 2008;299(18):2172–2179.
107. Expert Panel on Detection. Evaluation and Treatment of High Blood Cholesterol in Adults: Executive summary of the third report of the National Cholesterol Education Program (NCEP) expert panel on detection, evaluation, and treatment of high blood cholesterol in adults (Adult Treatment Panel III). JAMA. 2001;285(19):2486–2497.
108. Garg, A., Simha, V. Update on dyslipidemia. J Clin Endocrinol Metab. 2007;92(5):1581–1589.
109. Brunzell, J.D., et al. Lipoprotein management in patients with cardiometabolic risk: consensus conference report from the American Diabetes Association and the American College of Cardiology Foundation. J Am Coll Cardiol. 2008;51(15):1512–1524.
110. Glassberg, H., Rader, D.J. Management of lipids in the prevention of cardiovascular events. Annu Rev Med. 2008;59:79–94.
111. Grundy, S.M. Promise of low-density lipoprotein-lowering therapy for primary and secondary prevention. Circulation. 2008;117(4):569–573.
112. Tannock, L.R. Advances in the management of hyperlipidemia-induced atherosclerosis. Exp Rev Cardiovasc Ther. 2008;6(3):369–383.
113. Link, J.J., Rohatgi, A., de Lemos, J.A. HDL cholesterol: physiology, pathophysiology, and management. Curr Probl Cardiol. 2007;32(5):268–314.
114. Feig, J.E., Shamir, R., Fisher, E.A. Atheroprotective effects of HDL: beyond reverse cholesterol transport. Curr Drug Targets. 2008;9(3):196–203.
115. Yu, R., et al. Proatherogenic high-density lipoprotein, vascular inflammation, and mimetic peptides. Curr Atheroscler Rep. 2008;10(2):171–176.
116. Garcia, R.A. Pharmacological therapies for raising HDL cholesterol beyond synthetic small molecules. Curr Opin Invest Drugs. 2008;9(3):274–280.
117. Miller, M., Ginsberg, H.N., Schaefer, E.J. Relative atherogenicity and predictive value of non-high-density lipoprotein cholesterol for coronary heart disease. Am J Cardiol. 2008;101(7):1003–1008.
118. Schmieder, R.E., et al. Renin-angiotensin system and cardiovascular risk. Lancet. 2007;369(9568):1208–1219.
119. Rader, D.J., Daugherty, A. Translating molecular discoveries into new therapies for atherosclerosis. Nature. 2008;451(7181):904–913.
120. Basta, G. Receptor for advanced glycation endproducts and atherosclerosis: from basic mechanisms to clinical implications. Atherosclerosis. 2008;196(1):9–21.
121. Kashyap, S.R., Defronzo, R.A. The insulin resistance syndrome: physiological considerations. Diabetes Vasc Dis Res. 2007;4(1):13–19.
122. Farmer, J.A. Diabetic dyslipidemia and atherosclerosis: evidence from clinical trials. Curr Diabetes Rep. 2008;8(1):71–77.
123. Grundy, S.M. Metabolic syndrome pandemic. Arterioscler Thromb Vasc Biol. 2008;28(4):629–636.
124. Rader, D.J. Effect of insulin resistance, dyslipidemia, and intra-abdominal adiposity on the development of cardiovascular disease and diabetes mellitus. Am J Med. 2007;120(3 Suppl 1):S12–S18.
125. Fantuzzi, G., Mazzone, T. Adipose tissue and atherosclerosis: exploring the connection. Arterioscler Thromb Vasc Biol. 2007;27(5):996–1003.
126. Bays, H.E., et al. Pathogenic potential of adipose tissue and metabolic consequences of adipocyte hypertrophy and increased visceral adiposity. Exp Rev Cardiovasc Ther. 2008;6(3):343–368.
127. Steffens, S., et al. Adiponectin and adaptive immunity: linking the bridge from obesity to atherogenesis. Circ Res. 2008;102(2):140–142.
128. Singh, S.K., et al. The connection between C-reactive protein and atherosclerosis. Ann Med. 2008;40(2):110–120.
129. Virani, S.S., Polsani, V.R., Nambi, V. Novel markers of inflammation in atherosclerosis. Curr Atheroscler Rep. 2008;10(2):164–170.
130. Rossi, G.P., Seccia, T.M., Pessina, A.C. Homocysteine, left ventricular dysfunction and coronary artery disease: is there a link? Clin Chem Lab Med. 2007;45(12):1645–1651.
131. Mallika, V., Goswami, B., Rajappa, M. Atherosclerosis pathophysiology and the role of novel risk factors: a clinicobiochemical perspective. Angiology. 2007;58(5):513–522.
132. Lago, F., et al. The emerging role of adipokines as mediators of inflammation and immune responses. Cytokine Growth Factor Rev. 2007;18(3-4):313–325.
133. Selcuk, M.T., et al. Impact of plasma adiponectin levels to the presence and severity of coronary artery disease in patients with metabolic syndrome. Coron Artery Dis. 2008;19(2):79–84.
134. Komura, N., et al. Clinical significance of high-molecular weight form of adiponectin in male patients with coronary artery disease with metabolic syndrome. Circ J. 2008;72(1):23–28.
135. Wang, S.S., et al. Circulating Chlamydia pneumoniae DNA and advanced coronary artery disease. Int J Cardiol. 2007;118(2):215–219.
136. Vahdat, K., et al. Concurrent increased high sensitivity C-reactive protein and chronic infections are associated with coronary artery disease: a population-based study. Indian J Med Sci. 2007;61(3):135–143.
137. Madjid, M., et al. Systemic infections cause exaggerated local inflammation in atherosclerotic coronary arteries: clues to the triggering effect of acute infections on acute coronary syndromes. Texas Heart Inst J. 2007;34(1):11–18.
138. Foreman, R.D. Neurological mechanisms of chest pain and cardiac disease. Cleve Clin J Med. 2007;74(Suppl 1):S30–S33.
139. Li, J.J., et al. Inflammation in variant angina: is there any evidence? Med Hypotheses. 2007;68(3):635–640.
140. Yuksel, U.C., et al. Polymorphic ventricular tachycardia induced by coronary vasospasm: a malignant case of variant angina. Int J Cardiol. 2007;121(2):210–212.
141. D’Antono, B., et al. Silent ischemia: silent after all? Can J Cardiol. 2008;24(4):285–291.
142. Rozanski, A. Mental stress and the induction of silent myocardial ischemia in patients with coronary artery disease. N Engl J Med. 1988;318(16):1005–1012.
143. Kop, W.J., et al. Effects of acute mental stress and exercise on inflammatory markers in patients with coronary artery disease and healthy controls. Am J Cardiol. 2008;101(6):767–773.
144. Dimsdale, J.E. Psychological stress and cardiovascular disease. J Am Coll Cardiol. 2008;51(13):1237–1246.
145. Soufer, R., Burg, M.M. The heart-brain interaction during emotionally provoked myocardial ischemia: implications of cortical hyperactivation in CAD and gender interactions. Cleve Clin J Med. 2007;74(Suppl 1):S59–S62.
146. Beller, G.A. Noninvasive screening for coronary atherosclerosis and silent ischemia in asymptomatic type 2 diabetic patients: is it appropriate and cost-effective? J Am Coll Cardiol. 2007;49(19):1918–1923.
147. Madsen, J.K., et al. DANAMI study group. Revascularization compared to medical treatment in patients with silent vs. symptomatic residual ischemia after thrombolyzed myocardial infarction—the DANAMI study. Cardiology. 2007;108(4):243–251.
148. Cademartiri, F., et al. Non-invasive visualization of coronary atherosclerosis: state-of-art. J Cardiovasc Med. 2007;8(3):129–137.
149. Ben-Dor, I., Battler, A. Treatment of stable angina. Heart. 2007;93(7):868–874.
150. Fraker, T., Fihn, S. 2007 chronic angina. Focused update of the ACC/AHA 2002 guidelines for the management of patients with chronic stable angina: a report of the American College of Cardiology/American Heart Association Task Force on Practice Guidelines Writing Group to develop the focused update of the 2002 guidelines for the management of patients with chronic stable angina. J Am Coll Cardiol. 2007;50:2264–2274.
151. Akdim, F., et al. Pleiotropic effects of statins: stabilization of the vulnerable atherosclerotic plaque? Curr Pharm Des. 2007;13(10):1003–1012.
152. Clappers, N., et al. Antiplatelet treatment for coronary heart disease. Heart. 2007;93(2):258–265.
153. Kiernan, T.J., Prasad, A., Gersh, B.J. Current indications for percutaneous coronary intervention for chronic stable angina: implications of the COURAGE Trial. Rev Cardiovasc Med. 2007;8(4):234–239.
154. Bravata, D.M., et al. Systematic review: the comparative effectiveness of percutaneous coronary interventions and coronary artery bypass graft surgery. Ann Intern Med. 2007;147(10):703–716.
155. Klein, L.W. Clinical implications and mechanisms of plaque rupture in the acute coronary syndromes. Am Heart Hosp J. 2005;3(4):249–255.
156. Bhatheja, R., Mukherjee, D. Acute coronary syndromes: unstable angina/non-ST elevation myocardial infarction. Crit Care Clin. 2007;23(4):709–735.
157. Anderson, J., et al. ACC/AHA 2007 guidelines for the management of patients with unstable angina/non–ST-elevation myocardial infarction: a report of the American College of Cardiology/American Heart Association Task Force on Practice Guidelines. J Am Coll Cardiol. 2007;50:e1–e157.
158. Mukherjee, D., Eagle, K.A. The use of antithrombotics for acute coronary syndromes in the emergency department: considerations and impact. Prog Cardiovasc Dis. 2007;50(3):167–180.
159. Bavry, A.A., et al. Long-term benefit of statin therapy initiated during hospitalization for an acute coronary syndrome: a systematic review of randomized trials. Am J Cardiovasc Drugs. 2007;7(2):135–141.
160. Thygesen, K., Alpert, J., White, H. ESC/ACCF/AHA/WHF expert consensus document: universal definition of myocardial infarction. J Am Coll Cardiol. 2007;50:2173–2195.
161. Burke, A.P., Virmani, R. Pathophysiology of acute myocardial infarction. Med Clin North Am. 2007;91(4):553–572.
162. Bolli, R. Preconditioning: a paradigm shift in the biology of myocardial ischemia. Am J Physiol Heart Circ Physiol. 2007;292(1):H19–H27.
163. Camici, P., Prasad, S., Rimoldi, O. Stunning, hibernation and assessment of myocardial viability. Circulation. 2008;117:103–114.
164. Sun, Y. Oxidative stress and cardiac repair/remodeling following infarction. Am J Med Sci. 2007;334(3):197–205.
165. Palardy, M., Ducharme, A., O’Meara, E. Inhibiting the renin-angiotensin system with ACE Inhibitors or ARBs after MI. Curr Heart Fail Rep. 2007;4(4):190–197.
166. Antman, E., et al. Focused Update of the ACC/AHA 2004 Guidelines for the Management of Patients With ST-Elevation Myocardial Infarction. J Am Coll Cardiol. 2008;51:210–247.
167. Bogaty, P., et al. Primary PCI in ST-segment elevation myocardial infarction. N Engl J Med. 2008;358(16):1751–1752.
168. King, S., et al. 2007 focused update of the ACC/AHA/SCAI 2005 guideline update for percutaneous coronary intervention. J Am Coll Cardiol. 2008;51:172–209.
169. Vun Liew, T., Ray, K.K. Aggressive statin therapy for acute coronary syndromes. Curr Cardiol Rep. 2007;9(4):298–302.
170. Tingle, L.E., Molina, D., Calvert, C.W. Acute pericarditis. Am Fam Physician. 2007;76(10):1509–1514.
171. Hoit, B.D. Pericardial disease and pericardial tamponade. Crit Care Med. 2007;35(8 Suppl):S355–S364.
172. Marnejon, T., et al. The constricted heart. Postgrad Med. 2008;120(1):8–10.
173. Ariyarajah, V., Spodick, D.H. Acute pericarditis: diagnostic cues and common electrocardiographic manifestations. Cardiol Rev. 2007;15(1):24–30.
174. Imazio, M., Trinchero, R., Shabetai, R. Pathogenesis, management, and prevention of recurrent pericarditis. J Cardiovasc Med. 2007;8(6):404–410.
175. Saltzman, H., Weitz, H.H. Should all patients with acute pericarditis be treated with colchicine? Cleve Clin J Med. 2007;74(5):385–386.
176. Mullens, W., De Keyser, J., Herregods, M.C. Collapse of three cardiac chambers due to a pericardial effusion. Int J Cardiol. 2008;123(3):e62–e63.
177. Roy, C.L., et al. Does this patient with a pericardial effusion have cardiac tamponade? JAMA. 2007;297(16):1810–1818.
178. Wann, S., Passen, E. Echocardiography in pericardial disease. J Am Soc Echocardiogr. 2008;21(1):7–13.
179. Thai, V., Oneschuk, D. Malignant pericardial effusion treated with intrapericardial bleomycin. J Palliat Med. 2007;10(2):281–282.
180. Swanson, N., et al. Primary percutaneous balloon pericardiotomy for malignant pericardial effusion. Catheter Cardiovasc Interv. 2008;71(4):504–507.
181. Syed, F.F., Mayosi, B.M. A modern approach to tuberculous pericarditis. Prog Cardiovasc Dis. 2007;50(3):218–236.
182. Ohtsuki, I., Morimoto, S. Troponin: regulatory function and disorders. Biochem Biophys Res Commun. 2008;369(1):62–73.
183. Taha, M., Lopaschuk, G.D. Alterations in energy metabolism in cardiomyopathies. Ann Med. 2007;39(8):594–607.
184. Caforio, A.L., et al. Clinical implications of anti-heart autoantibodies in myocarditis and dilated cardiomyopathy. Autoimmunity. 2008;41(1):35–45.
185. Djousse, L., Gaziano, J.M. Alcohol consumption and heart failure: a systematic review. Curr Atheroscler Rep. 2008;10(2):117–120.
186. Esfandiarei, M., McManus, B.M. Molecular biology and pathogenesis of viral myocarditis. Annu Rev Pathol. 2008;3:127–155.
187. Strauer, B.E., Brehm, M., Schannwell, C.M. The therapeutic potential of stem cells in heart disease. Cell Prolif. 2008;41(Suppl 1):126–145.
188. Keren, A., Syrris, P., McKenna, W.J. Hypertrophic cardiomyopathy: the genetic determinants of clinical disease expression. Nat Clin Pract Cardiovasc Med. 2008;5(3):158–168.
189. van Spaendonck-Zwarts, K.Y., van den Berg, M.P., van Tintelen, J.P. DNA analysis in inherited cardiomyopathies: current status and clinical relevance. Pacing Clin Electrophysiol. 2008;31(Suppl 1):S46–S49.
190. Fifer, M.A., Vlahakes, G.J. Management of symptoms in hypertrophic cardiomyopathy. Circulation. 2008;117(3):429–439.
191. Stollberger, C., Finsterer, J. Extracardiac medical and neuromuscular implications in restrictive cardiomyopathy. Clin Cardiol. 2007;30(8):375–380.
192. Schoen, F.J. Cardiac valves and valvular pathology: update on function, disease, repair, and replacement. Cardiovasc Pathol. 2005;14(4):189–194.
193. Rahimtoola, S.H. The year in valvular heart disease. J Am Coll Cardiol. 2008;51(7):760–770.
194. Bosse, Y., Mathieu, P., Pibarot, P. Genomics: the next step to elucidate the etiology of calcific aortic valve stenosis. J Am Coll Cardiol. 2008;51(14):1327–1336.
195. Aronow, W.S. Aortic stenosis. Compr Ther. 2007;33(4):174–183.
196. Rosenhek, R., Baumgartner, H. Aortic sclerosis, aortic stenosis and lipid-lowering therapy. Exp Rev Cardiovasc Ther. 2008;6(3):385–390.
197. Ramaraj, R., Sorrell, V.L. Degenerative aortic stenosis. Br J Med. 2008;336(7643):550–555.
198. Goldbarg, S.H., Halperin, J.L. Aortic regurgitation: disease progression and management. Nat Clin Pract Cardiovasc Med. 2008;5(5):269–279.
199. Grau, J.B., et al. The genetics of mitral valve prolapse. Clin Genet. 2007;72(4):288–295.
200. Bonow, R.O., et al. ACC/AHA 2006 guidelines for the management of patients with valvular heart disease: a report of the American College of Cardiology/American Heart Association Task Force on Practice Guidelines (Writing Committee to Revise the 1998 Guidelines for the Management of Patients With Valvular Heart Disease). J Am Coll Cardiol. 2006;48:e1–e148.
201. Fedak, P.W., McCarthy, P.M., Bonow, R.O. Evolving concepts and technologies in mitral valve repair. Circulation. 2008;117(7):963–974.
202. Carapetis, J.R., McDonald, M., Wilson, N.J. Acute rheumatic fever. Lancet. 2005;366(9480):155–168.
203. Cunningham, M.W. Pathogenesis of group A streptococcal infections and their sequelae. Adv Exp Med Biol. 2008;609:29–42.
204. Guilherme, L., et al. T cell response in rheumatic fever: crossreactivity between streptococcal M protein peptides and heart tissue proteins. Curr Protein Pept Sci. 2007;8(1):39–44.
205. Weiner, S.G., Normandin, P.A. Sydenham chorea: a case report and review of the literature. Ped Emer Care. 2007;23(1):20–24.
206. Djani, A.S., et al. Guidelines for the diagnosis of rheumatic fever. Jones criteria, updated 1993. Circulation. 1993;87:302.
207. Ferrieri, P. Jones Criteria Working Group: Proceedings of the Jones criteria workshop. Circulation. 2002;106(19):2521–2523.
208. World Health Organization. Rheumatic fever and rheumatic heart disease: report of a WHO expert consultation. Geneva: World Health Organization; 2004.
209. Dale, J.B. Current status of group A streptococcal vaccine development. Adv Exp Med Biol. 2008;609:53–63.
210. Prendergast, M. The changing face of infective endocarditis. Heart. 2006;92:879–885.
211. Dandache, P., Aronow, W.S., Sakoulas, G. Clinical update on the diagnosis and treatment of bacterial endocarditis. Compr Ther. 2007;33(4):192–207.
212. Wilson, W., et al. Prevention of infective endocarditis: recommendations by the American Heart Association: a guideline from the American Heart Association Rheumatic Fever, Endocarditis and Kawasaki Disease Committee, Council on Cardiovascular Disease in the Young, and the Council on Clinical Cardiology, Council on Cardiovascular Surgery and Anesthesia, and the Quality of Care and Outcomes Research Interdisciplinary Working Group. Circulation. 2007;116:1736–1754.
213. Marrie, T.J. Osler’s nodes and Janeway lesions. Am J Med. 2008;121(2):105–106.
214. Durack, D.T., Lukes, A.S., Bright, D.K. New criteria for diagnosis of infective endocarditis: utilization of specific echocardiographic findings. Duke Endocarditis Service. Am J Med. 1994;96(3):200–209.
215. Habib, G. Management of infective endocarditis. Heart. 2006;92(1):124–130.
216. Kim, A., Keys, T. Infective endocarditis prophylaxis before dental procedures: new guidelines spark controversy. Cleve Clin J Med. 2008;75(2):89–92.
217. Pao, V., Lee, G.A., Grunfeld, C. HIV therapy, metabolic syndrome, and cardiovascular risk. Curr Atheroscler Rep. 2008;10(1):61–70.
218. Stein, J.H. Cardiovascular risks of antiretroviral therapy. N Engl J Med. 2007;356(17):1773–1775.
219. Nass, R.D., et al. Mechanisms of disease: ion channel remodeling in the failing ventricle. Nat Clin Pract Cardiovasc Med. 2008;5(4):196–207.
220. Chatterjee, K., Rame, J.E. Systolic heart failure: chronic and acute syndromes. Crit Care Med. 2008;36(1 Suppl):S44–S51.
221. Ashrafian, H., Frenneaux, M.P., Opie, L.H. Metabolic mechanisms in heart failure. Circulation. 2007;116(4):434–448.
222. Neubauer, S. The failing heart—an engine out of fuel. N Engl J Med. 2007;356(11):1140–1151.
223. Mudd, J.O., Kass, D.A. Tackling heart failure in the twenty-first century. Nature. 2008;451(7181):919–928.
224. Feldman, D.S., et al. Mechanisms of disease: detrimental adrenergic signaling in acute decompensated heart failure. Nat Clin Pract Cardiovas Med. 2008;5(4):208–218.
225. Osadchii, O.E. Cardiac hypertrophy induced by sustained beta-adrenoreceptor activation: pathophysiological aspects. Heart Fail Rev. 2007;12(1):66–86.
226. Pitt, B. Aldosterone blockade in patients with chronic heart failure. Cardiol Clin. 2008;26(1):15–21. [v].
227. Selektor, Y., Weber, K.T. The salt-avid state of congestive heart failure revisited. Am J Med Sci. 2008;335(3):209–218.
228. Rossi, J., Orlandi, C., Gheorghiade, M. Vasopressin antagonists in the management of heart failure. Exp Rev Cardiovasc Ther. 2007;5(2):323–330.
229. Lee, C.Y., Burnett, J.C., Jr. Natriuretic peptides and therapeutic applications. Heart Fail Rev. 2007;12(2):131–142.
230. Onwuanyi, A., Taylor, M. Acute decompensated heart failure: pathophysiology and treatment. Am J Cardiol. 2007;99(6B):2530.
231. Tavares, M., et al. New pharmacologic therapies for acute heart failure. Crit Care Med. 2008;36(1 Suppl):S112–S120.
232. Chen, D., et al. Cytokines and acute heart failure. Crit Care Med. 2008;36(1 Suppl):S9–S16.
233. Conraads, V.M., Hoymans, V.Y., Vrints, C.J. Heart failure and cachexia: insights offered from molecular biology. Front Biosci. 2008;13:325–335.
234. von Haehling, S., Doehner, W., Anker, S.D. Nutrition, metabolism, and the complex pathophysiology of cachexia in chronic heart failure. Cardiovasc Res. 2007;73(2):298–309.
235. Anand, I.S., Florea, V.G. Traditional and novel approaches to management of heart failure: successes and failures. Cardiol Clin. 2008;26(1):59–72. [vi].
236. Hunt, S.A., et al. ACC/AHA 2005 guideline update for the diagnosis and management of chronic heart failure in the adult: a report of the American College of Cardiology/American Heart Association Task Force on Practice Guidelines (Writing Committee to Update the 2001 Guidelines for the Evaluation and Management of Heart Failure) developed in collaboration with the American College of Chest Physicians and the International Society for Heart and Lung Transplantation, endorsed by the Heart Rhythm Society. Circulation. 2005;112:E154–E235.
237. Shin, D.D., et al. Review of current and investigational pharmacologic agents for acute heart failure syndromes. Am J Cardiol. 2007;99(2A):4A–23A.
238. McNamara, D.M. Pharmacogenomics for neurohormonal intervention in heart failure. Cardiol Clin. 2008;26(1):127–135. [viii].
239. Januzzi, J.L., Jr., Chen-Tournoux, A.A., Moe, G. Amino-terminal pro-B-type natriuretic peptide testing for the diagnosis or exclusion of heart failure in patients with acute symptoms. Am J Cardiol. 2008;101(3A):29–38.
240. Hildebrandt, P., Collinson, P.O. Amino-terminal pro-B-type natriuretic peptide testing to assist the diagnostic evaluation of heart failure in symptomatic primary care patients. Am J Cardiol. 2008;101(3A):25–28.
241. Bettencourt, P., Januzzi, J.L., Jr. Amino-terminal pro-B-type natriuretic peptide testing for inpatient monitoring and treatment guidance of acute destabilized heart failure. Am J Cardiol. 2008;101(3A):67–71.
242. Onwuanyi, A., Taylor, M. Acute decompensated heart failure: pathophysiology and treatment. Am J Cardiol. 2007;99(6B):2530.
243. Lehtonen, L., Poder, P. The utility of levosimendan in the treatment of heart failure. Ann Med. 2007;39(1):2–17.
244. Elkayam, U., et al. Vasodilators in the management of acute heart failure. Crit Care Med. 2008;36(1 Suppl):S95–S105.
245. Kazi, D., Deswal, A. Role and optimal dosing of angiotensin-converting enzyme inhibitors in heart failure. Cardiol Clin. 2008;26(1):1–14.
246. Rosenberg, J., Gustafsson, F. Bisoprolol for congestive heart failure. Exp Opin Pharmacother. 2008;9(2):293–300.
247. Ahmed, A., et al. Effects of digoxin at low serum concentrations on mortality and hospitalization in heart failure: a propensity-matched study of the DIG trial. Int J Cardiol. 2008;123(2):138–146.
248. Farmakis, D., et al. Anticoagulants, antiplatelets, and statins in heart failure. Cardiol Clin. 2008;26(1):49–58. [vi].
249. Lipinski, M.J., et al. Drug insight: statins for nonischemic heart failure—evidence and potential mechanisms. Nat Clin Pract Cardiovasc Med. 2007;4(4):196–205.
250. Anderson, L.J., et al. Patient selection and echocardiographic assessment of dyssynchrony in cardiac resynchronization therapy. Circulation. 2008;117(15):2009–2023.
251. Ailawadi, G., Kron, I.L. New strategies for surgical management of ischemic cardiomyopathy. Esp Rev Cardiovasc Ther. 2008;6(4):521–530.
252. Kale, P., Fang, J.C. Devices in acute heart failure. Crit Care Med. 2008;36(1 Suppl):S121–S128.
253. Leri, A., et al. Myocardial regeneration and stem cell repair. Curr Probl Cardiol. 2008;33(3):91–153.
254. Mishra, P.K. Bone marrow-derived mesenchymal stem cells for treatment of heart failure: is it all paracrine actions and immunomodulation? J Cardiovasc Med. 2008;9(2):122–128.
255. Sanderson, J.E. Heart failure with a normal ejection fraction. Heart. 2007;93(2):155–158.
256. Kumar, R., Gandhi, S.K., Little, W.C. Acute heart failure with preserved systolic function. Crit Care Med. 2008;36(1 Suppl):S52–S56.
257. Rapp, J.A., Gheorghiade, M. Role of neurohormonal modulators in heart failure with relatively preserved systolic function. Cardiol Clin. 2008;26(1):23–40. [vi].
258. Periasamy, M., Janssen, P.M. Molecular basis of diastolic dysfunction. Heart Fail Clin. 2008;4(1):13–21.
259. Greyson, C.R. Pathophysiology of right ventricular failure. Crit Care Med. 2008;36(1 Suppl):S57–S65.
260. Haddad, F., et al. Right ventricular function in cardiovascular disease, part II: pathophysiology, clinical importance, and management of right ventricular failure. Circulation. 2008;117(13):1717–1731.